
Heterochrony and developmental modularity of cranial osteogenesis in lipotyphlan mammals
Upload
khangminh22Category
view
0download
0
Istituto di Genetica Medica e di Gemellologia « Gregorio Mendel» - Roma Diret tore: Prof. L. Gedda
RARA OSSERVAZIONE DI "OSTEOGENESIS IMPERFECTA" RIGUARDANTE
LA MADRE E DUE FIGLIE GEMELLE MZ CONCORDANTI
di
Luigi Gedda e Raffaele Gentile
i . Introduzione
II valore del metodo gemellare nello studio della genetica umana consiste, fra l'altro, nel confermare la natura ereditaria di determinate malattie che tali appaiono attraverso I'indagine statistico-genealogica e nel consentire uno studio particolareggiato intorno ai meccanismi fenogenetici attraverso i quali la tara genotipica si manifesta. Infatti il criterio della concordanza intrageminale che, isolatamente considerato, non e sufficiente a stabilire l'origine genotipica di una particolare sindrome, affiancato a diligenti analisi, come le accennate, acquista un valore paragonabile a quello della prova del nove nel calcolo matematico, e tanto piu nello studio della genetica umana che non puo giovarsi dell'esperimento per le sue dimostrazioni.
Ora e certamente curioso il fatto che dagli studi ormai vasti che riguardano 1'osteoge-nesis imperfecta e che negli ultimi anni hanno portato una maggiore luce intorno a questa malattia, non siano emerse che poche e sommarie osservazioni gemellari.
Percio riteniamo che la coppia gemellare monozigotica affetta da osteogenesis imperfecta tarda e concordante che ci apprestiamo a descrivere non rappresenti soltanto un ulteriore contributo casistico alia conoscenza della malattia, ma un apporto significative, tanto piu che la madre delle gemelle si presenta anch'essa affetta da osteogenesis imperfecta come le figlie.
2. Bibliografia
L'osteogenesis imperfecta, fra i capitoli delle osteopatie rare, e forse quello che piu ha beneficiato di un approfondimento nosologico sulla base di una vasta casistica da lungo tempo raccolta.
Nel 1893, S. Miiller, a proposito del cosiddetto « rachitismo fetale », denominazione che comprendeva a quei tempi anche le forme osteopsatirotiche, osservava che fino a trent'anni prima erano scarse le ricerche riguardanti la malattia, ma soggiungeva che « da allora fino ad oggi, il problema del rachitismo fetale o congenito e stato l'oggetto di un numero straordinariamente grande di ricerche ».
Sono passati altri sessant'anni ed i lavori sono talmente aumentati in numero e qua-lita che ormai sarebbe necessario un libro, per riferirne convenientemente.
333
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiae
Per dare un'idea dell'apporto casistico alio studio dell'osteogenesis imperfecta basti l'accenno ad alcuni piu recenti lavori come quello di Fuss (1935) riguardante 89 fami-glie per un complesso di 515 ammalati, di Carriere, Delannoy e Huriez (1937) riguardante 5 famiglie con 34 ammalati, di Seedorff (1949) riguardante 55 famiglie danesi che comprendono 180 membri affetti, di Scott e Stiris (1953) riguardante 3 famiglie con 19 casi di osteogenesis imperfecta.
Fra i lavori italiani, piu cospicui, ricorderemo quello di Mastromarino (1938) che riferisce intorno ai 7 casi occorsi all'Istituto Rachitici di Milano e il contributo di Casuccio (1949) che riguarda la casistica dell'Istituto Rizzoli di Bologna, cioe 20 casi.
Pertanto il nostro compito dovra essere quello di scegliere, fra i molti possibili, il profilo genetico della malattia, trattandone da questo particolare punto di vista e, suc-cessivamente, quello di analizzare la casistica gemellare finora conosciuta.
Per l'osteogenesis imperfecta il punto di vista genetico ha un'importanza veramente esemplare non solo perche rappresenta il criterio eziologico essenziale trattandosi di una malattia ereditaria, ma anche perche, sulla base di questo criterio, e stato possibile conferire unita a svariate sindromi che si affacciavano (ed ancora vengono afFacciate) come indipendenti. Tale servizio reso alia nosografia e alia tassonomia nosologica, riguarda, per un lato, la reductio ad unitatem della osteogenesis imperfecta letale tipo Vrolik, della osteopsatirosis idiopatica tipo Lobstein, e della fragilitas ossium hereditaria, per l'altro il riassorbimento nella sindrome delle forme di transizione e delle microforme abortive e frustanee.
Per quanto anche oggi non manchino degli AA. preoccupati di tener distinta la malattia di Vrolik dalla malattia di Lobstein, pure la tendenza prevalente appare quella afFacciata precchio tempo fa da Looser (1905) ed oggi fortemente sostenuta dalla scuola svedese, essere l'osteogenesis imperfecta una malattia complessa che raggruppa tipi, sindromi e forme svariate e individuabili, ma sostanzialmente unitaria e ricostrui-bile tenendo conto, soprattutto, di possibili meccanismi genetici.
In realta la concezione unitaria della malattia che fece estendere anche all'osteopsa-tirosi il nome di osteogenesis imperfecta primitivamente riservata alia malattia di Vrolik, trasse la sua prima origine dalla constatazione del reperto osteoclasico che si ripeteva in modo analogo nei due casi, e dalla considerazione che quando la malattia insorge precocemente e con caratteri subletali cosi gravi da non concedere la vita e tanto meno la riproduzione, puo apparire sporadica, non ereditaria e, in complesso, assai differente da altra sindrome chiaramente ereditaria, non mortale, la quale si sviluppa fra la prima infanzia e i vent'anni, attraverso eta che impostano dei quadri altamente caratteristici come e la malattia di Lobstein. In sostanza, dunque, una differenza nei tempo d'insor-genza la quale determina due quadri apparentemente diversi.
Ma fu soprattutto la genetica a suggellare l'ipotesi delPunita nosologica attraverso alcune constatazioni. In primo luogo cadde l'opinione sostenuta ancora poco tempo fa dalla Vogelin (1943) che il tipo Vrolik non fosse ereditario. L'ereditarieta della forma congenita risulto da un attento esame degli alberi genealogici, a cominciare dal-l'osservazione di Glanzmann (1936) riguardante due fratelli colpiti nello stesso modo.
334
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
In secondo luogo emerse la constatazione della concordanza della malattia di Vrolik in coppie gemellari MZ che ne conferma l'eziologia ereditaria.
In terzo luogo apparve la possibilita di ricondurre alia sindrome osteopsatirotica, e di qui al piu vasto quadro dell'osteogenesis imperfecta, sulla base dei concetti di penetranza e di espressivita, determinate espressioni fenotipiche, come il sintomo sclerotiche blu, oppure la sindrome di Eddowes (sclerotiche + fragilita ossea), oppure la sindrome di van der Hoeve (sclerotiche + fragilita ossea + otosclerosi), come anche altre forme abortive o di transizione che sembrano collegare la sindrome di Vrolik a quella di Lobstein.
Infine e sembrato molto eloquente, per quanto raro, il riscontro della forma congenita e della forma tarda entrambe presenti in un medesimo albero genealogico.
Percio il criterio genetico congiunse cio che il criterio clinico aveva disgiunto e su questa base viene oggi sostenuta l'unita dell'osteogenesis imperfecta.
Per altro bisogna convenire che si tratta di un'unita molteplice, secondo il concetto latino delPunitas multiplex, vasto grembo nel quale e possibile distinguere fenotipizza-zioni diverse e numerose, secondo l'insorgenza, la forma clinica, l'oscillazione della manifestazione, ecc.
La classificazione interna dell'osteogenesis imperfecta piu accreditata e giustificata ci sembra quella di Seedorff e crediamo opportuno di accennarla anche perche ci atteniamo ad essa nella nostra esposizione.
Tipo I. Osteogenesis imperfecta congenita. Le fratture si verificano neWhabitat en-douterino, sono numerose e presenti alia nascita. La forma e letale e corrisponde per ogni altro aspetto alia sindrome di Vrolik.
Tipo II. Osteogenesis imperfecta tarda. Nella quale la prima frattura si verifica o durante il parto, o durante l'allattamento. Rapporto incongruo per eccesso fra agente ed evento traumatico. Anche 30-40 fratture prima della puberta in successione rapida e con guarigione difettosa cosi da determinare deformita a volte mostruose; vita per lo piu in asili per malati cronici. Questo tipo corrisponde, piu ancora del seguente, alia sindrome di Lobstein.
Tipo III. Osteogenesis imperfecta tarda levis. Caratterizzata dalla predisposizione alle fratture fra i 2-3 anni e i 15-25 anni. Numero delle fratture inferiore a quello del tipo II (massimo 10-12) e per motivi traumatizzanti sempre lievi ma meno futili che nel tipo II. Questi aa. non vengono appartati dalla vita sociale e la crisi effettiva della loro esistenza incomincia quando si presentano i sintomi secondari (sordita, osteomalacia sintomatica, diminuzione del peso e della statura [nanizzazione], dolori reumatici, insuf-ficienza cardiaca).
Affermata in questo modo la base unitaria e la fenotipizzazione molteplice dell'osteogenesis imperfecta, l'indagine genetica logicamente imposta i problemi relativi all'origine mutativa, al meccanismo ereditario e alia fenogenesi della malattia.
A proposito della mutazione che provoca l'inizio della malattia, ipotizzata, prima di ogni altro, da K. H. Bauer (1920), Seedorff ritiene che essa risponda ad una complessa
335
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiae
mutazione privativa e che abbia luogo nell'uno o nell'altro cromosoma di una coppia di autosomi.
Per quanto riguarda la forma congenita la mutazione sviluppa un fattore dominante che, anche in dose singola (eterozigotismo), ha un effetto subletale.
La mutazione che da origine alia forma tardiva di tipo 11 e meno grave di quella che origina il tipo I, ma gli individui che ne risultano si trovano raramente nella condizione di aver figli per cui questi casi appaiono solitari, « creando cosi I'impressione dell'esistenza di una forma non ereditaria della malattia». Quando il grado della mutazione e relativamente ancor meno grave ne risulta un individuo affetto dalla forma tardiva di tipo III. La frequenza della mutazione non e ancora precisabile.
Circa il meccanismo ereditario Bell (1928), riservatamente al sintomo della sclerotiche blu, ritiene trattarsi di una dominanza completa. Nel 1931 Holcomb descriveva una famiglia con 9 casi di osteopsatirosi in 5 generazioni; il reperto deponeva per la dominanza con qualche oscillazione, Pero Hanhart (1951) e Cocchi (1952) sostengono che Posteopsatirosi possa presentare tanto la trasmissione dominante quanto quella recessiva. D'altra parte tanto Key (1926) quanto Fuss (1935) sembrano considerare delle forme ereditarie e delle forme non ereditarie.
SeedorfF (1949) considera l'osteogenesis imperfecta come malattia ereditaria presen-tante una completa dominanza non legata al sesso (autosomatica). A sostegno del suo pensiero l'A., in un gruppo di 19 famiglie con presenza dell'osteogenesis imperfecta tarda in due o piu generazioni consecutive, calcola il rapporto tra fratelli tarati (117) e fratelli normali (110) e rileva che corrisponde assai bene al rapporto mendeliano (50% tarati: 50% normali) che si deve prevedere quando una malattia e di origine autosomatica, l'eredita dominante e l'uno o l'altro genitore eterozigote. Inoltre l'A. mette in rilievo un caso nel quale il padre tarato sposa successivamente due mogli avendo figli da cia-scuna di esse; le due fratellanze contengono tanto dei fratelli tarati, quanto dei fratelli normali.
Per quanto riguarda la penetranza essa e alta, ma inferiore al 100% (Holcomb, Hanhart, Cocchi). Dal punto di vista dei meccanismi fenogenetici a noi sembra che si debba tener presente l'ipotesi affacciata da K. H. Bauer il quale avanzo la teoria che l'osteogenesis imperfecta corrisponda ad un'inferiorita mesenchimale in quanto vengono colpiti svariati tessuti che prendono origine da questo foglietto embrionale (ossa, sclerotiche, dentina, tessuti vascolari).
II concetto di Bauer pud oggi essere ripreso e riconsiderato anche in base alle recenti osservazioni di Scott e Stiris i quali al ben noto quadro dell'osteogenesis imperfecta hanno aggiunto il sintomo delle cicatrici ipertrofiche dimostrando statistica-mente che il riscontro e significative Anche il tessuto cicatriziale in quanto derivato dal derma e di origine mesenchimale e percio a noi sembra che l'osteogenesis imperfecta possa veramente essere considerata come dovuta ad una compromissione del mesenchima per effetto dei fattori ereditari responsabili della malattia.
Con cio non e spiegato a sufficienza il meccanismo fenogenetico, ma sembra possi-bile individuare nella mesenchimosi un importante anello della catena che congiunge il genotipo morboso al fenotipo morboso.
336
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
Come e noto, per la conferma dell'ereditarieta di una malattia e per un approfondi-mento dei suoi caratteri, dei fattori che li condizionano, del meccanismo ereditario e del meccanismo fenogenetico, l'indagine gemellare riveste un'importanza di primo piano.
Ora e curioso osservare come nell'abbondante materiale finora raccolto riguardante le varie forme dell'osteogenesis imperfecta i casi gemellari siano molto rari, e cosi poco noti che talora financo si pensa che non esistano. In efFetti si tratta di pochi casi di cui alcuni molto antichi e spesso descritti in modo cosi succinto da rendere difficile l'uti-lizzazione. Percio riteniamo utile di passare in rassegna i casi di osteogenesis imperfecta in gemelli che abbiamo potuto riscontrare, esponendoli in ordine di data:
OSSERVAZIONE DI SIGFRID MULLER (1893)
Si tratta di un'a. di nome Anna nata per prima da parto gemellare che diede esito anche ad un maschio, il 17 gennaio 1892. II parto ebbe luogo spontaneamente cinque settimane prima della fine della gravidanza; entrambi i neonati furono partoriti in posizione cefalica. Anna e la settima figlia dei suoi genitori; il primo figlio mori all'eta di un anno; il secondo fu un aborto in 4° mese; il terzo partorito in 6° mese mori poco dopo la nascita; il quarto di quattro anni presenta scrofolosi; il quinto di tre anni e il sesto di un anno e mezzo sono sani. Anche il cogemello e sano e di costituzione rego-lare. Padre e madre sani, non luetici.
Cranio piccolo e molle; con suture abnormemente aperte e prolungate. Volto di aspetto senile. Volume della lingua aumentato.
Anna venne al mondo con gli arti storti quali furono descritti in occasione dell'e-same obiettivo.
Crepitio al terzo superiore del braccio destro. Avambraccio D fortemente curvo. Voluminoso callo a meta diafisi delPomero S. Ambo i femori e le ossa delle gambe fortemente incurvati.
Anna muore in quinta settimana e il reperto anatomo-patologico dimostra la presenza di bronchite capillare con focolai bronco-pneumonici ai lobi inferiori dei due polmoni. Le ossa vengono sottoposte a diligente esame istologico che conferma il reperto obiettivo.
Diagnosi clinica: osteogenesis imperfecta congenita. Diagnosi gemellologica: gemellanza dizigotica discordante.
