
Prevention of acetaminophen-induced hepatotoxicity by dimethyl sulfoxide
11
Toxicology, 52 (1988) 165--175 Elsevier ScientificPublishers Ireland Ltd. PREVENTION OF ACETAMINOPHEN-INDUCED HEPATOTOXICITY BY DIMETHYL SULPOXIDE YOUNGJA PARK, ROBIN D. SMITH, ALAN B. COMBSand JAMES P. KEHRER* Division of Pharmacology and Toxicology, College of Pharmacy, The University of Texas at Austi~ Austi~ TX 78715-107~ (U.S.A.) (Received October 12tb, 1987) (Accepted March 7th, 1988) SUMMARY Dimethyl sulfoxide (DMSO) has previously been shown to protect against acetaminophen (APAP)-induced hepatotoxicity, but the mechanism of this effect was not clear. Treatment of mice with 1 mg/kg DMSO 4 h before 250 mg/kg APAP resulted in significantly less hepatotoxicity than with APAP alone, as measured by serum glutamic pyruvic transaminase (SGPT) content 24 h after APAP. Protection was also evident when 1 ml/kg DMSO was given 4, but not 8 h after 250 mg/kg APAP. The APAP-induced depletion of liver glutathione was prevented in mice pretreated with DMSO, although DMSO alone had no effect on liver glutathione levels. The hepatic concentration of cytochrome P-450 (P450) 4 h after treatment of mice with 1 ml/kg DMSO, was significantly decreased compared to saline-treated animals. However, while this DMSO pretreatment significantly decreased the activity of cyto- chrome P-450-1inked aminopyrine-N-demethylase, it increased the activity of aniline hydroxylase. Covalent binding of [14C]APAP to hepatic protein in vivo was significantly decreased in mice pretreated with DMSO. Covalent binding of [14C]APAP to hepatic microsomal protein in vitro was not significantly altered after in vivo treatment with DMSO. However, the presence of DMSO in the in vitro incubation mixture significantly decreased covalent binding of [14C]APAP in a dose-dependent manner compared to microsomal fractions from untreated, saline-treated or DMSO pretreated animals. These data sug- gest that the DMSO-induced alterations in cytochrome P-450 content and activity may not be the cause of the observed protective action of this chemi- cal. The ability to competitively inhibit APAP bioactivation or to directly scavenge free radicals produced during APAP metabolism, including the *Address all correspondence and reprint requests to: Dr. James P. Kehrer, Division of Pharma- cology, College of Pharmacy, The University of Texas at Austin, Austin, TX 78712--1074, U.S.A. 0300-483X/88/$03.50 © Elsevier ScientificPublishers Ireland Ltd. Printed and Published in Ireland 165
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Prevention of acetaminophen-induced hepatotoxicity by dimethyl sulfoxide

Toxicology, 52 (1988) 165--175 Elsevier Scientific Publishers Ireland Ltd.
PREVENTION OF ACETAMINOPHEN-INDUCED HEPATOTOXICITY BY DIMETHYL SULPOXIDE
YOUNGJA PARK, ROBIN D. SMITH, ALAN B. COMBS and JAMES P. KEHRER* Division of Pharmacology and Toxicology, College of Pharmacy, The University of Texas at Austi~ Austi~ TX 78715-107~ (U.S.A.)
(Received October 12tb, 1987) (Accepted March 7th, 1988)
SUMMARY
Dimethyl sulfoxide (DMSO) has previously been shown to protect against acetaminophen (APAP)-induced hepatotoxicity, but the mechanism of this effect was not clear. Treatment of mice with 1 mg/kg DMSO 4 h before 250 mg/kg APAP resulted in significantly less hepatotoxicity than with APAP alone, as measured by serum glutamic pyruvic transaminase (SGPT) content 24 h after APAP. Protection was also evident when 1 ml/kg DMSO was given 4, but not 8 h after 250 mg/kg APAP. The APAP-induced depletion of liver glutathione was prevented in mice pretreated with DMSO, although DMSO alone had no effect on liver glutathione levels. The hepatic concentration of cytochrome P-450 (P450) 4 h after t reatment of mice with 1 ml/kg DMSO, was significantly decreased compared to saline-treated animals. However, while this DMSO pretreatment significantly decreased the activity of cyto- chrome P-450-1inked aminopyrine-N-demethylase, it increased the activity of aniline hydroxylase. Covalent binding of [14C]APAP to hepatic protein in vivo was significantly decreased in mice pretreated with DMSO. Covalent binding of [14C]APAP to hepatic microsomal protein in vitro was not significantly altered after in vivo t reatment with DMSO. However, the presence of DMSO in the in vitro incubation mixture significantly decreased covalent binding of [14C]APAP in a dose-dependent manner compared to microsomal fractions from untreated, saline-treated or DMSO pretreated animals. These data sug- gest that the DMSO-induced alterations in cytochrome P-450 content and activity may not be the cause of the observed protective action of this chemi- cal. The ability to competitively inhibit APAP bioactivation or to directly scavenge free radicals produced during APAP metabolism, including the
*Address all correspondence and reprint requests to: Dr. James P. Kehrer, Division of Pharma- cology, College of Pharmacy, The University of Texas at Austin, Austin, TX 78712--1074, U.S.A.
