
Prediction of signal recognition particle RNA genes
10
Prediction of signal recognition particle RNA genes Marco Regalia 1 , 2 , Magnus Alm Rosenblad 1 and Tore Samuelsson 1 , * 1 Department of Medical Biochemistry, Goteborg University, Box 440, SE-405 30 Goteborg, Sweden and 2 Department of General Physiology and Biochemistry, University of Milan, Via Celoria 20, Milan, I-20133, Italy Received April 8, 2002; Revised and Accepted June 17, 2002 ABSTRACT We describe a method for prediction of genes that encode the RNA component of the signal recogni- tion particle (SRP). A heuristic search for the strongly conserved helix 8 motif of SRP RNA is combined with covariance models that are based on previously known SRP RNA sequences. By screen- ing available genomic sequences we have identified a large number of novel SRP RNA genes and we can account for at least one gene in every genome that has been completely sequenced. Novel bacterial RNAs include that of Thermotoga maritima, which, unlike all other non-gram-positive eubacteria, is pre- dicted to have an Alu domain. We have also found the RNAs of Lactococcus lactis and Staphylococcus to have an unusual UGAC tetraloop in helix 8 instead of the normal GNRA sequence. An investi- gation of yeast RNAs reveals conserved sequence elements of the Alu domain that aid in the analysis of these RNAs. Analysis of the human genome reveals only two likely genes, both on chromosome 14. Our method for SRP RNA gene prediction is the first convenient tool for this task and should be useful in genome annotation. INTRODUCTION Although there are many efficient tools for the identification of protein genes in genome sequences, we lack tools for the analysis of many non-coding RNAs. As a consequence most of the finished genome sequences published today lack annota- tion information about these RNAs. One difficulty with the identification of non-coding RNAs is that sequence tends to be poorly conserved and, as a consequence, standard tools such as BLAST (1) may be used only in the identification of orthologs in fairly closely related organisms. On the other hand many non-coding RNAs tend to have conserved secondary structure elements that may be used for their identification. Methods that have been used so far include the ‘covariance models’ developed by Eddy and Durbin (2) as well as ‘pattern matching’, which is based on regular expression matching where base-pairing schemes are also taken into account. In the latter category there is PatScan (http://www-unix.mcs.anl. gov/compbio/PatScan/HTML/patscan.html) as well as the ‘rnabob’ tool of Sean Eddy (Washington University School of Medicine, St Louis, MO; http://www.genetics.wustl.edu/ eddy/software/#rnabob). In this work we have studied methods to identify the RNA component of the signal recognition particle (SRP). The SRP plays an important role in membrane targeting (3 and references therein). Some of its structural and functional features have been highly conserved during evolution and SRP has been identified in all three kingdoms of life. In eukaryotic cells, the SRP targets secretory proteins to the endoplasmic reticulum (ER) membrane. Eukaryotic SRP comprises one RNA molecule and six proteins, including SRP54, a GTPase. As the signal sequence of the secretory protein emerges from the ribosome it is recognized by the SRP54 protein. In the resulting ribosome–SRP complex, protein synthesis is arrested and the complex eventually docks to the SRP receptor at the ER membrane. Here, SRP is released, protein synthesis is resumed and translocation of the nascent chain in the ER membrane is initiated. The SRP receptor is composed of two subunits, SRa and SRb, which are both GTPase proteins. The SRP pathway in bacteria is similar to that in eukaryotes although the major substrates for the bacterial SRP seem to be integral membrane proteins (4–6). In Escherichia coli the SRP has only two components: a 4.5S RNA and the Ffh protein, homologous to SRP54. The bacterial FtsY protein is homologous to SRa. The overall design of SRP RNA is somewhat variable, as shown in Figure 1. However, they all share a common motif that will be referred to here as the helix 8 motif. The helix 8 region of the RNA binds to the M domain of the SRP54 protein, a domain that also harbors the signal sequence binding activity. Many studies show that the SRP RNA has an important biological role. For instance, the E.coli 4.5S RNA is necessary for viability (7) and in an in vitro system the SRa can stimulate the GTPase activity of SRP54 only when it is bound to the RNA (8). It has been shown that the E.coli 4.5S RNA plays an important role in the GTPase cycles of Ffh and FtsY (9,10). Furthermore, the three-dimensional structure of the helix 8/SRP54 domain of SRP has recently been elucidated (11) and this structure suggests that the signal sequence binding surface is composed not only of protein but also of RNA elements. The Alu domain is believed to be responsible for a translational arrest activity of SRP and contains the Alu domain of the RNA in complex with the heterodimer SRP9/ 14. Also this complex has been studied in structural detail (12). The SRP9/14 dimer is bound to a conserved core of the RNA Alu domain, which forms a U-turn that connects two *To whom correspondence should be addressed. Tel: +46 31 773 34 68; Fax: +46 31 41 61 08; Email: [email protected] 3368–3377 Nucleic Acids Research, 2002, Vol. 30 No. 15 ª 2002 Oxford University Press
-
Upload
udistrital -
Category
Documents
-
view
3 -
download
0
Transcript of Prediction of signal recognition particle RNA genes

Prediction of signal recognition particle RNA genesMarco Regalia1,2, Magnus Alm Rosenblad1 and Tore Samuelsson1,*
1Department of Medical Biochemistry, Goteborg University, Box 440, SE-405 30 Goteborg, Sweden and2Department of General Physiology and Biochemistry, University of Milan, Via Celoria 20, Milan, I-20133, Italy
Received April 8, 2002; Revised and Accepted June 17, 2002
ABSTRACT
We describe a method for prediction of genes thatencode the RNA component of the signal recogni-tion particle (SRP). A heuristic search for thestrongly conserved helix 8 motif of SRP RNA iscombined with covariance models that are based onpreviously known SRP RNA sequences. By screen-ing available genomic sequences we have identi®eda large number of novel SRP RNA genes and we canaccount for at least one gene in every genome thathas been completely sequenced. Novel bacterialRNAs include that of Thermotoga maritima, which,unlike all other non-gram-positive eubacteria, is pre-dicted to have an Alu domain. We have also foundthe RNAs of Lactococcus lactis and Staphylococcusto have an unusual UGAC tetraloop in helix 8instead of the normal GNRA sequence. An investi-gation of yeast RNAs reveals conserved sequenceelements of the Alu domain that aid in the analysisof these RNAs. Analysis of the human genomereveals only two likely genes, both on chromosome14. Our method for SRP RNA gene prediction is the®rst convenient tool for this task and should beuseful in genome annotation.
