
Immunosuppressive properties of purified immune t-interferon
Upload
independentCategory
view
4download
0
www.elsevier.com/locate/ybbrc
Biochemical and Biophysical Research Communications 328 (2005) 756–763
BBRC
RANK ligand and interferon gamma differentially regulatecathepsin gene expression in pre-osteoclastic cells
Manhui Pang, Ariel F. Martinez, Jay Jacobs, Wayne Balkan, Bruce R. Troen*
Geriatric Research, Education, and Clinical Center, and Research Service, Miami Veterans Affairs Medical Center, Miami, FL 33125, USA
Division of Gerontology and Geriatric Medicine, Department of Medicine, Miller School of Medicine, University of Miami, Miami, FL 33101, USA
Received 30 November 2004Available online 9 December 2004
Abstract
Receptor activator of NF-jB ligand (RANKL) and interferon gamma (IFN-c) are critical and opposing mediators of osteocl-astogenesis, exerting stimulatory and inhibitory effects, respectively. Cathepsin K (CTSK) is a secreted protease that plays an essen-tial role in osteoclastic bone resorption. We have examined the role of IFN-c in the regulation of CTSK expression in the murinemonocytic RAW 264.7 cell line, which can be readily differentiated to bone-resorbing osteoclasts upon RANKL treatment. Real-time RT-PCR reveals that RANKL stimulates CTSK mRNA expression in a dose- and time-dependent fashion, but that RANKLdoes not alter the expression of cathepsin L (CTSL) and cathepsin S (CTSS) mRNA. IFN-c stimulates both CTSL and CTSSexpression after 3 days, but fails to significantly alter CTSK expression. IFN-c markedly inhibits the stimulation of CTSK mRNAand protein by RANKL, whereas RANKL suppresses the stimulation of CTSL and CTSS mRNA by IFN-c. IFN-c also ablates theRANKL induced osteoclastic differentiation of RAW cells. In RAW cells stably transfected with a CTSK promoter-luciferase plas-mid containing the 1618 bp upstream of the transcription initiation site, IFN-c inhibits CTSK promoter activity and ablates itsinduction by RANKL. In conclusion, IFN-c and RANKL differentially regulate cathepsin K, S, and L gene expression in pre-os-teoclastic cells, and there appears to be significant cross talk between the signal transduction pathways mediating the responses toRANKL and IFN-c.Published by Elsevier Inc.
Keywords: Osteoclast; RANK ligand; Interferon-c; Cathepsin K; Cathepsin S; Cathepsin L
Cathepsins K, L, and S are acid activated cysteineproteinases that exhibit a broad spectrum of substratespecificity [1,2]. Earlier reports suggested the involve-ment of several cathepsins in bone resorption [3–7].More recent data, however, strongly implicate cathepsinK (CTSK) as the predominant effector in osteoclasticbone resorption [2]. Northern analysis showed that rab-bit cathepsin K mRNA was preferentially and highly ex-pressed in osteoclasts [8]. In situ hybridizationdemonstrated that cathepsin K is abundantly expressedin human osteoclasts, whereas cathepsins B, S, and Lwere not detected [9].
0006-291X/$ - see front matter. Published by Elsevier Inc.
doi:10.1016/j.bbrc.2004.12.005
* Corresponding author. Fax: +1 305 575 3365.E-mail address: [email protected] (B.R. Troen).
Cathepsin K is a potent collagenase and exhibitsbone-specific activity [10,11]. In actively resorbing oste-oclasts, cathepsin K is found at the ruffled border,whereas osteoblasts and osteocytes do not express eitherthe transcript or the protein [12]. Nonsense, missense,and stop codon mutations in the cathepsin K gene havebeen identified in humans with pycnodysostosis, whichis an autosomal recessive osteochondrodysplasia charac-terized by osteosclerosis and short stature [13–15]. Peo-ple completely lacking cathepsin K or lacking cathepsinK collagenolytic activity exhibit pycnodysostosis [16].Cathepsin K null mice develop osteopetrosis and displayfeatures characteristic of pycnodysostosis [17,18]. Ma-trix degradation is significantly diminished in these mice[18]. Furthermore, cathepsin K null osteoclasts exhibit

M. Pang et al. / Biochemical and Biophysical Research Communications 328 (2005) 756–763 757
impaired bone resorption in vitro [17]. Consequently,cathepsin K expression is required for normal skeletaldevelopment.
The regulation of bone resorption involves a compli-cated set of hormonal and/or cytokine interactions thatinitially stimulate osteoblasts and stromal cells, whichthen elaborate factors that signal osteoclasts to degradebone [19]. The receptor activator of NF-jB ligand(RANKL), which is a member of the TNF superfamilyof cytokines, stimulates the differentiation and subse-quent activation of osteoclast progenitor cells [20–22].RANKL activity is mediated by its cognate receptorRANK, which belongs to the TNF receptor familyand is expressed on the surface of osteoclast precursorsto provide the necessary signals for osteoclast differen-tiation [19]. RANKL stimulates cathepsin K mRNAand protein expression in osteoclasts derived from hu-man peripheral blood mononuclear cell precursors[23] and also enhances cathepsin K mRNA expressionin murine myeloid RAW 264.7 cells concomitant withinduction toward an osteoclastic phenotype [24]. Wehave also shown that RANKL acutely stimulatescathepsin K expression in mature osteoclasts isolatedfrom neonatal rat bones and in bone marrow derivedosteoclasts [25].
