
6Thioguanine damages mitochondrial DNA and causes mitochondrial dysfunction in human cells
Upload
khangminh22Category
view
2download
0
Mitochondrial genetics
Patrick Francis Chinnery and Gavin Hudson*
Institute of Genetic Medicine, International Centre for Life, Newcastle University, Central Parkway,Newcastle upon Tyne NE1 3BZ, UK
Introduction: In the last 10 years the field of mitochondrial genetics has widened,
shifting the focus from rare sporadic, metabolic disease to the effects of
mitochondrial DNA (mtDNA) variation in a growing spectrum of human disease. The
aim of this review is to guide the reader through some key concepts regarding
mitochondria before introducing both classic and emergingmitochondrial disorders.
Sources of data: In this article, a review of the current mitochondrial genetics
literature was conducted using PubMed (http://www.ncbi.nlm.nih.gov/pubmed/).
In addition, this review makes use of a growing number of publically available
databases including MITOMAP, a human mitochondrial genome database
(www.mitomap.org), the Human DNA polymerase GammaMutation Database
(http://tools.niehs.nih.gov/polg/) and PhyloTree.org (www.phylotree.org), a
repository of global mtDNAvariation.
Areas of agreement: The disruption in cellular energy, resulting from defects in
mtDNA or defects in the nuclear-encoded genes responsible for mitochondrial
maintenance, manifests in a growing number of human diseases.
Areas of controversy: The exact mechanisms which govern the inheritance of
mtDNA are hotly debated.
Growing points: Although still in the early stages, the development of in vitro
genetic manipulation could see an end to the inheritance of the most severe
mtDNA disease.
Keywords:mitochondria/genetics/mitochondrial DNA/mitochondrial disease/mtDNA
Accepted: April 16, 2013
Mitochondria
The mitochondrion is a highly specialized organelle, present in almost alleukaryotic cells and principally charged with the production of cellularenergy through oxidative phosphorylation (OXPHOS). In addition toenergy production, mitochondria are also key components in calcium sig-nalling, regulation of cellular metabolism, haem synthesis, steroid synthe-sis and, perhaps most importantly, programmed cell death (apoptosis).1
British Medical Bulletin 2013; 106: 135–159DOI:10.1093/bmb/ldt017
& The Author 2013. Published by Oxford University Press.This is an Open Access article distributed under the terms of the Creative Commons
Attribution License (http://creativecommons.org/licenses/by/3.0/), which permits unrestrictedreuse, distribution, and reproduction in any medium, provided the original work is properly cited.
*Correspondence address.Institute of Genetic
Medicine, InternationalCentre for Life, Newcastle
University, Central
Parkway, Newcastle uponTyne NE1 3BZ, UK. E-mail:

However, the simplistic elegance of biochemical ATP production belies a,complex, synergistic relationship between two genomes: the mitochon-drial genome (mtDNA) and the nuclear genome (nDNA). The aim of thisreview is to introduce these two genomes and shed light on the clinicalproblems arising when communication breaks down. The emphasis is onthe basic science underpinning mitochondrial diseases. Clinical aspectsare not considered in detail because they have recently been reviewedelsewhere in open-access publications.2–4
mtDNA
MtDNA is the only source of critical cellular proteins outside of the eu-karyotic nucleus. In the majority of eukaryotes, mtDNA is organizsed asa circular, double-stranded DNA molecule (Fig. 1).5 The strands are dis-tinguished by their nucleotide composition: heavy (H-strand) is guaninerich, compared with the cytosine-rich light strand (L-strand). The lengthvaries between species (15 000–17 000 bp), but is fairly consistent inhumans (∼16 569 bp).5 MtDNA is a multi-copy DNA, with cells contain-ing between 100 and 10 000 copies of mtDNA (dependent upon cellularenergy demand).
Fig. 1 Mitochondrial DNA. Schematic diagram of the 16.6-kb, circular, double-strandedmtDNA molecule, where the outer circle represents the heavy strand and the inner circle thelight strand. Shown are the genes encoding the mitochondrial RC: MTND1–6, MTCOI–II,MTATP6 and 8 and MTCYB; the two ribosomal RNAs (green boxes) and each of the 22 tRNAs(red spheres).
P. F. Chinnery and G. Hudson
136 British Medical Bulletin 2013;106

Structure
MtDNA contains 37 genes, 28 on the H-strand and 9 on the L-strand.Thirteen of the genes encode one polypeptide component of the mito-chondrial respiratory chain (RC), the site of cellular energy productionthrough OXPHOS. Twenty-four genes encode a mature RNA product:22 mitochondrial tRNA molecules, a 16 s rRNA (large ribosomalsubunit) and a 12 s rRNA (small ribosomal subunit).5 Unlike its nDNAcounterpart, mtDNA is extremely efficient with ∼93% representing acoding region. Unlike nDNA, mtDNA genes lack intronic regions andsome genes, notably MTATP6 and MTATP8, have overlapping regions.Most genes are contiguous, separated by one or two non-coding basepairs. mtDNA contains only one significant non-coding region, the dis-placement loop (D-loop).5 The D-loop contains the site of mtDNA repli-cation initiation (origin of heavy strand synthesis, OH) and is also the siteof both H-strand transcription promoters (HSP1 and HSP2).The mitochondrial genetic code differs slightly from nuclear DNA
(nDNA). MtDNA uses only two stop codons: ‘AGA’ and ‘AGG’6 (com-
pared with ‘UAA’, ‘UGA’ and ‘UAG’ in nDNA), conversely ‘UGA’encodes tryptophan. To compensate UAA codons have to be introducedat the post-transcriptional level. In addition ‘AUA’, isoleucine in nDNA,encodes for methionine in mtDNA.
Inheritance
Prevailing theory suggests that mtDNA is maternally inherited, withmtDNA nucleoids the unit of inheritance. During mammalian zygote for-mation, sperm mtDNA is removed by ubiquitination, likely occurringduring transport through the male reproductive tract.7 Consequently, themtDNA content of the zygote is determined exclusively by the previouslyunfertilized egg.To date only a single case of paternal transmission in humans has been
recorded.8 However, paternal transmission in other animals is bothcommon and recurring. Theory suggests that the lack of paternal inherit-ance is due to either (i) a dilution effect; sperm contain only 100 copies ofmtDNA, compared with 100 000 in the unfertilized egg, (ii) selective ubi-quitination of paternal mtDNA or (iii) the ‘mtDNA bottleneck’ excludesthe ‘minor’ paternal alleles.7 The advent of deep, next generation sequen-cing, allowing mtDNA can be sequenced at great depths (>20 000 fold)may enable researchers to revisit this phenomenon.
Homoplasmy and heteroplasmy
Cells contain thousands of molecules of mtDNA;9 and in the majority ofcases their sequence is identical, known as homoplasmy. However, an
Mitochondrial genetics
British Medical Bulletin 2013;106 137

inefficient mtDNA repair, a localized oxidative environment andincreased replication10 make mtDNA mutation frequent. The polyploidnature of mtDNA means that mutations often co-exist with their wild-type counterpart in various proportions (termed heteroplasmy). The pro-portion of mutant has important consequences in understanding mito-chondrial disease (discussed later).11
nDNA andmitochondrial function
According to recent data the mitochondrial proteome is estimated at∼1500 proteins.12 Mitochondria are dependent upon the nuclear genomefor the majority of the OXPHOS system and also for maintaining andreplicating mtDNA as well as organelle network proliferation anddestruction (Fig. 2).
OXPHOS system
To date, 92 structural OXPHOS subunit genes have been identified: 13encoded by mtDNA (Fig. 1) and 79 encoded by the nuclear genome.Briefly, complex I (NADH:ubiquinone oxidoreductase), the largest of theRC components, consists of 44 subunits: 14 enzymatic ‘core subunits’(7 from mtDNA and 7 from nDNA)13 and a further 30 nDNA accessorysubunits thought to maintain complex stability.14 Complex II (succinate:
Fig. 2 Interaction between nDNA and mtDNA. Cartoon demonstrating the complex interactionbetween genes encoded by nDNA and the processes they control in the mitochondrion.
P. F. Chinnery and G. Hudson
138 British Medical Bulletin 2013;106

ubiquinone oxidoreductase) is encoded entirely by nDNA (four subu-nits). Complex III (ubiquinol:cytochrome c oxidoreductase) contains 11subunits, 1 encoded by mtDNA (MTCYB) and 10 encoded by nDNA.15
Complex IV (cytochrome c oxidase) consists of three mtDNA-encodedsubunits and a further 11 nDNA-encoded subunits. Finally, complex V(F0F1-ATP synthase) comprises 19 subunits, 2 encoded by mtDNA andthe remaining 17 encoded by nDNA.In addition, nDNA encodes over 35 proteins required for the RC as-
sembly: complex I = 11 nDNA assembly factors,16 complex III = 2,15
complex IV = 1817 and complex V = 4.18
mtDNA replication
Unlike nDNA, mtDNA replication is not governed by the cell cycle(eukaryotic cell division) and is continuously recycled. MtDNA replica-tion and integrity maintenance is handled entirely by the nDNA. Ineukaryotes, mtDNA is replicated in a ‘replisome’ (a DNA/proteincomplex making up the replication machinery) by a trimeric proteincomplex composed of a catalytic subunit: polymerase gamma, a 140 kDaDNA polymerase encoded by POLG and two 55 kDa accessory subunits,encoded by POLG2.19 This enzyme complex performs three activities,DNA polymerase activity, 30-50 exonuclease/proofreading activity and a50dRP lyase activity (required for enzymatic DNA repair).In addition, the replisome also includes the mitochondrial single-
stranded binding protein (encoded bymtSSB), which is involved in stabiliz-ing single-stranded regions of mtDNA at replication forks, enhancing poly-merase gamma activity. Twinkle is a 50-30 DNA helicase, which unwindsdouble-stranded mtDNA, facilitating mtDNA synthesis, as well as actingas a mtDNA primase (an enzyme required to prime nucleotide synthesis).19
Several topoisomerases have been indentified in humans, including themitochondrial topoisomerases 1 (encoded by TOP1mt) and IIIα (encodedby TOP3a). Finally, the synergy between mitochondrial transcriptionfactor A (encoded by TFAM) and mtDNA copy number suggests thatTFAMmay act as an mtDNA chaperone (a protein that assists the functionof another protein) protecting it against oxidative damage.
mtDNA arrangement
Like its nDNA counterpart, mtDNA is also packaged in protein–DNAcomplexes, known as nucleoids.20 MtDNA nucleoids are associated withthe inner mitochondrial membrane, spaced evenly along the cristae. Inaddition to a single mtDNA molecule,21 mtDNA nucleoids contain anumber of proteins.20 Principally the site of mtDNA replication, it is
Mitochondrial genetics
British Medical Bulletin 2013;106 139

unsurprising that mtDNA nucleoids contain the protein machineryrequired for DNA replication, transcription, repair and packaging, in-cluding the mtDNA polymerase POLG, its accessory subunit POLG2,the activator of mtDNA transcription (encoded by TFAM) as well asmtDNA helicases and binding proteins (twinkle and mtSSB, respective-ly).20 In addition, mtDNA nucleoids contain chaperone proteins(HSP90-β and HSP70) required for mtDNA stability.
Transcription and translation
Transcription of mtDNA is ‘prokaryotic like’ and was thought of a two-component system involving a protein complex containing the mitochon-drial RNA polymerase (POLRMT) and two transcription factors(TFB1M and 2M).22,23 However, recent research indicates that TFB1Mdoes not modulate mtDNA transcription in the presence of TFB2M,rather it acts as a dimethyltransferase which stabilizes the small subunitof the mitochondrial ribosome. RNA transcription is regulated by mito-chondrial transcription factor A (TFAM).24
Briefly, each strand is transcribed as a polycistronic precursor mRNAmolecule (i.e. the mRNA contains all of the genes in one molecule).Light-strand transcription is initiated from the light-strand promoter;however, heavy-strand transcription initiates from two heavy strand pro-moters: HSP1 and HSP2 (Fig. 1).25 Transcript elongation is performed byPOLRMT, enhanced by both ‘transcription elongation factor mitochon-drial’ (TEFM) and termination of mature transcripts is carried out bymitochondrial termination factor 1 (MTERF1).25
Translation of the 13 mtDNA protein coding genes occurs in the mito-chondria. The mitoribosomes are partly coded by mtDNA (MTRNR1and MTRNR2, Fig. 1), but require a further 81 nDNA proteins.Translation is initiated by two mitochondrial initiation factors: mtIF1and mtIF3.26,27 mtIF3 begins initiation by dissociating the ‘mitoribo-some’ (the mitochondrial ribosomes) allowing assembly of the initiationcomplex.28 MRNA is then bound to the small subunit, aligning the startcodon to the peptidyl site of the mitoribosome. Peptide elongation is con-trolled by a number of nuclear-encoded genes, including mitochondrialelongation factor Tu (mtEFTu),29,30 which binds the tRNA to the mitori-bosome and mitochondrial elongation factor G1 (mtEFG1), required tomove the newly added amino acid along one position and allowingamino acid inclusion.31 Translation termination is carried out solely bymitochondrial release factor 1a (mtRF1a),32 which recognizes the stopcodons (UAA and UAG)33 and triggers hydrolysis of the bond betweenthe terminal tRNA and the nascent peptide.
P. F. Chinnery and G. Hudson
140 British Medical Bulletin 2013;106

Controlling mitochondrial network dynamics
Mitochondria are often depicted as distinct organelles; however, they ac-tually form a complex reticulum that is undergoing continual fusion andfission (Fig. 2).34 It is likely that fusion has evolved as a mechanism topromote intermictochondrial cooperation—allowing the sharing and dis-semination of mtDNA and mitochondrial proteins. Fission promotesmitochondrial compartmentalization,34 a mechanism that is needed todistribute mitochondria during cell division. Mitochondrial network dy-namics, much like mtDNA replication, is controlled completely bynDNA, although likely involves mtDNA–nDNA communication.34
Mitochondrial fusionThe principle player in mitochondrial fusion is mitofusin (Mfn) andmammalian mitochondria contain two similar mitofusin proteins: Mfn1and Mfn2 (Fig. 2),34 sharing 80% sequence homology. Studies indicatethat both Mfn1 and Mfn2 uniformly localize to the mitochondrial outermembrane and overexpression leads to peri-nuclear clustering on mito-chondria.34 Mitochondrial fusion is also dependent upon OPA1 expres-sion (Fig. 2),34 where inhibition of gene expression causes an increase inmitochondrial fragmentation, conversely the overexpression of OPA1breaks the network into spheres.
Mitochondrial fissionDNM1L, dynamin 1 like, controls mitochondrial fission in mammaliancells (Fig. 2).34 DNM1L codes for a principally cytosolic protein;however, it also localizes to fission sites on the mitochondria. Similar toMfn1, the overexpression of ‘mutant’ DNM1L results in a breakdown ofmitochondrial networks. Due to its dynamin similarity, two differentfunctions have been proposed forDNM1L. It has been hypothesized thatDNM1L may mechanically mediate membrane fission through GTP hy-drolysis; alternatively, it may act as a signalling molecule, conscriptingand activating separate fission enzymes such as Dnm1: the yeast homo-logue of Drp1.
Areas of agreement
Mitochondrial disease
An area where all mitochondrial researchers would agree is the capacityfor mitochondrial dysfunction to manifest as disease. Mitochondrialdisease is principally a chronic loss of cellular energy, where a failure tomeet cellular energy demand results in a clinical phenotype. The clinical
Mitochondrial genetics
British Medical Bulletin 2013;106 141
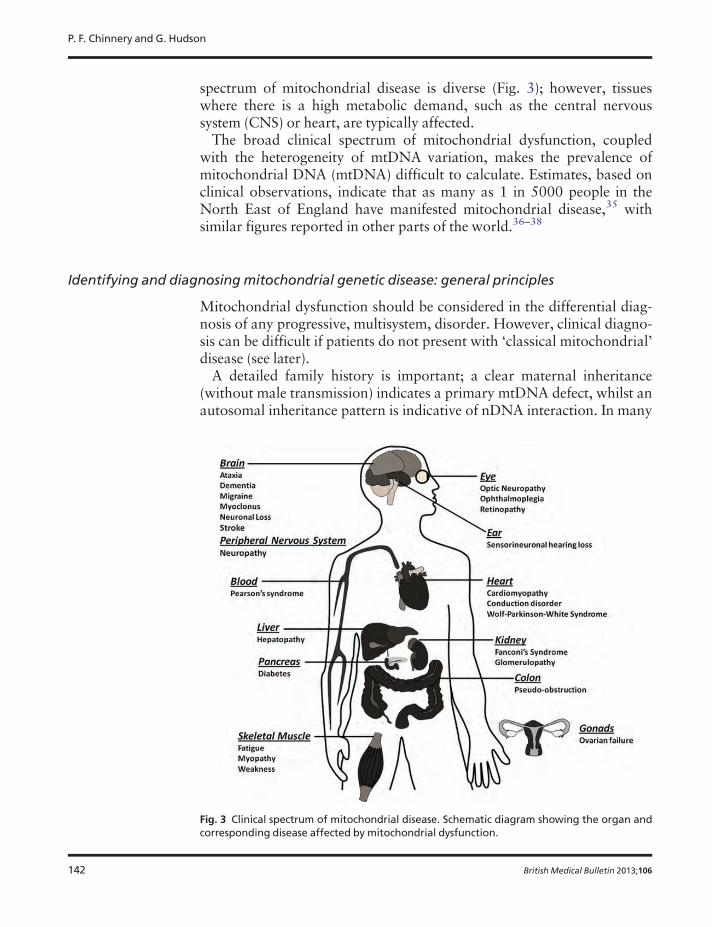
spectrum of mitochondrial disease is diverse (Fig. 3); however, tissueswhere there is a high metabolic demand, such as the central nervoussystem (CNS) or heart, are typically affected.The broad clinical spectrum of mitochondrial dysfunction, coupled
with the heterogeneity of mtDNA variation, makes the prevalence ofmitochondrial DNA (mtDNA) difficult to calculate. Estimates, based onclinical observations, indicate that as many as 1 in 5000 people in theNorth East of England have manifested mitochondrial disease,35 withsimilar figures reported in other parts of the world.36–38
Identifying and diagnosing mitochondrial genetic disease: general principles
Mitochondrial dysfunction should be considered in the differential diag-nosis of any progressive, multisystem, disorder. However, clinical diagno-sis can be difficult if patients do not present with ‘classical mitochondrial’disease (see later).A detailed family history is important; a clear maternal inheritance
(without male transmission) indicates a primary mtDNA defect, whilst anautosomal inheritance pattern is indicative of nDNA interaction. In many
Fig. 3 Clinical spectrum of mitochondrial disease. Schematic diagram showing the organ andcorresponding disease affected by mitochondrial dysfunction.
P. F. Chinnery and G. Hudson
142 British Medical Bulletin 2013;106

cases blood and/or CSF lactate concentration,39 neuroimaging,40,41
cardiac evaluation and muscle biopsy for histological or histochemicalevidence can indicate mitochondrial disease. However, establishing a mo-lecular genetic diagnosis is preferred.Molecular genetic testing can be carried out on DNA extracted from
blood (useful for the identification of some mtDNA and nDNA muta-tions),42,43 but DNA extracted from the affected tissue is preferred(as pathogenic mtDNA mutations are often not detectable in blood).44
Southern blot analysis can be used to identify mtDNA rearrangementsand ‘common’ mutations can be targeted by Sanger sequencing of eithermtDNA or nDNA.
The genetics of mitochondrial disease
The complex interaction between the two cellular genomes means mito-chondrial disease can arise through either (i) a primary mtDNA defect or(ii) a defect in a nuclear-encoded mitochondrial protein.
mtDNA and disease
Understanding mtDNAvariationmtDNA integrity is constantly attacked by mitochondrial reactive oxygenspecies (ROS) generated during cellular OXPHOS.45 ROS are potentgenotoxic agents, which cause mutagenic and cytotoxic effects. The prox-imity of mtDNA to the site of mitochondrial ROS production (principallycomplexes I and III of the RC) is the major cause of oxidative lesions andmtDNA instability and is directly responsible for the higher nucleotideinstability when compared with nDNA.Despite being packaged in mitochondrial nucleoids and possessing DNA
repair pathways evolved to cope with oxidative damage, including base ex-cision repair mechanisms,46 mtDNAmutation rates are significantly higherthan nDNA. Mutation creates two distinct classes of mtDNA variant:(i) single-base-pair variants and (ii) mtDNA rearrangements (deletions andinsertions). Single-base-pair variants are typically inheritable and are eithercommon in the populace (as proposed neutral variants) or enriched in indi-viduals with disease (as mtDNA mutations). Understanding the complexnature of mtDNAvariation is critical to understanding its affect on diseaseand there are a few key points that must be understood before assessing anmtDNAvariant.
Consequences of mtDNA heteroplasmyMtDNA heteroplasmy (described earlier) has a complex relationshipwith disease. The clinical expression of a heteroplasmic pathogenic
Mitochondrial genetics
British Medical Bulletin 2013;106 143

mtDNA mutation is directly correlatable with the relative proportion ofwild-type and mutant genomes.47 For common point mutations, a typicalthreshold of 80–90% mutant is required to manifest as disease at thecellular level,48,49 and tissue levels correlate loosely with the severity ofthe clinical phenotype. However, there is emerging evidence that muta-tion levels can change over time, increasing in post-mitotic tissues, suchas brain and muscle and decreasing in mitotic tissues including blood.This can present a challenge when interpreting some clinical moleculargenetic tests.44,50,51
Common mtDNAvariationEvolutionarily, common inherited mtDNA mutations have created stablepopulation subgroups separated by common sequence variation knownas haplogroups. Many of the major sub-divisions occurred over 10 000years ago, developing as humans migrated into new geographic areas.Over 95% of Europeans belong to 1 of 10 major haplogroups, H, J, T, U,K (a subgroup of U), M, I, V, W and X, with each haplogroup definedby specific sequence variants within the population.52 These common,inherited, mtDNA variants are usually not heteroplasmic, and due totheir selection neutrality have become fixed in the population. However,different haplogroups have been associated with a variety of human dis-eases, including primary mitochondrial disorders such as Leber’s heredi-tary optic neuropathy (LHON, an age-related loss of vision), wherebackground mitochondrial haplogroup has a direct, functional, effecton the RC protein complex assembly;53 but has expanded to includeage-related neurodegenerative disorders such as Parkinson’s disease(PD)54 Alzheimer’s disease55,56 and age-related macular degeneration.57
Rare mtDNAvariationRare, inherited, point mutations are a major cause of disease in humans,with an estimated incidence of 1 in 5000.58 They primarily occur inprotein coding and tRNA genes and ultimately result in a reduction ofcellular energy, through either a reduction in mitochondrial RC enzymeactivity or an impairment of mitochondrial protein synthesis.59 Unlikecommon inherited variants, rare point mutations are often heteroplasmic.In contrast to point mutations, primary mitochondrial rearrangements
of mtDNA are not inheritable; they are primarily, sporadic, large-scaledeletions, typically heteroplasmic and usually result in disease. To datearound 120 different mtDNA deletions have been identified in patientswith mitochondrial disease.60 Similarly to mtDNA point mutations, theratio of deleted versus ‘wild-type’ molecules is critical to disease aeti-ology, with mtDNA deletions manifesting disease at a lower hetero-plasmic threshold (∼50–60%).61 The exact mechanism of deletionformation is under debate and current research indicates two likely
P. F. Chinnery and G. Hudson
144 British Medical Bulletin 2013;106

models of deletion formation: (i) replication error and (ii) mtDNA repairinefficiency.62,63
‘Classical’mtDNA diseasesLHON is a common cause of inherited blindness that typically presentswith bilateral, painless, sub-acute visual failure in young adult males.LHON was the first maternally inherited disease to be associated with anmtDNA point mutation.64 Today, clinical diagnosis is usually confirmedby molecular genetic analysis for one of three ‘common’ mtDNA muta-tions, which all affect genes coding for complex I subunits of the RC:m.3460G>A, m.11778G>A and m14484T>C.65 Mitochondrial dysfunc-tion causes a specific loss of retinal ganglion cells,66 whilst preserving theremaining retinal layers. The optic nerve also shows characteristic degen-eration and an accumulation of mitochondria suggesting an impairmentof axoplasmic transport. LHON mutations are typically homoplasmic;however, not all patients harbouring a pathogenic LHON mtDNA muta-tion develop visual failure. Studies of LHON have identified commonmtDNA variants that may modulate LHON expression;67,68 additionallyenvironmental factors, such as cigarette smoke69 and oestrogen levelsmay play a role.70 However, the majority of research has focused on theidentification of a nuclear-encoded susceptibility allele.67,71–74
Non-syndromic and aminoglycoside-induced sensorineuronal hearingloss is associated with m.1555A>G, a homoplasmic point mutation in the12sRNA gene.75 The variant alters a highly conserved region of12sRNA, mutating the molecule to more closely resemble its bacterialhomologue. In vitro experiments on m.1555A>G mutant cell linesdemonstrated that exposure to aminoglycoside would impair growth;however, not all symptomatic individuals have been exposed to amino-glycoside.75
Surprisingly, given that they make up only 5% of mtDNA, the vastmajority of pathogenic mtDNA point mutations occur in the tRNA genes(Fig. 1).76,77 In addition, pathogenic tRNA mutations are typically het-eroplasmic.Mitochondrial encephalomyopathy, lactic acidosis and stroke-like epi-
sodes (MELAS) is typically a childhood, multisystem disorder. Patientscommonly manifest with generalized tonic-clonic seizures, recurrentheadaches, anorexia with recurrent vomiting and postlingual hearingloss,78–80 but can manifest with impaired: motor ability, vision andmental acuity due to the cumulative effect of multiple stroke-like epi-sodes. MELAS is commonly (80% of cases) caused by a A>G transitionat m.3243 in MTTL1,81 but is also associated with variants inMTND5.82 Biochemically, MELAS manifests as defects of complex I andIV activity; however, care must be taken when interpreting the findings asbiochemical results can often appear normal.
Mitochondrial genetics
British Medical Bulletin 2013;106 145

Myoclonus epilepsy with ragged red fibres (MERRF) is a neuromuscu-lar disorder primarily caused by m.8344A>G in MTTK.83 Clinically,patients with m.8344A>G present with myoclonus, epilepsy, muscleweakness, cerebellar ataxia and dementia, although neurological symp-toms can develop with age.83 Clinical severity is correlated with patientheteroplasmy with high levels of mutant mtDNA often causing, severecomplex I or IV deficiency and occasionally a combined complex I and IVdeficiency. Much like MELAS, the genotype–phenotype correlation ofm.8344A>G can be extended beyond MERRF. M.8344A>G has beenidentified is diverse mitochondrial phenotypes such as Leigh’s syndrome.m.7472insC, affecting MTTS (Fig. 1), was first identified in a large
Italian family presenting with hearing loss, ataxia and myoclonus. Thismutation was later found in several unrelated families, all showing a wideclinical spectrum, including isolated hearing loss, ataxia and MERRF.This mutation has been found at increasing frequencies in families pre-senting with maternally inherited hearing loss.Pathogenic rearrangements of mtDNA are typically large-scale dele-
tions and to date over 120 different pathogenic mtDNA deletions havebeen identified.60 As described previously, mtDNA deletions are typicallysporadic and not inheritable. Clinical severity is directly correlatable withthe level and tissue distribution of the rearrangement and mitochondrialdysfunction is simply a result of the removal of key mitochondrial genes.Homoplasmic tRNA gene loss is particularly detrimental as mitochon-dria cannot synthesize a functional OXPHOS system. mtDNA deletionsare associated with three main clinical phenotypes: Kearns–Sayre syn-drome (KSS),84 sporadic progressive external ophthalmoplegia (PEO)85
and Pearson’s syndrome.86
KSS is an early onset, sporadic, disorder characterized by PEO andpigmentary retinopathy; however, cases can also present with cerebel-lar syndrome, heart block, diabetes and shortness of stature.Mitochondrial dysfunction manifests as ragged red fibres (RRFs), anaccumulation of dysfunctional mitochondria in the sub-sarcolemmalregion of a muscle fibre (detectable when a muscle section is stainedwith Gomori trichrome stain).85
Large-scale deletions and duplications of mtDNA are a known cause ofPearson’s bone-marrow–pancreas syndrome, a rare infant disorder char-acterized by infantile sideroblastic anaemia and occasionally includingsevere exocrine pancreatic insufficiency.86
nDNAvariation and mitochondrial disease
Nuclear–mitochondrial disease can be classified into four distinct groups:(i) disorders resulting from a reduction in mtDNA stability; (ii) disorders
P. F. Chinnery and G. Hudson
146 British Medical Bulletin 2013;106

resulting from mutations in nuclear-encoded components or assemblyfactors of the OXPHOS system; (iii) disorders resulting from mutationsaffecting mitochondrial translation and (iv) disorders due to defects ingenes controlling mitochondrial network dynamics.
Disorders resulting from a reduction in mtDNA stabilityA growing number of disorders have become associated with mtDNA in-stability, primarily a result of impaired mtDNA replication. Mutationsin POLG, the gene encoding the only mtDNA polymerase, are by farthe commonest cause of mtDNA stability disorders. Mutations in thePOLG gene can cause either point mutations (through impaired mtDNAproofreading) or deletions (through impaired polymerase activity) inmtDNA.19 The first pathogenic mutations in POLG were identified infamilies with autosomal dominant PEO (adPEO); however, the spectrumof disease associated with POLG mutations has been expanded toinclude autosomal recessive PEO, adult onset ataxia, Alpers’ syndrome,parkinsonism and premature ovarian failure.87
adPEO, characterized by multiple mtDNA deletions, is caused by muta-tions in PEO1, which encodes ‘twinkle’ the putative mitochondrial heli-case.88 It is thought that twinkle mutations result in an accumulation ofreplication intermediates, causing replication stalling and eventually de-pletion. adPEO is also associated with mutations in ANT1,89 the genecoding adenine nucleotide translocase. Mutations in ANT1 impair ADP–ATP exchange through the mitochondrial membrane, causing a nucleo-tide imbalance (affecting replication) and a severe reduction in cellularenergy.In addition to structurally altering mtDNA, several disorders have been
identified that are caused by a reduction in mtDNA copy number.19
Alpers syndrome, characterized by diffuse and progressive cerebralatrophy,90 has been associated with mutations in POLG,91,92 whichcause impairment of the replicative machinery.93
Recessive mutations in thymidine phosphorylase cause mitochondrialneurogastrointestinal encephalopathy, characterized by mtDNA deple-tion, multiple deletions and point mutations. mtDNA depletion has alsobeen identified in early onset hypotonia with myopathy and hepaticinvolvement, caused by mutations in either thymidine kinase (TK2) ordeoxyguanosine kinase (DGUOK).94 Mutations in both of these genescause a reduction in the mtDNA nucleotide pooling, reducing replicationefficiency.
Disorders resulting frommutations in nuclear-encoded componentsor assembly factors of the OXPHOS systemIsolated complex I deficiency is by far the commonest biochemical defectfound in mitochondrial disorders; however, it is also the most complex
Mitochondrial genetics
British Medical Bulletin 2013;106 147

aetiology and clinical spectrum.95 Complex I deficiency is associated witha broad range of clinical phenotypes ranging from lethal neonatal diseaseto adult onset neurodegenerative disorders.96,97 A high level of geneticheterogeneity, coupled with weak genotype–phenotype correlations,makes it difficult to predict the genetic basis on pure clinical grounds.95
This is important because of the different inheritance patterns and differ-ent natural histories of the different genetic causes. However, somepatterns are starting to emerge.There are at least 46 nuclear-encoded subunits of complex I (compared
with 7 mtDNA encoded subunits) and so it is unsurprising that nDNAmutations have been identified in 14 of the structural subunits.Pathogenic mutations in NDUFS1,98 NDUFS3,95,99 NDUFS4,100
NDUFS7,101 NDUFS8,102 NDUFV1,98,103 NDUFA10,104 NDUFB395
and NDUFA2105 typically manifest as Leigh or Leigh-like syn-dromes.60,106 Conversely, mutations in NDUFS2,107 NDUFS6,108
NDUFV2,109 NDUFA1, NDUFA11110 and ACAD9111 are typicallyassociated with hypertrophic cardiomyopathy and encephalopathy. Inaddition, mutations in complex I assembly proteins can manifest asdisease: Leigh syndrome (NDUFAF2 and NDUFAF5),112,113 encephal-opathy (NDUFAF4)114 and cardioencephalomyopathy (NDUFAF1).115
Complex II is completely encoded by nDNA and is composed of fourpolypeptide subunits: SHD-A, -B, -C and -D. Mutations in SHD-A arerare, but are associated with Leigh’s syndrome. Surprisingly, mutations inSHD-B, -C and -D appear to be a common cause of inherited paragaglio-mas and phaeochromocytomas.116
Complex III deficiency typically causes a severe multisystem early onsetdisorder, which is recessively inherited and rare.117,118 identified mutationsin BCS1l, a complex III assembly protein, presenting with neonatal prox-imal tubulopathy, hepatic involvement and encephalopathy. Subsequently,a deletion in human ubiquinone–cytochrome c reductase binding proteinof complex III (UQCRB) was identified in a consanguineous family pre-senting with hypoglycaemia and lactic acidosis;119 and a missense muta-tion was identified in UQCRC, a ubiquinone-binding protein, in a largeconsanguineous Israeli-Bedoiun kindred.120 More recently, a mutation inTTC19 (a complex III structural subunit gene) was identified in individualswith a progressive neurodegenerative disorder in late infancy,121 expandingthe phenotype of complex mutations beyond early infant disorders.Mutations in complex IV result in severe, typically fatal, infantile disease
and to date mutations in four complex IV structural subunits have beenidentified. A homozygous mutation in COX6BI, identified in brothersfrom a consanguineous Saudi Arabian family, presented with gait instabil-ities visual disturbances, progressive neurological deterioration and leuko-dystrophic brain changes.122 Mutations in COX10, a homologue of yeasthaem A:farneslytransferase, are associated with Leigh syndrome123,124 and
P. F. Chinnery and G. Hudson
148 British Medical Bulletin 2013;106

a multisystem disorder.123 Atypically, mutations in COX7B125 are asso-ciated with facial dysmorphisms and congenital abnormalities,126 and asingle mutation in the structural subunit gene, COX4I2, was identified inadult Arab Muslim patients with exocrine pancreatic insufficiency, dysery-thropoietic anaemia and calvarial hyperostosis.127
In contrast, a number of mutations have been identified in complex IVassembly factors. Complex IV assembly gene disorders include SURF1(Surfeit locus protein 1), associated with Leigh Syndrome;128,129
C12ORF62 (chromosome 12 open reading frame 62), associated withfatal, neonatal, mitochondrial IV deficiency;130 COA5 (cytochrome coxidase assembly factor 5), associated with neonatal hypertrophic cardio-myopathy131 and FASTKD2, associated with cytochrome c oxidase-defective encephalomyopathy.132
Mutations in nDNA-encoded complex V subunit genes also appearvery rare. A mutation in ATP5E (ATP synthase, H+ transporting, mito-chondrial F1 complex, epsilon subunit) was identified in an Austrianwoman with complex V deficiency,133 and a single gene defect has beenidentified in the complex V assembly factor gene ATPAF2, resulting inimpaired complex V activity.134
Disorders resulting frommutations affecting mitochondrial translationSeveral nDNA mutations have been identified which influence the effi-ciency of mitochondrial translation. Mitochondrial ribosomal proteinS16 (MRPS16) and mitochondrial ribosomal protein S22 (MRPS22) arecomponents of the mitoribosome. Mutations in these genes are known tocause severe, infantile, lactic acidosis, developmental defects in the brain,and facial dysmorphisms (MRPS16) and fatal neonatal hypertrophiccardiomyopathy and kidney tubulopathy (MRPS22).135
Mutations in PUS1, peudorine synthase 1, have been shown to causemyopathy, lactic acidosis and sideroblastic anaemia.136 The mutation, inthe catalytic core of the protein, is thought to disrupt the conversion ofuridine to pseudouridine, required for tRNA synthesis.
Disorders due to defects in genes controlling mitochondrialnetwork dynamicsMutations in OPA1 are primarily a cause of optic atrophy,66 but add-itional phenotypes, such as deafness and neuromuscular disease, havealso been seen. Interestingly, mutations inOPA1 also appear to cause theformation of mtDNA deletions, indicating that Opa1 is also important tomtDNA maintenance.Much like OPA1, defects in MFN2 cause a disturbance of mtDNA
maintenance through impairment of mitochondrial network dynamics.66
Mutations in MFN2 are typically associated with Charcot-Marie-Tooth
Mitochondrial genetics
British Medical Bulletin 2013;106 149

disease (CMT2A) and hereditary motor and sensory neuropathy (CMTwith HMSN type VI).66
DNM1L (dynamin 1-like), another GTPase, is required for fission ofmitochondria.137 To date, only a singleDNM1L has been identified in aninfant presenting with both defective mitochondrial and peroxisomalfission.138 The patient presented in the first days of life with severe micro-cephaly, abnormal brain development, optic atrophy with hyperplasiaand lactic acidemia.138
Areas of controversy?
The mitochondrial bottleneck
Mutations in mtDNA are often heteroplasmic, with severity correlatingwith increasing percentage of mutant. Observations indicate that theamount of a variant inherited from a heteroplasmic mother varies betweenoffspring.139,140 This is important when investigating disease aetiology, asan asymptomatic mother, with a sub-clinical heteroplasmy level, can givebirth to children with significantly higher levels of an mtDNAmutation.The ‘mitochondrial bottleneck theory’ attempts to explain this phe-
nomenon.140 Briefly, the reduction of mtDNA during early development‘redistributes’ mtDNA to daughter cells (effectively sharing mtDNAcontent amongst daughter cells). Oocyte maturation is associated withthe rapid replication of mtDNA. This reduction-amplification leads toa purportedly random shift in mtDNA mutational load between cells.Researchers agree that the bottleneck is due to a rapid reduction inmtDNA levels during embryonic development; however, the exact mech-anism of segregation is hotly debated. There are currently three leadingtheories of the mtDNA bottleneck mechanism:140 (i) variation in hetero-plasmy is due to an unequal segregation of mtDNA during cell division,(ii) variation in heteroplasmy is due to an unequal segregation of mtDNAnucleoids during cell division and (iii) variation in heteroplasmy is due tothe selective replication of a specific sub-population of mtDNA.
Growing points
Assigning variant causality
Optimal mitochondrial function requires the synergistic cooperation ofboth mtDNA and nDNA; hence, the investigation of dysfunction requiresthe interrogation of both genomes. Correctly determining the pathogen-icity of potential mutants (in either genome) is critical to understanding
P. F. Chinnery and G. Hudson
150 British Medical Bulletin 2013;106

mitochondrial disease. This underpins the genetic counselling and subse-quent prenatal diagnosis of mitochondrial disorders.Despite the complexity of both mtDNA point mutations and deletions,
as well as the potential for heteroplasmy, assigning pathogenicity tomtDNA variants is analogous to nDNA mutations and is comprehensive-ly described by DiMauro and Schon.141 Briefly, the mutation must bepresent in cases significantly more than asymptomatic controls; if hetero-plasmic, the proportion of mutated mtDNA must be higher in patientscompared with controls (and subsequently higher in clinically affectedtissues compared with unaffected tissues). More importantly, the mutatedmtDNA must segregate with defined clinical outcome (described previ-ously). Other criteria, such as evolutionary conservation must be inter-preted with care, as very rare neutral variants (so-called ‘privatepolymorphisms’) or homoplasmic changes (such as in LHON) may bewrongly miss-classified using this approach.141 Assigning pathogenicityto tRNA mutations is slightly more challenging; tRNA variants arecommon; however, a small number of tRNA mutations are responsiblefor a disproportionate majority of mitochondrial disease.77 McFarlandet al.77 provide a comprehensive scoring system which can be used toaccurately determine tRNA mutation pathogenicity.Whole-exome sequencing (WES)142 has emerged as the preferred
method for identifying Mendelian disease genes, and is proving valuablein the diagnostic evaluation of phenotypically and genetically heteroge-neous disorders such as mitochondrial disease.95,143 Initially, candidatemutations can be identified by prioritizing known mitochondrial genes,such as the 1500 proposed in ‘MitoCarta’144 or Mitop2.145 Secondly,WES can drive the discovery of novel mitochondrial disease genes orprovide a link to previous disease genes that demonstrate an overlappingclinical phenotype.146–151 However, as with all new technologies, caremust be taken when interpreting WES data in novel disease genes.Variants identified in poorly characterized genes will require extensivebiochemical and functional laboratory analysis to assign causality.Additionally, WES is not wholly comprehensive, not capturing non-coding or regulatory regions and often failing to sequence large portionsof the exome.142,152 However, as technology improves and bioinformaticanalysis becomes streamlined, WES is likely to become a major facet inindentifying nuclear genes that affect mitochondrial function.
Managing mitochondrial disease
There are limited treatment options for patients with mitochondrialdiseases. The main emphasis is on disease prevention and the manage-ment of complications. Effective genetic counselling, especially given a
Mitochondrial genetics
British Medical Bulletin 2013;106 151

family history of mitochondrial disease, is crucial. However, the clinic-al variability, coupled with the unpredictable inheritance of a hetero-plasmic ‘mutant dose’ (through the bottleneck), makes a definitediagnosis difficult.153,154
Empiric recurrence risks are available for common homoplasmic muta-tions (i.e. for LHON), but genetic counselling for heteroplasmic mutationsis difficult because of the genetic bottleneck (described earlier). Increasedknowledge of the natural history of specific mitochondrial disorders hasinformed clinical practice. Particular attention to cardiac, ophthalmologic-al and endocrine complications (especially diabetes), can lead to promptsupportive management.155 However, there are no specific disease-modifying treatments at present, although some drugs show promise.156
An area that has had some in vitro and pre-clinical success is the develop-ment of ‘gene therapies’.157 There are currently three strategies for applyinggene therapy to mitochondrial disease: (i) the rescue of an RC defect by ex-pression of a ‘replacement’ gene product from the nucleus (so-called alloto-pic and xenotpoic expression,158,159 (ii) the rescue of a primarymitochondrial defect by importing ‘wild-type’ mtDNA into mitochondria(so-called mtDNA transfection) and (iii) manipulation of the heteroplasmicmtDNA balance (i.e. adjusting the wild-type:mutant type ratio), which canbe achieved by improving a patients exercise regime.160
More recently, and although in very early stages, allogenic haematopoi-etic stem cell therapy has been successfully used to treat mitochondrialneurogastrointestinal encephalomyopathy, but associated with high mor-tality.161 Similarly, liver transplants in patients (typically children) suffer-ing from MPV17-associated hepatocerebral mitochondrial depletionsyndrome have a poor prognosis.162
Pre-implantation genetic diagnosis can assist female heteroplasmicmtDNA mutation carriers in determining the risk to their offspring,assisting by preventing transmission of deleterious mtDNA.163,164
Briefly, embryos obtained after in vitro fertilization are analysed and onlythose with very low-level mutant levels are transferred to the uterus.However, these techniques are of little help to woman harbouringintermediate-level heteroplasmic mtDNA mutations, where uncertaintyregarding the clinical mutation threshold remains.163
Advances, harnessing ‘pro-nuclear transfer’, have made significant stepstowards treating primary mitochondrial disease at a mtDNA level.165
Briefly, the technique involves the transfer of nDNA from a donor zygote(from the mtDNA mutation carrier mother) to an enucleated recipientzygote via fusion. The new ‘reconstructed zygote’ retains the nDNA fromthe mother, but the mtDNA from a donor. More recently, a competinggroup has attempted a similar technique, utilizing ‘spindle transfer’ ofnDNA to an enucleated donor.166 Unlike pro-nuclear transfer, nDNA iso-lation occurs pre-fertilization, meaning once the technique is approved it
P. F. Chinnery and G. Hudson
152 British Medical Bulletin 2013;106

can be integrated into established in vitro fertilization techniques.However, caution is advised, as both pro-nuclear transfer and spindletransfer would only benefit a minority of female mtDNAmutation carriers,whereas prenatal diagnostic testing can be utilized for both all Mendelianmitochondrial disorders and the majority of mtDNAmutations.163,167
Funding
Funding to pay the Open Access publication charges for this article wasprovided by The Wellcome Trust.
References
1 van der GiezenM, Tovar J. Degenerate mitochondria. EMBORep 2005;6:525–30.2 Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic
mutations.Nat Rev Genet 2012;13:878–90.3 Schapira AH.Mitochondrial diseases. Lancet 2012;379:1825–34.4 Koopman WJ, Distelmaier F, Smeitink JA et al. OXPHOS mutations and neurodegeneration.
EMBO J 2013;32:9–29.5 Andrews RM, Kubacka I, Chinnery PF et al. Reanalysis and revision of the Cambridge refer-
ence sequence for human mitochondrial DNA.Nat Genet 1999;23:147.6 Temperley R, Richter R, Dennerlein S et al. Hungry codons promote frameshifting in human
mitochondrial ribosomes. Science 2010;327:301.7 Sutovsky P. Ubiquitin-dependent proteolysis in mammalian spermatogenesis, fertilization, and
sperm quality control: killing three birds with one stone.Microsc Res Tech 2003;61:88–102.8 Vissing JSaM. Paternal Inheritance if mitochondrial DNA.N Engl J Med 2002;2081–2.9 Miller FJ, Rosenfeldt FL, Zhang C et al. Precise determination of mitochondrial DNA copy
number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copynumber with age.Nucleic Acids Res 2003;31:e61.
10 Birky CW Jr. The inheritance of genes in mitochondria and chloroplasts: laws, mechanisms,and models. Annu Rev Genet 2001;35:125–48.
11 Payne BA, Wilson IJ, Yu-Wai-Man P et al. Universal heteroplasmy of human mitochondrialDNA.HumMol Genet 2013;22:384–90.
12 Calvo S, Jain M, Xie X et al. Systematic identification of human mitochondrial disease genesthrough integrative genomics.Nat Genet 2006;38:576–82.
13 Hirst J. Why does mitochondrial complex I have so many subunits? Biochem J 2011;437:e1–3.14 Angerer H, Zwicker K, Wumaier Z et al. A scaffold of accessory subunits links the peripheral
arm and the distal proton-pumping module of mitochondrial complex I. Biochem J2011;437:279–88.
15 Smith PM, Fox JL, Winge DR. Biogenesis of the cytochrome bc(1) complex and role of assem-bly factors. Biochim Biophys Acta 2012;1817:276–86.
16 Vogel RO, Smeitink JA, Nijtmans LG. Human mitochondrial complex I assembly: a dynamicand versatile process. Biochim Biophys Acta 2007;1767:1215–27.
17 Mick DU, Fox TD, Rehling P. Inventory control: cytochrome c oxidase assembly regulatesmitochondrial translation.Nat Rev Mol Cell Biol 2011;12:14–20.
18 Wang ZG, White PS, Ackerman SH. Atp11p and Atp12p are assembly factors for the F(1)-ATPase in human mitochondria. J Biol Chem 2001;276:30773–8.
19 Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit RevBiochemMol Biol 2012;47:64–74.
20 Bogenhagen DF. Mitochondrial DNA nucleoid structure. Biochim Biophys Acta2012;1819:914–20.
Mitochondrial genetics
British Medical Bulletin 2013;106 153

21 Kukat C, Wurm CA, Spahr H et al. Super-resolution microscopy reveals that mammalian mito-chondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. ProcNatl Acad Sci USA 2011;108:13534–9.
22 McCulloch V, Seidel-Rogol BL, Shadel GS. A human mitochondrial transcription factor isrelated to RNA adenine methyltransferases and binds S-adenosylmethionine. Mol Cell Biol2002;22:1116–25.
23 McCulloch V, Shadel GS. Human mitochondrial transcription factor B1 interacts with theC-terminal activation region of h-mtTFA and stimulates transcription independently of itsRNA methyltransferase activity.Mol Cell Biol 2003;23:5816–24.
24 Metodiev MD, Lesko N, Park CB et al. Methylation of 12S rRNA is necessary for in vivostability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab2009;9:386–97.
25 Gaspari M, Larsson NG, Gustafsson CM. The transcription machinery in mammalian mito-chondria. Biochim Biophys Acta 2004;1659:148–52.
26 Ma J, Farwell MA, Burkhart WA et al. Cloning and sequence analysis of the cDNA for bovinemitochondrial translational initiation factor 2. Biochim Biophys Acta 1995;1261:321–4.
27 Koc EC, Spremulli LL. Identification of mammalian mitochondrial translational initiationfactor 3 and examination of its role in initiation complex formation with natural mRNAs.J Biol Chem 2002;277:35541–9.
28 Christian B, Haque E, Spremulli L. Ribosome shifting or splitting: it is all up to the EF-G.Mol Cell 2009;35:400–2.
29 Hammarsund M, Wilson W, Corcoran M et al. Identification and characterization of twonovel human mitochondrial elongation factor genes, hEFG2 and hEFG1, phylogenetically con-served through evolution.HumGenet 2001;109:542–50.
30 Ling M, Merante F, Chen HS et al. The human mitochondrial elongation factor tu (EF-Tu)gene: cDNA sequence, genomic localization, genomic structure, and identification of a pseudo-gene.Gene 1997;197:325–36.
31 Smits P, Smeitink J, van den Heuvel L. Mitochondrial translation and beyond: processes impli-cated in combined oxidative phosphorylation deficiencies. J Biomed Biotechnol2010;2010:737385.
32 Zhang Y, Spremulli LL. Identification and cloning of human mitochondrial translationalrelease factor 1 and the ribosome recycling factor. Biochim Biophys Acta 1998;1443:245–50.
33 Soleimanpour-Lichaei HR, Kuhl I, Gaisne M et al. mtRF1a is a human mitochondrial transla-tion release factor decoding the major termination codons UAA and UAG. Mol Cell2007;27:745–57.
34 Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science2012;337:1062–5.
35 Schaefer AM, McFarland R, Blakely EL et al. Prevalence of mitochondrial DNA disease inadults. Ann Neurol 2008;63:35–9.
36 Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratorychain disorders in children. Brain 2003;126(Pt 8):1905–12.
37 Majamaa K, Moilanen JS, Uimonen S et al. Epidemiology of A3243G, the mutation for mito-chondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the muta-tion in an adult population. Am J HumGenet 1998;63:447–54.
38 Darin N, Oldfors A, Moslemi AR et al. The incidence of mitochondrial encephalomyopathiesin childhood: clinical features and morphological, biochemical, and DNA anbormalities.Ann Neurol 2001;49:377–83.
39 Magner M, Szentivanyi K, Svandova I et al. Elevated CSF-lactate is a reliable marker of mito-chondrial disorders in children even after brief seizures. Eur J Paediatr Neurol 2011;15:101–8.
40 Barkovich AJ, Good WV, Koch TK et al. Mitochondrial disorders: analysis of their clinical andimaging characteristics. AJNR Am J Neuroradiol 1993;14:1119–37.
41 Lin DD, Crawford TO, Barker PB. Proton MR spectroscopy in the diagnostic evaluation ofsuspected mitochondrial disease. AJNR Am J Neuroradiol 2003;24:33–41.
42 Leonard JV, Schapira AH. Mitochondrial respiratory chain disorders I: mitochondrial DNAdefects. Lancet 2000;355:299–304.
43 Leonard JV, Schapira AH. Mitochondrial respiratory chain disorders II: neurodegenerativedisorders and nuclear gene defects. Lancet 2000;355:389–94.
P. F. Chinnery and G. Hudson
154 British Medical Bulletin 2013;106

