
Recapitulation of Werner syndrome sensitivity to camptothecin by limited knockdown of the WRN...
14
RESEARCH ARTICLE Recapitulation of Werner syndrome sensitivity to camptothecin by limited knockdown of the WRN helicase/exonuclease Joseph L. E. Bird • Katrin C. B. Jennert-Burston • Marcus A. Bachler • Penelope A. Mason • Jill E. Lowe • Seok-Jin Heo • Judith Campisi • Richard G. A. Faragher • Lynne S. Cox Received: 31 December 2010 / Accepted: 10 May 2011 / Published online: 24 July 2011 Ó Springer Science+Business Media B.V. 2011 Abstract WRN is a RecQ helicase with an associ- ated exonuclease activity important in DNA metab- olism, including DNA replication, repair and recombination. In humans, deficiencies in WRN function cause the segmental progeroid Werner syndrome (WS), in which patients show premature onset of many hallmarks of normal human ageing. At the cellular level, WRN loss results in rapid replicative senescence, chromosomal instability and sensitivity to various DNA damaging agents includ- ing the topoisomerase inhibitor, camptothecin (CPT). Here, we investigate the potential of using either transient or stable WRN knockdown as a means of sensitising cells to CPT. We show that targeting WRN mRNA for degradation by either RNAi or hammerhead ribozyme catalysis renders human fibroblasts as sensitive to CPT as fibroblasts derived from WS patients, and furthermore, we find altered cell cycle transit and nucleolar destabilisation in these cells following CPT treatment. Such WS-like pheno- types are observed despite very limited decreases in total WRN protein, suggesting that levels of WRN protein are rate-limiting for the cellular response to camptothecin. These findings have major implica- tions for development of anti-WRN agents that may be useful in sensitising tumour cells to clinically relevant topoisomerase inhibitors. Keywords Werner syndrome WRN RecQ Camptothecin Topoisomerase RNAi Ribozyme Aging Cancer Introduction Werner syndrome (WS) is an autosomal recessive disorder in which many features of normal human ageing are accelerated (Rossi et al. 2010; Cox and Faragher 2007; Kipling et al. 2004). WS patient Joseph L. E. Bird and Katrin C. B. Jennert-Burston contributed equally to this study. J. L. E. Bird K. C. B. Jennert-Burston J. E. Lowe R. G. A. Faragher School of Pharmacy and Biomolecular Sciences, University of Brighton, Brighton, UK Present Address: J. L. E. Bird Wolfson Brain Imaging Centre, Department of Medicine, University of Cambridge, Addenbrooke’s Hospital, Hills Road, Cambridge CB2 2QQ, UK M. A. Bachler P. A. Mason L. S. Cox (&) Department of Biochemistry, University of Oxford, South Parks Road, Oxford OX13QU, UK e-mail: [email protected] S.-J. Heo Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, CA, USA J. Campisi Buck Institute for Research on Aging, 8001 Redwood Blvd, Novato, CA, USA 123 Biogerontology (2012) 13:49–62 DOI 10.1007/s10522-011-9341-8
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Recapitulation of Werner syndrome sensitivity to camptothecin by limited knockdown of the WRN...

RESEARCH ARTICLE
Recapitulation of Werner syndrome sensitivityto camptothecin by limited knockdown of the WRNhelicase/exonuclease
Joseph L. E. Bird • Katrin C. B. Jennert-Burston • Marcus A. Bachler •
Penelope A. Mason • Jill E. Lowe • Seok-Jin Heo • Judith Campisi •
Richard G. A. Faragher • Lynne S. Cox
Received: 31 December 2010 / Accepted: 10 May 2011 / Published online: 24 July 2011
� Springer Science+Business Media B.V. 2011
Abstract WRN is a RecQ helicase with an associ-
ated exonuclease activity important in DNA metab-
olism, including DNA replication, repair and
recombination. In humans, deficiencies in WRN
function cause the segmental progeroid Werner
syndrome (WS), in which patients show premature
onset of many hallmarks of normal human ageing. At
the cellular level, WRN loss results in rapid
replicative senescence, chromosomal instability and
sensitivity to various DNA damaging agents includ-
ing the topoisomerase inhibitor, camptothecin (CPT).
Here, we investigate the potential of using either
transient or stable WRN knockdown as a means of
sensitising cells to CPT. We show that targeting
WRN mRNA for degradation by either RNAi or
hammerhead ribozyme catalysis renders human
fibroblasts as sensitive to CPT as fibroblasts derived
from WS patients, and furthermore, we find altered
cell cycle transit and nucleolar destabilisation in these
cells following CPT treatment. Such WS-like pheno-
types are observed despite very limited decreases in
total WRN protein, suggesting that levels of WRN
protein are rate-limiting for the cellular response to
camptothecin. These findings have major implica-
tions for development of anti-WRN agents that may
be useful in sensitising tumour cells to clinically
relevant topoisomerase inhibitors.
Keywords Werner syndrome � WRN � RecQ �Camptothecin � Topoisomerase � RNAi � Ribozyme �Aging � Cancer
Introduction
Werner syndrome (WS) is an autosomal recessive
disorder in which many features of normal human
ageing are accelerated (Rossi et al. 2010; Cox and
Faragher 2007; Kipling et al. 2004). WS patient
Joseph L. E. Bird and Katrin C. B. Jennert-Burston contributed
equally to this study.
J. L. E. Bird � K. C. B. Jennert-Burston �J. E. Lowe � R. G. A. Faragher
School of Pharmacy and Biomolecular Sciences,
University of Brighton, Brighton, UK
Present Address:J. L. E. Bird
Wolfson Brain Imaging Centre, Department of Medicine,
University of Cambridge, Addenbrooke’s Hospital, Hills
Road, Cambridge CB2 2QQ, UK
M. A. Bachler � P. A. Mason � L. S. Cox (&)
Department of Biochemistry, University of Oxford,
South Parks Road, Oxford OX13QU, UK
e-mail: [email protected]
S.-J. Heo
Lawrence Berkeley National Laboratory, 1 Cyclotron
Road, Berkeley, CA, USA
J. Campisi
Buck Institute for Research on Aging, 8001 Redwood
Blvd, Novato, CA, USA
123
Biogerontology (2012) 13:49–62
DOI 10.1007/s10522-011-9341-8

fibroblasts show rapid replicative senescence, a
mutator phenotype and hypersensitivity to certain
DNA-damaging agents including the topoisomerase
poison camptothecin (CPT) (Christmann et al. 2008;
Lebel and Leder 1998; Ogburn et al. 1997; Okada
et al. 1998; Pichierri et al. 2000; Poot et al. 1999).
WS results from loss of function mutations in the
WRN gene (Yu et al., 1996), which encodes a RecQ-
like helicase with a unique 30–50 exonuclease domain
(Gray et al. 1997; Huang et al. 1998; Shen et al. 1998;
Suzuki et al. 1997). WRN acts in multiple DNA
transactions (reviewed in Cheng et al. 2007; Cox and
Faragher 2007; Kudlow et al. 2007), including the
suppression of recombination at stalled replication
forks during S phase (Franchitto and Pichierri 2004;
Rodriguez-Lopez et al. 2002; Sidorova et al. 2008).