INTRODUCTION
Although there are many ef®cient tools for the identi®cation ofprotein genes in genome sequences, we lack tools for theanalysis of many non-coding RNAs. As a consequence most ofthe ®nished genome sequences published today lack annota-tion information about these RNAs. One dif®culty with theidenti®cation of non-coding RNAs is that sequence tends to bepoorly conserved and, as a consequence, standard tools such asBLAST (1) may be used only in the identi®cation of orthologsin fairly closely related organisms. On the other hand manynon-coding RNAs tend to have conserved secondary structureelements that may be used for their identi®cation. Methodsthat have been used so far include the `covariance models'developed by Eddy and Durbin (2) as well as `patternmatching', which is based on regular expression matchingwhere base-pairing schemes are also taken into account. In thelatter category there is PatScan (http://www-unix.mcs.anl.gov/compbio/PatScan/HTML/patscan.html) as well as the`rnabob' tool of Sean Eddy (Washington University School
of Medicine, St Louis, MO; http://www.genetics.wustl.edu/eddy/software/#rnabob).
In this work we have studied methods to identify the RNAcomponent of the signal recognition particle (SRP). The SRPplays an important role in membrane targeting (3 andreferences therein). Some of its structural and functionalfeatures have been highly conserved during evolution andSRP has been identi®ed in all three kingdoms of life. Ineukaryotic cells, the SRP targets secretory proteins to theendoplasmic reticulum (ER) membrane. Eukaryotic SRPcomprises one RNA molecule and six proteins, includingSRP54, a GTPase. As the signal sequence of the secretoryprotein emerges from the ribosome it is recognized by theSRP54 protein. In the resulting ribosome±SRP complex,protein synthesis is arrested and the complex eventually docksto the SRP receptor at the ER membrane. Here, SRP isreleased, protein synthesis is resumed and translocation of thenascent chain in the ER membrane is initiated. The SRPreceptor is composed of two subunits, SRa and SRb, whichare both GTPase proteins.
The SRP pathway in bacteria is similar to that in eukaryotesalthough the major substrates for the bacterial SRP seem to beintegral membrane proteins (4±6). In Escherichia coli the SRPhas only two components: a 4.5S RNA and the Ffh protein,homologous to SRP54. The bacterial FtsY protein ishomologous to SRa.
The overall design of SRP RNA is somewhat variable, asshown in Figure 1. However, they all share a common motifthat will be referred to here as the helix 8 motif. The helix 8region of the RNA binds to the M domain of the SRP54protein, a domain that also harbors the signal sequence bindingactivity. Many studies show that the SRP RNA has animportant biological role. For instance, the E.coli 4.5S RNA isnecessary for viability (7) and in an in vitro system the SRacan stimulate the GTPase activity of SRP54 only when it isbound to the RNA (8). It has been shown that the E.coli 4.5SRNA plays an important role in the GTPase cycles of Ffh andFtsY (9,10). Furthermore, the three-dimensional structure ofthe helix 8/SRP54 domain of SRP has recently been elucidated(11) and this structure suggests that the signal sequencebinding surface is composed not only of protein but also ofRNA elements.
The Alu domain is believed to be responsible for atranslational arrest activity of SRP and contains the Aludomain of the RNA in complex with the heterodimer SRP9/14. Also this complex has been studied in structural detail(12). The SRP9/14 dimer is bound to a conserved core of theRNA Alu domain, which forms a U-turn that connects two
*To whom correspondence should be addressed. Tel: +46 31 773 34 68; Fax: +46 31 41 61 08; Email: [email protected]
3368±3377 Nucleic Acids Research, 2002, Vol. 30 No. 15 ã 2002 Oxford University Press
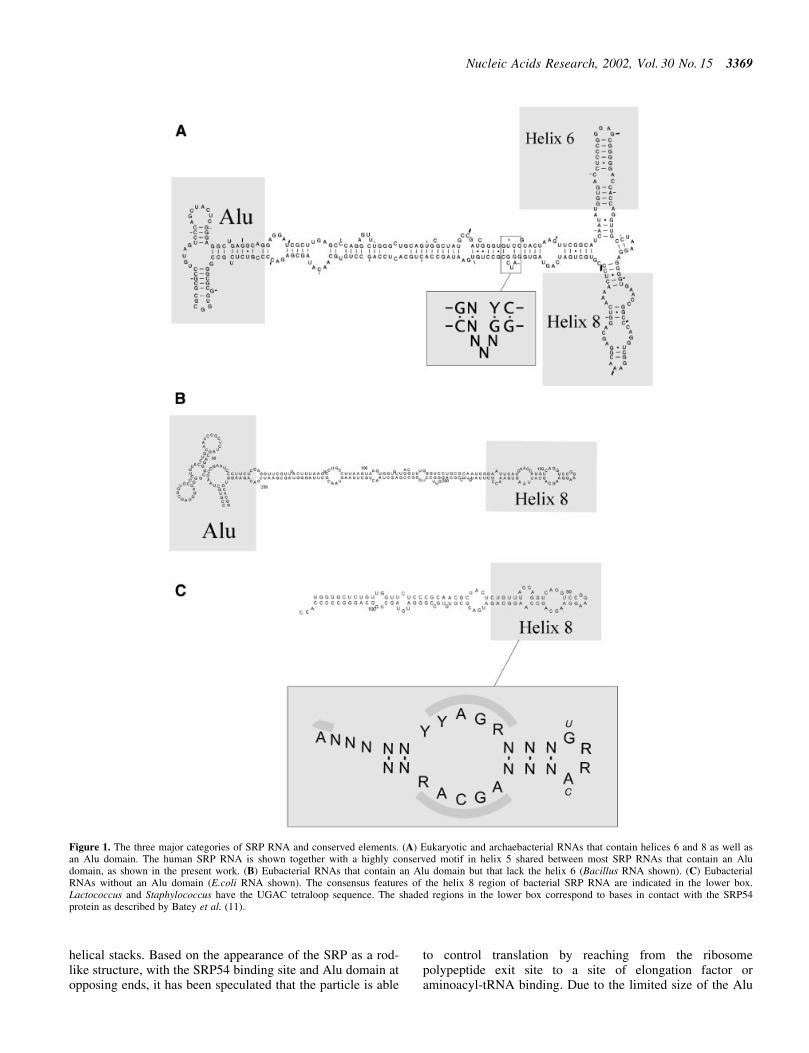
helical stacks. Based on the appearance of the SRP as a rod-like structure, with the SRP54 binding site and Alu domain atopposing ends, it has been speculated that the particle is able
to control translation by reaching from the ribosomepolypeptide exit site to a site of elongation factor oraminoacyl-tRNA binding. Due to the limited size of the Alu
Figure 1. The three major categories of SRP RNA and conserved elements. (A) Eukaryotic and archaebacterial RNAs that contain helices 6 and 8 as well asan Alu domain. The human SRP RNA is shown together with a highly conserved motif in helix 5 shared between most SRP RNAs that contain an Aludomain, as shown in the present work. (B) Eubacterial RNAs that contain an Alu domain but that lack the helix 6 (Bacillus RNA shown). (C) EubacterialRNAs without an Alu domain (E.coli RNA shown). The consensus features of the helix 8 region of bacterial SRP RNA are indicated in the lower box.Lactococcus and Staphylococcus have the UGAC tetraloop sequence. The shaded regions in the lower box correspond to bases in contact with the SRP54protein as described by Batey et al. (11).
