
Protective effect of vitamin E on ultraviolet B light-induced damage in keratinocytes
10
ARTICLES Protective Effect of Vitamin E on Ultraviolet B Light–Induced Damage in Keratinocytes Samar Maalouf, 1 Marwan El-Sabban, 2 Nadine Darwiche, 1 and Hala Gali-Muhtasib 1 * 1 Department of Biology, American University of Beirut, Beirut, Lebanon 2 Department of Human Morphology, American University of Beirut, Beirut, Lebanon Ultraviolet (UV) B radiation is the most common environmental factor in the pathogenesis of skin cancer. Exposure of human skin to UVB radiation leads to the depletion of cutaneous antioxidants, the activation of nuclear factor kappa B (NF-kB), and programmed cell death (apoptosis). Although antioxidant supplementation has been shown to prevent UVB-induced photooxidative damage, its effect on components of cell signaling pathways leading to gene expression has not been clearly established. In the present study, the effect of the antioxidant vitamin, a-tocopherol (a-T), and its acetate analog, a-tocopherol acetate (a-TAc), on UVB-induced damage in primary and neoplastic mouse keratinocytes was investigated. The ability of both vitamins to modulate UVB-induced apoptosis and activation of the transcription factor NF-kB were studied. Treatment of normal and neoplastic mouse epidermal keratinocytes (308 cells) with 30–60 mJ/cm 2 UVB markedly decreased viable cell number and was accompanied by DNA fragmentation. When both vitamins were applied to cells at times before and after UVB radiation, a significant increase in the percentage of viable cells and concomitant decrease in the number of apoptotic cells was noted, with vitamin pretreatment providing a better protection than posttreatment. Simultaneous posttreatment of irradiated cells with a-TAc abolished the cytotoxic effects of UVB and restored cell viability to control levels. In addition, simultaneous posttreatment of irradiated cells with a-T reduced the number of apoptotic cells by half, indicating a synergistic effect of two such treatments compared with any single one. Flow cytometry analysis indicated that vitamin treatment suppressed both an increase in pre-G 0 cells and a decrease in cycling cells by UVB exposure. In addition, NF-kB activation was detected 2 h after UV exposure and was maintained for up to 8 h. Pretreatment with vitamins significantly inhibited NF-kB activation at 4 and 8 h. These results indicate that vitamin E and its acetate analog can modulate the cellular response to UVB partly through their action on NF-kB activation. Thus, these antioxidant vitamins are potential drugs for the protection from or the reduction of UVB-associated epidermal damage. ß 2002 Wiley-Liss, Inc. Key words: antioxidants; photoprotection; nuclear factor kappa B; reactive oxygen species; skin INTRODUCTION Ultraviolet (UV) radiation is the major environ- mental cause of skin damage. Although only 0.5% of UVB radiation (290–320 nm) reaches the Earth, it is the main cause of sunburn and probably the most carcinogenic component of sunlight [1,2]. Exposure of mammalian skin to UV light impairs the enzymic and nonenzymic antioxidants [3,4] and increases the cellular levels of reactive oxygen species (ROS), which, in turn, damage lipids, proteins and nucleic acids in epidermal cells and are likely to contribute to the process of photocarcinogenesis and photo- aging [5,6]. The increase in ROS is accompanied by the activation of many ROS-sensitive signaling pathways and induced gene transcription [7]. In addition, exposure of cells to UVB radiation results in the loss of keratinocyte viability, an increase in membrane blebbing [8], cytoskeletal molecular changes [9–12], and apoptosis [13,14]. One important group of transcription factors induced by UV radiation is composed of members of the Rel/NF-kB family [15], which are known to play a major role in the transcriptional activation of many genes encoding inflammatory cytokines, adhesion molecules, and viral proteins. Five mem- bers of the NF-kB family have been identified so far in mammals: p50-NF-kB1, p52-NF-kB2, p65/RelA, MOLECULAR CARCINOGENESIS 34:121–130 (2002) ß 2002 WILEY-LISS, INC. *Correspondence to: Department of Biology, American University of Beirut, Beirut, Lebanon. E-mail: [email protected] Received 17 January 2002; Revised 16 April 2002; Accepted 23 April 2002 Abbreviations: UV, ultraviolet; ROS, reactive oxygen species; NF- kB, nuclear factor kappa B; a-T, a-tocopherol; a-TAc, a-tocopherol acetate; SMEM, Spinner modified Eagle’s medium; PBS, Dulbecco’s phosphate-buffered saline; PMK, primary mouse epidermal kerati- nocytes; DTT, dithiothreitol. DOI 10.1002/mc.10055
Transcript of Protective effect of vitamin E on ultraviolet B light-induced damage in keratinocytes

ARTICLES
Protective Effect of Vitamin E on Ultraviolet BLight–Induced Damage in Keratinocytes
Samar Maalouf,1 Marwan El-Sabban,2 Nadine Darwiche,1 and Hala Gali-Muhtasib1*1Department of Biology, American University of Beirut, Beirut, Lebanon2Department of Human Morphology, American University of Beirut, Beirut, Lebanon
Ultraviolet (UV) B radiation is the most common environmental factor in the pathogenesis of skin cancer. Exposureof human skin to UVB radiation leads to the depletion of cutaneous antioxidants, the activation of nuclear factorkappa B (NF-kB), and programmed cell death (apoptosis). Although antioxidant supplementation has been shown toprevent UVB-induced photooxidative damage, its effect on components of cell signaling pathways leading to geneexpression has not been clearly established. In the present study, the effect of the antioxidant vitamin, a-tocopherol(a-T), and its acetate analog, a-tocopherol acetate (a-TAc), on UVB-induced damage in primary and neoplastic mousekeratinocytes was investigated. The ability of both vitamins to modulate UVB-induced apoptosis and activation of thetranscription factor NF-kB were studied. Treatment of normal and neoplastic mouse epidermal keratinocytes (308cells) with 30–60 mJ/cm2 UVB markedly decreased viable cell number and was accompanied by DNA fragmentation.When both vitamins were applied to cells at times before and after UVB radiation, a significant increase in thepercentage of viable cells and concomitant decrease in the number of apoptotic cells was noted, with vitaminpretreatment providing a better protection than posttreatment. Simultaneous posttreatment of irradiated cells witha-TAc abolished the cytotoxic effects of UVB and restored cell viability to control levels. In addition, simultaneousposttreatment of irradiated cells with a-T reduced the number of apoptotic cells by half, indicating a synergistic effectof two such treatments compared with any single one. Flow cytometry analysis indicated that vitamin treatmentsuppressed both an increase in pre-G0 cells and a decrease in cycling cells by UVB exposure. In addition, NF-kBactivation was detected 2 h after UV exposure and was maintained for up to 8 h. Pretreatment with vitaminssignificantly inhibited NF-kB activation at 4 and 8 h. These results indicate that vitamin E and its acetate analog canmodulate the cellular response to UVB partly through their action on NF-kB activation. Thus, these antioxidantvitamins are potential drugs for the protection from or the reduction of UVB-associated epidermal damage.� 2002 Wiley-Liss, Inc.
