
Population Genetics Of Pseudacris Feriarum - Diginole: FSU's ...
72
Florida State University Libraries Electronic Theses, Treatises and Dissertations The Graduate School 2012 Population Genetics of Pseudacris Feriarum Moses J. Michelsohn Follow this and additional works at the FSU Digital Library. For more information, please contact [email protected]
-
Upload
khangminh22 -
Category
Documents
-
view
7 -
download
0
Transcript of Population Genetics Of Pseudacris Feriarum - Diginole: FSU's ...

Florida State University Libraries
Electronic Theses, Treatises and Dissertations The Graduate School
2012
Population Genetics of Pseudacris FeriarumMoses J. Michelsohn
Follow this and additional works at the FSU Digital Library. For more information, please contact [email protected]

THE FLORIDA STATE UNIVERSITY
COLLEGE OF ARTS AND SCIENCES
POPULATION GENETICS OF PSEUDACRIS FERIARUM
By
MOSES J. MICHELSOHN
A Thesis submitted to the
Department of Biological Science
in partial fulfillment of the
requirements for the degree of
Master of Science
Degree Awarded:
Spring Semester, 2012

ii
Moses J. Michelsohn defended this thesis on April 2, 2012.
The members of the supervisory committee were:
Emily Moriarty Lemmon
Professor Directing Thesis
Scott J. Steppan
Committee Member
Peter Beerli
Committee Member
The Graduate School has verified and approved the above-named committee members, and
certifies that the thesis has been approved in accordance with university requirements.

iii
This thesis is dedicated to all the Eckerd College folks that helped me get here:
Steven Denison, Elizabeth Forys, Yvonne King, William Szelistowski, & Peter Meylan

iv
ACKNOWLEDGEMENTS
I am very grateful for the guidance of my supervisory committee: Emily Lemmon, Scott
Steppan, and Peter Beerli. They have provided me with countless conversations, meaningful
suggestions, and the encouragement that was necessary for me to complete this project. This
work was supported by a Theodore Roosevelt Memorial Grant from the American Museum of
Natural History and by the Florida State University Department of Biological Science.
I am indebted to the many people who assisted me with specimen and tissue collection
including: L. N. Barrow, A. M. Heupel, A. R. Lemmon, E. M. Lemmon, D. B. Means, S. O.
Sunderman, A. R. Warwick. I greatly appreciate the laboratory help and training I received from
M. E. Bedwell, S. A. Emme, H. Ralicki, and S. Miller; as well as the help with analyses I
received from P. Beerli, K. E. Lotterhos, and M. Palczewski. I am especially thankful for the
encouragement, numerous manuscript revisions, and field assistance (including far too many
nights driving endlessly around Dothan) provided by Lisa Barrow.

v
TABLE OF CONTENTS
List of Tables ................................................................................................................................. vi
List of Figures ............................................................................................................................... vii
Abstract ........................................................................................................................................ viii
1. A TEST OF WALLACE'S RIVERINE BARRIER HYPOTHESIS: MODELING THE
EFFECTS OF THE APALACHICOLA-CHATTAHOOCHEE RIVER ON GENE MIGRATION
IN PSEUDACRIS FERIARUM ........................................................................................................1
1.1 Introduction ....................................................................................................................1
1.1.1 Wallace's Riverine Barrier Hypotheses .............................................................2
1.1.2 Oxbow Models ...................................................................................................5
1.1.3 Flood Model .......................................................................................................5
1.1.4 Additional Models .............................................................................................6
1.1.5 Study System .....................................................................................................6
1.2 Methods..........................................................................................................................7
1.3 Results ............................................................................................................................9
1.4 Discussion ....................................................................................................................10
1.4.1 Methods Used in Evaluating Riverine Effects .................................................12
1.4.2 Future Directions .............................................................................................15
2. CASCADING SPECIATION: EVIDENCE FOR POPULATION GENETIC EFFECTS OF
REPRODUCTIVE CHARACTER DISPLACEMENT IN PSEUDACRIS FERIARUM ..............17
2.1 Introduction ..................................................................................................................17
2.1.1 Study System ...................................................................................................19
2.2 Methods........................................................................................................................20
2.3 Results ..........................................................................................................................22
2.4 Discussion ....................................................................................................................22
TABLES ........................................................................................................................................25
FIGURES .......................................................................................................................................40
APPENDIX A SPECIMEN COLLECTION INFORMATION ...................................................44
APPENDIX B PROGRAM μSCOPE ...........................................................................................47
APPENDIX C 454-GENERATED PRIMER DEVELOPEMENT ..............................................49
APPENDIX D MULTIPLEX PCR PRIMER SETS & PROTOCOLS ........................................51
REFERENCES ..............................................................................................................................53
BIOGRAPHICAL SKETCH .........................................................................................................63

vi
LIST OF TABLES
1 A comparison of riverine barrier hypotheses ........................................................................25
2 Collection data for Pseudacris feriarum tissues ....................................................................26
3 Results of Hardy-Weinberg exact tests (p-values) and linkage disequilibrium probability
tests (p-values) conducted in Genepop v4.1 ..........................................................................31
4 Distance matrix of all inter-population distances used for creating geofiles ........................31
5 Bezier approximated marginal likelihoods (ln mL) of three models testing the effect of a
river on gene migration .........................................................................................................32
6 Parameter estimates for models of cross-river gene flow .....................................................32
7 Bezier approximated marginal likelihoods (ln mL) of three models testing riverine effects
on gene migration along a river .............................................................................................33
8 Parameter estimates for models of riverine gene flow ..........................................................34
9 Bezier approximated marginal likelihood (ln mL) results for tests of gene flow across a
region of reproductive character displacement ......................................................................35
10 Parameter estimates for models of gene flow across a region of reproductive character
displacement ..........................................................................................................................35
11 Microsatellite primer sequences developed from 454 reads .................................................36
12 Results of microsatellite primer amplification in a panel of Pseudacris feriarum selected
from across the range of the species ......................................................................................37
13 Primer sequences for sixteen microsatellite loci in Pseudacris feriarum ............................38
14 Multiplex composition used to genotype individuals for sixteen loci ...................................39

vii
LIST OF FIGURES
1 Models of cross-river gene migration ....................................................................................40
2 Models of riverine effects on gene migration ........................................................................40
3 Map of sampling localities ....................................................................................................42
4 Models of gene migration across a region of reproductive character displacement .............43

viii
ABSTRACT
Many genetic patterns observed within and between species are often attributed to
processes that affect interpopulation genetic exchange. These patterns are often taken as
evidence of the genetic processes without explicit tests of the population genetic dynamics
operating within species. The first chapter of this thesis uses a population genetic approach to
test Wallace's riverine barrier hypothesis, a 150-year-old theory that has largely been based on
interpretation of broad scale patterns rather than focused studies of the process. This work helps
clarify the definitions of many riverine hypotheses and uses a Bayesian model comparison
approach to test these hypotheses in Pseudacris feriarum, the upland chorus frog, along the
Apalachicola-Chattahoochee River using eleven microsatellite loci. A flood model of gene
migration best explains riverine effects in this species. This model is proposed as an alternative
way to think about riverine effects on gene flow that should be tested more broadly. The second
chapter of this thesis builds on previous observations of reproductive character displacement and
its effects on speciation. A Bayesian model testing approach is used to determine if predictions
based on female choice experiments lead to population differentiation. Eleven microsatellite loci
are used to model gene migration across a region of reproductive character displacement. This
approach provides evidence for the genetic consequences expected under a speciation cascade
model of taxon diversification.

1
CHAPTER ONE
A TEST OF WALLACE'S RIVERINE BARRIER HYPOTHESIS:
MODELING THE EFFECTS OF THE APALACHICOLA-
CHATTAHOOCHEE RIVER ON GENE MIGRATION IN
PSEUDACRIS FERIARUM
Introduction
Many geographic features are believed to act as genetic barriers, potentially capable of
leading to vicariant speciation (Mayr 1942; Cf. Coyne and Orr 2004). Rivers have often been
noted as barriers between lineages (reviewed in Colwell 2000), although the literature on this
subject often confuses observed patterns with the processes that create them. The interpretation
of Wallace's riverine barrier hypothesis is also muddled because the name has been applied to
many hypotheses concerning riverine effects on populations. Additionally, there has been a lack
of statistical rigor in the treatment of riverine effects (Knowles & Maddison 2002). This study
used a Bayesian model comparison approach (Beerli & Palczewski 2010) to investigate the
effects of the Apalachicola-Chattahoochee River on the population genetics of Pseudacris
feriarum, the upland chorus frog, as a model for evaluating population genetic processes that
lead to observed genetic patterns.
The phrase 'Wallace's riverine barrier hypothesis' has been used to describe a number of
related models describing the effects rivers have on their adjacent floras and faunas. These
models have often been poorly defined in the scientific literature, leading to inadequate tests of
their effects (but see Patton et al. 2004; Funk et al. 2007). In the following sections I will
summarize and define the tenets of each of these theories and present a few additional models
predicting the possible influence of rivers on gene movement.

2
Wallace's Riverine Barrier Hypotheses
Alfred Russel Wallace became the eponym of riverine barrier hypotheses due to
comments made in an 1852 address to the Zoological Society of London about Primates
observed in South America:
During my residence in the Amazon district I took every opportunity of determining the
limits of species, and I soon found that the Amazon, the Rio Negro and the Madeira
formed the limits beyond which certain species never passed.… On approaching the
sources of the rivers they cease to be a boundary, and most of the species are found on
both sides of them.
Wallace's description indicates a view that species' range limits are often bounded by major
rivers (Wallace 1876), but that the rivers' sources offer little impediment to movement, although
Wallace does not speculate whether this is an effect of river width or contiguous populations
above the headwaters. Wallace's confidence in this effect of rivers was so great that he proposed
the division of Amazonian fauna into four districts defined by major rivers (Wallace 1852). This
pattern of rivers acting as barriers in the Amazon was also noted to occur in Lepidoptera by
Wallace's fellow naturalist Henry Walter Bates (1863).
Over a century later, Hershkovitz (1977) expanded on Wallace's range demarcations of
primates noting that although not all species complexes share the same internal boundaries (e.g..,
Saguinus fuscicollis complex v. S. mystax complex), rivers divide "well-differentiated races" in
each case. A more recent study (Ayres & Clutton-Brock 1992) demonstrated a negative
correlation between annual river discharge and primate community similarity on opposite banks
of major Amazonian rivers. This first hypothesis addressing the effects of rivers represents a
view that rivers act as barriers that impede invasion by adjacent populations. These patterns of
subtaxa division along rivers are consistent with Wallace's (1876) statements and form the tenets
of what I will refer to as Wallace's Riverine Barrier Hypothesis (Table 1, WRBH).
Patton et al. (1994) reinterpreted the Riverine Barrier Hypothesis sensu Wallace (WRBH)
as a theory about gene movement. This hypothesis, which I will refer to as the Genetic Riverine
Barrier Hypothesis (Table 1, GRBH), states that genetic divergence between populations
spanning a river should be positively correlated with increasing river width. In this hypothesis,
rivers act as barriers to interpopulation dispersal within a species complex. Rivers may act as

3
either complete or incomplete barriers with the expectation that genetic similarity should
increase upstream where successful cross-river dispersal events are more common.
Funk et al. (2007) modified the GRBH with the additional condition that rivers must act
as regions of primary diversification within a lineage, a hypothesis I will refer to as the Primary
Diversification Riverine Barrier Hypothesis (Table 1, PDRBH). This definition is more
restrictive in that it assigns a river the role of initiating allopatric diversification rather than
simply acting to reduce gene migration between populations which may have already diversified
in isolation. Funk et al. (2007) outlines four testable predictions of the PDRBH:
(1) populations [that are] reciprocally monophyletic… will occur on opposite sides of
major… rivers after sufficient time has elapsed for lineage sorting
(2) genetic divergence between populations will be positively correlated with the
presence of intervening rivers after removing the effects of geographic distance
(3) genetic divergence between populations separated by a river will be greater in the
river's lower section than in the headwaters
(4) rivers will act as areas of primary differentiation rather than secondary contact of
lineages
Predictions (2) and (3) are consistent with the GRBH, while prediction (4) is unique to the
PDRBH. Prediction (1) describes a pattern (reciprocal monophyly of lineages on opposite
banks), which could only occur if all cross-river gene migration has ceased long enough to yield
reciprocally monophyletic lineages. In a coalescent population genetics framework, prediction
(1) precludes prediction (3) that genetic similarity will be greater upstream, although the
prediction could potentially be maintained if only testing statistical relationships rather than
using a genealogical approach. Prediction (1) made in Funk et al. (2007) referred specifically to
mtDNA haplotypes, a situation which could rectify predictions (1) and (3) if only male
individuals disperse and nuclear genes are included in analyses. In this situation mtDNA
lineages could diverge to reciprocal monophyly while the nuDNA continues to be exchanged
between cross-river populations. This situation does not represent a general case amongst all
organisms so I do not include prediction (1) in my definition of PDRBH (Table 1).
Haffer (1969) suggested a role of rivers as regions that help stabilize regions of secondary
contact. If rivers act to impede gene migration then secondary contact zones may drift to match

4
these regions of migration paucity (Barton & Hewitt 1985). Whether rivers act as primary or
secondary sites of speciation has no effect on the process of riverine barriers impeding gene
migration in proportion to river width. For this reason I focus on measuring the contemporary
effects of a riverine barrier using the GRBH as a model for gene migration. Under the GRBH
gene migration rates are expected to be (A) reduced in cross-river routes when compared to
routes that do not cross rivers after removing the effects of geographic distance; and (B) higher
near a river's headwaters than further downstream.
In this study I used a Bayesian model comparison approach to test the expectations of the
GRBH. Two sets of models were used to test this theory, one for each expectation. In addition I
tested other models of gene migration to investigate the gene flow processes that lead to the
patterns of genetic diversity observed along and across rivers. This model testing approach
emphasizes modeling of process rather than detection of pattern attempting to more accurately
reconstruct the effects on a river.
A series of models related to expectation (A) are presented in Figure 1. This first series
of models represents populations spanning a river exchanging migrants with one another. Each
arrow in the figure represents a mutation-scaled migration rate, M, while each circle represents a
population with a mutation-scaled population size, Θ. The Full Migration model (Fig. 1 A)
represents a parameter rich model that would be needed to explain a complicated history of gene
flow amongst these populations. The Equivalent Linear Migration model (Fig. 1 B) is a direct
violation of expectation (A) in that it reduces all M parameters to one shared rate. This model
represents a situation where rivers do not impede inter-population gene flow at any different rate
than regions without rivers spanning them. The Linear River Effect model (Fig. 1 C) restricts the
migration rate between populations without rivers between them to one M parameter and those
that do span a river to a separate set of M rates. This essentially models a situation where all
overland migration occurs at a constant rate, but cross-river migration rates are unrestricted by
the model. This first set of models will be used to determine if a river acts to reduce gene
migration between populations.
Figure 2 presents a series of models of riverine effects on population genetics. These
models are created based on the assumption that a river acts to reduce gene flow between
populations. The independent models presented each represent a different view on the specific
directional effects a river might have on cross-river genetic connectivity. The figure contains

5
two models of the GRBH that include expectation (B). The first model (Fig. 2 A) assumes that a
river acts as a complete barrier to gene migration and that cross-river dispersal events occur only
at the river's headwaters. The second model (Fig. 2 B) assumes that the river acts as an
incomplete barrier allowing gene flow along its length inversely proportional to river width.
Additional gene migration models that are inconsistent with the GRBH are described below.
Oxbow Models
River meandering and subsequent oxbow formation can act as a means of passive transfer
of biota across a river. A population that is passively transferred may form a genetic enclave on
an opposite bank. This population may interbreed with local populations leading to a genetic
pattern identical to that generated by individuals actively crossing the river (Hershkovitz 1983).
Rivers meander over low gradients which increase in frequency lower in a stream course. This
increased sinuosity downstream causes a directional permeability of a river that is counter to
river barrier hypotheses, i.e. cross-river genetic connectivity is expected to increase downstream.
An oxbow model of riverine effects still requires that a river must act as a barrier to gene
migration, an expectation that can be tested using cross-river population models (Fig. 1). Two
oxbow models are presented in Figure 2. The first model (Fig. 2 C) assumes that passive transfer
occurs only at the lowest regions of the river while the other model (Fig. 2 D) allows for either
passive or active gene migration along the length of the river with increased frequency
downstream.
Flood Model
Another alternative model of gene migration across rivers has been proposed for plants
by Jacquemyn et al. (2006). This model is based on periodic river flooding events causing long
range dispersal of progeny. Individuals that raft downstream have increased chances of crossing
to the other bank the further they travel. This model (Fig. 2 E) predicts genetic similarity in
cross-river downstream populations as a result of immigration from populations on both sides of
a river further upstream. This model has been used to explain genetic structuring in plants
(Jacquemyn et al. 2006; Van Looy et al. 2009) and should be considered as an alternative to
oxbow models in creating downstream genetic connectivity patterns.

