
Promotion of Rh catalyst interfaced with TiO2
6
Promotion of Rh catalyst interfaced with TiO 2 Elena A. Baranova, Gy€ orgy F oti * , Christos Comninellis Swiss Federal Institute of Technology, SB-ISP-UGEC, CH-1015 Lausanne, Switzerland Received 5 November 2003; received in revised form 17 November 2003; accepted 17 November 2003 Published online: 3 December 2003 Abstract The catalytic activity of Rh for the partial oxidation of methane to syngas can be markedly affected by interfacing polycrystalline Rh films with a dispersed TiO 2 interlayer deposited on YSZ. High CO selectivity (near 97%) can be reached in wide range of CH 4 :O 2 ratio at 550 °C. The observed modification of catalytic activity and selectivity towards CO and H 2 production over Rh interfaced with interlayer of TiO 2 is related to the lowered stability of surface Rh oxide against reduction to metallic Rh. The phenomenon may be interpreted either by electronic type strong metal–support interactions or by self-driven wireless electrochemical promotion mechanism. Ó 2003 Elsevier B.V. All rights reserved. Keywords: Catalyst promotion; Metal–support interactions; Rhodium catalyst; TiO 2 support; Selective methane oxidation 1. Introduction In the last decade, many efforts have been devoted to the catalytic partial oxidation of CH 4 with oxygen to achieve syngas formation according to [1,2]: CH 4 þð1=2ÞO 2 ! CO þ 2H 2 ð1Þ This process has some advantages over the conventional steam reforming, namely: it is more energy efficient and can produce the desired H 2 /CO molar ratio of 2 (instead of 3 in steam reforming) required for methanol or Fischer–Tropsch synthesis. However, this reaction has not been yet developed at industrial scale due to prob- lems related to catalyst deactivation mainly by carbon deposition and/or oxidation of the catalyst to the cor- responding oxide. Prettre et al. [3] were among the first to report for- mation of syngas by the partial oxidation of CH 4 using supported Ni catalyst in the temperature range 973– 1173 K. The reaction was reported to occur in two stages. In the first stage, methane is converted into CO 2 and water until complete conversion of oxygen is achieved according to CH 4 þ 2O 2 ! CO 2 þ 2H 2 O ð2Þ In the second stage, syngas is produced via secondary reactions such as the carbon dioxide and the steam- reforming reaction: CH 4 þ CO 2 ! 2CO þ 2H 2 ð3Þ CH 4 þ H 2 O ! CO þ 3H 2 ð4Þ It is generally agreed that the type of catalyst used can influence strongly the reaction pathway. In fact it has been reported that on Rh catalyst the partial oxidation of methane to CO and H 2 occurs in one step [Eq. (1)] without the formation of CO 2 intermediate [4] as was the case on Ni catalyst [3]. More recent studies have demonstrated that Rh is one of the most promising catalysts in the partial oxi- dation of methane, giving the highest selectivity toward H 2 production [4]. However, the problems related to the Rh catalyst deactivation, mainly due to carbon deposi- tion and/or Rh oxidation to Rh 2 O 3 have not been yet completely resolved. The aim of this work is to investigate the modification of the catalytic activity and stability toward deactivation Electrochemistry Communications 6 (2004) 170–175 www.elsevier.com/locate/elecom * Corresponding author. Tel.: +41-21-693-3673; fax: +41-21-693- 3190. E-mail address: gyorgy.foti@epfl.ch (G. F oti). 1388-2481/$ - see front matter Ó 2003 Elsevier B.V. All rights reserved. doi:10.1016/j.elecom.2003.11.009
Transcript of Promotion of Rh catalyst interfaced with TiO2

Electrochemistry Communications 6 (2004) 170–175
www.elsevier.com/locate/elecom
Promotion of Rh catalyst interfaced with TiO2
Elena A. Baranova, Gy€orgy F�oti *, Christos Comninellis
Swiss Federal Institute of Technology, SB-ISP-UGEC, CH-1015 Lausanne, Switzerland
Received 5 November 2003; received in revised form 17 November 2003; accepted 17 November 2003
Published online: 3 December 2003
Abstract
The catalytic activity of Rh for the partial oxidation of methane to syngas can be markedly affected by interfacing polycrystalline
Rh films with a dispersed TiO2 interlayer deposited on YSZ. High CO selectivity (near 97%) can be reached in wide range of CH4:O2
ratio at 550 �C. The observed modification of catalytic activity and selectivity towards CO and H2 production over Rh interfaced
with interlayer of TiO2 is related to the lowered stability of surface Rh oxide against reduction to metallic Rh. The phenomenon may
be interpreted either by electronic type strong metal–support interactions or by self-driven wireless electrochemical promotion
mechanism.