Interferon-c (IFN-c) suppressed osteoclast formationin a mouse osteoclast co-culture model and downregu-lated CTSK expression [26]. Since IFN-c can inhibitRANKL induced osteoclastogenesis in RAW cells [27],we have chosen to further investigate the regulation ofcathepsin K expression by RANKL and IFN-c in thesecells. We now demonstrate that while potently inhibitingthe stimulation of CTSK by RANKL, IFN-c concur-rently induces CTSL and CTSS gene expression. Fur-thermore, RANKL inhibits the IFN-c mediatedinduction of CTSL and CTSS. Consequently, there areboth differential regulation of cathepsin gene expressionand cross talk between the interferon and RANKL sig-naling pathways in pre-osteoclastic cells.
Experimental procedures
Cell culture. The murine RAW 264.7 cell line was kindly providedby Dr. A. Ian Cassady (University of Queensland, Australia). The cellswere maintained in high glucose DMEM supplemented with 10% heat-inactivated FBS, 100 lg/ml streptomycin, 100 U/ml penicillin G, andglutamine, in a humidified atmosphere of 5% CO2 at 37 �C. Sub-cul-turing was performed in nontissue-culture treated petri dishes pur-chased from Greiner. For all experiments, the cells were plated intotissue culture 12-well dishes.
TRAP staining. Cells were stained for tartrate resistant acidphosphatase (TRAP) expression using a leukocyte acid phosphatasekit from Sigma.
Transfection and luciferase assays. prKLPE1 is a rat cathepsin Kpromoter luciferase expression vector that contains 1618 bp of the 5 0
flanking region of the gene plus 27 bp of exon 1. A fragment wasamplified from rat genomic DNA using primers derived from the mouse
genomic CTSK sequence [28]: forward primer—50GGGTCTGTGCCAGGAATCCAGCTAG3 0, reverse primer—50GTCTGAAAAGAGCATAGACAACAG30. The amplified fragment was thencloned into the pCR2.1 vector (TA Cloning Kit, Invitrogen), subse-quently excised via an XhoI/SacI digest, and then cloned in the forwardorientation into the promoterless reporter plasmid pGL3-Basic (Pro-mega), which was previously digested with the same restriction en-zymes. The clone was verified by restriction enzyme analysis andsequencing. TransIT-Keratinocyte (Mirus) was used to transfect theRAW 264.7 cells according to the manufacturer�s instructions. TheprKLPE1 plasmid (0.7 lg) was co-transfected with the neomycin vectorpcDNA3.0 (Invitrogen) at a 3–1 ratio. After 24 h transfection themedium was replaced with fresh complete medium containing Geneti-cin (G-418 sulfate) to a final concentration of 400 lg/ml. The selectivemedia were changed every 2–3 days for 10 days and the cells fromdifferent wells were pooled together and split into noncoated petridishes. After cells were harvested, luciferase assays were performedaccording to the manufacturer�s instructions (Promega). Luciferaseactivity was normalized against total protein concentration.
RNA and protein isolation. Cells were harvested and lysed withTrizol reagent (Gibco-BRL) according to the manufacturer�sinstructions.
First-strand cDNA synthesis from total RNA. Reverse transcriptionusing 1 lg RNA was performed using the SuperScript First-strandSynthesis System for RT-PCR (Invitrogen) following the manufac-turer�s instructions: 50 ng of random hexamers, 10 mM dNTP mix andRNA, followed by incubation at 65 �C for 5 min. Thereafter, 10· RTbuffer, 25 mM MgCl2, 0.1 M DTT, 1 ll SuperScript II RT (50 U), andRNaseOUT recombinant ribonuclease inhibitor were added, and themixture was incubated at 42 �C for 50 min followed by 15 min at 70 �Cto stop the reaction. One microliter of RNase H was added followed byincubation at 37 �C for 20 min to remove RNA. The final reactionvolume was 20 ll.
Quantitative real-time PCR analysis. PCR amplification was per-formed using the LightCycler DNAMaster SYBR Green I kit (Roche)in a 20 ll reaction containing 0.5 lM of each primer, 3 mM MgCl2,and 2 ll cDNA. Primers are listed in Table 1. TaqStart antibody(Clontech) was systematically added to the amplification reactionmixture to block Taq DNA polymerase activity during the setup ofPCRs at room temperature. DNA amplification and detection werecarried out in the LightCycler as follows: the reaction mixture wasinitially incubated at 96 �C for 30 s to inactivate the TaqStart antibodyand to denature DNA.
Amplification was performed for 45 cycles of denaturation at 95 �Cfor 1 s, annealing at 62 �C for 10 s, and extension at 72 �C for 15 s.Ramp rate was 20 �C/s. The cumulative fluorescence for each ampliconwas normalized to that seen with GAPDH amplification using theDDCT (treatment DCT � control DCT) [29]. Results were expressed asthe fold increase, at each time point, over the respective GAPDHcontrols (calculated as 2DDCT). Means ± standard errors were calcu-lated from pooled data from up to three separate experiments, eachwith triplicate replicates. Amplified products were identified as distinctsingle bands on agarose gel electrophoresis.
