
Probing the molecular-level control of aluminosilicate dissolution: A sensitive solid-state NMR...
13
Probing the molecular-level control of aluminosilicate dissolution: A sensitive solid-state NMR proxy for reactive surface area Nancy M. Washton a,1 , Susan L. Brantley b , Karl T. Mueller a, * a Department of Chemistry, 104 Chemistry Building, Penn State University, University Park, PA 16802, USA b Department of Geosciences, 2217 Earth and Engineering Building, Penn State University, University Park, PA 16802, USA Received 7 March 2007; accepted in revised form 15 September 2008; available online 30 September 2008 Abstract For two suites of volcanic aluminosilicate glasses, the accessible and reactive sites for covalent attachment of the fluorine- containing (3,3,3-trifluoropropyl)dimethylchlorosilane (TFS) probe molecule were measured by quantitative 19 F nuclear magnetic resonance (NMR) spectroscopy. The first set of samples consists of six rhyolitic and dacitic glasses originating from volcanic activity in Iceland and one rhyolitic glass from the Bishop Tuff, CA. Due to differences in the reactive species present on the surfaces of these glasses, variations in the rate of acid-mediated dissolution (pH 4) for samples in this suite cannot be explained by variations in geometric or BET-measured surface area. In contrast, the rates scale directly with the surface density of TFS-reactive sites as measured by solid-state NMR. These data are consistent with the inference that the TFS-reactive M-OH species on the glass surface, which are known to be non-hydrogen-bonded Q 3 groups, represent loci accessible to and affected by proton-mediated dissolution. The second suite of samples, originating from a chronosequence in Kozushima, Japan, is com- prised of four rhyolites that have been weathered for 1.1, 1.8, 26, and 52 ka. The number of TFS-reactive sites per gram increases with duration of weathering in the laboratory for the ‘‘Icelandic’’ samples and with duration of field weathering for both ‘‘Icelandic’’ and Japanese samples. One hypothesis is consistent with these and published modeling, laboratory, and field obser- vations: over short timescales, dissolution is controlled by fast-dissolving sites, but over long timescales, dissolution is controlled by slower-dissolving sites, the surface density of which is proportional to the number of TFS-reactive Q 3 sites. These latter sites are not part of a hydrogen-bonded network on the surface of the glasses, and measurement of their surface site density allows predictions of trends in reactive surface area. The TFS treatment method, which is easily monitored by quantitative 19 F solid- state NMR, therefore provides a chemically specific and quantifiable proxy to understand the nature of how sites on dissolving silicates control dissolution. Furthermore, 27 Al NMR techniques are shown here to be useful in identifying clays on the glass surfaces, and these methods are therefore effective for quantifying concentrations of weathering impurities. Our interpretations offer a testable hypothesis for the mechanism of proton-promoted dissolution for low-iron aluminosilicate minerals and glasses and suggest that future investigations of reactive surfaces with high-sensitivity NMR techniques are warranted. Ó 2008 Elsevier Ltd. All rights reserved. 1. INTRODUCTION The ability to predict and model mineral and glass disso- lution rates is needed to advance our understanding of ele- ment cycles in the environment (Casey et al., 1993; Nugent et al., 1998; Oelkers, 2001; White and Brantley, 2003; White, 2008). To scale from laboratory observations to field systems requires that rates of interface-controlled dissolu- tion in the laboratory be extrapolated using the reactive surface area of the dissolving phase. Generally, reactive sur- face area (here defined as the surface density of the chemical species on a surface that are most directly involved in the chemical reaction of interest) is estimated through the measurement of the specific surface area using adsorption 0016-7037/$ - see front matter Ó 2008 Elsevier Ltd. All rights reserved. doi:10.1016/j.gca.2008.09.018 * Corresponding author. Fax: +1 814 863 8403. E-mail address: [email protected] (K.T. Mueller). 1 Present address: PPG Industries, Inc., 400 Guys Run Road, Cheswick, PA 15024, USA. www.elsevier.com/locate/gca Available online at www.sciencedirect.com Geochimica et Cosmochimica Acta 72 (2008) 5949–5961
Transcript of Probing the molecular-level control of aluminosilicate dissolution: A sensitive solid-state NMR...

Available online at www.sciencedirect.com
www.elsevier.com/locate/gca
Geochimica et Cosmochimica Acta 72 (2008) 5949–5961
Probing the molecular-level control of aluminosilicate dissolution:A sensitive solid-state NMR proxy for reactive surface area
Nancy M. Washton a,1, Susan L. Brantley b, Karl T. Mueller a,*
a Department of Chemistry, 104 Chemistry Building, Penn State University, University Park, PA 16802, USAb Department of Geosciences, 2217 Earth and Engineering Building, Penn State University, University Park, PA 16802, USA
Received 7 March 2007; accepted in revised form 15 September 2008; available online 30 September 2008
Abstract
For two suites of volcanic aluminosilicate glasses, the accessible and reactive sites for covalent attachment of the fluorine-containing (3,3,3-trifluoropropyl)dimethylchlorosilane (TFS) probe molecule were measured by quantitative 19F nuclearmagnetic resonance (NMR) spectroscopy. The first set of samples consists of six rhyolitic and dacitic glasses originating fromvolcanic activity in Iceland and one rhyolitic glass from the Bishop Tuff, CA. Due to differences in the reactive species present onthe surfaces of these glasses, variations in the rate of acid-mediated dissolution (pH 4) for samples in this suite cannot beexplained by variations in geometric or BET-measured surface area. In contrast, the rates scale directly with the surface densityof TFS-reactive sites as measured by solid-state NMR. These data are consistent with the inference that the TFS-reactive M-OHspecies on the glass surface, which are known to be non-hydrogen-bonded Q3 groups, represent loci accessible to and affected byproton-mediated dissolution. The second suite of samples, originating from a chronosequence in Kozushima, Japan, is com-prised of four rhyolites that have been weathered for 1.1, 1.8, 26, and 52 ka. The number of TFS-reactive sites per gram increaseswith duration of weathering in the laboratory for the ‘‘Icelandic’’ samples and with duration of field weathering for both‘‘Icelandic’’ and Japanese samples. One hypothesis is consistent with these and published modeling, laboratory, and field obser-vations: over short timescales, dissolution is controlled by fast-dissolving sites, but over long timescales, dissolution is controlledby slower-dissolving sites, the surface density of which is proportional to the number of TFS-reactive Q3 sites. These latter sitesare not part of a hydrogen-bonded network on the surface of the glasses, and measurement of their surface site density allowspredictions of trends in reactive surface area. The TFS treatment method, which is easily monitored by quantitative 19F solid-state NMR, therefore provides a chemically specific and quantifiable proxy to understand the nature of how sites on dissolvingsilicates control dissolution. Furthermore, 27Al NMR techniques are shown here to be useful in identifying clays on the glasssurfaces, and these methods are therefore effective for quantifying concentrations of weathering impurities. Our interpretationsoffer a testable hypothesis for the mechanism of proton-promoted dissolution for low-iron aluminosilicate minerals and glassesand suggest that future investigations of reactive surfaces with high-sensitivity NMR techniques are warranted.� 2008 Elsevier Ltd. All rights reserved.
1. INTRODUCTION
The ability to predict and model mineral and glass disso-lution rates is needed to advance our understanding of ele-
0016-7037/$ - see front matter � 2008 Elsevier Ltd. All rights reserved.
doi:10.1016/j.gca.2008.09.018
* Corresponding author. Fax: +1 814 863 8403.E-mail address: [email protected] (K.T. Mueller).
1 Present address: PPG Industries, Inc., 400 Guys Run Road,Cheswick, PA 15024, USA.
ment cycles in the environment (Casey et al., 1993; Nugentet al., 1998; Oelkers, 2001; White and Brantley, 2003;White, 2008). To scale from laboratory observations to fieldsystems requires that rates of interface-controlled dissolu-tion in the laboratory be extrapolated using the reactivesurface area of the dissolving phase. Generally, reactive sur-face area (here defined as the surface density of the chemicalspecies on a surface that are most directly involved in thechemical reaction of interest) is estimated through themeasurement of the specific surface area using adsorption

5950 N.M. Washton et al. / Geochimica et Cosmochimica Acta 72 (2008) 5949–5961
isotherms. Determination of surface area is most commonlyaccomplished through the Brunnauer, Emmett, Teller(BET) gas adsorption isotherm method, which provides avalue for the specific area (ABET). A second method ofapproximating reactive surface area (White and Peterzon,1990; Brantley et al., 1999; Brantley and Mellott, 2000) isthat of utilizing the calculated geometric surface area basedon particle size (AGeo). However, neither of these methodsprovide specific chemical information about the reactingsurface.
Chemical information specific to the surface of interest isneeded for further understanding of dissolution and precip-itation mechanisms and kinetics. In addition, the use ofeither ABET or AGeo as a proxy for reactive surface areahas come under scrutiny as some researchers have notedthe lack of correlation between dissolution rate and thesesurface area values (Helgeson et al., 1984; Holdren and Spe-yer, 1985; Anbeek, 1992; Van-Cappellen, 1996; Gautieret al., 2001; Metz et al., 2005). Therefore, to address thelack of an analytical technique capable of measuring reac-
tive surface area, we have investigated the use of NMR sen-sitive probe molecules to characterize the nature of surfacesites on complex oxide materials undergoing dissolution.Correlations between the number density of sites reactiveto the probe molecules and rates of dissolution (comparedon a common basis that is measurable directly, such asmass of sample) are then examined in the search for a proxyfor reactive surface area.
