
Asymptotic rejection of nonvanishing disturbances despite plant-model mismatch

Md
doi:10.1006/mcne.2000.0950, available online at http://www.idealibrary.com on
Molecular and Cellular Neuroscience 17, 500–513 (2001)MCN
Structural Diversity Despite Strong EvolutionaryConservation in the 5*-Untranslated Regionof the P-Type Dystrophin TranscriptHassan Abdulrazzak,* ,1 Nobuhiro Noro,† J. Paul Simons,*Geoffrey Goldspink,* Eric A. Barnard,‡ and Dariusz C. Gorecki* ,2
*Department of Anatomy and Developmental Biology, Division of Basic Medical Sciences,Royal Free and University College Medical School, Rowland Hill Street, London NW3 2PF,United Kingdom; †Department of Pharmacology, Tsukuba Research Laboratory,GlaxoWellcome, 43 Wadai, Tsukuba 300-4247, Japan; and ‡Department of Pharmacology,University of Cambridge, Tennis Court Road, Cambridge CB2 1QJ, United Kingdom
Analysis of the 5*-flanking regions of the Purkinje (P-)dystrophin genes and mRNAs in different species re-vealed strong sequence conservation but functional di-versity. Multiple transcription initiation sites were identi-fied in cerebella and muscles, tissues expressingP-dystrophin. The predominant initiation site was con-served, with another muscle-specific site located up-stream. Despite sequence homology, significant tissue-and species-specific structural diversity in the P-type 5*-ends exists, including alternative splicing within the5*-untranslated region combined with alternative splicingof intron 1. One amino terminus is conserved in mammalsand, to a lesser extent, in chicken. However, alternativeusage of ATG codons may result in a choice of N-terminior translation of short upstream ORFs in different species.Promoter activity of a fragment upstream of the cap sitewas shown by transient expression in myoblasts and invivo following intramuscular injection. It is tissue- anddevelopmentally regulated. Analysis of promoter dele-tions suggests the existence of negative regulatory ele-ments in the proximal region.
INTRODUCTION
Duchenne and Becker muscular dystrophies (DMD/BMD) are caused by mutations in the dystrophin gene
1 Current address: Cardiac Medicine, Imperial College School ofedicine, National Heart and Lung Institute, Dovehouse Street, Lon-
on SW3 6LY.2 To whom correspondence and reprint requests should be ad-
dressed at School of Pharmacy and Biomedical Sciences, University of
Portsmouth, St. Michael’s Building, White Swan Road, PortsmouthPO1 2DT, UK. Fax: 023-92-843-565. E-mail: [email protected].500
(Hoffman et al., 1987). As well as the muscle pathology,DMD is associated with a variable degree of mentalimpairment (Emery, 1993). The brain is the second ma-jor site of dystrophin gene expression and it is the brainwhich contains the highest number of dystrophin iso-forms (Gorecki et al., 1992; Gorecki and Barnard, 1995).They are expressed in specific areas associated withcognitive functions and in neurons, dystrophin is foundat post-synaptic densities (reviewed in Lidov, 1996).Alterations of expression of specific isoforms in hip-pocampal neurons in response to kainate (Gorecki et al.,1998) and in sympathetic neurons following axotomy(Zaccaria et al., 1998), as well as changes in synapticplasticity in dystrophic mice (Vaillend et al., 1999) sug-gest dystrophin involvement in assembly–disassemblyand/or preservation of the postsynaptic apparatus. Re-cently, the lack of full-length dystrophins was associ-ated with altered synaptic clustering of GABAA recep-tors (Knuesel et al., 1999).
A complex expression pattern of dystrophin is inagreement with complicated structure and regulation ofthis very large gene due to utilisation of alternativepromoters and generation of multiple mRNAs by alter-native splicing. At least eight distinct promoters controltissue-, cell-, and developmentally specific expressionof dystrophin in an independent and sometimes mutu-ally exclusive manner (reviewed in Sadoulet-Puccioand Kunkel, 1996). Three promoters control the expres-sion of alternative “full-length” (14 kb) mRNAs. Fourother, intragenic promoters, regulate expression of
truncated transcripts coding for 260-, 140-, 116-, and71-kDa protein products expressed predominantly in1044-7431/01 $35.00Copyright © 2001 by Academic Press
All rights of reproduction in any form reserved.

ApaeimqatmItcuicn
smtp3
501Regulation of the P-type Dystrophin Expression
different parts of the nervous system. Given the signif-icant and still not fully understood role of dystrophin inhuman health and disease, gaining an insight into thisintricate genetic regulation may have implications notonly for the molecular characterization of the dystro-phin gene, but may ultimately lead to therapies basedon modulation of expression or splicing in a subset ofpatients.
We have previously described one of the full-lengthtranscripts (P-type) that is expressed in the brain spe-cifically in cerebellar Purkinje cells (Gorecki et al., 1992).
n alternative promoter located between the M-typeromoter and the second exon of the dystrophin geneppears to drive its expression. The unique P-type 1stxon encodes an alternative N-terminus. To character-ze the P-type specific transcript and its putative pro-
oter we have searched phylogenetically related se-uences for conserved motifs and performed functionalnalysis of this genomic region. We have also studiedhe tissue distribution and the structure of P-type
RNA outside the brain and during development.dentification of mechanisms regulating expression ofhis transcript would expand our understanding of theomplex regulation of the dystrophin gene. This partic-lar form of dystrophin, in view of its distinctive local-
zation, could also provide a tool for identifying a spe-ific functional element in Purkinje cells, the majoreurons in the cerebellum.
RESULTS
Isolation and Characterization of the P-type5*-UTR and Promoter Region from DifferentSpecies
A cosmid library containing the 59-end of the humandystrophin gene (Nizetic et al., 1991) was screened witha probe specific for the P-type exon 1. Two positiveclones (4828 and 4829) were digested with HindIII (cut-ting once within the P-type exon 1) and the 1.78-kbhybridizing fragment found was subcloned and se-quenced. The integrity of the cloned region extending 59from the P-type exon 1 was confirmed by analysis of thecosmid clones and normal human genomic DNA byrestriction digestion, hybridization, and PCR.
Comparison of regulatory regions from evolutionar-ily distant organisms can reveal regulatory elementsthat are conserved and may be functionally relevant. Todelineate DNA elements of the P-type promoter, we
isolated the upstream regions from several species. Fig-ure 1 shows the sequence comparison between the cor-responding regions of six species using the Clustal Walignment program. The region encompassing the 59-end of the P-type transcript is highly conserved and anumber of conserved sequence motifs have been iden-tifed (Fig. 1). One notable difference was found in pigwhere a thymidine-rich 24-bp insert was present (nt2617 to 2641). The sequence divergence mirrors thephylogenetic relationships of the various species.
The P-type exon 1 sequence is highly conservedamongst the mammalian species examined (;90%). Themouse and rat sequences contain two upstream ATGcodons that are in an open reading frame that is con-tinuous with the rest of the dystrophin coding se-quence. All three rodent ATG codons are of equalweight with respect to the Kozak consensus sequence(Kozak, 1999). Translation initiated from the upstreamATG codons would add 11 or 27 amino acids to therodent P-type N-terminus. However, the upstreamATG codons in human and dog are not “in frame”and/or are followed by stop codons, resulting in shortupstream open reading frames.
The human and mouse P-type promoter regions andexons 1 have GC content of 34–44%, which is averagefor vertebrate sequences. No CpG islands could beidentified. In the human sequence there is a regionmatching the consensus TATA box sequence; however,this is not in the right context of the established cap sites(see below) and it is not conserved in all species. Severaltranscription factor-binding sites were detected in theP-type sequence, including two conserved E-box ele-ments (Fig. 1) and a cAMP-response element (in thehuman sequence).
