
Ultraviolet A-induced Modulation of Bcl-XL by p38 MAPK in Human Keratinocytes: POSTTRANSCRIPTIONAL...
12
Ultraviolet A-induced Modulation of Bcl-X L by p38 MAPK in Human Keratinocytes POST-TRANSCRIPTIONAL REGULATION THROUGH THE 3-UNTRANSLATED REGION* Received for publication, June 14, 2004, and in revised form, July 23, 2004 Published, JBC Papers in Press, August 2, 2004, DOI 10.1074/jbc.M406626200 Michael A. Bachelor and G. Timothy Bowden‡ From the Department of Cell Biology and Anatomy, Arizona Cancer Center, the University of Arizona, Tucson, Arizona 85724 We examined the effect of inhibiting p38 MAPK on UVA- irradiated HaCaT cells, a spontaneously immortalized hu- man keratinocyte cell line. Recent work from our labora- tory has shown that UVA (250 kJ/m 2 ) induces a rapid phosphorylation of p38 MAPK in the HaCaT cell line. Inhi- bition of p38 MAPK activity through the use of a specific inhibitor, SB202190, in combination with UVA treatment induced a rapid cleavage of caspase-9, caspase-8, and caspase-3, whereas UVA irradiation alone had no effect. Similarly, cleavage of the caspase substrate poly(ADP- ribose) polymerase was observed in UVA-irradiated HaCaT cells treated with SB202190 or in cells expressing a domi- nant-negative p38 MAPK. No effect of p38 MAPK inhibition upon caspase cleavage was observed in mock-irradiated HaCaT cells. In addition, increases in apoptosis were ob- served in UVA-irradiated cells treated with SB202190 by morphological analysis with no significant apoptosis occur- ring from UVA irradiation alone. Similar results were ob- tained by using normal human epidermal keratinocytes. UVA induced expression of the anti-apoptotic Bcl-2 family member, Bcl-X L , with abrogation of expression by using the p38 MAPK inhibitor SB202190. Overexpression of Bcl-X L prevented poly(ADP-ribose) polymerase cleavage induced by the combination of UVA and p38 MAPK inhibition. UVA enhanced the stability of Bcl-X L mRNA through increases in p38 MAPK activity. We determined that increases in UVA-induced expression of Bcl-X L occur through a post- transcriptional mechanism mediated by the 3-untrans- lated region (UTR). We used Bcl-X L 3-UTR luciferase con- structs to determine the mechanism by which UVA increased Bcl-X L mRNA stability. Additionally, RNA bind- ing studies indicate that UVA increases the binding of RNA- binding proteins to Bcl-X L 3-UTR mRNA, which can be decreased by using SB202190. In conclusion, p38 MAPK and Bcl-X L expression play critical roles in the survival of UVA- irradiated HaCaT cells. Programmed cell death, commonly referred to as apoptosis, is an essential process in the development of an organism, tissue remodeling, and immune system development (1). An imbalance between the processes of cell proliferation and death has been proposed to be a critical step in the pathogenesis of human tumors (2, 3). The process of apoptosis has also been described to play an important role in human epidermis, in- cluding controlling the size of its major cell population, the epidermal keratinocytes, maintaining epidermal barrier func- tion, and aging (1, 4). Ultraviolet radiation has been shown to trigger apoptosis in epidermal cells. With excessive UVB expo- sure, “sunburn cells” can be observed within the epidermis (5, 6). These cells are thought to be keratinocytes that have un- dergone apoptosis, selectively removing those cells that have sustained irreversible damage and that may pose a risk for malignant transformation (7). UV light has been described to affect directly the apoptotic response of keratinocytes; however, UVA-induced signaling leading to apoptotic susceptibility remains to be fully eluci- dated. In this report, we focus upon the effects of UVA (320 – 400 nm), which comprises the largest portion of solar radiation reaching the surface of the earth. It is estimated that 90 –99% of solar radiation reaching surface of the earth is comprised of UVA, with the remaining portion consisting of portions of UVB not filtered by the ozone layer (8, 9). The p38 mitogen-activated protein kinases (MAPKs) 1 and c-Jun N-terminal kinases are stress-regulated protein kinases belonging to the superfamily of MAPKs in addition to extracel- lular signal-regulated kinases (10). Induction of p38 MAPK activity has a variety of effects, including changes in transcrip- tion, protein synthesis, cell surface receptor expression, and cytoskeletal structure with the result ultimately leading to either cell survival or programmed cell death (10). Various extracellular stimuli have been shown to activate stress-regu- lated kinase cascades, including p38 MAPK. Our laboratory has shown that both UVA and UVB can activate p38 MAPK in the HaCaT cell line (11, 12). The role of p38 MAPK in apoptosis is controversial, appearing to be dependent upon cell type and stimuli. In some systems, activation of p38 MAPK results in apoptosis. Overexpression of MEKKs, specific for p38 MAPK activation, results in apoptosis in T cells and fibroblasts (13). In contrast, inhibition of p38 MAPK in Jurkat cells inhibited Fas and UV-induced apoptosis (14). Another complication of the role of p38 MAPK as it relates to the induction of apoptosis is that the induction or inhibition of apoptosis may be sensitive to the nature of p38 MAPK activation, i.e. the duration of activa- * This work was supported by National Institutes of Health Grants CA27502 and CA23074 and the Achievement Rewards for College Sci- entists Foundation (to M. A. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. ‡ To whom correspondence should be addressed: Arizona Cancer Cen- ter, the University of Arizona, 1515 N. Campbell Ave., Rm. 4999, Tucson, AZ 85724. Tel.: 520-626-6006; Fax: 520-626-4979; E-mail: [email protected]. 1 The abbreviations used are: MAPK, mitogen-activated protein ki- nase; PARP, substrate poly(ADP-ribose) polymerase; Z, benzyloxycar- bonyl; fmk, fluoromethyl ketone; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; UTR, untranslated region; EMSA, electrophoretic mo- bility shift assay; DMEM, Dulbecco’s modified Eagle’s medium; PBS, phosphate-buffered saline; NHEKs, normal human epidermal kerati- nocytes; AREs, adenylate/uridylate-rich elements; DNM, dominant- negative mutant. THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 279, No. 41, Issue of October 8, pp. 42658 –42668, 2004 © 2004 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. This paper is available on line at http://www.jbc.org 42658 by guest on October 21, 2016 http://www.jbc.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Ultraviolet A-induced Modulation of Bcl-XL by p38 MAPK in Human Keratinocytes: POSTTRANSCRIPTIONAL...

Ultraviolet A-induced Modulation of Bcl-XL by p38 MAPK inHuman KeratinocytesPOST-TRANSCRIPTIONAL REGULATION THROUGH THE 3�-UNTRANSLATED REGION*
Received for publication, June 14, 2004, and in revised form, July 23, 2004Published, JBC Papers in Press, August 2, 2004, DOI 10.1074/jbc.M406626200
Michael A. Bachelor and G. Timothy Bowden‡
From the Department of Cell Biology and Anatomy, Arizona Cancer Center, the University of Arizona,Tucson, Arizona 85724
We examined the effect of inhibiting p38 MAPK on UVA-irradiated HaCaT cells, a spontaneously immortalized hu-man keratinocyte cell line. Recent work from our labora-tory has shown that UVA (250 kJ/m2) induces a rapidphosphorylation of p38 MAPK in the HaCaT cell line. Inhi-bition of p38 MAPK activity through the use of a specificinhibitor, SB202190, in combination with UVA treatmentinduced a rapid cleavage of caspase-9, caspase-8, andcaspase-3, whereas UVA irradiation alone had no effect.Similarly, cleavage of the caspase substrate poly(ADP-ribose) polymerase was observed in UVA-irradiated HaCaTcells treated with SB202190 or in cells expressing a domi-nant-negative p38 MAPK. No effect of p38 MAPK inhibitionupon caspase cleavage was observed in mock-irradiatedHaCaT cells. In addition, increases in apoptosis were ob-served in UVA-irradiated cells treated with SB202190 bymorphological analysis with no significant apoptosis occur-ring from UVA irradiation alone. Similar results were ob-tained by using normal human epidermal keratinocytes.UVA induced expression of the anti-apoptotic Bcl-2 familymember, Bcl-XL, with abrogation of expression by using thep38 MAPK inhibitor SB202190. Overexpression of Bcl-XLprevented poly(ADP-ribose) polymerase cleavage inducedby the combination of UVA and p38 MAPK inhibition. UVAenhanced the stability of Bcl-XL mRNA through increasesin p38 MAPK activity. We determined that increases inUVA-induced expression of Bcl-XL occur through a post-transcriptional mechanism mediated by the 3�-untrans-lated region (UTR). We used Bcl-XL 3�-UTR luciferase con-structs to determine the mechanism by which UVAincreased Bcl-XL mRNA stability. Additionally, RNA bind-ing studies indicate that UVA increases the binding of RNA-binding proteins to Bcl-XL 3�-UTR mRNA, which can bedecreased by using SB202190. In conclusion, p38 MAPK andBcl-XL expression play critical roles in the survival of UVA-irradiated HaCaT cells.
Programmed cell death, commonly referred to as apoptosis,is an essential process in the development of an organism,tissue remodeling, and immune system development (1). Animbalance between the processes of cell proliferation and death
has been proposed to be a critical step in the pathogenesis ofhuman tumors (2, 3). The process of apoptosis has also beendescribed to play an important role in human epidermis, in-cluding controlling the size of its major cell population, theepidermal keratinocytes, maintaining epidermal barrier func-tion, and aging (1, 4). Ultraviolet radiation has been shown totrigger apoptosis in epidermal cells. With excessive UVB expo-sure, “sunburn cells” can be observed within the epidermis (5,6). These cells are thought to be keratinocytes that have un-dergone apoptosis, selectively removing those cells that havesustained irreversible damage and that may pose a risk formalignant transformation (7).