OSSERVAZIONE DI JULIUS H. HESS (1917)
In una casistica che annovera complessivamente quattro ammalati di osteogenesis imperfecta, Julius H. Hess riferisce intorno ad una bambina nata da parto gemellare spontaneo e ricoverata il 10 maggio 1914, all'eta di 16 mesi.
Anamnesi famigliare negativa. Allattata al seno fino a 3 mesi. Gastroenterite. A 9 mesi si reggeva seduta, ma non parlava e non camminava. A 14 mesi la madre noto l'instaurarsi di una malformazione a carico del rachide e poi spiccato deperimento e forte dolenzia alia palpazione degli arti.
Caput quadratum e craniotabe. Flessione completa degli arti inferiori. Scoliosi dorsale. Epifisi ingrandite.
337
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiae
Ai raggi notavansi molteplici fratture delle ossa lunghe (radio D, ulna D, radio S, ulna S, femore D, perone D, tibia S., perone S) con notevole formazione di calli.
Le ossa mostravano scarsa tendenza a piegarsi prima delle fratture. « La linea epi-fisaria era alquanto irregolare, segno di mutamenti rachitici».
Dopo cinque mesi, quando la paziente fu dimessa dall'ospedale, le fratture dimo-stravano uno spiccato miglioramento.
L'A. non fa nessuna menzione dell'altro membro della coppia gemellare. Diagnosi clinica: osteogenesis imperfecta tarda associata a segni di rachitismo. Diagnosi gemellologica: gemellanza a zigotismo ignoto, probabilmente discordante.
OSSERVAZIONE DI W. E. WELZE E B. L. LIEBERMANN (1927)
Due gemelle di razza negra (N° 1 e N° 2), presentate dagli AA. come uniovulari, nacquero il 13 novembre 1926 da madre che ebbe in antecedenza 4 gravidanze normali.
Negativo il gentilizio. Presentazione di testa; parto spontaneo. Poche ore dopo la nascita furono ricoverate nelPHerman Kiefer Hospital di Detroit perche presentavano numerose fratture degli arti e delle coste.
N° 1 mori dopo nove ore, N° 2 dopo quaranta ore. AlPesame radiografico quasi tutte le ossa degli arti presentano delle fratture in vario
grado di guarigione, cosi pure le coste dei due lati. Marcata deformazione dei femori dovuta alia guarigione in posizione viziata. II rachide appare indenne. Le clavicole sono fratturate in N° 1, indenni in N° 2. Le epifisi della tibia mostrano bilateralmente una linea orizzontale e densa di tessuto osseo.
AlPautopsia: N° 1 pesava gr. 1750, il suo timo gr. 11.85 (rapporto timo-corpo = 1 : 157), la sua tiroide gr. 4,85 (rapporto tiroide-corpo = 1 : 380); N° 2 pesava gr. 1900, il suo timo gr. 12 (rapporto timo-corpo = 1: 158), la sua tiroide gr. 5 (rapporto tiroide-corpo = 1 : 360).
La distribuzione, il tipo e il numero delle alterazioni scheletriche era essenzialmente lo stesso nei due feti.
Diagnosi clinica: 2 casi di ostegenesis imperfecta congenita. Diagnosi gemellologica: gemellanza monozigotica concordante.
OSSERVAZIONE DI J. N E W T O N SISK (1931)
Si tratta di due gemelle di 4 anni. La madre ha circa 40 anni ed e normale. 11 padre presenta sclerotiche bluastre, arcus senilis e otosclerosi progressiva bilaterale; ricorda che ebbe fratturata una gamba per un'evento traumatico di lieve entita. 1 genitori ebbero altri tre figli normali.
AU'esame obiettivo lo sviluppo generale delle gemelle corrisponde all'eta, all'infuori della statura modificata dalle fratture dei femori. Riescono a camminare con successo senza tuttavia impegnare le articolazioni dell'anca.
In entrambe arcus senilis e sclerotiche blu. AU'esame radiologico notansi in una gemella vecchie fratture al terzo superiore dei
femori, con mancato saldamento e notevole deformita. L'omero S e fratturato a meta
338
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
diafisi e saldato ad angolo re t to . L'ottava costa S e fratturata con mancato saldamento. Nella cogemella analoghe fratture dei femori, appaiono anche fratturate la settima,
ottava e decima costa S e l 'ottava D , tut te saldate. Queste fratture furono p rodo t t e separatamente d o p o la nascita, prima dei 2 anni di
eta e risultarono da t raumi cosi banali che « furono osservate solo qualche giorno dopo che si verificarono quando si comincio a notare la deformita ».
Diagnosi clinica: 2 casi di osteogenesis imperfecta tarda. Diagnosi gemellologica: gemellanza monozigotica concordante.
O S S E R V A Z I O N E D I N I L S F A X E N (1932)
Si tratta di una coppia di gemelle svedesi ricoverate nella Clinica Medica dell 'Ospe-dale Centrale di Vasteras e brevemente descritte da l l 'A . in una comunicazione alia Sezione Pediatrica di Stoccolma (17 aprile 1931).
Nate a termine, pesavano rispettivamente gr. 2.000 e gr. 2.400; furono nutr i te al seno fino a 5 mesi circa. II p r imo dente comparve all 'anno. Ammesse nella Clinica a 1 anno e 9 mesi pesavano gr. 8.580 e gr. 8.300 rispettivamente, erano pallide e avevano muscoli flaccidi tantoche non potevano mantenere la posizione eretta senza aiuto. Men-talmente ritardate, non parlavano ancora.
Leggero grado di anemia con valori ematologici identici. La circonferenza del cranio era di cm. 50 e cm. 49 rispettivamente, con sviluppo
sopranormale dei parietali; le fontanelle anteriori risultavano aperte. Le epifisi non ingrossate; gli arti incurvati e accorciati in piu luoghi. U n a gemella aveva 4 denti (2 dei quali comparsi recentemente) e l'altra solo 2.
Ai raggi X si rilevano notevoli trasformazioni scheletriche con diminuita compat-tezza della strut tura ossea e assottigliamento del cortex. Numerose fratture a differenti gradi di guarigione nelle ossa lunghe. Le ossa fratturate appaiono piu larghe, a struttura piu densa e accorciate.
Diagnosi clinica: 2 casi di osteogenesis imperfecta tarda. Diagnosi gemellologica: gemellanza monozigotica concordante.
O S S E R V A Z I O N E D I F . K N U T S S O N (1938)
Nel corso di una discussione svoltasi presso la Svenska Foren . Med. Radiol . Forhdl . in seguito a relazione di G. Zander sopra un caso di osteogenesis imperfecta tarda, viene riferito l ' intervento di F . Knutsson il quale durante la sua permanenza nella Clinica ortopedica di Stoccolma po te osservare 3 casi d i osteogenesis imperfecta. D u e di questi casi riguardavano una coppia di gemelli di 9 anni, concordanti .
In questi gemelli n o n vi era platispondilia come nell 'altro caso di monona to di 7 anni. Altrove le ossa presentavano fenomeni distrofici e abnorme fragilita. La pr ima frat-
tura ebbe luogo all 'eta di un anno e riguardava un avambraccio. Diagnosi clinica: 2 casi di osteogenesis imperfecta tarda. Diagnosi gemellologica: gemellanza monozigotica concordante .
339
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiae
O S S E R V A Z I O N E (a) D I K N U D S T A K E M A N N S E E D O R F F (1949)
Seedorff nel I I I Gruppo di osteogenesis imperfecta della sua casistica, che comprende i casi solitari di osteogenesis imperfecta tarda, descrive una famiglia nella quale e pre-sente una gemella MZ seconda nata, affetta da o.i.t. Dal padre e dalla madre che godono buona salute trasse origine, come prima, questa gravidanza gemellare a cui seguirono altri 5 figli monona t i ; l 'ultima e una femmina con spina bifida, mor ta in giovane eta.
La gravidanza gemellare si chiuse a termine il 9 gennaio 1922 con presentazioni di testa; alia nascita non si notavano fratture. Allattata per sei mesi e poi sottoposta a dieta mista l'a. fu sempre piut tosto debole e ritardata nello sviluppo fisico rispetto alia cogemella; questa si sviluppo in donna normale, solida e ben proporzionata . I genitori nota-rono che le estremita inferiori, per quanto l'a. non abbia mai imparato a reggersi sugli arti inferiori, erano diventate sempre piu ricurve col passare del tempo. L'a. ebbe nume-rose fratture manifeste dall 'eta di 2 anni fino ai 13 anni, e specialmente agli arti inferiori.
Si muove su sedia a rotelle non essendosi riscontrata nessuna possibility di corre-zione ortopedica.
Forma caratteristica della testa e della faccia. Buona acutezza di udi to . Sclerotiche blu-grigiastre. Estremita superiori leggermente piegate con mobilita limitata di entrambe le spalle. I gomiti sono deformati con difetti di estensione di circa 60°. Le anche difet-tano di 35° nell 'estensione totale e le ginocchia di 60°. I femori sono convessi lateral-mente e le gambe antero-convesse. I muscoli delle estremita inferiori atrofici, i piedi deformati e flaccidi; ipermobilita delle articolazioni delle mani.
Rapida nell 'apprendere, l'a. fece gli studi in casa e passa il t empo leggendo e in lavori di cuci to; gentile e allegra.
Diagnosi clinica: osteogenesis imperfecta tarda. Diagnosi gemellologica: gemellanza a zigotismo ignoto, discordante.
O S S E R V A Z I O N E (b ) D I K N U D S T A K E M A N N S E E D O R F F (1949)
Seedorff nel V gruppo della sua casistica, che comprende i casi di osteogenesis imperfecta tarda presenti in piu di 2 generazioni della medesima famiglia, descrive una famiglia che presenta o.i.t. in almeno 5 generazioni successive.
In sesta generazione notasi, fra l 'altro, una coppia di gemelli DZ bisesso indenne, e, in linea collaterale, una coppia di gemelli bimaschile discordante rispetto al carattere sclerotiche blu.
Nel ristretto gruppo famigliare di questa coppia gemellare si nota il padre con fre-quenti fratture delle estremita, con sclerotiche blu e sordita (anche la nonna e il bis-n o n n o e il t r i snonno dei gemelli presentavano questo t r ipode s intomatologico); una sorella monona ta piu anziana presenta anch'essa sclerotiche blu.
II gemello tarato, nato il 14 maggio 1943, non presenta fratture, ma cammina male. Caratteristica forma della testa e della faccia. Sclerotiche intensamente blu.
II cogemello e normale. Diagnosi clinica: osteogenesis imperfecta tarda. Diagnosi gemellologica: secondo Seedorff sembra trattarsi di gemellanza monozigotica
discordante; noi consideriamore la gemellanza a zigotismo ignoto, discordante.
340
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta» etc.
Oltre ai casi che abbiamo esposto, Seedorff fa menzione di una coppia gemellare di sesso ignoto e discordante rispetto alia forma congenita che sarebbe stata descritta da O. Schmidt nel 1893. Pero l'A. non fornisce riferimenti bibliografici e le nostre ricerche ci hanno permesso soltanto di ritrovare un Julius Schmidt che nel 1859 pubblicava un lavoro sulla fragilita congenita delle ossa in cui si parla di due neonati che non sono gemelli, ne fratelli e di cui uno soltanto puo essere considerato come affetto di osteogenesis imperfecta congenita. Puo darsi che Seedorff si riferisca ad altro Schmidt ma noi non lo abbiamo trovato. Ancora, da qualche AA. viene fatto cenno di una coppia MZ che Albrecht avrebbe descritto nel 1931, i membri della quale ammalarono con-temporaneamente di osteopsatirosi idiopatica e presentarono lo stesso decorso sebbene fossero vissuti in condizioni climatiche e d'alimentazione del tutto diverse. Ma anche il cosidetto caso di Albrecht sembra dovuto ad un'interpretazione inesatta.
3. Descrizione dei nostri casi
La nostra casistica di osteogenesis imperfecta riguarda un gruppo famigliare com-posto di una donna cinquantenne (Maria) e delle sue due figlie gemelle ventenni (Giovan-na e Ippolita) (cfr. fig. 1) dove tutti e tre i soggetti sono affetti dalla grave e rara osteopatia.
Trattandosi di un gentilizio unico, premettiamo l'analisi dell'albero genealogico (cfr. fig. 2) alio studio obiettivo delle tre ammalate.
REPERTO GENEALOGICO
L'albero genealogico costruito sulla base delle notizie che abbiamo potuto racco-gliere riguarda molto limitatamente gli ascendenti paterni delle nostre gemelle, preva-lentemente invece gli ascendenti ed i collaterali materni. Questo si deve al fatto che l'unione del padre e della madre era illegittima e percio le notizie riguardanti il padre e la di lui famiglia furono raccolte con estrema difftcolta.
Esponiamo ora le notizie che riguardano singoli individui compresi nell'albero genealogico:
I 1, morto apiu di 80 anni; I 2, non raggiunse la vecchiaia; I 3, morto a 88 anni di emorragia cerebrale; I 4, morta a 89 anni di broncopolmonite; II 2, sano; II 3, cieca da un occhio, morta a 64 anni per cancro dello stomaco; II 4, per lue contratta ebbe tre aborti (III 6, 7, 8) ; II 6, con deformita agli arti superiore e inferiore D, malato men-tale, mori a 39 anni; II 10, celibe, fucilato; II 11, malata mentale; II 13, negli ultimi 7 anni di vita immobilizzato da poliartrite cronica primaria anchilosante, morto a 68 anni; II 14, morte accidentale; III 9, deforme, morto a 5 anni; III 10, con un piede difettoso; III 13, normale; dalla nascita, con arti corti, vissuta pochi anni; III 14, osteogenesis imperfecta tarda; III 15, normale; III 17, normale, morto a 18 anni di tifo; III 18, normale, morta a 9 anni di peritonite tbc; III 19, morta a 3 anni per tetania; III 20, morta a 16 anni per cerebropatia; III 22, normale, a. di sifilide e blenorragia; III 24, normale; III 25, normale, morta in tenera eta per ustioni; III 26, normale, morte acci-
341
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiae
dentale in tenera eta; IV 4, mostro; IV 5, con due dita della mano S deformi; IV 6, normale; IV 9 e IV 10, gemelli morti nei primi giorni di vita; IV, 11, normale, morto a 7 anni di pleurite; IV 12, osteogenesis imperfecta tarda; IV 13, osteogenesis imperfecta tarda; IV 15, normale, morto a 4 mesi per tetania; IV 20, nata senza una mano,
Fig. 1. Maria F. e le sue due figlie gemelle Giovanna (a sinistra guardando) e Ippolita
morta a 4 anni; IV 26, nato senza un occhio; IV 32, muta e deforme; IV 33, mostro; IV 36, deforme; IV 37, deforme; IV 40, deforme; IV 41, deforme; V 2, con arti deformi; V 3, normale; V 4, normale; V 5, normale; V 6, normale; V 19, cerebropatico.