6
Additional Models
An equivalent migration model (Fig. 2 F) was specifically designed as a competitor to
both riverine barrier and oxbow models in that it restricts cross-river migration rates to be
equivalent along the entire river thus removing any directional effects. A nearest neighbor
model (Fig. 2 G) represents a complex alternative to all other models. This model is parameter
rich and should only be favored if true migration rates do not match any other hypothesized
model due to irregular patterns of gene flow in a system.
Study System
The genetic effects of rivers were tested in Pseudacris feriarum, the upland chorus frog,
along the Apalachicola-Chattahoochee River. P. feriarum is a wide-ranging species distributed
across the Eastern United States. P. feriarum is generally restricted to regions above the Fall
Line in the Southeastern United States, but its range extends deep into the Gulf Coastal Plain
along the Apalachicola River (Fig. 3). In this region P. feriarum breeds in ephemeral cypress or
gum swamps along the floodplain of the Apalachicola, while further north along the
Chattahoochee River it breeds in ditches, flooded fields, and open woods (Carr & Goin 1959;
Crenshaw & Blair, 1959; Mount 1975; Jensen et al. 2008). Pseudacris do not move far from a
breeding locality during the breeding season, less than 250 meters in P. triseriata (Kramer 1973),
and have generally small home ranges, less than two acres (Kramer 1974).
The region around the Apalachicola-Chattahoochee River system has been shown to be a
contact zone or range limit for numerous organisms (Blaney 1971; Means 1977; Engle &
Summers 2000; Marshall et al. 2000; Gonzales & Hamrick 2005; Swenson & Howard 2005;
Soltis et al. 2006; Pauly et al. 2007; Walker et al. 2009). The Apalachicola-Chattahoochee
River is one of a few Southeastern rivers originating above the coastal plain, completely
bisecting the coastal plain, thus presenting a potentially complete barrier to coastal plain species
(Pauly et al. 2007). The Apalachicola-Chattahoochee River flows through what was once an
extensive embayment that possibly extended all the way to the Fall Line (Neill 1957), thus the
lower Apalachicola-Chattahoochee River may act as a secondary contact zone between lineages
separated by a once greater saltwater barrier (Ellsworth et al. 1994; Burbrink et al. 2000; Church
et al. 2003). More recently, a large passive transfer of populations across the lower Apalachicola-
Chattahoochee River may have occurred during the Apalachicola stream capture events

7
(Randazzo & Jones 1997). These genetic consequences of these events may be teased apart by
other methods, but they are considered to be too distant in the past to leave signatures in high
mutation-rate markers such as microsatellites. More recent meandering of the Apalachicola-
Chattahoochee River may have led to passive transfer of populations across the river through
oxbowing events. Oxbowing and dispersal (either active or passive) are the only processes
expected to have affected gene migration during the contemporary history of the Apalachicola-
Chattahoochee River.
Many previous studies on the effects of rivers have suffered from inappropriate river or
organism choice (See Chapter 1 Discussion for a review of past studies). Pseudacris feriarum
along the Apalachicola-Chattahoochee River provides an ideal test of the genetic effects of rivers
because the history of the river has been relatively well characterized (Neill 1957; Randazzo &
Jones 1997) and the study organism is likely to experience the river as a major barrier to
dispersal due to its size and dispersal capabilities (Kramer 1973).
Methods
Pseudacris feriarum were collected from eight populations along the Apalachicola-
Chattahoochee River in Florida, Georgia and Alabama (Fig. 3). Tissue samples were obtained
from twenty individuals per population using either toe-clipping or dissection (Table 2;
Appendix A). Voucher specimens from each site were preserved in formalin. Populations were
sampled in two transects across the Apalachicola-Chattahoochee River: a Northern transect
consisting of populations in Macon and Russell Counties, Alabama, and Muscogee County,
Georgia; and a Southern transect consisting of populations in Houston County, Alabama, and
Early and Miller Counties, Georgia (Fig. 3). Two populations (Gulf and Liberty Counties, FL;
Fig. 3) were sampled from the southern extension of P. feriarum's range along the Apalachicola
River.
Genomic DNA was extracted from all 160 P. feriarum individuals using an E.Z.N.A. Gel
Extraction Kit (Omega Bio-Tek). Each individual was genotyped for a total of eleven
microsatellite loci; seven tetramer loci using published primer sets (Lemmon et al. 2011) and
primer sets developed using program μScope v0.0.6 (Appendix B) on previously generated 454
data (Lemmon & Lemmon unpublished; Appendix C), as well as for four dimer loci (Bedwell &
Lemmon unpublished; Ralicki & Lemmon unpublished). Details of primer development are

8
listed in Appendix C. Multiplex PCR reactions were set up using a Multiplex PCR Kit
(QIAGEN) and amplified using protocols described in Appendix D. Fragment analysis was
conducted on an Applied Biosystems 3730 Genetic Analyzer with Capillary Electrophoresis at
the Florida State University Core Research Facilities. Microsatellite fragment lengths were
measured using GeneMapper v4.1 (Applied Biosystems). Allele calling was done using tandem
v1.08 (Matschiner & Salzburger 2009). These allele calls were used to test for Hardy-Weinberg
equilibrium and linkage disequilibrium (Table 3) using Genepop v4.1 (Rousset 2008). Two loci
(D_C08b and A_E09d) exhibited linkage disequilibrium with the marker D_D12b, although not
with each other (Table 3). Lemmon et al. (2011) found no evidence of linkage disequilibrium
between D_D12b and D_C08b across 85 P. feriarum sampled from Macon County, AL. All
three markers were included in further analyses.
Fragment lengths were used to write a series of microsatellite data files for Migrate-n
v3.2.17 (Beerli 2009). Allele binning was performed by Migrate-n using the "#@M" command
in all data files. Migrate data files were written for the Northern transect and Southern transect
to test the effects of cross-river migration (Fig. 1). A third data file was written to test the
riverine effects on population genetics (Fig. 2). This data file included six populations along the
river: Liberty and Gulf Counties, FL; Houston and Russell Counties, AL; and Muscogee and
Early Counties, GA. (Fig. 3). Model parameters were set in Migrate-n using the user-specified
model options. All Migrate-n runs were conducted using a continuous Brownian-motion model
of microsatellite evolution (Felsenstein 2004). Locality information (Table 4) was used to scale
migration rate parameter estimates using interpopulation geographic distance. A strait line
distance matrix (geofile) was imported into Migrate-n to scale by these interpopulation
geographic distances.
Migrate-n run length was determined by setting up successive runs of the most parameter
rich model, the Nearest Neighbor model (Fig. 2 G). Run convergence and parameter prior
ranges were determined using posterior distribution plots of parameter values summed over ten
independent runs output by Migrate-n. A Uniform prior with a range of 0–400 was used for
estimating mutation-scaled migration rates, M, and a Uniform prior with a range of 0–100 was
used for estimating mutation-scaled population size, Θ. Parameter space for each Migrate-n
model was searched with a burn-in of 4,000,000 generations followed by 10,000 steps that were

9
sampled at iterations of every 100 generations to reduce autocorrelation for a grand total of
50,000,000 parameter value iterations explored per long chain. A Metropolis-coupled Markov
chain Monte Carlo (MCMCMC) approach (Geyer 1991) utilizing 4 adaptively heated chains was
implemented to better explore parameter space (Beerli 2006). Parameter values for each model
were sampled using 50 independent long chain samplings. Parameter value distributions were
summed over the fifty independent runs. The natural log marginal likelihood of each model
was estimated using Bézier quadrature (Beerli and Palczewski 2010), an alternative to the
harmonic mean which gives better estimates of the distribution when few heated chains are used.
The natural log marginal likelihood of each model was used to compute natural log Bayes factors
(BF: Kass & Raftery 1995) for model ranking and comparison.
Results
The comparison of cross-river gene migration models yielded favored support (>6,000
Bayes Factors (BF) for each transect) for models of restricted cross-river migration (Fig. 1 C
Linear River Effect) over migration models which held migration constant between populations
regardless of whether the migration route crossed the Apalachicola-Chattahoochee River (Fig. 1
B Equivalent Linear Migration). This same pattern of model support was observed in both the
Northern and Southern transects (Table 5). This supports the idea that rivers affect gene flow
differently from overland migration.
In each transect the Full Linear Migration model was favored overall by >3,300 BF
(Table 5), indicating support for a complex gene flow history in both transects. The model
parameter values estimated for this model in each transect indicate that there is directional skew
in overland migration. This skew may explain why the two models that restricted overland
migration rates (Fig. 1 B & C) received low model support values (Table 5). The cross-river M
parameter estimates in the favored Full Linear Migration model (Table 6) of both transects are
not recognizably lower than the overland M parameter estimates until they are converted to
numbers of migrants using the formula: ( ). In the Northern transect 120.9
individuals/generation migrate cross-river between populations while 2109.8
individuals/generation migrate between populations on the same side of the river. In the
Southern transect 251.5 individuals/generation migrate between cross-river populations while

10
271.5 individuals/generation migrate between same bank populations. This reduced number of
migrants per generation, especially in the Northern transect, indicates that the Apalachicola-
Chattahoochee does act to retard gene flow allowing for comparison of models testing the
directional effects of a river on gene migration.
The comparison of models explaining how rivers affect gene flow (Fig. 2) resulted in
overwhelming support (1,738 BF better than the second best model) for a flood model (Fig. 2 E)
of gene flow (Table 7). The worst riverine effects model was the Equal Migration model which
held cross-river migration constant along the entire river shed. This worst model exhibited far
less support (12,438 BF) than the most parameter rich model (Fig. 2 G). Parameter estimates for
all models are listed in Table 8.
Discussion
The first test of genetic effects of rivers matched the expectation that rivers act to retard
gene flow (Table 5; Funk et al. 2007). The Equivalent Linear Migration (Fig. 1 B) model
exhibited the lowest support in both transects (Table 5), while the Full Linear Migration (Fig. 1
A) model exhibited the highest support (Table 5). When the parameter estimates of the most
parameter rich model, the Full Linear Migration model (Fig. 1 A), are examined (Table 8) one
can see why this complicated model received high support. Asymmetry in migration rate
between populations on the same of the river is apparent (Table 6). This asymmetry is the likely
cause for preference of the Full Linear Migration model, the only model which does not restrict
overland migration to being symmetrical.
Another interesting pattern was observed from the estimated migration rates of the Full
Linear Migration models of both transects. In the Northern transect immigration East across the
river from the Russell County, AL into the Muscogee County, GA population occurs at
approximately twice the rate of the reverse (Table 6). The Southern transect shows a similar
pattern with immigration East from Houston County, AL, into Early County, GA, also at
approximately twice the rate of the reverse (Table 6). This cross-river directional gene flow
presents and interesting pattern, but this directionality is not expected to be caused by a riverine
effect.
The comparison of riverine effect models (Fig. 2) yielded higher support (greater than
1738 BF; Table 7) for the Flood model (Fig. 2 E) over all Oxbow and RBH models (Fig. 2 A–D).

11
The overwhelming support (1738.3 BF better than the second best model) for this model
conflicts with classic thinking about the influence of rivers on gene flow.
The Equal Migration model (Fig. 2 F) showed the least support in this study (5105.2 BF
worse than the best model; Table 7). This model specifically tests the assumption that a river has
a directional effect on gene migration. The asymmetric gene migration noted in the first set of
models may explain why this model faired so poorly. This cross-river asymmetry is accounted
for by the Nearest Neighbor model. In this model the cross-river migration rates are free to be
asymmetrical. The poor relative support for this model versus all oxbow or RBH models (at
least 1701.6 BF worse than any RBH or oxbow model) indicates that even when asymmetry is
allowed, the model is over parameterized by estimating six cross-river migration rates. This
lends support to the idea that rivers have a directional effect on gene migration.
Studies testing riverine barrier hypotheses have yielded mixed results on these effect
(Colwell 2000). One of the best known tests of the GRBH recovered a pattern of downstream
connectivity (Patton et al. 1994). The Flood model and both Oxbow models (Fig. 2 B, C, and E)
each describe processes that yield patterns of higher genetic connectivity downstream along a
river. There is a potential that the current framework in which riverine barrier hypotheses have
been evaluated is too restricted to test other processes. Although these studies have often been
used to cast doubt on the existence of a riverine barrier model (consistent with GRBH or
PDRBH), they have not been used to evaluate alternative models. The approach used in this
study that models process has the power to distinguish between the routes by which genetic
patterns may be created. This power to discriminate between models of process rather than
describe pattern allows for a better reconstruction of past events by directly modeling them.
The Flood model proposed in this research makes some intuitive sense; individuals of
small species often cross rivers through rafting on debris which transports individuals both
across a river and downstream. As debris floats downstream, the probability of the debris
landing on the opposite side increases with downstream distance until it has an approximately
equal chance of being deposited on either bank. Although I have termed this a Flood model
based its proposal by Jacquemyn et al. (2006), a flooding event is not necessary for this process
to operate. A direction of flow is the important part of this model, which may allow future
studies to test the effects of current speed, flow volume, turbidity, etc. on cross-river gene
migration.

