
Profound offense in secular democracies: An Australian perspective
Upload
independentCategory
view
0download
0
doi:10.1016/j.jmb.2005.02.044 J. Mol. Biol. (2005) 348, 1103–1112
Profound Flanking Sequence Preference of Dnmt3a andDnmt3b Mammalian DNA Methyltransferases Shape theHuman Epigenome
Vikas Handa1 and Albert Jeltsch2*
1Institut fur Biochemie, FB 08Heinrich-Buff-Ring 58Justus-Liebig-UniversitatGiessen, 35392 GiessenGermany
2International UniversityBremen, School of Engineeringand Science, Campus Ring 128759 Bremen, Germany
0022-2836/$ - see front matter q 2005 E
Abbreviations used: Dnmt1, DNAMTases, methyltransferases.E-mail address of the correspond
Mammalian DNA methyltransferases methylate cytosine residues withinCG dinucleotides. By statistical analysis of published data of the HumanEpigenome Project we have determined flanking sequences of up toGfourbase-pairs surrounding the central CG site that are characteristic of high(5 0-CTTGCGCAAG-3 0) and low (5 0-TGTTCGGTGG-3 0) levels of methyl-ation in human genomic DNA. We have investigated the influence offlanking sequence on the catalytic activity of the Dnmt3a and Dnmt3b denovo DNA methyltransferases using a set of synthetic oligonucleotidesubstrates that covers all possible G1 flanks in quantitative terms.Methylation kinetics experiments revealed a O13-fold difference betweenthe preferred (RCGY) and disfavored G1 flanking base-pairs (YCGR).In addition, AT-rich flanks are preferred over GC-rich ones. Theseexperimental preferences coincide with the genomic methylation patterns.Therefore, we have expanded our experimental analysis and found aO500-fold difference in the methylation rates of the consensus sequencesfor high and low levels of methylation in the genome. This result demon-strates a very pronounced flanking sequence preference of Dnmt3a andDnmt3b. It suggests that the methylation pattern of human DNA is due, inpart, to the flanking sequence preferences of the de novo DNA MTases andthat flanking sequence preferences could be involved in the origin of CGislands. Furthermore, similar flanking sequence preferences have beenfound for the stimulation of the immune system by unmethylated CGs,suggesting a co-evolution of DNA MTases and the immune system.
q 2005 Elsevier Ltd. All rights reserved.
Keywords: DNA methylation; CpG islands; flanking sequence preferences;epigenome; Dnmt3a
*Corresponding authorIntroduction
The cytosine-5 methylation in mammals is anepigenetic modification that plays an important rolein embryonic development, gene imprinting, X-chromosome inactivation, regulation of chromatinstructure, silencing of transposons and endogenousretroviruses, cancer biology and genetic diseases.1–6
In mammals, cytosine methylation takes placepredominantly at palindromic CG dinucleotides inboth strands of the DNA. The mammalian genomescontain w60 million CG dinucleotides and 70–80%of those are modified in a non-random pattern. The
lsevier Ltd. All rights reserve
methyltransferase 1;
ing author:
methylation pattern is inherited by daughter cellgenomes during DNA replication by the action ofDNA methyltransferase 1 (Dnmt1), which exhibitshigh preference for a hemimethylated DNA sub-strate.7–10
The genomic methylation pattern is set by de novoDNA methylation during gametogenesis in a sex-specific fashion and later, after extensive demethy-lation of the genome, during embryogenesis.5,11 Thede novo methylation is carried out by two de novoDNA methyltransferases (MTases), Dnmt3a andDnmt3b, which methylate unmethylated and hemi-methylated DNA.12,13 The role of Dnmt3a andDnmt3b in stage-specific de novo methylation ofmammalian genomes correlates with their highexpression in embryonic stem cells, early embryosand developing germ cells.12–15 The de novo methyl-ation activity of Dnmt3b is associated with methyl-ation of pericentromeric satellite regions.16–18
d.

† http://www.sanger.ac.uk/perl/MVP/mvp
1104 Flanking Sequence Preferences of Dnmt3 Enzymes
Dnmt3bK/K knockout mice die during lateembryonic stage and the embryos lack methylationin the pericentromeric repeat region.17 ICF (agenetic disorder resulting from mutations inDnmt3b) patients have low methylation in thepericentromeric satellite region of chromosome 1,9 and 16, leading to chromosome instability.19
Dnmt3a knockout mice show developmentalabnormalities and die a few weeks after birth.17
The enzyme has been associated with the methyl-ation of single copy genes and retrotransposons20–22
and it is critical to the establishment of the genomicimprint during germ cell development.23
In addition to their role in de novo methylation,Dnmt3a and Dnmt3b are involved in maintenanceof DNA methylation at later stages, as theycompensate for a lapse during conversion ofhemimethylated DNA to the fully methylatedstate by Dnmt1.24,25 This is evident from the findingthat Dnmt3aK/K/Dnmt3bK/K knockout embryo-nic stem cells lose genomic methylation gradually,although Dnmt1 is functional, but methylation canbe regained by episomal expression of the de novoMTases.25 In this manner a delicate balance betweende novo methylation and loss of methylation due toimperfect fidelity of Dnmt1 results in maintenanceof genomic methylation levels.
Unlike restriction modification system enzymesand transcription factors, mammalian cytosine-5DNA MTases have a short recognition sequence,CG, consisting of only 2 bases. There are interestingfindings on various aspects of the DNA substratesequence specificity of DNA MTases. Dnmt1 hasbeen found to have several-fold higher preferencefor hemimethylated CG when compared tounmethylated substrate.7–10 There is no preferencefor flanking sequences reported for Dnmt1,although highly GC-rich flanking sequences havebeen found to bind to the enzyme with higheraffinity.8 In mammalian genomes some non-CGcytosine residues have also been found to bemethylated. This observation is explained by thefinding that Dnmt3a methylates non-canonical sitesalso with a decreasing order of efficiency for CA, CTand CC dinucleotides.13,26,27 Although one of thetwo Dnmt3 enzymes, Dnmt3b, is processive innature there is no influence of CG density onmethylation activity.25
Flanking sequence preferences of Dnmt3a werefirst detected by Lin et al.,28 who found a strongpreference for a CG site flanked by pyrimidinebases and a loose consensus sequence of YNCGY.28
No such data are available for Dnmt3b. The resultsfor Dnmt3a were based on in vitro methylationexperiments in which plasmid DNA was methyl-ated by Dnmt3a followed by bisulfite sequencinganalysis. However, the influence of the flankingsequence on the rate of DNA methylation could notbe quantified in that study. In addition, the numberof different CG sites studied was too small to drawdefinite qualitative and quantitative conclusions onthe influence of flanking sequences on Dnmt3a. Ifone assumes an influence of only up to three bases
upstream and downstream of the central CG, thereare 4096 different flanks. Since one has to expect thatthe effect of each base at each position will dependon the nature of all other bases, there are only twoways to obtain statistically reliable information onflanking sequence preferences: (i) a large statisticalsurvey must be performed in order to integrate theeffects over many different flanking sequences; or(ii) synthetic substrates must be used in which oneor a few bases are changed while keeping theremaining parts of the flank constant.