UV light has been described to affect directly the apoptoticresponse of keratinocytes; however, UVA-induced signalingleading to apoptotic susceptibility remains to be fully eluci-dated. In this report, we focus upon the effects of UVA (320–400 nm), which comprises the largest portion of solar radiationreaching the surface of the earth. It is estimated that 90–99%of solar radiation reaching surface of the earth is comprised ofUVA, with the remaining portion consisting of portions of UVBnot filtered by the ozone layer (8, 9).
The p38 mitogen-activated protein kinases (MAPKs)1 andc-Jun N-terminal kinases are stress-regulated protein kinasesbelonging to the superfamily of MAPKs in addition to extracel-lular signal-regulated kinases (10). Induction of p38 MAPKactivity has a variety of effects, including changes in transcrip-tion, protein synthesis, cell surface receptor expression, andcytoskeletal structure with the result ultimately leading toeither cell survival or programmed cell death (10). Variousextracellular stimuli have been shown to activate stress-regu-lated kinase cascades, including p38 MAPK. Our laboratoryhas shown that both UVA and UVB can activate p38 MAPK inthe HaCaT cell line (11, 12). The role of p38 MAPK in apoptosisis controversial, appearing to be dependent upon cell type andstimuli. In some systems, activation of p38 MAPK results inapoptosis. Overexpression of MEKKs, specific for p38 MAPKactivation, results in apoptosis in T cells and fibroblasts (13). Incontrast, inhibition of p38 MAPK in Jurkat cells inhibited Fasand UV-induced apoptosis (14). Another complication of therole of p38 MAPK as it relates to the induction of apoptosis isthat the induction or inhibition of apoptosis may be sensitive tothe nature of p38 MAPK activation, i.e. the duration of activa-
* This work was supported by National Institutes of Health GrantsCA27502 and CA23074 and the Achievement Rewards for College Sci-entists Foundation (to M. A. B.). The costs of publication of this articlewere defrayed in part by the payment of page charges. This article musttherefore be hereby marked “advertisement” in accordance with 18U.S.C. Section 1734 solely to indicate this fact.
‡ To whom correspondence should be addressed: Arizona Cancer Cen-ter, the University of Arizona, 1515 N. Campbell Ave., Rm. 4999,Tucson, AZ 85724. Tel.: 520-626-6006; Fax: 520-626-4979; E-mail:[email protected].
1 The abbreviations used are: MAPK, mitogen-activated protein ki-nase; PARP, substrate poly(ADP-ribose) polymerase; Z, benzyloxycar-bonyl; fmk, fluoromethyl ketone; GAPDH, glyceraldehyde-3-phosphatedehydrogenase; UTR, untranslated region; EMSA, electrophoretic mo-bility shift assay; DMEM, Dulbecco’s modified Eagle’s medium; PBS,phosphate-buffered saline; NHEKs, normal human epidermal kerati-nocytes; AREs, adenylate/uridylate-rich elements; DNM, dominant-negative mutant.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 279, No. 41, Issue of October 8, pp. 42658–42668, 2004© 2004 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org42658
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

tion. For example, a transient early activation of p38 MAPK intumor necrosis factor-�-treated neutrophils prevents apoptosis,whereas late phase activation contributes to it (15). The oppo-site effect has also been reported in mouse erythroleukemiaSKT6 cells exposed to osmotic shock, where early activation ofp38 MAPK inhibits apoptosis and prolonged activation resultsin apoptosis (16).
UVA-induced p38 MAPK activity and its role in UVA-in-duced apoptosis has not been addressed in previous studies. Inthe studies described here, we demonstrate that UVA-inducedp38 MAPK activity plays a dramatic role in the survival ofkeratinocytes. We observed that inhibition of p38 MAPK de-creases expression of Bcl-XL and results in the mitochondrialpermeabilization through release of cytochrome c, cleavage ofinitiator caspases (caspase 8 and caspase 9), the effectorcaspase (caspase-3), as well as the apoptotic substrate poly-(ADP-ribose) polymerase (PARP). Morphological analysis re-vealed typical features of apoptosis in UVA-irradiated kerati-nocytes with inhibited p38 MAPK activity including nuclearcondensation and formation of apoptotic bodies. Apoptosis ob-served in these cells was determined to be caspase-dependent.We further demonstrate that UVA results in increases in theanti-apoptotic Bcl-2 family member, Bcl-XL, through a post-transcriptional mechanism involving the 3�-untranslated re-gion (3�-UTR). Overexpression of Bcl-XL demonstrates its func-tional role in preventing PARP cleavage in UVA-irradiatedHaCaTs where p38 MAPK activity has been inhibited. UVA-induced p38 MAPK activity and the subsequent increase inBcl-XL resulted in a resistance to UVA-induced apoptosis.
MATERIALS AND METHODS
Cells and UVA Treatment—HaCaT cells, a spontaneously immortal-ized human keratinocyte cell line, were cultured in DMEM supple-mented with 10% fetal bovine serum and 100 units/ml penicillin/strep-tomycin. They were grown to 90% confluence and placed in serum-freeDMEM for 24 h prior to irradiation. During irradiation, cells wereplaced in PBS supplemented with 0.01% MgCl2 and 0.01% CaCl2.Control cells were mock-irradiated in supplemented PBS in a separatetissue culture hood. Four F20T12/BL/HO PUVA bulbs (National Bio-logical Corp., Twinsburg, OH), providing a peak emission at 365 nm,were used. Plates were irradiated under 4-mm plate glass to filterwavelengths below 320 nm (described in Ref. 17). Irradiation doseswere determined using a UVX radiometer and UVX-36 sensor (UVP,Inc., Upland, CA). Cells were treated for 1 h prior to irradiation with thep38 MAPK inhibitor, SB202190, or its inactive analogue, SB202474.SB202190 or SB202474 was placed in medium following irradiationuntil the cells were harvested. Cells were treated with Z-VAD-fmk (100�M) (Calbiochem) for 30 min prior to UVA irradiation and were placedin serum-free medium containing Z-VAD-fmk following irradiation un-til the time of harvest. For message half-life studies, cells were pre-treated for 1 h prior to irradiation with actinomycin D (1 �g/ml) (Sigma)and again immediately following UVA irradiation.
HaCaT cells stably transfected with a tetracycline controlled Bcl-XL
construct were a kind gift from Dr. Ulrich Rodeck (Thomas JeffersonUniversity, Philadelphia) (18). Mock-transfected HaCaTs were used ascontrols in these experiments. Cells grown in the presence of tetracy-cline (1 �g/ml) repressed expression of Bcl-XL (Tet-Off). For experi-ments described here, cells were seeded at 4 � 104 cells/cm2, grown for48 h, and serum-starved for an additional 24 h prior to irradiation.
Normal human epidermal keratinocytes were obtained from individ-ual adult donors (Cambrex, Walkersville, MD) and used between pas-sages 2 and 4. Cells were maintained according to manufacturer’sprotocols. Briefly, normal human epidermal keratinocytes (NHEKs)were grown in KGM®-2 Bullet Kit® (containing bovine pituitary ex-tract, human epidermal growth factor, insulin, hydrocortisone, GA-1000 (gentamicin and amphotericin-B), epinephrine, and transferrin) to80% confluency and subcultured at 3500 cells/cm2.
Total Cellular Protein Extraction and Western Analysis—Cells werelysed in 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM
EGTA, 1% Triton X-100, 2.5 mM Na4P2O7, 1 mM �-glycerophosphate, 1mM Na3VO4, and 1 �g/ml leupeptin. Cells were scraped and centrifugedat 14,000 rpm for 5 min at 4 °C. Bio-Rad Dc reagent (Bio-Rad) was usedto determine protein concentration. For Western analysis, 40 �g of
protein were resolved on a 12.5% SDS-polyacrylamide gel. The proteinwas then transferred to a polyvinylidene difluoride membrane over-night at 4 °C. The membrane was then blocked with 5% milk in TBSTat room temperature for 1 h. Primary antibodies for caspase-8,caspase-9, caspase-3, PARP, Bcl-XL, MAPKAPK-2 (Thr-222) (Cell Sig-naling, Beverly, MA), and cytochrome c (Pharmingen) were used at1:1000 dilution and incubated at 4 °C overnight. The primary antibodyfor �-tubulin (Oncogene, La Jolla, CA) was used at 1:5000 and incu-bated at room temperature for 2 h. Membranes were incubated with theappropriate horseradish peroxidase secondary antibody in 5% milk/TBST for 1 h at room temperature. Goat anti-mouse secondary antibody(Santa Cruz Biotechnology, Santa Cruz, CA) was used for cytochrome cand �-tubulin Western blots at 1:5000, and anti-rabbit (Cell Signaling,Beverly, MA) was used at a 1:2000 dilution for all other antibodiesmentioned previously. Membranes were washed three times for 10 mineach in TBST between antibody incubations and were detected by usingECL Western blotting detection reagents (Amersham Biosciences).
Isolation of Cytoplasmic and Mitochondrial Fractions—Cells weretreated as indicated, harvested, and resuspended in cytosolic extractionbuffer (250 mM sucrose, 10 mM KCl, 1 mM EDTA, 20 mM Tris-HCl (pH7.2), 1 mM dithiothreitol, 1.5 mM MgCl2, 100 �M phenylmethylsulfonylfluoride, 10 �g/ml leupeptin, 2 �g/ml aprotinin). Cells were incubatedon ice for 5 min and centrifuged for 10 min at 4 °C at 14,000 rpm. Thesupernatant was saved as a cytosolic fraction. The remaining pellet wasresuspended in mitochondrial extraction buffer (50 mM Tris-HCl (pH7.4), 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.2% Triton X-100, 0.3%Nonidet P-40, 100 �M phenylmethylsulfonyl fluoride, 10 �g/ml leupep-tin, 2 �g/ml aprotinin), placed on ice for 5 min, and centrifuged at12,500 rpm for 10 min at 4 °C. The supernatant was saved as themitochondrial extract.