Under ambient conditions where surfaces are in contactwith water or water vapor, most unmodified oxide surfacesare terminated by hydroxyl groups, and many studies relatemineral dissolution rates to the concentration of these hy-droxyl groups (Iler, 1979; Davis and Kent, 1990; Heringand Stumm, 1990; Brantley, 2008). Surface densities of hy-droxyl groups have typically been quantified by exchange oftritium or deuterium (Davydov et al., 1964; Yates andHealy, 1975; Zhuravlev, 1987), by titration (Blum andLasaga, 1988; Schott, 1990; Stillings et al., 1995; Brantleyand Stillings, 1996), or by thermogravimetric analysis(James and Parks, 1982; Mueller et al., 2003). However,these methods do not distinguish between vicinal, geminal,or lone hydroxyls, nor do they differentiate sites bonded tothe crystal lattice by 1, 2 or 3 bridging oxygens (referred tohere as the Q1, Q2, and Q3 sites on silicates, see Fig. 1) (Da-vis and Kent, 1990). In addition, the species occupying thenext nearest coordination sphere is a critical issue in alumi-nosilicate dissolution, as various Qn[mAl] sites—silicatespecies with n oxygens bridging to m aluminum atomsand n � m Si atoms—will be present. As the identity ofthe particular type(s) of hydroxyl species implicated in
SiHO OH
SiHO
OHOH
Q1 Q2
Si
Q3
OH
Fig. 1. Representation of Qn sites, where Q = Si and n representsthe number of bonds to the bulk oxide. Bulk oxygens have beenomitted for clarity.
acid-mediated dissolution is still a matter of debate, a meth-od discriminating the nature of the hydroxyls that influencedissolution could therefore serve as a superior quantitativeproxy for reactive surface area.
We have previously shown that a sensitive and chemi-cally direct measure of hydroxyl reactivity for low surfacearea oxides is obtainable through the chemical process ofsilanization (Fry et al., 2003b), a surface modification tech-nique long used in the chromatographic industry to couplemolecules with desired functionality to high surface areamaterials (Van-Der-Voort and Vansant, 1996; Nawrocki,1997). Although silane coupling to oxides has typically beenutilized to modify the surface hydrophilicity, such modifica-tion often entails condensation of tri-functional organosi-lanes that can react with more than one surface hydroxylwhile also generating a cross-linked polymeric structurenear the surface via silane–silane reactions. However, thequantification of reactive surface sites using silanes requiresthe use of a well-understood chemical reaction that is selec-
tive for individual sites. In contrast to the multiple surfaceattachments of a tri-functional organosilane, a mono-func-tional organosilane attaches to a lone M-OH species on anoxide surface via a single covalent linkage.
As shown in Fig. 2 for a mono-functional organosilane,the organosilane-oxide condensation mechanism is com-prised of two steps. Step one consists of hydrolysis of thechlorosilane to form an organosilanol, followed by the sec-ond step wherein the organosilanol condenses with a single
reactive hydroxyl group present on the oxide surface. Afterthe coupling reactions are complete, quantitative spectro-scopic investigations are possible if the resulting surfacecomplexes are sufficiently abundant. However, silanizationproducts on low surface area natural and synthetic materi-als are especially challenging to study via spectroscopic, andtherefore chemically specific, analyses. Fry et al. (2003b)overcame the problem of low analyte concentrationthrough NMR observation of the highly sensitive 19F nucleicontained in a fluorinated organosilane. Using 3,3,3-(triflu-oropropyl)dimethylchlorosilane (TFS) probe molecules(Fig. 3), we demonstrated the viability of quantifying thenumber of reactive hydroxyls on low surface area silicateand alumino[boro]silicate glass fibers with specific surfaceareas of <0.25 m2/g (Fry et al., 2003a,b).
A similar silane coupling methodology, monitored by19F NMR, is applied here for the first time to two suitesof naturally and laboratory-weathered volcanic glasses.The first, a suite consisting of six Icelandic glasses andone ignimbritic sample from the Bishop Tuff, CA, was cho-sen to investigate the potential correlation between labora-tory-measured acid-mediated dissolution rates and thereactive surface area (as measured through the quantifica-tion of TFS-reactive hydroxyl sites). The second suite offour volcanic samples comprising a chronosequence fromKozushima, Japan, was acquired to investigate the effectsof field weathering on the abundance of reactive hydroxylspecies. With these samples, we tested whether the dissolu-tion rate varied with the surface density of the TFS-reactivehydroxyls. We then discuss how the chemistry controllingthe specificity of the chlorosilane condensation reactionelucidates the molecular-level details of the dissolution

RSi
Cl+ H2O -HCl
RSi
O
RSi
OH
H
+O
MO
OO
H
OMO
OO
HRSi
OH
OMO
OO
SiR
1.
2. SNi
Fig. 2. Mono-chlorosilane hydrolysis (1) and subsequent condensation with an oxide surface (2). The surface is represented by an M-OHgroup, with three oxygens bridging to the glass surface.
Cl
7.4 Å
H3C Si
FFF
CH3
Fig. 3. The probe molecule 3,3,3-(trifluoropropyldimethyl)chlro-rosilane (TFS) has a lateral extension (effective diameter) of 7.4 A.
A sensitive NMR proxy measuring reactive surface area for aluminosilicate dissolution 5951
process. Altogether, this approach allows an investigationof surface-specific chemical reactivity, the tie between reac-tive chemistry and proton-promoted dissolution, and thechange in surface reactivity as a function of weatheringacross temporal and spatial scales spanning from the labo-ratory to the field.
2. METHODS
2.1. Samples
For the first suite of samples, specimens of seven of therhyolitic and dacitic glasses reported in a previous publica-tion (Wolff-Boenisch et al., 2004) were obtained from theauthors as both field-weathered (but pre-dissolution exper-iment) samples and as post-dissolution experiment samples.We refer to this suite as the ‘‘Icelandic’’ suite because six ofthe glasses were of Icelandic origin while one was collectedfrom the Bishop Tuff, CA. The Icelandic samples wereweathered in the field for durations ranging from 800 to3500 years, while the Bishop Tuff sample was field-weath-ered for approximately 750,000 years. The weight percentSiO2 ranges from 62.76 to 72.62 in these glasses; chemicalcompositions and additional physicochemical propertiesare detailed in Wolff-Boenisch et al. (2004). The suite thatwe study constitutes a low-Fe subset of the 18 volcanicglasses that were studied by Wolff-Boenisch et al. (2004).Paramagnetic species present in some of the original 18samples (those with mol% Fe oxides >8%) precluded theirstudy utilizing NMR. Pre-dissolution samples were receivedas course-grained hand specimens. In addition, post-disso-lution samples were also provided as pre-sieved
(125–45 lm) fractions that had been dissolved at pH 4and 25 �C in mixed flow reactors for approximately 600 h,followed by storage in plastic vials under ambient condi-tions. No additional grinding or sieving was carried outon the post-dissolution samples.
A second suite of glasses consisted of four samples ofrhyolite collected from lava flows emplaced in Kozushima,Japan, between 1.1 and 52 thousand years ago (Yokoyamaand Banfield, 2002). The dissolution behaviors of theseglasses were investigated in laboratory dissolution experi-ments and described in detail by Yokoyama and Banfield(2002), from whom the four coarse grained block sampleswere obtained. These samples comprise a chronosequencebecause the four parent rhyolites share a common chemicalcomposition and the environmental conditions were consid-ered to be relatively constant over the 52,000 year span ofweathering (Yokoyama and Banfield, 2002).The SiO2
weight percent for the youngest (1.1 ka) sample is 76.75; fullchemical compositions and their change with weatheringtimes in the field are detailed in Oguchi et al. (1999) andYokoyama and Banfield (2002).
2.2. Sample preparation
Both the ‘‘Icelandic’’ and Kozushima hand specimenswere ground using an agate mortar and pestle, followedby dry sieving to the size fractions utilized in the respectivelaboratory dissolution studies: 125–45 lm for the ‘‘Icelan-dic’’ suite samples and 106–53 lm for the Kozushimasamples. Fine particles were removed using repetitiveultra-sonication in high purity acetone until the superna-tant was clear after 1 min stand time. Specific surface areasfor the Icelandic and Kozushima samples were measuredusing the eight-point BET method utilizing a MicromeriticsASAP 2000 with Kr gas.
The presence of weathering products on the oldest of theJapanese samples (52 ka), necessitated additional treatmentto remove reaction products and uncover the parent surfaceand thereby separate the effects of binding of probe mole-cules (TFS) to rhyolite surfaces or weathering products.Ultra-sonication was used to remove weathering products(inferred to be halloysite and allophane by Yokoyamaand Banfield (2002)). For comparison with weatheringproducts on the rhyolites, a sample of high defect kaoliniteKGa-2 (Washington County, GA) from the Source ClaysRepository was also studied. This sample was obtained

5952 N.M. Washton et al. / Geochimica et Cosmochimica Acta 72 (2008) 5949–5961
from J. Chorover (University of Arizona) after preparationas outlined in Chorover et al. (2003).
