
l-carnitine attenuates oxidant injury in HK-2 cells via ROS ...
Upload
independentCategory
view
0download
0
Yasushi Numaguchi, Hideo Matsui, Toyoaki Murohara and Kenji OkumuraRyotaro Takahashi, Toru Asai, Hisashi Murakami, Ryuichiro Murakami, Michitaka Tsuzuki,
Transporter Gene MutationPressure Overload-Induced Cardiomyopathy in Heterozygous Carrier Mice of Carnitine
Print ISSN: 0194-911X. Online ISSN: 1524-4563 Copyright © 2007 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Hypertension doi: 10.1161/HYPERTENSIONAHA.107.088609
2007;50:497-502; originally published online July 30, 2007;Hypertension.
http://hyper.ahajournals.org/content/50/3/497World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://hyper.ahajournals.org/content/suppl/2007/08/02/HYPERTENSIONAHA.107.088609.DC1.htmlData Supplement (unedited) at:
http://hyper.ahajournals.org//subscriptions/
is online at: Hypertension Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialHypertensionin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from

Pressure Overload–Induced Cardiomyopathy inHeterozygous Carrier Mice of Carnitine Transporter
Gene MutationRyotaro Takahashi, Toru Asai, Hisashi Murakami, Ryuichiro Murakami, Michitaka Tsuzuki,
Yasushi Numaguchi, Hideo Matsui, Toyoaki Murohara, Kenji Okumura
Abstract—Primary systemic carnitine deficiency is an autosomal recessive disorder caused by a decreased renal reabsorptionof carnitine because of mutations of the carnitine transporter OCTN2 gene, and hypertrophic cardiomyopathy is acommon clinical feature of homozygotes. Although heterozygotes for OCTN2 mutations are generally healthy withnormal cardiac performance, heterozygotes may be at risk for cardiomyopathy in the presence of additional risk factors,such as hypertension. To test this hypothesis, we investigated the effects of surgically induced pressure overload on thehearts of heterozygous mutants of a murine model of OCTN2 mutation, juvenile visceral steatosis mouse (jvs/�).Eleven-week-old jvs/� mice and age-matched wild-type mice were used. At baseline, there were no differences inphysical characteristics between wild-type and jvs/� mice. However, plasma and myocardial total carnitine levels injvs/� mice were lower than in wild-type mice. Both wild-type and jvs/� mice were subjected to ascending aorticconstriction with or without 1% L-carnitine supplementation for 4 weeks. At 4 weeks after ascending aortic constriction,jvs/� mice showed an exaggeration of cardiac hypertrophy and pulmonary congestion, further increased geneexpression of atrial natriuretic peptide in the left ventricles, further deterioration of left ventricular fractional shortening,reduced myocardial phosphocreatine:adenosine triphosphate ratio, and increased mortality compared with wild-typemice; L-carnitine supplementation prevented these changes in jvs/� mice subjected to ascending aortic constriction. Inconclusion, cardiomyopathy and heart failure with energy depletion may be induced by pressure overload in heterozygotesfor OCTN2 mutations and could be prevented by L-carnitine supplementation. (Hypertension. 2007;50:497-502.)
Key Words: cardiomyopathy � genes � heart failure � hypertension � hypertrophy
Mitochondrial �-oxidation of fatty acids is the mainsource of energy for the heart.1 Inborn errors in myocar-
dial fatty acid oxidation have been reported to be importantcauses of inherited cardiomyopathies, which are typicallyhypertrophic with diminished systolic function.2 Carnitine isessential for the transport of long-chain fatty acids into themitochondrial matrix for �-oxidation and plays an importantrole in cellular energy metabolism.3 Carnitine deficiencyleads to energy an metabolism disorder because of impairedfatty acid oxidation. Primary systemic carnitine deficiency(SCD; Online Mendelian Inheritance in Man 212140) is anautosomal recessive disorder caused by decreased renalreabsorption of carnitine because of mutations of the carnitinetransporter OCTN2 gene.4 Progressive cardiomyopathy is acommon clinical manifestation of SCD and could be pre-vented by high-dose L-carnitine supplementation.5,6
The juvenile visceral steatosis (JVS) mouse was estab-lished as an excellent murine model of SCD.7 SCD in JVS
mice is caused by decreased renal reabsorption of carnitinebecause of a spontaneous mutation in the OCTN2 gene, as hasbeen reported in human SCD as well.4 In homozygousmutants of JVS mice, carnitine levels in the myocardium aremarkedly reduced compared with the levels in wild-type(WT) mice.8 Homozygous JVS mice develop cardiomyopa-thy characterized by marked cardiac hypertrophy withoutfibrosis, myocardial energy metabolism disorder,8 and pro-gressive cardiac dysfunction.9
It was reported that myocardial carnitine levels were alsolower in heterozygous mutants of JVS mice compared withWT mice.10 Although there were no differences in the heartweight:body weight ratio and the mortality rate between WTmice and heterozygous JVS mice, heterozygous mice showedan age-associated increase in left ventricular myocyte diam-eters compared with WT mice.11 In humans, a geneticepidemiological study in Japan demonstrated that heterozy-gotes for OCTN2 mutations have consistently low serum
Received February 6, 2007; first decision February 19, 2007; revision accepted July 6, 2007.From the Department of Cardiology (R.T.), Chunichi Hospital, Nagoya, Japan; Department of Cardiology (T.A.), Aichi Cardiovascular and Respiratory
Center, Ichinomiya, Japan; Department of Cardiology (H.M., R.M., M.T., H.M., T.M.), Nagoya University Graduate School of Medicine, Nagoya, Japan;and the Medical Science of Proteases (Y.N.) and Cardiovascular Research Medicine (K.O.), Nagoya University School of Medicine, Nagoya, Japan.
Correspondence Kenji Okumura, Cardiovascular Research Medicine, Nagoya University School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya466-8550, Japan. E-mail [email protected]
© 2007 American Heart Association, Inc.
Hypertension is available at http://hyper.ahajournals.org DOI: 10.1161/HYPERTENSIONAHA.107.088609
497 by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from