Western blotting. Thirty micrograms of protein was run on aSDS–polyacrylamide gel and electro-transferred to Hybond-ECLnitrocellulose membranes (Amersham Bioscience). Membranes wereblocked in 5% nonfat milk in TBS-0.05% Tween 20 (TBS-T), over-night at 4 �C, and then incubated with rabbit antimouse CTSKpolyclonal antibody (kindly provided by Dr. Dieter Bromme) for 2 hat room temperature followed by incubation in secondary antibody(anti-rabbit IgG-HRP) for 1 h. Both antibodies were diluted inblocking buffer. Membranes were incubated in TBS-T 3 · 10 minbetween each step. Bands were detected by ECL plus Western Blot-ting Detection System (Amersham Bioscience). After exposure to X-ray film, membranes were washed and then exposed to anti-actinantibody (Sigma) and subsequently processed as above to assess actinexpression.
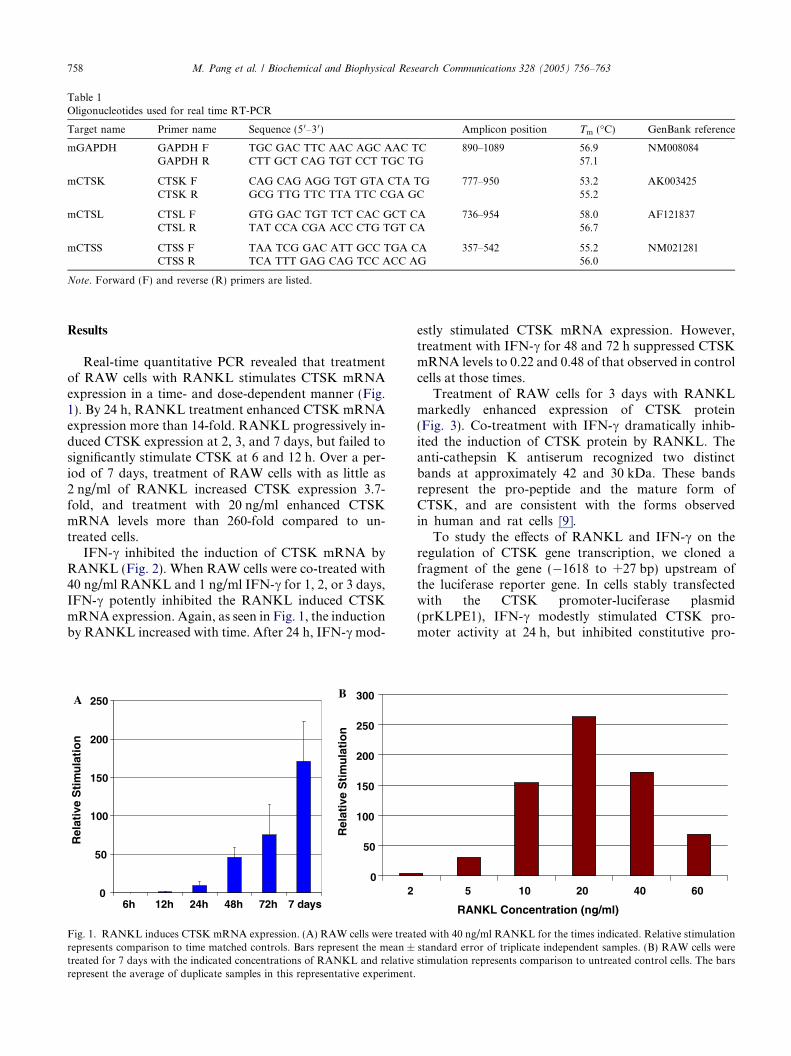
Table 1Oligonucleotides used for real time RT-PCR
Target name Primer name Sequence (5 0–30) Amplicon position Tm (�C) GenBank reference
mGAPDH GAPDH F TGC GAC TTC AAC AGC AAC TC 890–1089 56.9 NM008084GAPDH R CTT GCT CAG TGT CCT TGC TG 57.1
mCTSK CTSK F CAG CAG AGG TGT GTA CTA TG 777–950 53.2 AK003425CTSK R GCG TTG TTC TTA TTC CGA GC 55.2
mCTSL CTSL F GTG GAC TGT TCT CAC GCT CA 736–954 58.0 AF121837CTSL R TAT CCA CGA ACC CTG TGT CA 56.7
mCTSS CTSS F TAA TCG GAC ATT GCC TGA CA 357–542 55.2 NM021281CTSS R TCA TTT GAG CAG TCC ACC AG 56.0
Note. Forward (F) and reverse (R) primers are listed.