44 Rahman S, Poulton J, Marchington D et al. Decrease of 3243 A–>G mtDNA mutation fromblood in MELAS syndrome: a longitudinal study. Am J HumGenet 2001;68:238–40.
45 Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc 1972;20:145–7.46 HegdeML,Mantha AK, Hazra TK et al. Oxidative genome damage and its repair: implications
in aging and neurodegenerative diseases.Mech Ageing Dev 2012;133:157–68.47 Howell N. Origin, cellular expression, and cybrid transmission of mitochondrial CAP-R,
PYR-IND, and OLI-R mutant phenotypes. Somat Cell Genet 1983;9:1–24.48 Chinnery PF, Howell N, Lightowlers RN et al. Molecular pathology of MELAS and
MERRF. The relationship between mutation load and clinical phenotypes. Brain 1997;120:1713–21.
49 White SL, Collins VR, Wolfe R et al. Genetic counseling and prenatal diagnosis for the mito-chondrial DNAmutations at nucleotide 8993. Am J HumGenet 1999;65:474–82.
50 Weber K, Wilson JN, Taylor L et al. A new mtDNA mutation showing accumulation with timeand restriction to skeletal muscle. Am J HumGenet 1997;60:373–80.
51 Poulton J, O’Rahilly S, Morten KJ et al. Mitochondrial DNA, diabetes and pancreaticpathology in Kearns-Sayre syndrome.Diabetologia 1995;38:868–71.
52 Torroni A, Huoponen K, Francalacci P et al. Classification of European mtDNAs from ananalysis of three European populations.Genetics 1996;144:1835–50.
53 Pello R, Martin MA, Carelli V et al. Mitochondrial DNA background modulates the assemblykinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Hum Mol Genet2008;17:4001–11.
54 Pyle A, Foltynie T, Tiangyou W et al. Mitochondrial DNA haplogroup cluster UKJT reducesthe risk of PD. Ann Neurol 2005;57:564–7.
55 Santoro A, Balbi V, Balducci E et al. Evidence for sub-haplogroup h5 of mitochondrial DNA asa risk factor for late onset Alzheimer’s disease. PLoS One 2010;5:e12037.
56 Ridge PG, Maxwell TJ, Corcoran CD et al. Mitochondrial genomic analysis of late onsetAlzheimer’s disease reveals protective haplogroups H6A1A/H6A1B: the Cache County StudyonMemory in Aging. PLoS One 2012;7:e45134.
57 Udar N, Atilano SR, Memarzadeh M et al. Mitochondrial DNA haplogroups associated withage-related macular degeneration. Invest Ophthalmol Vis Sci 2009;50:2966–74.
58 Chinnery PF, Elliott HR, Hudson G et al. Epigenetics, epidemiology and mitochondrial DNAdiseases. Int J Epidemiol 2012;41:177–87.
59 Ghezzi D, Zeviani M. Assembly factors of human mitochondrial respiratory chain complexes:physiology and pathophysiology. Adv Exp Med Biol 2012;748:65–106.
60 MITOMAP. A human mitochondrial genome database, 2000. http://www.gen.emory.edu/mitomap.html.
61 Rossignol R, Faustin B, Rocher C et al. Mitochondrial threshold effects. Biochem J 2003;370(Pt 3):751–62.
62 Schon EA, Rizzuto R, Moraes CT et al. A direct repeat is a hotspot for large-scale deletion ofhuman mitochondrial DNA. Science 1989;244:346–9.
63 Krishnan KJ, Reeve AK, Samuels DC et al. What causes mitochondrial DNA deletions inhuman cells?Nat Genet 2008;40:275–9.
64 Wallace DC, Singh G, Lott MT et al. Mitochondrial DNA mutation associated with Leber’shereditary optic neuropathy. Science 1988;242:1427–30.
65 Carelli V, Ghelli A, Ratta M et al. Leber’s hereditary optic neuropathy: biochemical effect of11778/ND4 and 3460/ND1 mutations and correlation with the mitochondrial genotype.Neurology 1997;48:1623–32.
66 Carelli V, La Morgia C, Valentino ML et al. Retinal ganglion cell neurodegeneration in mito-chondrial inherited disorders. Biochim Biophys Acta 2009;1787:518–28.
67 Hudson G, Keers S, Yu Wai Man P et al. Identification of an X-chromosomal locus and haplo-type modulating the phenotype of a mitochondrial DNA disorder. Am J Hum Genet2005;77:1086–91.
68 Achilli A, Iommarini L, Olivieri A et al. Rare primary mitochondrial DNA mutations andprobable synergistic variants in Leber’s hereditary optic neuropathy. PLoS One 2012;7:e42242.
69 KirkmanMA, Yu-Wai-Man P, Korsten A et al. Gene-environment interactions in Leber heredi-tary optic neuropathy. Brain 2009;132(Pt 9):2317–26.
Mitochondrial genetics
British Medical Bulletin 2013;106 155

70 Hudson G, Carelli V, Spruijt L et al. Clinical expression of Leber hereditary optic neuropathy isaffected by the mitochondrial DNA-haplogroup background. Am J Hum Genet2007;81:228–33.
71 Carvalho MR, Muller B, Rotzer E et al. Leber’s hereditary optic neuroretinopathy and theX-chromosomal susceptability factor: no linkage to DXS7.HumHered 1992;42:316–20.
72 Chen JD, Cox I, Denton MJ. Prelimanary exclusion of an X-linked gene in Leber optic atrophyby linkage analysis.HumGenet 1989;82:302–7.
73 Abu-Amero K, Jaber M, Hellani A et al. Genome-wide expression profile of LHON patientswith the 11778 mutation. Br J Ophthalmol (2009/09/04 ed), 2009.
74 Phasukkijwatana N, Kunhapan B, Stankovich J et al. Genome-wide linkage scan and associ-ation study of PARL to the expression of LHON families in Thailand. Hum Genet (2010/04/22 ed).
75 Prezant TR, Agapian JV, Bohlman MC et al. Mitochondrial ribosomal RNA mutationassociated with both antibiotic-induced and non-syndromic deafness. Nat Genet 1993;4:289–94.
76 Zifa E, Giannouli S, Theotokis P et al. Mitochondrial tRNA mutations: clinical and functionalperturbations. RNA Biol 2007;4:38–66.
77 McFarland R, Elson JL, Taylor RW et al. Assigning pathogenicity to mitochondrial tRNAmutations: when ‘definitely maybe’ is not good enough. Trends Genet 2004;20:591–6.
78 Pavlakis SG, Phillips PC, DiMauro S et al. Mitochondrial myopathy, encephalopathy, lacticacidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol 1984;16:481–8.
79 Hirano M, Ricci E, Koenigsberger MR et al. Melas: an original case and clinical criteria fordiagnosis.Neuromuscul Disord 1992;2:125–35.
80 Manwaring N, Jones MM,Wang JJ et al. Population prevalence of the MELAS A3243G muta-tion.Mitochondrion 2007;7:230–3.
81 Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with theMELAS subgroup of mitochondrial encephalomyopathies.Nature 1990;348:651–3.
82 Santorelli FM, Tanji K, Kulikova R et al. Identification of a novel mutation in the mtDNAND5 gene associated with MELAS. Biochem Biophys Res Commun 1997;238:326–8.
83 Shoffner JM, Lott MT, Lezza AM et al. Myoclonic epilepsy and ragged-red fiber disease(MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 1990;61:931–7.
84 Zeviani M, Moraes CT, DiMauro S et al. Deletions of mitochondrial DNA in Kearns-Sayresyndorme.Neurology 1988;38:1339–46.
85 Moraes CT, DiMauro S, Zeviani M et al. Mitochondrial DNA deletions in progressive externalophthalmoplegia and Kearns-Sayre syndrome.N Engl J Med 1989;320:1293–9.
86 Rotig A, Cormier V, Blanche S et al. Pearson’s marrow-pancreas syndrome. A multisystemmitochondrial disorder in infancy. J Clin Invest 1990;86:1601–8.
87 Horvath R, Hudson G, Ferrari G et al. Phenotypic spectrum associated with mutations of themitochondrial polymerase gamma gene. Brain 2006;129(Pt 7):1674–84.
88 Spelbrink JN, Li FY, Tiranti V et al. Human mitochondrial DNA deletios associated with muta-tions in the gene encoding Twinkle, a phage T7 gene4-like protein localized in mitochondria.Nat Genet 2001;28:223–31.
89 Agostino A, Valletta L, Chinnery PF et al. Mutations of ANT1, Twinkle, and POLG1 in spor-adic progressive external ophthalmoplegia (PEO).Neurology 2003;60:1354–6.
90 Wolf A, Cowen D. The cerebral atrophies and encephalomalacias of infancy and childhood.Res Publ Assoc Res Nerv Ment Dis 1955;34:199–330.
91 Kollberg G, Moslemi AR, Darin N et al. POLG1 mutations associated with progressive enceph-alopathy in childhood. J Neuropathol Exp Neurol 2006;65:758–68.
92 Naviaux RK, Nguyen KV. POLG mutations associated with Alpers’ syndrome and mitochon-drial DNA depletion. Ann Neurol 2004;55:706–12.
93 Clayton DA. Replication of animal mitochondrial DNA. Cell 1982;28:693–705.94 Spinazzola A, Zeviani M. Disorders of nuclear-mitochondrial intergenomic communication.
Biosci Rep 2007;27:39–51.95 Haack TB, Haberberger B, Frisch EM et al. Molecular diagnosis in mitochondrial complex I
deficiency using exome sequencing. J Med Genet 2012;49:277–83.96 Loeffen JL, Smeitink JA, Trijbels JM et al. Isolated complex I deficiency in children: clinical,
biochemical and genetic aspects.HumMutat 2000;15:123–34.
P. F. Chinnery and G. Hudson
156 British Medical Bulletin 2013;106

97 Lebre AS, Rio M, Faivre d’Arcier L et al. A common pattern of brain MRI imaging in mito-chondrial diseases with complex I deficiency. J Med Genet 2011;48:16–23.
98 Benit P, Chretien D, Kadhom N et al. Large-scale deletion and point mutations of the nuclearNDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am J Hum Genet2001;68:1344–52.
99 Benit P, Slama A, Cartault F et al. Mutant NDUFS3 subunit of mitochondrial complex I causesLeigh syndrome. J Med Genet 2004;41:14–7.
100 van den Heuvel L, Ruitenbeek W, Smeets R et al. Demonstration of a new pathogenic mutationin human complex I deficiency: a 5-bp duplication in the nuclear gene encoding the 18-kD(AQDQ) subunit. Am J HumGenet 1998;62:262–8.
101 Smeitink J, van den Heuvel L. Human mitochondrial complex I in health and disease. Am JHumGenet 1999;64:1505–10.
102 Loeffen J, Smeitink J, Triepels R et al. The first nuclear-encoded complex I mutation in apatient with Leigh syndrome. Am J HumGenet 1998;63:1598–608.
103 Schuelke M, Smeitink J, Mariman E et al. Mutant NDUFV1 subunit of mitochondrial complexI causes leukodystrophy and myoclonic epilepsy.Nat Genet 1999;21:260–1.
104 Hoefs SJ, van Spronsen FJ, Lenssen EW et al. NDUFA10 mutations cause complex I deficiencyin a patient with Leigh disease. Eur J HumGenet 2011;19:270–4.
105 Hoefs SJ, Dieteren CE, Distelmaier F et al. NDUFA2 complex I mutation leads to Leighdisease. Am J HumGenet 2008;82:1306–15.
106 Shoubridge EA. Nuclear genetic defects of oxidative phosphorylation. Hum Mol Genet2001;10:2277–84.
107 Loeffen J, Elpeleg O, Smeitink J et al. Mutations in the complex I NDUFS2 gene of patientswith cardiomyopathy and encephalomyopathy. Ann Neurol 2001;49:195–201.
108 Kirby DM, Salemi R, Sugiana C et al. NDUFS6 mutations are a novel cause of lethal neonatalmitochondrial complex I deficiency. J Clin Invest 2004;114:837–45.
109 Benit P, Beugnot R, Chretien D et al. Mutant NDUFV2 subunit of mitochondrial complex Icauses early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat2003;21:582–6.
110 Berger I, Hershkovitz E, Shaag A et al. Mitochondrial complex I deficiency caused by a deleteri-ous NDUFA11 mutation. Ann Neurol 2008;63:405–8.
111 Haack TB, Danhauser K, Haberberger B et al. Exome sequencing identifies ACAD9 mutationsas a cause of complex I deficiency.Nat Genet 2010;42:1131–4.
112 Calvo SE, Tucker EJ, Compton AG et al. High-throughput, pooled sequencing identifiesmutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet 2010;42:851–8.
113 Gerards M, Sluiter W, van den Bosch BJ et al. Defective complex I assembly due to C20orf7mutations as a new cause of Leigh syndrome. J Med Genet 2010;47:507–12.
114 Saada A, Edvardson S, Rapoport M et al. C6ORF66 is an assembly factor of mitochondrialcomplex I. Am J HumGenet 2008;82:32–8.
115 Dunning CJ, McKenzie M, Sugiana C et al. Human CIA30 is involved in the early assemblyof mitochondrial complex I and mutations in its gene cause disease. EMBO J 2007;26:3227–37.
116 Baysal BE. Hereditary paraganglioma targets diverse paraganglia. J Med Genet 2002;39:617–22.
117 Petruzzella V, Tiranti V, Fernandez P et al. Identification and characterization of humancDNAs specific to BCS1, PET112, SCO1, COX15, and COX11, five genes involved in the for-mation and function of the mitochondrial respiratory chain.Genomics 1998;54:494–504.
118 de Lonlay P, Valnot I, Barrientos A et al. A mutant mitochondrial respiratory chain assemblyprotein causes complex III deficiency in patients with tubulopathy, encephalopathy and liverfailure.Nat Genet 2001;29:57–60.
119 Haut S, Brivet M, Touati G et al. A deletion in the human QP-C gene causes a complex III defi-ciency resulting in hypoglycaemia and lactic acidosis.HumGenet 2003;113:118–22.
120 Barel O, Shorer Z, Flusser H et al. Mitochondrial complex III deficiency associated with ahomozygous mutation in UQCRQ. Am J HumGenet 2008;82:1211–6.
121 Ghezzi D, Arzuffi P, Zordan M et al. Mutations in TTC19 cause mitochondrial complex IIIdeficiency and neurological impairment in humans and flies.Nat Genet 2011;43:259–63.
Mitochondrial genetics
British Medical Bulletin 2013;106 157

122 Massa V, Fernandez-Vizarra E, Alshahwan S et al. Severe infantile encephalomyopathy causedby a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am J HumGenet 2008;82:1281–9.
123 Antonicka H, Leary SC, Guercin GH et al. Mutations in COX10 result in a defect in mitochon-drial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associatedwith isolated COX deficiency.HumMol Genet 2003;12:2693–702.
124 Coenen MJ, van den Heuvel LP, Ugalde C et al. Cytochrome c oxidase biogenesis in a patientwith a mutation in COX10 gene. Ann Neurol 2004;56:560–4.
125 Indrieri A, van Rahden VA, Tiranti V et al. Mutations in COX7B cause microphthalmia withlinear skin lesions, an unconventional mitochondrial disease. Am J Hum Genet2012;91:942–9.
126 Zvulunov A, Kachko L, Manor E et al. Reticulolinear aplasia cutis congenita of the face andneck: a distinctive cutaneous manifestation in several syndromes linked to Xp22. Br JDermatol 1998;138:1046–52.
127 Shteyer E, Saada A, Shaag A et al. Exocrine pancreatic insufficiency, dyserythropoeitic anemia,and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am J Hum Genet2009;84:412–7.
128 Zhu Z, Yao J, Johns T et al. SURF1, encoding a factor involved in the biogenesis of cytochromec oxidase, is mutated in Leigh syndrome.Nat Genet 1998;20:337–43.
129 Tiranti V, Jaksch M, Hofmann S et al. Loss-of-function mutations of SURF-1 are specificallyassociated with Leigh syndrome with cytochrome c oxidase deficiency. Ann Neurol1999;46:161–6.
130 Weraarpachai W, Sasarman F, Nishimura T et al. Mutations in C12orf62, a factor that couplesCOX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am JHumGenet 2012;90:142–51.
131 Huigsloot M, Nijtmans LG, Szklarczyk R et al. A mutation in C2orf64 causes impaired cyto-chrome c oxidase assembly and mitochondrial cardiomyopathy. Am J Hum Genet2011;88:488–93.
132 Ghezzi D, Saada A, D’Adamo P et al. FASTKD2 nonsense mutation in an infantile mitochon-drial encephalomyopathy associated with cytochrome c oxidase deficiency. Am J Hum Genet2008;83:415–23.
133 Mayr JA, Havlickova V, Zimmermann F et al. Mitochondrial ATP synthase deficiency due to amutation in the ATP5E gene for the F1 epsilon subunit.HumMol Genet 2010;19:3430–9.
134 DeMeirleir L, Seneca S, LissensW et al. Respiratory chain complex V deficiency due to a muta-tion in the assembly gene ATP12. J Med Genet 2004;41:120–4.
135 Saada A, Shaag A, Arnon S et al. Antenatal mitochondrial disease caused by mitochondrialribosomal protein (MRPS22) mutation. J Med Genet 2007;44:784–6.
136 Zeharia A, Fischel-Ghodsian N, Casas K et al. Mitochondrial myopathy, sideroblastic anemia,and lactic acidosis: an autosomal recessive syndrome in Persian Jews caused by a mutation inthe PUS1 gene. J Child Neurol 2005;20:449–52.
137 Smirnova E, Shurland DL, Ryazantsev SN et al. A human dynamin-related protein controls thedistribution of mitochondria. J Cell Biol 1998;143:351–8.
138 Waterham HR, Koster J, van Roermund CW et al. A lethal defect of mitochondrial and peroxi-somal fission.N Engl J Med 2007;356:1736–41.
139 Cree LM, Samuels DC, de Sousa Lopes SC et al. A reduction of mitochondrial DNA moleculesduring embryogenesis explains the rapid segregation of genotypes. Nat Genet2008;40:249–54.
140 Carling PJ, Cree LM, Chinnery PF. The implications of mitochondrial DNA copy number regu-lation during embryogenesis.Mitochondrion 2011;11:686–92.
141 DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. Am J Med Genet2001;106:18–26.
142 Clark MJ, Chen R, Lam HY et al. Performance comparison of exome DNA sequencing tech-nologies.Nat Biotechnol 2011;29:908–14.
143 McCormick E, Place E, Falk MJ. Molecular Genetic Testing for Mitochondrial Disease: FromOne Generation to the Next.Neurotherapeutics 2013;10:251–61.
144 Pagliarini DJ, Calvo SE, Chang B et al. A mitochondrial protein compendium elucidatescomplex I disease biology. Cell 2008;134:112–23.
P. F. Chinnery and G. Hudson
158 British Medical Bulletin 2013;106

145 Prokisch H, Andreoli C, Ahting U et al. MitoP2: the mitochondrial proteome database–now in-cluding mouse data.Nucleic Acids Res 2006;34(database issue):D705–11.
146 Pierson TM, Adams D, Bonn F et al. Whole-exome sequencing identifies homozygous AFG3L2mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases.PLoS Genet 2011;7:e1002325.
147 Vedrenne V, Gowher A, De Lonlay P et al. Mutation in PNPT1, which encodes a polyribonu-cleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am J HumGenet 2012;91:912–8.
148 Janer A, Antonicka H, Lalonde E et al. An RMND1 Mutation causes encephalopathy asso-ciated with multiple oxidative phosphorylation complex deficiencies and a mitochondrialtranslation defect. Am J HumGenet 2012;91:737–43.
149 Casey JP, McGettigan P, Lynam-Lennon N et al. Identification of a mutation in LARS as anovel cause of infantile hepatopathy.Mol Genet Metab 2012;106:351–8.
150 Galmiche L, Serre V, Beinat M et al. Exome sequencing identifies MRPL3 mutation in mito-chondrial cardiomyopathy.HumMutat 2011;32:1225–31.
151 Gotz A, Tyynismaa H, Euro L et al. Exome sequencing identifies mitochondrial alanyl-tRNAsynthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet2011;88:635–42.
152 Pfeffer G, Elliott HR, Griffin H et al. Titin mutation segregates with hereditary myopathy withearly respiratory failure. Brain 2012;135(Pt 6):1695–713.
153 Chinnery PF, Thorburn DR, Samuels DC et al. The inheritance of mitochondrial DNA hetero-plasmy: random drift, selection or both? Trends Genet 2000;16:500–5.
154 Monnot S, Gigarel N, Samuels DC et al. Segregation of mtDNA throughout human embryofe-tal development: m.3243A>G as a model system.HumMutat 2011;32:116–25.
155 Pfeffer G, Majamaa K, Turnbull DM et al. Treatment for mitochondrial disorders. CochraneDatabase Syst Rev 2012;4:CD004426.
156 Klopstock T, Yu-Wai-Man P, Dimitriadis K et al. A randomized placebo-controlled trial ofidebenone in Leber’s hereditary optic neuropathy. Brain 2011;134:2677–86.
157 Kyriakouli DS, Boesch P, Taylor RW et al. Progress and prospects: gene therapy for mitochon-drial DNA disease.Gene Ther 2008;15:1017–23.
158 Nagley P, Farrell LB, Gearing DP et al. Assembly of functional proton-translocating ATPasecomplex in yeast mitochondria with cytoplasmically synthesized subunit 8, a polypeptide nor-mally encoded within the organelle. Proc Natl Acad Sci USA 1988;85:2091–5.
159 Bonnet C, Kaltimbacher V, Ellouze S et al. Allotopic mRNA localization to the mitochondrialsurface rescues respiratory chain defects in fibroblasts harboring mitochondrial DNA muta-tions affecting complex I or v subunits. Rejuvenation Res 2007;10:127–44.
160 Taivassalo T, Gardner JL, Taylor RW et al. Endurance training and detraining in mito-chondrial myopathies due to single large-scale mtDNA deletions. Brain 2006;129(Pt 12):3391–401.
161 Halter J, Schupbach WM, Casali C et al. Allogeneic hematopoietic SCT as treatment option forpatients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensusconference proposal for a standardized approach. Bone Marrow Transplant 2011;46:330–7.
162 Wong LJ, Brunetti-Pierri N, Zhang Q et al. Mutations in the MPV17 gene are responsible forrapidly progressive liver failure in infancy.Hepatology 2007;46:1218–27.
163 Hellebrekers DM, Wolfe R, Hendrickx AT et al. PGD and heteroplasmic mitochondrial DNApoint mutations: a systematic review estimating the chance of healthy offspring. Hum ReprodUpdate 2012;18:341–9.
164 Thorburn D, Wilton L, Stock-Myer S. Healthy baby girl born following pre-implantationGenetic diagnosis for mitochondrial DNA m.8993t>g Mutation. Mol Genet Metab2009;98:5–6.
165 Craven L, Tuppen HA, Greggains GD et al. Pronuclear transfer in human embryos to preventtransmission of mitochondrial DNA disease.Nature 2010;465:82–5.
166 Tachibana M, Amato P, Sparman M et al. Towards germline gene therapy of inherited mito-chondrial diseases.Nature 2013;493:627–31.
167 Poulton J, Oakeshott P. Nuclear transfer to prevent maternal transmission of mitochondrialDNA disease. BMJ 2012;345:e6651.
Mitochondrial genetics
British Medical Bulletin 2013;106 159

Mitochondrial disorders caused by mutations in respiratorychain assembly factors
Francisca Diaza,*, Heike Kotarskyb, Vineta Fellmanb,c, and Carlos T. Moraesa
aDepartment of Neurology, University of Miami Miller School of Medicine, Miami, Florida, USAbDepartment of Pediatrics, Clinical Sciences, Lund, Lund University, Lund, Sweden cDepartmentof Pediatrics, University of Helsinki, Helsinki, Finland
SummaryMitochondrial diseases involve the dysfunction of the oxidative phosphorylation (OXPHOS)system. This group of diseases presents with heterogeneous clinical symptoms affecting mainlyorgans with high energy demands. Defects in the multimeric complexes comprising the OXPHOSsystem have a dual genetic origin, mitochondrial or nuclear DNA. Although many nuclear DNAmutations involve genes coding for subunits of the respiratory complexes, the majority ofmutations found to date affect factors that do not form part of the final complexes. These assemblyfactors or chaperones have multiple functions ranging from cofactor insertion to proper assembly/stability of the complexes. Although significant progress has been made in the last few years in thediscovery of new assembly factors, the function of many remains elusive. Here, we describeassembly factors or chaperones that are required for respiratory chain complex assembly and theirclinical relevance.
KeywordsChaperones; Mitochondrial diseases; Newborn infant; Oxidative phosphorylation; Perinataldisorder; Respiratory chain deficiency
IntroductionMitochondrial diseases are usually referred to as disorders with defects in the oxidativephosphorylation (OXPHOS) system. They can be caused by mutations in the nuclear or inthe mitochondrial genome and therefore have distinct patterns of inheritance depending onthe genetic origin. The incidence of mitochondrial DNA (mtDNA) mutations causingdisease has been estimated to be 1 in 5000 children.1 This large number is accompanied bythe recent estimate that 1 in 200 people are carriers of pathogenic mtDNA mutations.2
© 2011 Elsevier Ltd. All rights reserved.*Corresponding author. Addresses: 1420 NW 9th Ave., TSL Building, Room 228, Miami, FL 33136, USA. Tel.: +1 305 243 7489;fax: +1 305 243 6955. [email protected] (F. Diaz).Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to ourcustomers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review ofthe resulting proof before it is published in its final citable form. Please note that during the production process errors may bediscovered which could affect the content, and all legal disclaimers that apply to the journal pertain.Conflict of interest statementNothing to declare.
NIH Public AccessAuthor ManuscriptSemin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
Published in final edited form as:Semin Fetal Neonatal Med. 2011 August ; 16(4): 197–204. doi:10.1016/j.siny.2011.05.004.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Mitochondrial diseases, being heterogeneous in nature and affecting single or multipleorgans, pose a challenge to develop effective treatments. In the last few years new strategieshave emerged and some are currently being tested in clinical trials.3–4 In this review, wefocus on mitochondrial diseases caused by mutations in the proteins with chaperone functionthat are essential for proper assembly and function but which do not form part of the finalcomplex. Mutations causing disease in the neonatal period are summarized in Table 1.
Complex IComplex I (CI), NADH:ubiquinone oxidoreductase or NADH dehydrogenase is the largestof the mitochondrial respiratory complexes comprising 45 subunits. Seven of its subunits areencoded by the mtDNA and the remaining by the nuclear DNA. CI catalyzes the transfer ofelectrons from NADH produced during the tricarboxylic acid cycle to coenzyme Q. Three-dimensional electron microscopy studies revealed that the structure of the complexresembles an ‘L’ with the presence of a peripheral hydrophilic arm (matrix arm) and ahighly hydrophobic membrane arm.5 The assembly of this enzyme is very elaborate, andalthough many steps have been elucidated by studies in mutants of Neurospora crassa andby studies of patients with CI defects, the complete sequence of events remains unknown.Several models have been proposed for the assembly of the holoenzyme that differ in thedetails but agree in inasmuch as the complex is assembled in several modules that go on toform the peripheral and the membrane arms.6,7 Patients with mutations of nuclear originleading to CI defects have severe manifestations during infancy and early childhood,frequently resulting in premature death.8 Numerous mutations in CI nuclear-and mtDNA-encoded subunits have been reported but only a fraction of the CI deficiency cases arecaused by mutations in the structural subunits.9 CI defects are also caused by mutations inauxiliary proteins that do not form part of the final holoenzyme but which assist in theassembly process. Here we describe recently discovered CI chaperones and the clinicalphenotypes associated with their mutations.
Complex I chaperonesNDUFAF1/CIA30—Studies in N. crassa led to the identification of two proteins associatedwith CI assembly intermediates CIA30 and CIA84 that were proposed to have chaperonefunction.10 The CIA30 human homolog NDUFAF1 gene located in chromosome 15q13.3 isubiquitously expressed with slightly increased levels in heart, lung, kidney and liver.11
Knockdown of NDUFAF1 by RNA interference (RNAi) resulted in decreased levels of thefully assembled complex and activity in HEK293 cells. Although NDUFAF1 was found tobe associated with two assembly intermediates of 600 and 700 kDa its exact function has notbeen elucidated.12
The first patient described with a mutation in NDUFAF1 had only 10% of control levels ofthis chaperone as well as a significant reduction of the levels of fully assembled CI. Thispatient was a compound heterozygote with c.1001A→C and c.1140A→G mutationspredicted to cause T207P and K253R amino acid substitutions in highly conservedresidues.13 This patient presented with failure to thrive, hypotonia, growth delay, lacticacidosis and cardiomyopathy after 11 months of age. At 3 years he was diagnosed withWolff–Parkinson–White syndrome and subsequently had visual cortex dysfunction andpigmentary retinopathy at 11 years of age. He developed osteoporosis and kyphoscoliosis atage 16 years and presented mild-to-moderate intellectual disability. At the time of the reportthe patient was 20 years old.13
Ecsit—Tandem affinity purification experiments identified Ecsit (evolutionary conservedsignaling intermediate in Toll pathway) as a binding partner of CIA30 in HEK293 cells.14 Aportion of Ecsit, previously described as a cytosolic protein required for embryonic
Diaz et al. Page 2
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

development and inflammatory signaling, was located in the mitochondria. Thismitochondrial isoform associated with CIA30 and it was found in assembly intermediates ofabout 460 and 830 kDa. Moreover, knockdown of Ecsit by RNAi caused a substantialreduction in the levels of NDUFAF1 and disturbed CI assembly, whereas knockdown ofNDUFAF1 slightly altered Ecsit levels.14 Although Ecsit and NDUFAF1 are found in thesame assembly intermediates and their function is unknown, previous results imply that thetwo proteins have independent functions and mitochondrial Ecsit might be required forNDUFAF1 stability.14
B17.2L/NDUFAF2—By whole genome comparison of fermentative and aerobic yeast, aputative CI assembly factor, B17.2L(NDUFAF2 or NDUFA12L),15 was identified. InYarrowia lipolytica B17.2 is a structural subunit located in the matrix arm of complex Iwhereas its human paralogue B17.2L is a putative assembly factor. The first patientdescribed was homozygous for the mutation (C182T) in B17.2L leading to a premature stopcodon (R45X).15 This infant presented with a progressive leukoencephalopathy withvanishing white matter and cortical and vermian atrophy. At 12 months, the patientpresented horizontal nystagmus, dysconjugated gaze and pale optic disc that eventuallyresulted in severe optic atrophy. She had myopathic facies, lethargy and acute ataxia. Brainlesions in the substantia nigra, mamillo and spinothalamic tracts were observed by magneticresonance imaging (MRI) at 3 years of age but did not affect her intellect. She suffered arespiratory failure at 8 years, remaining comatose and immobile, dying at 13 years of age.Analysis of brain tissue revealed extensive degeneration of white matter accompanied byglia scaring. Muscle biopsy obtained at 3 years of age did not reveal any histologicalabnormality, albeit the biochemical analysis of OXPHOS enzymes revealed a moderate CIdefect in muscle and a more severe defect in fibroblasts. The CI deficiency and the assemblydefect was rescued when B17.2L was expressed in the patient’s fibroblasts.15
B17.2L was associated mainly with a subassembly intermediate of 830 kDa in several CIpatients, in particular in patients with mutations in NDUFV1 and NDUFS4.15,16
Two other patients homozygous for a codon change of methionine (ATG) to leucine (TTG)in position 1 were described.17 The mutation in the first methionine caused the completeabsence of the chaperone and CI activity ranged from 24% to 36% of control in musclemitochondria and 53% in fibroblast.17 Hypotonia, nystagmus and optic atrophy at 8 monthsof age were the initial findings in a boy who later had apneic spells and died at 21 months ofage. MRI studies revealed lesions in the mamillothalamic and spinothalamic tracts, mediallemniscus and substantia nigra whereas cerebellum was only slightly affected and cortex andsubcortical regions were spared. The second patient presented first symptoms at 20 monthsof age with apnea and ophthalmoplegia followed by myoclonic seizures and encountered afatal apnea episode at 2 years of age. MRI studies revealed the same affected areas as in thefirst patient. Plasma and cerebrospinal fluid (CSF) lactate levels were normal in bothpatients.
More recently, Janssen et al.18 reported a patient with a chromosome 5 microdeletion thatencompassed three genes: NDUFAF2, ERCC8 and ELOVL7. ERCC8 encodes the CS-Aprotein that is required for DNA repair whereas ELOVL7 gene encodes an enzyme thatparticipates in the synthesis of the very long chain fatty acid pathway. Fibroblasts from thepatient with the microdeletion had 45% of control CI activity, and expression of NDUFAF2restored the activity to control levels. In the absence of this chaperone, fully assembled CIwas not completely absent and 380 and 480 kDa intermediates were accumulated.Furthermore, some of the levels of CI subunits (NDUFS3, NDUFA9 and NDUFB6) as wellas the chaperone NDUFAF1 were significantly reduced whereas the levels of NDUFAS2were only slightly decreased.18
Diaz et al. Page 3
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Thus, although the function of NDUFAF2/ B17.2L is still obscure, studies suggest that thischaperone might participate in late assembly steps, perhaps by stabilizing assemblyintermediates.
C20orf7—A lethal neonatal CI defect was mapped to the C20orf7 gene.19 The patient washomozygous for a missense mutation causing exchange of a conserved amino acid (L229P).The clinical findings were intrauterine growth restriction, ventricular septation, agenesis ofthe corpus callosum, lactic acidosis, and death by day 7.19
C20orf7 is a matrix protein associated with the inner mitochondrial membrane and itsknockdown resulted in a decrease in CI activity. Fibroblasts from this patient showed CIassembly intermediates of ~400 kDa that contained ND1 but not ND2 subunit, indicatingthat this chaperone is involved in early assembly steps.19
Another investigation of members of a consanguineous family with 11 children revealed thatthree of them presented with a very similar Leigh syndrome (LS) and progressive spasticitywith an onset at 3 years of age.20 At 5 years, increased lactate in CSF and slight atrophy andhypodensity in caudate nuclei and putamen were observed. A year later, extrapyramidalmovement disorder and delay of mental development were evident. At 23 years of agelesions in basal ganglia and brain stem were discovered. CI activity in both peripheral bloodlymphocytes and muscle was decreased to 36–48% and 6–33% respectively in two of thepatients. The cause of the disease was attributed to a mutation in the C20orf7 gene althoughthe patients also presented a mutation in the CRLS1 (cardiolipin synthase 1) gene.20
C6orf66/NDUFAF4—Five patients of a consanguineous family of Arab-Muslim originwith infantile encephalopathy and isolated CI deficiency were homozygous for a mutation inthe C6orf66 gene, later named NDUFAF4.21 The mutation was a T→C substitution atnucleotide 194 predicted to change a leucine for a proline at amino acid 65. The healthyfamily members were heterozygous for this mutation.21 The affected infants sufferedmetabolic acidosis soon after birth, and three of them died between 2 and 5 days of age. Onehad a severe cardiomyopathy. The other children survived for 18 months and 7 years of age(at the time of the report). They developed a severe encephalopathy with lactic acidosis inaddition to kyphosis and spastic contractures. MRI studies revealed severe atrophy of whiteand gray matter as well as demyelination. CI deficiency was evident in both muscle andfibroblast (5.5–17% and 32–67% of controls respectively) and introduction of wild-typeC6orf66 into patients’ fibroblasts restored CI defect.21 Although the exact function ofNDUFAF4 remains obscure, an interdependence of NDUFAF4 (C6orf66) and NDUFAF3(C3orf60) was discovered.21
C3orf60/NDUFAF3—Three families with different mutations in the C3orf60 (NDUFAF3)gene were recently reported.22 A homozygous c.229 G→C mutation that caused a G77Rchange was found in three siblings with heterozygous parents. In the second family, theinfant was homozygous for a c.365 G→C causing an R122P change. In the last family, theinfant was a compound heterozygous with a c.2 T→C and a c.365 G→C that resulted in adisruption of the starting codon (M1T) and a R122T change respectively. The patients fromthe first family presented with lactic acidemia and increased muscle tone. Their brain MRIwas normal as well as EEG and echocardiogram. They died at 3 months of age. The infantfrom the second family suffered from macrocephaly with wide anterior fontanelle andhypotonia. He also presented with elevated lactate and died aged 4 months. The third familypatient suffered from myoclonic seizures, had diffuse brain leukomalacia in MRI, and diedat 6 months of age. Muscle and fibroblast from the patients showed decreased CI activity.Analysis of CI assembly in the patient from the second family did not reveal any assembly
Diaz et al. Page 4
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

intermediates when probed for either ND1 or NDUFS2. The patient’s fibroblast CI defectwas complemented when expressing NDUFAF3-GFP protein.22
Experiments on RNAi of NDUFAF3 in HeLa cells showed decreased CI protein and activityas well as a significant reduction in the levels of the assembly factor NDUFAF4. Likewise,interference of NDUFAF4 resulted in decreased levels of NDUFAF3 protein. NDUFAF3and NDUFAF4 comigrate with the same CI assembly intermediates and interact with CIsubunits (NDUFS2, NDUFS3, NDUFA5 and NDUFS8).22
C8orf38—Using phylogenetic profiling, 19 other candidate genes were identified as CI-associated proteins.23 All these candidate genes were investigated as causes of CI deficiencyin two siblings presenting with lactic acidosis, ataxia, decreased muscle strength andrigidity. MRI revealed abnormalities consistent with LS. One sibling died at 34 months ofage and the other was 22 months at the time of the study. The defect was associated with amutation in the C8orf38 gene (c.296A→G) in a conserved residue.23 Further studies arerequired to validate the role of C8orf38 as a CI assembly factor, albeit knockdown ofC8orf38 resulted in reduced levels and activity of CI.23
Ind1/HuInd1—Studies in the Yarrowia lipolytica identified Ind1 as an iron–sulfur clusterbinding protein. Deletion of the Ind1 gene resulted in a marked decrease of CI levels andactivity in the aerobic yeast.24 Likewise, knockdown of the human version (HULND1) inHeLa cells showed that this protein is required for CI assembly and activity. Moreover,huInd1 appears to be specific for CI. Analysis of 55Fe incorporation into the OXPHOScomplexes showed a marked decrease in the iron content of CI but not in complex II orcomplex III in cells depleted of huInd1.25 The knockdown of huInd1 resulted in anintermediate of about 450 kDa that does not contain subunits forming the peripheral arm ofthe complex (NDUFS1, NDUFV1, NDUFA13 or NDUFA9) but contained one of thesubunits of the membrane arm (NDUFB6).25
Taken together, these results suggest that huInd1 is involved in the delivery of iron–sulfurclusters to CI subunits and in the absence of this protein, the prosthetic group cannot bedelivered and the subunits are unstable and degraded causing impairment in the assembly ofthe peripheral arm and accumulation of an assembly intermediate. To date, defects in thisgene have not been found in patients with CI deficiency.
ACAD9—Using tandem affinity purification ACAD9 was pulled down as a bindingcandidate for NDUFAF1 and Ecsit.26 ACAD9 belongs to the acyl-CoA dehydrogenasefamily and co-migrated with CI assembly intermediates of 500 and 800 kDa that alsocontained Ecsit and NDUFAF1. Knockdown of ACAD9 in HEK293 cells decreased thelevels and activity of CI as well as the levels of the two other chaperones. Likewise, thelevels of ACAD9 were decreased when knocking down Ecsit or NDUFAF1.26
The first published ACAD9 patient was homozygous for the mutation c.1553 G→A thatresulted in the change of a highly conserved residue (R518H). This infant suffered fromhypertrophic cardiomyopathy at 8 months of age with elevated lactate in blood and CSF.Disease progressed and the patient was 18 years old at the time of the study, when exerciseintolerance, mild hearing loss, and hypertrophic cardiomyopathy were noted. The secondpatient was a compound heterozygote affecting two highly conserved amino acids, and hadmoderate metabolic acidosis, anemia, progressive encephalopathy, and hypertrophiccardiomyopathy. The disease developed into multiple organ failure and death at 6 months ofage.
Diaz et al. Page 5
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Analysis of the levels of CI in both patients showed reduced levels of this complex as wellas low levels of the chaperones Ecsit and NDUFAF1, albeit ACAD9 was only detected inpatient 1. Expression of ACAD9 in the fibroblasts restored the defect.26
Other ACAD9 (R532W change) patients with later onset of disease were reported from adouble consanguineous family. Fatigue, vomiting, exercise intolerance, and lactic acidosisappeared at about 4 years age.27 The muscles were predominantly affected and showedmitochondrial proliferation in the subsarcolema. For the most part, brain appeared sparedwith the exception of a single stroke-like episode in two of the patients. Another patient withsimilar phenotype was a compound heterozygote (R127Q and R469W). The patients had areduction of CI enzymatic activity but not of protein levels. Interestingly, these patientsresponded to riboflavin treatment which improved their CI deficiency.27
Since none of the ACAD9 patients showed any evidence of β-oxidation, the function of thechaperone appears to be related to CI assembly. Haack et al. confirmed this by identifyingother mutations in the ACAD9 gene in five new patients with CI deficiency.28 The patientssuffered from hypertrophic cardiomyopathy, encephalopathy and lactic acidosis with fataloutcome varying from 45 days to 12 years of age. CI activity ranged from 13% to 26% inmuscle and from 32% to 38% in fibroblasts of control samples.28
Other factors—There remain CI defects of unknown cause, suggesting the existence ofother proteins involved in the assembly process. One of these is the apoptosis-inducingfactor AIF. Knockdown of AIF resulted in a reduction of the levels of certain CI subunits(NDUFA9, NDUFB6 and NDUFS7) and the fully assembled complex in vitro in HeLa cellsor in vivo in the Harlequine mouse suggesting a role as CI chaperone.29 A mutation-causingdeletion of an arginine (R201Δ) in the N-terminal FAD binding domain of AIF has beenidentified in X-linked mitochondrial encephalopathy.30 Analysis of patient fibroblastsrevealed CIII and CIV deficiencies whereas CI was only slightly influenced; analysis inmuscle revealed that CI, III and IV were affected. However, this was shown to be due tosevere mitochondrial DNA deficiency.30 Taken together, the role of AIF in mitochondrialfunction needs further investigation. Another newly discovered factor is FOXRED1.Mutations in this gene caused a severe CI deficiency in an infant leading to progressiveencephalomyopathy. The CI defect from fibroblast derived from the patient was rescued bylentiviral expression of the protein. Silencing the gene expression decreased the steady statelevels of CI in control fibroblasts. Although the exact function of FOXRED1 as a CIchaperone is unknown, its FAD-oxidoreductase domain suggests a functional role inelectrons transfer reactions.88
Complex IIComplex II (CII) or succinate dehydrogenase (SDH) is located in the inner membrane facingthe mitochondrial matrix where it links the respiratory chain with the Krebs cycle. SDHcatalyses the oxidation of succinate to fumarate, resulting in reduction of the FAD cofactorbound to SDH from which electrons are passed through three FeS centres to ubiquinone.31
CII is composed of four nuclear-encoded subunits (SDHA–D) of which subunits A and Bform the enzyme complex whereas subunits C and D anchor the complex to the innermitochondrial membrane.32 Mutations in either CII proteins or chaperones may cause LS(SDAH), infantile leukencephalopathy (SDAHF1) or familial paraganglioma syndrome(SDHB, C, D, SDHAF2). Several proteins assisting assembly of CII have been described,33
but only the two recently discovered factors SDHAF134 and SDHAF235 directly andspecifically aid CII assembly.
Diaz et al. Page 6
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Complex II chaperonesSDHAF1—SDHAF1 was first described in 2009 by Ghezzi et al. in two affected familieswith infantile leukencephalopathy with severe CII deficiency.34,36 By genome-wide linkageanalysis, two homozygous missense mutations were identified in an evolutionarily highlyconserved gene coding for a mitochondrial matrix protein. Transfection of patient fibroblastswith wild-type SDHAF1 resulted in full or partial recovery of structural and functionalassembled CII.34
SDHAF2—By investigation of uncharacterized but evolutionarily conserved mitochondrialproteins in yeast, Hao et al. identified a protein that co-purified with yeast SDH1 and namedit SDHAF2 (SDH5 in yeast).35 Deletion of yeast SDH5 resulted in loss of the SDH complexand activity, probably due to unstable assembly of SDH. Incorporation of the FAD cofactorwas impaired in the mutant. A human mutation in SDHAF2 was identified in a Dutch familywith hereditary paraganglioma.35,37
Other factors—In yeast, two additional factors indirectly modifying CII assembly havebeen described.
In respiratory-deficient yeast mutants, Flx1 was identified as FAD transporter necessary forFAD incorporation into CII.38 Later a more general role of Flx1 on post-transcriptionalexpression of Sdh1 was suggested.33,35,39
Tcm62 was initially shown to be required for yeast CII assembly40 but later experimentsindicate that it influences CII assembly more generally by affecting mitochondrial proteinstability under stress41 or protein-folding capacity within mitochondria.42
Complex IIIComplex III (CIII, or cytochrome bc1 complex; ubiquinol cytochrome c reductase) islocated in the mitochondrial inner membrane where it facilitates electron transfer fromreduced coenzyme Q to cytochrome c, a process coupled to simultaneous pumping ofprotons across the inner mitochondrial membrane. In mammals, CIII forms a homodimerthat can also be found in association with CI and CIV,43 forming supercomplexes that maybe stabilized by cardiolipin.44 Monomeric CIII is composed of three catalytic and eightstructural subunits that are encoded by 10 nuclear and one mitochondrial genes. Therespiratory subunits contain redox centres and include the nuclear-encoded cytochrome c1(CYC1), the Rieske iron–sulfur protein (RISP, UQCRFS1) and the mitochondrial-encodedcytochrome b (MT-CYB). The nuclear-encoded structural proteins are the core proteins 1and 2 (UQCRC1 and 2) and subunit 6 (UQCRH) involved in complex formation, subunits 7(UQCRB) and 8 (UQCRQ) involved in ubiquinone binding, subunit 9 (UQCR10) thatinteracts with cytochrome c1 and subunit 10 that may function as RISP protein-bindingfactor. In mammals, assembly of CIII is assisted by BCS1L45 and TTC19.46
CIII deficiencies are rare, but they cause a broad spectrum of symptoms and display tissuespecificity in humans.47 They are caused by mutations in catalytic and structural subunits aswell as in assembly factors. Mutations in the mitochondrial-encoded catalytic subunit,cytochrome b, have mainly been associated with myopathy;48 however, cytochrome bmutations are also the cause of Leber’s hereditary optic neuropathy (LHON),49
encephalomyopathy,50 encephalopathy51 cardiomyopathy47 and severe neonatalpolyvisceral failure.52 Structural genes as cause of CIII deficiency include UQCRB andUQCRQ. In one patient with hypoglycemia and lactic acidosis, re-arrangement of theUQCRB gene was identified.53 In another patient suffering from severe psychomotor
Diaz et al. Page 7
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

retardation with mildly elevated lactate levels, a single missense mutation was identified inUQCRQ.54
Complex III chaperonesBCS1L—CIII assembly has been extensively investigated in yeast where bcs1 wasidentified as a factor necessary for the expression of functional CIII.55 The protein Bcs1 actsas molecular chaperone assisting the incorporation of Rieske protein and Qcr10p into CIII.56
Bcs1 shares 50% amino acid identity with its human ortholog, BCS1L.57 In the BCS1Lprotein, three different domains, the N-terminal import domain, the BCS1L-specific domainand the C-terminal AAA ATPase domain can be identified. In humans, BCS1L mutationshave been found in all three domains, thereby causing CIII deficiency, resulting in a varietyof phenotypes. They range from the least severe Björnstad syndrome with neurosensoryhearing loss and pili torti,58,59 a neurologic disease with muscle weakness and opticatrophy60 to manifestations with encephalopathy, liver failure or tubulopathy, orcombinations thereof.45,61–65 Manifestations with neonatal onset usually present withhepatopathy (V. Fellman and H. Kotarsky, in this issue), the most severe being theGRACILE (growth restriction, aminoaciduria, cholestasis, iron overload, lactic acidosis andearly death) syndrome, caused by a homozygous missense mutation (S78G).66 Thephenotype is strikingly consistent,67 presenting with severe fetal growth restriction,lactacidosis and early death, further described in this issue (V. Fellman). Different BCS1Lmutations known to date are summarized in Figure 1.
TTC19—An additional CIII assembly factor has recently been identified in patients withCIII deficiency and a slowly progressive encephalopathy presenting with subtle motilitychanges, involuntary movements and regurgitations with an onset during the first months oflife.46 In these individuals, homozygous nonsense mutations were identified in the geneencoding tetratricopeptide 19 (TTC19) resulting in reduced expression of TTC19 mRNAand lack of TTC19 protein. TTC19 protein was localized to the inner mitochondrialmembrane where it is part of two high molecular weight complexes of which at least onecontains CIII intermediates. Accumulation of CIII assembly intermediates in affectedpatients suggests that TTC19 is important in early assembly. Knockout of TTC19 inDrosophila melanogaster results in low fertility and neurological abnormalities associatedwith CIII deficiency.
Complex IVComplex IV (CIV) or cytochrome c oxidase is the terminal enzyme of the electron transportchain that transfers electrons from cytochrome c to oxygen while it translocates protonsfrom the matrix into the intermembrane space. CIV is a homodimer of about 230 kDaconstituted by 13 subunits. Three of its subunits form the catalytic core and are encoded bythe mtDNA whereas the rest of the subunits are encoded by the nuclear genome. Due to itsdual origin the assembly process of this complex is also very complicated and highlyregulated. Most of what is known about CIV assembly has been elucidated from systematicstudies in mutants from the yeast Saccharomyces cerevisiae. Studies in yeast have revealedthe presence of about 40 assembly factors that aid numerous aspects of CIV biogenesis.68 Ofthe large number of assembly factors found in yeast, only some have human homologs.Below we focus on those factors with clinical relevance.
Complex IV chaperonesSurf1—Mutations in the Surf1 gene cause LS characterized by subacute neurodegenerationencompassing bilateral necrotic lesions in brain stem, thalamus and basal ganglia. Theselesions consist of neuronal loss, vascularization and demyelination and cause an early onset
Diaz et al. Page 8
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

of symptoms typically including ataxia, nystagmus, dystonia, ophthalmoparesis and opticatrophy. Affected individuals die between 6 months to 12 years. The majority of themutations reported in Surf1 create premature termination codons whereas fewer mutationsare missense mutations.69 The latter mutations have been associated with milderphenotypes.70 The CIV activity in patients with LS ranged from 10% to 25% of controlvalues. One puzzling fact is that not all mutations in Surf1 cause LS. The clinical symptomsof LS have also been associated with mutations in other genes located in the mtDNA, withCOX10, and with certain defects in pyruvate dehydrogenase, CI and CV activities.71 Theactual function of Surf1 remains obscure but it participates in early assembly of CIV. Recentstudies in bacteria point the function of two Surf1 homologs Surf1c and Surf1q as heme abinding proteins.72
LRPPRC—A less severe form of LS found exclusively in a region of Quebec (Charlevoixand Saguenay-Lac-St Jean) is the French-Canadian Leigh syndrome (FCLS) caused bymutations in the LRPPRC gene.73 In FCLS death also occurs early in life, between 3 and 10years and clinical presentations include mild psychomotor regression and lactic acidosis.The tissues with severe CIV deficiency in this disorder are brain and liver, whereas muscle,heart and kidney are relatively unaffected. The LRPPRC (leucine-rich pentatricopeptiderepeat cassette) protein homolog in yeast (Pet309) is required for stability and translation ofCOX1 mRNA (CIV catalytic subunit 1 encoded by the mtDNA) by presumably binding toits 5′UTR. The exact function of the mammalian LRPPRC is still unknown since there areno significant 5′UTR in mitochondrial mRNA, suggesting that the protein acts through adifferent mechanism. The levels of this chaperone as well as the levels of most of themitochondrial transcripts excluding the rRNA and tRNA were significantly decreased inLRPPRC patients.74 This decrease of mitochondrial mRNA was not proportional and thelevels of CIV transcripts were the most reduced. When decreasing the LRPPRC levels evenfurther, a severe decrease in all mitochondrial transcripts was observed with a generalizeddefect in all mitochondrially encoded OXPHOS complexes. LRPPRC was found to interactwith SLIRP and to bind mRNA. Although further studies are required, the authors proposethat these two proteins are post-transcriptional regulators of mitochondrial genes.74
TACO1—In a patient with late onset of LS, a mutation in the CCD44 gene was identifiedresulting in impaired COX1 protein synthesis.75 The gene was renamed TACO1 since itencodes a protein that serves as ‘translational activator of COX1’.75 In other patients withlate onset of LS and CIV deficiency, no mutations in TACO1 were found, suggesting thatmutations in this factor are rare or perhaps incompatible with life.76
SCO1 and SCO2—Complex IV requires copper in two catalytic centers, CuA and CuB,located in COX2 and COX1 subunits respectively. Several assembly factors (COX17,SCO1, SCO2 and COX11) are implicated in the delivery of this metal.77 Mutations in SCO1were described first in two neonate siblings suffering from severe metabolic acidosis andhepatic failure (microvesicular steatosis and hepatomegaly) and from metabolic acidosis andencephalopathy respectively. The first patient died at 2 months and the second at 5 days ofage; they were compound heterozygous for a 2 bp deletion in nucleotide 363–364 resultingin frame shift and stop codon, and for a C→T transition at nucleotide 520 resulting in anamino acid change of a conserved residue (P174L). The severe COX defect (less than 1% ofcontrol values) was observed in liver and muscle tissues in the first patient.78 Another SCO1case presented with hypertrophic cardiomyopathy, encephalopathy and hepatomegaly with afatal outcome at 6 months. The patient was homozygous for the missense mutation c.394G→A resulting in a G132s change.79
SCO2 mutations were first detected in three patients suffering from hypertrophiccardiomyopathy accompanied by encephalopathy manifested early in life.80 The brain
Diaz et al. Page 9
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

lesions differed from the typical LS. SCO2 brain abnormalities included spongiformneurodegeneration (midbrain, pons and medulla) and microglia proliferation (thalamus).They were all compound heterozygotes with different combinations.80 Since then, numerouscases of SCO2 mutations (at least 26 including deletions, insertions and premature stopcodons) have been reported with the most common mutation being the E140K mutation.Interestingly, patients that were homozygous for the E140K mutation presented with amilder phenotype and delayed onset of disease (2–4 months of age versus neonate onset forcompound heterozygous).81
COX10 and COX15—Another prosthetic group unique to CIV is heme a, which isassociated with the COX1 subunit. COX10 and COX15 functions are related to biosynthesisof heme a. COX10, a farnesyl transferase, catalyzes the first step of the heme a biosynthesisby incorporating a farnesyl group and converting heme b into heme o. Subsequently, heme ois converted to heme a by the action of COX15 and a monooxygenase (not characterized inhumans). Defects in this pathway result in clinical symptoms with early onset and fataloutcome from 2 months to 4 years of age. The first case of a COX10 mutation was anewborn infant with severe lactic acidemia, hypotonia, ataxia and pyramidal symptoms inaddition to leukodystrophy, and tubulopathy.82 A sibling presented with lactic acidosis afterbirth and died aged 5 days. Few other patients have been described suffering from anemia,sensorineural deafness and hypertrophic cardiomyopathy,83 anemia and LS83 and Leigh-likesyndrome.84,85 A newborn infant with hypotonia, seizures, lactic acidosis andcardiomyopathy leading to death at 1 month of age was the first case reported with a COX15mutation.83 Another infant had a similar but less dramatic clinical picture with hypotonia,motor regression, nystagmus, retinopathy, lactic acidosis and microcephaly by 7 months andLS by 1 year.86 A third case with a very slowly progressing LS survived at least 16 years.87
The CIV deficiency was 42% and 22% of control values in muscle and fibroblast,respectively, perhaps accounting for a milder phenotype.
Practice points
• Most individuals affected by complex I and complex IV deficiencies have anuneventful prenatal period and deficiencies are evident postnatally.
• Perinatal manifestations of BCS1L mutations present often with fetal growthrestriction and lactacidosis.
• Neonatal genetic screening of complex I, complex III, and complex IVdeficiencies should include genes coding for specific chaperones in addition tostructural subunits.
• Early diagnosis of origin of OXPHOS defect could aid in treatment.
• Treatments for OXPHOS defects address solely the symptoms.
Research directions
• Further studies are required to elucidate of the complete sequence of assemblysteps of complex I, complex III and complex IV and its regulation.
• A better understanding of the specific function of assembly chaperones and theirinteraction partners is required.
• Research focusing on understanding the genotype–clinical phenotype and theinfluence of genetic background and environmental factors is necessary foreffective treatments.
Diaz et al. Page 10
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