carnitine phenotypes, and the estimate of the overall preva-lence of heterozygotes was 1.01%.12 In this study, an echo-cardiographic analysis of the families of patients with SCDrevealed that heterozygotes for OCTN2 mutations were pre-disposed to late-onset cardiac hypertrophy with normal car-diac performance compared with the WT mice.
Heterozygotes for OCTN2 mutations may be at risk forcardiomyopathy in the presence of risk factors such ashypertension in addition to aging.13 To test this hypothesis,we investigated the effects of surgically induced pressureoverload on the hearts of heterozygous mutants of JVS mice.
MethodsAn expanded Materials and Methods section can be found in theonline supplemental data available at http://hyper.ahajournals.org.
Experimental Animals and Study ProtocolHeterozygous mutants of 11-week-old male JVS mice of the C3Hstrain were used. Heterozygous JVS mice (jvs/�) were created bymating homozygous mutants and heterozygous mutants of JVS mice.Heterozygous mutants were distinguished from homozygous mu-tants, which have swollen fatty livers recognized through theabdominal wall at 5 days after birth.7 Age-matched male WT mice ofthe C3H strain were used as controls. Both WT and jvs/� mice weredivided into the following 3 groups: (1) sham operation; (2)ascending aorta constriction (AAC); and (3) 1% L-carnitine supple-mentation (10 g of L-carnitine per kilogram of chow) for 4 weeksafter AAC. L-Carnitine was not included in the ingredients of thestandard chow. AAC was created by ligation of the ascending aortawith a 27-gauge needle using 7–0 silk suture in anesthetized (50mg/kg of pentobarbital IP) and ventilated mice. Sham-operated miceunderwent the same surgical procedure without the ligation of theaorta. All of the animals were studied 4 weeks after the operation.Animals were anesthetized with diethyl ether before sacrifice. Forsurvival analysis, cages were inspected daily for deceased animalsduring the study period.
This study conformed to the National Institutes of Health Guidefor the Care and Use of Laboratory Animals. All of the protocolsdescribed were approved by the Nagoya University Animal EthicsCommittee.
Echocardiographic andHemodynamic MeasurementsOn the day of sacrifice, the systolic blood pressure and heart ratewere determined in each animal. The tail-cuff method was used,employing a photoelectric tail cuff detection system, Softron BP-98A (Softron). Left ventricular function was evaluated by transtho-racic echocardiography using SONOS 7500 (Philips Medical Sys-tems) with a 10-MHz imaging transducer. To evaluate the degree ofstenosis, the pressure gradient across the constriction was assessedby Doppler echocardiography at 3 days after operation.
Plasma and Myocardial Total Carnitine LevelsPlasma and myocardial total carnitine levels were determined by anenzymatic method described elsewhere.14
Histological AnalysisHeart tissue was examined by means of light microscopy. The tissuewas fixed in 10% formaldehyde in phosphate buffer and wasembedded in paraffin. Transverse sections (4-�m thickness) at thelevel of papillary muscle were stained with hematoxylin/eosin orwith Masson’s trichrome. The myocyte cross-sectional area and theextent of fibrosis were evaluated with the use of the image analysissoftware WinROOF (Mitani Corp).
Real-Time Quantitative RT-PCRTotal RNA was extracted from the left ventricle, and reversetranscription was performed by a method described previously.15 Toexamine the relative mRNA expression of atrial natriuretic peptide,quantitative RT-PCR analysis was performed using the StratageneMx3000P QPCR System (Stratagene) and the Power SYBR GreenPCR Master Mix (Applied Biosystems).
Myocardial High-Energy Phosphate ContentAnimals were anesthetized with diethyl ether, and the hearts wererapidly excised and snap-frozen with liquid nitrogen. The frozentissue was homogenized in 0.5 mol/L of ice-cold perchloric acid fordeproteinization and then neutralized with 2 mol/L of K2CO3. Aftercentrifugation, the supernatant was filtered, and an aliquot of 20 �Lwas used for measurement. Phosphocreatinine (PCr) and adenosinetriphosphate (ATP) contents were measured by high-performanceliquid chromatography, as described previously.16
StatisticsAll of the values are expressed as mean�SEM. The survival analysiswas performed by the Kaplan-Meier method, and between-groupdifferences in survival were tested by the log-rank test. Between-group comparisons were assessed by 1-way ANOVA followed bythe Bonferroni posthoc test. A value of P�0.05 was considered to bestatistically significant.
Results
Baseline Characteristics of Heterozygous JVS MiceTable S1 demonstrates the characteristics of jvs/� mice atbasal condition. At baseline, there were no differences inthe gravimetric, hemodynamic, and echocardiographicdata between WT and jvs/� mice. However, the plasmaand myocardial total carnitine levels in jvs/� mice werelower than in WT mice (52.8�0.9 �mol/L versus70.2�1.1 �mol/L, P�0.01; 349.2�25.9 nmol/g versus642.3�30.1 nmol/g, P�0.01, respectively).
Assessment of Cardiac Hypertrophy, PulmonaryCongestion, and HemodynamicsGravimetric and hemodynamic data at 4 weeks after AAC areshown in the Table. jvs/�-AAC mice showed a further increasein the heart weight:body weight ratio, the lung weight:bodyweight ratio, and the wet lung weight:dry lung weight ratiocompared with WT-AAC mice. Systolic blood pressure wasfurther reduced in jvs/�-AAC mice compared with WT-AACmice. L-Carnitine supplementation prevented these changes injvs/�-AAC mice. There was no difference in heart rate amongall of the groups.
Echocardiographic ExaminationEchocardiographic data are demonstrated in Figure 1.There was no difference in aortic pressure gradient amongall of the groups at 3 days after AAC (Figure 1E). Fourweeks after operation, jvs/�-AAC mice showed a furtherincrease in left ventricular end-systolic dimension and afurther deterioration of left ventricular fractional shorten-ing compared with WT-AAC mice (Figure 1C and 1D).L-Carnitine supplementation prevented these changes injvs/�-AAC mice. However, there was no difference in leftventricular end-diastolic dimension between WT-AAC andjvs/�-AAC mice (Figure 1B).
498 Hypertension September 2007
by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from