758 M. Pang et al. / Biochemical and Biophysical Research Communications 328 (2005) 756–763
Results
Real-time quantitative PCR revealed that treatmentof RAW cells with RANKL stimulates CTSK mRNAexpression in a time- and dose-dependent manner (Fig.1). By 24 h, RANKL treatment enhanced CTSK mRNAexpression more than 14-fold. RANKL progressively in-duced CTSK expression at 2, 3, and 7 days, but failed tosignificantly stimulate CTSK at 6 and 12 h. Over a per-iod of 7 days, treatment of RAW cells with as little as2 ng/ml of RANKL increased CTSK expression 3.7-fold, and treatment with 20 ng/ml enhanced CTSKmRNA levels more than 260-fold compared to un-treated cells.
IFN-c inhibited the induction of CTSK mRNA byRANKL (Fig. 2). When RAW cells were co-treated with40 ng/ml RANKL and 1 ng/ml IFN-c for 1, 2, or 3 days,IFN-c potently inhibited the RANKL induced CTSKmRNA expression. Again, as seen in Fig. 1, the inductionby RANKL increased with time. After 24 h, IFN-cmod-
Fig. 1. RANKL induces CTSK mRNA expression. (A) RAW cells were treatrepresents comparison to time matched controls. Bars represent the mean ±treated for 7 days with the indicated concentrations of RANKL and relativerepresent the average of duplicate samples in this representative experiment.
estly stimulated CTSK mRNA expression. However,treatment with IFN-c for 48 and 72 h suppressed CTSKmRNA levels to 0.22 and 0.48 of that observed in controlcells at those times.
Treatment of RAW cells for 3 days with RANKLmarkedly enhanced expression of CTSK protein(Fig. 3). Co-treatment with IFN-c dramatically inhib-ited the induction of CTSK protein by RANKL. Theanti-cathepsin K antiserum recognized two distinctbands at approximately 42 and 30 kDa. These bandsrepresent the pro-peptide and the mature form ofCTSK, and are consistent with the forms observedin human and rat cells [9].
To study the effects of RANKL and IFN-c on theregulation of CTSK gene transcription, we cloned afragment of the gene (�1618 to +27 bp) upstream ofthe luciferase reporter gene. In cells stably transfectedwith the CTSK promoter-luciferase plasmid(prKLPE1), IFN-c modestly stimulated CTSK pro-moter activity at 24 h, but inhibited constitutive pro-
ed with 40 ng/ml RANKL for the times indicated. Relative stimulationstandard error of triplicate independent samples. (B) RAW cells werestimulation represents comparison to untreated control cells. The bars

Fig. 3. IFN-c inhibits stimulation of CTSK protein expression byRANKL. RAW cells were treated for 72 h with 40 ng/ml RANKL,1 ng/ml IFN-c, or RANKL plus IFN-c.
Fig. 4. IFN-c suppresses CTSK promoter activity in stably transfected RARANKL, 25 ng/ml IFN-c, or RANKL plus IFN-c. Luciferase specific activBars represent the mean ± standard error of a representative experiment witRANKL (RL), 1 ng/ml IFN-c, or RANKL plus IFN-c. Relative stimularepresent the mean ± standard error of triplicate independent samples.
Fig. 2. IFN-c inhibits stimulation of CTSK mRNA expression byRANKL. RAW cells were treated for the indicated times with 40 ng/ml RANKL (RL), 1 ng/ml IFN-c, or RANKL plus IFN-c. Relativestimulation reflects comparison to untreated cells. Bars represent themean ± standard error of triplicate independent samples.
M. Pang et al. / Biochemical and Biophysical Research Communications 328 (2005) 756–763 759
moter activity at 48 and 72 h (Fig. 4A). At all times,IFN-c suppressed the stimulation of CTSK promoteractivity by RANKL. IFN-c also markedly suppressedthe stimulation of endogenous CTSK mRNA expressionby RANKL in these stably transfected cells (Fig. 4B).
In order to confirm that IFN-c inhibits osteoclast for-mation, RAW cells were treated for 5 days withRANKL and RANKL plus IFN-c (Fig. 5). RANKLalone induced the formation of numerous tartrate resis-tant acid phosphatase (TRAP) positive multinucleatedcells. Consistent with its effects upon CTSK expression,IFN-c abrogated the RANKL induced osteoclastogene-sis. In cultures treated with IFN-c, the RAW cells as-sumed a stellate morphology.
In order to investigate more fully gene expressionduring osteoclast differentiation, we assessed the effectsof RANKL upon cathepsin S (CTSS), cathepsin L(CTSL), in addition to CTSK (Fig. 6). RANKL treat-ment for 7 days failed to significantly alter expressionof either CTSS or CTSL, resulting in levels of 0.4 and1.13 times that observed in untreated cells, respectively.However, as seen in the previous experiments, RANKLmarkedly stimulated expression of CTSK mRNA tomore than 160-fold of that observed in untreated cells.We then assessed the impact of IFN-c upon CTSS andCTSL expression (Fig. 6). As before, IFN-c dramati-cally suppressed the stimulation of CTSK mRNA byRANKL after 3 days of treatment. In contrast, treat-ment with IFN-c alone significantly enhanced bothCTSS and CTSL mRNA expression. Interestingly,RANKL suppressed the stimulatory effect of IFN-cupon the expression of the CTSS and CTSL genes.