0300-483X/88/$03.50 © Elsevier Scientific Publishers Ireland Ltd. Printed and Published in Ireland
165

activated species which covalently binds to protein, may account for the hepatoprotection afforded by DMSO.
Key words: Acetaminophen; Liver; DMSO; Covalent binding; Hepatotoxicity
INTRODUCTION
The hepatotoxicity of APAP is mediated by the P450 dependent production of the quantitatively minor electrophilic metabolite N-acetyl-p- benzoquinone imine which covalently binds to macromolecules both in vitro and in vivo [i-5]. Following therapeutic doses of APAP, the reactive meta- bolite appears to be detoxified, at least in part, by its covalent interaction with tissue reduced glutathione (GSH) [6,7]. The toxic metabolite reacts with the nucleophilic sulfhydryl of GSH, either directly, or by an enzymatic reac- tion catalyzed by the glutathione S-transferases. When APAP is present in excess, however, the GSH level becomes depleted allowing the toxic meta- bolite to react extensively with other tissue molecules. This may result in liver necrosis, since the degree of cellular damage correlates with the amount of covalent binding to tissue protein of radiolabel derived from APAP [2]. Alternatively, the 1-electron oxidation of acetaminophen by P450 may generate reactive oxygen species [8,9] and the subsequent depletion of soluble and protein-bound thiols via oxidation may lead to disturbances of calcium homeostasis and cell death [10].
Numerous drugs and chemicals have been found to alter APAP-induced liver damage. Inducers of P450 such as phenobarbital or 3-methylcho- lanthrene potentiate the toxicity of APAP. In contrast, inhibitors of/)450 such as piperonyl butoxide, SKF 525A, and cimetidine decrease the incidence and severity of this toxicity [1,11], although the mechanism of cimetidine has been questioned [12]. Sulfhydryl compounds such as N-acetylcysteine also provide protection against APAP toxicity [7,11]. This is probably due to the ability of N-acetylcysteine to provide precursors for GSH biosynthesis [13- 16], and not to direct interactions with the reactive APAP metabolite.
DMSO is the most prominent member of a family of polar, but aprotic substances endowed with unique physical and chemical properties that make them useful as vehicles to dissolve various drugs and chemicals. DMSO has been used as a vehicle for APAP both in vivo [17] and in vitro [18] despite data showing that it blocks APAP-induced hepatotoxicity [19,20]. The mecha- nism of this protection is not known. The present study indicates that the ability of DMSO to prevent covalent binding of APAP is unrelated to changes in mixed-function oxidase activities, and requires the direct pres- ence of this solvent. This supports the concept that DMSO protects against APAP hepatotoxicity either by directly scavenging reactive species such as the acetaminophen and superoxide free radicals or the N-acetyl-p-benzoqui- none imine formed during the oxidative metabolism of this drug [4,9,21], or by virtue of competitively inhibiting the bioactivation of this drug.
166

MATERIALS AND METHODS
Experimental animals and materials Male BALB/c mice, 24--30 g (9--14 weeks of age), bred and maintained
with a 12 h light/dark cycle in the Animal Resources Center at University of Texas at Austin, were used in this study. Acetaminophen, SGPT assay kits, Folin's reagent, aminopyrine, aniline, Triton N-101, NADPH, glycerol, EDTA, sodium cholate, butylated hydroxytoluene, acetylacetone and p-ami- nophenol were purchased from Sigma Chemical Company (St. Louis, MO). 14C-Labeled acetaminophen {19.5 Ci/~mol, 980/0 pure by thin-layer chromatogra- phy) was obtained from Amersham (Arlington Heights, IL). All other chemi- cals used were of reagent grade.
Treatments DMSO and APAP were dissolved in isotonic saline and given i.p. to mice
previously fasted for 12 h. The concentrations of each drug were adjusted so that all animals received 10 ml/kg body weight. Control mice received an equal volume of saline.
Preparation of microsomes Hepatic microsomes were prepared 4 h after t rea tment by a standard dif-
ferential centrifugation procedure [22]. Tissues were perfused free of blood, washed with 1.15O/o KC1 and homogenized in 4 volumes of 0.1 M Tris-acetate (pH 7.4) containing 0.1 M KC1, 1 mM EDTA and 20 ~M BHT [to prevent lipid peroxidation] using a teflon-glass tissue homogenizer. The homogenate was centrifuged at 10 000 g for 20 min and the resultant supernatant at 100 000 g for 60 min. The microsomal pellet was washed by suspending it in 0.1 M potassium pyrophosphate (pH 7.4), containing 1 mM EDTA and 20 ~M BHT and then centrifuging again at 100 000 g for 60 min. This pellet was resus- pended at a concentration of 2 mg protein/ml in 10 mM Tris-acetate (pH 7.4) containing 1 mM EDTA and 200/0 {w/v) glycerol. Protein concentrations were measured by the micro-biuret method [23] using bovine serum albumin as the standard.