Real Time PCR—Total RNA was isolated from HaCaT cells by usingthe RNeasy mini kit (Qiagen, Carlsbad, CA). 1 �g of total RNA was usedto generate random primed hexamer cDNA by using Omniscript re-verse transcription kit (Qiagen). Primers and Taqman® probes forBcl-XL and human GAPDH were purchased from Assay-on-Demand(Applied Biosystems, Foster City, CA). The real time PCR was per-formed using the Taqman® Universal PCR Master Mix and the ABIPRISM® 7700 Sequence Detection System (Applied Biosystems, FosterCity, CA). The PCR conditions are as follows: 50 °C for 2 min, 95 °C for10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Therelative CT method was used to calculate mRNA levels of Bcl-XL nor-malized to GAPDH in each sample as described in User Bulletin 2, ABIPRISM® 7700 Sequence Detection system.
Flow Cytometry—Apoptosis was measured by determining the DNAcontent of cells by propidium iodide staining and flow cytometry. Cellswere trypsinized and stained with Modified Krishan Buffer (0.1% so-dium citrate, 0.2 mg/ml RNase, 0.3% Nonidet P-40, and 0.05 mg/mlpropidium iodide (pH 7.4)) before being analyzed. Cells with DNAcontent less than the G1 amount of untreated cells were consideredapoptotic as determined by using the ModFit LT software program.
Morphological Analysis—After treatment, cells were collected (ad-herent and floating) from each condition. Cytospins were prepared byusing a Shandon Cytospin 2. Slides were fixed in 100% methanol for 3min and then stained with a modified Giemsa stain (1:10 dilution of0.4% w/v (pH 6.9)) in methanol (Sigma). Cells were examined under100� oil immersion lens and evaluated for apoptotic features (con-densed chromatin, nuclear fragmentation, cell shrinkage, and apoptoticbody formation).
Generation of p38 MAPK Dominant-negative HaCaT Cell Line—Ap38 DNM construct was obtained from Dr. Zigang Dong (Hormel Insti-tute, University of Minnesota, Austin). The plasmid expresses a domi-nant-negative form of p38 MAPK containing a double point mutation ofThr-Gly-Tyr to Ala-Gly-Phe at the activating sites of phosphorylation(threonine 180 and tyrosine 182). The p38 MAPK dominant-negativeconstruct described above was cloned into to HpaI and BamHI restric-tion sites within the multiple cloning site of pLXSN (pLXSN-p38DNM).PT67 packaging cells were stably transfected with pLXSN-p38DNM orpLXSN alone and selected for antibiotic resistance by using 500 �g/mlG418. Colonies were selected and viral titer was determined by usingNIH3T3 cells per the manufacturer’s recommendations. HaCaT cellswere infected with pLXSN (HaCaT-empty vector) or pLXSN-p38 (Ha-CaT-p38 DNM) and stable clones selected using 800 �g/ml G418.
Luciferase Reporter Construction—The 3�-untranslated region ofBcl-XL was cloned from genomic DNA isolated from HaCaT cells usingthe DNeasy tissue kit (Qiagen, Valencia, CA). An EagI site was insertedinto the PCR primers to facilitate cloning downstream of the luciferasegene in the pRL-TK vector (Promega, Madison, WI). A nested PCR wasperformed by using the following primers in the first amplification:
Regulation of UVA-induced Bcl-XL Expression 42659
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

5�-TCGGCGGCCGCCAGACACTGACCATCCACT-3� and 5�-GAACTG-CACTTTCACCTTCACA-3�. A second round of PCR was performed byusing the following primer (EagI restriction site underlined): 5�-GGGGCGGCCGCATCAGGCCGTCCAATCT-3�. The 3�-UTR of Bcl-XL
was inserted into the EagI site of the expression vector pRL-TK Renillaluciferase (Promega, Madison, WI), the region adjacent to the codingsequence. The portion of the Bcl-XL 3�-UTR cloned into the pRL-TKvector corresponds to base pairs 1130–2396 of Bcl-XL (GenBankTM
accession number NM_138578). The DNA construct was analyzed byrestriction mapping and DNA sequencing.
Transient Transfections—HaCaT cells were transiently transfectedby using LipofectAMINE PLUS (Invitrogen) according to the manufac-turer’s protocol. Briefly, cells were plated in 6-well plates 1 day prior totransfection and grown to 95% confluence. Plasmid DNA was preparedin 100 �l of serum-free DMEM and incubated with 3 �l of PLUS reagentat room temperature for 15 min, followed by 2.5 �l of LipofectAMINE inan additional 100 �l of DMEM. The mixture was incubated for another15 min and then added to the well containing 1 ml of serum-freeDMEM. Transfections were allowed to proceed for 6 h. A pGL2 fireflyluciferase construct was co-transfected along with the Renilla luciferasereporter to control for transfection efficiency. The cells were thenwashed two times with serum-free DMEM and incubated for an addi-tional 18 h in DMEM before UVA treatment.
Luciferase Assay—After treatments, cells were washed two timeswith PBS and lysed in passive lysis buffer (dual luciferase assay,Promega). All treatments were done in triplicate for each experiment.Luciferase activity was measured by using the dual luciferase reporterassay system (Promega, Madison, WI) and a TD-20/20 luminometer(Turner Designs, Sunnydale, CA). Luciferase activity is expressed asthe ratio of Renilla/firefly.
RNA-EMSA—The region of the Bcl-XL 3�-UTR corresponding to basepairs 1258–1419 were cloned into pBluescript and used in in vitrotranscription incorporating [32P]ATP (50 �Ci) into sense RNAs usingRiboprobe® system-T7 (Promega, Madison, WI). Unlabeled RNA tran-scripts were used for cold competition controls. Following mock or UVAirradiation with or without SB202190 treatment, cells were washedtwice with cold phosphate-buffered saline and lysed (25 mM Tris-HCl(pH 7.5), 0.5% Triton X-100). Cells were scraped and centrifuged at14,000 � g for 10 min at 4 °C. Protein concentration was determinedusing Bio-Rad Dc reagent (Bio-Rad). RNA-EMSAs were performed asreported previously by Dixon et al. (19). Briefly, 10 �g of protein wasincubated with radiolabeled RNA in binding buffer (20 mM HEPES (pH7.5), 3 mM MgCl2, 30 mM KCl, 1 mM dithiothreitol, 5% glycerol) in a totalvolume of 20 �l for 20 min. Heparin was added at 5 mg/ml and incu-
bated for an additional 20 min. The samples were resolved on 4%polyacrylamide gels (60:1 acrylamide/bisacrylamide) in 1� TBE. Thegel was dried and visualized using a PhosphorImager (AmershamBiosciences).
RESULTS
UVA in Combination with SB202190 Induces Caspase Cleav-age and PARP Cleavage—HaCaT cells were UVA-irradiated(250 kJ/m2) or mock-irradiated with or without treatment withSB202190, a specific inhibitor of p38� and p38�1, and collectedat various time points. Our laboratory has previously shownSB202190 to inhibit p38 MAPK activity at 0.5 and 2.0 �M bydemonstrating inhibition of phosphorylation of a downstreamtarget, MAPKAPK-2 (12, 20). Caspase activation resultingfrom a cleavage of the inactive pro-form to the active cleavedform was analyzed by Western analysis. Beginning at 1 hpost-irradiation, increases in the cleaved forms of caspase-9and caspase-8 were observed in UVA-irradiated HaCaTs
FIG. 1. Caspase cleavage induced by UVA and SB202190 treatment. HaCaT cells were grown to 95% confluency and serum-starved for 24 hprior to UVA irradiation (250 kJ/m2). Cells were treated with SB202190 (0.5 or 2.0 �M) (Calbiochem) or vehicle control (V) Me2SO for 1 h prior toirradiation. Whole cell lysates were collected at various time points post-irradiation. Proteins were resolved by SDS-PAGE, and Western analysiswas performed for caspase-9, caspase-8, and caspase-3 and PARP cleavage (Cell Signaling, Beverly, MA). The antibodies used recognized both theinactive pro-form and active cleaved forms of the caspase and PARP.
FIG. 2. SB202474, an inactive analogue of SB202190, does notinduce PARP cleavage in combination with UVA irradiation.HaCaT cells were grown to 95% confluency and serum-starved for 24 hprior to UVA irradiation. Cells were treated with the active p38 MAPKinhibitor SB202190 (Calbiochem), the inactive analogue SB202474(Calbiochem), or with vehicle Me2SO for 1 h prior to irradiation. Cellswere UVA-irradiated, placed in medium containing SB202190 orSB202474, and collected at 1 h post-irradiation. Protein was resolved bySDS-PAGE, and Western analysis was performed by using a PARPantibody (Cell Signaling, Beverly, MA).
Regulation of UVA-induced Bcl-XL Expression42660
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
treated with SB202190 at 2 �M (Fig. 1). By 2 h post-irradiation,the pro-forms of caspases-9, -8, and -3 and PARP were con-verted to the active cleaved forms. Increases in caspase-3 andthe apoptotic substrate PARP were also observed under theseconditions. Lower doses of the p38 MAPK inhibitor (0.5 �M) incombination with UVA showed cleavage of caspases-9, -8, and-3 and PARP at 6 h post-irradiation. The observed effects ofcaspase activation were dose-dependent, with an increase incleavage occurring with increasing dose of p38 MAPK inhibitorin combination with UVA. UVA alone (250 kJ/m2) had no effecton caspases-8, -9, and -3 or PARP cleavage at any time point.