12
Methods Used in Evaluating Riverine Effects
Many approaches have been used to investigate the effects of rivers on organisms. Most
of these methods suffer from modeling rivers as impermeable barriers while claiming to test a
riverine hypothesis that defines them as permeable. Tests that model rivers as permeable tend to
lack reasonable alternative models in their analyses. This problem leads to a situation where data
can reject a riverine hypothesis, but is not used to support a riverine model over competing
alternatives beyond panmixia. I have summarized many of the methods previously used to
address riverine effects on populations below and evaluate their effectiveness when compared to
the model testing framework used in this study.
Very few attempts have been made to directly measure riverine effects on dispersal. Klee
et al. (2004) directly assessed the strength of rivers as dispersal barriers through relocation
experiments. This study counted the number of marked individuals found crossing a river after
being relocated to the opposite side. All other evidence for riverine barrier processes comes
from interpreting the patterns left behind.
A few pattern based approaches have been used to test riverine hypotheses without
genetic data, a concept more in line with WRBH (Table 1). Ayres & Clutton-Brock (1992)
interpreted patterns of species similarity along rivers as evidence for riverine barrier effects.
That work used river characterizations, including size, and compared it to a species similarity
index. Jaccard's similarity index (Ludwig & Reynolds 1988) has also been used to make
pairwise comparisons of community similarity at upriver and downriver sites (Gascon et al.
2000). Hayes & Sewlal (2004) used chi-squared tests to determine if range limits within avian
families were influenced by riverine barriers. All these studies are designed to evaluate the
patterns left behind by riverine barrier effects but suffer from a lack of power to identify
causality due to the vast scope of other factors influencing species' range borders (e.g.,
interspecies dependencies).
Another series of pattern based approaches use morphology to look for intraspecies
differences along a river. One of the greatest problems with these morphological studies is that
they cannot include large datasets of homologous characters, such as genetic data, which limits
their power to detect process. Differences in morphological traits and karyotypes have been

13
observed to correspond with rivers in some taxa (Willey & Willey 1967; Lamborot 1991),
although these studies are only able to detect that rivers act as impediments to dispersal, not a
directional effect particular to rivers. Pounds & Jackson (1981) used canonical analysis (Gould
& Johnston 1972) to evaluate phenetic differences in lizard populations within interfluvia
between major rivers. ANOVA and MANOVA analyses have also been used to test if
populations separated by a river differ significantly in morphometric characters (Lamborot &
Eaton 1997; Lamborot et al. 2003; Pauly et al. 2006). These relationships indicate that rivers can
act as breaks in types, but do not directly demonstrate the processes of a river as a directionally
permeable barrier.
Some explicit tests of genetic patterns have been used to show that rivers act as
impediments to gene flow. Bates et al. (2004) compared sequence divergence between same-
bank and cross-river individuals. Slatkin's (Slatkin 1993) has been used to compare pair-wise
population genetic differentiation between same-bank and cross-river populations (Gascon et al.
1998). Additionally, Gascon et al. (1998) used randomization tests to compare distributions of
arc-cosine genetic distances (Cavalli-Sforza & Edwards 1967) generated under panmixia or river
segregation simulations to determine if rivers acted as barriers to gene flow. These studies once
again are useful in confirming a pattern, but do not explicitly model a process.
Clustering analyses have also been used to demonstrate genetic breaks at rivers. Avise et
al. (1979) utilized cluster analysis using estimates of p (Upholt 1977) to determine if natural
genetic subdivisions corresponded to river regions. Principal component analysis of
morphological characters (Lamborot & Eaton 1997) and genetic data (Mylecraine et al. 2004)
has also been used to cluster populations and look for corresponding river barriers. Population
assignment methods (Pritchard et al. 2000; Piry et al. 2004; Guillot 2008) have been used to
determine if genetic populations correspond to river features (Newman & Rissler 2011; Dίaz-
Muñoz 2012). These assignment methods are largely concerned with grouping panmictic units,
but are insufficient for measuring population processes such as gene migration (Palsbøll et al.
2007). They also lack the power to determine process from pattern.
Many studies have used summary statistics to determine if phylogenetic patterns
consistent with a riverine barrier hypothesis exist. Patton et al. (1994) compared cross-bank
sequence divergence at multiple sites along the course of a river to determine if upstream
populations had a higher genetic similarity than downstream populations. Mantel tests (Mantel

14
1967), partial Mantel tests (Smouse et al. 1986), and partial regression analysis (Smouse et al.
1986) have also been used by many authors to evaluate effects of rivers in a population genetics
context (Gascon et al. 1996; Gonzales & Hamrick 2005; Pellegrino et al. 2005; Cabanne et al.
2007; Funk et al. 2007; Lemmon et al. 2007). Gascon et al. (1996) used inbreeding coefficient
analysis to determine hierarchical differences between within-site, same-bank, and total genetic
variances in populations spanning a river. AMOVA has also been used to attribute genetic
variation to rivers by comparing genetic and geographic distances (Davis et al. 2002; Aleixo
2004; Eriksson et al. 2004; Pauly et al. 2006; Cabanne et al. 2007; Zhang et al. 2007; Jalil et al.
2008; Colombi 2010; Dίaz-Muñoz 2012). These methods evaluate whether a pattern consistent
with a riverine barrier hypothesis is observed, but they do not test the underlying process against
other processes that might yield similar patterns of cross-river differentiation.
A number of researchers have used phylogenetic methods to evaluate patterns left by
riverine barrier effects. Unweighted pair group method with arithmetic mean has been used to
construct dendrograms which are then compared to sample localities spanning a river (Gascon et
al. 1998; Mylecraine et al. 2004; Gonzales & Hamrick 2005). Hall & Harvey (2002) used a
morphological parsimony tree to determine if bifurcations corresponded to rivers. Many studies
have generated phylogenetic trees that are then used to evaluate if rivers correspond to
bifurcations (Ellsworth et al. 1994; Chesser 1999; Collins & Dubach 1999; Burbrink et al. 2000;
Donovan et al. 2000; Church et al. 2003; Aleixo 2004; Liu et al. 2005; Kozak et al. 2006; Pauly
et al. 2006; Fu & Zeng 2008, Li et al. 2009). Although these studies often yield robust trees they
lack a means to test the effects of riverine barriers beyond post-hoc attributions of taxonomic
splits to river regions.
Very few studies have used statistically rigorous tests of riverine barriers in a
phylogenetic context. Lougheed et al. (1999) used topology-dependent permutation tail
probability tests (Faith & Cranston 1991) to compare trees constrained to exhibit patterns under a
riverine barrier to trees that were unconstrained. Constraint tree filtering (Miller et al. 2002) was
used by Newman & Rissler (2011) to compare constrained and unconstrained trees. Although
these studies make explicit tests of lineage histories, they assume that a model of a riverine
barrier hypothesis is equivalent to a reciprocal monophyly constraint on each side of a river, i.e.
the river acts as an impermeable barrier. Funk et al. (2007) used parametric bootstrap tests
(Goldman et al. 2000) to evaluate the genetic effects of rivers at different sites along their

15
courses, but still used reciprocal monophyly as the constraint. These methods all use a
phylogenetic context to test a river as a barrier, but do not model the directional effects
consistent with riverine barrier theory. Reciprocal monophyly as a constraint is also a model
inconsistent with the permeable barrier model of a river.
Although many studies have attempted to find patterns consistent with a riverine barrier
hypothesis model, this study is the first to explicitly model the process rather than describe the
pattern. Although the effects of rivers are certain to leave a phylogenetic signal, this pattern can
be obscured due to the dynamic nature of rivers (Patton & da Silva 1998). Problems with
assumptions of the model versus its expected pattern have lead to many incongruous tests of
riverine hypotheses. Tests of riverine effects in this population genetics context are more
appropriate because they attempt to capture the process while it is operating in a system that still
shares genetic exchange, at least in part, across a riverine barrier. Bayesian inference in Migrate-
n (Beerli & Palczewski 2010) offers a statistically rigorous method for discriminating between
models of gene flow across permeable barriers, such as rivers. These methods also allow for
discrimination between competing generalized models of historical gene flow allowing more
confidence in a general conclusion than previous tests that only offer an opportunity to reject a
conclusion without an opportunity to measure model support. These methods are also
advantageous because they explicitly test the process that leads to observed patterns rather than
simply measuring summary statistics of current genetic distributions.
Future Directions
Although this test was only performed on one species, on one river, it provides an
example of the utility of Bayesian model testing in discovering the processes underlying
observed genetic patterns. The Flood model which was favored in this study provides a
promising alternative model of how rivers affect the gene flow of species. This model should be
tested, along with all other hypothesized models of gene flow, across a wide panel of rivers and
taxa. This study provides a framework for reevaluating previous tests of riverine barrier
hypotheses with an emphasis on the contemporary processes that determine genetic patterns
observed around rivers. Some previously collected datasets may be reinterpreted using these
methods to determine the processes generating the previously detected patterns. The powerful

16
analytical approach used in this study should be applied broadly to determine if rivers generate
phylogeographic effects consistent with the proposed Flood model.

17
CHAPTER TWO
CASCADING SPECIATION: EVIDENCE FOR POPULATION
GENETIC EFFECTS OF REPRODUCTIVE CHARACTER
DISPLACEMENT IN PSEUDACRIS FERIARUM
Introduction
Processes of speciation have historically been classified based on geographic pattern.
The majority of speciation events have been categorized as allopatric or peripatric (Futuyma &
Mayer 1980). Under these scenarios speciation occurs through the accumulation of reproductive
barriers in geographically and genetically isolated populations (reviewed in Coyne & Orr 2004).
Sympatric speciation occurs when genetic isolation evolves in the absence of geographic
isolation. This has been well documented in many systems; e.g.., African Cichlidae (Kocher
2004), Rhagoletis pomonella (Filchak et al. 2000); although it is not considered to be a prevalent
mode of speciation (Phillimore et al. 2008). Parapatric speciation, in which peripheral
populations diverge with gene flow, has long been accepted as a theoretical possibility (Fisher
1930; Balkau & Feldman 1973), although conclusive examples in nature are sparse (reviewed in
Coyne & Orr 2004 and Berner et al. 2009). Conclusive examples of parapatric speciation are
difficult to demonstrate due to the difficulties in reconstructing historical rates of gene flow
(Pinho & Hey 2010), but parapatric speciation may represent an underreported, yet frequent
mode of diversification (Gavrilets et al. 1998; Coyne & Orr 2004).
Hybrid zones are often thought of as regions of secondary contact between allopatric
lineages (Barton & Hewitt 1985), although they can be generated by geographic differences in
selective pressures along a gradient—even in the face of constant gene flow (Endler 1977;
Schilthuizen 2000). Simulation studies that model a series of populations distributed linearly
along an environmental gradient have generated evidence that such a regime can lead to
population differentiation and even 'speciation' (Gavrilets 1999; Gavrilets et al. 2000). Models
that yield peripheral population genetic isolation and 'speciation', i.e. a threshold number of
differing loci between individuals, assume that few mutations are required for reproductive

18
isolation. These models emphasize environmental selection gradients but establish a framework
that is useful for thinking about trait and population divergence along sexual selection gradients.
Character displacement (Brown & Wilson 1956) often occurs when two related species
overlap in geographic range. Reproductive character displacement is a mechanism in which
reproductive isolation between related species is strengthened through trait divergence in
sympatric regions (Brown & Wilson 1956; Crozier 1974). A species that exhibits overlap in a
reproductive trait and partial overlap in geographic range with a related species is composed of a
series of populations that exist along a sexual selection gradient analogous to the Gavrilets
(1999) model. Neural network simulation studies have demonstrated that this sexual selection
gradient can result in reproductive character displacement in multiple dimensions in separate
sympatric regions (Pfennig & Ryan 2006; Pfennig & Ryan 2007). Lemmon (2009) provided an
empirical example where a reproductive signal trait diverged in a unique fashion in at least three
regions of sympatry.
Reinforcement is one process that can exert directional selection on a reproductive trait in
sympatry to yield reproductive character displacement (Howard 1993). Under reinforcement,
hybrid offspring between related species exhibit reduced fitness. This fitness reduction leads to
selection on prezygotic isolating mechanisms in the parental species to avoid hybridization. If a
single trait acts as both a signal of interspecies identification and as a signal of intraspecies mate
quality (Rand et al. 1992), then reinforcement can generate a reproductive character
displacement that both reduces interspecies hybridization and alters population gene flow within
the species exhibiting the reproductive character displacement. Alternatively, ecological
speciation may also generate the reproductive character displacement that alters interspecies and
intraspecies gene flow. These processes have been demonstrated in studies of peripatric
speciation followed by secondary contact with the parent lineage (Hoskin et al. 2005) and in
intraspecies primary hybrid zones (Pfennig 2000; Lemmon 2009).
The downstream effects of reproductive character displacement may offer insight into the
process of parapatric speciation. Reproductive character displacement driven by sympatry with a
related species can lead to increased genetic isolation between sympatric and allopatric
populations within species (Higgie & Blows 2008). This process may be repeated independently
in multiple interspecies range overlap zones leading to multiple peripheral populations exhibiting
genetic isolation from both the allopatric range of the species and each other sympatric isolate

19
(Howard 1993). This series of isolating events stemming from range overlap between related
species is termed a 'speciation cascade' (Pfennig & Ryan 2006) due to its potential as a process
leading to rapid speciation events. Ortiz-Barrientos et al. (2009) further implicate reinforcement
as the driving force in this burst of speciation in their 'cascade reinforcement hypothesis'.
Numerous studies have now demonstrated that reproductive character displacement can
lead to divergent trait preferences in sympatric and allopatric populations (e.g.., Pfennig 2000;
Lemmon 2009). Very few studies, however, have evaluated the population genetic
consequences of these trait preferences to evaluate their potential for leading to reproductive
isolation (but see Rice & Pfennig 2010). In this work, I will test the population genetic
predictions of previous female choice experiments (Lemmon 2009) to evaluate support that
range overlap can ultimately lead to reproductive isolation and eventual speciation as in the
speciation cascade model (Pfennig & Ryan 2006).
Study System
Pseudacris feriarum, the upland chorus frog, provides an ideal case study for parapatric
speciation in progress. This species ranges across most of the Eastern United States above the
Fall Line (Fig. 3). The species' range extends into the coastal plain along a few major drainages
of the Atlantic and Gulf coasts. These range extensions bring P. feriarum into sympatry with P.
nigrita, the southern chorus frog, a coastal plains congener (Fig. 3). The acoustic signal of P.
feriarum in these sympatric extensions has undergone reproductive character displacement
(Fouquette 1975; Lemmon 2009) due to reinforcement (Lemmon & Lemmon 2010). This
selective gradient between allopatry and sympatry is repeated along numerous drainages
representing at least three independent events resulting in interspecies hybrid zones between call
types (Lemmon 2009). Each of these hybrid zones have resulted in unique displaced signal
characters (Lemmon 2009).
Lemmon (2009) demonstrated, through female preference experiments, that sympatric P.
feriarum males express a call type that is favored by adjacent allopatric P. feriarum females
when compared to local males along the Apalachicola-Chattahoochee River. Conversely,
allopatric P. feriarum males express a call type that is rejected by adjacent sympatric P. feriarum
females when compared to local males. This preference sets up a case in which a selective

20
gradient is established whereby males that disperse toward the central range of this species into
allopatry are expected to have high fitness while males dispersing into the sympatric peripheral
range are expected to have low fitness. This fitness scenario should lead to increased isolation of
the peripheral populations consistent with models of parapatric speciation (Nosil et al. 2005).
This study used a Bayesian model comparison method to test the predictions of the
female choice experiments in Lemmon (2009). The contemporary gene flow between
populations along the sympatric/allopatric selective gradient was modeled (Fig. 4) to determine
if this system exemplifies a case of increased genetic isolation in sympatric populations
potentially leading to parapatric speciation.
A series of migration models were created to be compared to determine if gene flow
reflects the predictions of Lemmon (2009). The Full Migration model (Fig. 4 A) estimates
independent migration rates in both directions between adjacent populations. This is a parameter
rich model that would explain a complex gene migration pattern. The Equivalent Migration
model (Fig. 4 B) restricts all migration rates, M, to be equivalent, representing no difference in
migration rate caused by reproductive character displacement. The Sympatric Isolation model
(Fig. 4 C) is based on the predictions of Lemmon (2009) that allopatric males dispersing into
sympatry will be less fit than local males. In this model migration from allopatry into sympatry
is not estimated as it is assumed to be a parameter that has little effect on the data and represents
over parameterization.
Methods
Pseudacris feriarum were collected from six populations spanning the region of
reproductive character displacement along the Apalachicola-Chattahoochee River in Florida,
Georgia, and Alabama (Fig. 3). Tissue samples were obtained from twenty individuals per
population using either toe-clipping or dissection (Table 2; Appendix A). Voucher specimens
from each site were preserved in formalin. Populations were sampled in two transects across the
region of reproductive character displacement: an Eastern transect composed of populations
from Russell and Houston Counties, Alabama, and Gulf County, Florida; and a Western transect
composed of populations from Muscogee and Early Counties, Georgia, and Liberty County,
Florida (Fig. 3).