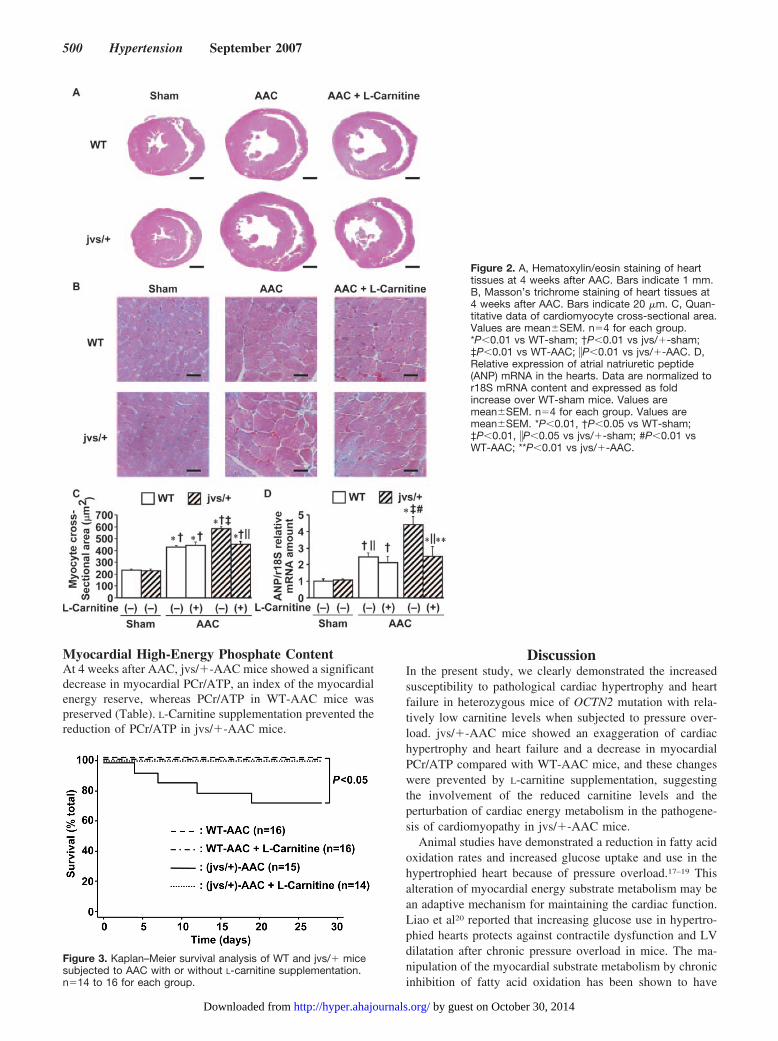
Histopathologic Analysis and Relative AtrialNatriuretic Peptide mRNA Expression inthe HeartTransverse sections of the hearts are shown in Figure 2A.jvs/�-AAC mice showed an exaggeration of cardiomegalycompared with WT-AAC mice. L-Carnitine supplementationprevented the cardiomegaly in jvs/�-AAC mice. jvs/�-AACmice showed an exaggeration of cardiomyocyte hypertrophycompared with WT-AAC mice (Figure 2B and 2C).L-Carnitine supplementation also prevented the cardiomyo-cyte hypertrophy in jvs/�-AAC mice. The relative mRNAexpression of atrial natriuretic peptide, a molecular marker ofcardiac hypertrophy, in the left ventricles of jvs/�-AAC miceincreased more than in WT-AAC mice; L-carnitine supple-
mentation prevented this change in jvs/�-AAC mice (Figure2D). The extent of fibrosis was similar between WT-AACand jvs/�-AAC mice (Figure S1A and S1B).
Survival StudyThe acute mortality rate within 72 hours after AAC was notdifferent among all of the mice that were subjected to AAC(16% of WT-AAC mice, 16% of WT-AAC mice treated withL-carnitine, 17% of jvs/�-AAC mice, and 18% of jvs/�-AAC mice treated with L-carnitine; P value was not signifi-cant for all). None of the mice died after the sham operation.In mice that survived the acute perioperative period afterAAC, 27% of jvs/�-AAC mice (4 of 15) died in thefollowing 4 weeks, whereas none of the mice in the othergroups died during the same period (Figure 3).
Gravimetric and Hemodynamic Measurements and Myocardial PCr/ATP at 4 Weeks After AAC
Operation and Mice/Parameter
Sham AAC
WT jvs/� WT WT�CAR jvs/� jvs/��CAR
BW, g 24.5�1.3 24.3�0.7 24.6�0.7 23.8�0.9 22.7�0.9 23.2�0.9
HW, mg 99.5�4.7 95.4�2.8 152.9�3.7*† 147.2�5.3*† 161.9�6.7*† 143.3�3.6*†§
HW/BW, mg/g 4.07�0.05 3.93�0.06 6.22�0.07*† 6.20�0.15*† 7.14�0.13*†‡ 6.24�0.17*†§
Wet LW/BW, mg/g 5.21�0.16 5.16�0.04 9.39�0.86*† 9.61�1.73*† 16.73�0.89*†‡ 11.43�0.77*†§
Wet LW/dry LW 4.55�0.03 4.63�0.03 4.78�0.03* 4.67�0.05 5.11�0.09*†‡ 4.80�0.04*§
Systolic BP, mm Hg 106�2 105�1 88�2*† 91�1*† 75�2*†‡ 87�2*†§
HR, bpm 584�11 572�10 558�23 547�2 559�14 557�4
Myocardial PCr/ATP 1.89�0.33 1.94�0.23 1.84�0.31 1.80�0.11 1.05�0.05� 1.82�0.24
Data are mean�SEM. n�6 to 8 in each group for gravimetric and hemodynamic measurements. n�5 in each group for myocardial PCr/ATP. CARindicates L-carnitine; BW, body weight; HW, heart weight; LW, lung weight; LVEDD, left ventricular end-diastolic dimension; LVESD, left ventricularend-systolic dimension; LVFS, left ventricular fractional shortening; BP, blood pressure; HR, heart rate.
*P�0.01 vs WT-sham; †P�0.01 vs jvs/�-sham; ‡P�0.01 vs WT-AAC; §P�0.01 vs jvs/�-AAC; �P�0.05 vs all other groups.
Figure 1. A, Representative M-mode echocardiograms in WT and jvs/� mice at 4 weeks after AAC. B, C, D, and E, Summary data forechocardiographic measurements. B, Left ventricular end-diastolic dimension (LVEDD), (C) left ventricular end-systolic dimension(LVESD), (D) left ventricular fractional shortening (LVFS) at 4 weeks after AAC, and (E) aortic pressure gradient at 3 days after AAC areshown. Values are mean�SEM. n�6 for each group. *P�0.01 vs WT-sham; †P�0.01 vs jvs/�-sham; ‡P�0.01 vs WT-AAC; �P�0.01 vsjvs/�-AAC.
Takahashi et al Cardiomyopathy in Heterozygotes for OCTN2 Mutation 499
by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from
Myocardial High-Energy Phosphate ContentAt 4 weeks after AAC, jvs/�-AAC mice showed a significantdecrease in myocardial PCr/ATP, an index of the myocardialenergy reserve, whereas PCr/ATP in WT-AAC mice waspreserved (Table). L-Carnitine supplementation prevented thereduction of PCr/ATP in jvs/�-AAC mice.
DiscussionIn the present study, we clearly demonstrated the increasedsusceptibility to pathological cardiac hypertrophy and heartfailure in heterozygous mice of OCTN2 mutation with rela-tively low carnitine levels when subjected to pressure over-load. jvs/�-AAC mice showed an exaggeration of cardiachypertrophy and heart failure and a decrease in myocardialPCr/ATP compared with WT-AAC mice, and these changeswere prevented by L-carnitine supplementation, suggestingthe involvement of the reduced carnitine levels and theperturbation of cardiac energy metabolism in the pathogene-sis of cardiomyopathy in jvs/�-AAC mice.
Animal studies have demonstrated a reduction in fatty acidoxidation rates and increased glucose uptake and use in thehypertrophied heart because of pressure overload.17–19 Thisalteration of myocardial energy substrate metabolism may bean adaptive mechanism for maintaining the cardiac function.Liao et al20 reported that increasing glucose use in hypertro-phied hearts protects against contractile dysfunction and LVdilatation after chronic pressure overload in mice. The ma-nipulation of the myocardial substrate metabolism by chronicinhibition of fatty acid oxidation has been shown to have
Figure 2. A, Hematoxylin/eosin staining of hearttissues at 4 weeks after AAC. Bars indicate 1 mm.B, Masson’s trichrome staining of heart tissues at4 weeks after AAC. Bars indicate 20 �m. C, Quan-titative data of cardiomyocyte cross-sectional area.Values are mean�SEM. n�4 for each group.*P�0.01 vs WT-sham; †P�0.01 vs jvs/�-sham;‡P�0.01 vs WT-AAC; �P�0.01 vs jvs/�-AAC. D,Relative expression of atrial natriuretic peptide(ANP) mRNA in the hearts. Data are normalized tor18S mRNA content and expressed as foldincrease over WT-sham mice. Values aremean�SEM. n�4 for each group. Values aremean�SEM. *P�0.01, †P�0.05 vs WT-sham;‡P�0.01, �P�0.05 vs jvs/�-sham; #P�0.01 vsWT-AAC; **P�0.01 vs jvs/�-AAC.
Figure 3. Kaplan–Meier survival analysis of WT and jvs/� micesubjected to AAC with or without L-carnitine supplementation.n�14 to 16 for each group.
500 Hypertension September 2007
by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from