Discussion
Our data demonstrate that RANKL and interferonexert specific and opposite effects upon cathepsin gene
W cells. (A) Cells were treated for 24, 48, and 72 h with 100 ng/mlity was normalized against total protein concentration of the sample.h triplicate independent samples. (B) Cells were treated with 40 ng/mltion of CTSK mRNA expression compared to untreated cells. Bars

Fig. 5. IFN-c suppresses osteoclastogenesis. RAW cells were treated for 5 days with (A) RANKL (40 ng/ml, 10· magnification), (B) RANKL plusIFN-c (1 ng/ml, 20·), and (C) IFN-c alone (20·). Cells were then stained for TRAP. OC, osteoclasts. Representative fields are shown.
Fig. 6. RANKL and IFN-c effects upon CTSS and CTSL gene expression. (A) RAW cells were treated for 7 days with 40 ng/ml RANKL. Relativestimulation reflects comparison to untreated cells. The bars represent the average of duplicate samples in this representative experiment. (B) RAWcells were treated for 3 days with 1 ng/ml IFN-c, 40 ng/ml RANKL (RL), or IFN-c plus RANKL. Relative stimulation reflects comparison withuntreated cells. Bars represent the mean ± standard error of a representative experiment with triplicate independent samples.
760 M. Pang et al. / Biochemical and Biophysical Research Communications 328 (2005) 756–763
expression. We confirm the previous reports thatRANKL stimulates cathepsin K mRNA expression[23–25] and also show stimulation of CTSK proteinexpression. RANKL can stimulate CTSK promoteractivity by enhancing the binding of microphthalmiatranscription factor (MITF) [30], by activating the nu-clear factor of activated T cells (NFAT-2) [31], and by
enhancing NFAT, MITF, and PU.1 interaction andbinding [32]. We now show that in stably transfectedcells, the 1618 bp promoter region immediately up-stream of the rat CTSK gene transcription start sitecan mediate the stimulatory response to RANKL. Tran-sient transfections of RAW cells reveal that as few as238 bp upstream of the transcription initiation site can

M. Pang et al. / Biochemical and Biophysical Research Communications 328 (2005) 756–763 761
confer RANKL responsiveness, and this small regioncontains potential MITF, NFAT, and AP-1 (activatorprotein-1) binding motifs [33].
IFN-c inhibits CTSKmRNAexpression in osteoclastsderived in a murine coculture system [26]. We now buildupon these findings by demonstrating that IFN-c sup-presses the stimulation of CTSK gene expression byRANKL. IFN-c induces both the ubiquitination ofTRAF6 and PA28-mediated proteasome 20S activation[34]. These actions accelerate degradation of TRAF6.Since TRAF6 plays a critical role in RANKL signalingduring osteoclastogenesis [35], and TRAF6 overexpres-sion can stimulate CTSK promoter activity [33], it islikely that IFN-c inhibits the RANKL induction ofCTSK expression by accelerating the degradation ofTRAF6. While IFN-c appears to be acting over the shortterm (1–3 days) to inhibit the induction of CTSK byRANKL, we have also confirmed previous work [36] thatIFN-c inhibits the formation of multinucleated TRAPpositive cells. IFN-c also appears to suppress RANKLinduced osteoclastogenesis via its effects upon TRAF6[36]. Our data help to explain why mice that overexpressIFN-c exhibit an osteochondrodysplasia due, in part, toaltered osteoclast function [37]. IFN-c can also exert neg-ative regulatory effects upon transcription via IFN regu-latory factor 2 (IRF2) and/or IFN consensus sequencebinding protein (ICSBP) binding to the Gamma IFNactivated site (GAS) [38–40]. The rat CTSK promotercontains multiple GAS motifs that are potential media-tors of the response to IFN-c.
There are multiple reports that IFN-c regulates CTSSand CTSL expression. IFN-c induced CTSS mRNA andprotein expression in a human keratinocyte cell line, butdid not increase CTSL and CTSB expression [41]. IFN-c induced CTSL synthesis and secretion in synovial fibro-blast-like cells [42], stimulated CTSL andCTSS in humanmonocyte/macrophages [43], whereas IFN-c inhibitedCTSL mRNA expression in murine macrophages [44].IFN-c markedly stimulated CTSS mRNA expression inhuman smooth muscle cells, but exerted no effect uponCTSK mRNA [45]. Our data demonstrate for the firsttime that IFN-c induces both CTSS and CTSL mRNAexpression in preosteoclastic RAWcells. Since the humanCTSS promoter contains an IFN-stimulated response ele-ment (ISRE) that is critical for IFN-c-induced gene tran-scription in epithelial cells [46], it is possible that the sameelement mediates the response in RAW cells.
Perhaps more importantly, we also report the novelfinding that RANKL inhibits the stimulation of bothCTSS and CTSL gene expression by IFN-c, while at thesame time it profoundly induces CTSK expression.Hence, IFN-c suppresses both osteoclast differentiationand osteoclast specific gene expression, while concur-rently enhancing the expression of genes that may berepresentative of an inflammatory response. Therefore,IFN-c and RANKL can both inhibit each other�s specific
stimulatory effects upon gene expression. Unlike the inhi-bition of the response to RANKL by IFN-c, it is unclearwhat mechanism underlies suppression of CTSL andCTSS induction by IFN-c. However, our data furthersupport the importanceof cross talkbetween theRANKLand IFN-c signaling pathways, and strongly suggest thatan interplay between osteoclastogenic and inflammatoryinfluences plays a critical role in bone resorption.