21
Genomic DNA was extracted from 120 P. feriarum individuals using an E.Z.N.A. Gel
Extraction Kit (Omega Bio-Tek). Each individual was genotyped for a total of eleven
microsatellite loci; seven tetramer loci using published primer sets (Lemmon et al. 2011) and
primer sets developed using program μScope v0.0.6 (Appendix B) on previously generated 454
data (Lemmon & Lemmon unpublished; Appendix C), as well as for four dimer loci (Bedwell &
Lemmon unpublished; Ralicki & Lemmon unpublished). Details of primer development are
listed in Appendix C. Multiplex PCR reactions were set up using a Multiplex PCR Kit
(QIAGEN) and amplified using protocols described in Appendix D. Fragment analysis was
conducted on an Applied Biosystems 3730 Genetic Analyzer with Capillary Electrophoresis at
the Florida State University Core Research Facilities. Microsatellite fragment lengths were
measured using GeneMapper v4.1 (Applied Biosystems). Allele calling was done using tandem
v1.08 (Matschiner & Salzburger 2009). These allele calls were used to test for Hardy-Weinberg
equilibrium and linkage disequilibrium (Table 3) using Genepop v4.1 (Rousset 2008). Two loci
(D_C08b and A_E09d) exhibited linkage disequilibrium with the marker D_D12b, although not
with each other (Table 3). Lemmon et al. (2011) found no evidence of linkage disequilibrium
between D_D12b and D_C08b across 85 P. feriarum sampled from Macon County, AL. All
three markers were included in further analyses.
Fragment lengths were used to write a series of microsatellite data files for Migrate-n
v3.2.17 (Beerli 2009). Allele binning was performed by Migrate-n using the "#@M" command
in all data files. Migrate data files were written for the Eastern transect and Western transect to
test the effects of reproductive character displacement on gene migration (Fig. 3). Model
parameters were set in Migrate-n using the user-specified model options. All Migrate-n runs
were conducted using a continuous Brownian-motion model of microsatellite evolution
(Felsenstein 2004). Locality information (Table 4) was used to scale migration rate parameter
estimates using interpopulation geographic distance. A strait line distance matrix (geofile) was
imported into Migrate-n to scale by these interpopulation geographic distances.
A Uniform prior with a range of 0–400 was used for estimating mutation-scaled
migration rates, M, and a Uniform prior with a range of 0–100 was used for estimating mutation-
scaled population size, Θ. Parameter space for each Migrate-n model was searched with a burn-
in of 4,000,000 generations followed by 10,000 steps that were sampled at iterations of every
100 generations to reduce autocorrelation for a grand total of 50,000,000 parameter value

22
iterations explored per long chain. A Metropolis-coupled Markov chain Monte Carlo
(MCMCMC) approach (Geyer 1991) utilizing 4 adaptively heated chains was implemented to
better explore parameter space (Beerli 2006). Parameter values for each model were sampled
using 50 independent long chain samplings. Parameter value distributions were summed over
the fifty independent runs. The natural log marginal likelihood of each model was estimated
using Bézier quadrature (Beerli and Palczewski 2010), an alternative to the harmonic mean
which gives better estimates of the distribution when few heated chains are used. The natural log
marginal likelihood of each model was used to compute natural log Bayes factors (BF: Kass &
Raftery 1995) for model ranking and comparison.
Results
The comparison of gene flow across regions of reproductive character displacement
resulted in patterns that differed between the Eastern and Western transects. In the Eastern
transect the sympatric isolation model (Fig. 4 C) was favored (181 BF better than the second best
model; Table 9). In the Western transect a full migration model (Fig. 4 A) was favored (393 BF
better than the second best model; Table 9).
The individual parameter estimates for all models are listed in Table 10. Migration rate
(M) parameter estimates for migration into allopatry are the lowest of all M parameters for the
Western transect in the best supported model. The Sympatric Isolation model (Fig. 4 C) is
favored in the Eastern transect. In this transect the M parameter estimate for the Full Migration
model (Table 10) is lowest for migration from allopatry into sympatry which is consistent with
the idea that it is a nuisance parameter that is best left out of the model as in the Sympatric
Isolation model.
Discussion
This research evaluated the plausibility of the speciation cascade hypothesis and provides
some of the first genetic evidence supporting this model. In the Eastern transect the Sympatric
Isolation model (Fig. 4 C) was strongly favored over all other models (180.5 BF compared to the
second best model; Table 9). This model matches the expected outcome based on previous work
(Lemmon 2009) and suggests that female preferences may drive interpopulation gene migration.
Analysis of the Western transect, however, favors a Full Migration model (Fig. 4 A) over all

23
other models (392.9 BF compared to the second best model; Table 9), leading to the conclusion
that reproductive character displacement may not be affecting all populations in this region of
sympatry equivalently.
Reproductive character displacement is known to occur on the Western side of the
Apalachicola-Chattahoochee River (Fouquette 1975; Lemmon personal communication). The
female preference tests conducted by Lemmon (2009) used sympatric females from populations
on the Eastern side of the river where the best supported migration model matches the female
choice experiment expectations. Although the acoustic signal of P. feriarum is displaced on the
West side of the Apalachicola-Chattahoochee River, this may represent a case of parallel
evolution. Previous phylogenetic work (Lemmon & Lemmon 2008) provides some evidence of
independent invasions of the Coastal Plain on either side of this river. In this scenario, sympatry
with P. nigrita in the current study may actually represent two independently derived patterns of
reproductive character displacement. It is plausible that female choice experiment conducted
using P. feriarum from west of the Apalachicola-Chattahoochee River may yield differing results
from Lemmon (2009).
One of the problems that must be overcome in a speciation cascade model is the lack of
genetic isolation sympatric and allopatric regions. If a reproductive signal diverges in sympatry
into a less fit intraspecies trait space then isolation of peripheral populations is not likely to occur
due to dispersing allopatric individuals attaining reproductive success within the sympatric
region (e.g. Pfennig 2000). In P. feriarum the divergent signal is actually favored in allopatry
and sympatry over the allopatric signal (Lemmon 2009). This situation, represented by the
Sympatric Isolation model (Fig. 4 C), could generate the isolation necessary for speciation to
occur under a speciation cascade model (Howard 1993). Current evidence for species cascade
models largely consist of descriptions of reproductive character displacement (Howard 1993;
Pfennig & Ryan 2006). This current study provides the first evidence of documented
reproductive character displacement leading to genetic isolation due to female preferences,
which has the potential to generate parapatric speciation under a cascading speciation model.
This research provides a first step in determining the nature of parapatric speciation
within Pseudacris feriarum. This system provides the opportunity to test speciation cascade
theory in multiple independent regions of secondary contact between P. feriarum and P. nigrita
where reproductive character displacement has been observed (Lemmon 2009). Female choice

24
experiments can be used to generate models for gene flow at each of these contact zones to
evaluate a broader pattern of preference effects. This study has laid the groundwork for future
studies that can incorporate multiple contact zones and evaluate them with similar methods
resulting in replicated studies of the effects of reproductive character displacement on
contemporary gene flow.

25
Table 1. A comparison of riverine barrier hypotheses.
Hypothesis Wallace's Riverine
Barrier Hypothesis
Genetic Riverine
Barrier Hypothesis
Primary
Diversification
Riverine Barrier
Hypothesis
Abbreviation WRBH GRBH PDRBH
Process
Rivers impede species
range expansion.
Rivers impede
dispersal by
individuals.
Rivers impede
dispersal by
individuals.
Rivers initiate
allopatric
diversification.
Expected
Pattern
Species ranges are
bordered by rivers.
Communities on
opposite banks become
more dissimilar as a
river becomes wider.
Genetic divergence
between cross-bank
populations within a
species is positively
correlated with river
width.
Genetic divergence
between cross-bank
populations within a
species is positively
correlated with river
width.
Populations do not
show evidence of
recent expansion into
regions around a river
(secondary contact).

26
Table 2. Collection data for Pseudacris feriarum tissues.
Specimen
Number Collection Date Collection Locality Latitude Longitude
ECM0936 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0937 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0938 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0939 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0940 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0941 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0942 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0944 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0945 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0946 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0947 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0948 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0949 3-1-2004 Macon, AL 32.44951 -85.65438
ECM0977 3-3-2004 Macon, AL 32.44951 -85.65438
ECM0978 3-3-2004 Macon, AL 32.44951 -85.65438
ECM0979 3-3-2004 Macon, AL 32.44951 -85.65438
ECM0980 3-3-2004 Macon, AL 32.44951 -85.65438
ECM0981 3-3-2004 Macon, AL 32.44951 -85.65438
ECM0982 3-3-2004 Macon, AL 32.44951 -85.65438
ECM1244 2-3-2005 Macon, AL 32.44411 -85.65521
MJM00603 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00604 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00605 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00606 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00607 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00608 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00609 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00610 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00611 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00612 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00613 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00614 12-2-2009 Liberty, FL 30.21116 -85.06274
MJM00633 12-3-2009 Liberty, FL 30.19702 -85.07587
MJM00634 12-3-2009 Liberty, FL 30.19702 -85.07587
MJM00635 12-3-2009 Liberty, FL 30.19702 -85.07587

27
Table 2 (continued).
Specimen
Number
Collection
Date Collection Locality Latitude Longitude
MJM00636 12-3-2009 Liberty, FL 30.19702 -85.07587
MJM00637 12-3-2009 Liberty, FL 30.19702 -85.07587
MJM00638 12-3-2009 Liberty, FL 30.19702 -85.07587
MJM00647 12-3-2009 Liberty, FL 30.19702 -85.07587
MJM00648 12-3-2009 Liberty, FL 30.19702 -85.07587
MJM00685 1-16-2010 Gulf, FL 29.98787 -85.11545
MJM00689 1-16-2010 Gulf, FL 29.98253 -85.10759
MJM00692 1-16-2010 Gulf, FL 29.97876 -85.10328
MJM00693 1-16-2010 Gulf, FL 29.96974 -85.09278
MJM00694 1-16-2010 Gulf, FL 29.97025 -85.08444
MJM00697 1-16-2010 Gulf, FL 29.97023 -85.09402
MJM00745 1-21-2010 Miller, GA 31.1272 -84.69959
MJM00746 1-21-2010 Miller, GA 31.1272 -84.69959
MJM00747 1-21-2010 Miller, GA 31.1272 -84.69959
MJM00748 1-21-2010 Miller, GA 31.1272 -84.69959
MJM00749 1-21-2010 Miller, GA 31.1272 -84.69959
MJM00750 1-21-2010 Miller, GA 31.1272 -84.69959
MJM00751 1-21-2010 Miller, GA 31.13693 -84.70488
MJM00752 1-21-2010 Miller, GA 31.13693 -84.70488
MJM00756 1-21-2010 Miller, GA 31.1689 -84.75686
MJM00757 1-21-2010 Miller, GA 31.1689 -84.75686
MJM00758 1-21-2010 Miller, GA 31.1689 -84.75686
MJM00761 1-21-2010 Miller, GA 31.16536 -84.76899
MJM00776 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00777 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00778 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00779 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00780 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00781 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00782 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00783 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00784 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00785 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00786 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00787 1-22-2010 Houston, AL 31.06472 -85.04583

28
Table 2 (continued).
Specimen
Number
Collection
Date Collection Locality Latitude Longitude
MJM00788 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00789 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00790 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00791 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00792 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00793 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00794 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00795 1-22-2010 Houston, AL 31.06472 -85.04583
MJM00981 1-1-2011 Gulf, FL 30.034 -85.17026
MJM00983 1-1-2011 Gulf, FL 30.02963 -85.16807
MJM00985 1-1-2011 Gulf, FL 30.00083 -85.14157
MJM00986 1-1-2011 Gulf, FL 30.00083 -85.14157
MJM00987 1-1-2011 Gulf, FL 30.00083 -85.14157
MJM00988 1-1-2011 Gulf, FL 29.99604 -85.12428
MJM00989 1-1-2011 Gulf, FL 29.99604 -85.12428
MJM00994 1-1-2011 Gulf, FL 29.9969 -85.12465
MJM00995 1-1-2011 Gulf, FL 29.9969 -85.12465
MJM00998 1-1-2011 Gulf, FL 30.00093 -85.13302
MJM01000 1-1-2011 Gulf, FL 30.00087 -85.13306
MJM01001 1-1-2011 Gulf, FL 30.00087 -85.13306
MJM01002 1-1-2011 Gulf, FL 30.00087 -85.13306
MJM01006 1-1-2011 Gulf, FL 29.96949 -85.09112
MJM01017 1-31-2011 Miller, GA 31.17422 -84.74257
MJM01018 1-31-2011 Miller, GA 31.17422 -84.74257
MJM01019 1-31-2011 Miller, GA 31.17422 -84.74257
MJM01020 1-31-2011 Miller, GA 31.17422 -84.74257
MJM01021 1-31-2011 Miller, GA 31.17422 -84.74257
MJM01022 1-31-2011 Miller, GA 31.17422 -84.74257
MJM01023 1-31-2011 Miller, GA 31.17422 -84.74257
MJM01024 1-31-2011 Miller, GA 31.17422 -84.74257
MJM01028 3-1-2011 Early, GA 31.36524 -84.94428
MJM01029 3-1-2011 Early, GA 31.36524 -84.94428
MJM01030 3-1-2011 Early, GA 31.36524 -84.94428
MJM01031 3-1-2011 Early, GA 31.36524 -84.94428
MJM01032 3-1-2011 Early, GA 31.36524 -84.94428

29
Table 2 (continued).
Specimen
Number
Collection
Date Collection Locality Latitude Longitude
MJM01033 3-1-2011 Early, GA 31.36524 -84.94428
MJM01034 3-1-2011 Early, GA 31.36524 -84.94428
MJM01035 3-1-2011 Early, GA 31.36524 -84.94428
MJM01036 3-1-2011 Early, GA 31.36524 -84.94428
MJM01037 3-1-2011 Early, GA 31.36524 -84.94428
MJM01038 3-1-2011 Early, GA 31.36524 -84.94428
MJM01039 3-1-2011 Early, GA 31.36524 -84.94428
MJM01040 3-1-2011 Early, GA 31.36524 -84.94428
MJM01041 3-1-2011 Early, GA 31.36524 -84.94428
MJM01042 3-1-2011 Early, GA 31.36524 -84.94428
MJM01043 3-1-2011 Early, GA 31.36524 -84.94428
MJM01044 3-1-2011 Early, GA 31.36524 -84.94428
MJM01045 3-1-2011 Early, GA 31.36524 -84.94428
MJM01046 3-1-2011 Early, GA 31.36524 -84.94428
MJM01047 3-1-2011 Early, GA 31.36524 -84.94428
MJM01067 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01068 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01069 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01070 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01071 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01072 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01073 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01074 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01075 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01076 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01077 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01078 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01079 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01080 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01081 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01082 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01083 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01084 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01085 3-6-2011 Muscogee, GA 32.5527 -84.87294
MJM01086 3-6-2011 Muscogee, GA 32.5527 -84.87294

