
Predicting the Kinetics of Heap Leaching with Unsteady-State ...
175
University of Nevada Reno /1«U5 IhiM X)53 Predicting the Kinetics of Heap Leaching with Unsteady-State Models A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Metallurgical Engineering by David G. Dixon in Dr. James L. Hendrix, Dissertation Advisor November 1992
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of Predicting the Kinetics of Heap Leaching with Unsteady-State ...

University of Nevada
Reno
/1«U5
I h i MX )5 3
Predicting the Kinetics of Heap Leaching with Unsteady-State Models
A dissertation submitted in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Metallurgical Engineering
by
David G. Dixonin
Dr. James L. Hendrix, Dissertation Advisor November 1992

The dissertation of David G. Dixon is approved:
Dtssertatissertation Advisor
jDean, Graduate School
University of Nevada Reno
November 1992

Copyright by David Grant Dixon 1992 All Rights Reserved

11
A bstract
An investigation has been conducted into the kinetics of heap leaching of one or
several solid reactants by a single rate-controlling reagent which is a component of the
liquid phase only, and not a dissolved gas. A particle-scale model was derived in
unsteady-state which simulates the leaching of one or more solid reactants from an inert,
porous, spherical pellet. This model was then incorporated into an unsteady-state heap-
scale model which assumes ideal plug flow for the lixiviant. Numerical solutions of the
model equations were obtained with implicit finite difference approximations. Batch and
column leaching experiments were conducted which validate both models.
The first part of the research concerned the derivation and testing of the particle-
scale leaching model. The effects of diffusion rate, chemical reaction rate, apparent
reaction order, and competition between multiple solid reactants were investigated using
the concept of the effectiveness factor. The importance of deposits of solid reactants on
the pellet surface was ascertained. It was shown that the model is capable of simulating
both "zone-wise" and "homogeneous" kinetics, depending on the choice of two
dimensionless parameters. Also, an approach was developed for simulating leaching
from particles with a distribution of sizes by using average parameters and the Gates-
Gaudin-Schuhmann distribution function. A series of batch leaching tests was performed
on manufactured ore agglomerates made of silica sand, Portland cement and pure silver
powder. Pellets of several sizes and various amounts of silver were leached continuously
with a circulating solution of sodium cyanide and sodium hydroxide. These experiments
demonstrated the usefulness of the model in general, and validated the concept of the
variable reaction order.

The second part of the research involved the derivation and testing of the heap-
scale leaching model. The effects of kinetics at the particle-scale, as well as the lixiviant
flowrate, heap height, particle size distribution and competition between multiple solid
reactants were examined using the concept of the heap effectiveness factor. It was shown
that the kinetic behavior of heaps is analogous to that of individual particles, operating
either in a zone-wise manner, or reacting throughout the heap simultaneously depending
mostly on the value of a single dimensionless parameter. Column leaching tests using
the same pellets as for the batch tests showed that the model is fundamentally correct.
However, the value of the heap-scale parameter could only be determined empirically,
depending on the degree of contact effectiveness between the ore pellets and the lixiviant
solution. A rough correlation relating the contact effectiveness with Reynolds number
was generated from the results of computer simulation of the column tests.
iii

IV
Acknowledgem ents
Many people have helped and encouraged me, both directly and indirectly, during
the course of my graduate study. I am most deeply indebted to my advisor, Dean James
L. Hendrix, for his unswerving support; technical, financial and moral. It was his
guidance, encouragement and friendship which made this work possible. I would also
like to thank the other members of my examining committee: Professors Philip Altick,
Manoranjan Misra, Ross Smith, Dhanesh Chandra, and especially John Nelson, for their
many helpful comments and suggestions, and for the painstaking care with which they
read and corrected this dissertation.
I owe a special debt of gratitude to Tom Carnahan of the U.S. Bureau of Mines.
For encouraging me to pursue a doctorate, and for having faith in me and concern for
my professional future, I will always be grateful.
For help with chemical analyses, setting up experiments, and being called upon
to take the odd inconvenient sample, I wish to thank members of the MMRRI technical
support staff, past and present: Chuck Gemmell, Mojtaba Ahmadiantehrani, Dave
Castillo, Tim Burchett, Paul Wilmot, Charles Hess, Shannon Rogan, Cindy Evans and
Dave Kashuba. One couldn’t wish for a better, more friendly bunch of people to work
with. Also, a special thanks to Jim Sjoberg and assistants at the USBM Reno Research
Center Mineralogy Lab for their efforts on my behalf.
For their intelligence, wit, good humor, and superior cnbbage skills, I wish to
thank my friends Emil Milosavljevic, Ljiljana Solujic, and Scott Rader. For moral
support, I especially wish to thank Carl Nesbitt and Bobby Varelas, whose friendship I
shall always cherish.

Lastly, I wish to express my deepest gratitude to my family. To my wonderful
parents, Joan and Nelson, for their neverending love, support and encouragement, which
words can never repay. To my parents-in-law, Jim and Betty Hulse, for making me so
much a part of their family. But most profoundly, to my beautiful wife, Jane, whose
support and self-sacrifice are deeply appreciated. With love, courage and understanding
she shared the burden of my graduate study, and it is to her that I dedicate this work.

VI
Table of Contents
ABSTRACT ............................................................................................................... ii
ACKNOWLEDGEMENTS ...................................................................................... iv
LIST OF FIGURES ................................................................................................... viii
LIST OF TABLES ..................................................................................................... xiii
INTRODUCTION
CHAPTER ONE ....................................................................................................... 4A General Model for Leaching of One or More Solid Reactants from Porous Ore Particles
I. Introduction ......................................................................II. Model Development ........................................................
a. Derivation of Model Equations ............................b. Model Equations in Dimensionless Form ...........c. Important Model Functionals ...............................
III. Experimental ...................................................................a. Experimental Procedures and Equipment ............b. Artificial Ore Preparation ....................................
IV. Results and Discussion - Theoretical ...........................a. Computer Simulation: One Solid Reactant .........b. Computer Simulation: Competing Solid Reactantsc. An Approach for Particle Size Distributions .....
V. Results and Discussion - Experimental .......................VI. Conclusions ...................................................................
477
101214141718 18 30 34 40 56
CHAPTER TWO .................................................................... . • .................. ; ...... "A Mathematical Model for Heap Leaching of One or More Solid Reactants from PorousOre Pellets
I. Introduction ......................................................................II. Model Development ........................................................
a. The Model in Dimensionless Form .....................b. Important Model Functionals ...............................
III. Experimental ............................... ....................................IV. Results and Discussion -- Theoretical ...........................
a. Computer Simulation: One Solid Reactant .........b. Computer Simulation: Competing Solid Reactants
5760636567696979

c. An Approach for Particle Size Distributions ........................... 87V. Results and Discussion — Experimental ............................................. 90VI. Conclusions ............................................................................................ 104
SUMMARY ................................................................................................................ 106Conclusions and Recommendations
APPENDIX ONE ...................................................................................................... 108A Theoretical Basis for the Variable Order Assumption in the Kinetics of Leaching of Discreet Reactant Grains
APPENDIX TWO .................................................................................................... 120Numerical Methods
APPENDIX THREE ................................................................................................ 124Experimental Data
APPENDIX FOUR ................................................................................................... 129Nomenclature
APPENDIX FIVE .................................................................................................... 132FORTRAN Computer Program Source Code
APPENDIX SIX .................................................................................................... 152Analytical Solutions to the Pseudo-Steady-State Two-Reactant Problem
vii
BIBLIOGRAPHY 157

List of Figures
viii
FIGURE 1-1 ............................................................................................................ 8Schematic diagram of a porous, spherical ore particle with concentration gradients.
FIGURE 1-2 ............................................................................................................ 15Schematic diagram of the batch leaching test apparatus.
FIGURE 1-3 ............................................................................................................ 20Solutions to the simplified continuity equations for reagent A (a) and one solid reactant(O p ).
FIGURE 1-4 ............................................................................................................ 22Fractional conversion (X) vs. dimensionless reaction time (kp0t) for the nine continuity equation solutions shown in Figure 1-3.
FIGURE 1-5 ............................................................................................................ 23Effectiveness factor (rj) vs. fractional conversion (X) for the continuity equation solutions shown in Figure 1-3.
FIGURE 1-6 ...................................... ; .................................................................... 25Solutions to the continuity equations for reagent A (a) and one solid reactant (crp,ffs) given a surface fraction (X) of 0.10.
FIGURE 1-7 ............................................................................................................ 27Fractional conversion (X) vs. dimensionless diffusion time (r) given various surface fractions (X).
FIGURE 1-8 ............................................................................................................ 28Effectiveness factor (17) vs. fractional conversion (X) given various surface fractions (X).
FIGURE 1-9 ............................................................................................................. 29Fractional conversion (X) vs. dimensionless reaction time (/Cp0r) given various reaction orders (4>p) at kv = 1 and 100, 0 = 1.
FIGURE 1-10............................................................................................................. 31Effectiveness factor (rj) vs. fractional conversion (X) given various reaction orders (<f>p) at kp = 1 and 100, 0 = 1.
FIGURE 1-11 ............................................................................................................. 32Solutions to the continuity equations for reagent A (a) and solid reactant 1 (crpl), alone (n = l) and in the presence of a second reactant (n=2). ^ = 1, 0j = 1, <£pl = 2/3, kp2 = 10, 02 = 0.01, 0p2 = 0, Ar = 0.25.

IX
FIGURE 1-12............................................................................................................ 35Fractional conversion of solid reactant 1 (Xj) vs. dimensionless diffusion time (r) for the systems shown in Figure 1-11.
FIGURE 1-13............................................................................................................ 36Effectiveness factor fo) vs. fractional conversion of solid reactant 1 (X,) for the systems shown in Figure 1-11.
FIGURE 1-14............................................................................................................ 38The Gates-Gaudin-Schuhmann normalized weight distribution function (w(E)) vs. dimensionless particle size (S) at various values of the distribution parameter m.
FIGURE 1-15............................................................................................................ 42Fractional conversion for various particle sizes (X(E)) (solid curves), and for the entire size distribution (XT) (dotted curves) vs. reference diffusion time (7) for various values of the distribution parameter m.
FIGURE 1-16............................................................................................................ 43Effectiveness factor (77) vs. fractional conversion for various particle sizes (X(H)) at various particle sizes (S).
FIGURE 1-17................................................................................... 47Results of batch test 1.
FIGURE 1-18............................................................................................................ 48Results of batch test 2.
FIGURE 1-19............................................................................................................ 49Results of batch test 3.
FIGURE 1-20............................................................................................................. 50Results of batch test 4.
FIGURE 1-21............................................................................................................. 53Calculated effectiveness factor (77) vs. Ag conversion (X) for the batch leaching tests.
FIGURE 1-22............................................................................................................ 54Scanning electron micrograph (SEM) of pure silver powder used in the preparation of the artificial ore pellets. Magnification: lOOOx.
FIGURE 1-23 ............................................................................................................. 55Grain size distribution data from statistical image analysis of the silver powder sample shown in Figure 1-22.

FIGURE 2-1 ............................................................................................................ 61Schematic diagram of a heap.
FIGURE 2-2 ............................................................................................................ 68Schematic diagram of the experimental apparatus.
FIGURE 2-3 ............................................................................................................ 71Solutions to the simplified continuity equations for reagent A in the bulk solution (ab) and the fractional conversion (X) as functions of heap depth.
FIGURE 2-4 ............................................................................................................ 72Effluent concentration (xb(l,0)), fractional conversion (X) and extraction (E) vs. dimensionless flow time (6) given various values of /3 and co.
FIGURE 2-5 ............................................................................................................ 74Fractional conversion (X) vs. reaction time (kp(Sp<j:(6-1)/3), given various values of go.
FIGURE 2-6 ............................................................................................................. 76Heap effectiveness factor (r?) vs. fractional conversion (X) for the continuity equation solutions shown in Figure 2-3.
FIGURE 2-7 ............................................................................................................ 77Fractional conversion (X) vs. dimensionless flow time (0) given various surface fractions(X).
FIGURE 2-8 ............................................................................................................. 78Heap effectiveness factor (17) vs. dimensionless flow time (0) given various surface fractions (X).
FIGURE 2-9 ............................................................................................................. 80Fractional conversion (X) vs. dimensionless flow time (0) given various orders of reaction (</>p).
FIGURE 2-10............................................................................................................. 81Heap effectiveness factor (rj) vs. dimensionless flow time (0) given various orders of reaction (<f>p).
FIGURE 2-11 ............................................................................................................. 82Solutions to the continuity equations for reagent A (ab) and fractional conversion of reactant 1 (Xj), alone (n= l) and in the presence of a second reactant (n=2). = 1,f t = 1, </>„, = 2/3, kp2 = 10, 02 = 0.01, ^ = 0, co = 1, 1; = 1, Ad = 1.
FIGURE 2-12............................................................................................................. 85Fractional conversion of solid reactant 1 (Xj) vs. dimensionless flow time (0) for the systems shown in Figure 2-11.

XI
FIGURE 2-13............................................................................................................ 86Heap effectiveness factor (77,) vs. fractional conversion of solid reactant 1 (X,) for the systems shown in Figure 2-11.
FIGURE 2-14............................................................................................................ 89Effluent concentration (xb(M )) vs. dimensionless flow time (0) given various values of a), for a single particle size (m=oo) and a GGS distribution (m =l).
FIGURE 2-15............................................................................................................ 91Fractional conversion (X) vs. dimensionless flow time (6) for the simulations shown in Figure 2-14.
FIGURE 2-16............................................................................................................ 92Heap effectiveness factor (77) vs. fractional conversion (X) for the simulations shown in Figure 2-14.
FIGURE 2-17............................................................................................................ 94Effluent concentration and extraction data from column leaching test 1.
FIGURE 2-18............................................................................................................ 95Effluent concentration and extraction data from column leaching test 2.
FIGURE 2-19............................................................................................................. 96Effluent concentration and extraction data from column leaching test 3.
FIGURE 2-20............................................................................................................ 97Effluent concentration and extraction data from column leaching test 4.
FIGURE 2-21 ............................................................................................................. 98Effluent concentration and extraction data from column leaching test 5.
FIGURE 2-22............................................................................................................. 100Heap effectiveness factor (77) vs. fractional conversion (X) for the model simulations of the experimental column leaching tests.
FIGURE 2-23 ............................................................................................................. 103Contact effectiveness (coapp/upr) vs. Reynolds number (NRe) from the experimental column leaching tests.

FIGURE A. 1-1 .................................The log-normal distribution function.
xii
115
FIGURE A. 1 -2 ........................................................................................................... 116Collective mass fraction remaining (a) vs. reaction time (r) for various grain weight distributions.
FIGURE A. 1-3 ........................................................................................................... 117Apparent reaction order (&) vs. collective mass fraction remaining (a) for various grain weight distributions.
FIGURE A. 1-4 ........................................................................................................... 118Average apparent reaction order (<£avc) vs. the standard deviation of the log-normal grain weight distribution (s).
FIGURE A. 1-5 ........................................................................................................... 119Collective mass fraction remaining (a) vs. reaction time (r) as calculated by equations (A. 1-6) (solid curves) and (A. 1-1) (dashed curves).

List of Tables
TABLE 1-1............................................................................................................... 41Parameter values for the size distribution model solution.
TABLE l - I I .............................................................................................................. 46Parameter values for the batch leaching tests.
TABLE 2-1 .............................................................................................................. 93Parameter values for the column leaching tests.
TABLE A.3-I ........................................................................................................... 125Data from batch leaching tests.
TABLE A.3-II .......................................................................................................... 126Data from column leaching test 1.
TABLE A.3-III ......................................................................................................... 128Data from column leaching tests 2 through 5.
TABLE A.6-I ........................................................................................................... 153Bessel functions to use in equation (A.6-3).
xiii

1
Introduction
Heap and dump leaching have become the dominant modes of treatment for low-
grade, oxidized ores of gold, silver, copper and uranium. These coarse-ore leaching
methods offer many economic and environmental advantages over conventional milling,
including smaller capital investment, lower operating costs and energy requirements, and
adaptability to any sized mining operation.
The idea of using heaps for basic cyanide leaching of low-grade precious metal
ores was first suggested by researchers at the U.S. Bureau of Mines in 1967.1 Since that
time, rising gold prices have precipitated dramatic advances in heap leaching technology,
and an explosion in domestic gold production. Between 1987 and 1989 alone, gold
production in the United States increased by 67 percent, and at Newmont Gold Company,
the largest domestic producer of gold, the share of gold production from heap leaching
increased from 19 percent in 1984 to 30 percent in 1990, while total tons of ore treated
increased nearly twenty-four-fold.2
Coarse-ore leaching has a much longer history for copper ores than for gold
ores.1,3 As early as the sixteenth century, miners in Hungary were recycling copper
bearing solutions through sulfidic waste heaps, and acid heap leaching of copper oxides
was employed on the banks of the Rio Tinto in Spain around 1752. By 1900, the use
of copper oxide heaps was widespread, and techniques to enhance recovery such as
leach/rest cycles were already being employed. Dump leaching of low-grade copper
sulfides is currently practiced worldwide. In addition, acid and alkaline heap leaching
of uranium ores has been common practice for the last forty years.
Though heap leaching has been a boon to the mining industry, the process still

2
suffers from its share of problems and uncertainties. In precious metals leaching,
especially, the rate of extraction from a heap may not coincide with the predictions made
on the basis of laboratory column tests, which creates confusion as to what would be the
proper cutoff grade where milling the ore becomes more economical than heap leaching.
Depending on the mode of heap construction and the degree of mineral liberation prior
to leaching, total extraction from heaps may be as low as 50 percent. On the other hand,
heaps often outperform column tests in total extraction of metal values. The president
of Newmont Gold Company reports that many mature heaps are yielding recoveries much
higher than estimated from laboratory tests, and suggests that the construction of several
expensive mills could have been avoided.2 Even refractory ores — those not amenable
to heap leaching due to the encapsulation of metal values within impervious grains of
sulfide — may undergo natural bio-degradation within the heap environment to such an
extent that the ores are rendered non-refractory over a period of months or years, similar
to copper sulfides in dumps. Hence, it is obvious that a sophisticated approach to heap
design and the ability to predict leaching performance in many different situations could
have great economic importance.
The purpose of the present study was to develop a mathematical model for the
kinetics of heap leaching of low-grade ores, in dimensionless form, which would
accommodate direct scale-up from column tests, taking into account such factors as ore
grade, heap depth, lixiviant flowrate and particle size. In any such endeavor, one must
begin at the microscale and build up to the macroscale. Hence, the first chapter is
devoted to the development of a particle-scale leaching model based on unsteady-state
diffusion and dissolution of reactant grains distributed within a porous matrix. Suitability

3
of this model is shown experimentally in batch leaching tests of manufactured pellets
containing known amounts of pure silver powder.
In the second chapter, the particle leaching model is incorporated into an
unsteady-state heap-scale convection model. The results of column tests involving the
artificial ore pellets compare favorably with model simulations. An assumption
concerning the order of reaction within the model is examined in detail in the first
appendix, and a population balance model for a distribution of reactant grain sizes is
developed which clarifies the assumption.
Throughout the study, all model equations are solved with a digital computer
using numerical methods, since the complexity of those equations and the time- and
space-dependent nature of their boundary conditions generally precludes all possibility
of analytical solution. The specific mathematical techniques employed, and the
FORTRAN source code for the computer programs are reported in the second and fifth
appendices, respectively. Tabulated data from all batch and column leaching experiments
are reported in the third appendix. A full list of pertinent nomenclature is presented in
the fourth appendix. Finally, solutions to the model equations for a special case
involving the presence of a reactive gangue material are obtained in the sixth appendix.

Chapter OneA General Model for Leaching of One or More
Solid Reactants from Porous Ore Particles
I. Introduction
Many coarse-ore leaching processes involve the dissolution of one or more solid
reactants from an inert porous matrix. In recent years, the modeling of such phenomena
has received much attention, especially as it applies to the large-scale leaching processes
of copper sulfide ores. Braun et al.4 derived a model for leaching of primary sulfide ores
which assumed steady-state diffusion of dissolved oxygen as the rate controlling factor,
resulting in the formation of a narrow "reaction zone" which moves slowly toward the
center of the ore fragments, similar to the familiar "shrinking core" model for gas-solid
reactions.5 This model, which incorporates empirical rate enhancement parameters in
order to account for particle degradation under leach conditions, was shown to fit
experimental leaching data over a wide range of particle sizes and shapes,4,6 and has
subsequently been used as a basis for modeling large-scale in situ leaching processes.7
The model has recently been extended to the leaching of rocks containing more than one
copper sulfide mineral, each with its own specific rate constant.8
Models of copper sulfide leaching have not been limited to the shrinking core
type. A more general model involving the unsteady-state continuity equation for
dissolved oxygen in spherical coordinates was devised by Bartlett9 to describe the
leaching kinetics of low-grade chalcopyrite ores. In a low-grade ore, the rate of
diffusion is often fast compared to the reaction rate, so that reaction occurs
homogeneously throughout the particle. Bartlett’s model assumes a log-normal size
distribution of spherical copper sulfide grains, distributed evenly throughout the pore
4

5
structure. A modified version of this model was later verified experimentally in
autoclave leaching tests.10 A similar model, which assumes the steady-state diffusion of
ferric ion through the porous matrix to be rate controlling, was derived by Madsen and
Wadsworth,11 and included separate rate terms for a number of different copper sulfide
minerals, as well as empirical rate enhancement parameters.
While most of the attention has been focused on sulfides, several models have
been developed to describe acid leaching of copper oxide ores. Roman et al.,12 and
Shafer et al.13 were able to simulate the results of column leaching tests of a copper oxide
ore with the shrinking core model in unaltered form. More recently, Chae and
Wadsworth14 have incorporated the reaction zone model of Braun et al. into an in situ
copper oxide leaching simulation, accounting also for the consumption of acid by gangue
minerals, and the initial flushing of copper oxides from the external surfaces of ore
fragments.
Recently, Box and Prosser15 have attempted to derive a general model for the
leaching of several minerals with several reagents which requires no prior laboratory
testwork. In this model, reactions which occur at similar rates are lumped into cohesive
groups, and then each group is solved as a single reaction by the shrinking core model.
The procedure for solution of the model, however, is cumbersome, and an attempt at
simulating the experimental data of Hicban and Gray16 achieved only limited success.
Prosser17 has applied a modified version of the model to precious metal leaching with
basic cyanide solution, again with only limited success.
Bartlett18 was able to describe the leaching of oxidized gold ores with an unsteady-
state extraction model, solved by Crank in spherical coordinates,19 by assuming the actual

6
dissolution reaction to be near-instantaneous relative to the diffusion of dissolved species
out of the porous ore particle. A critical review of this and many of the other models
listed above has recently been published by Bartlett.20
In this chapter, a general model is derived which simulates the leaching of several
solid reactants from a porous particle with a common reagent. Like Bartlett’s sulfide
leaching model,9 the present model is based on the unsteady-state continuity equation for
a reagent species in spherical coordinates. Unlike Bartlett’s model, however, the
dissolution rate of each solid reactant is represented by a variable-order, power-law type
rate expression, a common practice in the modeling of non-catalytic gas-solid
reactions.21,22 Also, the model equations are made dimensionless for the purpose of
identifying the important design and scale-up parameters.
With this model it is possible to simulate not only reaction zone type, diffusion
controlled leaching, but also more homogeneous, reaction controlled leaching, as well
as those situations which fall somewhere in between, depending on the choice of
parameters. Also, any rate enhancement effects due to the presence of solid reactants
on the particle surface are easily determined, as well as the effects of different solid
reactant grain configurations, and of two or more solid reactants competing for the same
reagent.

n . Model Development
Derivation of Model Equations
Consider a porous, structurally uniform, spherical ore particle of radius R which
is submerged in a lixiviant solution, and which contains small amounts of solid reactants
evenly distributed along the pore walls, as shown in Figure 1-1. These solid reactants,
B;, are dissolved by a single reagent, A, according to the formula
7
n
A + b jB i - d i s s o l v e d p r o d u c t s ( 1- 1 )2=1
where b; are the stoichiometric numbers and n is the total number of solid reactants. At
any particle radius r, assuming that the dissolution of each solid reactant is first order in
the concentration of one rate-controlling reagent, and variable order in its own solid
concentration (see Appendix 1 for an in-depth discussion of the reasoning behind this
assumption), the rate of dissolution of solid reactant i may be expressed
d c ■pid t
= — js- r&P1 (i KpiCP2 CA ( 1- 2 )
where Cpi is the solid concentration of solid reactant i at the pore walls at particle radius
r, kpj is the reaction rate constant expressed per unit particle mass, CA is the reagent
concentration at particle radius r, and <f>pi is the reaction order in the solid concentration
of solid reactant i. Equation (1-2) has the initial condition
Cp i ( r , 0 ) = Cpi0 ( 1 - 3 )
Since every reaction within the particle involves the consumption of reagent A,
the mass balance for reagent A within the porous sphere takes the form of a continuity

8
Pore network
ore reactant deposits
O Particle radius, r R
CsO
(J)c0)r>o>Q.<X>■oOO)
Cs ‘
FIGURE 1-1Schematic diagram of a porous, spherical ore particle with concentration gradients.