Holliday junctions (HJ) are thought to result from
replication fork collapse, for example following
attempts to replicate a template containing campto-
thecin-blocked topoisomerase, and WRN exonucle-
ase activity is implicated in their resolution (Machwe
et al. 2007). The importance of HJ resolution in
overcoming the effects of camptothecin-mediated
arrest has been demonstrated using a Holliday
junction endonuclease RusA, that reduces the hyper-
sensitivity of WS cells to CPT (Rodriguez-Lopez
et al. 2007). The hypersensitivity of WS patient cells
to CPT thus presumably occurs because WS cells are
defective in processing and/or restarting stalled or
collapsed replication forks (Franchitto and Pichierri
2004; Machwe et al. 2007; Rodriguez-Lopez et al.
2002; Sidorova et al. 2008), since WRN usually
prevents conversion of single strand to double strand
DNA breaks (Christmann et al. 2008).
Whilst first identified as an important factor in
preventing premature ageing, WRN has also more
recently been recognised as a significant tumour
suppressor. WS patients show high tumour incidence,
especially sarcomas (Goto et al. 1996). Epigenetic
inactivation by methylation of CpG islands in the
WRN promoter is associated with tumorigenesis in
many common tumour types of both epithelial and
mesenchymal origin (Agrelo et al. 2006), while
reactivation of WRN expression leads to reduction
of tumour growth in a mouse xenograft model
(Agrelo et al. 2006). Since loss of WRN predisposes
to CPT sensitivity, it is an interesting proposition to
exploit WRN inactivation in cancer chemotherapy
(Futami et al. 2007, 2008), as many different tumour
cell types require WRN for survival (Opresko et al.
2007) and CPT analogues are currently in use in the
cancer clinic. Furthermore, treatments that induce
transient WRN loss should not kill normal cells—at
worst, those cells most affected by treatment may
become senescent, a recognised tumour suppressive
mechanism (Campisi 2005), resulting in selective
killing of cancer cells. In order to pursue this goal of
exploiting the ageing-associated WRN as a therapeu-
tic target in cancer, it is necessary to characterise
different possible methods of ablating WRN expres-
sion and determining the degree to which WRN loss
is required in order to obtain the required phenotype
of CPT sensitivity.
Here we test two different strategies (short hairpin
interfering RNAs and hammerhead ribozymes,
(Brummelkamp et al. 2002; Citti et al. 1999; Citti
and Rainaldi 2005; Paddison et al. 2002)) designed to
permit either transient or stable WRN knockdown, and
measure both degree of WRN protein loss and CPT
sensitivity. The transfected human cell lines we report
here in which WRN is targeted for degradation possess
isogenic partner controls, allowing precise analysis of
the contribution of WRN to any phenotype observed,
and so present a useful research resource. We demon-
strate that targeting WRN by these approaches gives
levels of sensitivity to camptothecin equivalent to
those observed in WS patient-derived fibroblasts, even
when very minor decreases in WRN protein levels are
detected, suggesting that WRN levels are rate-limiting
for the cellular response to camptothecin. Moreover,
marked changes in nucleolar integrity were observed
following camptothecin treatment of the ‘knockdown’
cells. These findings are discussed in the context of
recent developments in cancer chemotherapy based on
WRN status.
Materials and methods
shRNA & ribozyme design and plasmid
constructs
Two different anti-WRN shRNA retroviral vectors
were constructed: MSCV-WRN shRNA KJ1 and
MSCV siWRN4, which target nucleotides (nt)
121–150 and 3724–3742 relative to the ATG start
codon, respectively. An anti-WRN hammerhead
ribozyme expression vector, MSCV-WRN Rib3 was
50 Biogerontology (2012) 13:49–62
123

also created, targeting nt 3297–3327. All targets had
at least 8 nt differences from any other human gene as
determined by BLAST searches. An anti-firefly
luciferase shRNA (pSHAG-Ff1) (Paddison et al.
2002) and an ineffective hairpin targeting nt -3 to
?16 of the special AT-rich sequence binding protein
1 (SATB1) were used as negative shRNAi controls,
whilst the insertless pMSCV-puromycin retroviral
vector (Clontech) served as a further control.
The KJ1 hairpin sequence (Table 1) encodes
inverted repeats of 29 base pairs (bp) separated by a
4-nt spacer. A U6 promoter expression cassette was
generated by PCR (Hemann et al. 2003) and cloned
into pENTR/TOPO-D (Invitrogen). siWRN4 and
siSATB1 hairpins contain 19 bp inverted repeats
separated by a 9 bp spacer. These were synthesised
as complementary 64-mer oligonucleotides, annealed
and initially cloned into pSuper (Brummelkamp et al.
2002).
The anti-WRN hammerhead ribozymes were cre-
ated using two complementary 54-mer oligonucleo-
tides containing a 24 bp consensus hammerhead core
sandwiched between 14 nt of complementary
sequence 50 of the target and 15 nt of complementary
sequence 3’ of the target (Table 1). These were
annealed and cloned into pCR2.1-TOPO (Invitrogen)
and subcloned into pU1-ribozyme (Montgomery and
Dietz 1997) to create a U1 expression cassette for a
U1/ribozyme hybrid where the ribozymes are stabi-
lised by U1 stem loop structures. All expression
cassettes were subcloned into the pMSCV-puromycin
retroviral vector and high titre virus was produced in
CRIP amphotropic retroviral packaging cells.
Cell culture and generation of stable recombinant
cell lines
1BR.3neo and WV1 are SV40 T antigen-immorta-
lised fibroblasts from wt or WS donors, respectively,
and were cultured in DMEM (Invitrogen) containing
10% heat-inactivated foetal calf serum (Biowest),
2 mM L-glutamine, 100 U/ml penicillin and 100 lg/ml
streptomycin (Invitrogen) in a humidified incubator
(37�C, 5% CO2).
All RNA-mediated WRN knock-down and con-
trol cells were generated from 1BR.3neo by retro-
viral infection with the appropriate vector. Clones or
bulk cultures were selected using puromycin at
5 lg/ml, and cultures were maintained in 2.5 lg/ml
puromycin.
Table 1 Oligonucleotides use in this study
Construct name Oligonucleotide sequence
Rib1 Ribozyme: 3’AtcgtcgcctttaCACAAAGCAGGAGTGCCTGAGTAGTCgacttacctacttag 5’
Antisense: 5’agcagcggaaatGTGTTTCGTCCTCACGGACTCATCAGctgaatggatgaatcA 3’
Rib2 Ribozyme: 3’AaacgagaaaggaCACAAAGCAGGAGTGCCTGAGTAGTCtcttctataatcgta 5’
Antisense: 5’ttgctctttcctGTGTTTCGTCCTCACGGACTCATCAGagaagatattagcatA 3’
Rib3 Ribozyme: 3’AatgtctcttcttCACAAAGCAGGAGTGCCTGAGTAGTCattgaacctcttcaa 5’
Antisense: 5’tacagagaagaaGTGTTTCGTCCTCACGGACTCATCAGtaacttggagaagttA 3’
WRN4 Forward primer:5’gatccccCAGATGACGAGTCAGGTAGAGttcaagagaCTACCTGACTCGTCATCTGtttttggaaa 3’(linker/anti-sense/loop/sense/linker)
Reverse primer:5’ agcttttccaaaaaCAGATGACGAGTCAGGTAGtctcttgaaCTACCTGACTCGTCATCTGggg 3’ (linker/sense/loop/anti-sense/linker)
KJ1 5’ggaattcAAAAAGTGAATTCTAAGAAGAGGAGGCCACCTCCccaaGAAGATGACCTCCCCTTCTTAGAATTCACTagtatatgtgctgccgaagc 3’
(HindIII site/U6 terminator/sense/loop/anti-sense/3’ end of U6 promoter)
KJ2 5’ggaattcAAAAAGTACTTAATCCAACTAGTACTCGATTACAccaaTATAATCAAGTACCAGTTGAATTAAGTACTagtatatgtgctgccgaagc 3’
(HindIII site/U6 terminator/sense/loop/anti-sense/3’ end of U6 promoter)
U6 forward 5’ caccaaggtcgggcaggaaga 3’
U1 forward 5’ atacttacctggcaggggagat 3’
U1 reverse 5’ tccactgtaggattaacaactaag 3’
Biogerontology (2012) 13:49–62 51
123

RT-PCR
Reverse-transcription PCR to detect expression of the
U1 cassette was conducted using One-step RT-PCR
(Qiagen) according to the manufacturer’s instructions.