To address the influence of flanking sequence onDNA methylation we have used two differentapproaches in combination. We analyzed themethylation pattern in human epigenomic data inthe context of different flanking sequences aroundmethylated CG dinucleotides. Here, 390 methylatedCG sites were analyzed, allowing us to drawstatistically relevant conclusions for longer flanks.In addition, biochemical experiments were per-formed using oligonucleotide substrates for meth-ylation kinetics under single turnover conditionsusing different de novo MTases. Surprisingly, wefound strong correlation of the flanking sequencepreferences of Dnmt3a and Dnmt3b and theaverage methylation level of CG sites in thehuman genome.
Results
Human epigenomic data analysis
First, high-throughput data on the methylationpattern of the human genome were collected in thehuman epigenome pilot project mostly for CpGislands, promoters and coding regions of genes andthe results published recently.29–32 Using the Epi-genome WEB site†, we analyzed the methylationlevels at various CG sites in the context of theirrespective flanking sequences, looking for someregular pattern in the sequences in relation tomethylation levels. A set of 390 methylated CGsites spanning a 220 kb region of human chromo-some 6 were analyzed with respect to the meanmethylation levels determined from various tissuesamples for each site (Figure 1). The data setconsisted of a heterogeneous population of CGswith methylation levels varying from zero to 100%.The 220 kb sequence belongs to the major histo-compatibility gene locus and was found to havehigh gene density containing 23 genes, 11 CpGislands and a region comprising two Alu and oneL1 repeat sequences. We were interested in the 8 bpflanking the central CG sites, four in the upstreamand four in the downstream direction. The flankingsequences were arranged in the order of averagemethylation levels of corresponding CG sites.
Using these data we wanted to look for consensusin the flanking sequences of those CGs that have
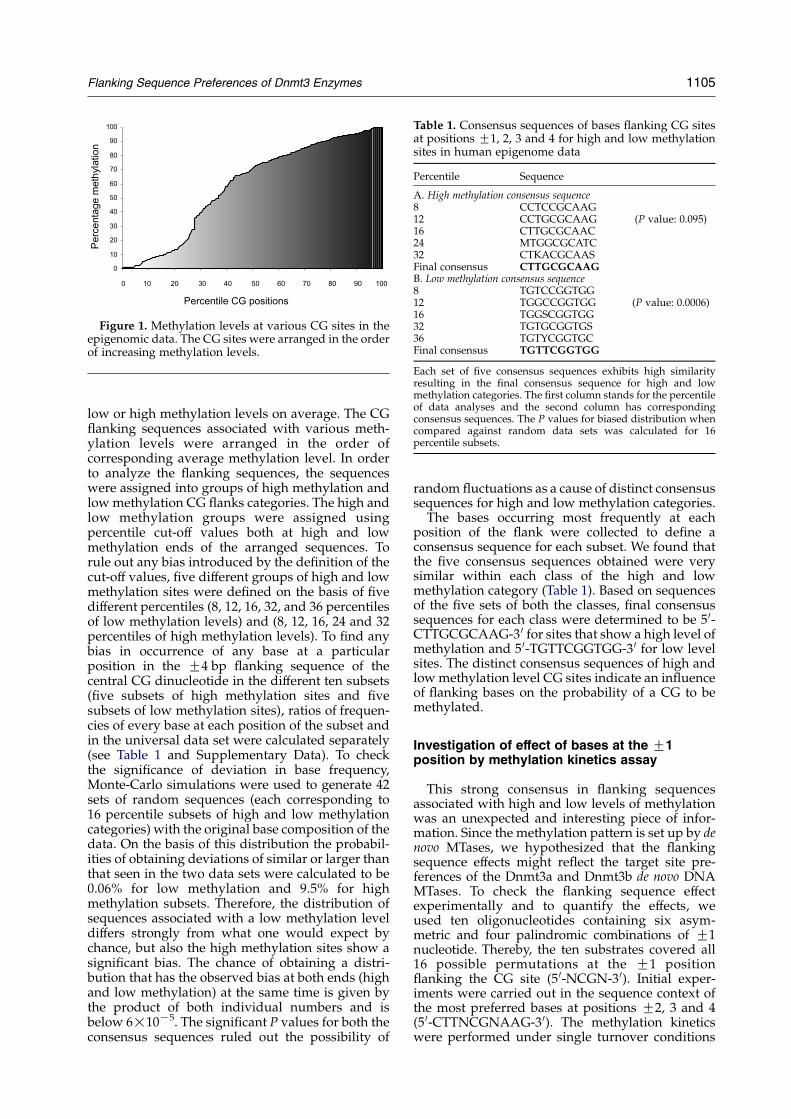
Figure 1. Methylation levels at various CG sites in theepigenomic data. The CG sites were arranged in the orderof increasing methylation levels.
Table 1. Consensus sequences of bases flanking CG sitesat positions G1, 2, 3 and 4 for high and low methylationsites in human epigenome data
Percentile Sequence
A. High methylation consensus sequence8 CCTCCGCAAG12 CCTGCGCAAG (P value: 0.095)16 CTTGCGCAAC24 MTGGCGCATC32 CTKACGCAASFinal consensus CTTGCGCAAGB. Low methylation consensus sequence8 TGTCCGGTGG12 TGGCCGGTGG (P value: 0.0006)16 TGGSCGGTGG32 TGTGCGGTGS36 TGTYCGGTGCFinal consensus TGTTCGGTGG
Each set of five consensus sequences exhibits high similarityresulting in the final consensus sequence for high and lowmethylation categories. The first column stands for the percentileof data analyses and the second column has correspondingconsensus sequences. The P values for biased distribution whencompared against random data sets was calculated for 16percentile subsets.