The high homology observed in mammals lead us toquestion whether the P-type dystrophin might bepresent in more distant species. In zoo blots the P-typeprobe hybridized to a ;2.5-kb band in HindIII-digestedchicken genomic DNA (not shown). Amplification ofchicken cerebellar RNA with a primer complementaryto chicken exon 2–3 junction (Lemaire et al., 1988) and aecond primer based on the consensus mouse and hu-an P-type sequences gave a specific product. In this,
he sequence upstream from exon 2 of chicken dystro-hin shows 53.6% nucleotide sequence homology to the9end of P-type exon 1 in human and mouse. The pre-
dicted amino acid sequence of the chicken P-type N-terminus is: Met-Ser-Gly-Asp-Leu-Leu-Asp (mam-malian: Met-Ser-Glu-Val-Ser-Ser-Asp) and its length isidentical to that found in mammals, with 3 out of 7amino acids being conserved.
Our analysis of two human genomic sequences
and two more sequences recently deposited inthe database (Accession Nos. AB037493 and
q
E
iaasv
taoisrPmMs
e sha
502 Abdulrazzak et al.
AL049643.12|HSJ672M15) did not confirm the se-quence variation within the human coding regionreported by Holder et al. (1996). In fact, their se-
uence is identical with the mouse P-type sequence.
xpression Pattern of the P-Type Dystrophin
Hybridisation of RNA with a P-type specific probedentified a single band of ;14 kb in adult mouse brainnd skeletal muscle samples (Fig. 2A). This size is ingreement with other full-length dystrophin tran-
FIG. 1. Clustal W alignment of the P-type dystrophin first exon andin frame with the rest of dystrophin are shown in bold. Stop codons icerebellum is underlined. Putative transcription factor binding sites arin cerebellum and muscle.
cripts. S1 nuclease protection confirmed earlier obser-ations (Holder et al., 1996; Gorecki and Barnard, 1997)
oc
hat the P-type transcript is expressed in skeletal andlso cardiac muscle, albeit at low levels. No expressionf this mRNA has been found in other tissues tested,
.e., spleen, kidney, lungs, stomach, and liver (data nothown). RT-PCR analysis of different mouse musclesevealed expression in all muscle groups tested. RT-CR of RNA isolated from individual human striateduscle fibers showed that the P-type transcript, like-type, is present in all muscle fiber types (data not
hown).Analysis of P-type expression during mouse devel-
king genomic sequences from various species. Potential start codonsme are italicised. The 93-bp region alternatively spliced in the mouseded. The arrows indicate the major transcription initiation sites found
flann fra
pment was performed in outline using PCR amplifi-ation of RNA isolated from embryos as well as from

Con
503Regulation of the P-type Dystrophin Expression
neonatal and postnatal mice (Fig. 2B). Expression wasalready detectable in brains of 12.5 d.p.c. mouse em-bryos, corresponding well with the in situ hybridizationdata of Houzelstein et al. (1992).
Identification of Transcription Initiation Sites
Primer extension using mouse cerebellar RNA gaveseveral products: a predominant band of 284 bp andtwo smaller bands of 256 and 254 bp were observed. Inhuman cerebellar RNA, a single clear 287 bp band wasdetected (Fig. 3). These results suggest that in Purkinje
FIG. 1—
cells the P-type dystrophin transcript is initiated at thesame site in both human and mouse (arrow in Fig. 1). In
muscle, primer extension was not conclusive due to lowlevels of this transcript.
Rapid amplification of cDNA ends (59-RACE) wasemployed to isolate the 59-ends of the P-type transcriptsin human and mouse cerebella and skeletal muscle (Fig.4). In the mouse cerebellum three products were iden-tified, cloned and sequenced (A1, A2, and A3 in Fig.4A). Product A1, the most upstream-extending, alsolacked a 93-bp region that we infer to have beenspliced-out (as determined by comparison to the mousegenomic DNA sequence). This novel alternative splic-ing is discussed in more detail below. Two other prod-
tinued
ucts were also identified. The longer of the two, starts atnt 2266 with respect to the Met-1 codon and is com-

wtw
n9qa1ptwra
tHcAttRphdNTv
504 Abdulrazzak et al.
patible with the 284-bp product obtained by primerextension. The second 59RACE product starts at nt 2241and is compatible with the 256-bp primer extensionproduct.
In human cerebellum 59RACE gave two products.The longer of the two (B1) starts at nucleotide 2268
ith respect to the human Met-1. It is compatible with
FIG. 2. Expression of the P-type transcript. (A) RNA-gel hybridiza-ion. Lane B, mouse brain RNA; Lane M, mouse skeletal muscle RNA.
ybridization was with the P-type exon 1-specific probe; arrow indi-ates specific bands of ;14 kb in skeletal muscle and the cerebellum.
GAPDH-specific probe was used as a positive control and to showhe relative RNA loading in each lane. (B) Developmental analysis ofhe P-type mRNA expression in the mouse brain. Southern blot ofT-PCR products obtained from mouse embryonic, neonatal, andostnatal brains with the P-type specific primer set (SEQ/ECO3) andybridized with internal probe M2nd (nt 266–292 relative to humanystrophin sequence). Lanes: (1) Embryonic day 12.5; (2) E15.5; (3)eonatal; (4) Postnatal day 6; (5) P16; (6) P24; (7) No cDNA control.wo bands representing splice variants of the P-type transcript areisible.
he 289-bp primer extension product and is comparableith that from mice. A shorter product (B2) initiating at
nt 2128 was also identified. The most 59 site for tran-scription initiation of the P-type transcript in humancerebellum predicts the size of exon 1 as 282 bp, ofwhich 263 bp (93.3%) is noncoding.
In the mouse muscle 59RACE gave two major prod-ucts (Figs. 4A and 4B). The 59-end of the predominantone was at nt 2259, seven nucleotides short of the majorcap site found in mouse and human cerebellum. Thistype constituted about 70% of RACE clones. However,a minority of clones extended further upstream to nt2745, predicting an alternative transcription initiationsite and a much longer P-type exon 1 in muscle.