Briefly 0.5 lg total RNA extracted from transfected
cells using RNeasy (Qiagen) was incubated with U1
primers as listed in Table 1, and under the following
conditions: 50�C 30 min, 95�C 15 min, followed by 40
cycles of 94�C 30 s, 50�C 30 s, 72�C 60 s, and one
final extension step 72�C for 10 min in a Thermo PX2
PCR block. Products were analysed on 1% agarose gels
in 19 TBE with 1 lg/ml ethidium bromide and
visualised under UV illumination at 280 nm.
Single-cell gel electrophoresis (COMET) assay
Single-cell gel electrophoresis assay was performed as
described (Clingen et al. 2000; Lowe et al. 2004).
Following treatment of proliferating cells with 10 lM
camptothecin for 1 h, COMET tail lengths were
quantified using CASys software (Comet Analysis
Synoptics) from microscopic images taken using a
Nikon microscope UVP videocapture system. 50 nuclei
were scored from two slides for each experiment, and
each experiment was repeated at least 5 times.
Flow cytometry analysis
Cells from a 10 cm tissue culture dish were washed in
PBS, harvested by incubating with trypsin, collected,
trypsin neutralised by addition of foetal calf serum
then cells collected by centrifugation, washed in PBS,
repelleted and fixed by gradual addition of 1 ml
methanol with mixing. Fixed cells were treated with
RNase then DNA was stained with propidium iodide
and processed for flow cytometry using a Becton–
Dickinson FACS machine with Facscalibur software.
SDS-PAGE and western blotting
Cellular proteins were extracted in 50 mM Tris–HCl
pH 7.5, 150 mM NaCl, 1% (v/v) NP40, EDTA-free
protease inhibitors (Roche) with or without 25 U/ml
benzonase (Merck). Equal protein quantities were
solubilised in 62.5 mM Tris–HCl pH 6.8, 10% (v/v)
glycerol, 2% (w/v) SDS, 0.00125% (w/v) bromophe-
nol blue, 200 mM DTT, separated by SDS-PAGE, and
transferred to PVDF or nitrocellulose membranes
(Sigma). The membranes were blocked in 5% (w/v)
non-fat milk powder (NFMP), 0.4% (v/v) Tween 20,
PBS pH 7.5 overnight at 4�C, then incubated with anti-
WRN antibodies (monoclonal, BD Biosciences, 1:250
or polyclonal Ab200, AbCam, at 1:1000), mouse anti-
GAPDH monoclonal antibody (Biogenesis), anti-
tubulin, anti-actin or anti-MRE11, diluted in 0.1%
(w/v) NFMP in PBS. After washing, membranes were
incubated with HRP-conjugated goat anti-mouse or
swine anti-rabbit immunoglobulins (Dako) (1:2000) in
1% (w/v) NFMP. After further washing, immune
complexes were visualised using enhanced chemilu-
minescence (Amersham Pharmacia Biotech) and
X-ray film detection. Signals were measured using a
MultiImageTM transilluminator (Alpha Innotech) and
IOD values quantified using dedicated software.
Statistical analysis
All data are given as mean ± SD. The Student t-test
was used to determine statistical significance.
Immunofluorescence
Transfected cell lines were cultured on coverslips
until approximately 50% confluent. After washing in
PBS, the cells were fixed in buffer containing 4%
(w/v) paraformaldehyde (Sigma-Aldrich), permeabilised
with 0.1% Triton X-100 in PBS then incubated with
mouse anti-WRN monoclonal antibody (BD Biosci-
ences) or rabbit anti-WRN (Ab200, Abcam) in 0.1%
Tween 20 in PBS. Where relevant, anti-nucleolin
antibody (AbCam) was also used. Non-specific
binding was prevented by incubation with 5% (w/v)
non-fat milk powder in PBS or 10% donkey serum in
PBS. After further washing in PBS, the cells were
incubated with FITC horse anti-mouse (Vector Labs)
in 5% non-fat milk powder in PBS or FITC donkey
anti-rabbit IgG conjugated with fluorescein iso-
thiocyanate (Jackson Laboratories). For double
staining, WRN was visualized with a FITC-conju-
gated secondary antibody and nucleolin with a
rhodamine-conjugated secondary antibody (Jackson
Laboratories). Finally, after washing in PBS, cover-
slips were mounted in Vectorshield mounting med-
ium containing DAPI (Vector Labs). Mounted cells
were viewed using a Zeiss Axioskop 2 microscope
and images were captured with a CCD camera using
Axiovision software.
52 Biogerontology (2012) 13:49–62
123

Results
WRN knockdown on transient transfection
with shRNAi and ribozyme constructs
To achieve transient knockdown of WRN, we
generated various short hairpin interfering RNAs
(shRNAi) or ribozymes designed against the
sequence of WRN mRNA (Fig. 1; Table 1), and
transfected these constructs and appropriate controls
(see ‘‘Materials and Methods’’ section) into SV40-
transformed fibroblasts 1Br.3neo.
The degree of WRN knockdown achieved with the
shRNA or ribozyme constructs was assessed by
western blotting at 48 or 72 h following transfection
(Fig. 2a, b, respectively), normalising the intensity of
the WRN protein band against GAPDH, an internal
loading control. We observed no appreciable differ-
ence in WRN protein levels between cells transfected
with anti-WRN shRNA constructs KJ1 and KJ2, or
the ribozyme Rib2, and the negative controls (lipo-
fectamine alone [lipo], insertless vector pMSCV,
firefly luciferase shRNAi [FF1] or SATB1 shRNAi).
By contrast, the ribozyme targeted towards the 30 end
of WRN mRNA (Rib3) did appear to lead to limited
decrease in WRN protein levels at 48 h (Fig. 2a) and
greater reduction at 72 h (Fig. 2b).