Premesso che dei 128 individui compresi nell'albero genealogico noi abbiamo potuto studiarne soltanto 3 (III 14, IV 12 e 13) e che per tutti gli altri le notizie furono desunte da un lungo e ripetuto interrogatorio della madre (III 14) abbiamo pensato di contrassegnare con colore rosso i predetti casi di « ostogenesis imperfecta » obiettivamente ac-certati, di contrassegnare con il colore verde i soggetti che sulla base dell'aramnesi presentano forse la medesima malattia (II 6, III 9, III 13, IV 4, IV 32, IV 33, IV 36, IV 37, IV 40, IV 41) e di contrassegnare con una raggera verde i soggetti che presentano altre anomalie (III 10 piede difettoso, senza una mano; IV 5 due dita della mano S deformi; IV 20 nata senza una mano; IV 26 nata senza un occhio).
342
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta» etc.
•^LS^I^O
P • 1
Figg. 3 e 4. Saggi di calligrafia di Giovanna e Ippolita F.
0
343
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
Acta Geneticae Medicae et Gemellologiae
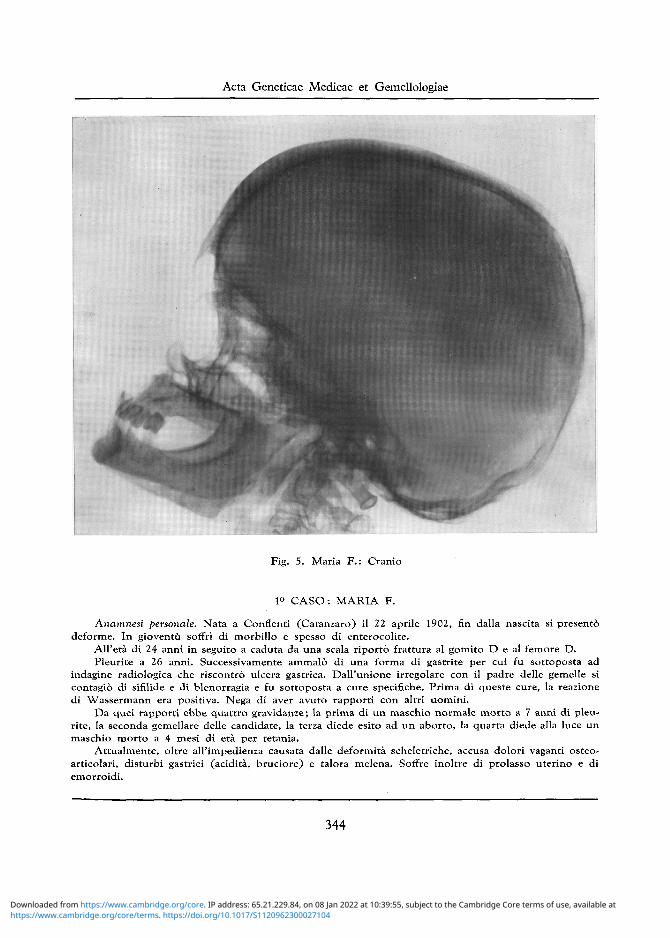
Fig. 5. Maria F . : Cranio
1° C A S O : M A R I A F.
Anamnesi personale. Nata a Conflenti (Catanzaro) il 22 aprile 1902, fin dalla nascita si presento deforme. In gioventu soffri di morbillo e spesso di enterocolite.
All'eta di 24 anni in seguito a caduta da una scala riporto frattura al gomito D e al femore D. Pleurite a 26 anni. Successivamente ammalo di una forma di gastrite per cui fu sottoposta ad
indagine radiologica che riscontro ulcera gastrica. Dall 'unione irregolare con il padre delle gemelle si contagio di sifilide e di blenorragia e fu sottoposta a cure specifiche. Prima di queste cure, la reazione di Wassermann era positiva. Nega di aver avuto rapporti con altri uomini.
Da quei rapporti ebbe quattro gravidanze; la prima di un maschio normale morto a 7 anni di pleurite, la seconda gemellare delle candidate, la terza diede esito ad un aborto, la quarta diede alia luce un maschio morto a 4 mesi di eta per tetania.
Attualmente, oltre all'impedienza causata dalle deformita scheletriche, accusa dolori vaganti osteo-articolari, disturbi gastrici (acidita, bruciore) e talora melena. Soffre inoltre di prolasso uterino e di emorroidi.
344
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
L. Gedda e R. Gentile: Rara osservazione di Osteogenesis imperfecta etc.
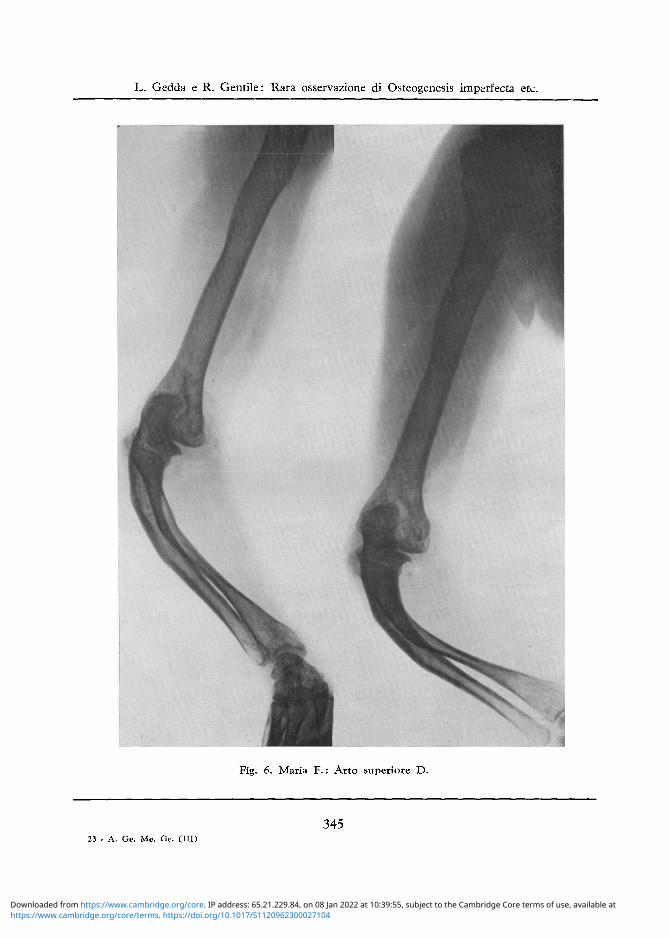
Fig. 6. Maria F . : Arto superiore D.
345 23 - A. Ge. Me. Ge. (I l l )
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae A4edicae et Gemellologiae
Fig. 7. Maria F . : Mano D .
Esame obiettivo. Soggetto di costituzione scheletrica irregolare. Statura cm. 116. Peso Kg. 34 La testa ha un aspetto idrocefaloide con frontea mpia e naso alquanto schiacciato; i denti sono in gran parte mancanti.
II torace e tozzo. Gli arti non hanno sempre le stesse misure nei segmenti omologhi, come appare dalla tabella 1. I piedi sono in posizione equino-vara. L'addome, trattabile, e dolente nelle regioni annessiali. Fegato alquanto aumentato di volume.
Milza palpabile. Aia gastrica ingrandita. La deambulazione e possibile in quanto l'a. con I'aiuto di un doppio sostegno compie dei piccoli
passi. Udi to normale. Sclerotiche di colore grigio-blu. L'intelligenza e normale e sviluppata in modo
proporzionale all'ambiente. Reazione di Wasserman = negativa.
Reperto radiografico :
Cranio: Assenza di alterazioni a carico dei tavolati ossei. Sella turcica con introitus ampio e pavi-mento poco approfondito. Seno sfenoidale piuttosto ampio ed alquanto opacato (cfr. fig. 5).
346
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di «Osteogenesis imperfecta» etc.
Fig. 8. Maria F . : Ar to superiore S.
347
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
Acta Geneticae Medicae et Gemellologiae
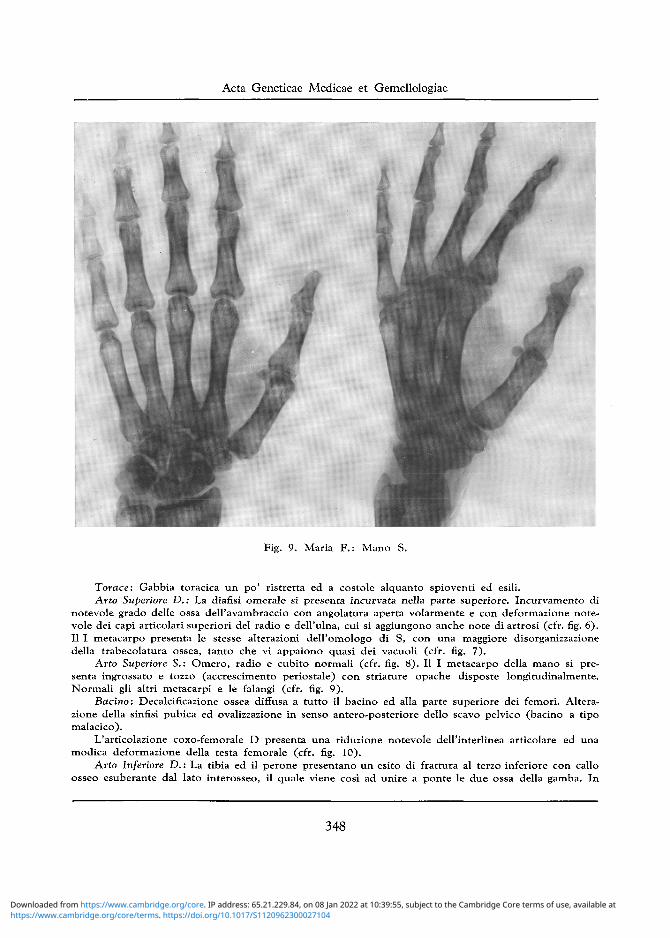
Fig. 9. Maria F . : Mano S.
Torace: Gabbia toracica un po ' ristretta ed a costole alquanto spioventi ed esili. Arto Superiore D.: La diafisi omerale si presenta incurvata nella parte superiore. Incurvamento di
notevole grado delle ossa dell'avambraccio con angolatura aperta volarmente e con deformazione note-vole dei capi articolari superiori del radio e dell'ulna, cui si aggiungono anche note di artrosi (cfr. fig. 6). II I metacarpo presenta le stesse alterazioni dell'omologo di S, con una maggiore disorganizzazione della trabecolatura ossea, tanto che vi appaiono quasi dei vacuoli (cfr. fig. 7).
Arto Superiore S.: Omero, radio e cubito normali (cfr. fig. 8) . II I metacarpo della mano si presenta ingrossato e tozzo (accrescimento periostale) con striature opache disposte longitudinalmente. Normali gli altri metacarpi e le falangi (cfr. fig. 9) .
Bacino: Decalcificazione ossea diffusa a tutto il bacino ed alia parte superiore dei femori. Altera-zione della sinfisi pubica ed ovalizzazione in senso antero-posteriore dello scavo pelvico (bacino a tipo malacico).
L'articolazione coxo-femorale D presenta una riduzione notevole dell'interlinea articolare ed una modica deformazione della testa femorale (cfr. fig. 10).
Arto Inferiore D.: La tibia ed il perone presentano un esito di frattura al terzo inferiore con callo osseo esuberante dal lato interosseo, il quale viene cosi ad unire a ponte le due ossa della gamba. In
348
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
Fig. 10. Maria F . : Bacino
questa regione la tibia presenta anche un'immagine ovalare trasparente contornata da un orlo di tessuto osseo sclerotico da riferire a focolaio osteomielitico spento, insediatosi sul luogo di frattura (cfr. fig. 11).
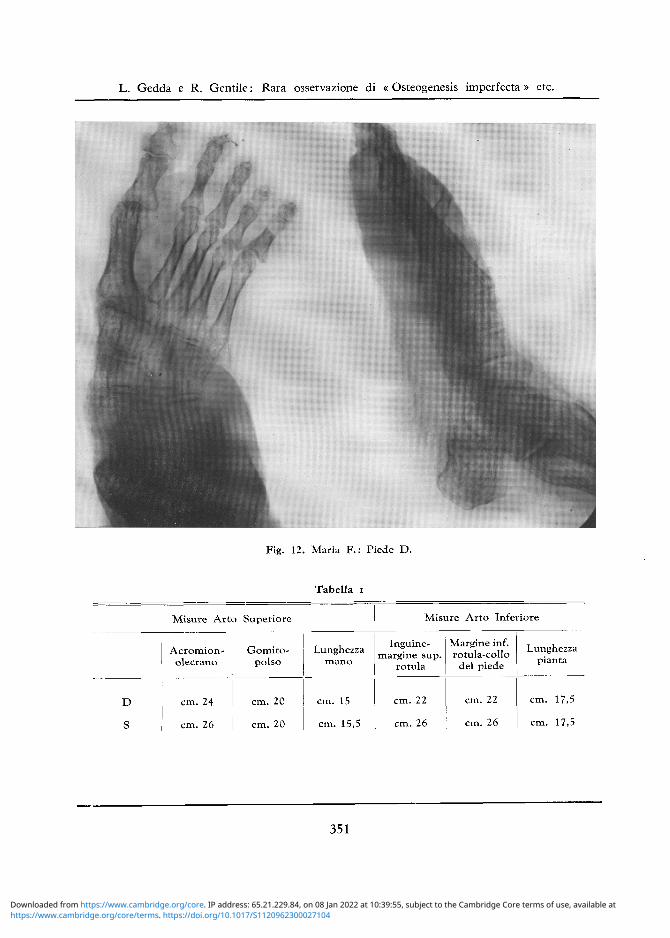
La decalcificazione e meno intensa che nel lato opposto sia a carico dei capi articolari del ginoc-chio che delle ossa del piede (e sempre ben visibile la rarefazione a larghe maglie della spugnosa). (cfr. fig. 12).
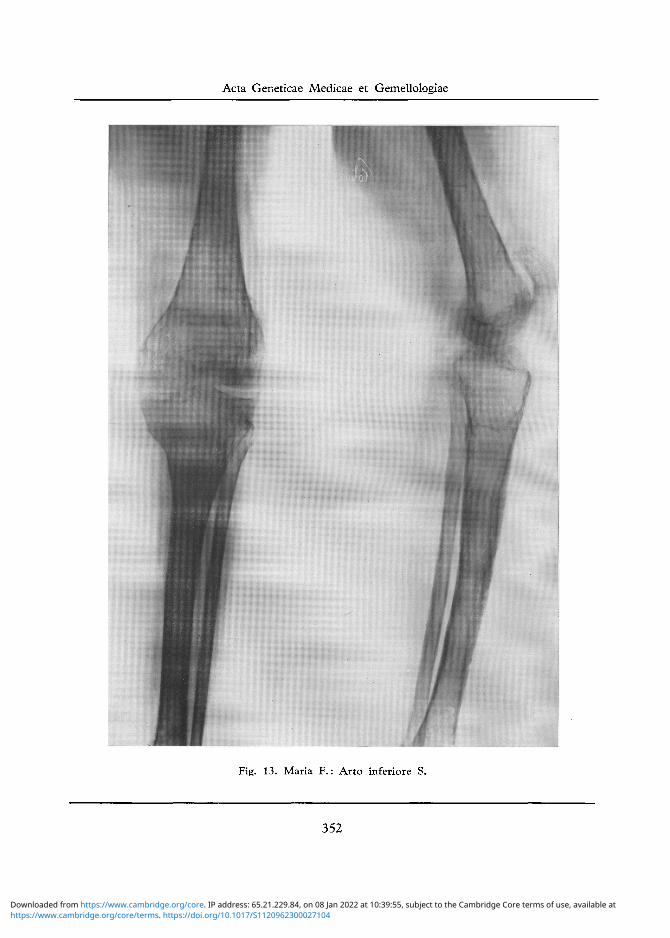
Arto Inferiore S.: Diffusa decalcificazione ossea ben visibile specie a carico dei capi articolari del ginocchio e della cresta della tibia che appare cosparsa di piccole aree di rarefazione ossea (cfr. fig. 13).