� 2003 Elsevier B.V. All rights reserved.
Keywords: Catalyst promotion; Metal–support interactions; Rhodium catalyst; TiO2 support; Selective methane oxidation
1. Introduction
In the last decade, many efforts have been devoted to
the catalytic partial oxidation of CH4 with oxygen toachieve syngas formation according to [1,2]:
CH4 þ ð1=2ÞO2 ! COþ 2H2 ð1ÞThis process has some advantages over the conventional
steam reforming, namely: it is more energy efficient and
can produce the desired H2/CO molar ratio of 2 (instead
of 3 in steam reforming) required for methanol or
Fischer–Tropsch synthesis. However, this reaction has
not been yet developed at industrial scale due to prob-
lems related to catalyst deactivation mainly by carbondeposition and/or oxidation of the catalyst to the cor-
responding oxide.
Prettre et al. [3] were among the first to report for-
mation of syngas by the partial oxidation of CH4 using
supported Ni catalyst in the temperature range 973–
1173 K. The reaction was reported to occur in two
stages. In the first stage, methane is converted into CO2
* Corresponding author. Tel.: +41-21-693-3673; fax: +41-21-693-
3190.
E-mail address: [email protected] (G. F�oti).
1388-2481/$ - see front matter � 2003 Elsevier B.V. All rights reserved.
doi:10.1016/j.elecom.2003.11.009
and water until complete conversion of oxygen is
achieved according to
CH4 þ 2O2 ! CO2 þ 2H2O ð2ÞIn the second stage, syngas is produced via secondary
reactions such as the carbon dioxide and the steam-reforming reaction:
CH4 þ CO2 ! 2COþ 2H2 ð3Þ
CH4 þH2O ! COþ 3H2 ð4ÞIt is generally agreed that the type of catalyst used can
influence strongly the reaction pathway. In fact it has
been reported that on Rh catalyst the partial oxidation
of methane to CO and H2 occurs in one step [Eq. (1)]
without the formation of CO2 intermediate [4] as was
the case on Ni catalyst [3].
More recent studies have demonstrated that Rh isone of the most promising catalysts in the partial oxi-
dation of methane, giving the highest selectivity toward
H2 production [4]. However, the problems related to the
Rh catalyst deactivation, mainly due to carbon deposi-
tion and/or Rh oxidation to Rh2O3 have not been yet
completely resolved.
The aim of this work is to investigate the modification
of the catalytic activity and stability toward deactivation

E.A. Baranova et al. / Electrochemistry Communications 6 (2004) 170–175 171
of Rh catalyst, for the partial oxidation of methane, byinterfacing Rh catalyst with TiO2 particles supported on
YSZ solid electrolyte (Rh/TiO2/YSZ). In this first part
of the work, the promoting effect of TiO2 will be studied
under open-circuit conditions where the presence of the
YSZ support is not considered to play any role in the
observed phenomena but it is used to enable potential
measurements. The results will be discussed on the base
of the phenomenon of strong metal–support interaction(SMSI) [5] and electrochemical promotion of catalysis
(NEMCA) [6]. Investigation of the same systems under
closed-circuit conditions is the subject of a next paper.
2. Experimental
The ring-type electrochemical cell used in the presentwork is shown in Fig. 1(a) and has been described in
detail elsewhere [7,8]. With the aim to measure catalyst
potential an ionic (O2�) conductive support was chosen
in this work. The catalyst film was deposited on the
inner surface, while the gold reference electrode was
deposited on the outer surface of an yttria-stabilized
zirconia (YSZ 8 mol%) (Technox 802 from Dynamic
Ceramic) ring (OD 20 mm, ID 17 mm) of 10 mm height,
YSZRh catalyst
VWR
gold
O2CH4 He
FRCs
R
G/PT
NDIR CO CO2 CH4
GC H2 O2 CO CH4
(a)
(b)
Fig. 1. (a) Ring-type electrochemical cell; (b) schematic representation
of the experimental setup. FRCs, flow rate controllers; R, reactor; T,
temperature control; G/P, galvano-potentiostat; GC, gas chromato-
graph; NDIR, infrared analyzers.
providing a geometrical catalyst surface of about 5 cm2.Two catalytic systems have been investigated:
Rh/YSZ system: prepared by deposition of 30 ll/cm2
of Rh paste (Engelhard Rh8826; 10% Rh) diluted 3
times (by mass) with THF on YSZ support. Three
consecutive layers were deposited. Each layer was fired
at 400 �C/10 min; final heat treatment was made at 550
�C/1 h (estimated loading 0.54 mg Rh/cm2).