therapeutic potential as a treatment for heart failure in animalmodels and in humans.21,22 However, in the present study,heterozygous JVS mice with reduced carnitine levels showedan exaggeration of cardiac hypertrophy and heart failure andimpaired myocardial energy metabolism when subjected topressure overload. Previously, we have demonstrated thathomozygous JVS mice are hypoglycemic,9,15,23 which indi-cates the energy substrate metabolism shift toward glucosemetabolism because of fatty acid oxidation disorder. Al-though plasma glucose levels or myocardial energy substratemetabolism were not assessed in the present study, themyocardial energy substrate metabolism may shift from fattyacid oxidation toward glucose metabolism also in heterozy-gous JVS mice because of the relatively low carnitine levels.In heterozygous JVS mice, the glucose metabolism may befurther enhanced in the heart when subjected to pressureoverload. The exaggeration of heart failure and the reductionof PCr/ATP observed in jvs/�-AAC mice suggest that thisincrease in glucose metabolism may be insufficient to supportthe myocardial energy metabolism. The results of our studyindicate that the presence of substantial levels of carnitine tosupport the fatty acid oxidation capacity may be required tomaintain the cardiac function in mice subjected to pressureoverload for 4 weeks. However, myocardial energy substratemetabolism was not assessed in the present study. Furtherinvestigation is required to elucidate the mechanism under-lying the energy depletion in heterozygous JVS mice sub-jected to pressure overload.
Although several early studies had initially suggested thatmyocardial high-energy phosphate contents were unaltered inheart failure,24,25 more recent studies have provided evidencethat there is an energy deficit in the failing heart and that thefailing heart is “energy starved.”26,27 In terms of animalmodels of pressure-overloaded hearts, myothermal measure-ments demonstrated that heat production was lower inpressure-overloaded hearts than in normal hearts in rabbits.28
In mice subjected to chronic pressure overload (8 weeks afterAAC), myocardial PCr/ATP was significantly reduced com-pared with sham-operated mice.20 In our study, WT micesubjected to AAC for 4 weeks did not show any decrease inmyocardial PCr/ATP. However, heterozygous JVS miceshowed a decrease in myocardial PCr/ATP and an exacerba-tion of cardiac dysfunction and heart failure when subjectedto AAC for 4 weeks; these changes were prevented byL-carnitine supplementation. Whether heart failure or cardiacremodeling is caused by a defect in energy production or adeficit in energy use has been a continuing debate. During thecompensatory stage in cardiac remodeling, modifications inthe cellular apparatus that are responsible for energy produc-tion could account for further impairment of myocardialfunction.29 The results of our study support this hypothesisand suggest that the reduction in carnitine leading to myo-cardial energy depletion plays a causal role for the exacerba-tion of cardiac dysfunction and heart failure in pressure-overloaded hearts.
A human study using echocardiography showed that vari-ations in the peroxisome proliferator-activated receptor-�, akey transcriptional regulator of the expression of fatty acidoxidation pathways genes, influence the hypertrophic growth
in response to both exercise and hypertension.30 It was alsoreported that the myocardial fatty acid metabolism is anindependent predictor of left ventricular mass in hypertensionand in left ventricular dysfunction in humans.31 These obser-vations provide evidence that the perturbation of myocardialfatty acid oxidation may play a causal role in the pathogenesisand/or modulation of the hypertrophic phenotype because ofhypertension. Consistently, the present study suggest that thereduction in carnitine, possibly leading to impaired myocar-dial fatty acid oxidation, may have been involved in theexaggeration of cardiac hypertrophy and heart failure inheterozygous JVS mice subjected to pressure overload.
We have reported previously that homozygous JVS micedevelop pathological cardiac hypertrophy with vast lipidaccumulation and that the increase in myocardial 1,2-diacylglycerol, a lipid second messenger, and the alteration offatty acid composition of 1,2-diacylglycerol were implicatedin the pathogenesis of cardiac hypertrophy in homozygousJVS mice.9,15,23,32 In the present study, we have also carriedout myocardial lipid analysis in heterozygous JVS micesubjected to pressure overload (Table S2). In contrast tohomozygous JVS mice, jvs/�-sham mice showed only amodest but significant increase in myocardial triacylglyceroland 1,2-diacylglycerol levels compared with WT-sham mice.However, in mice subjected to AAC, the myocardial triacyl-glycerol level was significantly reduced to identical levels inboth WT and jvs/� mice, and the 1,2-diacylglycerol level injvs/� mice was significantly reduced to WT mice levels. Incontrast to the dramatic alteration of the fatty acid composi-tion of myocardial 1,2-diacylglycerol in homozygous JVSmice as demonstrated in our previous reports,9,15,23,32 jvs/�-sham mice did not show significant alteration in the fatty acidcomposition of 1,2-diacylglycerol. Although mice subjectedto AAC showed changes in fatty acid composition of myo-cardial 1,2-diacylglycerol compared with sham-operatedmice, there was no difference between WT-AAC and jvs/�-AAC mice. L-Carnitine supplementation did not alter themyocardial triacylglycerol or 1,2-diacylglycerol levels andalso did not change the fatty acid composition of 1,2-diacylglycerol in both WT-AAC and jvs/�-AAC mice. Theseresults suggest that myocardial lipid moieties may not beinvolved in the pathogenesis of cardiomyopathy and heartfailure in heterozygous JVS mice subjected to pressureoverload.
PerspectivesA genetic epidemiological study in the Akita prefecture ofJapan demonstrated that the estimate of the overall preva-lence of heterozygotes for OCTN2 mutations was 1.01%, andan echocardiographic analysis revealed that heterozygoteswere predisposed to late-onset cardiac hypertrophy withnormal cardiac performance.12 Although heterozygotes ofSCD are generally healthy without cardiac complications, ithas been proposed that heterozygotes may be at risk forcardiomyopathy in the presence of additional risk factors,such as hypertension.13 The results of our study support thishypothesis and suggest that L-carnitine supplementation maybe a potential therapy for preventing cardiomyopathy andheart failure in heterozygotes for OCTN2 mutations with
Takahashi et al Cardiomyopathy in Heterozygotes for OCTN2 Mutation 501
by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from