Acknowledgments
This study was supported in part by the Departmentof Veterans Affairs (Merit Review) and the Indian TrailFoundation. We appreciate the excellent technical assis-tance of Linjian Zhu and Isabel Fernandez. We alsoappreciate the support of Drs. Bernard Roos and GuyHoward, the Miami VA GRECC, and the Gerontologyand Geriatrics Division at the University of MiamiSchool of Medicine.
References
[1] B.R. Troen, Cathepsin genes: structure, expression and regulation,in: A. Ouali, D.I. Demeyer, F.J.M. Smulders (Eds.), Expression oftissue proteinases and regulation of protein degradation as relatedto meat quality, Audet Tijdschriften bv, Nijmegen, The Nether-lands, 1995, pp. 29–46.
[2] B.R. Troen, The role of cathepsin K in normal bone resorption,Drug News Perspect. 17 (2004) 19–28.
[3] J.M. Delaisse, P. Ledent, G. Vaes, Collagenolytic cysteineproteinases of bone tissue. Cathepsin B, (pro)cathepsin L anda cathepsin L-like 70 kDa proteinase, Biochem. J. (1991) 167–174.
[4] S.S. Dong, G.I. Stransky, C.H. Whitaker, S.E. Jordan, P.H.Schlesinger, J.C. Edwards, H.C. Blair, Avian cathepsin B cDNA:sequence and demonstration that mRNAs of two sizes areproduced in cell types producing large quantities of the enzyme,Biochim. Biophys. Acta 1251 (1995) 69–73.
[5] T. Goto, T. Tsukuba, T. Kiyoshima, Y. Nishimura, K. Kato, K.Yamamoto, T. Tanaka, Immunohistochemical localization ofcathepsins B, D and L in the rat osteoclast, Histochemistry 99(1993) 411–414.
[6] S. Goto, Molecular biological approaches to aging in Japan: anoverview, Arch. Gerontol. Geriatr. 19 (1994) 159–170.
[7] T. Sasaki, M.E. Ueno, Cysteine-proteinase localization in osteo-clasts: an immunocytochemical study, Cell Tissue Res. 271 (1993)177–179.
[8] K. Tezuka, Y. Tezuka, A. Maejima, T. Sato, K. Nemoto, H.Kamioka, Y. Hakeda, M. Kumegawa, Molecular cloning of apossible cysteine proteinase predominantly expressed in osteo-clasts, J. Biol. Chem. 269 (1994) 1106–1109.
[9] F.H. Drake, R.A. Dodds, I.E. James, J.R. Connor, C. Debouck,S. Richardson, E. Lee-Rykaczewski, L. Coleman, D. Rieman, R.Barthlow, G. Hastings, M. Gowen, Cathepsin K, but notcathepsins B, L, or S, is abundantly expressed in humanosteoclasts, J. Biol. Chem. 271 (1996) 12511–12516.
[10] P. Garnero, O. Borel, I. Byrjalsen, M. Ferreras, F.H. Drake,M.S. McQueney, N.T. Foged, P.D. Delmas, J.M. Delaisse, Thecollagenolytic activity of cathepsin K is unique among mamma-lian proteinases, J. Biol. Chem. 273 (1998) 32347–32352.

762 M. Pang et al. / Biochemical and Biophysical Research Communications 328 (2005) 756–763
[11] Z. Li, W.S. Hou, C.R. Escalante-Torres, B.D. Gelb, D. Bromme,Collagenase activity of cathepsin K depends on complex formationwith chondroitin sulfate, J. Biol. Chem. 277 (2002) 28669–28676.
[12] A. Littlewood-Evans, T. Kokubo, O. Ishibashi, T. Inaoka, B.Wlodarski, J.A. Gallagher, G. Bilbe, Localization of cathepsin Kin human osteoclasts by in situ hybridization and immunohisto-chemistry, Bone 20 (1997) 81–86.
[13] B.D. Gelb, G.P. Shi, H.A. Chapman, R.J. Desnick, Pycnodysos-tosis a, lysosomal disease caused by cathepsin K deficiency,Science 273 (1996) 1236–1238.
[14] M.R. Johnson, M.H. Polymeropoulos, H.L. Vos, R.I. Ortiz deLuna, C.A. Francomano, A nonsense mutation in the cathepsin Kgene observed in a family with pycnodysostosis, Genome Res. 6(1996) 1050–1055.
[15] W.S. Hou, D. Bromme, Y. Zhao, E. Mehler, C. Dushey, H.Weinstein, C.S. Miranda, C. Fraga, F. Greig, J. Carey, D.L.Rimoin, R.J. Desnick, B.D. Gelb, Characterization of novelcathepsin K mutations in the pro and mature polypeptide regionscausing pycnodysostosis, J. Clin. Invest. 103 (1999) 731–738.