Rh/TiO2/YSZ system: The only difference from theRh/YSZ system was that the YSZ support was covered
with a thin film of TiO2. In order to achieve this, a slurry
of 5% TiO2 (0.25 g TiO2 Degussa P25 in 5 ml
EtOH:H2O 1:1) was applied onto the inner surface of
the YSZ ring. Several layers were applied until reaching
the desired loading (1.7 mg TiO2/cm2). For the purpose
of catalyst potential measurement during the catalytic
experiments, a gold film has been deposited onto theouter surface of the ring using a gold paste (Gwent
C70219R4); the deposition temperature was 550 �C.Prior to measurements both systems Rh/YSZ and
Rh/TiO2/YSZ were pre-treated in H2 at 550 �C/1 h.
The experimental set-up used in this study consists of
the gas flow system, the reactor, and the gas analysis
unit (Fig. 1(b)) [8]. Reactants were Carbagas certified
standards of CH4 (99.95%) and O2 (99.95%) supplied as20% mixtures in He (99.996). The balance was helium.
The experiments were performed in the temperature
range of 400–600 �C using an atmospheric pressure
flow-through reactor made of quartz (300 mm length,
ID 25 mm). Typical flow rate was 200 ml/min STP.
The CO selectivity, S(CO), of the methane oxidation
reaction was calculated with the following equation:
SðCOÞ ¼ rCOrCO þ rCO2
; ð5Þ
where rCO and rCO2are the rate of CO and CO2 product
formation.
3. Results
The catalytic activity of Rh/YSZ and Rh/TiO2/YSZ
catalysts was investigated by cyclic change in the
CH4:O2 molar ratio in the feed, starting from a maxi-
mum ratio (39:1) down to a minimum value (1:2) (for-
ward run), then back from the minimal molar ratio (1:2)
to the maximum value 39:1 (reverse run). All measure-
ments were made at stationary conditions; the typicalwaiting time after changing the inlet composition was
20–30 min. A typical example of results obtained at 550
�C is shown in Fig. 2. This figure shows that a freshly
prepared Rh/YSZ catalyst is active toward CH4 partial
oxidation at a methane/oxygen molar ratio 39:1 (Fig. 2,
left). However, during the forward run (from the ratio
39:1 to 1:2) the Rh/YSZ catalytic activity towards CO
production decreases rapidly and fells to almost zero at

0.0
0.2
0.4
0.6
0.8
1.0
0 10 20 30 40
ratio CH4 / O2
S (C
O) Rh/TiO2/YSZ
Rh/YSZ
Fig. 3. Oxidation of methane over Rh/YSZ (diamonds) and Rh/TiO2/
YSZ (squares) catalysts at T ¼ 550 �C. Selectivity of CO formation,
S(CO) [Eq. (5)], as a function of the CH4/O2 molar ratio. Feed com-
position, symbols and lines as in Fig. 2.
Rh/YSZ Rh/TiO2/YSZ
CO
0 10 20 30
ratio CH4/O2
40
CO2
0.0
0.1
0.2
0.3
0.4
0.5
rm
olC
O s-1
mgR
h-1
CO
0.0
0.1
0.2
0.3
0.4
0.5
0 10 21 30 40
ratio CH4/O2
r /
/ µ
µm
olC
O 2
s-1
mgR
h-1
CO2
Fig. 2. Oxidation of methane over Rh/YSZ (left) and Rh/TiO2/YSZ (right) catalysts at T ¼ 550 �C. Specific formation rate, r, of CO (circles) and CO2
(squares) as a function of the CH4/O2 molar ratio. Feed composition: CH4(variable)/O2(0.5 kPa). Open symbols and narrow lines: forward scan; full
symbols and wide lines: reverse scan.
172 E.A. Baranova et al. / Electrochemistry Communications 6 (2004) 170–175
CH4:O2 molar ratio of about three. During the reverse
run, when increasing the CH4:O2 ratio from 1:2 to 39:1,
the specific reaction rate of CO formation remains al-most zero while the CO2 formation rate slightly in-
creases with increasing CH4/O2 molar ratio.