Flanking Sequence Preferences of Dnmt3 Enzymes 1105
low or high methylation levels on average. The CGflanking sequences associated with various meth-ylation levels were arranged in the order ofcorresponding average methylation level. In orderto analyze the flanking sequences, the sequenceswere assigned into groups of high methylation andlowmethylation CG flanks categories. The high andlow methylation groups were assigned usingpercentile cut-off values both at high and lowmethylation ends of the arranged sequences. Torule out any bias introduced by the definition of thecut-off values, five different groups of high and lowmethylation sites were defined on the basis of fivedifferent percentiles (8, 12, 16, 32, and 36 percentilesof low methylation levels) and (8, 12, 16, 24 and 32percentiles of high methylation levels). To find anybias in occurrence of any base at a particularposition in the G4 bp flanking sequence of thecentral CG dinucleotide in the different ten subsets(five subsets of high methylation sites and fivesubsets of low methylation sites), ratios of frequen-cies of every base at each position of the subset andin the universal data set were calculated separately(see Table 1 and Supplementary Data). To checkthe significance of deviation in base frequency,Monte-Carlo simulations were used to generate 42sets of random sequences (each corresponding to16 percentile subsets of high and low methylationcategories) with the original base composition of thedata. On the basis of this distribution the probabil-ities of obtaining deviations of similar or larger thanthat seen in the two data sets were calculated to be0.06% for low methylation and 9.5% for highmethylation subsets. Therefore, the distribution ofsequences associated with a low methylation leveldiffers strongly from what one would expect bychance, but also the high methylation sites show asignificant bias. The chance of obtaining a distri-bution that has the observed bias at both ends (highand low methylation) at the same time is given bythe product of both individual numbers and isbelow 6!10K5. The significant P values for both theconsensus sequences ruled out the possibility of
random fluctuations as a cause of distinct consensussequences for high and low methylation categories.The bases occurring most frequently at each
position of the flank were collected to define aconsensus sequence for each subset. We found thatthe five consensus sequences obtained were verysimilar within each class of the high and lowmethylation category (Table 1). Based on sequencesof the five sets of both the classes, final consensussequences for each class were determined to be 5 0-CTTGCGCAAG-3 0 for sites that show a high level ofmethylation and 5 0-TGTTCGGTGG-3 0 for low levelsites. The distinct consensus sequences of high andlowmethylation level CG sites indicate an influenceof flanking bases on the probability of a CG to bemethylated.
Investigation of effect of bases at the G1position by methylation kinetics assay
This strong consensus in flanking sequencesassociated with high and low levels of methylationwas an unexpected and interesting piece of infor-mation. Since the methylation pattern is set up by denovo MTases, we hypothesized that the flankingsequence effects might reflect the target site pre-ferences of the Dnmt3a and Dnmt3b de novo DNAMTases. To check the flanking sequence effectexperimentally and to quantify the effects, weused ten oligonucleotides containing six asym-metric and four palindromic combinations of G1nucleotide. Thereby, the ten substrates covered all16 possible permutations at the G1 positionflanking the CG site (5 0-NCGN-3 0). Initial exper-iments were carried out in the sequence context ofthe most preferred bases at positions G2, 3 and 4(5 0-CTTNCGNAAG-3 0). The methylation kineticswere performed under single turnover conditions
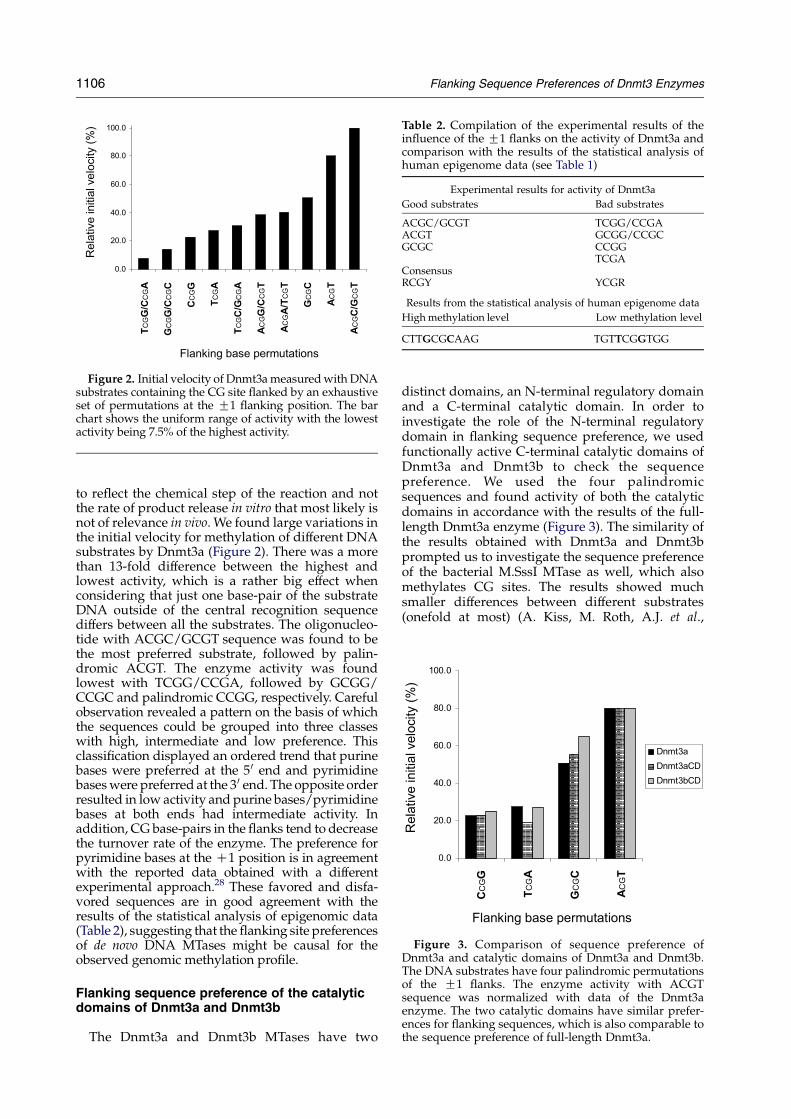
Table 2. Compilation of the experimental results of theinfluence of the G1 flanks on the activity of Dnmt3a andcomparison with the results of the statistical analysis ofhuman epigenome data (see Table 1)
Experimental results for activity of Dnmt3a
Good substrates Bad substrates
ACGC/GCGT TCGG/CCGAACGT GCGG/CCGCGCGC CCGG
TCGAConsensusRCGY YCGR
Results from the statistical analysis of human epigenome data
Highmethylation level Low methylation level
CTTGCGCAAG TGTTCGGTGG
Figure 3. Comparison of sequence preference ofDnmt3a and catalytic domains of Dnmt3a and Dnmt3b.
Figure 2. Initial velocity of Dnmt3ameasuredwithDNAsubstrates containing the CG site flanked by an exhaustiveset of permutations at the G1 flanking position. The barchart shows the uniform range of activity with the lowestactivity being 7.5% of the highest activity.