Alternative Splicing at the 5* End of the P-TypeDystrophin Transcript
The 59RACE in mouse cerebellum revealed an alter-ative 59UTR sequence of the P-type mRNA, lacking a3-bp fragment. Typical donor/acceptor splice site se-uences present in the genomic sequence suggest thatlternative splicing occurred (Horowitz and Krainer,994). Since no corresponding band was detected usingrimer extension, further experiments were conducted
o rule out a RACE artefact. Amplification with a for-ard primer upstream of the 93-bp sequence and a
everse primer spanning exons 2/3 border gave severalmplification products (Fig. 5). These were sequenced
FIG. 3. Primer extension analysis of P-type transcripts. (A) Sche-matic representation of the primer (SI) used for primer extension withrespect to the 59-end of the P-type dystrophin. (B) Left panel, mousecerebellar RNA. Increasing amounts of the final reaction were run ineach lane. Right panel, human cerebellar RNA. The sizes of the main
extension products are indicated (calculated from the sequencingreaction run as a size marker).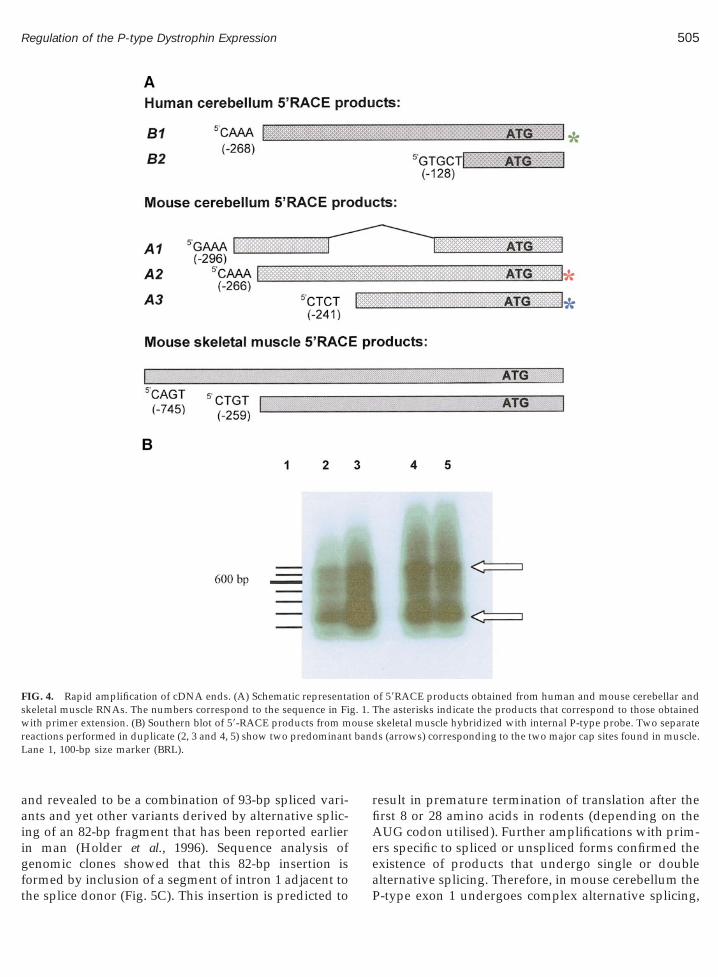
gft
t band
505Regulation of the P-type Dystrophin Expression
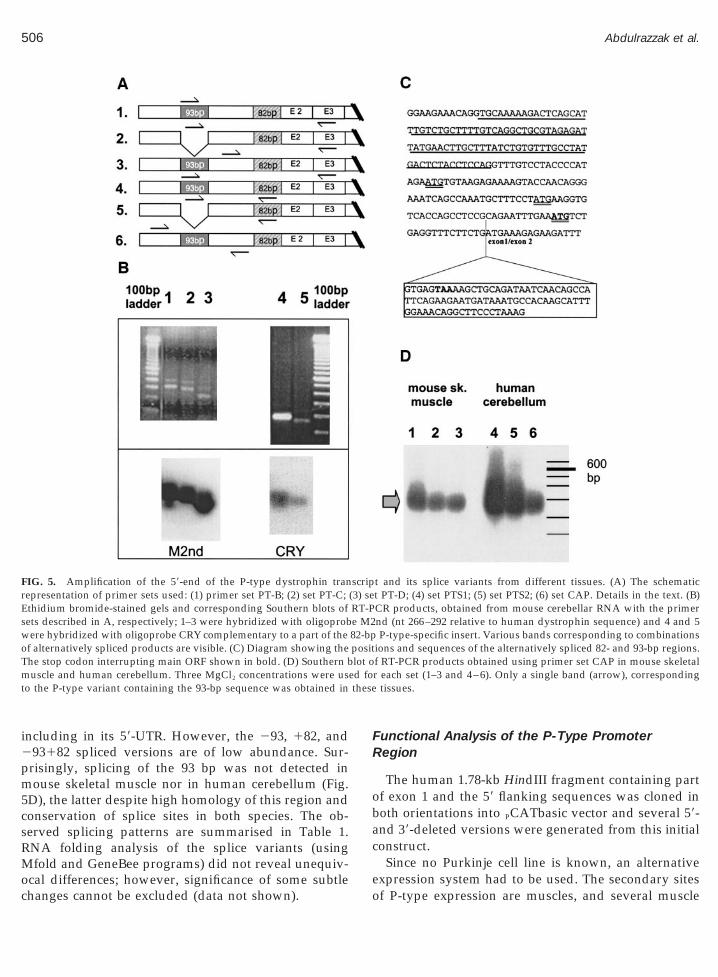
and revealed to be a combination of 93-bp spliced vari-ants and yet other variants derived by alternative splic-ing of an 82-bp fragment that has been reported earlierin man (Holder et al., 1996). Sequence analysis of
enomic clones showed that this 82-bp insertion is
FIG. 4. Rapid amplification of cDNA ends. (A) Schematic representaskeletal muscle RNAs. The numbers correspond to the sequence in Fiwith primer extension. (B) Southern blot of 59-RACE products from mreactions performed in duplicate (2, 3 and 4, 5) show two predominanLane 1, 100-bp size marker (BRL).
ormed by inclusion of a segment of intron 1 adjacent tohe splice donor (Fig. 5C). This insertion is predicted to
result in premature termination of translation after thefirst 8 or 28 amino acids in rodents (depending on theAUG codon utilised). Further amplifications with prim-ers specific to spliced or unspliced forms confirmed theexistence of products that undergo single or double
of 59RACE products obtained from human and mouse cerebellar andThe asterisks indicate the products that correspond to those obtainedskeletal muscle hybridized with internal P-type probe. Two separates (arrows) corresponding to the two major cap sites found in muscle.
tiong. 1.ouse
alternative splicing. Therefore, in mouse cerebellum theP-type exon 1 undergoes complex alternative splicing,
pm5csRMoc
FR
ob
ed fothese
506 Abdulrazzak et al.
including in its 59-UTR. However, the 293, 182, and293182 spliced versions are of low abundance. Sur-
risingly, splicing of the 93 bp was not detected inouse skeletal muscle nor in human cerebellum (Fig.
D), the latter despite high homology of this region andonservation of splice sites in both species. The ob-erved splicing patterns are summarised in Table 1.NA folding analysis of the splice variants (usingfold and GeneBee programs) did not reveal unequiv-
FIG. 5. Amplification of the 59-end of the P-type dystrophin tranrepresentation of primer sets used: (1) primer set PT-B; (2) set PT-C;Ethidium bromide-stained gels and corresponding Southern blots ofsets described in A, respectively; 1–3 were hybridized with oligoprobwere hybridized with oligoprobe CRY complementary to a part of theof alternatively spliced products are visible. (C) Diagram showing theThe stop codon interrupting main ORF shown in bold. (D) Southern bmuscle and human cerebellum. Three MgCl2 concentrations were usto the P-type variant containing the 93-bp sequence was obtained in
cal differences; however, significance of some subtlehanges cannot be excluded (data not shown).
unctional Analysis of the P-Type Promoteregion
The human 1.78-kb HindIII fragment containing partf exon 1 and the 59 flanking sequences was cloned inoth orientations into PCATbasic vector and several 59-
and 39-deleted versions were generated from this initialconstruct.