Our laboratory has published data previously (20) showingthat inhibition of UVA-induced p38 MAPK by SB202190 isspecific and does not alter extracellular signal-regulated kinaseor c-Jun N-terminal kinase phosphorylation. To confirm fur-ther the specificity of the apoptotic effects we observed with thecombination of UVA and p38 MAPK inhibition usingSB202190, we examined effects of PARP cleavage using aninactive analogue of SB202190, SB202474. As shown in Fig. 2,PARP cleavage was only observed in combination with UVA incells treated with the active p38 MAPK inhibitor, SB202190.Treatment with UVA alone or in combination with SB202474had no effect on PARP cleavage indicating the effects on apo-ptosis observed with UVA irradiation and p38 MAPK inhibi-tion are specific.
By using a retroviral vector, we established a HaCaT cell linethat expressed a dominant-negative p38 MAPK to confirm the
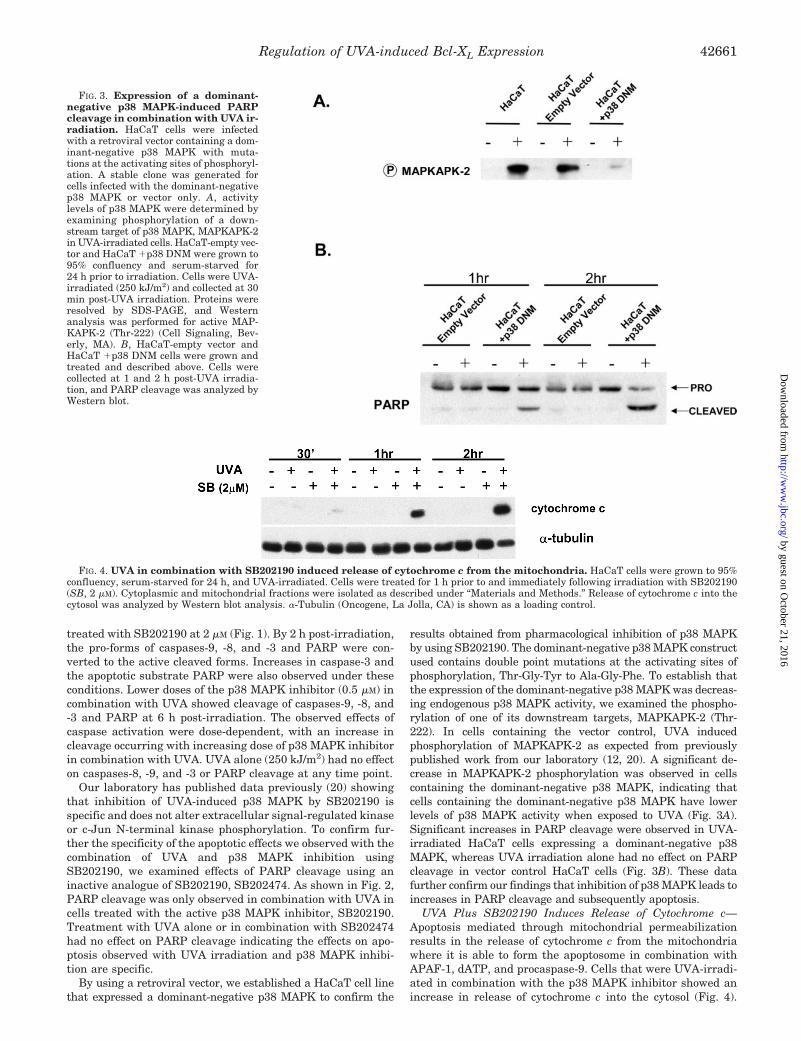
results obtained from pharmacological inhibition of p38 MAPKby using SB202190. The dominant-negative p38 MAPK constructused contains double point mutations at the activating sites ofphosphorylation, Thr-Gly-Tyr to Ala-Gly-Phe. To establish thatthe expression of the dominant-negative p38 MAPK was decreas-ing endogenous p38 MAPK activity, we examined the phospho-rylation of one of its downstream targets, MAPKAPK-2 (Thr-222). In cells containing the vector control, UVA inducedphosphorylation of MAPKAPK-2 as expected from previouslypublished work from our laboratory (12, 20). A significant de-crease in MAPKAPK-2 phosphorylation was observed in cellscontaining the dominant-negative p38 MAPK, indicating thatcells containing the dominant-negative p38 MAPK have lowerlevels of p38 MAPK activity when exposed to UVA (Fig. 3A).Significant increases in PARP cleavage were observed in UVA-irradiated HaCaT cells expressing a dominant-negative p38MAPK, whereas UVA irradiation alone had no effect on PARPcleavage in vector control HaCaT cells (Fig. 3B). These datafurther confirm our findings that inhibition of p38 MAPK leads toincreases in PARP cleavage and subsequently apoptosis.
UVA Plus SB202190 Induces Release of Cytochrome c—Apoptosis mediated through mitochondrial permeabilizationresults in the release of cytochrome c from the mitochondriawhere it is able to form the apoptosome in combination withAPAF-1, dATP, and procaspase-9. Cells that were UVA-irradi-ated in combination with the p38 MAPK inhibitor showed anincrease in release of cytochrome c into the cytosol (Fig. 4).
FIG. 3. Expression of a dominant-negative p38 MAPK-induced PARPcleavage in combination with UVA ir-radiation. HaCaT cells were infectedwith a retroviral vector containing a dom-inant-negative p38 MAPK with muta-tions at the activating sites of phosphoryl-ation. A stable clone was generated forcells infected with the dominant-negativep38 MAPK or vector only. A, activitylevels of p38 MAPK were determined byexamining phosphorylation of a down-stream target of p38 MAPK, MAPKAPK-2in UVA-irradiated cells. HaCaT-empty vec-tor and HaCaT �p38 DNM were grown to95% confluency and serum-starved for24 h prior to irradiation. Cells were UVA-irradiated (250 kJ/m2) and collected at 30min post-UVA irradiation. Proteins wereresolved by SDS-PAGE, and Westernanalysis was performed for active MAP-KAPK-2 (Thr-222) (Cell Signaling, Bev-erly, MA). B, HaCaT-empty vector andHaCaT �p38 DNM cells were grown andtreated and described above. Cells werecollected at 1 and 2 h post-UVA irradia-tion, and PARP cleavage was analyzed byWestern blot.
FIG. 4. UVA in combination with SB202190 induced release of cytochrome c from the mitochondria. HaCaT cells were grown to 95%confluency, serum-starved for 24 h, and UVA-irradiated. Cells were treated for 1 h prior to and immediately following irradiation with SB202190(SB, 2 �M). Cytoplasmic and mitochondrial fractions were isolated as described under “Materials and Methods.” Release of cytochrome c into thecytosol was analyzed by Western blot analysis. �-Tubulin (Oncogene, La Jolla, CA) is shown as a loading control.
Regulation of UVA-induced Bcl-XL Expression 42661
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

These changes were only observed in cells that were UVA-irradiated in combination with SB202190 and did not resultfrom UVA irradiation alone.
UVA Plus SB202190 Induces Morphological Changes Typi-cal of Apoptosis and Increases in Cells with Sub-G0 DNA Con-tent—The morphology of cells was examined under 100� oil
immersion lens and evaluated for apoptotic features (con-densed chromatin, nuclear fragmentation, cell shrinkage, andapoptotic body formation) (Fig. 5A). These hallmarks of apo-ptosis were observed only in UVA-irradiated cells treated withSB202190. The effects were again shown to be dose-dependentwith increases in the number of cells undergoing apoptosis with
FIG. 5. Apoptosis induced by UVAand p38 MAPK inhibition is caspase-dependent. A, HaCaT cells were grownand treated as described previously. Cellswere treated with SB202190 (SB, 0.5 or2.0 �M) for 1 h prior to and immediatelyfollowing irradiation with or without thegeneral-caspase inhibitor Z-VAD-fmk(100 �M) (Calbiochem). Cells were col-lected at 4 h post-UVA irradiation follow-ing 1 h of pretreatment with various con-centrations of SB202190, and cytospinswere prepared to evaluate the morpholog-ical features of apoptosis. Shown are im-ages obtained under a 100� oil immer-sion lens. B, HaCaT cells were UVA-irradiated with vehicle control, SB202190treatment with or without Z-VAD-fmkpretreatment. SB202190 with or withoutZ-VAD-fmk was added to medium follow-ing UVA irradiation. Cells were collectedat 6 h post-UVA irradiation, stained witha modified Krishan Buffer, and analyzedfor increases in sub-G0 DNA content us-ing ModFitLT. C, cells were treated asdescribed above and harvested at 1 hpost-UVA irradiation. Western blot anal-ysis was performed for PARP cleavage.
Regulation of UVA-induced Bcl-XL Expression42662
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

increasing amounts of SB202190. In Fig. 5A, the arrows indi-cate the appearance of apoptotic bodies. We analyzed sub-G0
DNA content in cells to quantitate the levels of apoptosis thatoccurred. Approximately 4.2% of cells showed increases insub-G0 DNA content at 6 h post-irradiation, whereas higherdoses of SB202190 in UVA-irradiated cells showed 70.3%sub-G0 DNA content (Fig. 5B). It is important to note that nomorphological change or increase in sub-G0 content was ob-served in cells receiving UVA irradiation alone. These observedincreases in sub-G0 DNA are consistent with the pattern ofcaspase activation and morphological analysis shown in Figs. 1and 5A.