WRN4 shRNA and Rib3 ribozyme expression
induces camptothecin hypersensitivity
and aberrant cell cycle response to CPT
Cells lacking functional WRN are hypersensitive to
the topoisomerase inhibitor camptothecin (CPT)
(Lebel and Leder 1998; Ogburn et al. 1997; Okada
et al. 1998; Pichierri et al. 2000; Poot et al. 1999), so
we next tested CPT sensitivity of stable transfectants
expressing either WRN4 shRNA, Rib3 ribozyme or
various control constructs, using a modified COMET
assay to allow the quantification of DNA strand
breaks in individual cells (Clingen et al. 2000). We
have previously shown this to be a sensitive but
robust differentiator between cells expressing wild-
type or mutant WRN following treatment with
camptothecin (Lowe et al. 2004). The increase in
COMET tail length due to CPT treatment (deter-
mined by subtracting the mean tail length without
drug exposure from the mean tail length after CPT
exposure) was plotted (Fig. 3).
Interestingly, whilst the KJ1 construct designed
against WRN resulted in little protein knockdown
even on transient transfection (e.g. see Fig. 2), this
construct gave significantly elevated CPT-dependent
COMET tail lengths equivalent to those observed for
WV1 cells on CPT treatment (Fig. 3a), that were
*2.5 fold longer than those detected for the control
1Br.3neo parental cells or 1Br.3neo stably transfected
with the vector backbone (pMSCV), or shRNAi
targeted against firefly luciferase (FF). This effect
was not dependent on a particular clone of cells
expressing the KJ1 construct as 2 separate samples
from 3 independent clones (numbered 4, 8 and 16) of
the KJ1-transfected cells all showed marked increases
in CPT-induced COMET tail length (Fig. 3a).
Furthermore, stable expression of WRN4 shRNA
in 1BR.3neo cells also resulted in hypersensitivity to
camptothecin (Fig. 3b), with an increase in CPT-
dependent COMET tail length equivalent to that
observed for WS-patient derived cell line WV1
lacking functional WRN protein (P \ 0.05), and
Fig. 1 WRN-directed shRNA and ribozyme constructs.
shRNA retroviral vectors were constructed containing WRN-
targeting KJ1 or WRN4 inserts (see Table 1 for sequence)
under the control of the U6 promoter. Alternatively, WRN-
directed ribozyme constructs were generated for expression
from the U1 promoter (see ‘‘Materials and Methods’’ section
for details)
Biogerontology (2012) 13:49–62 53
123

significantly longer (P \ 0.01) than COMET tails
observed on CPT treatment of the 1Br.3neo parental
cell line (Fig. 3b; Table 2). No significant campto-
thecin sensitivity was observed in 1BR.3neo fibro-
blasts transfected with an shRNA targeting the human
SATB1 mRNA (Fig. 3b; Table 2), suggesting that
enhanced CPT sensitivity is due to expression of
WRN4 shRNAi specifically, rather than a general
effect of shRNAi expression on cells.
As an alternative to WRN inactivation by RNAi,
we tested the impact of stable expression of the
hammerhead ribozyme Rib3 targeted against WRN,
which in transient transfection experiments led to a
drop in WRN protein levels (see Fig. 2). 1BR.3neo
fibroblasts transduced with the Rib3 ribozyme
showed levels of camptothecin sensitivity that were
statistically indistinguishable from those seen in
WV1 fibroblasts lacking functional WRN (Fig. 3c;
Table 2).
Box plots with quartiles marked show the COMET
tail length measured in both untreated and CPT-
related cell lines (Fig. 3d, e), further confirming that
the CPT-dependent increase in tail length in cells
with full or partial knockdown of WRN is genuine
and not due to aberrant outliers impacting dispropor-
tionately on the mean tail length measured.
Expression of the U1 backbone cassette containing
the Rib3 ribozyme was verified by RT-PCR (Fig. 3f),
which demonstrated expression of U1-containing
RNA species in all cells, as expected for expression
of endogenous U1 RNA (asterisk in Fig. 3f), but also
showing production of a slightly larger U1-containing
RNA in Rib3-transfected cells, consistent with the
size of the Rib3 construct (arrow in Fig. 3f). (The
absence of a band representing endogenous U1 RNA
in the Rib3-cells is suggestive of very high levels of
Rib3 RNA expression, out-competing endogenous
U1 for primers in RT-PCR). We also verified that U1-
containing RNA species were not diminished in the
transfected cells following CPT treatment (data not
shown).
COMET tails result from breakage of DNA. Given
that WRN is implicated in resolving or restarting
stalled replication forks (Rodriguez-Lopez et al.
2002, 2007; Sidorova et al. 2008), the increased
COMET tail lengths we observe using the WRN
knockdown constructs may reflect cells that have
been unable to repair DSBs arising from DNA
replication over a CPT-blocked topoisomerase
(which transforms a ss DNA break to a DSB). To
test for this, and to further analyse the response of the
transfected cell lines to CPT, we conducted cell cycle
Fig. 2 Rib3 ribozyme reduces WRN protein levels in tran-
siently transfected 1Br.3neo cells. 1Br.3neo cells were
transiently transfected with various constructs designed to
express shRNA against WRN (KJ1), firefly luciferase (FF1),
special AT-rich sequence binding protein 1 (SATB), or vector
without insert (pMSCV), and protein levels compared with
patient-derived WV-1 fibroblasts and parental cell line
1Br.3neo at 48 h (a) or 72 h (b) following transfection, on
western blots probed for WRN (upper panel) or loading control
GAPDH (middle panel). Band intensity for WRN was
normalised against GAPDH (bottom panel)
54 Biogerontology (2012) 13:49–62
123
analysis of the cell lines with and without CPT
treatment by flow cytometry. WV1 cells showed a
higher S phase fraction than 1Br3neo cells that are
wild type for WRN, and this increased S phase
fraction, which was also observed for Rib3-express-
ing cells (data not shown), did not alter on CPT
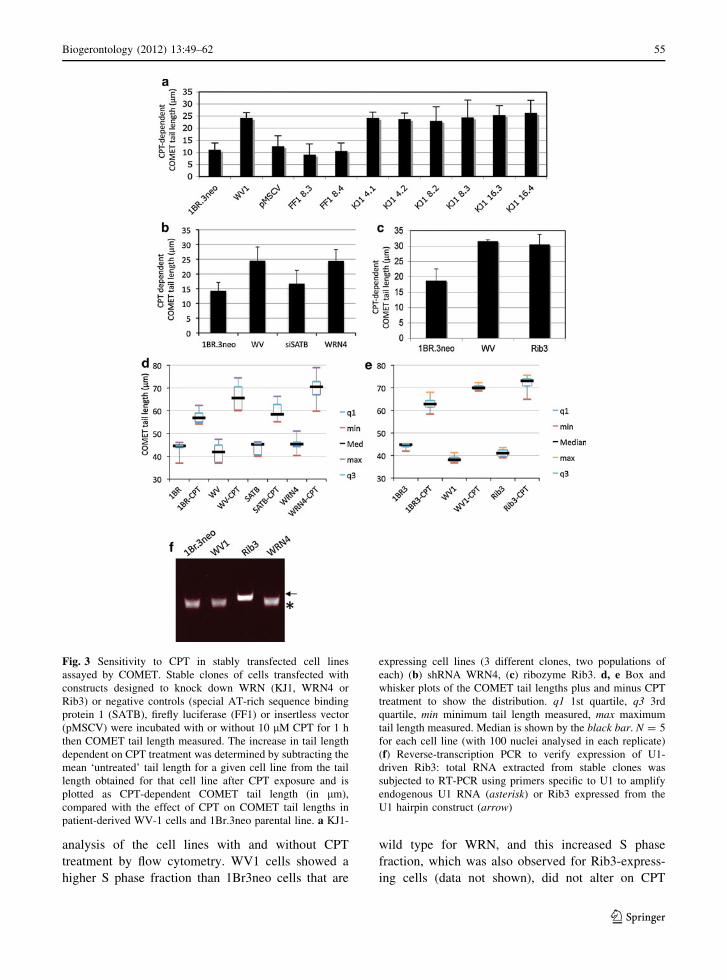
Fig. 3 Sensitivity to CPT in stably transfected cell lines
assayed by COMET. Stable clones of cells transfected with
constructs designed to knock down WRN (KJ1, WRN4 or
Rib3) or negative controls (special AT-rich sequence binding
protein 1 (SATB), firefly luciferase (FF1) or insertless vector
(pMSCV) were incubated with or without 10 lM CPT for 1 h
then COMET tail length measured. The increase in tail length
dependent on CPT treatment was determined by subtracting the
mean ‘untreated’ tail length for a given cell line from the tail
length obtained for that cell line after CPT exposure and is
plotted as CPT-dependent COMET tail length (in lm),
compared with the effect of CPT on COMET tail lengths in