1106 Flanking Sequence Preferences of Dnmt3 Enzymes
to reflect the chemical step of the reaction and notthe rate of product release in vitro that most likely isnot of relevance in vivo. We found large variations inthe initial velocity for methylation of different DNAsubstrates by Dnmt3a (Figure 2). There was a morethan 13-fold difference between the highest andlowest activity, which is a rather big effect whenconsidering that just one base-pair of the substrateDNA outside of the central recognition sequencediffers between all the substrates. The oligonucleo-tide with ACGC/GCGT sequence was found to bethe most preferred substrate, followed by palin-dromic ACGT. The enzyme activity was foundlowest with TCGG/CCGA, followed by GCGG/CCGC and palindromic CCGG, respectively. Carefulobservation revealed a pattern on the basis of whichthe sequences could be grouped into three classeswith high, intermediate and low preference. Thisclassification displayed an ordered trend that purinebases were preferred at the 50 end and pyrimidinebaseswere preferred at the 30 end. The opposite orderresulted in low activity and purine bases/pyrimidinebases at both ends had intermediate activity. Inaddition, CG base-pairs in the flanks tend to decreasethe turnover rate of the enzyme. The preference forpyrimidine bases at the C1 position is in agreementwith the reported data obtained with a differentexperimental approach.28 These favored and disfa-vored sequences are in good agreement with theresults of the statistical analysis of epigenomic data(Table 2), suggesting that the flanking site preferencesof de novo DNA MTases might be causal for theobserved genomic methylation profile.
The DNA substrates have four palindromic permutationsof the G1 flanks. The enzyme activity with ACGTsequence was normalized with data of the Dnmt3aenzyme. The two catalytic domains have similar prefer-ences for flanking sequences, which is also comparable tothe sequence preference of full-length Dnmt3a.
Flanking sequence preference of the catalyticdomains of Dnmt3a and Dnmt3b
The Dnmt3a and Dnmt3b MTases have two
distinct domains, an N-terminal regulatory domainand a C-terminal catalytic domain. In order toinvestigate the role of the N-terminal regulatorydomain in flanking sequence preference, we usedfunctionally active C-terminal catalytic domains ofDnmt3a and Dnmt3b to check the sequencepreference. We used the four palindromicsequences and found activity of both the catalyticdomains in accordance with the results of the full-length Dnmt3a enzyme (Figure 3). The similarity ofthe results obtained with Dnmt3a and Dnmt3bprompted us to investigate the sequence preferenceof the bacterial M.SssI MTase as well, which alsomethylates CG sites. The results showed muchsmaller differences between different substrates(onefold at most) (A. Kiss, M. Roth, A.J. et al.,

Flanking Sequence Preferences of Dnmt3 Enzymes 1107
unpublished results). Furthermore, the ACGC andTCGG substrates, which are the extreme cases withDnmt3a, are modified at the same rate by M.SssI.This observation confirms that the similar flankingsequence preferences of the catalytic domains ofDnmt3a, CD-Dnmt3b and full-length Dnmt3a arenot due to an experimental artifact. We concludethat the N-terminal domain of Dnmt3a does notplay an important role in the flanking sequencepreference as already suggested by the finding thatthe isolated catalytic domains are of comparableactivity as the full-length enzymes.33,34 In additionour results demonstrate that both the Dnmt3enzymes share similar flanking sequence prefer-ences, which may be explained by the high aminoacid sequence similarity of the C-terminal regionsshared by the two de novo enzymes.
Figure 4. Enzyme activity of Dnmt3a in context withthe G2, 3 and 4 positions of flanking bases in DNAsubstrate. The underlined and overlined bases indicatelow and high preference sequences, respectively. The firsttwo sequences are the most preferred permutation at theG1 position flanked by most and least preferred flanks atthe G2, 3 and 4 positions, respectively and the next twosequences are the least preferred permutation at the G1position flanked by most and least preferred flanks at theG2, 3 and 4 positions, respectively. In both data sets, theenzyme activity drops sharply when the G2, 3 and 4positions of the flanks were changed from most preferredto least preferred bases.
Influence of the G2, 3 and 4 position bases
The O13-fold difference in activity of Dnmt3aafter variation of just 1 bp in front of and followingthe CG site is a very interesting observation, as itclearly shows a pronounced influence of flankingbases on enzyme catalysis. However, so far all ourexperiments were performed in the context of thehigh-methylation flanking sequences at the G2, 3and 4 positions. Next, we wanted to investigate ifthe bases at the G2, 3 and 4 positions have anyinfluence on the enzyme activity. An exhaustiveexperimental evaluation of the influence of fartherflanks would require a very large number ofsubstrate sequences. In lieu of this practically nearlyimpossible approach, we designed an experimentbased on the information obtained from theepigenome data analysis. We designed two oligo-nucleotides with ACGC/GCGT (most preferred)and TCGG/CCGA (least preferred) sequencesflanked by least preferred sequence at positionG2, 3 and 4 (5 0-TGTNCGNTGG-3 0) as determinedfrom epigenomic data analysis results. We foundthat there was a nearly fourfold drop in activity forthe ACGC/GCGT site in the context of theunfavorable outer flanking sequence. Furthermore,the enzyme activity at the TCGG/CCGA sequenceapproached zero when flanking sequences werechanged from most preferred to least preferred atthe G2, 3 and 4 positions (Figure 4). Whencomparing best and worst overall flanks, weobserved a very wide range of enzyme activities.When taking into account the detection limit in ourexperiment we conclude there is O500-fold differ-ence in the rates of methylation at 5 0-CTTACG-CAAG-3 0 versus 5 0-TGTTCGGTGG-3 0 sites. Thisresult indicates that the G2, 3 and 4 positionshave a pronounced influence on the catalyticactivity of the Dnmt3 de novo MTases. Again, weobserve that preferred flanks from statistical anal-ysis of methylation levels closely correlate with theenzymatic activities of Dnmt3a and Dnmt3b,suggesting that the sequence preferences ofDnmt3a and Dnmt3b have a major influence on
shaping the methylation pattern of the humangenome.