Key words: antioxidants; photoprotection; nuclear factor kappa B; reactive oxygen species; skin
INTRODUCTION
Ultraviolet (UV) radiation is the major environ-mental cause of skin damage. Although only 0.5%of UVB radiation (290–320 nm) reaches the Earth, itis the main cause of sunburn and probably the mostcarcinogenic component of sunlight [1,2]. Exposureof mammalian skin to UV light impairs the enzymicand nonenzymic antioxidants [3,4] and increasesthe cellular levels of reactive oxygen species (ROS),which, in turn, damage lipids, proteins and nucleicacids in epidermal cells and are likely to contributeto the process of photocarcinogenesis and photo-aging [5,6]. The increase in ROS is accompanied bythe activation of many ROS-sensitive signalingpathways and induced gene transcription [7]. Inaddition, exposure of cells to UVB radiation resultsin the loss of keratinocyte viability, an increase inmembrane blebbing [8], cytoskeletal molecularchanges [9–12], and apoptosis [13,14].
One important group of transcription factorsinduced by UV radiation is composed of membersof the Rel/NF-kB family [15], which are known toplay a major role in the transcriptional activation ofmany genes encoding inflammatory cytokines,adhesion molecules, and viral proteins. Five mem-bers of the NF-kB family have been identified so farin mammals: p50-NF-kB1, p52-NF-kB2, p65/RelA,
MOLECULAR CARCINOGENESIS 34:121–130 (2002)
� 2002 WILEY-LISS, INC.
*Correspondence to: Department of Biology, American Universityof Beirut, Beirut, Lebanon. E-mail: [email protected]
Received 17 January 2002; Revised 16 April 2002; Accepted 23April 2002
Abbreviations: UV, ultraviolet; ROS, reactive oxygen species; NF-kB, nuclear factor kappa B; a-T, a-tocopherol; a-TAc, a-tocopherolacetate; SMEM, Spinner modified Eagle’s medium; PBS, Dulbecco’sphosphate-buffered saline; PMK, primary mouse epidermal kerati-nocytes; DTT, dithiothreitol.
DOI 10.1002/mc.10055

c-Rel, and RelB [reviewed in 16]. Typically NF-kBconsists of a dimer composed of the p50 and p65subunits. UVB-induced activation of NF-kB is medi-ated by the oxidative state of the cell [17,18] andoccurs in nuclear-free extracts only if the membranefraction is present [19], suggesting that the oxidativemodification of membrane components is impor-tant in UV-dependent NF-kB activation [17–19].This activation involves the phosphorylation of theNF-kB inhibitory subunit, which is then degradedby proteasomes [20]. The NF-kB complex is thenfree to translocate into the nucleus, bind to targetDNA sequences, and activate genes involved inimmune and inflammatory responses, cell growth,and apoptosis [21].
Recently, emphasis has been placed on identify-ing cancer chemopreventive agents, such as vita-mins, that can protect against UV-induced skindamage [22–24]. Vitamin E is a potent antioxidantthat can protect against free-radical damage causedby skin exposure to UVB light [25–27]. Althoughvitamin E is known to protect against free-radicaldamage by both acting as a free radical scavengerand a stabilizer of the cell surface and subcellularmembranes [28,29], its exact cellular and molecularmechanisms of action have not been well investi-gated. Since many antioxidants are known to elicitphotoprotection through modulation of NF-kBactivation [30], we aimed at studying whethervitamin E is capable of protecting against thecytotoxic effects of UVB radiation by inhibitingUVB-induced apoptosis and by modulating NF-kBactivation. Here we show that vitamin E and itsacetate analogue protected against UVB-associatedepidermal damage, increased cellular viability, andreduced the number of apoptotic cells in irradiatedprimary and neoplastic mouse keratinocytes. Thedrugs’ efficacy as photo-protectants may partlyresult from their ability to inhibit the activation ofNF-kB. These results demonstrate a new rationalefor the use of vitamin E to prevent UVB-inducedepidermal damage.
MATERIALS AND METHODS
Reagents and Drugs
Vitamin E (a-T) was purchased from SigmaChemical Company (St. Louis, MO). Its acetateanalog (a-TAc) was kindly provided by Dr. HoussamEl-Sabban (Nature Made, Mission Hills, CA.).Hoechst 33342. Prolong Antifade and propidiumiodide were purchased from Molecular Probes(Eugene, OR). RNase and trypan blue were obtainedfrom Sigma Chemical Co. Spinner modified Eagle’smedium (SMEM), Dulbecco’s phosphate-bufferedsaline (PBS), fetal bovine serum, dispase, sodiumbicarbonate, and penicillin-streptomycin were allobtained from Gibco BRL Life Technologies (GrandIsland, NY).
Cell Cultures
The 308 neoplastic epidermal cells and primarymouse epidermal keratinocytes (PMK) were used inexperiments. The 308 cells (gift from Dr. S.H. Yuspa,NIH, Bethesda, MD) are a papilloma-derived cell linefrom adult BALB/c mouse skin initiated in vivo with7,12-dimethylbenz[a]anthracene [31]. Normal mousekeratinocytes were isolated from newborn BALB/cmice. One- to two-day-old neonatal BALBc micewere killed on ice and their skins floated in 0.25%dispase in SMEM overnight at 48C. The epidermiswas peeled from the dermis and incubated in 0.25%trypsin for 15 min at 378C, and the cell suspensionpassed through a 40-mm cell strainer. Cells werecultured in a humidified incubator (378C) in 95%air and 5% CO2 and were maintained in SMEMsupplemented with L-glutamine (2 mM), penicillin(100 U/ml) streptomycin (100 mg/mL), and 8%dialyzed fetal bovine serum. In all experiments,308 cells were used at low passages and seeded at adensity of 0.8� 105 cells in 35-mm dishes or0.4� 106 cells in 100-mm dishes. Normal mousekeratinocytes were seeded at a density of two mouseequivalents per 100 mm dish and used at 3 d inculture.