patient-derived WV-1 cells and 1Br.3neo parental line. a KJ1-
expressing cell lines (3 different clones, two populations of
each) (b) shRNA WRN4, (c) ribozyme Rib3. d, e Box and
whisker plots of the COMET tail lengths plus and minus CPT
treatment to show the distribution. q1 1st quartile, q3 3rd
quartile, min minimum tail length measured, max maximum
tail length measured. Median is shown by the black bar. N = 5
for each cell line (with 100 nuclei analysed in each replicate)
(f) Reverse-transcription PCR to verify expression of U1-
driven Rib3: total RNA extracted from stable clones was
subjected to RT-PCR using primers specific to U1 to amplify
endogenous U1 RNA (asterisk) or Rib3 expressed from the
U1 hairpin construct (arrow)
Biogerontology (2012) 13:49–62 55
123

treatment. WRN4 cells showed a profile intermediate
between the wt 1Br3neo and the patient-derived WV1
cells lacking WRN. An increase in the S phase
fraction may reflect a failure to complete S phase and
suggests an inability to respond appropriately to DNA
damage when WRN protein is limiting. Note that
there was no marked increase in a sub-G1 population
on CPT treatment of any of the cell lines (data not
shown) suggesting that the CPT treatment was not
sufficiently severe to invoke an apoptotic response.
Thus the long COMET tails observed in WV1 and in
cells expressing the WRN knockdown constructs are
more consistent with DNA breakage during DNA
replication over an unrepaired template rather than
apoptotic cell death.
WRN protein levels by western blotting
are not significantly decreased in stably
transfected ‘knock-down’ cells
Since expression of the WRN-directed ribozyme
Rib3 and shRNA WRN4 resulted in significant CPT
sensitivity (Fig. 3) and alterations in cell cycle
profiles, it was reasonable to assume that these
phenotypes resulted from decreases in WRN protein
levels in the stably transfected clones compared with
the negative controls. This was tested by western
blotting for WRN protein against an extensive range
of loading controls and using several different protein
extraction conditions.
As expected, WRN protein was undetectable in
WV1 patient-derived cells (Fig. 4a–c), but a band at
*162 kDa was observed using an anti-WRN mono-
clonal antibody in cell lysates of the control cell lines
(pMSCV, FF1 and SATB). Unexpectedly, given the
CPT sensitivity and cell cycle phenotypes observed
(Fig. 3), WRN protein levels (normalised against a
tubulin internal loading control) did not appear
significantly diminished in cells stably transfected
with the WRN knockdown constructs Rib3 and
WRN4, compared with the negative controls
(Fig. 4a).
WRN has been reported to be present in different
pools in the cell with at least one fraction associated
with chromatin, so in order to extract both soluble
and chromatin-associated WRN, the potent nuclease
benzonase was used during protein preparation from
cells. While more WRN was extracted on nuclease
treatment, the ratio of WRN to a further loading
control GAPDH was not significantly decreased in
the WRN ‘knockdown’ lines (WRN4, Rib 3 or KJ1)
compared with controls (Fig. 4b).
As this lack of WRN protein reduction in the
‘knockdown’ cell lines was unexpected, the analysis
was repeated several times in our three separate
laboratories using the same cells lines but different
antibodies and different extraction procedures to
extract either soluble protein, chromatin-bound pro-
tein, or total protein using boiling SDS-dyes. Addi-
tional loading controls including Ponceau S staining
of blots and Coomassie blue quantification of protein
levels were also conducted. In all cases, very little
decrease in WRN protein levels was observed in the
WRN4 or Rib3-expressing cells (data not shown)
even though these cell lines showed a highly
significant CPT hypersensitivity phenotype.
Because WRN is a DNA repair protein and hence
its levels and/or activity may alter on DNA damage
(such as treatment with 10 lM CPT for 1 h), we also
compared WRN protein levels with another chroma-
tin-associated DNA repair protein, MRE11, with and
without CPT treatment (Fig. 4c). Note that WRN
physically associates with the MRN complex which
contains MRE11 (Franchitto and Pichierri 2004).
MRE11 protein levels (normalised against actin, a
further loading control) remained stable across all
cell lines analysed with and without CPT treatment.
By contrast, WRN:actin ratios appeared to rise
slightly following 1 h treatment with 10 lM CPT in
Table 2 Statistical comparison of CPT-dependent COMET
tail length
1Br3neo WV1 SATB WRN4
1Br3neo – 0.002 0.193 0.011
WV1 0.002 – 0.025 0.975
SATB 0.193 0.025 – 0.011
WRN4 0.011 0.975 0.011 –
1Br3neo WV1 Rib3
1Br3neo – 0.005 0.0004
WV1 0.005 – 0.516
Rib3 0.0004 0.516 –
Pairwise comparisons between CPT-dependent mean COMET
tail length of 5 independent experiments (100 nuclei for each
cell line in each replicate) for each cell line, assuming a two-
tailed distribution of paired samples, using the Student t-test.
P values are shown
56 Biogerontology (2012) 13:49–62
123

the parental 1Br3.neo cells but decreased in cells
bearing the WRN ‘knockdown’ constructs Rib3 and
WRN4. This resulted in a decrease in WRN:MRE11
ratios in ‘knockdown’ cells (Fig. 4c, graph).
WRN knockdown is observed
by immunofluorescence
The low level of reduction in WRN protein observed
on western blots of lysates of Rib3 and WRN4-
expressing cells was highly unexpected given their
marked sensitivity to CPT. To test whether the small
reduction was due to a mild knockdown in all or most
cells, or a marked knockdown in a few cells with
little or no knockdown in others (i.e. clonal effects),
we studied WRN levels in the transfected cells by
immunofluorescence microscopy. As can be seen in
Fig. 5, WRN protein was not detectable in patient-
derived WV-1 cells while a nuclear WRN signal was
detected in 1Br3.neo cells with a strong nucleolar
signal (Fig. 5), as expected. Compared with controls
(1Br3.neo parental line or cells transfected with
vector alone (pMSCV), shRNA against firefly lucif-
erase (FF1) or special AT-rich sequence binding
protein 1 (SATB1)), the majority of cells bearing the
WRN4 and Rib3 constructs showed slight but con-
sistent reductions in levels of WRN protein as
detected by immunofluorescence (Fig. 5), though
we never observed total loss of signal. Thus the low
levels of WRN protein reduction observed by western
blotting probably represents the case for the majority
of cells in culture, rather than being due to clonal
variation in which only some cells show marked
levels of knockdown while others do not display
protein loss.