Discussion
It has been the purpose of this study to determinethe flanking sequence preferences of the Dnmt3aand Dnmt3b enzymes and investigate their poten-tial biological implications. During the last year,first results of high-throughput methylation ana-lysis of human DNA have been published.29–32
Using available epigenomic data we discoveredthat there is a clear relationship between thetendency of a CG site to undergo methylation andits flanking sequence. There are distinct and statisti-cally significant consensus sequences flanking CGsites that induce different levels of methylation (50-CTTGCGCAAG-3 0 forhighand50-TGTTCGGTGG-30
for low methylation) (Table 1). Although there arereports of recruitment of de novomethyltransferasesby transcription factors that bind to DNA in asequence-specific manner,35 this hardly explainssuch methylation bias at a global level.In order to understand this bias, we supposed it
might reflect the intrinsic preferences of the de novo

1108 Flanking Sequence Preferences of Dnmt3 Enzymes
MTases for certain flanking sequences. To checkthe flanking sequence effect on the methylationactivity of de novo DNA MTases, oligonucleotideDNA substrates were designed and subjected tomethylation kinetic studies with Dnmt3a andDnmt3b. The inner G1 flanks were investigated inan unbiased way using all possible flanks in anidentical sequence context. Outer flanks (G2, 3 and4) were checked using the consensus sequencesassociated with a high and low level of methylationin the epigenome data. These experiments revealeda more than 500-fold difference in the methylationrates observed at the best and worst substrate sitesthat correlates almost completely with the resultsfrom the statistical analysis of epigenome data. Thisfinding strongly suggests that the flanking sequencepreferences of Dnmt3a and Dnmt3b have a pro-nounced influence on the methylation pattern ofhuman genomic DNA. Furthermore, we observethat C and G-rich flanks tend to reduce the activityof Dnm3a and Dnmt3b. This property could berelated to the fact, that DNA segments containingmany CG dinucleotides in a highly GC-richsequence environment (CG islands) usually remainunmethylated during the wave of de novo methyl-ation in the early embryo.5 Therefore, the flankingsequence preferences of Dnmt3a and Dnmt3b couldhave been one driving force in the evolution of CGislands.
It should be mentioned that this agreement ofgenomic methylation pattern and enzymatic prop-erties of the de novo DNA MTases is of remarkablesignificance, keeping in mind that several factorsmay be involved in diluting the effect of sequencepreference of enzymes on genomic methylation.De novo methylation of a region may depend onadditional factors such as availability of free DNA,local chromatin structure, inhibition or recruitmentof MTases by specific DNA-binding factors, inter-action of Dnmt3 enzymes with histones, etc. Inaddition, flanking sequence preferences of Dnmt1could affect genomic methylation levels. Never-theless, we show here that the inherent properties ofthe Dnmt3 enzymes to prefer certain sequencesover others play an important role in shaping thegenomic methylation pattern. The very low activityof Dnmt3a and Dnmt3b at 5 0-TGTTCGGTGG-3 0
sites is particularly interesting, as it shows thatthere are some CG sites in the genome that arehighly discriminated by the de novo enzymes andmethylation might take place only under specialcircumstances such as involvement of some recruit-ing factors or factors like Dnmt3L that stimulatesde novo MTases.20,21,36–38
Our experimental data at position K1 and C1can be compared to a previous report on thesequence preference of Dnmt3a.28 Similar to us,Lin et al. found a strong preference for pyrimidine atposition C1. However, there are differences at theK1 position, because Lin et al. did not detect apreference for any base at this position whereas wehave found strong preference for purine bases. Thisdifference can be explained by the different
experimental approaches. Lin et al. have investi-gated methylation of random DNA and analyzedthe methylation levels of CpG sites in the context ofdifferent flanking sequences. The conclusion drawnfrom this approach on the preferences at the G1position may be biased due to the influence of outerflanks, because outer flank influence is not statisti-cally averaged. Therefore, it is possible that Lin et al.have missed a possible contribution of a purine atthe K1 position. For example, it is feasible thatmany RCG sites in their data set are positionedwithin unfavorable outer flanks, or many YCG sitesare within favorable outer flanks. In contrast, weinvestigated all possible G1 flanks in an identicalsequence context. In this manner we could dissectout the effects associated with individual permu-tations at position G1 very accurately. Our result issupported by the finding of a complementaryconsensus sequence for poor substrates that has apyrimidine at K1 and a purine at C1 position. Inaddition, our results correlate well with the statisti-cal analysis of human epigenome data that is basedon a large data set of 390 CG sites, thereby ensuringsufficient averaging of outer flank effects. Anagreement of Dnmt3a and Dnmt3b catalytic effi-ciencies and genomic methylation levels was alsoobserved in experiments directly investigating theouter flank effects, where O500-fold difference inthe methylation rates of different oligonucleotidesubstrates was found to be correlated with thegenomic methylation level of the correspondingflanking sequences. Our finding that the flankingsequence preferences of Dnmt3a and Dnmt3b arereflected by the human epigenome data indicates animportant role for Dnmt3a and Dnmt3b in settinginitial patterns of DNA methylation. Furthermore,since insufficient maintenance methylation byDnmt1 is counteracted by low level of de novomethylation, the Dnmt3a and Dnmt3b also play arole in the preservation of methylation levels.24,25,39
In the human epigenome data one frequentlyobserves methylation patterns in which one highlymethylated site is embedded into a low or inter-mediate methylation region, or in which a lowmethylation site is surrounded by high methylationsites. These can be explained by selective targetingof theMTase to a highmethylation site and blockingof methylation by other proteins at a low methyl-ation site. However, we demonstrate here thatanother explanation that also should be consideredis that the flanking sequences of the site arecontributory to the effect.
In a recent report, CG islands have been classifiedinto methylation-prone and resistant categories.The results were based on overexpression ofDnmt1 followed by detection of methylation levelsin CG islands of various genes.40 Based on flankingsequence preference information of de novo MTases,we analyzed sequences of methylation-prone andresistant CG islands. However, there was nosignificant difference found in the two sets of CGislands, indicating that Dnmt1 sequence preference

Flanking Sequence Preferences of Dnmt3 Enzymes 1109
is not related to the flanking sequence preference ofDnmt3 enzymes (data not shown).
The biological implications of the sequencepreferences of Dnmt3a and Dnmt3b de novo MTasesmight extend beyond the mere methylation level ofhuman DNA. DNA containing unmethylated CGdinucleotide sequences is immunogenic in mam-mals. Unmethylated CG sites stimulate B cells toproduce IL-6 and IL-12, CD41 Tcells to produce IL-6and IFN-g, and NK cells to produce IFN-g.41,42 Inseveral reports it has been shown that DNA withCG flanked by purine at the 5 0 end and pyrimidineat the 3 0 end induces a higher immunogenicresponse when compared to other sequences.41,43
This consensus sequence is identical with the highpreference consensus sequence for DNA MTasesfound by us. It is an interesting observation that theflanking sequence that renders high immunogeni-city to unmethylated CG dinucleotide sites belongsto the most preferred consensus sequence for denovo DNA methyltransferases. Therefore, thesequences with highest immunogenicity have thelowest probability to be unmethylated in the humanDNA, which minimizes the risk of an autoimmuneresponse generated from self DNA. This obser-vation indicates co-evolution of de novo DNAMTases and immune system in the context of CGdinucleotides and the flanking sequences.