Since no Purkinje cell line is known, an alternative
t and its splice variants from different tissues. (A) The schematict PT-D; (4) set PTS1; (5) set PTS2; (6) set CAP. Details in the text. (B)CR products, obtained from mouse cerebellar RNA with the primernd (nt 266–292 relative to human dystrophin sequence) and 4 and 5P-type-specific insert. Various bands corresponding to combinations
ions and sequences of the alternatively spliced 82- and 93-bp regions.f RT-PCR products obtained using primer set CAP in mouse skeletalr each set (1–3 and 4–6). Only a single band (arrow), corresponding
tissues.
scrip(3) seRT-Pe M282-bppositlot o
expression system had to be used. The secondary sitesof P-type expression are muscles, and several muscle

vt(
507Regulation of the P-type Dystrophin Expression
cell lines are available. We have analyzed rat L6 andmouse C2C12 lines and found that C2C12 cells (Yaffeand Saxel, 1977) expressed the P-type dystrophin upondifferentiation into myotubes in vitro, but not as undif-ferentiated myoblasts (results not shown).
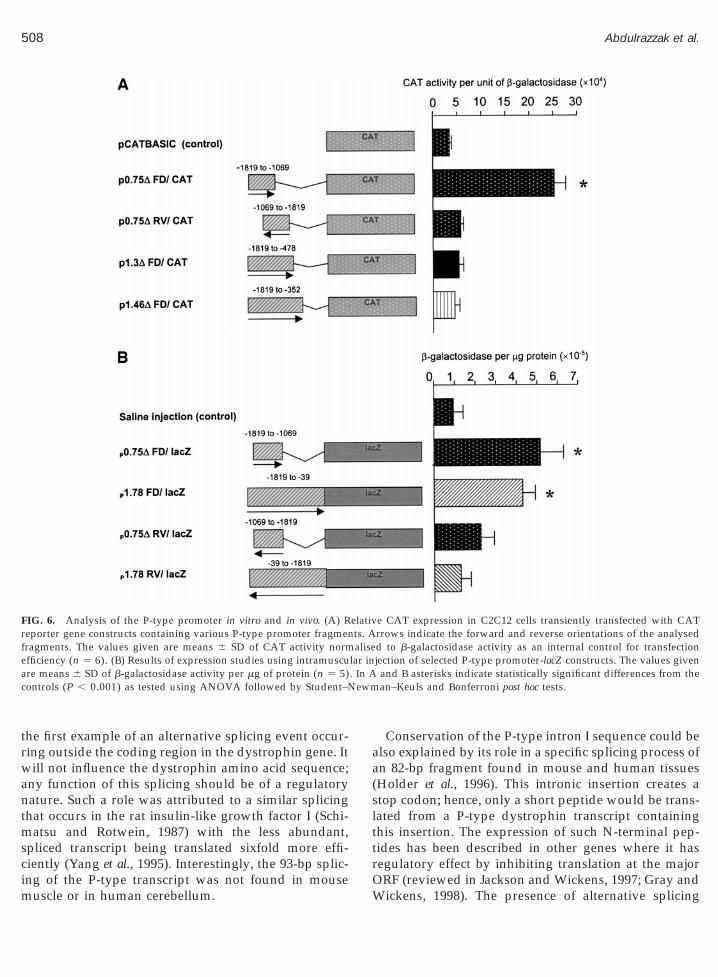
C2C12 myoblasts were transfected with specific con-structs (containing the putative promoter regionslinked with the CAT reporter gene) and were differen-tiated into myotubes. A b-galactosidase expression vec-tor was cotransfected with the CAT constructs, andb-gal activity was used to correct for transfection effi-ciency. Surprisingly, constructs containing the full-length 1.78-kb P-type fragment in either forward orreverse orientation failed to produce significantly moreCAT activity than the controls. This was also the casefor 59-deleted constructs, however, a 39-deleted, 0.75-kbfragment (nt 21819 to 21069) in forward orientationdrove CAT expression in myotubes (Fig. 6A). The samefragment in reverse orientation, as well as longer, 39-deleted constructs (p1.3FD/CAT and p1.46FD/CAT)failed to produce significantly more CAT activity thanthe control plasmid. Therefore, the P-type region ofnucleotides 21819 to 21069 is capable of driving thereporter gene expression in an orientation-specific man-ner, suggesting that this region contains core regulatoryelement(s) active in differentiated myotubes.
The P-type promoter activity was confirmed in vivousing direct muscle injection of constructs containingfragments upstream of the cap site ligated to anotherreporter gene (lacZ). In this system constructs contain-ing the whole 1.78-kb HindIII fragment as well as the0.75-kb fragment produced significant levels of b-galac-tosidase (Fig. 6B).
DISCUSSION
The DMD gene exhibits a structural arrangementsimilar to a number of other genes in which multiple
TABLE 1
Alternative Splicing at the 59 End of the P-Type DystrophinTranscript Found in Mouse and Human Tissues
Splicingtype
Mousecerebellum
Mousemuscle
Humancerebellum
Humanmuscle
293 bp Yes No No No182 bp Yes Yes Yes Yes
promoters generate numerous transcripts that differ intheir first exons, e.g., BDNF, nNOS, dystrobrevin (Holz-
Pt
feind et al., 1999; Nanda and Mack, 1998; Wang et al.,1999). In the present study we have characterized thedystrophin P-type promoter region. Like other dystro-phin promoters it shows tissue-selectivity rather thantissue-specificity: It is active, albeit at different levels inneurons and muscle and may be developmentally reg-ulated (see also Holder et al., 1996; Torelli et al., 1999).
The region upstream of the 59UTRs exhibited none ofthe consensus features typical for tissue-specific pro-moters. The presence of multiple transcriptional startsites is consistent with the lack of conserved TATA boxelements. Thus, the P-type is similar to the C-type dys-trophin promoter, which also lacks a TATA box and isactive in neurons and at a low level in muscle. Unlikehousekeeping genes, both P- and C-types have low GCcontents (Hugnot et al., 1993). Although an increasingnumber of TATA-less, nonhousekeeping promoters isbecoming recognized, little is known of the basis oftheir function (Smale, 1997).
The P-type first exon is mainly untranslated (93.3%),similarly to other dystrophin first exons (Klamut et al.,1990; Makover et al., 1991; Fracasso and Patarnello,1998). The conservation of the entire P-type region ishigh. There is no large open reading frame (ORF) oneither DNA strand in this region, which excludes con-servation due to the existence of another, overlappinggene. The high degree of homology extends into intron1 in human and mouse. This nucleotide preservationmay signify that the 59UTR may effect translationalcontrol of P-dystrophin biosynthesis via specific mech-anisms of which two can be relevant here.
First, in ;5–10% of vertebrate mRNAs, the first AUGis not the translational initiation site of the major openreading frame. Some of these upstream AUGs can dra-matically inhibit translation at major start sites (re-viewed in: Jackson and Wickens, 1997). Interestingly,despite the high overall P-type homology, there are twoadditional in-frame AUG sites in rodents, which inhuman and dog can only create small upstream ORFsdue to the existence of downstream stop codons. Such ashort ORF within the 59-UTR can regulate translationwhile sequences downstream and upstream of the shortORF can affect reinitiation at a downstream site(Ayoubi and Van de Ven, 1996; Jackson and Wickens,1997).
A second mechanism of translational control involvessecondary structures within the 59UTR. They can se-
erely inhibit translation, which is in agreement withhe scanning hypothesis of mRNA translation initiationKozak, 1999). In this context another feature of the
-type transcript may be relevant, namely an alterna-ive splicing of a 93 bp region within the 59UTR. This is
im
aa(
rf
a . In Ac ewm
508 Abdulrazzak et al.
the first example of an alternative splicing event occur-ring outside the coding region in the dystrophin gene. Itwill not influence the dystrophin amino acid sequence;any function of this splicing should be of a regulatorynature. Such a role was attributed to a similar splicingthat occurs in the rat insulin-like growth factor I (Schi-matsu and Rotwein, 1987) with the less abundant,spliced transcript being translated sixfold more effi-ciently (Yang et al., 1995). Interestingly, the 93-bp splic-
FIG. 6. Analysis of the P-type promoter in vitro and in vivo. (A) Reporter gene constructs containing various P-type promoter fragmenragments. The values given are means 6 SD of CAT activity norm
efficiency (n 5 6). (B) Results of expression studies using intramuscure means 6 SD of b-galactosidase activity per mg of protein (n 5 5)ontrols (P , 0.001) as tested using ANOVA followed by Student–N
ng of the P-type transcript was not found in mouseuscle or in human cerebellum.