Biochemical assays Cytochrome P-$50 content: The concentration of P450 in the microsomal
preparations was determined by the carbon monoxide difference spectrum method of Omura and Sato [24]. Microsomes (2 mg protein/ml) were added to 0.1 M potassium phosphate (pH 7.4) containing 1 mM EDTA, 20% (w/v) gly- cerol, 0.50/0 (w/v) sodium cholate, and 0.4% (w/v) Triton N-101. Spectra were recorded until the 450 nm peak became maximal. P450 content was calcu- lated as f o l l o w s (A450_490)ob . . . . . d - - (A450-490)b~e l lne /0"091 ---- nmol P450/ml [22].
Enzymatic activities Aminopyrine N-demethylase activity was assessed by measuring the production of formaldehyde (HCH0) formed by the deme- thylation of aminopyrine [25]. The reaction mixture contained 1 mg microso- mal protein, 50 mM potassium phosphate, and 2.0 mM aminopyrine in a total
167

volume of 1.5 ml. This mixture was pre-incubated for 3 min at 37 °C and 0.5 mM NADPH was added to initiate the reaction. After 10 min at 37°C the reaction was stopped by the addition of 0.5 ml 17% HCIO 4 and cooled to 4°C. After centrifugation at 1000 g for 5 min, 1 ml of the supernatant was mixed with 0.4 ml Nash reagent (2.89 M ammonium acetate, 50 mM glacial acetic acid, 20 mM acetylacetone). The tubes were then capped and heated at 60 °C for 20 min. After cooling in tap water, the absorbance was read at 412 nm versus a water blank. Absorbance values for blanks lacking aminopyrine and blanks lacking NADPH were similar and were subtracted from the experi- mental values. A standard curve was prepared using authentic HCHO.
Aniline hydroxylase activity was assayed by determining p-aminophenol formation from aniline [22]. The basic incubation system was the same as described above, except that 5 mM aniline was used as the substrate. The reaction was initiated by the addition of 0.5 mM NADPH. After shaking for 10 min at 37°C, the reaction was terminated by the addition of 0.75 ml of 20% trichloroacetic acid (TCA). This mixture was centrifuged at 1000 g for 5 min and 1 ml of the supernatant added to 1 ml of 0.5 M NaOH containing 1% phenol. After mixing, 1 ml of 1 M Na2CO s was added. After 20 min at room temperature the absorbance was read at 630 nm against a water blank. Val- ues were corrected for blanks lacking aniline and NADPH, and compared to values obtained from authentic p aminophenol. SGPT activity was assessed at 25°C, using assay vials obtained from Sigma Chemical Company, by mea- suring the decrease in absorbance produced by serum samples at 340 nm as NADH was oxidized to NAD ÷.
Measurement of covalent binding to microsomal protein in vitro. The incubation mixture contained 5 mg microsomal protein in 2 ml of 0.3 mM potassium phosphate (pH 7.4). APAP (5.0 mM + 5.0 ~Ci ['4C]APAP) was added, mixed, and the mixture incubated for 2 min at 37°C. The reaction was initiated by adding 0.5 ml of 1.15% KC1 containing 5 mM NADPH. Con- trol vessels received 0.5 ml of 1.15°/o KC1. The reaction was carried out at 37°C for 60 rain, and was stopped by adding 0.8 ml of 3 M TCA. This mix- ture was centrifuged for 15 rain at 2000 g. The supernatant was discarded and the protein precipitate was washed twice in 3.0 ml of 0.6 M TCA. Precip- itated protein was extracted with 3 ml CH3OH/water (4.'1) until no further radioactivity could be removed {usually 6 extractions). The extracted protein was incubated for 1 h at 50°C with 1 ml Soluene-350 {Beckman) and the resulting solution was neutralized with acetic acid. An aliquot was added to Beckman Readysolv MP scintillation fluid and counted in a liquid scintillation counter.
Measurement of covalent binding of A P A P to hepatic protein in vivo. Mice were given 1 ml/kg DMSO or saline 4 h prior to ['4C]acetaminophen (250 mg/kg + 5.0 ~Ci). Livers were removed 4 h after injection of APAP and homogenized as described above. One-milliliter aliquots of each homogenate were added to 2 ml of 0.9 M TCA to precipitate proteins. After the addition of TCA, the reaction mixture was centrifuged at 2000 g for 15 min at room temperature. The resultant supernatant was discarded, and the precipitate
168

was washed twice wi th 3.0 ml of 0.6 M TCA. The final pel le t was r e s u s p e n d e d in 3.0 ml of 80% CH~OH/water (4:1) and washed until no more rad ioac t iv i ty could be r e m o v e d (usually 6 extract ions) , then frozen, lyophilized, and weighed. This pel le t was dissolved and counted as descr ibed above.