2.3. Surface modification
The air-dried volcanic glass powders were placed in avacuum oven at 160 �C and approximately 25 torr for12 h prior to treatment with (3,3,3-trifluoropropyl)dimeth-ylchlorosilane (Gelest, Inc.). The surfaces of the powderswere reacted with probe molecules by adding 1 ml of TFSto 300 mg of sample in an oven-dried 100 ml Schlenk flaskcontaining 25 ml anhydrous toluene, followed by evacua-tion and subsequent Ar back-filling. Mixtures were gentlystirred at room temperature for 72 h. To remove unreactedTFS, the powders were rinsed with approximately 250 mlreagent grade toluene (VWR, Inc.) followed by drying ina glass vial for 1 h at 120 �C under 25 torr of pressure.The boiling point of pure TFS under standard conditionsis 118 �C, and therefore excess TFS should be completelyremoved from the surface under these conditions (Washton,2007).
2.4. Nuclear magnetic resonance
Nuclear magnetic resonance experiments were per-formed on a homebuilt, three-channel NMR spectrometerequipped with a 9.4 T Oxford magnet and utilizing a Tec-mag Libra pulse programming and data acquisition system.19F direct polarization data were acquired using a 4 mmCPMAS Chemagnetics double resonance probe with thehigh-frequency channel (nominally at 300 MHz) tuned tothe 19F resonance frequency (376.346 MHz) at 9.4 T. Cali-brated p/2 pulses of 3 ls, spinning frequencies of 13–14 kHz, and 5 s pulse delays were used in all experiments.The number of time-averaged acquisitions ranged from256 to 48,000 depending on signal strength. Time domainfree induction decays were apodized with exponential func-tions corresponding to 100 Hz of Lorentzian line broaden-ing prior to Fourier transformation. Quantitative hydroxylmeasurements were obtained by comparing the total inte-grated spectral areas of the 19F NMR signals from a knownquantity of sodium 3,3,3-trifluoroacetate to the 19F NMRsignals from measured quantities of the TFS-treatedpowders. After Fourier transformation to the frequencydomain, each NMR spectrum was analyzed using theline-fitting routine in the NUTS data analysis environment(Acorn NMR). User-guided fittings of the resonance lineswere accomplished, allowing variation of resonance shift,signal intensity, line width, and percent Gaussian character.Overall resonance areas, including any measurable MASsidebands, were tabulated and normalized by number ofscans and sample mass. The reproducibility of these mea-surements is reported elsewhere (Washton, 2007) and isfound to be better than 5% across a range of TFS densities.
27Al direct polarization NMR data were acquired at9.4 T using a 4 mm CPMAS Chemagnetics double reso-nance probe tuned to 104.227 MHz. Calibrated p/20 pulsesof 0.6 ls, spinning frequencies of 13–14 KHz, and a 1 spulse delay were used to acquire 256 or 512 time-averagedscans. Time domain free induction decays were apodized
with exponential functions corresponding to 100 Hz ofLorentzian line broadening prior to Fourier transforma-tion. After Fourier transformation, each NMR spectrumwas analyzed using the line-fitting routine in the NUTSdata analysis environment (Acorn NMR). User-guided fit-tings of the 27Al resonance lines corresponding to AlIV
and AlVI were accomplished, allowing variation of reso-nance shift, signal intensity, line width, and percent Gauss-ian character.
3. RESULTS
3.1. Observations from the ‘‘Icelandic’’ suite (including
Bishop Tuff)
One of the primary conclusions of Wolff-Boenisch et al.(2004) was that BET-normalized dissolution rates exhibitedless of a correlation to bulk glass SiO2 wt% than rates nor-malized to geometric surface area. For example, R2 valuesfor linear fits of 0.60 versus 0.75 were calculated after nor-malization of dissolution rates by BET and geometric sur-face area, respectively, for 18 natural glass samples ofvarious compositions. In fact, data taken from the Wolff-Boenisch study show that for the seven samples consideredhere, variation in the mass-normalized dissolution ratescannot be explained satisfactorily by either the BET or geo-metric surface area of pre-dissolution samples (Fig. 4). Sucha lack of correlation between dissolution rate and BET sur-face area has also been observed in other studies, such asthose investigating the dissolution behavior of feldspars(Holdren and Speyer, 1985; Holdren, 1987), quartz (Gau-tier et al., 2001), and biogenic silica (Van-Cappellen,1996). Although Wolff-Boenisch et al. (2004) attributed thisvariation in dissolution rate to variations in bulk chemicalcomposition, we have investigated the degree to which thesedifferences can be attributed to variations in surface sitespeciation as described below.
Based on the findings by Fry et al. (2003a,b) that amono-functional chlorosilane (TFS) was useful as a probefor oxide reactivity, TFS was chemisorbed to the surfaceof the six low-Fe Icelandic glasses, as well as the BishopTuff sample. Total surface reactivity with TFS was thenmeasured using NMR. The number of TFS molecules,which are interpreted to equal the number of TFS-reactivehydroxyl groups on the oxide surface, are listed in Table 1on a per gram basis, as well as on a per nm2 basis for com-parison (using BET surface area for total surface area). Forall of the pre-dissolution samples, the measured TFS-reac-tive hydroxyl densities are significantly below the valuesof 2–6 OH/nm2 inferred to be present on hydroxylated sil-icate and aluminosilicate surfaces (Iler, 1979; Zhuravlev,1987; Davis and Kent, 1990). If a large number of TFS mol-ecules are chemisorbed onto the sample, steric limitationsimposed by this probe molecule could limit the maximumnumber of chemisorption sites measured by these methods;however, based on the calculated lateral extension of thetwo methyl groups attached to the Si of TFS (Fig. 3), thetotal area required for free rotation of the TFS moleculewhen attached to an oxide surface allows up to three TFSmolecules per nm2 (Van-Der-Voort et al., 1993). Since

Fig. 4. Mass-normalized rates of dissolution of ‘‘Icelandic’’ volcanic glasses cannot be predicted from geometric (a, R2 = 0.23) or BET (b,R2 = 0.11) specific surface area measured on pre-dissolution powders. Symbol labels are explained in Table 1, and error bars shown ongeometric surface areas are ±5%. Data are from Wolff-Boenisch et al. (2004).
Table 1BET surface area, hydroxyl concentrations, and dissolution rates of glass and clay samples
Sample Composition Location BETa,b
(m2/g)OH/gc
(�1017)OH/nm2 c Dissolution rated
(�10�12) (mol Si g�1 s�1)
BT Rhyolite Bishop Tuff 3.12a 1.81 0.16 9.42BTae Rhyolite Bishop Tuff 2.28b 9.42 0.41 9.42O62 Rhyolite Oraefajokull 0.43a 0.603 0.17 4.40O62ae Rhyolite Oraefajokull 0.254b 5.67 2.2 4.40H1 Rhyolite Hekla 0.56a 0.986 0.19 5.11H1ae Rhyolite Hekla 0.401b 7.18 1.8 5.11H3W Rhyolite Hekla 1.08a 3.48 0.68 10.5H3Wae Rhyolite Hekla 0.596b 14.0 2.4 10.5H3B Dacite Hekla 1.21a 4.68 0.72 23.1H3Bae Dacite Hekla 0.623b 20.5 3.3 23.1HZOf Dacite Hekla 0.62a 2.19 0.35 8.56SLNf Dacite Silk 2.65a 2.31 0.14 15.51.1 ka Rhyolite Tenjyo-san 0.24j 0.16b 0.274 0.17 NA1.8 ka Rhyolite Kobe-yama 0.30j 0.19b 0.370 0.19 NA26 ka Rhyolite Ohsawa-yama 2.57j 0.185b 2.80 1.5 NA52 ka Rhyolite Awanomikoto-yama 3.73j 1.36b,g 0.52k 18.5 0.5g 1.4k 3.5k,i NAClay and finesfrom 52 ka
Clay and ka52 rhyolite Awanomikoto-yama 34.8 36.0 0.10 NA
KGa-2 Kaolinite Washington County,GA
23.2h 4.18i 20.8 0.089 0.44i NA
a Values obtained by Wolff-Boenisch et al. (2004).b Values obtained in this study.c Hydroxyl concentrations measured via TFS treatment followed by quantitative NMR.d Rates of dissolution measured at pH 4.0 reported by Wolff-Boenisch et al. (2004).e The letter ‘a’ suffixed to the sample name denotes post-dissolution sample.f Post-dissolution samples are not included for HZO and SLN due to the presence of a 19F containing contaminant.g This value is prior to correcting for clay impurities.h Value taken from Chorover et al. (2003).i Values calculated using edge-site surface area of approximately 18% of the total surface area of the clay (Bickmore et al., 2002).j Value measured on a non-ground cylinder of rhyolite (Yokoyama and Banfield, 2002).
k Extrapolated value based on power law analysis.
A sensitive NMR proxy measuring reactive surface area for aluminosilicate dissolution 5953
our values are much lower than three adsorbed moleculesper nm2, the low TFS loadings cannot be accounted forsimply by steric limitations. Review of the presumedchemisorption mechanism (Corriu and Guerin, 1980,1982; Holmes, 1990) as shown in Fig. 2 does not help clarify
the reason for low TFS loadings compared to the expectednumber of chemisorption sites for oxide glasses. Since thereis no indication that these glass samples have a significantlylower number of total surface hydroxyl groups than a typ-ical glass that has 2 to 6 hydroxyl groups per nm2, chemical
5954 N.M. Washton et al. / Geochimica et Cosmochimica Acta 72 (2008) 5949–5961
specificity is inferred to play a role in the chemisorption ofTFS to the oxide surface. This specificity is discussed belowto illuminate acid-mediated dissolution mechanisms forthese systems.