9
equation with a summed consumption term
DAeP c Ad r 2
2 dC.r d r ~ P o d - e 0) £
T1 Tr-
2=1^ p i ^ p l A _ dC, 5 --- -
° a t( 1 - 4 )
where DAe is the effective diffusivity of reagent A within the particle pores, p0 is the
specific gravity of the ore matrix , and e0 is the porosity of the particle. In the interest
of simplicity, it is assumed that all of these parameters remain unchanged with time and
position. Equation (1-4) has initial and boundary conditions
CA( r , 0 )
Ca (R, t)
= 0
= c.Ab
dcAd r
(o , t) = 0
( 1 - 5 )
( 1 - 6 )
( 1 - 7 )
where CAb is the concentration of reagent A in the bulk solution external to the particle.
Most naturally occurring porous ore particles consist of tightly bound grains of
material. It is generally along the boundaries of these grains that the deposits of solid
reactants are found, and hence, these boundaries serve as the "pores" for intraparticular
diffusional processes. When ore is crushed for the purpose of exposing solid reactants
to the lixiviant solution, many of the grain boundary surfaces which would have been
accessible only via porous diffusion into the particle become accessible at the particle
surface. Therefore, it is necessary to account for the dissolution of these "surface
deposits" in addition to those within the pores of the particle. Of the previous
investigators, only Chae and Wadsworth14 have accounted for the leaching of surface
deposits in a large-scale coarse ore leaching model. However, they assumed the deposits
to be completely dissolved before any reagent enters the particle pores, i.e., that the two

10
dissolution processes (surface and intraparticular) occur in series. Here it is assumed that
the two processes occur in parallel.
Making the same assumptions concerning reaction order as for the intraparticular
reaction, the rate of dissolution of solid reactant i at the particle surface is written
dCsi 3 k r*'-1 r‘-'Ab ( 1 - 8 )d t R p0 ( i - e 0)
where Csi is the solid concentration of solid reactant i at the particle surface, is the
reaction rate constant expressed per unit particle surface area, and <£si is the reaction
order in the solid concentration of solid reactant i. Equation (1-8) has an initial condition
Cs i (0) = Csio ( 1 - 9 )
Model Equations in Dimensionless Form
For the purpose of finding the important parameters of the model, all of the above
equations are recast here in dimensionless form.
Defining the set of dimensionless variables
CA“ = CU AO b C° AO
0 • " Cpl p i r
CT . - ° s iS I (-1usiO
$ = Rx = DAet
e aR2
where CA0 is some reference reagent concentration, and recasting the model in
dimensionless terms allows one to define a set of dimensionless parameters:

11
e o^i<'A0 X -c .^SlO
Po ( l _eo) ^Eio CEio
Po( l ~ Go) kplCpf0R2 3 k gic tf0RKsi
where
c . = c . + c .
represents the total extractable solid reactant i in the ore particles, and where ft is a
dimensionless stoichiometric ratio which indicates reagent strength relative to the grade
of solid reactant i; \ is the fraction of solid reactant i residing on particle surfaces
initially; and /cpi and Ksi are the ratios of the reaction rate of solid reactant i within particle
pores and on the particle surface, respectively, to the porous diffusion rate of reagent A,
and correspond to Damkohler numbers of the second type.
The rate of dissolution of solid reactant i along the pore walls of a particle,
equation (1-2), is rewritten
do pidx
* p iP i (1 - ^ )
VPi
with initial condition
op i (Z,0) = 1
The continuity equation for reagent A, equation (1-4), becomes
( i - i o )
( i - i i )
d2a3£2
2 _8oc _ v-v$ 8$ 1=1
■pi°iiadec0X
( 1 - 1 2 )
with initial and boundary conditions

12
a ( 5 , 0 ) = 0 ( 1 - 1 3 )
a ( 1 , t ) = ( 1 - 1 4 )
I f l O . t ) = 0 ( 1 - 1 5 )
Finally, the rate of dissolution of solid reactant i at the surface of a particle,
equation (1-8), is written
d °si _ Ks i P i , A i „~ d T " " T T *
with initial condition
° Si ( 0 ) = 1
( 1 - 1 6 )
( 1 - 1 7 )
Important Model Functionals
While the dimensionless model equations allow one to simulate the leaching
process with the fewest number of parameters, the solutions to these equations, in
concentration vs. particle radius and time, are useless for design purposes. Functionals,
or specially defined functions of other functions, provide a means of transforming the
results of the model into terms which are useful from a technical as well as an economic
standpoint. For our purposes, these functionals include the fractional conversion of solid
reactant X; and the effectiveness factor 77;.
Fractional conversion, which represents the fraction of extractable solid reactant
which has been dissolved at any given time, is expressed

13
l= 3 ( 1 - A i ) f ( l - api)Vdl + ^ ( 1 - a si ) ( 1 - 1 8 )
0
and assumes values from zero, at the beginning of the leach cycle, to one, at the end of
it. By ’extractable’ is meant only that reactant which is accessible to lixiviant solution,
and is therefore neither encapsulated within non-porous crystallites, nor residing within
inaccessible pores. Unfortunately, the amount of reactant within any given ore which
is amenable to leaching may only be determined (at present) by actual leaching
experiments, although at least one attempt has been made to derive a model by which
extractable grade might be predicted based on purely geometric considerations.23
While fractional conversion provides a meaningful way to interpret model results,
it gives little indication of the relative importance of model parameters. For this
purpose, the concept of the effectiveness factor is employed. In heterogeneous catalysis,
the effectiveness factor is the total rate of reaction occurring within a catalyst particle
taken as a fraction of the "ideal" rate which would result if there were no diffusion
limitation into the particle, i.e., if the entire volume of the particle contained reagent at
the same concentration as at the surface of the particle. The effectiveness factor for non-
catalytic gas-solid reactions was introduced by Ishida and Wen.24 Here, their formulation
is extended to each solid reactant and incorporates the rates of reaction occurring along
the external surface of the particle. The effectiveness factor rit, then, is defined as the
total rate of dissolution of solid reactant i throughout the entire particle, including the
external surface, taken as a fraction of the total rate which would result from no diffusion
limitation into the particle, and is expressed

14
3Kp i / afeioCS2dS +T|i = ------ £------------------------------- d - 1 9 )
3 KP i f ai i “ i ,z2 d z + < s i a t i a b0
This functional is bounded between the limits of zero and one, and can be thought of as
the ratio of the chemical reaction resistance to the total resistance due to diffusion and
chemical reaction.
HI. Experimental
Experimental Procedures and Equipment
Four batch cyanidation leaching tests were performed on artificial pellets
containing pure silver powder, mostly to provide parameters for the column leaching
studies discussed in chapter two. The pertinent chemical reactions include
2 A g° - 2 A g + + 2 e "
2Ag* + 4 CN~ - 2Ag{CN)'2
- | 02 + HzO + 2 e~ - 2 OH~
It is well known that oxygen gas as well as cyanide ion is necessary for the cyanidation
leaching of precious metals. The experiments in this study were open to the atmosphere,
and cyanide ion concentrations were maintained at or below 10‘3 M concentrations, which
is low enough to ensure that cyanide is the rate controlling reagent, and not dissolved
oxygen gas.25
All four tests were performed in an identical manner with the circulating-flow
apparatus shown schematically in Figure 1-2. This apparatus consisted of a column made

► OUT TO PUMP
polypropylene screen
glass wool
artificial ore pellets
__ threaded rod
plastic balls
tube barb
IN FROM FLASK
FIGURE 1-2Schematic diagram of the batch leaching test apparatus.

16
from a 15 cm piece of transparent acrylic pipe 2.5 cm in diameter, sealed at each end
with a plexiglas plate fitted with a tube barb. The plate on the bottom end was
permanently cemented onto the end of the column, while the top plate was held tightly
in place by three threaded rods, as per the diagram. Sections of 0.95 cm Tygon tubing
were attached to the top and bottom of the column assembly. To the top section was
connected a Masterflex peristaltic pump fitted with #18 Masterflex tubing, and the bottom
section extended into a 2-liter Erlenmeyer flask. A third section of Tygon tubing went
from the flask to the pump, thus completing the circuit. A small teflon impeller operated
by an adjustable speed motor was submerged into the flask.
The column was filled in a certain manner in order to prevent the movement of
fine material through the pump, and to prevent the formation of stagnant zones within
the ore charge. First, a small circle of polypropylene screen was dropped into the
column. This was followed by enough 1 cm diameter plastic balls to fill approximately
4 cm of the column, in order to force the solution into plug flow before it reached the
ore charge. This was topped with a small patch of glass wool, followed by the ore
charge, another patch of glass wool, enough plastic balls to fill the column, and another
bit of screen to keep the plastic balls out of the exit hole. Finally, the column was
sealed.
After charging with 79 g of pellets, the column was flooded with distilled water
and allowed to drain in order to saturate the pellet pores. The Erlenmeyer flask was
filled with 2000 mL of lixiviant solution prepared from distilled water, and containing
sodium cyanide and sodium hydroxide, both at 10-3 M concentration. At time zero, the
impeller was activated, and the pump was engaged so that solution flowed from the flask

17
up through the column at a fast flowrate. The residence time of the column was only a
few seconds, and the tests were run over a period of days, so perfect mixing could be
assumed.
Samples were taken at increasing intervals over the course of each test, and
analyzed with a Perkin-Elmer Model 3100 Atomic Absorption Spectrometer (AAS).
While the readings were within the range of the prepared standard solutions, the samples
were aspirated directly to the AAS from the Erlenmeyer flask. Otherwise, 1 mL was
pipetted from the flask and diluted 10:1 in dilution tubes with stock cyanide solution.
Since only a few samples were taken from each test, no special precautions were taken
to replace the solution which was removed.
The AAS was fitted with a silver lamp under a current of 10 mA. The slit width
was 7 nm, and the wavelength was 338.3 nm, giving a linear calibration range of about
10 ppm silver. Standards used were 0 (blank), 1, 5, and 10 ppm silver, and were
prepared from a 1000 ppm silver-nitric acid standard solution and the stock cyanide
solution from the batch tests. The AAS output was set to zero for the blank standard,
and was read in Absorbance units in continuous mode. Cyanide ion concentration was
not analyzed during the experiments.
Artificial Ore Preparation
Two batches of artificial silver ore pellets were prepared from a mixture of 95
wt-% assay-grade silica sand, 2/3 left coarse and 1/3 pulverized fine; 5 wt-% Type II
Portland cement, and small amounts of 99.9% pure silver powder, supplied by Johnson-
Matthey Electronics, and measuring 4 yum to 7 /im diameter. In one batch, 34.2 g of

18
silver powder was added to 50 kg ore, resulting in a grade of 6.34 x Id 6 mol Ag/g ore,
or 20 Troy oz Ag/ton. In the other batch, 0.206 g of silver powder was added to 3 kg
ore, resulting in a grade of 6.34 x 10'7 mol Ag/g ore, or 2 Troy oz/ton.
Each batch was mixed for several hours in a cement mixer fitted with a plastic
insert, then agglomerated with tapwater on a laboratory disk pelletizer, resulting in
pellets ranging from about 0.5 to 2 cm in diameter. The wet pellets were allowed to
cure under cover for several weeks. They became strong enough not to break when
dropped on the floor, but were prone to abrasion, necessitating quite careful handling.
IV. Results and Discussion — Theoretical
Computer Simulation: One Solid Reactant
In order to examine its general behavior, the following restrictions are applied
temporarily to the model: 1) only one solid reactant (n = 1), 2) first-order rate dependence
in the solid reactant concentration (</>p = 1), 3) no surface deposits of solid reactant (A=0),
and 4) constant bulk reagent concentration (ab= l). The continuity equation for reagent
A, equation (1-12), now becomes
w = -|5-d i2 i di p p a-c(1-2 0)
and the rate expression for disappearance of the solid reactant, equation (1-10),
dap _
dx = ~ KpPapa (1-2 1 )
These equations have initial and boundary conditions

19
a (5,0) = 0 a ( l , x ) = 1
(1-2 2 )( 1 - 2 3 )
( 1 - 2 4 )
ap ($,0) = 1 ( 1 - 2 5 )
and require the specification of only two parameters for their unique solution, kp and (3.
Concentration profiles of reagent A (solid curves) and one solid reactant (dotted
curves) are presented in Figure 1-3 and for all combinations of ^ = 0.01, 1, and 100,
and /3 = 0.01, 1, and 100. At kp = 0.01, where the particles are small and contain a
minute amount of solid reactant, reaction occurs at a fairly uniform rate throughout the
particle, regardless of the value of /?. This is as expected since a low value of np
suggests fast diffusion relative to reaction, and hence, negligible diffusion resistance.
Only at very high values of jS are any reagent concentration gradients established within
the particle, and even then the phenomenon is transient. Hence, at low values the
reaction zone assumption does not apply, since the rate of reaction is homogeneous
throughout the particle. In the absence of diffusion resistance, equation (1-20) is
unnecessary, and the reaction may be adequately described by equation (1-21), setting
a = ab.
For moderate values of kp, the type of kinetics depends primarily on the value of
/3, and hence, on the concentration of reagent A relative to solid reactant. At ^ = 1,
non-transient reagent gradients are established at all values of 13, but only at very high
(3 are they steep enough to result in diffusion controlled, zone-wise kinetics. At high ^
values, zone-wise kinetics prevail regardless of the value of (3, and the zones only
become narrower and more distinct with increasing /?. Therefore, for kp > 100, the

20
/? = 0.01
/? = 1
jS = 100
/cp = 0.01 Ka = 1 Kp = 100
At = 100
0.0 0.2 0.4 0.6 0.8 1.0
At = 0.01
0.0 0.2 0.4 0.6 0.8 1.0
At = 0.001
FIGURE 1-3Solutions to the simplified continuity equations for reagent A (a) and one solid reactant Op)-

21
reaction zone assumption becomes valid regardless of the reagent concentration, and the
steady-state approximation may safely be applied to equation (1-20).
The parameter /3 represents the ratio of stoichiometric equivalents of reagent to
solid reactant. In a particle of given porosity, a value of ft = 1 signifies that the pores
of the particle would contain just enough reagent to dissolve all extractable solid reactant
i present in the particle, if the pores could be completely filled with reagent at
concentration a = 1. Hence, it should be noted that for most applications of interest,
/3 will assume a value less than one.
Figure 1-4 shows the relation between fractional conversion and relative reaction
time for the nine model solutions in Figure 1-3. As is expected, the relative conversion
rates are essentially identical in the chemical reaction controlled situations, but decrease
dramatically with increasing diffusion resistance.
The relative magnitudes of diffusion and reaction resistance are most conveniently
illustrated in semi-log plots of effectiveness factor vs. conversion, as shown in Figure
1-5. At kp = 0.01, rj rapidly approaches unity at all values of /3. At kp = 1, the shape
of the r?-X curve changes dramatically between /3 = 10 and j3 = 100, signifying a shift
from reaction controlled to diffusion controlled kinetics. However, all rj-X curves
approach unity near complete conversion, which is the case for all one-reactant systems.
Finally, at kp = 100, the diffusion controlled behavior is well established at all 13 values.
After attaining a peak due to the initial transient influx of reagent into the barren pores,
the effectiveness factor decreases to a minimum. This corresponds to the reaction zone
moving toward lower reagent concentrations near the center of the particle. Once the
leading edge of the reaction zone reaches the center of the particle, the tj-X curves turn

Fra
ctio
nal
conv
ersi
on
22
FIGURE 1-4Fractional conversion (X) vs. dimensionless reaction time (3t) for the nine continuity equation solutions shown in Figure 1-3.

23
FIGURE 1-5Effectiveness factor (tj) v s . fractional conversion (X) for the continuity equation solutions shown in Figure 1-3.

24
up toward unity, similar to the effectiveness factor plots reported by Ishida and Wen for
a zero-order solid-gas reaction system.24
Relaxing the restriction of no surface deposits (X^O), and also assuming first-
order rate dependence at the surface (<£s= 1), the surface rate expression, equation (1-16),
becomes
( 1 - 2 6 )
Note that since the bulk reagent concentration is constant (ab= l) it need not appear in
the equation. Equation (1-21) is modified to
«pP(l-X) a ap
and these equations have initial conditions
( 1 - 2 7 )
®p ( 5 , 0) = 1 d - 2 8 )
CTs (0 ) = 1 ( 1 - 2 9 )
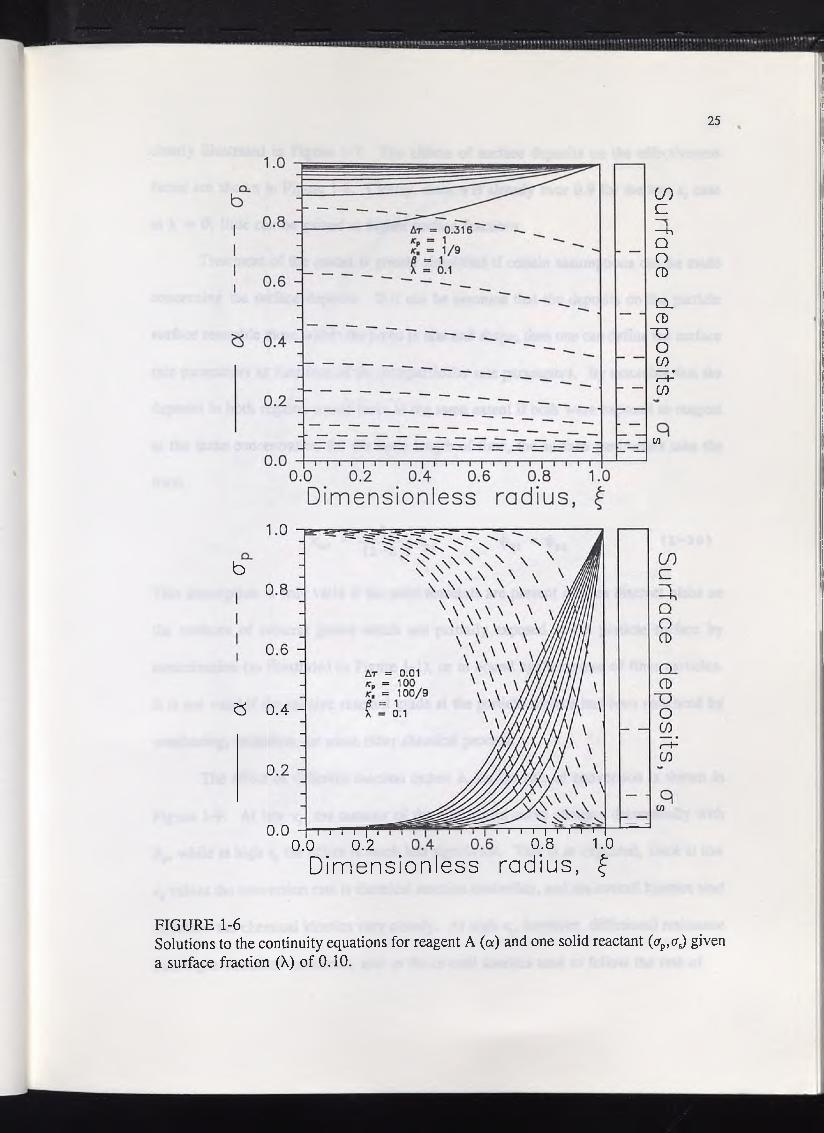
Figure 1-6 shows the results of two particle model simulations with X = 0.1, /3
= 1, kp = 1 and 100, and ks = Xkp/(1-X). These plots clearly show that, when the
particle undergoes zone-wise reaction, at a high value of kv, the solid reactant inside the
particle reacts largely in series with that at the particle surface, as predicted by Chae and
Wadsworth.14 However, when the particle is reacting in a more homogeneous manner,
at a low value of kp, the two regions react in parallel. The result is that, in the
homogeneous reaction mode, the presence of solid reactants on the particle surface has
little or no effect on the conversion rate, while in the less efficient zone-wise reaction
mode, these deposits can significantly enhance the rate of conversion. This result is
25
— - 0 3
LDc—hQOCD
Q_ CD
"O O CD ^i—HCO
b
b 0.4 -
i i i ~rp 0.0 0.2
i—i—|—rn i r 0.8 1.0
Dimensionless radius, f
FIGURE 1-6Solutions to the continuity equations for reagent A (a) and one solid reactant (ap,a^ given a surface fraction (X) of 0.10.

26
clearly illustrated in Figure 1-7. The effects of surface deposits on the effectiveness
factor are shown in Figure 1-8. Clearly, since rj is already over 0.9 for the low /cp case
at X = 0, little can be gained at higher surface fractions.
Treatment of the model is greatly simplified if certain assumptions can be made
concerning the surface deposits. If it can be assumed that the deposits on the particle
surface resemble those within the pores in size and shape, then one can define the surface
rate parameters as functions of the intraparticular rate parameters. By assuming that the
deposits in both regions would react to the same extent if both were exposed to reagent
at the same concentration for the same length of time, the surface parameters take the
form
Ksi - tsi - 4>pi ( 1 - 3 0)
This assumption is only valid if the solid reactants are present only as discreet blebs on
the surfaces of mineral grains which are partially exposed at the particle surface by
comminution (as illustrated in Figure 1-1), or in bound agglomerates of finer particles.
It is not valid if the relative reactant grade at the particle surface has been enhanced by
weathering, oxidation, or some other chemical process.
The effect of different reaction orders <£p on the rate of conversion is shown in
Figure 1-9. At low kp, the contour of the conversion curve changes dramatically with
<j) while at high Kj, the effect is much less significant. This is as expected, since at low
K values the conversion rate is chemical reaction controlled, and the overall kinetics tend
to follow the chemical kinetics very closely. At high kp, however, diffusional resistance
outweighs chemical resistance, and so the overall kinetics tend to follow the rate of

27
Dimensionless diffusion time, r
Dimensionless diffusion time, t
FIGURE 1-7Fractional conversion (X) vs. dimensionless diffusion time (r) given various surface fractions (X).

28
oocoCO<DcCD>+•>o0)
LU
FIGURE 1-8Effectiveness factor (rj) vs. fractional conversion (X) given various surface fractions (X).

29
FIGURE 1-9Fractional conversion (X) vs. dimensionless reaction time (k t) given various reaction orders (<£p) at kp = 1 and 100, 13 = 1.