AcknowledgmentsFunding sources
The research group of C.T. Moraes was funded by The James and Esther King Biomedical Research Program,Florida Department of Health (FD) and PHS grants NS041777, AG AG036871, CA085700 and EY10804 (CTM).The research group of V. Fellman was funded by Lund University funds, Swedish Royal Physiografic Society,Swedish Research Council 2008–2898, and Regional ALF funds.
References1. Schaefer AM, Taylor RW, Turnbull DM, et al. The epidemiology of mitochondrial disorders – past,
present and future. Biochim Biophys Acta. 2004; 1659:115–120. [PubMed: 15576042]2. Cree LM, Samuels DC, Chinnery PF. The inheritance of pathogenic mitochondrial DNA mutations.
Biochim Biophys Acta. 2009; 1792:1097–1092. [PubMed: 19303927]3. Kerr DS. Treatment of mitochondrial electron transport chain disorders: a review of clinical trials
over the past decade. Molec Genet Metab. 2010; 99:246–255. [PubMed: 20060349]4. Wenz T, Williams SL, Bacman SR, et al. Emerging therapeutic approaches to mitochondrial
diseases. Dev Disabil Res Rev. 2010; 16:219–229. [PubMed: 20818736]5. Clason T, Ruiz T, Schagger H, et al. The structure of eukaryotic and prokaryotic complex I. J Struct
Biol. 2010; 169:81–88. [PubMed: 19732833]6. McKenzie M, Ryan MT. Assembly factors of human mitochondrial complex I and their defects in
disease. IUBMB Life. 2010; 62:497–502. [PubMed: 20552642]7. Perales-Clemente E, Fernandez-Vizarra E, Acin-Perez R, et al. Five entry points of the
mitochondrially encoded subunits in mammalian complex I assembly. Molec Cell Biol. 2010;30:3038–3047. [PubMed: 20385768]
8. Kirby DM, Crawford M, Cleary MA, et al. Respiratory chain complex I deficiency: anunderdiagnosed energy generation disorder. Neurology. 1999; 52:1255–1264. [PubMed: 10214753]
9. Distelmaier F, Koopman WJ, van den Heuvel LP, et al. Mitochondrial complex I deficiency: fromorganelle dysfunction to clinical disease. Brain. 2009; 132:833–842. [PubMed: 19336460]
10. Kuffner R, Rohr A, Schmiede A, et al. Involvement of two novel chaperones in the assembly ofmitochondrial NADH:Ubiquinone oxidoreductase (complex I). J Mol Biol. 1998; 283:409–417.[PubMed: 9769214]
11. Janssen R, Smeitink J, Smeets R, et al. CIA30 complex I assembly factor: a candidate for humancomplex I deficiency? Hum Genet. 2002; 110:264–270. [PubMed: 11935339]
12. Vogel RO, Janssen RJ, Ugalde C, et al. Human mitochondrial complex I assembly is mediated byNDUFAF1. FEBS J. 2005; 272:5317–5326. [PubMed: 16218961]
13. Dunning CR, McKenzie M, Sugiana C, et al. Human CIA30 is involved in the early assembly ofmitochondrial complex I and mutations in its gene cause disease. 2007; 26:3227–3237.
14. Vogel RO, Janssen RJ, van den Brand MA, et al. Cytosolic signaling protein Ecsit also localizes tomitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly.Genes Dev. 2007; 21:615–624. [PubMed: 17344420]
15. Ogilvie I, Kennaway NG, Shoubridge EA. A molecular chaperone for mitochondrial complex Iassembly is mutated in a progressive encephalopathy. J Clin Invest. 2005; 115:2784–2792.[PubMed: 16200211]
16. Vogel RO, van den Brand MA, Rodenburg RJ, et al. Investigation of the complex I assemblychaperones B17.2L and NDUFAF1 in a cohort of CI deficient patients. Molec Genet Metab. 2007;91:176–182. [PubMed: 17383918]
17. Barghuti F, Elian K, Gomori JM, et al. The unique neuroradiology of complex I deficiency due toNDUFA12L defect. Molec Genet Metab. 2008; 94:78–82. [PubMed: 18180188]
18. Janssen RJ, Distelmaier F, Smeets R, et al. Contiguous gene deletion of ELOVL7, ERCC8 andNDUFAF2 in a patient with a fatal multisystem disorder. Hum Molec Genet. 2009; 18:3365–3374.[PubMed: 19525295]
Diaz et al. Page 11
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

19. Sugiana C, Pagliarini DJ, McKenzie M, et al. Mutation of C20orf7 disrupts complex I assemblyand causes lethal neonatal mitochondrial disease. Am J Hum Genet. 2008; 83:468–478. [PubMed:18940309]
20. Gerards M, Sluiter W, van den Bosch BJ, et al. Defective complex I assembly due to C20orf7mutations as a new cause of Leigh syndrome. J Med Genet. 2010; 47:507–512. [PubMed:19542079]
21. Saada A, Edvardson S, Rapoport M, et al. C6ORF66 is an assembly factor of mitochondrialcomplex I. Am J Hum Genet. 2008; 82:32–38. [PubMed: 18179882]
22. Saada A, Vogel RO, Hoefs SJ, et al. Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4(C6ORF66)-interacting complex I assembly protein, cause fatal neonatal mitochondrial disease.Am J Hum Genet. 2009; 84:718–727. [PubMed: 19463981]
23. Pagliarini DJ, Calvo SE, Chang B, et al. A mitochondrial protein compendium elucidates complexI disease biology. Cell. 2008; 134:112–123. [PubMed: 18614015]
24. Bych K, Kerscher S, Netz DJ, et al. The iron-sulphur protein Ind1 is required for effective complexI assembly. EMBO J. 2008; 27:1736–1746. [PubMed: 18497740]
25. Sheftel AD, Stehling O, Pierik AJ, et al. Human ind1, an iron-sulfur cluster assembly factor forrespiratory complex I. Molec Cell Biol. 2009; 29:6059–6073. [PubMed: 19752196]
26. Nouws J, Nijtmans L, Houten SM, et al. Acyl-CoA dehydrogenase 9 is required for the biogenesisof oxidative phosphorylation complex I. Cell Metab. 2010; 12:283–294. [PubMed: 20816094]
27. Gerards M, van den Bosch BJ, Danhauser K, et al. Riboflavin-responsive oxidativephosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene.Brain. 2011; 134:210–219. [PubMed: 20929961]
28. Haack TB, Danhauser K, Haberberger B, et al. Exome sequencing identifies ACAD9 mutations asa cause of complex I deficiency. Nat Genet. 2010; 42:1131–1134. [PubMed: 21057504]
29. Vahsen N, Cande C, Briere JJ, et al. AIF deficiency compromises oxidative phosphorylation.EMBO J. 2004; 23:4679–4689. [PubMed: 15526035]
30. Ghezzi D, Sevrioukova I, Invernizzi F, et al. Severe X-linked mitochondrial encephalomyopathyassociated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010; 86:639–649.[PubMed: 20362274]
31. Hagerhall C. Succinate: quinone oxidoreductases. Variations on a conserved theme. BiochimBiophys Acta. 1997; 1320:107–141. [PubMed: 9210286]
32. Sun F, Huo X, Zhai Y, et al. Crystal structure of mitochondrial respiratory membrane proteincomplex II. Cell. 2005; 121:1043–1057. [PubMed: 15989954]
33. Rutter J, Winge DR, Schiffman JD. Succinate dehydrogenase – assembly, regulation and role inhuman disease. Mitochondrion. 2010; 10:393–401. [PubMed: 20226277]
34. Ghezzi D, Goffrini P, Uziel G, et al. SDHAF1, encoding a LYR complex-II specific assemblyfactor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet. 2009; 41:654–656.[PubMed: 19465911]
35. Hao HX, Khalimonchuk O, Schraders M, et al. SDH5, a gene required for flavination of succinatedehydrogenase, is mutated in paraganglioma. Science. 2009; 325:1139–1142. [PubMed:19628817]
36. Bugiani M, Lamantea E, Invernizzi F, et al. Effects of riboflavin in children with complex IIdeficiency. Brain Dev. 2006; 28:576–581. [PubMed: 16737791]
37. van Baars F, Cremers C, van den Broek P, et al. Genetic aspects of nonchromaffin paraganglioma.Hum Genet. 1982; 60:305–309. [PubMed: 6286462]
38. Tzagoloff A, Jang J, Glerum DM, et al. FLX1 codes for a carrier protein involved in maintaining aproper balance of flavin nucleotides in yeast mitochondria. J Biol Chem. 1996; 271:7392–7397.[PubMed: 8631763]
39. Bafunno V, Giancaspero TA, Brizio C, et al. Riboflavin uptake and FAD synthesis inSaccharomyces cerevisiae mitochondria: involvement of the Flx1p carrier in FAD export. J BiolChem. 2004; 279:95–102. [PubMed: 14555654]
40. Dibrov E, Fu S, Lemire BD. The Saccharomyces cerevisiae TCM62 gene encodes a chaperonenecessary for the assembly of the mitochondrial succinate dehydrogenase (complex II). J BiolChem. 1998; 273:32042–32048. [PubMed: 9822678]
Diaz et al. Page 12
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

41. Klanner C, Neupert W, Langer T. The chaperonin-related protein Tcm62p ensures mitochondrialgene expression under heat stress. FEBS Lett. 2000; 470:365–369. [PubMed: 10745098]
42. Vander Heiden MG, Choy JS, VanderWeele DJ, et al. Bcl-x(L) complements Saccharomycescerevisiae genes that facilitate the switch from glycolytic to oxidative metabolism. J Biol Chem.2002; 277:44870–44876. [PubMed: 12244097]
43. Schagger H. Respiratory chain supercomplexes of mitochondria and bacteria. Biochim BiophysActa. 2002; 1555:154–159. [PubMed: 12206908]
44. McKenzie M, Lazarou M, Thorburn DR, et al. Mitochondrial respiratory chain supercomplexes aredestabilized in Barth syndrome patients. J Molec Biol. 2006; 361:462–469. [PubMed: 16857210]
45. de Lonlay P, Valnot I, Barrientos A, et al. A mutant mitochondrial respiratory chain assemblyprotein causes complex III deficiency in patients with tubulopathy, encephalopathy and liverfailure. Nat Genet. 2001; 29:57–60. [PubMed: 11528392]
46. Ghezzi D, Arzuffi P, Zordan M, et al. Mutations in TTC19 cause mitochondrial complex IIIdeficiency and neurological impairment in humans and flies. Nat Genet. 2011; 43:259–263.[PubMed: 21278747]
47. Benit P, Lebon S, Rustin P. Respiratory-chain diseases related to complex III deficiency. BiochimBiophys Acta. 2009; 1793:181–185. [PubMed: 18601960]
48. Andreu AL, Hanna MG, Reichmann H, et al. Exercise intolerance due to mutations in thecytochrome b gene of mitochondrial DNA. N Engl J Med. 1999; 341:1037–1044. [PubMed:10502593]
49. Johns DR, Neufeld MJ. Cytochrome b mutations in Leber hereditary optic neuropathy. BiochemBiophys Res Commun. 1991; 181:1358–1364. [PubMed: 1764087]
50. Blakely EL, Mitchell AL, Fisher N, et al. A mitochondrial cytochrome b mutation causing severerespiratory chain enzyme deficiency in humans and yeast. FEBS J. 2005; 272:3583–3592.[PubMed: 16008558]
51. Keightley JA, Anitori R, Burton MD, et al. Mitochondrial encephalomyopathy and complex IIIdeficiency associated with a stop-codon mutation in the cytochrome b gene. Am J Hum Genet.2000; 67:1400–1410. [PubMed: 11047755]
52. Fragaki K, Procaccio V, Bannwarth S, et al. A neonatal polyvisceral failure linked to a de novohomoplasmic mutation in the mitochondrially encoded cytochrome b gene. Mitochondrion. 2009;9:346–352. [PubMed: 19563916]
53. Haut S, Brivet M, Touati G, et al. A deletion in the human QP-C gene causes a complex IIIdeficiency resulting in hypoglycaemia and lactic acidosis. Hum Genet. 2003; 113:118–122.[PubMed: 12709789]
54. Barel O, Shorer Z, Flusser H, et al. Mitochondrial complex III deficiency associated with ahomozygous mutation in UQCRQ. Am J Hum Genet. 2008; 82:1211–1216. [PubMed: 18439546]
55. Nobrega FG, Nobrega MP, Tzagoloff A. BCS1, a novel gene required for the expression offunctional Rieske iron-sulfur protein in Saccharomyces cerevisiae. EMBO J. 1992; 11:3821–3829.[PubMed: 1327750]
56. Cruciat CM, Hell K, Folsch H, et al. Bcs1p, an AAA-family member, is a chaperone for theassembly of the cytochrome bc(1) complex. EMBO J. 1999; 18:5226–5233. [PubMed: 10508156]
57. Petruzzella V, Tiranti V, Fernandez P, et al. Identification and characterization of human cDNAsspecific to BCS1, PET112, SCO1, COX15, and COX11, five genes involved in the formation andfunction of the mitochondrial respiratory chain. Genomics. 1998; 54:494–404. [PubMed:9878253]
58. Hinson JT, Fantin VR, Schonberger J, et al. Missense mutations in the BCS1L gene as a cause ofthe Björnstad syndrome. N Engl J Med. 2007; 356:809–819. [PubMed: 17314340]
59. Selvaag E. Pili torti and sensorineural hearing loss. A follow-up of Björnstad’s original patientsand a review of the literature. Eur J Dermatol. 2000; 10:91–97. [PubMed: 10694305]
60. Tuppen HA, Fehmi J, Czermin B, et al. Long-term survival of neonatal mitochondrial complex IIIdeficiency associated with a novel BCS1L gene mutation. Molec Genet Metab. 2010; 100:345–348. [PubMed: 20472482]
Diaz et al. Page 13
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

61. Blazquez A, Gil-Borlado MC, Moran M, et al. Infantile mitochondrial encephalomyopathy withunusual phenotype caused by a novel BCS1L mutation in an isolated complex III-deficient patient.Neuromuscul Disord. 2009; 19:143–146. [PubMed: 19162478]
62. De Meirleir L, Seneca S, Damis E, et al. Clinical and diagnostic characteristics of complex IIIdeficiency due to mutations in the BCS1L gene. Am J Med Genet A. 2003; 121:126–131.[PubMed: 12910490]
63. Fernandez-Vizarra E, Bugiani M, Goffrini P, et al. Impaired complex III assembly associated withBCS1L gene mutations in isolated mitochondrial encephalopathy. Hum Mol Genet. 2007;16:1241–1252. [PubMed: 17403714]
64. Gil-Borlado MC, Gonzalez-Hoyuela M, Blazquez A, et al. Pathogenic mutations in the 5′untranslated region of BCS1L mRNA in mitochondrial complex III deficiency. Mitochondrion.2009; 9:299–305. [PubMed: 19389488]
65. Morris AA, Taylor RW, Birch-Machin MA, et al. Neonatal Fanconi syndrome due to deficiency ofcomplex III of the respiratory chain. Pediatr Nephrol. 1995; 9:407–411. [PubMed: 7577396]
66. Visapaa I, Fellman V, Vesa J, et al. GRACILE syndrome, a lethal metabolic disorder with ironoverload, is caused by a point mutation in BCS1L. Am J Hum Genet. 2002; 71:863–876.[PubMed: 12215968]
67. Fellman V, Lemmela S, Sajantila A, et al. Screening of BCS1L mutations in severe neonataldisorders suspicious for mitochondrial cause. J Hum Genet. 2008; 53:554–558. [PubMed:18386115]
68. Barrientos A, Gouget K, Horn D, et al. Suppression mechanisms of COX assembly defects in yeastand human: insights into the COX assembly process. Biochim Biophys Acta. 2009; 1793:97–107.[PubMed: 18522805]
69. Pecina P, Houstkova H, Hansikova H, et al. Genetic defects of cytochrome c oxidase assembly.Physiol Res. 2004; 53:S213–S223. [PubMed: 15119951]
70. Piekutowska-Abramczuk D, Magner M, Popowska E, et al. SURF1 missense mutations promote amild Leigh phenotype. Clin Genet. 2009; 76:195–204. [PubMed: 19780766]
71. Rahman S, Blok RB, Dahl HH, et al. Leigh syndrome: clinical features and biochemical and DNAabnormalities. Ann Neurol. 1996; 39:343–351. [PubMed: 8602753]
72. Bundschuh FA, Hannappel A, Anderka O, et al. Surf1, associated with Leigh syndrome in humans,is a heme-binding protein in bacterial oxidase biogenesis. J Biol Chem. 2009; 284:25735–25741.[PubMed: 19625251]
73. Mootha VK, Lepage P, Miller K, et al. Identification of a gene causing human cytochrome coxidase deficiency by integrative genomics. Proc Natl Acad Sci USA. 2003; 100:605–610.[PubMed: 12529507]
74. Sasarman F, Brunel-Guitton C, Antonicka H, et al. LRPPRC and SLIRP interact in aribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria.Molec Biol Cell. 2010; 21:1315–1323. [PubMed: 20200222]
75. Weraarpachai W, Antonicka H, Sasarman F, et al. Mutation in TACO1, encoding a translationalactivator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. NatGenet. 2009; 41:833–837. [PubMed: 19503089]
76. Seeger J, Schrank B, Pyle A, et al. Clinical and neuropathological findings in patients with TACO1mutations. Neuromuscul Disord. 2010; 20:720–724. [PubMed: 20727754]
77. Leary SC. Redox regulation of SCO protein function: controlling copper at a mitochondrialcrossroad. Antioxid Redox Signal. 2010; 13:1403–1416. [PubMed: 20136502]
78. Valnot I, Osmond S, Gigarel N, et al. Mutations of the SCO1 gene in mitochondrial cytochrome coxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am J Hum Genet.2000; 67:1104–1109. [PubMed: 11013136]
79. Stiburek L, Vesela K, Hansikova H, et al. Loss of function of Sco1 and its interaction withcytochrome c oxidase. Am J Physiol Cell Physiol. 2009; 296:C1218–C1226. [PubMed: 19295170]
80. Papadopoulou LC, Sue CM, Davidson MM, et al. Fatal infantile cardioencephalomyopathy withCOX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet. 1999; 23:333–337.[PubMed: 10545952]
Diaz et al. Page 14
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

81. Joost K, Rodenburg R, Piirsoo A, et al. A novel mutation in the SCO2 gene in a neonate withearly-onset cardioencephalomyopathy. Pediatr Neurol. 2010; 42:227–230. [PubMed: 20159436]
82. Valnot I, von Kleist-Retzow JC, Barrientos A, et al. A mutation in the human hemeA:farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum Molec Genet.2000; 9:1245–1249. [PubMed: 10767350]
83. Antonicka H, Leary SC, Guercin GH, et al. Mutations in COX10 result in a defect in mitochondrialheme A biosynthesis and account for multiple, early-onset clinical phenotypes associated withisolated COX deficiency. Hum Molec Genet. 2003; 12:2693–2702. [PubMed: 12928484]
84. Coenen MJ, Smeitink JA, Pots JM, et al. Sequence analysis of the structural nuclear encodedsubunits and assembly genes of cytochrome c oxidase in a cohort of 10 isolated complexIVdeficient patients revealed five mutations. J Child Neurol. 2006; 21:508–511. [PubMed:16948936]
85. Coenen MJ, van den Heuvel LP, Ugalde C, et al. Cytochrome c oxidase biogenesis in a patientwith a mutation in COX10 gene. Ann Neurol. 2004; 56:560–564. [PubMed: 15455402]
86. Oquendo CE, Antonicka H, Shoubridge EA, et al. Functional and genetic studies demonstrate thatmutation in the COX15 gene can cause Leigh syndrome. J Med Genet. 2004; 41:540–544.[PubMed: 15235026]
87. Bugiani M, Tiranti V, Farina L, et al. Novel mutations in COX15 in a long surviving Leighsyndrome patient with cytochrome c oxidase deficiency. J Med Genet. 2005; 42:28.
88. Fassone E, Duncan AJ, Taanman JW, et al. FOXRED1, encoding an FAD-dependentoxidoreductase complex-I-specific molecular chaperone, is mutated in infantileonsetmitochondrial encephalopathy. Hum Molec Genet. 2010; 19:4837e47. [PubMed: 20858599]
Diaz et al. Page 15
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 1.BCS1L is encoded by eight exons with the start codon in exon 2 and the stop codon in exon8. Mutations in the 5′UTR region causing complex III deficiency are shown. In the BCS1Lprotein three functional domains, the import domain, the BCS1L_N-specific domain and theAAA_ATPase domain can be identfied. BCS1L mutations are indicated by their respectiveamino acid exchange. Mutations occur in all domains of the protein but cause a widespectrum of symptoms ranging from mild complex III deficiency and Björnstad syndrome(shown above the BCS1L protein) to mutations causing mainly hepatopathy (bold, italic),mainly encephalopathy (underlined) or hepatopathy and encephalopathy (bold, italic andunderlined) (shown below the BCS1L protein). BCS1L mutations are indicated by theirrespective amino acid exchange. Patients identified so far are either homozygous formutations (P99L, S277N, S78G, T50A, G129R) or compound heterozygotes (R45C/R56ter,V327A/R56ter, G35R/184C, S78G/144Q, R73C/F368I, R155P/V353M).
Diaz et al. Page 16
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Diaz et al. Page 17
Tabl
e 1
Mut
atio
ns in
ass
embl
y fa
ctor
s as c
ause
of n
eona
tal m
itoch
ondr
ial d
isor
der
Ass
embl
yfa
ctor
Mut
atio
nIU
GR
Lac
ticac
idos
isL
eigh
synd
rom
eC
ardi
om
yopa
thy
Liv
erdi
seas
eO
ther
Ref
.
ND
UFA
2ΔE
xon2
++
+15
R45
X
ML1
C20
orf7
L229
P+
++
19–2
0
159F
C3o
rf60
G77
R+
Wea
k m
uscl
e22
ND
UFA
F3R
122P
R12
2T
C6o
rf66
L65P
++
21
ND
UFA
F4
AC
AD
9V
ario
us+
++
26–2
8
BC
S1L
Var
ious
++
++
+W
eak
mus
cle
45,4
8,60
–66
SUR
F1V
ario
us+
+69
SCO
1P1
74L
++
++
78,7
9
G13
2S
Stop
SCO
2V
ario
us+
+80
,81
CO
X10
P225
L+
++
Wea
k m
uscl
e82
,83,
85
T204
K
T196
K
CO
X15
R21
7W+
+83
S344
P
H15
2X
IUG
R, i
ntra
uter
ine
grow
th re
stric
tion.
Semin Fetal Neonatal Med. Author manuscript; available in PMC 2012 August 1.

Mitochondrial DNA and disease
SALVATORE DIMAURO & GUIDO DAVIDZON
Department of Neurology, Columbia University Medical Center, New York, NY, U S A
AbstractThe small circle of mitochondrial DNA (mtDNA) present in all human cells has proven to be a veritable Pandora’s box ofpathogenic mutations and rearrangements. In this review, we summarize the distinctive rules of mitochondrial genetics(maternal inheritance, mitotic segregation, heteroplasmy and threshold effect), stress the relatively high prevalence ofmtDNA-related diseases, and consider recent additions to the already long list of pathogenic mutations (especiallymutations affecting protein-coding genes). We then discuss more controversial issues, including the functional orpathological role of mtDNA haplotypes, the pathogenicity of homoplasmic mutations and the still largely obscurepathophysiology of mtDNA mutations.
Key words: Haplotypes, heteroplasmy, homoplasmy, maternal inheritance, mitochondrial DNA, mtDNA
Introduction
Mitochondrial DNA (mtDNA) is a fossil molecule
proving that endosymbiosis did occur, when – about
1.5 billion years ago – protobacteria populated
primordial eukaryotic cells and took permanent
residence in the new environment (1). Unlike a
fossil, however, mtDNA has lost its independence
but not its life and it keeps functioning (sometimes
malfunctioning, which is the reason for this article)
under the overarching control of the nuclear
genome.
Human mtDNA (Figure 1) is a 16,569-kb circu-
lar, double-stranded molecule, which contains 37
genes: 2 rRNA genes, 22 tRNA genes, and 13
structural genes encoding subunits of the mitochon-
drial respiratory chain, which is the ‘business end’ of
oxidative metabolism, where ATP is generated (2).
Reducing equivalents produced in the Krebs cycle
and in the ß-oxidation spirals are passed along a
series of protein complexes embedded in the inner
mitochondrial membrane (the electron transport
chain), which consists of four multimeric complexes
(I to IV) plus two small electron carriers, coenzyme
Q (or ubiquinone) and cytochrome c (Figure 2).
The energy generated by the reactions of the
electron transport chain is used to pump protons
from the mitochondrial matrix into the space
between the inner and outer mitochondrial mem-
branes. This creates an electrochemical proton
gradient, which is utilized by complex V (or ATP
synthase), a tiny rotary machine that generates ATP
as protons flow back into the matrix through its
membrane-embedded F0 portion, the rotor of the
turbine (3).
Starting in 1988, when mutations in mtDNA were
first associated with human disease (4,5), the circle
of mtDNA has become crowded with pathogenic
mutations, and three principles of mitochondrial
genetics should, therefore, be familiar to the practi-
cing physician.
1. Heteroplasmy and threshold effect. Each cell
contains hundreds or thousands of mtDNA copies,
which, at cell division, distribute randomly among
daughter cells. In normal tissues, all mtDNA
molecules are identical (homoplasmy). Deleterious
mutations of mtDNA usually affect some but not all
mtDNAs within a cell, a tissue, an individual
(heteroplasmy), and the clinical expression of a
pathogenic mtDNA mutation is largely determined
by the relative proportion of normal and mutant
genomes in different tissues. A minimum critical
number of mutant mtDNAs is required to cause
mitochondrial dysfunction in a particular organ or
tissue and mitochondrial disease in an individual
(threshold effect).
2. Mitotic segregation. At cell division, the proportion
of mutant mtDNAs in daughter cells may shift and
the phenotype may change accordingly. This
Correspondence: Salvatore DiMauro, MD, 4-420 College of Physicians & Surgeons, 630 West 168th Street, New York, NY 10032, U S A. Fax: 212-305-
3986. E-mail: [email protected]
Annals of Medicine. 2005; 37: 222–232
ISSN 0785-3890 print/ISSN 1365-2060 online # 2005 Taylor & Francis Ltd
DOI: 10.1080/07853890510007368
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
VU
B V
rije
Uni
vers
ity B
russ
els
on 1
0/03
/13
For
pers
onal
use
onl
y.

phenomenon, called mitotic segregation, explains
how certain patients with mtDNA-related disorders
may actually shift from one clinical phenotype to a
different one as they grow older.
3. Maternal inheritance. At fertilization, all mtDNA
derives from the ovum. Therefore, the mode of
transmission of mtDNA and of mtDNA point
mutations (single deletions of mtDNA are usually
sporadic events) differs from Mendelian inheritance.
A mother carrying a mtDNA point mutation will
pass it on to all her children (males and females), but
only her daughters will transmit it to their progeny.
A disease expressed in both sexes but with no
evidence of paternal transmission is strongly
suggestive of a mtDNA point mutation.
Over 150 point mutations and innumerable large-
scale rearrangements have been associated with a
bewildering variety of diseases (Figure 1). This is
not surprising when one considers that mitochondria
are ubiquitous organelles and all human tissues, in
isolation or in various combinations, can be affected
by mtDNA mutations (6). This concept is illustrated
in Table I, a compilation of symptoms and signs
described in mitochondrial encephalomyopathies
due to single rearrangements (KSS, PS, and PEO),
to point mutations in genes affecting protein synth-
esis in toto (MELAS; MERRF), and to a protein-
coding gene (NARP and MILS). This table
highlights the clinical features of the most common
syndromes, which are difficult to miss in typical
patients. It is also a reminder that any combination
of these symptoms and signs ought to alert the astute
clinician to the possibility of a mtDNA-related
disorder. A third function of the table is to serve as
a concise descriptor of the six most common
mtDNA-related syndromes, which will not be
described here in any more detail. If more details
are needed, the reader is referred to textbook reviews
(6).
As shown in Table II, mtDNA-related disorders
fall into two major groups: 1) those due to mutations
in genes involved in mitochondrial protein synthesis,
and 2) those due to mutations in genes encoding
individual proteins of the respiratory chain. Clinical
features are not terribly useful in differentiating the
two groups, but laboratory results, muscle biopsy,
and muscle biochemistry are better discriminators
and offer useful clues for a targeted molecular
analysis. In defects of protein synthesis, lactic
acidosis is seen more consistently and tends to be
higher, and muscle biopsies almost invariably show
RRF, which are characteristically COX-negative
(except in typical MELAS, where RRF stain with
the COX reaction, though not intensely so). Not
Key messages
N Mitochondrial DNA (mtDNA) is a
Pandora’s box of pathogenic mutations
potentially affecting every organ of the
body. mtDNA-related diseases are usually
transmitted by non-Mendelian, maternal
inheritance.
N Pathogenic mutations of mtDNA can be
divided into two main groups, those affecting
mitochondrial protein synthesis in toto, and
those affecting individual proteins of the
respiratory chain.
N The functional and possible pathogenic
significance of mtDNA haplotypes is under
intense investigation. Homoplasmy does not
rule out pathogenicity.
N Although we have learnt a lot about mtDNA
mutations, the pathophysiology of mtDNA-
related diseases remains largely unknown.
Abbreviations
ATP adenosine triphosphate
CK creatine kinase
COX cytochrome c oxidase
cyt b cytochrome b
KSS Kearns-Sayre syndrome
LHON Leber hereditary optic
neuropathy
LS Leigh syndrome
MELAS mitochondrial
encephalomyopathy, lactic
acidosis, stroke-like episodes
MERRF myoclonus epilepsy and ragged-
red fibers
MILS maternally inherited Leigh
syndrome
MRS nuclear magnetic resonance
spectroscopy
MtDNA mitochondrial DNA
NARP neuropathy, ataxia, retinitis
pigmentosa
ND NADH dehydrogenase
PCR polymerase chain reaction
PEO progressive external
ophthalmoplegia
PS Pearson syndrome
rRNA ribosomal RNA
RRF ragged-red fibers
tRNA transfer RNA
SDH succinate dehydrogenase
Mitochondrial DNA and disease 223
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
VU
B V
rije
Uni
vers
ity B
russ
els
on 1
0/03
/13
For
pers
onal
use
onl
y.

Figure 1. Morbidity map of the human mitochondrial genome. The map of the 16,569 bp mtDNA shows differently colored areas
representing the protein-coding genes for the seven subunits of complex I (ND), the three subunits of cytochrome c oxidase (CO),
cytochrome b (cyt b), and the two subunits of ATP synthase (A6 and A8), the 12S and 16S rRNAs (12S, 16S), and the 22 tRNAs identified
by one-letter codes for the corresponding amino acids. See list of abbreviations. FBSN5familial bilateral striatal necrosis.
Figure 2. Schematic representation of the respiratory chain. Subunits encoded by mtDNA are in blue and subunits encoded by nuclear
DNA are in red. Electrons (e-) flow along the electron transport chain, and protons (H+) are pumped from the matrix to the intermembrane
space through complexes I, III, and IV, then back into the matrix through complex V, producing ATP. Coenzyme Q (CoQ) and
cytochrome c are electron carriers.
224 S. DiMauro & G. Davidzon
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
VU
B V
rije
Uni
vers
ity B
russ
els
on 1
0/03
/13
For
pers
onal
use
onl
y.
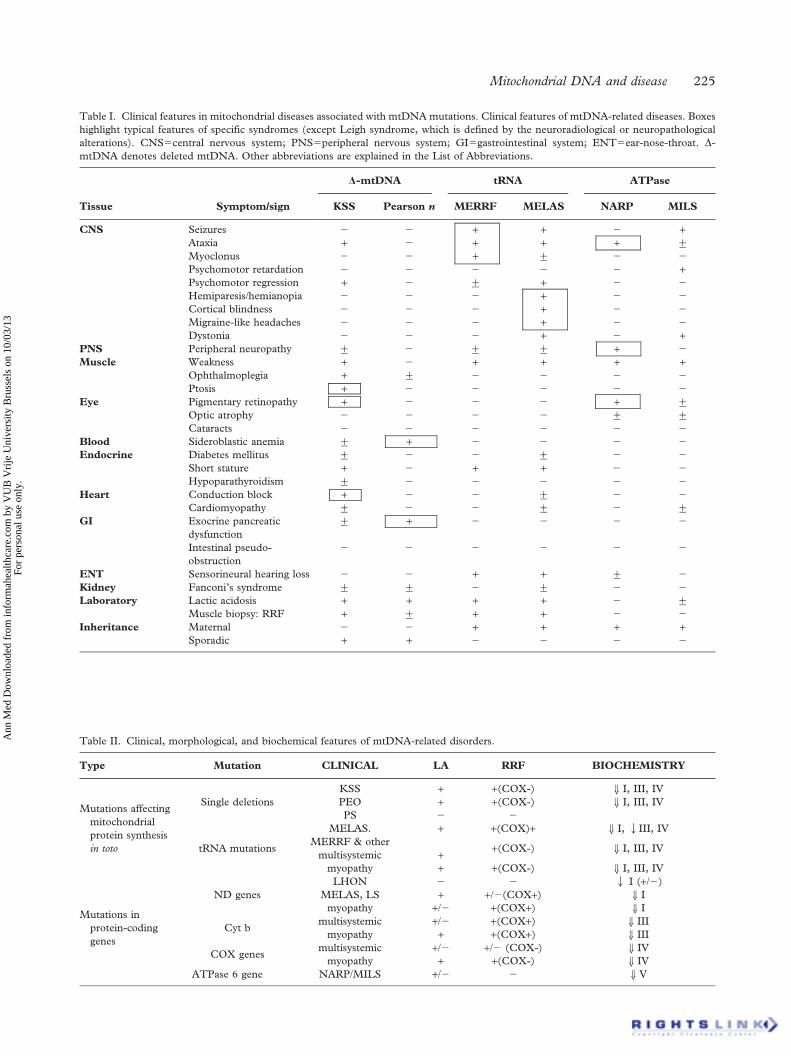
Table I. Clinical features in mitochondrial diseases associated with mtDNA mutations. Clinical features of mtDNA-related diseases. Boxes
highlight typical features of specific syndromes (except Leigh syndrome, which is defined by the neuroradiological or neuropathological
alterations). CNS5central nervous system; PNS5peripheral nervous system; GI5gastrointestinal system; ENT5ear-nose-throat. D-
mtDNA denotes deleted mtDNA. Other abbreviations are explained in the List of Abbreviations.
Tissue Symptom/sign
D-mtDNA tRNA ATPase
KSS Pearson n MERRF MELAS NARP MILS
CNS Seizures 2 2 + + 2 +Ataxia + 2 + + + ¡
Myoclonus 2 2 + ¡ 2 2
Psychomotor retardation 2 2 2 2 2 +Psychomotor regression + 2 ¡ + 2 2
Hemiparesis/hemianopia 2 2 2 + 2 2
Cortical blindness 2 2 2 + 2 2
Migraine-like headaches 2 2 2 + 2 2
Dystonia 2 2 2 + 2 +PNS Peripheral neuropathy ¡ 2 ¡ ¡ + 2
Muscle Weakness + 2 + + + +Ophthalmoplegia + ¡ 2 2 2 2
Ptosis + 2 2 2 2 2
Eye Pigmentary retinopathy + 2 2 2 + ¡
Optic atrophy 2 2 2 2 ¡ ¡
Cataracts 2 2 2 2 2 2
Blood Sideroblastic anemia ¡ + 2 2 2 2
Endocrine Diabetes mellitus ¡ 2 2 ¡ 2 2
Short stature + 2 + + 2 2
Hypoparathyroidism ¡ 2 2 2 2 2
Heart Conduction block + 2 2 ¡ 2 2
Cardiomyopathy ¡ 2 2 ¡ 2 ¡
GI Exocrine pancreatic
dysfunction
¡ + 2 2 2 2
Intestinal pseudo-
obstruction
2 2 2 2 2 2
ENT Sensorineural hearing loss 2 2 + + ¡ 2
Kidney Fanconi’s syndrome ¡ ¡ 2 ¡ 2 2
Laboratory Lactic acidosis + + + + 2 ¡
Muscle biopsy: RRF + ¡ + + 2 2
Inheritance Maternal 2 2 + + + +Sporadic + + 2 2 2 2
Table II. Clinical, morphological, and biochemical features of mtDNA-related disorders.
Type Mutation CLINICAL LA RRF BIOCHEMISTRY
Mutations affecting
mitochondrial
protein synthesis
in toto
Single deletions
KSS + +(COX-) Y I, III, IV
PEO + +(COX-) Y I, III, IV
PS 2 2
tRNA mutations
MELAS. + +(COX)+ Y I, QIII, IV
MERRF & other
multisystemic + +(COX-) Y I, III, IV
myopathy + +(COX-) Y I, III, IV
Mutations in
protein-coding
genes
ND genes
LHON 2 2 Q I (+/2)
MELAS, LS + +/2(COX+) Y I
myopathy +/2 +(COX+) Y I
Cyt bmultisystemic +/2 +(COX+) Y III
myopathy + +(COX+) Y III
COX genesmultisystemic +/2 +/2 (COX-) Y IV
myopathy + +(COX-) Y IV
ATPase 6 gene NARP/MILS +/2 2 Y V
Mitochondrial DNA and disease 225
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
VU
B V
rije
Uni
vers
ity B
russ
els
on 1
0/03
/13
For
pers
onal
use
onl
y.

surprisingly, given the impaired synthesis of all 13
mtDNA-encoded subunits, biochemical analysis of
muscle shows combined defects of all complexes
containing such subunits, which are easily demon-
strable for complexes I, III, and IV, but more
difficult to document for complex V, which is not
measured routinely. In contrast, the biochemical
finding of a specific respiratory chain deficiency is a
strong indicator that a mutation may be present in
one of the mtDNA genes encoding subunits of that
particular complex. Until a few years ago, it was
believed that RRF were not seen in disorders due to
mutations in protein-coding genes. This belief was
based on our experience with LHON (due to
mutations in three ND genes) and NARP/MILS
(due to mutations in the ATPase 6 gene), but was
proven erroneous by studies of patients with myo-
pathy or multisystem disorders and mutations in
ND, cyt b, or COX genes. In contrast to the COX-
negative RRF typical of defects of protein synthesis,
the RRF in these patients stain intensely both with
the SDH stain and with the COX stain (7), except,
of course, for patients with mutations in COX genes
(8).
There have been some practical improvements in
our diagnostic armamentarium, such as the observa-
tion that urinary sediment cells – obtainable more
easily and less invasively than blood samples – are
more sensitive than blood cells for the detection
and quantitation of the A3243G mutation in
oligosymptomatic MELAS patients (9,10). This is
almost certainly true for other mtDNA mutations,
although large series are not yet available. Another
useful test in patients with MELAS and in their
oligosymptomatic or asymptomatic relatives is pro-
ton magnetic resonance spectroscopy (MRS) of the
brain, which allows the measurement of cerebro-
spinal fluid (CSF) lactate non-invasively. In a large
cohort, there was a good correlation between
cerebral lactic acidosis – estimated by ventricular
MRS lactate levels – and severity of neurological
and neuropsychological impairment (11). The rest
of this review will be devoted to new and con-
troversial issues regarding the pathology of mtDNA
mutations.
Frequency of mtDNA-related diseases
A flurry of epidemiological studies in recent years
(reviewed by Schaefer et al. (12)) has confirmed the
notion that mitochondrial diseases – long considered
of purely academic interest – are, in fact, among the
most common genetic disorders and a major burden
for society. When studies in children and adults are
combined and both nuclear DNA and mtDNA
mutations are considered, the minimum prevalence
is at least 1 in 5,000 (12). To focus on patients with
mtDNA mutations, a study of adult patients in
northern England had shown an overall prevalence
of 6.57/100,000 (13), with a particularly high
prevalence of LHON (3.22/100,000) (14). An
apparent discrepancy between the high prevalence
of the MELAS-3243G-related symptoms in north-
ern Finland (5.71/100,000) (15) and a much lower
prevalence (0.95/100,000) in northern England
(13), was apparently due to an underestimation of
the affected English population (12). Two epide-
miological studies of affected children have been
conducted in antipodal countries, Sweden (16) and
Australia (17). The results were remarkably similar,
both in terms of overall prevalence of mitochon-
drial diseases (about 5/100,000) and in terms of
prevalence of mtDNA-related disorders, which
accounted for about 15% of the total.
Inheritance of single mtDNA deletions
By and large, single mtDNA deletions are neither
inherited from the mother nor transmitted to the
progeny and disorders due to single mtDNA
deletions, KSS, PEO, and PS, are almost always
sporadic. This phenomenon is attributed to a
‘bottleneck’ between the ovum (where the giant
deletion probably occurs) and the embryo, such that
only a small minority of maternal mtDNA populates
the fetus (2). However, a few cases of maternal
transmission have been reported (18–21), raising the
question of how reassuring can we be in counseling
carriers of single mtDNA deletions. A recent multi-
center retrospective study of 226 families has
confirmed that the risk of a carrier woman to have
affected children is very small, but finite (1 in 24
births) and has disproved the idea that the risk
increases with maternal age (22).
Pathogenic mutations in mtDNA: Recent
arrivals
Although the small circle of mtDNA is getting
saturated with pathogenic mutations, it is true now –
as it was 5 years ago (23) – that we are not yet
scraping the bottom of the barrel. Novel mutations
are still being reported, especially in protein-coding
genes, albeit at a slower pace. A first flurry of new
mutations in protein-coding genes was associated
with the all-too-frequent but often elusive syndrome
of exercise intolerance (with or without episodic
myoglobinuria). In retrospect, it was surprising that
these symptoms, long associated with defects in the
226 S. DiMauro & G. Davidzon
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
VU
B V
rije
Uni
vers
ity B
russ
els
on 1
0/03
/13
For
pers
onal
use
onl
y.

utilization of the two major ‘muscle fuels’, glycogen
and fatty acids, had not been seen in defects of the
respiratory chain, the quintessential energy-yielding
pathway (24). Within a few years, numerous patients
with exercise intolerance were found (or rediscov-
ered) to have mutations in genes encoding subunits
of complex I (25–27), complex IV (8,28,29) and
especially in the one mtDNA gene for complex III,
cyt b (7,30–36). One reason that delayed the
identification of these patients is that these muta-
tions are de novo events occurring in myoblasts or
myoblast precursors after germ-layer differentiation:
thus, contrary to the ‘rules’ of mitochondrial
genetics, these patients are sporadic and the disease
is confined to skeletal muscle (7). As an unfortunate
clinical consequence, these patients are often mis-
diagnosed as having chronic fatigue syndrome,
fibromyalgia rheumatica or conversion syndrome.
As a matter of curiosity, one of these mutations, a
microdeletion in the ND2 gene (27), generated a
furor in mitochondrial circles not in and by itself, but
because it led to the discovery that most skeletal
muscle mtDNA in this patient was of paternal
origin. This revolutionary finding was shown to be
the proverbial exception that confirms the rule (37–
39) but the unique coexistence of paternal and
maternal mtDNA in the same tissue made it possible
to document that mtDNA molecules can recombine
(40).
More recently, another protein-coding gene has
been the object of much attention and has reached
‘hotspot’ status, ND5. Several mutations have been
described, all associated with maternally inherited
multisystemic disorders (Table III). One mutation
(G13513A) seems to be particularly common and
causes MELAS (41,42), LS (43–45) or MELAS/
LHON overlap (46). Five other mutations in the
same gene have been associated with MELAS
(47,48), MELAS/LS (49), MELAS/MERRF (50)
and even a three-way overlap syndrome, MELAS/
LS/LHON (48). Muscle biopsy in these patients
occasionally shows RRF or pre-RRF (fibers with
subsarcolemmal rims of hyperintense SDH stain)
that are invariably COX-positive (Table III).
Two recently reported mutations are of special
interest because of their unusual clinical phenotypes.
The first, G12147A in tRNAHis, was described in a
previously normal 19-year-old man, who had an
unusually dramatic presentation, with stroke follow-
ing a seizure and complicated by severe brain edema
leading to emergency temporal lobectomy (51). The
stroke was preceded by rhabdomyolysis (serum CK
37, 880 IU/L) and further complicated by Reye-like
liver failure. The take-home message here is that
some mtDNA mutations can debut in a very stormy
fashion, suggesting, as in this young man, venous
infarction or encephalitis. The second mutation,
G611A in tRNAPhe, is unusual in that it presented in
a young woman with the clinical phenotype of
MERRF (52), which thus far has been associated
only with mutations in the tRNALys gene (53).
The question of homoplasmy
Because mtDNA mutates spontaneously at a high
rate and most changes are neutral polymorphisms, a
situation exploited in forensic medicine (54,55), a
set of canonical rules has been established to prove
the pathogenicity of a novel mtDNA mutation. First,
the mutation should not be present in normal
individuals of the same ethnic group. Second, it
should alter a site conserved in evolution and,
therefore, functionally important. Third, it should
cause single or multiple respiratory chain enzyme
deficiencies in affected tissues or defects of mito-
chondrial protein synthesis and respiration demon-
strable in cybrid cell lines. Fourth, there should be a
correlation between degree of heteroplasmy and
clinical severity as well as a correlation between
Table III. Clinical syndromes, abundance of RRF, and complex I deficiency in patients with mutations in the ND5 gene. B, brain; L, liver;
M, muscle.
Mutation Syndrome RRF Complex I Reference
G13513A MELAS +(COX+) 46% (B); 52% (L) (41)
MELAS/LHON 5% (COX+) ‘isolated defect’ (M) (46)
MELAS 1% (COX+) 42% (M) (42)
LS 0 35% (M) (43)
LS n.d. partial defect’’ (M) (44)
LS 0 25% (M) (45)
A13514G MELAS 0-several 40%–60% (M) (47)
A13084T MELAS/LS 0 85% (M) (49)
A12770G MELAS 0 normal(48)
A13045C MELAS/LHON/LS 0 ‘mildly reduced’ (M)
G13042A MELAS/MERRF 0 15%–57% (M) (50)
Mitochondrial DNA and disease 227
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
VU
B V
rije
Uni
vers
ity B
russ
els
on 1
0/03
/13
For
pers
onal
use
onl
y.

degree of heteroplasmy and cell pathology (best
documented by single fiber PCR).
As the last criterion implies, a corollary of these
guidelines is that pathogenic mutations are usually
heteroplasmic whereas neutral polymorphisms are
homoplasmic. In general this is true, but there are
many exceptions and an increasing awareness of the
possible or documented pathogenicity of homoplas-
mic mutations. In fact, the first point mutation
(G11778A in ND4) associated with a human
disease, LHON, was homoplasmic (5), as are other
mutations causing LHON (56). Similarly, most non-
syndromic forms of deafness are due to homoplas-
mic mutations, including A15555G in the 12S
rRNA gene (57), and two mutations in the
tRNASer(UCN) gene, A7455G (58) and T7511C
(59). A homoplasmic mutation (A4300G) in
tRNAIle caused maternally inherited hypertrophic
cardiomyopathy in two families (60), and a homo-
plasmic mutation (C1624T) in tRNAVal caused
multiple neonatal deaths and LS in the offspring of
an also homoplasmic but mildly affected woman
(61). The T14709C mutation in the tRNAGlu gene,
typically associated with myopathy and diabetes
(62), attained homoplasmy in several members of
one large family, some of whom – strangely – were
asymptomatic (63). The latest ‘cause celebre’ of
homoplasmy regards a large pedigree in which the
‘metabolic syndrome’ (syndrome X or dyslipidemic
hypertension), which includes various combinations
of central obesity, atherogenic dyslipidemia, hyper-
tension, hypomagnesemia, and insulin resistance,
was transmitted maternally and attributed to a
homoplasmic mutation (T4291C) in the tRNAIle
(64). Because the metabolic syndrome afflicts about
one fourth of the US population, this finding did not
go unnoticed (65). The challenge with this, as with
other homoplasmic mutations, is to go beyond the
association and to document a deleterious functional
effect. Nuclear magnetic resonance spectroscopy
(MRS) and muscle biopsy were performed in a
single 55-year-old member of the family with the
metabolic syndrome. MRS showed decreased ATP
production and a few pre-RRF were seen in the
biopsy – rather meager evidence of mitochondrial
dysfunction, especially considering the age of the
patient studied.
Most of the canonical criteria for pathogenicity
listed above do not apply to homoplasmic mutations,
except for biochemical evidence of respiratory chain
dysfunction or direct evidence that the expression of
mutated genes is impaired. Defective ATP produc-
tion due to distinct respiratory chain dysfunctions
has been documented in cybrid cells harboring
different LHON mutations (66,67), and high
resolution Northern blots have shown decreased
steady-state levels of tRNAIle in the heart of patients
with the A4300G mutation (60), and decreased
levels of tRNAGlu in muscle of patients with the
T14709C mutation (63). Similar studies are neces-
sary to document a pathogenic role of the T4291C
mutation in patients with the metabolic syndrome.
Another major question underlying the patho-
genic mechanism of homoplasmic mtDNA muta-
tions is why they are expressed in some family
members but not in others and why they can result
in different clinical phenotypes. At least four factors
can influence phenotypic expression: environmental
factors, mtDNA haplotype, nuclear DNA back-
ground, and tissue-specific expression of interacting
genes. The importance of environmental factors is
exemplified by the deleterious effect of aminoglyco-
side exposure in triggering deafness in carriers of the
A1555G mutation (57,68). The influence of
mtDNA haplotype was shown by the higher pene-
trance of the A7455G mutation in a family that also
harbored three ‘secondary’ LHON mutations (69).
The influence of nuclear background was illustrated
by the different severity of complex I deficiency in
two cybrid cell lines, both homoplasmic for the
A3460G LHON mutation, but derived from differ-
ent rho0 cells (osteosarcoma and lung) (70).
The importance of mtDNA haplotypes
In their migration out of Africa, human beings have
accumulated distinctive variations from the mtDNA
of our ancestral ‘mitochondrial Eve’, resulting in
several haplotypes characteristic of different ethnic
groups (71). It has been suggested that different
mtDNA haplotypes may modulate oxidative phos-
phorylation, thus influencing the overall physiology
of individuals and predisposing them to, or protect-
ing them from, certain diseases (71). Among the
functional characteristics reportedly influenced by
mtDNA haplotypes are intelligence quotient (IQ)
(72), spermatozoa swiftness (73) and adaptation to
cold climates (74). Among the diseases, cardiomyo-
pathy (75), Alzheimer disease and dementia with
Lewy bodies (76), and multiple sclerosis (77) have
been associated with specific mtDNA haplotypes.
Also, patients with LHON and certain mtDNA
haplogroups have a higher risk of developing
blindness (78), whereas no link has been established
between the variable phenotypic expression of
MELAS-3243 and mtDNA haplotypes (79). In fact,
a recent retrospective multicenter study of patients
with MELAS and the A3243G mutation (80) has
also failed to confirm a previously reported associa-
tion between a polymorphic variant (A12308G) and
228 S. DiMauro & G. Davidzon
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
VU
B V
rije
Uni
vers
ity B
russ
els
on 1
0/03
/13
For
pers
onal
use
onl
y.

increased risk of stroke (81). Clearly, much work
remains to be done to better define both the
pathogenic role of homoplasmic mutations and the
modulatory role of haplotypes in health and disease.
Pathogenesis
A recent review article by Neil Howell on mitochon-
drial diseases was aptly subtitled: ‘answering ques-
tions and questioning answers’ (78). Seventeen years
after the discovery of pathogenic mutations in
mtDNA, we have lots of answers, i.e. molecular
causes, but precious little understanding of how the
different molecular defects cause different syn-
dromes. In fact, it is surprising that mtDNA
mutations should cause different syndromes in the
first place. If, as conventional wisdom dictates,
mtDNA rearrangements and point mutations in
rRNA or tRNA genes impair mitochondrial protein
synthesis and ATP production, it would be logical to
expect a clinical swamp of ill-defined and over-
lapping symptoms and signs, as was originally
predicted by the ‘lumpers’ (82). Although clinical
overlap does occur in mtDNA-related diseases (see
above), it is fair to say that the ‘splitters’ won the day
in that most mutations result in well defined and
rather stereotypical syndromes.
One major obstacle to functional studies of
mtDNA mutations is the lack of animal models
due to the still unsurmountable problem of introdu-
cing mtDNA into the mitochondria of mammalian
cells. An alternative approach has been to use cybrid
cells, that is, established human cell lines first
depleted of their own mtDNA then repopulated
with various proportions of mutated genomes (83).
This technique has largely confirmed the prediction
that single deletions or mutations in tRNA genes
impair respiration and protein synthesis and
decrease ATP production (84). Elegant as it is, the
in vitro cybrid system cannot replace animal models
in understanding clinical expression. To be sure, two
transmitochondrial mice or ‘mito-mice’ were
obtained through clever, if circuitous, stratagems,
one harboring mtDNA deletions (85) and the other
harboring a point mutation for cloramphenicol
resistance (86). Although these animal ‘prototypes’
are important proofs of principle (87), there has
been no recent progress in this area.
In an attempt to explain the distinctive brain
symptoms in patients with KSS, MERRF, and
MELAS, the different mutations have been
‘mapped’ indirectly through immunohistochemical
techniques. Consistent with clinical symptomatology
and laboratory data, immunocytochemical evidence
suggests that the 3243-MELAS mutation is
abundant in the walls of cerebral arterioles (Tanji
and Bonilla, unpublished), the 8344-MERRF muta-
tion is abundant in the dentate nucleus of the
cerebellum (88), and both the MELAS mutation
and single deletions abound in the choroid plexus
(89). However, these data do not explain what
‘directs’ each mutation to a particular area of the
brain.
Finally, mutations in different tRNA genes may
have different mechanisms of action, as suggested by
the selective tissue vulnerability associated with
mutations in certain tRNAs: for example, cardio-
pathy is often associated with mutations in tRNAIle;
diabetes is a frequent manifestation of the T14709C
mutation in tRNAGlu (62) and multiple lipomas
have been reported only in patients with mutations
in tRNALys (53). But, again, these are associations,
not explanations. It is fair to conclude that the
pathogenesis of mtDNA-related disorders is still
largely terra incognita.
This review does not exhaust the subject of the
title, ‘mitochondrial DNA and disease’. We have
only considered disorders whose primary causes are
well-defined mtDNA mutations, but presumably
secondary mtDNA alterations are also involved in
aging and neurodegenerative disorders (90) – a vast
chapter that deserves a separate review. Nor have we
considered therapy of mtDNA-related disorders,
which is still woefully inadequate and largely
palliative. However, at least at an experimental level,
interesting strategies are being developed, mostly
aimed at shifting downward the percentage of
mutant mtDNAs in affected tissues (91,92).
Because the pathogenic threshold of most mtDNA
mutations is both high and steep, a small shift in
heteroplasmy may result in a disproportionate
clinical benefit. There is a glimmer of light at the
end of the present therapeutic tunnel.
Acknowledgements
Part of the work described here was supported by
NIH grants NS11766 and HD32062, by a grant
from the Muscular Dystrophy Association, and by
the Marriott Mitochondrial Disorders Clinical
Research Fund (MMDCRF).
References
1. Schon EA. The mitochondrial genome. In: Rosenberg RN,
Prusiner SB, DiMauro S, Barchi RL, Nestler EJ, editors. The
Molecular and Genetic Basis of Neurological and Psychi-
atric Disease. Boston: Butterworth Heinemann. 2003:
p. 179–87.
2. DiMauro S, Schon EA. Mitochondrial respiratory-chain
diseases. New Engl J Med. 2003;348:2656–68.
Mitochondrial DNA and disease 229
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
VU
B V
rije
Uni
vers
ity B
russ
els
on 1
0/03
/13
For
pers
onal
use
onl
y.