hypertension. The prevalence of heterozygotes for OCTN2mutations seems to be higher than expected, suggesting thatheterozygotes are often overlooked clinically. Further exam-ination and epidemiological studies are necessary to stratifythe risk for cardiomyopathy in hypertensive patients withOCTN2 mutations. The identification of OCTN2 mutations inhypertensive patients and a prospective study to investigatethe efficacy of L-carnitine supplementation may contribute tothe development of novel strategies for the prevention andtreatment of cardiomyopathy and heart failure in thesesubjects.
AcknowledgmentsWe thank Dr Masahiko Nishimura of the Division for Research ofLaboratory Animals, Center for Research of Laboratory Animals andMedical Research Engineering, Nagoya University Graduate Schoolof Medicine, for caring for the animals.
Source of FundingThis work was supported in part by a grant from the Ministry ofEducation, Culture, Sports, Science, and Technology of Japan (No.14570655, to K.O.).
DisclosuresNone.
References1. Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate
and lipids. Prog Cardiovasc Dis. 1972;15:289–329.2. Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med.
1994;330:913–919.3. Bremer J. Carnitine metabolism and functions. Physiol Rev. 1983;63:
1420–1480.4. Nezu J, Tamai I, Oku A, Ohashi R, Yabuuchi H, Hashimoto N, Nikaido
H, Sai Y, Koizumi A, Shoji Y, Takada G, Matsuishi T, Yoshino M, KatoH, Ohura T, Tsujimoto G, Hayakawa J, Shimane M, Tsuji A. Primarysystemic carnitine deficiency is caused by mutations in a gene encodingsodium ion-dependent carnitine transporter. Nat Genet. 1999;21:91–94.
5. Stanley CA, DeLeeuw S, Coates PM, Vianey-Liad C, Divry P, BonnefontJP, Saudubray JM, Haymond M, Trefz FK, Breningstall GN, WappnerRS, Byrd DJ, Sansaricq C, Tein I, Grover W, Valle D, Rutledge SL,Treem WR. Chronic cardiomyopathy and weakness or acute coma inchildren with a defect in carnitine uptake. Ann Neurol. 1991;30:709–716.
6. Lamhonwah AM, Olpin SE, Pollitt RJ, Vianey-Saban C, Divry P, GuffonN, Besley GTN, Onizuka R, De Meirleir LJ, Cvitanovic-Sojat L, Baric I,Dionisi-Vici C, Fumic K, Maradin M, Tein I. Novel OCTN2 mutations:no genotype-phenotype correlations: early carnitine therapy prevents car-diomyopathy. Am J Med Genet. 2002;111:271–284.
7. Kuwajima M, Kono N, Horiuchi M, Imamura Y, Ono A, Inui Y, KawataS, Koizumi T, Hayakawa J, Saheki T, Tarui S. Animal model of systemiccarnitine deficiency: analysis in C3H-H-2o strain of mouse associatedwith juvenile visceral steatosis. Biochem Biophys Res Commun. 1991;174:1090–1094.
8. Kuwajima M, Lu K, Sei M, Ono A, Hayashi M, Ishiguro K, Ozaki K,Hotta K, Okita K, Murakami T, Miyagawa J, Narama I, Nikaido H,Hayakawa J, Nakajima H, Namba M, Hanafusa T, Matsuzawa Y, ShimaK. Characteristics of cardiac hypertrophy in the juvenile visceral steatosismouse with systemic carnitine deficiency. J Mol Cell Cardiol. 1998;30:773–781.
9. Takahashi R, Okumura K, Asai T, Hirai T, Murakami H, Murakami R,Numaguchi Y, Matsui H, Ito M, Murohara T. Dietary fish oil attenuatescardiac hypertrophy in lipotoxic cardiomyopathy due to systemic car-nitine deficiency. Cardiovasc Res. 2005;68:213–223.
10. Lahjouji K, Elimrani I, Wu J, Mitchell GA, Qureshi IA. A heterozygotephenotype is present in the jvs �/- mutant mouse livers. Mol GenetMetab. 2002;76:76–80.
11. Xiaofei E, Wada Y, Dakeishi M, Hirasawa F, Murata K, Masuda H,Sugiyama T, Nikaido H, Koizumi A. Age-associated cardiomyopathy in
heterozygous carrier mice of a pathological mutation of carnitine trans-porter gene, OCTN2. J Gerontol. 2002;57A:B270–B278.
12. Koizumi A, Nozaki J, Ohura T, Kayo T, Wada Y, Nezu J, Ohashi R,Tamai I, Shoji Y, Takada G, Kibara S, Matsuishi T, Tsuji A. Geneticepidemiology of the carnitine transporter OCTN2 gene in a Japanesepopulation and phenotypic characterization in Japanese pedigrees withprimary systemic carnitine deficiency. Hum Mol Genet. 1999;8:2247–2254.
13. Tein I. Carnitine transport: pathophysiology and metabolism of knownmolecular defects. J Inherit Metab Dis. 2003;26:147–169.
14. McGarry JD, Foster, DW. Free and esterified carnitine. Radiometricmethod. In: Bergmeyer J, Gra�l M, eds. Methods of Enzymatic Analysis.New York, NY: Academic Press; 1981:474–488.
15. Asai T, Okumura K, Takahashi R, Matsui H, Numaguchi Y, Murakami H,Murakami R, Murohara T. Combined therapy with PPAR� agonist andL-carnitine rescues lipotoxic cardiomyopathy due to systemic carnitinedeficiency. Cardiovasc Res. 2006;70:566–577.
16. Volonte MG, Yuln G, Quiroga P, Consolini AE. Development of anHPLC method for determination of metabolic compounds in myocardialtissue. J Pharm Biomed Anal. 2004;35:647–653.
17. Kagaya Y, Kanno Y, Takeyama D, Ishide N, Maruyama Y, Takahashi T,Ido T, Takishima T. Effects of long-term pressure overload on regionalmyocardial glucose and free fatty acid uptake in rats. Circulation. 1990;81:1353–1361.
18. Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD.Contribution of oxidative metabolism and glycolysis to ATP productionin hypertrophied hearts. Am J Physiol. 1994;267:H742–H750.
19. Christe ME, Rodgers RL. Altered glucose and fatty acid oxidation inhearts of the spontaneously hypertensive rat. J Mol Cell Cardiol. 1994;26:1371–1375.
20. Liao R, Jain M, Cui L, D’Agostino J, Aiello F, Luptak I, Ngoy S,Mortensen RM, Tian R. Cardiac-specific overexpression of GLUT1prevents the development of heart failure attributable to pressure overloadin mice. Circulation. 2002;106:2125–2131.
21. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metab-olism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129.
22. Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P,Williams L, Ashrafian H, Horowitz J, Fraser AG, Clarke K, FrenneauxM. Metabolic modulation with perhexiline in chronic heart failure: arandomized, controlled trial of short-term use of a novel treatment.Circulation. 2005;112:3280–3288.
23. Takahashi R, Okumura K, Matsui H, Saburi Y, Kamiya H, Matsubara K,Asai T, Ito M, Murohara T. Impact of �-tocopherol on cardiac hypertro-phy due to energy metabolism disorder: the involvement of 1,2-diacylglycerol. Cardiovasc Res. 2003;58:565–574.
24. Olson RE. Abnormalities of myocardial metabolism. Circ Res. 1964;14/15(suppl II):II109–II115.
25. Pool PE, Chandler EH, Sonnenblick, Braunwald E. Integrity of energystores in cat papillary muscle. Circ Res. 1968;22:213–219.
26. Katz AM. Cardiomyopathy of overload. A major determinant ofprognosis in congestive heart failure. N Engl J Med. 1990;322:110–111.
27. Ingwall JS, Weiss RG. Is the failing heart energy starved? On usingchemical energy to support cardiac function. Circ Res. 2004;95:135–145.
28. Alpert NR, Mulieri LA, Hasenfuss G. Myocardial chemo-mechanicalenergy transduction. In: Fozzard HA, Haber E, Katz AM, Jennings R,Morgan H, eds. The Heart and Cardiovascular System. New York, NY:Raven Press; 1992:111–128.
29. Swynghedauw B. Molecular mechanisms of myocardial remodeling.Physiol Rev. 1999;79:215–262.
30. Jamshidi Y, Montgomery HE, Hense HW, Myerson SG, Torra IP, StaelsB, World MJ, Doering A, Erdmann J, Hengstenberg C, Humphries SE,Schunkert H, Flavell DM. Peroxisome proliferators-activated receptor �gene regulates left ventricular growth in response to exercise and hyper-tension. Circulation. 2002;105:950–955.
31. de las Fuentes L, Herrero P, Peterson LR, Kelly DP, Gropler RJ,Davilla-Roman VG. Myocardial fatty acid metabolism: independent pre-dictor of left ventricular mass in hypertensive heart disease.Hypertension. 2003;41:83–87.
32. Saburi Y, Okumura K, Matsui H, Hayashi K, Kamiya H, Takahashi R,Matsubara K, Ito M. Changes in distinct species of 1,2-diacylglycerol incardiac hypertrophy due to energy metabolic disorder. Cardiovasc Res.2003;57:92–100.
502 Hypertension September 2007
by guest on October 30, 2014http://hyper.ahajournals.org/Downloaded from