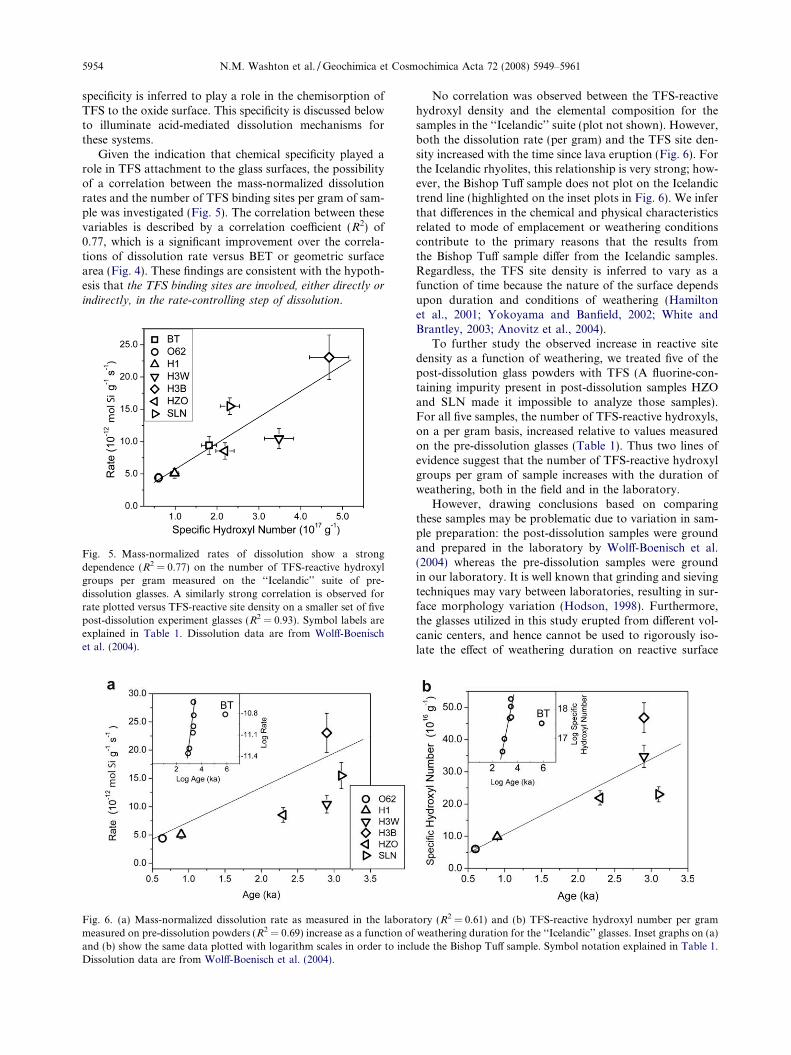
Given the indication that chemical specificity played arole in TFS attachment to the glass surfaces, the possibilityof a correlation between the mass-normalized dissolutionrates and the number of TFS binding sites per gram of sam-ple was investigated (Fig. 5). The correlation between thesevariables is described by a correlation coefficient (R2) of0.77, which is a significant improvement over the correla-tions of dissolution rate versus BET or geometric surfacearea (Fig. 4). These findings are consistent with the hypoth-esis that the TFS binding sites are involved, either directly or
indirectly, in the rate-controlling step of dissolution.
Fig. 5. Mass-normalized rates of dissolution show a strongdependence (R2 = 0.77) on the number of TFS-reactive hydroxylgroups per gram measured on the ‘‘Icelandic’’ suite of pre-dissolution glasses. A similarly strong correlation is observed forrate plotted versus TFS-reactive site density on a smaller set of fivepost-dissolution experiment glasses (R2 = 0.93). Symbol labels areexplained in Table 1. Dissolution data are from Wolff-Boenischet al. (2004).
Fig. 6. (a) Mass-normalized dissolution rate as measured in the laborameasured on pre-dissolution powders (R2 = 0.69) increase as a function ofand (b) show the same data plotted with logarithm scales in order to inclDissolution data are from Wolff-Boenisch et al. (2004).
No correlation was observed between the TFS-reactivehydroxyl density and the elemental composition for thesamples in the ‘‘Icelandic’’ suite (plot not shown). However,both the dissolution rate (per gram) and the TFS site den-sity increased with the time since lava eruption (Fig. 6). Forthe Icelandic rhyolites, this relationship is very strong; how-ever, the Bishop Tuff sample does not plot on the Icelandictrend line (highlighted on the inset plots in Fig. 6). We inferthat differences in the chemical and physical characteristicsrelated to mode of emplacement or weathering conditionscontribute to the primary reasons that the results fromthe Bishop Tuff sample differ from the Icelandic samples.Regardless, the TFS site density is inferred to vary as afunction of time because the nature of the surface dependsupon duration and conditions of weathering (Hamiltonet al., 2001; Yokoyama and Banfield, 2002; White andBrantley, 2003; Anovitz et al., 2004).
To further study the observed increase in reactive sitedensity as a function of weathering, we treated five of thepost-dissolution glass powders with TFS (A fluorine-con-taining impurity present in post-dissolution samples HZOand SLN made it impossible to analyze those samples).For all five samples, the number of TFS-reactive hydroxyls,on a per gram basis, increased relative to values measuredon the pre-dissolution glasses (Table 1). Thus two lines ofevidence suggest that the number of TFS-reactive hydroxylgroups per gram of sample increases with the duration ofweathering, both in the field and in the laboratory.
However, drawing conclusions based on comparingthese samples may be problematic due to variation in sam-ple preparation: the post-dissolution samples were groundand prepared in the laboratory by Wolff-Boenisch et al.(2004) whereas the pre-dissolution samples were groundin our laboratory. It is well known that grinding and sievingtechniques may vary between laboratories, resulting in sur-face morphology variation (Hodson, 1998). Furthermore,the glasses utilized in this study erupted from different vol-canic centers, and hence cannot be used to rigorously iso-late the effect of weathering duration on reactive surface
tory (R2 = 0.61) and (b) TFS-reactive hydroxyl number per gramweathering duration for the ‘‘Icelandic’’ glasses. Inset graphs on (a)
ude the Bishop Tuff sample. Symbol notation explained in Table 1.
250 200 150 100 50 0 -50 -100 -150Frequency (ppm from 0.1 M Al+3(aq))
a
b
c
d
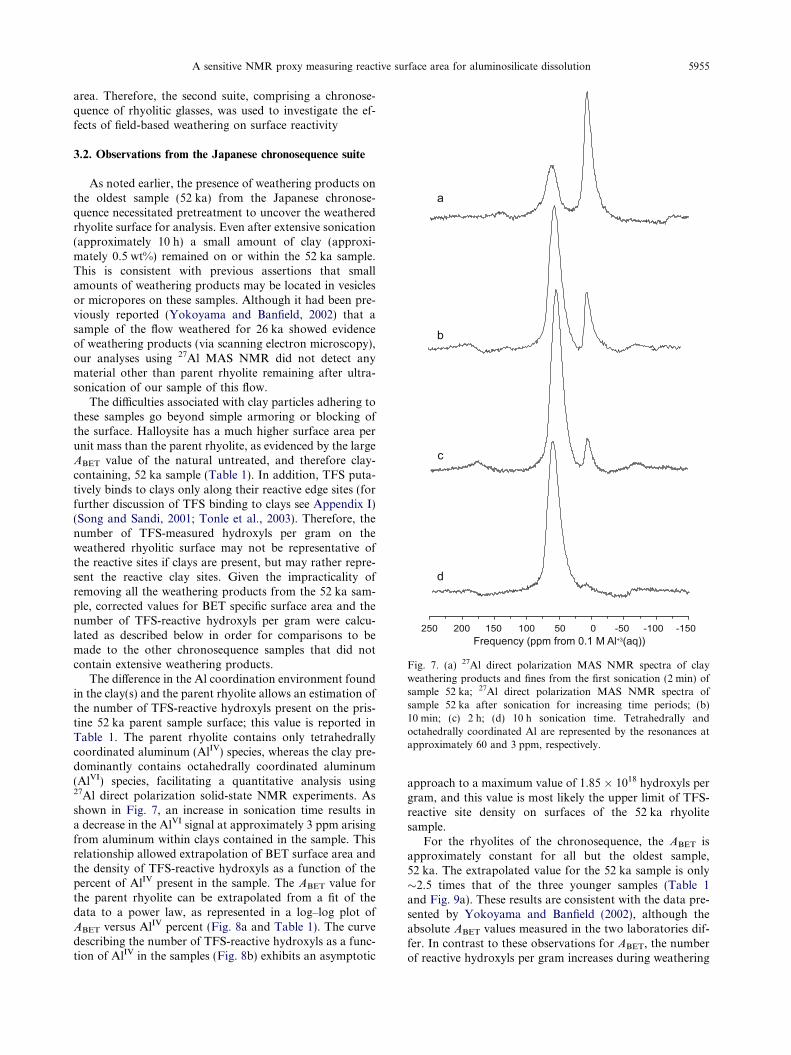
Fig. 7. (a) 27Al direct polarization MAS NMR spectra of clayweathering products and fines from the first sonication (2 min) ofsample 52 ka; 27Al direct polarization MAS NMR spectra ofsample 52 ka after sonication for increasing time periods; (b)10 min; (c) 2 h; (d) 10 h sonication time. Tetrahedrally andoctahedrally coordinated Al are represented by the resonances atapproximately 60 and 3 ppm, respectively.