Conclusions
Wehave studied the flanking sequence preferencesofDnmt3aandDnmt3bextendingapioneering studyby Lin et al. on Dnmt3a.28 We have studied theinfluence ofG1 flanks on the activity of Dnmt3a andDnmt3b by determining the methylation rate of allpossible sites within the same sequence context inquantitative terms.On thebasis of ourdata,wedefinea consensus both for favored and disfavoredsequences which match each other reasonably well.The Dnmt3a and Dnmt3b enzymes have very similarflanking sequence preferences. Our results show thatde novo mammalian DNA MTases exhibit profoundpreference for bases flanking a CG site (50-CTTACG-CAAG-30 consensus sequence) on one hand andshow almost no activity for some flanking sequences(50-TGTTCGGTGG-30 consensus sequence) on theother hand. The effects are so strong that certain CGsites are almost refractory to methylation in vitro. Wehave employed a bioinformatics approach to analyzethe DNA methylation patterns of human DNA. Wefound that the significant positive and negativeflanking sequence bias of de novo MTases is reflectedin genomic DNA methylation levels. This findingdemonstrates that the in vitro properties of Dnmt3aand Dnmt3b observed here are of clear relevance invivo. Our results suggest the intrinsic sequencepreference of de novoMTases could be one parameterthat influences the generation of the DNA methyl-ation patterns ofmammalian genomes, a process thatis largely not understood so far. In addition,we founda preference of Dnmt3a and Dnmt3b for AT-rich G1
flanks that could be correlated to the origin of CGislands, which are usually unmethylated in the germline. The preferred flanking sequences have also beenfound to be correlated to the immune responseelicited by an unmethylated CG motif containingDNA depending on the preceding and succeedingbases, indicating a co-evolution of DNAMTases andthe immune system.
Materials and Methods
Nomenclature
Throughout this work, the bases flanking the centralCG site are designated as illustrated below.
K4 K3 K2 K1 C1 C2 C3 C45 0-N N N N N CG N N N N N-30
Oligodeoxynucleotides
HPLC-purified oligodeoxynucleotides were purchasedfrom MWG (Ebersberg, Germany). The quality of theoligonucleotide synthesis was confirmed by denaturingpolyacrylamide gel electrophoresis, demonstrating that alloligonucleotides had the expected length and were pure toO95%. The concentrations of oligodeoxynucleotides solu-tions were determined spectroscopically using E260 valuesprovided by the supplier. Duplex oligodeoxynucleotideswere prepared by adding equimolar amounts of com-plementary strands, heating to 95 8C and slow-cooling toroom temperature. Complete annealing was confirmed bynative polyacrylamide gel electrophoresis, demonstratingthe absence of detectable amounts of single-stranded DNAafter the annealing process. Following are the sequences ofall the oligonucleotide substrates used, where Bt denotesbiotin. Every oligonucleotide has the same sequence, exceptfor four altered bases flanking CG on either side.
s1CG 50 Bt- gaagctgggacttccggaaggagagtgcaa -30
a1CG 50 - ttgcactctccttccggaagtcccagcttc -30
s1AA 50 Bt- gaagctgggacttacgaaaggagagtgcaa -30
a1AA 50 - ttgcactctcctttcgtaagtcccagcttc -30
s1AT 50 Bt- gaagctgggacttacgtaaggagagtgcaa -30
a1AT 50 - ttgcactctccttacgtaagtcccagcttc -30
s1AG 50 Bt- gaagctgggacttacggaaggagagtgcaa -30
a1AG 50 - ttgcactctccttccgtaagtcccagcttc -30
s1AC 50 Bt- gaagctgggacttacgcaaggagagtgcaa -30
a1AC 50 - ttgcactctccttgcgtaagtcccagcttc -30
s1TA 50 Bt- gaagctgggactttcgaaaggagagtgcaa -30
a1TA 50 - ttgcactctcctttcgaaagtcccagcttc -30
s1TC 50 Bt- gaagctgggactttcgcaaggagagtgcaa -30
a1TC 50 - ttgcactctccttgcgaaagtcccagcttc -30
s1TG 50 Bt- gaagctgggactttcggaaggagagtgcaa -30
a1TG 50 - ttgcactctccttccgaaagtcccagcttc -30
s1GC 50 Bt- gaagctgggacttgcgcaaggagagtgcaa -30
a1GC 50 - ttgcactctccttgcgcaagtcccagcttc -30
s1GG 50 Bt- gaagctgggacttgcggaaggagagtgcaa -30
a1GG 50 - ttgcactctccttccgcaagtcccagcttc -30
s2AC 50 Bt- gaagctgggatgtacgctgggagagtgcaa -30
a2AC 50 - ttgcactctcccagcgtacatcccagcttc -30
s2TG 50 Bt- gaagctgggatgttcggtgggagagtgcaa -30
a2TG 50 - ttgcactctcccaccgaacatcccagcttc -30

ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffis KnGexpÞ
2
1110 Flanking Sequence Preferences of Dnmt3 Enzymes
Epigenomic data analysis
The human epigenomic data were collected from theweb site†.29,30 For our analysis we used all available data ofa continuous stretch of DNA on the human chromosome 6between positions 31570047 and 31789835, which waschosen arbitrarily and is of sufficient size to represent thewhole data set. Methylation data were extracted forsequences comprising 10 bp containing the CG motif inthe center. We used the percentage of methylation for eachCG site deposited in the data-base. The arithmetic mean ofthe percentage of methylation among different samplesand different tissues was calculated for every CG site.Thereby every CG site was used just once and differentnumbers of tissues analyzed for the different sites did notinfluence our results. The mean percentage methylationvalues were aligned against corresponding flankingsequences of the CG sites in an increasing order. Thedata set was divided into low and highmethylation classesin many overlapping subsets using different cut-off values.The relative frequency for each base at each flankingpositionwas calculated by taking the ratio of frequencies ofthe subset and the universal set. This was used to find thebase with maximum occurrence at a particular position.To determine the significance of bias of the frequency of
occurrence of each base, we first calculated a factor (Bi) thatdescribes the deviation of found base distribution at eachposition (i) from the distribution expected on the basis ofthe overall frequencies of all four bases at each position inthe overall data set:
Bi ZffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiðnAobs KnAexpÞ
2 C ðnTobs KnTexpÞ2 C ðnCobs KnCexpÞ
2 C ðnGob
q
where nXobs and nXexp denote the number of base X observedand expected at the respective flanking position. Theoverall bias (B) was defined as the sum of the individualbiases for all eight flanking positions:
BZX4
iZK4
Bi
To estimate the significance of this number, a Monte-Carlo simulation was performed. A randomized set ofsequences with the same overall distribution of all bases ateach position as in the experimental set was generated.Using this set, random B-values were determined to obtainthe average B-value and its standard deviation. Therandom B-values showed a Gaussian distribution. Usingthese numbers, the probability of obtaining the observeddeviation by chance alone was calculated by standardstatistical procedures.