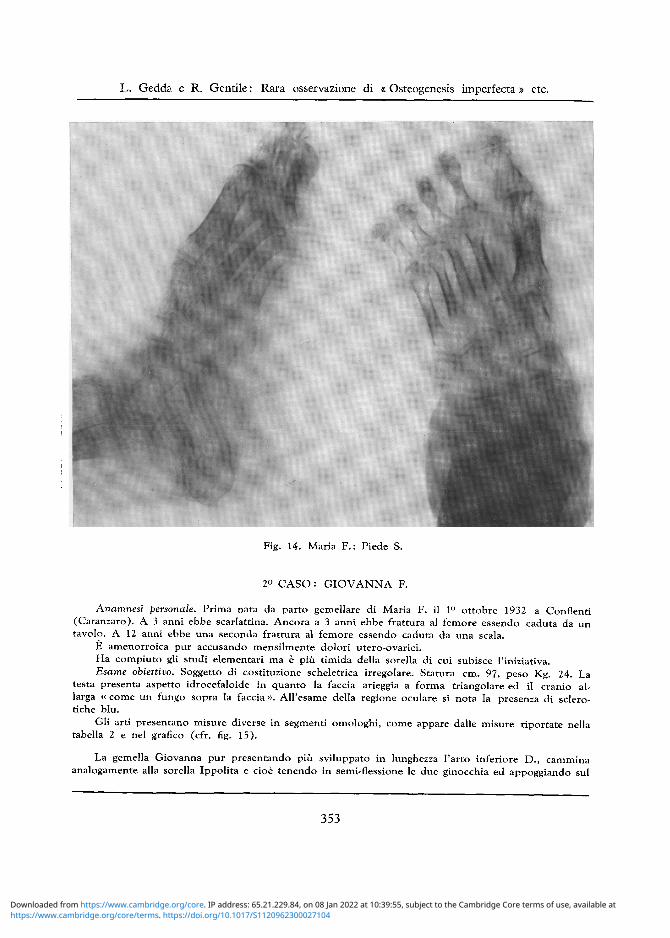
Decalcificazione ossea anche di tutte le ossa del tarso ove e riconoscibile una trama a larghe maglie, mentre le falangi appaiono come vacuolizzate (cfr. fig. 14).
349
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiae
Fig. 11. Maria F . : Arto inferiore D.
350
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
Fig. 12. Maria F.: Piede D.
Tabella i
D
S
Misure Arto Superiore
Acromion-olecrano
cm. 24
cm. 26
Gomito-polso
cm. 20
cm. 20
Lunghezza mano
cm. 15
cm. 15,5
Misure Arto Inferiore
Inguine-margine sup.
rotula
cm. 22
cm. 26
Margine inf. rotula-collo
del piede
cm. 22
cm. 26
Lunghezza pianta
cm. 17,5
cm. 17,5
351
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
Acta Geneticae Medicae et Gemellologiae
Fig. 13. Maria F.: Arto inferiore S.
352
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
L. Gedda e R. Gentile: Rara osservazione di «Osteogenesis imperfecta» etc.
Fig. 14. Maria F . : Piede S.
2° CASO : G I O V A N N A F.
Anamnesi personate. Prima nata da parto gemellare di Maria F. il 1° ottobre 1932 a Conflenti (Catanzaro) A 3 anni ebbe scarlattina. Ancora a 3 anni ebbe frattura al femore essendo caduta da un tavolo. A 12 anni ebbe una seconda frattura al femore essendo caduta da una scala.
E amenorroica pur accusando mensilmente dolori utero-ovarici. Ha compiuto gli studi elementari ma e piu timida della sorella di cui subisce Tiniziativa Esame obiettivo. Soggetto di costituzione scheletrica irregolare. Statura cm. 97, peso Kg 24 La
testa presenta aspetto idrocefaloide in quanto la faccia arieggia a forma triangolare ed il cranio al-larga « come un fungo sopra la faccia ». All'esame della regione oculare si nota la presenza di sclero-tiche blu.
Gli arti presentano misure diverse in segmenti omologhi, come appare dalle misure riportate nella tabella 2 e nel grafico (cfr. fig. 15).
La gemella Giovanna pur presentando piu sviluppato in lunghezza 1'arto inferiore D., cammina analogamente alia sorella Ippolita e cioe tenendo in semi-flessione le due ginocchia ed appoggiando sul
353
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiae
terreno la pianta del piede D e il ginocchio S (cfr. fig. 1); pertanto gamba e piede S vengono trasci-nati durante la deambulazione.
Udito normale; voce bene intonata. Denti normali e sani di colore leggermente giallo. Reazione di Wassermann = negativa.
D. S.
'4
11
24
IPPOLITA
25
<+5
K
11 G/QV4A/AA4
I—1= SG4LJ /:/0
012 34 5 \o 15 2o 25 vm.
Fig. 15.
Reperto Radiografico
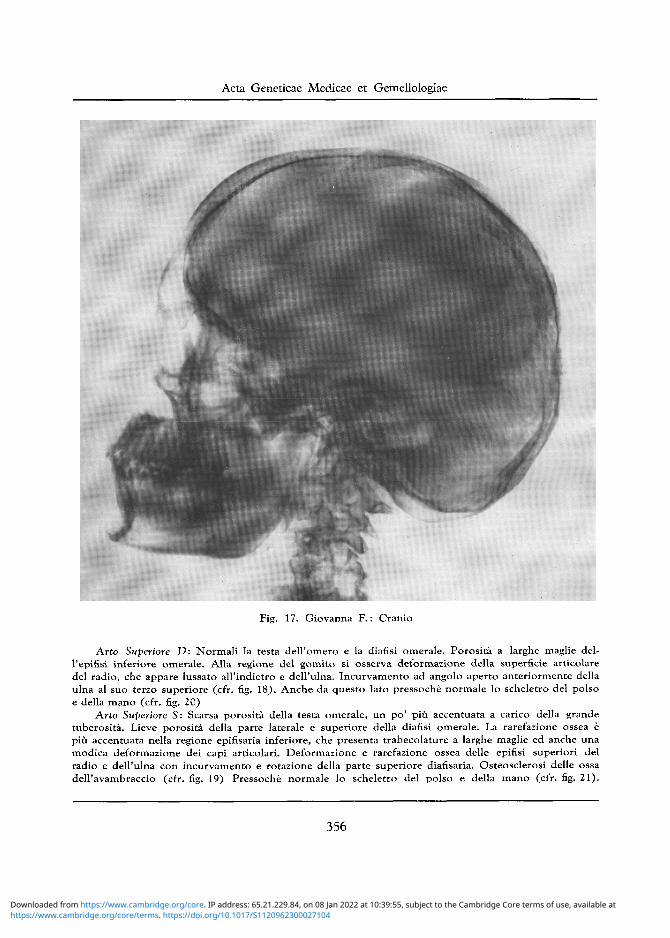
Cranio: Aumento del diametro trasverso bitemporale e profilo «a grondaia» (cfr. figg. 16 e 17). Zone osteoporotiche poco estese e non bene delimitate a carico del tavolato esterno della regione superiore parietooccipitale. Sella turcica svasata, con introitus molto ampio e pavimento pochissimo approfondito. Apofisi clinoidee anteriori appuntite e lamina quadrilatera con lievi irregolarita del dorsum. II seno sfenoidale e situato anteriormente alia sella.
Torace: Gabbia toracica fortemente svasata alia base e con costole molto piu esili che nella cogemella.
354
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta» etc.
*Sfl(-,Wff«H
Fig. 16. Giovanna F.: Cranio
355
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
Acta Geneticae Medicae et Gemellologiae
Fig. 17. Giovanna F . : Cranio
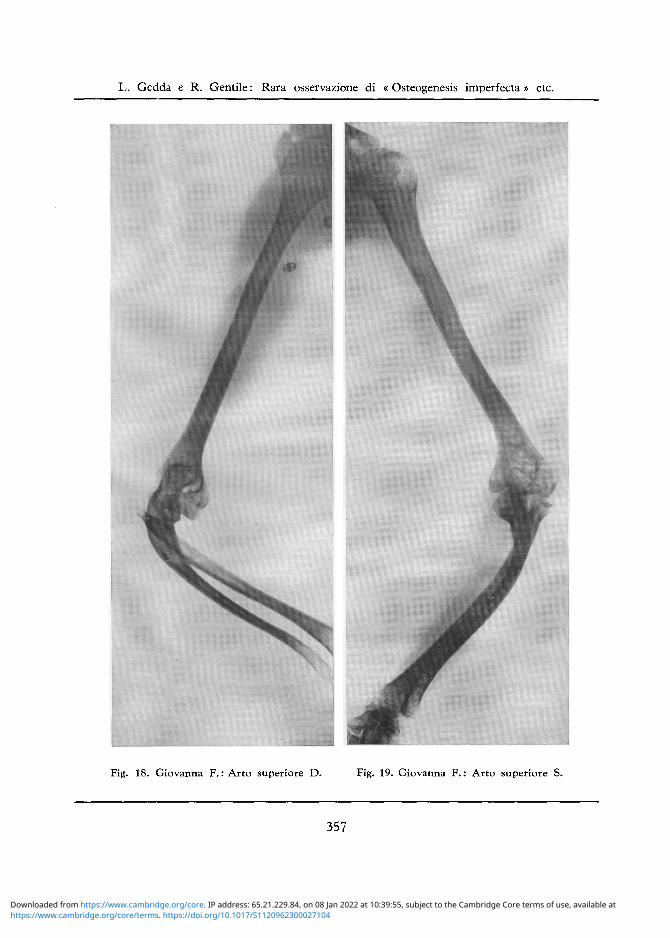
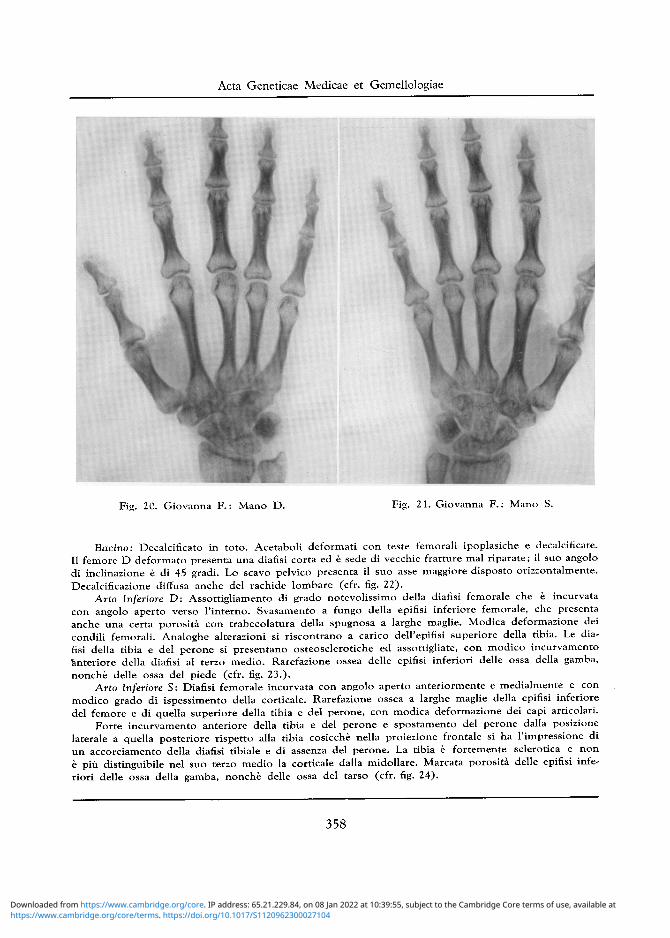
Arto Superiore D: Normali la testa dell 'omero e la diafisi omerale. Porosita a larghe maglie del-l'epifisi inferiore omerale. Alia regione del gomito si osserva deformazione della superficie articolare del radio, che appare lussato all'indietro e dell'ulna. Incurvamento ad angolo aperto anteriormente della ulna al suo terzo superiore (cfr. fig. 18). Anche da questo lato pressoche normale lo scheletro del polso e della mano (cfr. fig. 20)
Arto Superiore S: Scarsa porosita della testa omerale, un po ' piu accentuata a carico della grande tuberosita. Lieve porosita della parte laterale e superiore della diafisi omerale. La rarefazione ossea e piu accentuata nella regione epifisaria inferiore, che presenta trabecolature a larghe maglie ed anche una modica deformazione dei capi articolari. Deformazione e rarefazione ossea delle epifisi superiori del radio e dell'ulna con incurvamento e rotazione della parte superiore diafisaria. Osteosclerosi delle ossa dell'avambraccio (cfr. fig. 19) Pressoche normale lo scheletro del polso e della mano (cfr. fig. 21).
356
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
Fig. 18. Giovanna F.: Arto superiore D. Fig. 19. Giovanna F.: Arto superiore S.
357
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
Acta Geneticae Medicae et Gemellologiae
Fig. 20. Giovanna F . : Mano D. Fig. 21. Giovanna F . : Mano S.
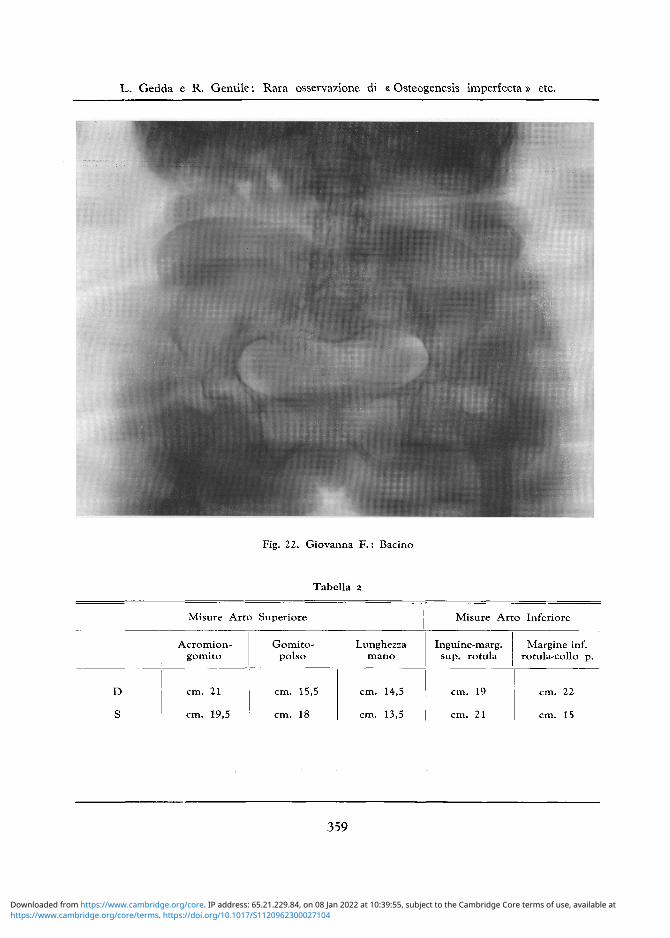
Bacino: Decalcificato in toto. Acetaboli deformati con teste femorali ipoplasiche e decalcificate. II femore D deformato presenta una diafisi corta ed e sede di vecchie fratture mal riparate; il suo angolo di inclinazione e di 45 gradi. Lo scavo pelvico presenta il suo asse maggiore disposto orizzontalmente. Decalcificazione diffusa anche del rachide lombare (cfr. fig. 22).