A completely different behavior during methane
partial oxidation was observed with the Rh catalyst in-
terfaced with TiO2 (Fig. 2, right). In this case both
forward and reverse runs are characterized by high
catalytic activity towards CO and H2 formation. During
the forward run on Rh/TiO2/YSZ the specific rate ofCO2 production increases with decreasing CH4/O2 mo-
lar ratio and reaches a maximum value at CH4/O2 ¼ 1:1
molar ratio, meanwhile at this gas composition CO
production rate is almost completely vanished. Fur-
thermore, the reverse run on Rh/TiO2/YSZ is charac-
terized by smaller reaction rate of CO2 formation and
higher catalytic activity towards CO formation as
compared to the forward run.Fig. 3 shows the selectivity of CO formation [Eq. (5)]
as a function of the CH4/O2 molar ratio for Rh/YSZ
and Rh/TiO2/YSZ. In all cases, Rh film supported on
TiO2 interlayer has much higher selectivity towards CO
formation than the Rh/YSZ catalyst. However, the
main effect of the TiO2 support is the fact that the Rh/
TiO2/YSZ catalyst deactivated in oxidizing feed
(CH4:O2 < 2:1) could be easily reactivated by exposure
to a reducing gas composition. The cycling of feed
composition can be repeated many times without any
indication of activity loss. This is not the case with the
Rh/YSZ catalyst, which can not be reactivated by ex-
posure to a reducing gas composition. In fact, the re-verse run was always characterized by zero CO
production, hence, also zero CO selectivity (Fig. 3).
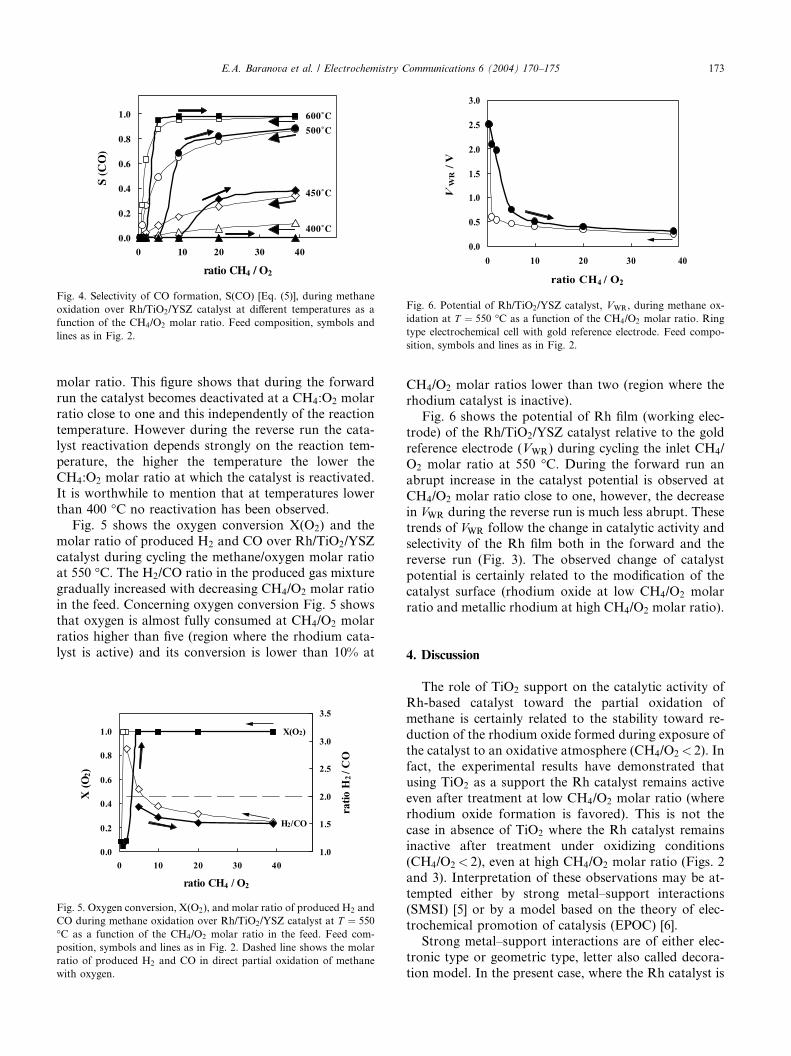
Fig. 4 shows the influence of temperature on the CO
selectivity of Rh/TiO2/YSZ catalysts during cycling the
feed composition from high to low methane/oxygen
0.0
0.2
0.4
0.6
0.8
1.0
0 10 20 30 40
ratio CH4 / O2
S (C
O)
600˚C
450˚C
500˚C
400˚C
Fig. 4. Selectivity of CO formation, S(CO) [Eq. (5)], during methane
oxidation over Rh/TiO2/YSZ catalyst at different temperatures as a
function of the CH4/O2 molar ratio. Feed composition, symbols and
lines as in Fig. 2.