Glutathione assay: Hepa t i c GSH conten t was d e t e r m i n e d using a spec t ropho tom e t r i c a s say whe re GSH is r eac t ed with 5,5'-dithiobis(2-nitro- benzoic acid) to give a compound which abso rbs a t 412 nm [26]. The assay was conducted on the d e p r o t e i n a t e d 1000 g s u p e r n a t a n t f ract ions of l iver homogenized in 5% TCA containing 1 mM EDTA. While not specific for GSH, o the r non-prote in su l fhydry l s con t r ibu te less than 5% of the to ta l [27].
Statistical analysis: The da ta a re e x p r e s s e d as means _+ S.E. The da ta we re analyzed by one way analys is of var iance and mult ip le compar isons b e t w e e n g roups w e r e p e r f o r m e d with S t u d e n t - N e w m a n - K e u r s method. A P-va lue of less than 0.05 was cons idered significant.
RESULTS
As assessed by S G P T levels a f t e r 24 h (Table 1), s ignif icant p ro tec t ion f rom the hepa to tox ic effects of 250 mg/kg A P A P could be obta ined when mice were t r e a t e d with 1 ml/kg DMSO f rom 4 h before to 4 h a f t e r A P A P . If DMSO t r e a t m e n t was de layed until 8 h a f t e r A P A P , the p ro tec t ive effect was no longer ev iden t (Table I}. Mice t r e a t e d with DMSO alone exhibi ted no increases in S G P T ac t iv i ty com pa red to sa l ine- t rea ted controls . T ime course s tudies of S G P T levels following the admin i s t r a t ion of A P A P plus DMSO indicated the p ro t ec t i ve ef fec t was not due s imply to a delay in S G P T re lease (data not shown). The p ro t ec t ive effect of DMSO was fu r t he r re f lec ted in a normal hepat ic con ten t of GSH in compar i son to mice given A P A P wi thou t DMSO (Fig. 1}. The 100% mor ta l i t y seen in mice g iven doses
TABLE I
EFFECTS OF DMSO ON ACETAMINOPHEN I HEPATOTOXICITY
Treatment SGPT b
Saline 33 ± 12 {5) DMSO alone (1 ml/kg) 48 ± 31 (4) Acetaminophen alone 4352 ± 604 (27)* Acetaminophen + DMSO (0.5 ml/kg 4 h before APAP) 87 ± 30 (5) Acetaminophen + DMSO (1.0 ml/kg 4 h before APAP) 51 ± 7 (10) Acetaminophen + DMSO (1.0 ml/kg 4 h after APAP) 458 ± 242 (4) Acetaminophen + DMSO (1.0 ml/kg 8 h after APAP) 4537 ± 2577 (4)*
°Acetaminophen was administered i.p. at 250 mg/kg. bSGPT data are expressed as international units/L ± S.E. and were determined 24 h after injec- tion of acetaminophen. Values in parentheses = n.
*Significantly different from groups without asterisks (P < 0.05).
169
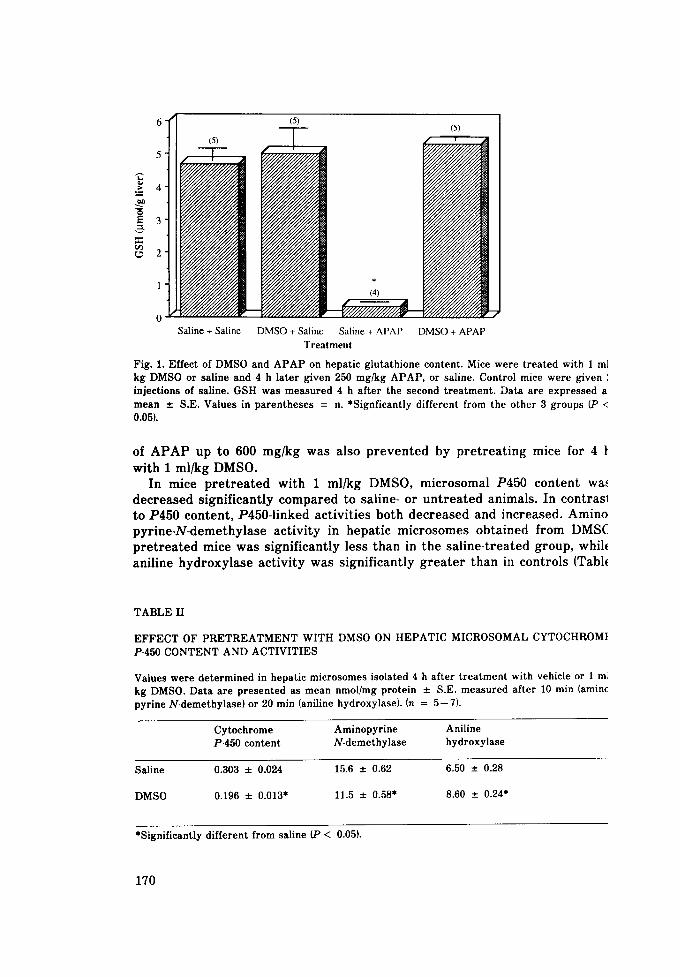
6 (5)
(4)
o Saline + Saline DMSO + Saline Saline + APAP DMSO + APAP
Treatment
Fig. 1. Effect of DMSO and A P A P on hepatic glutathione content. Mice were treated with 1 m] kg DMSO or saline and 4 h later given 250 mg/kg APAP, or saline. Control mice were given ', injections of saline. GSH was measured 4 h after the second treatment. Data are expressed a: mean __. S.E. Values in parentheses = n. *Signfieantly different from the other 3 groups (P < 0.05).