Irradiation Procedure
UV radiation was generated using a parallel bankof 2 FS40T12 Fluorescent sunlamps (Phillips) loca-lized in a sterile hood emitting a continuousspectrum between 280 and 320 nm with a peakemission at 312 nm. The UVB output in mW/cm2
was measured with a UVX digital radiometerequipped with a UVX-31 midrange sensor and fittedwith a UVB filter sensitive to radiation between 280and 340 nm. The total energy output was calibratedby adjusting the time of exposure to the lightsource. Under these conditions, the UVB radiantflux to 308 cells was 60 mJ/cm2, while the flux tonormal keratinocytes was restricted to 30 mJ/cm2
due to the greater sensitivity of these normal cells tothe damaging effects of UVB light. Prior to irradia-tion, the cell-culture dishes containing keratino-cytes were washed with PBS, placed without coversin a thin layer of PBS, and irradiated at a distance of60 cm from the center of the lamps. After irradia-tion, the cells were replenished with medium andleft for 24 h in the incubator before harvest.
Vitamin E Treatments
Cells were treated with a-T or a-TAc at doses of100 and 60 mg/mL, respectively. Vitamins weredissolved in absolute ethanol such as the concentra-tion of ethanol did not exceed 0.1%. Vitamins wereadded to 308 cells following different protocols: (1)cells were pre-incubated with vitamins for 48 hbefore exposure to UVB; (2) cells were treated withvitamins 0, 3, 6 or 12 h after irradiation; (3) cells
122 MAALOUF ET AL.

were pre-incubated with vitamins for 48 h beforeexposure and replenished 0, 3, 6 or 12 h after ir-radiation. The vitamins were not present duringexposure itself. All control cells were treated with0.1% ethanol. Cells were observed using an invertedmicroscope 24 h after UVB treatment.
Cell Viability Assays
Cells seeded in six-well plates were treated witha-T (100 mg/mL) or a-TAc (60 mg/mL) 24 h afterplating, irradiated with UVB and harvested 24 hfollowing exposure. Counting cells at predeter-mined intervals using trypan blue exclusion assaysrevealed cell viability. Each experiment was per-formed in three replicates and repeated twice.
Apoptosis and Cell Cycle Analysis
Apoptosis was scored either by assessing thefraction of cells with a sub-G0/G1 DNA content byflow cytometry or by estimating the extent of apop-totic bodies using nuclear staining techniques. Forflow cytometry studies, cells were seeded in 100-mmdishes, incubated, allowed to grow to 60% con-fluency, and then treated as described earlier. Cellswere then harvested, washed twice with PBS,permeabilized with 70% ethanol at 08C, treatedwith 0.2 mg/mL RNase, and finally stained with1 mg/mL propidium iodide. Distribution of cell-cycle phases with different DNA contents was readin a FACScan flow cytometer (Becton-Dickinson,San Jose, CA) using the CellQuest program. Datawere analyzed using ModFit software. For DNAstaining studies, cells were plated on autoclavedglass coverslips in six-well culture plates and treatedas previously mentioned. The medium was thenaspirated and cells were washed twice with warmPBS. Cellular DNA was stained with 1 mL Hoechst33342 stain (2 mg/mL) for 20 min in the dark and atroom temperature. Cells were then washed withPBS, fixed in 4% formaldehyde for 4 h at roomtemperature, mounted on slides using ProlongAntifade, and subsequently stored at 48C untilanalysis. Nuclear chromatin condensation wasobserved under fluorescence microscopy (LSM 410,Zeiss, Germany).
Nuclear Protein Extraction
Cells were washed with ice-cold PBS and scrapedoff in 70 mL of buffer containing 10 mM HEPESpH 7.9, 10 mM KCl, 1.5 mM MgCl2, and 1 mMdithiothreitol (DTT). Following centrifugation at4000 rpm for 10 min at 48C, the pellet wasresuspended in 15 mL of buffer containing 20 mMHEPES pH 7.9, 0.4 M NaCl, 1.5 mM MgCl2, 25%(vol/vol) glycerol, 0.2 mM EDTA, 1 mM DTT, and0.5 mM phenylmethylsulfonyl fluoride. The nucleiwere extracted by gentle mixing for 30 min at 48Cfollowed by centrifugation at 14 000 rpm for 20 min
(48C). The supernatant was then diluted with 30 mLof buffer containing 20 mM HEPES pH 7.9, 50 mMKCl, 20% glycerol, 0.2 mM EDTA, 1 mM DTT, and0.5 mM phenylmethylsulfonyl fluoride and storedat �708C until analysis. Protein concentration wasdetermined using DC Protein assay kit (BioRad,Hertfordshire, England).
Western Blot Analysis
Equal amounts of nuclear extracts (30 mg ofprotein) were resolved on 10% polyacrylamide gelsunder denaturing conditions. After electrophoresis,proteins were transferred to nitrocellulose mem-brane. Membranes were then blocked in a washbuffer (100 mM Tris-HCl buffer, pH 7.5, 150 mMNaCl, and 0.05% Tween 20) with 5% nonfat driedmilk. The membranes were further incubated inrabbit polyclonal antiserum to NF-kB subunit RelA(C20) (Santa Cruz Biotechnology, Santa Cruz, CA),and washed three times for 10 min each time toremove unbound antiserum. Anti-RelA was diluted1:1000 in wash buffer with 5% nonfat dried milkand incubated for 1 h at room temperature. Blotswere rinsed in wash buffer and incubated for 45 minat room temperature with peroxidase-conjugatedsecondary antibodies from rabbit (Santa CruzBiotechnology) diluted in blocking buffer. Boundantibody was then visualized using an ECL chemi-luminescence detection kit (Amersham, Bucking-hamshire, England) following the manufacturer’sinstructions. Equal protein loading was verifiedusing Comassie protein stain.