Apoptosis Induced by UVA and SB202190 Treatment IsCaspase-dependent—Both caspase-dependent and -independ-ent pathways leading to apoptosis have been described. Weinvestigated whether the apoptotic effects induced by UVA incombination with SB202190 were dependent on caspase activ-ity. By using the general caspase inhibitor Z-VAD-fmk, weshowed that PARP cleavage is completely inhibited with UVAand SB202190 treatment (Fig. 5C). We further showed that thecharacteristic features of apoptosis are not observed in UVA-irradiated cells treated with SB202190 and Z-VAD-fmk.Sub-G0 DNA content analysis revealed a complete reversal ofobserved sub-G0 DNA content with Z-VAD-fmk treatment aswell (Fig. 5B). The primary caspase-independent mechanismdescribed leading to apoptosis involved the release of apo-ptosis-inducing factor from the mitochondria resulting in DNAfragmentation and chromosomal condensation (21). In additionto the aforementioned experiments, we observed no release ofapoptosis-inducing factor from the mitochondria in cellstreated with UVA alone or with SB202190 treatment (data notshown). We conclude from these experiments that the apoptosisinduced by the combination of UVA and p38 MAPK inhibitionrequires the activity of caspases.
Effects of UVA and p38 MAPK Inhibition on PARP Cleavagein Normal Human Epidermal Keratinocytes—As an extensionof the studies performed in HaCaT cells, we investigatedwhether inhibition of p38 MAPK induced apoptosis in UVA-irradiated NHEKs as observed through PARP cleavage. Inhi-bition of p38 MAPK caused a dose-dependent increase in PARP
cleavage in UVA-irradiated cells (Fig. 6A). No effect on PARPcleavage was observed in cells that received UVA irradiationalone, consistent with those observations made in HaCaT cells.We determined that the PARP cleavage induced by UVA incombination with p38 MAPK inhibition was caspase-depend-ent by using the general-caspase inhibitor Z-VAD-fmk (Fig.6B). Treatment with Z-VAD-fmk completely reversed the ob-served cleavage of PARP induced by UVA and SB202190 treat-ment. Both morphological analysis and sub-G0 DNA contentwere performed concomitantly. The results shown with PARPcleavage were in accordance with both the morphological statusof the cell as well as any change in sub-G0 DNA content (datanot shown). The experiment was performed with NHEKs fromthree individual donors. Fig. 6, A and B, is representative of theobservations made from three independent experiments.
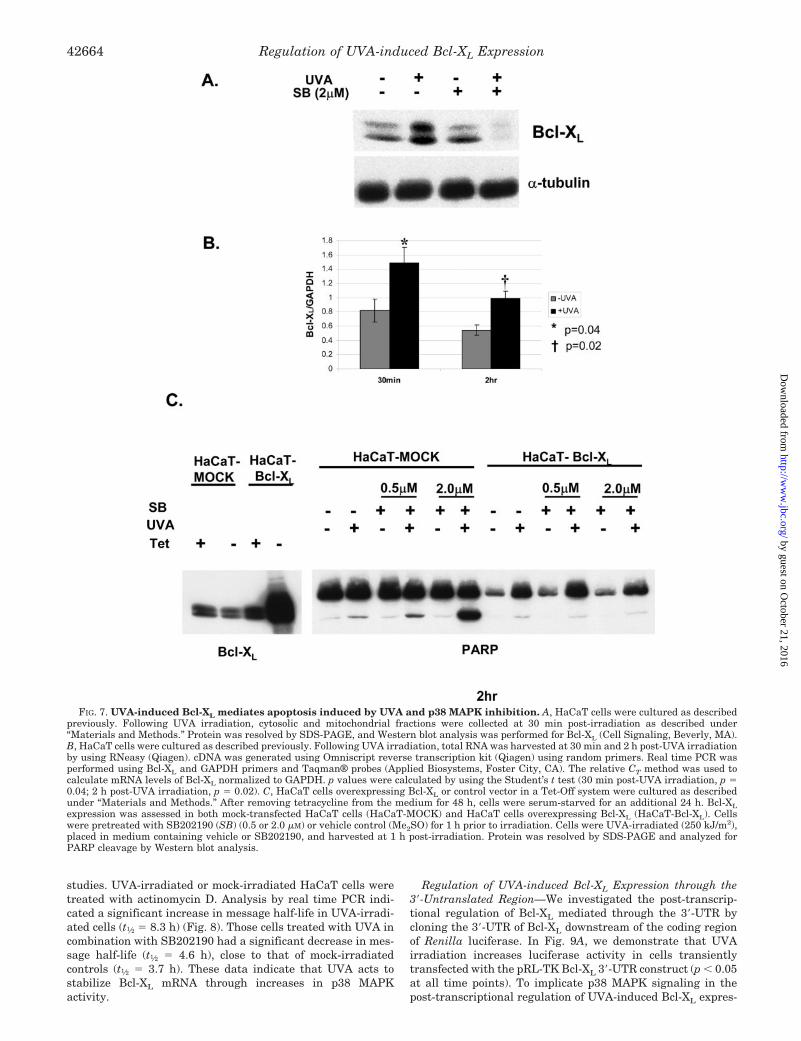
Bcl-XL Expression Modulates the Apoptotic Effects of UVA inCombination with p38 MAPK Inhibition—In Fig. 7A, we dem-onstrate that UVA (250 kJ/m2) increases expression of Bcl-XL
at the protein level. Most interestingly, inhibition of p38 MAPKresulted in decreased Bcl-XL expression in UVA-irradiated Ha-CaTs. We also confirmed UVA induced steady-state levels byusing real time PCR for Bcl-XL. Statistically significant in-creases in Bcl-XL were observed at both 30 min (p � 0.04) and2 h (p � 0.02) post-UVA irradiation (Fig. 7B). To establish thatmodulations in Bcl-XL expression played a functional role inthe observed apoptosis, we used HaCaT cells that overex-pressed Bcl-XL in a Tet-Off System (HaCaT-Bcl-XL) (18). Cellswere plated 72 h prior to irradiation at 40,000 cells/cm2 intetracycline-free medium. After 48 h, the medium was changedto serum-free medium and was cultured for an additional 24 h.Under these conditions, Bcl-XL was significantly overexpressedas observed by Western analysis (Fig. 7C). HaCaT-MOCK cellswere treated in a similar manner and used as the control cellline for these experiments. Overexpression of Bcl-XL inhibitedPARP cleavage induced by UVA and SB202190 treatment in-dicating a functional role of Bcl-XL expression in the observedapoptotic response (Fig. 7C).
UVA-induced Bcl-XL Expression Is Post-transcriptionallyRegulated—To investigate the possibility that Bcl-XL was post-transcriptionally regulated, we performed mRNA half-life
FIG. 6. UVA and SB202190 treatment induces caspase-dependent apoptosis in NHEK. A, NHEKs were cultured to 95% confluency inKGM®-2 Bullet Kit® (Cambrex, Walkersville, MD). NHEKs were serum-starved in KGM®-2 without supplementation of growth factors for 24 hprior to UVA irradiation. Cells were pretreated with SB202190 (0.5 or 2.0 �M) or vehicle (Me2SO) for 1 h prior to irradiation. Cells wereUVA-irradiated (250 kJ/m2), placed in KGM®-2 with SB202190, and collected at various time points. Protein was resolved by SDS-PAGE, andWestern blot analysis was performed by using a PARP antibody (Cell Signaling, Beverly, MA). B, NHEKs were treated as described above.Z-VAD-fmk was added 1 h prior to and immediately following irradiation in addition to SB202190 (SB). Cells were UVA-irradiated (250 kJ/m2) andcollected at 4 h post-irradiation. Protein was resolved by SDS-PAGE, and Western blot analysis was performed by using a PARP antibody (CellSignaling, Beverly, MA).
Regulation of UVA-induced Bcl-XL Expression 42663
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
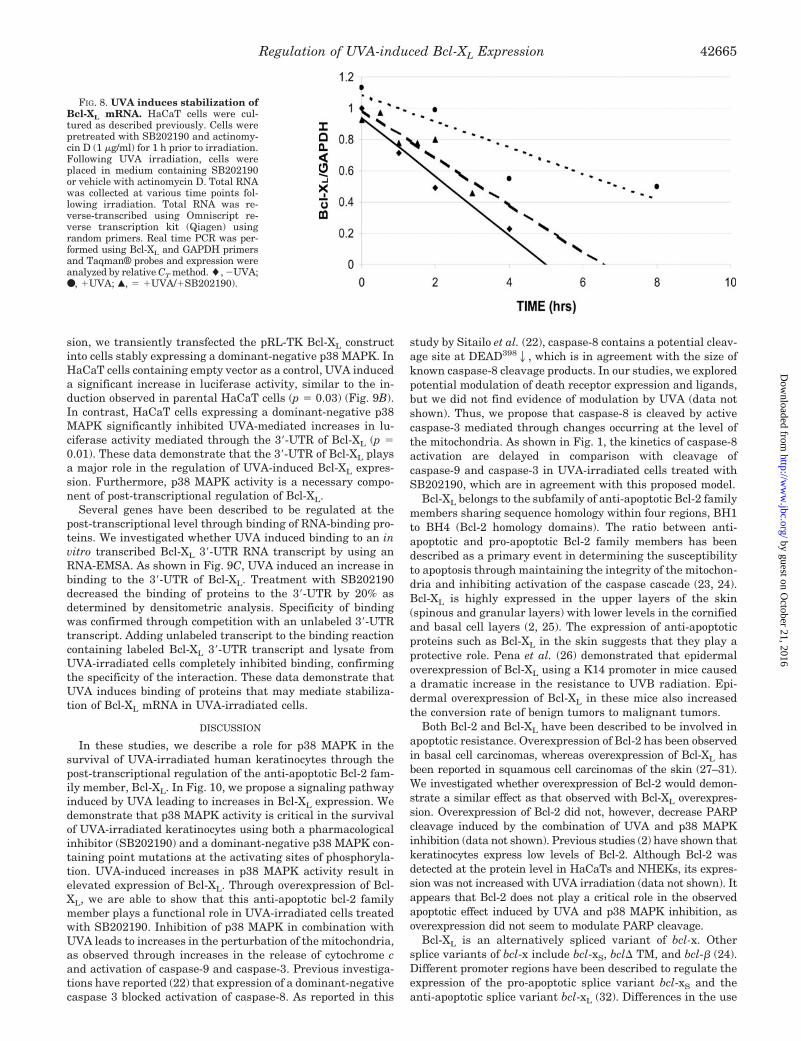
studies. UVA-irradiated or mock-irradiated HaCaT cells weretreated with actinomycin D. Analysis by real time PCR indi-cated a significant increase in message half-life in UVA-irradi-ated cells (t1⁄2 � 8.3 h) (Fig. 8). Those cells treated with UVA incombination with SB202190 had a significant decrease in mes-sage half-life (t1⁄2 � 4.6 h), close to that of mock-irradiatedcontrols (t1⁄2 � 3.7 h). These data indicate that UVA acts tostabilize Bcl-XL mRNA through increases in p38 MAPKactivity.