of A P A P u p t o 600 m g / k g w a s a l s o p r e v e n t e d b y p r e t r e a t i n g m i c e fo r 4
w i t h 1 m l / k g D M S O . In m i c e p r e t r e a t e d w i t h 1 m l / k g D M S O , m i c r o s o m a l P 4 5 0 c o n t e n t wa~,
d e c r e a s e d s i g n i f i c a n t l y c o m p a r e d t o s a l i n e - o r u n t r e a t e d a n i m a l s . I n c o n t r a s l
t o P 4 5 0 c o n t e n t , P 4 5 0 - 1 i n k e d a c t i v i t i e s b o t h d e c r e a s e d a n d i n c r e a s e d . A m i n o
p y r i n e - N - d e m e t h y l a s e a c t i v i t y in h e p a t i c m i c r o s o m e s o b t a i n e d f r o m D M S (
p r e t r e a t e d m i c e w a s s i g n i f i c a n t l y l e s s t h a n in t h e s a l i n e - t r e a t e d g r o u p , whi le
a n i l i n e h y d r o x y l a s e a c t i v i t y w a s s i g n i f i c a n t l y g r e a t e r t h a n in c o n t r o l s (Tabl~
TABLE II
EFFECT OF PRETREATMENT WITH DMSO ON HEPATIC MICROSOMAL CYTOCHROMt P-450 CONTENT AND ACTIVITIES
Values were determined in hepatic microsomes isolated 4 h after treatment with vehicle or 1 m] kg DMSO. Data are presented as mean nmol/mg protein ± S.E. measured after 10 rain (aminc pyrine N-demethylase) or 20 min (aniline hydroxylase). (n = 5--7}.
Cytochrome Aminopyrine Aniline P-450 content N-demethylase hydroxylase
Saline 0.303 ± 0.024 15.6 ± 0.62 6.50 ± 0.28
DMSO 0.196 ± 0.013" 11.5 ± 0.58* 8.60 ± 0.24*
*Significantly different from saline (P < 0.05).
170
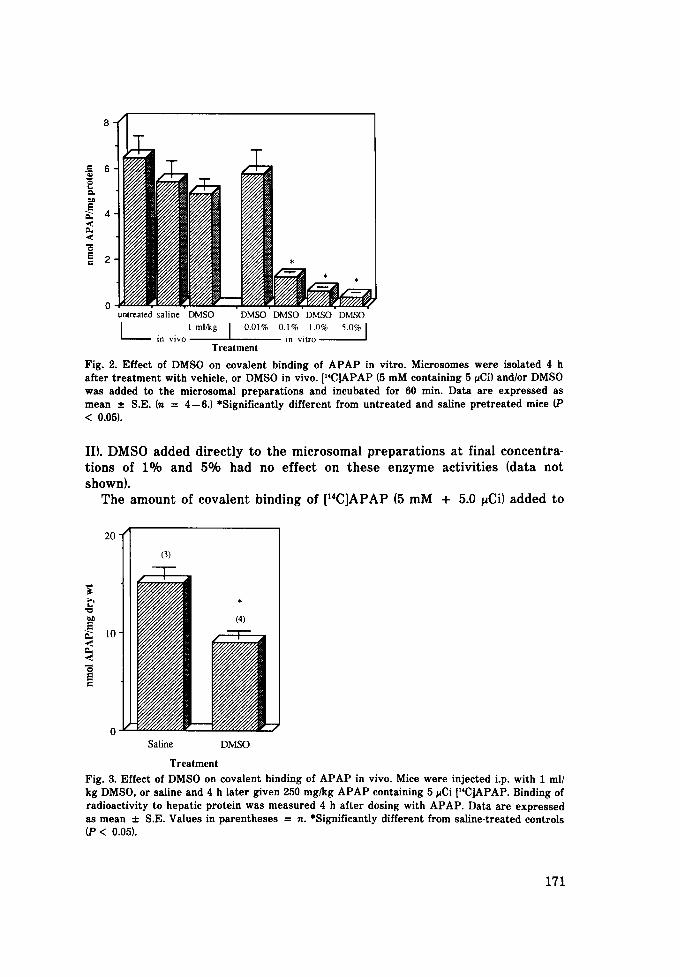
8
• i °
0 . . . . . . . . untreated saline DMSO DMSO DMSO DMSO DMSO l 1 ml/kg [ 0.01% 0.1% 1.0% 5.0% I
in vivo in vitro Treatment
Fig. 2. Effect of DMSO on covalent binding of APAP in vitro. Microsomes were isolated 4 h after treatment with vehicle, or DMSO in vivo. p'C]APAP (5 mM containing 5 ~Ci) and/or DMSO was added to the microsomal preparations and incubated for 60 rain. Data are expressed as mean :t: S.E. (n = 4--6.) *Significantly different from untreated and saline pretreated mice (P < 0.05).