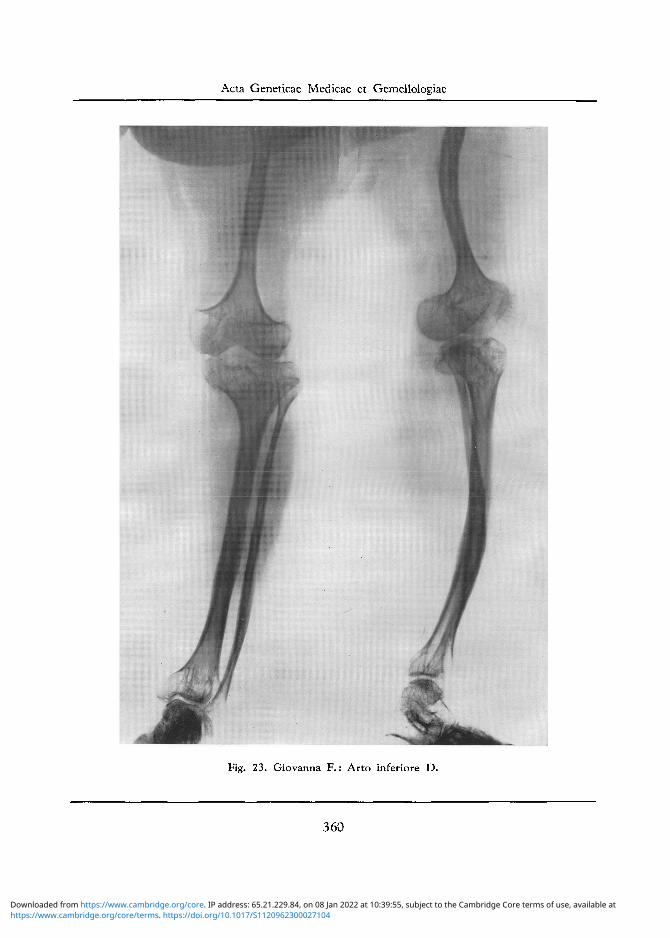
Arto Inferiore D: Assottigliamento di grado notevolissimo della diafisi femorale che e incurvata con angolo aperto verso I'interno. Svasamento a fungo della epifisi inferiore femorale, che presenta anche una certa porosita con trabecolatura della spugnosa a larghe maglie. Modica deformazione dei condili femorali. Analoghe alterazioni si riscontrano a carico dell'epifisi superiore della tibia. Le diafisi della tibia e del perone si presentano osteosclerotiche ed assottigliate, con modico incurvamento 'anteriore della diafisi al terzo medio. Rarefazione ossea delle epifisi inferiori delle ossa della gamba, nonche delle ossa del piede (cfr. fig. 23.).
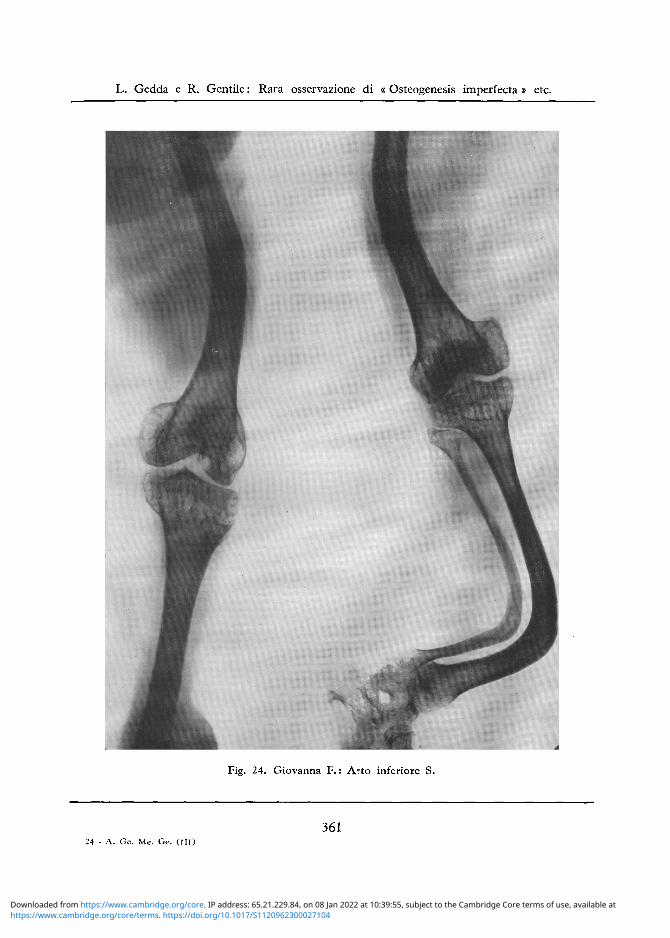
Arto Inferiore S: Diafisi femorale incurvata con angolo aperto anteriormente e medialmente e con modico grado di ispesslmento della corticale. Rarefazione ossea a larghe maglie della epifisi inferiore del femore e di quella superiore della tibia e del perone, con modica deformazione dei capi articolari.
Forte incurvamento anteriore della tibia e del perone e spostamento del perone dalla posizione laterale a quella posteriore rispetto alia tibia cosicche nella proiezione frontale si ha I'impressione di un accorciamento della diafisi tibiale e di assenza del perone. La tibia e fortemente sclerotica e non e piu distinguibile nel suo terzo medio la corticale dalla midollare. Marcata porosita delle epifisi inferiori delle ossa della gamba, nonche delle ossa del tarso (cfr. fig. 24).
358
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
Fig. 22. Giovanna F.: Bacino
Tabella 2
Misure Arto Superiore
D
S
Acromion-gomito
cm. 21
cm. 19,5
Gomito-polso
cm. 15,5
cm. 18
Lunghezza mano
cm. 14,5
cm. 13,5
Misure Arto Inferiore
Inguine-marg. sup. rotula
cm. 19
cm. 21
Margine inf. rotula-collo p.
cm. 22
cm. 15
359
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
Acta Geneticae Medicae et Gemellologiae
Fig. 23. Giovanna F.: Arto inferiore D.
360
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
L. Gedda e R. Gentile: Rara osservazione di «Osteogenesis imperfecta» etc.
Fig. 24. Giovanna F.: Arto inferiore S.
361 24 - A. Ge. Me. Ge. (HI)
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
Acta Geneticae Medicae et Gemellologiae
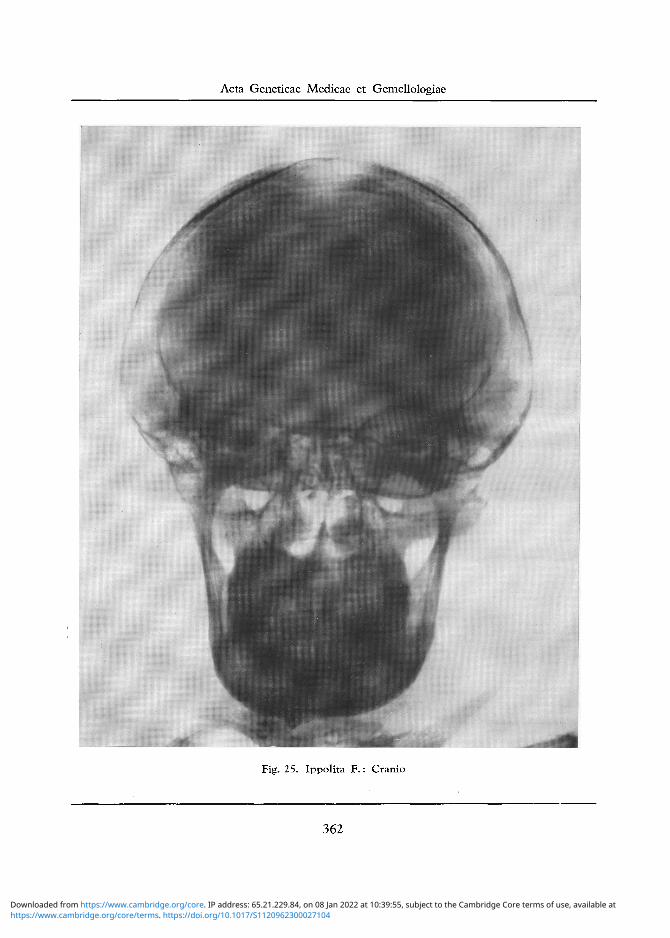
Fig. 25. Ippolita F . : Cranio
362
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
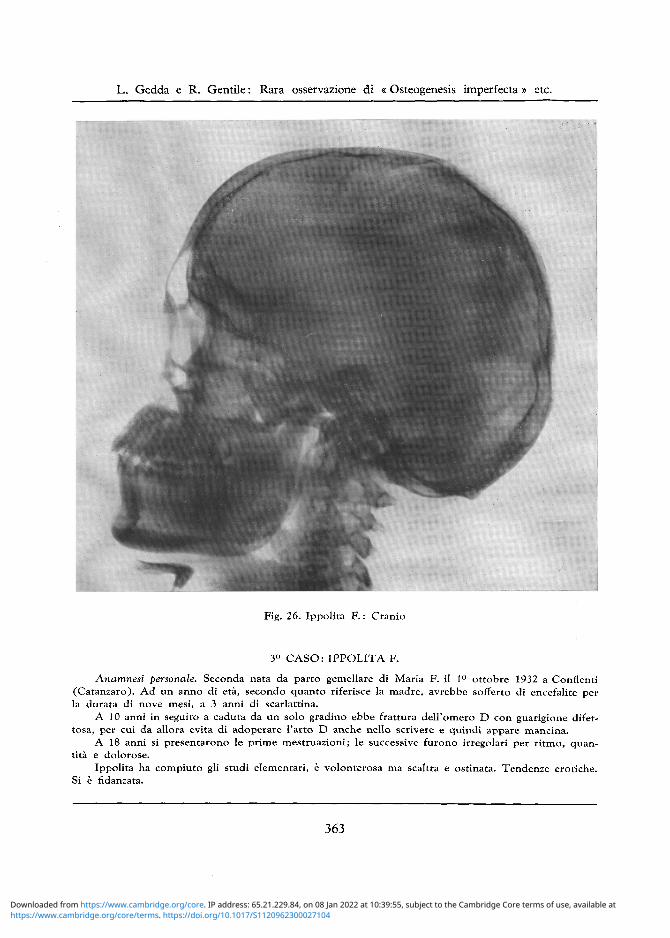
Fig. 26. Ippolita F . : Cranio
3° C A S O : IPPOLITA F.
Anamnesi personale. Seconda nata da parto gemellare di Maria F. il 1° ottobre 1932 a Conflenti (Catanzaro). Ad un anno di eta, secondo quanto riferisce la madre, avrebbe sofferto di encefalite per la durata di nove mesi, a 3 anni di scarlattina.
A 10 anni in seguito a caduta da un solo gradino ebbe frattura deU'omero D con guarigione difet-tosa, per eui da allora evita di adoperare l 'arto D anche nello scrivere e quindi appare mancina.
A 18 anni si presentarono le prime mestruazioni; le successive furono irregolari per ri tmo, quan-tita e dolorose.
Ippolita ha compiuto gli studi elementari, e volonterosa ma scaltra e ostinata. Tendenze erotiche. Si e fidanzata.
363
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
Acta Geneticae Medicae et Gemellologiae
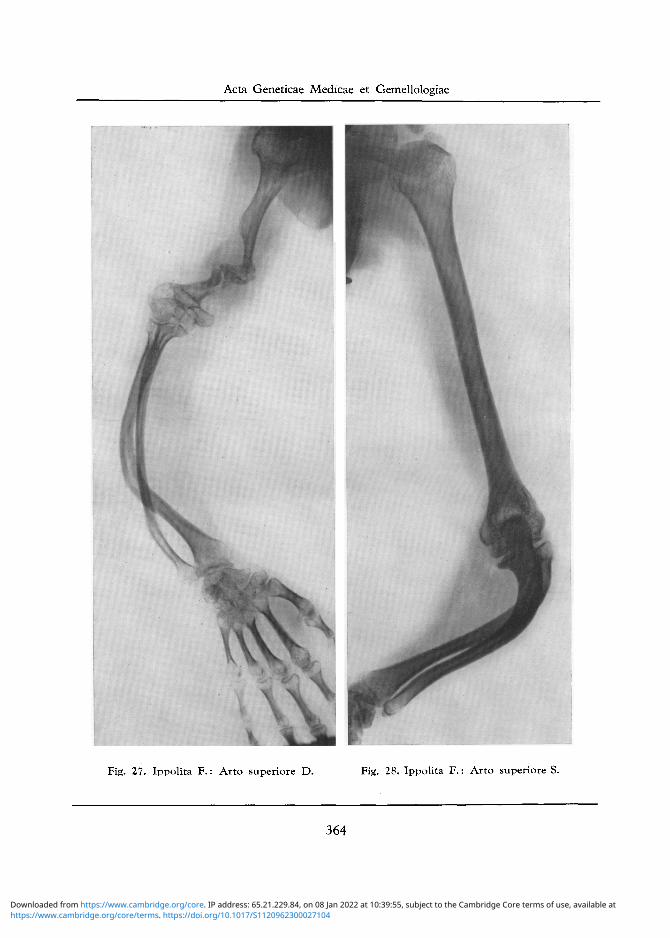
Fig. 27. Ippolita F.: Arto superiore D. Fig. 28. Ippolita F.: Arto superiore S.
364
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
Fig. 29. Ippolita F . : Mano D. Fig. 30. Ippolita F . : Mano S.
Esame obiettivo. Soggetto di costituzione scheletrica irregolare. Statura cm. 113 Peso Kg. 30. La testa presenta l'aspetto idrocefaloide descritto a proposito della sorella.
AH'esame della regione oculare si nota la presenza di sclerotiche blu. Gli arti presentano misure diverse in segmenti omologhi, come appare dalle misure riportate nella
seguente tabella e dal grafico (cfr. fig. 15).
La gemella Ippolita che presenta piu sviluppati in lunghezza gli arti di S e specialmente 1'arto infe-riore S, cammina tenendo in semiflessione le due ginocchia ed appoggiando sul pavimento la pianta del piede D e il ginocchio S, pressappoco nella posizione ritratta nella fig. 1. Pertanto gamba e piede S vengono trascinati durante la deambulazione,
Udito normale; voce bene intonata. Denti normali e sani di colore leggermente giallo.