30
reagent diffusion into the particle, which is far less dependent on reaction order. As
shown in Figure 1-10, at kv = 100, the shape of the tj-X curves changes with changing
4>P, but not necessarily the overall magnitude.
Computer Simulation: Competing Solid Reactants
In the presence of one or more reactants competing for reagent A, the kinetics of
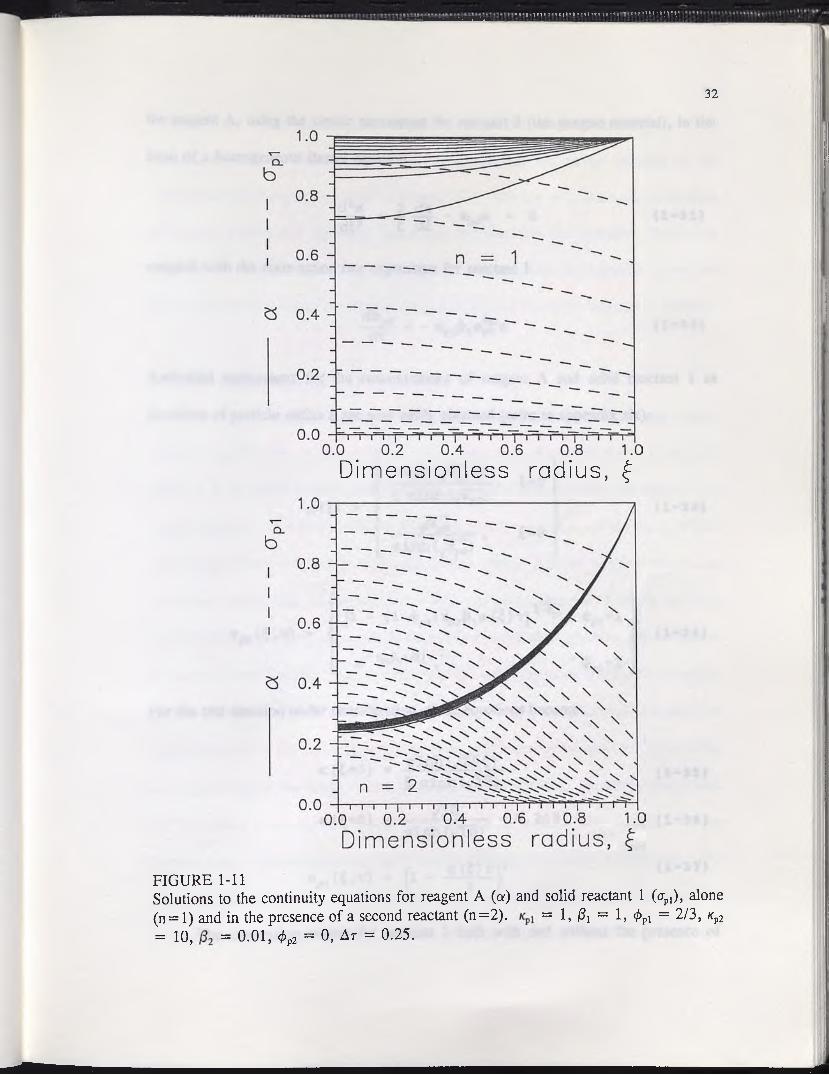
conversion of a valuable mineral component can be significantly altered. Figure 1-11
shows the concentration gradients of reagent A and one solid reactant with ^ — 1, /3a
= 1, and $pl = 2/3, and no surface deposits. A value of (£pl = 2/3 implies that solid
reactant 1 is present as discreet spherical blebs of uniform size (see Appendix 1). In one
simulation the reactant is alone, and in the other it is accompanied by a reactive "gangue"
material with *p2 = 10, /?2 = 0.01, and </>p2 = 0, also with no surface deposits. At these
parameter values it is assumed that there is one hundred times more solid reactant 2 than
1, and its intrinsic reaction rate is ten times slower. Also, a value of </>p2 = 0 implies
that the gangue material forms an even coating over the pore walls of the particle, or
could even be the ore matrix material itself.
By itself, solid reactant 1 reacts effectively throughout the particle, and the
reagent concentration gradients are not significant. In the presence of solid reactant 2,
however, the reaction shifts more toward the particle surface, and the reagent gradients
are steep, with a < 0.3 at the particle center. Also, the reagent gradients do not change
significantly with time, since the reagent demand of reactant 2 does not vary over the
entire course of leaching of reactant 1. Hence, the conversion kinetics of reactant 1
could be approximately described in this special case by a steady state continuity equation

31
ooCOCOCDc0>u0
LlJ
FIGURE 1-10 . ., .Effectiveness factor ft) v s . fractional conversion (X) given various reaction orders (4>P)at kp = 1 and 100, j3 = 1.
» » . y • ? n > i * 1 1 j * »* * 1 1 \ »• * ; s • * i \ t ' .* ■ * ? * ! ' * ! \ * V
32
Dimensionless radius, £
FIGURE 1-11Solutions to the continuity equations for reagent A (a) and solid reactant 1 (apl), alone (n = l) and in the presence of a second reactant (n=2). = 1, ft = 1, <£pl = 2/3, /cp2= 10, = 0.01, </>p2 = 0, AT = 0.25.

33
for reagent A, using the kinetic parameters for reactant 2 (the gangue material), in the
form of a homogeneous Bessel equation
d 2a ciad$2 5 dl ~ *P2a 0
coupled with the mass-action rate expression for reactant 1
( 1 - 3 1 )
dadx
pi -= - K’PiPl°VplPi a ( 1 - 3 2 )
Analytical expressions for the concentrations of reagent A and solid reactant 1 as
functions of particle radius £ are now easily obtained (refer to appendix six):
a ( 0 =
s i n h ( ^ $ )£ s i n h
5=0p 2
s i n h ( y / k ^ )
( 1 - 3 3 )
api ( 5 / t ) = •
[1 - ( lHfrpjjKpiPiCC ( 5 ) X] ^ P1 , 4>p l * l
KpiP 4>pi =i
For the test situation under consideration, these equations become
( 1 - 3 4 )
a(5*0)
a (£=0)
- (i -
s i n h ( / 1 0 £ )
£ s i n h ( / T o )
y/To s i n h ( v T O )
a(£) t \ 3
= 0 . 2 6 8
( 1 - 3 5 )
( 1 - 3 6 )
( 1 - 3 7 )
The conversion curves for reactant 1 both with and without the presence of

34
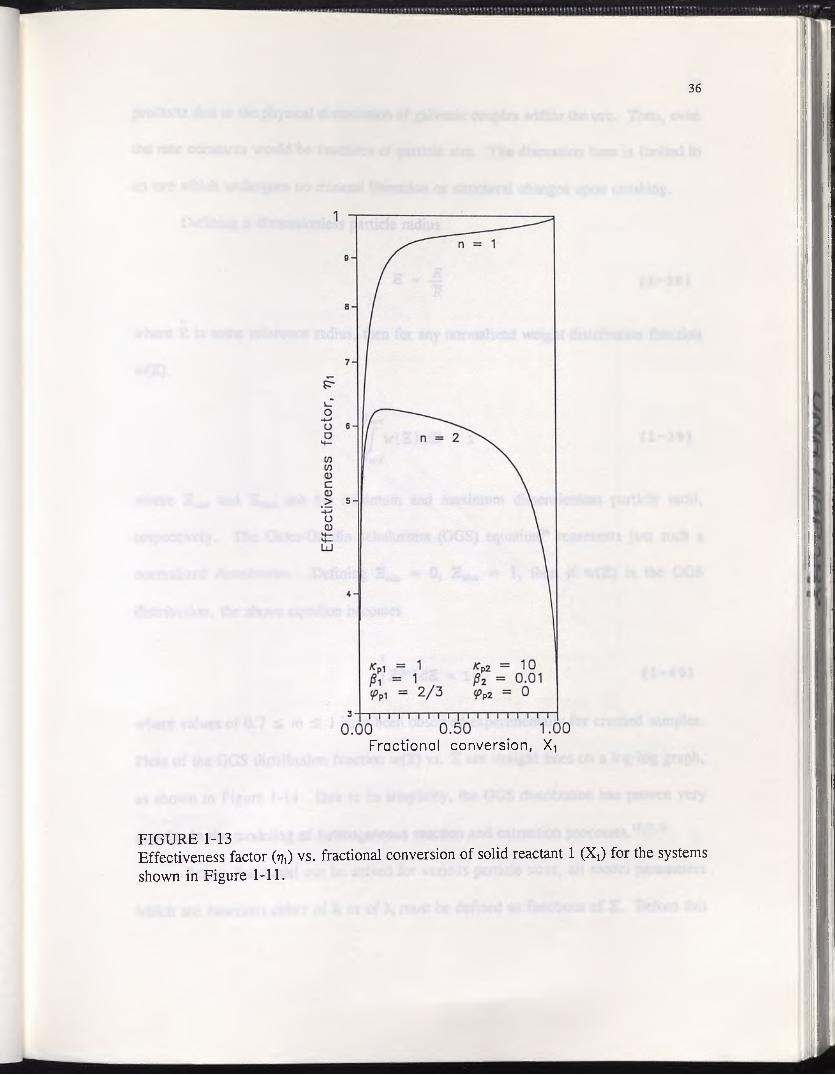
reactant 2 are shown in Figure 1-12. As expected, the conversion when n = 2 is
significantly delayed relative to n = 1. The effect of the second reactant on the
effectiveness factor r\x is illustrated in Figure 1-13. When n = 2, the curve does
not approach unity near complete conversion, but does just the opposite. This is the
norm for all minor constituents of multi-reactant systems, since the reagent is maintained
at low concentrations near the center of the particle until the major reactant is depleted.
An Approach for Particle Size Distributions
In order to apply a leaching model based on the particle scale to any real coarse-
ore leaching situation, it must be solved for a distribution of particle sizes. In order to
do this it is necessary to solve the model for a number of different sizes and integrate the
results together. This may present serious difficulties because it is not always clear how
process parameters will change with particle size. Much depends on the structure and
mineralogy of the ore. If, for instance, a highly porous low-grade oxide ore has been
crushed prior to leaching, there may be little difference between the various size fractions
in terms of their leaching characteristics, and the only parameter that would change is
particle size itself. However, if the ore is not very porous, such that solid reactants are
inaccessible to the lixiviant solution, then these reactants may be liberated by crushing
to varying degrees depending on the particle size. In that case, the extractable grades
of the various solid reactants would be a function of particle size, as well. In the
extreme case, leaching chemistry itself may be a strong function of particle size. If a
copper sulfide ore consists of cemented grains of pyrite and chalcopyrite bound within
an inert gangue material, different size fractions may actually yield different reaction

Fra
ctio
nal
conv
ersi
on,
35
FIGURE 1-12Fractional conversion of solid reactant 1 (X,) vs. dimensionless diffusion time (r) for the systems shown in Figure 1-11.
36
Fractional conversion, X-[
FIGURE 1-13Effectiveness factor (17,) vs. fractional conversion of solid reactant 1 (Xj) for the systems shown in Figure 1-11.

37
products due to the physical dissociation of galvanic couples within the ore. Then, even
the rate constants would be functions of particle size. The discussion here is limited to
an ore which undergoes no mineral liberation or structural changes upon crushing.
Defining a dimensionless particle radius
where R is some reference radius, then for any normalized weight distribution function
w(E)
where Emin and EMax are the minimum and maximum dimensionless particle radii,
respectively. The Gates-Gaudin-Schuhmann (GGS) equation26 represents just such a
normalized distribution. Defining = 0, EMax = 1, then if w(E) is the GGS
distribution, the above equation becomes
where values of 0.7 < m < 1 have been observed experimentally for crushed samples.
Plots of the GGS distribution function w(E) vs. E are straight lines on a log-log graph,
as shown in Figure 1-14. Due to its simplicity, the GGS distribution has proven very
18 27 28popular in the modeling of heterogeneous reaction and extraction processes.
Before the model can be solved for various particle sizes, all model parameters
which are functions either of R or of \ must be defined as functions of E. Before this
RR
( 1 - 3 8 )
( 1 - 3 9 )
l( 1 - 4 0 )

0.01 . , ° - 1 ,Dimensionless particle size, a
FIGURE 1-14 . . . . . . . ,-vT h e G ates-G audin-Schuhm ann normalized w eig h t distribution function (w (a )) vs.dimensionless particle size (S) at various values of the distribution parameter m.

39
can be done, various assumptions must be made concerning the effect of comminution
on the ore. For purposes of analysis, let us assume the following: (1) Only the surface
fraction X; is affected by crushing, and not the total extractable reactant grade CEi0, or
any other parameter, and (2) X; is inversely proportional to particle size (i.e.,
proportional to the ratio of particle area to particle volume). Hence, the surface fraction
X; of any given particle is defined relative to the reference particle thus
1 a
where \ is the surface fraction of the reference particles. Assuming that the intrinsic
kinetics of solid reactant deposits are not affected by comminution, the following rate
parameters are defined as functions of particle size
Kpi =( B- Xj )
1 - * i ,iSK'Pi KSi =
where xpi and Ksi are rate constants of the reference particles. Also, the diffusion time
r is defined in terms of a reference time r
x =1T72
T■32
These relationships allow us to solve the particle model for a range of particle sizes g
based on parameters from the reference size, S = 1.
The fractional conversion results of each particle size are combined via a
quadrature solution of the total fractional conversion integral:

40
aMX 1Xn = / Xi (Z)w(E)dZ = mfxi (S)B*-1d3 ( 1 - 4 1 )
min 0
where X;(2) is the fractional conversion for particles of size 2.
Figure 1-15 shows the conversion curves of various particle sizes (solid curves)
for a single reactant system, as well as the total conversion (dashed curves) for GGS
distributions at various values of m. Parameter values for each particle size are given
in Table 1-1. The total conversion curves are quite different in shape from the size-
dependent conversion curves, but are fairly insensitive to the distribution parameter m.
Figure 1-16 shows the rj-X plots for the various particle sizes. The largest three fractions
are undergoing zone-wise reaction while the smallest three are reacting homogeneously.
The approach taken above is only valid if there is no change in the total reactant
grade with comminution. If any liberation of previously unextractable material were to
occur during crushing, by the exposure of inaccessible diffusion channels, the unlocking
of encapsulated solid reactant, or otherwise, one would have to express the liberation as
a function of particle size, 1,(2), and include it the integrand of equation (1-41) thus
^ Max 1
XTi= f x I (3)l1(3)w(3)dS = mfx1(S)li (E)S--1dS ( 1 - 4 2 )
srain 0
making the requisite changes in Kpi and * si as well.
V. Results and Discussion ~ Experimental
In order to interpret the results of the batch leaching experiments, a model must
be derived for the reactor which relates the data (in this case, the concentration of

41
TABLE IT: Parameter values for the size distribution model solution.
2 *p 0 4>P X Ka &
1 100 1 2/3 0.01 1.01 2/3
0.8 63.8 1 2/3 0.0125 0.808 2/3
0.6 35.8 1 2/3 0.0167 0.606 2/3
0.4 15.8 1 2/3 0.025 0.404 2/3
0.2 3.84 1 2/3 0.05 0.202 2/3
0.01 - 1 - 1 0.0101 2/3

42
FIGURE 1-15 . . tFractional conversion for various particle sizes (X(S)) (solid curve^), and for the entire size distribution (Xx) (dotted curves) vs. reference diffusion time (t) for various values of the distribution parameter m.

43
5
oD
ww0c0>o0
UJ
FIGURE 1-16 E ffe c t iv e n e ss factor (y) v s . v a r io u s p artic le s izes ( a ) .
fractional conversion for various particle sizes (X(a)) at

, r -
dicyanoargentate ion, Ag(CN)2', in the batch lixiviant) to the general leaching model
derived in section II.
First, one needs an equation which represents the mass balance of reagent A
within the batch lixiviant: consumption by ore pellets = accumulation in batch lixiviant.
This is written, in dimensionless form
44
- 3 ( |f ) = v-3a,dx
and has an initial condition
a , ( 0 ) = l
where v is redefined thus
( 1 - 4 3 )
( 1 - 4 4 )
V =e0d “ ei)
and where eb is the ratio of the volume of batch lixiviant to the total volume of lixiviant
plus ore pellets. Since pellet sizes were kept fairly uniform in each batch leaching test,
the size distribution integral has been left out of equation (1-43).
Next, if one ignores the dissolved reactant species held up within the pellet pores
(legitimate if v is very large), then the fractional conversion may be defined in terms of
the batch lixiviant concentration of dissolved reactant species thus
* i = PivXii, ( 1 - 4 5 )
where
Xi*'ib
^i^AO —

45
and where Cib is the concentration of dissolved reactant species in the batch lixiviant.
Data from the experimental batch tests can now be simulated with the general model.
Now one can see why 79 g of pellets were used in all of the tests. It is because
this resulted in integer values for the product fiv. These values were: 2 in the high-grade
pellet tests, and 20 in the low-grade pellet test. Careful inspection will show that this
product actually represents the stoichiometric excess of rate-controlling reagent in the
batch at time zero. Thus, there was twice as much cyanide as was needed for full
conversion in the high-grade tests, and 20 times as much in the low-grade test.
Results of the batch tests are shown in Figures 1-17 through 1-20, and Table l-II
summarizes the parameters involved. The leaching tests of the high-grade particles
(Figures 1-17 through 1-19) provided an estimate of the effective diffusivity, DAe, based
on the apparently (by inspection of the datacurve shapes) diffusion-controlled kinetics of
all three situations. The low-grade ore leaching test (Figure 1-20) provided the estimate
of the variable reaction order, </>p, due to the apparently reaction-controlled kinetics
manifested in that test.
It is interesting to note that, if the effective diffusivity is assumed to obey
DjuP o (1-46)u ke T*'o
where DAB is the bulk diffusivity and r0 the tortuosity of the porous medium, then the ore
pellets have a tortuosity of r0 = 1.58. The theoretical tortuosity of a bed of packed
spheres is r0 = tt/2 = 1.57, which is ratio of the length of the shortest path around a
spherical obstacle to its diameter. The literature suggests taking a value for the tortuosity
of Tq = 2 in all leaching simulations,20 but for pellets made up of packed particles, or

46
TABLE l-II: Parameter values for the batch leaching tests. (All units cgs. Concentrations based on gmols.)
Parameters Batch test 1 Batch test 2 Batch test 3 Batch test 4
Ore mass 79 79 79 79
Soln. volume 2000 2000 2000 2000
b 0.5 0.5 0.5 0.5
n > o i o -6 IO’6 i o -6 IO'6
Cpo 6.34 -IO'7 6.34-IO'6 6.34-IO'6 6.34-IO'6
o Ae* 2.35 • IO6 2.35 • IO6 2.35-IO-6 2.35 • 10'6
k p * 3.76-107 3.76-IO7 3.76-IO7 3.76-IO7
R 0.68 0.86 0.60 0.62
0 0.094 0.0094 0.0094 0.0094
eb 0.977 0.977 0.977 0.977
e0 0.20 0.20 0.20 0.20
Kp 10 1600 780 830
V 213 213 213 213
Po 2.1 2.1 2.1 2.1
2 2 2 2
^Determined from the experimental results.* Calculated from experimentally determined values.

Convers
ion
47
FIGURE 1-17Results of batch test 1.

48
FIGURE 1-18Results of batch test 2.

49
00
0.8 -
0.6 -
0.4 -
0 .0
—^ aa
a
/ a/ a / • Batch test 3:
/ • Kp = 780<P? = 2|S = 0.0094
i i i i i—i—i—i i n i i i i i i i i i i0 10 15 20
Dimensionless diffusion time, r
FIGURE 1-19Results of batch test 3.

50
FIGURE 1-20 Results of batch test 4.

agglomerates of fine material, perhaps r0 = ir/2 is a more realistic assumption.
Since the pellets had a high porosity, and were known to be homogeneous, it was
assumed that no significant surface deposits were present. Thus, only the parameters kp,
4>p, and the ratio r/t had to be determined for the general leaching model, j8 and v being
given from direct measurements and the operating conditions of the tests. The ratio r/t
was obtained from matching model curves to the data from the three high-grade leaching
tests. This number was then used to non-dimensionalize the data from the low-grade
leaching test, after which kp was adjusted until the initial slopes of the model curve and
the data curve matched. Only then was <t>p adjusted until the most satisfactory fit of the
entire curve was obtained. Finally, the value of <f>p was used to calculate kp for the high-
grade tests. Thus, even though the ratio of grades between the two pellet batches was
only 1:10, since ^ is proportional to the ore grade taken to the </>p power, an order of 4>p
= 2 results in a 1:100 ratio in kp values. Of course, this increase is enough to transform
kinetics from the homogeneous regime to the zone-wise regime (refer to Figure 1-3,
above). The validity of this approach is borne out by comparing the results of the low-
grade test in Figure 1-17 to the results of the large-diameter high-grade test in Figure 1-
18, where kp was chosen strictly on the basis of the model fit of the low-grade test.
Figures 1-19 and 1-20 show the results of duplicate tests of medium-diameter
high-grade pellets. The model fits are excellent up to about X = 0.4, after which the
model overshoots the data by a consistent AX of about 0.1. This phenomenon is difficult
to explain. It may not be purely coincidental that X = 0.4 corresponds to Cb » 10 ppm,
the top of the linear calibration range, and therefore marks the spot where the samples
were no longer aspirated directly from the reactor flask, but had to be pipetted out and

52
diluted 1:10. How much error was introduced into the data as a result of dilution is,
however, impossible to ascertain. Whatever the cause, the important thing to notice from
the plots is how accurately the model simulated the rate of reaction (i.e., the slope of the
data) with time, irrespective of the dislocation in X.
Figure 1-21 shows the 73-X plots resulting from the model fits of all four batch
tests. As is expected, the low-grade pellets from test 1 react at high effectiveness while
the higher grade pellets are much less effectively leached, the largest pellets even less
so than the others.
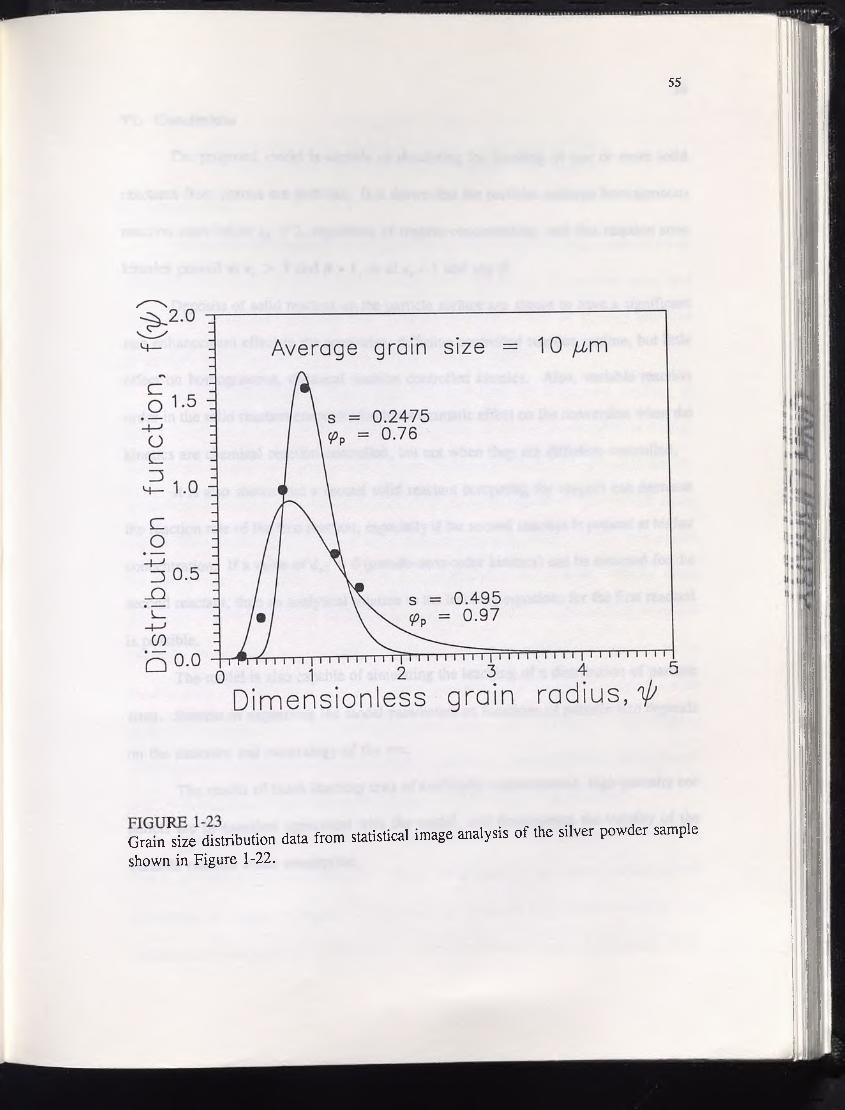
Finally, for the sake of completeness, a scanning electron micrograph (SEM) of
the silver powder used in the artificial pellets is shown in Figure 1-22, and the
corresponding size distribution from statistical image analysis is shown in Figure 1-23.
(See Appendix One for details concerning the interpretation of the Figure.) Based on the
variable order argument posed in Appendix One, one would expect the dissolution
reaction to be roughly first-order or lower. However, since the average grain size from
the image analysis (10 /xm) is more than twice that reported by the supplier of the silver
powder, and also judging from the wide variability of shapes and sizes, and especially
the degree of particle overlap apparent from the micrograph, results derived from the
statistical data from the image analysis cannot be taken as conclusive.