II). DMSO added directly to the microsomal preparations at final concentra- tions of 1% and 5% had no effect on these enzyme activities (data not shown).
The amount of covalent binding of [14C]APAP (5 mM + 5.0 ~Ci) added to
"~ 20 t (3)
(4) E .~ / - T - A
N m o
0 Saline DMSO
Treatment Fig. 3. Effect of DMS0 on covalent binding of APAP in vivo. Mice were injected i.p. with 1 ml/ kg DMSO, or saline and 4 h later given 250 mg/kg APAP containing 5 ~Ci [uC]APAP. Binding of radioactivity to hepatic protein was measured 4 h after dosing with APAP. Data are expressed as mean =l: S.E. Values in parentheses = n. *Significantly different from saline-treated controls (P < 0.05).
171

the microsomal protein isolated from DMSO pretreated mice was not signifi- cantly different from that in the saline pretreated group (Fig. 2). However, the covalent binding of radiolabeled APAP to microsomes obtained from untreated mice was inhibited in a dose-dependent manner by DMSO added directly to the incubation mixture (Fig. 2). In in vivo studies, the covalenl binding of [14C]APAP (250 mg/kg) to hepatic proteins in mice treated wit~ DMSO (1 ml/kg) 4 h earlier was significantly less than saline pretreated animals (Fig. 3).
DISCUSSION
DMSO can be visualized in terms of a tetrahedron containing a centraJ sulfur atom. The sulfur atom plays an important role in the physiological and pharmacological properties of DMSO [28,29]. These properties include: (1 penetrant carrier activity; (2) radical scavenging activity; (3) anti-inflamma tory activity; and (4) inhibition or activation of various enzymes.
The combination of APAP and DMSO has previously been found to be significantly less toxic than APAP alone [19,20,30]. The current study con firms these findings. Three possible mechanisms of this protective effect are (1) competitive or non-competitive inhibition of mixed function oxidase activ ity which reduces the amount of reactive metabolite produced; (2) direct scavenging of the reactive metabolite; or (3) increasing the availability oJ GSH or other pathways involved in removing the reactive APAP metabolite Mercaptan contaminants found in commercial DMSO could play some role ir this third mechanism. However, the DMSO used in the current study con tained only 68 nmol of sulfhydryl equivalents/ml DMSO, as measured by reactivity with 5,5'-dithiobis(2-nitrobenzoic acid). The in vivo and in vitr( doses used in the current s tudy contained, therefore, 10 000 times less sulfhy dryl equivalents than have been reported necessary to prevent liver damage or covalent binding [15,16,31]. Thus, it seems unlikely this contaminan| played any role in the protective action of DMSO.
The effects of DMSO on xenobiotic metabolism are generally believed to bc minor. DMSO appears to specifically inhibit sulfoxide metabolism [32], bul more general tests of drug-metabolizing activity such as hexobarbital sleep ing times are not altered [33]. Furthermore, other studies suggest DMSC may induce certain drug-metabolizing enzymes [34]. Our data support these findings regarding the minimal effects of DMSO on the mixed-function oxi dase system. Although DMSO trea tment decreased total hepatic P450 con tent significantly, 2 different mixed-function oxidase activities, which arc mediated primarily by different isozymes [35], showed opposite changes. Ir addition, up to 5% DMSO in the assay mixture failed to affect either of th~ P450 associated activities which were measured. Together, these result,, suggested that DMSO was unlikely to significantly affect APAP metabolism However, it remains possible that a competitive inhibition of APAP bio activation, similar to that proposed with ethanol [36] may occur.
172

The same conclusions about the effects of DMSO on APAP metabolism are also supported by the finding that the covalent binding of APAP was no different between hepatic microsomes obtained from mice pretreated with DMSO or saline. In contrast, when DMSO was added simultaneously with APAP to microsomes obtained from untreated mice, there was significantly less binding than when APAP was added alone. Complete inhibition of [14C]APAP binding could be obtained with as little as 0.1% DMSO. This does not appear to be a competitive effect of DMSO on APAP metabolism, how- ever, since this solvent had no effect on P450-associated activities when added in vitro at levels 50 times greater. These data show that DMSO must be present at time of exposure to protect against covalent binding of APAP, and they suggest that the primary effect of DMSO may be scavenging of the APAP reactive metabolite.
DMSO pretreatment in vivo significantly decreased in vivo APAP cova- lent binding to hepatic protein in comparison to saline-teated mice. Again, this effect is most likely due to a direct interaction of the reactive species of APAP with DMSO since DMSO alone did not alter GSH content, although changes in metabolism can not be ruled out. Although covalent binding may not be the event which mediated cell death, and does not always correlate with chemical toxicity [37], the relationship between such binding and APAP hepatotoxicity has been well established.