[16] G. Motyckova, D.E. Fisher, Pycnodysostosis: role and regulationof cathepsin K in osteoclast function and human disease, Curr.Mol. Med. 2 (2002) 407–421.
[17] P. Saftig, E. Hunziker, O. Wehmeyer, S. Jones, A. Boyde, W.Rommerskirch, J.D. Moritz, P. Schu, K. von Figura, Impairedosteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice, Proc. Natl. Acad. Sci. USA 95 (1998) 13453–13458.
[18] M. Gowen, F. Lazner, R. Dodds, R. Kapadia, J. Feild, M.Tavaria, I. Bertoncello, F. Drake, S. Zavarselk, I. Tellis, P.Hertzog, C. Debouck, I. Kola, Cathepsin K knockout micedevelop osteopetrosis due to a deficit in matrix degradation butnot demineralization, J. Bone Miner. Res. 14 (1999) 1654–1663.
[19] B.R. Troen, Molecular mechanisms underlying osteoclast forma-tion and activation, Exp. Gerontol. 38 (2003) 605–614.
[20] D.L. Lacey, E. Timms, H.L. Tan, M.J. Kelley, C.R. Dunstan, T.Burgess, R. Elliott, A. Colombero, G. Elliott, S. Scully, H. Hsu,J. Sullivan, N. Hawkins, E. Davy, C. Capparelli, A. Eli, Y.X.Qian, S. Kaufman, I. Sarosi, V. Shalhoub, G. Senaldi, J. Guo, J.Delaney, W.J. Boyle, Osteoprotegerin ligand is a cytokine thatregulates osteoclast differentiation and activation, Cell 93 (1998)165–176.
[21] W.S. Simonet, D.L. Lacey, C.R. Dunstan, M. Kelley, M.S.Chang, R. Luthy, H.Q. Nguyen, S. Wooden, L. Bennett, T.Boone, G. Shimamoto, M. DeRose, R. Elliott, A. Colombero,H.L. Tan, G. Trail, J. Sullivan, E. Davy, N. Bucay, L.Renshaw-Gegg, T.M. Hughes, D. Hill, W. Pattison, P. Campbell,W.J. Boyle, et al., Osteoprotegerin: a novel secreted proteininvolved in the regulation of bone density (see comments), Cell 89(1997) 309–319.
[22] H. Yasuda, N. Shima, N. Nakagawa, K. Yamaguchi, M.Kinosaki, S. Mochizuki, A. Tomoyasu, K. Yano, M. Goto, A.Murakami, E. Tsuda, T. Morinaga, K. Higashio, N. Udagawa,N. Takahashi, T. Suda, Osteoclast differentiation factor is aligand for osteoprotegerin/osteoclastogenesis-inhibitory factorand is identical to TRANCE/RANKL, Proc. Natl. Acad. Sci.USA 95 (1998) 3597–3602.
[23] V. Shalhoub, J. Faust, W.J. Boyle, C.R. Dunstan, M. Kelley, S.Kaufman, S. Scully, G. Van, D.L. Lacey, Osteoprotegerin andosteoprotegerin ligand effects on osteoclast formation fromhuman peripheral blood mononuclear cell precursors, J. Cell.Biochem. 72 (1999) 251–261.
[24] H. Hsu, D.L. Lacey, C.R. Dunstan, I. Solovyev, A. Colombero,E. Timms, H.L. Tan, G. Elliott, M.J. Kelley, I. Sarosi, L. Wang,X.Z. Xia, R. Elliott, L. Chiu, T. Black, S. Scully, C. Capparelli,S. Morony, G. Shimamoto, M.B. Bass, W.J. Boyle, Tumornecrosis factor receptor family member RANK mediates osteo-clast differentiation and activation induced by osteoprotegerinligand, Proc. Natl. Acad. Sci. USA 96 (1999) 3540–3545.
[25] S. Corisdeo, M. Gyda, M. Zaidi, B.S. Moonga, B.R. Troen, Newinsights into regulation of cathepsin K gene expression byosteoprotegerin ligand, Biochem. Biophys. Res. Commun. 285(2001) 335–339.
[26] S. Kamolmatyakul, W. Chen, Y.P. Li, Interferon-gamma down-regulates gene expression of cathepsin K in osteoclasts andinhibits osteoclast formation, J. Dent. Res. 80 (2001) 351–355.
[27] W. Huang, R.J. O�Keefe, E.M. Schwarz, Exposure to receptor–activator of NFkappaB ligand renders pre-osteoclasts resistant toIFN-gamma by inducing terminal differentiation, Arthritis Res.Ther. 5 (2003) R49–59.
[28] Y.P. Li, W. Chen, Characterization of mouse cathepsin K gene,the gene promoter, and the gene expression, J. Bone Miner. Res.14 (1999) 487–499.
[29] M.W. Pfaffl, A new mathematical model for relative quantifica-tion in real-time RT-PCR, Nucleic Acids Res. 29 (2001) e45.
[30] G. Motyckova, K.N. Weilbaecher, M. Horstmann, D.J. Rieman,D.Z. Fisher, D.E. Fisher, Linking osteopetrosis and pycnodysos-tosis: regulation of cathepsin K expression by the microphthalmiatranscription factor family, Proc. Natl. Acad. Sci. USA 98 (2001)5798–5803.