30
Table 2 (continued).
Specimen
Number
Collection
Date Collection Locality Latitude Longitude
MJM01090 3-9-2011 Russell, AL 32.45618 -85.19571
MJM01091 3-9-2011 Russell, AL 32.45618 -85.19571
MJM01092 3-9-2011 Russell, AL 32.45618 -85.19571
MJM01093 3-9-2011 Russell, AL 32.45618 -85.19571
MJM01094 3-9-2011 Russell, AL 32.45618 -85.19571
MJM01095 3-9-2011 Russell, AL 32.45618 -85.19571
MJM01096 3-9-2011 Russell, AL 32.45602 -85.19704
MJM01097 3-9-2011 Russell, AL 32.45602 -85.19704
MJM01098 3-9-2011 Russell, AL 32.45602 -85.19704
MJM01099 3-9-2011 Russell, AL 32.45602 -85.19704
MJM01100 3-9-2011 Russell, AL 32.45602 -85.19704
MJM01101 3-9-2011 Russell, AL 32.45602 -85.19704
MJM01102 3-9-2011 Russell, AL 32.45602 -85.19704
MJM01103 3-9-2011 Russell, AL 32.45602 -85.19704
MJM01104 3-10-2011 Russell, AL 32.45602 -85.19704
MJM01105 3-10-2011 Russell, AL 32.45602 -85.19704
MJM01106 3-10-2011 Russell, AL 32.45602 -85.19704
MJM01107 3-10-2011 Russell, AL 32.45602 -85.19704
MJM01108 3-10-2011 Russell, AL 32.45602 -85.19704
MJM01109 3-10-2011 Russell, AL 32.45602 -85.19704

31
Table 3. Results of Hardy-Weinberg exact tests (p-values) and linkage disequilibrium
probability tests (p-values) conducted in Genepop v4.1. Loci that significantly deviated from
Hardy-Weinberg equilibrium across all sampled populations and loci that exhibited significant
linkage disequilibrium are highlighted in blue.
D6 0.0000
D_D12b 0.0000 0.857
D_C08b 0.0413 0.691 0.004
D_D10 0.208 0.987 0.840 0.972
P_fer_57550 0.866 0.752 0.959 0.274 0.491
P_fer_46999 0.0000 0.131 0.996 0.998 0.213 0.987
P_fer_101070 0.315 0.986 0.859 0.997 0.389 0.181 0.937
A_C08d 0.0000 0.766 0.999 0.991 0.982 0.641 0.995 1.000
A_E09d 0.0014 0.291 0.002 0.500 0.935 0.989 0.995 1.000 1.000
A_B04d 0.631 0.066 0.747 0.724 1.000 0.588 0.716 0.959 0.912 0.555
P_fer_DBNXL 0.266 0.221 0.812 0.957 0.144 0.686 0.994 0.993 0.800 1.000 0.213
HW
P V
alu
e
D6
D_D
12b
D_C
08b
D_D
10
P_fe
r_57550
P_fe
r_46999
P_fe
r_101070
A_C
08d
A_E
09d
A_B
04d
Linkage Disequilibrium
Table 4. Distance matrix of all inter-population distances used for creating geofiles. All
distances are recorded in km.
Russell 42.23
Muscogee 74.50 32.16
Houston 163.48 154.94 165.80
Early 137.23 123.28 131.85 34.70
Miller 165.21 148.48 153.35 31.37 28.59
Gulf 279.45 175.80 287.10 121.42 155.32 137.67
Liberty 255.17 250.74 261.90 96.24 130.13 112.95 25.20
Macon Russell Muscogee Houston Early Miller Gulf

32
Table 5. Bezier approximated marginal likelihoods (ln mL) of three models testing the effect of
a river on gene migration. Model rankings are indicated, with the best supported model
highlighted in each transect. Bayes factors are computed for each model compared to the best
model in each transect. The Northern transect consists of populations collected in Macon Co.,
AL, Russell Co., AL, and Muscogee Co., GA; the Southern transect consists of populations
collected in Houston Co., AL, Early Co., GA, and Miller Co., GA
Northern Transect Southern Transect
Model ln mL Rank BF ln mL Rank BF
Full Linear Migration -13814.8 1 -15481.3 1
Equivalent Linear Migration -24156.9 3 10342.2 -29073.0 3 13591.7
Linear River Effect -17142.5 2 3327.7 -19145.3 2 3663.9
Table 6. Parameter estimates for models of cross-river gene flow. The mode of the probability
distribution for each parameter is reported. The model with the highest support (Table 5) is
highlighted in blue.
Parameter
Full
Linear
Migration
Equivalent
Linear
Migration
Linear
River
Effect
Nort
her
n T
ran
sect
ΘMacon 98.100 13.966 96.233
ΘRussell 4.233 7.833 3.566
ΘMuscogee 2.833 4.033 2.366
MMacon → Russell 328.133 392.667 334.530
MRussell → Macon 71.867 392.667 28.933
MRussell → Muscogee 111.867 392.667 392.667
MMuscogee → Russell 39.333 392.667 392.667
Sou
ther
n T
ran
sect
ΘMiller 8.7 4.366 8.966
ΘEarly 1.966 2.766 1.833
ΘHouston 11.566 7.833 6.7
MMiller → Early 203.6 383.867 244.4
MEarly → Miller 78.8 383.867 54
MEarly → Houston 63.867 383.867 392.667
MHouston → Early 135.867 383.867 392.667
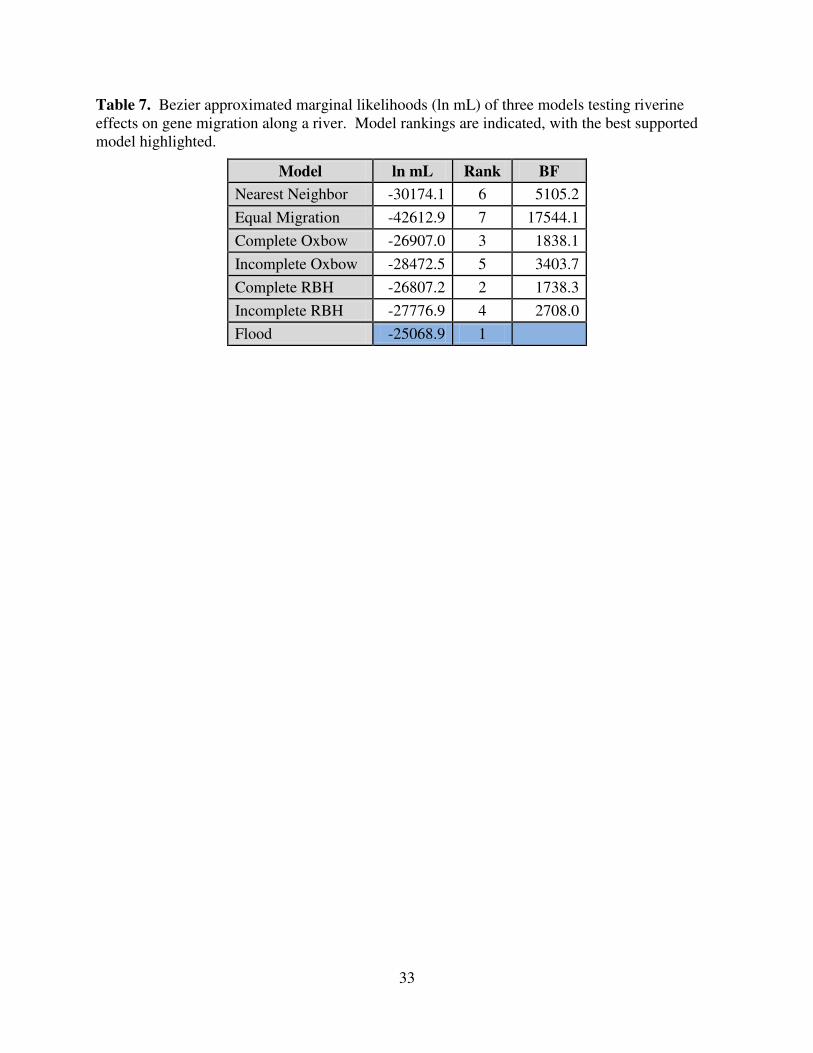
33
Table 7. Bezier approximated marginal likelihoods (ln mL) of three models testing riverine
effects on gene migration along a river. Model rankings are indicated, with the best supported
model highlighted.
Model ln mL Rank BF
Nearest Neighbor -30174.1 6 5105.2
Equal Migration -42612.9 7 17544.1
Complete Oxbow -26907.0 3 1838.1
Incomplete Oxbow -28472.5 5 3403.7
Complete RBH -26807.2 2 1738.3
Incomplete RBH -27776.9 4 2708.0
Flood -25068.9 1

34
Table 8. Parameter estimates for models of riverine gene flow. The mode of the probability
distribution for each parameter is reported. The model with the highest support (Table 7) is
highlighted in blue.
Parameter
Nearest
Neighbor
Equal
Migration
Complete
Oxbow
Incomplete
Oxbow
Complete
RBH
Incomplete
RBH Flood
ΘRussell 17.433 6.033 10.660 20.766 17.433 13.900 14.833
ΘHouston 4.566 4.633 7.433 3.366 8.233 5.300 5.233
ΘGulf 5.500 5.900 3.900 5.500 8.233 10.433 7.100
ΘMuscogee 2.166 2.966 4.833 7.033 1.833 2.100 5.366
ΘEarly 1.966 1.966 4.033 1.766 4.100 1.966 2.633
ΘLiberty 6.700 5.433 3.966 6.033 13.900 15.700 10.100
MRussell → Houston 8.933 0.133 66.800 14.800 386.800 0.133 0.133
MHouston → Russell 17.733 48.933 74.267 99.600 24.933 59.867 45.200
MHouston → Gulf 387.867 62.533 258.267 386.000 392.133 392.133 392.133
MGulf → Houston 379.067 387.333 390.267 190.800 48.133 94.533 214.267
MMuscogee → Early 37.200 20.933 0.133 13.200 158.800 31.867 0.133
MEarly → Muscogee 11.333 6.267 81.200 95.600 23.333 4.933 50.000
MEarly → Liberty 389.467 380.933 310.000 378.267 392.133 389.200 62.267
MLiberty → Early 158.000 382.533 387.867 231.333 18.800 91.333 96.133
MRussell → Muscogee 11.600 392.667 49.467 11.867
MMuscogee → Russell 0.133 392.667 49.733 0.133
MHouston → Early 0.133 392.667 7.330 16.400
MEarly → Houston 0.133 392.667 7.867 380.400
MGulf → Liberty 9.733 392.667 381.733 45.200
MLiberty → Gulf 0.133 392.667 13.467 76.933
MRussell → Early 227.333
MMuscogee → Houston 34.800
MHouston → Liberty 392.133
MEarly → Gulf 383.600

35
Table 9. Bezier approximated marginal likelihood (ln mL) results for tests of gene flow across a
region of reproductive character displacement. The best supported model is highlighted in each
model set.
Western Transect Eastern Transect
Model ln mL Rank BF ln mL Rank BF
Full Migration -10668.9 1 -11044.7 2 180.5
Equivalent Migration -12056.4 3 1387.5 -12487.3 3 1623.1
Sympatric Isolation -11061.8 2 392.9 -10864.2 1
Table 10. Parameter estimates for models of gene flow across a region of reproductive character
displacement. The mode of the probability distribution for each parameter is reported. The
model with the highest support (Table 9) is highlighted in blue.
Parameter
Full
Migration
Equivalent
Migration
Sympatric
Isolation
Wes
tern
Tra
nse
ct
ΘRussell 13.167 12.500 10.700
ΘHouston 7.100 8.767 13.766
ΘGulf 9.967 8.233 9.166
MRussell → Houston 384.667 393.200
MHouston → Russell 56.133 393.200 248.933
MHouston → Gulf 392.400 393.200 385.733
MGulf → Houston 383.867 393.200 317.733
East
ern
Tra
nse
ct
ΘMuscogee 6.966 5.500 5.766
ΘEarly 3.567 5.766 4.566
ΘLiberty 22.900 18.366 28.966
MMuscogee → Early 15.867 392.667
MEarly → Muscogee 51.600 392.667 131.330
MEarly → Liberty 392.400 392.667 75.067
MLiberty → Early 80.400 392.667 211.067

36
Table 11. Microsatellite primer sequences developed from 454 reads (M-13 tag not included). R
ever
se P
rim
er S
equ
ence
AA
AA
CC
AG
AA
AC
CC
CA
TA
GT
AA
AT
C
AA
TG
TT
GT
TC
TA
AA
TA
GA
CG
TG
AT
GA
A
AG
TC
AC
TT
TC
AT
GT
TT
CT
CT
TG
TT
CA
T
TA
TT
AA
GT
GA
CC
AC
TG
AT
CC
TA
CC
TC
TT
AG
TC
TA
GT
TT
CC
CT
CA
GG
TT
AA
AT
AT
C
GC
TG
AC
CA
GT
AT
AC
TT
TG
TT
TA
AT
GC
TT
AT
CT
GA
GT
AG
TA
AA
TG
GA
AA
TG
TT
AA
GG
AA
GT
GG
TA
TT
TC
AA
AA
AC
AG
AC
CT
AT
C
GT
TA
GG
CT
CA
TT
CT
CA
CT
TA
CA
AT
TT
C
TG
TT
AG
TA
AC
TA
TT
TA
TT
AT
GT
GC
GA
CA
GT
AT
AA
TT
TT
CA
GC
AC
TC
TT
CT
AT
GA
CA
TC
AA
AG
TA
AT
AA
AA
AT
TT
AG
CT
CA
TG
GA
TT
CT
AA
AA
TT
TG
TG
TT
AT
GG
AT
AT
AG
AG
CA
TC
GT
AC
AT
TT
CT
TC
TA
AT
CC
TT
TT
TG
GA
A
TT
AA
TG
TT
GT
AT
CA
CA
CG
TT
CA
TA
AT
AA
AG
TT
TT
GT
CT
TC
TT
TT
TG
AA
CT
CT
GC
AG
TT
TA
CT
CA
TT
TT
GT
TC
CT
TG
GT
CT
TA
AA
TA
CT
AA
AG
GT
GG
GG
AA
TA
TC
AA
GT
AA
CT
TG
AT
TT
CA
GC
AT
CT
TG
TT
TC
T
AG
AC
TT
TG
CA
GA
TT
CA
AA
AG
TC
AT
T
GT
GT
AA
GG
TT
AA
AA
AC
TA
TA
AG
GT
GA
CA
AA
Forw
ard
Pri
mer
Seq
uen
ce
TA
TT
GC
TC
TT
CA
TT
AA
AA
AT
TT
CT
TT
CT
C
CT
TG
TC
AT
GT
AA
TT
CT
CA
TC
TT
CT
TA
AT
G
AT
TA
CT
TC
TG
AC
AA
AT
AA
CG
TA
AT
GC
TA
AG
GA
TC
TC
AG
AA
CC
AG
AT
CA
TA
CT
CA
TT
AC
CA
TA
AT
TA
TT
AG
AC
CT
GA
AC
AG
AT
TC
TC
AC
CA
GT
GT
AA
AG
CC
TT
TT
AT
TT
GA
A
AA
TT
TA
TT
AA
GT
GC
TC
AA
TG
CC
AG
T
AA
AA
TT
GA
CA
CA
AA
AA
TG
TC
TA
TA
GG
G
AA
CC
AG
AT
AT
TT
TG
CA
TT
GC
TA
TC
AC
TG
GA
AG
GC
GG
TG
TC
TG
GA
AT
AG
TA
AA
GC
AG
CA
AG
TA
TT
GA
CC
TA
AG
TA
CT
GA
GT
AT
TT
AG
TG
CA
AT
TT
CC
AA
AT
TT
GT
GG
TT
TA
GA
CA
GC
AT
GA
AA
TT
AT
TC
GT
AA
AG
AA
CT
AC
TG
GT
AG
AG
AC
AC
CC
AA
AC
AC
AT
TT
TC
TT
AG
CA
TT
GT
CA
TC
TT
CA
TG
GT
AT
AT
TT
TG
TG
AT
GT
T
GA
GA
AT
TA
CT
GA
CT
CG
CT
AA
GG
GT
A
TA
AT
CA
GT
GT
TC
AT
CT
CC
TG
AT
AA
TT
G
CA
AA
CG
AC
AA
AA
AT
AA
AA
TA
TG
TC
AC
TA
AT
AC
TG
GA
AA
GA
AG
AA
TG
CA
AA
GA
T
GA
CT
AT
GA
CA
GA
AT
AT
TA
AC
CT
AA
TG
GA
AA
Moti
f
AA
AT
AC
AT
AA
AG
AG
AT
AC
GG
AA
AT
AA
GT
AG
AT
AC
GG
AC
AT
AG
AT
AA
AT
AA
AT
AA
AT
AA
AT
AA
AT
AA
GT
AG
GG
AA
AC
AC
CG
AA
AT
Locu
s N
am
e
P_fe
r_64
64
P_fe
r_92
51
P_fe
r_26
421
P_fe
r_32
589
P_fe
r_34
263
P_fe
r_44
482
P_fe
r_46
724
P_fe
r_46
999
P_fe
r_52
033
P_fe
r_52
281
P_fe
r_57
550
P_fe
r_64
901
P_fe
r_69
274
P_fe
r_70
269
P_fe
r_73
827
P_fe
r_83
490
P_fe
r_10
0489
P_fe
r_10
1070
P_fe
r_10
5253
P_fe
r_11
3902
P_fe
r_11
6964