The protective effect of DMSO given 4 h after APAP, when GSH is aleady depleted and covalent binding is maximal, was unexpected. However, since covalent binding may not be the direct cause of APAP toxicity [9], it is possible that lethal tissue damage continues to occur after 4 h. The adminis- tration of DMSO may have been able to stop this additional damage.
The results of the current s tudy clearly show that DMSO is an excellent preventative agent for APAP-induced liver damage in mice. The ability of DMSO to protect against tissue injury by virtue of its radical scavenging activity has previously been postulated to explain the protective effect of this chemical against acute lung injury after thermal trauma to the skin [38]. Based on the direct ability of DMSO to prevent covalent binding, the absence of any effects of DMSO on hepatic GSH content, and the minor effects of DMSO on mixed-function oxidase activity, this hepato-protective effect appears to be due to the interaction of DMSO with free radical or reactive APAP metabolites before they can bind to GSH or macromolecules and damage the tissue.
ACKNOWLEDGEMENTS
This work was supported by grant CH-263A from the American Cancer Society and by BRSG grants RR05849 and RR07091 awarded to the College of Pharmacy and the University of Texas at Austin. JPK is the recipient of RCDA HL01435 from the National Heart , Lung and Blood Institute and holds the Gustavus Pfeiffer Centennial Fellowship in Pharmacology.
173

REFERENCES
1 J.R. Mitchell, D.J. Jollow, W.Z. Potter, D.C. Davis, J.R. Gillette and B.B. Brodie, Acetaminc phen-induced hepatic necrosis: I. Role of drug metabolism. J. Pharmacol. Exp. Ther., 18 ' (1973} 185.
2 D.J. Jollow, J.R. Mitchell, W.Z. Potter, D.C. Davis, J.R. Gillette and B.B. Brodie, Acetaminc phen-induced hepatotoxicity: II. Role of covalent binding in vivo. J. Pharmacol. Exp. Ther 187 (1973) 195.
3 W.Z. Potter, D.C. Davis, J.R. Mitchell, D.J. Jollow, J.R. Gillette and B.B. Brodie, Acetamin¢ phen-induced hepatic necrosis: III. Cytochrome P-450 mediated covalent binding in vitro. Pbarmacol. Exp. Ther., 187 (1973} 203.
4 D.C. Dahlin, G.T. Miwa, A.Y.H. Lu and S.D. Nelson, N-acetyl-p-benzoquinone imine: A cytc chrome P-450-mediated oxidation product of acetaminophen. Proc. Natl. Acad. Sci. U.S.A 81 (1984) 1327.
5 E. Albano, M. Rundgren, P.J. Harvison, S.D. Nelson and P. MoldSus, Mechanisms of ,~ acetyl-p-benzoquinone imine cytotoxicity. Mol. Pharmacol., 28 (1985) 306.
6 J.R. Mitchell, D.J. Jollow, W.Z. Potter, J.R. Gillette and B.B. Brodie, Acetaminophex induced hepatic necrosis. IV. Protective role of glutathione. J. Pharmacol. Exp. Ther., 18 {1973) 211.
7 J.A. Hinson, L.R. Pohl, T.J. Monks and J.R. Gillette, Acetaminophen-induced hepatotoxi~ ity. Life Sci., 29 (1981) 107.
8 M.E. Kyle, S. Miccadei, D. Nakae and J.L. Farber, Superoxide dismutase and catalase pr~ tect cultured hepatocytes from the cytotoxicity of acetaminophen. Biochem. Biophys. Re~ Commun., 149 (1987} 889.
9 G.M. Rosen, W.V. Singletary Jr., E.J. Rauckman and P.G. Killenberg, Acetaminophen hep~ totoxicity. An alternative mechanism. Biochem. Pharmacol., 32 (1983) 2053.
10 M. Moore, H. Thor, G. Moore, S. Nelson, P. Mold~us and S. Orrenius, The toxicity of acq taminophen and N-acetyl-p-benzoquinoneimine in isolated hepatocytes is associated wit thiol depletion and increased cytosolic Ca 2.. J. Biol. Chem., 260 (1985) 13035.
11 L.F. Prescott and J.A.J.H. Critchley, The treatment of acetaminophen poisoning. Anm Rev. Pharmacol. Toxicol., 23 (1983) 87.
12 J.O. Miners, R. Drew and D.J. Birkett, Effect of cimetidine on paracetamol activation i mice. Biochem. Pharmacol., 33 (1984) 1996.
13 J.O. Miners, R. Drew and D.J. Birkett, Mechanisms of action of paracetamol protectiv agents in mice in vivo. Biochem. Pharmacol., 33 (1984) 2995.
14 B.H. Lauterburg, G.B. Corcoran and J.R. Mitchell, Mechanism of action of N-acetylcystein in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J. Clil Invest., 71 (1983) 980.