Electrophoretic Mobility Shift Assay andSupershift Analysis
NF-kB consensus oligonucleotide (Santa CruzBiotechnology) was end-labeled with [g-32P]ATPusing T4 polynucleotide kinase. Hybridization reac-tion was performed using 10 mg of nuclear proteinextract, 1 mg of poly(dIdC), 1 mg of poly(dN6) asnonspecific competitor, and 10 mg of bovine serumalbumin in 20 mM HEPES, 50 mM KCl, 1 mM EDTA,and 5 mM DTT. This reaction was diluted with waterbefore the labeled probe was added. The mixturewas incubated for 30 min and then stopped byadding 6 mL of 15% Ficoll. The reaction mixture(20 mL of each sample) was electrophoresed on a 4%non-denaturing polyacrylamide gel, which wasdried at 708C under vacuum for 2 h on Whatmanfilter paper, and processed for autoradiography at�708C overnight. Specific NF-kB bands were deter-mined by competition experiments using a mutantoligonucleotide that has lost its ability to bind to thetranscription factor. Finally, to assay for the speci-ficity of the subunit, 1 mg of polyclonal antibodiesagainst different subunits of NF-kB (Santa CruzBiotechnology), known to supershift NF-kB com-plexes, was incubated with nuclear proteins for10 min prior to addition of 32P-labeled NF-kB probe.
PHOTOPROTECTION BY VITAMIN E 123

RESULTS
UVB and Vitamin E Effects on Cell Morphology
Using light microscopy, we investigated theeffects of UVB radiation on the morphological fea-tures of keratinocytes and the capability of vita-min E to protect the cells from injury (Figure 1). Themorphology of primary keratinocytes and of 308cells did not change upon treatment with 100 mg/mL a-T (not shown) or 60 mg/mL a-TAc (Figure 1).Treatment of 308 cells with 60 mJ/cm2 UVB or PMKwith 30 mJ/cm2 UVB induced wide alterations in themorphology of cells as seen by the loss of squamous-like appearance and rounding of cells. Pretreatmentwith a-TAc protected both keratinocyte cultures(better protection in 308 cells) from the above-mentioned UV-induced lesions as demonstrated bythe overall higher density of normal appearing cells(Figure 1). Protection against UV-induced damage ofcell morphology was also observed when bothvitamins were given after UVB irradiation of cells(not shown).
Protective Effects of Vitamin E on UVB Cytotoxicity
In an attempt to further analyze the observedeffects of UVB radiation on epidermal cell morphol-
ogy, a quantitative evaluation of live and dead cellswas performed (Figure 2). Treatment with eithervitamin at concentrations up to 200 mM had noeffect on the viability of 308 cells, while PMK wereslightly more sensitive to vitamin treatment (Figure2a). When challenged by UVB exposure, 308 cellsexhibited dose-dependent decrease in cell viability(Figure 2b). Pretreatment of 308 cells with a-T(Figure 2c) or a-TAc (Figure 2d) increased theviability of UVB-irradiated cells by 30–60%, withthe acetate analog being more effective. Posttreat-ment with a-TAc immediately after irradiation (0 h)improved cell viability to levels significantly greaterthan those observed in delayed posttreatments(Figure 2d). When a-TAc was applied before andimmediately after UVB, the cytotoxic effects of UVBradiation on epidermal keratinocytes were abol-ished and cell viability was restored to controlnonirradiated cells (Figure 2c and d), indicative ofcomplete protection from UVB irradiation.
Protective Effects of Vitamin E onUVB-Induced Apoptosis
Exposure of 308 cells to 60 mJ/cm2 UVB radiationincreased the number of Hoechst positive apoptoticcells up to 40% (Figure 3). We then investigated the
Figure 1. Morphological effects of UVB and/or a-TAc on PMK and neoplastic 308 cells. Cells grown understandard conditions to approximately 60% confluency were either left untreated or irradiated with 60 mJ/cm2 or30 mJ/cm2 UVB for 24 h or pretreated with 60 mg/mL a-TAc for 48 h prior to irradiation and photographed 24 hafter exposure. All cells were treated with vehicle (ethanol) at a concentration of 0.1%.
124 MAALOUF ET AL.

protective effects of vitamins on UVB-inducedapoptosis. Simultaneous pretreatment and post-treatment of irradiated cells with a-T was the mosteffective at reducing the number of apoptotic cellsby 50% (Figure 3), indicating a synergistic effect oftwo treatments compared with a single one.
UVB and Vitamin E Effects onCell Cycle Progression
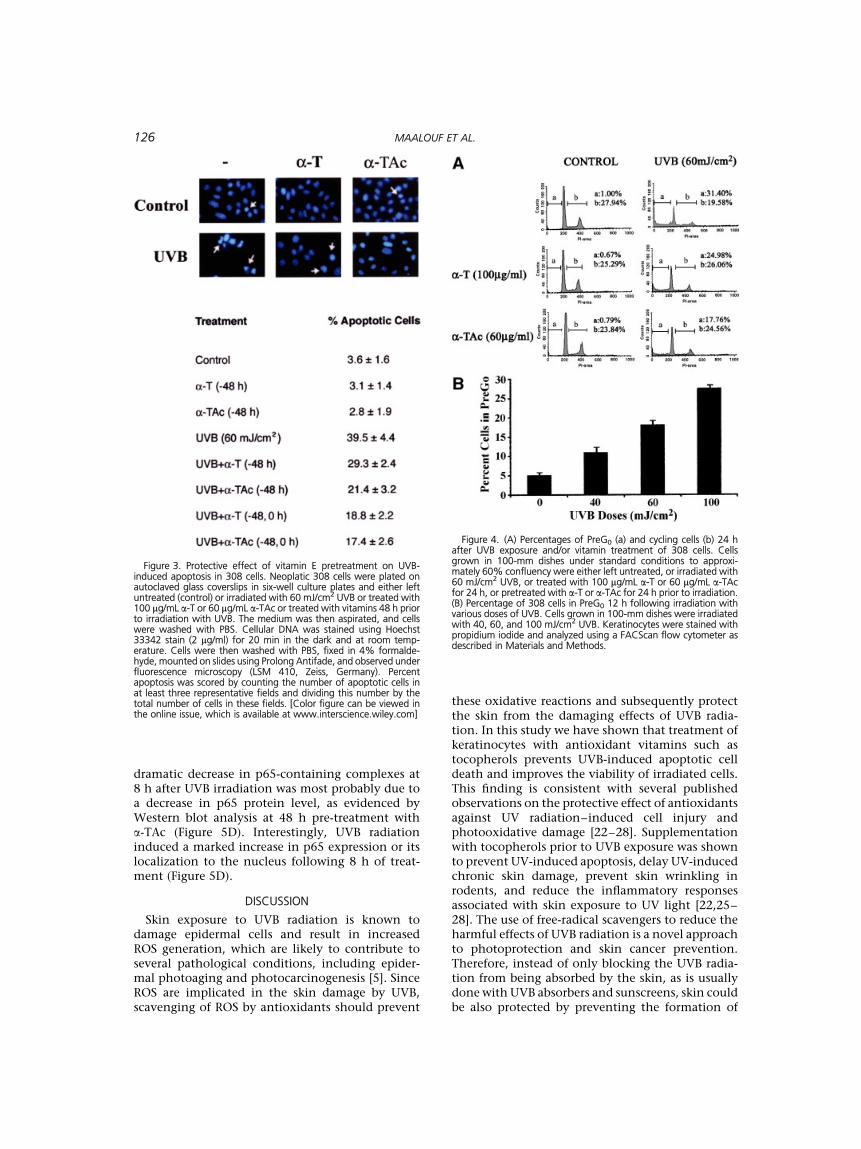
To study the role of UVB and vitamin treatmentson cell-cycle progression, we examined cell cycledistribution profiles by flow cytometry using propi-diumiodidestainingofDNAcontent(Figure4).Treat-ment with UVB doses of 40, 60, and 100 mJ/cm2 for12 h prevented G1 to S phase progression and causeda respective increase of 120, 260, and 450% in pre-G0 (apoptotic) cells with respect to control un-treated cells (Figure 4B). All tested doses of eithervitamin did not affect cell-cycle progression. Theprotective effects of a 48-h pretreatment with a-T orits acetate analog were demonstrated by their abilityto reduce the percentage of apoptotic cells from40% to 29% and 21%, respectively (Figure 3). Thisdecrease in apoptotic pre-G0 cells was accompaniedby a 20–25% increase in the proportion of cyclingcells (Figure 4A).
Vitamin E Modulation of UVB-InducedNF-kB Activation