3. Schon EA, Santra S, Pallotti F, Girvin ME. Pathogenesis of
primary defects in mitochondrial ATP synthesis. Cell
Develop Biol. 2001;12:441–8.
4. Holt IJ, Harding AE, Morgan Hughes JA. Deletions of muscle
mitochondrial DNA in patients with mitochondrial myo-
pathies. Nature. 1988;331:717–9.
5. Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG,
Lezza A, et al. Mitochondrial DNA mutation associated
with Leber’s hereditary optic neuropathy. Science. 1988;242:
1427–30.
6. DiMauro S, Bonilla E. Mitochondrial encephalomyopathies.
In: Engel AG, Franzini-Armstrong C, editors. Myology. New
York: McGraw-Hill. 2004: p. 1623–62.
7. Andreu AL, Hanna MG, Reichmann H, Bruno C, Penn AS,
Tanji K, et al. Exercise intolerance due to mutations in the
cytochrome b gene of mitochondrial DNA. New Engl J Med.
1999;341:1037–44.
8. Karadimas CL, Greenstein P, Sue CM, Joseph JT, Tanji K,
Haller RG, et al. Recurrent myoglobinuria due to a nonsense
mutation in the COX I gene of mtDNA. Neurology.
2000;55:644–9.
9. Shanske S, Pancrudo J, Kaufmann P, Engelstad K, Jhung S,
Lu J, et al. Varying loads of the mitochondrial DNA A3243G
mutation in different tissues: Implications for diagnosis.
Am J Med Genet. 2004;130A:134–7.
10. McDonnell MT, Schaefer AM, Blakely EL, McFarland R,
Chinnery PF, Turnbull DM, et al. Noninvasive diagnosis of
the 3243AwG mitochondrial DNA mutation using urinary
epithelial cells. Eur J Hum Genet. 2004;12:778–81.
11. Kaufmann P, Shungu D, Sano MC, Jungh G, Engelstad K,
Mitsis E, et al. Cerebral lactic acidosis correlates with
neurological impairment in MELAS. Neurology. 2004;62:
1297–302.
12. Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The
epidemiology of mitochondrial disorders - past, present and
future. Biochim Biophys Acta. 2004;1659:115–20.
13. Chinnery PF, Wardell TM, Singh-Kler R, Hayes C,
Johnson MA, Taylor RW, et al. The epidemiology of
pathogenic mitochondrial DNA mutations. Ann Neurol.
2000;48:188–93.
14. Man PY, Griffiths PG, Brown DT, Howell N, Turnbull DM,
Chinnery PF. The epidemiology of Leber hereditary optic
neuropathy in the Northeast of England. Am J Hum Genet.
2003;72:333–9.
15. Majamaa K, Moilanen JS, Uimonen S, Remes AM,
Salmela PI, Karppa M, et al. Epidemiology of A3243G,
the mutation for mitochondrial encephalomyopathy,
lactic acidosis, and stroke-like episodes: Prevalence of the
mutation in an adult population. Am J Hum Genet.
1998;63:447–54.
16. Darin N, Oldfors A, Moslemi A-R, Holme E, Tulinius M.
The incidence of mitochondrial encephalomyopathies in
childhood: Clinical features and morphological, biochemical,
and DNA abnormalities. Ann Neurol. 2001;49:377–83.
17. Skladal D, Halliday J, Thorburn DR. Minimum birth
prevalence of mitochondrial respiratory chain disorders in
children. Brain. 2003;126:1905–12.
18. Shanske S, Tang Y, Hirano M, Nishigaki Y, Tanji K,
Bonilla E, et al. Identical mitochondrial DNA deletion in a
woman with ocular myopathy and in her son with Pearson
syndrome. Am J Hum Genet. 2002;71:679–83.
19. Poulton J, Deadamn ME, Ramacharan S. Germ-line dele-
tions of mtDNA in mitochondrial myopathy. Am J Hum
Genet. 1991;48:649–53.
20. Bernes SM, Bacino C, Prezant TR, Pearson MA, Wood TS,
Fournier P, et al. Identical mitochondrial DNA deletion in
mother with progressive external ophthalmoplegia and son
with Pearson marrow-pancreas syndrome. J Pediat. 1993;
123:598–602.
21. Puoti G, Carrara F, Sampaolo S, De Caro M, Vincitorio CM,
Invernizzi F, et al. Identical large scale rearrangement of
mitochondrial DNA causes Kearns-Sayre syndrome in a
mother and her son. J Med Genet. 2003;40:858–63.
22. Chinnery PF, DiMauro S, Shanske S, Schon EA, Zeviani M,
Mariotti C, et al. Risk of developing a mitochondrial DNA
deletion disorder. Lancet. 2004;364:592–5.
23. DiMauro S, Andreu AL. Mutations in mtDNA: Are we
scraping the bottom of the barrel? Brain Pathology.
2000;10:431–41.
24. DiMauro S. Exercise intolerance and the mitochondrial
respiratory chain. It J Neurol Sci. 1999;20:387–93.
25. Andreu AL, Tanji K, Bruno C, Hadjigeorgiou GM, Sue CM,
Jay C, et al. Exercise intolerance due to a nonsense
mutation in the mtDNA ND4 gene. Ann Neurol. 1999;45:
820–3.
26. Musumeci O, Andreu AL, Shanske S, Bresolin N, Comi GP,
Rothstein R, et al. Intragenic inversion of mtDNA: A new
type of pathogenic mutation in a patient with mitochondrial
myopathy. Am J Hum Genet. 2000;66:1900–4.
27. Schwartz M, Vissing J. Paternal inheritance of mitochondrial
DNA. New Engl J Med. 2002;347:576–80.
28. Keightley JA, Hoffbuhr KC, Burton MD, Salas V,
Johnston WSW, Penn AMW, et al. A microdeletion in
cytochrome c oxidase (COX) subunit III associated with
COX deficiency and recurrent myoglobinuria. Nat Genet.
1996;12:410–15.
29. Rahman S, Taanman J-W, Cooper M, Nelson I, Hargreaves I,
Meunier B, et al. A missense mutation of cytochrome oxidase
subunit II causes defective assembly and myopathy. Am J Hum
Genet. 1999;65:1030–39.
30. Dumoulin R, Sagnol I, Ferlin T, Bozon D, Stepien G,
Mousson B. A novel gly290asp mitochondrial cytochrome b
mutation linked to a complex III deficiency in progres-
sive exercise intolerance. Mol Cell Probes. 1996;10:
389–91.
31. Keightley JA, Anitori R, Burton MD, Quan F, Buist NRM,
Kennaway NG. Mitochondrial encephalomyopathy and
complex III deficiency associated with a stop-codon mutation
in the cytochrome b gene. Am J Hum Genet. 2000;67:
1400–10.
32. Andreu AL, Bruno C, Shanske S, Shtilbans A, Hirano M,
Krishna S, et al. Missense mutation in the mtDNA
cytochrome b gene in a patient with myopathy. Neurology.
1998;51:1444–7.
33. Andreu AL, Bruno C, Dunne TC, Tanji K, Shanske S,
Sue CM, et al. A nonsense mutation (G15059A) in the
cytochrome b gene in a patient with exercise intolerance and
myoglobinuria. Ann Neurol. 1999;45:127–30.
34. Lamantea E, Carrara F, Mariotti C, Morandi L, Tiranti V,
Zeviani M. A novel nonsense mutation (Q352X) in the
mitochondrial cytochrome b gene associated with a combined
defect of complexes I and III. Neuromusc Disord.
2002;12:49–52.
35. Mancuso M, Filosto M, Stevens JC, Patterson M, Shanske S,
Krishna S, et al. Mitochondrial myopathy and complex III
deficiency in a patient with a new stop-codon mutation
(G339X) in the cytochrome b gene. J Neurol Sci. 2003;209:
61–3.
36. Legros F, Chatzoglou E, Frachon P, Ogier De Baulny H,
Laforet P, Jardel C, et al. Functional characterization of novel
mutations in the human cytochrome b gene. Eur J Hum
Genet. 2001;9:510–18.
230 S. DiMauro & G. Davidzon
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
VU
B V
rije
Uni
vers
ity B
russ
els
on 1
0/03
/13
For
pers
onal
use
onl
y.

37. Taylor RW, McDonnell MT, Blakely EL, Chinnery PF,
Taylor GA, Howell N, et al. Genotypes from patients indicate
no paternal mitochondrial contribution. Ann Neurol.
2003;54:521–24.
38. Filosto M, Mancuso M, Vives-Bauza C, Vila MR, Shanske S,
Hirano M, et al. Lack of paternal inheritance of muscle
mitochondrial DNA in sporadic mitochondrial myopathies.
Ann Neurol. 2003;54:524–6.
39. Schwartz F, Vissing J. No evidence of paternal inheritance of
mtDNA in patients with sporadic mtDNA mutations.
J Neurol Sci. 2004;218:99–101.
40. Kraytsberg Y, Schwartz M, Brown TA, Ebralidse K,
Kunz WS, Clayton DA, et al. Recombination of human
mitochondrial DNA. Science. 2004;304:981.
41. Santorelli FM, Tanji K, Kulikova R, Shanske S, Vilarinho L,
Hays AP, et al. Identification of a novel mutation in the
mtDNA ND5 gene associated with MELAS. Biochem
Biophys Res Comm. 1997;238:326–8.
42. Penisson-Besnier I, Reynier P, Asfar P, Douay O, Sortais A,
Dubas F, et al. Recurrent brain hematomas in MELAS
associated with an ND5 gene mitochondrial mutation.
Neurology. 2000;55:317–8.
43. Kirby DM, Boneh A, Chow CW, Ohtake A, Ryan MT,
Thyagarajan D, et al. Low mutant load of mitochondrial
DNA G13513A mutation can cause Leigh’s disease. Ann
Neurol. 2003;54:473–8.
44. Chol M, LebonS, Benit P, Chretien D,de LonlayP, Cormier V,
et al. The mtDNA G-A mutation at nt 13513, peviously
reported in MELAS syndrome, is a frequent cause of
Leigh disease. Am J Hum Genet. 2002;71(Suppl.):Abstract #
1494.
45. Petruzzella V, Di Giacinto G, Scacco S, Piemonte F,
Torraco A, Carrozzo R, et al. Atypical Leigh syndrome
associated with the D393N mutation in the mitochondrial
ND5 subunit. Neurology. 2003;61:1017–8.
46. Pulkes T, Eunson L, Patterson V, Siddiqui A, Wood NW,
Nelson IP, et al. The mitochondrial DNA G13513A
transition in ND5 is associated with a LHON/MELAS
overlap syndrome and may be a frequent cause of MELAS.
Ann Neurol. 1999;46:916–9.
47. Corona P, Antozzi C, Carrara F, D’Incerti L, Lamantea E,
Tiranti V, et al. A novel mtDNA mutation in the ND5
subunit of complex I i two MELAS patients. Ann Neurol.
2001;49:106–10.
48. Liolitsa D, Rahman S, Benton S, Carr LJ, Hanna MG. Is the
mitochondrial complex I ND5 gene a hot-spot for MELAS
causing mutations? Ann Neurol. 2003;53:128–32.
49. Crimi M, Galbiati S, Moroni I, Bordoni A, Perini MP,
Lamantea E, et al. A missense mutation in the mitochondrial
ND5 gene associated with a Leigh-MELAS overlap syn-
drome. Neurology. 2003;60:1857–61.
50. Naini A, Lu J, Kaufmann P, Bernstein RA, Mancuso M,
Bonilla E, et al. Novel mtDNA ND5 mutation in a patient
with clinical features of MELAS and MERRF. Arch Neurol.
2005;64:539–41.
51. Taylor RW, Schaefer AM, McDonnell MT, Petty RKH,
Thomas AM, Blakely EL, et al. Catastrophic presentation of
mitochondrial disease due to a mutation in the tRNAHis
gene. Neurology. 2004;62:1420–23.
52. Mancuso M, Filosto M, Mootha VK, Rocchi A, Pistolesi S,
Murri L, et al. A novel mitochondrial tRNAPhe mutation
causes MERRF syndrome. Neurology. 2004;62:2119–21.
53. DiMauro S, Hirano M, Kaufmann P, Tanji K, Sano M,
Shungu DC, et al. Clinical features and genetics of myoclonic
epilepsy with ragged red fibers. In: Fahn S, Frucht SJ, editors.
Myoclonus and Paroxysmal Dyskinesia. Philadelphia:
Lippincott Williams & Wilkins. 2002: p. 217–29.
54. Gill P, Ivanov PL, Kimpton C, Piercy R, Benson N, Tully G,
et al. Identification of the remains of the Romanov family by
DNA analysis. Nat Genet. 1994;6:130–5.
55. Anslinger K, Weichhold G, Keil W, Bayer B. Identification of
the skeletal remains of Martin Bormann by mtDNA analysis.
Int J Legal Med. 2001;114:194–6.
56. Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial
dysfunction as a cause of optic neuropathies. Progr Retin Eye
Res. 2004;23:53–89.
57. Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S,
Qiu W-Q, et al. Mitochondrial ribosomal RNA mutation
associated with both antibiotic-induced and non-syndromic
deafness. Nat Genet. 1993;4:289–93.
58. Sue CM, Tanji K, Hadjigeorgiou G, Andreu AL, Nishino I,
Krishna S, et al. Maternally inherited hearing loss in a large
kindred with a novel mutation in the mitochondrial DNA
tRNASer(UCN) gene. Neurology. 1999;52:1905–8.
59. Li X, Fischel-Ghodsian N, Schwartz F, Yan Q, Friedman RA,
Guan MX. Biochemical characterization of the mitochondrial
tRNASer(UCN) T7511C mutation associated with nonsyn-
dromic deafness. Nucleic Acids Res. 2004;32:867–77.
60. Taylor RW, Giordano C, Davidson MM, d’Amati G,
Bain H, Hayes CM, et al. A homoplasmic mitochondrial
transfer ribonucleic acid mutation as a cause of maternally
inherited cardiomyopathy. J Am Coll Cardiol. 2003;41:
1786–96.
61. McFarland R, Clark KM, Morris AAM, Taylor RW,
Macphail S, Lightowlers RN, et al. Multiple neonatal deaths
due to homoplasmic mitochondrial DNA mutation. Nat
Genet. 2002;30:145–6.
62. Mancuso M, Ferraris S, Nisjigaki Y, Azan G, Mauro A,
Sammarco P, et al. Congenital or late-onset myopathy with
the T14709C mtDNA mutation. J Neurol Sci. 2005;228:
93–7.
63. McFarland R, Schaefer AM, Gardner A, Lynn S, Hayes CM,
Barron MJ, et al. Familial myopathy: New insights into the
T14709C mitochondrial tRNA mutation. Ann Neurol.
2004;55:478–84.
64. Wilson FH, Hariri A, Farhi A, Zhao H, Petersen KF,
Toka HR, et al. A cluster of metabolic defects caused by
mutation in a mitochondrial tRNA. Science. 2004;306:
1190–4.
65. Hampton T. Mitochondrial defects may play a role in the
metabolic syndrome. JAMA. 2004;292:2823–4.
66. Baracca A, Solaini G, Sgarbi G, Lenaz G, Baruzzi A,
Schapira AHV, et al. Severe impairment of complex I-driven
ATP synthesis in Leber’s hereditary optic neuropathy cybrids.
Arch Neurol. 2005. In press.
67. Hirano M, DiMauro S. Leber’s hereditary optic neuropathy:
biochemical lights in a blurry scenario. Arch Neurol. 2005. In
press.
68. Estivill X, Govea N, Barcelo’ A, Perello’ E, Badenas C,
Romero E, et al. Familial progressive sensorineural deafness is
mainly due to the mtDNA A1555G mutation and is
enhanced by treatment with aminoglycosides. Am J Hum
Genet. 1998;62:27–35.
69. Fischel-Ghotsian N. Mitochondrial mutations and hearing
loss: Paradigm for mitochondrial genetics. Am J Hum Genet.
1998;62:15–9.
70. Cock HR, Tabrizi SJ, Cooper JM, Schapira AHV. The
influence of nuclear background on the biochemical expres-
sion of 3460 Leber’s hereditary optic neuropathy. Ann
Neurol. 1998;44:187–93.
Mitochondrial DNA and disease 231
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
VU
B V
rije
Uni
vers
ity B
russ
els
on 1
0/03
/13
For
pers
onal
use
onl
y.

71. Wallace DC, Brown MD, Lott MT. Mitochondrial DNA
variation in human evolution and disease. Gene. 1999;238:
211–30.
72. Skuder P. A polymorphism in mitochondrial DNA associated
with IQ? Intelligence. 1995;21:1–11.
73. Ruiz-Pesini E, Lapena AC, Diez-Sanchez C, Perez-Martos A,
Montoya J, Alvarez E, et al. Human mtDNA haplogroups
associated with high or reduced spermatozoa motility.
Am J Hum Genet. 2000;67:682–96.
74. Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V,
Wallace DC. Effects of purifying and adaptive selection on
regional variation in human mtDNA. Science. 2004;303:
223–226.
75. Shin WS, Tanaka M, Suzuki J-i, Hemmi C, Toyo-oka T. A
novel homoplasmic mutation in mtDNA with a single
evolutionary origin as a risk factor for cardiomyopathy.
Am J Hum Genet. 2000;67:1617–20.
76. Chinnery PF, Taylor GA, Howell N, Andrews RM,
Morris CM, Taylor RW, et al. Mitochondrial DNA haplo-
groups and susceptibility to AD and dementia with Lewy
bodies. Neurology. 2000;55:302–4.
77. Kalman B, Li S, Chatterjee D, O’Connor J, Voehl MR,
Brown MD, et al. Large scale screening of the mitochondrial
DNA reveals no pathogenic mutations but a haplotype
associated with multiple sclerosis in Caucasians. Acta
Neurol Scand. 1999;99:16–25.
78. Howell N. Human mitochondrial diseases: answering ques-
tions and questioning answers. Int Rev Cytol. 1999;186:
49–116.
79. Torroni A, Campos Y, Rengo C, Sellitto D, Achilli A,
Magri C, et al. Mitochondrial DNA haplogroups do not play
a role in the variable phenotypic presentation of the A3243G
mutation. Am J Hum Genet. 2003;72:1005–12.
80. Deschauer M, Chinnery PF, Schaefer AM, Turnbull DM,
Taylor RW, Zierz S, et al. No association of the mitochon-
drial DNA A12308G polymorphism with increased risk of
stroke in patients with the A3243G mutation. J Neurol
Neurosurg Psychiat. 2004;75:1204–5.
81. Pulkes T, Sweeney MG, Hanna MG. Increased risk of stroke
in patients with the A12308G polymorphism in mitochon-
dria. Lancet. 2000;356:2068–9.
82. Rowland LP. Mitochondrial encephalomyopathies: Lumping,
splitting, and melding. In: Schapira AHV, DiMauro S,
editors. Mitochondrial disorders in neurology. Oxford:
Butterworth-Heinemann. 1994: p. 116–29.
83. King MP, Attardi G. Human cells lacking mtDNA: repopu-
lation with exogenous mitochondria by complementation.
Science. 1989;246:500–3.
84. Pallotti F, Baracca A, Hernandez-Rosa E, Walker WF,
Solaini G, Lenaz G, et al. Biochemical analysis of respiratory
function in cybrid cell lines harbouring mitochondrial DNA
mutations. Biochem J. 2004;384:287–93.
85. Inoue K, Nakada K, Ogura A, Isobe K, Goto Y-i, Nonaka I,
et al. Generation of mice with mitochondrial dysfunction by
introducing mouse mtDNA carrying a deletion into zygotes.
Nat Genet. 2000;26:176–81.
86. Sligh JE, Levy SE, Waymire KG, Allard P, Dillehay DL,
Nusinowitz S, et al. Maternal germ-line transmission of
mutant mtDNAs from embryonic stem cell-derived chimeric
mice. Proc Nat Acad Sci U S A. 2000;97:14461–6.
87. Hirano M. Transmitochondrial mice: Proof of principle and
promises. Proc Nat Acad Sci U S A. 2001;98:401–3.
88. Sparaco M, Schon EA, DiMauro S, Bonilla E. Myoclonic
epilepsy with ragged-red fibers (MERRF): An immunohisto-
chemical study of the brain. Brain Pathol. 1995;5:125–33.
89. Tanji K, Schon EA, DiMauro S, Bonilla E. Kearns-Sayre
syndrome: oncocytic transformation of choroid plexus
epithelium. J Neurol Sci. 2000;178:29–36.
90. Schon EA, Manfredi G. Neuronal degeneration and mito-
chondrial dysfunction. J Clin Invest. 2003;111:303–12.
91. Schon EA, DiMauro S. Medicinal and genetic approach to
the treatment of mitochondrial disease. Curr Medicinal
Chem. 2003;10:2523–33.
92. Taylor RW, Turnbull DM. Current and future prospects for
the treatment of mitochondrial disorders. In: Schapira AHV,
DiMauro S, editors. Mitochondrial Disorders in Neurology 2.
Boston: Butterworth-Heinemann. 2002: p. 213–27.
232 S. DiMauro & G. Davidzon
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
VU
B V
rije
Uni
vers
ity B
russ
els
on 1
0/03
/13
For
pers
onal
use
onl
y.

HUMAN MUTATION 15:7�12 (2000)
© 2000 WILEY-LISS, INC.
MDI SPECIAL ARTICLE
Mutation Nomenclature Extensions and Suggestionsto Describe Complex Mutations: A DiscussionJohan T. den Dunnen1* and Stylianos E. Antonarakis2*1MGC-Department of Human and Clinical Genetics, Leiden University Medical Center, Leiden, The Netherlands2Division of Medical Genetics, University of Geneva Medical School, Geneva, Switzerland
Consistent gene mutation nomenclature is essential for efficient and accurate reporting, testing,and curation of the growing number of disease mutations and useful polymorphisms being discov-ered in the human genome. While a codified mutation nomenclature system for simple DNAlesions has now been adopted broadly by the medical genetics community, it is inherently difficultto represent complex mutations in a unified manner. In this article, suggestions are presented forreporting just such complex mutations. Hum Mutat 15:7–12, 2000. © 2000 Wiley-Liss, Inc.
KEY WORDS: complex mutation; mutation detection; mutation database; nomenclature; MDI
INTRODUCTION
Recently, a nomenclature system has been sug-gested for the description of changes (mutationsand polymorphisms) in DNA and protein se-quences [Antonarakis et al., 1998]. These nomen-clature recommendations have now been largelyaccepted and stimulated the uniform and un-equivocal description of sequence changes. How-ever, current rules do not yet cover all types ofmutations, nor do they cover more complex mu-tations. The goal of this article is to make sugges-tions for additional mutation nomenclaturerecommendations and to stimulate discussions asto how far the rules should go regarding the de-scription of complex mutations. Discussions re-garding the advantages and disadvantages of thesuggestions are necessary in order to continuouslyimprove the designation of sequence changes. Theconsensus of the discussions will be posted on theWorld Wide Web (http://www.dmd.nl/mutnomen.html). We also invite investigators to communi-cate with us complicated cases with a suggestionof how to describe these (send e-mail to: [email protected] and [email protected]); anoverview will be listed at the same WWW-address.Ultimately, this list of examples for the descriptionof complicated cases may evolve into a uniformlyaccepted reference for mutation nomenclature.
The nomenclature needs to be accurate andunambiguous, but flexible, and the nucleotidechange must always be included in the original
report. The items missing in the current nomen-clature recommendations which will be coveredin this article include simple omissions (changesin mitochondrial DNA), simple mutations (dupli-cations, inversions), and more complex mutations(mutations in recessive disease, insertion/deletions,changes inside codons, frame shifts). It should benoted that when describing a mutation as a “dele-tion,” “insertion,” or “other,” one should alwaysindicate which level is described; a substitution atthe DNA level may cause an insertion at the RNAlevel and a nonsense mutation at the protein level.Furthermore, for descriptions at protein and RNAlevels, which are mostly derived from the DNA se-quence with only rarely experimental proof, it shouldbe clear that recommendations cover a descriptionof the consequence rather than the nature of the se-quence change. It can be debated whether this shouldbe done, but we believe it should.
A weak element in many reports is often thedescription at the level of mature or processedmessenger RNA. Many reports fail to mentionclearly, and discriminate in tabular listings, atwhich level the sequence variations reported were
Received 7 September 1999; accepted revised manuscript 4October 1999.
*Correspondence to: Johan T. den Dunnen, Dept. of Humanand Clinical Genetics, LUMC, Wassenaarseweg 72, 2333 ALLeiden, The Netherlands. E-mail: [email protected]; or Stylianos E. Antonarakis, Division of MedicalGenetics, University of Geneva Medical School, 9 Avenue deChampel, 1211 Geneva, Switzerland. E-mail: [email protected]

8 DEN DUNNEN AND ANTONARAKIS
analyzed. Consequently, it often remains unclearwhether a change might have an effect on theRNA level or whether the suggested effect on RNAhas been proven or not. That this is not trivial isexemplified by several cases where changes wereoriginally reported as silent or missense (based onDNA data), although they actually representedchanges affecting mRNA processing [see, e.g., Ri-chard and Beckmann, 1995]. Several of the rec-ommendations below are specifically made toclarify this issue.
Nomenclature recommendations by themselvesdo not safeguard against mistakes. A clear exampleis the recommendation to use the one-letter aminoacid code for descriptions at the protein level. Sev-eral examples from recent publications show that itis not rare that mistakes are made. Codes are mixedup easily for amino acids which have the same initialletter (e.g., Alanine, Arginine, Asparagine, and As-partic acid or Glycine, Glutamine, and Glutamicacid). Hopefully, electronic tools for submission ofsequence changes, with automatic error checks, willhelp to avoid these problems (see http://ariel.ucs.unimelb.edu.au:80/~cotton/entry.htm).
SUMMARY OF PUBLISHED
RECOMMENDATIONS
General [from Antonarakis et al., 1998]
• Sequence variations are best described at theDNA level.
• The accession number in primary sequencedatabases (Genbank, EMBL, DDJB, SWISS-PROT) should be mentioned in the publica-tion/database submission. When available,the genomic reference sequence is preferred.For each gene, a reference sequence needs tobe established.
• To avoid confusion, the nucleotide number ispreceded by “g.” when a genomic or by “c.”when a cDNA reference sequence is used.
• For genomic DNA and cDNA sequences, theA of the ATG of the initiator Methioninecodon is denoted nucleotide +1 (there is nonucleotide zero). The nucleotide 5′ to +1 isnumbered –1.
• For variations in single nt (or amino acid)stretches or tandem repeats, the most 3′ copyis arbitrarily assigned to have been changed(e.g., ATGTGCA to ATGCA is described as4-5delTG).
• Two mutations in the same allele are listedwithin brackets, separated by a semicolon; e.g.[1997G>T; 2001A>C] (see below).
• A unique identifier should be obtained for eachmutation. Preferably, locus-specific databasecurators should assign unique identifiers. Alter-natively, the OMIM unique identifier or theHGMD entry can be used as a reference sourcefor previously cataloged mutations.
Description at the DNA Level
• Nucleotide changes start with the nucleotidenumber and the change follows this number;{nucleotide interval}{sequence changednucleotide}{type of change}{sequence newnucleotide}.
• Substitutions are designated by “>”; 1997G>Tdenotes that at nt 1997 of the reference se-quence a G is changed to a T.
• Deletions are designated by “del” after the de-leted interval (followed by the deleted nts).1997-1999del (alternatively 1997-1999-delTTC) denotes a TTC deletion from nts 1997to 1999. A TG deletion in the sequenceACTGTGTGCC (A is nt 1991) is designatedas 1997-1998del (or 1997-1998delTG).
• Insertions are designated by “ins,” followedby the inserted nts. 1997-1998insT denotesthat a T was inserted between nts 1997 and1998. A TG insertion in the TG-tandem re-peat sequence of ACTGTGTGCC (A is nt1991) is described as 1998-1999insTG (where1998 is the last G of the TG-repeat).
• Variability of short sequence repeats, e.g., inACTGTGTGCC (A is nt 1991), is designatedas 1993(TG)3-22 with nt 1993 containing thefirst TG-dinucleotide which is found repeated3 to 22 times in the population.
• Intron mutations are designated by the intronnumber (preceded by “IVS”) or cDNA posi-tion; positive numbers starting from the G ofthe GT splice donor site, negative numbersstarting from the G of the AG splice acceptorsite. IVS4-2A>C (1998-2A>C) denotes theA to C substitution at nt –2 of intron 4, atthe cDNA level positioned between nucle-otides 1997 and 1998. IVS4+1G>T (1997+1G>T) denotes the G to T substitution at nt+1 of intron 4. When the full-length genomicsequence is known, the mutation is best des-ignated by the nt number of the genomic ref-erence sequence.
Description at the Protein Level
Note that recommendations for sequence varia-tions at the protein level describe the deducedconsequence and not the nature of the mutation.

MUTATION NOMENCLATURE 9
Furthermore, it should be avoided to reportchanges at the amino acid level without includingthe description at the nucleotide level.
• The codon for the initiator Methionine iscodon 1.
• Stop codons are designated by X. R97X de-notes a change of Arginine 96 to a termina-tion codon.
• The single letter amino acid code is recom-mended, three letter code is acceptable.
• Amino acid changes are described in the for-mat {code first amino acid changed}{aminoacid interval}{code new amino acid or typeof change}. Y97S denotes Tyrosine97 is sub-stituted by a Serine.
• Deletions are designated by “del” after theamino acid interval. T97-C102del (or T97-C102del6) denotes that amino acid Threo-nine97 to Cysteine102 are deleted.
• Insertions are designated by “ins” after theamino acid interval, followed by the insertedamino acids or the number of amino acidsinserted. T97-W98insLQS (or T97-W98ins3)denotes that three amino acids are insertedbetween amino acids 97 and 98 (i.e., afterThreonine97).
EXTENSIONS AND ADDITIONAL
SUGGESTIONS
Sequence Variations in Mitochondrial DNASuggestion (from David Fung, Camperdown,
TABLE 2. Sequence Variation Description at the Level of Mature or Processed Messenger RNA
Type of variation Description Alternative Remark
r.15c>u change described in relation to a RNAsequence
as for DNA but lower case lettersSplice variants [16g>t + 16_112del] mutation affecting splicing, producing two
mRNAs, one normal and one with adeletion of nucleotides 16 to 112 (exon 2)
Complex depends on change examples at http://www.dmd.nl/mutnomen.html
Suggestions from this article in italics. Sample sequence 5′-CAUGC AUG CAU GCA UGU UUC GUC-3′ with nucleotides-CAUGC- being the 5′ untranslated region in exon 1 and -AUG- being the translation initiation codon.
TABLE 1. Sequence Variation Description at the DNA Level
Type of variation Description Alternative Remark
g.12T>A change described in relation to a genomicsequence
c.12T>A change described in relation to a cDNAsequence
m.12T>A change described in relation to amitochondrial sequence
[6T>C + 13_14del] one allele containing two changes[13_14del] + [?] mutations in the 2 alleles of a recessive
disease, one being unknown (=?)Substitution 12T>A
15+1G>C IVS1+1G>C change in intronic sequence (splice donorsite)
93-2A>G IVS1-2A>G change in intronic sequence (spliceacceptor site)
Deletion 13_14delTT 13_14delIVS1_IVS5 15+?_645+?del genomic deletion of exons 2 to 5
or EX2_EX5delDuplication 10_11dupTG 10_11dupInsertion 14_15insTInversion 4_15invInsertion/deletion 112_117delAGGTCAinsTG 112_117delinsTG also known as ‘‘indel’’
or 112_117>TGComplex depends on change examples at http://www.dmd.nl/mutno
men.html
Suggestions from this article in italics. Sample sequence 5′-CATGC ATG CAT GCA TGT TTC GTGAGTATATC-3′ with nucleotides-CATGC- being the 5′ untranslated region in exon 1, -ATG-being the translation site and -GTGAGT...- being the first nucleotidesin intron 1.

10 DEN DUNNEN AND ANTONARAKIS
Australia): when a mitochondrial reference se-quence is used, the nucleotide number is precededby “m.” (e.g., m.1203A>T).
Sequence Variations in Protein Sequences
Suggestion: when a protein reference sequenceis used, the amino acid is preceded by “p.” (e.g.,p.K93W).
Descriptions of a Range
Due to the fact that the “–” symbol is used for twodifferent purposes, i.e., to indicate a range (R12-W13)as well as to indicate a negative distance (77-2A>G),current recommendations to describe sequencechanges starting and or ending in intronic sequencesmay easily cause confusion. For example, does 5-77-77del describe a deletion from nt 5-77 to 77 or fromnt 5 to 77-77? It would be better not to use the “–”symbol for two purposes. Since for intronic positionsboth the “–” and “+” symbols are used, a changeshould involve the “–” symbol separating the firstfrom the last affected nt.
Suggestion: the “_” symbol (underscore) shouldbe used to separate the first from the last affectednt (or amino acid), e.g., 85_86delAG. On the pro-tein level as K23_W29.
Duplications
No recommendations have been made to describeduplications. Although they can be seen as a specifictype of insertion, and could be described as such,they often originate through other mutational mecha-nisms. We therefore prefer to provide a distinctivedesignation of this type of sequence change
Suggestion: duplications are described as1992_1994dupCTG (or 1992_1994dup). On theprotein level as L78dup.
As a consequence, duplicating insertions in
short tandem repeats (or single nucleotidestretches) can also be described as a duplication,e.g., 1997_1998dupTG (now 1998_1999insTG).
Inversions
Recommendations for inversions are missing butcan be rather straightforward using the existingrules.
Suggestion: inversions are described as 203_506inv (or 203_506inv304) indicating that the 304nt’s from position 203 to 506 have been inverted.
Mutations in Recessive Diseases
The current suggestion for the description ofrecessive mutations is [1997G>T + 2001A>G],indicating the substitution of nt 1997 on one al-lele and of nt 2001 on the other allele. The de-scription should be given the status of arecommendation, which is important for two rea-sons, 1) it makes clear whether a mutation wasfound on both alleles, and 2) it ensures that re-searchers show which mutations were identifiedin which combinations. The latter is importantsince severity might depend on the combinationof mutations present, while other combinationsmight not be deleterious at all. However, the cur-rent description is not completely straightforwardand might cause confusion with that for the de-scription of two mutations in one allele, currentlylike [1997G>T; 2001A>C].
Suggestion: two variations in one allele aredescribed as [1997G>T + 2001A>C] whilevariations in different alleles, e.g., in recessivediseases, are designated as [1997G>T] +[2001A>G]. In homozygous cases the formatis [1997G>T] + [1997G>T]. When only onemutated allele has been identified, the formatis [1997G>T] + [?]. On the protein level, the
TABLE 3. Sequence Variation Description at the Protein Level
Type of variation Description Alternative Remark
p.R5S change described in relation to a proteinsequence
others see Table ISubstitution R5S
W4X change to stop codonDeletion L3_W4delDuplication L3_W4dupInsertion W4_R5insKInversion – not relevantFrame shift W4fsX8 W4fs frame shift causing a translational stop 5 codons
downstreamComplex depends on change examples at http://www.dmd.nl/mutnomen.
html
Suggestions from this article in italics. Sample sequence M-I-L-W-R-R-C, with amino acid M representing the translation initiat-ing Methionine.

MUTATION NOMENCLATURE 11
designation is likewise, e.g., two variations inone allele [R175X + C305S] versus [R175X]+ [C305S] for variations in different alleles.
Mutations Analyzed at Which Level?
Many reports fail to mention clearly, and dis-criminate in tabular listings, at which level (DNA,RNA, and/or protein) the sequence variations re-ported were analyzed and whether experimentalproof was obtained regarding the descriptions pro-vided at the RNA and protein level.
Suggestion: In tabular listings of the sequencevariations identified, it should be stated clearlywhich variation was analyzed at which level, i.e.,DNA, RNA, and/or protein.
Description of Mutations at the RNA Level
When RNA has been analyzed, the effect is of-ten not described properly and recommendationsfor its description are lacking. Based on the no-menclature rules to describe mutations on theDNA level, suggestions can be simply copied whenthree additions are made.
Suggestion 1: An “r.” is used to indicate that achange is described at the RNA level.
Suggestion 2: To discriminate between descrip-tions at the DNA and protein levels, lower caseletters and the “u” for Uracil are used to describechanges at the RNA level.
Suggestion 3: When one change affects RNA-processing, yielding two or more transcripts, theseare described between brackets, separated by a “+”symbol, e.g., [r.76a>c + r.70_77del], i.e., the ntchange c.76A>C causes the appearance of twoRNA molecules, one carrying the variation onlyand one containing a deletion of nucleotides 70 to77 (shift of splice site).
It should be noted here that the designations atthe RNA level, similar to that on the protein level,describe the consequence and not the nature ofthe mutation.
Exon Deletions Detected at the
Genomic Level
In diseases such as Duchenne Muscular Dys-trophy (DMD), many mutations are found whichdelete (sets of) whole exons, detected on South-ern blot using cDNA probes or using exon specificPCR-tests. Current rules for mutation descriptiondo not cover these, with the consequence thateverybody uses their own system.
Suggestion: exonic deletions are described asIVS2_IVS5del (alternatives 77+?_923+? orEX3_5del), indicating a deletion starting at an
unknown position in intron 2 (after base 77) andending at an unknown position in intron 5 (afterbase 923).
Insertion-Deletions
The occurrence of a combination of a deletionand insertion, sometimes named “indel,” is notrare. Recommendations for their description havenot yet been made. Based on existing terminol-ogy, a suggestion can be rather straightforward.
Suggestion: a combination of a deletion andinsertion at the same site is described as 112_117delAGGTCAinsTG (alternatively 112_117delinsTG or 112_117>TG). On the proteinlevel, likewise as W33_K35delinsR.
Mutations in the Translation Initiation Site
Currently, mutations in the translation initiat-ing Methionine (M1) are mostly described as asubstitution, e.g., M1V. We would like to note thatthis is not correct; either no protein is producedor the translation initiation site moves up- ordownstream. Unless experimental proof is avail-able, it is probably best to report the effect on pro-tein level as “unknown.” When experimental datashow that no protein is made, the description “p.0”might be most appropriate.
Sequence Variation in Codons
Current recommendations do not fully cover thedescription of amino acid substitutions andchanges inside codons that do not alter the read-ing frame.
Suggestion 1: C28_V29delinsW denotes a 3 bpdeletion affecting the codons for Cysteine28 andValine29, changing them to a codon for Tryp-tophan.
Suggestion 2: C28delinsWV denotes a 3 bp in-sertion in the codon for Cysteine28, generatingcodons for Tryptophan and Valine.
Frame-Shifting Mutations
At the protein level, no recommendations havebeen made regarding the description of frame-shift-ing mutations. Although it is probably not usefulto add much detail in this description, it might besensible, e.g., in the case of C-terminal mutations,to include the length of the new, shifted readingframe.
Suggestion 1: Frame-shifting mutations are des-ignated by “fs.” R97fs denotes a frame-shiftingchange with Arginine97 as the first affected aminoacid. The length of the shifted open reading framemay be described using the format R97fsX121, in-

12 DEN DUNNEN AND ANTONARAKIS
dicating a frame-shifting change with Arginine97as the first affected amino acid and the new read-ing frame being open for 23 amino acids.
REFERENCES
Antonarakis SE, the Nomenclature Working Group. 1998. Rec-ommendations for a nomenclature system for human genemutations. Hum Mutat 11:1–3.
Beaudet AL, the Ad Hoc Committee on Mutation Nomencla-ture. 1996. Update on nomenclature for human gene muta-tions. Hum Mutat 8:197–202.
Beutler E, McKusick VA, Motulsky A, Scriver CR, HutchinsonF. 1996. Mutation nomenclature: nicknames, systematicnames and unique identifiers. Hum Mutat 8:203–206.
Richard I, Beckmann JS. 1995. How neutral are synonymouscodon mutations? Nat Genet 10:259.

© 2013 The Society for Investigative Dermatology www.jidonline.org 1
research Techniques made simple
Next-Generation Sequencing: Methodology and ApplicationAyman Grada1 and Kate Weinbrecht2
Journal of Investigative Dermatology (2013) 133, e11; doi:10.1038/jid.2013.248
inTrOducTiOnNucleic acid sequencing is a method for determining the exact order of nucleotides present in a given DNA or RNA molecule. In the past decade, the use of nucleic acid sequencing has increased exponentially as the ability to sequence has become accessible to research and clini-cal labs all over the world. The first major foray into DNA sequencing was the Human Genome Project, a $3 billion, 13-year-long endeavor, completed in 2003. The Human Genome Project was accomplished with first-generation sequencing, known as Sanger sequencing. Sanger sequenc-ing (the chain-termination method), developed in 1975 by Edward Sanger, was considered the gold standard for nucleic acid sequencing for the subsequent two and a half decades (Sanger et al., 1977).
Since completion of the first human genome sequence, demand for cheaper and faster sequencing methods has increased greatly. This demand has driven the develop-ment of second-generation sequencing methods, or next-generation sequencing (NGS). NGS platforms perform massively parallel sequencing, during which millions of fragments of DNA from a single sample are sequenced in unison. Massively parallel sequencing technology facili-tates high-throughput sequencing, which allows an entire genome to be sequenced in less than one day. In the past decade, several NGS platforms have been developed that provide low-cost, high-throughput sequencing. Here we highlight two of the most commonly used platforms in research and clinical labs today: the LifeTechnologies Ion Torrent Personal Genome Machine (PGM) and the Illumina MiSeq. The creation of these and other NGS platforms has made sequencing accessible to more labs, rapidly increas-ing the amount of research and clinical diagnostics being performed with nucleic acid sequencing.
OVerVieW OF The meThOdOlOGYAlthough each NGS platform is unique in how sequenc-ing is accomplished, the Ion Torrent PGM and the Illumina MiSeq have a similar base methodology that includes tem-plate preparation, sequencing and imaging, and data analysis (Metzker, 2010). Within each generalized step, the individual
platforms discussed have unique aspects. An overview of the sequencing methodologies discussed is provided in Figure 1.
WHAT NGS DOES• NGS provides a much cheaper and higher-
throughput alternative to sequencing DNA than traditional Sanger sequencing. Whole small genomes can now be sequenced in a day.
• High-throughput sequencing of the human genome facilitates the discovery of genes and regulatory elements associated with disease.
• Targeted sequencing allows the identification of disease-causing mutations for diagnosis of pathological conditions.
• RNA-seq can provide information on the entire transcriptome of a sample in a single analysis without requiring previous knowledge of the genetic sequence of an organism. This technique offers a strong alternative to the use of microarrays in gene expression studies.
LIMITATIONS • NGS, although much less costly in time and money
in comparison to first-generation sequencing, is still too expensive for many labs. NGS platforms can cost more than $100,000 in start-up costs, and individual sequencing reactions can cost upward of $1,000 per genome.
• Inaccurate sequencing of homopolymer regions (spans of repeating nucleotides) on certain NGS platforms, including the Ion Torrent PGM, and short-sequencing read lengths (on average 200–500 nucleotides) can lead to sequence errors.
• Data analysis can be time-consuming and may require special knowledge of bioinformatics to garner accurate information from sequence data.
1Department of Dermatology, Boston University School of Medicine, Boston, Massachusetts, USA and 2School of Forensic Sciences, Center for Health Sci-ences, Oklahoma State University, Tulsa, Oklahoma, USA
Correspondence: Ayman Grada, Department of Dermatology, Boston University School of Medicine, 609 Albany Street, Boston, Massachusetts 02118, USA. E-mail: [email protected]

research Techniques made simple
2 Journal of Investigative Dermatology (2013), Volume 133 © 2013 The Society for Investigative Dermatology
research Techniques made simple
data analysisOnce sequencing is complete, raw sequence data must undergo several analysis steps. A generalized data analysis pipeline for NGS data includes preprocessing the data to remove adapter sequences and low-quality reads, mapping of the data to a reference genome or de novo alignment of the sequence reads, and analysis of the compiled sequence. Analysis of the sequence can include a wide variety of bio-informatics assessments, including genetic variant calling for detection of SNPs or indels (i.e., the insertion or deletion of bases), detection of novel genes or regulatory elements, and assessment of transcript expression levels. Analysis can also include identification of both somatic and germline mutation events that may contribute to the diagnosis of a disease or genetic condition. Many free online tools and software pack-ages exist to perform the bioinformatics necessary to success-fully analyze sequence data (Gogol-Döring and Chen, 2012).
applicaTiOnsThe applications of NGS seem almost endless, allowing for rapid advances in many fields related to the biological sci-ences. Resequencing of the human genome is being per-formed to identify genes and regulatory elements involved in pathological processes. NGS has also provided a wealth of knowledge for comparative biology studies through whole-genome sequencing of a wide variety of organisms. NGS is applied in the fields of public health and epidemiology through the sequencing of bacterial and viral species to facili-tate the identification of novel virulence factors. Additionally, gene expression studies using RNA-Seq (NGS of RNA) have begun to replace the use of microarray analysis, providing researchers and clinicians with the ability to visualize RNA expression in sequence form. These are simply some of the broad applications that begin to skim the surface of what NGS can offer the researcher and the clinician. As NGS con-tinues to grow in popularity, it is inevitable that there will be additional innovative applications.
nGs in pracTiceWhole-exome sequencingMutation events that occur in gene-coding or control regions can give rise to indistinguishable clinical presenta-tions, leaving the diagnosing clinician with many possible causes for a given condition or disease. With NGS, clini-cians are provided a fast, affordable, and thorough way to determine the genetic cause of a disease. Although high-throughput sequencing of the entire human genome is pos-sible, researchers and clinicians are typically interested in only the protein-coding regions of the genome, referred to as the exome. The exome comprises just over 1% of the genome and is therefore much more cost-effective to sequence than the entire genome, while providing sequence information for protein-coding regions.
Exome sequencing has been used extensively in the past several years in gene discovery research. Several gene dis-covery studies have resulted in the identification of genes that are relevant to inherited skin disease (Lai-Cheong and McGrath, 2011). Exome sequencing can also facilitate the
Template preparationTemplate preparation consists of building a library of nucleic acids (DNA or complementary DNA (cDNA)) and amplify-ing that library. Sequencing libraries are constructed by frag-menting the DNA (or cDNA) sample and ligating adapter sequences (synthetic oligonucleotides of a known sequence) onto the ends of the DNA fragments. Once constructed, libraries are clonally amplified in preparation for sequencing. The PGM utilizes emulsion PCR on the OneTouch system to amplify single library fragments onto microbeads, whereas the MiSeq utilizes bridge amplification to form template clus-ters on a flow cell (Berglund et al., 2011; Quail et al., 2012).
sequencing and imagingTo obtain nucleic acid sequence from the amplified librar-ies, the Ion Torrent PGM and the MiSeq both rely on sequencing by synthesis. The library fragments act as a template, off of which a new DNA fragment is synthesized. The sequencing occurs through a cycle of washing and flooding the fragments with the known nucleotides in a sequential order. As nucleotides incorporate into the grow-ing DNA strand, they are digitally recorded as sequence. The PGM and the MiSeq each rely on a slightly different mechanism for detecting nucleotide sequence information. The PGM performs semiconductor sequencing that relies on the detection of pH changes induced by the release of a hydrogen ion upon the incorporation of a nucleotide into a growing strand of DNA (Quail et al., 2012). By contrast, the MiSeq relies on the detection of fluorescence generated by the incorporation of fluorescently labeled nucleotides into the growing strand of DNA (Quail et al., 2012).
Figure 1. next-generation sequencing methodology.