A sensitive NMR proxy measuring reactive surface area for aluminosilicate dissolution 5955
area. Therefore, the second suite, comprising a chronose-quence of rhyolitic glasses, was used to investigate the ef-fects of field-based weathering on surface reactivity
3.2. Observations from the Japanese chronosequence suite
As noted earlier, the presence of weathering products onthe oldest sample (52 ka) from the Japanese chronose-quence necessitated pretreatment to uncover the weatheredrhyolite surface for analysis. Even after extensive sonication(approximately 10 h) a small amount of clay (approxi-mately 0.5 wt%) remained on or within the 52 ka sample.This is consistent with previous assertions that smallamounts of weathering products may be located in vesiclesor micropores on these samples. Although it had been pre-viously reported (Yokoyama and Banfield, 2002) that asample of the flow weathered for 26 ka showed evidenceof weathering products (via scanning electron microscopy),our analyses using 27Al MAS NMR did not detect anymaterial other than parent rhyolite remaining after ultra-sonication of our sample of this flow.
The difficulties associated with clay particles adhering tothese samples go beyond simple armoring or blocking ofthe surface. Halloysite has a much higher surface area perunit mass than the parent rhyolite, as evidenced by the largeABET value of the natural untreated, and therefore clay-containing, 52 ka sample (Table 1). In addition, TFS puta-tively binds to clays only along their reactive edge sites (forfurther discussion of TFS binding to clays see Appendix I)(Song and Sandi, 2001; Tonle et al., 2003). Therefore, thenumber of TFS-measured hydroxyls per gram on theweathered rhyolitic surface may not be representative ofthe reactive sites if clays are present, but may rather repre-sent the reactive clay sites. Given the impracticality ofremoving all the weathering products from the 52 ka sam-ple, corrected values for BET specific surface area and thenumber of TFS-reactive hydroxyls per gram were calcu-lated as described below in order for comparisons to bemade to the other chronosequence samples that did notcontain extensive weathering products.
The difference in the Al coordination environment foundin the clay(s) and the parent rhyolite allows an estimation ofthe number of TFS-reactive hydroxyls present on the pris-tine 52 ka parent sample surface; this value is reported inTable 1. The parent rhyolite contains only tetrahedrallycoordinated aluminum (AlIV) species, whereas the clay pre-dominantly contains octahedrally coordinated aluminum(AlVI) species, facilitating a quantitative analysis using27Al direct polarization solid-state NMR experiments. Asshown in Fig. 7, an increase in sonication time results ina decrease in the AlVI signal at approximately 3 ppm arisingfrom aluminum within clays contained in the sample. Thisrelationship allowed extrapolation of BET surface area andthe density of TFS-reactive hydroxyls as a function of thepercent of AlIV present in the sample. The ABET value forthe parent rhyolite can be extrapolated from a fit of thedata to a power law, as represented in a log–log plot ofABET versus AlIV percent (Fig. 8a and Table 1). The curvedescribing the number of TFS-reactive hydroxyls as a func-tion of AlIV in the samples (Fig. 8b) exhibits an asymptotic
approach to a maximum value of 1.85 � 1018 hydroxyls pergram, and this value is most likely the upper limit of TFS-reactive site density on surfaces of the 52 ka rhyolitesample.
For the rhyolites of the chronosequence, the ABET isapproximately constant for all but the oldest sample,52 ka. The extrapolated value for the 52 ka sample is only�2.5 times that of the three younger samples (Table 1and Fig. 9a). These results are consistent with the data pre-sented by Yokoyama and Banfield (2002), although theabsolute ABET values measured in the two laboratories dif-fer. In contrast to these observations for ABET, the numberof reactive hydroxyls per gram increases during weathering
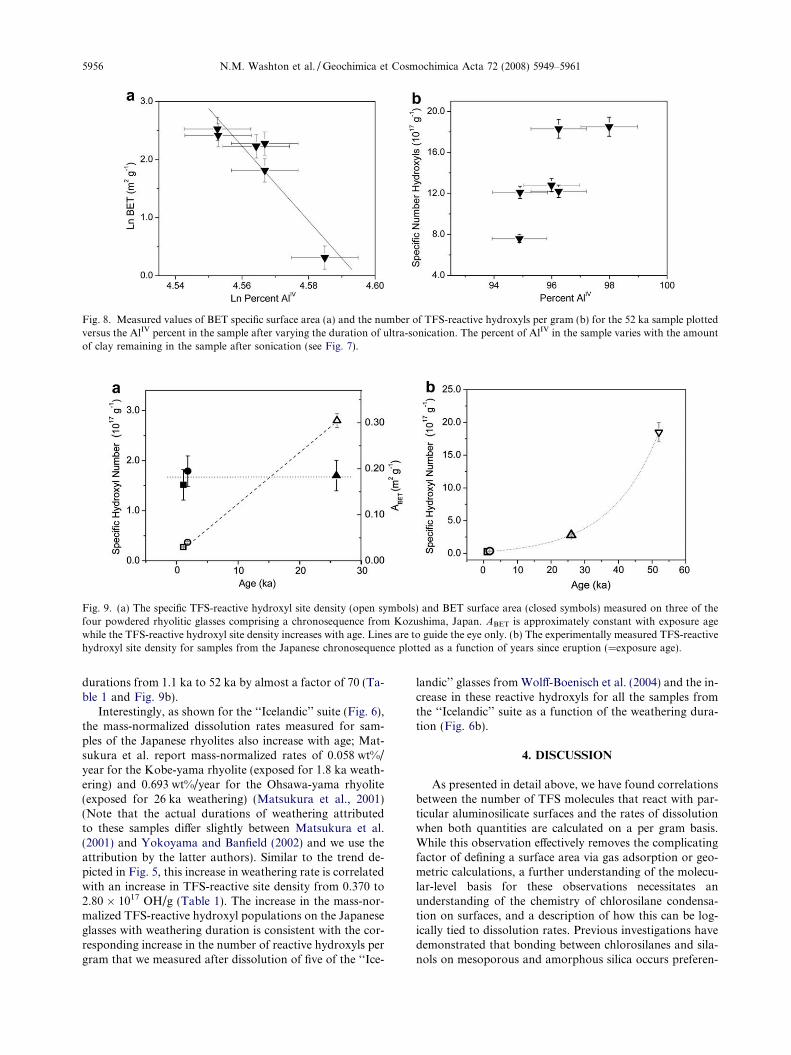
Fig. 8. Measured values of BET specific surface area (a) and the number of TFS-reactive hydroxyls per gram (b) for the 52 ka sample plottedversus the AlIV percent in the sample after varying the duration of ultra-sonication. The percent of AlIV in the sample varies with the amountof clay remaining in the sample after sonication (see Fig. 7).
Fig. 9. (a) The specific TFS-reactive hydroxyl site density (open symbols) and BET surface area (closed symbols) measured on three of thefour powdered rhyolitic glasses comprising a chronosequence from Kozushima, Japan. ABET is approximately constant with exposure agewhile the TFS-reactive hydroxyl site density increases with age. Lines are to guide the eye only. (b) The experimentally measured TFS-reactivehydroxyl site density for samples from the Japanese chronosequence plotted as a function of years since eruption (=exposure age).
5956 N.M. Washton et al. / Geochimica et Cosmochimica Acta 72 (2008) 5949–5961
durations from 1.1 ka to 52 ka by almost a factor of 70 (Ta-ble 1 and Fig. 9b).
Interestingly, as shown for the ‘‘Icelandic’’ suite (Fig. 6),the mass-normalized dissolution rates measured for sam-ples of the Japanese rhyolites also increase with age; Mat-sukura et al. report mass-normalized rates of 0.058 wt%/year for the Kobe-yama rhyolite (exposed for 1.8 ka weath-ering) and 0.693 wt%/year for the Ohsawa-yama rhyolite(exposed for 26 ka weathering) (Matsukura et al., 2001)(Note that the actual durations of weathering attributedto these samples differ slightly between Matsukura et al.(2001) and Yokoyama and Banfield (2002) and we use theattribution by the latter authors). Similar to the trend de-picted in Fig. 5, this increase in weathering rate is correlatedwith an increase in TFS-reactive site density from 0.370 to2.80 � 1017 OH/g (Table 1). The increase in the mass-nor-malized TFS-reactive hydroxyl populations on the Japaneseglasses with weathering duration is consistent with the cor-responding increase in the number of reactive hydroxyls pergram that we measured after dissolution of five of the ‘‘Ice-
landic’’ glasses from Wolff-Boenisch et al. (2004) and the in-crease in these reactive hydroxyls for all the samples fromthe ‘‘Icelandic’’ suite as a function of the weathering dura-tion (Fig. 6b).
4. DISCUSSION
As presented in detail above, we have found correlationsbetween the number of TFS molecules that react with par-ticular aluminosilicate surfaces and the rates of dissolutionwhen both quantities are calculated on a per gram basis.While this observation effectively removes the complicatingfactor of defining a surface area via gas adsorption or geo-metric calculations, a further understanding of the molecu-lar-level basis for these observations necessitates anunderstanding of the chemistry of chlorosilane condensa-tion on surfaces, and a description of how this can be log-ically tied to dissolution rates. Previous investigations havedemonstrated that bonding between chlorosilanes and sila-nols on mesoporous and amorphous silica occurs preferen-

A sensitive NMR proxy measuring reactive surface area for aluminosilicate dissolution 5957
tially at non-hydrogen-bonded lone Q3 silanols (Lochmul-ler and Kersey, 1988; Van-Der-Voort et al., 1990; Kawaiand Tsutsumi, 1998, 1999; Zhao and Lu, 1998). In fact,Zhao and Lu (1998) hypothesized that intra-surface hydro-gen bonding between Qn groups functions as a barrier toreaction with mono-functional organosilanes. Supportingthis hypothesis, Kawai and Tsutsumi (1998, 1999) showedthat areas of extensive hydrogen-bonding on aluminum-containing zeolites created ‘‘hydroxy nests’’ that were unre-active to condensation with chlorosilanes. Based on all ofthese observations, it is most likely that TFS attaches onthe volcanic glasses to accessible non-hydrogen-bonded sil-anol or aluminol species that lie outside of the orderedhydrogen-bonding surface network that forms where hy-droxyl groups are neighboring (Du et al., 1994; Schlegelet al., 2002; Yang et al., 2004, 2005).