Interestingly, we noted a slight but consistent
increase in nuclear size in the cells stably transfected
with the WRN knockdown constructs, and a decrease
in the WRN nucleolar signal (Fig. 5, Rib3 and
WRN4). To investigate this further, we employed
an antibody against the nucleolar protein, nucleolin,
as an additional marker for nucleolar integrity. In
1Br3.neo cells, WRN is observed with the usual
nuclear/nucleolar distribution that alters to nucleo-
plasmic following exposure of cells to 10 lM CPT
for 1 h (Fig. 6a), consistent with WRN responding to
DNA damage and its role in resolving stalled
replication forks e.g. at CPT-induced lesions (Rodri-
guez-Lopez et al. 2002, 2007; Sidorova et al. 2008).
As expected, there was no detectable WRN signal in
Fig. 4 Anti-WRN
ribozyme and shRNAs do
not significantly reduce
total WRN protein levels.
a Protein extracts of stable
cell clones were analysed
by immunoblotting, probing
for WRN, using tubulin as
an internal loading control.
b Cell extracts were
prepared with (?) or
without (-) benzonase
nuclease treatment and
immunoblotted for WRN
and GAPDH (loading
control). c Protein samples
from cell lines exposed for
1 h to 10 lM CPT or
DMSO control were probed
for WRN, MRE11 and actin
(loading control). The
following protein ratios
were determined under both
conditions for each cell
line—MRE11:actin;
WRN:actin; WRN:MRE11
(graph)
Biogerontology (2012) 13:49–62 57
123
WV1 cells, but we also observed that the nucleolin
signal was diminished, and that this decreased further
on CPT treatment (Fig. 6b), suggesting that WRN
may be important for stabilising the nucleolus on
DNA damage. (Note that the nucleolus contains
highly repetitive rDNA in tandem arrays, with DNA
replication fork barriers to promote unidirectional
DNA synthesis, and that yeast cells lacking the WRN
homologue Rqh1, show marked rDNA array insta-
bility (Ahn et al. 2005)). Interestingly, a low nucle-
olin signal that decreased further on CPT treatment
was also detected in cells expressing WRN4 shRNAi
(Fig. 6d), though cells transfected with the Rib3
ribozyme had a robust nucleolin signal with or
without CPT treatment (Fig. 6c). Notably, WV1 (no
functional WRN protein) and Rib3 and WRN4 cells
(slight knockdown of WRN) consistently showed
increased nuclear size compared with the wild type
1Br3.neo parental line.
Discussion
Progeroid Werner syndrome patient cells show
hypersensitivity to camptothecin, a drug that inhibits
topoisomerase activity, and derivatives of which are
widely used in the cancer clinic. Exploiting loss of
the WRN protein, which normally acts to stabilise the
genome particularly in response to genotoxic insults
that impact on DNA replication, may be a powerful
route to improve cancer treatments, and here we have
created molecular tools (shRNA and ribozymes)
designed to deplete WRN in cells. We demonstrate
using the COMET assay (a robust screening tool for
lack of functional WRN protein in cells (Lowe et al.
2004)) that fibroblasts carrying these constructs are as
sensitive to camptothecin as fibroblasts from WS
patients, and that this sensitivity is dependent upon
the anti-WRN component of the constructs since no
abnormal camptothecin sensitivity was apparent in
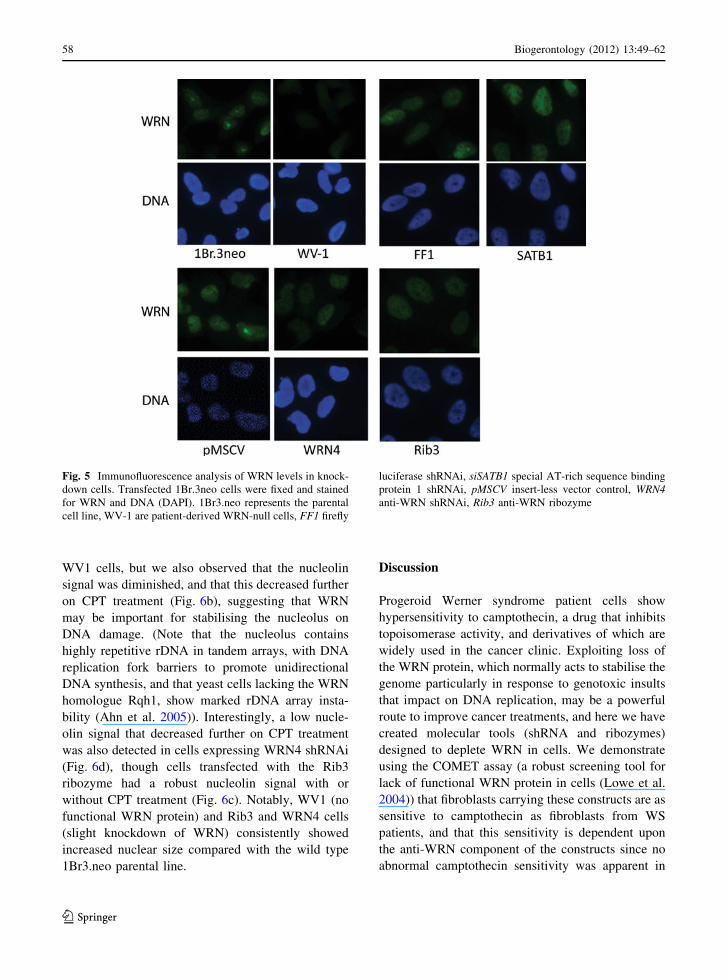
Fig. 5 Immunofluorescence analysis of WRN levels in knock-
down cells. Transfected 1Br.3neo cells were fixed and stained
for WRN and DNA (DAPI). 1Br3.neo represents the parental
cell line, WV-1 are patient-derived WRN-null cells, FF1 firefly
luciferase shRNAi, siSATB1 special AT-rich sequence binding
protein 1 shRNAi, pMSCV insert-less vector control, WRN4anti-WRN shRNAi, Rib3 anti-WRN ribozyme
58 Biogerontology (2012) 13:49–62
123

fibroblasts transduced with control constructs (hair-
pins that target firefly luciferase or SATB1, or insert-
less vector controls).
Since RNAi can produce off-target effects (Sledz
and Williams 2004), we also created a unique
hammerhead ribozyme, Rib3, which acts indepen-
dently of the endogenous RISC silencing machinery.