Several studies have shown that UVB radiationcauses NF-kB activation that is subject to regulationby cellular redox status [17–19,30]. Therefore, weexamined whether a-TAc could modulate UVB-induced NF-kB DNA binding protein complexes.Treatment of 308 cells with 60 mJ/cm2 UVBactivated a factor that bound to a radiolabeled kBconsensus oligonucleotide, and this activation wasmaximal 8 h postirradiation (Figure 5A). Thespecificity of binding was demonstrated as excessof labeled kB consensus oligonucleotide competedfor binding but not an excess of unlabeled mutantoligonucleotide (Figure 5B). In order to determinethe subunit composition of these putative NF-kBcomplexes, we used Rel-A (p65)- or p50-specificantibodies to induce a supershift. Treating nuclearprotein extracts with the antibodies specific to thep65 but not to the p50 subunit of NF-kB retarded theelectrophoretic migration of the factor that isactivated by UVB radiation (Figure 5C). Treatmentof cells with a-TAc alone did not significantly alterNF-kB complex formation while pre-treatment ofirradiated cells with vitamin reduced NF-kB DNAbinding at 4 and 8 h after UVB treatment. The
Figure 2. Effects of UVB, vitamin E, and their combination onthe viability of 308 cells (a, b, c, and d) or PMK (a and b). Cells seededin six-well plates at approximately 105 cells per well were eithertreated for 48 h with different doses of vitamins ranging from 0 to200 mM (a), irradiated with various doses of UVB (0, 30, 40, 60, and
100 mJ/cm2) for 24 h (b), treated with a-T (c) or a-TAc (d) fordifferent times before (�48 h), after (0, 6, and 12 h) or before andafter (�48 and 0 h) irradiation with 60 mJ/cm2 UVB. The valuesdepict an average (�SD) of quadruplicate measurements of at leasttwo independent experiments.
PHOTOPROTECTION BY VITAMIN E 125
dramatic decrease in p65-containing complexes at8 h after UVB irradiation was most probably due toa decrease in p65 protein level, as evidenced byWestern blot analysis at 48 h pre-treatment witha-TAc (Figure 5D). Interestingly, UVB radiationinduced a marked increase in p65 expression or itslocalization to the nucleus following 8 h of treat-ment (Figure 5D).
DISCUSSION
Skin exposure to UVB radiation is known todamage epidermal cells and result in increasedROS generation, which are likely to contribute toseveral pathological conditions, including epider-mal photoaging and photocarcinogenesis [5]. SinceROS are implicated in the skin damage by UVB,scavenging of ROS by antioxidants should prevent
these oxidative reactions and subsequently protectthe skin from the damaging effects of UVB radia-tion. In this study we have shown that treatment ofkeratinocytes with antioxidant vitamins such astocopherols prevents UVB-induced apoptotic celldeath and improves the viability of irradiated cells.This finding is consistent with several publishedobservations on the protective effect of antioxidantsagainst UV radiation–induced cell injury andphotooxidative damage [22–28]. Supplementationwith tocopherols prior to UVB exposure was shownto prevent UV-induced apoptosis, delay UV-inducedchronic skin damage, prevent skin wrinkling inrodents, and reduce the inflammatory responsesassociated with skin exposure to UV light [22,25–28]. The use of free-radical scavengers to reduce theharmful effects of UVB radiation is a novel approachto photoprotection and skin cancer prevention.Therefore, instead of only blocking the UVB radia-tion from being absorbed by the skin, as is usuallydone with UVB absorbers and sunscreens, skin couldbe also protected by preventing the formation of
Figure 3. Protective effect of vitamin E pretreatment on UVB-induced apoptosis in 308 cells. Neoplatic 308 cells were plated onautoclaved glass coverslips in six-well culture plates and either leftuntreated (control) or irradiated with 60 mJ/cm2 UVB or treated with100 mg/mL a-T or 60 mg/mL a-TAc or treated with vitamins 48 h priorto irradiation with UVB. The medium was then aspirated, and cellswere washed with PBS. Cellular DNA was stained using Hoechst33342 stain (2 mg/ml) for 20 min in the dark and at room temp-erature. Cells were then washed with PBS, fixed in 4% formalde-hyde, mounted on slides using Prolong Antifade, and observed underfluorescence microscopy (LSM 410, Zeiss, Germany). Percentapoptosis was scored by counting the number of apoptotic cells inat least three representative fields and dividing this number by thetotal number of cells in these fields. [Color figure can be viewed inthe online issue, which is available at www.interscience.wiley.com]
Figure 4. (A) Percentages of PreG0 (a) and cycling cells (b) 24 hafter UVB exposure and/or vitamin treatment of 308 cells. Cellsgrown in 100-mm dishes under standard conditions to approxi-mately 60% confluency were either left untreated, or irradiated with60 mJ/cm2 UVB, or treated with 100 mg/mL a-T or 60 mg/mL a-TAcfor 24 h, or pretreated with a-T or a-TAc for 24 h prior to irradiation.(B) Percentage of 308 cells in PreG0 12 h following irradiation withvarious doses of UVB. Cells grown in 100-mm dishes were irradiatedwith 40, 60, and 100 mJ/cm2 UVB. Keratinocytes were stained withpropidium iodide and analyzed using a FACScan flow cytometer asdescribed in Materials and Methods.