© 2013 The Society for Investigative Dermatology www.jidonline.org 3
research Techniques made simple research Techniques made simple
identification of disease-causing mutations in pathogenic presentations where the exact genetic cause is not known.
Figure 2 (Cullinane et al., 2011) demonstrates the direct effect that NGS can have on the correct diagnoses of a patient. It summarizes the use of homozygosity mapping followed by whole-exome sequencing to identify two dis-ease-causing mutations in a patient with oculocutaneous albinism and congenital neutropenia (Cullinane et al., 2011). Figure 2a and 2b display the phenotypic traits common to oculocutaneous albinism type 4 and neutropenia observed in this patient. Figure 2c is a pedigree of the patient’s family, both the affected and unaffected individuals. The idiogram (graphic chromosome map) in Figure 2d highlights the areas of genetic homozygosity. These regions were identi-fied by single-nucleotide-polymorphism array analysis and were considered possible locations for the disease-causing mutation(s). Figures 2e and 2f display chromatograms for the two disease-causing mutations identified by whole-exome sequencing. Figure 2e depicts the mutation in SLC45A2, and Figure 2f depicts the mutation in G6PC3. This case portrays the valuable role that NGS can play in the correct diagno-sis of an individual patient who displays disparate symptoms with an unidentified genetic cause.
Targeted sequencingAlthough whole-genome and whole-exome sequencing are possible, in many cases where a suspected disease or con-dition has been identified, targeted sequencing of specific genes or genomic regions is preferred. Targeted sequencing is more affordable, yields much higher coverage of genomic regions of interest, and reduces sequencing cost and time (Xuan et al., 2012). Researchers have begun to develop sequencing panels that target hundreds of genomic regions that are hotspots for disease-causing mutations. These pan-els target only desired regions of the genome for sequenc-ing, eliminating the majority of the genome from analysis. Targeted sequencing panels can be developed by researchers
1. The basic methodological steps of nGs include the following:
A. Template preparation, emulsion PCR, sequencing, data analysis.
B. Template preparation, sequencing and imaging, data analysis.
C. Template amplification, sequencing and imaging, data analysis.
D. Template preparation, sequencing and imaging, alignment to a reference genome.
E. DNA fragmentation, sequencing, data analysis.
2. advantages of targeted sequencing as opposed to full-genome, exome, or transcriptome sequencing include the following:
A. Affordable and efficient for quickly interrogating particular genomic regions of interest.
B. Provides a deeper coverage of genomic regions of interest.
C. Can be utilized in deciding a therapeutic plan of action for both germline and somatic cancers.
D. Detects and quantifies low-frequency variants such as rare drug-resistant viral mutations (e.g., HIV, hepatitis B virus, or microbial pathogens).
E. All of the above.
3. applications of nGs in medicine include the following:
A. Detecting mutations that play a role in diseases such as cancer.
B. Identifying genes responsible for inherited skin diseases.
C. Determining RNA expression levels.
D. Identifying novel virulence factors through sequencing of bacterial and viral species.
E. All of the above.
QUESTIONSAnswers are available as supplementary material online and at http://www.scilogs.com/jid/.
or clinicians to include specific genomic regions of interest. In addition, sequencing panels that target common regions of interest can be purchased for clinical use; these include pan-els that target hotspots for cancer-causing mutations (Rehm, 2013). Targeted sequencing—whether of individual genes or whole panels of genomic regions—aids in the rapid diagno-sis of many genetic diseases. The results of disease-targeted sequencing can aid in therapeutic decision making in many diseases, including many cancers for which the treatments can be cancer-type specific (Rehm, 2013).
Figure 2. clinical application of whole-exome sequencing in the detection of two disease-causing mutations. Reprinted from Cullinane et al., 2011.

research Techniques made simple
4 Journal of Investigative Dermatology (2013), Volume 133 © 2013 The Society for Investigative Dermatology
research Techniques made simple
CONFLICT OF INTERESTThe authors state no conflict of interest.
SUPPLEMENTARY MATERIALAnswers and a PowerPoint slide presentation appropriate for journal club or other teaching exercises are available at http://dx.doi.org/10.1038/jid.2013.248.
REFERENCESBerglund EC, Kiialainen A, Syvänen AC (2011) Next-generation sequencing
technologies and applications for human genetic history and forensics. Invest Genet 2:23
Cullinane AR, Vilboux T, O’Brien K et al. (2011) Homozygosity mapping and whole-exome sequencing to detect SLC45A2 and G6PC3 mutations in a single patient with oculocutaneous albinism and neutropenia. J Invest Dermatol 131:2017–25
Gogol-Döring A, Chen W (2012) An overview of the analysis of next generation sequencing data. Methods Mol Biol 802:249–57
Lai-Cheong JE, McGrath JA (2011) Next-generation diagnostics for inherited skin disorders. J Invest Dermatol 131:1971–3
Metzker ML (2010) Sequencing technologies—the next generation. Nat Rev Genet 11:31–46
Quail MA, Smith M, Coupland P et al. (2012) A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom 13:341
Rehm HL (2013) Disease-targeted sequencing: a cornerstone in the clinic. Nat Rev Genet 14:295–300
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–7
Xuan J, Yu Y, Qing T et al. (2012) Next-generation sequencing in the clinic: promises and challenges. Cancer Lett; e-pub ahead of print 19 November 2012

review article
T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 366;12 nejm.org march 22, 20121132
Mechanisms of Disease
Monogenic Mitochondrial Disorders
Werner J.H. Koopman, Ph.D., Peter H.G.M. Willems, Ph.D., and Jan A.M. Smeitink, Ph.D.
From the Department of Biochemistry, Nijmegen Center for Molecular Life Sci-ences (W.J.H.K., P.H.G.M.W.), and the De-partment of Pediatrics, Nijmegen Center for Mitochondrial Disorders (J.A.M.S.) — both at Radboud University Medical Cen-ter, Nijmegen, the Netherlands. Address reprint requests to Dr. Smeitink at Nijme-gen Center for Mitochondrial Disorders, Radboud University Nijmegen Medical Center, Geert Grooteplein Zuid 10, P.O. Box 9101, 6500 HB Nijmegen, the Neth-erlands, or at [email protected].
N Engl J Med 2012;366:1132-41.Copyright © 2012 Massachusetts Medical Society.
T o function normally, human cells require energy in the form of ATP. In many cell types, ATP is primarily generated by mitochondria, which are also key players in other important cellular processes, such as
adaptive thermogenesis,1 ion homeostasis,2 innate immune responses,3 production of reactive oxygen species,4 and programmed cell death (apoptosis).5 Mitochondria contain their own DNA (mtDNA), which encodes 13 mitochondrial proteins, 2 ribo-somal RNAs (rRNAs), and 22 transfer RNAs (tRNAs). The replication, transcription, translation, and repair of mtDNA are controlled by proteins encoded by nuclear DNA (nDNA).6,7
Mitochondrial dysfunction is not only observed in monogenic mitochondrial disorders but is also associated with more common pathologic conditions, such as Alzheimer’s disease,8 Parkinson’s disease,9 cancer,10 cardiac disease,11 diabetes,12 epilepsy,13 Huntington’s disease,14 and obesity.15 In addition, a progressive decline in the expression of mitochondrial genes is a central feature of normal human aging.16 It is not entirely clear whether the changes in expression that occur with aging have positive or negative effects on life span. Given the aging of the population in devel-oped societies and the increasing prevalence of conditions such as Alzheimer’s dis-ease and diabetes, the investigation of mitochondrial processes and diseases may make a timely contribution to our understanding of the human aging process and its relationship with the above conditions.
Other sources of interference in mitochondrial function include the off-target ef-fects of environmental toxins (e.g., rotenone) and frequently used drugs (e.g., amiodarone, biguanides, haloperidol, statins, valproic acid, zidovudine), anesthetics (e.g., halothane), antibiotics (e.g., chloramphenicol), chemotherapeutic agents (e.g., doxorubicin), and even aspirin (acetylsalicylic acid).17,18 Certain drugs may lead more frequently to adverse reactions and side effects in patients with mitochondrial dis-orders than in healthy persons.17,19 The term “mitochondrial medicine”20 refers to approaches that have been developed to manage mitochondrial dysfunction and, directly or indirectly, its consequences.21
In the past decade, the analysis of monogenic mitochondrial diseases has consid-erably advanced our understanding of the cellular pathophysiology of mitochondrial dysfunction. Here we summarize these insights and explain how they can contribute to the rational design of intervention strategies for mitochondrial dysfunction; we present our ongoing research on human mitochondrial complex I deficiency as an example of mitochondrial medicine. This information is preceded by a brief primer on the basics of mitochondrial structure and ATP generation.
The New England Journal of Medicine Downloaded from nejm.org at KU LEUVEN BIOMEDICAL LIBRARY on September 11, 2012. For personal use only. No other uses without permission.
Copyright © 2012 Massachusetts Medical Society. All rights reserved.

Mechanisms of Disease
n engl j med 366;12 nejm.org march 22, 2012 1133
In ter na l a nd E x ter na l S TRUC T UR E A ND ATP GENER ATION
Structure
Mitochondrial internal and external structure var-ies with cell type and metabolic state and often becomes altered during mitochondrial dysfunction (Fig. 1A and 1B).22 Net mitochondrial morphology depends on the balance between mitochondrial motility, fission, and fusion. This equilibrium is regulated by dedicated proteins and plays a role in metabolic energy dissipation, turnover of dam-aged mitochondria, distribution of mitochondria throughout the cell, induction of apoptosis, mito-chondrial inheritance during cell division, and de-fense against aging.23 Mitochondria consist of outer and inner membranes that envelop an aqueous compartment, the mitochondrial matrix (Fig. 1C and 1D). The space between the inner and outer membranes is referred to as the intermembrane space. The outer membrane is relatively smooth, whereas the inner membrane contains many ma-trix-protruding folds (or cristae) that increase the surface area of the inner membrane and have a dy-namic structure.22 These structural dynamics may serve to regulate mitochondrial metabolism.24
ATP Generation
The best-known mitochondrial function is the production of ATP. For this purpose, glucose is converted to pyruvate through the glycolysis path-way in the cytosol (Fig. 1E). Pyruvate can be con-verted into lactate10 or taken up by mitochondria, where it is converted to acetyl coenzyme A (CoA). Acetyl CoA enters the tricarboxylic acid cycle that generates the reduced forms of NADH and flavin adenine dinucleotide (FADH2). Alternatively, fatty acids25 and glutamine10 can be used as substrates. NADH and FADH2 fuel the oxidative-phosphory-lation system, which consists of five functionally coupled mitochondrial protein complexes.26 NADH and FADH2 are oxidized at mitochondrial com-plexes I and II, respectively (Fig. 1F). The released electrons are transported to complex III by coen-zyme Q10 (CoQ10) and then to complex IV by cy-tochrome c, where they are donated to molecular oxygen to form water. The energy released by electron transport is used at complexes I, III, and IV to expel protons (H+) from the mitochondrial matrix, which establishes a proton gradient across the mitochondrial inner membrane. The lat-ter is associated with an inside-negative inner-
membrane potential (Δψ) and a matrix-directed proton-motive force. The proton-motive force is used at complex V to generate ATP, which is transported to the cytosol by the adenine nucleo-tide translocator. The proton-motive force also sustains many other mitochondrial functions (e.g., ion transport, metabolite exchange, protein import, and mitochondrial fusion).27 As a conse-quence, defects in oxidative phosphorylation of-ten induce multiple cellular aberrations.
MONO GENIC CELL MODEL S OF MI T O CHONDR I A L DYSFUNC TION
Primary and Secondary Disorders
With the use of mitochondrial proteome analysis,28 approximately 1000 genes encoding mitochondrial proteins have been identified in humans (MitoCarta human inventory, Broad Institute). Mitochondrial dysfunction can arise from a mutation in one of these genes (causing a primary mitochondrial dis-order) or from an outside influence on mitochon-dria (causing a secondary mitochondrial disorder). Causes of secondary disorders include viral infec-tions29 and off-target drug effects.17,18 To date, mutations in 228 protein-encoding nDNA genes and 13 mtDNA genes have been linked to a human disorder (Fig. 2). (For a list of these genes and their associated disease phenotypes, see Table 1 in the Supplementary Appendix, available with the full text of this article at NEJM.org.) Given the preponderance of nDNA-encoded mutations, this review focuses mainly on these genes. A detailed discussion of mtDNA mutations and their associ-ated pathology is provided elsewhere.30-35
Mutations and Phenotype
Although it is evident that mutations in mito-chondrial proteins induce mitochondrial dysfunc-tion, it is less clear how specific genetic defects are linked to dysfunction at the level of cells, or-gans, and the whole organism. One major diffi-culty in determining the effects of specific defects is that the consequences of mitochondrial dysfunc-tion are affected by the action of compensatory stress–response pathways. At the cellular level, these responses include mitochondrial biogenesis, increased expression of oxidative phosphorylation proteins, a switch to a more glycolytic mode of ATP production, and the removal of dysfunctional mitochondria by quality-control systems.9,36-39 To complicate matters even further, the substrate sup-
The New England Journal of Medicine Downloaded from nejm.org at KU LEUVEN BIOMEDICAL LIBRARY on September 11, 2012. For personal use only. No other uses without permission.
Copyright © 2012 Massachusetts Medical Society. All rights reserved.
T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 366;12 nejm.org march 22, 20121134
ply, the mode of ATP generation, the level of de-mand for ATP, mitochondrial dynamics, and the rate of oxidative phosphorylation differ among cell types and tissue types.
These factors increase the likelihood that the various types of tissue are not equally sensitive to mitochondrial dysfunction.27,40 At the level of the
organism, genetic background — both mitochon-drial and nuclear — also modifies the phenotype of human mitochondrial diseases (e.g., as reported by Bénit et al.41). In this sense, mutations in mi-tochondrial proteins can evoke pathologic pheno-types specific to a cell, tissue, organ, or patient that are manifested only when a certain threshold
1
Longo
3/02/12
AUTHOR PLEASE NOTE:Figure has been redrawn and type has been reset
Please check carefully
Author
Fig #
Title
ME
DEArtist
Issue date
COLOR FIGURE
Draft 4Koopman
Knoper
3/22/12
A
C D
E F
Normal fibroblast B Patient fibroblast (Leigh’s disease)
Low ∆ψ High ∆ψ Low ∆ψ High ∆ψ
Glucose
Glycolysis
Cyt
osol
Cytosol
Mito
chon
drio
n
Mitochondrion
Matrix
Matrix
Cristae IMS
MIMMOM
MOMIMSMIM
Pyruvate
Pyruvate
Acetyl CoA
Fatty acids
GIutamine
Glutamate
TCA cycle
OXPHOS
Lactate
ATP
ATP
ATPATP
NADH NADH
NADH FADH2
FADH2
CoQ10
O2
O2
H+
H+
H+
H+
VDAC
VDAC
VDAC
VDAC
VDAC
CIV
CV
+++
---
Cytochrome c
The New England Journal of Medicine Downloaded from nejm.org at KU LEUVEN BIOMEDICAL LIBRARY on September 11, 2012. For personal use only. No other uses without permission.
Copyright © 2012 Massachusetts Medical Society. All rights reserved.

Mechanisms of Disease
n engl j med 366;12 nejm.org march 22, 2012 1135
of mitochondrial dysfunction or cellular demand on mitochondrial metabolism is exceeded. This phenomenon is exemplified by the broad variety of clinical presentations associated with mutations in nDNA-encoded mitochondrial genes (see Table 1 in the Supplementary Appendix).
Developing a Rational Intervention Strategy
Mitochondrial function is often studied in mito-chondrial suspensions that have been isolated from patient cells or tissues with the use of detergent
treatment.42 However, mitochondrial metabolism in situ is closely linked to that of the cell and is generally associated with submaximal rates of mi-tochondrial enzyme activity. In addition, the cel-lular milieu provides for communication among mitochondria, cytosol, and other organelles, and mitochondrial structure is greatly altered during isolation. As a consequence, the functional prop-erties of isolated mitochondria differ considerably from those within the cell.43-45
Mitochondrial function can also be investigated in intact (patient) cells.22,24,25,27,42,46-48 Here we summarize our ongoing research on complex I deficiency to illustrate how live-cell analysis can be integrated into an approach to the development of treatment strategies (Fig. 3).
Using biopsy specimens from pediatric patients carrying nDNA-encoded complex I mutations, we have cultured primary fibroblast cell lines, which are maintained under standardized conditions.47 Cellular and mitochondrial physiology is assessed with the use of fluorescent reporter molecules that are introduced into the cells and analyzed by means of live-cell microscopy (Fig. 4). The results are then compared with those in cells from healthy children.49,52-57 Statistical evaluation of 26 phys-iological variables in 14 control and 24 patient cell lines has revealed that complex I mutations lead to a reduction in the expression of complex I, depo-larization of mitochondrial membrane potential, the accumulation of NADH, increased levels of reactive oxygen species, aberrations in mitochon-drial structure, and aberrations in the homeostasis of cytosolic and mitochondrial Ca2+ and ATP.58 The extent of these aberrations has been corre-lated with the age of the patient at disease onset and with death from the disease.58
These results are currently being used in dis-ease modeling and drug-target prediction to de-sign an intervention strategy focused on the above aberrations. Once a lead compound has been iden-tified, the dependence of its effects on time and concentration can be evaluated at the cellular level to assess structure–activity relationships and its properties of absorption, distribution, metabolism, and excretion (or toxicity).59 In the case of complex I deficiency, lead compounds will then be evaluat-ed in an animal model.57,60 Animal models are also available for several other primary and second-ary mitochondrial disorders.31,32,56,61,62 Once the outcome scores in the animal model are satisfac-tory, the effects of any compounds that are de-veloped can be evaluated in clinical trials.
Figure 1 (facing page). Mitochondrial Structure and ATP Generation.
Panel A shows elongated mitochondria (filaments) in a living skin fibroblast from a healthy person (staining with the mitochondria-specific cation tetramethylrho-damine methyl ester [TMRM] and visualized with fluo-rescence microscopy). The more intense (red) the TMRM signal, the more negative the mitochondrial membrane potential (Δψ). Panel B shows the fragment-ed mitochondrial structure in living skin fibroblasts from a patient with a mitochondrial disorder (Leigh’s disease) caused by a mutation in NDUFS2, which en-codes a subunit of complex I. TMRM staining is less in-tense (i.e., membrane potential is less negative) than in healthy fibroblasts. In the electron micrograph shown in Panel C, individual mitochondrial filaments (arrows) can be seen in the cytosol of a fixed primary-skin fibro-blast from a healthy person. The dark line is the mem-brane of the cell nucleus. Panel D shows mitochondrial ultrastructure and compartmentalization, highlighting the mitochondrial outer membrane (MOM), mitochon-drial inner membrane (MIM), and intermembrane space (IMS). Panel E summarizes the main ATP production pathway in human cells. In the cytosol, glucose is con-verted into pyruvate, which is subsequently processed in the mitochondrion to acetyl coenzyme A (CoA) and the reduced forms of NADH and flavin adenine dinu-cleotide (FADH2). In addition to using pyruvate as a substrate, the mitochondrion can also use fatty acids and glutamine. Mitochondrial ATP production is car-ried out by the oxidative phosphorylation (OXPHOS) system, shown at the bottom of Panel E. This system is embedded in the MIM and consists of five multiprotein complexes (CI through CV), as shown in Panel F. NADH and FADH2 are oxidized at CI and CII, and the electrons released are transported to CIII by coenzyme Q10 (CoQ10). The electrons are then transferred to cyto-chrome c and delivered to CIV, where they react with molecular oxygen (O2). TCA denotes tricarboxylic acid. The energy released during electron transport is used to expel protons (H+) from the mitochondrial matrix across the MIM into the IMS. This creates a proton gradient across the MIM, which is associated with an inside-negative potential. At CV, the energy stored in the proton gradient is used to drive ATP production by allowing the reentry of H+ into the matrix. Because of the presence of voltage-dependent anion channels (VDACs), the MOM is ion-permeable.
The New England Journal of Medicine Downloaded from nejm.org at KU LEUVEN BIOMEDICAL LIBRARY on September 11, 2012. For personal use only. No other uses without permission.
Copyright © 2012 Massachusetts Medical Society. All rights reserved.

T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 366;12 nejm.org march 22, 20121136
IN TERV EN TION S TR ATEGIES
In a primary mitochondrial disorder, the mutation directly affects the expression level of the corre-sponding protein, its function, or both, inducing primary cell consequences and secondary cell con-sequences (Fig. 5).27,63 The secondary consequenc-es often constitute part of the adaptive signaling mechanisms.
Adaptations include the partial mitigation of reduced mitochondrial ATP production by in-creased glycolysis, increases in the cellular levels of mitochondrial proteins and mitochondrial func-tion through mitochondrial biogenesis, and up-regulation of the detoxification of reactive oxygen species, triggered by changes in the redox state
that were induced by reactive oxygen species. It appears that primary and secondary mitochondrial disorders have similar consequences at the cellular level (Fig. 5).27,58,64,65
Four intervention strategies for mitochondrial dysfunction have been described: genetic therapy, the use of small molecules to target mitochondrial dysfunction, metabolic manipulation, and diet and exercise (Fig. 5, and Table 2 in the Supplemen-tary Appendix). Genetic therapy, which is carried out at the preclinical level and focuses mainly on mtDNA-associated disorders, is discussed else-where.19,63,64,66,67 Small-molecule therapies, met-abolic manipulation, and dietary and exercise regi-mens generally aim to increase the capacity for ATP synthesis, bypass the mitochondrial defect,
2
Longo
2/10/12
AUTHOR PLEASE NOTE:Figure has been redrawn and type has been reset
Please check carefully
Author
Fig #
Title
ME
DEArtist
Issue date
COLOR FIGURE
Draft 6Koopman
Knoper
3/22/12
HLCSLRPPRCLRRK2MFN2MLYCDNFU1 PARK2PARK7SACSSPG20WWOX
GLRX5HOGA1MMABMMADHCPDSS2
AFG3L2COQ2COQ6COQ9GLDC
PNKDPUS1REEP1STARTMEM126A
Mitochondrialmatrix
Submitochondriallocation
Partiallylocalized
Mitochondrial inner membrane
Mitochondrial outer membrane
Intermembranespace
AARS2ACAD8ACAD9ACADMACADSACADSBACAT1ALAS2ALDH2ALDH4A1ALDH6A1AMTATPAF2AUHBCAT2BCKDHABCKDHBBCS1LC8orf38C10orf2C12orf65C20orf7COA5COX10COX15CPS1D2HGDHDARS2DBTDECR1DGUOKDLDDLATDMGDHETFAETFBETFDHFOXRED1
FHGCDHGCSHGFM1GLUD1HADHHARS2HIBCHHMGCS2HMGCLHSD17B10HSPD1IDH2IDH3BISCUIVDKARSMCCC1MCCC2MCEEME2MRPS16MRPS22MTFMTMTPAPMUTNAGSNDUFAF1NDUFAF2NDUFAF3NDUFAF4NUBPLOATOGDHOTCOXCT1PCPCCA
PCCBPCK2PDHA1PDHBPDHXPDP1POLGPOLG2PYCR1RARS2RMRPSARDHSARS2SCO1SCO2SDHAF1SDHAF2SOD2SUCLA2SUCLG1SURF1TACO1TK2TMEM70TRMUTSFMTTC19TUFMUNGXPNPEP3YARS2
ABCB7ACADVLADCK3AGKATP5EC12orf62COX4I2COX6B1CPT2CRATCYCSCYP11A1CYP11B1CYP11B2CYP24A1CYP27A1CYP27B1
DHODHDNAJC19FASTKD2GPD2HADHAHADHBHCCSL2HGDHMMAAMPV17NDUFA1NDUFA2NDUFA9NDUFA10NDUFA11NDUFA12NDUFA13
NDUFB3NDUFB9 NDUFV1NDUFV2NDUFS1NDUFS2NDUFS3NDUFS4NDUFS6NDUFS7NDUFS8OPA1OPA3PDSS1SDHASDHBSDHC
SDHDSLC25A3SLC25A4SLC25A12SLC25A13SLC25A15SLC25A19SLC25A20SLC25A22SLC25A38SPG7TIMM8AUCP1UCP2UCP3UQCRBUQCRQ
ARMS2BCL2CPT1ADNM1LGCKGKKIF1BMAOAPINK1
AK2DIABLOGATMGFERHTRA2PANK2PPOX
MTND1MTND2MTND3MTND4MTND4LMTND5MTND6MTCYBMTCO1MTCO2MTCO3MTATP6MTATP8
97 81
22
15107
AIFM1AKAP10AMACRAPTXBAXBOLA3CYB5R3ETHE1FXNGDAP1HK1
Figure 2. Genes of Mitochondria-Localized Proteins Linked to Disease in Humans.
This list summarizes the currently identified mtDNA-encoded (red) and nDNA-encoded (black) genes associated with mitochondrial diseases in humans when mutated. The genes are categorized according to the predicted localization of their corresponding proteins in the mitochondrial matrix (107 genes), the outer membrane (9 genes), the intermembrane space (7 genes), and the inner membrane (81 genes). Also included are gene products of unknown submitochondrial location (15 genes) and gene products that partially localize to mitochondria (22 genes). Detailed information about the genes and their associated disease phenotypes is provided in Table 1 in the Supplementary Appendix.
The New England Journal of Medicine Downloaded from nejm.org at KU LEUVEN BIOMEDICAL LIBRARY on September 11, 2012. For personal use only. No other uses without permission.
Copyright © 2012 Massachusetts Medical Society. All rights reserved.

Mechanisms of Disease
n engl j med 366;12 nejm.org march 22, 2012 1137
stimulate mitochondrial biogenesis, or reduce lev-els of reactive oxygen species.66 Targeting reactive oxygen species may not be entirely beneficial, however, since they can act as signaling mole-cules,4,27,48,53 in which case their elimination might also have detrimental effects.
Mitochondria-Targeted Small Molecules
In contrast with membrane-permeable small molecules, which exert their effect throughout the cell, novel therapeutic strategies involve the specific targeting of mitochondria by small mol-ecules (referred to as “cargo”).65,68-74 Targeting can be achieved by coupling the cargo to a delo-calized lipophilic cation, leading to its accumula-tion in mitochondria. Several antioxidants have been successfully targeted using the cation ap-proach (e.g., plastoquinones and CoQ variants). Protein-based cargo can be coupled to a mitochon-drial targeting sequence recognized by the mito-chondrial protein import machinery. Another strat-egy involves the use of mitochondria-penetrating peptides, engineered cell-penetrating peptides that target mitochondria on the basis of their mem-brane potential and lipophilicity (e.g., the antioxi-dant Szeto–Schiller peptide, SS-31). There are also vesicle-based transporters that target mitochon-dria through macropinocytosis, endosomal es-cape, and membrane fusion (e.g., DQAsomes and
MITO-porters). Recently, in a mouse model of mitochondrial lipoamide dehydrogenase (LAD) deficiency, mitochondrial enzyme-replacement therapy mediated by TAT (a transactivating tran-scriptional activator of human immunodeficiency virus 1) was used to introduce a TAT-LAD fusion protein.75
Treatment of Mitochondrial Disorders
In 2006, a large-scale review of published clinical trials of various treatments for primary mitochon-drial disorders revealed no evidence supporting the use of any intervention.76 More recently, the results of several trials in which a variety of treatments for mitochondrial disease were studied, including dichloroacetate, vitamins, and a cocktail of spe-cific food components (nutraceuticals), were re-viewed.77 Although some of these trials showed a positive effect on one or more clinical or biochem-ical primary end points, none led to the filing of a New Drug Application with the Food and Drug Administration.
The drug idebenone (a CoQ10 variant) has been approved for the treatment of Friedreich’s atax-ia.41 A large multicenter, randomized, placebo-controlled trial of idebenone in patients with Leber’s hereditary optic neuropathy revealed that patients with discordant visual acuities were the most likely to benefit from this treatment.78 In
PatientPrimary
cells
Fluorescentreportermolecule
Live-cellmicros-
copy
Imageanalysis
Statisticalevaluation
AutomatedAutomated
ManualManual
Automated
Manual
Chemical
Protein
Cellulartarget
Leadcompound
Chemicalmodification
SARSADME–Tox
Leadoptimi-zation
Animalmodel
Outcomescores
Clinicaltrials
Administration – Dosage – Duration
Administration – Dosage – Duration
High content, high throughput, or bothCulturing Outcomescores
Figure 3. Proposed Approach to the Development of Intervention Strategies through Microscopical Analysis of Cells from Patients with Mitochondrial Diseases.
This schematic flow chart illustrates how the analysis of patient-derived primary cells by means of fluorescent reporter molecules and live-cell microscopy can be used to determine the cellular consequences of a mitochondrial dysfunction. This strategy can be used to extract the maximal amount of information from individual images (high content), test a large number of conditions (high throughput), or both. The information obtained can provide the basis for the discovery of lead compounds with pharmacologic or biologic activity. The chemi-cal structure of the lead compounds is then modified to improve their potency, selectivity, or pharmacokinetic characteristics (a process known as lead optimization). These optimized compounds can then be tested in cells (red line) and, if this step is successful, in a suit-able animal model and clinical trials. The blue boxes signify the administration of test agents at various concentrations and for various durations. SARS denotes structure–activity relationships, and ADME–Tox absorption, distribution, metabolism, excretion, and toxicity.
The New England Journal of Medicine Downloaded from nejm.org at KU LEUVEN BIOMEDICAL LIBRARY on September 11, 2012. For personal use only. No other uses without permission.
Copyright © 2012 Massachusetts Medical Society. All rights reserved.

T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 366;12 nejm.org march 22, 20121138
one study, investigators found that a ketogenic diet was effective in preventing epileptic seizures in children with electron-transport-chain defects.79 The findings in another study indicated that nu-tritional modulation affects mitochondrial func-tion, suggesting that it may be worthwhile to pursue nutritional treatment strategies.64
A successful treatment strategy has been de-veloped for patients with a secondary mitochon-drial disorder involving Ullrich’s congenital mus-cular dystrophy and Bethlem’s myopathy.80-82 These myopathies, caused by mutations in the extracellular-matrix protein collagen VI, are as-sociated with mitochondrial dysfunction and muscle-cell apoptosis. The pathogenic mechanism involves inappropriate opening of the mitochon-drial permeability transition pore. This action was prevented in patients treated with a permeability-transition-pore desensitizer, cyclosporin A. An-other study showed that cyclosporin A reduced the size of myocardial infarcts during cardiac re-perfusion.83 Other published intervention strate-gies are summarized in Table 2 in the Supple-mentary Appendix.
An overview of current clinical trials can be found online (www.clinicaltrials.gov/ct2/results?
term-mitochondrial+disease); these include stud-ies of CoQ10 for the treatment of muscle weakness and mitochondrial diseases, dietary supplements for MELAS (mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes), EPI-743 for mitochondrial diseases, human growth hor-mone for obesity, nutritional therapy for diabetes, pioglitazone for diabetes, idebenone for MELAS, and vitamin E for mitochondrial trifunctional protein deficiency.
FU T UR E PER SPEC TI V ES
A field that began more than 50 years ago, when a physician detected a mitochondrial disorder in a single patient with hypermetabolism,84 has now evolved into the discipline of mitochondrial med-icine. Lessons learned from studies of rare diseases have implications for a broad range of medical disciplines. Within the next few years, the appli-cation of new technologies such as whole-exome sequencing can be expected to result in a huge expansion of the number of known causative nu-clear gene defects in patients with mitochondrial disease (which is currently diagnosed by means of biochemical methods, mtDNA and nDNA se-
A Morphology and Mem-brane Potential
B NADH
E Cytosolic Free ATPF Mitochondrial Free Ca2+
C ROS D Redox State G Size-Based Mitogram
Figure 4. Typical Cell Readouts in Primary Fibroblasts from Human Skin.
Mitochondrial morphology and membrane potential can be visualized with the use of TMRM staining (Panel A),46 NADH levels with auto-fluorescence (Panel B),49 levels of reactive oxygen species with the chemical reporter CM-H2DCFDA (Panel C),48 mitochondrial redox state with the mitochondria-targeted protein-based sensor Mit-roGFP149 (400-nm image shown) (Panel D), the cytosolic free ATP con-centration with the protein-based sensor ATeam (Panel E),50 and levels of mitochondrial free Ca2+ in a cell stimulated with the hormone bradykinin and the calcium-sensitive cation Rhod-2 (Panel F).51 A mitogram shows all mitochondrial structures in the cell, sorted on the basis of size (Panel G).52
The New England Journal of Medicine Downloaded from nejm.org at KU LEUVEN BIOMEDICAL LIBRARY on September 11, 2012. For personal use only. No other uses without permission.
Copyright © 2012 Massachusetts Medical Society. All rights reserved.

Mechanisms of Disease
n engl j med 366;12 nejm.org march 22, 2012 1139
quencing, and candidate-gene studies). In the near future, the challenge will be to increase our under-standing of the consequences of mitochondrial dysfunction at all levels of complexity in order to drive the development of rational treatment strat-egies. Although gene therapy has been proved beneficial, its use is still limited by the sheer number of potential mitochondrial gene defects. Direct enzyme-replacement therapy may be feasi-ble in addressing single-protein enzymes, such as those in the tricarboxylic acid cycle, and perhaps mitochondrial multiprotein enzyme complexes. Another promising approach may be indirect enzyme-replacement therapy, which bypasses electron-transport defects with the use of alter-native dehydrogenases or oxidases (see Table 2 in the Supplementary Appendix). Although more ex-perimental data are required, exogenous stimula-tion of endogenous cellular adaptation programs
may also be useful to at least partially counter-balance the consequences of mitochondrial dys-function.
Given the metabolic individuality in humans,85 we do not expect monotherapeutic metabolic ma-nipulation strategies to be a magic bullet but pre-dict that the next step in treatment development will be the use of combinations of manipulation strategies applied in an individualized way. In the meantime, efforts must be made on a global scale to genetically categorize patient cohorts, monitor them in a standardized way by means of prognostic scoring systems,86 and develop new biomarkers to allow for proper monitoring of the effect of intervention strategies.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
We thank Dr. J. Fransen, Department of Cell Biology, Radboud University Nijmegen Medical Center, for providing electron-microscopical images.
Primarymitochondrial
disorder
Secondary mitochondrial disorderMutation
ExpressionFunction
∆ΨReactive oxygen
speciesDynamicsSubstrate accu-
mulation
ATP productionGlycolysisRedox statePTP openingMitophagyApoptosisIonic balanceSignalingMitochondrial
biogenesis
BrainEyesHeartLiverKidneyMuscleNerve
Mitochondrialprotein defect
Primary cellconsequences
Secondary cellconsequences
Disease
Geneticstrategies
Geneticstrategies
Treatment withsmall molecules
Metabolicmanipulation
Adaptation
Diet andexercise
Figure 5. Consequences of Mitochondrial Dysfunction and Potential Intervention Strategies.
Mitochondrial dysfunction can have primary and secondary pathologic consequences. Several cellular feedback mechanisms exist that can counteract the effects of the mitochondrial dysfunction (adaptation). For instance, the increased production of reactive oxygen species (ROS) caused by mitochondrial dysfunction can induce the expres-sion of antioxidant systems. Possible intervention strategies (indicated by dotted lines) may intervene at the level of the protein defect, the primary consequences, the secondary consequences, or the adaptive responses. The symbol Δψ denotes change in mitochondrial membrane potential, and PTP permeability transition pore.
The New England Journal of Medicine Downloaded from nejm.org at KU LEUVEN BIOMEDICAL LIBRARY on September 11, 2012. For personal use only. No other uses without permission.
Copyright © 2012 Massachusetts Medical Society. All rights reserved.

T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 366;12 nejm.org march 22, 20121140
References
1. Azzu V, Brand MD. The on-off switch-es of the mitochondrial uncoupling pro-teins. Trends Biochem Sci 2010;35:298-307.2. Cardoso AR, Queliconi BB, Kowal-towski AJ. Mitochondrial ion transport pathways: role in metabolic diseases. Bio-chim Biophys Acta 2010;1797:832-8.3. West AP, Shadel GS, Ghosh S. Mito-chondria in innate immune responses. Nat Rev Immunol 2011;11:389-402.4. Murphy MP, Holmgren A, Larsson NG, et al. Unraveling the biological roles of reactive oxygen species. Cell Metab 2011; 13:361-6.5. Spencer SL, Sorger PK. Measuring and modeling apoptosis in single cells. Cell 2011;144:926-39.6. Scarpulla RC. Transcriptional para-digms in mammalian mitochondrial bio-genesis and function. Physiol Rev 2008; 88:611-38.7. Shutt TE, Shadel GS. A compendium of human mitochondrial gene expression machinery with links to disease. Environ Mol Mutagen 2010;51:360-79.8. Coskun P, Wyrembak J, Schriner S, et al. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta 2011 August 16 (Epub ahead of print).9. McCoy MK, Cookson MR. Mitochon-drial quality control and dynamics in Par-kinson’s disease. Antioxid Redox Signal 2011 July 7 (Epub ahead of print).10. Cairns RA, Harris IS, Mak TW. Regu-lation of cancer cell metabolism. Nat Rev Cancer 2011;11:85-95.11. Hoppel CL, Tandler B, Fujioka H, Riva A. Dynamic organization of mitochon-dria in human heart and in myocardial disease. Int J Biochem Cell Biol 2009; 41:1949-56.12. Sleigh A, Raymond-Barker P, Thack-ray K, et al. Mitochondrial dysfunction in patients with primary congenital insulin resistance. J Clin Invest 2011;121:2457-61.13. Lönnqvist T, Paetau A, Valanne L, Pihko H. Recessive twinkle mutations cause se-vere epileptic encephalopathy. Brain 2009; 132:1553-62.14. Mochel F, Haller RG. Energy deficit in Huntington disease: why it matters. J Clin Invest 2011;121:493-9.15. Tseng YH, Cypess AM, Kahn CR. Cel-lular bioenergetics as a target for obesity therapy. Nat Rev Drug Discov 2010;9:465-82.16. Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive de-cline. Nature 2010;464:529-35.17. Finsterer J, Segall L. Drugs interfering with mitochondrial disorders. Drug Chem Toxicol 2010;33:138-51.18. Cohen BH. Pharmacologic effects on mitochondrial function. Dev Disabil Res Rev 2010;16:189-99.19. Dimauro S, Rustin P. A critical ap-proach to the therapy of mitochondrial respiratory chain and oxidative phosphor-
ylation diseases. Biochim Biophys Acta 2009;1792:1159-67.20. Luft R. The development of mitochon-drial medicine. Proc Natl Acad Sci U S A 1994;91:8731-8.21. Swerdlow RH. Mitochondrial medicine and the neurodegenerative mitochondri-opathies. Pharmaceuticals (Basel) 2009;2: 150-67.22. Benard G, Rossignol R. Ultrastruc-ture of the mitochondrion and its bearing on function and bioenergetics. Antioxid Redox Signal 2008;10:1313-42.23. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 2010;11:872-84.24. Dieteren CEJ, Gielen SCAM, Nijtmans LGJ, et al. Solute diffusion is hindered in the mitochondrial matrix. Proc Natl Acad Sci U S A 2011;108:8657-62.25. Bastin J, Lopes-Costa A, Djouadi F. Exposure to resveratrol triggers pharmaco-logical correction of fatty acid utilization in human fatty acid oxidation-deficient fibroblasts. Hum Mol Genet 2011;20:2048-57.26. Smeitink JAM, van den Heuvel L, DiMauro S. The genetics and pathology of oxidative phosphorylation. Nat Rev Genet 2001;2:342-52.27. Koopman WJH, Nijtmans LGJ, Diet-eren CEJ, et al. Mammalian mitochondri-al complex I: biogenesis, regulation, and reactive oxygen species generation. Anti-oxid Redox Signal 2010;12:1431-70.28. Pagliarini DJ, Calvo SE, Chang B, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008;134:112-23.29. Wang C, Liu X, Wei B. Mitochondrion: an emerging platform critical for host an-tiviral signaling. Expert Opin Ther Targets 2011;15:647-65.30. Dimauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci 2008;31:91-123.31. Tyynismaa H, Suomalainen A. Mouse models of mitochondrial DNA defects and their relevance for human disease. EMBO Rep 2009;10:137-43.32. Wallace DC, Fan W. The pathophysiol-ogy of mitochondrial disease as modeled in the mouse. Genes Dev 2009;23:1714-36.33. Gredilla R, Bohr VA, Stevnsner T. Mi-tochondrial DNA repair and association with aging — an update. Exp Gerontol 2010;45:478-88.34. Swerdlow RH. Does mitochondrial DNA play a role in Parkinson’s disease? A review of cybrid and other supportive evi-dence. Antioxid Redox Signal 2011 May 25 (Epub ahead of print).35. Yarham JW, Elson JL, Blakely EL, McFarland R, Taylor RW. Mitochondrial tRNA mutations and disease. Wiley Inter-discip Rev RNA 2010;1:304-24.36. Erol A. Deciphering the intricate reg-ulatory mechanisms for the cellular choice
between cell repair, apoptosis or senes-cence in response to damaging signals. Cell Signal 2011;23:1076-81.37. Kourtis N, Tavernarakis N. Cellular stress response pathways and ageing: intri-cate molecular relationships. EMBO J 2011; 30:2520-31.38. Twig G, Shirihai OS. The interplay be-tween mitochondrial dynamics and mi-tophagy. Antioxid Redox Signal 2011;14: 1939-51.39. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12:9-14.40. Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochon-drial threshold effects. Biochem J 2003; 370:751-62.41. Bénit P, El-Khoury R, Schiff M, Sain-sard-Chanet A, Rustin P. Genetic back-ground influences mitochondrial func-tion: modeling mitochondrial disease for therapeutic development. Trends Mol Med 2010;16:210-7.42. Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Bio-chem J 2011;435:297-312.43. Cortassa S, Aon MA, Winslow RL, O’Rourke B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys J 2004;87:2060-73.44. Juhaszova M, Zorov DB, Kim SH, et al. Glycogen synthase kinase-3 beta medi-ates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 2004;113: 1535-49.45. Picard M, Taivassalo T, Ritchie D, et al. Mitochondrial structure and function are impaired by standard isolation meth-ods. PLoS One 2011;6(3):e18317.46. Distelmaier F, Koopman WJH, Testa ER, et al. Life cell quantification of mito-chondrial membrane potential at the sin-gle organelle level. Cytometry A 2008;73: 129-38.47. Distelmaier F, Koopman WJH, van den Heuvel LP, et al. Mitochondrial com-plex I deficiency: from organelle dysfunc-tion to clinical disease. Brain 2009;132: 833-42.48. Forkink M, Smeitink JAM, Brock R, Willems PH, Koopman WJ. Detection and manipulation of mitochondrial reactive oxygen species in mammalian cells. Bio-chim Biophys Acta 2010;1797:1034-44.49. Verkaart S, Koopman WJH, Cheek J, et al. Mitochondrial and cytosolic thiol re-dox state are not detectably altered in iso-lated human NADH:ubiquinone oxidore-ductase deficiency. Biochim Biophys Acta 2007;1772:1041-51.50. Imamura H, Nhat KP, Togawa H, et al. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encod-ed indicators. Proc Natl Acad Sci U S A 2009;106:15651-6.
The New England Journal of Medicine Downloaded from nejm.org at KU LEUVEN BIOMEDICAL LIBRARY on September 11, 2012. For personal use only. No other uses without permission.
Copyright © 2012 Massachusetts Medical Society. All rights reserved.

Mechanisms of Disease
n engl j med 366;12 nejm.org march 22, 2012 1141
51. Koopman WJH, Distelmaier F, Esseling JJ, Smeitink JA, Willems PH. Computer-assisted live cell analysis of mitochondrial membrane potential, morphology and cal-cium handling. Methods 2008;46:304-11.52. Koopman WJH, Visch HJ, Verkaart S, van den Heuvel LW, Smeitink JA, Willems PH. Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am J Physiol Cell Physiol 2005;289:C881-C890.53. Koopman WJH, Verkaart S, Visch HJ, et al. Human NADH:ubiquinone oxidore-ductase deficiency: radical changes in mi-tochondrial morphology? Am J Physiol Cell Physiol 2007;293:C22-C29.54. Koopman WJH, Verkaart S, van Emst-de Vries SE, et al. Mitigation of NADH:ubiquinone oxidoreductase defi-ciency by chronic Trolox treatment. Bio-chim Biophys Acta 2008;1777:853-9.55. Koene S, Smeitink JAM. Metabolic manipulators: a well founded strategy to combat mitochondrial dysfunction. J In-herit Metab Dis 2010;34:315-25.56. Valsecchi F, Koopman WJH, Manjeri GR, Rodenburg RJ, Smeitink JA, Willems PH. Complex I disorders: causes, mecha-nisms, and development of treatment strategies at the cellular level. Dev Disabil Res Rev 2010;16:175-82.57. Roestenberg P, Manjeri GR, Valsecchi F, Smeitink JA, Willems PH, Koopman WJ. Pharmacological targeting of mitochon-drial complex I deficiency: the cellular level and beyond. Mitochondrion 2012;12:57-65.58. Blanchet L, Buydens LMC, Smeitink JAM, et al. Isolated mitochondrial com-plex I deficiency: explorative data analysis of patient cell parameters. Curr Pharm Des 2011;17:4023-33.59. Zanella F, Lorens JB, Link W. High content screening: seeing is believing. Trends Biotechnol 2010;28:237-45.60. Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD. Mice with mitochondrial complex I defi-ciency develop a fatal encephalomyopathy. Cell Metab 2008;7:312-20.61. Koene S, Willems PHGM, Roestenberg P, Koopman WJ, Smeitink JA. Mouse mod-els for nuclear DNA-encoded mitochon-drial complex I deficiency. J Inherit Metab Dis 2011;34:293-307.
62. Schmidt S, Linnartz B, Mendritzki S, et al. Genetic mouse models for Parkinson’s disease display severe pathology in glial cell mitochondria. Hum Mol Genet 2011; 20:1197-211.63. Wallace DC, Fan W, Procaccio V. Mi-tochondrial energetics and therapeutics. Annu Rev Pathol 2010;5:297-348.64. Schiff M, Bénit P, Coulibaly A, Loublier S, El-Khoury R, Rustin P. Mitochondrial response to controlled nutrition in health and disease. Nutr Rev 2011;69:65-79.65. Camara AKS, Lesnefsky EJ, Stowe DF. Potential therapeutic benefits of strategies directed to mitochondria. Antioxid Redox Signal 2010;13:279-347.66. Wenz T, Williams SL, Bacman SR, Moraes CT. Emerging therapeutic ap-proaches to mitochondrial diseases. Dev Disabil Res Rev 2010;16:219-29.67. Craven L, Elson J, Irving L, et al. Mito-chondrial DNA diseases: new options for prevention. Hum Mol Genet 2011;20:R168-R174.68. Yousif LF, Stewart KM, Horton KL, Kelley SO. Mitochondria-penetrating pep-tides: sequence effects and model cargo transport. Chembiochem 2009;10:2081-8.69. Chacko BK, Reily C, Srivastava A, et al. Prevention of diabetic nephropathy in Ins2(+/)− (AkitaJ) mice by the mitochon-dria-targeted therapy MitoQ. Biochem J 2010;432:9-19.70. Shokolenko IN, Alexeyev MF, LeDoux SP, Wilson GL. The approaches for ma-nipulating mitochondrial proteome. Envi-ron Mol Mutagen 2010;51:451-61.71. Zhang E, Zhang C, Su Y, Cheng T, Shi C. Newly developed strategies for multi-functional mitochondria-targeted agents in cancer therapy. Drug Discov Today 2011; 16:140-6.72. Skulachev MV, Antonenko YN, Anisi-mov VN, et al. Mitochondrial-targeted plastoquinone derivatives: effect on senes-cence and acute age-related pathologies. Curr Drug Targets 2011;12:800-26.73. Smith RA, Hartley RC, Murphy MP. Mitochondria-targeted small molecule ther-apeutics and probes. Antioxid Redox Sig-nal 2011;15:3021-38.74. Won YW, Lim KS, Kim YH. Intracel-lular organelle-targeted non-viral gene delivery systems. J Control Release 2011; 152:99-109.75. Rapoport M, Salman L, Sabag O, Patel
MS, Lorberboum-Galski H. Successful TAT-mediated enzyme replacement therapy in a mouse model of mitochondrial E3 defi-ciency. J Mol Med (Berl) 2011;89:161-70.76. Chinnery P, Majamaa K, Turnbull D, Thorburn D. Treatment for mitochondrial disorders. Cochrane Database Syst Rev 2006;1:CD004426.77. Stacpoole PW. Why are there no prov-en therapies for genetic mitochondrial diseases? Mitochondrion 2011;11:679-85.78. Klopstock T, Yu-Wai-Man P, Dimitriadis K, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary op-tic neuropathy. Brain 2011;134:2677-86.79. Kang HC, Lee YM, Kim HD, Lee JS, Slama A. Safe and effective use of the ke-togenic diet in children with epilepsy and mitochondrial respiratory chain complex defects. Epilepsia 2007;48:82-8.80. Irwin WA, Bergamin N, Sabatelli P, et al. Mitochondrial dysfunction and apop-tosis in myopathic mice with collagen VI deficiency. Nat Genet 2003;35:367-71.81. Angelin A, Tiepolo T, Sabatelli P, et al. Mitochondrial dysfunction in the patho-genesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc Natl Acad Sci U S A 2007;104:991-6.82. Merlini L, Angelin A, Tiepolo T, et al. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in pa-tients with collagen VI myopathies. Proc Natl Acad Sci U S A 2008;105:5225-9.83. Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 2008;359:473-81.84. Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B. A case of severe hypermetabo-lism of nonthyroid origin with a defect in the maintenance of mitochondrial respi-ratory control: a correlated clinical, bio-chemical, and morphological study. J Clin Invest 1962;41:1776-804.85. Suhre K, Shin SY, Petersen AK, et al. Human metabolic individuality in bio-medical and pharmaceutical research. Nature 2011;477:54-60.86. Morava E, van den Heuvel L, Hol F, et al. Mitochondrial disease criteria: diag-nostic applications in children. Neurolo-gy 2006;67:1823-6.Copyright © 2012 Massachusetts Medical Society.
The New England Journal of Medicine Downloaded from nejm.org at KU LEUVEN BIOMEDICAL LIBRARY on September 11, 2012. For personal use only. No other uses without permission.
Copyright © 2012 Massachusetts Medical Society. All rights reserved.