This anti-WRN ribozyme produced camptothecin
sensitivity equivalent to that seen in WS-derived
fibroblasts and indistinguishable from that observed
with two different shRNAs, KJ1 and WRN4. Con-
structs designed to knock down WRN expression by
different biochemical pathways therefore resulted in
a WS-like phenotype of CPT hypersensitivity, but
puzzlingly, without much decrease in WRN protein
levels. Although we cannot at this stage completely
rule out the possibility that production of the dsRNA
from our various shRNA or ribozyme constructs is
triggering a DNA damage response (for example
activation of apoptosis through an interferon response
to dsRNA), we believe this to be unlikely since
expression of dsRNA from the control shRNAi
constructs (SATB and FF) did not result in increased
COMET tail length (Fig. 3) nor in increased sub-G1
cell fractions in flow cytometry (data not shown). We
therefore believe that the phenotype of CPT sensi-
tivity observed is due to the impact of the WRN-
directed constructs on WRN, and not via off-target
effects. It would be somewhat surprising if three
different constructs using two different silencing
approaches all resulted in the same off-target effect of
inducing CPT sensitivity without acting through their
intended target WRN, the loss of which does indeed
induce CPT sensitivity.
Our observations of limited WRN knockdown
cannot be explained by simple negative selection
against clones with high levels of transgene expres-
sion. Although primary fibroblasts from WS individ-
uals tend to proliferate more slowly and with very
limited replicative potential compared with those
from normal individuals, these differences are
almost completely removed by SV40-immortalisation
(Huschtscha et al. 1986). Since we have used SV40
immortalised cell lines, any negative selection for
loss of WRN in terms of loss of proliferative capacity
would be negligible. WRN loss has been shown to
result in apoptosis of a range of different transformed
cell lines (Opresko et al. 2007). However, it is highly
Fig. 6 Increased nuclear size and loss of nucleolar integrity in
cells lacking WRN. Cells were treated with 10 lM CPT for
1 h, or DMSO vehicle control, then fixed and stained for WRN
(FITC), DNA (DAPI) and nucleolin (rhodamine). 1Br
represents the parental cell line 1Br3.neo, WV-1 are patient-
derived WRN-null cells, WRN4 anti-WRN shRNAi, Rib3 anti-
WRN ribozyme
Biogerontology (2012) 13:49–62 59
123

improbable that the DNA damage we measure as
increased COMET tail lengths is indicative of
apoptosis in cells actively expressing the constructs,
since our microscopy (Figs. 5, 6) and FACS obser-
vations do not support the idea of mass apoptosis.
Moreover, RT-PCR detection of the U1-Rib3 RNA
product (Fig. 3) does not support the possibility that
the bulk of cells have avoided knockdown and hence
apoptosis by deleting or otherwise silencing the
‘knockdown’ constructs. Instead, we suggest that our
data indicate that phenotypic assays such as COMET
analysis of CPT-induced DNA damage can be
considerably more sensitive than the simple analysis
of total protein levels.
It is known that WRN exists in cells in different
pools with different solubility and that this can alter
on DNA damage (Karmakar and Bohr 2005); since
RNAi and ribozyme treatments lead to destruction of
cognate mRNA transcripts, it is possible that the
camptothecin sensitivity we observe reflects a need
for newly synthesised WRN to respond to acute DNA
damage. While we see little or no loss of WRN in
western blotting, a small but consistent decrease in
WRN signal is detected by immunofluorescence.
Detection of WRN in microscopy can be affected by
epitope masking, for example when WRN is phys-
ically associated with a protein partner or tightly
associated with chromatin (such associations should
be disrupted in western blotting hence all WRN
protein should be detected). Our results are consistent
with the hypothesis that a specific subset of WRN
(e.g. newly synthesised WRN that has not yet been
recruited into multiprotein complexes and thus in
which epitopes are still available for immunological
detection) might be necessary for the cellular
response to CPT, perhaps because this WRN fraction
can be readily mobilised to sites where it is required.
Loss of this subset of WRN (by RNAi or ribozyme
routes) may have as severe an impact as total loss of
WRN for the cellular response to transient DNA
damage such as that induced by CPT treatment.
It is thus probable that WRN is a rate-limiting
enzyme in the damage response to camptothecin and
would be a good target for chemical inhibition.
Furthermore, by generating WRN knockdown con-
structs suitable for stable expression, it should be
possible to assess the long-term phenotypic effects of
low levels of WRN knockdown in any cell type
desired, of significant value since only fibroblastic
and lymphoblastoid lines are currently available from
WS patients.
Recent reports of dose-dependent effects of short
interfering RNAs against WRN following transient
transfection (Futami et al. 2007) are compatible with
our findings. While long-term loss of WRN results in
significant genomic instability which predisposes to
cancer development, tumours in which WRN is
inactive can be more readily killed using low dose
CPT (Agrelo et al. 2006) or clinically relevant
analogues such as etoposide. Transient low-level
reductions in WRN in surrounding normal cells
would not be expected to have significant detrimental
effects. If the requirement for only minor decreases in
WRN protein levels in order to induce profound CPT
sensitivity are borne out by further experimental
studies, this bodes well for the use of WRN
knockdown or inhibition in combination with topo-
isomerase inhibitors as an anti-cancer strategy.
Acknowledgments We thank Mrs Christine Borer for
technical support to LSC and MAB. This work was funded
by the BBSRC grants [107/EGH16152 and 107/ERA16270] to
RGAF, JLEB, KJ-B and JL, BBSRC grants [BB/E000924/1]
and [43/ERA16310] and ESRC programme grant [ES/
G037086/1] (under the cross-council New Dynamics of
Ageing initiative) to LSC, and NIH grant AG024399 to JC.
Conflicts of interest The authors state no conflicts of
interest.