53
FIGURE 1-21Calculated effectiveness factor (77) vs. Ag conversion (X) for the batch leaching tests.

55
Grain^size distribution data from statistical image analysis of the silver powder sample shown in Figure 1-22.

VI. Conclusions
The proposed model is capable of simulating the leaching of one or more solid
reactants from porous ore particles. It is shown that the particles undergo homogeneous
reaction rates below kp = 1, regardless of reagent concentration, and that reaction zone
kinetics prevail at *p > 1 and j3 » 1, or at kp » 1 and any 0.
Deposits of solid reactant on the particle surface are shown to have a significant
rate enhancement effect in the zone-wise, diffusion controlled reaction regime, but little
effect on homogeneous, chemical reaction controlled kinetics. Also, variable reaction
order in the solid reactant concentration has a dramatic effect on the conversion when the
kinetics are chemical reaction controlled, but not when they are diffusion controlled.
It is also shown that a second solid reactant competing for reagent can decrease
the reaction rate of the first reactant, especially if the second reactant is present at higher
concentration. If a value of 4>p2 = 0 (pseudo-zero-order kinetics) can be assumed for the
second reactant, then an analytical solution to the leaching equations for the first reactant
is possible.
The model is also capable of simulating the leaching of a distribution of particle
sizes. Success in expressing the model parameters as functions of particle size depends
on the structure and mineralogy of the ore.
The results of batch leaching tests of artificially agglomerated, high-porosity ore
pellets are in excellent agreement with the model, and demonstrate the validity of the
variable reaction order assumption.

Chapter TwoA Mathematical Model for Heap Leaching of One or More Solid Reactants from Porous Ore Pellets
I. Introduction
There is an abundance of literature referring to the kinetics of large-scale leaching
processes, but many of the articles deal only with kinetics at the particle scale, and
completely ignore the larger process. Those which do not can be divided into two
categories: sulfide leaching models and oxide leaching models.
Several mathematical models have been developed for large-scale leaching of
coarse sulfide ores, including heap and dump leaching, as well as in-situ solution mining.
Perhaps the earliest was published in 1942 by Taylor and Whelan,29 who found that the
extraction of copper from sulfidic dumps could be described by an exponential decay
curve. Their model was of little value for scale-up, however, since it could only predict
the future recovery from a dump whose past performance was known. Nearly thirty
years later, Harris30 proposed the first truly predictive model for dump leaching of copper
sulfides. He assumed that the entire process was simply an extension of leaching at the
particle scale, the rate-controlling step being the diffusion of oxygen through the void
space of the dump. While this "pseudo-particle model" was definitely an improvement
over the earlier model, difficulty in identifying parameters from experiment and a general
lack of mathematical rigor rendered it impractical for engineering purposes.
Certainly the most rigorous, and complex, model of sulfide dump leaching to date
is that of Cathles and co-workers.31,32 Their model assumes the unsteady-state thermal
convection of oxygen through the void space of the dump to be rate-controlling, and
includes both oxygen and heat balances. While the earliest version, published in 1975,
57

58
was restricted to one-dimensional air and solution flow, this was later extended to two-
dimensional air flow with the aid of stream functions and potential flow. The model also
accounts for the presence of bacterial catalysis, intermittent solution flow, and seasonal
ambient temperature changes, and was validated experimentally in both large column
tests and small dumps.31'33
More recently, a model for in-situ solution mining of copper sulfides has been
developed by Gao, et al.,7,34 which also includes unsteady-state oxygen and heat balances
in one dimension. Unlike dump leaching, in-situ solution mining involves the forcing
of lixiviant solutions through underground cracks and fissures. As a result, the void
spaces are mostly saturated with solution, and no thermally driven convection is possible.
Thus, the variability of temperature only affects the equilibrium concentration of
dissolved oxygen in solution, and only one fluid phase need be considered.
With the exception of the earliest models, all of the large-scale sulfide leaching
models assume particle level kinetics of the shrinking-core or the reaction zone type,
as derived by Braun, et al.4 To this author’s knowledge, the unsteady-state model for
the leaching of sulfide particles derived by Bartlett9 has never been incorporated into any
simulation of a large-scale leaching operation.
Concerning the acid leaching of copper oxide ores, Roman and co-workers12,35
developed a model which accounts for the gradual consumption of sulfuric acid in the
lixiviant solution as it trickles through the heap. To achieve this end, the heap is
conceived as a number of adjacent columns, or "unit heaps" which in turn consist of
several unit volumes stacked one atop another. By solving for the acid consumption
within each unit volume, the total concentration gradient within the heap may be pieced

59
together. Chae and Wadsworth14 have recently developed a similar model for large scale
leaching of copper oxides, which is based explicitly on the "maximum gradient," or plug-
flow, reactor. Both of these models are for the one-dimensional, isothermal case, and
assume pseudo-steady-state, "shrinking-core" type kinetics at the particle level. The
latter model, however, also accounts for consumption of acid by gangue materials, and
initial enhancement of the leaching rate due to the flushing of minerals on particle
surfaces.
Much less attention has been focused on modeling the kinetics of precious metal
heap leaching. To this author’s knowledge, only one previous attempt has been
published to date, that of Prosser.17 Prosser and Box36 derived a general model for the
heap leaching of several minerals with several reagents, but assumed that transport of
reagent through the heap to the ore particles, chemical reaction, and transport of
dissolved products out of the heap all occurred very fast compared to diffusion of reagent
into the particle pores. Hence, the model was reduced to nothing more than the modified
shrinking core model of Box and Prosser15 discussed in chapter one.
In this study, a general model is derived for application to the heap leaching of
any porous, low-grade ore when the rate-controlling reagent is strictly in the aqueous
phase, as in acid leaching of copper oxides, or basic cyanide leaching of oxidized
precious metals ores. The general model fully developed in chapter one is assumed at
the particle level, and provides both intraparticular and particle surface leaching rate
terms for a global heap-scale model based on the one-dimensional, plug-flow reactor at
unsteady state. Reduction of the model to dimensionless form facilitates the identification
of important scale-up parameters, and the analysis of various leaching rate regimes.

60
II. Model Development
The heap leaching operation is a heterogeneous, non-catalytic, fixed-bed reactor
operating under unsaturated flow conditions. Lixiviant solution enters at the top of the
heap and trickles through the interstices of the ore particles. Reagents diffuse into the
pores and fissures of the ore particles, and are gradually consumed by reaction with one
or more solid reactants. These solids, in turn, are gradually dissolved throughout the
heap. Based on this physical picture, shown schematically in Figure 2-1, a global heap
model is derived with the following assumptions:
1. The heap leach reactor is essentially an unsteady-state plug flow reactor, i.e., flow of reagents and dissolved products occurs only by axial convection with no short-circuiting.
2. All physical parameters within the heap remain uniform and constant throughout the leach cycle.
3. No inhibition of diffusion into or out of the particles occurs due to dense packing or a stagnant boundary layer.
4. The heap operates isothermally.
At the particular level, the following assumptions are made:
1. The particles are spherical, and of uniform size, density and porosity.
2. All effective diffusivities are constant and uniform.
3. Reactive solids are evenly distributed throughout the pore surfaces of the particle, and their total relative volume is insignificant.
4. Dissolution reactions are irreversible, first order in the concentration of one rate-controlling reagent, and variable order in the solid concentration of the reactant.
5. Intraparticular processes are not at steady state.
These assumptions at the particle-scale result in the formulation of the general model

M E T A L D E P O S IT S :
ALONG PORE WALLS
ON PARTICLE SURFACES
FIGURE 2-1Schematic diagram of a heap.
ORE PARTICLES v = ( l - ejvh
ENTRAINED AIRV = (e„ - eJV„
BULK LIXIVLANTV = e,V*
ORE MATRIX V = (1 - t J Q - eJVh
PORE STRUCTURE V = ( 1 - eJe0V,
ON

62
derived in chapter one. In addition to equations (1-1) through (1-9), it is useful to
account for the diffusion of dissolved products from ore particles and into the bulk
lixiviant of the heap. Assuming a one-to-one correspondence between moles of solid
reactant i and its dissolved species, a mass balance for the dissolved species within the
porous sphere is written
Died2C0 r :
i , 2 dCj r d r
+ p 0 ( l £0) kp icpi CA e a t(2-1)
where Q is the concentration of dissolved species i, Die is the effective diffusivity of the
solute within the particle pores, and all other terms are as previously defined. Equation
(2-1) has initial and boundary conditions
C ^ r ^ ) = 0 (2-2 )Ci t ) = Cib ( 2 - 3 )
( 0 , t ) = 0 ( 2 - 4 )d r
where Cib is the concentration of dissolved species i in the bulk lixiviant solution external
to the ore particle.
Consider a packed bed of porous ore particles of radius R through which is passed
a lixiviant solution at a constant flowrate. Assuming ideal plug flow through the bed,
and ideal mass transfer from the bulk solution into the pellet pores, the following mass
balance may be written for dissolved species i in the bulk lixiviant which accounts for
both the intraparticular and particle surface reactions

63
8C.- Ub i b 3 ( i - e „ )
dz Ry c^slC — P ‘K s i ' - s i b > ie V dr Z = R
= e. dCibd t
( 2 - 5 )
where us is the superficial velocity of lixiviant flow through the bed, eh is the void
fraction of the bed (not including particle pores), and eb is the relative volume of the bed
occupied by bulk lixiviant (generally a small number). Equation (2-5) has initial and
boundary conditions
Ci b ( z , 0) = 0 ( 2 - 6 )
Ci b ( 0 , t ) = 0 ( 2 - 7 )
An analogous mass balance equation for reagent A in the bulk lixiviant may be
written when reagent A is strictly a component of the aqueous phase
- u.dC ^ 3 ( l - e h)dz R
n v r^Biny> K si^si uAb + £>i = l b i 4 % ,
dC.= e. Ab
d t(2-8 )
with initial and boundary conditions
0 ) = 0
^Ab ® ' t) ~ CaO
( 2 - 9 )
(2-1 0)
The Model in Dimensionless Form
As in chapter one, the above equations are recast in dimensionless form in order
to determine the important design parameters. In addition to the dimensionless variables
defined in chapter one for the particular model, the following are defined for the global
heap model:
C i - Cib C = — 0 =Z l = i Z ■ b t CMZ ebZ

64New dimensionless parameters for the global heap model include:
6, - DV =
Ae e 0 ( l - e A)0) = 3 ( 1 - e h)DAeZ
UsR‘
where is a diffusivity ratio of dissolved species i to reagent A, v is the volume ratio
of bulk lixiviant to solution held up in particle pores, and w is the ratio of the porous
diffusion rate of reagent A to the axial convection rate of lixiviant in the heap and
corresponds to the inverse of the Peclet dimensionless group for mass transfer.
In addition to equations (1-10) through (1-17), which describe the particular
leaching model in dimensionless terms, the mass balance for dissolved species i within
the pores of a particle is written
d2Xi ^ 2 dXid ? 5 K + V ° P i
5Xi _ 3 3Xidx v o ) 5 0
( 2 - 1 1 )
with initial and boundary conditions
X ( 5 , 0 ) = 0
X (l,r) = %ib
% (0,T) = 0
(2-1 2)( 2 - 1 3 )
( 2 - 1 4 )
In all of the particular model equations, as in equation (2-11), the dimensionless diffusion
time r must be replaced with the dimensionless bulk flow time 9 so that the particular
and global heap models may be solved with a common time variable.
In the bulk lixiviant of the heap, the mass balance for dissolved species i is
written

65
. Ks i a ^ i „ _ 5X ii ( 2 - 1 5 )_ U e 7* U , v ZT}T—
with initial and boundary conditions
Xxb(C# 0) = 0
xijb(o,0) = 0( 2 - 1 6 )
( 2 - 1 7 )
The analogous balance equation for a strictly aqueous-phase reagent A in the bulk
lixiviant is written
with initial and boundary conditions
Important Model Functionals
As in chapter one, while the dimensionless model equations involve the fewest
number of parameters, the solutions to these equations, in concentration vs. heap depth
and time, are of little use for designing heaps. The functionals which will prove the
most useful include fractional conversion (integrated here over the entire depth of the
heap), extraction, and heap effectiveness factor.
Once again, fractional conversion represents the fraction of extractable solid
reactant which has been dissolved at a given time, and is simply equation (1-18)
integrated over the total depth of the heap. For solid reactant i, this is expressed
a ^ C / 0) = 0
<**(0,0) = l
( 2 - 1 9 )
(2-20)

66
3 ( U i ) / ( l - opi ) ¥ d t + ^ ( 1 - a al ) dC (2-21)
As in the particle model, X; takes values from zero to one.
Extraction of reactant i from the heap represents the total fraction of extractable
reactant i which has issued from the heap at any given time. Except for very short
leaching times, or in heaps with an unusually high holdup, extraction takes approximately
the same value as conversion as a function of time, but may be written simply as a
function of the bulk concentration of dissolved species i at the bottom of the heap thus
(2-22)
Unlike conversion, extraction may be evaluated directly from chemical analysis of heap
effluent and is therefore a far more convenient measure of heap performance. It must
be re-emphasized that these functionals are based on only that amount of reactant which
can be obtained from the heap, and not the total grade of the ore.
The heap effectiveness factor is defined, analogous to the particle effectiveness
factor of chapter one, as the reaction rate of the entire heap taken as a fraction of the rate
which would obtain if there were 1) no diffusion limitation into the ore particles, and 2)
no convection limitation through the heap, i.e., if the heap were a stirred tank reactor
with reagent concentration maintained at CA0. As such, the heap effectiveness factor may
be written by simply integrating (separately) both the numerator and the denominator of
equation (1-19). For solid reactant i, the resulting expression takes the form

67
( 2 - 2 3 )
and, like the particle effectiveness factor, assumes values between zero and one.
HI. Experimental
Figure 2-2 shows a schematic diagram of the experimental leaching columns. The
tall column was comprised of six 61 cm (2 ft) sections made of transparent acrylic pipe
of 9.53 cm (3.75 in) ID, and the short column comprised only one section 30 cm (12 in)
long and 6.35 cm (2.5 in) ID. All column sections were fitted with plexiglas perforated
plates and funnels with hoses at the bottom to collect and distribute lixiviant.
Approximately two centimeters of glass wool were packed into the bottom of each
column section to prevent the movement of any fine solids from one section to another.
All column sections were open at the top. Pumping from the lixiviant tank to the
column, and from section three to section four in the tall column, was achieved with
Masterflex peristaltic pumps fitted with #13 Masterflex tubing.
Initially, the column sections were flushed with water to wash out any fine
material which may have resulted from packing the ore pellets, and to fill the pellet pores
with water. Then, water was pumped into the column, and the flowrate was adjusted to
3.8 mL/min in the tall column, and 4.1 mL/min in the short column, corresponding to
superficial velocities of 8.9 x 10r* cm/s and 2.2 x 10‘3 cm/s, respectively. Once the flow
was steady, lixiviant was introduced. The lixiviant was a solution containing 10 M
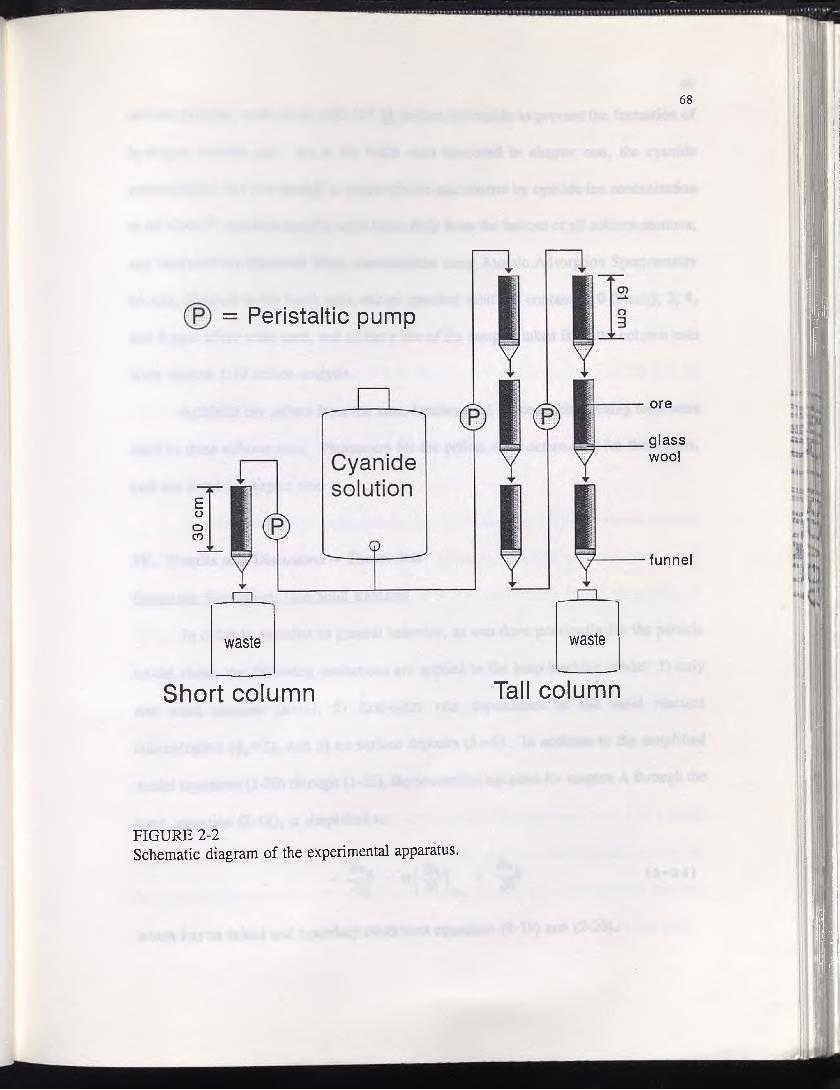
30 cm
68
= Peristaltic pump
oreglasswool
funnel
waste waste
— _____ _
Short column Tall column
FIGURE 2-2Schematic diagram of the experimental apparatus.

sodium cyanide, made basic with Id3 M sodium hydroxide to prevent the formation of
hydrogen cyanide gas. As in the batch tests discussed in chapter one, the cyanide
concentration was low enough to ensure kinetic rate control by cyanide ion concentration
at all times.25 Solution samples were taken daily from the bottom of all column sections,
and analyzed for dissolved silver concentration using Atomic Adsorption Spectrometry
(AAS), identical to the batch tests, except standard solutions containing 0 (blank), 2, 4,
and 6 ppm silver were used, and all but a few of the samples taken from the column tests
were diluted 1:10 before analysis.
Artificial ore pellets from the same batches used in the batch leaching tests were
used in these column tests. Parameters for the pellets were determined for those tests,
and are listed in chapter one.
IV. Results and Discussion -- Theoretical
Computer Simulation: One Solid Reactant
In order to examine its general behavior, as was done previously for the particle
model alone, the following restrictions are applied to the heap leaching model: 1) only
one solid reactant (n = l), 2) first-order rate dependence in the solid reactant
concentration (</>„=1), and 3) no surface deposits (\=0). In addition to the simplified
model equations (1-20) through (1-25), the convection equation for reagent A through the
heap, equation (2-18), is simplified to
( 2 - 2 4 )
which has as initial and boundary conditions equations (2-19) and (2-20).

70
The simplified heap model requires the specification of four parameters in order to obtain
an unique solution, kp, fi, v and u.
Heap concentration profiles of reagent A (solid curves) and fractional conversion
profiles of one solid reactant (dotted curves) are presented for all combinations of «p =
1, 10, and 100, and the product /cpu = 1, 10, and 100, at /3 and v = 1 in Figure 2-3.
At KpW = 1, which represents a short heap operating at a high lixiviant flowrate, reaction
occurs at a fairly uniform rate throughout the heap, regardless of the value of This
is as expected since a low value of kpco suggests a fast convection rate relative to
reaction, and thus, negligible convection resistance. Hence, at low values of kpgj, the
global model of the heap is largely unnecessary, and only the particle model is needed
to simulate the leaching process.
As co is increased at any value of kp, the convection limitation becomes more
significant. At kpco = 10, the reagent concentration profiles are monotonic, and
convection through the heap may or may not be the rate-limiting factor, depending on
the actual value of co. At kpco = 100, the reagent profiles are sigmoidal, with only a
narrow region of the heap under active leach at any given time, and the degree of
reactant conversion at any depth parallels the propagation of reagent to that depth. At
these parameter values, heap leaching is completely convection controlled, and one is left
with what might be called "shrinking-heap" kinetics.
Figure 2-4 shows plots of the effluent complex concentration, with 6 = 1 (solid
curves), overall fractional conversion (large dashes) and extraction (small dashes) as
functions of flow time for k, = 1. At 0 = 0.01, the number of residence times required
for complete reaction is large, and with so much fluid being passed through the heap
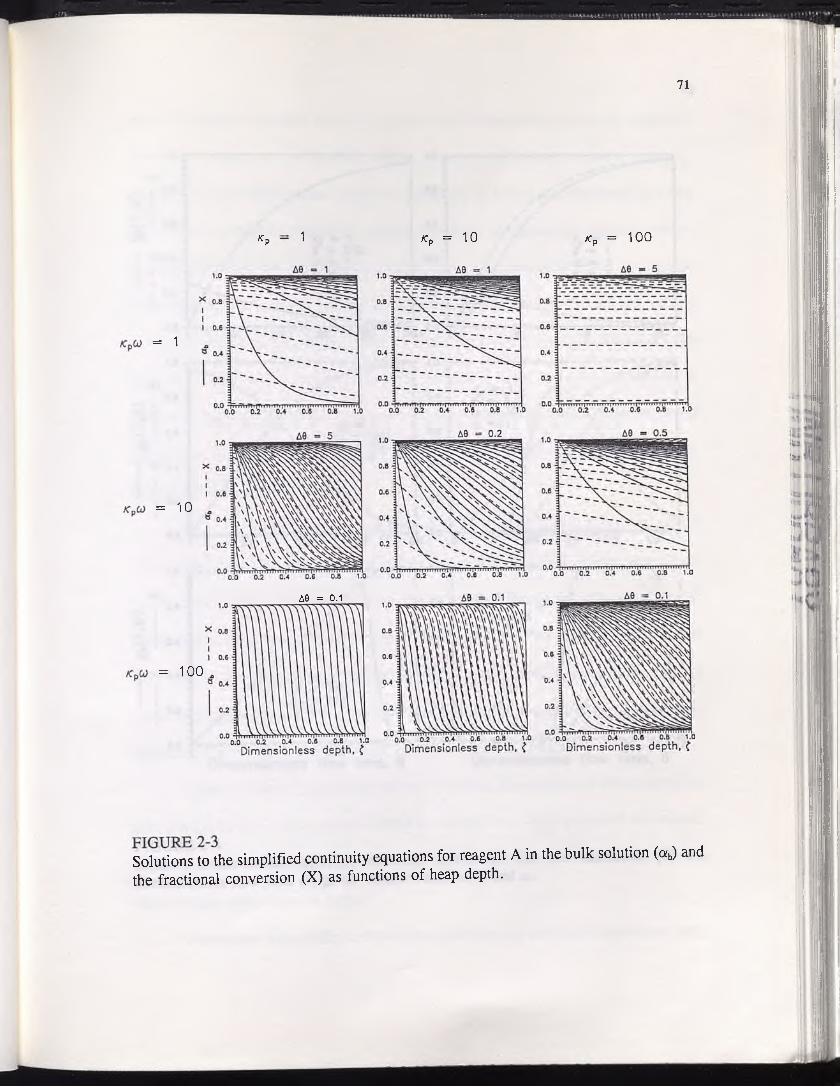
71
Kp — 1 /Cp = 10 Kp = 100
= 1
= 10
A0 = 0.1
= 100
Solutions to the simplified continuity equations for reagent A in the bulk solution (ab) and the fractional conversion (X) as functions of heap depth.