TRENDS IN MOLECULAR MEDICINE
Mechanisms of mitochondrial diseases
EMIL YLIKALLIO 1 & ANU SUOMALAINEN 1,2
1 Research Programs Unit, Molecular Neurology, Biomedicum-Helsinki, University of Helsinki, Finland, and 2 Department of Neurology, Helsinki University Central Hospital, Helsinki, Finland
Abstract Mitochondria are essential organelles with multiple functions, the most well known being the production of adenosine triphos-phate (ATP) through oxidative phosphorylation (OXPHOS). The mitochondrial diseases are defi ned by impairment of OXPHOS. They are a diverse group of diseases that can present in virtually any tissue in either adults or children. Here we review the main molecular mechanisms of mitochondrial diseases, as presently known. A number of disease-causing genetic defects, either in the nuclear genome or in the mitochondria ’ s own genome, mitochondrial DNA (mtDNA), have been identifi ed. The most classical genetic defect causing mitochondrial disease is a mutation in a gene encoding a structural OXPHOS subunit. However, mitochondrial diseases can also arise through impaired mtDNA maintenance, defects in mitochondrial transla-tion factors, and various more indirect mechanisms. The putative consequences of mitochondrial dysfunction on a cellular level are discussed.
Key words: Genetic diseases , inborn , mitochondria , mitochondrial diseases
Mitochondrial diseases are among the most common of the inherited disorders of metabolism (reviewed in (1)). For the diseases caused by mutations in mito-chondrial DNA (mtDNA), a population prevalence of at least 9.2 per 100,000 people was measured in the working-age population of north-east England. In addition, 16.2 per 100,000 people were identifi ed as asymptomatic mtDNA mutation carriers who are at risk of developing mitochondrial disease (2). These fi gures did not include mitochondrial diseases caused by nuclear gene defects, and thus the total prevalence of mitochondrial diseases is thought to be higher, at least in the order of 1 in 5,000 people.
The mitochondrial diseases may cause symptoms in any organ and present at any age (reviewed in (3,4)). A central function of mitochondria is produc-tion of the cellular energy currency adenosine triphos-phate (ATP) through oxidative phosphorylation (OXPHOS). OXPHOS is carried out by fi ve enzyme complexes in the inner mitochondrial membrane (IMM) (Figure 1). Apart from energy conversion,
mito chondria take part in a number of other pro-cesses including heme biosynthesis, steroid hormone biosynthesis, calcium homeostasis, and apoptosis (reviewed in (5)). Mitochondrial diseases are gener-ally understood as diseases where one or more of the OXPHOS complexes are dysfunctional. This review focuses on the various molecular mechanisms behind these diseases.
Mitochondria are surrounded by two membranes. The outer mitochondrial membrane (OMM) con-tains large amounts of porin (also known as voltage-dependent anion channel or VDAC), which allows passage of small hydrophilic molecules such as sug-ars, ions, and amino acids. The IMM is impermeable to most solutes. It has numerous folds called cristae, and it encloses the mitochondrial matrix. Mitochon-dria are dynamic organelles that can move, fuse, and divide according to the needs of the cell. In fact, in many cell types, it is often more appropriate to regard the mitochondria as a dynamic network rather than multiple discrete organelles (reviewed in (5)).
Correspondence: Emil Ylikallio, University of Helsinki, Room C519b, Haartmaninkatu 8, Helsinki 00290, Finland. Fax: � 358 9 4717 1964. E-mail: [email protected]
(Received 2 April 2011 ; accepted 9 June 2011 )
Annals of Medicine, 2012; 44: 41–59
ISSN 0785-3890 print/ISSN 1365-2060 online © 2012 Informa UK, Ltd.DOI: 10.3109/07853890.2011.598547
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

42 E. Ylikallio & A. Suomalainen
Genetics of mitochondrial disease
Mitochondrial diseases can be inherited by maternal, autosomal, or X-chromosomal transmission. This exceptional genetic variability is due to the unique genetic control of the organelle, which depends on two separate genomes. Mitochondria contain their own genome, called mtDNA. Human mtDNA is an intron-less multi-copy, circular 16,569 bp molecule that is exclusively inherited via the maternal line (6). Mito-chondrial DNA encodes 13 proteins, each of which is an essential subunit of one of the OXPHOS com-plexes (Figure 1). In addition, mtDNA encodes 22 tRNAs and 2 rRNAs required for the translation of these proteins (6). Primary or secondary defects of mtDNA are an important cause of mitochondrial dis-ease, often leading to one of several classical mtDNA syndromes (Table I). The mtDNA syndromes are sporadic or maternally inherited, when the primary defect is a mutation or deletion in mtDNA, and autosomally inherited when the mtDNA defect is secondary to a mutation in a nuclear gene that is required for mtDNA maintenance. However, most of the mitochondrial proteins, including the majority of OXPHOS com-plex subunits and assembly factors, are encoded by the nuclear DNA (nDNA) (7). Therefore, the major-ity of the mitochondrial diseases are caused by defects in nuclear genes and are inherited according to their chromosomal location. A mitochondrial disease may involve isolated dysfunction of only one OXPHOS complex or combined dysfunction of several com-plexes. The latter results for instance from defects in mtDNA maintenance, mitochondrial protein synthesis, or mitochondrial protein importation.
Mitochondrial DNA genetics
Inheritance of mtDNA mutations is different from that of nDNA mutations because mtDNA is a multi-copy genome with hundreds or thousands of copies per cell, and because mtDNA is only inherited from the mother. In most cases, all of the mtDNA molecules
Key messages
Mitochondrial diseases can manifest in any organ •at any age. They are caused by mutations in the genes of mitochondrial DNA or nuclear DNA, and their inheritance can be autosomal-dominant or recessive, X-linked, or maternal. Many of the genes that are mutated in mito- •chondrial diseases encode proteins that are structural components or assembly factors of the OXPHOS complexes. Others encode proteins involved in mtDNA maintenance or mitochondrial protein synthesis. In addi-tion, OXPHOS dysfunction can arise through indirect mechanisms. The metabolic consequences of mitochondrial •dysfunction are incompletely understood, both on the level of the cell and the level of the organism, and cannot be explained by mere ATP defi ciency.
Abbreviations
ad autosomal-dominant ar autosomal-recessive AS Alpers syndrome ATP adenosine triphosphate CI complex I CII complex II CIII complex III CIV complex IV CV complex V CoQ 10 coenzyme Q COX cytochrome c oxidase dATP deoxyadenosine triphosphate dCTP deoxycytidine triphosphate dGK deoxyguanosine kinase dGTP deoxyguanosine triphosphate dNTP deoxynucleoside triphosphate dTTP thymidine triphosphate GRACILE growth retardation, amino aciduria, cholestasis,
iron overload, lactic acidosis, and early death IMM inner mitochondrial membrane IMS inter-membrane space IOSCA Infantile-onset spinocerebellar ataxia KSS Kearns – Sayre syndrome LHON Leber ’ s hereditary optic neuropathy LS Leigh syndrome LSBL leukoencephalopathy with brain-stem and
spinal cord involvement and lactate elevation MDS mitochondrial DNA depletion syndrome MELAS mitochondrial encephalopathy with lactic acidosis
and stroke-like episodes MERRF myoclonic epilepsy with ragged-red fi bers MIRAS mitochondrial recessive ataxia syndrome MLASA mitochondrial myopathy, lactic acidosis, and
sideroblastic anemia MNGIE mitochondrial neurogastrointestinal
encephalopathy MRP mitochondrial ribosomal protein mtDNA mitochondrial DNA NARP neurogenic weakness, ataxia, retinitis pigmentosa nDNA nuclear DNA OMM outer mitochondrial membrane OXPHOS oxidative phosphorylation PEO progressive external ophthalmoplegia PS Pearson ’ s syndrome Q ubiquinone (oxidized) QH 2 ubiquinone (reduced) RC respiratory chain RNR ribonucleotide reductase ROS reactive oxygen species SANDO sensory ataxic neuropathy, dysarthria, and
ophthalmoparesis SCA-E spinocerebellar ataxia with epilepsy TK2 thymidine kinase 2 TP thymidine phosphorylase
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.
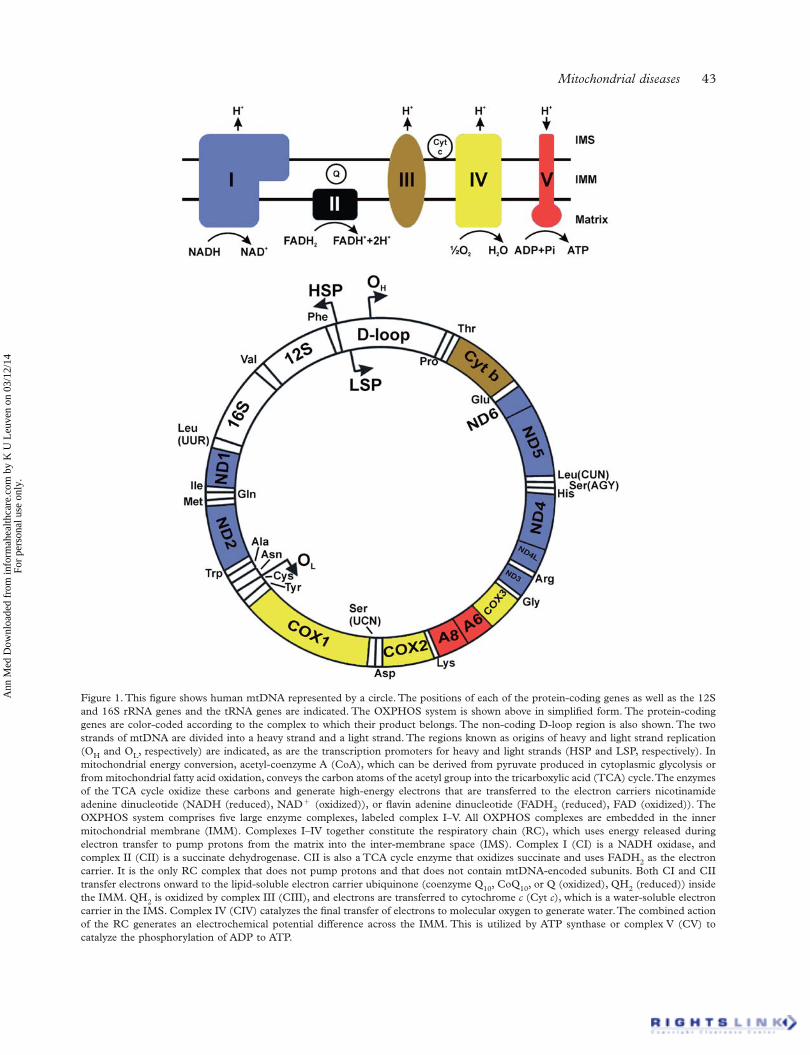
Mitochondrial diseases 43
Figure 1. This fi gure shows human mtDNA represented by a circle. The positions of each of the protein-coding genes as well as the 12S and 16S rRNA genes and the tRNA genes are indicated. The OXPHOS system is shown above in simplifi ed form. The protein-coding genes are color-coded according to the complex to which their product belongs. The non-coding D-loop region is also shown. The two strands of mtDNA are divided into a heavy strand and a light strand. The regions known as origins of heavy and light strand replication (OH and OL, respectively) are indicated, as are the transcription promoters for heavy and light strands (HSP and LSP, respectively). In mitochondrial energy conversion, acetyl-coenzyme A (CoA), which can be derived from pyruvate produced in cytoplasmic glycolysis or from mitochondrial fatty acid oxidation, conveys the carbon atoms of the acetyl group into the tricarboxylic acid (TCA) cycle. The enzymes of the TCA cycle oxidize these carbons and generate high-energy electrons that are transferred to the electron carriers nicotinamide adenine dinucleotide (NADH (reduced), NAD � (oxidized)), or fl avin adenine dinucleotide (FADH2 (reduced), FAD (oxidized)). The OXPHOS system comprises fi ve large enzyme complexes, labeled complex I–V. All OXPHOS complexes are embedded in the inner mitochondrial membrane (IMM). Complexes I–IV together constitute the respiratory chain (RC), which uses energy released during electron transfer to pump protons from the matrix into the inter-membrane space (IMS). Complex I (CI) is a NADH oxidase, and complex II (CII) is a succinate dehydrogenase. CII is also a TCA cycle enzyme that oxidizes succinate and uses FADH2 as the electron carrier. It is the only RC complex that does not pump protons and that does not contain mtDNA-encoded subunits. Both CI and CII transfer electrons onward to the lipid-soluble electron carrier ubiquinone (coenzyme Q10, CoQ10, or Q (oxidized), QH2 (reduced)) inside the IMM. QH2 is oxidized by complex III (CIII), and electrons are transferred to cytochrome c (Cyt c), which is a water-soluble electron carrier in the IMS. Complex IV (CIV) catalyzes the fi nal transfer of electrons to molecular oxygen to generate water. The combined action of the RC generates an electrochemical potential difference across the IMM. This is utilized by ATP synthase or complex V (CV) to catalyze the phosphorylation of ADP to ATP.
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

44 E. Ylikallio & A. Suomalainen
random segregation of mtDNA species as the germ cells divide (9). Alternatively, the bottle-neck may be caused by replication of only a subset of mtDNAs in primary oocytes that undergo folliculogenesis (10). Because of the bottle-neck, heteroplasmy levels can shift rapidly between generations. Therefore, members of the same family may exhibit striking clinical variabil-ity. Evidence from mouse models of mtDNA disease suggests that severely defective mtDNA molecules can be selected against during germ cell or oocyte development (11,12). This purifying selection may explain why moderately pathogenic mtDNA mutations that do not cause complete inhibition of OXPHOS and lead to late-onset disease are more common than highly deleterious mtDNA mutations (11).
Mitochondrial DNA mutations are also known to occur sporadically in somatic tissues. The mutations may undergo clonal expansion, eventually reaching the threshold level and causing mitochondrial dysfunc-tion at a later age. Accumulating mtDNA mutations, and subsequent progressive mitochondrial dysfunc-tion, are thought to contribute to the normal aging process (reviewed in (13)).
within the cells of an individual are identical. This situation is known as homoplasmy . However, in mtDNA diseases, the patients are often heteroplasmic , mean-ing that they have two different mtDNA populations. In general, disease severity correlates with the relative proportion of mutated to intact mtDNA. A heteroplas-mic person will only develop the disease if the mutant load exceeds a certain threshold. The threshold may be different for different mutations and different tissues.
The uniparental inheritance pattern of mtDNA means that it is in theory possible that mutations accumulate slowly over successive generations until, eventually, a threshold mutant level is reached that manifests as a disease (reviewed in (8)). Therefore, a mechanism is required to prevent the transmission of defective mtDNA to offspring. This protection has been attributed to a genetic bottle-neck of mtDNA in developing primordial germ cells (reviewed in (8)), meaning that only a small subset of the mtDNAs of the original oocyte populate the germ cells that will become the next generation. The bottle-neck may result from a strong reduction of mtDNA copy number in primordial germ cells, followed by amplifi cation and
Table I. This table lists the common mitochondrial syndromes and the main organs that are affected in each case. The syndromes are grouped according to their typical molecular etiology. MELAS, MERRF, NARP, and LHON are caused by mtDNA point mutations. KSS, PS, and PEO are caused by single mtDNA deletions that are either sporadic or maternally inherited; ad/ar PEO, MDS, MNGIE, MIRAS, and AS are caused by defective mtDNA due to mutations in nuclear genes responsible for mtDNA maintenance. LS is typically caused by mutations in mitochondrial or nuclear genes encoding structural proteins or assembly factors of the OXPHOS complexes. The inheritance of LS may thus be maternal, autosomal, or X-linked. Adapted from reference (4).
Abbreviated name Full name Typical molecular etiology Principal affected organs
MELAS Mitochondrial encephalopathy, lactic acidosis, stroke-like episodes
Mutation in tRNA or protein-coding gene of mtDNA
Skeletal muscle, brain, heart, endocrine pancreas, ear, gut
MERRF Myoclonus epilepsy with ragged-red fi bers
Mutation in tRNA gene of mtDNA
Skeletal muscle, brain, heart
NARP Neuropathy, ataxia, and retinitis pigmentosa
Mutation in protein-coding gene of mtDNA
Brain, eye
LHON Leber ’ s hereditary optic neuropathy
mutation in protein-coding gene of mtDNA
Eye, heart
KSS Kearns – Sayre syndrome Single mtDNA deletion Skeletal muscle, brain, heart, eyePS Pearson ’ s syndrome Single mtDNA deletion Bone-marrow, exocrine pancreas,
skeletal muscle, brainPEO Progressive external
ophthalmoplegiaSingle mtDNA deletion Skeletal muscle (especially
extra-ocular), heartad/ar PEO Autosomal-dominant/
recessive PEOMultiple mtDNA deletions Skeletal muscle (especially
extra-ocular), heartMDS mtDNA depletion syndrome mtDNA depletion Skeletal muscle, brain, liver,
kidney; typically tissue-specifi cMNGIE Mitochondrial
neurogastrointestinal encephalopathy
mtDNA depletion, multiple mtDNA deletions and point mutations
Gut, skeletal muscle, brain, peripheral nerve
MIRAS a Mitochondrial recessive ataxia syndrome
mtDNA deletions and subtle depletion
Brain, peripheral nerve
AS Alpers syndrome mtDNA depletion Brain, liverLS Leigh syndrome Mutation in protein coding
gene of mtDNA or nDNABrain, eye, skeletal muscle,
peripheral nerve
a MIRAS is also known as SCA-E (spinocerebellar ataxia with epilepsy) or SANDO (sensory ataxic neuropathy, dysarthria, and ophthalmoparesis). ad � autosomal-dominant; ar � autosomal-recessive.
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

Mitochondrial diseases 45
Defects of structural OXPHOS subunits and assembly factors
The OXPHOS complexes are made up of at least 89 structural protein subunits encoded by either mtDNA or nDNA. In addition, a number of nDNA-encoded assembly factors are needed for the intricate assem-bly processes of the complexes. Defects in a number of structural and assembly proteins are known to cause mitochondrial disease (reviewed in (14)). The diseases present with either isolated or combined com-plex defi ciencies, and their clinical manifestations are diverse (Table II).
Complex I defi ciency
Isolated complex I defi ciency is among the most com-mon forms of mitochondrial disease. Complex I, also known as NADH dehydrogenase or NADH:quinone reductase, is a ∼ 980 kDa structure composed of 45 subunits, of which 7 are encoded by mtDNA and 38 by nDNA (15). The complex is L-shaped, with a hydro-phobic arm embedded in the IMM and a perpen-dicular hydrophilic peripheral arm that protrudes into the matrix. The conserved structures of both subunits from bacteria and yeast have been charac-terized recently (16 – 19). CI has three functional
Table II. This table lists the disease-associated genes that encode either structural or assembly factors of the OXPHOS complexes. Note that structural proteins are encoded by either nDNA or mtDNA, whereas assembly factors, i.e. proteins needed to make the complex but that are not part of the fi nal complex, are only encoded by nDNA.
Genetic defect Genes Major clinical manifestations Reference
CI mtDNA-encoded structural gene
ND1 ND2 ND3 ND4 ND4L ND5 ND6
LHON, MELAS, LS (98)
nDNA-encoded structural gene
NDUFS1 NDUFS2 NDUFS3 NDUFS4 NDUFS6 NDUFS7 NDUFS8 NDUFV1 NDUFV2 NDUFA1 NDUFA2 NDUFA11
LS, encephalopathy, cardiomyopathy
(20,123,124)
nDNA-encoded assembly factor
NDUFAF1 NDUFAF2 C6orf66 C20orf7ACAD9 C8ORF38
LS, cardioencephalomyopathy, infantile lactic acidosis
CII nDNA-encoded structural gene
SDH-A SDH-B SDH-C SDH-D
LS, paraganglioma, pheochromocytoma
(123)
nDNA-encoded assembly factor
SDHAF1 SDHAF2 Infantile leukoencephalopathy, paraganglioma
(125,126)
CIII mtDNA-encoded structural gene
CYTB LHON, encephalopathy, cardiomyopathy, myopathy
(31)
nDNA-encoded structural gene
UQCRB UQCRQ Hypoglycemia, lactic acidosis or psychomotor retardation with extrapyramidal signs
(31,127)
nDNA-encoded assembly factor
BCS1L TTC19 Encephalopathy, liver failure, tubulopathy, GRACILE syndrome, Björnstad syndrome
(31,128)
CIV mtDNA-encoded structural gene
COX1 COX2 COX3 Encephalopathy, myopathy, sideroblastic anemia, myoglobinuria, MELAS
(37)
nDNA-encoded structural gene
COX6B1 COX4I2 Infantile encephalomyopathy, or exocrine pancreatic insuffi ciency and anemia
(37,129)
nDNA-encoded assembly factor
SURF1 SCO1 SCO2 COX10 COX15 LRPPRC C2orf64
LS, French-Canadian LS, encephalopathy, cardiomyopathy, myopathy, liver failure, tubulopathy
(37,130)
CV mtDNA-encoded structural gene
ATP6 ATP8 LS, NARP, cardiomyopathy (98,131)
nDNA-encoded structural gene
ATP5E 3-methylglutaconic aciduria, lactic acidosis, mild mental retardation, peripheral neuropathy
(48)
nDNA-encoded assembly factor
ATP12 TMEM70 Encephalopathy, lactic acidosis, cardioencephalomyopathy
(47,132)
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

46 E. Ylikallio & A. Suomalainen
modules: a dehydrogenase module that oxidizes NADH, a hydrogenase module that reduces ubiquinone, and a proton-transferring module that pumps protons. The latter module constitutes much of the membrane arm and also contains all of the mtDNA-encoded subunits. Electron transfer in the hydrophilic arm induces conformational changes that push an α -helix that is positioned in a parallel orientation within the membrane arm, and whose movement tilts other heli-ces in the membrane arm to allow the passage of pro-tons from the matrix side to the inter-membrane space (IMS) (17,18).
CI defi ciency is associated with various phenotypes both in children and adults. Many patients have Leigh syndrome (LS), which is characterized clinically by a severe symptom constellation including develop-mental delay and failure to thrive, and radiologically or histologically by typical necrotic lesions in the basal ganglia and brain-stem (reviewed in (4)). Others present with fatal infantile lactic acidosis, neonatal cardiomyopathy, leukoencephalopathy, myopathy, com-bined tubulopathy and hepatopathy, or less well defi ned phenotypes (reviewed in (20)). Another classi-cal CI-related disorder is Leber ’ s hereditary optic neuropathy (LHON), which is an adult-onset disease affecting almost exclusively the optic nerve. The fi rst disease-causing mtDNA mutations were described in LHON patients, in the gene encoding the ND4 sub-unit of CI (21). Children with CI-related disease tend to have normal prenatal development, which may refl ect a relatively low demand for mitochondrial func-tion during embryonic life, and up-regulation of OXPHOS following birth (reviewed in (20)).
Pathogenic mutations have been reported in all of the mtDNA-encoded CI subunits and a number of nDNA-encoded subunits (Table II). Some genetic defects interfere with the insertion of the polypeptide into the complex, while others allow complex assem-bly but impair the enzymatic activity of the fi nal com-plex (22). In the former, complex assembly intermediates accumulate depending on which stage of the assem-bly process the defective subunit is normally inserted in (23).
Two different models of human CI assembly have been suggested. In the fi rst model, the membrane arm and peripheral arms are fi rst assembled individually and then attached to each other. This is reminiscent of the well studied CI assembly process in the fungus Neurospora crassa (23). In the second model, the sub-units from both arms form subcomplexes that come together already before the process is fully fi nished. The second model is supported by the accumulated assembly intermediates that have been described in patients (14,22,24). Proteins that are needed for the assembly of CI, but that are not themselves com-ponents of the fi nal structure, are referred to as CI
assembly factors. Several such proteins exist, and disease-causing mutations have so far been found in the genes encoding six of them (Table II). Further, in many cases where CI fails to assemble, the respon-sible gene defects have not been found. Therefore, it is expected that more assembly factor mutations will be found in the future. Also, more information will be needed on the precise mechanisms of CI assembly.
The tissue-selectivity of clinical manifestations in CI defi ciency may depend on differences in the resid-ual level of CI activity in different tissues. The cases of CI defi ciency that are caused by mtDNA mutations exhibit greater variation in severity, which can be par-tially accounted for by the relative load of mutant mtDNA (20). Finally, CI defi ciency may be impor-tant in the pathogenesis of ataxia, having been found in the mitochondrial ataxia syndromes (described under disorders of mtDNA maintenance), and a CI-defi cient mouse model (25).
Complex II defi ciency
Succinate-coenzyme Q reductase, or CII, is different from the other OXPHOS complexes since all of its four subunits, SDH-A , SDH-B , SDH-C , and SDH-D , are encoded by nDNA. Furthermore, CII provides a physical link between OXPHOS and the citric acid cycle, as it catalyzes the transfer of electrons from the citric acid cycle intermediate succinate to the OXPHOS electron carrier ubiquinone. Homozygous or com-pound heterozygous mutations in the SDH-A gene cause LS (26). Mutations in the other three genes predispose to tumors, specifi cally paragangliomas of the carotid bodies and pheochromocytomas (27 – 29). Tumor predisposition is inherited in an autosomal-dominant manner. The mechanisms by which citric acid cycle or OXPHOS defects lead to tumor forma-tion are not known. One possibility is that changes in the cell ’ s metabolic state are sensed as hypoxia, leading to the activation of growth-promoting transcriptional pathways (reviewed in (30)).
Complex III defi ciency
Isolated CIII defects are among the least common respiratory chain (RC) disorders (reviewed in (31)). The full name of complex III is coenzyme Q:cytochrome c oxidoreductase. It consists of 11 subunits, of which only cytochrome b is encoded by mtDNA. Electron transfer from QH 2 to cytochrome c is achieved by a stepwise process known as the Q cycle (reviewed in (32)). The Q cycle involves a partially oxidized semi-quinone intermediate that is prone to auto-oxidation, i.e. transfer of one electron to oxygen to make super-oxide. Therefore, CIII is a major source of reactive oxygen species (ROS), which can damage many
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

Mitochondrial diseases 47
cellular macromolecules. Increased ROS may thus be an important feature in diseases caused by CIII defi ciency (reviewed in (31)).
Almost all of the known CIII subunit mutations are in the gene encoding cytochrome b . Most of the pathogenic amino acid changes are located outside of the transmembrane domains of the protein, and they affect the catalytic activity and/or assembly of CIII (reviewed in (31)). The clinical consequences of defec-tive cytochrome b are diverse: predominant symptoms may arise in optic nerve, heart, brain, or muscle. Some cytochrome b amino acid changes lead to CI defi -ciency, which supports the notion that CI and CIII function as a supercomplex and that supercomplex formation is dependent on intact cytochrome b (33). Furthermore, supercomplex stabilization appears to depend on cardiolipin, which is a phospholipid that is unique to the IMM. Deficiency of cardio-lipin causes Barth syndrome (cardioskeletal myopa-thy, neutropenia, and 3-methylglutaconic aciduria), probably through destabilization of respiratory supercomplexes (34).
Disease-causing mutations have been identifi ed in two nDNA genes encoding structural CIII subunits and in the BCS1L gene, which encodes an essential CIII assembly factor (Table II). At least 20 BCS1L mutations have been reported, and they underlie a remarkably wide range of phenotypes ranging from the GRACILE syndrome (growth retardation, amino aciduria, cholestasis, iron overload, lactic acidosis, and early death) (35) to the Bj ö rnstad syndrome that includes sensorineural hearing loss and hair changes known as pili torti (36). The function of the BCS1L gene product is to insert the Rieske iron-sulfur pro-tein during the late stages of complex assembly (31). Accordingly, BCS1L gene mutations have been found to cause the accumulation of an almost fully assem-bled CIII that is severely lacking in enzymatic activity. Mutations causing more severe phenotypes have been found to induce greater ROS production, which may account for variable disease severity and organ involvement (36).
Complex IV defi ciency
Complex IV, or cytochrome c oxidase (COX), has 13 subunits, of which the 3 large core subunits (COX1, COX2, and COX3) that are responsible for electron transfer are mtDNA-encoded. The 10 nDNA-encoded subunits are not directly involved in catalysis but are apparently needed for stability and regulation of the complex (reviewed in (14)).
Similar to the other RC complexes, COX-defi ciency is responsible for a diverse number of clinical pre-sentations. Mutations in the COX1-3 genes of mtDNA cause single-organ or multi-organ manifestations,
such as myopathy or MELAS (mitochondrial enceph-alopathy with lactic acidosis and stroke-like episodes), respectively. The mutations have been found to alter the assembly or stability of the complex. The regula-tion of COX activity is complex, involving tissue-specifi c isozymes, phosphorylation, and allosteric regulation. Therefore, tissue variability of regulatory factors is a potential explanation for the observed clinical diversity (reviewed in (37)).
The assembly of CIV is a stepwise process that is estimated to require more than 20 nuclear-encoded factors that are not part of the fi nal holoenzyme (reviewed in (14)). Defects in a number of these fac-tors are associated with variable diseases including LS or Leigh-like diseases, cardiomyopathy, and liver failure (Table II). Two disease-associated assembly factors, COX10 and COX15, are needed for the syn-thesis of the heme a component of CIV, and their defects caused loss of CIV holoenzyme (38,39). Another example of an essential CIV assembly factor is the 30 kDa Surf1 protein (40). The function of this protein is not completely understood, but it may be involved in the insertion of heme a into COX1 (41).
A new mechanism of CIV defi ciency was found in a patient with slowly progressive LS, who had a mutation in a gene encoding a protein of previously unknown function. This protein, named TACO1, was found to be needed for the activation of transla-tion of COX1 (42). TACO1 was suggested to be a member of a group of translational activators needed for the activation of mitochondrial mRNAs, which in contrast to cytoplasmic mRNAs lack 5 ’ untrans-lated regions. Furthermore, CIV defi ciency and mitochondrial encephalomyopathy was found in patients with mutations in the gene FASTKD2 , which encoded a mitochondrial protein potentially involved in apoptosis (43). However, the exact mechanism of CIV defi ciency had not been fully elucidated in the setting of FASTKD2 mutation.
Complex V defi ciency
Mitochondrial complex V (CV) couples proton cur-rent to the phosphorylation of ADP. Known also as ATP synthase, the complex has a membrane part F 0 and a peripheral part F 1 . Two F 0 subunits, ATP6 and ATP8, are mtDNA-encoded, whereas all of the F 1 subunits are nuclear-encoded. Mutations in the ATP6 gene of mtDNA are well known to cause disease. The T8993G and T8993C mutations in this gene cause disease depending on mutant load: a low load is asymptomatic, an intermediate load causes NARP (neurogenic weakness, ataxia, retinitis pigmentosa) (44), and a high load causes maternally inherited LS, potentially through impaired CV assembly (reviewed in (45)).
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

48 E. Ylikallio & A. Suomalainen
Like the other OXPHOS complexes, CV has asse-mbly factors encoded by nuclear genes. Atp12 and the related Atp11 have been characterized as essen-tial factors required for the formation of the F 1 com-ponent of CV in both yeast and humans (46). Mutations in the ATP12 gene were found in a child with early-onset encephalopathy and lactic acidosis, and the amount of assembled CV was strongly decreased (47). Furthermore, a number of other patients with apparent defects in CV assembly, but no mutations in the known disease genes, have been described. A mutation in a nDNA-encoded structural gene has also been identifi ed recently (Table II) (48). The nuclear gene defects leading to CV defi ciency generally lead to different phenotypes than NARP or LS (reviewed in (45)).
Disorders of mtDNA maintenance
Defects in the nuclear-encoded proteins responsible for mtDNA maintenance lead to decreased mtDNA copy number, mtDNA point mutations, multiple mtDNA deletions, or combinations thereof. The dis-orders of mtDNA maintenance form a diverse group that ranges from severe infant-onset multi-system disease, where mtDNA copy number is severely depleted in one or more tissues (mtDNA depletion syndrome, MDS), to mild adult-onset autosomal-dominant (ad) or autosomal-recessive (ar) progres-sive external ophthalmoplegia (PEO). The genetic causes are heterogeneous (reviewed in (49)). In PEO, mtDNA deletions gradually accumulate in patients ’ tissues, but mtDNA copy number usually remains normal. The disease affects particularly external eye muscles, but more generalized myopa-thy with exercise intolerance is often observed, as well as additional sym ptoms that include cardiomy-opathy, mild ataxia, Parkinsonism and depression (4,49,50). Furthermore, we recently found that a very high mtDNA copy number was associated with a number of detrimental effects on mtDNA mainte-nance and expression in transgenic mice (51). These results suggested that mitochondrial disease could also be caused by an abnormally high mtDNA copy number. However, this possibility remains unexplored in humans.
Most mtDNA maintenance diseases are caused by mutations in genes encoding proteins with well defi ned roles in mtDNA replication or mitochondrial deoxy-nucleoside triphosphate (dNTP) pool maintenance (Table III). Two proteins that are intimately involved in mtDNA replication have particularly strong asso-ciations with human disease: the mtDNA polymerase γ (Pol γ ), and the replicative helicase Twinkle (reviewed in (52)). Mitochondrial dNTP pools are regulated by a complex system of interlinked pathways localized
both in the cytosol and mitochondria (reviewed in (53)). Interestingly, defects in one mtDNA mainte-nance protein can cause several different types of syn-dromes. Thus, the clini cal picture is not always a good guide to fi nding the responsible gene.
The mtDNA replisome in disease
A large number of disease-causing mutations have been found in the POLG1 gene, which encodes the catalytic subunit of Pol γ (Pol γ A) (reviewed in (54)). The spectrum of Pol γ -related clinical manifestations is likewise large. The three main Pol γ -related phenotypes are: infant- or childhood-onset hepatocerebral MDS (known as Alpers syndrome), ataxia-neuropathy disor-ders such as MIRAS (mitochondrial recessive ataxia syndrome) that has a median age of onset of 28 years (range 5 – 41), and PEO with multiple mtDNA dele-tions, which can be either dominant or recessive and which rarely presents before 20 years of age (49,55,56). In addition, Pol γ defects have been associated with a number of other phenotypes including Parkinsonism and premature menopause (50).
The positions of POLG1 mutations show correla-tion with the clinical outcome. Pol γ A has a C-terminal polymerase domain and an N-terminal proof-reading exonuclease domain, separated by a spacer region involved in DNA binding and interaction with the accessory subunit Pol γ B. The latter is encoded by the gene POLG2 , and it confers increased DNA bind-ing, processivity, and polymerization rate to the holoen-zyme (reviewed in (52)). In general, polymerase domain mutations tend to cause dominant disease, as the DNA binding is not impaired and the mutant can compete with the wild-type protein. Recessive muta-tions cluster in the spacer and exonuclease regions, often causing reductions in DNA binding ability and accessory subunit interaction (54).
The Pol γ crystal structure was particularly per-tinent to the understanding of the spacer regions muta-tions, since this region does not share homology with the related DNA polymerase of the T7 phage in the manner the polymerase domain does (57). One of the most common Pol γ amino acid changes, A467T, affects a residue that is located in the thumb domain of Pol γ A. This domain was found to be involved in both template binding and the interaction with Pol γ B. Thus, the position of the mutated residue in the enzyme ’ s tertiary structure explained the results from earlier biochemical experiments that the mutation impairs both polymerase activity and processivity (57). Mutation in the POLG2 gene has been associ-ated with PEO in two patients so far (58,59). POLG2 defects may be a rare cause of PEO, but more patients need to be found before the position of POLG2 as a disease gene is fi rmly established.
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

Mitochondrial diseases 49
Twinkle is the mitochondrial replicative helicase. It was found based on its similarity to the T7 phage helicase primase gp4, and was found to be defective in several families with adPEO and multiple mtDNA deletions in skeletal muscle (60). Recessive Twinkle mutations cause either hepatocerebral MDS that is reminiscent of Alpers syndrome (61,62), or a syndrome of intermediate severity termed infantile-onset spi-nocerebellar ataxia (IOSCA) (63). IOSCA is part of the Finnish disease heritage. It manifests between ages 1 and 2 years and is slowly progressive with ataxia, neuropathy, ophthalmoplegia, and seizures. Brains from IOSCA patients showed mtDNA depletion but no deletions or point mutations (63). Mitochondrial DNA depletion in IOSCA may be more pronounced in particular neuronal populations, for instance cer-ebellar Purkinje cells, which could account for the restricted clinical findings and longer life-span compared to other forms of MDS (63).
The mechanisms by which Pol γ and Twinkle mutations cause mtDNA deletions and instability have been studied in various systems, including cul-tured cells, in-vitro biochemical assays, and a trans-genic mouse model (reviewed in (52)). The Twinkle holoenzyme is a homohexamer, and most of the dom-inant mutations impair subunit interaction (52). PEO mutations are dominant because the presence of just one defective subunit can impair the assembly and loading of the entire holoenzyme. Transgenic expres-sion of Twinkle with a linker region duplication of amino acids 353 – 364, which was originally described in a Finnish adPEO family (60), caused progressive mtDNA deletion accumulation in skeletal muscle and brain, similar to the patients (64). These so-called Deletor mice provided an ideal tool for studying the in-vivo pathogenic mechanisms of the dominant Twinkle defect. Expression of PEO-related mutants of both Twinkle and Pol γ in human cultured cells
Table III. The genes listed in this table encode proteins that are essential for mtDNA maintenance. Mutations in these genes cause mtDNA disease through different mechanisms, as indicated.
GeneGene product and its
function Molecular consequence Clinical consequence Ref
mtDNA replication: POLG Pol γ A, the catalytic
subunit of the mtDNA polymerase
Multiple mtDNA deletions ad/arPEO (56)mtDNA depletion Alpers-type MDS or
MIRAS(55)
POLG2 Pol γ B, the accessory subunit of the mtDNA polymerase
Multiple mtDNA deletions ad PEO (58)
C10Orf2 Twinkle, the replicative mtDNA helicase
Multiple mtDNA deletions adPEO (60)mtDNA depletion Hepatocerebral MDS or
IOSCA(61)
dNTP pool maintenance: DGUOK dGK, intramitochondrial
salvage of purine nucleosides
mtDNA depletion Hepatocerebral MDS (71)
TK2 TK2, intramitochondrial salvage of pyrimidine nucleosides
mtDNA depletion Myopathic MDS (72)
TYMP TP, cytoplasmic breakdown of thymidine nucleosides
mtDNA depletion, deletions, and point mutations
MNGIE (76)
RRM2B p53R2, constitutively expressed small subunit of ribonucleotide reductase
mtDNA depletion or mtDNA deletions
Multi-system MDS or adPEO
(73,74)
SLC25A4 ANT1, adenine nucleotide translocator in the IMM
Multiple mtDNA deletions adPEO (133)
SUCLG1SUCLA2
Subunits of succinyl CoA synthetase, citric acid cycle and nucleoside salvage
mtDNA depletion and succinyl-CoA accumulation
Encephalomyopathy (134,135)
Other or unknown: OPA1 Protein required for IMM
fusionMultiple mtDNA deletions adPEO (136)
MPV17 IMM protein of unknown function
mtDNA depletion Hepatic MDS (137)
ad � autosomal-dominant; ar � autosomal-recessive; PEO � progressive external ophthalmoplegia; MDS � mtDNA depletion syndrome; MIRAS � mitochondrial recessive ataxia syndrome; IOSCA � infantile-onset spinocerebellar ataxia; MNGIE � mitochondrial neurogastrointestinal encephalopathy; IMM � inner mitochondrial membrane.
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

50 E. Ylikallio & A. Suomalainen
induced signifi cant stalling of the mtDNA replisome (65,66). Replication stalling was also observed in the tissues of Deletor mice (65). Stalling could lead to double-strand DNA breaks, which in turn are known to induce deletion formation through double-strand break repair pathways (67). Importantly, replication stalling in Deletor mice was evident already at 6 weeks of age, whereas deletions were detectable starting at ∼ 12 months age (64,65). This suggested that the under-lying replication defect in PEO is present throughout life and that deletions accumulate from initially very small levels, until they eventually reach the threshold that causes disease. Deletion accumulation is enhanced by clonal expansion of deleted molecules within indi-vidual muscle cells (68). Clonal expansion may be enhanced by the deleted molecules having a replica-tive advantage due to their shorter length compared to wild-type mtDNA (69).
Mitochondrial dNTP pool maintenance and disease
The four dNTPs, deoxyadenosine-, deoxyguanosine-, deoxycytidine-, and thymidine triphosphate (dATP, dGTP, dCTP, and dTTP, respectively), are the build-ing blocks of DNA. A suffi ciently large dNTP pool is required for replication and repair of both nDNA and mtDNA. The relative proportions of the four dNTPs also need to be balanced in order to avoid mutagenesis (reviewed in (70)). Nuclear DNA rep-licates only in S-phase of the cell cycle, during which dNTP synthesis is heavily up-regulated. Mitochon-drial DNA replicates independently of the cell cycle in replicative cells and continues to be replicated in post-mitotic cells, for instance in neurons and skeletal muscle cells. Therefore, dNTP synthesis is required also in non-proliferative cells. There are two main pathways to supply dNTPs: in the de-novo pathway ribonucleotides, derived from the purine and pyrim-idine biosynthetic pathways, are converted into the corresponding deoxyribonucleotides by the cytoplas-mic enzyme ribonucleotide reductase (RNR); in the salvage pathway, deoxynucleosides undergo sequen-tial phosphorylations into deoxynucleoside mono- and diphosphates, which are fi nally phosphorylated into dNTPs (reviewed in (53)).
The de-novo pathway for deoxynucleotide synthe-sis is located in the cytoplasm, but mitochondria con-tain their own salvage pathway. Two mitochondrial kinases, thymidine kinase 2 (TK2) and deoxyguanos-ine kinase (dGK), are able to phosphorylate all four deoxynucleosides and thus form the basis of the mito-chondrial deoxynucleoside salvage pathway. Muta-tions in the genes encoding TK2 or dGK in humans were found to cause recessively inherited, muscle-specifi c or hepatocerebral MDS, respectively (71,72). This proved the importance of deoxynucleoside
salvage for mtDNA maintenance. For some time it was thought that post-mitotic cells almost completely lack RNR, and that mtDNA replication in non-cycling cells relies solely on dNTPs derived from the salvage pathway. However, this view changed when it was found that RNR is in fact also essential for mtDNA maintenance in post-mitotic tissues. Inacti-vating mutations in the RRM2B gene, encoding the p53-inducible small subunit of RNR (p53R2), caused multi-system, recessively inherited MDS (73). Fur-thermore, we recently found that a heterozygous muta-tion of RRM2B , that leads to truncation of the p53R2 protein, causes adPEO with multiple mtDNA deletions (74). Therefore, both salvage and de-novo production of dNTPs is necessary for mtDNA replication.
The patients with homozygous or compound hete-ro zygous defects in the anabolic enzymes of deoxy-nucleotide synthesis, TK2, dGK, or p53R2, are usually normal at birth, but the MDS manifests abruptly in specifi c tissues during the fi rst months or years of life. Reasons for the time of onset and tissue selectivity of these diseases are unknown. A recent study on a mouse model of TK2 defi ciency suggested that disease onset coincides with down-regulation of the cyto-plasmic salvage enzyme TK1 (75). This TK1 down-regulation essentially unmasked a latent TK2 defi ciency. The same study also suggested that increased tran-scription of mtDNA could partially compensate for mtDNA depletion in skeletal muscle (75). Therefore, it is conceivable that tissue-specifi cities of MDS may be determined by different capacities to compensate for the various genetic defects.
The importance of proper dNTP pool balance is illustrated by the disease MNGIE (mitochondrial neurogastrointestinal encephalomyopathy). This disease is caused by deficiency of the catabolic enzyme thymidine phosphorylase (TP) that degrades thymidine (76). TP defi ciency leads to thymidine excess and probably to elevations of the mitochon-drial dTTP pool in relation to the other dNTPs. This dNTP imbalance causes mtDNA deletions and point mutations in addition to mtDNA deple-tion (reviewed in (77)). Furthermore, we recently studied a mouse model over-expressing more than one RNR subunit. The RNR over-expression caused dNTP pool imbalance and led to mtDNA depletion in skeletal muscle, but it did not cause mtDNA dele-tions or point mutations (78). Therefore, it is clear that mtDNA replication is sensitive to the dNTP pool balance. However, the exact mechanisms by which dNTP pool imbalances cause mtDNA insta-bility remain incompletely understood. It is also unknown why certain changes to the enzymatic pathways regulating dNTP pools lead to mtDNA depletion only, while other changes cause point mutations or deletions.
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

Mitochondrial diseases 51
Disorders of mitochondrial protein synthesis
Mitochondrial translation is more reminiscent of prokaryotic translation than eukaryotic cytoplasmic translation. However, several signifi cant differences exist, and the mitochondrial translation machinery is incompletely characterized to date (reviewed in (79)). Mitochondria use a genetic code that is slightly different from the universal genetic code of other sys-tems. Furthermore, the mitochondrial mRNAs also lack 5 ’ -end untranslated regions and ‘ cap ’ structures, and the same tRNA-Met is used in both translation initiation and elongation. In addition to the 22 tRNAs and 2 rRNAs that are encoded by mtDNA, mito-chondrial translation requires a wealth of nDNA-encoded proteins. Translation initiation requires the initiation factors mtIF2 and mtIF3, whereas elonga-tion of the polypeptide is dependent on the elongation factors mtEFTu, mtEFTs, mtEFG1, and mtEFG2 (reviewed in (80)). The release factors mtRF1 and mtRF1a are needed to terminate translation at stop codons. The mitochondrial ribosomes have particularly high protein content, with up to 81 mitochondrial ribosomal proteins (MRPs). Finally, aminoacylation of mitochondrial tRNAs is performed by mitochon-drial aminoacyl-tRNA synthetases, of which 19 have been described (reviewed in (81)).
Defects in nuclear-encoded mitochondrial translation factors
Disease-causing mutations have been identifi ed in several of the nDNA-encoded mitochondrial transla-tion factors (Table IV). In general, the associated phenotypes are severe, affecting the synthesis of all fi ve OXPHOS complexes, and causing early death. However, the organ manifestations are diverse, even among patients who carry the same mutation. Under-standing the molecular basis of the tissue specifi city is challenging, given that all factors are involved in the same molecular machinery.
Mutations in the PUS1 gene were found in patients with the disease MLASA (mitochondrial myopathy, lactic acidosis, and sideroblastic anemia), which affects bone-marrow, skeletal muscle, and the central nervous system but shows clinical variability (82). PUS1 encodes pseudouridine synthase 1 (Pus1), which is not directly involved in translation but is required for the post-translational modifi cation of tRNAs both in the cytoplasm and in mitochondria (79). It was suggested that modifi cation of the expres-sion level of ribosomal proteins could compensate for the phenotype and account for the variability in phenotypes between tissues and between individuals (83). In addition, Pus1 could have several non-tRNA substrates (83).
Defects in another enzyme involved in tRNA mod-ifi cation, tRNA 5-methylaminomethyl-thiouridylate (Trmu), are also associated with mitochondrial dis-ease. Trmu modifi es wobble base positions of three mitochondrial tRNAs: tRNA-Lys, tRNA-Gln, and tRNA-Glu. Several mutations in the TRMU gene caused acute liver failure and were found to render the tRNAs hypomodifi ed, leading to defective mito-chondrial protein synthesis (84). One TRMU muta-tion was not associated with liver failure but caused aggravation of the deafness phenotype associated with mitochondrial 12S rRNA mutations (85).
Mutations in the mitoribosomal proteins MRPS16 and MRPS22 impair assembly and stability of the small ribosomal subunit (86,87). Clinically, the MRPS16 defect led to agenesis of the corpus callosum, dysmor-phism, and fatal neonatal lactic acidosis (86). The MRPS22 defect led to antenatal skin edema, hypotonia, cardiomyopathy, and tubulopathy (87).
Disease-causing mutations have been identifi ed in genes encoding translational elongation factors but not in those encoding initiation factors. The syn-dromes associated with elongation factor mutations exhibit remarkable tissue selectivity. All of these muta-tions are associated with defective translation of most
Table IV. Nuclear-encoded mitochondrial translation factors involved in disease.
Function Gene/protein Affected tissue Reference
tRNA modifi cation PUS1 Skeletal muscle, bone-marrow (82)TRMU Liver, ear (84,85)
Elongation factor GFM1/mtEFG1 Brain, liver (88)TUFM/mtEFTu Brain (89)TSFM/mtEFTs Heart or skeletal muscle and brain (91)
Ribosomal protein MRPS16 Brain (86)MRPS22 Heart, kidney (87)
Aminoacyl-tRNA synthetase RARS2 Brain (92)DARS2 Brain (93)YARS2 Skeletal muscle, bone-marrow (94)SASR2 Kidney, pulmonary circulation (95)HARS2 Ovary, ear (97)AARS2 Heart (96)
Peptide release factor C12orf65 Skeletal muscle, brain (138)
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