- 1 -
Expanded Materials and Methods
Experimental Animals and Study Protocol
Heterozygous mutants of 11-week-old male JVS mice of the C3H strain were used.
Heterozygous JVS mice (jvs/+) were created by mating homozygous mutants and
heterozygous mutants of JVS mice. Heterozygous mutants were distinguished from
homozygous mutants, which have swollen fatty livers recognized through the abdominal wall
at 5 days after birth.1 Age-matched male WT mice of the C3H strain were used as controls.
Both WT and jvs/+ mice were divided into the following 3 groups: 1) sham operation; 2)
ascending aorta constriction (AAC); 3) 1% L-carnitine supplementation (10 g L-carnitine per
kg chow) for 4 weeks after AAC. L-Carnitine was not included in the ingredient of the
standard chow. AAC was created by ligation of the ascending aorta with a 27-gauge needle
using 7-0 silk suture in anesthetized (pentobarbital 50 mg/kg IP) and ventilated mice.
Sham-operated mice underwent the same surgical procedure without the ligation of the aorta.
All animals were studied 4 weeks after the operation. Animals were anesthetized with diethyl
ether before sacrifice. For survival analysis, cages were inspected daily for deceased animals
during the study period.
This study conformed to the Guide for the Care and Use of Laboratory Animals published
by the National Institutes of Health (NIH Publication No. 85-23, revised 1996). All protocols
described were approved by the Animal Ethics Committee of Nagoya University, Nagoya,
Japan.
Echocardiographic and Hemodynamic Measurements
On the day of sacrifice, the systolic blood pressure and heart rate were determined in each
animal. The tail-cuff method was used, employing a photoelectric tail cuff detection system,
Softron BP-98A (Softron). Left ventricular function was evaluated by transthoracic