Reperto radiografico:
Cranio: Si nota un aumento del diametro trasverso bitemporale e profilo " a grondaia " (cfr. figg. 25, 26). Trabecolatura ossea non omogenea ma alquanto porotica nella regione occipitale, con irrego lare assottigliamento del tavolato osseo esterno. Obliquita del pavimento della fossa cranica anteriore. Sella turcica svasata, con introitus molto grande e scarso approfondimento del pavimento. Apofisi clinoidee appuntite.
II seno sfenoidale e situato piuttosto anteriormente alia sella.
365
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
Acta Geneticae Medicae et Gemellologiac
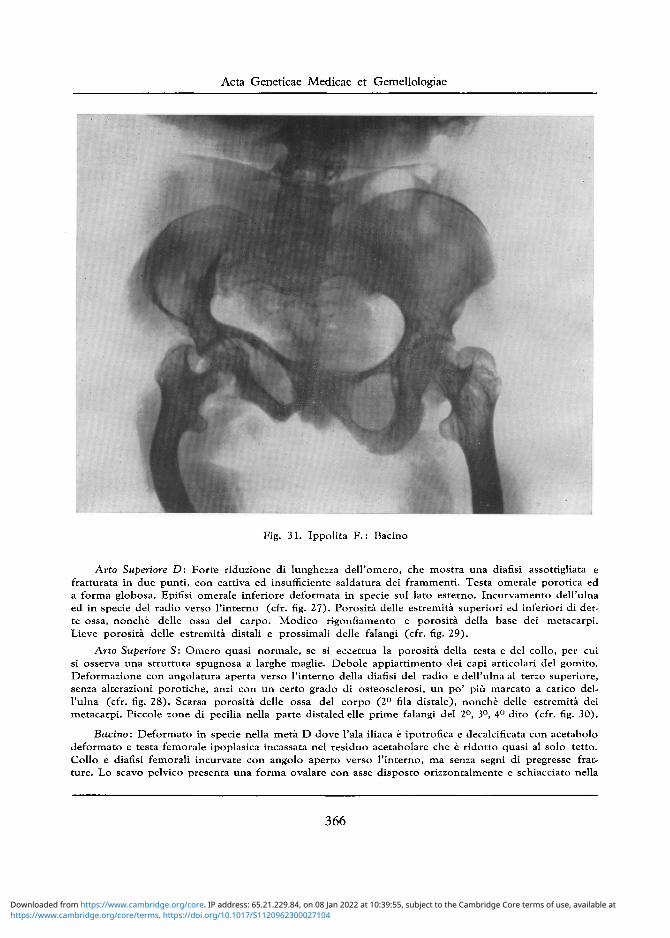
Fig. 31. Ippolita F . : Bacino
Arto Superiore D: Forte riduzione di lunghezza dell 'omero, che mostra una diafisi assottigliata e fratturata in due punti, con cattiva ed insufficiente saldatura dei frammenti. Testa omerale porotica ed a forma globosa. Epifisi omerale inferiore deformata in specie sul lato esterno. Incurvamento dell'ulna ed in specie del radio verso l'interno (cfr. fig. 27). Porosita delle estremita superiori ed inferiori di det-te ossa, nonche delle ossa del carpo. Modico rigonfiamento e porosita della base dei metacarpi. Lieve porosita delle estremita distali e prossimali delle falangi (cfr. fig. 29).
Arto Superiore S: Omero quasi normale, se si eccettua la porosita della testa e del collo, per cui si osserva una struttura spugnosa a larghe maglie. Debole appiattimento dei capi articolari del gomito. Deformazione con angolatura aperta verso l'interno della diafisi del radio e dell'ulna al terzo superiore, senza alterazioni porotiche, anzi con un certo grado di osteosclerosi, un po ' piu marcato a carico dell'ulna (cfr. fig. 28). Scarsa porosita delle ossa del corpo (2° fila distale), nonche delle estremita dei metacarpi. Piccole zone di pecilia nella parte distaled elle prime falangi del 2°, 3°, 4° dito (cfr. fig. 30).
Bacino: Deformato in specie nella meta D dove 1'ala iliaca e ipotrofica e decalcificata con acetabolo deformato e testa femorale ipoplasica incassata nel residuo acetabolare che e ridotto quasi al solo tetto. Collo e diafisi femorali incurvate con angolo aperto verso l 'interno, ma senza segni di pregresse frat-ture. Lo scavo pelvico presenta una forma ovalare con asse disposto orizzontalmente e schiacciato nella
366
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
Fig. 32. Ippolita F.: Arto inferiore D.
367
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
Acta Geneticae Medicae et Gemellologiae
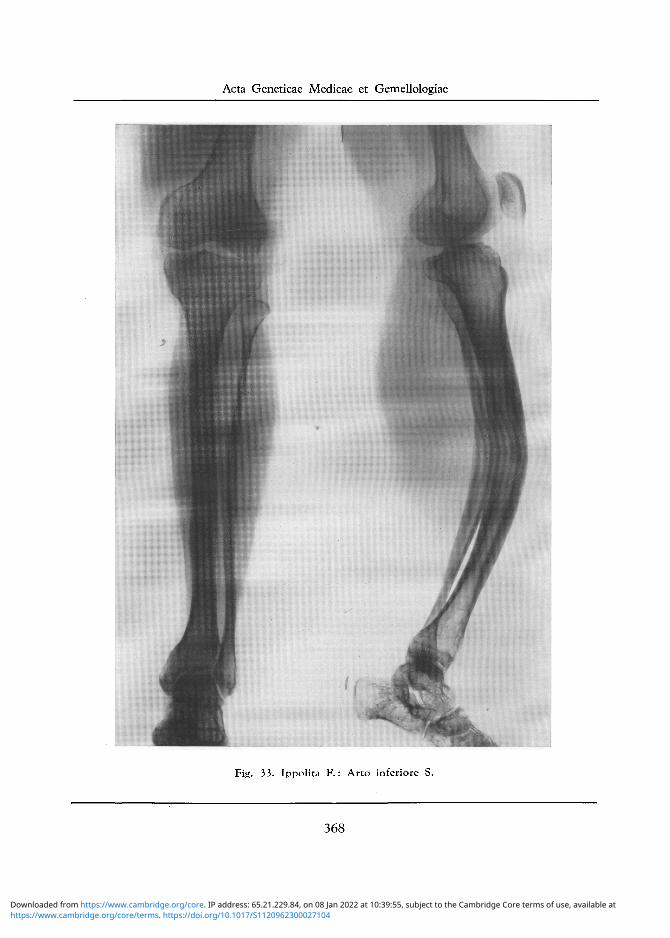
Fig. 33. Ippolita F . : Arto inferiore S.
368
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta» etc.
meta D. Anche il forame otturatorio D presenta una forma ovalare secondo un asse orizzontale. I corpi vertebrali lombari appaiono decalcificati e ruotati (cfr. fig. 31).
Arto Inferiore D: Accentuatissimo incurvamento della diafisi femorale ad angolo aperto verso il davanti e l 'interno, con assottigliamento della diafisi che appare molto esile. Alterata struttura trabe-colare della regione condiloidea, cui si contrappongono analoghe alterazioni delPestremo superiore della tibia, con risultante deformita del ginocchio. Forte incurvamento delle diafisi della tibia e del perone verso il davanti con lievi note di osteosclerosi (cfr. fig. 32).
Arto Inferiore S: II tratto visibile del femore appare normale, se si eccettua una debole porosita del condilo interno. Lieve obliquita della rima articolare del ginocchio, che e un po ' ridotta di altezza sul lato esterno. Modica porosita del capitello tibiale e della testa del perone. Incurvamento della diafisi tibiale e peroneale verso il davanti, con osteosclerosi della parte anteriore della corticale della tibia stessa.
Decalcificazione ossea si nota invece aU'estremo inferiore della tibia ed a carico di tutte le ossa del tarso (cfr. fig. 33).
Tabella 3
Misure Arto Superiore
D
S
Acromion-gomito
cm. 22
cm. 23
Gomito-polso
cm. 14
cm. 15
Lunghezza Mano
cm. 13,5
cm. 14,5
Misure Arto Inferiore
Inguine-marg. sup. rotula
cm. 19
cm. 32
Margine inf. rotula-collo p.
cm. 24
cm. 27,5
Diagnosi clinica: 3 casi di nanismo disarmonico da osteogenesis imperfecta tarda. Diagnosi gemellologica: lo studio sistematico dei caratteri ereditari somatici secondo
i principi e la tecnica della diagnosi di rassomiglianza di Siemens-v.Verschuer, intorno al quale studio per economia di spazio non ci intratteniamo, dimostra un tale grado di concordanza da rendere indubbia la diagnosi di coppia gemellare monozigotica. Inoltre depongono in questo senso la cospicua concordanza nel fenotipo della malattia pres-so le due gemelle ed i caratteri a disposizione speculare che si riscontrano in alcuni segmenti degli arti come nella lunghezza delle mani le quali presentano asimmetria controlaterale nella singola gemella e concordanza speculare intrageminale assoluta; altrove la concordanza speculare e relativa come nella lunghezza degli arti inferiori dove le misure non sono identiche, ma egualmente significative (maggiore lunghezza dell'arto inferiore D di Giovanna e dell'arto inferiore S di Ippolita).
Dal punto di vista psicologico si nota una notevole diversita caratteriologica sotto-lineata da un diverso grado di iniziativa per cui tanto nei rapporti intrageminali, quanto nei rapporti esterni l'iniziativa, il comando e la rappresentanza appartengono ad Ippolita (gemello'guida).
Anche la calligrafia presenta delle cospicue differenze intrageminali (cfr. figg. 3 e 4), ma bisogna tener conto del fatto che Ippolita scrive con la sinistra a motivo dell'impe-dienza funzionale dell'arto superiore D conseguente a fratture, come sopra si e detto.
369
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiac
4. Discussione
La diagnosi della malattia di cui soffrono le nostre tre candidate non appare ardua dal punto di vista differenziale ma piuttosto interessante per ulteriori considerazioni riguardanti la malattia, di cui diremo.
Nella discussione diagnostica della osteogenesis imperfecta si parte di solito dal sintomo della fragilita ossea (Grunert, Eliason, Seedorff) prendendo in considerazione la fragilita dovuta a modificazioni locali (neoplasmi, malattie infettive croniche e necrosi ossea aset-tica), oppure a malattie sistemiche neurogene, ematiche, metaboliche, professional!, eredi-tarie). Quando, come nei nostri casi, trattasi di un'affezione sistematizzata, la discri-minante diagnostica fondamentale riguarda il carattere ereditario o non ereditario della malattia. Se il carattere ereditario e dimostrato, cadono le discussioni relative alle malattie neurogene (tabe dorsale, siringomielia, paralisi), alle malattie del sangue (linfogra-nulomatosi maligna, morbo di Gaucher), alle malattie metaboliche (osteomalacia, osteo-porosi senile, atrofia post-traumatica da inattivita, sindrome di Milkmann, avitaminosi, malattia di Recklinghausen, morbo di Paget), alle malattie professional! (avvelenamento da mesotorio, acido pirogallico, malattia dei lavoratori dei fiammiferi e delle perle) e rimangono in discussione solo le malattie ereditarie costituzionali del sistema osseo e cioe, praticamente, l'osteogenesis imperfecta di fronte all'osteosclerosi di Albers-Schonberg.
Siccome la dimostrazione dell'ereditarieta della malattia di cui trattiamo non potrebbe essere piu manifesta, sia per il rapporto genealogico (madre-figlie), sia per la condizione gemellare (concordanza in gemelle MZ), non ci rimarrebbe che il compito di discutere la diagnosi differenziale nei confronti dell'osteosclerosi.
Nei caso nostro ai noti motivi che impongono la diagnosi differenziale fra l'osteogenesis imperfecta e il morbo di Albers-Schonberg (essere le due malattie ereditarie e caratterizzate da facilita alle fratture) se ne aggiunge un altro e cioe la constatazione che nello scheletro delle nostre AA. esistono delle zone di addensamento che presentano opacita ai raggi aumentata, cosi da poter essere considerate come zone di osteosclerosi (cfr. radio S, ulna S, tibia D, perone D, tibia S, in entrambe le gemelle).
Bisogna pero dire subito che nei nostri casi per un lato manca la sistematizzazione del processo osteosclerotico caratteristica del morbo di Albers-Schonberg, mentre pre-domina l'osteoporosi e l'osteomalacia del tessuto. Anche nella malattia di Albers-Schonberg l'osteoporosi puo essere presente ma sotto quella forma detta « vello di zebra» e cioe a striscie alternate di zone sclerotiche e di zone porotiche, che nei nostri casi non si verifica. Quanto alle deformazioni osteomalaciche, essendo queste sconosciute nel-l'osteosclerosi ereditaria mentre sono di grossolana evidenza nelle nostre tre ammalate, rappresentano il sintomo che, di primo acchito, permette di scartare l'ipotesi del morbo di Albers-Schonberg dispensandoci dal discuterne i sintomi minori.
Detto questo circa l'osteoclerosi ereditaria, e doveroso osservare che nei nostri casi il sintomo osteopsatirotico, e cioe la fragilita ossea, pur esistendo e meno evidente del sintomo osteomalacico della deformabilita e percio ci sembra opportuno mettere in con-fronto i nostri casi con le altre condizioni osteomalaciche, non certo a deroga ma in
370
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
aggiunta al criterio fondamentale dell'eredita che negli altri casi di osteomalacia o di asteoporosi non e presente.
Sotto questo profilo ci sembra che sia opportuno accennare alia diagnosi differen-ziale, in sede di esame obiettivo, nei confronti dell'osteomalacia e del rachitismo.
Osteomalacia. A parte le considerazioni relative alia sostanziale differenza patoge-netica per cui nelPosteomalacia si tratta di una decalcificazione di tessuto osseo gia pre-cedentemente costruito come di norma, mentre nell'osteogenesis imperfecta si tratta di una costruzione difettosa dell'osso, resta il fatto che nei due casi si puo riscontrare un aumento di plasticita delle ossa, con tutte le conseguenze relative, fra cui la diminu-zione dell'altezza. Financo una certa colorazione bluastra delle sclerotiche puo essere talora presente nell'osteomalacia. Percio i criteri di discriminazione, oltre a quello fondamentale dell'ereditarieta, sono dati dalla conformazione del cranio a grondaia carat-teristica dell'osteogenesis imperfecta e presente nelle nostre due gemelle, dai criteri radiografici di un buon rilievo della struttura ossea quale si riscontra nei nostri radio-grammi e che e caratteristico dell'osteogenesis imperfecta, (mentre nell'osteomalacia e peculiare il risalto dei contorni essendo ultimo il cortex a perdere il suo contenuto in calcio), e dalle zone di addensamento osseo che non sono presenti nell'osteomalacia:
Ancora, l'osteomalacia puo produrre delle fenocopie analoghe alle malformazioni che abbiamo riscontrato nei torace e nei bacino delle nostre ammalate, ma ordinaria-mente rispetta gli arti superiori e inferiori che qui sono notevolmente e grossolanamente interessati.