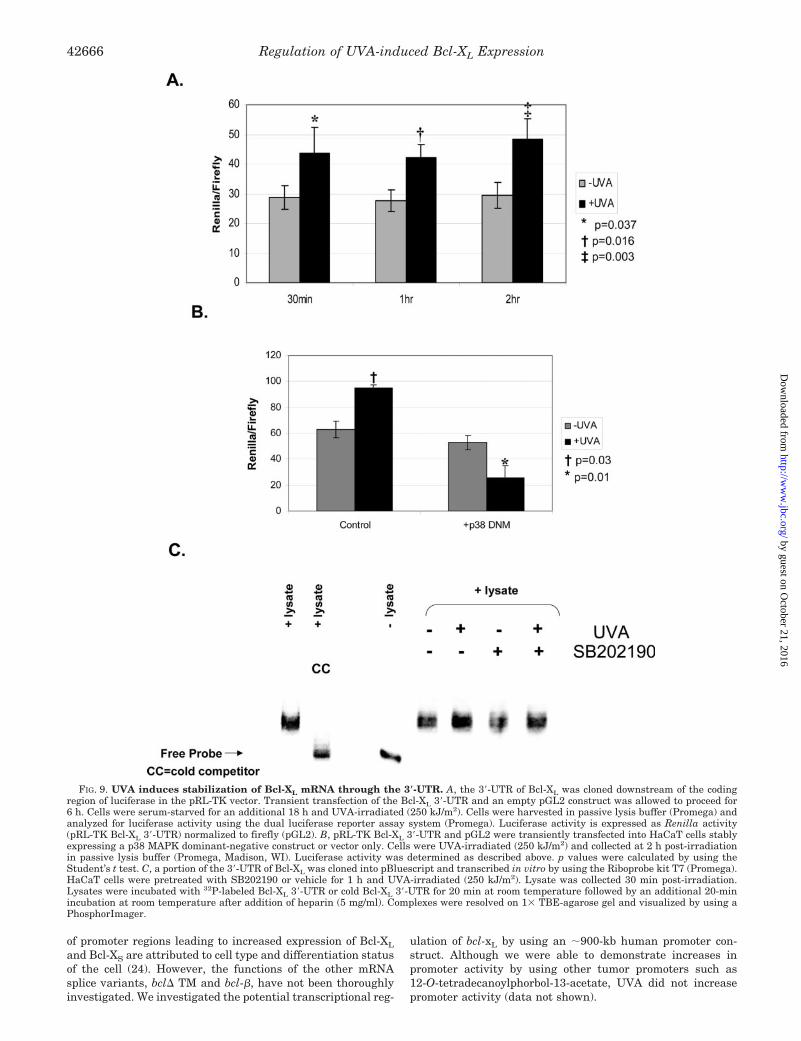
Regulation of UVA-induced Bcl-XL Expression through the3�-Untranslated Region—We investigated the post-transcrip-tional regulation of Bcl-XL mediated through the 3�-UTR bycloning the 3�-UTR of Bcl-XL downstream of the coding regionof Renilla luciferase. In Fig. 9A, we demonstrate that UVAirradiation increases luciferase activity in cells transientlytransfected with the pRL-TK Bcl-XL 3�-UTR construct (p � 0.05at all time points). To implicate p38 MAPK signaling in thepost-transcriptional regulation of UVA-induced Bcl-XL expres-
FIG. 7. UVA-induced Bcl-XL mediates apoptosis induced by UVA and p38 MAPK inhibition. A, HaCaT cells were cultured as describedpreviously. Following UVA irradiation, cytosolic and mitochondrial fractions were collected at 30 min post-irradiation as described under“Materials and Methods.” Protein was resolved by SDS-PAGE, and Western blot analysis was performed for Bcl-XL (Cell Signaling, Beverly, MA).B, HaCaT cells were cultured as described previously. Following UVA irradiation, total RNA was harvested at 30 min and 2 h post-UVA irradiationby using RNeasy (Qiagen). cDNA was generated using Omniscript reverse transcription kit (Qiagen) using random primers. Real time PCR wasperformed using Bcl-XL and GAPDH primers and Taqman® probes (Applied Biosystems, Foster City, CA). The relative CT method was used tocalculate mRNA levels of Bcl-XL normalized to GAPDH. p values were calculated by using the Student’s t test (30 min post-UVA irradiation, p �0.04; 2 h post-UVA irradiation, p � 0.02). C, HaCaT cells overexpressing Bcl-XL or control vector in a Tet-Off system were cultured as describedunder “Materials and Methods.” After removing tetracycline from the medium for 48 h, cells were serum-starved for an additional 24 h. Bcl-XLexpression was assessed in both mock-transfected HaCaT cells (HaCaT-MOCK) and HaCaT cells overexpressing Bcl-XL (HaCaT-Bcl-XL). Cellswere pretreated with SB202190 (SB) (0.5 or 2.0 �M) or vehicle control (Me2SO) for 1 h prior to irradiation. Cells were UVA-irradiated (250 kJ/m2),placed in medium containing vehicle or SB202190, and harvested at 1 h post-irradiation. Protein was resolved by SDS-PAGE and analyzed forPARP cleavage by Western blot analysis.
Regulation of UVA-induced Bcl-XL Expression42664
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
sion, we transiently transfected the pRL-TK Bcl-XL constructinto cells stably expressing a dominant-negative p38 MAPK. InHaCaT cells containing empty vector as a control, UVA induceda significant increase in luciferase activity, similar to the in-duction observed in parental HaCaT cells (p � 0.03) (Fig. 9B).In contrast, HaCaT cells expressing a dominant-negative p38MAPK significantly inhibited UVA-mediated increases in lu-ciferase activity mediated through the 3�-UTR of Bcl-XL (p �0.01). These data demonstrate that the 3�-UTR of Bcl-XL playsa major role in the regulation of UVA-induced Bcl-XL expres-sion. Furthermore, p38 MAPK activity is a necessary compo-nent of post-transcriptional regulation of Bcl-XL.
Several genes have been described to be regulated at thepost-transcriptional level through binding of RNA-binding pro-teins. We investigated whether UVA induced binding to an invitro transcribed Bcl-XL 3�-UTR RNA transcript by using anRNA-EMSA. As shown in Fig. 9C, UVA induced an increase inbinding to the 3�-UTR of Bcl-XL. Treatment with SB202190decreased the binding of proteins to the 3�-UTR by 20% asdetermined by densitometric analysis. Specificity of bindingwas confirmed through competition with an unlabeled 3�-UTRtranscript. Adding unlabeled transcript to the binding reactioncontaining labeled Bcl-XL 3�-UTR transcript and lysate fromUVA-irradiated cells completely inhibited binding, confirmingthe specificity of the interaction. These data demonstrate thatUVA induces binding of proteins that may mediate stabiliza-tion of Bcl-XL mRNA in UVA-irradiated cells.
DISCUSSION
In these studies, we describe a role for p38 MAPK in thesurvival of UVA-irradiated human keratinocytes through thepost-transcriptional regulation of the anti-apoptotic Bcl-2 fam-ily member, Bcl-XL. In Fig. 10, we propose a signaling pathwayinduced by UVA leading to increases in Bcl-XL expression. Wedemonstrate that p38 MAPK activity is critical in the survivalof UVA-irradiated keratinocytes using both a pharmacologicalinhibitor (SB202190) and a dominant-negative p38 MAPK con-taining point mutations at the activating sites of phosphoryla-tion. UVA-induced increases in p38 MAPK activity result inelevated expression of Bcl-XL. Through overexpression of Bcl-XL, we are able to show that this anti-apoptotic bcl-2 familymember plays a functional role in UVA-irradiated cells treatedwith SB202190. Inhibition of p38 MAPK in combination withUVA leads to increases in the perturbation of the mitochondria,as observed through increases in the release of cytochrome cand activation of caspase-9 and caspase-3. Previous investiga-tions have reported (22) that expression of a dominant-negativecaspase 3 blocked activation of caspase-8. As reported in this
study by Sitailo et al. (22), caspase-8 contains a potential cleav-age site at DEAD3982, which is in agreement with the size ofknown caspase-8 cleavage products. In our studies, we exploredpotential modulation of death receptor expression and ligands,but we did not find evidence of modulation by UVA (data notshown). Thus, we propose that caspase-8 is cleaved by activecaspase-3 mediated through changes occurring at the level ofthe mitochondria. As shown in Fig. 1, the kinetics of caspase-8activation are delayed in comparison with cleavage ofcaspase-9 and caspase-3 in UVA-irradiated cells treated withSB202190, which are in agreement with this proposed model.