Combining this inference for the nature of the TFS-reac-tive sites with the observations described in this paper leadsto the conclusion that, as the volcanic glasses weather, thesurface density of non-hydrogen-bonded Q3 (TFS-reactive)sites increases. Three lines of evidence from our paper sup-port this: (i) the data presented in Fig. 6b; (ii) the observa-tion that TFS-reactive site density is higher for post-dissolution as compared to pre-dissolution surfaces of the‘‘Icelandic’’ glasses (Table 1); and (iii) the observation thatthe TFS-reactive site density of the Japanese rhyolites in-crease with duration of weathering of the surfaces (Table1 and Fig. 9b). In addition, the dissolution rates of boththe ‘‘Icelandic’’ and Japanese samples also increased withTFS-reactive site density. These lines of evidence could beexplained either because non-hydrogen-bonded Q3 sitesare rate-controlling for dissolution (i.e. causation) or be-cause the densities of these sites covary with the densitiesof the rate-controlling site for dissolution (i.e. correlation).
As described here and in the next paragraph, correlation
is a better explanation for our observations than causation.First, in contrast to the increase in dissolution rate observedfor volcanic glasses with increasing exposure age over thou-sands of years, Yokoyama and Banfield (2002) observedthat the dissolution rates of powdered rhyolites from theJapanese chronosequence decreased with duration of disso-lution time in the laboratory. Similar decreases in dissolu-tion rates observed in the laboratory with experimentalduration have been noted by many researchers for many[alumino]silicate powders (Brantley, 2003; White andBrantley, 2003) and were noted for the laboratory-ground‘‘Icelandic’’ samples (Wolff-Boenisch et al., 2004). In con-trast to the decreasing dissolution rate during laboratorytimeframes observed for these latter samples, the surfacedensity of TFS-reactive sites increased between pre- andpost-dissolution (Table 1). Since dissolution rate decreasedbut TFS-reactive site density increased in the laboratory,the TFS-reactive sites are not the rate-controlling sites overthese timescales.
Furthermore, much evidence has been summarized inthe literature to suggest that the rate-controlling site for dis-solution is Qn where n 6 2 rather than Q3 (Oelkers, 2001;Teng et al., 2001; Brantley, 2003, 2008). Oelkers (2001) ar-gues that the rate-determining species are those that are‘‘partially freed’’ from the surface through their adjacency
to previously exchanged metal sites, and he uses this modelto present a general dissolution model for multioxide min-erals and glasses. From a theoretical basis, analysis of dis-solution of Si clusters (Q3) has documented that thecalculated activation energy (120 kJ/mol) for the proton-promoted hydrolysis of Q3 species is larger than the exper-imentally determined Ea (67–88 kJ/mol) for aluminosilicatedissolution (Criscenti et al., 2006). The most likely site onthe silica surface to control dissolution based on that workis the Qn site where n 6 2. Pelmenschikov and coworkers(Pelmenschikov et al., 1997) also calculated activation ener-gies for different Qn species and, based on comparison withexperimental data, concluded that Qn sites (n 6 2) are mostlikely rate-controlling.
A working hypothesis therefore emerges that is consis-tent with modeling, laboratory, and field observations: overshort timescales, the silicon dissolution rates of groundsamples are controlled by fast-dissolving sites (which aremost logically Q1 sites that are attached to the surface bya single bridging oxygen), but over long timescales, dissolu-tion is controlled by slower-dissolving sites, the surface den-sity of which is proportional to the number of TFS-reactiveQ3 sites. The simplest explanation for this could be that therate-determining (‘‘partially freed’’ or, most likely, Q2) sitescovary with the density of Q3 sites. For such a case, mea-surement of the surface site density of non-hydrogen-bonded Q3 sites for a suite of samples using TFS andNMR allows prediction of the relative dissolution rates be-cause the densities of TFS-reactive Q3 sites correlate di-rectly with the reactive surface area.
This line of argument also can explain why TFS-reactivesite density apparently increases on volcanic glass surfaceswith increasing time of exposure to water regardless ofwhether dissolution is decreasing over short timescales(e.g. in the laboratory as observed for the ‘‘Icelandic’’ sam-ples) or increasing over long timescales (e.g. in the field asobserved for the Japanese samples). This apparent contra-diction can be explained if the decrease in dissolution rateobserved for laboratory-ground samples is due largely toa decrease in Q1 sites while the increase in weathering rateof the samples in the field is explained largely by an increasein Qn sites (with n > 1).
Such evolution in surface site density has been recentlysimulated for dissolution of model crystals with 4-coordi-nate atoms (Bandstra and Brantley, 2008). These simula-tions show that the relative distribution of Q1, Q2, and Q3
sites evolves and coavaries with dissolution, eventuallyreaching a steady state distribution that is a function ofthe chemical affinity of the dissolving solution and the rel-ative reaction rates of the Q1, Q2, and Q3 sites. In them-selves, the model simulations cannot predict the time tosteady state during dissolution.
The observed increase in TFS-reactive site densities andchanges in dissolution rates even after long durations ofweathering (Figs. 6 and 9) are consistent with lack of steadystate for dissolving volcanic glass surfaces over long timeperiods. Such long transients in attaining steady state mayindicate that laboratory experiments have not attained stea-dy state for such glasses or that a steady state is not possiblefor dissolution of these glasses. Long transients could be re-

5958 N.M. Washton et al. / Geochimica et Cosmochimica Acta 72 (2008) 5949–5961
lated to the lack of periodicity in the atomic structure of theglasses, or to leaching of Al, micro-cracking, spalling, for-mation of hydrated surface layers, or other such processesthat were not included in the model simulations of dissolu-tion (Bandstra and Brantley, 2008). Multiple lines of evi-dence have documented such processes occurring on boththe ‘‘Icelandic’’ and Japanese samples (Oguchi et al.,1999; Matsukura et al., 2001; Wolff-Boenisch et al., 2004;Yokoyama and Matsukura, 2006). For the varying compo-sitions of the ‘‘Icelandic’’ suite, chemical compositionwould presumably also exert a control on the steady statesurface site distribution and the time to steady state: it iswell known that Al atoms are preferentially leached fromglass surfaces at low pH. Such surface leaching could affectthe surface site density of Qn (or more specifically, Qn[mAl])sites.
A broader picture then also emerges that is consistentwith the correlative observations and hypotheses presentedhere. We do know that the species being counted by TFSchemisorption are those Q3 groups that are the most ex-posed on a glass surface, in that they are not part of ahydrogen-bonding network, and therefore represent theareas on the surface where exchange reactions can occurthat begin the process of acid-mediated dissolution. Theseprocesses include the exchange of protons for metal speciessuch as aluminum, and therefore lead directly into the dis-solution process (Oelkers, 2001). While attack of the oxy-gens that are bridging from a non-hydrogen-bonded Si-OH, and the subsequent breaking of the first Si–O–Si orSi–O–Al bond may not be the rate determining step for dis-solution, this process must occur for the ultimate removalof silicate species from the surface. Therefore, scaling ofreaction rates by this proxy for reactive surface area shouldindeed lead to the behavior demonstrated in Fig. 5: the dis-solution rate of these glasses, on a per gram basis, demon-strates a linear dependence on the number of TFS-reactivehydroxyls present on the surface. These hydroxyls are notpart of an extensive hydrogen-bonding network, and there-fore represent an area (or, a reactive surface area) of acces-sible sites for surface reactions.
In summary, we suggest that the TFS-reactive site den-sity of surfaces of volcanic silicates may be a good predictorfor relative dissolution rates because the surface site densi-ties of TFS-reactive Q3 sites varies directly with the rate-controlling precursor complexes on these glass surfaces.Many questions and opportunities for future research re-main for further exploration. We have given no explanationas to why the TFS site densities that characterize the steadystate dissolving surfaces vary significantly on samples suchas H3B and H3W although they were sampled from lavaflows of similar age. Compositional differences in theglasses or the weathering solutions may explain why thesurfaces are dominated by differing concentrations of lonenon-hydrogen-bonded silanol sites, and to address these is-sues we have begun studies of surface chemistry and TFSreactivity on model aluminosilicate gel systems (Washton,2007). Better understanding of controls on the Q-speciationand the nature of hydrogen-bonding on glass and mineralsurfaces will clarify both reaction mechanisms and how topredict mineral weathering.