Nucleic Acids Research, 2002, Vol. 30 No. 15 3369

domain it could well enter into a tRNA or elongation factorbinding site.
There are three major classes of SRP RNA with respect toits domain organization (Fig. 1). Archaebacterial RNAs areeukaryote-like with an Alu domain as well as a helix 6domain. A group of gram-positive bacteria, includingBacillus, has an Alu domain but lacks the helix 6 region.The remaining eubacteria-like proteobacteria have a simplerRNA that lacks the Alu domain as well as the helix 6 domain.
The great diversity in terms of SRP RNA structure hashampered the development of convenient methods for theiridenti®cation in genome sequences. In the context of theSignal Recognition Particle Database (SRPDB) (13) we havepreviously attempted to identify SRP RNA genes using acombination of pattern searches and sequence similaritysearches. However, we have now improved our searchstrategy considerably and developed an automated methodthat is based on pattern matching and the COVE programs.Using this method we have systematically searched for SRPRNA genes in public sequence data and have discovered anumber of novel SRP RNAs as well as novel features of theseRNAs.
MATERIALS AND METHODS
Public sequence data were downloaded from the NCBI andEBI ftp sites. Finished sequence data as well as HTG sectionswere retrieved. Human expressed sequence tag (EST)sequences were downloaded and used to obtain evidence ofexpressed SRP RNA genes. Genomes of microorganisms werealso obtained from the TIGR CMR database at http://www.tigr.org/tigr-scripts/CMR2/CMRGenomes.spl and sitescited there. Sequences of Candida albicans were downloadedfrom the Stanford Genome Technology Center websiteat http://www-sequence.stanford.edu/group/candida (sequenc-ing of C.albicans was accomplished with the support of theNIDR and the Burroughs Wellcome Fund). Neurosporasequences were from the Neurospora Sequencing Project,Whitehead Institute/MIT Center for Genome Research (http://www-genome.wi.mit.edu). The data set used in these studieswas neurospora_1.fasta.
Rnabob. Rnabob source code was obtained from http://www.genetics.wustl.edu/eddy/software/#rnabob. Rnabob descrip-tors were constructed for the different categories: (i) archae-bacteria and eubacteria with GRRA loop; (ii) eubacteria withTRRC loop; (iii) plants; (iv) yeasts; and (v) metazoans. Theyare listed and described in more detail at the website http://bio.lundberg.gu.se/srpscan/. The rnabob source code wasslightly modi®ed to produce output with sequences ¯ankingthe descriptor pattern and to be able to produce sequenceoutput in fasta format. It was used to scan the sequencedatabases with the appropriate descriptors for the differentclasses of SRP RNA genes and to produce outputs in fastaformat using the -f option. The -s (skip) option was used toavoid non-signi®cant hits in regions of `N' repeats.
COVE. The COVE programs (version 2.4.2) were obtainedfrom http://www.genetics.wustl.edu/eddy/software/#cove.COVE statistical models were built from the SRP RNAgenes alignments available on the SRPDB website at http://
bio.lundberg.gu.se/dbs/SRPDB/srprna.html using the -aoption of the covet (train) program. The same sequences,unaligned, were used as initial training data for the models.The -m (maximum likelihood) option was used to give morereliable results. Statistical models were built for the followingcategories: (i) prokaryotes without Alu domain; (ii) pro-karyotes with Alu domain; (iii) yeasts; and (iv) non-yeasteukaryotes. Each model was trained on the subset ofsequences in the SRPDB belonging to organisms of thatparticular category. Once novel sequences were found, modelswere rebuilt on the new set of sequences to obtain a higheraccuracy. The covels (local score) program was used to searchrnabob fasta outputs for SRP RNA gene candidates. A list wasproduced with the sequences with the best match to thestatistical model used. The -w (window) option was set to avalue slightly larger than the expected maximum size for anSRP RNA. Alignments of the sequences were constructedusing the covea (align) program and the appropriate statisticalmodel for each category. The -o (out®le) and -s (score®le)options were used to produce ®le outputs with alignments andscores of similarity to the model. SRP RNA genes alignmentsand COVE covariance statistical models are available at thewebsite http://bio.lundberg.gu.se/srpscan/.
Mfold. Mfold version 3.1 was generously provided byDr M. Zuker (Rensselaer Polytechnic Institute, Troy, NY)(14). It was used to predict the secondary structure ofpredicted SRP RNAs. To obtain a folding consistent withthe secondary structure constraints implicit in the rnabobdescriptors or in the COVE models, we used these constraintsas input to mfold in many instances.
Automated procedure. Perl scripts were used to produce anautomatic procedure where genomic sequences are searchedusing rnabob with descriptors of the helix8 motif in combin-ation with the COVE programs. As an alternative, rnabob maybe used alone, where one makes use of descriptors for helix8as well as for the Alu domain to obtain candidates for the fullRNA sequence.