126 MAALOUF ET AL.

photooxidants that result in radical damage andcutaneous pathology.
The greatest overall protection by a-Ts as deter-mined by the maintenance of normal cell morphol-ogy, improved cell viability, and reduced apoptosisin irradiated epidermal cells, was mostly observedwhen these cells were treated with vitamins beforeand after UVB irradiation, indicating that combin-ing two vitamin treatments may be more beneficialfor skin photoprotection. Furthermore, addingvitamins to cells immediately after irradiation wasmore effective than subsequent delayed treatments.This is perhaps due to the ability of these com-pounds to scavenge free radicals induced at stagesimmediately after UVB irradiation. Normal mousekeratinocytes were more sensitive than the neoplas-tic epidermal cells to the toxic effects of UVBradiation and to UV-induced apoptosis. Toxicityeffects of irradiation in normal cells were notedupon the use of half the doses of UVB than thoseused in neoplastic cells. Perhaps these papillomacells have acquired mutations leading to their grea-ter resistance to UVB radiation than primary mouseepidermal keratinocytes. Contrary to neoplasticcells, vitamin treatment of normal keratinocytesdid not significantly protect against UV-inducedtoxicity (data not shown). The higher toxic effect of
UVB light on primary cells may explain whyvitamin treatment failed to significantly inhibitUV-induced effects in these cells. Previous studieshave shown that UVB irradiation of human kerati-nocyte HaCaT cells leads to phosphorylation ofp38 mitogen-activated protein kinase and c-jun N-terminal protein kinase and also a significant acti-vation of caspase-3 [29]. Furthermore, pretreatmentof these irradiated keratinocytes with mitogen-activated protein kinase and caspase-3 inhibitorsresults in 60% reduction of apoptosis, while variousantioxidants including a vitamin E analog (Trolox;Aldrich Chemical Co., Milwaukee, WI) did notaffect the caspase activation induced by UVB and,consequently, cell death. The greater sensitivity ofPMK to UV-induced damage and the lack of vitaminE protection in our normal mouse keratinocytescompared with neoplastic ones is noteworthy. Itwould be interesting to determine whether normalkeratinocytes rely less on ROS-mediated effects,since they possess normal apoptotic pathways thatare caspase-3 dependent, while their neoplasticcounterparts rely more on ROS-mediated mechan-isms to induce apoptosis following UVB damagebecause they lack non–ROS-mediated apoptoticpathways. However, Trolox was recently shown todecrease the rate of cell death induced by UVB
Figure 5. Effects of UVB, a-TAc, and their combination on NF-kBsignaling. Neoplastic 308 cells were left untreated (control) orirradiated with 60 mJ/cm2 UVB or treated with 100 mg/mL a-T or60 mg/mL a-TAc or treated with vitamins 24 h prior to irradiationwith UVB and harvested at 0, 2, 4 and 8 h after irradiation formeasurement of the activation of the transcription factor NF-kB byelectrophoretic mobility shift assay using consensus oligonucleotideprobes for NF-kB (A) as described in Materials and Methods. Specific
NF-kB bands were determined by competition experiments using amutant oligonucleotide that has lost its ability to bind to thetranscription factor (B). The specificity of the subunit was determinedby using 1 mg of polyclonal antibodies against the NF-kB componentsp50 and p65/RelA, which were incubated with nuclear proteins for10 min prior to addition of 32P-labeled NF-kB probe (C). Effects ofUVB, a-TAc, and their combination on the expression of p65/RelAprotein (D).
PHOTOPROTECTION BY VITAMIN E 127

irradiation in human keratinocytes [32]. Due to thedifferential sensitivity of PMK and 308 cells to UVBand protection by a-TAc, 308 cells rather than PMKwere used in later experiments to study the pro-tective effects of vitamin E on UV-induced apoptosisand the possible involvement of NFkB activation.Although the exact mechanism of protectionagainst cellular damage by vitamin E is not wellunderstood, insights could be gained from earlierfindings that vitamin E increases the stability of cellmembranes, prevents DNA photodamage, and pro-tects cells from intracytoplasmic and/or membranealterations induced by UV [28,33,34]. This vitamin Eprotective mechanism could be associated with anoxidative modification of membrane lipids, whichin turn may contribute to the maintenance ofcytoskeletal component assembly and integrity[8–12,33].
Exogenous supplementation of antioxidants hasbeen shown to prevent UVB-induced oxidativedamage, yet their effects on components of signal-ing pathways leading to gene expression are notwell understood. The NF-kB transcription factor isknown to be activated in response to UV radiationand to be regulated by the redox state of the cell[17]. This factor is also implicated in the inducibleexpression of a wide variety of genes that areinvolved in oxidative stress and cellular responsemechanism [35]. The regulation of NF-kB geneexpression by oxidants and antioxidants has emerg-ed as a novel area that has promising therapeuticimplication [35]. We observed an upregulation ofNF-kB activation in irradiated neoplastic epidermalcells at a UVB dose of 60 mJ/cm2, which is inagreement with many previous reports indicating alink between UV irradiation, oxidative stress, andNF-kB activation [15,17–19,30,35–37]. It appearsthat the UVB dose necessary to activate NF-kB differsaccording to the cell type and the keratinocytemodel. Although 15 mJ/cm2 UVB was enough toactivate NF-kB in human epidermoid carcinoma A-431 cells [19], the mouse keratinocyte cell line Balb/MK requires a dose of 50 mJ/cm2 for activation [36].In human skin fibroblasts, a minimal UVB dose of750 mJ/cm2 is required to activate NF-kB [37]. Whileusing a solar UV simulator, Saliou et al. [30] foundthat a dose of 150 mJ/cm2 UV is required for thenuclear translocation of NF-kB in the humankeratinocyte cell line HaCaT [30]. The inconsisten-cies reported in the various studies described areperhaps due to differences in the UV sources as wellas to varying cell-type sensitivities towards NF-kBactivation by UVB radiation. Our attempts toconfirm the identity of the DNA-binding activecomplex using supershift assays with antibodiesagainst p65 and p50 demonstrated a minor con-tribution of the p50 component, suggesting thatp65-p65 homodimers or p65-crel and p65-p52heterodimers may be the active complexes. We
have seen the same pattern in other cell systems wehave used such as Mod-K intestinal epithelial cells.