Infine i nostri casi non presentano dati anamnestici o sintomi che possano far pen-sare a un'osteomalacia achilica, epatica, acidotica, tireotica, renale, gravidica o puer-perale, senile, ecc. ne hanno reagito favorevolmente alia terapia causale (calcio e vitamina D) .
Rachitismo. Per quanto riguarda la diagnosi differenziale nei confronti della rachitis tarda vale specialmente il reperto clinico del mancato ingrossamento della epifisi e l'assenza del reperto radiografico caratteristico dato dalle linee epifisarie allargate e irre-golari, dalle metafisi concave e slabbrate. Inoltre anche qui la terapia specifica non ha condotto a risultati di sorta.
Giunti in questo modo, per esclusione, alia diagnosi di osteogenesis imperfecta tarda nei tre casi che abbiamo descritto, dobbiamo fermarci a discutere i sintomi cardinali e secondari della malattia, come si presentano nelle nostre ammalate.
Criterio di fondamentale importanza per stabilire una diagnosi di osteogenesis imperfecta, come si e accennato, e quello del processo ereditario dell'affezione il quale, nella nostra casistica, gode di una particolare evidenza dimostrativa.
Gia nei gruppo famigliare che abbiamo direttamente studiato appare eloquente la concordanza dell'affezione nella madre e nelle figlie e non solo per il fatto del rapporto generativo fra la prima e le seconde, ma anche per il fatto che queste seconde non sono figlie mononate, ma gemelle MZ concordanti, come appunto di solito si verifica per le malattie ereditarie che colpiscono lo zigote gemellogenetico e che sono dotate di una sufficiente penetranza.
Se poi passiamo dal caratteristico gruppo famigliare immediato delle tre persone
371
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiae
da noi esaminate, agli altri gruppi famigliari e stipiti di derivazione dell ' intero albero genealogico, quale fu possibile ricostruire, emergono altri dati di notevole interesse.
La paucita delle notizie riguardanti lo stipite pa terno delle gemelle e alquanto con-fortata dall'assenza di tare significative presso gli zii ed i nonni paterni, ma soprat tut to dal fatto che lo stipite materno presenta invece un 'abbondante messe di rilievi. In que-sto stipite, infatti, si possono sorprendere dei fenomeni patologia di valore fondamentale che r iguardano:
a) il gruppo famigliare della madre ; fc>) lo stipite famigliare della nonna materna; c) la famiglia del n o n n o materno.
Nel commentare Palbero genealogico consideriamo « p r o b a n d e » le due gemelle e non terremo conto, nell 'applicare la terminologia al parentado, della madre Maria quale probanda.
a ) Nel gruppo famigliare della madre. Per cominciare da un fenomeno a significato non patologico, osserviamo la presenza di due cugini di p r imo grado gemelli ( IV 9 e 10) mor t i in tenera eta, per cui n o n sappiamo se MZ o DZ, ma sufficienti per supporre che l'eredita del fattore G (o fattore gemellogenetico) sia provenuto alio zigote delle nostre gemelle da parte materna.
Dal pun to di vista patologico, nel gruppo famigliare della madre osserviamo, oltre alia presenza della medesima ( I I I 14) per la quale abbiamo precisato obiettivamente la dia-gnosi di osteogenesis imperfecta, la presenza di una sorella della madre (III 13) anche essa « deforme, con arti corti, vissuta pochi anni » dietro alia quale descrizione puo celarsi un altro caso di osteogenesis imperfecta.
b) Nello stipite famigliare della nonna materna. Abb iamo accertato che fra la famiglia della nonna materna (II 3 ) e quella del n o n n o materno (I I 13) non esistono rap-port i di consanguineita. Cio premesso, osserviamo che nella famiglia della nonna materna delle gemelle t roviamo un complesso padre-figlio di cui l 'anamnesi dice a proposi to del padre ( I I 6) « con deformita agli arti superiore e inferiore D » ed a proposi to del figlio ( I II 9) « deforme, mor to a 5 anni ». D i un altro figlio del medesimo genitore ( III 10) viene riferito « con un piede difettoso »; inoltre fra i discendenti della medesima famiglia si trova « un mos t ro » ( IV 4) del quale non viene indicato neppure il sesso, e una femmina (V 2) « con arti deformi ». D u n q u e quat t ro soggetti per i quali si p u o sospettare la presenza di un processo di osteogenesis imperfecta congenita o tarda.
c) Nello stipite famigliare del nonno materno. Nella famiglia del n o n n o materno, e precisamente fra i discendenti di un secondo matr imonio del medesimo nonno , not iamo la presenza di 6 individui caratterizzati dalle seguenti no te anamnestiche: femmina « muta e deforme » ( IV 32 ) ; « mos t ro » ( IV 3 3 ) ; maschio « deforme » ( IV 36) ; femmina « deforme » ( IV 3 7 ) ; maschio « deforme » ( IV 4 0 ) ; maschio « deforme » ( IV 41) . Anche per questi sei casi p u o essere sospettata la presenza di un processo di osteogenesis imperfecta o, quanto meno, di un processo osteopatico.
Constatato 1'accumulo nello stipite materno delle gemelle di numerosi casi dove talvolta certamente (come nella madre ) , e talvolta eventualmente, trattasi di osteogenesis
372
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta » etc.
imperfecta, cerchiamo di precisare il meccanismo attraverso il quale tali osteopatie possono essersi trasmesse.
Nelle osservazioni dei paragrafi a ) e b) che riguardano il gruppo famigliare della madre e lo stipite famigliare della nonna materna assistiamo due volte ad un fenomeno di trasmissione diretta di tipo dominante dalla madre alle due figlie gemelle (III 14, IV 12 e 13) e dal padre al figlio (II 6, III 9), ma altrove il meccanismo di trasmissione non e facilmente interpretabile (IV 4, V 2).
Nelle osservazioni del paragrafo c) che riguardano lo stipite famigliare del nonno materno i sei casi di deformita non presentano dei precedenti nelPascendenza e la natura ereditaria viene sospettata soltanto per l'accumulo dei casi nella generazione. Certo e che se le deformita dei soggetti IV 32, IV 33, IV 36, IV 37, IV 40, IV 41 si devono ad un processo di osteogenesis imperfecta, e non gia ad un processo di altra natura (per es. a rachitismo) sarebbe necessario ammettere che la madre delle nostre gemelle puo aver ricevuto una doppia eredita morbosa riguardante il sistema schele-trico, da due philum non legati fra loro da vincoli di consanguineita.
Lasciando le altre considerazioni nel rango delle ipotesi, ci sembra di poter affer-mare che il processo di osteogenesis imperfecta di cui soffrono i nostri tre soggetti e di natura ereditaria a tipo dominante nell'ambito della nostra osservazione obiettiva, e forse a tipo irregolarmente dominante quando si tenga conto degli ascendenti e dei collateral! materni.
Passiamo ora a considerare il dato morboso piu importante che colpisce all'ispe-zione e che domina gli accertamenti clinici ulteriori nello studio di Maria F. e delle sue due figlie gemelle Giovanna e Ippolita, rappresentato dalla notevole compromissione e della conseguente profonda anomalia dell'apparato scheletrico.
Bisogna anzittutto tener conto del carattere sistematico dell'osteopatia la quale colpisce vastamente ed irregolarmente la diafisi delle ossa lunghe, ma anche altre ossa ed altre parti dei segmenti ossei, come appare dall'esame delle radiografie del cranio, del rachide, del bacino, ecc.
II processo osteopatico nel momento attuale appare stabilizzato con esiti di notevole deformita e non e facile stabilirne l'inizio ne il periodo di massima floridezza, anche perche nei nostri casi la componente « fragilita ossea » e presente in misura limitata, mentre appare preponderante la componente « deformabilita ossea ». Le deformazioni sono meno improvvise delle fratture e quindi non lasciano nell'anamnesi tracce altret-tanto cospicue. Una storia di fratture esiste indubbiamente sia nell'anamnestico della madre (due fratture del femore), quanto della gemella Giovanna (frattura del femore). e della gemella Ippolita (frattura dell'omero D ) Oltre a questi avvenimenti denunciati dall'anamnesi, si possono trovare all'esame radiografico i segni di altre fratture, come quella della tibia e perone D nella madre Maria, di deformazioni articolari come quella del gomito D nella gemella Giovanna e dell'articolazione coxofemorale D nella gemella Ippolita.
Percio il carattere osteopsatirotico e presente, benche si debba osservare che il rap-porto fra evento traumatizzante ed efFetto traumatico risente di quella sproporzione di modico grado descritta da Seedorff per il tipo III, e non di quella sproporzione asso-lutamente paradossale che distingue l'osteogenesis imperfecta di tipo I e II.
373
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiae
D'altra parte, che l'avvenimento osteoclasico non sia da mettersi soltanto in rapporto con la causa occasionale ma soprattutto con il processo osteopatico in atto risulta, per esempio, dall'esame delle due fratture che interessano 1'omero D di Ippolita anche per le evidenti deficienze del callo osseo.
Inoltre si puo osservare con certezza che la fragilita ossea delle nostre tre ammalate nel periodo florido della malattia non toccava quell'alto grado che merito alia malattia il nome di « osteopsatirosi» formulato da Lobstein e tanto meno quello d i « ossa di vetro » formulato da Apert.
II sintomo osteopatico che domina il quadro e invece quello della « deformabilita » la quale interessa soprattutto la diafisi delle ossa lunghe, in tutti e tre i casi, ma non soltanto queste ossa come appare dallo studio del cranio delle gemelle dove i diametri trasversi sono concordemente aumentati ed il profilo del cranio nelle proiezioni orto-gonali riproduce con evidenza l'immagine « a grondaia » caratteristica dell'osteogenesis imperfecta. Questo sintomo e meno evidente nel cranio della madre Maria F. Ancora nel cranio delle gemelle si nota concordemente svasamento della sella turcica con introitus ampio e pavimento pochissimo approfondito. Anche le radiografie del bacino della madre e delle figlie rivelano delle profonde anomalie che denunciano un notevole grado di malacia del tessuto osseo.
Dal punto di vista della struttura, il tessuto osseo colpito dall'affezione rivela due processi di trasformazione ben distinti per quanto presenti in diversa misura. Anzittutto e soprattutto un vasto processo di osteoporosi a grosse maglie che colpisce specialmente i capi articolari (epifisi); in secondo luogo si nota qua e la un processo di addensamento del tessuto osseo, per es. nelle ossa lunghe degli arti inferiori della gemella Giovanna.
Concludiamo le osservazioni riguardanti il sistema scheletrico notando come le molteplici alterazioni osteoclasiche, osteomalaciche ed osteoporotiche, sommandosi ed accentuandosi, abbiano contribuito a quel processo di nanizzazione delle nostre aa., e cioe di progressiva diminuzione staturale e ponderale che e stato descritto nei con-fronti dell'osteo-genesis imperfecta e che produce un nanismo secondario e disarmonico.
Per quanto riguarda gli altri sintomi dell'osteogenesis imperfecta notiamo la pre-senza di sclere blu nelle due figlie gemelle e di sclere grigio-blu nella madre.
Questo reperto nella madre puo essere interpretato in base all'osservazione di qualche AA. che, con il procedere dell'eta, il colorito blu delle sclere puo diminuire, talora persistere sotto forma di sfumature grigiastre od anche scomparire del tutto. Ad ogni modo la presenza delle sclere blu ha un'importanza clinica in quanto corri-sponde ad un carattere molto importante della malattia, ed anche un'importanza pa-togenetica perche si collega all'interpretazione di Bauer dell'inferiorita mesenchimale, nell'osteogenesis imperfecta. Sclere e tessuto osseo derivano dal medesimo foglietto embrionario e sotto questo aspetto viene giustificata la simpatia morbosa dei due, cosi differenti, organi. L'altro sintomo di mesenchimosi consistente in anomalie del tessuto cicatriziale (Scott e Stiris) non ha potuto essere messo in evidenza nei nostri casi per assenza di cicatrici.
II sintomo dell'otosclerosi e assente nei tre casi di nostra osservazione e questo coincide con l'osservazione di parecchi AA. circa la relativa assenza di questo carattere.
374
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di « Osteogenesis imperfecta» etc.
Vero e che, almeno per le gemelle, non puo del tu t to escludersi che I'otosclerosi appaia in piu tarda eta.
Presente l'iperlassabilita dei legamenti articolari specie alle mani delle due gemelle. La diagnosi clinica converge dunque , come la differenziale, sulPosteogenesis imper
fecta tarda. Se vogliamo attenerci alia classificazione di Seedorff possiamo aggiungere che si
tratta di una forma di t ipo I I I in quanto insorta ad una certa distanza dalla nascita, con
deafness
" t>lue solerotica "
fragility
- van der Hoeves Syndrom.
J = o.i.t.- 2. J = o.i.t. -A = o . i . c
Fig. 34.
modica sintomatologia osteoclasica, e per il fatto che non ha escluso la madre dalla vita sociale avendo essa avuto persino dei figli.
Sotto il profilo tipologico ci sembra interessante r iportare la rappresentazione grafica che Seedorff ha costruito per sviluppare la sua concezione teorica in torno al grado di muta-zione riguardante le varie forme dell 'osteogenesis imperfecta (cfr. fig. 34) . Egli ha raf-figurato un c romosoma tarato mappando in esso i geni responsabili dei sintomi cardi-nali i quali vengono indicati a sinistra guardando, mentre a destra vengono precisate le varie sindromi dell 'osteogenesis imperfecta.
Circa i sintomi si not i che la fragilita viene concepita come comprendente 3 geni « essendo ognuno importante per il normale sviluppo del tessuto osseo (a, b , c) ma nulla di certo puo essere affermato in to rno alle proprieta specifiche che ognuno rap-presenta ». Altri geni furono disegnati per le sclerotiche blu e per la sordita (deafness).
Circa le s indromi abbiamo gia esposto che cosa Seedorff intenda con i tipi I, I I e I I I ; dobbiamo soggiungere che egli enuclea dalla osteogenesis imperfecta tarda di I I I t ipo la cosidetta s indrome di van der Hoeve (fragilita ossea + sordita + sclere blu + eredita) perche spesso accade di riscontrare casi di osteogenesis imperfecta tarda di I I I t ipo senza sordita, e questo appunto si verifica nei nostr i casi.
Passando ad alcune considerazioni nosologiche, osserviamo che lo studio di questi
375
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiae
casi ci ha confermato nella convinzione che l'inserimento dell'osteopsatirosi di Lobstein, delle forme afHni e di transizione, in un quadro piu ampio come quello dell'osteogenesis imperfecta amplificata rispetto alia primitiva concezione della osteogenesis imperfecta letale di Vrolik, ci appare giustificato perche il quadro morboso presenta un'espressi-vita cosi proteiforme che appare illogico fissarne qualche piu frequente aspetto come malattia per se stante, mentre appare logico abbracciare queste diverse forme in un quadro unico riportandone l'origine al motivo patogenetico fondamentale e cioe alia di-fettosa formazione del tessuto osseo, se non addirittura al difetto ereditario che col-pisce un determinate foglietto embrionario.