RESULTS AND DISCUSSION
A protocol for identi®cation of SRP RNA genes
As many other non-coding RNAs, SRP RNA is poorlyconserved with respect to sequence and effective methods forRNA identi®cation have to incorporate secondary structureinformation. The region most highly conserved in SRP RNA isthe helix 8 region, which is characterized by a speci®c base-pairing scheme in combination with primary sequenceelements, as indicated in Figure 1. It is interesting to notethat there is a strong correlation between the consensusfeatures and the sites of protein interaction as revealed by thestructure of an SRP54±RNA complex (Fig. 1C) (11).
We have developed a procedure to identify potential SRPRNA genes in genome sequences where the ®rst step relies onthe identi®cation of the helix 8 motif. We search for this motifusing a slightly modi®ed version of the RNA motif ®nderrnabob (http://www.genetics.wustl.edu/eddy/software). Thehelix 8 motif is retrieved together with appropriate ¯ankingsequences. As a second step, the sequence(s) retrieved in the
3370 Nucleic Acids Research, 2002, Vol. 30 No. 15

®rst step may be further analyzed using rnabob with a moredetailed descriptor that also takes into account the Alu domainmotif. An example of such a descriptor for plant RNAs isshown in Figure 2. However, a more accurate, althoughslower, method is to use the probabilistic covariance modelsimplemented in the COVE programs (2) to analyze thesequences retrieved in the ®rst pattern matching step where amodel is produced on the basis of previously known SRP RNAsequences. The COVE programs were also highly useful inproducing multiple alignments of SRP RNA sequences, toidentify conserved motifs in these RNAs and to predict the 5¢and 3¢ ends of the RNA, as will be described in more detailbelow. It should be noted that COVE is computationallydemanding and therefore it is not feasible to analyze completegenome sequences without a ®rst heuristic step. It is possiblethat the pattern-matching approach as a ®rst step will excludesequences that would obtain a high score from COVE, but wehave tried to minimize this problem by using relativelydegenerate descriptors in the rnabob search.
As SRP RNAs are very different in different taxonomicgroups, it was not possible to use a single helix 8 descriptor for
rnabob or a single COVE model for an ef®cient identi®cationof all SRP RNAs. For instance, the helix 8 motif is different inplants and certain yeasts that have a 6 nt (GYUUCA) loopinstead of the normal tetraloop sequence, and a few bacteriahave a UGAC tetraloop (see below). Therefore, for theproblem of gene identi®cation we had to consider differentclasses of SRP RNA such as archaebacteria, eubacteria withand without Alu domain, plants, yeasts and metazoans.
A web server for the prediction of SRP RNA genes isavailable at http://bio.lundberg.gu.se/srpscan/. These pagesalso present more detailed information on the work describedhere. The web service also presents the potential secondarystructure of the predicted RNA sequence according to mfold(14). In this procedure the folding is constrained by the basepairing as speci®ed by the rnabob descriptor or by the COVEmodel used.
Identifying the helix 8 motif in bacterial genomesequences
As mentioned above, the ®rst step of our SRP RNA gene®nding procedure is one where we search for the helix 8 motif.As a starting point we designed a pattern based on allpreviously known SRP RNA sequences as collected from theSRPDB (13). A helix 8 motif NNAGG±xxx±GNRA±yyy±AGCAG (where xxx pairs with yyy) was derived fromavailable SRP RNA genes. We then used rnabob to identifythis pattern in genome sequence data. From a majority ofbacterial genomes, this motif identi®ed one gene that could besuccessfully folded into a hairpin structure characteristic ofSRP RNAs.
However, in a few genomes, such as in Vibrio cholerae andLactococcus lactis, no SRP RNA could be identi®ed using thismotif. To resolve this problem we made BLAST (1) orFASTA (15) searches using SRP RNA sequence from aclosely related organism. Hence, the E.coli sequence was usedas a query in a BLAST or FASTA search of the V.choleraegenome. This result revealed one homologous sequence inVibrio that could be folded into a hairpin structure and that hadthe consensus features of the helix 8, except that in one of thebulges it had NNAGA instead of the normal NNAGG. Wethen made a rnabob search in bacterial genomic sequenceswith a revised pattern, NNAGR±xxx±GNRA±yyy±AGCAG.As expected, this search revealed the SRP RNA genecandidate in Vibrio but also in Deinococcus radiodurans andBuchnera spp., two other strains for which we previouslyfailed to identify an SRP RNA.
The pattern as described still failed to identify an RNA inthe strains Staphylococcus aureus and L.lactis. A FASTAsearch of the Lactococcus genome using the Bacillus subtilisSRP RNA revealed a homolog that had the characteristics ofSRP RNA but, instead of the normal GGAA tetraloop, it hadthe unusual tetraloop sequence UGAC (Fig. 3). This unex-pected ®nding led us to search for a helix 8 pattern where wehad substituted the GNRA tetraloop for UGAC. Using rnabobwith such a pattern also revealed SRP RNA candidates inS.aureus (Fig. 3) and Staphylococcus equi. Thus, bothLactococcus and Staphylococcus have the helix 8 motif butwith the UGAC tetraloop. Even though this is a very unusualtetraloop sequence we believe that we have identi®ed the trueSRP RNA gene homologs. This is because the sequencesimilarity between these RNAs and SRP RNAs of other
Figure 2. Characteristics of plant SRP RNAs. Consensus features of helix 8and Alu domains are shown schematically, as well as descriptor for rnabobbased on these features. N represents any nucleotide and an asterisk indi-cates an optional nucleotide. The numbers refer to allowed number of basesof Alu domain loops and spacers between the Alu and helix 8 domains.
Nucleic Acids Research, 2002, Vol. 30 No. 15 3371

closely related bacteria is highly signi®cant. Furthermore,there are no other obvious SRP RNA gene candidates inLactococcus and Staphylococcus. Finally, the SRP RNA genecandidates with the UGAC loop are not part of any knownprotein-coding regions.