References
Agrelo R, Cheng WH, Setien F, Ropero S, Espada J, Fraga MF,
Herranz M, Paz MF, Sanchez-Cespedes M, Artiga MJ,
Guerrero D, Castells A, von Kobbe C, Bohr VA et al
(2006) Epigenetic inactivation of the premature aging
Werner syndrome gene in human cancer. Proc Natl Acad
Sci USA 103(23):8822–8827
Ahn JS, Osman F, Whitby MC (2005) Replication fork
blockage by RTS1 at an ectopic site promotes recombi-
nation in fission yeast. EMBO J 24(11):2011–2023
Brummelkamp TR, Bernards R, Agami R (2002) Stable sup-
pression of tumorigenicity by virus-mediated RNA inter-
ference. Cancer Cell 2(3):243–247
Campisi J (2005) Suppressing cancer: the importance of being
senescent. Science 309(5736):886–887
Cheng WH, Muftuoglu M, Bohr VA (2007) Werner syndrome
protein: functions in the response to DNA damage and
replication stress in S-phase. Exp Gerontol 42(9):871–878
Christmann M, Tomicic MT, Gestrich C, Roos WP, Bohr VA,
Kaina B (2008) WRN protects against topo I but not topo
II inhibitors by preventing DNA break formation. DNA
Repair (Amst) 7(12):1999–2009
60 Biogerontology (2012) 13:49–62
123

Citti L, Rainaldi G (2005) Synthetic hammerhead ribozymes as
therapeutic tools to control disease genes. Curr Gene Ther
5(1):11–24
Citti L, Eckstein F, Capecchi B, Mariani L, Nevischi S, Poggi
A, Rainaldi G (1999) Transient transfection of a synthetic
hammerhead ribozyme targeted against human MGMT
gene to cells in culture potentiates the genotoxicity of the
alkylation damage induced by mitozolomide. Antisense
Nucleic Acid Drug Dev 9(2):125–133
Clingen PH, Lowe JE, Green MHL (2000) Measurement of
DNA damage and repair capacity as a function of age
using the Comet assay. In: Barnett YA, Barnett CR (eds)
Methods in molecular medicine, vol 38: ageing methods
and protocols. Human Press, Totowa, NJ, pp 143–157
Cox LS, Faragher RG (2007) From old organisms to new
molecules: integrative biology and therapeutic targets in
accelerated human ageing. Cell Mol Life Sci 64(19–20):
2620–2641
Franchitto A, Pichierri P (2004) Werner syndrome protein and
the MRE11 complex are involved in a common pathway
of replication fork recovery. Cell Cycle 3(10):1331–1339
Futami K, Takagi M, Shimamoto A, Sugimoto M, Furuichi Y
(2007) Increased chemotherapeutic activity of campto-
thecin in cancer cells by siRNA-induced silencing of
WRN helicase. Biol Pharm Bull 30(10):1958–1961
Futami K, Ishikawa Y, Goto M, Furuichi Y, Sugimoto M
(2008) Role of Werner syndrome gene product helicase in
carcinogenesis and in resistance to genotoxins by cancer
cells. Cancer Sci 99(5):843–848
Goto M, Miller RW, Ishikawa Y, Sugano H (1996) Excess of
rare cancers in Werner syndrome (adult progeria). Cancer
Epidemiol Biomarkers Prev 5(4):239–246
Gray MD, Shen JC, Kamath-Loeb AS, Blank A, Sopher BL,
Martin GM, Oshima J, Loeb LA (1997) The Werner
syndrome protein is a DNA helicase. Nat Genet 17(1):
100–103
Hemann MT, Fridman JS, Zilfou JT, Hernando E, Paddison PJ,
Cordon-Cardo C, Hannon GJ, Lowe SW (2003) An epi-
allelic series of p53 hypomorphs created by stable RNAi
produces distinct tumor phenotypes in vivo. Nat Genet
33(3):396–400
Huang S, Li B, Gray MD, Oshima J, Mian IS, Campisi J (1998)
The premature ageing syndrome protein, WRN, is a
30 ? 50 exonuclease. Nat Genet 20(2):114–116
Huschtscha LI, Thompson KV, Holliday R (1986) The sus-
ceptibility of Werner’s syndrome and other human skin
fibroblasts to SV40-induced transformation and immor-
talization. Proc R Soc Lond B Biol Sci 229(1254):1–12
Karmakar P, Bohr VA (2005) Cellular dynamics and modu-
lation of WRN protein is DNA damage specific. Mech
Ageing Dev 126(11):1146–1158
Kipling D, Davis T, Ostler EL, Faragher RG (2004) What can
progeroid syndromes tell us about human aging? Science
305(5689):1426–1431
Kudlow BA, Kennedy BK, Monnat RJ Jr (2007) Werner and
Hutchinson-Gilford progeria syndromes: mechanistic
basis of human progeroid diseases. Nat Rev Mol Cell Biol
8(5):394–404
Lebel M, Leder P (1998) A deletion within the murine Werner
syndrome helicase induces sensitivity to inhibitors of
topoisomerase and loss of cellular replicative capacity.
Proc Natl Acad Sci USA 95:13097–13102
Lowe J, Sheerin A, Jennert-Burston K, Burton D, Ostler EL,
Bird J, Green MH, Faragher RG (2004) Camptothecin
sensitivity in Werner syndrome fibroblasts as assessed by
the COMET technique. Ann N Y Acad Sci 1019:256–259
Machwe A, Xiao L, Lloyd RG, Bolt E, Orren DK (2007)
Replication fork regression in vitro by the Werner syn-
drome protein (WRN): Holliday junction formation, the
effect of leading arm structure and a potential role for
WRN exonuclease activity. Nucleic Acids Res
35(17):5729–5747
Montgomery RA, Dietz HC (1997) Inhibition of fibrillin 1
expression using U1 snRNA as a vehicle for the presen-
tation of antisense targeting sequence. Hum Mol Genet
6(4):519–525
Ogburn CE, Oshima J, Poot M, Chen R, Hunt KE, Gollahon
KA, Rabinovitch PS, Martin GM (1997) An apoptosis-
inducing genotoxin differentiates heterozygotic carriers
for Werner helicase mutations from wild-type and
homozygous mutants. Hum Genet 101(2):121–125
Okada M, Goto M, Furuichi Y, Sugimoto M (1998) Differen-
tial effects of cytotoxic drugs on mortal and immortalized
B-lymphoblastoid cell lines from normal and Werner’s
syndrome patients. Biol Pharm Bull 21(3):235–239
Opresko PL, Calvo JP, von Kobbe C (2007) Role for the
Werner syndrome protein in the promotion of tumor cell
growth. Mech Ageing Dev 128(7–8):423–436
Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS
(2002) Short hairpin RNAs (shRNAs) induce sequence-
specific silencing in mammalian cells. Genes Dev
16(8):948–958
Pichierri P, Franchitto A, Mosesso P, Palitti F (2000) Werner’s
syndrome cell lines are hypersensitive to camptothecin-
induced chromosomal damage. Mutat Res 456(1–2):45–57
Poot M, Gollahon KA, Rabinovitch PS (1999) Werner syn-
drome lymphoblastoid cells are sensitive to camptothecin-
induced apoptosis in S-phase. Hum-Genet 104(1):10–14
Rodriguez-Lopez AM, Jackson DA, Iborra F, Cox LS (2002)
Asymmetry of DNA replication fork progression in
Werner’s syndrome. Aging Cell 1(1):30–39
Rodriguez-Lopez AM, Whitby MC, Borer CM, Bachler MA,
Cox LS (2007) Correction of proliferation and drug sen-
sitivity defects in the progeroid Werner’s Syndrome by
Holliday junction resolution. Rejuvenation Res
10(1):27–40
Rossi ML, Ghosh AK, Bohr VA (2010) Roles of Werner
syndrome protein in protection of genome integrity. DNA
repair 9(3):331–344
Shen JC, Gray MD, Oshima J, Kamath Loeb AS, Fry M, Loeb
LA (1998) Werner syndrome protein. I. DNA helicase and
DNA exonuclease reside on the same polypeptide. J Biol
Chem 273(51):34139–34144
Sidorova JM, Li N, Folch A, Monnat RJ, Jr (2008) The RecQ
helicase WRN is required for normal replication fork
progression after DNA damage or replication fork arrest.
Cell Cycle 7(6):796–807
Sledz CA, Williams BR (2004) RNA interference and double-
stranded-RNA-activated pathways. Biochem Soc Trans
32(Pt 6):952–956
Biogerontology (2012) 13:49–62 61
123

Suzuki N, Shimamoto A, Imamura O, Kuromitsu J, Kitao S,
Goto M, Furuichi Y (1997) DNA helicase activity in
Werner’s syndrome gene product synthesized in a bacu-
lovirus system. Nucleic Acids Res 25(15):2973–2978
Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R,
Matthews S, Nakura J, Miki T, Ouais S, Martin GM,
Mulligan J, Schellenberg GD (1996) Positional cloning of
the Werner’s syndrome gene. Science 272(5259):258–262
62 Biogerontology (2012) 13:49–62
123


