37
Table 12. Results of microsatellite primer amplification in a panel of Pseudacris feriarum
selected from across the range of the species. A 'Y' indicates that a sample amplified with at
least one detectable allele, while an 'N' indicates that no alleles amplified in that sample. The
total observed alleles (NA) within the 9 individuals genotyped is recorded. The three loci
accepted for use in this thesis are highlighted in blue.
Locus Name 454 R
epea
t N
um
ber
EC
M388 M
aco
n, A
L
EC
M7041 H
eard
, G
A
EC
M389 M
aco
n, A
L
EC
M7
042 H
eard
, G
A
EC
M7143 P
rin
ce E
dw
ard
, V
A
MJM
00603 L
iber
ty, F
L
MJM
00782 H
ou
ston
, A
L
MJM
00981 G
ulf
, F
L
MJM
01017 M
ille
r, G
A
NA
P_fer_6464 4 N N
P_fer_9251 4 N N
P_fer_26421 4 N N
P_fer_32589 14 Y N
P_fer_34263 5 N N
P_fer_44482 5 Y Y Y Y Y Y Y Y Y 3
P_fer_46724 4 Y Y Y Y Y Y Y Y Y 1
P_fer_46999 10 Y Y Y Y Y Y N Y Y 9
P_fer_52033 4 N N
P_fer_52281 4 N N
P_fer_57550 4 Y Y N Y Y Y N Y Y 4
P_fer_64901 5 N N
P_fer_69274 4 N N
P_fer_70269 4 Y Y Y N Y Y Y N N 2
P_fer_73827 4 N N
P_fer_83490 4 Y Y N N N N N N N
P_fer_100489 4 Y N N N N N N N N
P_fer_101070 4 Y Y Y N Y Y Y Y Y 11
P_fer_105253 5 N N
P_fer_113902 4 N Y N N N N N N N
P_fer_116964 6 N N

38
Table 13. Primer sequences for sixteen microsatellite loci in Pseudacris feriarum. Reverse
primer sequences are highlighted in blue.
Locus Name X-mer Primer Sequence
D6 Tetramer CTGCTGTGATATTTTTGTG
GGTGTCGTGAGCTAAGTGTT
D_D12b Tetramer TATAACATGTAACTGGGCTAACA
AGGAGAAGAGCCATTTCCTG
D_C08b Tetramer CTTACACAGCTCCATAGAATATGACA
ACAAACCTACAGGAGCTGATAGAAT
D_D10 Tetramer CTCTACATACATTTACCTTCTACCTTC
GCTGTCTACTGAATTTATACTGTAAGG
P_fer_57550 Tetramer GAATAGTAAAGCAGCAAGTATTGACCTA
ATAATTTTCAGCACTCTTCTATGACATC
P_fer_46999 Tetramer AAAATTGACACAAAAATGTCTATAGGG
AAGTGGTATTTCAAAAACAGACCTATC
P_fer_101070 Tetramer TAATCAGTGTTCATCTCCTGATAATTG
TAAATACTAAAGGTGGGGAATATCAAGT
A_C08d Dimer GGTAATAAACCAATCTTAAATTAGTAACACAA
CTCATGGATACTACAACTCTCGACAGTAAC
A_E09d Dimer ATTACATGTAGCTTTTAAGGATATATTGTTGC
TAAATGCTTTATCTCTGTTACATCTGTATAGG
P_fer_G79VC Dimer ACCAGGACTAGCTATATAAGAACATAGACTTT
CCGATACCTCTCTTGCATGTGT
A_B04d Dimer AGTGACTTCCAATATCCTATAGTCCTCTT
ATGGACTGTAGCAGTTACCATGTGTAGT
P_fer_DJURT Dimer AGAAGTATTCTTGCACCCTAGGAAT
GACACATTATTTCATTCTGTAACTAAAAAGTA
P_fer_A7NK3_2 Dimer GAATATTTTAGTTTTCCCTTTTCAATAAATC
TGTGATAAATACGGTATATGTATATGTGTCTG
P_fer_A3PXV Dimer TAATAAACAAAACACCTTTGTAGGTGAAAATA
ATCTGATTTAGTAGTCAGTTGTGCAGTAAAAG
P_fer_CIMKE Dimer CCGTGTTGTGTATTAACTATAAGGAAAAA
ACTAGTATCACCACACAAAACAAACAA
P_fer_DBNXL Dimer ATCAATAGAGGAGGTGGGAGCTG
ACATATAGAGCAATAACTGAGCTGTGAAT

39
Table 14. Multiplex composition used to genotype individuals for sixteen loci. Five loci
amplified poorly, inconsistently, or exhibited primer nonspecificity; these loci are highlighted in
blue and were not included in data analysis. The total observed alleles (NA) within the 160
individuals genotyped for this project, the observed shortest and longest fragment lengths, and
the fluorophore used are recorded. Recorded fragment lengths include the primers and use the
fluorophores indicated.
Mu
ltip
lex
Pri
mer
Set
Na
me
Dev
elop
er
NA
Sh
ort
est
Fra
gm
ent
Len
gth
L
on
ges
t F
ragm
ent
Len
gth
Flu
oro
ph
ore
1
D6 Lemmon et al. (2011) 25 100 372 NED
D_D12b Lemmon et al. (2011) 28 268 532 FAM
D_C08b Lemmon et al. (2011) 16 227 445 HEX
2
D_D10 Lemmon et al. (2011) 6 152 398 NED
P_fer_57550 Michelsohn (Appendix C) 7 248 267 FAM
P_fer_46999 Michelsohn (Appendix C) 23 184 270 PET
P_fer_101070 Michelsohn (Appendix C) 10 191 237 VIC
3
A_C08d Bedwell & Lemmon (unpublished) 19 300 300 NED
A_E09d Bedwell & Lemmon (unpublished) 27 216 216 FAM
P_fer_G79VC Ralicki & Lemmon (unpublished) N/A 135 168 PET
A_B04d Bedwell & Lemmon (unpublished) 24 138 138 VIC
4
P_fer_DJURT Ralicki & Lemmon (unpublished) N/A 190 202 NED
P_fer_A7NK3_2 Ralicki & Lemmon (unpublished) N/A 237 260 FAM
P_fer_A3PXV Ralicki & Lemmon (unpublished) N/A 107 165 PET
P_fer_CIMKE Ralicki & Lemmon (unpublished) N/A 155 180 VIC
N/A P_fer_DBNXL Ralicki & Lemmon (unpublished) 18 152 186 NED

40
Figure 1. Illustrations of gene flow models used to evaluate the effect of a river on gene
migration compared to overland migration. The vertical blue bar represents the Apalachicola-
Chattahoochee River; solid black arrows represent independent M parameters; dashed red arrows
represent M parameters that are restricted to be equivalent estimates within a model (1 M
parameter for all equivalent rates); circles represent independent Θ parameters. These models
were tested in both a Northern (1- Macon, AL; 2- Russell, AL; 3- Muscogee, GA) and a
Southern transect (1- Miller, GA; 2- Early, GA; 3- Houston, AL). The total number of
parameters simulated in each model is noted in parentheses after the model name.
Figure 2 (next page). Illustrations of gene flow models used to evaluate the effects of rivers on
population genetics. The vertical blue arrow represents the Apalachicola-Chattahoochee River
and its direction of flow; solid black arrows represent independent M parameters; dashed red
arrows represent M parameters that are restricted to be equivalent estimates within a model (1 M
parameter for all equivalent rates); circles represent Θ parameters with subscripts denoting the
counties where populations were sampled (R- Russell, AL; H- Houston, AL; G- Gulf, FL; M-
Muscogee, GA; E- Early, GA; L- Liberty, FL). The total number of parameters simulated in
each model is noted in parentheses after the model name.

41
A. Complete RBH (16) B. Incomplete RBH (18) C. Complete Oxbow ( 16)
@ @ @ @ @ @
ll ll ll ll ll ll
@ @ @ @ @ @
ll ll ll ll ll ll
@ @ @ @ @ @
D. Incomplete Oxbow (18) E. Flood (18) F. Equal Migration (15)
@ @ @ @ ` セZ@ ]セ `@
ll ll ᆪ Q セ@ ll ll
@ @ ` セZ@ Zセ Z `@
ll ll ll ll ll ll
@ @ @ @ ` セZ@ ]セ `@
G. Nearest Neighbor (20)

42
Figure 3. Map depicting sampling localities used in this study. The Apalachicola-
Chattahoochee River is indicated by the blue line; the dashed white line represents the
approximate region of the reproductive character displacement cline. County names are
indicated on the sampling localities in the inset map.

43
Figure 4. Illustrations of gene flow models used to evaluate the effect of reproductive character
displacement (RCD) on gene migration. The vertical green bar represents the region of RCD;
solid black arrows represent independent M parameters; dashed red arrows represent M
parameters that are restricted to be equivalent estimates within a model (1 M parameter for all
equivalent rates); circles represent independent Θ parameters for sympatric populations of P.
feriarum; squares represent independent Θ parameters for allopatric populations of P. feriarum.
These models were tested in both an Eastern (1- Liberty, FL; 2- Early, GA; 3- Muscogee, GA)
and a Western transect (1- Gulf, FL; 2- Houston, AL; 3- Russell, AL). The total number of
parameters simulated in each model is noted in parentheses after the model name.

44
APPENDIX A
SPECIMEN COLLECTION INFORMATION
All specimens used in this work (Table 2) were collected under the following permits
issued to E. M. Lemmon or M. J. Michelsohn:
Florida: LSSC-09-0362
Georgia: 22378
Alabama: 2010000037468680
All live animals were collected, tissue sampled, and preserved following protocols
established in the Animal Care and Use Committee (ACUC) protocol 0905 for the E. M.
Lemmon lab at Florida State University.

45

46

47
APPENDIX B
PROGRAM μSCOPE
The program μScope is a python script that was developed in this research to search for
candidate microsatellite loci using 454 sequence reads. This program detects and classifies
sequence reads that contain microsatellites. These sequence reads are then output for use in
primer design. Individual microsatellites are classified by motif length as x-mers where x
represents the motif length (e.g.. a tetramer is represented as 4-mer).
The program requires five user selected input values to run, as well as at least one data
file in FASTA format. These input values include:
threshold_value - This input value is used to determine the number of times an individual
x-mer sequence must be repeated to be detected as a microsatellite. The program
uses the following inequality to determine if a repeated sequence is considered to
occur more often than expected by chance.
In this inequality x is the number of nucleotides in an x-mer motif, and j is an
integer value specifying a number of repeats. The integer value of j is increased
sequentially until the inequality is true. The smallest integer value of j for which
the inequality is true is set as the significance_value for all x-mers of length x.
All x-mer sequences repeated equal to or more times than the significance_value
for that x-mer are considered to be microsatellites.
max_motif_length - This input value is used to determine the largest x-mer length that
will be detected as a microsatellite by the program. In μScope v0.0.6 this input is
limited to integer values in the range 1–6.
file_extension - This input value allows the user to specify which data files will be used in
the search. All data files in the current working directory with the extension
specified will be read into the program. These data files are required to be in
FASTA format but can have any extension specified.
min_intemotif_length - This input value sets the nucleotide distance between any detected
microsatellites within a sequence read that is considered to be interacting.

48
minimum_repeats - This input value determines the minimum number of x-mer repeats
within a microsatellite region required for the containing sequence read to be
included within an output file of this program.
The program classifies detected microsatellites based on their proximity to other detected
microsatellites (specified using the input min_intemotif_length). The microsatellite regions
within a read are classified as one of four types:
Perfect - Microsatellites that contain a single motif x-mer separated from all other
microsatellites by a distance of min_intermotif_length or greater.
Imperfect Monomer - Microsatellite regions that contain a single motif x-mer but also
include non-repetitive insertions.
Imperfect Multimer - Microsatellite regions that contain multiple motifs of the same x-
mer. These may also include non-repetitive insertions.
Compound - Microsatellite regions that contain multiple motifs, some of which do not
have the same x-mer. These regions may also include non-repetitive insertions.
After searching and sorting all microsatellite regions from all the imported data, the sequence
reads containing the microsatellite regions that match the user inputs are output in a series of
FASTA formatted files in the current working directory. An output file is generated for each
microsatellite type in each x-mer.
Run times vary depending on the number and length of imported sequence reads, number
of microsatellites contained within each sequence read, as well as user input values. While the
program is running a rough estimate search completeness is displayed. When a search is
completed, summary values are displayed that list the numbers of sequence reads containing
microsatellite regions of each type and x-mer.

49
APPENDIX C
454-GENERATED PRIMER DEVELOPMENT
The program μScope (Appendix B) was used to search through 130,222 previously
generated 454 sequence reads (Lemmon & Lemmon unpublished). The search used a
threshold_value of 0.5 which requires dimers to be repeated twice (e.g.. ACACAC) and all other
x-mers to be repeated only once before they are counted as microsatellites. The search used a
max_motif_length of 6 and a min_intemotif_length of 50. Fifty-five reads containing at least four
consecutive repeats (minimum_repeats = 4) of a perfect tetramer microsatellite were imported
into BioEdit v7.0.9.0 (Hall 1999). The NCBI BLAST v2.0 (Altschul et al. 1997) plug-in was
used to identify overlapping reads. All overlapping reads were assembled into aligned contigs
by using the NCBI BLAST results. Thirty-three non-overlapping reads and aligned contigs were
imported into Primer3 v0.4.0 (Rozen & Skaletsky 2000) for primer design. Designed primers
were checked by eye to avoid placement in regions that contained homopolymers due to the risk
of 454 sequencing error (Margulies et al. 2005).
M-13 tagged primer sets (Schuelke 2000) were designed for twenty-one loci (Table 11).
These primer sets were first tested using gDNA samples from two P. feriarum, ECM388 and
ECM7041 (Table 12). Genomic DNA was extracted from tissue samples using an E.Z.N.A. Gel
Extraction Kit (Omega Bio-tek). Microsatellites were amplified in 10μL reactions of 1μL 10×
NH4 Reaction Buffer (Biolase), 0.8μL 10mM dNTPs, 0.6μL 50mM MgCl2, 6.5μL H2O, 0.05μL
Biolase Taq, 0.1μL 2.5mM M-13 tagged forward primer, 0.4μL 10mM reverse primer, 0.1μL
2.5mM FAM tagged M-13 tag, and 0.5μL 15–20ng/μL template gDNA on a DNA Engine Tetrad
2 thermal cycler using the following PCR program: (1) 94°C for 3 min, (2) 94°C denaturation
for 30 s, (3) 54°C annealing for 30 s, (4) 72°C extension for 45 s, repeat steps 2–4 for 25 cycles,
(5) 94°C denaturation for 30 s, (6) 53°C annealing for 30 s, (7) 72°C extension for 45 s, repeat
steps 5–7 for 8 cycles, (8) 72°C extension for 10 min. Fragment analysis of PCR products was
conducted on an Applied Biosystems 3730 Genetic Analyzer with Capillary Electrophoresis
using Genescan 500 ROX size standard. Loci were genotyped using GeneMapper v4.1 (Applied
Biosystems). Primers pairs that exhibited clear amplification in at least one individual were
tested across a wider panel of seven more P. feriarum individuals (Table 12).