XbO
.e)
------
-Xb(
i.0)
------
-Xb(
i.e)
72
FIGURE 2-4 .Effluent concentration ( X b ( M ) ) > fractional conversion ( X )
dimensionless flow time (0) given various values of /3 and co.and extraction (E) vs.

73
over the course of the leach cycle, the total holdup of dissolved products is negligible.
Hence, the conversion and extraction curves largely coincide at all values of kp and w.
At kp(j) = 1, the effluent concentration curve consists of a short peak followed by a long
tail, and the conversion curves are roughly the shape of exponential response curves,
signifying the dominant role of particle kinetics. As kpw is increased, the Xb peak grows
taller until, at kpw = 100, it reaches its maximum value of one and flattens out. Above
a certain value of co, the dimensionless flow time for complete reaction decreases to its
limit, d = M(3v, which is simply the time required in order to feed a stoichiometric
amount of reagent to the heap. The conversion curve is a straight line, since the rate of
propagation of the reaction zone through the heap is constant at dfx/d0 ~ ftp (e.g., the
bottom left comer of Figure 2-3).
At 13 = 1, the overall flow times are much shorter, and a "chromatographic
effect" due to the high diffusion driving force at the beginning of the leach cycle results
in significant holdup. This effect, which causes the characteristic delay in attaining peak
concentration in all heap leaching operations, is only significant when the ore grade is
very low, as in precious metal leaching operations. At kpu = 100 and kp — 1, both the
conversion and extraction curves are linear, yet nearly half of the solid reactant has been
converted before any dissolved complex is seen in the heap effluent.
Figure 2-5 shows the relation between fractional conversion and relative reaction
time for the same particles at three different values of to. As is expected, the relative
conversion rate is much slower in the high to, "shrinking-heap" situation than in the
shorter heaps with faster flowrates.
The relative magnitudes of convection, diffusion and reaction resistance are most

74
FIGURE 2-5 .Fractional conversion (X) vs. reaction time (y3w(0-l)/3), given various values of w.

75
conveniently illustrated in semi-log plots of heap effectiveness factor vs. fractional
conversion, as shown in Figure 2-6. At low values of w, the heap effectiveness factor
approaches the particle effectiveness factor as a limiting value. As co is increased, the
effectiveness drops until, at about kpgo = 100, the ineffective character of the bulk flow
process dominates, obscuring any effects at the particle level. This happens irrespective
of the reagent concentration of the incoming fluid.
Relaxing the restriction of no surface deposits and also assuming first-order rate
dependence at the surface, the surface rate expression becomes
and the intraparticular rate expression becomes
Since it was already determined in chapter one that surface deposits have little or
no effect on the kinetics of particles with low Kp, only the case when this parameter is
high, and particle kinetics are diffusion controlled, will be considered here. Figure 2-7
shows the effect of various surface fractions X on the rate of reactant conversion from
the same particles in two different heap simulations. Higher surface fractions result in
much faster conversion rates at co = 0.1, but have little impact at u = 1. These results
are illustrated in the tj-X plot shown in Figure 2-8. At low u, kinetics at the particle
( 2 - 2 6 )
These equations have initial conditions
op i t , 0) = 1
os ( f , 0 ) = l
( 2 - 2 7 )
( 2 - 2 8 )

Hea
p ef
fect
iven
ess
fact
or,
m a a m sm
76
FIGURE 2-6 r vHeap effectiveness factor (rj) vs. fractional conversion (X) for the continuity equationsolutions shown in Figure 2-3.

M W W U H m iH llH IIH lU iM I
Fractional conversion (X) vs. dimensionless flow time (0) given various surface fractions(X).

78
FIGURE 2-8 „ . . ,Heap effectiveness factor (v) vs. dimensionless flow time (i9) given various surfacefractions (X).

79
level control the entire process, so the leaching rate is strongly enhanced by higher
surface fractions. At the higher u, however, the characteristic contours of the particle
kinetics all but disappear as the rate of bulk flow through the heap becomes rate
controlling.
Similar results are obtained by varying the reaction order <£p, as shown in Figure
2-9. For these plots, a low value of for the particles is assumed since reaction order
is only an important factor in systems under chemical reaction control, as shown in
chapter one. Again, the effect is greater at low values of o, especially at the beginning
of the leach cycle. However, the suppression of particle kinetics at the higher value of
cj is less pronounced since a higher reaction order tends to widen the active reaction zone
in the heap, Afx, resulting in a net increase of heap effectiveness with reaction order, as
shown in Figure 2-10.
Computer Simulation: Competing Solid Reactants
In chapter one it was shown that the presence of one or more competing reactants
within porous ore particles can significantly alter the kinetics of leaching. In particular,
computer simulation of the particle model showed how the presence of a reactive gangue
material may push the otherwise highly effective reaction of a valuable component into
a much less effective regime, and how the overall process may approach a near-steady
state. Figure 2-11 shows the concentration gradient of reagent A and the fractional
conversion of one solid reactant as functions of heap depth, for the same hypothetical
particles as in Figure 1-11, in a heap at a = 1 and v = 1. In one simulation, reactant
1 is alone, and in the other it is accompanied by the reactive gangue material, exactly as

■MM—M— n
nI t
rHi
Fractional conversion (X) vs. dimensionless flow time (0) given various orders of reaction (<£p).

1
oowwCDCCD>(JCD
M—M—CD
Q_DCD
X
0.1
In
FIGURE 2-10 . ^ „ ,Heap effectiveness factor (77) vs. dimensionless flow time (0) given various orders ofreaction (cpp).

FIGURE 2-11 J . . ,Solutions to the continuity equations for reagent A (ab) and fractional conversion ofreactant 1 (X,), alone (n = l) and in the presence of a second reactant (n=2). *pi - U j8j = 1, * pl = 2/3, *p2 = 10, |S 2 = 0 .01, </>p2 = 0, u = 1, ? = 1, ^

83
in Figure 1-11.
Alone, reactant 1 dissolves effectively throughout the heap, and the reagent
gradients are insignificant, analogous to the particle alone. In the presence of solid
reactant 2, however, the reaction shifts more toward the top of the heap, and the reagent
gradients are steep, with ab < 0.12 at the bottom. In addition, the reagent gradients
within the heap approach a steady state, just as they do within the pores of the individual
pellets. Hence, the conversion kinetics of reactant 1 could be approximately described
in this case by a steady state solution for reagent A, using only the parameters for
reactant 2. As before, the diffusion equation for reagent A becomes the Bessel equation
(1-31) coupled with the mass-action rate expression for reactant 1
An analytical expression for the concentration of reagent A as a function of particle
radius and heap depth is now easily obtained (refer to appendix six):
KpiPiVO)--- 3
( 2 - 2 9 )
Additionally, the heap convection equation for reagent A becomes
= 0 ( 2 - 3 0 )
s i n he " “ YC 5 * 0
<x(5,0 =5 sin h (^ ic^ ) ( 2 - 3 1 )
e " “ YC 5=0s in h ( 0 ^ )
where

84
= V p2 ~ t a n h ( ^ ) tanh(^ ic^)
The concentration of solid reactant 1 is expressed
o ( $ , C , 6 ) =i -
-.V PlV(J t (i,0 6a 3
W 1
4>Pi =1
( 2 - 3 2 )
For the test situation, these equations become
octC/S^O) = s i n h (yT O $ )y/T? - tanh(-y/l?) ^
tanh (%/T5)$ s i n h ( v / T o )
s i n h ( 7 l O $ ) e ~ 2 . 1 7c1 1 . 8 $
( 2 - 3 3 )
a ( C , $ =0) = yTos i n h (y T o ')
v/10 ” tanh (\/10) tanh (v/lfr) = 0 . 2 6 8 e " 2 ‘17C
«b(C=1) = ey/I? - tanh(y/l5')
tanh(v/Tff) = 0 . 1 1 4
( 2 - 3 4 )
( 2 - 3 5 )
opl ( $ , C , 0) = (1 -a ($,C) 0 \3 ( 2 - 3 6 )
The conversion curves for reactant 1 both within and without the presence of
reactant 2 are shown in Figure 2-12. As expected, the conversion when n = 2 is
significantly delayed relative to when n = 1. The effect of the second reactant on the
heap effectiveness factor of the first reactant m is illustrated in Figure 2-13. When n =
2, the curve does not approach unity near complete conversion, identical to the
particle model alone. However, in a very ineffective (high co) heap, this would not
necessarily be the case, since the second reactant would have to be depleted at every
depth before the reaction zone could move downward into fresh ore. Under these

85
Fractional conversion of solid reactant 1 (X,) vs. dimensionless flow time (0) for the systems shown in Figure 2-11.

86
FIGURE 2-13Heap effectiveness factor (j]x) vs. systems shown in Figure 2-11.
fractional conversion of solid reactant 1 (Xj) for the

87
extreme circumstances, the time for total dissolution of reactant 1 (or any other minor
constituent) would be 6 ~ 1/(32p.
Computer Simulation: An Approach for Particle Size Distributions
In chapter one, the particular leaching model was solved for a distribution of
particle sizes based on the Gates-Gaudin-Schuhmann distribution function. In this
chapter, those results are applied to the global heap model, but the discussion is limited
to ore with no significant mineral deposits exposed on external particle surfaces.
From the definition of dimensionless particle radius in equation (1-38), and the
normalized weight fraction distribution in equation (1-39), assuming the simplest case of
no mineral liberation upon comminution, as in chapter one, and also ignoring surface
deposits, then the rate constant /cpi, the diffusion time r, and the global heap parameter
co will vary as functions of particle size thus:
Kpi - - V 22 2 2
where ~icpi, r and u apply to the reference particles. Substitution of these altered
parameters into the particular leaching equations allows one to solve the particle model
for a range of particle sizes S based on the parameters from the reference size, a — 1.
In order to gauge the effect that a distribution of particles sizes will have on the heap
leaching process as a whole, the convection equations must be altered to

88
dxib 3X±ba HcX
‘ - “ /I dZ (2-37)30 3C l 3W?=i S 2ainin
dab dab Max
= - G> f 1 W(S) ds (2-38)30 3( J 1i 3 U ?=1 S 2
Employing the GGS distribution, these take the form
d X i b - - oil r n Z m ~2 dE (2 -39)3 0 3C J '
d a b +- - « r i m Z m ~2 d Z (2 -40)
30 3C J\\fth-1
where m is a parameter of the GGS distribution, and the initial and boundary conditions
are the same as before. Likewise, the fractional conversion becomes
i i iX. = 3 f f f w - 0piH 2d$dEdC (2-41)
0 0 0
and the heap effectiveness factor, in the absence of surface deposits, is written
i i i
„ _ 0 0 0 111
(2-42)
fffmE^olfVdtdEdC0 0 0
Figure 2-14 shows plots of effluent dissolved species concentration vs. dimensionless
flow time calculated for both a particle size distribution (m = l, dashed curves), and the
reference size alone (m=oo, solid curves). As would be expected, the difference
between the two situations is larger at kp = 100 than at xp = 10, since particle size has
more bearing on the reaction rate at larger kp values. The effect is obscured at high co

89
F IG U R E 2 -1 4 . ,E ffluent concentration (Xb( M ) ) vs. dim ensionless flow tim e (6) g iven various values o fw, for a single particle size (m=°°) and a GGS distribution (m — 1).

90
values, since the effluent concentration is already at or near its upper limit. At low w
values, the X b(M ) curves are simply a little higher in the size distributed system than in
the isometric one. The most drastic difference is at moderate u values, where the entire
shape of the effluent response may be dramatically altered. These trends are also to be
observed in the fractional conversion plots of Figure 2-15. As shown in Figure 2-16, the
effect of the particle size distribution is to increase the overall heap effectiveness, to a
greater degree at higher xp values and lower w values.
V. Results and Discussion - Experimental
Table 2-1 summarizes all of the parameters for five column leaching tests, the
results of which are shown in Figures 2-17 through 2-21. All of these parameters were
either measured directly or calculated from known values, unless otherwise noted.
Values of DAe, kp, and 4>p were known from the batch kinetics tests discussed in chapter
one. The surface fraction X was taken as zero. The value of the GGS distribution
parameter m was based on a random sample of 100 pellets from column test 2, and
assumed valid for the other tests. In all of the tests, u is treated as an empirical
parameter, chosen on the basis of the best fit of the experimental data. This value is
denoted coapp, and the value predicted from theory for comparison is denoted wpr.
The ’expected’ values of CA0 and Cp0 in Table 2-1 are the products of what was
added to the lixiviant solution and the ore pellets, respectively, while the ’observed
values are the result of integrating the effluent responses of the individual column tests.
Hence, for the model fits, 100% extraction was based on the observed values of Cp0 in
order that the extraction integrals of the data and the simulations be equal. ’Actual’

91
FIGURE 2-15Fractional conversion (X) vs. Figure 2-14.
dimensionless flow time (0) for the simulations shown in

Hea
p ef
fect
iven
ess
fact
or

93
TABLE 2-1: Parameter values for the column leaching tests. (All units cgs. Concentrations based on gmols.)
b = 0.5 eh = 0.39DAe = 2.35 • 10-6 e0 = 0.20k„ = 3.76* 107 v ■= 0.246m = 6 Po = 2.15 = 1 (assumed) eb = 0.03
4>r = 2
Parameters Column test 1
Column test 2
Column test 3
Column test 4
Column test 5
CA0-106 (exp) 1.0 1.0 1.0 1.0 1.0
CAO-106 (obs) 0.91 1.0 1.0 1.0 1.0
Cp0* 10s (exp) 6.34 6.34 6.34 6.34 0.634
Cp0* 106 (obs) 5.13 5.60 5.26 6.05 0.623
n Rc 0.117 0.288 0.358 0.218 0.288
R 0.655 0.655 0.814 0.496 0.655
us • 103 0.89 2.2 2.2 2.2 2.2
Z 366 30 30 30 30
q** 0.0106 0.0106 0.0113 0.0098 0.0955
IS ^ 930 930 1430 530 9.3
app 1.1 0.10 0.071 0.13 0.10
^pr 4.12 0.137 0.089 0.238 0.137
*Based on expected values.**Based on observed values.

Xb(
C.Q
)
94
Column test 1
............... ( = 1 / 6ooooo 1 /3■ ■ ■ ■ ■ 1 / 2□ □□□□ 2/3▲ ▲ ▲ ▲ ▲ 5/6A A A A A 1------ Model fit:
/cp = 930, p = 0.0106 p p = 2, i/ = 0.246 w = 1.1, m = 6
1.0
0.8 -
0.6 -
0.4 J
0.2 -
FIGURE 2-17Effluent concentration and extraction data from column leaching test 1.
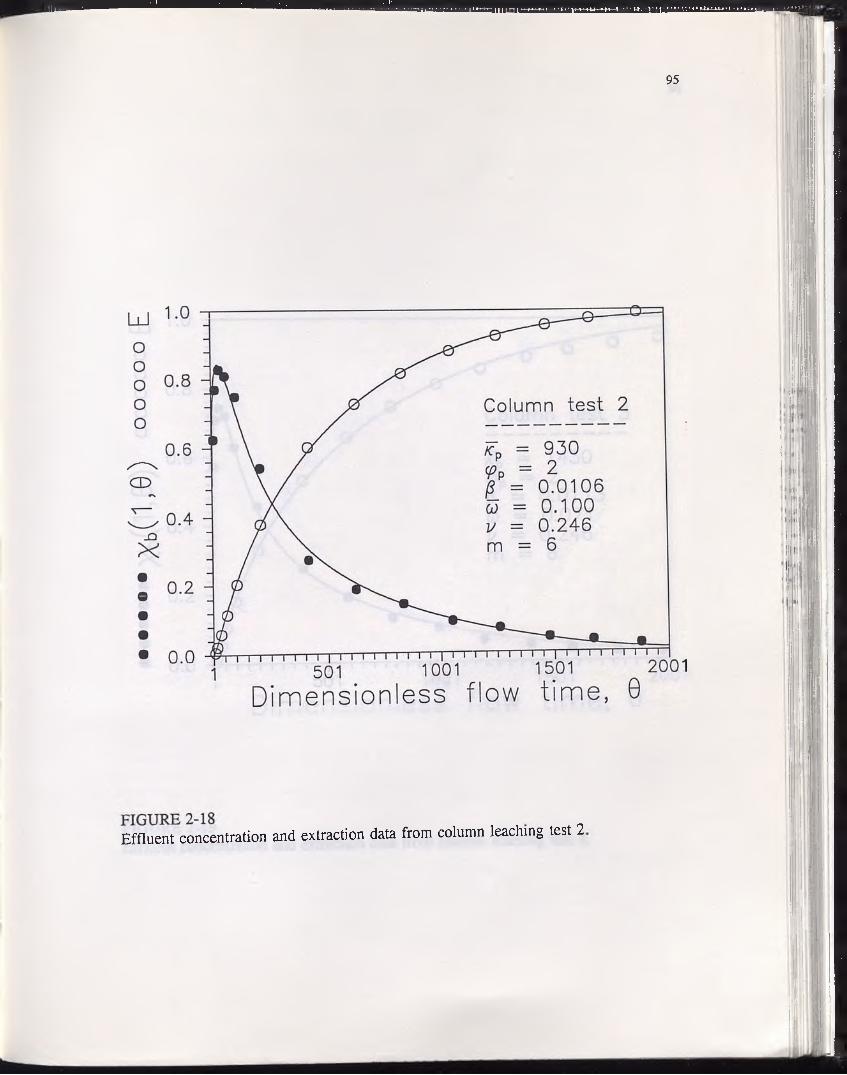
95
Effluent concentration and extraction data from column leaching test 2
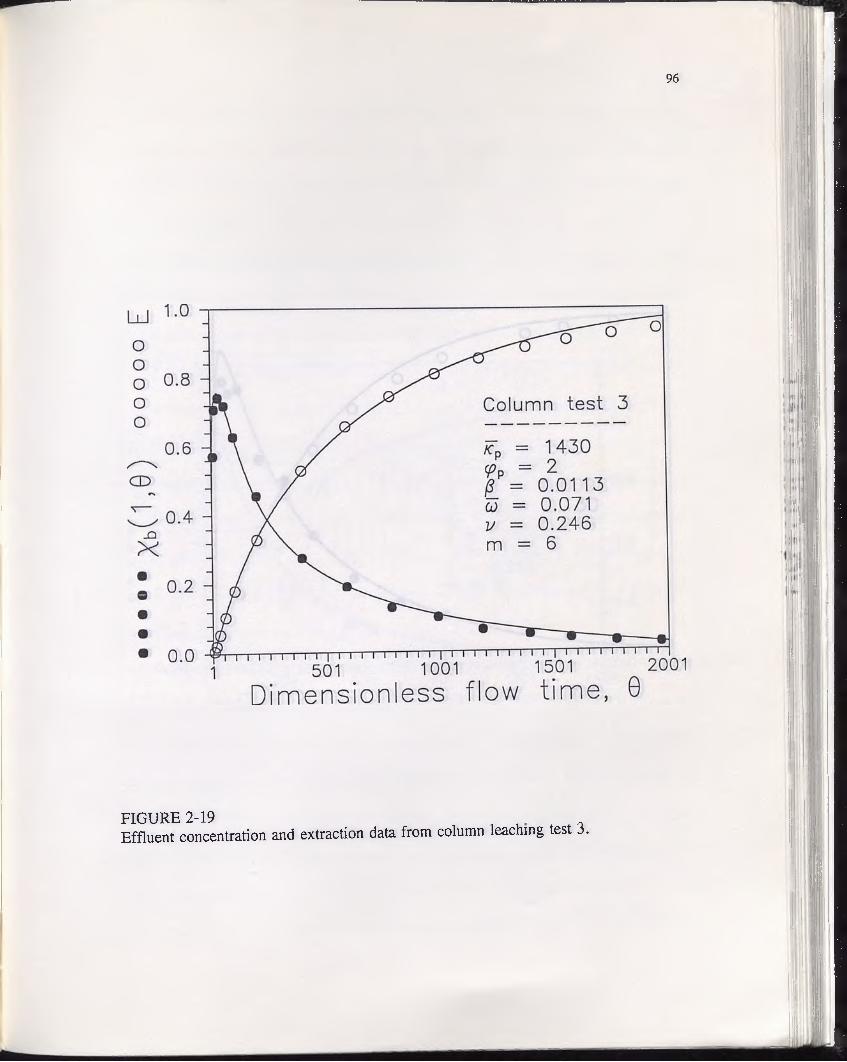
96
FIGURE 2-19 , _Effluent concentration and extraction data from column leaching test 3.

Xb
O
>0
)
97
LU
OOOoo
1.0
0.6
0.4 -
0.2
0 .0
G
__C
l
•V / o
10 \ / Co lum n te s t 4
» / /cp = 530 = 2
/3 = 0 .0098co = 0 .130 1/ = 0 .246 m = 6
•------m____ 1
501 1001 1501 2001
Dimensionless flow time, 0
FIGURE 2-20Effluent concentration and extraction data from column leaching test 4.

Xb
O.e
)
BIIUW IUHHUHHHM HHHHW M I
98
FIGURE 2-21Effluent concentration and extraction data from column leaching test 5.

99
extractions ranged from 80.9% in the tall column (test 1) to 98.3% in the low-grade
pellet test (test 5), and were apparent functions of pellet grade and particle size. The
parameter /3 was calculated based on the ’observed’ values, while kp was based on the
’expected’ values.
Effluent profiles from the tall column test (test 1) are shown in Figure 2-17. This
column simulates an ineffective heap (see Figure 2-22), since the effluent response is at
the maximum value of Xb(M) = 1 over most of the leach cycle. The apparent value of
u is 1.1, while the predicted value is 4.12. This large disparity is most likely due to a
combination of non-ideal flow patterns (i.e., rivulet flow, stagnant zones) within the
column, and to the blinding of pellet surfaces due to close packing. The former would
have the effect of increasing the local bulk fluid velocity in some regions, leaving others
accessible only by dispersion along pellet interstices. The latter would cause a decrease
in the effective particle size. Both would account for the observed deficiency in co,
resulting in apparently higher column effectiveness while obscuring the effects of kinetics
at the particle level.
Figures 2-18 through 2-20 show effluent response and extraction curves for three
short column tests (tests 2-4). Each involve the same grade of ore pellets as test 1, but
at three different particle sizes. As shown in Figure 2-22, these columns simulate fairly
effective heaps, based on the shape of the effluent response curves. Again, however, the
apparent values of a fall short of the predicted values, but not by as large a margin as
in the tall column test. Finally, the results of a short column test with low-grade pellets
(test 5) are shown in Figure 2-21. At the same u value as for test 2, the model fits the
data quite well, even though the kinetics within the pellets of test 5 are mostly reaction

100
FIGURE 2-22 . .Heap effectiveness factor (rj) vs. fractional conversion (X) for the model simulations ofthe experimental column leaching tests.