Although our present study is the first showingthat vitamin E inhibited NF-kB activation inducedby UVB in 308 mouse neoplastic keratinocytes,growing evidence indicates a role for antioxidants inmodulating UV-induced NF-kB activation. The freeradical scavengers a-lipoic acid, N-acetyl-L-cysteine,and silymarin have been shown to inhibit NF-kBactivation induced by UVR in HaCaT keratinocytes[30]. The interest in vitamin E as a photoprotectantstems from the fact that this vitamin is common inthe diet and displays remarkable in vitro and in vivoantioxidant activity that is a basis for its variousbeneficial biological properties [22–28]. Althoughthe exact mechanism by which vitamin E inhibitsNF-kB activation is not known, four possibilitiesexist. First, by scavenging ROS, vitamin E mayinhibit certain oxidant-sensitive protein kinasessuch as NF-kB–inducing kinase, the activation ofwhich results in the phosphorylation of NF-kB in-hibitory subunit kinases and the subsequent trans-location of NF-kB to the nucleus. Second, reducedthioredoxin, an important cellular protein oxidor-eductase with antioxidant functions, may reduceactivated NF-kB proteins, thus facilitating nucleartranslocation. Third, reduced thioredoxin maypromote in vitro DNA binding of NF-kB. Fourth,vitamin E may inhibit NF-kB transactivation. Theindications from clinical studies that vitamin Ederivatives are useful in the treatment of a widevariety of diseases associated with skin photodam-age [34] provide a strong rationale for subjectingthese antioxidants to further testing to unveil themolecular basis of their action.
Several reports have demonstrated the involve-ment of NF-kB in a variety of human diseases. Forexample, the upregulation of NF-kB has been shownto be associated with rheumatoid arthritis, asthma,and arteriosclerosis [38]. Furthermore, more re-cently it has been shown that NF-kB is activated byseveral oncoproteins, including ras, and NF-kBactivation is required for these oncoproteins toinduce cellular transformation, while NF-kB inhibi-tion blocks cell transformation and, in turn, blockstumor formation [39]. The ability of vitamin E toinhibit NF-kB activation points to the importance ofdeveloping this drug for potential pharmacologicalintervention in diseases associated with the dysre-gulation or activation of NF-kB, including chemo-prevention of UVB-induced skin carcinogenesis. It isworth mentioning that many dietary compoundsthat have been found to block the initiation ofcancer are also known to inhibit NF-kB activation[40].
Taken together, these results reconfirm the linkbetween exposure to UV radiation, generation ofROS, and NF-kB activation. The acute upregulationof NF-kB activation via UVB irradiation suggests that
128 MAALOUF ET AL.

its expression could be a potential pharmacologicaltarget mediating UV-induced skin tumor develop-ment. The results clearly point to a key role forantioxidants as skin photoprotectants and modula-tors of UVB-induced NF-kB–dependent gene expres-sion. Because NF-kB signaling impacts a number ofintracellular cascades governing the cell cycle,differentiation, apoptosis, cytokine release, andoncogenic pathways, it is not clear exactly howantioxidant vitamins modulate UVB-induced NF-kBactivation. Future studies will focus on the mechan-ism by which vitamin E minimizes the activation ofNF-kB by UVB light.
ACKNOWLEDGMENTS
This work was supported by funds from theUniversity Research Board of the American Uni-versity of Beirut and the Lebanese National Councilfor Scientific Research.
REFERENCES
1. De Gruijl FR, Sterenborg HJ, Forbes PD, et al. Thewavelength dependence of skin cancer induction byultraviolet irradiation of albino hairless mice. Cancer Res1993;53:53–60.
2. Vander Leun JC, De Gruijl FR. Influence of ozone depletionon human and animal health. In: Tevini M, editor. UV-Bradiation and ozone depletion: effects on humans, animals,plants, microorganisms and materials. Boca Raton, FL: LewisPublishers; 1993. p 95–123.
3. Fuchs J, Huflejt ME, Rothfuss LM, Wilson DS, Carcamo G,Packer L. Impairment of enzymic and nonenzymic antiox-idants in skin by UVB radiation. J Invest Dermatol 1989;93:769–773.
4. Shindo Y, Witt E, Han D, Packer L. Dose-response effects ofacute ultraviolet irradiation on antioxidants and molecularmarkers of oxidation in murine epidermis and dermis. JInvest Dermatol 1994;102:470–475.
5. Scharffetter-Kochanek K, Wlaschek M, Brenneisen P,Schauen M, Blaudschun R, Wenk J. UV-induced reactiveoxygen species in photocarcinogenesis and photoaging. BiolChem 1997;378(11):1247–1257.
6. Podda M, Traber MG, Weber C, Yan LJ, Packer L. UV-irradiation depletes antioxidants and causes oxidativedamage in a model of human skin. Free Radic Biol Med1998;24:55–65.
7. Schulze-Osthoff K, Bauer M, Vogt M. Reactive oxygenintermediates as primary signals and second messengers inthe activation of transcription factors. In: Forman DJ,Cadenas E, editors. Oxidative stress and signal transduction.New York: International Thomson Publishing; 1997. p 239–259.
8. Malorni W, Donelli G, Straface E, Santini MT, Paradisi S,Giacomoni PU. Both ultraviolet A and B induce cytoskeletal-dependent surface blebbing in epidermoid cells. J Photo-chem Photobiol B: Biol 1994;26:265–270.
9. Zamarski GB, Perrino GB, Chou IN. Disruption of cytoplas-mic microtubules by ultraviolet radiation. Exp Cell Res 1991;195:269–273.