Expression and purification of enzymes
Recombinant expression of full-length Dnmt3a, and thecatalytic domains of Dnmt3a and Dnmt3b was carried outin BL21 E. coli cells, using pETDnmt3a, pETDnmt3aCDand pETDnmt3bCD plasmids. Transformed cells weregrown at 37 8C in 500 ml of LB medium containing75 mg/ml of kanamycin. Protein expression was inducedat a cell density of 0.3 A600 nm by addition of 1 mM IPTGand cells were grown for an additional one hour at 37 8C.Protein purification was carried out by Ni-NTA affinitychromatography as described.33
† http://www.sanger.ac.uk/perl/MVP/mvp
Methylation kinetics
DNA methylation assays using double-stranded oligo-deoxynucleotide substrate was carried out in a microtitreplate as described.44 The DNA substrate and enzymeconcentration was 0.5 mM each in methylation buffer(20 mM Hepes (pH 7.0), 1 mM EDTA) at 37 8C for periodsof 1, 2, 4, 8, 12, 16, 24 and 40 minutes. Labeled S-[methyl-3H]adenosyl-L-methionine (3048 GBq/mmol,NEN) was used at 0.76 mM. All methylation experimentswere carried out at least in triplicate and the resultsaveraged. Standard deviations of the average methylationrates were below G20%.
Acknowledgements
This work has been supported by grants from theBMBF (BioFuture programme), DFG (JE 252/1 andJE 252/4) and the Fonds der Chemischen Industrie.Thanks are due to H. Gowher for providing purifiedCDDnmt3b, and to M. Roth and A. Kiss forproviding data on M.SssI kinetics prior to publi-cation. We thank the Human Epigenome Consor-tium (http://www.epigenome.org/) for open accessand pre-publication release of data.
Supplementary data
Supplementary data associated with this articlecan be found, in the online version, at doi:10.1016/j.jmb.2005.02.044
References
1. Ehrlich, M. (2003). Expression of various genes iscontrolled by DNA methylation during mammaliandevelopment. J. Cell. Biochem. 88, 899–910.
2. Bird, A. (2002). DNA methylation patterns andepigenetic memory. Genes Dev. 16, 6–21.
3. Jeltsch, A. (2002). Beyond Watson and Crick: DNAmethylation and molecular enzymology of DNAmethyltransferases. ChemBiochem, 3, 274–293.
4. Jones, P. A. & Takai, D. (2001). The role of DNAmethylation in mammalian epigenetics. Science, 293,1068–1070.
5. Li, E. (2002). Chromatin modification and epigeneticreprogramming in mammalian development. NatureRev. Genet. 3, 662–673.
6. Feinberg, A. P. & Tycko, B. (2004). The history of cancerepigenetics. Nature Rev. Cancer, 4, 143–153.
7. Zucker, K. E., Riggs, A. D. & Smith, S. S. (1985).Purification of human DNA (cytosine-5-)-methyltrans-ferase. J. Cell. Biochem. 29, 337–349.
8. Flynn, J., Azzam, R. & Reich, N. (1998). DNA bindingdiscrimination of the murine DNA cytosine-C5 meth-yltransferase. J. Mol. Biol. 279, 101–116.
9. Fatemi, M., Hermann, A., Pradhan, S. & Jeltsch, A.(2001). The activity of the murine DNA methyltrans-ferase Dnmt1 is controlled by interaction of the

Flanking Sequence Preferences of Dnmt3 Enzymes 1111
catalytic domain with the N-terminal part of theenzyme leading to an allosteric activation of theenzyme after binding to methylated DNA. J. Mol.Biol. 309, 1189–1199.
10. Pradhan, S., Bacolla, A., Wells, R. D. & Roberts, R. J.(1999). Recombinant human DNA (cytosine-5) meth-yltransferase. I. Expression, purification, and compari-son of de novo and maintenance methylation. J. Biol.Chem. 274, 33002–33010.
11. Meehan, R. R. (2003). DNA methylation in animaldevelopment. Semin. Cell. Dev. Biol. 14, 53–65.
12. Okano, M., Xie, S. & Li, E. (1998). Cloning andcharacterization of a family of novel mammalianDNA (cytosine-5) methyltransferases. Nature Genet.19, 219–220.
13. Gowher, H. & Jeltsch, A. (2001). Enzymatic propertiesof recombinant Dnmt3a DNA methyltransferase frommouse: the enzyme modifies DNA in a non-processivemanner and also methylates non-CpG (correction ofnon-CpA) sites. J. Mol. Biol. 309, 1201–1208.
14. Huntriss, J., Hinkins, M., Oliver, B., Harris, S. E.,Beazley, J. C., Rutherford, A. J. et al. (2004). Expressionof mRNAs for DNA methyltransferases and methyl-CpG-binding proteins in the human female germ line,preimplantation embryos, and embryonic stem cells.Mol. Reprod. Dev. 67, 323–336.
15. Chen, T., Ueda, Y., Xie, S. & Li, E. (2002). A novelDnmt3a isoform produced from an alternative pro-moter localizes to euchromatin and its expressioncorrelates with active de novomethylation. J. Biol. Chem.277, 38746–38754.
16. Hansen, R. S., Wijmenga, C., Luo, P., Stanek, A. M.,Canfield, T. K., Weemaes, C. M. & Gartler, S. M. (1999).The DNMT3BDNAmethyltransferase gene ismutatedin the ICF immunodeficiency syndrome. Proc. NatlAcad. Sci. USA, 96, 14412–14417.
17. Okano, M., Bell, D. W., Haber, D. A. & Li, E. (1999).DNA methyltransferases Dnmt3a and Dnmt3b areessential for de novo methylation and mammaliandevelopment. Cell, 99, 247–257.
18. Xu, G. L., Bestor, T. H., Bourc’his, D., Hsieh, C. L.,Tommerup, N., Bugge, M. et al. (1999). Chromosomeinstability and immunodeficiency syndrome caused bymutations in a DNA methyltransferase gene. Nature,402, 187–191.
19. Ehrlich, M. (2003). The ICF syndrome, a DNAmethyltransferase 3B deficiency and immunodefi-ciency disease. Clin. Immunol. 109, 17–28.