0.0
0.5
1.0
1.5
2.0
2.5
3.0
0 10 20 30
ratio CH4 / O2
VW
R /
V
40
Fig. 6. Potential of Rh/TiO2/YSZ catalyst, VWR, during methane ox-
idation at T ¼ 550 �C as a function of the CH4/O2 molar ratio. Ring
type electrochemical cell with gold reference electrode. Feed compo-
sition, symbols and lines as in Fig. 2.
E.A. Baranova et al. / Electrochemistry Communications 6 (2004) 170–175 173
molar ratio. This figure shows that during the forward
run the catalyst becomes deactivated at a CH4:O2 molar
ratio close to one and this independently of the reaction
temperature. However during the reverse run the cata-lyst reactivation depends strongly on the reaction tem-
perature, the higher the temperature the lower the
CH4:O2 molar ratio at which the catalyst is reactivated.
It is worthwhile to mention that at temperatures lower
than 400 �C no reactivation has been observed.
Fig. 5 shows the oxygen conversion X(O2) and the
molar ratio of produced H2 and CO over Rh/TiO2/YSZ
catalyst during cycling the methane/oxygen molar ratioat 550 �C. The H2/CO ratio in the produced gas mixture
gradually increased with decreasing CH4/O2 molar ratio
in the feed. Concerning oxygen conversion Fig. 5 shows
that oxygen is almost fully consumed at CH4/O2 molar
ratios higher than five (region where the rhodium cata-
lyst is active) and its conversion is lower than 10% at
0.0
0.2
0.4
0.6
0.8
1.0
0 10 20 30 40
ratio CH4 / O2
X (O
2)
1.0
1.5
2.0
2.5
3.0
3.5
ratio
H2 /
CO
X(O2)
H2/CO
Fig. 5. Oxygen conversion, X(O2), and molar ratio of produced H2 and
CO during methane oxidation over Rh/TiO2/YSZ catalyst at T ¼ 550
�C as a function of the CH4/O2 molar ratio in the feed. Feed com-
position, symbols and lines as in Fig. 2. Dashed line shows the molar
ratio of produced H2 and CO in direct partial oxidation of methane
with oxygen.
CH4/O2 molar ratios lower than two (region where therhodium catalyst is inactive).
Fig. 6 shows the potential of Rh film (working elec-
trode) of the Rh/TiO2/YSZ catalyst relative to the gold
reference electrode (VWR) during cycling the inlet CH4/
O2 molar ratio at 550 �C. During the forward run an
abrupt increase in the catalyst potential is observed at
CH4/O2 molar ratio close to one, however, the decrease
in VWR during the reverse run is much less abrupt. Thesetrends of VWR follow the change in catalytic activity and
selectivity of the Rh film both in the forward and the
reverse run (Fig. 3). The observed change of catalyst
potential is certainly related to the modification of the
catalyst surface (rhodium oxide at low CH4/O2 molar
ratio and metallic rhodium at high CH4/O2 molar ratio).
4. Discussion
The role of TiO2 support on the catalytic activity of
Rh-based catalyst toward the partial oxidation of
methane is certainly related to the stability toward re-
duction of the rhodium oxide formed during exposure of
the catalyst to an oxidative atmosphere (CH4/O2 < 2). In
fact, the experimental results have demonstrated thatusing TiO2 as a support the Rh catalyst remains active
even after treatment at low CH4/O2 molar ratio (where
rhodium oxide formation is favored). This is not the
case in absence of TiO2 where the Rh catalyst remains
inactive after treatment under oxidizing conditions
(CH4/O2 < 2), even at high CH4/O2 molar ratio (Figs. 2
and 3). Interpretation of these observations may be at-
tempted either by strong metal–support interactions(SMSI) [5] or by a model based on the theory of elec-
trochemical promotion of catalysis (EPOC) [6].