50
Six primer sets amplified well across the test panel (Table 12). Three primers showed
low levels of allelic diversity (NA < 4 across 9 individuals) and were excluded from further use
due to budget constraints. Three primers (P_fer_46999, P_fer_57550, and P_fer_101070) were
considered viable for use in this thesis.

51
APPENDIX D
MULTIPLEX PCR PRIMER SETS & PROTOCOLS
Multiplex sets were designed to keep dimer and tetramer microsatellite primers (Table
13) together. Fragment length ranges from previous M-13 tagged genotyping (Appendix C;
Lemmon unpublished; Bedwell & Lemmon unpublished; Ralicki & Lemmon unpublished) were
used to group primers into minimally overlapping groups to minimize pull-up peaks in analysis.
Multiplex set 1 used 3 fluorophore chemistry (NED, FAM, HEX) while sets 2, 3, and 4 used 4
fluorophore chemistry (NED, FAM, PET, VIC). Primer P_fer_DBNXL was tagged with NED
but not included within a multiplex.
PCR reaction mixtures for all multiplexes were similar except for the content of the
primer mix. 10× multiplex primer mix was made for each multiplex with fluorescent tagged
forward primers and reverse primers all mixed at a concentration of 100μM. 10μL reactions of
5μL 2× Qiagen Multimix, 3μL dsH20, 1μL 15–20ng/μL template gDNA, and 1μL 10× multiplex
primer mix. This protocol yielded a 10μM concentration of each primer in the final reaction
mixtures. The primer set for P_fer_DBNXL was diluted as though it were in a multiplex;
otherwise reactions were set up the same for this locus.
The following PCR protocol was used for multiplex 1: (1) 95°C for 15 min, (2) 94°C
denaturation for 30 s, (3) 54°C annealing for 90 s, (4) 72°C extension for 90 s, repeat steps 2–4
for 35 cycles, (5) 72°C extension for 7 min. The PCR protocol used for multiplexes 2 & 4 were
the same: (1) 95°C for 15 min, (2) 94°C denaturation for 30 s, (3) 54°C annealing for 90 s, (4)
72°C extension for 60 s, repeat steps 2–4 for 35 cycles, (5) 60°C extension for 30 min. The
following PCR protocol was used for multiplex 3: (1) 95°C for 15 min, (2) 94°C denaturation
for 30 s, (3) 48°C annealing for 90 s, (4) 72°C extension for 90 s, repeat steps 2–4 for 35 cycles,
(5) 72°C extension for 7 min. The PCR protocol used for M-13 tagged primer tests in Appendix
C was used for amplification in P_fer_DBNXL.
Fragment analysis of PCR products was conducted on an Applied Biosystems 3730
Genetic Analyzer with Capillary Electrophoresis using Genescan 500 ROX size standard for
multiplex 1 and P_fer_DBNXL and Genescan 500 LIZ size standard for multiplexes 2, 3, & 4.
Loci were genotyped using GeneMapper v4.1 (Applied Biosystems). The primer set
P_fer_G79VC as well as all primers in multiplex 4 yielded poor quality genotype data. Most of

52
these five loci exhibited multiple peaks likely resulting from primer nonspecificity. Additionally
many loci amplified inconsistently across individuals when PCR and fragment analysis was
duplicated. Some of these primers were designed using a separate panel of test individuals
(Lemmon & Ralicki unpublished) which exhibited better results. All of these loci showed
geographic regional inconsistencies in amplification indicating that these loci may be useful for
fine scale studies in some areas. All resulting genotypes from these five loci were excluded from
analysis in this thesis due to their poor quality across the sampled range (Fig. 3). Fragment
length data for the eleven included loci was imported into tandem v1.08 (Matschiner &
Salzburger 2009) for automated binning to determine observed alleles for each locus (NA, Table
14).

53
REFERENCES
Aleixo, A. 2004. Historical diversification of a terra-firme forest bird superspecies: a
phylogeographic perspective on the role of different hypotheses of Amazonian
diversification. Evolution 58: 1303–1317.
Altschul, S. F., T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller, & D. J. Lipman.
1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs. Nucleic Acids Research 25: 3389–3402.
Avise, J. C. 1992. Molecular population structure and the biogeographic history of a regional
fauna: a case history with lessons for conservation biology. Oikos 63: 62–76.
Avise, J. C., C. Giblin-Davidson, J. Laerm, J. C. Patton, & R. A. Lansman. 1979. Mitochondrial
DNA clones and matriarchal phylogeny within and among geographic populations of the
pocket gopher, Geomys pinetis. Proceedings of the National Academy of Sciences 76:
6694–6698.
Ayres, J. M. & T. H. Clutton-Brock. 1992. River boundaries and species range size in
Amazonian Primates. The American Naturalist 140 (3): 531–537.
Balkau, B. J. & M. M. Feldman. 1973. Selection for migration modification. Genetics 74:
171–174.
Barton, N. H., & G. M. Hewitt. 1985. Analysis of hybrid zones. Annual Review of Ecology
and Systematics 16: 113–148.
Bates, H. W. 1863. The naturalist on the river Amazons; a record of adventures, habits of
animals, sketches of Brazilian and Indian life, and aspects of nature under the equator,
during eleven years of travel. 2 volumes. John Murray: London.
Bates, J. M., J. Haffer, & E. Grismer. 2004. Avian mitochondrial DNA sequence divergence
across a headwater stream of the Rio Tapajós, a major Amazonian river. Journal of
Ornithology 145: 199–205.
Beerli, P. 2006. Comparison of Bayesian and maximum likelihood inference of population
genetic parameters. Bioinformatics 22: 341–345.
Beerli, P. 2009. How to use migrate or why are markov chain monte carlo programs difficult to
use? In Bertorelle, G., M. W. Bruford, H. C. Hauffe, A. Rizzoli, & C. Vernesi eds.
Population genetics for animal conservation (pp. 42–79). Cambridge University Press:
Cambridge.
Beerli, P. & M. Palczewski. 2010. Unified framework to evaluate panmixia and migration
direction among multiple sampling locations. Genetics 185: 313–326.

54
Berner, D., A. Grandchamp, & A. P. Hendry. 2009. Variable progress toward ecological
speciation in parapatry: stickleback across eight lake-stream transitions. Evolution 63:
1740–1753.
Blaney, R. M. 1971. An annotated check list and biogeographic analysis of the insular
herpetofauna of the apalachicola region, Florida. Herpetologica 27: 406–430.
Brown, W. L., & E. O. Wilson. 1956. Character displacement. Systematic Zoology 5: 49–64.
Burbrink, F. T., R. Lawson, & J. B. Slowinski. 2000. Mitochondrial DNA phylogeography of
the polytypic North American rat snake (Elaphe obsoleta): A critique of the subspecies
concept. Evolution 54: 2107–2118.
Cabanne, G. S., F. R. Santos, & C. Y. Miyaki. 2007. Phylogeography of Xiphorhynchus fuscus
(Passerirormes, Dendrocolaptidae): vicariance and recent demographic expansion in
southern Atlantic forest. Biological Journal of the Linnean Society 91: 73–84.
Carr, A. & C. J. Goin. 1959. Guide to the reptiles, amphibians, and fresh-water fishes of
Florida. University of Florida Press: Gainesville.
Chesser, R. T. 1999. Molecular systematic of the Rhinocryptid genus Pteroptochos. The
Condor 101: 439–446.
Church, S. A., J. M. Kraus, J. C. Mitchell, D. R. Church, D. R. Taylor. 2003. Evidence for
multiple Pleistocene refugia in the postglacial expansion of the eastern tiger salamander,
Ambystoma tigrinum tigrinum. Evolution 57: 372–383.
Collins, A. C. & J. M. Dubach. 1999. Biogeographic and ecological forces responsible for
speciation in Ateles. International Journal of Primatology 21: 421–444.
Colombi, V. H., S. R. Lopes, & V. Fagundes. 2010. Testing the Rio Doce as a riverine barrier
in shaping the Atlantic rainforest population divergence in the rodent Akdon cursor.
Genetics and Molecular Biology 33: 785–789.
Colwell, R. K. 2000. A barrier runs through it... or maybe just a river. Proceedings of the
National Academy of Science 97: 13470–13472.
Coyne, J. A., & H. A. Orr. 2004. Speciation. Sinauer Associates: Sunderland, MA.
Crenshaw, J. W., & W. F. Blair. 1959. Relationships in the Pseudacris nigrita complex in
Southwestern Georgia. Copeia 1959: 215–222.
Crozier, R. H. 1974. Niche shape and genetic aspects of character displacement. American
Zoologist 14: 1151–1157.

55
Davis, L. M., T. C. Glenn, D. C. Strickland, L. J. Guillette Jr., R. M. Elsey, W. E. Rhodes, H. C.
Dessauer, & R. H. Sawyer. 2002. Microsatellite DNA analyses support an East-West
phylogeographic split of American alligator populations. Journal of Experimental
Zoology 294: 352–372.
Dίaz-Muñoz, S. L. 2012. Role of recent and old riverine barriers in fine-scale population
genetic structure of Geoffroy's tamarin (Saguinus geoffroyi) in the Panama Canal
watershed. Ecology and Evolution 2: 298–309.
Donovan, M. F., R. D. Semlitsch, & E. J. Routman. 2000. Biogeography of the Southeastern
United States: a comparison of salamander phylogeographic studies. Evolution 54:
1449–1456.
Ellsworth, D. L., R. L. Honeycutt, N. J. Silvy, J. W. Bickham, & W. D. Klimstra. 1994.
Historical biogeography and contemporary patterns of mitochondrial DNA variation in
white-tailed deer from the Southeastern United States. Evolution 48: 122–136.
Endler, J. A. 1977. Geographic variation, speciation, and clines. Princeton University Press:
Princeton, NJ.
Engle, V. D. & J. K. Summers. 2000. Biogeography of benthic macroinvertebrates in estuaries
along the Gulf of Mexico and western Atlantic coasts. Hydrobiologia 436: 17–33.
Eriksson, J., G. Hohmann, C. Boesch, & L. Vigilant. 2004. Rivers influence the population
genetic structure of bonobos (Pan paniscus). Molecular Ecology 13: 3425–3435.
Faith, D. P., & P. S. Cranston. 1991. Could a cladogram this short have arisen by chance
alone?: on permutation tests for cladistic structure. Cladistics 7: 1–28.
Felsenstein, J. 2004. Inferring Phylogenies. Sinauer Associates: Sunderland, Massachusetts.
Filchak, K. E., J. B. Roethele, & J. L. Feder. 2000. Natural selection and sympatric divergence
in the apple maggot Rhagoletis pomonella. Nature 407: 739–742.
Fisher, R. A. 1930. The genetical theory of natural selection. Clarendon Press: Oxford,
England.
Fouquette, M. J. 1975. Speciation in chorus frogs. I. reproductive character displacement in the
Pseudacris nigrita complex. Systematic Zoology 24: 16–23.
Fu, J. & X. Zeng. 2008. How many species are in the genus Batrachuperus? a
phylogeographical analysis of the stream salamanders (family Hynobiidae) from
southwestern China. Molecular Ecology 17: 1469–1488.

56
Funk, W. C., J. P. Caldwell, C. E. Peden, J. M. Padial, I. De la Riva, & D. Cannatella. 2007.
Tests of biogeographic hypotheses for diversification in the Amazonian forest frog,
Physalaemus petersi. Molecular Phylogenetics and Evolution 44: 825–37.
Futuyma, D. J. & G. C. Mayer. 1980. Non-allopatric speciation in animals. Systematic Zoology
29: 254–271.
Gascon, C., J. R. Malcom, J. L. Patton, M. N. F. da Silva, J. P. Bogart, S. C. Lougheed, C. A.
Peres, S. Neckel, & P. T. Boag. 2000. Riverine barriers and the geographic distribution
of Amazonian species. Proceedings of the National Academy of Sciences 97: 13672–13677.
Gascon, C., S. C. Lougheed, & J. P. Bogart. 1996. Genetic and morphological variation in
Vanzolinius discodactylus: a test of the river hypothesis of speciation. Biotropica 28:
376–387.
Gascon, C., S. C. Lougheed, & J. P. Bogart. 1998. Patterns of genetic population differentiation
in four species of Amazonian frogs: a test of the riverine barrier hypothesis. Biotropica
30: 104–119.
Gavrilets, S. 1999. A dynamical theory of speciation on holey adaptive landscapes. The
American Naturalist 154: 1–22
Gavrilets, S., H. Li, & M. D. Vose. 1998. Rapid parapatric speciation on holey adaptive
landscapes. Proceedings of the Royal Society B 265: 1483–1489.
Gavrilets, S., H. Li, & M. D. Vose. 2000. Patterns of parapatric speciation. Evolution 54:
1126–1134.
Geyer, C. J. 1991. Markov chain Monte Carlo maximum likelihood. In Keramidas (ed.)
Computing Science and Statistics: Proceedings of the 23rd Symposium on the Interface.
Interface Foundation, Fairfax Station, pp. 156–163.
Goldman, N., Anderson, J. P., & Rodrigo, A. G. 2000. Likelihood-based tests of topologies in
phylogenetics. Systematic Biology 49: 652–670.
Gonzales, E. & J. L. Hamrick. 2005. Distribution of genetic diversity among disjunct
populations of the rare forest understory herb, Trillium reliquum. Heredity 95: 306–314.
Guillot, G. 2008. Inference of structure in subdivided populations at low levels of genetic
differentiation–the correlated allele frequencies model revisited. Bioinformatics 24:
2222–2228.
Haffer, J. 1969. Speciation in Amazonian forest birds. Science 165: 131–166

57
Hall, T. A. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis
program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95–98.
Hayes, F. E. & J. N. Sewlal. 2004. The Amazon river as a dispersal barrier to passerine birds:
effects of river width, habitat and taxonomy. Journal of Biogeography 31: 1809–1818.
Hershkovitz, P. 1977. Living new world monkeys (Platyrrhini); with an introduction to
Primates Vol. 1. University of Chicago Press: Chicago.
Hershkovitz, P. 1983. Two species of night monkeys, genus Aotus (Cebidae, Platyrrhini): a
preliminary report on Aotus taxonomy. American Journal of Primatology 4: 209–243.
Higgie, M., & M. W. Blows. 2008. The evolution of reproductive character displacement
conflicts with how sexual selection operates within a species. Evolution 62: 1192–1203.
Hoskin, C. J., M. Higgie, K. R. McDonald, & C. Moritz. 2005. Reinforcement drives rapid
allopatric speciation. Nature 437: 1353–1356.
Howard, D. J. 1993. Reinforcement: origin, dynamics, and fate of an evolutionary hypothesis.
In Harrison, R. G. (Ed.) Hybrid Zones and the Evolutionary Process. Oxford University
Press: New York, NY.
Jacquemyn, H., O. Honnay, K. Van Looy, & P. Breyne. 2006. Spatiotemporal structure of
genetic variation of a spreading plant metapopulation on dynamic riverbanks along the
Meuse River. Heredity 96: 471–8.
Jalil, M. F., J. Cable, J. Sinyor, I. Lackman-Ancrenaz, M. Ancrenaz, M. W. Bruford, & B.
Goossens. 2008. Riverine effects on mitochondrial structure of Bornean ogang-utans
(Pongo pygmaeus) at two spatial scales. Molecular Ecology 17: 2898–2909.
Jensen, J. B., C. D. Camp, W. Gibbons, M. J. Elliott. 2008. Amphibians and reptiles of Georgia.
University of Georgia Press: Athens.
Kass, R. E. & A. E. Raftery. 1995. Bayes factors. Journal of the American Statistical
Association 90: 773–795.
Klee, R. V., A. C. Mahoney, C. C. Christopher, & G. W. Barrett. 2004. Riverine peninsulas: an
experimental approach to homing in white-footed mice (Peromyscus leucopus).
American Midland Naturalist 151: 408–413.
Knowles, L. L. 2001. Did the Pleistocene glaciations promote divergence? tests of explicit
refugial models in montane grasshoppers. Molecular Ecology 10: 691–701.
Knowles, L. L., & W. P. Maddison. 2002. Statistical phylogeography. Molecular Ecology 11:
2623–2635.