15 G.B. Corcoran and B.K. Wong, Role of glutathione in prevention of acetaminophen-induce hepatotoxicity by N-acetyl-L-cysteine in vivo; Studies with N-acetyl-D-cysteine in mice. , Pharmacol. Exp. Ther., 238 (1986) 54.
16 G.A. Hazelton, J.J. Hjelle and C.D. Klaassen, Effects of cysteine pro-drugs on acetamin phen-induced hepatotoxicity. J. Pharmacol. Exp. Ther., 237 (1986} 341.
17 L.G. Costa and S.D. Murphy, Interaction between acetaminophen and organophosphates i mice. Res. Commun. Chem. Pathol. Pharmacol., 44 (1984) 389.
18 R.J. Gerson, A. Casini, D. Gilfor, A. Serroni and J.L. Farber, Oxygen-mediated cell injury : the killing of cultured hepatocytes by acetaminophen. Biochem. Biophys. Res. Commun., 1; (1985} 1129.
19 A.N. E1-Hage, E.H. Herman and V.J. Ferrans, Examination of the protective effect of ICR: 187 and dimethyl sulfoxide against acetaminophen-induced hepatotoxicity in Syrian gold~ hamsters. Toxicology, 28 (1983} 295.
20 C-P. Siegers, Antidotal effects of dimethyl sulfoxide against paracetamol-, bromobenzem and thioacetamide-induced hepatotoxicity. J. Pharmacol. Pharm., 30 (1978} 375.
21 A.V. Fischer, P.R. West, S.D. Nelson, P.J. Harvison and R.P. Mason, Formation of 4-amin phenoxyl free radical from the acetaminophen metabolite N-acetyl-p-benzoquione imine. Biol. Chem., 260 (1985} 11446.
174

22 F.P. Guengerieh, Microsomal enzymes involved in toxicology analysis and separation, in A.W. Hayes (Ed.), Principles and Methods of Toxicology, Raven Press, New York, 1982, p. 609.
23 R. Itzhaki and D.M. Gill, A micro-biuret method for estimating proteins. Anal. Biochem., 9 (1964) 401.
24 T. Omura and R. Sato, The carbon monoxide-binding pigment of liver microsomes. J. Biol. Chem., 239 (1964) 2370.
25 T. Nash, The colorimetric estimation of formaldehyde by means of the Hantzsch reaction. Biochem. J., 55 (1953) 416.
26 R.J. Richardson and S.D. Murphy, Effect of glutathione depletion on tissue deposition of methylmercury. Toxicol. Appl. Pharmacol., 31 (1975) 505.
27 K.-H. Summer and H. Greim, Detoxification of chloroprene (2-chloro-l,3-butadienel with glu- tathione in the rat. Biochem. Biophys. Res. Commun., 96 (1980) 566.
28 N. Kharasch and B.S. Thyagarajan, Structural basis for biological activities of dimethyl sul- foxide. Ann. N.Y. Acad. Sci., 411 (1983) 391.
29 C.F. Brayton, Dimethyl sulfoxide. Cornell Vet., 76 (1986) 61. 30 E. Jeffery and W.M. Haschek, Protection by dimethylsulfoxide against acetaminophen-
induced hepatic, but not respiratory toxicity in the mouse. Toxicol. Appl. Pharmacol., 93 (1988) 452.
31 J.A. Hinson, Biochemical toxicology of acetaminophen, in E. Hodgson, J.R. Bend and R.M. Philpot (Eds.t, Reviews in Biochemical Toxicology, Vol. 2, Elsevier, New York, 1980, p. 103.
32 B.N. Swanson, Mojaverian and V.K. Boppana, Inhibition of sulindac metabolism by dimethyl sulfoxide in the rat. J. Toxicol. Environ. Health, 12 (1983) 213.
33 R.E. Mancini and J.J. Kocsis, Dimethylsulfoxide increases the lethality of CCI 4 in rats but decreases its hepatotoxicity. Toxicol. Appl. Pharmacol., 27 (1974) 206.
34 J.J. Kocsis, S. Harkaway, M.C. Santoyo and R. Snyder, Dimethyl sulfoxide: Interactions with aromatic hydrocarbons. Science, 160 (1968} 427.
35 T. Omura and R. Sato, Cytochrome P-450, Academic Press, New York. 1978, p. 38. 36 C. Sato, M. Nakano and C.S. Lieber, Prevention of acetaminophen-induced hepatotoxicity by
acute ethanol administration in the rat: Comparison with carbon tetrachloride-induced hepa- totoxicity. J. Pharmacol. Exp. Ther., 218 (1981) 805.
37 J.R. Gillette, A perspective on the role of chemically reactive metabolites of foreign com- pound in toxicity. Biochem. Pharmacol., 23 (1974) 2785.
38 G.O. Till, J.R. Hatherill, W.W. Tourtellotte, M.J. Lutz and P.A. Ward, Lipid peroxidation and acute lung injury after thermal trauma to skin. Am. J. Patbol., 119 (1985) 376.
175




