Strong metal–support interactions are of either elec-
tronic type or geometric type, letter also called decora-
tion model. In the present case, where the Rh catalyst is

Fig. 7. Self-driven electrochemical promotion of CH4 partial oxidation
on Rh interfaced with TiO2 with internal short-circuiting.
174 E.A. Baranova et al. / Electrochemistry Communications 6 (2004) 170–175
not highly dispersed but it is rather a film deposited totitania, an electronic effect may be effective only if long-
range interactions are present. Modifications in the
electronic structure of the metal by long-range effect are
changes in the d-band population, changes in the density
of states at the Fermi level, and shift of the Fermi level
due to the different work function of metal and support.
In the case of Rh, the first two effects are less important
because of the high density of unoccupied d states abovethe Fermi level, whereas shifting of the Fermi level may
have a great importance depending on the nature of the
support. In fact, the metal–semiconductor boundary
layer theory states that, at thermodynamic equilibrium,
the Fermi energy level of electrons of the two solids in
contact is equal. Upon contact of two solids of different
Fermi level (work function), charge must be transported
from one material to the other until the Fermi level atthe interface is equilibrated. Obviously, the solid of
lower work function gets charged positively and the
other negatively. The work function of stoichiometric,
unreduced TiO2 is 5.5 eV and that of Rh is 5.1 eV
therefore, upon contact, negative charge is transported
from the Rh catalyst to the TiO2 support. The resulting
increase in the catalyst work function causes a weak-
ening of the catalyst–oxygen bond strength and, conse-quently, a decrease in the stability of rhodium oxide.
Modification of the catalytic activity of Rh on TiO2
support has been already reported in the case of highly
dispersed Rh catalyst [9,10]. In the present work, similar
results have been obtained using a Rh film catalyst de-
posited on TiO2. This demonstrates that electronic type
SMSI may be a long-range interaction affecting the
whole metal catalyst.The observed modification of activity and selectivity
of Rh catalyst supported on TiO2 might be also related
to the partial reduction of TiO2 (formation of TiOx) by
hydrogen produced during the reaction [Eq. (1)]. In fact,
TiO2 can be reduced by hydrogen at relatively moderate
temperature (T > 400 �C) [5]. When one atom of oxygen
is lost from the surface, two electrons are left in the
oxygen vacancy to maintain electrical neutrality. Oneelectron is trapped by a neighboring Ti4þ to give Ti3þ,whereas the other electron is essentially free. This in-
creases the surface free electron concentration, hence,
causes a decrease in work function [11,12]. TiOx is a
degenerated semiconductor with very high carrier con-
centration, and the high electrical resistivity of TiO2
(1010 ohm cm) dramatically decreases with partial re-
duction to give 10 and 10�2 ohm cm at x ¼ 1:9995 and1.75, respectively [13]. At the surface of a dispersed TiO2
support partly covered with Rh catalyst, a spatially non-
uniform surface oxidation is expected resulting in the
coexistence of regions where TiO2 remains unreduced
and others with variable value of x depending on the
accessibility of the hydrogen gas to the TiO2 surface [14].
At the temperature of our experiments the suboxide
(TiOx) species may well diffuse towards the metal anddecorate its surface. Nevertheless, in spite of the largely
different electrical properties of TiO2 and TiOx, the es-
sential role of the TiOx entities migrated to the metal
surface remains to render this latter partially inaccessi-
ble to chemisorption and to catalytic action [15]. Hence,
it is believed that the geometric decoration model of
SMSI is inadequate to interpret the present experimental
findings.An alternative explanation of the effect of TiO2 sup-
port on the stability of rhodium oxide toward reduction
to metallic Rh is based on the theory of electrochemical
promotion of catalysis (EPOC). The model of electro-
chemical promotion regards the phenomenon as catal-
ysis in presence of an electrically controlled double layer
formed by ion backspillover mechanism at the gas-
exposed catalyst surface. As in the previous case ofelectronic type SMSI, the thermodynamic driving force
for electron transport is the difference of the Fermi level
of the two solids in contact. Since TiO2 is a semicon-
ductor of mixed (electron and O2� ion) conductiv-
ity, the positive charge on Rh built up by the electronic
effect will induce migration of O2� ions to the catalyst/
support interface, the driving force being the difference
in the electrochemical potential of oxide ions in the twooppositely charged solids. The O2� ions, accompanied
by their mirror charge in the metal, may then spread out
the whole gas-exposed catalyst surface by backspillover
mechanism thus forming an overall neutral electric
double layer. Population of the surface with dipoles
increases the surface potential, and the concomitant
increase in work function affects the binding strength of
chemisorbed species. Namely, the adsorption energy ofelectron donors is strengthened, while that of electron
acceptors (e.g., atomic O) and also the stability of Rh
oxide against reduction are weakened.