[31] H. Hirotani, N.A. Tuohy, J.T. Woo, P.H. Stern, N.A. Clipstone,The calcineurin/nuclear factor of activated T cells signalingpathway regulates osteoclastogenesis in RAW264.7 cells, J. Biol.Chem. 279 (2004) 13984–13992.
[32] M. Matsumoto, M. Kogawa, S. Wada, H. Takayanagi, M.Tsujimoto, S. Katayama, K. Hisatake, Y. Nogi, Essential role ofp38 mitogen-activated protein kinase in cathepsin K gene expres-sion during osteoclastogenesis through association of NFATc1and PU.1, J. Biol. Chem. 279 (2004) 45969–45979.
[33] A.F. Martinez, M. Pang, J. Jacobs, W. Balkan, B.R. Troen,RANK ligand stimulates cathepsin K expression via TRAF6 andc-Jun, J. Bone Miner. Res. 19 (Suppl. 1) (2004) S297.
[34] H. Takayanagi, K. Ogasawara, S. Hida, T. Chiba, S. Murata, K.Sato, A. Takaoka, T. Yokochi, H. Oda, K. Tanaka, K. Nakam-ura, T. Taniguchi, T-cell-mediated regulation of osteoclastogen-esis by signalling cross-talk between RANKL and IFN-gamma,Nature 408 (2000) 600–605.
[35] Y. Kadono, T. Akiyama, T. Ogata, I. Nakamura, H. Oda, K.Nakamura, S. Tanaka, TRAF6 expression promoted byRANKL/RANK signaling plays an important role in osteoclastdifferentiation, J. Bone Miner. Res. 16 (Suppl. 1) (2001) S176.
[36] J.M. Fakruddin, J. Laurence, HIV envelope gp120-mediatedregulation of osteoclastogenesis via receptor activator of nuclearfactor kappa B ligand (RANKL) secretion and its modulation bycertain HIV protease inhibitors through interferon-gamma/RANKL cross-talk, J. Biol. Chem. 278 (2003) 48251–48258.
[37] A. Nii, D.A. Reynolds, H.A. Young, J.M. Ward, Osteochondro-dysplasia occurring in transgenic mice expressing interferon-gamma, Vet. Pathol. 34 (1997) 431–441.
[38] J.E. Darnell Jr., I.M. Kerr, G.R. Stark, Jak-STAT pathways andtranscriptional activation in response to IFNs and other extracel-lular signaling proteins, Science 264 (1994) 1415–1421.
[39] T. Yamagata, J. Nishida, S. Tanaka, R. Sakai, K. Mitani, M.Yoshida, T. Taniguchi, Y. Yazaki, H. Hirai, A novel interferonregulatory factor family transcription factor, ICSAT/Pip/LSIRF,that negatively regulates the activity of interferon-regulated genes,Mol. Cell. Biol. 16 (1996) 1283–1294.
[40] S.J. Gobin, M. van Zutphen, A.M. Woltman, P.J. van den Elsen,Transactivation of classical and nonclassical HLA class I genesthrough the IFN-stimulated response element, J. Immunol. 163(1999) 1428–1434.
[41] G. Schwarz, W.H. Boehncke, M. Braun, C.J. Schroter, T. Burster,T. Flad, D. Dressel, E. Weber, H. Schmid, H. Kalbacher,Cathepsin S activity is detectable in human keratinocytes and isselectively upregulated upon stimulation with interferon-gamma,J. Invest. Dermatol. 119 (2002) 44–49.

M. Pang et al. / Biochemical and Biophysical Research Communications 328 (2005) 756–763 763
[42] R. Lemaire, G. Huet, F. Zerimech, G. Grard, C. Fontaine, B.Duquesnoy, R.M. Flipo, Selective induction of the secretion ofcathepsins B and L by cytokines in synovial fibroblast-like cells,Br. J. Rheumatol. 36 (1997) 735–743.
[43] H. Schmid, R. Sauerbrei, G. Schwarz, E. Weber, H. Kalbacher,C. Driessen, Modulation of the endosomal and lysosomaldistribution of cathepsins B, L and S in human monocytes/macrophages, Biol. Chem. 383 (2002) 1277–1283.
[44] C. Beers, K. Honey, S. Fink, K. Forbush, A. Rudensky,Differential regulation of cathepsin S and cathepsin L in
interferon gamma-treated macrophages, J. Exp. Med. 197(2003) 169–179.
[45] G.K. Sukhova, G.P. Shi, D.I. Simon, H.A. Chapman, P. Libby,Expression of the elastolytic cathepsins S and K in humanatheroma and regulation of their production in smooth musclecells, J. Clin. Invest. 102 (1998) 576–583.
[46] K. Storm van�s Gravesande, M.D. Layne, Q. Ye, L. Le, R.M.Baron, M.A. Perrella, L. Santambrogio, E.S. Silverman, R.J.Riese, IFN regulatory factor-1 regulates IFN-gamma-dependentcathepsin S expression, J. Immunol. 168 (2002) 4488–4494.
Copyright © 2022 FDOKUMEN




