101
rate-controlled, while those of test 2 are diffusion-controlled. The simulated extraction
curve ends up significantly lower than the data, but this is probably as much the result
of experimental error as of any physical phenomenon.
Overall, the experimental results attest to the validity of the plug-flow modeling
approach. However, in order to predict the performance of actual heap leaching
operations from column tests, some sort of correlation between the theoretical plug-flow
model and the actual flow patterns within the heap is necessary. Roman37 modeled the
flow pattern within the heap as a collection of rivulets separated by stagnant zones across
which dispersion is the only transport mechanism. In his model, the distances from one
rivulet to the next are assumed to conform to some distribution function. This is
probably a fairly accurate picture of the flow within most heaps. Unfortunately, Roman
included no physical parameters in his model, rendering it impractical for prediction
purposes.
Assuming any correlation between convective and diffusive transport to be a
function of such factors as bulk flowrate, particle size distribution, reagent diffusivity,
surface tension, and so on, it may take the general form
o app = f ( u pI , NRe, NSclNWe, m, . . . ) ( 2 - 4 3 )
where NRe, NSc and NWe are Reynolds, Schmidt and Weber numbers, respectively, m is
a parameter of the particle size distribution, and so on. In the present study, only the
effect of Reynolds number can be accounted for, since the same ore type and reagent
species were used in all of the tests. Satterfield,38 in a review of the literature on the
performance of catalytic trickle-bed reactors, defines the ’contact effectiveness’ as the

102
ratio of the apparent to the predicted mass transfer coefficient, and suggests a
relationship, derived by Bondi,39 of the form
where the k’s are mass transfer coefficients, L is the mass flowrate, and A’ and b are
constants. Modifying this general form to accommodate the present parameters, and
defining the Reynolds number as
_ 2Rusp (2-4 5)
where p and p are the solution density and viscosity, respectively, the experimental
results may be approximated by
^JPP = 1 - 0 . 0 6 6 NrI1,15 ( 2 - 4 6 )03 p r
This result is shown graphically in Figure 2-23. Obviously, a value of one on the
ordinate represents a situation in which all of the assumptions which went into the
derivation of the global heap model are valid. Correspondingly, a heap with a serious
short-circuiting problem would approach an ordinate value of zero in Figure 2-23. Thus,
it makes sense for the curve to approach an ordinate value of one asymptotically with
increasing Reynolds number, since faster flowrates (uj coupled with smaller specific
particle area (3/R) would facilitate a more even distribution of lixiviant over particle
surfaces. While certainly, not enough data have been analyzed here to develop an
empirical correlation with a wide range of applicability, at least the method of approach
suggests an excellent avenue for future study.

103
FIGURE 2-23Contact effectiveness (coapp/wpr) vs. leaching tests.
Reynolds number (NRc) from the experimental column

104
VI. Conclusions
The proposed model is capable of simulating the heap leaching of one or more
solid reactants from porous ore particles. It is shown that the heap undergoes
homogeneous, high effectiveness kinetics for co « 1, zone-wise "shrinking-heap" kinetics
for oj » 1, and one or the other when oj ~ 1, depending on the value of *p.
Factors which affect kinetics at the particle level, such as deposits of solid
reactants on the external ore surfaces and the variable order of reaction, are shown to
have a significant effect on the rate of heap leaching only in heaps with low co values.
All kinetic resistances at the particle level are overshadowed by the bulk convection
resistance in high co heaps.
It is also shown that a second solid reactant competing for reagent can decrease
the reaction rate of the first reactant, and if a zero-order reaction may be assumed for
a major reactant (i.e., a gangue material) then an analytical, steady-state solution to the
model equations for any minor reactant is straightforward.
The model is also capable of simulating the heap leaching of particles with a
distribution of sizes. Particle size distribution has the greatest effect in heaps with low
to moderate co values, and only when the kinetics at the particle level are diffusion
controlled.
Results of experimental column leaching tests over a range of parameter values
confirm the appropriateness of the plug-flow approach to the global heap model, even
though the underlying assumptions might not be completely valid. The model provides
an excellent fit to the experimental data if to is treated as an empirical parameter. It is
shown that the apparent and predicted values of co may be correlated with Reynolds

105
number, indicating that the deviations from ideality are largely a matter of fluid
mechanics.

106
SummaryConclusions and Recommendations
I have endeavored in the first part of this work to give credence to what might
be taken as the primary maxim for anyone serious about mathematical modeling. To
paraphrase the Austrian engineer-tumed-philosopher Ludwig Wittgenstein,
"Our fundamental principle is that every question which can be decided at all by logic (mathematics) can be decided without further machinery.(And if we get into a situation where we need to answer such a problem by looking at the world, this shows that we are on a fundamentally wrong track.)"
(§5.551, Tractatus Logico-philosophicus)
Indeed, I have shown that most of the overriding dynamic factors of heap leaching can
be identified by manipulating a few basic equations of transport phenomena, without any
recourse to empirical considerations. With only mass balances, one can predict the
effects of particle size and ore grade on the rate of leaching at the particle scale, and the
corresponding effects of heap height, lixiviant flowrate, and the degree of particle size
distribution at the heap scale.
The most important aspect of such a theoretical investigation is defining the
limiting cases of a process, and how they manifest themselves in terms of certain
parameter values. It is well known that reaction systems such as heap leaching are really
several processes occurring in series. First, reagent must be transported with the bulk
solution to the particle surfaces, then it must diffuse into the particles, and only then can
it dissolve the solid reactants within the particle. Depending upon the specific resistances
of these individual steps, the rate of leaching may be controlled by any one, or to some
extent by all of them. The real value of parameters which result from the non-
dimensionalization of rate equations is that each represents an actual ratio of resistances

107
of any two interconnected processes. Thus, depending on the value of *p, particle
leaching kinetics will be either reaction controlled, diffusion controlled, or a combination
of both. By the same token, the value of w determines whether processes at the particle
scale or the convection of reagent through the heap voids will control the rate of heap
leaching.
The ultimate value of mathematical modeling is that fundamental insights about
the process are gained, even though some or all of the assumptions built into the model -
- not only to simplify the model equations, but also the interpretation of simulation
results — may not be completely valid. I have shown that the unsteady-state plug-flow
approach, coupled with an appropriate particle-scale leaching model, is adequate for
describing the kinetics of heap leaching. However, before the model can actually be
used to predict the outcome of real leaching situations, the relative validity of the model
assumptions must be determined.
In my estimation, any future research in this area should be geared toward
providing a concrete form for equation (2-43). I was able to show only crudely the
effect that Reynolds number might have on the contact efficiency between the bulk
lixiviant and the particle surfaces. However, a thorough study of the dependence of the
rate parameters on factors such as lixiviant flowrate, particle size distribution, and
especially surface tension is called for. Many processes of importance to the mining
industry, including heap pretreatment of refractory ores, heap leaching of carbonaceous
gold ores, decommissioning of spent heaps, and even the remediation of contaminated
soil, would benefit from a better knowledge of the interaction between solids and
solutions in unsaturated flow.

Appendix OneA Theoretical Basis for the Variable Order Assumption in the Kinetics of Leaching of Discreet Reactant Grains
While the assumption of spherical micrograins of solid reactant in the previous
development of the particular leaching model results in a reaction order exponent value
of cj) = 2/3, one important question is left unanswered by this reasonable and ostensibly
harmless supposition: Given that those micrograins will most likely possess a range of
different sizes, how will that affect the 2/3-order assumption?
This question was effectively avoided by Bartlett,9 who assumed a log-normal
weight distribution of spherical grains which would give a pseudo-first-order response,
and incorporated the result into his leaching model. Bartlett’s method, while
circumventing the problem, is rather limited in its range of application. Thus, it would
seem desirable to estimate the effect of grain size distribution on the reaction order a
priori in order to judge whether the issue warrants further consideration.
Beginning with the previously developed expression for the rate of dissolution of
grains all within the same environment of reagent concentration
do_ _ _ 0<i>a (A. 1-1)dr
where a represents the collective mass fraction of grains remaining at time t, and
X = kCtfo C-Aot
where k = a rate constant and CM0 = the initial apparent concentration of solid reactant,
the "shrinking-particle model" for each individual grain may be expressed
108

109
d y _ _ y2/3g d x ' ^
(A. 1-2)
where y is mass fraction remaining of the individual grain, \]/ is the initial normalized
radius of the grain relative to some reference grain radius, and
T/ = 2Mk'CA0 tR gP
where M = the molecular weight of the reactant, k’ = an intrinsic rate constant, Rg =
some reference grain radius, and p = the reactant specific gravity. Equations (A. 1-1)
and (A. 1-2) have initial conditions
0 (x=0) = 1 y (ijr, x=0) = 1
If a grain weight distribution function f(^) is defined such that
(A. 1-3)
(A. 1-4)
jf f (i|0 cft|r = 1 (A. 1-5)
then the collective mass fraction of solid reactant remaining may be expressed in terms
of the individual grains with a volume average over each individual grain size fraction
thus
o = | f (\|r) Y ('I'/*7) d^rnin
(A.1-6)
D ifferen tia tin g th is equation w ith resp ect to tim e b y L e ib n itz ru le g iv es

110
max
min(A. 1-7)
Defining K = t ’/t , equation (A. 1-2) becomes
dy = _ Ky2/3a (A. 1-8)d-z i|f
and combining equations (A. 1-1), (A. 1-7) and (A. 1-8) gives
(A. 1-9)
At time r = 0, applying the initial conditions (A. 1-3) and (A. 1-4) to equation (A. 1-9)
one obtains
(A. 1-10)
from which the constant K is recognized as the inverse of the (-l)th l/'-moment
distribution
K =
/f ( t )
(A.1-11)
Substituting equation (A. 1-11) into equation (A. 1-9) gives

Ill
dodr
raaxa j* ^ y2'3 (ty> t7) cfy
\|r ■“min ________max
U lL L d *min
o^a (A.1-12)
Finally, from equations (A. 1-6) and (A. 1-12) the effective reaction order as a function
of reaction time is obtained:
I n
I n ( a * )
^max
/ - ^ P - y 2/2 ( t ^ 7)^min
- I n
^max
^min
I n ( a )I n
max
J f ( i | 0 Y ( ^ / t 7) c ty^min
(A. 1-13)
For purposes of discussion, it is assumed that the reagent concentration is constant
at a = 1. This being the case, the exact solution of equation (A. 1-2) is straightforward,
giving
Y (ijf/t') 1 -
where u(\j/,T') — a unit step function such that
u (i|f, r 7)f 1 , ?7 <; 3 iJj)1 0, t' > 3\lrj
(A.1-14)
Hence, equation (A. 1-13) becomes
I n
^max / / N2
di|i - I n
^max/ m i d ,
^min<l>
I n
^max| f (v |0 u (ilr , x 7)
^min3 -
(A.1-15)

112
While the choice of a distribution function in the absence of experimental data is
an arbitrary one, the log-normal distribution40 allows easy comparison with the results
of Bartlett. In dimensionless form, this function is
f ( * , . ex p f-2 s 2
(A.1-16)v/2"rcs\|f
where s = the standard deviation of the distribution. Integrating f(i/0 between the limits
(0 < ty < o°) results in a definite integral which satisfies the criterion of equation (A. 1-5):
maxf f ( t y ) d t y
min? -1— e x p f- ( ln f - - W{ y/2nsty l 2 s 2 )
i — f e x p f - ( l n ^ 2) d ( l n t|t) ■ n s V 2 s )
(A.1-17)
y/2ns
= 1
In addition, the log-normal (-i)th ^-moment distribution is readily solved:
4*max
/minf 1 1 1 1 a y = f ----------1 — - e x p
J i|r J0 y f2 n s ty 2
^ ^ ) d t y(A.1-18)
e x p
Hence, the final expression for the reaction order as a function of reaction time is
I n
<t> =I 1
y in s ty 2
In / e x p - | u ( * , t') 1 - - U d*2 S' 3t|r,
(A. 1-19)
This equation involves only one parameter for its unique solution; namely, the standard

113
deviation, s, of the log-normal distribution.
Solutions to equation (A. 1-16) are plotted in Figure A. 1-1 for six values of the
standard deviation. These values were chosen, after Bartlett, according to
resulting in the distribution properties summarized in Table A. 1-1.
Semi-log plots of a vs. r according to equation (A. 1-6) for the six values of s are
shown in Figure A. 1-2. These curves show qualitatively how wider distributions of grain
size result in higher apparent orders of reaction. This observation is confirmed
quantitatively in Figure A. 1-3, where plots of 4> as calculated by equation (A. 1-19) are
plotted as functions of a. It should be noted that these curves are completely independent
of actual time of reaction. The dashed lines represent least-squares zero-order
polynomial fits of the 0-c curves to give "average" reaction orders which, as shown in
Figure A. 1-4, bear a near-perfect quadratic relationship to the standard deviation.
Returning to Figure A. 1-3, obviously, the range of 4> over the course of leaching
increases dramatically with increasing s. However, as shown in Figure A. 1-5, when the
values of <£ave are used in the solution of equation (A. 1-1), the results (dashed curves) fall
surprisingly close to the actual <r-r curves as calculated by equation (A. 1-6) (solid
curves), even at high s values. Thus, one may conclude that the assumption of a single
value for the overall order of reaction in the concentration of a solid reactant in equation
(A. 1-1) is acceptable over a fairly wide range of grain size distributions. Furthermore,
4> may possess a wide range of values greater than or equal to 2/3, but not less than 2/3,
when the individual reacting grains are roughly spherical.
(A.1-20)

114
TABLE A.l-I: Properties of the log-normal distributions represented in Figure A. 1-1.
Curve s '/'oW'/'o.oi '/'o.oi ’/'0.99
1 0 10° 1 1
2 0.2475 10l/2 0.562 1.78
3 0.495 101 0.316 3.16
4 0.99 102 0.1 10
5 1.485 103 0.0316 31.6
6 1.98 104 0.01 100—

115
FIGURE A. 1-1The log-normal distribution function.

116
F IG U R E A. 1-2C o llective m ass fraction remaining (cr) v s.
distributions.
reaction tim e (r) for various grain w eight


118
FIGURE A. 1-4Average apparent reaction order (<£ave) vs. weight distribution (s).
the standard deviation of the log-normal grain

119
Collective mass fraction remaining (a) vs. reaction time (r) as calculated by equations (A. 1-6) (solid curves) and (A. 1-1) (dashed curves).

120
Appendix TwoNumerical methods
The variable-order dissolution rate expressions are ordinary differential equations
which are solved by the classical four-point Runge-Kutta technique:41
dx
K =
k 2 = f ( x j + \ h , uj + \ h k j
k 2 = f ( x j + j h lUj + \ h k 2)
Jc4 = f (xj + h, Uj + hkz)
Uj+i = uj + (-^i + 2k2 + 2kz + k4.)
The particle model equations are solved numerically by fully implicit finite
difference approximations of the form42
a2; du 1 - { ( i+ l) ui+lfJtldt i ( 6 £ ) :2 J
= 6( 8 0 2
( Ul , j + 1 “ U0,J+1
du _ dx , j + 1 ~ U i . j ^
K U = KUi , j +1
where iis the particle radius index and j is the time index. Since all of the variables in
they* time level are known, and those of the (/+l)th time level are unknown, except
at the particle surface, a system of N equations for N unknowns is generated, each with
variable in each of the (i-l)th, ith, and (i+l)th radius levels. So, for the diffusionone
equation in spherical coordinates

121
3 ^ * | | H ♦ KU =d \2 l 3$ 3x
th e N a lg eb ra ic equation s form a tri-d iagon al c o e ffic ien t m atrix equation
1 *€r N. ii o 6 \ i uo,j+i ~ u 0 , j
0 ~ V i * 0 2p ~ui .3
2 r ~ V i * 0 2 r U2, j+1—
~U2.j
n-1 ^ ~ V i * 0
E Z l nn
- 2 - nn -1 r
- V i t o Un, j+1 r u n , j - ( ^ Kw h ic h is e a s ily so lv ed b y G aussian e lim in a tio n , 4 3 and w h ere
\|ri=0 = 6p - k ( 6t ) + 1
= 2 \i - k ( 8t ) + 1
and u b is th e su rface boundary con d ition for the (/ + l ) th tim e in d ex .
T h e g lo b a l heap m o d el P D E ’s are transform ed in to ordinary d ifferen tia l equation s
th rou gh th e d efin itio n o f the substantial tim e, or m aterial d er iv a tiv e , 4 4 ex p ressed in
d im e n s io n le ss coord in ates
JD_ _ _d_ + _3_D0 00 0C
T h is tran sform ation facilita tes the m athem atical treatm ent o f the g lo b a l heap m o d e l from
th e L a p la c ia n v iew p o in t, i . e . , v ia the tracking o f flu id e lem en ts (m aterial v o lu m es)
through th e h eap . S im p le backw ard fin ite d ifferen ce approxim ations are em p lo y ed to
s o lv e th e c o n v e c tio n eq u ation s e x p lic it ly .

I m m m
122
DuD0 6C
(uk.j ~ U;
KU = KUJc-l.j
where £ is the heap depth index and 7 is the time index.
All integrals are evaluated by compound quadrature formulae.45 Since the number
of space increments can always be chosen as an even number, space and phase space
variables, including fractional conversion, effectiveness factor, and particle size
distribution are integrated by Simpson’s rule:
f f ( x ) d x * s M = - | [ f ( x 0) + 4 £ { x j ) + 2 f [ x 2) + 4 J f ( x 3) + • * *
+ 2 f ( x M_2) + 4 f ( x w ) + f U M) ]
where
h = —----- - f x , = a + j h , (0 <. j <■ M)M
and M is any even integer. Simpson’s rule is an order of magnitude more accurate than
the Trapezoid rule:
j*f ( x ) d x ~ t N = - | [ f ( x 0) + 2 f [ x 1) + • • • + 2 f ( x N_1) + f ( Xj j ) ]a
which is reserved for integrating extraction, the only time variable, and where
h = b ~ — , x , = a + j h , (0 <, j <■ N)N J
and N is any integer.
The order of solution of the model equations is as follows. First, all initial
conditions are defined. Then, starting at time 9 = 0 and heap depth f = 0, the particle

123
surface solid reactant concentrations crsi are determined. Next, the radial concentration
profile of reagent A, a(£), is solved given a value of the time step 59, and these values
are used to solve for the concentration profile of the dissolved complexes, Xi(£)- Then,
the intraparticular solid reactant concentrations crpi are updated. After the particular
model equations have been solved, the particle surface concentration gradients are
calculated, and these and the surface reactant concentrations are used to solve the global
heap model equations, given a depth increment 5f, for the bulk solution concentrations
of reagent A, ab, and the dissolved complexes, Xib> at f + 5f.
The above procedure is repeated until f = 1, then the model functionals are
calculated, and the entire process is reiterated at the new time, 9 + 59.

124
Appendix ThreeExperimental Data
Experimental data from the batch leaching tests are presented in Table A.3-1.
Data from column leaching test 1 are presented in Table A.3-II, and from tests 2 through
5 in Table A.3-III. Parameters and operating variables for all experimental leaching tests
are given in chapters one and two.

125
TABLE A.3-1: Data from batch leaching tests.
Batch test 1 Batch test 2 Batch test 3 Batch test 4
Time(min)
^Ag |(ppm) |
Time(min)
^Ag(ppm)
Time(min)
^Ag |(ppm) 1
Time(min)
C-Ag(ppm)
30 0.05 15 0.20 5 0.01 30 0.87
60 0.07 30 0.48 10 0.13 60 1.79
90 0.11 60 1.04 20 0.36 90 2.72
180 0.33 130 2.16 80 2.22 120 3.42
270 0.48 220 3.35 140 3.78 240 5.23
360 0.58 360 4.68 200 4.72 410 7.03
610 0.90 600 6.16 260 5.94 770 9.38
1140 1.34 1290 9.29 360 6.98 1380 10.9
1620 1.59 1770 10.9 540 8.30 1830 12.4
2595 2.02 2910 12.8 640 9.08 2880 14.6
4080 2.22 4530 16.5 1320 11.3 4200 17.1
5460 2.39 5820 18.3 1680 12.5 5640 18.8
6900 2.52 - - 2160 14.3 - -
_ _ - 2760 15.1 - -
_ - - 3250 16.8 - -
_ - - 4200 18.2 - -
_ - - 5820 19.4 - -
. _ - 7170 20.5 - -
_ - - 8910 22.7 - -
- - - - 9960 22.6 - -

■ W M W M W I W — — — — — — — — — — — — —
126
TABLE A.3-II: Data from column leaching test 1.
Time Effluent silver concentration, CAg(Z,t) (ppm)(hr)
Z = T Z = 4’ Z = 6’ Z = 8’ Z = 10’ Z = 12’ |
6 20.7 13.4 4.9 2.3 1.7 0.4
24 33.1 34.9 29.9 24.6 18.2 12.4
48 33.5 43.0 43.3 38.7 36.0 32.1
73 38.7 47.8 44.3 43.3 42.0 39.2
96 34.6 46.8 45.4 46.2 46.0 46.5
122 28.2 45.3 49.7 47.7 46.3 47.4
145 20.5 44.7 47.5 47.6 45.7 45.6
168 22.5 46.7 47.3 47.7 47.4 47.5
191 20.7 47.1 49.2 48.1 47.9 46.7
219 17.3 45.6 48.0 49.3 49.4 49.3
241 18.4 47.7 47.8 50.7 49.9 48.9
266 13.8 43.5 48.6 48.7 49.1 49.3
291 11.7 40.1 45.9 48.3 47.5 48.3
313 10.8 35.6 45.4 46.8 46.1 47.0
340 11.2 34.9 48.5 48.9 48.6 47.8
364 12.8 29.7 46.7 48.5 47.3 48.1
384 6.2 27.2 46.9 47.7 48.0 48.3
410 4.8 21.8 46.4 48.1 47.5 48.3
434 6.1 20.5 46.5 47.6 48.3 48.5
457 4.4 15.4 44.6 48.0 48.6 48.5
481 2.6 12.5 46.4 46.6 48.2 48.1
509 3.9 11.0 41.1 47.3 47.0 47.0
533 2.6 9.1 37.1 46.9 47.5 49.3
552 4.0 7.8 36.1 46.9 47.5 48.1
580 7.1 9.0 34.2 47.4 48.6 48.5

127
Time 2’ 4’ 6’ 8’ 10’ 12’
602 1.8 5.3 30.0 45.2 48.1 48.6 1
633 5.1 6.4 24.9 43.1 47.4 47.3
652 1.8 3.8 22.0 40.9 47.7 47.2
674 1.1 3.2 22.2 42.1 46.5 47.2 1
697 1.2 2.9 20.7 37.6 50.8 50.7
723 2.8 4.0 16.5 33.8 48.4 49.8
746 1.9 3.4 16.4 32.2 49.9 50.0
769 1.1 2.3 12.6 28.6 49.0 49.2
798 1.5 2.6 9.8 25.2 49.0 49.1
821 1.8 3.4 9.1 23.7 49.7 49.3 1
842 1.7 2.9 9.6 22.4 48.8 49.0
870 1.8 2.5 8.3 21.7 50.5 51.3
889 1.3 2.3 7.4 20.0 47.4 51.2
914 1.4 2.6 6.8 17.9 45.6 50.3
938 1.2 2.3 6.1 16.5 42.1 49.8
963 0.4 1.4 4.8 17.0 39.1 48.8
984 0.6 1.5 4.1 19.6 41.5 48.6
1013 0.9 2.1 4.5 19.3 36.0 49.7
1032 0.9 1.6 4.1 17.2 34.4 48.9
1062 0.6 1.7 3.6 15.2 31.3 47.7
1108 0.2 1.2 2.6 12.8 26.6 46.0
1159 1.2 2.0 3.3 13.0 25.5 48.0
1198 0.7 1.6 2.8 11.9 21.8 45.9
1246 0.7 1.2 2.1 9.3 18.9 43.5
1300 0.6 1.3 2.0 8.3 17.0 35.3
1350 0.4 1.3 2.0 7.4 16.3 30.6
1398 0.3 0.7 1.6 6.3 11.3 24.6
i

128
TABLE A.3-III: Data from column leaching tests 2 through 5.
Column test 2 Column test 3 | Column test 4 Column test 5
Time C-Ag Time ^Ag j Time C-Ag Time ^Ag(hr) (ppm) (hr) (ppm) | (hr) (ppm) (hr) (ppm)
1 33.6 1 30.8 1 31.0 1 3.94
2 41.5 2 38.2 2 40.3 2 6.65
4 44.6 4 40.0 4 42.5 4 8.38
7 43.6 7 38.7 7 40.7 7 8.14
12 40.3 12 33.8 12 41.3 12 7.37
24 29.1 24 24.6 23 30.2 23 4.19
48 14.7 48 14.8 36 27.0 36 3.10
72 10.0 72 10.3 51 18.4 51 1.20
96 7.63 96 6.96 72 11.6 72 0.64
121 4.92 121 5.40 96 7.85 96 0.27
145 3.81 145 3.42 120 5.64 120 0.16
170 2.34 170 2.65 164 2.71 164 0.08
192 1.82 192 2.04 184 1.96 184 0.04
216 1.23 216 1.52 210 1.38 210 0.02
240 1.15 240 1.23 236 1.01 236 0.02
265 0.79 265 0.93 262 0.85 262 0.02
290 0.57 290 0.72 283 0.66 283 0.00
313 0.50 313 0.64 310 0.48 - -
342 0.35 342 0.43 335 0.31 - -
360 0.35 360 0.35 - - - -