Conservation of the P-type intron I sequence could belso explained by its role in a specific splicing process ofn 82-bp fragment found in mouse and human tissuesHolder et al., 1996). This intronic insertion creates a
stop codon; hence, only a short peptide would be trans-lated from a P-type dystrophin transcript containingthis insertion. The expression of such N-terminal pep-tides has been described in other genes where it hasregulatory effect by inhibiting translation at the major
e CAT expression in C2C12 cells transiently transfected with CATrrows indicate the forward and reverse orientations of the analysedd to b-galactosidase activity as an internal control for transfectionjection of selected P-type promoter-lacZ constructs. The values given
and B asterisks indicate statistically significant differences from thean–Keuls and Bonferroni post hoc tests.
elativts. Aalise
lar in
ORF (reviewed in Jackson and Wickens, 1997; Gray andWickens, 1998). The presence of alternative splicing

tptes
ocftTts
509Regulation of the P-type Dystrophin Expression
events that create stop codons occurs in other dystro-phin mRNAs too (Surono et al., 1997). Translation ofhese mRNAs may re-initiate downstream: in DMDatients with deletions of exons 3–7, a dystrophin pro-
ein is produced that lacks the sequences encoded byxons 1–7, probably due to reinitiation at a downstreamite in exon 8 (Winnard et al., 1995).
Both the 93-bp splicing and the 82-bp insertion canccur in combination with one another and show spe-ies- and tissue-specificity (Table 1). The predominantorm of the cerebellar P-type mRNA is one that containshe 93-bp 59UTR sequence and lacks the 82-bp insertion.he least abundant combination is a transcript lacking
he 93-bp 59UTR sequence and retaining the 82-bp in-ertion. This complex pattern of 59-alternative splicing
may represent a novel mechanism of posttranscrip-tional regulation of P-type dystrophin where gene ex-pression would be regulated by tissue- and/or devel-opmentally specific splicing factors. Therefore,alternative splicing of dystrophin may play a wider rolethan previously appreciated not only in producing var-ious truncated dystrophins with possible unique func-tions but it may also provide a regulatory mechanism.
Noteworthy, the P-type posttranscriptional regula-tion may involve distinct mechanisms in mouse andhuman. These are upstream short ORF but no 59-UTRsplicing present in human, and 59-UTR splicing but noupstream short ORF sequences in mouse dystrophinmRNAs.
The functional analysis of the P-type promoter waslimited here by the lack of a Purkinje cell line. However,based upon the finding that skeletal muscle constitutesa secondary site of P-type expression, we used a myo-genic cell line to study the promoter and found that theregion spanning nt 21819 to 21069 is capable of driv-ing the expression of the reporter gene in an orienta-tion-specific manner in differentiated myotubes in vitro.The expression decreased with increased length of thisfragment towards its 39-end and this effect was strongerin myotubes in vitro than in muscle in vivo. This sug-gests the presence of one or more negative regulatoryelements in the proximal part of this region and that itsactivity may change with muscle differentiation.
Interestingly, the Purkinje-cell specific promoter,pcp-2 (L7) is up- and down-regulated by homeodomainproteins (Sanlioglu et al., 1998). It contains a series ofTAAT homeodomain protein binding motifs adjacent toa consensus E-box and lying roughly 60 bp upstream of
the TATA box. We found an almost identical configu-ration of TAAT, E-box, and TATA motifs in the P-typesequence. However, several findings in this study, e.g.,lack of TATA box conservation, multiple transcriptionstart sites (characteristic of TATA-less promoters), andlack of promoter activity with constructs containing thisTATA box suggest that a TATA-less promoter drivesthe P-type transcription. However, the coexistence ofTATA-dependent and TATA-independent transcrip-tion regulation resembling one found in the iNOS gene(Chu et al., 1995) cannot be excluded. The same pro-moter can bind different regulatory factors dependingon the tissue in which it is used (Ayoubi and Van deVen, 1996). Moreover, it is becoming increasingly clearthat dystrophin regulatory DNA sequences drivingtranscription in tissue culture are often different fromthose that direct correct tissue specificity and temporo-spatial regulation in vivo. Extrapolation of our results toPurkinje cells would, in any case, need verification invivo, e.g., in a transgenic system, as proved by recentdata on dystrophin M-type and C-type promoters. Inthese cases, the genomic regions showing strong pro-moter activity in cultures were active only in the rightventricle (M-type) or cerebral cortex (C-type) of trans-genic mice (Kimura et al., 1997a and 1997b).
The expression pattern of P-type dystrophin duringdevelopment has not been studied in depth. In situhybridization experiments using a probe detecting allfull-length mRNAs revealed dystrophin expression inthe developing mouse cerebellum at 13.5 days (Houzel-stein et al., 1992). This is in agreement with our RT-PCRresults showing P-type dystrophin as early as 12.5 daysin mouse embryonic brain. Purkinje cells are reportedto arise between days 11 and 13 of mouse embryogen-esis (Miale and Sidman, 1961). Thus, P-type dystrophinmay serve as a marker for Purkinje cells early in devel-opment in an analogous manner to calbindin. More-over, we have found increasing levels of the P-typemRNA in differentiating myotubes and developmentalexpression of the P-type transcript has been reported inhuman cerebral cortex (Holder et al., 1996). Since DMD-associated mental retardation, unlike muscle disease, isnonprogressive the developmental regulation of ex-pression of dystrophin may be critical and requiresfurther investigations.
The physiological role of the P-type isoform is as yetunknown. In birds and mammals the cerebellar anat-omy and the output are organised in the same way,with Purkinje cells having the central role (Voogd andGlickstein, 1998). Therefore, the existence of a specific
isoform in these cells in both man and chicken suggestsa significant role. The altered synaptic clustering of
m
hifitttttimd(
aaapmpoac
pitsdipm
510 Abdulrazzak et al.
GABAA receptors recently reported in mdx neuronsay be relevant (Knuesel et al., 1999).The functional significance of the numerous, often
ighly similar dystrophins remains unclear. Whilentragenic promoters produce specific protein iso-orms with clearly altered properties (Dp116, Dp71),t is unknown if the short alternative N-termini of thehree full-length dystrophins are functionally dis-inct. On the one hand, in all mammalian speciesested the amino acid sequence of the major P-typeerminus was conserved. Its length was also main-ained in chicken, with three of seven amino acidsdentical. However, in rodents the P-type N-terminus
ay be longer, as is the N-terminus of the M-typeystrophin in chicken when compared to mammals
Lemaire et al., 1988). This variability within an iso-form may imply the lack of functional significance ofthe amino-termini of full-length dystrophins but thisremains to be confirmed experimentally. An alterna-tive explanation for near-identical dystophins couldinvolve redundant expression. It is uncertain, how-ever, if any genes or transcripts are truly redundantas they may increase fitness in subtle ways (Cooke etal., 1997). Therefore, the relevance of multiple dystro-phins may lie in tissue- and spatiotemporal-specific-ity of expression and/or varying levels of expression(transcriptional and translational control).