It is interesting to note that the UGAC tetraloop is found intwo species that both have close relatives with the normal loopsequence. Thus, Staphylococcus is most closely related toBacillus, and Lactococcus is most closely related toStreptococcus. A phylogenetic tree with these sequences isshown in Figure 4. It would therefore seem as if the changefrom GGAA to UGAC loop sequence occurred at least twiceduring evolution. This suggests that the UGAC loop confers
some biological advantage in these gram-positive bacteria.Recently, evidence has been presented that the tetraloopsequence of E.coli SRP RNA (4.5S RNA) is important for theinteraction of FtsY, the receptor for bacterial SRP (16). In thecontext of this evidence it will be interesting to test thefunction of the UGAC tetraloop experimentally as well as tostudy its structural role in the helix 8 domain.
For every prokaryotic genome we examined we identi®edonly one possible SRP RNA gene candidate. When we usedrnabob alone for prediction there was sometimes more thanone gene candidate to one genome, but most often falsepositives could be identi®ed because they did not fold into thehairpin structure characteristic of SRP RNAs or because they
Figure 3. Comparison of SRP RNAs with GGAA and UGAC tetraloops. Streptococcus pyogenes and Lactococcus are closely related in sequence andsecondary structure but differ with respect to their tetraloop sequence. Similarly, the Bacillus and Staphylococcus RNAs, both with an Alu domain, are closelyrelated but differ in their tetraloop sequence. Also shown is the predicted RNA of T.maritima, which was unexpectedly found to have an Alu domain like theBacillus group of RNAs.
3372 Nucleic Acids Research, 2002, Vol. 30 No. 15

were part of protein-coding regions. At the same time, genepredictions for bacterial genomes with the COVE models werealways very speci®c, i.e. for each genome tested we observedonly one gene candidate. When we used a COVE model withthe helix 8 domain only to score potential SRP RNA genecandidates obtained from an rnabob search we sometimesobserved 16S rRNA genes with a relatively high score.However, these hits are not likely to be signi®cant as theregions in 16S RNA identi®ed by COVE are not the same inthe different RNAs.
Bacterial Alu domains
Archaebacteria and certain eubacteria have SRP RNAs withdomains that seem to be analogous to the Alu domains ofeukaryotic SRP RNAs. Such eubacterial RNAs with Aludomains have been identi®ed in Bacillus, Listeria andClostridium. We now wanted to know which RNAs that weidenti®ed using rnabob and the helix 8 motif descriptor alsocontain an Alu domain. We used rnabob with a descriptoraccounting for consensus features of the Alu domain togetherwith a variable spacer between the helix 8 and the Alu domain.The features of the Alu domain descriptor were based on acareful inspection of available Bacillus, Listeria andClostridium sequences. The resulting descriptor was used toanalyze all the sequences that we identi®ed using the helix 8motif as described above. In addition to the Bacillus, Listeriaand Clostridium sequences that we expected to ®nd, we alsoidenti®ed an Alu domain in the RNA of Thermotoga maritima(Fig. 3). A similar result was obtained when we used theCOVE programs with gram-positive Alu domain RNAs in themodel. This was a somewhat unexpected ®nding asThermotoga is clearly distinct from the Firmicutes group.The Thermotoga RNA does not contain a helix 6 region, and inthis respect it belongs to the Bacillus group of RNAs. This®nding raises interesting questions as to the relationshipbetween Thermotoga and the Bacillus branch. One possibleexplanation for the presence of an Alu domain RNA inThermotoga is a horizontal gene transfer event. However, we®nd no evidence of such an event, as phylogenetic trees based
on SRP RNA sequences are very similar to trees based on 16SrRNA sequences (data not shown).
In a search among the archaebacterial genomes we found asexpected an RNA with an Alu domain in all of the speciesexamined, also in the recently sequenced Thermoplasmavolcani and Thermoplasma acidophilum. All the sequences weidenti®ed also had a helix 6 region according to the structurepredicted by mfold. The Aeropyrum pernix RNA differs fromthe other archaebacterial RNAs in that it seems to have a helixinserted in the Alu domain between the helices 3 and 4 (http://bio.lundberg.gu.se/srpscan).
We noted that the sequence from Sulfolobus solfataricusreported by Kaine (17) is very different from the sequencepredicted from the genome sequence of the same bacterium(18). In fact, it is as distant to the genomic sequence as to thegene predicted from the genome of Sulfolobus tokodaii (19).Unless there are very large differences between differentisolates of S.solfataricus, it seems likely that there is a mistakeas to the identity of the strain.
Prediction of 5¢ and 3¢ ends
We want our method to predict as accurately as possible the 5¢and 3¢ ends of the RNA gene product. For the RNAs with anAlu domain we expect that the ends should be close to the Aludomain. For the eubacterial RNAs without an Alu domain theprediction of ends cannot be guided by such an element, andanother dif®culty is that there is a variation in size betweendifferent species. A few such RNAs have been sequenced,such as those of E.coli (20), a number of mycoplasmas(21±23) and Pseudomonas aeruginosa (24), with a variation insize from 77 to 114 nt. However, analysis by COVE can alsomake use of the fact that a majority of genes have a T-richsequence immediately downstream of the 3¢ end of the maturesequence. Figure 5 shows an alignment of a number ofsequences of RNAs without Alu domain that was produced byCOVE and where the starting point was an alignment wherethe T-rich regions had been manually aligned. However, asimilar result was obtained when unaligned sequences wereused as input to the program. In spite of the variation in sizebetween these bacterial RNAs, it seems that the COVE modelcan predict the 5¢ and 3¢ ends reasonably well. For the RNAsthat have been sequenced and for which we have informationabout the correct ends, these have the predicted site <2±3 bpfrom the correct one. Of course we cannot exclude thepossibility that the ends of a few RNAs are incorrectlypredicted because these RNAs have a structure which isdifferent from the expected hairpin structure.
The T-rich sequence downstream of the gene is highlightedin Figure 5. The hairpin structure of the mature RNA sequencein combination with the T-rich sequence is highly reminiscentof the rho-independent transcription termination signal. Itseems likely that this signal is the actual transcriptiontermination signal and that very little, if any, 3¢ end processingis needed to obtain the mature 3¢ end.