52 E. Ylikallio & A. Suomalainen
mitochondrial transcripts, but bicistronic transcripts (e.g. the one encoding the ATP6 and ATP8 genes) can be translated at normal or elevated levels. The mtEFG1 defi ciency caused a severe hepato(encephalo)pathy, but the heart was spared despite a high nor-mal expression level of the affected protein in this tissue (88 – 90). Mutation of mtEFTu in turn was associated with a rapidly progressive encephalopathy (89). However, a single homozygous mutation in mtEFTs caused hypertrophic cardiomyopathy in one patient and encephalomyopathy in another (91). The relatively benign phenotype in some tissues is apparently related to more favorable relative expres-sion levels of the translation factors, as well as adap-tive changes in these ratios. For instance, the heart of patients with mtEFG1 mutation had an up-regula-tion of the mtEFTu:mtEFTs ratio, whereas the reverse was true for the liver (90). In addition, there may be other genetic modifi ers that are responsible for tailoring mitochondrial translation to tissue-specifi c needs. The mtEFTs mutant could be com-pensated for by over-expression of mtEFTu, probably because the mutation interferes with the formation of the complex between these two proteins and renders both proteins unstable. Bringing in additional mtEFTu thus helps stabilize the complex (91). It is possible that this type of compensatory mechanism accounted for the difference between the two mtEFTs patients.
Mutations in two of the aminoacyl-tRNA syn-thetases affected the brain. RARS2 (encoding mito-chondrial arginyl-tRNA synthase) mutations caused pontocerebellar hypoplasia (92), whereas the DARS2 (encoding mitochondrial aspartyl-tRNA synthetase) mutations led to LSBL (leukoencephalopathy with brain-stem and spinal cord involvement and lactate elevation) (93). The mutations were in introns and affected splice sites. Higher expression of splicing fac-tors in extracerebral tissues was suggested as a rea-son for brain specifi city of the phenotypes in RARS2 and DARS2 mutations (92). The RARS2 mutation caused a decrease in the total abundance of mito-chondrial tRNA-Arg, probably because the unloaded tRNA is unstable (92). However, the DARS2 muta-tion did not lead to a translation defi cit or OXPHOS dysfunction in patient fi broblasts, even though the aminoacylation activity was strongly reduced (93). The mechanism by which mutation in this gene causes disease is therefore unclear. It is possible that only a subset of neurons is affected, meaning that studies on whole-brain lysates or cultured fi broblasts are not informative (93).
A mutation in YARS2 , which encodes the mito-chondrial tyrosyl-tRNA, synthetase was found to cause MLASA with no apparent effects on brain structure or function (94). Since the phenotype was highly
similar to that caused by PUS1 mutation, a common mechanism was invoked. The dysfunctional Pus1 may cause disease through hypomodifi cation of tRNA-Tyr, leading to decreased amino acid charging rem-iniscent of the situation in YARS2 defi ciency (94). However, it remains unclear why the brain is not affected. Three further mitochondrial aminoacyl-tRNA synthetases, encoded by SARS2 , HARS2 , and AARS2 , have been associated with disease very recently (95 – 97). The involved organs appear to be strikingly diverse and specifi c for each gene (Table IV).
Mitochondrial tRNA and rRNA mutations and single mtDNA deletions
Most human mtDNA point mutations occur in tRNA genes (www.mitomap.org). The clinical manifesta-tions of tRNA mutations are manifold. Two of the best studied mitochondrial tRNA mutations are the A3243G mutation in tRNA-Leu(UUR) and the A8344G mutation in tRNA-Lys. The former has a population prevalence of as high as 5.7 per 100,000 and causes a wide range of phenotypes of which MELAS is prominent, and the latter is associated with MERRF (myoclonic epilepsy with ragged-red fi bers) (reviewed in (98)). Mutations in other tRNA genes can lead to distinct phenotypes, e.g. deafness due to mutations in tRNA-Ser(UCN) or cardiomyo-pathy due to mutations in tRNA-Ile (reviewed in (99,100)). A number of explanations have been put forth to explain the clinical heterogeneity of tRNA mutations, including variations in heteroplasmy or mtDNA segregation, different OXPHOS complexes being differently affected depending on their amino acid composition, tissue-specifi c toxicities of prema-turely terminated translation products, or other tissue-specifi c modifi ers that affect mitochondrial protein synthesis or function (100).
Mutations in the mitochondrial rRNA genes are associated with non-syndromic sensorineural deaf-ness or aminoglycoside-induced deafness. The most common mutation is A1555G in the 12S rRNA gene (101). The mutation alters the structure of the rRNA such that it more closely resembles the bacterial 16S rRNA. This facilitates interaction with aminoglyco-sides, leading to translation defect. Not all people who carry this mutation develop deafness. In addition to aminoglycoside exposure, the penetrance depends on mtDNA haplotype and nuclear modifi er genes (79). This is a good example of how a mitochondrial disease is modifi ed by genetic and environmental factors.
The mtDNA deletion syndromes are caused by large-scale (usually several thousand base pairs) dele-tions of mtDNA, which are usually sporadic (102). In these disorders, the patient tissues typically harbor a single partially deleted species together with the
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

Mitochondrial diseases 53
wild-type mtDNA. These diseases are denoted single mtDNA deletion diseases to distinguish them from the multiple deletion diseases, in which deletions of differ-ent sizes are present and which are caused by defects in mtDNA maintenance. Deletions usually involve sev-eral genes including at least one tRNA, meaning that mitochondrial protein synthesis as a whole will be affected. Clinically, the mtDNA deletion syndromes form a spectrum that ranges from relatively benign adult-onset disorders such as PEO to severe infantile-onset disorders such as Pearson ’ s syndrome (Table I).
Disorders of mitochondrial dynamics
Mitochondrial fusion and fi ssion, collectively mito-chondrial dynamics, are essential for mitochondrial distribution and function. Mitochondria form an inter-connected network that is constantly reorganized (reviewed in (5)). Defective fusion or fi ssion underlies a number of neurological diseases in humans. Mito-chondrial fusion requires three proteins: Mitofusins 1 and 2 (Mfn1 and Mfn2) are needed for outer mem-brane fusion, and Opa1 has been suggested to be involved in inner membrane fusion (reviewed in (103)). Mutations in the MFN2 gene cause the autosomal-dominant Charcot – Marie – Tooth disease type 2A, which is a peripheral neuropathy caused by degeneration of axons in long sensory and motor nerves of the dis-tal extremities (104). The selective manifestation of Mfn2 mutations in neurons has been attributed to a low relative level of Mfn1 expression. Mfn1 is able to complement Mfn2 functionally (105), and tissues with high endogenous Mfn1 expression may thus be resis-tant to Mfn2 defects. In addition, peripheral neurons may be particularly susceptible to aberrant mitochon-drial dynamics due to the extreme length of their axons (reviewed in (103)).
OPA1 mutations cause autosomal-dominant optic atrophy (106,107). In most cases, this disease specifi -cally involves degeneration of retinal ganglion cells. However, more diverse OPA1 -related phenotypes have been described recently, and they include disorders of mtDNA maintenance (Table III) (103).
Disorders where OXPHOS is indirectly affected
In most of the disorders described so far, mitochondrial dysfunction arises as a direct consequence of a defect in genes involved in OXPHOS or in the synthesis of OXPHOS subunits. However, mitochondrial dysfunction can arise also through indirect mechanisms.
Ubiquinone or coenzyme Q (CoQ 10 ) shuttles electrons from CI and CII to CIII. It consists of a benzoquinone ring and a tail of typically 10 isoprenyl subunits that anchors it to membranes (reviewed in (108)). Reduced ubiquinone (QH 2 ) also functions as an antioxidant. Disorders of CoQ 10 biosynthesis form a group of rare autosomal-recessive disorders. Clinical manifestations are reminiscent of other mitochondrial disorders and are divided into fi ve groups: encephalomyopathy, severe infantile multi-systemic disease, cerebellar ataxia, isolated myopa-thy, and nephritic syndrome (Table V) (108). Correct diagnosis of these patients is essential, as ubiquinone Q10 supplementation may improve the condition of some patients dramatically.
The import of nuclear-encoded proteins into mitochondria requires specialized multi-subunit machineries in the IMM, IMS, and OMM. Defects in the protein translocation machinery have been found to cause disease by impairing the importation of proteins needed for OXPHOS. The consequence of mutation in the deafness/dystonia protein 1/translocase
Table V. Disease-associated genes invloved in the biosynthesis of coenzyme Q (CoQ10). Derived from reference (108).
Gene Protein function/disease mechanism Phenotype
ADCK3/CABC1 Encodes a putative mitochondrial kinase involved in the regulation of CoQ10 biosynthesis
Encephalomyopathy, juvenile-onset cerebellar ataxia
APTX Encodes aprataxin, which is involved in DNA repair. Secondary CoQ10 defi ciency
Ataxia-oculomotor apraxia 1
COQ2 Encodes para-hydroxybenzoate-polyprenyl transferase, direct role in CoQ10 synthesis
Infantile-onset multi-system syndrome or renal disease
PDSS2 Polyprenyl diphosphate synthase, direct role in CoQ10 biosynthesis
Nephrotic syndrome and LS
PDSS1 Polyprenyl diphosphate synthase, direct role in CoQ10 biosynthesis
Infantile-onset multi-system syndrome
COQ9 Direct role in CoQ10 biosynthesis Infantile-onset multi-system syndromeETFDH Encodes electron-transferring-fl avoprotein
dehydrogenase. Also associated with glutaric aciduria type II in which CoQ10 levels in muscle are normal
Myopathy
BRAF Involved in mitogen-activated protein kinase pathway, CoQ10 defi ciency by indirect mechanism
Cardiofaciocutaneous syndrome
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

54 E. Ylikallio & A. Suomalainen
of mitochondrial inner membrane 8a (DDP1/TIMM8a) is Mohr – Tranebjaerg syndrome/deafness-dystonia syndrome, which is an X-linked disorder characterized by progressive sensorineural deafness, cortical blindness, dystonia, dysphagia, and paranoia (109,110). Certain cysteine-rich proteins are imported into the IMS with the aid of a disulfi de relay system, which catalyzes the oxidative folding of these proteins. The disulfi de relay system donates electrons to oxygen via cytochrome c and CIV, thus connecting it to the RC. Accordingly, mutation in the gene GFER , which encodes one of two key com-ponents of the disulfi de relay system, was found to cause dysfunction of multiple RC complexes and a progressive mitochondrial myopathy (111).
Another cause of OXPHOS defect was found in the dysfunction of the protein paraplegin in patients with hereditary spastic paraplegia (112). Paraplegin is a subunit of a ubiquitous mitochondrial protease that has a quality control role, suggesting that the patients have problems with mitochondrial protein turn-over (113). Recently, mutation in the X-chromosomal gene AIFM1 , encoding the apoptosis-inducing fac-tor (AIF), was found to be the cause of mitochon-drial encephalomyopathy in two male infants (114). AIF is an IMS-targeted protein whose function is incompletely understood. The protein can be released into the cytoplasm and nucleus where it induces frag-mentation of nuclear DNA, leading to a form of pro-grammed cell death that is independent of caspase. Patient muscle displayed increased sensitivity to this form of programmed cell death, in addition to mtDNA depletion and OXPHOS failure. The latter may have been due to destabilization of the IMM (114).
Finally, severe lack of CIV activity was found in patients with ethylmalonic encephalopathy and mutation in the gene ETHE1 . The gene encodes a mitochondrial dioxygenase, and the mutation caused accumulation of sulfi de, which is a potent inhibitor of CIV (115). Together, these studies showed that mitochondrial dysfunction can result from a multi-tude of mechanisms other than direct impairment of OXPHOS.
Consequences of OXPHOS defects
Our understanding of the genetic etiology of mito-chondrial diseases is expanding rapidly. However, relatively little is known of the molecular conse-quences of impaired OXPHOS that account for the development of symptoms in specifi c tissues. Given the multitude of functions that mitochondria take part in and the universal need of ATP by all cells, the pathogenic mechanisms are likely to be complex. Potential consequences of OXPHOS impairment include metabolite build-up, defi ciency of ATP,
aberrant calcium handling, and ROS production (reviewed in (116)).
Mitochondria produce ROS as a side product of normal respiration. The production of ROS can be considerably increased in malfunctioning mitochon-dria, and the effect of ROS is believed to be toxic, since they can irreversibly modify many cellular macromol-ecules. There are various ROS detoxifying enzymes to mitigate these effects. Moreover, a limited produc-tion of ROS also may play an important signaling role, by acting as a retrograde signal that infl uences gene expression in the nucleus in response to alterations of mitochondrial membrane potential or redox state (reviewed in (117)).
One consequence of OXPHOS dysfunction is that NADH produced by the citric acid cycle cannot be fed forward, leading to elevation of the NADH:NAD � ratio. The level of pyruvate, produced through glyco-lysis, is increased due to an inhibition of the citric acid cycle. Thus, the equilibrium of the NADH-dependent lactate dehydrogenase is shifted towards the produc-tion of lactate from pyruvate. Lactate is released to the blood-stream, which causes a systemic drop in pH. Indeed, lactic acidosis is among the most important symptoms of mitochondrial disease (reviewed in (3)).
ATP defi ciency is one of the most obvious con-sequences of OXPHOS defi ciency. Decreased rates of ATP production have been found in mitochondria from CI-defi cient patient muscle, but ATP defi ciency may not be the most crucial metabolic derangement responsible for cellular dysfunction in these disor-ders (116). ATP defi ciency and aberrant calcium sig-naling have been proposed as interlinked pathways leading to cell dysfunction in mitochondrial disease: fi rst, because mitochondrial ATP production is needed to fuel calcium pumps in the endoplasmic reticulum, and, second, because mitochondria themselves can buffer calcium and have their function regulated by the infl ux of calcium (reviewed in (118)). This model could account for the frequent involvement of mus-cle and nerve tissues in mitochondrial diseases, since these cells rely heavily on ATP and on fl uctuating levels of activity mediated by Ca 2 � pulses. However, as different mitochondrial disorders are highly tissue-specifi c, ATP defi ciency cannot be the sole underlying factor in mitochondrial diseases.
Finally, OXPHOS dysfunction in individual tis-sues is likely to have metabolic consequences on the systemic level. For instance, we recently reported that skeletal muscle in mice reacts to OXPHOS dys-function by developing a starvation-like state that includes up-regulating the expression of the fasting-related hormone FGF21, which is likely to induce the mobilization of energy stores in liver and fat for use in the starved skeletal muscle (119). The starva-tion-like state could also entail increased turn-over
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

Mitochondrial diseases 55
of mitochondria by the process known as mitochon-drial autophagy, or mitophagy. Selective removal of dysfunctional mitochondria by mitophagy is likely to be a central mechanism by which cells maintain a healthy mitochondrial population (120,121). Intrigu-ingly, defective mitophagy has been implicated in the development of Parkinson ’ s disease (120). Hence, mod-ifi cation of the physiological responses to OXPHOS dysfunction could provide novel therapeutic tools to combat mitochondrial and neurodegenerative dis-eases. For instance, we found that high-fat feeding of mice considerably improved the ultrastructural changes of mitochondria in skeletal muscle of late-onset mito-chondrial myopathy mice (122). These results sug-gested the following: 1) Mitochondrial biogenesis is boosted upon lipid feeding. The mitochondria are the major lipid-oxidizing organelle, whereas glucose can be used for ATP production both by mitochondria and by cytoplasmic glycolysis. Use of lipids requires active mitochondria. 2) If glucose is available and glycolytic ATP production is possible, even mild dys-function in mitochondria may cause their active down-regulation, leading to cells relying on glycolysis. 3) By lipid-feeding mitochondria are forced to be active, and poorly working mitochondria may actually be more beneficial for cells than down-regulated mitochondria, as they produce more ATP than mere glycolysis. Therefore, correct composition of nutri-tion may be crucial for mitochondrial patients.
Conclusion
The fi rst description of genetic mutations in mito-chondrial disease came in 1988 (21,102), and since then the fi eld has expanded exponentially. The molec-ular pathways required for the synthesis and assem-bly of functional OXPHOS machinery are remarkably complex, requiring over a thousand genes in two sep-arate genomes. New techniques for gene searching, such as whole-exome sequencing, are likely to expand rapidly the list of genes implicated in mitochondrial diseases, including those with strictly tissue-specifi c manifestation. However, the real challenge will be to explain the molecular mechanisms responsible for diversity of phenotypes, both on the level of the individual cell and the entire organism.
Declaration of interest: The authors state no con-fl ict of interest and have received no payment in preparation of this manuscript.
References
Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The 1. epidemiology of mitochondrial disorders — past, present and future. Biochim Biophys Acta. 2004;1659:115 – 20.
Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, 2. Taylor RW, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35 – 9. Debray FG, Lambert M, Mitchell GA. Disorders of mito-3. chondrial function. Curr Opin Pediatr. 2008;20:471 – 82. McFarland R, Taylor RW, Turnbull DM. The neurology of 4. mitochondrial DNA disease. Lancet Neurol. 2002;1:343 – 51. Chan DC. Mitochondria: dynamic organelles in disease, aging, 5. and development. Cell. 2006;125:1241 – 52. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, 6. Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457 – 65. Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, 7. Ong SE, et al. A mitochondrial protein compendium eluci-dates complex I disease biology. Cell. 2008;134:112 – 23. Poulton J, Chiaratti MR, Meirelles FV, Kennedy S, Wells D, 8. Holt IJ. Transmission of mitochondrial DNA diseases and ways to prevent them. PLoS Genet. 2010;6:pii:e1001066. Cree LM, Samuels DC, de Sousa Lopes SC, Rajasimha HK, 9. Wonnapinij P, Mann JR, et al. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet. 2008;40:249 – 54. Wai T, Teoli D, Shoubridge EA. The mitochondrial DNA 10. genetic bottleneck results from replication of a subpopula-tion of genomes. Nat Genet. 2008;40:1484 – 8. Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE, 11. et al. A mouse model of mitochondrial disease reveals germ-line selection against severe mtDNA mutations. Science. 2008;319:958 – 62. Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, 12. Trifunovic A, et al. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 2008;6:e10. Greaves LC, Turnbull DM. Mitochondrial DNA mutations 13. and ageing. Biochim Biophys Acta. 2009;1790:1015 – 20. Fernandez-Vizarra E, Tiranti V, Zeviani M. Assembly of the 14. oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim Biophys Acta. 2009;1793:200 – 11. Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, 15. Walker JE. Bovine complex I is a complex of 45 different subunits. J Biol Chem. 2006;281:32724 – 7. Berrisford JM, Sazanov LA. Structural basis for the mechanism 16. of respiratory complex I. J Biol Chem. 2009;284:29773 – 83. Efremov RG, Baradaran R, Sazanov LA. The architecture of 17. respiratory complex I. Nature. 2010;465:441 – 5. Hunte C, Zickermann V, Brandt U. Functional modules and 18. structural basis of conformational coupling in mitochondrial complex I. Science. 2010;329:448 – 51. Sazanov LA, Hinchliffe P. Structure of the hydrophilic 19. domain of respiratory complex I from Thermus thermophilus. Science. 2006;311:1430 – 6. Distelmaier F, Koopman WJ, van den Heuvel LP, Rodenburg RJ, 20. Mayatepek E, Willems PH, et al. Mitochondrial complex I defi ciency: from organelle dysfunction to clinical disease. Brain. 2009;132(Pt 4):833 – 42. Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, 21. Lezza AM, et al. Mitochondrial DNA mutation associated with Leber ’ s hereditary optic neuropathy. Science. 1988;242:1427 – 30. Antonicka H, Ogilvie I, Taivassalo T, Anitori RP, Haller RG, 22. Vissing J, et al. Identifi cation and characterization of a com-mon set of complex I assembly intermediates in mitochon-dria from patients with complex I defi ciency. J Biol Chem. 2003;278:43081 – 8. Ugalde C, Vogel R, Huijbens R, Van Den Heuvel B, Smeitink J, 23. Nijtmans L. Human mitochondrial complex I assembles
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

56 E. Ylikallio & A. Suomalainen
through the combination of evolutionary conserved mod-ules: a framework to interpret complex I defi ciencies. Hum Mol Genet. 2004;13:2461 – 72. Vogel RO, Dieteren CE, van den Heuvel LP, Willems PH, 24. Smeitink JA, Koopman WJ, et al. Identifi cation of mitochon-drial complex I assembly intermediates by tracing tagged NDUFS3 demonstrates the entry point of mitochondrial subunits. J Biol Chem. 2007;282:7582 – 90. Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, 25. Hurd RE, Frankel WN, et al. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367 – 74. Bourgeron T, Rustin P, Chretien D, Birch-Machin M, 26. Bourgeois M, Viegas-Pequignot E, et al. Mutation of a nuclear succinate dehydrogenase gene results in mitochon-drial respiratory chain defi ciency. Nat Genet. 1995;11:144 – 9. Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, 27. et al. Gene mutations in the succinate dehydrogenase subu-nit SDHB cause susceptibility to familial pheochromocy-toma and to familial paraganglioma. Am J Hum Genet. 2001;69:49 – 54. Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, 28. Myssiorek D, Bosch A, et al. Mutations in SDHD, a mito-chondrial complex II gene, in hereditary paraganglioma. Sci-ence. 2000;287:848 – 51. Niemann S, Muller U. Mutations in SDHC cause autosomal 29. dominant paraganglioma, type 3. Nat Genet. 2000;26:268 – 70. Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for 30. mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer. 2003;3:193 – 202. Benit P, Lebon S, Rustin P. Respiratory-chain diseases 31. related to complex III defi ciency. Biochim Biophys Acta. 2009;1793:181 – 5. Crofts AR. The cytochrome bc1 complex: function in the 32. context of structure. Annu Rev Physiol. 2004;66:689 – 733. Acin-Perez R, Bayona-Bafaluy MP, Fernandez-Silva P, 33. Moreno-Loshuertos R, Perez-Martos A, Bruno C, et al. Res-piratory complex III is required to maintain complex I in mammalian mitochondria. Mol Cell. 2004;13:805 – 15. McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mito-34. chondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol. 2006;361:462 – 9. Visapaa I, Fellman V, Vesa J, Dasvarma A, Hutton JL, Kumar V, 35. et al. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am J Hum Genet. 2002;71:863 – 76. Hinson JT, Fantin VR, Schonberger J, Breivik N, Siem G, 36. McDonough B, et al. Missense mutations in the BCS1L gene as a cause of the Bjornstad syndrome. N Engl J Med. 2007;356:809 – 19. Diaz F. Cytochrome c oxidase defi ciency: patients and ani-37. mal models. Biochim Biophys Acta. 2010;1802:100 – 10. Antonicka H, Leary SC, Guercin GH, Agar JN, Horvath R, 38. Kennaway NG, et al. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for mul-tiple, early-onset clinical phenotypes associated with isolated COX defi ciency. Hum Mol Genet. 2003;12:2693 – 702. Antonicka H, Mattman A, Carlson CG, Glerum DM, 39. Hoffbuhr KC, Leary SC, et al. Mutations in COX15 pro-duce a defect in the mitochondrial heme biosynthetic path-way, causing early-onset fatal hypertrophic cardiomyopathy. Am J Hum Genet. 2003;72:101 – 14. Zhu Z, Yao J, Johns T, Fu K, De Bie I, Macmillan C, et al. 40. SURF1, encoding a factor involved in the biogenesis of cyto-chrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20:337 – 43.
Smith D, Gray J, Mitchell L, Antholine WE, Hosler JP. 41. Assembly of cytochrome-c oxidase in the absence of assem-bly protein Surf1p leads to loss of the active site heme. J Biol Chem. 2005;280:17652 – 6. Weraarpachai W, Antonicka H, Sasarman F, Seeger J, 42. Schrank B, Kolesar JE, et al. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase defi ciency and late-onset Leigh syndrome. Nat Genet. 2009;41:833 – 7. Ghezzi D, Saada A, D ’ Adamo P, Fernandez-Vizarra E, 43. Gasparini P, Tiranti V, et al. FASTKD2 nonsense mutation in an infantile mitochondrial encephalomyopathy associated with cytochrome c oxidase defi ciency. Am J Hum Genet. 2008;83:415 – 23. Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new 44. mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428 – 33. Houstek J, Pickova A, Vojtiskova A, Mracek T, Pecina P, 45. Jesina P. Mitochondrial diseases and genetic defects of ATP synthase. Biochim Biophys Acta. 2006;1757:1400 – 5. Wang ZG, White PS, Ackerman SH. Atp11p and Atp12p are 46. assembly factors for the F(1)-ATPase in human mitochon-dria. J Biol Chem. 2001;276:30773 – 8. De Meirleir L, Seneca S, Lissens W, De Clercq I, Eyskens F, 47. Gerlo E, et al. Respiratory chain complex V defi ciency due to a mutation in the assembly gene ATP12. J Med Genet. 2004;41:120 – 4. Mayr JA, Havlickova V, Zimmermann F, Magler I, Kaplanova V, 48. Jesina P, et al. Mitochondrial ATP synthase defi ciency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Hum Mol Genet. 2010;19:3430 – 9. Copeland WC. Inherited mitochondrial diseases of DNA 49. replication. Annu Rev Med. 2008;59:131 – 46. Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, 50. Chalmers RM, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clini cal and molecular genetic study. Lancet. 2004;364:875 – 82. Ylikallio E, Tyynismaa H, Tsutsui H, Ide T, Suomalainen A. 51. High mitochondrial DNA copy number has detrimental effects in mice. Hum Mol Genet. 2010;19:2695 – 705. Wanrooij S, Falkenberg M. The human mitochondrial rep-52. lication fork in health and disease. Biochim Biophys Acta. 2010;1797:1378 – 88. Rampazzo C, Miazzi C, Franzolin E, Pontarin G, Ferraro P, 53. Frangini M, et al. Regulation by degradation, a cellular defense against deoxyribonucleotide pool imbalances. Mutat Res. 2010 28;703:2 – 10. Chan SS, Copeland WC. DNA polymerase gamma and mito-54. chondrial disease: understanding the consequence of POLG mutations. Biochim Biophys Acta. 2009;1787:312 – 9. Hakonen AH, Heiskanen S, Juvonen V, Lappalainen I, 55. Luoma PT, Rantamaki M, et al. Mitochondrial DNA polymer-ase W748S mutation: a common cause of autosomal reces-sive ataxia with ancient European origin. Am J Hum Genet. 2005;77:430 – 41. Van Goethem G, Dermaut B, Lofgren A, Martin JJ, 56. Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28:211 – 2. Lee YS, Kennedy WD, Yin YW. Structural insight into proces-57. sive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell. 2009;139:312 – 24. Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, 58. Taylor RW, et al. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmo-plegia. Am J Hum Genet. 2006;78:1026 – 34.
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

Mitochondrial diseases 57
Walter MC, Czermin B, Muller-Ziermann S, Bulst S, 59. Stewart JD, Hudson G, et al. Late-onset ptosis and myopa-thy in a patient with a heterozygous insertion in POLG2. J Neurol. 2010;257:1517 – 23. Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, 60. et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28:223 – 31. Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen 61. A, Lonnqvist T. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain. 2007;130(Pt 11):3032 – 40. Sarzi E, Goffart S, Serre V, Chretien D, Slama A, Munnich A, 62. et al. Twinkle helicase (PEO1) gene mutation causes mito-chondrial DNA depletion. Ann Neurol. 2007;62:579 – 87. Hakonen AH, Goffart S, Marjavaara S, Paetau A, Cooper H, 63. Mattila K, et al. Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum Mol Genet. 2008;17:3822 – 35. Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, 64. Ylikallio E, Jalanko A, et al. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. 2005;102:17687 – 92. Goffart S, Cooper HM, Tyynismaa H, Wanrooij S, 65. Suomalainen A, Spelbrink JN. Twinkle mutations associated with autosomal dominant progressive external ophthalmo-plegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum Mol Genet. 2009;18:328 – 40. Wanrooij S, Luoma P, van Goethem G, van Broeckhoven C, 66. Suomalainen A, Spelbrink JN. Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 2004;32:3053 – 64. Srivastava S, Moraes CT. Double-strand breaks of mouse 67. muscle mtDNA promote large deletions similar to multiple mtDNA deletions in humans. Hum Mol Genet. 2005;14:893 – 902. Moslemi AR, Melberg A, Holme E, Oldfors A. Clonal 68. expansion of mitochondrial DNA with multiple deletions in autosomal dominant progressive external ophthalmoplegia. Ann Neurol. 1996;40:707 – 13. Fukui H, Moraes CT. Mechanisms of formation and accu-69. mulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet. 2009;18:1028 – 36. Reichard P. Interactions between deoxyribonucleotide and 70. DNA synthesis. Annu Rev Biochem. 1988;57:349 – 74. Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, 71. et al. The deoxyguanosine kinase gene is mutated in indi-viduals with depleted hepatocerebral mitochondrial DNA. Nat Genet. 2001;29:337 – 41. Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. 72. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29:342 – 4. Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, et al. 73. Mutation of RRM2B, encoding p53-controlled ribonucle-otide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776 – 80. Tyynismaa H, Ylikallio E, Patel M, Molnar MJ, Haller RG, 74. Suomalainen A. A heterozygous truncating mutation in RRM2B causes autosomal-dominant progressive external oph-thalmoplegia with multiple mtDNA deletions. Am J Hum Genet. 2009;85:290 – 5. Dorado B, Area E, Akman HO, Hirano M. Onset and 75. organ specificity of Tk2 deficiency depends on Tk1
down-regulation and transcriptional compensation. Hum Mol Genet. 2010;20:155 – 64. Nishino I, Spinazzola A, Hirano M. Thymidine phosphory-76. lase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689 – 92. Hirano M, Nishigaki Y, Marti R. Mitochondrial neurogas-77. trointestinal encephalomyopathy (MNGIE): a disease of two genomes. Neurologist. 2004;10:8 – 17. Ylikallio E, Page JL, Xu X, Lampinen M, Bepler G, Ide T, 78. et al. Ribonucleotide reductase is not limiting for mitochon-drial DNA copy number in mice. Nucleic Acids Res. 2010;38:8208 – 18. Smits P, Smeitink J, van den Heuvel L. Mitochondrial trans-79. lation and beyond: processes implicated in combined oxida-tive phosphorylation defi ciencies. J Biomed Biotechnol. 2010;2010:737385. Spremulli LL, Coursey A, Navratil T, Hunter SE. Initiation 80. and elongation factors in mammalian mitochondrial protein biosynthesis. Prog Nucleic Acid Res Mol Biol. 2004;77:211 – 61. Smits P, Smeitink JA, van den Heuvel LP, Huynen MA, 81. Ettema TJ. Reconstructing the evolution of the mitochon-drial ribosomal proteome. Nucleic Acids Res. 2007;35:4686 – 703. Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-82. Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet. 2004;74:1303 – 8. Bykhovskaya Y, Mengesha E, Fischel-Ghodsian N. 83. Pleiotropic effects and compensation mechanisms deter-mine tissue specifi city in mitochondrial myopathy and sideroblastic anemia (MLASA). Mol Genet Metab. 2007;91:148 – 56. Zeharia A, Shaag A, Pappo O, Mager-Heckel AM, Saada A, 84. Beinat M, et al. Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet. 2009;85:401 – 7. Guan MX, Yan Q, Li X, Bykhovskaya Y, Gallo-Teran J, Hajek P, 85. et al. Mutation in TRMU related to transfer RNA modifi ca-tion modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am J Hum Genet. 2006;79:291 – 302. Miller C, Saada A, Shaul N, Shabtai N, Ben-Shalom E, 86. Shaag A, et al. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol. 2004;56:734 – 8. Saada A, Shaag A, Arnon S, Dolfi n T, Miller C, Fuchs-Telem D, 87. et al. Antenatal mitochondrial disease caused by mitochon-drial ribosomal protein (MRPS22) mutation. J Med Genet. 2007;44:784 – 6. Coenen MJ, Antonicka H, Ugalde C, Sasarman F, Rossi R, 88. Heister JG, et al. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation defi ciency. N Engl J Med. 2004;351:2080 – 6. Valente L, Tiranti V, Marsano RM, Malfatti E, 89. Fernandez-Vizarra E, Donnini C, et al. Infantile encepha-lopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet. 2007;80:44 – 58. Antonicka H, Sasarman F, Kennaway NG, Shoubridge EA. 90. The molecular basis for tissue specifi city of the oxidative phosphorylation defi ciencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet. 2006;15:1835 – 46. Smeitink JA, Elpeleg O, Antonicka H, Diepstra H, Saada A, 91. Smits P, et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am J Hum Genet. 2006;79:869 – 77.
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

58 E. Ylikallio & A. Suomalainen
Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, 92. Einbinder T, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857 – 62.
Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, 93. Sissler M, Smet J, et al. Mitochondrial aspartyl-tRNA syn-thetase defi ciency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534 – 9.
Riley LG, Cooper S, Hickey P, Rudinger-Thirion J, 94. McKenzie M, Compton A, et al. Mutation of the mitochon-drial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia — MLASA syndrome. Am J Hum Genet. 2010;87:52 – 9.
Belostotsky R, Ben-Shalom E, Rinat C, Becker-Cohen R, 95. Feinstein S, Zeligson S, et al. Mutations in the mitochon-drial seryl-tRNA synthetase cause hyperuricemia, pul-monary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88:193 – 200.
Gotz A, Tyynismaa H, Euro L, Ellonen P, Hyotylainen T, 96. Ojala T, et al. Exome sequencing identifi es mitochondrial alanyl-tRNA synthetase mutations in infantile mitochon-drial cardiomyopathy. Am J Hum Genet. 2011;88:635 – 42.
Pierce SB, Chisholm KM, Lynch ED, Lee MK, Walsh T, 97. Opitz JM, et al. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sen-sorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci U S A. 2011;108:6543 – 8.
DiMauro S, Davidzon G. Mitochondrial DNA and disease. 98. Ann Med. 2005;37:222 – 32.
Scaglia F, Wong LJ. Human mitochondrial transfer RNAs: 99. role of pathogenic mutation in disease. Muscle Nerve. 2008;37:150 – 71.
Jacobs HT. Disorders of mitochondrial protein synthesis. 100. Hum Mol Genet. 2003;12 Spec No 2:R293 – 301.
Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, 101. Qiu WQ, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. 1993;4:289 – 94.
Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of 102. muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717 – 9.
Detmer SA, Chan DC. Functions and dysfunctions of mito-103. chondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870 – 9.
Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, 104. Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropa-thy type 2A. Nat Genet. 2004;36:449 – 51.
Detmer SA, Chan DC. Complementation between mouse 105. Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol. 2007;176:405 – 14.
Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, 106. Moore A, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211 – 5.
Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, 107. Belenguer P, et al. Nuclear gene OPA1, encoding a mito-chondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207 – 10.
Quinzii CM, Hirano M. Coenzyme Q and mitochondrial 108. disease. Dev Disabil Res Rev. 2010;16:183 – 8.
Jin H, May M, Tranebjaerg L, Kendall E, Fontan G, Jackson J, 109. et al. A novel X-linked gene, DDP, shows mutations in fam-ilies with deafness (DFN-1), dystonia, mental defi ciency and blindness. Nat Genet. 1996;14:177 – 80.
Roesch K, Curran SP, Tranebjaerg L, Koehler CM. Human 110. deafness dystonia syndrome is caused by a defect in assem-bly of the DDP1/TIMM8a-TIMM13 complex. Hum Mol Genet. 2002;11:477 – 86.
Di Fonzo A, Ronchi D, Lodi T, Fassone E, Tigano M, 111. Lamperti C, et al. The mitochondrial disulfi de relay system protein GFER is mutated in autosomal-recessive myopathy with cataract and combined respiratory-chain defi ciency. Am J Hum Genet. 2009;84:594 – 604.
Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, 112. Fernandez P, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973 – 83.
Rugarli EI, Langer T. Translating m-AAA protease function 113. in mitochondria to hereditary spastic paraplegia. Trends Mol Med. 2006;12:262 – 9.
Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, 114. D ’ Adamo P, et al. Severe X-linked mitochondrial encepha-lomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010;86:639 – 49.
Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, 115. Tiveron C, et al. Loss of ETHE1, a mitochondrial dioxy-genase, causes fatal sulfi de toxicity in ethylmalonic enceph-alopathy. Nat Med. 2009;15:200 – 5.
Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT. Mito-116. chondrial medicine: a metabolic perspective on the pathol-ogy of oxidative phosphorylation disorders. Cell Metab. 2006;3:9 – 13.
Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, 117. and aging. Cell. 2005;120:483 – 95.
Willems PH, Valsecchi F, Distelmaier F, Verkaart S, Visch HJ, 118. Smeitink JA, et al. Mitochondrial Ca2 � homeostasis in human NADH:ubiquinone oxidoreductase defi ciency. Cell Calcium. 2008;44:123 – 33.
Tyynismaa H, Carroll CJ, Raimundo N, Ahola-Erkkila S, 119. Wenz T, Ruhanen H, et al. Mitochondrial myopathy induces a starvation-like response. Hum Mol Genet. 2010;19:3948 – 58.
Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is 120. recruited selectively to impaired mitochondria and pro-motes their autophagy. J Cell Biol. 2008;183:795 – 803.
Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, 121. Walzer G, et al. Fission and selective fusion govern mito-chondrial segregation and elimination by autophagy. EMBO J. 2008;27:433 – 46.
Ahola-Erkkil ä S, Carroll CJ, Peltola-Mjosund K, Tulkki V, 122. Mattila I, Seppanen-Laakso T, et al. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum Mol Genet. 2010;19:1974 – 84.
Zhu X, Peng X, Guan MX, Yan Q. Pathogenic mutations 123. of nuclear genes associated with mitochondrial disorders. Acta Biochim Biophys Sin (Shanghai). 2009;41:179 – 87.
Haack TB, Danhauser K, Haberberger B, Hoser J, Strecker 124. V, Boehm D, et al. Exome sequencing identifi es ACAD9 mutations as a cause of complex I defi ciency. Nat Genet. 2010;42:1131 – 4.
Ghezzi D, Goffrini P, Uziel G, Horvath R, Klopstock T, 125. Lochmuller H, et al. SDHAF1, encoding a LYR complex-II specifi c assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet. 2009;41:654 – 6.
Hao HX, Khalimonchuk O, Schraders M, Dephoure N, 126. Bayley JP, Kunst H, et al. SDH5, a gene required for fl a-vination of succinate dehydrogenase, is mutated in paragan-glioma. Science. 2009;325:1139 – 42.
Barel O, Shorer Z, Flusser H, Ofi r R, Narkis G, Finer G, 127. et al. Mitochondrial complex III defi ciency associated with a homozygous mutation in UQCRQ. Am J Hum Genet. 2008;82:1211 – 6.
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

Mitochondrial diseases 59
Ghezzi D, Arzuffi P, Zordan M, Da Re C, Lamperti C, 128. Benna C, et al. Mutations in TTC19 cause mitochondrial complex III defi ciency and neurological impairment in humans and fl ies. Nat Genet. 2011;43:259 – 63.
Shteyer E, Saada A, Shaag A, Al-Hijawi FA, Kidess R, 129. Revel-Vilk S, et al. Exocrine pancreatic insuffi ciency, dys-erythropoeitic anemia, and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am J Hum Genet. 2009;84:412 – 7.
Huigsloot M, Nijtmans LG, Szklarczyk R, Baars MJ, van 130. den Brand MA, Hendriksfranssen MG, et al. A mutation in c2orf64 causes impaired cytochrome C oxidase assembly and mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:488 – 93.
Jonckheere AI, Hogeveen M, Nijtmans LG, van den 131. Brand MA, Janssen AJ, Diepstra JH, et al. A novel mitochon-drial ATP8 gene mutation in a patient with apical hyper-trophic cardiomyopathy and neuropathy. J Med Genet. 2008;45:129 – 33.
Cizkova A, Stranecky V, Mayr JA, Tesarova M, Havlickova V, 132. Paul J, et al. TMEM70 mutations cause isolated ATP syn-thase defi ciency and neonatal mitochondrial encephalocar-diomyopathy. Nat Genet. 2008;40:1288 – 90.
Kaukonen J, Juselius JK, Tiranti V, Kyttala A, 133. Zeviani M, Comi GP, et al. Role of adenine nucleotide
translocator 1 in mtDNA maintenance. Science. 2000;289:782 – 5.
Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, 134. Bondi-Rubinstein G, Rahman S, et al. Defi ciency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA deple-tion. Am J Hum Genet. 2005;76:1081 – 6.
Ostergaard E, Christensen E, Kristensen E, Mogensen B, 135. Duno M, Shoubridge EA, et al. Defi ciency of the alpha subunit of succinate-coenzyme A ligase causes fatal infan-tile lactic acidosis with mitochondrial DNA depletion. Am J Hum Genet. 2007;81:383 – 7.
Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, 136. Bornstein B, Boissiere A, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘ plus ’ phenotypes. Brain. 2008;131(Pt 2):338 – 51.
Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, 137. D ’ Adamo P, Calvo S, et al. MPV17 encodes an inner mito-chondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38:570 – 5.
Antonicka H, Ostergaard E, Sasarman F, Weraarpachai W, 138. Wibrand F, Pedersen AM, et al. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet. 2010;87:115 – 22.
Ann
Med
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
K U
Leu
ven
on 0
3/12
/14
For
pers
onal
use
onl
y.

ORIGINAL COMMUNICATION
The m.3243A>G mitochondrial DNA mutation and relatedphenotypes. A matter of gender?
Michelangelo Mancuso • Daniele Orsucci • Corrado Angelini • Enrico Bertini • Valerio Carelli •
Giacomo Pietro Comi • Alice Donati • Carlo Minetti • Maurizio Moggio • Tiziana Mongini •
Serenella Servidei • Paola Tonin • Antonio Toscano • Graziella Uziel • Claudio Bruno • Elena Caldarazzo Ienco •
Massimiliano Filosto • Costanza Lamperti • Michela Catteruccia • Isabella Moroni • Olimpia Musumeci •
Elena Pegoraro • Dario Ronchi • Filippo Maria Santorelli • Donato Sauchelli • Mauro Scarpelli •
Monica Sciacco • Maria Lucia Valentino • Liliana Vercelli • Massimo Zeviani • Gabriele Siciliano
Received: 13 November 2013 / Revised: 16 December 2013 / Accepted: 17 December 2013
� Springer-Verlag Berlin Heidelberg 2013
Abstract The m.3243A[G ‘‘MELAS’’ (mitochondrial
encephalopathy with lactic acidosis and stroke-like epi-
sodes) mutation is one of the most common point muta-
tions of the mitochondrial DNA, but its phenotypic
variability is incompletely understood. The aim of this
study was to revise the phenotypic spectrum associated
with the mitochondrial m.3243A[G mutation in 126 Italian
carriers of the mutation, by a retrospective, database-based
study (‘‘Nation-wide Italian Collaborative Network of
Mitochondrial Diseases’’). Our results confirmed the high
clinical heterogeneity of the m.3243A[G mutation.
Hearing loss and diabetes were the most frequent clinical
features, followed by stroke-like episodes. ‘‘MIDD’’
(maternally-inherited diabetes and deafness) and ‘‘PEO’’
(progressive external ophthalmoplegia) are nosographic
terms without any real prognostic value, because these
patients may be even more prone to the development of
multisystem complications such as stroke-like episodes and
heart involvement. The ‘‘MELAS’’ acronym is convincing
and useful to denote patients with histological, biochemical
and/or molecular evidence of mitochondrial disease who
experience stroke-like episodes. Of note, we observed for
M. Mancuso (&) � D. Orsucci � E. C. Ienco � G. Siciliano
Neurological Clinic, University of Pisa, Via Roma 67,
56100 Pisa, Italy
e-mail: [email protected]
C. Angelini � E. Pegoraro
Neurological Clinic, University of Padova, Padua, Italy
C. Angelini � E. Pegoraro
IRCCS S. Camillo, Venice, Italy
E. Bertini � M. Catteruccia
Laboratory of Molecular Medicine, Bambino Gesu Hospital,
Rome, Italy
V. Carelli � M. L. Valentino
IRCCS Istituto Delle Scienze Neurologiche di Bologna,
Bologna, Italy
V. Carelli � M. L. Valentino
Department of Biomedical and Neuromotor Sciences, University
of Bologna, Bologna, Italy
G. P. Comi � D. Ronchi
Neuroscience Section, Neurology Unit, Dino Ferrari Centre,
Department of Pathophysiology and Transplantation (DEPT),
IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico,
University of Milan, Milan, Italy
A. Donati
A. Meyer Children’s Hospital, University of Florence, Florence,
Italy
C. Minetti � C. Bruno
Neuropediatric and Muscle Disorders Unit, University of Genoa,
G. Gaslini Institute, Genoa, Italy
M. Moggio � M. Sciacco
Neuromuscular Unit, Dino Ferrari Centre, Fondazione IRCCS
Ca’ Granda Ospedale Maggiore Policlinico, University of Milan,
Milan, Italy
T. Mongini � L. Vercelli
Department of Neuroscience, University of Turin, Turin, Italy
S. Servidei � D. Sauchelli
Institute of Neurology, Catholic University, Rome, Italy
P. Tonin � M. Scarpelli
Neurological Clinic, University of Verona, Verona, Italy
A. Toscano � O. Musumeci
Department of Neurosciences, University of Messina, Messina,
Italy
123
J Neurol
DOI 10.1007/s00415-013-7225-3

the first time that male gender could represent a risk factor
for the development of stroke-like episodes in Italian
m.3243A[G carriers. Gender effect is not a new concept in
mitochondrial medicine, but it has never been observed in
MELAS. A better elucidation of the complex network
linking mitochondrial dysfunction, apoptosis, estrogen
effects and stroke-like episodes may hold therapeutic
promises.
Keywords A3243G � MELAS � MIDD � Mitochondrial
DNA � PEO � Stroke-like episodes
Introduction
The m.3243A[G mutation is one of the most common
point mutations of the mitochondrial genome (mtDNA).
The clinical syndromes that have been historically ascribed
to this substitution include mitochondrial encephalopathy
with lactic acidosis and stroke-like episodes (MELAS),
maternally inherited deafness and diabetes (MIDD), and
progressive external ophthalmoplegia (PEO) [1], but het-
erogeneity is high [2]. Other reported features include
isolated myopathy, cardiomyopathy, seizures, migraine,
ataxia, cognitive impairment, gastro-intestinal dysmotility,
and short stature [1]. This phenotypic variability is
incompletely understood; at least in part it may be due to
heteroplasmy, with varying proportions of mutant and
wild-type mtDNA molecules in different tissues [2].
However, other factors, environmental and/or genetic, must
be involved.
Here, we revise the phenotypic spectrum associated with
the m.3243A[G mutation based on a cohort of 126 carriers
of the mutation from the database of the ‘‘Nation-wide
Italian Collaborative Network of Mitochondrial Diseases’’,
in order to answer the following questions: (1) Is the dis-
tinction in three phenotypes (MELAS; PEO; MIDD) fully
justified or somehow arbitrary? (2) Are some of the most
frequent clinical features mutually associated? (3) Which
factors can explain the marked clinical heterogeneity of
these patients? and (4) Which clinical features may direct
clinicians toward this molecular diagnosis?
Methods
We reviewed the clinical, morphological, and laboratory
features of all carriers of the m.3243A[G substitution
enrolled in the database of the ‘‘Nation—wide Italian
Collaborative Network of Mitochondrial Diseases’’. In
those patients, the diagnosis of mitochondrial disease was
supported by clinical, histological, biochemical, and/or
molecular findings [3].
Data were expressed as mean ± standard deviation. The
numeric data were compared by unpaired, two-tailed
t testing (or one-way ANOVA when the groups were [2)
or correlated by Spearman’s coefficient of rank correlation.
Proportions were compared by Fisher’s exact test. Cate-
gorical data were associated by means of the Fisher’s exact
test. Bonferroni’s correction for multiple tests was utilized
when appropriate. A P value \0.05 was considered as
significant. Data analysis was carried out using MedCalc�
ans SPSS�.
Standard protocol approval, registration, and patient
consent
The establishment of the database, and its use for scientific
purposes, was approved by the local Ethical Committees of
all the involved centers, who obtained written consent from
each patient, in accordance with the ethical standards of the
1964 Declaration of Helsinki. The database creation was
supported by a Telethon grant (GUP09004).
Results
Relative frequency of m.3243A[G mutation in our
database
A total of 133 patients harboring the m.3243A[G mutation
were identified among the 1,160 mitochondrial patients
(11.5 %) enlisted in our database (last accession July 1st,
2013). They represented the 36.9 % (133/360) of all indi-
viduals with mtDNA point mutations. The clinical features
were not available for seven patients, who therefore were
not considered for the following analysis, performed on the
remaining 126 individuals.
Patients’ mean age at the last evaluation was
36.0 ± 20.7 years. Men were 65/126 (51.6 %). Eight of
126 (6.3 %) subjects were asymptomatic carriers (age at
last evaluation was 28.4 ± 27.9 years; range 1–77).
G. Uziel � I. Moroni
Child Neurology Unit, The Foundation ‘‘Carlo Besta’’ Institute
of Neurology, IRCCS, Milan, Italy
M. Filosto
Neurological Clinic, University Hospital Spedali Civili, Brescia,
Italy
C. Lamperti � M. Zeviani
Unit of Molecular Neurogenetics, The Foundation ‘‘Carlo Besta’’
Institute of Neurology, IRCCS, Milan, Italy
F. M. Santorelli
IRCCS Stella Maris, Pisa, Italy
J Neurol
123

Clinical features
Table 1 and Fig. 1 summarize the clinical features at onset
and at the last evaluation. The mean age at onset of the 118
symptomatic patients was 23.4 ± 15.6 years, with 44.1 %
of the cases having a childhood (\16 years) onset. Hearing
loss was the most common complaint at the onset, followed
by generalized seizures and diabetes (see Table 1).
At the last evaluation, mean age and disease duration
were 36.0 ± 20.7 (range 1–77) and 18.4 ± 16.6 years,
respectively. Considering our 126 patients, at the mean age
of &36 years, the clinical picture was characterized by the
following signs/symptoms, starting from the most frequent:
hearing loss ([50 % of patients); diabetes, stroke-like
episodes (40–49.9 %); muscle weakness, generalized sei-
zures, exercise intolerance, heart disease (30–39.9 %);
ptosis/ophthalmoparesis, migraine, cognitive impairment,
muscle wasting (20–29.9 %); ataxia, increased CK levels,
failure to thrive/short stature (15–19.9 %); gastrointestinal
dysmotility, hypotonia, pyramidal signs, peripheral neu-
ropathy, muscle pain, retinopathy (10–14.9 %).
Seventeen patients, all with central nervous system
(CNS) involvement (stroke-like episodes reported in 12
cases) died at a mean age of 37.2 ± 22.3 years (range
6–76) because of cardiocirculatory failure (four cases),
stroke (three cases), status epilepticus or intractable lactic
acidosis (each in one case). The cause of death remained
unknown for eight patients.
Stroke-like episodes
Stroke-like episodes are crucial for the syndromic diag-
nosis of MELAS. Here, we define as ‘‘MELAS’’ the group
of patients with clinical and or radiological evidence of
stroke-like episodes; on the other hand, ‘‘non-MELAS’’
patients are defined as individuals carrying the
m.3243A[G who have not experienced stroke-like
episodes.
Fifty-one out of 126 (40.5 %) of m.3243A[G carriers,
at the mean age of 36 years, had suffered stroke-like epi-
sodes (see Table 1). Age at onset, age at last control, and
disease duration were not different between ‘‘MELAS’’ and
‘‘non-MELAS’’ groups. Furthermore, heteroplasmy levels
could not predict stroke-like episodes, and no difference in
the proportion of patients with ragged red fibers and COX-
negative fibers was observed between the two groups.
Lactate increase was strongly associated with MELAS
(P = 0.0003), confirming the validity of the acronym.
Table 1 Clinical features of the m.3243A[G carriers (n = 126)
Onset
23.4 ± 15.6
yearsa (%)
Last evaluation
36.0 ± 20.7
years (%)
Hearing loss 39 (31.0) 73 (57.9)
Generalized seizures 23 (18.3) 47 (37.3)
Diabetes 22 (17.5) 51 (40.5)
Ptosis/ophthalmoparesis 16 (12.7) 35 (27.8)
Migraine 16 (12.7) 35 (27.8)
Stroke-like episodes 14 (11.1) 51 (40.5)
Muscle weakness 14 (11.1) 50 (39.7)
Exercise intolerance 14 (11.1) 41 (32.5)
Heart disease 11 (8.7) 40 (31.7)
Cognitive involvement 11 (8.7) 31 (24.6)
Failure to thrive/short st. 11 (8.7) 21 (16.7)
Increased CK 7 (5.6) 25 (19.8)
Muscle pain 7 (5.6) 13 (10.3)
Muscle wasting 6 (4.8) 29 (23.0)
Vomiting 5 (4.0) 7 (5.6)
Gastrointestinal dysmotil. 4 (3.2) 16 (12.7)
Neuropathy 4 (3.2) 13 (10.3)
Ataxia 3 (2.4) 25 (19.8)
Hypotonia 3 (2.4) 15 (11.9)
Retinopathy 3 (1.8) 13 (10.3)
Myoclonus 3 (2.4) 8 (6.3)
Hypothyroidism 2 (1.6) 5 (4.0)
Psychiatric involvement 1 (0.8) 8 (6.3)
Optic neuropathy 1 (0.8) 6 (4.8)
Hypogonadism 1 (0.8) 5 (4.0)
Pyramidal signs – 14 (11.1)
Respiratory impairment – 6 (4.8)
Status epilepticus – 5 (4.0)
The ten most frequent features are shown in bold. Swallowing
impairment, tremor, anemia, cataract, dyskinesia, hepatopathy, kid-
ney inv. \3 %a The symptomatic patients
0
10
20
30
40
50
60
70
80
90
100
Hea
ring
loss
Sei
zure
sD
iabe
tes
Pto
sis/
PE
OM
igra
ine
Str
oke-
like
ep.
Mus
cle
wea
kn.
Exe
rcis
e in
tol.
Hea
rt d
isea
seC
ogni
tive
inv.
Thri
ve fa
il./s
hort
st.
Incr
ease
d C
KM
uscl
e pa
inM
uscl
e w
astin
gG
astr
oint
dys
mot
il.N
euro
path
yA
taxi
aH
ypot
onia
Ret
inop
athy
Pyr
amid
al s
igns
%
Fig. 1 Clinical features of the m.3243A[G patients. In the figure are
shown the features present in C10 % of patients at the last evaluation.
White onset; black last evaluation. For details, see text and Table 1
J Neurol
123