Bcl-XL belongs to the subfamily of anti-apoptotic Bcl-2 familymembers sharing sequence homology within four regions, BH1to BH4 (Bcl-2 homology domains). The ratio between anti-apoptotic and pro-apoptotic Bcl-2 family members has beendescribed as a primary event in determining the susceptibilityto apoptosis through maintaining the integrity of the mitochon-dria and inhibiting activation of the caspase cascade (23, 24).Bcl-XL is highly expressed in the upper layers of the skin(spinous and granular layers) with lower levels in the cornifiedand basal cell layers (2, 25). The expression of anti-apoptoticproteins such as Bcl-XL in the skin suggests that they play aprotective role. Pena et al. (26) demonstrated that epidermaloverexpression of Bcl-XL using a K14 promoter in mice causeda dramatic increase in the resistance to UVB radiation. Epi-dermal overexpression of Bcl-XL in these mice also increasedthe conversion rate of benign tumors to malignant tumors.
Both Bcl-2 and Bcl-XL have been described to be involved inapoptotic resistance. Overexpression of Bcl-2 has been observedin basal cell carcinomas, whereas overexpression of Bcl-XL hasbeen reported in squamous cell carcinomas of the skin (27–31).We investigated whether overexpression of Bcl-2 would demon-strate a similar effect as that observed with Bcl-XL overexpres-sion. Overexpression of Bcl-2 did not, however, decrease PARPcleavage induced by the combination of UVA and p38 MAPKinhibition (data not shown). Previous studies (2) have shown thatkeratinocytes express low levels of Bcl-2. Although Bcl-2 wasdetected at the protein level in HaCaTs and NHEKs, its expres-sion was not increased with UVA irradiation (data not shown). Itappears that Bcl-2 does not play a critical role in the observedapoptotic effect induced by UVA and p38 MAPK inhibition, asoverexpression did not seem to modulate PARP cleavage.
Bcl-XL is an alternatively spliced variant of bcl-x. Othersplice variants of bcl-x include bcl-xS, bcl� TM, and bcl-� (24).Different promoter regions have been described to regulate theexpression of the pro-apoptotic splice variant bcl-xS and theanti-apoptotic splice variant bcl-xL (32). Differences in the use
FIG. 8. UVA induces stabilization ofBcl-XL mRNA. HaCaT cells were cul-tured as described previously. Cells werepretreated with SB202190 and actinomy-cin D (1 �g/ml) for 1 h prior to irradiation.Following UVA irradiation, cells wereplaced in medium containing SB202190or vehicle with actinomycin D. Total RNAwas collected at various time points fol-lowing irradiation. Total RNA was re-verse-transcribed using Omniscript re-verse transcription kit (Qiagen) usingrandom primers. Real time PCR was per-formed using Bcl-XL and GAPDH primersand Taqman® probes and expression wereanalyzed by relative CT method. �, �UVA;●, �UVA; Œ, � �UVA/�SB202190).
Regulation of UVA-induced Bcl-XL Expression 42665
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from
of promoter regions leading to increased expression of Bcl-XL
and Bcl-XS are attributed to cell type and differentiation statusof the cell (24). However, the functions of the other mRNAsplice variants, bcl� TM and bcl-�, have not been thoroughlyinvestigated. We investigated the potential transcriptional reg-
ulation of bcl-xL by using an �900-kb human promoter con-struct. Although we were able to demonstrate increases inpromoter activity by using other tumor promoters such as12-O-tetradecanoylphorbol-13-acetate, UVA did not increasepromoter activity (data not shown).
FIG. 9. UVA induces stabilization of Bcl-XL mRNA through the 3�-UTR. A, the 3�-UTR of Bcl-XL was cloned downstream of the codingregion of luciferase in the pRL-TK vector. Transient transfection of the Bcl-XL 3�-UTR and an empty pGL2 construct was allowed to proceed for6 h. Cells were serum-starved for an additional 18 h and UVA-irradiated (250 kJ/m2). Cells were harvested in passive lysis buffer (Promega) andanalyzed for luciferase activity using the dual luciferase reporter assay system (Promega). Luciferase activity is expressed as Renilla activity(pRL-TK Bcl-XL 3�-UTR) normalized to firefly (pGL2). B, pRL-TK Bcl-XL 3�-UTR and pGL2 were transiently transfected into HaCaT cells stablyexpressing a p38 MAPK dominant-negative construct or vector only. Cells were UVA-irradiated (250 kJ/m2) and collected at 2 h post-irradiationin passive lysis buffer (Promega, Madison, WI). Luciferase activity was determined as described above. p values were calculated by using theStudent’s t test. C, a portion of the 3�-UTR of Bcl-XL was cloned into pBluescript and transcribed in vitro by using the Riboprobe kit T7 (Promega).HaCaT cells were pretreated with SB202190 or vehicle for 1 h and UVA-irradiated (250 kJ/m2). Lysate was collected 30 min post-irradiation.Lysates were incubated with 32P-labeled Bcl-XL 3�-UTR or cold Bcl-XL 3�-UTR for 20 min at room temperature followed by an additional 20-minincubation at room temperature after addition of heparin (5 mg/ml). Complexes were resolved on 1� TBE-agarose gel and visualized by using aPhosphorImager.
Regulation of UVA-induced Bcl-XL Expression42666
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

Several genes have been reported to be regulated at the post-transcriptional level through the 3�-untranslated region, includ-ing c-fos, cyclooxygenase-2, granulocyte-macrophage colony-stim-ulating factor, interleukin-3, �- interferon, c-jun, junB, and c-myc(19, 33–41). One mechanism by which stabilization of mRNA canoccur is through adenylate/uridylate-rich elements (AREs)within the 3�-untranslated region, consisting of the pentamercore AUUUA (42). We identified three previously unidentifiedAU-rich elements within the 3�-UTR of Bcl-XL. RNA-bindingproteins belonging to the embryonic lethal abnormal vision fam-ily of proteins have remained of interest as trans-acting factorsregulating mRNA turnover. Other RNA-binding protein familiesshown to bind to AREs include AUF1 (ARE/poly-(U)-binding/degradation factor 1), TIA-1 (T cell restricted intracellular anti-gen and TIAR (TIA-related protein), FBP (far upstream sequenceelement binding protein), TTP (tristetraprolin), hnRNP (hetero-geneous nuclear ribonucleoprotein) A0, and poly(A)-binding pro-tein. In our investigation, we report increases in binding of pro-teins to the 3�-UTR in UVA-irradiated cells with a subsequentdecrease through inhibition of p38 MAPK activity. We observeda 20% inhibition of overall binding to the Bcl-XL 3�-UTR in cellstreated with UVA and SB202190 compared with UVA-irradiatedcells alone. RNA-binding proteins have been reported to act asboth positive and negative regulators of message stability (43).The observed change could be the result of a change in thecomposition of the complex itself or a change in its ability toregulate message stability through post-translational modifica-tions as a result of decreased p38 signaling as demonstrated inprevious investigations (44, 45). It will be of interest to identifythose proteins that bind to the 3�-UTR of Bcl-XL in response toUVA irradiation.
Inhibition of apoptosis is of interest because of the connectionbetween this process and skin carcinogenesis (24, 46). UVB hasbeen shown to induce apoptosis in keratinocytes, whereas therole of UVA in modulating expression of mitochondrial proteinshas remained largely unexplored. Decreases in Bcl-XL throughantisense treatment has been shown to sensitize cells to UVB-induced apoptosis (2). These studies, however, did not demon-strate that UVB induced Bcl-XL expression but more directlyimplicated Bcl-XL in mediating resistance to UVB-induced apo-
ptosis. p53 also plays a role in UVB-induced apoptosis throughphosphorylation on Ser-15 by p38 MAPK (47). Phosphorylationof p53 by p38 MAPK stabilizes cytoplasmic p53 in normalkeratinocytes (47). UVB irradiation and p38 MAPK activity didnot alter distribution or expression of p53 in HaCaT cells,presumably due the fact that it contains two mutant p53 alle-les. Although these experiments suggest a role for p53 in UVB-induced apoptosis, our data suggest that apoptosis induced byUVA and p38 MAPK inhibition is p53-independent as ourfindings of UVA-induced protection through increased p38MAPK activity were consistent in both normal keratinocytesand HaCaT cells.
Most interestingly, UVA irradiation alone did not induceapoptosis in HaCaT cells or NHEKs as determined biochemi-cally through caspase and PARP cleavage, sub-G0 analysis, andmorphological status. Contradictory reports (48–50) have beenpublished regarding UVA-induced apoptosis, described as bothinducing apoptosis and having no significant effect in humankeratinocytes by using various UVA sources. In our experi-ments, any contaminating UVB emission was removed throughfiltering. UVA spectra with and without filtering have beenpublished previously (17) by our laboratory. It is of interest tonote that UVA is of lower energy than UVB, thus penetratinginto deeper layers of skin. Pourzand et al. (51) reported UVA-induced apoptosis through antioxidant properties of Bcl-2 inRat 6 fibroblasts. Studies by Bernerd et al. (52) demonstratethat UVA induces apoptosis of fibroblasts in the dermis with-out significant effects on kertinocytes residing in the epidermisby using an in vitro reconstructed skin model. These datasuggest a differential effect of UVA on sensitivity to apoptosisdepending upon cell type. Our data confirm the findings thatUVA does not induce apoptosis of keratinocytes at a physiolog-ically relevant dose and further extend these observations tosuggest a mechanism by which these cells become resistant toapoptosis through modulation of Bcl-XL.