Analysis of eukaryotes
Whereas prokaryotic genomes have only one SRP RNA gene,many eukaryotes contain multiple genes as listed in Table 1.As for the analysis of prokaryotes, it was necessary to de®nedifferent descriptors and cove models for different taxonomic
Figure 4. Relationship of gram-positive SRP RNAs with unusual UGACtetraloop. Shown in the ®gure is a part of a phylogenetic tree based on analignment with COVE of all available SRP RNA sequences from this study.The tree was based on UPGMA and was drawn using Phylodraw (33). Asindicated in the ®gure, the Lactococcus RNA is more closely related to theStreptococcus RNA than to the Staphylococcus RNA, and the Staphylo-coccus RNA is more closely related to the Bacillus RNA than to theLactococcus RNA. Furthermore, the Staphylococcus/Bacillus RNAs have anAlu domain.
Nucleic Acids Research, 2002, Vol. 30 No. 15 3373

groups. For COVE analysis, we divided eukaryotes into yeastsand non-yeasts.
Yeasts. The yeast SRP RNA genes are dif®cult to identify andanalyze as they show a low degree of conservation betweendifferent yeast species, and because they are distinct fromother eukaryotic RNAs. Previously, SRP RNAs have beenidenti®ed in Yarrowia (25), Schizosaccharomyces pombe (26)and Saccharomyces cerevisiae (27). The S.cerevisiae se-quence is unusual in that the total length of the RNA is 519 nt,whereas most eukaryotic RNAs are ~300 nt. All the yeast
sequences have a helix 8 motif like the metazoan counterparts.However, the Alu domain is very different from the bacterialand metazoan counterparts. It has been suggested that theS.pombe, S.cerevisiae and Yarrowia lipolytica RNAs have asimpli®ed Alu domain that has the consensus featuresindicated in Figure 6 (28).
We now also analyzed un®nished genome sequence data ofNeurospora crassa (http://biology.unm.edu/biology/ngp/home.html) and C.albicans (http://www-sequence.stanford.edu/group/candida/) and were able to use our method to detectSRP RNA candidates in these genomes. This was possible by
Table 1. Eukaryotic SRP RNA genes as predicted from genomic sequences
Chromosomallocation
Number of genes GenBank accessionnumbers
Neurospora crassa Unknown 1 ±Candida albicans Unknown 1 ±Arabidopsis thaliana Chromosome 1 2 AC011807
AC013453Chromosome 2 2 AC005662
AC005311Chromosome 4 3 AC005275
AF069442Chromosome 5 1 (+ 2 pseudogenes?) ATF17I14
AB026661Drosophila Chromosome 3R 2 AC095014Caenorhabditis elegans Chromosome 3 4 CEB0285
U00064U23515CEB0284
Chromosome 5 1 (pseudogene?) U41993Homo sapiens Chromosome 14 2 CNS01DX7
AC020711
The Drosophila genes are identical, in an opposite orientation, and separated by ~2500 nt. The genes onC.elegans chromosome 3 have 5¢ start positions 3172520, 3162782, 3822816 and 4011623, respectively.
Figure 5. Alignment of eubacterial RNAs without Alu domain. All eubacterial SRP RNAs without an Alu domain identi®ed in this study were aligned usinga COVE model. The ®gure shows selected sequences from this alignment. The starting point for the alignment was one where T-rich regions at the 3¢ endwere manually aligned to each other, but a similar result was obtained when unaligned sequences were used as input. The T-rich region is highlighted as wellas conserved elements of the helix 8 region (see also Fig. 1).
3374 Nucleic Acids Research, 2002, Vol. 30 No. 15

using in the ®rst screen relatively degenerate helix 8 descrip-tors for rnabob. In the case of Neurospora it has a 6 nt loop inthe helix 8 part like S.cerevisiae.
Interestingly, both C.albicans and N.crassa RNAs have asequence that is consistent with the simpli®ed Alu motif,suggesting that this motif is highly conserved in yeasts.Figure 6 shows a tentative alignment with the yeast sequencesobtained by COVE. It highlights the homology in the Aludomain as well as a predicted structural homology of part ofthe molecule with the helices 6 and 8. On the basis of thisalignment it seems likely that yeast RNAs basically have thesame folding as other other SRP RNAs although there arelarge insertions in the S.cerevisiae RNA with unknownfolding. It must be noted that it is dif®cult to predict the 5¢and 3¢ ends of the yeast RNAs and experimental studies willhave to be carried out in order to determine these as well as tofully understand the folding of the different yeast RNAs.
Plants. The plants tend to have fairly conserved SRP RNAs.The predictions of plant RNAs might therefore be consideredmore reliable than for the other categories of eukaryotes. InArabidopsis thaliana we have identi®ed eight different genecandidates, as shown in Figure 7. Of these, only two werepreviously reported (13,29). It will require experimental workto see if all these genes are actually expressed or if some ofthem are pseudogenes. It has previously been shown that twoArabidopsis SRP RNA genes contain promoter elements, USEand TATA, upstream of the coding region that are identical tothe promoters of pol III-speci®c plant U-snRNA RNA genes(30) and that are important for transcriptional activity. It hasbeen proposed that the plant SRP RNA transcription mainly
relies on these elements and that internal promoter elementsplay a minor role. These upstream elements are indicated inFigure 7 and it is interesting to note that they are found invirtually all the novel genes. We therefore speculate that themajority of these genes are not pseudogenes. However, two ofthe genes on chromosome 5 do not have these elements (Fig. 7)and may therefore be pseudogenes.
Metazoans. In Drosophila and Caenorhabditis elegans wefound two and ®ve genes, respectively (Table 1). The codingsequences of the Drosophila genes are identical and the genesare located in an opposite orientation, separated by ~2500 nt.In C.elegans the genes on chromosome 3 were identi®edbefore whereas the one on chromosome 5 is a novel ®nding.However, it might be a pseudogene as the promoter sequenceis very different from the genes on chromosome 3. As with theDrosophila genes, the genes on chromosome 3 are in pairswith the genes in opposite orientation (Table 1).