Che ci siano delle cospicue varieta di fenotipizzazione del fondamentale disturbo osteogenetico viene dimostrato anche dai nostri casi perche in tutte e tre le nostre ammalate il carattere « fragilita » e nettamente superato dal carattere « deformabilita ».
Si puo pensare che la fase delle deformazioni della malattia sia nettamente isolabile dalla fase delle fratture, non solo perche nel caso nostro essa appare prevalente, ma perche da un punto di vista meccanico la plasticita male si accorda con la frattura secondo l'antico proverbio « frangar non flectar» che, nel caso nostro, potrebbe essere rove-sciato cosi: «flectar non frangar». In ogni caso un'osteogenesi difettosa puo essere responsabile tanto della mancante elasticita e della struttura che predispone alle fratture, quanto della malacia ossea che predispone alia deformazione di determinati segmenti scheletrici. Che prevalga l'una forma o l'altra, oppure che esse vadano alternandosi nel tempo, puo dipendere da varieta famigliari del genotipo morboso dell'osteogenesis imperfecta.
Nello stesso senso puo essere commentata l'assensa dell'otoscherosi nelle nostre tre ammalate.
Ancor piu sulle generali, si puo confermare quanto osserva Cocchi sulla buona pro-gnosi « quoad vitam » dell'osteogenesis imperfecta tarda poiche nelle nostre tre ammalate il processo, pur fissato in esiti gravemente deformanti, appare clinicamente spento e compatibile con un proseguimento della vita.
Inline dobbiamo considerare il nostro riscontro gemellare nel quadro della casistica omogenea che abbiamo dettagliatamente esposto, per quanto ci fu possibile rilevare dai testi spesso lacunosi, nel capitolo della bibliografia.
Nella tabella 4 riassumiamo questa casistica la quale, salvo errori od omissioni, com-prende 9 coppie gemellari interessate dall'osteogenesis imperfecta di cui 2 casi (di Miil-ler, Welz e Libermann) sono di osteogenesis imperfecta congenita, e 7 casi (Hess, Sisk, Faxen, Knutsson, SeedorfF (a), SeedorfF (b), Gedda e Gentile) sono di osteogenesis imperfecta tarda.
Nel primo gruppo l'antica coppia bisesso di Miiller e sicuramente DZ e discordante, mentre quella delle gemelle negre di Welz e Liebermann e sicuramente MZ e concordante.
Nel secondo gruppo la coppia femminile di Sisk e MZ e concordante, la coppia maschile di Knutsson e MZ e concordante, la coppia femminile di Gedda e Gentile e MZ e concordante; invece la coppia di Hess, la coppia bifemminile di Seedorff (a) e la coppia bimaschile di Seedorff (b) sono a zigotismo ignoto e tutte discordanti. In
376
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L. Gedda e R. Gentile: Rara osservazione di «Osteogenesis imperfecta» etc.
realta SeedorfF rappresenta la coppia (b) con il simbolo delle coppie identiche, ma non fornisce elementi di giudizio. Scorrendo i protocolli di Hess e di SeedorfF si ritrae l'impressione che si tratti di coppie DZ, ma questa non e altro che un'impressione. Certo e che nei due gruppi non conosciamo nessuna coppia sicuramente MZ che sia discordante.
Per quanto la casistica delle coppie MZ sia cosi ristretta (una coppia per l'osteoge-nesis imperfecta congenita e tre coppie per l'osteogenesis imperfecta tarda) sulla base di questo materiale si puo affacciare l'ipotesi che l'osteogenesis imperfecta possegga una penetranza di alto grado. Osservazioni ulteriori potranno confermare questa ipo-tesi. Dal punto di vista dell'espressivita della malattia non e possibile pronunciarsi sulla base dei reperti gemellari in quanto, pur mancando coppie gemellari con sindrome fru-stanea o con microforme, conosciamo assai bene dalla casistica della malattia in soggetti mononati la possibilita di forme abortive e percio riteniamo che la casistica gemellare non permetta finora di giungere ad affacciare un'ipotesi attendibile.
Ad ogni modo i nostri casi non solo portano nuove conferme al carattere ereditario dell'osteogenesis imperfecta, ma indicano che anche determinati particolari della sindrome, come il prevalente sviluppo della malacia nei confronti della fragilita, dipendono dal genotipo e cioe sono ereditariamente e famigliarmente condizionati.
Dal punto di vista gemellologico si deve anche rilevare che le zone di addensamento osseo riscontrabili nelle radiografie le quali possono essere considerate come zone di osteosclerosi localizzata secondaria al processo dell'osteogenesis imperfecta, presentano una collocazione esattamente corrispondente nello scheletro delle due gemelle e con cio inducono a pensare che alia base di questi fenomeni sclerotici vi siano delle com-ponenti regolate dal genotipo.
Tabella 4
1 2 3 4 5 6 7 8 9
Autore
Miiller Hess Welz e Liebermann Sisk Faxen Knutsson Seedorff (a) Seedorff (b) Gedda e Gentile
Anno
1893 1917 1927 1931 1932 1938 1949 1949 1953
Sesso
9 ( d * ) * 9 (0 9 9 9 9 9 9 d* a* 9 (9)* i * ( d * ) *
9 9
Eta
5 settimane 21 mesi ia_2a giornata 4 anni 9 mesi 9 anni 2 7 anni 6 anni 20 anni
Diagnosi gemell.
DZ 7
MZ MZ MZ MZ 7
7
MZ
Diagnosi clinica
o.i.c. o.i.t. o.i.c. o.i.t. o.i.t.** o.i.t. o.i.t. o.i.t. o.i.t.
X
X X GO
GO
0 0
oc
X X oc
Fra parentesi il sesso del gemello non ammalato. Seedorff considera il caso di Faxen come o.i.c.
377 25 - A. Ge. Me. Ge. (I l l )
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

Acta Geneticae Medicae et Gemellologiae
5. Riassunto
Gli AA. riferiscono dettagliatamente intorno ad una rara osservazione famigliare dell'osteogenesis imperfecta tarda, la quale interessa una donna cinquantenne e le sue due figlie gemelle MZ di vent'anni. II nuovo apporto casistico conferma la natura ere-ditaria della malattia ed il suo meccanismo dominante; inoltre depone per un'alta pene-tranza e per la possibility di un'impronta famigliare che potrebbe consistere, come in questo caso, in un prevalente sviluppo della plasticita sulla fragilita ossea e nell'assenza dell'otosclerosi.
Nello scheletro delle gemelle, inoltre, a differenza della madre, notansi zone di addensamento osseo (osteosclerosi localizzata secondaria) in posizioni analoghe, il che fa pensare ad un condizionamento genotipico anche di questo sintomo non consueto.
6. Bibliografia
BAUER, K. H . : Ueber Osteogenesis imperfecta. Dtsch. Zschr. Chir. 154: pp. 166-211 (1920). — Ueber Identitat und Wesen der sogennanten Osteopsathyrosis idiopathica und Osteogenesis imper
fecta. Dtsch. Zschr. Chir. 160: pp. 289-351 (1920). — Erbkonstitutionelle « Systemerkrankungen » und Mesenchym. Klin. Wschr. Jg. 2: pp. 624-626 (1923). BELL, J.: Blue Sclerotics and Fragility of Bone. Treasury of Human Inheritance. Vol. II, Part III, Uni
versity of London, Cambridge University Press, Cambridge and London, 1928. C A R R I E R E , G., E. D E L A N N O Y et M. H U R I E Z : A propos de 5 families dont 34 membres sur 86 sont
atteints de Maladie de Lobstein. Presse Med. 55: 1023 (1937). CASUCCIO C : Osteopatie rare. Edizioni Scientifiche Istituto Rizzoli, 1949. C O C C H I , U . : Malattie ereditarie con alterazione delle ossa; in Schinz: Trattato di Roentgendiagnostica,
Roma, Abruzzini, 1952. E D D O W E S , A. : Dark sclerotics and fragilitas ossium. Brit. med. J. 1900 (II) p . 222. F R E R K I N G , H . W. and O. C. Z I N K : A case of osteogenesis imperfecta diagnosed in utero. American J.
Roentgenology, 67: 1, pp. 103-105, 1952. Fuss , H . : Die erbliche Osteopsathyrosis. Dtsch. Zschr. Chir. 245-279, 1935. G A U T I E R , P., G U I N A R D - D O N I O L , J.: U n syndrome nouveau: La maladie de Lobstein associee a la
thrombasthenie familiale et hereditaire de Glanzmann. Journal Suisse de Medecine 82: 15,407 (1952). G E D D A , L U I G I : La carcinosi osteoplastica. Archivio Italiano di Anatomia e Istologia patologica, Anno II,
n. 3 (maggio-giugno 1931). — Semeiotica e diagnostica dell'apparato locomotore; in Sisto P., Semeiotica e Diagnostica Medica.
Torino, Minerva Medica, 1939. — Studio dei gemelli, Roma, Orizzonte Medico, 1951. GLANZMANN, E D U A R D : Familiare Osteogenesis imperfecta (Typus Vrolik) und ihre Behandlung mit
Vitamin D - Stoss. Bulletin der Schweizerischen Akademie der Medizinischen Wissenschaften 1: 3 (1945).
H A N H A R T , E.: Ueber eine neue Form von Osteopsathyrosis congenita mit einfach-rezessivem, sowie 4 neue Lippen mit dominantem Erbgang und die Frage der Vererbung der sog. Osteogenesis imperfecta. Archiv der Julius Klaus Stiftung 26: 3/4, pp. 426-437 (1951).
H E S S , JULIUS H . : Osteogenesis imperfecta; The Archives of Internal Medicine 19 :2 , pp. 163-93 (1917). HoEVE, J. (van der) 6k A. DE KLEYN : Blaue Sclerae, Knochenbriichigkeit und Schwerhorigkeit. Arch.
f. Ophthalm 95: pp. 81-93 (1918). H O L C O M B , D . Y. : A fragile-boned family. Hereditary Fragilitas Ossium. Journal of Heredity 22: 3,
pp. 104-115 (1931).
378
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at

L . Gedda e R. Gen t i l e : Rara osservazione di « Osteogenesis imperfecta » etc.
KEY, J. A. : Brittle Bones and Blue Sclerae. Arch. Surg. 13: pp. 523-567 (1926). K N U T S S O N , F . : Intervento dopo la comunicazione di Zander G.: Fall av osteogenesis imperfecta tarda
med ovaniiga kotkroppsforandringar alia Svenska Foren. Med. Radiol. Forhdl. (3 . XII . 1938). Nord. Med. 1: p . 802 (1939).
L O B S T E I N : Lehrb. d. pathol. Anat. 2, 179, 1834. L O O S E R : Zur Kenntnis der Osteogenesis imperfecta congenita und tarda, Mitt. a. d. Grenzgeb. d. Med.
u. Chir. 1905, 15, 161. Ueber Osteogenesis imperfecta tarda, Verhandl. d. deutsch. path. Gesellsch., 1905, p . 239.
M A S T R O M A R I N O , A . : Osteogenesi imperfetta congenita precoce e tardiva. Archivio ortop., 53, 729-805 (1937).
MuLLER, SlGFRlD: Periostale Aplasie mit Osteopsathyrosis unter dem Bilde der sogenannten fotalen Rhachitis. Miinchener medicinische Abhandlungen, 1893.
S C H M I D T , J U L I U S : Angeborene Knochenbriichigkeit bei einem neugebornen Kinde. Mschr. Geburtsk. u. Frauenkr. 14: 426-436 (1859).
S C O T T D A G and S T I R I S G A B R I E L : Osteogenesis imperfecta tarda. Acta Medica Scandinavica. Vol. CXLV Fas. IV, p. 237 (1953).
S E E D O R F F , K N U D S T A K E M A N N : Osteogenesis imperfecta Universitetsforlaget, Arhus 1949. S I R K , J. N E W T O N : Fragilitas Ossium in Twins ; Case Report . The Wisconsin Medical Journal 30: 272-
273 (1931). V O E G E L I N , M A R G A R E T H A . : Zur pathologischen Anatomie der Osteogenesis imperfecta Typus Lobstein.
Radiologia Clinica, Vol. XII (1943). V R O L I K , W . : Tabul. ad illustrandam embryogenesis hominis et mammalium, Amstelodami, 1845, Tab . 91 .
RESUME
Les auteurs referent en detail relativement a une rare observation familiale de l'osteogenesis imperfecta tardive concernant une femme agee d'une cinquan-taine d'annees et ses deux filles jumelles MZ agees de vingt ans. Cette etude confirme la nature hereditaire de la maladie et son mecanisme dominant; elle depose en outre en faveur d'une haute penetration et de la possi-bilite d'une empreinte familiale qui pourrait consister, comme dans ce cas, dans un preponderant developpement de la plas-ticite sur la fragilite osseuse et dans l'absence de l'otosclerose.
En outre, a l'encontre de ce que Ton peut constater chez la mere, on note dans le squelette des jumelles des zones de condensation osseuse (osteosclerose localisee secondaire) dans des positions analogues, ce qui fait songer a un conditionnement ge-notypique meme de ce symptome non habituel.
SUMMARY
Die Verfasser berichten aus-fiihrlich iiber einen seltenen Fa-milienbefund einer osteogenesis imperfecta tarda bei einer fiinf-zigjahrigen Frau und ihren bei-den Zwillingstochtern, MZ, im Alter von zwanzig Jahren. Die-ser neue veroffendichte Fall be-statigt die Erblichkeit der Krankheit und ihren dominan-ten Charakter; ausserdem zeugt er von einem hohen Durchdrin-gungsvermogen und der M6g-lichkeit eines Familienmerkmals, welches — vie im vorliegenden Falle —• in einem Vorwiegen der Plastizitat iiber die Zerbrechlich-keit der Knochen und in feh-lender Otosklerose besteht.
Beim Skelett der Zwillinge bemerkt man ausserdem — im Gegensatz zur Mutter — Zo-nen von Knochenverdichtung (lokalisierte, sekundare Osteo-sklerose); und zwar an ahnlichen Stellen, was darauf schliessen lasst, dass sie genotypisch be-dingt sind, obwohl dieses ein ungewohnliches Symptom ist.
ZUSAMMENFASSUNG
The Authors give a detailed description of a rare familial observation of osteogenesis imperfecta tarda in a fifty year old woman and in her two M Z twin daughters twenty years of age. Further cases confirmed the hereditary nature of the disease and its dominant mecchanism; moreover, a high degree of penetration and the possibility of familial transmission is suggested, this possibility consisting, such as in this case, in a greater development of the plasticity and a lesser one of the fragility of the bony tissue, and in the absence of otosclerosis.
The skeleton of each twin, unlike that of the mother, shows bony protuberances (secondary localised osteosclerosis) at similar points, which makes one think of a genotypical conditioning even in this uncommon symptom.
379
https://www.cambridge.org/core/terms. https://doi.org/10.1017/S1120962300027104Downloaded from https://www.cambridge.org/core. IP address: 65.21.229.84, on 08 Jan 2022 at 10:39:55, subject to the Cambridge Core terms of use, available at
Copyright © 2022 FDOKUMEN




