Appendix FourNomenclature
Roman Letters
b; stoichiometry number, mol i/mol A
CA concentration of reagent A, mol A/cmf3
CAb concentration of reagent A external to particle, mol A/cmf3
CEi0 initial extractable grade of solid reactant i, mol i/g ore
q concentration of dissolved species i, mol i/cmf3
q b bulk concentration of dissolved species i, mol i/cmf3
Cpi grade of solid reactant i within particle, mol i/g ore
Cpi0 initial grade of solid reactant i within particle, mol i/g ore
Csi grade of solid reactant i on particle surface, mol i/g ore
q i0 initial grade of solid reactant i on particle surface, mol if,g ore
DAc effective pore diffusivity of reagent A, cmf3/cmp s
Die effective pore diffusivity of dissolved species i, cmf3/cmp s
E; extraction of dissolved species i, dimensionless
kpj rate constant of solid reactant i within particle, mol i/g ore s [Cpf [CA]
k*; rate constant of solid reactant i on particle surface, mol i/cny s [CJ* [CA]
1; liberation function of solid reactant i, dimensionless
m Gates-Gaudin-Schuhmann size distribution parameter, dimensionless
n number of solid reactants
NRe pellet flow Reynolds number, dimensionless
r radius, cm

130
R p artic le radius, cm
t tim e , s
u s su p erfic ia l b u lk f lo w v e lo c ity , cm f3 /cm h 2 s
w G ates-G audin-Schuhm ann size d istribution fu n ction , d im en sio n less
X; fraction a l co n v ersio n o f so lid reactant i , d im en sio n less
z dep th , cm
Z heap dep th , cm
G reek L etters
a d im en sio n less con cen tration o f reagent A
a b d im en sio n less con cen tration o f reagent A external to p article
/3 ; reagen t strength param eter relative to so lid reactant i , d im en sio n less
7 m od u lu s in stead y-sta te m odel
5 . ratio o f d iffu s iv ity o f d isso lv ed sp ec ies i to reagen t A , d im en sio n less
eb b u lk so lu tio n v o lu m e fraction
eh heap v o id fraction
eQ o re p o ro sity , cm f3 /c m p 3
f d im e n sio n less depth
,qi e ffe c tiv e n e ss factor for so lid reactant i , d im en sio n less
9 d im e n sio n le ss f lo w tim e
K D a m k o h ler II num ber fo r so lid reactant i w ith in p artic le , d im en sio n less
Ksi D am k d h ler II num ber for so lid reactant i on p artic le su rface , d im en sio n less
X; su rface fraction o f so lid reactant i, d im en sio n less

ratio o f v o lu m e o f b u lk flu id to flu id in particle p ores, d im en sio n less
d im e n sio n le ss radius
d im e n sio n le ss p artic le radius
o re d en s ity , g o re /cm 3 ore
d im e n sio n le ss grade o f so lid reactant i w ith in particle
d im e n sio n le ss grad e o f so lid reactant i on particle surface
d im e n sio n le ss d iffu s io n tim e
reaction order fo r so lid reactant i w ith in p article, d im en sio n less
rea ctio n order fo r so lid reactant i on particle surface, d im en sio n less
d im e n sio n less con cen tration o f d isso lved sp ecies i
d im e n sio n le ss b u lk concentration o f d isso lved sp ec ies i
id ea l co lu m n param eter (in verse P ec le t num ber), d im en sio n less

O U
A ppendix FiveFORTRAN Computer Program Source Code
$DEBUGPROGRAM BATCHDIM
David G. Dixon
C This program calculates the batch heap leaching model C in dimensionless form.C ----------------------------------------------------------------------------------------------------
D IM E N S IO N A (1 0 1 ) ,B (4 ) ,C (4 ,1 0 1 ) ,C B (4 ) ,D (4 ) ,D C D R (4 ) ,E (4 ) D IM E N S IO N F A (4 ) ,F A C S (4 ) ,F P (4 ) ,F S (4 ) ,H (4 ) ,H K (4 ) ,H D (1 0 1 ) ,H N (1 0 1 ) D IM E N S IO N O K P (4 ),O K S ( 4 ) ,O L A (4 ) ,S P (4 ,1 0 1 ) ,S S (4) ,X (4 ) ,X F (1 0 1 )
C O M M O N /V A R /A ,C ,D T B ,IR ,N M ,O L A ,S P C O M M O N /P A R /B ,D ,F P ,O K P ,O N ,Q ,U ,V ,W ,Y
E X T E R N A L SR K E X T E R N A L G R A D
W R IT E C *,*)’ C yan id e con tro l (not 1) or constan t reagen t (1 )? ’ R E A D (* ,* )N C O N
W R IT E (* ,* )’ Input output freq u en cy in d ex for p r o f ile s : ’
R E A D (* ,* )N O F IW R IT E (* ,* )’ Input output freq u en cy in d ex for b a tch :’
R E A D (* ,* )N B F I
W R IT E (* ,* )’ Input datafile: ’R E A D (1 0 ,* )N M ,I R R E A D (1 0 ,* )V IF (N M .N E .O ) T H E N
R E A D (1 0 ,’i:) (B (M ) ,M = : l ,N M )R E A D (1 0 ,* ) (D (M ) ,M = 1 ,N M )R E A D (1 0 ,* )(O K P (M ),M = 1 ,N M )R E A D (1 0 ,* ) (F P (M ) ,M = 1 ,N M )R E A D ( 1 0 ,* ) (O L A (M ) ,M = 1 , N M )R E A D (1 0 ,* ) (O K S (M ) ,M = 1 ,N M )R E A D (1 0 ,* ) (F S (M ) ,M = 1 ,N M )
E N D IF W = 3 ./V W R IT E (* ,* )

CALL INITCON(0.)
DO 1 M=1,NMIF(OLA(M).EQ.O. .OR. OKS(M).EQ.O.) THEN
H(M) = 0.SS(M) = 0.
ELSEH(M) = l ./( l . + OKP(M)/OKS(M))SS(M) = 1.
ENDIFMW = 100 + MW RITE(V)’ T-R-A-C-S Output file, metal \M
60 FORMAT(A,Il)DO 2 I=1,IR+1
RIND = FLOAT (I-1 )/FLO AT (IR)WRITE(MW,51)0.,RIND,0. ,0., 1.
51 FORMAT(E11.4,F6.3,3E11.4)2 CONTINUE
WRITE(MW,51)0., 1. ,0. ,0. ,0.WRITE(MW, 51)0., 1.05,0. ,0.,0.WRITE(MW,51)0., 1.05,0. ,0. ,SS(M)WRITE(MW,51)0., 1.15,0. ,0. ,SS(M)WRITE(MW,51)0. ,1.15,0. ,0. ,0.WRITE(MW,51)0. ,0. ,0. ,0. ,0.WRITE(*,*)WRITE(*,60)’ T-C-X-E-H-A Output file, metal ’,M IF(H(M).EQ.0.) H(M) = l.E-6 WRITE(M,52)0. ,0. ,0. ,0. ,H(M), 1.WRITE(*,*)
52 F ORM AT (6E11.4)1 CONTINUE
T = 0.AB = 1.JOFI = 1 JBFI = 1
100 WRITE(*,*)’ Enter DT (Taus): ’READ(*,*)DT DTB = DTWRITE(*,*)’ Enter number of time increments:’READ(*,*)NTNTOT = NTOT + NT
DO 3 J=1,NT

JINC = NTOT - NT + J IF(JINC/NBFI.EQ.JBFI) THEN
WRITE(*,*)WRITE(*,61)’ On increment ’,JINC
61 FORMAT(A,I5)ENDIF T = T + DT
CALL PARTICLE(AB, CB)
DADR = GRAD(A(IR-1),A(IR),A(IR+ 1),IR)FAC1 = 0.DO 4 M=1,NM
DCDR(M) = GRAD(C(M,IR-1),C(M,IR),C(M,IR+1),IR) IF(SS(M).GT.O.) THEN
FAC = OKS (M) *SS (M) * *FS (M) * AB/V ELSE
FAC = 0.ENDIFFAC1 = FAC1 - FACCB(M) = CB(M) + DT*(FAC - 3.*D(M)*DCDR(M)/V) IF(OLA(M). NE. 0.) THEN
SFAC = -OKS (M) *B (M) * AB/ OLA (M)SS(M) = SRK(SS(M),SFAC,DT,FS(M))
ENDIF4 CONTINUE
IF(NCON.EQ.l) THEN AB = 1.
ELSEAB = AB + DT*(FAC1 - 3.*DADR/V)
ENDIF
DO 5 M=1,NM MW = 100 + M DO 6 I=1,IR+1
RIND = FLOAT (I-1 )/FLO AT (IR) IF(JINC/NOFI.EQ.JOFI) THEN
WRITE(M W, 51) T, RIND ,A(I),C(M,I),SP (M, I)ENDIFXF(I) = 1. - SP(M,I)IF(SP(M,I).GT.O.) THEN
HN(I) = OKP(M)*SP(M,I)**FP(M)*A(I)HD (I) = OKP(M)*SP(M,I)**FP(M)*A(IR+1)
ELSE

135
HN(I) = 0.HD (I) = 0.
ENDIF6 CONTINUE
IF(JINC/NOFI.EQ.JOFI) THEN WRITE(MW,51)T, 1. ,0. ,0. ,0.WRITE(MW,51)T, 1.05,0. ,0. ,0.WRITE(MW,51)T, 1.05,0. ,0. ,SS(M)WRITE(MW,51)T, 1.15,0. ,0. ,SS(M)WRITE(MW,51)T, 1.15,0. ,0. ,0.WRITE(MW,51)T,0. ,0. ,0. ,0.IF(M.EQ.NM) JOFI = JOFI + 1
ENDIFCALL SIMPSON(IR,XF, 1 ,XW)CALL SIMPSON(IR,HN, 1 ,HNW)CALL SIMPSON(IR,HD, 1 ,HDW)X(M) = (1. - OLA(M))*XW + OLA(M)*(l. - SS(M)) IF(SS(M).GT.O.) THEN
HNW = HNW + OKS (M) *S S (M) * *FS (M) * A (IR +1) HDW = HDW + OKS(M)*SS(M)**FS(M)*A(IR+1)
ENDIFIF(HDW.GT.O.) THEN
H(M) = HNW/HDW ELSE
H(M) = 1.ENDIFE(M) = B(M)*V*CB(M)IF(JINC/NBFI.EQ.JBFI) THEN
WRITE(M, 52)T, CB(M), X(M) ,E(M) ,H(M), AB IF(M.EQ.NM) JBFI = JBFI + 1
ENDIF5 CONTINUE
3 CONTINUEWRITE(*,*)WRITE(*,62)’ Time (tau) = ’,T
62 FORMAT(A,F11.4)WRITE(*,*)
DO 7 M=1,NMWR1TE(*,53)’ Extraction of metal ’,M,’ = ’,E(M) WRITE(*,53)’ Conversion of metal ’,M,’ = ’,X(M) WRITE(*,53)’ Effectiveness of metal \M ,’ = ’,H(M)
53 F0RMAT(A,I2,A,F6.3)WRITE(*,*)

136
7 CONTINUE
WRITE(*,*)’ To continue, enter "1" ’ READ (*, *)NREP IF(NREP.EQ.l) GOTO 100
END
$INCLUDE: TNITCON.FOR’$INCLUDE: ’PARTICLE. FOR’$INCLUDE: ’ ASOLVE.FOR’SINCLUDE: ’CSOLVE.FOR’$INCLUDE: ’TRI.FOR’$INCLUDE: ’SPSOLVE.FOR’$INCLUDE: ’SRK.FOR’ $INCLUDE:’SIMPSON.FOR’$INCLUDE: ’GRAD.FOR’

0 U
$DEBUGPROGRAM HEAPGGS
David G. Dixon
C This program calculates the heap leaching model C in dimensionless form for a distribution of particle sizes. c --------------------------------------------------------------------
DIMENSION A(17,65,9),B(2),C(2,17,65,9),CB(2),CBOLD(2),D(2) DIMENSION E(2) ,FACS(2) ,FP(2) ,FS(2) ,H(2) ,HD(2,65) DIMENSION HN(2,65) ,HDF(17) ,HDL(65) ,HNF(17) ,HNL(65) DIMENSION HTD (2) ,HTN(2), OKP(2), OKS (2), OLA (2) DIMENSION SP(2,17,65,9),SS(2,65),WC(2),X(2,65),XF(17) DIMENSION XI(9),XL(65),XT(2)
COMMON /VAR/A,C,DTB,IR,NK,NL,NM,SP,SS COMMON /PAR/B,D,FP,FS,OKP,OKS,OLA,V,W,XI
EXTERNAL GRAD EXTERNAL SRK
WRITE(*,*)’ Enter profile output frequency index:’ READ(*,*)NOFIWRITE(*,*)’ Enter effluent output frequency index:’READ(*,*)NEFIJOFI = 0JEFI = 0
WRITE(*,*)’ Input datafile: ’READ(10,*)NM,IR,NL,NKREAD(10,*)V,W,XI(1)IF(NM.NE.O) THEN
READ(10,*)(B(M),M=1,NM)READ(10,*)(D(M),M=1,NM)READ(10,*)(OKP(M),M=1,NM)READ(10,*)(FP(M),M=1,NM)READ(10,*)(OLA(M),M=1,NM)READ (10,*) (OKS (M), M = 1, NM)READ(10,*)(FS(M),M=1,NM)
ENDIF
DZ = l./FLOAT(NL)GM = LOG (0.01 )/LOG (XI (1))

DO 1 K=2,NKXI(K) = (0.01 + 0.99 *FLO AT (K-1) /FLOAT (NK)) * * (1. / GM)
1 CONTINUE XI(NK+1) = 1.IF(GM.EQ.l) THEN
FIRM = LOG(l./XI(l))ELSE
FIRM = (1. - XI(1)**(GM - l.))*GM/(GM - 1.)ENDIF
WRITE(*,*)WRITE(*,*)’ Z-T-AB-CB Output file:’WRITE(11,*)WRITE(*,*)
WRITE(*,*)WRITER,*)’ Z-T-XL Output file:’WRITE(12,*)WRITE(*,*)
DO 2 M=1,NMWRITE(*,61)’ T-C-X-E-H Output file, metal ’,M WRITE(M,*)WRITE(*,*)
2 CONTINUE61 FORMAT(A,Il)
CALL INITCON(0.)
100 WRITE(*,*)’ Enter DTB (Thetas):’READ(*,*)DTBWRITE(*,*)’ Enter number of time increments:’READ(*,*)NTNTOT = NTOT + NTIF(NREP.EQ. 1) NT = NT-1
-------------------------------------------------------------------------------------
DO 3 J = 1,NT+1 JINC = NTOT-NT-l+J IF (JINC/NEFI. EQ. JEFI) THEN
WRITE(*,*)WRITER,62)’ On increment’,JINC
ENDIF62 FORM AT(A, 15)
IF(JINC.NE.O) T = T + DTB

139
DO 4 L =1,N L +1 IF(L.EQ.l) AB = 1.FAC = 0.DO 5 M=1,NM
IF(L.EQ.l) CB(M) = 0.IF(SS(M,L).GT.O. .AND. OLA(M).GT.O.) THEN
FACS(M) = OKS(M)*SS(M,L)**FS(M)*AB/3.SFAC = - OKS (M) *B(M) * V *W* AB/ (3. * OL A(M))SS(M,L) = SRK(SS(M,L),SFAC,DTB,FS(M))
ELSEFACS(M) = 0.SS(M,L) = 0.
ENDIFFAC = FAC + FACS(M)
5 CONTINUE
WAW = 0.DO 6 K=1,NK+1
CALL ASOLVE(AB,L,K)DADR = GRAD(A(IR-1,L,K),A(IR,L,K),A(IR+1,L,K),IR)GA = (DADR*GM*XI(K)**(GM - 3.))/0.99 IF(K.GT.l) THEN
WAW = WAW + (GA + GAOLD)*(XI(K) - XI(K-l))/2.ENDIFGAOLD = GA
6 CONTINUEWA = WAW + FAC*FIRM/0.99
DO 7 M=1,NM WCW = 0.DO 8 K=1,NK+1
CALL CSOLVE(CB(M),M,L,K)DCDR = GRAD(C(M,IR-1,L,K),C(M,IR,L,K),C(M,IR+1,L,K),IR) GC = (D(M)*DCDR*GM*XI(K)**(GM - 3.))/0.99 IF(K.GT.l) THEN
WCW = WCW + (GC + GCOLD)*(XI(K) - XI(K-l))/2.ENDIFGCOLD = GC
8 CONTINUEWC(M) = WCW - FACS (M) *FIRM/0.99
7 CONTINUE
DO 9 K=1,NK+1 CALL SSOLVE(L,K)
9 CONTINUE

140
ZL = FLOAT (L- l)/FLOAT (NL)TL = T + ZLIF(JINC/NOFI.EQ.JOFI) WRITE(11,50)ZL,TL,AB,(CB(M),M = 1 ,NM) IF(JINC/NEFI.EQ. JEFI) THEN
IF(JINC.EQ.O .OR. L.EQ.NL+1) THENWRITE(*,50)ZL,TL,AB,(CB(M),M=1,NM)
ENDIFENDIF
50 FORMAT(F6.3,F9.3,5E11.4)
DO 10 M=1,NM IF (JINC/NEFI. EQ. JEFI) THEN
X(M,L) = 0.HN(M,L) = 0.HD(M,L) = 0.DO 11 K=1,NK+1
DO 12 I=1,IR+1 XF(I) = 1. - SP(M,I,L,K)HNF(I) = SP(M,I,L,K)**FP(M)*A(I,L,K)HDF(I) = SP(M,I,L,K)**FP(M)
12 CONTINUECALL SIMPSON(IR,XF, 1 ,XW)CALL SIMPSON(IR,HNF, 1 ,HNW)CALL SIMPSON(IR,HDF, 1 ,HDW)FACK = XI(K) - OLA(M)XW = FACK*XW*GM*XI(K)**(GM - l.)/0.99 FACK = FACK/(1. - OLA(M))HNW = HNW*FACK*OKP(M)*GM*XI(K)**(GM - l.)/0.99 HDW = HDW*FACK*OKP(M)*GM*XI(K)**(GM - l.)/0.99 IF(K.NE. 1) THEN
X(M,L) = X(M,L) + (XW + XWOLD)*(XI(K) - XI(K-l))/2. HN(M,L) = HN(M,L) + (HNW + HNWO)*(XI(K)-XI(K-l))/2. HD(M,L) = HD(M,L) + (HDW + HDWO)*(XI(K)-XI(K-l))/2.
ENDIFX W O L D = X W H N W O = H N W H D W O = H D W
11 C O N T IN U EX(M,L) = X(M,L) + OLA(M)5i:(l. - SS(M,L))HN(M,L) = HN(M,L) + OKS(M)*SS(M,L)^FS(M)*AB HD(M,L) = HD(M,L) + OKS(M)*SS(M,L)**FS(M)
ENDIFIF(L.NE.NL+1) THEN
CB(M) = CB(M) - W*WC(M)*DZ IF(CB(M).LT.l.E-30) CB(M) = 0.

141
ENDIF10 CONTINUE
IF(JINC/NOFLEQJOFI)WWTE(12,50)ZL,TL,(X(M,L),M=1,NM)
IF(L.NE.NLEl) THEN AB = AB - W*WA*DZ IF(AB.LT. 1 .E-30) AB = 0.
ENDIF
4 CONTINUE
DO 13 M=1,NM IF(JINC.NE.O) THEN
E(M) = E(M) + V *B (M) * (CBOLD (M) + CB(M))*DTB/2.ELSE
E(M) = 0.ENDIF
13 CONTINUE
IF(JINC/NEFI.EQ JEFI) THEN DO 14 M=1,NM
DO 15 L=1,NL+1 XL(L) = X(M,L)HNL(L) = HN(M,L)HDL(L) = HD(M,L)
15 CONTINUECALL SIMPSON(NL,XL,0,XT(M))CALL SIMPSON(NL,HNL,0,HTN(M))CALL SIMPSON(NL,HDL,0,HTD(M))IF(HTD(M).NE.O.) THEN
H(M) = HTN (M) /HTD (M)WRITE(M,52)T +1. ,CB(M),XT(M),E(M),H(M)
ELSEWRITE(M,52)T +1. ,CB(M),XT(M),E(M)
ENDIF52 F0RMAT(F8.2,4E11.4)14 CONTINUE
JEFI = JEFI + 1ENDIF
DO 16 M=1,NM CBOLD(M) = CB(M)
16 CONTINUE

142
IF(JINC/NOFI.EQ.JOFI) THEN WRITE(11,50) 1. ,TL, 1. ,(0. ,M = 1 ,NM) WRITE(12,50)1.,TL,(0.,M=1,NM) WRITE(12,50)0. ,TL,(0. ,M = 1,NM)JOFI = JOFI + 1
ENDIF
3 CONTINUEWRITE(*,*)
DO 17 M=1,NMWRITE(*,63)’ Extraction of metal ’,M,’ = ’,E(M)
63 FORMAT(A,I2,A,F6.3)WRITER,*)
17 CONTINUE
WRITER,*)’ To continue, enter "1" ’ READ(*,*)NREP IF(NREP.EQ.l) GOTO 100
300 END
$INCLUDE: TNITCON.FOR’SINCLUDE: ’ ASOLVE.FOR’$INCLUDE:’CSOLVE.FOR’$INCLUDE: ’TRI.FOR’$INCLUDE:’SSOLVE.FOR’$INCLUDE: ’SRK.FOR’$INCLUDE: ’SIMPSON.FOR’$INCLUDE: ’GRAD.FOR’

SUBROUTINE INITCON(AO)
c ----------------------------------------------------------------------------------------------------C This subroutine sets initial conditions for all variables.c ----------------------------------------------------------------------------------------------------
DIMENSION A(17,65,9),C(2,17,65,9),SP(2,17,65,9) DIMENSION SS(2,65)
COMMON /VAR/A,C,DTB,IR,NK,NL,NM,SP,SS
DO 1 K=1,NK+1 DO 2 L=1,NL+1
DO 3 I=1,IR+1 A(I,L,K) = AO
3 CONTINUE DO 4 M=1,NM
SS(M,L) = 1.DO 5 I = 1,IR+1
SP(M,I,L,K) = 1.C(M,I,L,K) = 0.
5 CONTINUE4 CONTINUE2 CONTINUE1 CONTINUE
RETURNEND
SUBROUTINE AS OLVE(AB,L,K)
This subroutine solves the intraparticle reagent concentration profile by an iterative finite difference approximation.
D IM E N S IO N A (1 7 ,6 5 ,9 ) ,B (2 ) ,B L (1 7 ) ,B M (1 7 ) ,B R (1 7 ) ,C (2 ,1 7 ,6 5 ,9 )
D IM E N S IO N D (2 ), F P (2 ) ,F S (2 ) , O K P (2 ), O K S (2 ), O L A (2 ) ,R ( 17) D IM E N S IO N S P (2 ,1 7 ,6 5 ,9 ) ,S S (2 ,6 5 ) ,X I (9 ) ,A W (1 7 )
COMMON /VAR/A,C,DTB,IR,NK,NL,NM,SP,SS COMMON /PAR/B,D,FP,FS,OKP,OKS,OLA,V,W,XI