The prediction of SRP RNA genes in the human genome iscomplicated by the presence of a large number of Alu repeatsevolutionarily related to SRP RNA. As a result there is a largebackground from hits to Alu repeats in the list of high-scoringhits from COVE. The strongest candidate genes are threegenes on chromosome 14 (one gene in CNS01DX7 and twogenes in AC020711; see Table 1) and one on chromosome 10(in GenBank accession no. AC091810, not shown in Table 1).The predicted RNAs of these genes can all be folded into thestructure characteristic of eukaryotic RNAs. The genes onchromosome 14 have similar sequences immediately upstreamof the coding sequence whereas the gene on chromosome 10 isdifferent in this region. We also carried out BLAST searches
Figure 6. Tentative alignment of yeast RNAs. RNAs shown are from S.cerevisiae, S.pombe and Y.lipolytica as well as RNAs predicted in this study fromgenome sequence data of N.crassa and C.albicans. The alignment was produced by COVE. COVE had dif®culties in correctly aligning helix 8 of theS.cerevisiae RNA to the corresponding part of the other RNAs, using the unaligned sequences as a starting point. Therefore, the starting point for the COVEalignment shown in the ®gure was one where the helix 8 domain of the S.cerevisiae RNA had been manually aligned with the helix 8 domain of the otherRNAs. A conserved motif at the 5¢ end is shown with the consensus sequence RCUGURAUGGY, with base pairing as indicated by the arrows. The `~'symbols indicate insertions in the S.cerevisiae RNA relative to the other RNAs.
Nucleic Acids Research, 2002, Vol. 30 No. 15 3375

of human EST database sequences to obtain evidence ofexpression of these genes. The results show that the majorspecies in EST databases are CNS01DX7 (~50%) and one ofthe genes in AC020711 (~50%). These two variants alsocorrespond to the sequences reported by Ullu et al. (31). Thefact that only one gene on the AC020711 segment is expressedalso has support from previous experimental studies (32). Inconclusion, the likely predominating human SRP RNA genesare the one contained in CNS01DX7 and one of the genes inAC020711 on chromosome 14.
When COVE was used to produce an alignment of all ourpredicted eukaryotic SRP RNAs, including those of yeasts, theUGU consensus sequence of the predicted Alu domain of theyeast RNAs, as indicated in Figure 6, aligned well with thecorresponding sequence in the Alu domains of the metazoans.This gives further support to the structural relationshipbetween the metazoan and yeast Alu domains. The result ofthis alignment also shows that it is possible to use only oneCOVE model for the prediction and analysis of all eukaryoticSRP RNAs, with the possible exception of the S.cerevisiaeRNA. This model will now be very useful in the identi®cationof SRP RNA genes in genomes that become available in thefuture.
Alignment of all the eukaryotic SRP RNAs also revealedconserved elements. In addition to the helix 8 and Alu domainmotifs already referred to, the most prominent one is the motifshown in Figure 1, which is located in helix 5 near the helices6 and 8. This region of the RNA is involved in the interactionwith the SRP68/72 protein dimer. Possibly, the conservedmotif is an important recognition element for these proteins,just as the conserved parts of helix 8 and the Alu domain arerecognized by the SRP9/14 dimer and the SRP54 protein,respectively.
Conclusion
Non-coding RNA genes vary in their degree of primary andsecondary structure conservation. It is obvious that the lower
the degree of conservation, the more dif®cult is the predictionof such genes from genomic sequence information. CertainRNAs, like rRNAs, are so conserved that they may beeffectively detected by primary sequence homology only.Other RNAs, like tRNA and SRP RNA, have to incorporate asearch for secondary structure features. With both tRNA andSRP RNA, the degree of conservation in terms of combinedprimary and secondary structure features is suf®cient todevelop an accurate prediction method. However, there aremore dif®cult cases, like the yeast SRP RNAs, that quitestrongly diverge from a consensus structure. The predictionsof yeast RNAs will therefore have to be veri®ed by experi-mental studies. Experimental work is also required to deter-mine the exact location of the 5¢ and 3¢ ends of mature RNAsand to distinguish pseudogenes from genes that give rise tofunctional RNAs.
Nevertheless, the methods described are in general veryef®cient in identifying SRP RNA gene candidates. In thismethod we combine a heuristic pattern screen for theconserved helix 8 and Alu domain motifs with the COVEprograms. We account for SRP RNA genes in all organismsthat have been completely sequenced. We have detected anumber of novel RNAs as well as novel features of SRP RNAssuch as the unusual tetraloop sequence of the Lactococcus andStaphylococcus RNAs. In particular, our method is verysensitive and speci®c in the prediction of bacterial RNA genes.For the ®rst time, we now have an automated procedure todetect SRP RNA genes that may be used as a routine toolduring genome annotation.
REFERENCES
1. Altschul,S.F., Madden,T.L., Schaffer,A.A., Zhang, J., Zhang,Z.,Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: anew generation of protein database search programs. Nucleic Acids Res.,25, 3389±3402.
2. Eddy,S.R. and Durbin,R. (1994) RNA sequence analysis usingcovariance models. Nucleic Acids Res., 22, 2079±2088.
Figure 7. Predicted genes in Arabidopsis. Multiple alignment of predicted SRP RNA genes in A.thaliana was produced by COVE using unaligned genes asinput. Only the 5¢ portion of mature RNA together with upstream sequences are shown. Promoter elements that were previously shown to be important fortranscription of SRP RNA genes (USE and TATA) in Arabidopsis are indicated (30). Two of the genes on chromosome 4 were previously identi®ed.Asterisks indicate possible pseudogenes.
3376 Nucleic Acids Research, 2002, Vol. 30 No. 15

3. Keenan,R.J., Freymann,D.M., Stroud,R.M. and Walter,P. (2001) Thesignal recognition particle. Annu. Rev. Biochem., 70, 755±775.
4. Ulbrandt,N.D., Newitt,J.A. and Bernstein,H.D. (1997) The E. coli signalrecognition particle is required for the insertion of a subset of innermembrane proteins. Cell, 88, 187±196.
5. Newitt,J.A., Ulbrandt,N.D. and Bernstein,H.D. (1999) The structure ofmultiple polypeptide domains determines the signal recognition particletargeting requirement of Escherichia coli inner membrane proteins.J. Bacteriol., 181, 4561±4567.
6. Lee,H.C. and Bernstein,H.D. (2001) The targeting pathway ofEscherichia coli presecretory and integral membrane proteins is speci®edby the hydrophobicity of the targeting signal. Proc. Natl Acad. Sci. USA,98, 3471±3476.