One consequence of tissue- and developmental-specific expression of alternative transcripts might bethe way a dystrophin mutation can express a differ-ent phenotype in skeletal muscle, heart, and CNS.Our data, as well as others (Muntoni et al., 1995a andb; Nakamura et al., 1997; Holder et al., 1996; Torelli etl., 1999), indicate that the P-type transcript displaysbroader complexity of expression than previously
ppreciated. The mRNA levels of the P-type dystro-hin can be increased in skeletal but not cardiacuscle of some patients with mutations of M-type
romoter associated with X-linked dilated cardiomy-pathy (XLDCM) (Muntoni et al., 1993, 1995; Ferlini etl., 1999) and in BMD (Nakamura et al., 1997), indi-ating compensatory mechanisms.
In conclusion, both the gene structure and the ex-ressional regulation of the DMD gene are exceed-
ngly elaborate. These properties present it as one ofhe most complex genes described to date. Given theignificant role of dystrophin in human health andisease, understanding this intricate regulation is an
mportant goal and further studies of selectively ex-
ressed isoforms (such as the P-type) can be infor-ative.EXPERIMENTAL METHODS
Isolation, Characterization, and Sequencing ofGenomic Clones
The human cosmid clones containing the dystrophinP-type exon 1 and putative promoter region werekindly supplied by Drs A. P. Monaco and A. P. Walker.The positive hybridizing ;1.8-kb HindIII fragment wassubcloned into pBluescript (Stratagene). The mousegenomic clone was isolated from a genomic library asdescribed previously (Gorecki et al., 1992). The pig,sheep, and dog genomic fragments were obtained byPCR amplification of genomic DNA using primersbased on conserved regions of human and mouse se-quences and high-fidelity DNA polymerase. The dogsequence was compared to a dog sequence recentlypublished by Schatzberg et al. (1999) and found to beessentially identical. Rat and chicken P-type first exonswere amplified by RT-PCR. Both manual (using Seque-nase, USB kit) and automated sequencing of doublestranded plasmid DNA were performed using vector-and gene-specific primers.
The sequences were searched for regulatory regionsusing the EUKPROM and FINDPATTERNS programs(GCG package) against the Eukaryotic Promoter Data-base (EPD). The TFSEARCH software Version 1.3 wasalso used to search for highly correlated sequence frag-ments versus TFMATRIX transcription factor bindingsite profile database. The zoo blots containing genomicDNA from chicken, carp, xenopus, and the aforemen-tioned human cosmid clone, digested with HindIII,were hybridized with a 1195-bp mouse P-type probea-32P-labeled. Hybridizations were performed at 45–50°C in the Church buffer (Sambrook et al., 1989), filterswere washed (final wash: 0.16 M NaHPO4, pH 7.2; 1mM EDTA; 1% SDS at 50°C) and autoradiographed.
Primer Extension
Primer extension was performed using total humanand mouse cerebellar and muscle RNA. Primer SI com-plementary to nt 222 to 118 of the human sequencewas end-labeled with [g-32P]ATP and T4 PNK (NEB).RNA mixed with 10 pmol of labeled primer was pre-cipitated and resuspended in hybridisation buffer. Twotypes of buffers and conditions were used, S1 buffer(80% formamide, 40 mM Pipes, 400 mM NaCl, 1 mMEDTA), hybridized at 45°C, and aqueous buffer (1 MNaCl, 0.167 M Hepes, 0.3 mM EDTA), hybridized at
30°C (both for 18–30 h). Following hybridization, mix-tures were precipitated, pellets resuspended in water,
fG
p
511Regulation of the P-type Dystrophin Expression
and the hybridized primers extended using MMLV re-verse transcriptase at 37°C for 90 min. After phenol/chloroform extraction and precipitation, the extensionproducts were analyzed on 6% polyacrylamide-urea geland autoradiographed. Sequencing reactions were usedas size markers.
5*-RACE, RT-PCR, and RNA-Gel Analysis
The 59-RACE with mouse or human cerebellum andmouse muscle were carried out using the 59RACE PCRkit from GIBCO-BRL, the SMART RACE kit from Clon-tech (both according to manufacturer’s instructions), oras described in Gorecki et al. (1992). Total RNA ormRNA were reverse transcribed using primer SI. Firstround PCR amplifications were performed with an ex-ternal anchor primer and P-type specific primer W-18(59-CCTCAGACATTTCCAATTCTGCGG-39). Secondround PCR was performed with primer SR (59-CACCT-TCATAGGAAAGCTTTTGGCT-39) and an internal an-chor primer. Controls included the use of non-anchoredfirst strands, the exclusion of reverse transcriptase andthe use of a single primer for amplification. The prod-ucts were run on agarose gels, purified (QIAquick kit,QIAGEN), digested with HindIII and SpeI, and clonedinto pBluescript (KS) or cloned directly into pGEMvector (Promega). For each RACE method, clones fromseveral PCR reactions were sequenced using the T7sequencing kit (Pharmacia).
Alternative splicing at the 59 end of the P-type dys-trophin mRNA was studied using RT-PCR and primersets: PT-A: forward: [CAPF] 59-GTTGGCAAAAT-GCTGTCTGTGAAGC-39 and reverse: [EX2/3] 59-TA-GATCGATGTGCATTTATCCATTTRGTGAATG-39;PT-B: forward: [INTR-1] 59-CTTTATCTGTGTTTGC-CTATGACTC-39; reverse: [ECO3] complementary tont 406 – 435 of the human dystrophin sequence; PT-C:forward: [SPL-1] 59-GTGGAAGAAACAGGTTTGTC-CTACC-39 and reverse: [ECO3]; PT-D: forward:[SEQ] nt 251 to 228 and reverse: [EX2/3]. PTS1:orward [INTR-1] and reverse [W-6] 59-GAATGGCT-TTGATTATCTGCAGCT-39, complementary to a se-
quence within the 82-bp insertion. PTS2: forward[SPL-1] and reverse [W-6]. CAP: forward [CAPF] andreverse: [W-18]. Relative primer positions are sum-marised in Fig. 5A.
Typically, PCR amplification was carried out in afinal volume of 50 ml, 1.5 mM MgCl2, 0.5 mM dNTP, 20
M of each individual primer and 2 Units of Taq poly-merase with hot start. Cycles were: 94°C for 1 min, 60°C
for 1 min, 72°C for 1.2 min. PCR products were ana-lyzed on agarose gels, Southern blotted and hybridizedwith internal probes.
RNA-gel hybridization was performed as describedpreviously (Gorecki et al., 1991).
Construction of Promoter-Reporter FusionPlasmids, Myoblast Transfection, and ReporterGene Assays
The human 1.78-kb genomic fragment was single- ordouble-digested with restriction enzymes PstI and XbaIand resulting fragments cloned in forward and reverseorientation into pCATbasic vector (Promega). Furtherconstructs were made by PCR amplification (with PfuDNA polymerase) using a primer containing SalI site,followed by digestion and cloning into the reportervector.
C2C12 myoblasts (0.5 3 106) were seeded 2 days priorto the transfection in 100-mm culture plates. Ten micro-grams of a test plasmid and 4 mg of pSV-b-galactosidase(control for transfection efficiency) complexed with 70mg of LipofectAMINE (GIBCO-BRL) in Opti-MEM me-dium were used for transfection. The mixture wasadded and cells were incubated with the complexes at37°C for 6 h. Following addition of growth mediumcells were incubated for a further 24 h, after which theywere switched to differentiation medium (DMEM con-taining 5% horse serum) to induce myotube formation.Myotubes were harvested 10 days postdifferentiationand protein extracts were used for chloramphenicolacetyltransferase (CAT) and b-galactosidase assays. Al-ternatively, cells were maintained as myoblasts ingrowth medium and harvested after 24 or 48 h.