- 2 -
echocardiography using SONOS 7500 (Philips Medical Systems) with a 10-MHz imaging
transducer. Briefly, each animal was slightly sedated with pentobarbital (20 mg/kg IP).
M-mode images of the left ventricle (LV) were recorded, and left ventricular dimensions at
end-diastole (LVEDD) and end-systole (LVESD) were measured by the leading edge method.
For each measurement, data from three cardiac cycles were averaged, and left ventricular
fractional shortening (LVFS) was calculated as LVFS (%) = (LVEDD-LVESD)/LVEDD x 100.
To evaluate the degree of stenosis, the pressure gradient across the constriction was assessed
by Doppler echocardiography at 3 days after operation. The transducer was placed at the apex
and oriented toward the proximal ascending aorta. The peak velocity (m/s) was measured, and
the maximum instantaneous gradient (mmHg) was calculated by use of the Bernoulli equation:
pressure gradient = 4 x (velocity)2.
Plasma and Myocardial Total Carnitine Levels
Plasma and myocardial total carnitine levels were determined by an enzymatic method
described elsewhere.2
Histological Analysis
Heart tissue was examined by means of light microscopy. The tissue was fixed in 10%
formaldehyde in phosphate buffer and was embedded in paraffin. Transverse sections (4 µm
thickness) at the level of papillary muscle were stained with hematoxylin-eosin (H-E) or with
Masson’s trichrome. The myocyte cross-sectional area of LV was measured from myocytes
that were cut transversely and exhibited both a nucleus and an intact cell membrane. At least
200 myocytes per LV were assessed and the average value was used for analysis. The extent of
fibrosis was evaluated by calculating the ratio of Masson’s trichrome-stained fibrotic area to
the total area of the myocardium with the use of the image analysis software WinROOF
(Mitani Corp.).

- 3 -
Real-Time Quantitative RT-PCR
Total RNA was extracted from the left ventricle and reverse transcription was performed by a
previously described method.3 To examine the relative mRNA expression of atrial natriuretic
peptide (ANP), quantitative reverse transcription (RT)-polymerase chain reaction (PCR)
analysis was performed using the Stratagene Mx3000PTM QPCR System (Stratagene) and
Power SYBR® Green PCR Master Mix (Applied Biosystems). Sequences of the sense and
antisense primers used for amplification were: ANP, 5’-CGTCTTGGCCTTTTGGCT-3’ and
5’-CCAGGTGGTCTAGCAGGTTCTT-3’, product size: 104 bp; r18S,
5’-GTAACCCGTTGAACCCCATT-3’ and 5’-CCATCCAATCGGTAGTAGCG-3’, product
size: 151 bp. Standard curves were generated by full sequence plasmids of known
concentrations. Gene expression of ANP mRNA was normalized to r18S and was expressed as
fold increase over sham-operated WT mice.
Myocardial High-Energy Phosphate Content
Animals were anesthetized with diethyl ether and the hearts were rapidly excised and
snap-frozen with liquid nitrogen. The frozen tissue was homogenized in ice-cold 0.5 mol/L
perchloric acid for deproteinization, and then neutralized with 2 mol/L K2CO3. After
centrifugation, the supernatant was filtered and an aliquot of 20 µL was used for measurement.
Phosphocreatinine (PCr) and adenosine triphosphate (ATP) contents were measured by
high-performance liquid chromatography (HPLC), as previously described.4 Briefly, HPLC
was performed with the use of the Hewlett Packard Model 1100 HPLC system consisting of an
1100 quaternary pump, variable wavelength UV-VIS detector, Hypersil ODS C18
reversed-phase column (250 mm x 2.1 mm i.d., particle size 5 µm), and ChemStation software.
The mobile phase was composed of 215 mmol/L potassium dihydrogen phosphate, 2.3
mmol/L tetrabutyl ammonium hydrogen sulfate, 4% acetonitrile, and 4 mmol/L potassium
hydroxide, and the flow rate was 300 µL/min. The absorbance was monitored at 220 nm.

- 4 -
Myocardial Lipid Analysis
The lyophilized tissue samples were homogenized in 5 mL of a chilled chloroform-methanol
mixture (2:1, v/v) containing 0.01% butylated hydroxytoluene as an antioxidant and
cholesteryl acetate as an internal standard. Simultaneous quantitation of 1,2-diacylglycerol
and triacylglycerol was performed by the TLC–FID method, as previously described.5 In brief,
1 µL of a lipid extract solution containing ceramides, neutral lipids, and free fatty acids was
dissolved in chloroform and applied carefully to silica gel using 75-µm precoated thin-layer
rods (Chromarod-SIII, Iatron Lab., Inc.). The first development was carried out in a solvent
system of chloroform–methanol–H2O (57:12:0.6, v/v). The second and the third developments
were carried out in 1,2-dichloroethane–chloroform–acetic acid (46:6:0.05, v/v). The fourth
development was carried out in n-hexane–diethyl ether–acetic acid (98:1:1, v/v). The
Chromarods were then scanned in an Iatroscan MK-5 analyzer (Iatron Lab., Inc.). The fatty
acid composition of 1,2-diacylglycerol obtained by TLC was determined by gas
chromatography (model GC 14-A, Shimadzu) using an HR-SS-10 fused silica capillary
column (30 m x 0.25 mm internal diameter, Shinwakakoh), as previously described.6
Statistics
All values are expressed as mean±SEM. The survival analysis was performed by the
Kaplan-Meier method, and between-group differences in survival were tested by the log-rank
test. Between-group comparisons were assessed by one-way ANOVA, followed by the
Bonferroni post hoc test. A value of P<0.05 was considered to be statistically significant.