7. Brown,S. and Fournier,M.J. (1984) The 4.5 S RNA gene of Escherichiacoli is essential for cell growth. J. Mol. Biol., 178, 533±550.
8. Miller,J.D., Wilhelm,H., Gierasch,L., Gilmore,R. and Walter,P. (1993)GTP binding and hydrolysis by the signal recognition particle duringinitiation of protein translocation. Nature, 366, 351±354.
9. Peluso,P., Herschlag,D., Nock,S., Freymann,D.M., Johnson,A.E. andWalter,P. (2000) Role of 4.5S RNA in assembly of the bacterial signalrecognition particle with its receptor. Science, 288, 1640±1643.
10. Peluso,P., Shan,S.O., Nock,S., Herschlag,D. and Walter,P. (2001) Roleof SRP RNA in the GTPase cycles of Ffh and FtsY. Biochemistry, 40,15224±15233.
11. Batey,R.T., Rambo,R.P., Lucast,L., Rha,B. and Doudna,J.A. (2000)Crystal structure of the ribonucleoprotein core of the signal recognitionparticle. Science, 287, 1232±1239.
12. Weichenrieder,O., Wild,K., Strub,K. and Cusack,S. (2000) Structure andassembly of the Alu domain of the mammalian signal recognitionparticle. Nature, 408, 167±173.
13. Gorodkin,J., Knudsen,B., Zwieb,C. and Samuelsson,T. (2001) SRPDB(Signal Recognition Particle Database). Nucleic Acids Res., 29, 169±170.
14. Zuker,M. (1989) On ®nding all suboptimal foldings of an RNA molecule.Science, 244, 48±52.
15. Pearson,W.R. (2000) Flexible sequence similarity searching with theFASTA3 program package. Methods Mol. Biol., 132, 185±219.
16. Jagath,J.R., Matassova,N.B., de Leeuw,E., Warnecke,J.M., Lentzen,G.,Rodnina,M.V., Luirink,J. and Wintermeyer,W. (2001) Important role ofthe tetraloop region of 4.5S RNA in SRP binding to its receptor FtsY.RNA, 7, 293±301.
17. Kaine,B.P. (1990) Structure of the archaebacterial 7S RNA molecule.Mol. Gen. Genet., 221, 315±321.
18. She,Q., Singh,R.K., Confalonieri,F., Zivanovic,Y., Allard,G.,Awayez,M.J., Chan-Weiher,C.C., Clausen,I.G., Curtis,B.A.,De Moors,A. et al. (2001) The complete genome of the crenarchaeonSulfolobus solfataricus P2. Proc. Natl Acad. Sci. USA, 98, 7835±7840.
19. Kawarabayasi,Y., Hino,Y., Horikawa,H., Jin-no,K., Takahashi,M.,Sekine,M., Baba,S., Ankai,A., Kosugi,H., Hosoyama,A. et al. (2001)Complete genome sequence of an aerobic thermoacidophiliccrenarchaeon, Sulfolobus tokodaii strain7. DNA Res., 8, 123±140.
20. Hsu,L.M., Zagorski,J. and Fournier,M.J. (1984) Cloning and sequenceanalysis of the Escherichia coli 4.5 S RNA gene. J. Mol. Biol., 178,509±531.
21. Muto,A., Andachi,Y., Yuzawa,H., Yamao,F. and Osawa,S. (1990) Theorganization and evolution of transfer RNA genes in Mycoplasmacapricolum. Nucleic Acids Res., 18, 5037±5043.
22. Samuelsson,T. and Guindy,Y. (1990) Nucleotide sequence of aMycoplasma mycoides RNA which is homologous to E. coli 4.5S RNA.Nucleic Acids Res., 18, 4938.
23. Simoneau,P. and Hu,P.C. (1992) The gene for a 4.5S RNA homolog fromMycoplasma pneumoniae: genetic selection, sequence, and transcriptionanalysis. J. Bacteriol., 174, 627±629.
24. Toschka,H.Y., Struck,J.C. and Erdmann,V.A. (1989) The 4.5S RNAgene from Pseudomonas aeruginosa. Nucleic Acids Res., 17, 31±36.
25. He,F., Yaver,D., Beckerich,J.M., Ogrydziak,D. and Gaillardin,C. (1990)The yeast Yarrowia lipolytica has two, functional, signal recognitionparticle 7S RNA genes. Curr. Genet., 17, 289±292.
26. Brennwald,P., Liao,X., Holm,K., Porter,G. and Wise,J.A. (1988)Identi®cation of an essential Schizosaccharomyces pombe RNAhomologous to the 7SL component of signal recognition particle. Mol.Cell. Biol., 8, 1580±1590.
27. Felici,F., Cesareni,G. and Hughes,J.M. (1989) The most abundant smallcytoplasmic RNA of Saccharomyces cerevisiae has an importantfunction required for normal cell growth. Mol. Cell. Biol., 9,3260±3268.
28. Strub,K., Fornallaz,M. and Bui,N. (1999) The Alu domain homolog ofthe yeast signal recognition particle consists of an Srp14p homodimerand a yeast-speci®c RNA structure. RNA, 5, 1333±1347.
29. Marques,J.P., Gualberto,J.M. and Palme,K. (1993) Sequence of theArabidopsis thaliana 7SL RNA gene. Nucleic Acids Res., 21, 3581.
30. Heard,D.J., Filipowicz,W., Marques,J.P., Palme,K. and Gualberto,J.M.(1995) An upstream U-snRNA gene-like promoter is required fortranscription of the Arabidopsis thaliana 7SL RNA gene. Nucleic AcidsRes., 23, 1970±1976.
31. Ullu,E., Murphy,S. and Melli,M. (1982) Human 7SL RNA consists of a140 nucleotide middle-repetitive sequence inserted in an alu sequence.Cell, 29, 195±202.
32. Ullu,E. and Tschudi,C. (1984) Alu sequences are processed 7SL RNAgenes. Nature, 312, 171±172.
33. Choi,J.H., Jung,H.Y., Kim,H.S. and Cho,H.G. (2000) PhyloDraw: aphylogenetic tree drawing system. Bioinformatics, 16, 1056±1058.
Nucleic Acids Research, 2002, Vol. 30 No. 15 3377




