In this study, we report several novel findings. First, UVA-induced p38 MAPK activity regulates Bcl-XL expression andsusceptibility to apoptosis in UVA-irradiated keratinocytes.Previous findings have not demonstrated a role for p38 MAPKin Bcl-XL expression. Second, p38 MAPK regulates Bcl-XL ex-
FIG. 10. Proposed signaling path-way through p38 MAPK leading toBcl-XL expression and resistance toUVA-induced apoptosis. UVA-inducedincreases in p38 MAPK activity lead toincreases in expression of Bcl-XL. Inhibi-tion of p38 MAPK decreases Bcl-XL ex-pression and leads to perturbation of themitochondria as observed through releaseof cytochrome c, caspase-9, and caspase-3activation and finally PARP cleavage.Cleaved caspase-3 has been reported toactivate caspase-8 (22), which can thencleave Bid, a known substrate ofcaspase-8, and lead to further increases incaspase-3 cleavage.
Regulation of UVA-induced Bcl-XL Expression 42667
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

pression at the post-transcriptional level. Although previousinvestigations have elucidated transcriptional regulation ofBcl-XL, this is the first report, to our knowledge, that demon-strates post-transcriptional regulation of Bcl-XL. Finally, thepost-transcriptional regulation of Bcl-XL is mediated throughthe 3�-UTR and is dependent upon p38 MAPK activity. AsBcl-XL is overexpressed in squamous cell carcinoma (27) andpotentially other cancer types, new insights into mechanismsregulating its expression are of great interest. In summary, wedemonstrate modulation of Bcl-XL expression by UVA-inducedp38 MAPK activity providing protection from apoptosis.
Acknowledgement—We thank Dr. Ulrich Rodeck (Thomas JeffersonUniversity, Philadelphia) for the kind gift of the Tet-Off Bcl-XL HaCaTcell line.
REFERENCES
1. Zhuang, L., Wang, B., and Sauder, D. N. (2000) J. Interferon Cytokine Res. 20,445–454
2. Taylor, J. K., Zhang, Q. Q., Monia, B. P., Marcusson, E. G., and Dean, N. M.(1999) Oncogene 18, 4495–4504
3. Oltvai, Z. N., and Korsmeyer, S. J. (1994) Cell 79, 189–1924. Polakowska, R. R., Piacentini, M., Bartlett, R., Goldsmith, L. A., and Haake,
A. R. (1994) Dev. Dyn. 199, 176–1885. Schwarz, A., Bhardwaj, R., Aragane, Y., Mahnke, K., Riemann, H., Metze, D.,
Luger, T. A., and Schwarz, T. (1995) J. Investig. Dermatol. 104, 922–9276. Casciola-Rosen, L. A., Anhalt, G., and Rosen, A. (1994) J. Exp. Med. 179,
1317–13307. Ziegler, A., Jonason, A. S., Leffell, D. J., Simon, J. A., Sharma, H. W., Kim-
melman, J., Remington, L., Jacks, T., and Brash, D. E. (1994) Nature 372,773–776
8. Matsui, M. S., and DeLeo, V. A. (1991) Cancer Cells 3, 8–129. de Gruijl, F. R. (2000) Methods Enzymol. 319, 359–366
10. Obata, T., Brown, G. E., and Yaffe, M. B. (2000) Crit. Care Med. 28, 67–7711. Chen, W., and Bowden, G. T. (1999) Oncogene 18, 7469–747612. Bachelor, M. A., Silvers, A. L., and Bowden, G. T. (2002) Oncogene 21,
7092–709913. Huang, S., Jiang, Y., Li, Z., Nishida, E., Mathias, P., Lin, S., Ulevitch, R. J.,
Nemerow, G. R., and Han, J. (1997) Immunity 6, 739–74914. Nemoto, S., Xiang, J., Huang, S., and Lin, A. (1998) J. Biol. Chem. 273,
16415–1642015. Roulston, A., Reinhard, C., Amiri, P., and Williams, L. T. (1998) J. Biol. Chem.
273, 10232–1023916. Nagata, Y., and Todokoro, K. (1999) Blood 94, 853–86317. Silvers, A. L., and Bowden, G. T. (2002) Photochem. Photobiol. 75, 302–31018. Jost, M., Gasparro, F. P., Jensen, P. J., and Rodeck, U. (2001) J. Investig.
Dermatol. 116, 860–86619. Dixon, D. A., Kaplan, C. D., McIntyre, T. M., Zimmerman, G. A., and Prescott,
S. M. (2000) J. Biol. Chem. 275, 11750–11757
20. Silvers, A. L., Bachelor, M. A., and Bowden, G. T. (2003) Neoplasia 5, 319–32921. van Loo, G., Saelens, X., van Gurp, M., MacFarlane, M., Martin, S. J., and
Vandenabeele, P. (2002) Cell Death Differ. 9, 1031–104222. Sitailo, L. A., Tibudan, S. S., and Denning, M. F. (2002) J. Biol. Chem. 277,
19346–1935223. Tsujimoto, Y. (1998) Genes Cells 3, 697–70724. Sevilla, L., Zaldumbide, A., Pognonec, P., and Boulukos, K. E. (2001) Histol.
Histopathol. 16, 595–60125. Krajewski, S., Krajewska, M., Shabaik, A., Wang, H. G., Irie, S., Fong, L., and
Reed, J. C. (1994) Cancer Res. 54, 5501–550726. Pena, J. C., Fuchs, E., and Thompson, C. B. (1997) Cell Growth & Differ. 8,
619–62927. Nickoloff, B. J., Qin, J. Z., Chaturvedi, V., Bacon, P., Panella, J., and Denning,
M. F. (2002) J. Investig Dermatol. Symp. Proc. 7, 27–3528. Cerroni, L., and Kerl, H. (1994) J. Cutan. Pathol. 21, 398–40329. Morales-Ducret, C. R., van de Rijn, M., LeBrun, D. P., and Smoller, B. R. (1995)
Arch. Dermatol. 131, 909–91230. Rodriguez-Villanueva, J., Greenhalgh, D., Wang, X. J., Bundman, D., Cho, S.,
Delehedde, M., Roop, D., and McDonnell, T. J. (1998) Oncogene 16, 853–86331. Wrone-Smith, T., Bergstrom, J., Quevedo, M. E., Reddy, V., Gutierrez-Steil, C.,
and Nickoloff, B. J. (1999) J. Dermatol. Sci. 19, 53–6732. Grillot, D. A., Gonzalez-Garcia, M., Ekhterae, D., Duan, L., Inohara, N., Ohta,
S., Seldin, M. F., and Nunez, G. (1997) J. Immunol. 158, 4750–475733. Peppel, K., Vinci, J. M., and Baglioni, C. (1991) J. Exp. Med. 173, 349–35534. Jones, T. R., and Cole, M. D. (1987) Mol. Cell. Biol. 7, 4513–452135. Treisman, R. (1985) Cell 42, 889–90236. Stoecklin, G., Hahn, S., and Moroni, C. (1994) J. Biol. Chem. 269, 28591–2859737. Chen, C. Y., and Shyu, A. B. (1994) Mol. Cell. Biol. 14, 8471–848238. Shaw, G., and Kamen, R. (1986) Cell 46, 659–66739. Cok, S. J., and Morrison, A. R. (2001) J. Biol. Chem. 276, 23179–2318540. Dixon, D. A., Tolley, N. D., King, P. H., Nabors, L. B., McIntyre, T. M.,
Zimmerman, G. A., and Prescott, S. M. (2001) J. Clin. Investig. 108,1657–1665
41. Lasa, M., Mahtani, K. R., Finch, A., Brewer, G., Saklatvala, J., and Clark, A. R.(2000) Mol. Cell. Biol. 20, 4265–4274
42. Chen, C. Y., and Shyu, A. B. (1995) Trends Biochem. Sci. 20, 465–47043. Dean, J. L., Sully, G., Clark, A. R., and Saklatvala, J. (2004) Cell. Signal. 16,
1113–112144. Rousseau, S., Morrice, N., Peggie, M., Campbell, D. G., Gaestel, M., and Cohen,
P. (2002) EMBO J. 21, 6505–651445. Mahtani, K. R., Brook, M., Dean, J. L., Sully, G., Saklatvala, J., and Clark,
A. R. (2001) Mol. Cell. Biol. 21, 6461–646946. Kamb, A. (1994) Nature 372, 730–73147. Chouinard, N., Valerie, K., Rouabhia, M., and Huot, J. (2002) Biochem. J. 365,
133–14548. He, Y. Y., Huang, J. L., Ramirez, D. C., and Chignell, C. F. (2003) J. Biol.
Chem. 278, 8058–806449. He, Y. Y., Huang, J. L., Gentry, J. B., and Chignell, C. F. (2003) J. Biol. Chem.
278, 42457–4246550. Didier, C., Emonet-Piccardi, N., Beani, J. C., Cadet, J., and Richard, M. J.
(1999) FASEB J. 13, 1817–182451. Pourzand, C., Rossier, G., Reelfs, O., Borner, C., and Tyrrell, R. M. (1997)
Cancer Res. 57, 1405–141152. Bernerd, F., and Asselineau, D. (1998) Cell Death Differ. 5, 792–802
Regulation of UVA-induced Bcl-XL Expression42668
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from

Michael A. Bachelor and G. Timothy Bowden-UNTRANSLATED REGION
′Keratinocytes: POST-TRANSCRIPTIONAL REGULATION THROUGH THE 3 by p38 MAPK in HumanLUltraviolet A-induced Modulation of Bcl-X
doi: 10.1074/jbc.M406626200 originally published online August 2, 20042004, 279:42658-42668.J. Biol. Chem.
10.1074/jbc.M406626200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/279/41/42658.full.html#ref-list-1
This article cites 52 references, 21 of which can be accessed free at
by guest on October 21, 2016
http://ww
w.jbc.org/
Dow
nloaded from




