20. Hata, K., Okano, M., Lei, H. & Li, E. (2002). Dnmt3Lcooperates with the Dnmt3 family of de novo DNAmethyltransferases to establish maternal imprints inmice. Development, 129, 1983–1993.
21. Bourc’his, D. & Bestor, T. H. (2004). Meiotic catastropheand retrotransposon reactivation in male germ cellslacking Dnmt3L. Nature, 431, 96–99.
22. Bourc’his, D., Xu, G. L., Lin, C. S., Bollman, B. & Bestor,T. H. (2001). Dnmt3L and the establishment ofmaternalgenomic imprints. Science, 294, 2536–2539.
23. Kaneda, M., Okano, M., Hata, K., Sado, T., Tsujimoto,N., Li, E. & Sasaki, H. (2004). Essential role for de novoDNA methyltransferase Dnmt3a in paternal andmaternal imprinting. Nature, 429, 900–903.
24. Riggs, A. D. & Xiong, Z. (2004). Methylation andepigenetic fidelity. Proc. Natl Acad. Sci. USA, 101, 4–5.
25. Chen, T., Ueda, Y., Dodge, J. E.,Wang, Z. & Li, E. (2003).Establishment and maintenance of genomic methyl-ation patterns in mouse embryonic stem cells byDnmt3a and Dnmt3b. Mol. Cell. Biol. 23, 5594–5605.
26. Dodge, J. E., Ramsahoye, B. H., Wo, Z. G., Okano, M. &
Li, E. (2002).De novomethylation ofMMLV provirus inembryonic stem cells: CpG versus non-CpG methyl-ation. Gene, 289, 41–48.
27. Ramsahoye, B. H., Biniszkiewicz, D., Lyko, F., Clark, V.,Bird, A. P. & Jaenisch, R. (2000). Non-CpGmethylationis prevalent in embryonic stem cells and may bemediated by DNA methyltransferase 3a. Proc. NatlAcad. Sci. USA, 97, 5237–5242.
28. Lin, I. G., Han, L., Taghva, A., O’Brien, L. E. & Hsieh,C. L. (2002). Murine de novomethyltransferase Dnmt3ademonstrates strand asymmetry and site preference inthemethylation of DNA in vitro.Mol. Cell. Biol. 22, 704–723.
29. Eckhardt, F., Beck, S., Gut, I. G. & Berlin, K. (2004).Future potential of the human epigenome project.Expert Rev. Mol. Diagn. 4, 609–618.
30. Rakyan, V. K., Hildmann, T., Novik, K. L., Lewin, J.,Tost, J., Cox, A. V. et al. (2004). DNA methylationprofiling of the human major histocompatibility com-plex: a pilot study for the human epigenome project.PLoS Biol. 2, e405.
31. Novik, K. L., Nimmrich, I., Genc, B., Maier, S.,Piepenbrock, C., Olek, A. & Beck, S. (2002). Epige-nomics: genome-wide study of methylation phenom-ena. Curr. Issues Mol. Biol. 4, 111–128.
32. Beck, S., Olek, A. & Walter, J. (1999). From genomics toepigenomics: a loftier view of life.Nature Biotechnol. 17,1144.
33. Gowher, H. & Jeltsch, A. (2002). Molecular enzymologyof the catalytic domains of the Dnmt3a and Dnmt3bDNA methyltransferases. J. Biol. Chem. 277, 20409–20414.
34. Reither, S., Li, F., Gowher, H. & Jeltsch, A. (2003).Catalytic mechanism of DNA-(cytosine-C5)-methyl-transferases revisited: covalent intermediate formationis not essential for methyl group transfer by themurineDnmt3a enzyme. J. Mol. Biol. 329, 675–684.
35. Di Croce, L., Raker, V. A., Corsaro, M., Fazi, F., Fanelli,M., Faretta, M. et al. (2002). Methyltransferase recruit-ment and DNA hypermethylation of target promotersby an oncogenic transcription factor. Science, 295, 1079–1082.
36. Chedin, F., Lieber, M. R. & Hsieh, C. L. (2002). TheDNA methyltransferase-like protein DNMT3L stimu-lates de novo methylation by Dnmt3a. Proc. Natl Acad.Sci. USA, 99, 16916–16921.
37. Suetake, I., Shinozaki, F., Miyagawa, J., Takeshima, H.& Tajima, S. (2004). DNMT3L stimulates the DNAmethylation activity of Dnmt3a and Dnmt3b through adirect interaction. J. Biol. Chem. 279, 27816–27823.
38. Gowher, H., Liebert, K., Hermann, A., Xu, G. & Jeltsch,A. (2005). Mechanism of stimulation of catalyticactivity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J. Biol. Chem. In thepress.
39. Pfeifer, G. P., Steigerwald, S. D., Hansen, R. S., Gartler,S. M. & Riggs, A. D. (1990). Polymerase chain reaction-aided genomic sequencing of an X chromosome-linkedCpG island: methylation patterns suggest clonalinheritance. CpG site autonomy, and an explanationof activity state stability. Proc. Natl Acad. Sci. USA, 87,8252–8256.
40. Feltus, F. A., Lee, E. K., Costello, J. F., Plass, C. &Vertino, P. M. (2003). Predicting aberrant CpG islandmethylation. Proc. Natl Acad. Sci. USA, 100, 12253–12258.
41. Krieg, A. M. (2002). CpG motifs in bacterial DNA andtheir immune effects. Annu. Rev. Immunol. 20, 709–760.
42. Rui, L., Vinuesa, C. G., Blasioli, J. & Goodnow, C. C.

1112 Flanking Sequence Preferences of Dnmt3 Enzymes
(2003). Resistance to CpG DNA-induced autoimmu-nity through tolerogenic B cell antigen receptor ERKsignaling. Nature Immunol. 4, 594–600.
43. Klinman, D.M., Yi, A. K., Beaucage, S. L., Conover, J. &Krieg, A. M. (1996). CpG motifs present in bacteriaDNA rapidly induce lymphocytes to secrete
interleukin 6, interleukin 12, and interferon gamma.Proc. Natl Acad. Sci. USA, 93, 2879–2883.
44. Roth, M. & Jeltsch, A. (2000). Biotin–avidin microplateassay for the quantitative analysis of enzymaticmethylation of DNA by DNA methyltransferases.Biol. Chem. 381, 269–272.
Edited by J. Karn
(Received 24 January 2005; received in revised form 18 February 2005; accepted 18 February 2005)
Copyright © 2022 FDOKUMEN




