CAT assays were carried out using the liquid scintil-lation method (Seed and Sheen, 1988). Cell extractsadjusted to equal protein concentrations were heated at65°C for 10 min to inactivate endogenous acetylases,cooled, and made up to a final reaction volume of 125 mlcontaining 5 ml of [14C]chloroamphenicol and n-butyrylcoenzyme A (5 mg/ml). The reaction was terminatedby addition of 300 ml mixed xylenes. Following phaseseparation, 240 ml of the upper phase was transferred tofresh tubes and a back extraction step using 100 ml offresh 0.25 M Tris–HCl, pH 8.0, was incorporated toreduce background counts. Two hundred microliters ofthe xylene extract was counted in a liquid scintillation
counter. b-galactosidase assay was performed as de-scribed in Sambrook et al. (1989).
B
E
F
F
G
G
G
G
G
G
H
H
H
H
H
H
H
J
K
K
K
K
512 Abdulrazzak et al.
Analysis of the Promoter Region in SkeletalMuscle in Vivo
Selected fragments identified in vitro were cloned intothe lacZ reporter gene vector (kindly provided by Drs.E. Asante and D. Wells; Asante et al., 1994). Purified,pyrogen-free plasmids (20 mg in saline) or saline only(control) were injected into tibialis anterior muscle of4-week-old male C57Bl6 mice. The muscles were pre-treated with barium chloride (1.2%) 5 days before DNAinjection in order to increase plasmid uptake. One weekafter injection the muscles were removed, protein ex-tracts prepared and assayed for b-galactosidase activityas described before.
ACKNOWLEDGMENTS
This work was supported by the Wellcome Trust (D.C.G. is arecipient of a Wellcome Trust Career Development Fellowship). Theauthors thank Dr. J. A. A. Sant’Ana Pereira for samples of singlemuscle fibers, Drs. A. Walker and A. P. Monaco for cosmid clones, Dr.A. Jacks for help with RNA folding analysis, and Dr. J. Brown forsuggestions regarding statistical analysis.
REFERENCES
Asante, E. A., Boswell, J. M., Burt, D. W., and Bulfield, G. (1994).Tissue specific expression of an alpha-skeletal actin-lacZ fusiongene during development in transgenic mice. Transgenic Res. 3:59–66.
Ayoubi, T. A., and Van De Ven, W. J. (1996). Regulation of geneexpression by alternative promoters. FASEB J. 10: 453–460.
ucher, P. (1990). Weight matrix descriptions of four eukaryotic RNApolymerase II promoter elements derived from 502 unrelated pro-moter sequences. J. Mol. Biol. 212: 563–578.
Chu, S. C., Wu, H. P., Banks, T. C., Eissa, N. T., and Moss, J. (1995).Structural diversity in the 59-untranslated region of cytokine-stim-ulated human inducible nitric oxide synthase mRNA. J. Biol. Chem.270: 10625–10630.
Cooke, J., Nowak, M. A., Boerlijst, M., and Maynard-Smith, J. (1997).Evolutionary origins and maintenance of redundant gene expres-sion during metazoan development. Trends Genet. 13: 360–364.
mery, A. E. (1993). Duchenne Muscular Dystrophy. Oxford Univ.Press, Oxford.
erlini, A., Sewry, C., Melis, M. A., Mateddu, A., and Muntoni, F.(1999). X-linked dilated cardiomyopathy and the dystrophin gene.Neuromusc. Dis. 9: 339–346.
racasso, C., and Patarnello, T. (1998). Evolution of the dystrophinmuscular promoter and 59 flanking region in primates. J. Mol. Evol.46: 168–179.orecki, D. C., and Barnard, E. A. (1997). Expression of the dystro-phin complex in the brain. In Dystrophin: Gene, Protein and Function(S. Brown and J. Lucy, Ed.), pp. 105–138, Cambridge Univ. Press,Cambridge.
orecki, D. C., and Barnard, E. A. (1995). Specific expression ofG-dystrophin (Dp71) in the brain. NeuroReport 6: 893–896.orecki, D., Geng, Y., Thomas, K., Hunt, S. P., Barnard, E. A., andBarnard, P. J. (1991). Expression of the dystrophin gene in mouseand rat brain. Neuroreport 2: 773–776.orecki, D. C., Monaco, A. P., Derry, J. M., Walker, A. P., Barnard,E. A., and Barnard, P. J. (1992). Expression of four alternativedystrophin transcripts in brain regions regulated by different pro-moters. Hum. Mol. Genet. 1: 505–510.orecki, D. C., Lukasiuk, K., Szklarczyk, A., and Kaczmarek, L.(1998). Kainate-evoked changes in dystrophin messenger RNA lev-els in the rat hippocampus. Neuroscience 84: 467–477.ray, N. K., and Wickens, M. (1998). Control of translation initiationin animals. Annu. Rev. Cell Dev. Biol. 14: 399–458.offman, E. P., Brown, R. H., Jr., and Kunkel, L. M. (1987). Dystro-phin: the protein product of the Duchenne muscular dystrophylocus. Cell 51: 919–928.offman, E. P., Hudecki, M. S., Rosenberg, P. A., Pollina, C. M., andKunkel, L. M. (1988). Cell and fiber-type distribution of dystrophin.Neuron 1: 411–420.older, E., Maeda, M., and Bies, R. D. (1996). Expression and regu-lation of the dystrophin Purkinje promoter in human skeletal mus-cle, heart, and brain. Hum. Genet. 97: 232–239.olzfeind, P. J., Ambrose, H. J., Newey, S. E., Nawrotzki, R. A., Blake,D. J., and Davies, K. E. (1999). Tissue-selective expression of alpha-dystrobrevin is determined by multiple promoters. J. Biol. Chem.274: 6250–6258.orowitz, D. S., and Krainer, A. R. (1994). Mechanisms for selecting59 splice sites in mammalian pre-mRNA splicing. Trends Genet. 10:100–106.ouzelstein, D., Lyons, G. E., Chamberlain, J., and Buckingham, M. E.(1992). Localization of dystrophin gene transcripts during mouseembryogenesis. J. Cell Biol. 119: 811–821.ugnot, J. P., Gilgenkrantz, H., Jeanpierre, M., Chelly, J., Kaplan, J. C.,and Kahn, A. (1993). Striking conservation of the brain-specificregion of the dystrophin gene. Mamm. Genome 4: 393–396.
ackson, R. J., and Wickens, M. (1997). Translational controls imping-ing on the 59-untranslated region and initiation factor proteins.Curr. Opin. Genet. Dev. 7: 233–241.
imura, S., Abe, K., Suzuki, M., Ogawa, M., Yoshioka, K., Yamamura,K., and Miike, T. (1997). 2.1 kb 59-flanking region of the brain typedystrophin gene directs the expression of lacZ in the cerebralcortex, but not in the hippocampus. J. Neurol. Sci. 147: 13–20.
imura, S., Abe, K., Suzuki, M., Ogawa, M., Yoshioka, K., Kaname, T.,Miike, T., and Yamamura, K. (1997). A 900 bp genomic region fromthe mouse dystrophin promoter directs lacZ reporter expressiononly to the right heart of transgenic mice. Dev. Growth Differ. 39:257–265.
lamut, H. J., Gangopadhyay, S. B., Worton, R. G., and Ray, P. N.(1990). Molecular and functional analysis of the muscle-specificpromoter region of the Duchenne muscular dystrophy gene. Mol.Cell Biol. 10: 193–205.
nuesel, I., Mastrocola, M., Zuellig, R. A., Bornhauser, B., Schaub,M. C., and Fritschy, J. M. (1999). Altered synaptic clustering ofGABAA receptors in mice lacking dystrophin. Eur. J. Neurosci. 12:4457–4462.