5. CONCLUSIONS
The laboratory-measured dissolution rates of a suite ofvolcanic glasses have been shown to correlate with the sur-face site density of hydroxyl groups as measured by TFSchemisorption followed by quantitative 19F NMR. Thesedata are consistent with the conclusion that variations inthe rate of dissolution of naturally weathered samples is re-lated to the density of non-hydrogen-bonded Q3 surfacesites that increase in concentration during dissolution.Our data show that the surface density of non-hydrogen-bonded Q3 sites (i.e. the TFS-reactive sites) increase as afunction of both laboratory- and field-induced weatheringas the more reactive Q1 site is removed over time. A betterunderstanding of the steady state surface densities of TFS-reactive sites (lone non-hydrogen-bonded hydroxls) as wellas other surface sites will lead to predictive models for dis-solution of glasses.
The novel analytic approach of TFS chemisorption fol-lowed by analysis with high-resolution 19F NMR provides afunctional and easily measurable proxy for surface sites onlow-iron natural and synthetic aluminosilicate glasses. Weexpect that other silicate and aluminosilicate systems mayshow the same dissolution rate correlation with TFS-reac-tive surface species, revealing insights into the ultimate con-trol of the dissolution process with respect to reactivehydroxyl density. We propose that our method will alsobe useful for predicting dissolution rates of such mineralswhere sufficient surface area is available for quantitative19F measurements. Alternatively, performing the 19FNMR measurements on ultra high-field instruments, suchas the 21.14 T system currently available at the PacificNorthwest National Lab High-Field NMR User Facility,should increase sensitivity and allow the investigation ofmaterials with very low surface areas. Of additional impor-tance is our observation that TFS chemisorption, coupledwith quantitative 27Al MAS NMR spectroscopy, can beused to determine the extent of contamination of primaryminerals with secondary clay minerals for weathered sam-ples. Further research into the potential of TFS chemisorp-tion for elucidating weathering reactions should bepursued.
ACKNOWLEDGMENTS
The authors wish to thank D. Wolff-Boenisch, T. Yokoyama,and J. Chorover for kindly supplying samples for analysis. Wefurther thank J. Kubicki and Associate Editor E. Odkers for valu-able scientific discussions as well as a suite of anonymous review-ers for their useful advice. This material is based upon worksupported by the National Science Foundation under GrantNo. CHE-0431328 for the Center for Environmental KineticsAnalysis.
APPENDIX I. CLAY REACTIVITY TO
SILANIZATION
A brief consideration of the use of NMR analysis ofreactive surface area for clays is presented. This analysiswas necessitated by the presence of extensive weathering

A sensitive NMR proxy measuring reactive surface area for aluminosilicate dissolution 5959
products on the 52 ka Japanese sample. Clay reactivity isdominated by edge sites, where dissolution and precipita-tion are known to occur in many clays (Casey and Bunker,1990; Fitzgerald et al., 1997; Nagy et al., 1999; Bickmoreet al., 2001) Therefore, the use of total surface area mea-surements as proxies for reactive surface area on clays isless than optimal given that edge-specific surface area istypically 10–35% of the total surface area (Bickmoreet al., 2002; Metz et al., 2005). This is consistent with ourfindings of increased surface area and decreased reactivehydroxyl density as a function of weathering product con-tent on the 52 ka sample from the Japanese chronosequence(Table 1 and Fig. 8b). The basal planes of many clays (e.g.kaolinite, smectite, montmorillonite, sepiolite) are knownto be unreactive to silane condensation under the experi-mental conditions utilized here, thus explaining the de-crease in the number of measured TFS-reactive hydroxylsper gram for the 52 ka sample when weathering productsare present (Song and Sandi, 2001; Tonle et al., 2003) andpresumably armoring the surface. Therefore, to furtherquantify the contribution made by weathering products tothe measurements made on the 52 ka chronosequence sam-ple and to assess the viability of our 19F NMR techniquefor measuring clay reactivity, the number of reactivehydroxyls were also measured on TFS-modified kaolinite(Table 1; KGa-2, Washington County, GA).
To estimate the number of reactive hydroxyls per nm2
present on kaolinite, a scaled value for the BET surface areawas used (Table 1) per the observations by Bickmore et al.(2002) that kaolinite edge-site area is approximately 18% ofthe total surface area. Normalizing the number of hydrox-yls per nm2 of edge-site surface area gives a value of 0.44OH/nm2, which is similar to the values obtained for sam-ples within both volcanic glass suites. In addition, the Siedge species are predominantly isolated Q3 groups (Fitzger-ald et al., 1997). These preliminary data are consistent withthe interpretation that TFS is binding to clay on the lone Q3
silicate groups, similar to our inferences for the aluminosil-icate glasses. However, a significant difference between theglasses in this study and kaolinite is the presence of octahe-drally coordinated Al in the layer structure. Although TFSdoes bind to sites containing octahedrally coordinated Al(Washton, 2007), it is unclear if the chemisorption mecha-nism is the same for AlVI-OH as for Si-OH binding. Furtherstudies are underway to determine the exact chemical spe-cies involved in TFS binding to clay edge-sites, and theinvolvement of these species in dissolution events.
REFERENCES
Anbeek C. (1992) Surface roughness of minerals and implicationsfor dissolution studies. Geochim. Cosmochim. Acta 56, 1461–
1469.
Anovitz L. M., Elam J. M., Riciputi L. R. and Cole D. R. (2004)Isothermal time-series determination of the rate of diffusion ofwater in pachuca obsidian. Archaeometry 46, 301–326.
Bandstra J. Z. and Brantley S. L. (2008) Mineral surface evolutioninvestigated with a binary random Markov field model.Geochim. Cosmochim. Acta 72, 2587–2600.
Bickmore B. R., Bosbach D., Hochella M. F., Charlet L. and RufeE. (2001) In situ atomic force microscopy study of hectorite and
nontronite dissolution: implications for phyllosilicate edgesurface structures and dissolution mechanisms. Am. Mineral.
86, 411–423.
Bickmore B. R., Nagy K. L., Sandlin P. E. and Crater T. S. (2002)Quantifying surface areas of clays by atomic force microscopy.Am. Mineral. 87, 780–783.
Blum A. E. and Lasaga A. C. (1988) Role of surface speciation inthe low-temperature dissolution of minerals. Nature 331, 431–
433.
Brantley S., White A. F. and Hodson M. (1999) Surface area ofprimary silicate minerals. In Kongsberg Seminar, 11th (eds. P.Meakin and B. Jamtveit). Kluwer Academic Publishing, Kon-
gsberg, Norway.
Brantley S. L. (2003) Reaction kinetics of primary rock-formingminerals under ambient conditions. In Treatise on Geochemistry
(eds. J. I. Drever, K. K. Turekian and H. D. Holland).Pergamon Press, Oxford.
Brantley S. L. (2008) Kinetics of Mineral Dissolution. Springer–Kluwer, New York.
Brantley S. L. and Mellott N. P. (2000) Surface area and porosityof primary silicate minerals. Am. Mineral. 85, 1767–1783.
Brantley S. L. and Stillings L. L. (1996) Feldspar dissolution at25 �C and low pH. Am. J. Sci. 296, 101–127.
Casey W. H., Banfield J. F., Westrich H. R. and Mclaughlin L.(1993) What do dissolution experiments tell us about naturalweathering. Chem. Geol. 105, 1–15.
Casey W. H. and Bunker B. (1990) Leaching of mineral and glasssurfaces during dissolution. In Mineral-Water Interface Geo-
chemistry (eds. M. F. Hochella and A. F. White). The
Mineralogical Society of America, Washington, DC.
Chorover J., Choi S., Amistadi M. K., Karthikeyan K. G., CrossonG. and Mueller K. T. (2003) Linking cesium and strontiumuptake to kaolinite weathering in simulated tank waste leach-ate. Environ. Sci. Technol. 37, 2200–2208.
Corriu R. J. P. and Guerin C. (1980) Nucleophilic displacement atsilicon stereochemistry and mechanistic implications. J. Orga-
nomet. Chem. 198, 231–320.
Corriu R. J. P. and Guerin C. (1982) Nucleophilic displacement atsilicon—recent developments and mechanistic implications.Adv. Organomet. Chem. 20, 265–312.
Criscenti L. J., Kubicki J. D. and Brantley S. L. (2006) Silicate glassand mineral dissolution: calculated reaction paths and activa-tion energies for hydrolysis of a Q(3) Si by H3O+ using ab initiomethods. J. Phys. Chem. A110, 198–206.
Davis J. A. and Kent D. B. (1990) Surface complexation modelingin aqueous geochemistry. In Mineral-Water Interface Geochem-
istry (eds. F. Michael, J. Hochella and A. F. White). Miner-
alogical Society of America, Washington, DC.
Davydov V. Y., Kiselev A. V. and Zhuravlev L. T. (1964) Study ofthe surface and bulk hydroxyl groups of silica by infra-redspectra and D2O exchange. Trans. Faraday Soc. 60, 2254–2264.
Du Q., Freysz E. and Shen Y. R. (1994) Vibrational spectra ofwater molecules at quartz/water interfaces. Phys. Rev. Lett. 72,
238–241.
Fitzgerald J. J., Hamza A. I., Bronnimann C. E. and Dec S. F.(1997) Studies of the solid/solution ‘‘ineterfacial’’ dealumina-tion of kaolinite in HCl(aq) using solid-state 1H CRAMPS andSP/MAS 29Si NMR spectroscopy. J. Am. Chem. Soc. 119,
7105–7113.
Fry R., Pantano C. G. and Mueller K. T. (2003a) Effect of boron-oxide on surface hydroxyl coverage of aluminoborosilicate glassfibers: a 19F solid-state NMR study. Phys. Chem. Glasses 44,
64–68.