10. Zamarski GB, Chou IN. Disruption of keratin intermediatefilaments by ultraviolet radiation in cultured human kera-tinocytes. Photochem Photobiol 1990;52:903–906.
11. Zamarski GB, Chou IN. Environmental wavelengths ofultraviolet light induced cytoskeletal damage. J InvestDermatol 1987;89:603–606.
12. Moll I, Bohnert E, Treib U, Jung ED. Effects of ultraviolet Bradiation on cytoskeletal and adhesion molecules in humanepidermis. Photodermatol Photoimmunol Photomed 1994;10:26–32.
13. Godar DE. Preprogrammed and programmed cell deathmechanisms of apoptosis: UV-induced immediate and de-layed apoptosis. Photochem Photobiol 1996;63:825–830.
14. Shindo Y, Hashimoto T. Ultraviolet B-induced cell death infour cutaneous cell lines exhibiting different enzymaticantioxidant defences: involvement of apoptosis. J DermatolSci 1998;17(2):140–150.
15. Li N, Karin M. Ionizing radiation and short wavelength UVactivate NF-kB through two distinct mechanisms. Proc NatlAcad Sci USA 1998;95:13012–13017.
16. Baurle PA, Baltimore D. NF-kB: Ten years after. Cell 1996;87:13–20.
17. Flohe L, Brigelius-Flohe R, Saliou C, Traber MG, Pecker L.Redox regulation of NF-Kappa B activation. Free Radic BiolMed 1997;22(6):1115–1126.
18. Schreck R, Albermann K, Baeuerle PA. Nuclear factor KappaB: an oxidative stress-responsive transcription factor ofeukaryotic cells (a review). Free Radic Res Commun 1992;17(4):221–237.
19. Simon MM, Aragane Y, Schwarz A, Luger TA, Schwarz T.UVB light induces nuclear factor kappa B (NF kappa B)activity independently from chromosomal DNA damage incell free cytosolic extracts. J Invest Dermatol 1994;102:422–427.
20. Baldwin AS. The NF-kB and IkB proteins: New discoveriesand insights. Annu Rev Immunol 1996;14:649–681.
21. Ghosh S, May M, Kopp E. NFkB and Rel proteins: Evolu-tionarily conserved mediators of the immunce response.Annu Rev Immunol 1998;16:225–260.
22. Bisset DL, Chattrjee R, Hannon DP. Photoprotective effect ofsuperoxide-scavenging antioxidants against ultraviolet radia-tion-induced chronic skin damage in the hairless mouse.Photodermatol Photoimmunol Photomed 1990;7:56–62.
23. Khettab N, Amory MC, Briand G, et al. Photoprotectiveeffect of vitamins A and E on polyamine and oxygenatedfree radical metabolism in hairless mouse epidermis.Biochimie 1988;70:1709–1713.
24. Bissett DL, Chatterjee R, Hannon DP. Chronic ultravioletradiation-induced increase in skin iron and the photopro-tective effects of topically applied iron chelators. PhotochemPhotobiol 1991;54:215–223.
25. Record IR, Dreosti LE, Konstantinopoulos M, Buckley RA.The influence of topical and systemic vitamin E on ultravioletlight-induced skin damage in hairless mice. Nutr Cancer1991;16:219–225.
26. Fryer M. Evidence for the photoprotective effects of vitaminE. Photochem Photobiol 1993;58:304–312.
27. Jurkiewicz BA, Bisset DL, Buettner GR. Effect of topicallyapplied tocopherol on ultraviolet radiation-mediated freeradical damage in skin. J Invest Dermatol 1995;104:484–488.
28. Forrest VJ, Kang Y-H, McClain DE, Robinson DH, Ramak-rishnan N. Oxidative stress-induced apoptosis prevented byTrolox. Free Radical Biol Med 1994;16:675–684.
29. Shimizu H, Banno Y, Sumi N, Naganawa T, Kitajima Y,Nozawa Y. Activation of p38 mitogen-activated proteinkinase and caspases in UVB-induced apoptosis of humankeratinocyte HaCaT cells. J Invest Dermatol 1999;112:769–774.
30. Saliou C, Kitazawa M, McLaughlin L, et al. Antioxidantsmodulate acute solar ultraviolet radiation-induced NF-kappa-B activation in a human keratinocytes cell line. FreeRad Biol Med 1999;26:174–183.
31. Yuspa SH, Hennings H, Tucker RW, Jaken S, Kilkenny AE,Roop DR. Signal transduction for proliferation and differ-entiation in keratinocytes. Ann NY Acad Sci 1988;548:191–196.
PHOTOPROTECTION BY VITAMIN E 129

32. Peus D, Meves A, Pott M, Beyerle A, Pittelkow MR. Vitamin Eanalog modulates UVB-induced signaling pathway activa-tion and enhances cell survival. Free Radic Biol Med 2001;30:425–432.
33. Malorni W, Starface E, Donelli G, Glacomoni PU. UV-induced cytoskeletal damage, surface blebbing and apop-tosis are hindered by a-tocopherol in cultured humankeratinocytes. Eur J Dermatol 1996;6:414–420.
34. McVean M, Liebler DC. Prevention of DNA photodamage byvitamin E compounds and sunscreens: Roles of ultravioletabsorbance and cellular uptake. Mol Carcinog 1999;24:169–176.
35. Sen CK, Packer L. Antioxidant and redox regulation of genetranscription. FASEB J 1996;10:709–720.
36. Tobin D, Nilsson M, Toftgard R. Ras-independent activationof Rel-family transcription factors by UVB and TPA incultured keratinocytes. Oncogene 1996;12:785–793.
37. Vile GF, Tanew-Ilitschew A, Tyrell RM. Activation of NF-kappa B in human skin fibroblasts by the oxidative stressgenerated by UVA radiation. Photochem Photobiol 1995;62:463–468.
38. Baldwin AS Jr. The transcription factor NF-kB and humandisease. J Clin Invest 2001;107(1):3–6.
39. Rayet B, Gelinas C. Aberrant Rel/NF-kB genes and activity inhuman cancer. Oncogene 1999;18:6948–6958.
40. Holmes-McNary M, Baldwin A. Chemopreventive propertiesof trans-resveratrol are associated with inhibition of the IkBkinase. Cancer Res 2000;60:3477–3483.
130 MAALOUF ET AL.




