Ou
uu
2
144
DTR = DTB*V*W/(3.*XI(K)**2)DR = l./FLOAT(IR)DM = DTR/DR**2
DO 1 1=1,IR F = 0.DO 2 M=1,NM
IF(SP(M,I,L,K).GT.O.) THEN FXI = XI(K)*(XI(K) - OLA(M))/(l. - OLA(M))F = F + FXI*OKP(M)*SP(M,I,L,K)**FP(M)*DTR
ENDIF CONTINUE IF(I.EQ.l) THEN
BL(I) = 0.BM(I) = 6.*DM + F + 1.BR(I) = -6.*DM
ELSEBL(I) = FLOAT (1 -I) *DM/FLO AT (I)BM(I) = 2.*DM + F + 1.BR(I) = FLOAT (I) *DM/FLO AT (1 -I)
ENDIF
1R(I) = A(I,L,K)
CONTINUER(IR) = R(IR) - BR(IR)*AB
CALL TRI(IR,BL,BM,BR,R,AW)
3
DO 3 1=1,IR A(I,L,K) = AW(I)
CONTINUE A(IR+1,L,K) = AB
RETURNEND
SUBROUTINE CSOLVE(CB,M,L,K)
This subroutine solves the intraparticle complex concentration profile by an iterative finite difference approximation.
DIMENSION A(17,65,9),B(2),BL(17),BM(17),BR(17),C(2,17,65,9) DIMENSION D(2),FP(2),FS (2), OKP(2), OKS (2), OLA(2),R( 17)

noon
DIMENSION SP(2,17,65,9),SS(2,65),XI(9),CW(17)
COMMON /VAR/A,C,DTB,IR,NK,NL,NM,SP,SS COMMON /PAR/B,D,FP,FS,OKP,OKS,OLA,V,W,XI
DTR = DTB*V*W*D(M)/(3.*XI(K)**2)DR = l./FLOAT(IR)DM = DTR/DR**2
DO 1 1=1,IRFXI = XI(K)*(XI(K) - OLA(M))/(l. - OLA(M))F = FXI*OKP(M)*SP(M,I,L,K)**FP(M)*A(I,L,K)*DTR/D(M) IF(SP(M,I,L,K).LE.O.) F = 0.IF(I.EQ.l) THEN
BL(I) = 0.BM(I) = 6.*DM + 1.BR(I) = -6.*DM
ELSEBL(I) = FLOAT (1 -I) *DM/FLO AT (I)BM(I) = 2.*DM + 1.BR(I) = FLOAT (I) *DM/FLO AT (1 -I)
ENDIFR(I) = C(M,I,L,K) + F
1 CONTINUER(IR) = R(IR) - BR(IR)*CB
CALL TRI(IR,BL,BM,BR,R,CW)
DO 2 1=1,IR C(M,I,L,K) = CW(I)
2 CONTINUE C(M,IR+1,L,K) = CB
RETURNEND
SUBROUTINE TRI(N,BL,BM,BR,R,U)
This subroutine solves a tri-diagonal matrix by Gaussian elimination.
DIMENSION B L (1 0 1 ),B M (1 0 1 ),B R (1 0 1 ),R (1 0 1 ),U (1 0 1 )

0 o
u u
T r r « T i iT j - i j a a u '. r 3 r r r r r t - i w ti t i t ’
146DO 1 1=2,N
E = BL(I-1)/BM(I-1)BM(I) = BM(I) - E*BR(I-1)R(I) = R(I) - E*R(I-1)
1 CONTINUE
U(N) = R(N)/BM(N)DO 2 1=1,N-l
U(N-I) = (R(N-I) - BR(N-I)*U(N-I+ 1))/BM(N-I)2 CONTINUE
RETURNEND
SUBROUTINE SSOLVE(L,K)
This subroutine solves the intraparticle solid reactant concentration profile by a classical Runge-Kutta technique.
DIMENSION A(17,65,9),B(2),C(2,17,65,9),D(2),FP(2),FS(2) DIMENSION OKP(2),OKS(2),OLA(2),SP(2,17,65,9),SS(2,65),XI(9)
COMMON /VAR/A,C,DTB,IR,NK,NL,NM,SP,SS COMMON /PAR/B,D,FP,FS,OKP,OKS,OLA,V,W,XI
EXTERNAL SRK
DTR = DTB*V*W/3.DO 1 M=1,NM
DO 2 I=1,IR+1IF(SP(M,I,L,K).GT.O. .AND. OLA(M).LT. 1.) THEN
FAC = -OKP(M)*B(M)*A(I,L,K)/(l. - OLA(M))SP(M,I,L,K) = SRK(SP(M,I,L,K),FAC,DTR,FP(M)) IF(SP(M,I,L,K).LT.O.) SP(M,I,L,K) = 0.
ENDIF2 CONTINUE1 CONTINUE
RETURNEND

FUNCTION SRK(S,FAC,DT,F)
C ---------------------------------------------------------------------------------------------------C This function subprogram solves the solid reactant dissolution C rate expression by a classical Runge-Kutta technique.C ---------------------------------------------------------------------------------------------------
FI = FAC*S**F G = S + 0.5*DT*F1 IF(G.LT.O.) GOTO 100 F2 = FAC*G**F G = S + 0.5*DT*F2 IF(G.LT.O.) GOTO 100 F3 = FAC*G**F G = S + DT*F3 IF(G.LT.O.) GOTO 100 F4 = FAC*G**FSRK = S + DT*(F1 + 2.*F2 + 2.*F3 + F4)/6. IF(SRK.LE.0.) SRK = 0.GOTO 200
100 SRK = 0.
200 RETURN END
SUBROUTINE SIMPSON(N,X,INDEX,U)
C--------------------------------------------------------------------C This subroutine performs numerical integration by compound C quadrature techniques (Trapezoid and Simpson’s rules).C--------------------------------------------------------------------
DIMENSION X(101)N1 = N/2 N2 = N - N/2 IF(N1.NE.N2) GOTO 100
IF (INDEX. EQ.l) F = X(N+1)IF (INDEX. NE.l) F = X(l) + X(N+1)
DO 1 I=3,N-1,2IF (INDEX. EQ. 1) F = F + 2.*X(I)*(FLOAT(I-l)/FLOAT(N))**2 IF(INDEX.NE.l) F = F + 2.J,:X(I)
1 CONTINUE

nn
nn
2
148
DO 2 1=2,N,2IF (INDEX. EQ.l) F = F + 4.*X(I)*(FLOAT(I-l)/FLOAT(N))**2 IF(INDEX.NE. 1) F = F + 4.*X(I)
CONTINUE
IF(INDEX.EQ.l) U = F/FLOAT(N)IF(INDEX.NE.l) U = F/(3.*FLOAT(N))RETURN
100 IF (INDEX. EQ.l) F = X(N+1) IF(INDEX.NE. 1) F = X(l) + X(N+1)
3
C—ccc —-
DO 3 1=2,NIF(INDEX.EQ.l) F = F + 2.*X(I)*(FLOAT(I-l)/FLOAT(N))**2 IF(INDEX.NE. 1) F = F + 2.*X(I)
CONTINUE
IF(INDEX.EQ. 1) U = 3.*F/(2.*FLOAT(N))IF (INDEX. NE.l) U = F/(2.*FLOAT(N))RETURN
END
FUNCTION GRAD(F 1 ,F2 ,F3 ,IR)
This function subprogram solves the intraparticle concentration gradients at R by a three-point cubic interpolation spline.
DR = l./FLOAT(IR)C = 3.*(F3 - 2.*F2 + F1)/(4.*DR**2)B = (F3 - F2)/DR - 2.*DR*C/3.D = -C/(3.*DR)GRAD = B + 2.*C*DR + 3.*D*DR**2
RETURNEND

0 U
$LARGE$DEBUG
PROGRAM ORDER
David G. Dixon
C This program uses a log-normal grain weight distributionC function to test the variable order assumption of heterogeneousC kinetics of the dissolution of spherical mineral grains.C ----------------------------------------------------------------------------------------------------
DIMENSION PSI(2001),F(2001),FAC(2001),FACP(2001)
PI = 4.*ATAN(1.)WRITER,*)’ T-S-PHI Output file:’WRITE(1,*)0.,1.WRITE(*,*)W RITE(V)’ F-PSI Output file:’WRITE(11,*)0.,0.WRITE(*,*)WRITE(*,*)’ Enter the log-normal standard deviation:’READ(*,*)SWRITE(*,*)F(l) = 0.PSI(l) = 0.DO 1 1=2,1001
PSI(I) = FLOAT (I-1) * 1. E-3F(I) = EXP(-(LOG(PSI(I))**2/(2*S**2)))/(SQRT(2*PI)*S*PSI(I))
1 CONTINUECALL SIMPTRAP(1000,F,1,1.E-3,FI1)
100 WRITE(*,*)’ Enter psi increment to try after median:’READ(*,*)DPSI DO 2 1=1002,2001
PSI(I) = 1. + FLOAT (I-1001) *DPSIF(I) = EXP(-(LOG(PSI(I))**2/(2*S**2)))/(SQRT(2*PI)*S*PSI(I))
2 CONTINUECALL SIMPTRAP(1000,F, 1001 ,DPSI,FI2)FI = FI1 + FI2WRITE(*,*)’ Integral of f(psi) = ’,FI WRITE(*,*)WRITE(*,*)’ If this is unsatisfactory, type "1":’READ(*,*)NREP IF(NREP.EQ.l) GOTO 100 DO 3 1=2,2001

WRITE(11,*)PSI(I),F(I) 3 CONTINUE
FPIC = EXP(S**2/2)AK = l./FPIC WRITE(*,*)
200 WRITE(*,*)’ Enter DT (taus):’READ(*,*)DTWRITE(*,*)’ Enter number of time increments:’READ(*,*)NTWRITE(*,*)
DO 4 J=1,NT JTOT = JTOT + 1 WRITE(*,*)’ On increment: ’,JTOT T = T + DT TP = AK*T FAC(l) = 0.FACP(l) = 0.DO 5 1=2,2001
TFAC = 1. - TP/(3*PSI(I))IF(TFAC.LT.O.) TFAC = 0.FAC(I) = F(I)*TFAC**3 FACP(I) = F(I)*TFAC**2/PSI(I)
5 CONTINUECALL SIMPTRAP(1000,FAC, 1,1 .E-3,GI1)CALL SIMPTRAP(1000,FAC, 1001 ,DPSI,GI2)CALL SIMPTRAP(1000,FACP,1,1.E-3,GPI1)CALL SIMPTRAP(1000,FACP, 1001 ,DPSI,GPI2)H = GI1 + GI2HP = (GPI1 + GPI2)/FPICIF(H.GT.0. .AND. HP.GT.0.) PHI = LOG(HP)/LOG(H) WRITE(1,*)T,H,PHI WRITE(*,*)T,H,PHI
4 CONTINUE
W RITE(V)WRITE(*,*)’ To continue, type "1":’READ (*, *)NREP IF(NREP.EQ.l) GOTO 200
300 END
$INCLUDE: ’SIMPTRAP.FOR’

uu
uu
151
SUBROUTINE SIMPTRAP(N,X,NS,D,U)
This subroutine performs numerical integration by compound quadrature techniques for ORDER.
DIMENSION X(2001)N1 = N/2 N2 = N - N/2 IF(N1.NE.N2) GOTO 100
F = X(NS) + X(N+NS)DO 1 I=NS+2,N+NS-2,2
F = F + 2.*X(I)1 CONTINUE
DO 2 I=NS + 1,N+NS-1,2 F = F + 4.*X(I)
2 CONTINUE
U = F*D/3.RETURN
100 F = (X(NS) + X(N+NS))/2.DO 3 I=NS + 1,N+NS-1
F = F + X(I)3 CONTINUE
U = F*D RETURN
END

152
Appendix SixAnalytical Solutions to the Pseudo-Steady-State Two-Reactant Problem
The generalized differential equation
X 2 & y + X (a 2b x ' ) - & d x 2 dx ( A . 6 - 1 )
+ [c + d x 2s - b ( l - a - r ) x x + b 2x 2z] y
may be reduced to Bessel’s equation
= o
+ X *Z + ( x 2 - p 2) y = 0 d x 2 dx
( A . 6 - 2 )
by proper transformation of variables. The solution may then be obtained in terms of
Bessel functions.46 The generalized solution of equation (A.6-1) is then
y _ j r (1-a) / 2 g - ( b x 1 / r)
where
/ d X s + c,z.2 -p\yf\d
/j( A . 6 - 3 )
p = 7 N l" T -l - a \2 - c
Cl and c2 are constants, and Zp and Z p are Bessel functions of orders p and -p,
respectively. Table A.6-1 summarizes which Bessel functions are involved in the solution
depending on the conditions of the Bessel function arguments and orders.
Multiplying equation (1-31) by | 2 gives
' K*>V a ’ 0( A . 6 - 4 )
Comparing equations (A.6-4) and (A.6-1), and noting that y = a and x - £, the
following identities are observed:

153
TABLE A.6-1: Bessel functions to use in equation (A.6-3)
Conditions V'd/s is real V'd/s is imaginary
p ^ 0 or Z p = J P z P = iPan integer n Z p = J - p
Z.p — I.p
p = 0 or Zp = jn Zp = I„an integer n z.p = Yn Z.p = Kn

154
a = 2 £> = 0 c = 0
d = “ Kp2 s = 1
and r is undefined. Thus, equation (A.6-3) takes the form
a « + c2z .pVv 5)] <&- 6 - 5 >
where p = 1/2. Since p is neither zero nor an integer, and Vd/s is imaginary, then
according to Table A.6-1, Zp and Z.p are Bessel functions of the first kind of order (1/2)
and (-1/2); I1/2 and I.1/2, respectively. Furthermore, these Bessel functions satisfy the
following identities:
Xi/2 (x)
J - l /2 (x)
— s i n h ( x )TZ X
—— cosh(x)7 I X
Thus, equation (A.6-5) becomes
. s i n h ( ^ C C ) / c o s h ( 0 c^£) a = c \ --------- - ± r f ------ + c '2---------— —-------
where c \ and c’2 are constants. Since
( A . 6 - 6 )
cosh (x) l im ------------x-0 x
oo
c’2 must equal zero. Therefore,
c , s i n h ( 0 c ^ g ) (a . 6 - 7 )
1 ^
Applying boundary condition (1-14) to this equation, remembering that a b can be taken
as a constant since the system is at steady-state, one obtains a solution for the constant

155
c \ = -----------------absinM ^ic~)
Substituting equation (A.6-8) into (A.6-7) gives the final result:
« ( « ) ■ q ,$ sinhCyK )At £ = 0, the solution is obtained by noting that
limx-o
s i n h ( . x )x
= 1
Therefore, at £ = 0,
( A . 6 - 8 )
( A . 6 - 9 )
a (0) = --------------------ajb ( A. 6 - 1 0 )s i n h ( v/K^)
Since a is not a function of time, solution of equation (1-32) involves only a
straightforward integration to give (1-34), and therefore is not shown here.
Differentiating equation (A.6-9) with respect to £, evaluating the result at £ = 1,
and substituting the result into equation (2-30) gives
+ y « h = 0 ( A . 6 - 1 1 )dC *
where
~ t a n M ^ )Y t a n h ( ^ k^ )
Equation (A.6-11) may be integrated directly, and combined with boundary condition (2-
20) to give

a b (C) = e ~ “YC ( A . 6 - 1 2 )
which, when combined with equations (A.6-9) and (A.6-10), results in the complete
solution to the pseudo-steady-state two-reactant problem, equation (2-31).
156

Bibliography
157
1. Heinen, H.J., Peterson, D.G., and Lindstrom, R.E., "Processing Gold Ores Using Heap Leach-Carbon Adsorption Methods," (Report IC 8770, U.S. Bureau of Mines, 1978).
2. Philip, T.P., "To Mill or To Leach?," World Gold ’91. (Parkville, Victoria, Australia: The Australasian Inst. Min. Metall., 1991), pp. 1-2.
3. Dorey, R ., Van Zyl, D ., and Kiel, J ., "Overview of Heap Leaching Technology," Introduction to Evaluation. Design and Operation of Precious Metal Heap Leaching Projects, eds. Van Zyl, K., Hutchison, I., and Keil, J., (Littleton, CO: SME-AIME, 1988), pp. 3-22.
4. Braun, R.L., Lewis, A.E., and Wadsworth, M.E., "In-Place Leaching of Primary Sulfide Ores: Laboratory Leaching Data and Kinetics Model," Metall. Trans.. 5 (1974), pp. 1717-1726.
5. Levenspiel, O., Chemical Reaction Engineering. 2nd ed. (New York, NY: John Wiley & Sons, 1976), pp. 357-408.
6. Madsen, B.W., Wadsworth, M.E., and Groves, R.D., "Application of a Mixed Kinetics Model to the Leaching of Low Grade Copper Sulfide Ores," Trans. SME-AIME. 258 (1975), pp. 69-74.
7. Gao, H.W., Sohn, H.Y., and Wadsworth, M.E., "A Mathematical Model for the Solution Mining of Primary Copper Ore: Part I. Leaching by Oxygen-Saturated Solution Containing No Gas Bubbles," Metall. Trans. B. 14B (1983), pp. 541- 551.
8. Lin, H.K., and Sohn, H.Y., "Mixed-Control Kinetics of Oxygen Leaching of Chalcopyrite and Pyrite from Porous Primary Ore Fragments," Metall. Trans. B, 18B (1987), pp. 497-504.
9. Bartlett, R.W., "A Combined Pore Diffusion and Chalcopyrite Dissolution Kinetics Model for In Situ Leaching of a Fragmented Copper Porphyry," International Symposium on Hvdrometallurgy, ed. Evans , D.J.I., and Shoemaker, R.S., (New York, NY: AIME, 1973), pp. 331-374.
10. Braithwaite, J.W., (Ph. D. dissertation, University of Utah, 1976).
11. Madsen B.W., and Wadsworth, M.E., "A Mixed Kinetics Dump Leaching Model for Ores Containing a Variety of Copper Sulfide Minerals," (Report RI 8547, U.S. Bureau of Mines, 1981).

12. Roman, R.J., Benner, B.R., and Becker, G.W., "Diffusion Model for Heap Leaching and Its Application to Scale-Up," Trans. SME-ATMF,. 256 (1974), pp. 247-252.
13. Shafer, J.L., White, M.L., and Caenepeel, C.L., "Application of the Shrinking Core Model for Copper Oxide Leaching," Min. Engng.. 31 (2) (1979), pp. 165- 171.
14. Chae, D.G., and Wadsworth, M.E., "The Leaching of Dilute Copper Oxide Ore Phases," In Situ Recovery of Minerals, ed. Coyne K.R., and Hiskey, J.B., (New York, NY: Engineering Foundation, 1989), pp. 1-21.
15. Box, J.C., and Prosser, A.P., "A General Model for the Reaction of Several Minerals and Several Reagents in Heap and Dump Leaching," Hvdrometallurgy. 16 (1986), pp. 77-92.
16. Hicban, W .T., and Gray, P.M. J ., "Cost Minimization in the Extraction of Copper from an Oxide Ore," Extraction Metallurgy ’81. ed. Jones, M.J., (London: Inst. Min. Metall., 1981), pp. 165-172.
17. Prosser, A.P., "Simulation of Gold Heap Leaching as an Aid to Process Development," Precious Metals ’89. ed. Jha M.C., and Hill, S.D., (Warrendale, PA: The Minerals, Metals & Materials Society, 1988), pp. 121-135.
18. Bartlett, R.W., "Pore Diffusion-Limited Metallurgical Extraction from Ground Ore Particles," Metall. Trans.. 3 (1972), pp. 913-917.
19. Crank, J., The Mathematics of Diffusion. 2nd ed., (Oxford: Clarendon Press, 1975), p. 91.
20. Bartlett, R. W ., "Simulation of Ore Heap Leaching Using Deterministic Models," Hvdrometallurgy. 29 (1992), pp. 231-260.
21. Wen, C.Y., "Noncatalytic Heterogeneous Solid Fluid Reaction Models," Ind. Eng. Chem.. 60 (9) (1968), pp. 34-54.
22. Ramachandran, P.A., and Doraiswamy, L.K., "Modeling of Noncatalytic Gas- Solid Reactions," AIChE J .. 28 (6) (1982), pp. 881-900.
23. Roach, G.I.D., and Prosser, A.P., "Prediction of Rates of Chemical Processes for Treatment of Low-Grade Materials: Theory and Tests for Mass-Transfer Controlled Reactions," Trans. Inst. Min. Metall.. C87 (1978), pp. C129-C138.
24. Ishida, M., and Wen, C.Y., "Comparison of Kinetic and Diffusional Models for Solid-Gas Reactions," AIChE J ., 14 (2) (1968), pp. 311-317.
158

159
25. Habashi, F., "Kinetics and Mechanism of Gold and Silver Dissolution in Cyanide Solution," (Bulletin 59, Montana College of Mineral Science and Technology, 1967).
26. Schuhmann, R., "Energy Input and Size Distribution in Comminution," Trans. SME-AIME. 217 (1960), pp. 22-25.
27. Bartlett, R.W., "Conversion and Extraction Efficiencies for Ground Particles in Heterogeneous Process Reactors," Metall. Trans.. 2 (1971), pp. 2999-3006.
28. Evans, J.W., and Song, S., "A Model for the Design of Gas-Solid Reactors," Metall. Trans.. 4 (1973), pp. 1701-1707.
29. Taylor, J., and Whelan, P.T., "The Leaching of Cuprous Pyrites and the Precipitation of Copper at Rio Tinto, Spain," Inst, Min. Metall. Bull.. 457 (1942), pp. 1-36.
30. Harris, J.A., "Development of a Theoretical Approach to the Heap Leaching of Copper Sulphide Ores," Proc. Aust. Inst. Min. Metall.. 230 (1969), pp. 81-92.
31. Cathles, L.M., and Apps, J.A., "A Model of the Dump Leaching Process that Incorporates Oxygen Balance, Heat Balance, and Air Convection," Metall. Trans. B, 6B (1975), pp. 617-624.
32. Cathles, L.M., and Schlitt, W.J., "A Model of the Dump Leaching Process that Incorporates Oxygen Balance, Heat Balance, and Two Dimensional Air Convection," Leaching and Recovering Copper From As-Mined Materials, ed. Schlitt, W.J., (Littleton, CO: SME-AIME, 1980), pp. 9-27.
33. Cathles, L.M., and Murr, L.E., "Evaluation of an Experiment Involving Large Column Leaching of Low Grade Copper Sulfide Waste: A Critical Test of a Model of the Waste Leaching Process," Leaching and Recovering Copper From As-Mined Materials, ed. Schlitt, W.J., (Littleton, CO: SME-AIME, 1980), pp. 29-48.
34. Gao, H.W., Sohn, H.Y., and Wadsworth, M.E., "A Mathematical Model for the Solution Mining of Primary Copper Ore: Part II. Leaching by Solution Containing Oxygen Bubbles," Metall. Trans. B. 14B (1983), pp. 553-558.
35. Roman, R.J., and Olsen, C., "Theoretical Scale-Up of Heap Leaching," Solution Mining Symposium, ed. Apian, F.F., McKinney, W.A., and Pemichele, A.D., (New York, NY: AIME, 1974), pp. 211-229.

160
36. Prosser, A.P., and Box, J.C., "Simulation of the Mineralogical and Chemical Aspects of Heap and Dump Leaching as an Aid to Ore-Process Evaluation," Computers in Mining — 1983. (Parkville, Victoria, Australia: Australasian Inst. Min. Metall., 1983), pp. 171-178.
37. Roman, R.J., "Solution Channeling in Leach Dumps," Trans. SME-AIME. 262 (1977), pp. 73-74.
38. Satterfield, C.N., "Trickle-Bed Reactors," AIChEJ.. 21 (2) (1975), pp. 209-228.
39. Bondi, A., "Handling Kinetics from Trickle-Phase Reactors," Chem. Tech.. (3) (1971), p. 185.
40. Aitcheson, J., and Brown, J.A.C., The Lognormal Distribution. (Cambridge: Cambridge University Press, 1957).
41. Yakowitz, S., and Szidarovsky, F., An Introduction to Numerical Computations. (New York, NY: Macmillan, 1989), p. 372.
42. Crank, p. 149.
43. Smith, G.D., Numerical Solution of Partial Differential Equations. (London: Oxford University Press, 1965), pp. 17-23.
44. Bird, R.B., Stewart, W.E., and Lightfoot, E.N., Transport Phenomena. (New York, NY: John Wiley and Sons, 1960), pp. 73-74.
45. Yakowitz and Szidarovsky, pp. 197-214.
46. Mickley, H.S., Sherwood, T.K., and Reed, C.E., Applied Mathematics in Chemical Engineering. (New York, NY: McGraw-Hill Book Company, 1957), p. 174.
iM




