- 5 -
Results of Myocardial Lipid Analysis
Table S2 shows the results of myocardial lipid analysis at 4 weeks after operation. In
sham-operated mice, the myocardial triacylglycerol level was 1.6-fold higher and the
1,2-diacylglycerol level was 1.7-fold higher in jvs/+ mice than in WT mice. However, in mice
subjected to AAC, the myocardial triacylglycerol level was significantly reduced to identical
levels in both WT and jvs/+ mice, and the 1,2-diacylglycerol level in jvs/+ mice was
significantly reduced to WT mice levels. L-Carnitine supplementation did not alter the
myocardial triacylglycerol or 1,2-diacylglycerol levels in both WT and jvs/+ mice subjected to
AAC.
In sham-operated mice, the fatty acid composition of myocardial 1,2-diacylglycerol showed
that C14:0 and C18:0 were modestly reduced, and C18:2n-6 was modestly increased in jvs/+
mice compared to WT mice. However, these changes were not significant. In mice subjected
to AAC, C18:2n-6 and C20:1n-9 were significantly reduced in both WT and jvs/+ mice
compared to sham-operated groups. C14:0 and C18:0 were significantly increased in AAC
groups compared to sham-operated jvs/+ mice. L-Carnitine supplementation did not alter the
fatty acid composition of myocardial 1,2-diacylglycerol in both WT and jvs/+ mice subjected
to AAC.

- 6 -
References
1. Kuwajima M, Kono N, Horiuchi M, Imamura Y, Ono A, Inui Y, Kawata S, Koizumi T,
Hayakawa J, Saheki T, Tarui S. Animal model of systemic carnitine deficiency: analysis
in C3H-H-2o strain of mouse associated with juvenile visceral steatosis. Biochem Biophys
Res Commun. 1991;174:1090-1094.
2. McGarry JD, Foster, DW. Free and esterified carnitine. Radiometric method. In:
Bergmeyer J, Graßl M, eds. Methods of Enzymatic Analysis. New York: Academic Press,
1981:474-488.
3. Asai T, Okumura K, Takahashi R, Matsui H, Numaguchi Y, Murakami H, Murakami R,
Murohara T. Combined therapy with PPARα agonist and L-carnitine rescues lipotoxic
cardiomyopathy due to systemic carnitine deficiency. Cardiovasc Res. 2006;70:566-577.
4. Volonté MG, Yuln G, Quiroga P, Consolini AE. Development of an HPLC method for
determination of metabolic compounds in myocardial tissue. J Pharm Biomed Anal.
2004;35:647-653.
5. Okumura K, Hayashi K, Morishima I, Murase K, Matsui H, Toki Y, Ito T. Simultaneous
quantitation of ceramides and 1,2-diacylglycerol in tissues by Iatroscan thin-layer
chromatography–flame-ionization detection. Lipids. 1998;33:529-532.
6. Takahashi R, Okumura K, Matsui H, Saburi Y, Kamiya H, Matsubara K, Asai T, Ito M,
Murohara T. Impact of α-tocopherol on cardiac hypertrophy due to energy metabolism
disorder: the involvement of 1,2-diacylglycerol. Cardiovasc Res. 2003;58:565-574.

- 7 -
Figure Legend for Figure S1
Figure S1. A, Masson’s trichrome staining of the hearts at 4 weeks after ascending aorta
constriction (AAC). Bars indicate 100 µm. B, Quantitative data of the area of fibrosis in the
hearts. Values are mean±SEM. n=4 each group. *P<0.01 vs WT-sham; †P<0.01 vs
(jvs/+)-sham.

- 8 -
TABLE S1. Gravimetric, Hemodynamic, and Echocardiographic Measurements, and Total
Carnitine Levels at Baseline
Mice/Parameter WT jvs/+
BW, g 23.1±0.1 23.1±0.4
HW, mg 94.8±2.2 94.5±2.0
HW/BW, mg/g 4.10±0.10 4.09±0.13
Wet LW/BW, mg/g 5.18±0.14 5.46±0.04
Wet LW/dry LW 4.63±0.03 4.70±0.05
Systolic BP, mmHg 117±1 116±4
HR, bpm 561±4 561±7
LVEDD, mm 2.9±0.1 2.8±0.1
LVESD, mm 1.2±0.1 1.2±0.1
LVFS, % 56.5±0.4 55.7±0.9
Total carnitine levels
Plasma, µmol/L 70.2±1.1 52.8±0.9*
Myocardium, nmol/g 642.3±30.1 349.2±25.9*
Data are mean±SEM. n=6 to 8 for each group. BW indicates body weight; HW, heart weight;
LW, lung weight; BP, blood pressure; HR, heart rate; LVEDD, left ventricular end-diastolic
dimension; LVESD, left ventricular end-systolic dimension; LVFS, left ventricular fractional
shortening.
*P<0.01 vs WT.

- 9 -
TABLE S2. Myocardial Lipid Analysis at 4 Weeks After AAC
Sham AAC Operation and mice/Parameter
WT jvs/+ WT WT+CAR jvs/+ (jvs/+)+CAR
Triacylglycerol, µg/mg dry weight 10.0±1.2 15.9±2.1* 2.6±0.2*† 2.5±0.3*† 2.6±0.1*† 3.3±0.3*†
1,2-Diacylglycerol, ng/mg dry weight 218±27 383±83* 195±64† 211±34† 181±41† 184±72†
Fatty acid composition of 1,2-diacylglycerol, % of total fatty acids
C14:0 10.9±1.3 9.1±0.8 13.0±1.1† 12.8±0.4† 13.7±1.8† 14.3±1.0†
C16:0 23.2±1.8 24.2±1.2 23.1±1.2 26.1±0.5 24.7±0.9 25.1±0.7
C18:0 10.9±1.5 8.7±0.5 13.0±2.0† 13.1±1.4† 14.9±1.2† 13.8±0.8†
C18:1n-7,9 16.2±1.3 16.7±0.8 13.9±0.7 16.7±1.8 14.0±0.7 16.7±0.6
C18:2n-6 19.1±2.9 23.5±1.8 11.5±2.2*† 10.9±1.4*† 10.9±2.5*† 9.5±1.3*†
C20:1n-9 1.7±0.2 1.6±0.1 1.1±0.1*† 0.8±0.1*† 1.1±0.2*† 0.9±0.1*†
C20:4n-6 2.3±0.1 2.0±0.2 2.0±0.2 2.3±0.2 1.9±0.1 2.0±0.2
C24:1n-9 1.6±0.3 1.7±0.2 1.8±0.2 1.4±0.2 1.7±0.4 1.8±0.1
Data are mean±SEM. n=4 to 6 for each group. AAC indicates ascending aorta constriction; CAR, L-carnitine.
*P<0.05 vs WT-sham; †P<0.05 vs (jvs/+)-sham.

- 10 -
Figure S1
WT
jvs/+
Sham AAC AAC + L-CarnitineA
B
02468
1012
Are
a of
fibr
osis
(%)
∗† ∗† ∗† ∗†
L-Carnitine (–) (–) (–) (+) (–) (+)Sham AAC
WT jvs/+
Copyright © 2022 FDOKUMEN




