58
Kocher, T. D. 2004. Adaptive evolution and explosive speciation: the cichlid fish model.
Nature Reviews Genetics 5: 288–298.
Kozak, K. H., R. A. Blaine, & A. Larson. 2006. Gene lineages and eastern North American
paleodrainage basins: phylogeography and speciation in salamanders of the Eurycea
bislineata species complex. Molecular Ecology 15: 191–207.
Kramer, D. C. 1973. Movements of western chorus frogs Pseudacris triseriata triseriata tagged
with Co60. Journal of Herpetology 7: 231–235.
Kramer, D. C. 1974. Home range of the western chorus frog Pseudacris triseriata triseriata.
Journal of Herpetology 8: 245–246.
Lamborot, M. 1991. Karyotypic variation among populations of Liolaemus monticola
(Tropiduridae) separated by riverine barriers in the Andean range. Copeia 1991: 1044–1059.
Lamborot, M. & L. Eaton. 1997. The Maipo river as a biogeographical barrier to Liolaemus
monticola (Tropiduridae) in the mountain ranges of central Chile. Journal of Zoological
Systematics and Evolutionary Research 35: 105–111.
Lamborot, M., L. Eaton, & B. A. Carrasco. 2003. The Aconcagua river as another barrier to
Liolaemus monticola (Sauria: Iguanidae) chromosomal races of central Chile. Revista
Chilena de Historia Natural 76: 23–34.
Lemmon, E. M. 2009. Diversification of conspecific signals in sympatry: geographic overlap
drives multi-dimensional reproductive character displacement in frogs. Evolution 63:
1155–1170.
Lemmon, A. R., & E. M. Lemmon. 2008. A likelihood framework for estimating
phylogeographic history on a continuous landscape. Systematic Biology 57: 544–561.
Lemmon, E. M., & A. R. Lemmon. 2010. Reinforcement in chorus frogs: lifetime fitness
estimates including intrinsic natural selection and sexual selection against hybrids.
Evolution 64: 1748–1761.
Lemmon, E. M., A. R. Lemmon, & D. C. Cannatella. 2007. Geological and climatic forces
driving speciation in the continentally distributed trilling chorus frogs (Pseudacris).
Evolution 61: 2086–2103Lemmon, E. M., M. Murphy, & T. E. Juenger. 2011.
Identification and characterization of nuclear microsatellite loci for multiple species of
chorus frogs (Pseudacris) for population genetic analyses. Conservation Genetics
Resources 3: 233–237.
Lemmon, E. M., M. Murphy, & T. E. Juenger. 2011. Identification and characterization of
nuclear microsatellite loci for multiple species of chorus frogs (Pseudacris) for
population genetic analyses. Conservation Genetics Resources 3: 233–237.

59
Li, R., W. Chen, L. Tu, & J. Fu. 2009. Rivers as barriers for high elevation amphibians: a
phylogeographic analysis of the alpine stream frog of the Hengduan mountains. Journal
of Zoology 277: 309–316.
Liu, F. R., P. E. Moler, & M. M. Miyamoto. 2005. Phylogeography of the salamander genus
Pseudobranchus in the southeastern United States. Molecular Phylogenetics and
Evolution 39: 149–159.
Lougheed, S. C., C. Gascon, D. A. Jones, J. P. Bogart, & P. T. Boag. 1999. Ridges and rivers: a
test of competing hypotheses of Amazonian diversification using a dart-poison frog
(Epipedobates femoralis). Proceedings of the Royal Society B 266: 1829–1835.
Ludwig, J. A., & J. F. Reynolds. 1988. Statistical ecology: a primer on methods and
computing. John Wiley & Sons: USA.
Mantel, N. 1967. The detection of disease clustering and a generalized regression approach.
Cancer Research 27: 209–220.
Margulies, M., M. Egholm, W. E. Altman, S. Attiya, J. S. Bader, L. A. Bemben, J. Berka, M. S.
Braverman, Y. Chen, Z. Chen, S. B. Dewell, L. Du, J. M. Fierro, X. V. Gomes, B. C.
Godwin, W. He, S. Helgesen, C. H. Ho, G. P. Irzyk, S. C. Jando, M. L. I. Alenquer, T. P.
Jarvie, K. B. Jirage, J. Kim, J. R. Knight, J. R. Lanza, J. H. Leamon, S. M. Lefkowitz, M.
Lei, J. Li, K. L. Lohman, H. Lu, V. B. Makhijani, K. E. McDade, M. P. McKenna, E. W.
Myers, E. Nickerson, J. R. Nobile, R. Plant, B. P. Puc, M. T. Ronan, G. T. Roth, G. J.
Sarkis, J. F. Simons, J. W. Simpson, M. Srinivasan, K. R. Tartaro, A. Tomasz, K. A.
Vogt, G. A. Volkmer, S. H. Wang, Y. Wang, M. P. Weiner, P. Yu, R. F. Begley, & J. M.
Rothberg. 2005. Genome sequencing in microfabricated high-density picolitre reactors.
Nature 437: 376–380.
Marshall, S. D., W. R. Hoeh, & M. A. Deyrup. 2000. Biogeography and conservation biology
of Florida's Geolycosa wolf spiders: threatened spiders in endangered ecosystems.
Journal of Insect Conservation 4: 11–21.
Matschiner, M., & W. Salzburger. 2009. TANDEM: integrating automated allele binning into
genetics and genomics workflows. Bioinformatics 25: 1982–1983.
Mayr, E. 1942. Systematics and the origin of species from the viewpoint of a zoologist.
Columbia University Press: New York.
Means, D. B. 1977. Aspects of the significance to terrestrial vertebrates of the apalachicola
river drainage basin, Florida. Florida Marine Research Publications 26: 37–67.
Miller, R. E., T. R. Buckley, & P. S. Manos. 2002. An examination of the monophyly of
morning glory taxa using Bayesian phylogenetic inference. Systematic Biology 51:
740–753.

60
Mount, R. H. 1975. The reptiles and amphibians of Alabama. University of Alabama Press:
Tuscaloosa.
Mylecraine, K. A., J. E. Kuser, P. E. Smouse, & G. L. Zimmermann. 2004. Geographic
allozyme variation in Atlantic white-cedar, Chamaecyparis thyoides (Cupressaceae).
Canadian Journal of Forest Research 34: 2443–2454.
Neill, W. T. 1957. Historical biogeography of present-day Florida. Bulletin of the Florida State
Museum 2: 175–220
Newman, C. E. & L. J. Rissler. 2011. Phylogeographic analyses of the southern leopard frog:
the impact of geography and climate on the distribution of genetic lineages vs.
subspecies. Molecular Ecology 20: 5295–5312.
Nosil, P., T. H. Vines, & D. J. Funk. 2005. Reproductive isolation caused by natural selection
against immigrants from divergent habitats. Evolution 59: 705–719.
Ortiz-Barrientos, D., A. Grealy, & P. Nosil. 2009. The genetics and ecology of reinforcement:
implications for the evolution of prezygotic isolation in sympatry and beyond. The Year
in Evolutionary Biology 2009. Annals of the New York Academy of Sciences 1168 156–182.
Palsbøll, P. J., M. Bérubé, & F. W. Allendorf. 2006. Identification of management units using
population genetic data. Trends in Ecology and Evolution 22: 11–16.
Patton, J. L., M. N. F. da Silva, & J. R. Malcolm. 1994. Gene genealogy and differentiation
among arboreal spiny rats (Rodentia: Echimyidae) of the Amazon Basin: a test of the
riverine barrier hypothesis. Evolution 48: 1314–1323.
Pauly, G. B., O. Piskurek, & H. B. Shaffer. 2007. Phylogeographic concordance in the
southeastern United States: the flatwoods salamander, Ambystoma cingulatum, as a test
case. Molecular Ecology 16: 415–29.
Pellegrino, K. C. M., M. T. Rodrigues, A. N. Waite, M. Morando, Y. Y. Yassuda, & J. Sites Jr.
2005. Phylogeography and species limits in the Gymnodactylus darwinii complex
(Gekkonidae, Squamata): genetic structure coincides with river systems in the Brazilian
Atlantic forest. Biological Journal of the Linnean Society 85: 13–26.
Pfennig, K. S. 2000. Female spadefoot toads compromise on mate quality to ensure conspecific
matings. Behavioral Ecology 11: 220–227.
Pfennig, K. S., & M. J. Ryan. 2006. Reproductive character displacement generates
reproductive isolation among conspecific populations: an artificial neural network study.
Proceedings of the Royal Society B 273: 1361–1368.

61
Pfennig, K. S., & M. J. Ryan. 2007. Character displacement and the evolution of mate choice:
an artificial neural network approach. Philosophical Transactions of the Royal Society B
362: 411–419.
Phillimore, A. B., C. D. L. Orme, G. H. Thomas, T. M. Blackburn, P. M. Bennett, K. J. Gaston,
& I. P. F. Owens. 2008. Sympatric speciation in birds is rare: insights from range data
and simulations. The American Naturalist 171: 646–657.
Pinho, C., & J. Hey. 2010. Divergence with gene flow: models and data. Annual Review of
Ecology, Evolution, and Systematics 2010 41: 215–230.
Piry, S., A. Alapetite, J. M. Cornuet, D. Paetkau, L. Baudouin, & A. Estoup. 2004.
GENECLASS2: a software for genetic assignment and first-generation migrant
detection. Journal of Heredity 95: 536–539.
Pounds, J. A. & J. F. Jackson. 1981. Riverine barriers to gene flow and the differentiation of
fence lizard populations. Evolution 35: 516–528.
Rand, S. A., M. J. Ryan, & W. Wilczynski. 1992. Signal redundancy and receiver
permissiveness in acoustic mate recognition by the Túngara frog, Physalaemus
pustulosus. American Zoologist 32: 81–90.
Randazzo, A. F. & D. S. Jones eds. 1997. The geology of Florida. University Press of Florida:
Gainesville.
Rice, A. M., & D. W. Pfennig. 2010. Does character displacement initiate speciation? evidence
of reduced gene flow between populations experiencing divergent selection. Journal of
Evolutionary Biology 23: 854–865.
Rousset, F. 2008. Genepop'007: a complete reimplementation of the Genepop software for
Windows and Linux. Molecular Ecology Resources 8: 103–106.
Rozen, S. & H. J. Skaletsky. 2000. Primer3 on the WWW for general users and for biologist
programmers. In Krawetz S, Misener S (eds.) Bioinformatics Methods and Protocols:
Methods in Molecular Biology. Humana Press, Totowa, NJ, pp 365–386.
Schilthuizen, M. 2000. Ecotone: speciation-prone. Trends in Ecology & Evolution 15: 130–131.
Schuelke, M. 2000. An economic method for the fluorescent labeling of PCR fragments.
Nature Biotechnology 18: 233–234.
Slatkin, M. 1993. Isolation by distance in equilibrium and non-equilibrium populations.
Evolution 47: 264–279.

62
Smouse, P. E., J. C. Long, & R. R. Sokal. 1986. Multiple regression and correlation extensions
of the Mantel test of matrix correspondence. Systematic Zoology 35: 627–632.
Soltis, D. E., A. B. Morris, J. S. McLachlan, P. S. Manos, & P. S. Soltis. 2006. Comparative
phylogeography of unglaciated eastern North America. Molecular Ecology 15: 4261–4293.
Swenson, N. G. & D. J. Howard. 2005. Clustering of contact zones, hybrid zones, and
phylogeographic breaks in North America. The American Naturalist 166: 581–591.
Upholt, W. B. 1977. Estimation of DNA sequence divergence from comparison of restriction
endonuclease digests. Nucleic Acids Research 4: 1257–1265.
Van Looy, K., H. Jacquemyn, P. Breyne, & O. Honnay. 2009. Effects of flood events on the
genetic structure of riparian populations of the grassland plant Origanum vulgare.
Biological Conservation 142: 870–8.
Wallace, A. R. 1852. On the monkeys of the Amazon. Proceedings of the Zoological Society
of London 20: 107–10.
Wallace, A. R. 1876. The geographical distribution of animals; with a study of the relations of
living and extinct faunas as elucidation the past changes of the Earth's surface. 2
volumes. Harper & Brothers: New York.
Walker, M. J., A. K. Stockman, P. E. Marek, & J. E. Bond. 2009. Pleistocene glacial refugia
across the Appalachian Mountains and coastal plain in the millipede genus Narceus:
evidence from population genetic, phylogeographic, and paleoclimatic data. BMC
Evolutionary Biology 9: 25.
Willey, R. B. & R. L. Willey. 1967. Barriers to gene flow in natural populations of
grasshoppers I. the Black Canyon of the Gunnison River and Arphia conspera. Psyche
74: 42–57.
Zhang, Z., X. Zheng, & S. Ge. 2007. Population genetic structure of Vitex negundo
(Verbenaceae) in the Three-gorge area of the Yangtze river: the riverine barrier to seed
dispersal in plants. Biochemical Systematics and Ecology 35: 506–516.

63
BIOGRAPHICAL SKETCH
Moses James Michelsohn
Moses was born on August 16, 1985 in Roswell, New Mexico. He grew up watching
birds and often dragged his family to Bitter Lake National Wildlife Refuge to see the geese and
cranes. In 2003 Moses left his desert home to attend Eckerd College in St. Petersburg, Florida,
where he first learned about cladistics from Peter Meylan. He graduated with a Bachelor of
Science degree in Biology in 2007, then spent a few years working as a herpetologist across the
United States and in Costa Rica. He joined Emily Lemmon's lab at Florida State University in
2009 where he began working on Pseudacris. There he became increasingly fascinated by
coalescent theory and Bayesian methods with the encouragement of Peter Beerli. Following
graduation, he hopes to continue working in phylogenetics and one day return to a more arid
climate.




