Fry R. A., Tsomaia N., Pantano C. G. and Mueller K. T. (2003b)19F MAS NMR quantification of accessible hydroxyl sites onfiberglass surfaces. J. Am. Chem. Soc. 125, 2378–2379.

5960 N.M. Washton et al. / Geochimica et Cosmochimica Acta 72 (2008) 5949–5961
Gautier J.-M., Oelkers E. H. and Schott J. (2001) Are quartzdissolution rates proportional to B.E.T. surface area? Geochim.
Cosmochim. Acta 65, 1059–1070.
Hamilton J. P., Brantley S. L., Pantano C. G., Criscenti L. J. andKubicki J. D. (2001) Dissolution of nepheline, jadeite and albiteglasses: toward better models for aluminosilicate dissolution.Geochim. Cosmochim. Acta 65, 3683–3702.
Helgeson H. C., Murphy W. M. and Aagaard P. (1984) Thermo-dynamic and kinetic constraints on reaction rates amongminerals and aqueous solutions: II. Rate constants, effectivesurface area, and the hydrolysis of feldspar. Geochim. Cosmo-
chim. Acta 48, 2405–2432.
Hering J. G. and Stumm W. (1990) Oxidative and reductivedissolution. In Mineral-Water Interface Geochemistry (eds. F.Michael, J. Hochella and A. F. White). Mineralogical Society of
America, Washington, DC.
Hodson M. (1998) Micropore surface area variation with grain sizein unweathered alkali feldspars: implications for surfaceroughness and dissolution studies. Geochim. Cosmochim. Acta
62, 3429–3435.
Holdren G. R. (1987) Reaction rate–surface area relationshipsduring the early stages of weathering: II. Data on eightadditional feldspars. Geochim. Cosmochim. Acta 51, 2311–
2318.
Holdren G. R. and Speyer P. M. (1985) Reaction rate–surface arearelationships during the early stages of weathering: I. Initialobservation. Geochim. Cosmochim. Acta 49, 675–681.
Holmes R. R. (1990) The stereochemistry of nucleophilic substi-tution at tetracoordinated silicon. Chem. Rev. 90, 17–30.
Iler R. K. (1979) The Chemistry of Silica: Solubility, Polymeriza-
tion, Colloid and Surface Properties, and Biochemistry of Silica.Wiley-Interscience, New York.
James R. O. and Parks G. A. (1982) Characterization of aqueouscolloids by their electrical double layer and intrinsic surfacechemical properties. Surf. Colloid Sci. 12, 119–216.
Kawai T. and Tsutsumi K. (1998) Reactivity of silanol groups onzeolite surfaces. Colloid Polym. Sci. 276, 992–998.
Kawai T. and Tsutsumi K. (1999) A study on the surface silanolgroups developed by hydrothermal and acid treatment offaujasite type zeolites. J. Colloid Interface Sci. 212, 310–316.
Lochmuller C. H. and Kersey M. T. (1988) Effect of thermalpretreatment on the surface reactivity of amorphous silica.Langmuir 4, 572–578.
Matsukura Y., Hirose T. and Oguchi C. T. (2001) Rates ofchemical weathering of porous rhyolites: 5-year measurementsusing the weight-loss method. Catena 43, 341–347.
Metz V., Raanan H., Pieper H., Bosbach D. and Ganor J. (2005)Towards the establishment of a reliable proxy for the reactivesurface area of smectite. Geochim. Cosmochim. Acta 69, 2581–
2591.
Mueller R., Kammler H. K., Wegner K. and Pratsinis S. E. (2003)OH surface density of SiO2 and TiO2 by thermogravimetricanalysis. Langmuir 19, 160–165.
Nagy K. L., Cygan R. T., Hanchar J. M. and Sturchio N. C. (1999)Gibbsite growth kinetics on gibbsite, kaolinite, and muscovitesubstrates: atomic force microscopy evidence for epitaxy and anassessment of reactive surface area. Geochim. Cosmochim. Acta
63, 2337–2351.
Nawrocki J. (1997) The silanol group and its role in liquidchromatography. J. Chromatogr. 779, 29–71.
Nugent M. A., Brantley S. L., Pantano C. G. and Maurice P. A.(1998) The influence of natural mineral coatings on feldsparweathering. Nature 395, 588–591.
Oelkers E. H. (2001) General kinetic description of multioxidesilicate mineral and glass dissolution. Geochim. Cosmochim.
Acta 65, 3703–3719.
Oguchi C. T., Hatta T. and Matsukura Y. (1999) Weathering ratesover 40,000 years based on changes in rock properties of porousrhyolite. Phys. Chem. Earth 24, 861–870.
Pelmenschikov A. G., Morosi G. and Gamba A. (1997) Adsorptionof water and methanol on silica hydroxyls: ab initio energy andfrequency calculations. J. Phys. Chem. A 101, 1178–1187.
Schlegel M. L., Nagy K. L., Fenter P. and Sturchio N. C. (2002)Structures of quartz (1010)- and (1011)-water interfacesdetermined by X-ray reflectivity and atomic force microscopyof natural growth surfaces. Geochim. Cosmochim. Acta 66,
3037–3054.
Schott J. (1990) Modeling of the Dissolution of Strained and
Unstrained Multiple Oxides: The Surface Speciation Approach.John Wiley & Sons, Inc., New York.
Song K. and Sandi G. (2001) Characterization of montmorillonitesurfaces after modification by organosilane. Clay Clay Miner.
49, 119–125.
Stillings L. L., Brantley S. L. and Machesky M. (1995) Protonadsorption at an adularia feldspar surface. Geochim. Cosmo-
chim. Acta 59, 1473–1482.
Teng H. H., Fenter P., Cheng L. and Sturchio N. C. (2001)Resolving orthoclase dissolution processes with atomic forcemicroscopy and X-ray reflectivity. Geochim. Cosmochim. Acta
65, 3459–3474.
Tonle I. K., Ngameni E., Njopwouo D., Carteret C. and WalcariusA. (2003) Functionalization of natural smectite-type clays bygrafting with organosilanes: physico-chemical characterizationand application to mercury(II) uptake. Phys. Chem. Chem.
Phys. 5, 4951–4961.
Van-Cappellen P. (1996) Reactive surface area control of thedissolution kinetics of biogenic silica in deep-sea sediments.Chem. Geol. 132, 125–130.
Van-Der-Voort P., Gillis-D’Hamers I. and Vansant E. F. (1990)Estimation of the distribution of surface hydroxyl groups onsilica gel, using chemical modification with trichlorosilane. J.
Chem. Soc., Faraday Trans. 86, 3751–3755.
Van-Der-Voort P. and Vansant E. F. (1996) Silylation of the silicasurface: a review. J. Liquid Chromatogr. Relat. Technol. 19,
2723–2752.
Van-Der-Voort P., Vercauteren S., Peeters K. and Vansant E. F.(1993) Some precautions when determining the silanol number,using chemical modification with methylchlorosilanes. J. Col-
loid Interface Sci. 157, 518–519.
Washton N. M. (2007) Determination of oxide surface reactivityvia solid-state nuclear magnetic resonance and ab initiomethods. Ph.D., Penn State University.
White A. F. (2008) Quantitative Approaches to Characterizing
Natural Chemical Weathering Rates. Springer, New York.White A. F. and Brantley S. L. (2003) The effect of time on the
weathering of silicate minerals: why do weathering rates differin the laboratory and field? Chem. Geol. 202, 479–506.
White A. F. and Peterzon M. L. (1990) Role of reactive surfacearea characterization in geochemical models. In Chemical
Modeling of Aqueous Systems II (eds. D. L. Melchior and R.L. Bassett). Am. Chem. Soc. Symp. Ser.
Wolff-Boenisch D., Gislason S. R., Oelkers E. H. and Putnis C. V.(2004) The dissolution rates of natural glasses as a function oftheir composition at pH 4 and 10.6, and temperatures from 25to 74 oC. Geochim. Cosmochim. Acta 68, 4843–4858.
Yang J., Meng S., Xu L. and Wang E. G. (2005) Water adsorptionon hydroxylated silica surfaces studied using the densityfunctional theory. Phys. Rev. B 71, 035413-1–035413-12.
Yang J., Meng S., Xu L. F. and Wang E. G. (2004) Ice tessellationon a hydroxylated silica surface. Phys. Rev. Lett. 92, 146102-1–
146102-4.

A sensitive NMR proxy measuring reactive surface area for aluminosilicate dissolution 5961
Yates D. E. and Healy T. W. (1975) Surface charge and tritiumexchange studies of the iron oxide (goethite)-solution interface.In Proceedings of the International Conference of Colloid and
Surface Science (ed. E. Wolfram). Akad. Kiado, Budapest,
Hungary.
Yokoyama T. and Banfield J. F. (2002) Direct determinations ofthe rates of rhyolite dissolution and clay formation over 52,000years and comparison with laboratory measurements. Geochim.
Cosmochim. Acta 66, 2665–2681.
Yokoyama T. and Matsukura Y. (2006) Field and laboratoryexperiments on weathering rates of granodiorite: separation ofchemical and physical processes. Geology 34, 809–812.
Zhao X. S. and Lu G. Q. (1998) Modification of MCM-41 bysurface silylation with trimethylchlorosilane and adsorptionstudy. J. Phys. Chem. B 102, 1556–1561.
Zhuravlev L. T. (1987) Concentration of hydroxyl groups on thesurface of amorphous silicas. Langmuir 3, 316–318.
Associate editor: Eric H. Oelkers




