The close analogy between electrochemical promo-
tion of catalysis (EPOC) and strong metal–support in-
teraction (SMSI) is obvious, and they may be considered
as two functionally identical and only operationally
different phenomena [16]. In the present case, when nopotential or current is applied, even the operational
difference vanishes under steady state reaction condi-
tions. As shown in Fig. 7, the promoting O2� species are
slowly consumed in the catalytic reaction occurring at
the catalyst surface (O2� is a sacrificial promoter) while
spent O2� is continuously replenished via reduction of

E.A. Baranova et al. / Electrochemistry Communications 6 (2004) 170–175 175
gaseous O2 at the TiO2/gas (or TiOx/gas) interface: self-driven wireless EPOC mechanism.
5. Conclusion
In this paper, the partial oxidation of methane over
Rh catalyst supported on YSZ containing thin layer of
TiO2 was investigated. Under working conditions rho-dium catalyst has two well distinguished surface states:
active and inactive. According to the catalyst potential
these two states correspond to rhodium metal (active
state) and rhodium oxide (inactive state).
The remarkable increase in catalytic activity and se-
lectivity towards CO and H2 production when Rh
contacts TiO2 is related to the lowered stability of sur-
face Rh oxide against reduction to metallic Rh. Thephenomenon may be interpreted either by electronic
type strong metal–support interactions or by self-driven
wireless electrochemical promotion mechanism. In both
cases, the ultimate cause of promotion is the different
work function of catalyst and support. Equilibration of
the Fermi level causes weakening of the Rh–O chemi-
sorptive bond and makes reduction of oxidized surface
sites easier.
Acknowledgements
Financial supports from the Fonds National Suisse de
la Recherche Scientifique and from the Office F�ed�eral
Suisse de l’�Education et de la Science are gratefully ac-knowledged.
References
[1] M.A. Pena, J.P. Gomez, J.L.G. Fierro, Appl. Catal. A 144
(1996) 7.
[2] S.C. Tsang, J.B. Claridge, M.L.H. Green, Catal. Today 23
(1995) 3.
[3] M. Prettre, C. Eichner, M. Perrin, J. Chem. Soc. Faraday Trans.
43 (1946) 335.
[4] D.A. Hickman, E.A. Haupfear, L.D. Schmidt, Catal. Lett. 17
(1993) 223.
[5] S.J. Tauster, S.C. Fung, R.L. Garten, JACS 100 (1978) 170.
[6] C.G. Vayenas, S. Bebelis, C. Pliangos, S. Brosda, D. Tsiplakides,
Electrochemical Activation of Catalysis. Promotion, Electrochem-
ical Promotion, and Metal–Support interaction, Kluwer Aca-
demic/Plenum Publishers, New York, 2001.
[7] G. F�oti, I. Bolzonella, J. Eaves, Ch. Comninellis, CHIMIA 56
(2002) 137.
[8] G. F�oti, S. Wodiunig, Ch. Comninellis, Curr. Top. Electrochem.
7 (2000) 1.
[9] F. Solymosi, I. Tombacz, M. Kocsis, J. Catal. 75 (1982) 78.
[10] P. M�eriaudeau, O.H. Ellestad, M. Dufaux, C. Naccache, J. Catal.
75 (1982) 243.
[11] Y.W. Chung, W.J. Lo, G.A. Somorjai, Surf. Sci. 64 (1977)
588.
[12] T. Ioannides, X.E. Verykios, J. Catal. 161 (1996) 560.
[13] Gmelin�s Handbook Part 24, Weinheim, 1951.
[14] F. Pesty, H.P. Steinruck, T.E. Madey, Surf. Sci. 339 (1995) 83.
[15] G.L. Haller, D.E. Resasco, Adv. Catal. 36 (1989) 173.
[16] J. Nicole, D. Tsiplakides, C. Pliangos, X.E. Verykios, Ch.
Comninellis, C.G. Vayenas, J. Catal. 204 (2001) 23.




