Kozak, M. (1999). Initiation of translation in prokaryotes and eu-karyotes. Gene 234: 187–208.
Lemaire, C., Heilig, R., and Mandel, J. L. (1988). The chicken dystro-phin cDNA: Striking conservation of the C-terminal coding and 39untranslated regions between man and chicken. EMBO J. 7: 4157–4162.
Lidov, H. G. (1996). Dystrophin in the nervous system. Brain Pathol. 6:63–77.

M
M
N
N
N
Q
S
S
S
S
S
S
S
S
T
V
V
W
W
Y
Y
Z
513Regulation of the P-type Dystrophin Expression
Makover, A., Zuk, D., Breakstone, J., Yaffe, D., and Nudel, U. (1991).Brain-type and muscle-type promoters of the dystrophin gene dif-fer greatly in structure. Neuromusc. Disord. 1: 39–45.
Miale, I., and Sidman, R. (1961). An autoradiographic analysis ofhistogenesis in the mouse cerebellum. Exp. Neurology 4: 277–296.
Muntoni, F., Wilson, L., Marrosu, G., Marrosu, M. G., Cianchetti, C.,Mestroni, L., Ganau, A., Dubowitz, V., and Sewry, C. (1995). Amutation in the dystrophin gene selectively affecting dystrophinexpression in the heart. J. Clin. Invest. 96: 693–699.untoni, F., Melis, M. A., Ganau, A., and Dubowitz, V. (1995). Tran-scription of the dystrophin gene in normal tissues and in skeletalmuscle of a family with X-linked dilated cardiomyopathy. Am. J.Hum. Genet. 56: 151–157.untoni, F., Gobbi, P., Sewry, C., Sherratt, T., Taylor, J., Sandhu, S. K.,Abbs, S., Roberts, R., Hodgson, S. V., and Bobrow, M., et al. (1994).Deletions in the 59 region of dystrophin and resulting phenotypes.J. Med. Genet. 31: 843–847.akamura, A., Ikeda, S., Yazaki, M., Yoshida, K., Kobayashi, O.,Yanagisawa, N., and Takeda, S. (1997). Up-regulation of the brainand Purkinje-cell forms of dystrophin transcripts, in Becker mus-cular dystrophy. Am. J. Hum. Genet. 60: 1555–1558.anda, S., and Mack, K. J. (1998). Multiple promoters direct stimulusand temporal specific expression of brain-derived neurotrophicfactor in the somatosensory cortex. Brain Res. Mol. Brain Res. 62:216–219.izetic, D., Zehetner, G., Monaco, A. P., Gellen, L., Young, B. D., andLehrach, H. (1991). Construction, arraying, and high-densityscreening of large insert libraries of human chromosomes X and 21:Their potential use as reference libraries. Proc. Natl. Acad. Sci. USA88: 3233–3237.uandt, K., Frech, K., Karas, H., Wingender, E., and Werner, T. (1995).MatInd and MatInspector—New fast and versatile tools for detec-tion of consensus matches in nucleotide sequence data. NucleicAcids Res. 23: 4878–4884.
adoulet-Puccio, H. M., and Kunkel, L. M. (1996). Dystrophin and itsisoforms. Brain Pathol. 6: 25–35.
ambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning.A Laboratory Manual. Cold Spring Harbor Laboratory Press, ColdSpring Harbor, NY.
anlioglu, S., Zhang, X., Baader, S. L., and Oberdick, J. (1998). Regu-lation of a Purkinje cell-specific promoter by homeodomain pro-teins: Repression by engrailed-2 vs. synergistic activation by Hoxa5and Hoxb7. J. Neurobiol. 36: 559–571.
chatzberg, S., Olby, N., Steingold, S., Keene, B., Atkins, C., Meurs, K.,Solomon, G., Goedegebuure, S. A., Wilton, S., and Sharp, N. (1999).
A polymerase chain reaction screening strategy for the promoter ofthe canine dystrophin gene. Am. J. Vet. Res. 60: 1040–1046.
eed, B., and Sheen, J. Y. (1988). A simple phase-extraction assay forCAT activity. Gene 67: 271–277.
himatsu, A., and Rotwein, P. (1987). Sequence of two rat insulin-likegrowth factor I mRNAs differing within the 59 untranslated region.Nucleic Acids Res. 15: 7196.
male, S. T. (1997). Transcription initiation from TATA-less promoterswithin eukaryotic protein-coding genes. Biochim. Biophys. Acta.1351: 73–88.
urono, A., Takeshima, Y., Wibawa, T., Pramono, Z. A., and Matsuo,M. (1997). Six novel transcripts that remove a huge intron rangingfrom 250 to 800 kb are produced by alternative splicing of the 59region of the dystrophin gene in human skeletal muscle. Biochem.Biophys. Res. Commun. 239: 895–899.
orelli, S., Ferlini, A., Obici, L., Sewry, C., and Muntoni, F. (1999).Expression, regulation and localisation of dystrophin isoforms inhuman foetal skeletal and cardiac muscle. Neuromusc. Dis. 9: 541–551.
aillend, C., Ungerer, A., and Billard, J. M. (1999). Facilitated NMDAreceptor-mediated synaptic plasticity in the hippocampal CA1 areaof dystrophin-deficient mice. Synapse 33: 59–70.
oogd, J., and Glickstein, M. (1998). The anatomy of the cerebellum.Trends Neurosci. 21: 370–375.ang, Y., Newton, D. C., and Marsden, P. A. (1999). Neuronal NOS:Gene structure, mRNA diversity, and functional relevance. Crit.Rev. Neurobiol. 13: 21–43.innard, A. V., Mendell, J. R., Prior, T. W., Florence, J., and Burghes,A. H. (1995). Frameshift deletions of exons 3-7 and revertant fibersin Duchenne muscular dystrophy: Mechanisms of dystrophin pro-duction. Am. J. Hum. Genet. 56: 158–166.
affe, D., and Saxel, O. (1977). Serial passaging and differentiation ofmyogenic cells isolated from dystrophic mouse muscle. Nature 270:725–727.
ang, H., Adamo, M. L., Koval, A. P., McGuinness, M. C., Ben-Hur,H., Yang, Y., LeRoith, D., and Roberts, C. T., Jr. (1995). Alternativeleader sequences in insulin-like growth factor I mRNAs modulatetranslational efficiency and encode multiple signal peptides. Mol.Endocrinol. 9: 1380–1395.
accaria, M. L., De Stefano, M. E., Properzi, F., Gotti, C., Petrucci,T. C., and Paggi, P. (1998). Disassembly of the cholinergic postsyn-aptic apparatus induced by axotomy in mouse sympathetic neu-rons: The loss of dystrophin and beta-dystroglycan immunoreac-
tivity precedes that of the acetylcholine receptor. J. Neuropathol. Exp.Neurol. 57: 768–779.Received September 19, 2000Revised November 27, 2000Accepted December 7, 2000
Published online February 21, 2001
Copyright © 2022 FDOKUMEN




