Among the other nine most frequent clinical features (see
Table 1, in bold—significance level after Bonferroni’s
correction: 0.0056), stroke-like episodes were strongly
associated with generalized seizures (‘‘MELAS’’ with sei-
zures 35/51, 68.6 %; ‘‘non-MELAS’’ with seizures 12/75,
16.0 %; P \ 0.000001), cognitive involvement
(‘‘MELAS’’ with cognitive impairment 20/51, 39.2 %;
‘‘non-MELAS’’ with cognitive impairment 11/75, 14.7 %;
P = 0.0028) and hearing loss (‘‘MELAS’’ with hearing
loss 38/51, 74.5 %; ‘‘non-MELAS’’ with hearing loss
35/75, 46.7 %; P = 0.0030). Stroke-like episodes were not
associated with migraine.
Is there a gender effect in MELAS?
The ‘‘MELAS’’ group was enriched in males (33/51,
64.7 %) if compared with the ‘‘non-MELAS’’ group (men
32/75, 42.7 %). This difference, which is statistically sig-
nificant with a P = 0.019 (Table 2), was not explained by
possible confounding factors such as onset age, age at the
last control, disease duration, and levels of heteroplasmy in
tissues (muscle, blood, and urine), which did not signifi-
cantly differ between the two groups.
‘‘MIDD syndrome’’
Hearing loss and diabetes were associated with a high
statistical significance (P = 0.0002) in m.3243A[G
patients (Table 3), thus confirming the validity of the
acronym MIDD at least from this point of view.
Apart from stroke-like episodes and diabetes, among the
other seven most frequent clinical features (see Table 1, in
bold—significance level after Bonferroni’s correction:
0.0071) hearing loss (‘‘HL’’) was significantly associated
with heart disease (‘‘HL’’ with heart disease 32/73, 43.8 %;
‘‘non-HL’’ with heart disease 8/53, 15.1 %; P = 0.00086)
and exercise intolerance (‘‘HL’’ with exercise intolerance
32/73, 43.8 %; ‘‘non-HL’’ with exercise intolerance 9/53,
17.0 %; P = 0.0019). A non-significant trend was
observed for the association with cognitive involvement
(P = 0.012) and muscle weakness (P = 0.015).
On the contrary, diabetes did not result associated with
any of the most frequent clinical features (except hearing
loss).
Noteworthy, ‘‘pure’’ MIDD was very rare in our cohort
(3/126, 2.4 %). Therefore, ‘‘MIDD’’ may be useful to
denote the very strong association between diabetes and
hearing loss, but less to represent a well-defined, pure
clinical picture in m.3243A[G patients.
Progressive external ophthalmoplegia (PEO)
As already seen, ptosis/ophthalmoparesis was not associ-
ated with stroke-like episodes, hearing loss, or diabetes.
Among the other six most common clinical features (see
Table 1—significance level after Bonferroni’s correction
0.0083), PEO was significantly associated with heart dis-
ease (‘‘PEO’’ with heart disease 18/35, 51.4 %; ‘‘non-
PEO’’ with heart disease 22/91, 24.2 %; P = 0.0051). A
non-significant trend was observed for the association with
muscle weakness (P = 0.044).
As was the case for the pure MIDD, ‘‘pure’’ PEO was
rare in our cohort (4/126, 3.2 %).
Comparison with the m.8344A[G mutation
Table 4 shows the clinical features of our patients with the
m.3243A[G mutation compared with the subjects harbor-
ing the m.8344A[G ‘‘MERRF’’ mutation (the second most
common encephalomyopathy-associated mtDNA muta-
tion), which were recently reported [4]. Noteworthy,
stroke-like episodes and diabetes (and to a lesser extent,
hearing loss, migraine, and cognitive impairment) strongly
support the molecular diagnosis of the m.3243A[G
mutation, whereas multiple lipomatosis and less stringently
myoclonus and increased CK levels are all predictors of the
m.8344A[G mutation.
Neuroradiological features
Recent (\5 years before the last examination) magnetic
resonance imaging (MRI) data were available for 84
patients with the m.3243A[G mutation. Scans were normal
in eleven patients (13.1 %, significantly different from the
58.8 % of m.8344A[G patients; [4], P = 0.01).
Stroke-like episodes were observed in 46/84 (54.8 %)
m.3243A[G mutation carriers. Cortical (36/84, 42.9 %),
Table 2 Male gender is associated with stroke-like episodes in
m.3243A[G carriers
‘‘MELAS’’ ‘‘non-MELAS’’ Total
Males 33 32 65
Females 18 43 61
51 75
n = 126; P = 0.01856 (Fisher’s exact test)
Table 3 Diabetes and hearing loss are strictly associated in
m.3243A[G carriers
Diabetes: yes Diabetes: no Total
Hearing loss: yes 40 33 73
Hearing loss: no 11 42 53
51 75
n = 126; P = 0.000119 (Fisher’s exact test)
J Neurol
123

subcortical (18/84, 21.4 %) and cerebellar atrophy (20/84,
23.8 %) were also common. White matter abnormalities
were observed in 22/84 (26.2 %) patients and calcifications
in 17/84 (20.2 %) patients.
Histological findings
Muscle biopsy findings were available for 65 patients.
Ragged red fibers (RRFs) were present in 60 cases
(92.3 %), COX-negative fibers in 53 (81.54 %). Lipid
storage was noted in 22 biopsies (33.8 %). Strongly suc-
cinate dehydrogenase-reactive blood vessels (SSVs) were
seen in ten patients (15.4 %): five of them had a MELAS
phenotype whereas the other five did not have stroke-like
episodes (not significant).
Heteroplasmy levels
Heteroplasmy levels of the m.3243A[G mutation were
quantified in muscle tissue for 39 patients (mutation load
was on average 61.9 ± 18.6 %), in peripheral blood for 66
patients (mutation load 27.0 ± 19.9 %), and in urinary
sediment for 29 patients (mutation load 42.9 ± 27.7 %),
being lower in blood than in muscle and urine (P \ 0.01).
Heteroplasmy levels in blood and urinary sediment were
directly correlated (q = 0.782, P \ 0.0001), whereas no
significant correlation was observed between the levels in
muscle and/or urinary sediment (significance level after
Bonferroni’s correction 0.017).
A negative correlation was observed between age at
onset and mutation load in blood (q = -0.447,
P = 0.0034), meaning that subjects with earlier onset had
higher mutation levels in blood. No significant correlation
was observed with muscle and urinary levels (significance
level after Bonferroni’s correction 0.017).
Heteroplasmy levels could not predict stroke-like epi-
sodes; the same for other clinical signs such as hearing loss,
diabetes, and PEO.
Discussion
Our study presents a retrospective characterization of 126
Italian carriers of the mtDNA m.3243A[G mutation. We
showed that the m.3243A[G-associated phenotypes are a
continuum from asymptomatic to lethal multisystem dis-
eases; between these extremes lie all the possible expres-
sivity degrees.
Stroke-like episodes and diabetes (and, to a lesser
extent, hearing loss, migraine, and cognitive involvement)
support the molecular diagnosis of the m.3243A[G
mutation, whereas multiple lipomatosis, and to a lesser
extent myoclonus and high CK levels, were associated with
the m.8344A[G mutation [4]. A normal MRI supports the
latter [4], whereas it is a rare finding in m.3243A[G
patients.
Heteroplasmy levels could not predict stroke-like epi-
sodes nor other clinical features such as hearing loss, dia-
betes, and PEO. Subjects with earlier onset had higher
mutation levels in blood. Mutation levels were lower in
blood than in muscle and urine, confirming that the latter two
tissues are more useful for informative genetic analysis [5].
Table 4 Clinical features of the m.3243A[G (n = 126) and m.8344A[G (n = 39) carriers
m.3243A[G
36.0 ± 26.7 years (%)
m.8344A[G
45.6 ± 15.3 years (%)
P
Hearing loss 73 (57.9) 12 (30.8) 0.0034
Generalized seizures 47 (37.3) 12 (30.8) n.s.
Diabetes 51 (40.5) 4 (10.3) 0.0004
Ptosis/ophthalmoparesis 35 (27.8) 10 (25.6) n.s.
Migraine 35 (27.8) 3 (7.7) 0.009
Stroke-like episodes 51 (40.5) 1 (2.6) 0.000001
Muscle weakness 50 (39.7) 20 (51.3) n.s.
Exercise intolerance 41 (32.5) 15 (38.5) n.s.
Heart disease 40 (31.7) 6 (15.4) n.s.
Cognitive involvement 31 (24.6) 3 (7.7) 0.02
Increased CK 25 (19.8) 15 (38.5) 0.03
Muscle wasting 29 (23.0) 7 (17.9) n.s.
Ataxia 25 (19.8) 8 (20.5) n.s.
Myoclonus 8 (6.3) 8 (20.5) 0.02
Multiple lipomatosis 0 11 (28.2) \0.000001
The table shows only the clinical features present in C20 % of m.3243A[G and/or m.8344A[G patients. Significance levels after Bonferroni’s
correction: P = 0.0033. Significant values are italicized, non-significant trends are in bold
J Neurol
123

An earlier revision [6] reported 111 published subjects,
with the following distribution for clue clinical features:
myopathy 53 %; epilepsy 50 %; recurrent strokes 48 %;
deafness 44 %; CPEO 28 %; dementia 27 %; ataxia 24 %;
diabetes mellitus 15 %; pigmentary retinopathy 15 %;
short stature 15 %; myoclonus 8 %; neuropathy 5 %;
gastrointestinal involvement, cardiomyopathy, extrapyra-
midal features 3 % each; lipomatosis 1 %; optic atrophy
1 %. Our results partly diverge from these data. For
instance, about 6 % of our m.3243A[G-positive subjects
were asymptomatic (mean age at last evaluation 28 years),
and hearing loss and diabetes (two common clinical sig-
natures of mitochondrial disorders) [5] were the most fre-
quent clinical features, followed by stroke-like episodes.
As somehow expected, CNS involvement (and especially
stroke-like episodes) could represent a negative prognostic
factor, since it was a feature shared by the deceased
patients. Heart involvement occurred in more than 30 % of
cases, confirming the need for complete cardiac screening
in these patients.
Three recent surveys are also available [1, 2, 7]. Ka-
uffman and coworkers studied 123 matrilineal relatives
from 45 families, including 45 fully symptomatic patients
with MELAS and 78 carrier relatives. Mutation carriers’
clinical and laboratory results were frequently abnormal,
confirming that the m.3243A[G mutation carriers have
multiple medical problems [2]. De Laat and coworkers [7]
studied 34 m.3243A[G families, observing a wide variety
of signs and symptoms, MIDD being far more prevalent
than MELAS [7]. Finally, Nesbitt and coworkers [1]
recently reported that 10 % of patients exhibited a classical
MELAS phenotype, 30 % had MIDD, 6 % MELAS/
MIDD, 2 % MELAS/PEO and 5 % MIDD/PEO overlap
syndromes; 6 % had PEO and other features of mito-
chondrial disease not consistent with another recognized
syndrome, whereas isolated sensorineural hearing loss
occurred in 3 % of the cases [1]. Many patients harboring
this mutation exhibited a clinical phenotype that did not
fall within accepted criteria for the currently recognized
classical mitochondrial syndromes [1]; our data reinforce
this concept. Furthermore, whereas the ‘‘MELAS’’ acro-
nym is convincing and useful to denote patients with his-
tological, biochemical, and/or molecular evidence of
mitochondrial disease who experience stroke-like episodes,
pure MIDD and PEO are very rare in our cohort of patients
with the m.3243A[G mutation.
Association analyses of the different clinical features
were not attempted in the above considered studies [1, 2, 6,
7], nor in other previous series [8–11]. Moreover, in order
to confirm our observation of male prevalence in MELAS,
we have tried to meta-analyze our data with those of the
large surveys mentioned above [1, 2, 6, 7]. Unfortunately,
this was not possible, because in those series the reported
data were not specifically designed to explore associations
between gender and phenotypes.
Here, we observed that hearing loss and diabetes were
mutually associated with a high statistical significance.
Therefore, the acronym MIDD may be useful to describe
the strong association between hearing loss and diabetes in
these patients. However, pure MIDD is very rare and this
acronym should not be used in contrast to MELAS: in fact,
hearing loss was associated with the presence of stroke-like
episodes and may be a clinical marker of multisystem
impairment. PEO was associated with heart involvement
and, less, with muscle weakness. Therefore, ‘‘MIDD’’ and
‘‘PEO’’ should not be considered reassuring phenotypic
labels, because in fact these patients may develop multi-
system complications.
Stroke-like episodes were present, at the mean age of
36 years, in about 40 % of our patients, who were therefore
defined ‘‘MELAS’’. Stroke-like episodes were strongly
associated with generalized seizures, cognitive involve-
ment, and hearing loss; therefore, these features might be
considered possible adjunctive manifestations of MELAS.
Lactate increase was associated with MELAS, confirming
the validity of the acronym. Heteroplasmy levels, muscle
histology (including the presence of SSVs), and other
clinical features such as migraine could not predict stroke-
like episodes. There is strong need for a better under-
standing of the genetic background and environmental
factors influencing the clinical picture and the prognosis in
mtDNA mutation carriers.
We observed, for the first time, that male gender could
represent a risk factor for the development of stroke-like
episodes in m.3243A[G patients in the Italian population.
Possible confounding factors (onset age, age at the last
control, disease duration, heteroplasmy levels) were
excluded. Even if this finding must be confirmed by spe-
cifically designed longitudinal studies, gender effect is not
a new concept in mitochondrial medicine. The higher
prevalence of Leber hereditary optic neuropathy (LHON)
in males is known [12]. This was recently ascribed to
multilayer cytoprotective effects induced by estrogens,
which could reduce the production of reactive oxygen
species, restore membrane potential, limit apoptotic cell
death, activate mitochondrial biogenesis, and improve
energetic metabolism in LHON cybrids [12]. Estrogen
receptor b is localized to the mitochondrial network of
human retinal ganglion cells with the unmyelinated portion
of their axons in the retinal nerve fiber layer, and their
expression is retained in the surviving retinal ganglion cells
of patients with LHON [12]. Interestingly, in some
instances MELAS is also associated with point mutations
in ND subunits of complex I, as with LHON, and the
complex I defect seems to be a prevalent feature in
MELAS, as recently shown in induced pluripotent stem
J Neurol
123

cells derived from fibroblasts carrying different hetero-
plasmic loads of m.3243A[G [13]. Noteworthy, in
MELAS patients, the highest proportion of mutated
mtDNA was observed in the walls of the leptomeningeal
and cortical blood vessels (in all brain regions), supporting
the hypothesis of vascular mitochondrial dysfunction in the
pathogenesis of stroke-like episodes [14]. Estrogen recep-
tors have been localized to multiple cell types within the
CNS, including neurons, astrocytes, microglia, and endo-
thelial cells [15]. Estrogens, particularly 17b-estradiol, are
powerful neuroprotective agents in animal models of
cerebral ischemia [15]. A better elucidation of the complex
network linking mitochondrial dysfunction, apoptosis,
estrogen effects, and stroke-like episodes may hold prom-
ises for a therapeutic use for estrogen-like molecules in this
devastating disorder.
Acknowledgments This work was supported by Telethon (grant
number GUP09004). The authors are grateful to the Italian patient
association ‘‘Mitocon’’ for the informatic support. The work of the
‘‘Besta’’ group of Milan was supported by the Pierfranco and Luisa
Mariani Foundation Italy, Ricerca 2000; Fondazione Giuseppe
Tomasello ONLUS and Fondazione Telethon-Italy grants GGP07019
and GPP10005. The work of the University of Milan group was
supported by Associazione Amici del Centro Dino Ferrari, by the
Telethon project GTB07001ER, and by a Telethon grant ‘‘Network of
Genetic Biobanks’’ no GTB07001.
Conflicts of interest The authors declare no conflicts of interest.
References
1. Nesbitt V, Pitceathly RD, Turnbull DM, Taylor RW, Sweeney
MG, Mudanohwo EE, Rahman S, Hanna MG, McFarland R
(2013) The UK MRC mitochondrial disease patient cohort study:
clinical phenotypes associated with the m.3243A[G mutation—
implications for diagnosis and management. J Neurol Neurosurg
Psychiatry 84:936–938
2. Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Bat-
tista V, Koenigsberger DY, Pascual JM, Sano M, Hirano M,
DiMauro S, Shungu DC, Mao X, De Vivo DC (2009) Protean
phenotypic features of the A3243G mitochondrial DNA mutation.
Arch Neurol 66:85–91
3. Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA,
Thorburn DR (2002) Diagnostic criteria for respiratory chain
disorders in adults and children. Neurology 59:1406–1411
4. Mancuso M, Orsucci D, Angelini CI, Bertini E, Carelli V, Comi
GP, Minetti C, Moggio M, Mongini T, Servidei S, Tonin P,
Toscano A, Uziel G, Bruno C, Caldarazzo Ienco E, Filosto M,
Lamperti C, Martinelli D, Moroni I, Musumeci O, Pegoraro E,
Ronchi D, Santorelli FM, Sauchelli D, Scarpelli M, Sciacco M,
Spinazzi M, Valentino ML, Vercelli L, Zeviani M, Siciliano G
(2013) Phenotypic heterogeneity of the 8344A[G mtDNA
‘‘MERRF’’ mutation. Neurology 80:2049–2054
5. Mancuso M, Orsucci D, Coppede F, Nesti C, Choub A, Siciliano
G (2009) Diagnostic approach to mitochondrial disorders: the
need for a reliable biomarker. Curr Mol Med 9:1095–1107
6. Chinnery PF, Howell N, Lightowlers RN, Turnbull DM (1997)
Molecular pathology of MELAS and MERRF. The relationship
between mutation load and clinical phenotypes. Brain 120(Pt
10):1713–1721
7. de Laat P, Koene S, van den Heuvel LP, Rodenburg RJ, Janssen
MC, Smeitink JA (2012) Clinical features and heteroplasmy in
blood, urine and saliva in 34 Dutch families carrying the
m.3243A[G mutation. J Inherit Metab Dis 35:1059–1069
8. Chae JH, Hwang H, Lim BC, Cheong HI, Hwang YS, Kim KJ
(2004) Clinical features of A3243G mitochondrial tRNA muta-
tion. Brain Dev 26:459–462
9. Jeppesen TD, Schwartz M, Frederiksen AL, Wibrand F, Olsen
DB, Vissing J (2006) Muscle phenotype and mutation load in 51
persons with the 3243A[G mitochondrial DNA mutation. Arch
Neurol 63:1701–1706
10. Karppa M, Herva R, Moslemi AR, Oldfors A, Kakko S, Majamaa
K (2005) Spectrum of myopathic findings in 50 patients with the
3243A[G mutation in mitochondrial DNA. Brain 128:1861–1869
11. Koga Y, Akita Y, Takane N, Sato Y, Kato H (2000) Heteroge-
neous presentation in A3243G mutation in the mitochondrial
tRNA(Leu(UUR)) gene. Arch Dis Child 82:407–411
12. Giordano C, Montopoli M, Perli E, Orlandi M, Fantin M, Ross-
Cisneros FN, Caparrotta L, Martinuzzi A, Ragazzi E, Ghelli A,
Sadun AA, d’Amati G, Carelli V (2011) Oestrogens ameliorate
mitochondrial dysfunction in Leber’s hereditary optic neuropa-
thy. Brain 134:220–234
13. Hamalainen RH, Manninen T, Koivumaki H, Kislin M, Oto-
nkoski T, Suomalainen A (2013) Tissue- and cell-type-specific
manifestations of heteroplasmic mtDNA 3243A[G mutation in
human induced pluripotent stem cell-derived disease model. Proc
Natl Acad Sci USA 110:E3622–E3630
14. Betts J, Jaros E, Perry RH, Schaefer AM, Taylor RW, Abdel-All
Z, Lightowlers RN, Turnbull DM (2006) Molecular neuropa-
thology of MELAS: level of heteroplasmy in individual neurones
and evidence of extensive vascular involvement. Neuropathol
Appl Neurobiol 32:359–373
15. Schreihofer DA, Ma Y (2013) Estrogen receptors and ischemic
neuroprotection: who, what, where, and when? Brain Res
1514:107–122
J Neurol
123

Phenotypic heterogeneity of the8344A.G mtDNA “MERRF” mutation
ABSTRACT
Objectives: Myoclonic epilepsy with ragged-red fibers (MERRF) is a rare mitochondrial syndrome,mostly caused by the 8344A.G mitochondrial DNA mutation. Most of the previous studies havebeen based on single case/family reports or series with few patients. The primary aim of this studywas the characterization of a large cohort of patients with the 8344A.G mutation. The second-ary aim was revision of the previously published data.
Methods: Retrospective, database-based study (Nation-wide Italian Collaborative Network ofMitochondrial Diseases) and systematic revision.
Results: Forty-two patients carrying the mutation were identified. The great majority did not havefull-blownMERRF syndrome. Myoclonus was present in 1 of 5 patients, whereas myopathic signsand symptoms, generalized seizures, hearing loss, eyelid ptosis, and multiple lipomatosis repre-sented the most common clinical features. Some asymptomatic mutation carriers have also beenobserved. Myoclonus was more strictly associated with ataxia than generalized seizures in adult8344A.G subjects. Considering all of the 321 patients so far available, including our datasetand previously published cases, at the mean age of approximately 35 years, the clinical picturewas characterized by the following signs/symptoms, in descending order: myoclonus, muscleweakness, ataxia (35%–45% of patients); generalized seizures, hearing loss (25%–34.9%);cognitive impairment, multiple lipomatosis, neuropathy, exercise intolerance (15%–24.9%);and increased creatine kinase levels, ptosis/ophthalmoparesis, optic atrophy, cardiomyopathy,muscle wasting, respiratory impairment, diabetes, muscle pain, tremor, migraine (5%–14.9%).
Conclusions: Our results showed higher clinical heterogeneity than commonly thought. Moreover,MERRF could be better defined as a myoclonic ataxia rather than a myoclonic epilepsy.Neurology� 2013;80:2049–2054
GLOSSARYCK5 creatine kinase;MELAS5mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes;MERRF5myoclonicepilepsy with ragged-red fibers; mtDNA 5 mitochondrial DNA.
Myoclonic epilepsy with ragged-red fibers (MERRF) is a mitochondrial syndrome, originallydescribed in 19801 and mostly characterized by generalized seizures, myoclonus, and ataxia, withvariable onset and associated with various mitochondrial DNA (mtDNA) point mutations, themost frequent of which is the 8344A.G change in the gene encoding transfer RNA lysine.2 Earlyreports showed that the 8344A.G transition was highly correlated to the MERRF phenotype,3
although it can also be associated with other phenotypes, for instance Leigh disease, or myopathywith truncal lipomas.4 The absence of the mutation in some typical MERRF patients suggested
Michelangelo Mancuso,MD, PhD
Daniele Orsucci, MDCorrado Angelini, MD,
PhDEnrico Bertini, MD, PhDValerio Carelli, MD, PhDGiacomo Pietro Comi,
MDCarlo Minetti, MDMaurizio Moggio, MD,
PhDTiziana Mongini, MD,
PhDSerenella Servidei, MD,
PhDPaola Tonin, MD, PhDAntonio Toscano, MD,
PhDGraziella Uziel, MD, PhDClaudio Bruno, MD,
PhDElena Caldarazzo Ienco,
MDMassimiliano Filosto,
MD, PhDCostanza Lamperti, MDDiego Martinelli, MDIsabella Moroni, MDOlimpia Musumeci, MDElena Pegoraro, MDDario Ronchi, PhDFilippo Maria Santorelli,
MDDonato Sauchelli, MDMauro Scarpelli, MD
Author list continued on next page
From the Neurological Clinic (M. Mancuso, D.O., E.C.I., G.S.), University of Pisa; Neurological Clinic (C.A., E.P., M. Spinazzi), University ofPadova, and IRCCS S. Camillo, Venice; Bambino Gesù Hospital (E.B., D.M.), Rome; IRCCS Istituto di Scienze Neurologiche and Departmentof Biomedical and Neuromotor Sciences (V.C., M.L.V.), University of Bologna; Dino Ferrari Centre (G.P.C., D.R.), Neuroscience Section,Department of Pathophysiology and Transplantation (DEPT), University of Milan, Neurology Unit, IRCCS Foundation Ca’ Granda OspedaleMaggiore Policlinico, Milan; Neuropediatric and Muscle Disorders Unit (C.M., C.B.), University of Genoa and G. Gaslini Institute, Genoa;Neuromuscular Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico (M. Moggio, M. Sciacco), Milano, Dino Ferrari CentreUniversity of Milan; Department of Neuroscience (T.M., L.V.), University of Turin; Institute of Neurology (S.S., D.S.), Catholic University,Rome; Neurological Clinic (P.T., M. Scarpelli), University of Verona; Department of Neurosciences (A.T., O.M.), University of Messina; ChildNeurology Unit (G.U., I.M.) and Unit of Molecular Neurogenetics (C.L., M.Z.), The Foundation “Carlo Besta” Institute of Neurology–IRCCS,Milan; University Hospital Spedali Civili (M.F.), Neurological Clinic, Brescia; and IRCCS Stella Maris (F.M.S.), Pisa, Italy.
Go to Neurology.org for full disclosures. Funding information and disclosures deemed relevant by the authors, if any, are provided at the end of the article.
© 2013 American Academy of Neurology 2049

that other mutations may produce the samephenotype,4 including the 8356T.C, whichalso affects the transfer RNA lysine gene.5
MERRF is very rare, as is the 8344A.Gmutation (,0.5/100,000, 4% of all pathogenicmtDNA mutations).6,7 Therefore, most of theprevious studies describing the clinical featuresof MERRF patients and/or subjects carryingthe 8344A.G mutation have been based onsingle case/family reports8 or case series withvery few patients.9 Some early reviews of liter-ature are also available,10 but recent data arelacking, and clinical heterogeneity is probablyhigher than previously suspected.11
The primary aim of this study was the clin-ical characterization of a cohort of mitochon-drial patients with the 8344A.G mutation,registered in the database of the Nation-wideItalian Collaborative Network of Mitochon-drial Diseases. The secondary aim was thethorough revision of previously reported clin-ical data about this mutation.
METHODS We reviewed the clinical, morphologic, and labo-
ratory features of all the A8344A.G-positive cases contained in
the database of the Nation-wide Italian Collaborative Network of
Mitochondrial Diseases.
A systematic revision of previously reported data has also been
performed. Individuals harboring the 8344A.G mutation were
identified through a PubMed search of the scientific literature
(September 2012). Additional articles were identified in other
databases such as OMIM (http://www.ncbi.nlm.nih.gov/omim)
and MITOMAP (http://www.mitomap.org). Studies with an
incomplete clinical description, as well as those reporting Italian
patients who could be included in our cohort, were excluded.
Through this approach, we identified 75 articles (shown in the
e-references on the Neurology® Web site at www.neurology.org).
Data were expressed as means 6 SD. The numeric data were
compared by unpaired 2-tailed Student t test, or correlated by
Spearman rank correlation test; proportions were compared using
the x2 test. Categorical data were associated by means of the Fisher
exact test. Bonferroni correction for multiple tests was utilized when
appropriate. A p value ,0.05 was considered as significant. Data
analysis was performed using MedCalc version 7.3.0.1.
Standard protocol approvals, registrations, and patientconsents. The establishment of the database, and its use for scien-
tific purposes, was approved by the local ethical committees of all
the involved centers, which obtained written consent from each
patient, in accordance with the ethical standards of the 1964 Dec-
laration of Helsinki.
RESULTS The 8344A>G mutation in our database. Atotal of 42 patients harboring the 8344A.Gmutation(25 patients from 10 families and 17 other subjects)were identified among the 1,086 mitochondrial pa-tients (3.9%) present in our database (May 1, 2012).They represented the 12.4% (42/340) of all indi-viduals with mtDNA point mutations. Twenty-two
of 42 (52.4%) were male, and 15 of 42 patients(35.7%) had childhood onset (younger than 16 years).Of note, among the 17 subjects without molecularlyconfirmed relatives, only 5 had a family history poten-tially indicative of mtDNA-related disease.
The clinical picture was not available for 3 patients,who have not been considered in the following analysis.Four of 39 subjects (10.3%) were asymptomatic muta-tion carriers (age at last evaluation 37.0 6 9.9 years;range 26–50). Heteroplasmy levels of asymptomaticcarriers were 56.7% 6 11.5% in blood and 55.8% 6
25.1% in urine, which were not different from thesymptomatic patients.
At the last evaluation, 38 of 39 subjects were adult(older than 16 years). The cardinal clinical featureof MERRF syndrome is deemed to be “myoclonicepilepsy.”12 However, Fisher exact test revealed noassociation between myoclonus and generalized seiz-ures in the 38 adult 8344A.Gmutation carriers withknown clinical picture; contrariwise, myoclonus andcerebellar ataxia were associated (table 1).
The mean age at onset of the 35 symptomatic pa-tients was 30.16 18.4 years (range 0–66; 11/35 withclinical onset before age 16). Table e-1 summarizes theclinical features at onset.
Disease progression. At the last evaluation, mean ageand disease duration were 45.6 6 15.3 years (range5–72), and 21.6 6 15.3 years (range 2–59), respec-tively. A 5-year-old boy had a complex syndromecharacterized by consciousness disturbance, cognitiveimpairment, pyramidal signs, tremor, swallowingimpairment, lactic acidosis, facial dysmorphism, andcataract. The clinical features of the 34 adult patientsare summarized in the figure and described below.
Monica Sciacco, MDMarco Spinazzi, MDMaria Lucia Valentino,
MDLiliana Vercelli, MDMassimo Zeviani, MD,
PhDGabriele Siciliano, MD,
PhD
Correspondence toDr. Mancuso:[email protected]
Supplemental data atwww.neurology.org
Table 1 Is myoclonus associated with epilepticseizure and/or ataxia in 8344A>Gadult carriers?
Myoclonus:no (n 5 31)
Myoclonus:yes (n 5 7) Total
Seizuresa
No 23 3 26
Yes 8 4 12
Ataxiab
No 27 3 30
Yes 4 4 8
aNo association between myoclonus and epilepsy in adultsubjects with the 8344A.G mutation (including asymp-tomatic subjects). Total n 5 38, p 5 0.176 (Fisher exacttest), which is not significant (significance level 0.025 afterBonferroni correction).b Relationship between myoclonus and ataxia in adultsubjects with the 8344A.G mutation. Total n 5 38, p 5
0.0245 (Fisher exact test), which is significant after thecorrection (significance level 0.025).
2050 Neurology 80 May 28, 2013

Multiple lipomatosis was present in 11 of 34 patients(32.4%), only 2 of whom had isolated lipomatosis withno myopathic complaints (6 cases) and/or CNS involve-ment (5 cases). Of the 4 patients with isolated lipomato-sis at onset, 2 developed myopathy but none had CNSsymptoms (mean follow-up 20.0 years, range 3–48).
Nineteen of 34 patients (55.9%) showed clinicalCNS involvement at the last evaluation (generalized seiz-ures 12/34 [35.3%], cerebellar ataxia 8/34 [23.5%],myoclonus 8/34 [23.5%], cognitive involvement 2/34[5.9%], stroke-like episodes 1/34 [2.9%]). Fourteen ofthese 19 patients also had myopathic complaints. Nei-ther age at onset, age at last evaluation, nor disease dura-tion could predict the development of CNS symptoms.
Signs and symptoms of neuromuscular dysfunctionwere present in 26 of 34 patients (76.5%): muscle weak-ness 20/34 (58.8%), exercise intolerance 15/34 (44.1%),increased creatine kinase (CK) levels 15/34 (44.1%),eyelid ptosis 10/34 (29.4%) with ophthalmoparesis in2 cases (5.9%), muscle wasting 7/34 (20.6%), muscle
pain 6/34 (17.6%), cardiomyopathy with arrhythmia4/34 (11.8%), arrhythmia without cardiomyopathy2/34 (5.9%), peripheral neuropathy 5/34 (14.7%),swallowing impairment 1/34 (2.9%), and respiratory(ventilatory) impairment 1/34 (2.9%). Neither age atonset, age at last evaluation, nor disease duration sig-nificantly differed between patients with or withoutclinically evident myopathic involvement.
Moreover, at the last clinical evaluation, 12/34 pa-tients had hearing loss (35.3%), 4/34 diabetes mellitus(11.8%), 3/34 migraine (8.8%), 2/34 hypothyroidism(5.9%), 1/34 cataract, 1/34 isolated hypogonadism,1/34 psychiatric involvement, and 1/34 gastrointes-tinal dysmotility (2.9% each).
Some patients with mild phenotypes were reported,including a 50-year-old patient with eyelid ptosis, 263- and 28-year-old subjects with isolated multiplelipomatosis, and a 27-year-old patient with isolatedhypogonadism.
Four patients, all with CNS involvement (includ-ing generalized seizures in all cases and myoclonus in2 cases), are deceased (mean age 37.5 6 18.5 years,range 20–54; disease duration 9.8 6 4.6 years, range5–15) because of heart failure (2 cases), respiratoryfailure, or intractable lactic acidosis (1 case each).
Basal lactate levels were increased in 65.0% of thecases, but neither CNS involvement nor other clinicalfeatures, including age at onset, were related to thislaboratory marker.
The 8344A>G mutation: Neurodiagnostic features.
Recent (,5 years before the last examination)MRI datawere available for 17 patients with the 8344A.Gmuta-tion, and were normal in 10 (58.8%). White matterabnormalities were present in 4 of 17 patients (23.5%),one of whom had no clinically evident CNS involve-ment; 2 patients also had cortical/subcortical atrophy.Other MRI findings included periaqueductal lesions,cysts or vacuolated lesions, brainstem atrophy, andstroke-like lesions (1/17 [5.9%] each). No definitivedata regarding proton spectroscopy MRI are available.
The 8344A>G mutation: Histologic findings. Musclebiopsy findings were available for 27 patients. Ragged-red fibers were present in 26 patients (96.3%), including4 patients with no clinically evident myopathic involve-ment (one of whomwas last seen at age 37). Cytochromec oxidase-negative fibers were present in 20 cases(74.1%), but failed to predict neuromuscular involve-ment. Lipid storage was noted in 5 biopsies (18.5%),and was neither associated with multiple lipomatosis norwith other clinical features. Strongly succinate dehydro-genase-reactive blood vessels, more typical of MELAS(mitochondrial encephalomyopathy, lactic acidosis, andstroke-like episodes) syndrome, were found in one8344A.G patient with myopathy, eyelid ptosis, andmigraine, but not in the patient with stroke-like episodes.
Figure Clinical features of the symptomatic8344A>G patients older than 16 yearswhen evaluated
For details, see text. CK 5 creatine kinase; imp 5 impair-ment; intol 5 intolerance.
Neurology 80 May 28, 2013 2051

Heteroplasmy levels. Heteroplasmy levels of the8344A.G were available in muscle for 22 patients(mutation load 67.0% 6 16.1%), in blood for 16patients (mutation load 57.6% 6 16.1%), and inurinary sediment for 11 patients (mutation load59.4% 6 16.3%). No correlation was observedbetween these levels and age at onset. The hetero-plasmy levels in muscle, blood, and urinary sedimentwere not mutually related, but a positive trend wasobtained by comparing the levels in blood and uri-nary sediment (r 5 0.734, p 5 0.052 [significancelevel after Bonferroni correction 0.017]).
Only a nonsignificant trend was obtained by asso-ciating clinically evident myopathic involvement andmuscle mutation load (69.6%6 15.0% in 17 patientswith clinical myopathy vs 58.0% 6 17.9% in 5 pa-tients without clinically evident myopathy [p5 0.16]).
Systematic revision of the literature. Table e-2 shows theclinical data of our 39 mutation carriers with knownclinical features, as well as those of 282 previouslyreported 8344A.G-positive individuals (male/femaleratio 0.85; mean age at the last evaluation 34.26 19.1years) (see e-references). The asymptomatic subjectswere included for both groups.
Considering all 321 patients so far available, includ-ing our dataset and previously published cases, at themean age of approximately 35 years, the clinical picturewas characterized by the following signs/symptoms, indescending order: myoclonus, muscle weakness, ataxia(35%–45% of patients); generalized seizures, hearing loss(25%–34.9%); cognitive impairment, multiple lipoma-tosis, neuropathy, exercise intolerance (15%–24.9%);and increased CK levels, ptosis/ophthalmoparesis, opticatrophy, cardiomyopathy, muscle wasting, respiratoryimpairment, diabetes, muscle pain, tremor, migraine(5%–14.9%).
Increased CK levels and exercise intolerance weremore frequently reported in our patients than in thepublished cases (p , 0.0001 and p , 0.0005, respec-tively). However, in the great majority of the previousreports, CKwas only mentioned when increased, but itis unclear in how many cases this test was performedand found normal or was not performed at all. More-over, exercise intolerance is a subtle symptom thatcould be missed if not specifically assessed. Again, itis unclear whether this sign was systematically searchedfor in the previous studies. Ataxia and myoclonus wereless frequent in our own series (p , 0.005).
DISCUSSION We present here a retrospective studyon 42 8344A.G carriers registered in the Italiannationwide database of mitochondrial disease patients.Although most of the previous studies were based onsingle or small series of cases/families,8,9 an early system-atic literature revision10 reported 55 published subjects,
with the following distribution for clue clinical features:recurrent strokes 1%, ophthalmoparesis 6%, diabetesmellitus 3%, deafness 39%, dementia 25%, epilepsy43%, myopathy 70%, lipomatosis 8%, optic atrophy13%, neuropathy 24%, ataxia 50%, and myoclonus61%. Our results partly diverge from these data. Forinstance, 10% of our 8344A.G-positive subjects wereasymptomatic (mean age at last evaluation 37 years),and myoclonus, which was deemed as the most fre-quent feature (61%),10 was only about 20% in ourseries. Other “typical” features, such as ataxia and gen-eralized epileptic seizures, were found in approximately20% and 30% in our cases. Generalized epilepsy seemsto be a negative prognostic factor because it was a fea-ture common to our 4 deceased patients. The frequencyof cognitive impairment and stroke-like episodes waslower in our series than in previous studies.
Clinically evident myopathy was previously reportedin approximately 70% of 8344A.G carriers,10 and thiswas confirmed in our study. The most common fea-tures were muscle weakness, exercise intolerance, and,unexpectedly, increased CK levels, which is differentfrom what is commonly thought for the great majorityof mitochondrial diseases.13 Eyelid ptosis occurred in25% of our cases, whereas ophthalmoparesis was rarer(5%). Heart involvement occurred in about 15% ofcases, confirming the need for complete cardiac screen-ing in these patients. Peripheral neuropathy was lessfrequent in our cohort (,15%) than previouslyobserved for the 8344A.G mutations10 or for othermitochondrial diseases.14,15 Multiple lipomatosis wascommon (30%) in our cohort, far more than previ-ously reported,10 as were hearing loss and diabetesmellitus, 2 common clinical signatures of mitochon-drial multisystemic disorders.13
The MERRF acronym suggests that myoclonus andepilepsy are tightly associated, occurring together in thesame individual in most of the cases.12 However, in ouradult 8344A.G carriers “myoclonus” was more fre-quently linked to “ataxia” than to “generalized seizures,”supporting the idea that the cerebellum may have animportant role in the genesis of cortical reflex myoclo-nus.16 Of note, Purkinje cells and neurons of the den-tate nucleus and inferior olivary nuclei seem to have aspecial vulnerability to the effects of the A8344A.Gmutation (and to POLGmutations)17 as compared withother mtDNA mutations (i.e., 3243A.G).17 Based onour results, MERRF could be better defined as a myo-clonic ataxia16 rather than a myoclonic epilepsy. Thisconclusion could not be reached in previous studiesbecause the clinical picture of each and every case wasnot available in each and every report. Furthermore, insome cases the clinical features were unclear; for in-stance, the term myoclonic epilepsy can be confoundingif a detailed clinical description is missing (i.e., gener-alized seizures are present or not, etc.).
2052 Neurology 80 May 28, 2013

We failed to find clear predictive factors, includingage at onset, disease duration, age at last evaluation, lac-tate levels, biopsy findings, or heteroplasmy levels, forthe development of CNS or myopathic symptoms andsigns in our cohort, possibly reflecting the well-knownvariability of the mtDNA-related diseases. Further-more, heteroplasmy levels of asymptomatic carrierswere not significantly different from the symptomaticpatients. This on one hand precluded further analysis(e.g., threshold estimates), and on the other highlightsthe strong need for a better understanding of thegenetic background and environmental factors capableof influencing the clinical picture and the prognosis inmtDNA mutation carriers. Useful biomarkersare strongly needed as well.13 Ragged-red fibers werepresent in most of our patients as were cytochrome coxidase-negative fibers. Lipid storage was not associatedwith multiple lipomatosis, disproving the idea that pro-liferation of adipose tissue and lipid storage myopathyare correlated.18 There was only a nonsignificant trendfor the association between clinically evident myopathicinvolvement and muscle mutation load. Importantly,differently from what has been previously reported in aseries of 9 patients,19 muscle and blood heteroplasmylevels did not correlate.
Herein we showed that 8344A.G phenotypesare a continuum from asymptomatic to lethal mul-tisystem diseases; between these extremes there areall the possible expressivity degrees. This multicenterstudy provides new information about clinical phe-notype, natural history, and outcome of 8344A.Gcarriers that can be useful in their counseling. Inter-estingly, the majority of our patients (approximately80%) did not show a full-blown MERRF clinicalpicture. Myoclonus was present in 1 of 5 patients,whereas common clinical features included myopathicsigns and symptoms, generalized seizures, hearing loss,eyelid ptosis, and multiple lipomatosis. Interestingly,in adult patients, myoclonus was linked with ataxia
more than with generalized epilepsy, so that theMERRFacronym could well be replaced by MARRF. Althoughlarger prospective studies are needed to obtain a completepicture of the natural history of this disease, the datapresented in table e-2 led us to propose a new flowchartfor the follow-up of 8344A.G patients (table 2).
AUTHOR CONTRIBUTIONSAll the authors made substantive intellectual contributions to the data-
base development, data collection, and interpretation.
ACKNOWLEDGMENTThe authors are grateful to the Italian patient association Mitocon for the
informatic support. The work of the Besta group of Milan was supported
by the Pierfranco and Luisa Mariani Foundation Italy, Ricerca 2000; Fon-
dazione Giuseppe Tomasello ONLUS and Fondazione Telethon–Italy
grants GGP07019 and GPP10005. The work of the group of University
of Milan was supported by Associazione Amici del Centro Dino Ferrari, by
the Telethon project GTB07001ER, and by a Telethon grant “Network of
Genetic Biobanks” no. GTB07001.
STUDY FUNDINGThis work was supported by Telethon (grant GUP09004).
DISCLOSUREThe authors report no disclosures relevant to the manuscript. Go to
Neurology.org for full disclosures.
Received December 10, 2012. Accepted in final form February 15, 2013.
REFERENCES1. Fukuhara N, Tokiguchi S, Shirakawa K, Tsubaki T. Myoc-
lonus epilepsy associated with ragged-red fibres (mitochon-
drial abnormalities): disease entity or a syndrome? Light- and
electron-microscopic studies of two cases and review of liter-
ature. J Neurol Sci 1980;47:117–133.
2. Noer AS, Sudoyo H, Lertrit P, et al. A tRNA(Lys) muta-
tion in the mtDNA is the causal genetic lesion underlying
myoclonic epilepsy and ragged-red fiber (MERRF) syndrome.
Am J Hum Genet 1991;49:715–722.
3. Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW,
Wallace DC. Myoclonic epilepsy and ragged-red fiber disease
(MERRF) is associated with a mitochondrial DNA tRNA
(Lys) mutation. Cell 1990;61:931–937.
Table 2 Suggested follow-up scheme for the 8344A>G patients
Time interval between assessments
Neurologic examination with CK and lactate 6 mo
Cardiologic evaluation with echocardiography and ECG 12 mo (or 3–6 mo after the development of heart disease)
Ophthalmologic evaluation with fundoscopy 18 mo (or less in symptomatic patients)
Respiratory function tests 24 mo (or 3–6 mo after the development of respiratory involvement)
Swallowing evaluation 24 mo (or 6 mo after the development of swallowing impairment)
Thyroid hormones, glucose, liver and kidney function,complete blood count
24–36 mo (or less in some instances, e.g., treatment with newantiepileptic drugs)
Audiologic assessment 24–36 mo (or less in symptomatic patients)
Psychiatric evaluation 24–36 mo (or less in patients with clinically apparent psychiatricinvolvement)
Neuropsychological tests 36–48 mo (or less in patients with clinically apparent cognitiveimpairment)
Neurology 80 May 28, 2013 2053

4. Silvestri G, Ciafaloni E, Santorelli FM, et al. Clinical fea-
tures associated with the A/G transition at nucleotide
8344 of mtDNA (“MERRF mutation”). Neurology 1993;
43:1200–1206.
5. Silvestri G, Moraes CT, Shanske S, Oh SJ, DiMauro S. A
new mtDNA mutation in the tRNA(Lys) gene associated
with myoclonic epilepsy and ragged-red fibers (MERRF).
Am J Hum Genet 1992;51:1213–1217.
6. Remes AM, Karppa M, Moilanen JS, et al. Epidemiology of
the mitochondrial DNA 8344A.G mutation for the myoc-
lonus epilepsy and ragged red fibres (MERRF) syndrome.
J Neurol Neurosurg Psychiatry 2003;74:1158–1159.
7. Schaefer AM, McFarland R, Blakely EL, et al. Prevalence of
mitochondrial DNA disease in adults. Ann Neurol 2008;63:
35–39.
8. Wiedemann FR, Bartels C, Kirches E, Mawrin C,
Wallesch CW. Unusual presentations of patients with
the mitochondrial MERRF mutation A8344G. Clin Neu-
rol Neurosurg 2008;110:859–863.
9. Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Silvado CE,
Werneck LC. MERRF: clinical features, muscle biopsy
and molecular genetics in Brazilian patients. Mitochon-
drion 2011;11:528–532.
10. Chinnery PF, Howell N, Lightowlers RN, Turnbull DM.
Molecular pathology of MELAS and MERRF: the rela-
tionship between mutation load and clinical phenotypes.
Brain 1997;120(pt 10):1713–1721.
11. Graf WD, Sumi SM, Copass MK, et al. Phenotypic het-
erogeneity in families with the myoclonic epilepsy and
ragged-red fiber disease point mutation in mitochondrial
DNA. Ann Neurol 1993;33:640–645.
12. DiMauro S, Hirano M, Kaufmann P, et al. Clinical fea-
tures and genetics of myoclonic epilepsy with ragged red
fibers. Adv Neurol 2002;89:217–229.
13. Mancuso M, Orsucci D, Coppede F, Nesti C, Choub A,
Siciliano G. Diagnostic approach to mitochondrial disorders:
the need for a reliable biomarker. Curr Mol Med 2009;9:
1095–1107.
14. Girlanda P, Toscano A, Nicolosi C, et al. Electrophysiological
study of neuromuscular system involvement in mitochondrial
cytopathy. Clin Neurophysiol 1999;110:1284–1289.
15. Mancuso M, Piazza S, Volpi L, et al. Nerve and muscle
involvement in mitochondrial disorders: an electrophysio-
logical study. Neurol Sci 2012;33:449–452.
16. Shibasaki H, Thompson PD. Milestones in myoclonus.
Mov Disord 2011;26:1142–1148.
17. Lax NZ, Hepplewhite PD, Reeve AK, et al. Cerebellar
ataxia in patients with mitochondrial DNA disease: a molec-
ular clinicopathological study. J Neuropathol Exp Neurol
2012;71:148–161.
18. Munoz-Malaga A, Bautista J, Salazar JA, et al. Lipomatosis,
proximal myopathy, and the mitochondrial 8344 mutation:
a lipid storage myopathy? Muscle Nerve 2000;23:538–542.
19. Larsson NG, Tulinius MH, Holme E, et al. Segregation
and manifestations of the mtDNA tRNA(Lys) A/G
(8344) mutation of myoclonus epilepsy and ragged-red
fibers (MERRF) syndrome. Am J Hum Genet 1992;51:
1201–1212.
Neurology® Launches Subspecialty Alerts by E-mail!Customize your online journal experience by signing up for e-mail alerts related to your subspecialtyor area of interest. Access this free service by visiting http://www.neurology.org/site/subscriptions/etoc.xhtml or click on the “E-mail Alerts” link on the home page. An extensive list of subspecialties,methods, and study design choices will be available for you to choose from—allowing you priorityalerts to cutting-edge research in your field!
Commenting Online is Easier Now with WriteClick™
Have a comment on a recent Neurology® article you would like to share? Now it is easier and moreconvenient. Neurology.org has launched WriteClick on the home page and sidebars of each article toencourage remarks and debate among users.
WriteClick is restricted to comments about studies published in Neurology within the last eightweeks.
Learn more at http://www.neurology.org/letters
2054 Neurology 80 May 28, 2013

Mitochondria — from the Greek mitos (thread-like) and khondros (grain or granule) — are bacterium-sized organelles found in all nucleated cells. They have a myr-iad of functions, but it is fair to say that what sets them apart is their role in energy production. To reiterate an oft-used cliché, mitochondria are the ‘powerhouses of the cell’ because they generate more than 90% of a typical cell’s ATP, which is the ‘energy currency’ of the cell. Mitochondria contain their own DNA (mitochon-drial DNA (mtDNA)), which is linked to their origin as eukaryotic endosymbionts1. Unlike nuclear DNA (nDNA), mtDNAs are maternally inherited and are present in multiple copies per cell; the number varies according to the bioenergetic needs of each particular tissue and can range over three orders of magnitude, depending on the cell type2. As such, mitochondrial dis-eases caused by mutations in mtDNA follow the laws of population genetics. This is a crucial distinction from diseases that follow Mendelian genetics and underlies many of the unique, and often perplexing, features of these disorders.
Over the past 25 years, the identification of patho-genic mtDNA mutations has evolved in parallel with advances in sequencing and cell biology technologies. The first decade was mainly devoted to identifying new pathogenic mutations associated with various syndromes (BOX 1) using standard sequencing tech-niques. This effort, which is now based on automated microarray-based sequencing and high-throughput sequencing strategies, was aided enormously by the use of cybrid technology (BOX 2). Beginning in 1995, mutations in nuclear genes affecting oxidative phos-phorylation (OXPHOS) function were identified at an
escalating pace initially on the basis of linkage analysis and homozygosity mapping, then on monochromosomal transfer techniques, gene complementation studies and sequencing of candidate genes, and more recently on scanning entire patient genomes through whole-exome sequencing3. These studies have revealed that mitochondrial disorders can result from mutations not only in genes encoding subunits of the OXPHOS complexes but also in genes required for their trans-lation and assembly, for providing the proper lipid milieu and for determining the spatial and temporal relationships of the organelles to their subcellular envi-ronment (‘mitodynamics’). However, as mtDNA muta-tions are now being linked to several complex traits, we need to improve our understanding of mitochondrial genetics at the same time as learning more about the nuclear genome.
In addition to being of interest to scientists, mtDNA-related diseases may pose a public health concern. Epidemiological studies of mtDNA mutations in adults have shown a minimum prevalence of approximately 1 in 5,000, making mitochondrial diseases among the most common genetic disorders and a major burden for society4.
There have been several recent excellent reviews describing the role of mtDNA mutations in disease5–7, and so we discuss primary pathogenic mtDNA mutations only in broad outline to demonstrate some of the unique aspects of mitochondrial genetics and func-tion. We focus on more speculative roles of mtDNA mutations — both germline and somatic — in complex phenotypes, such as ageing, neurodegenerative diseases and cancer.
1Department of Neurology, Columbia University Medical Center, 630 West 168th Street, New York, New York 10032, USA.2Department of Genetics and Development, Columbia University Medical Center, 701 West 168th Street, New York, New York 10032, USA.Correspondence to E.A.S. e‑mail: [email protected]:10.1038/nrg3275
Microarray-based sequencingDNA sequencing based on the ability of the target DNA to hybridize to an ordered set of defined oligonucleotides immobilized on a chip.
Monochromosomal transferTypically, the transfer of individual human chromosomes (or parts of chromosomes) to rodent cells in order to identify and map human genes responsible for disease.
MitodynamicsThe morphological properties of mitochondria (for example, fusion, fission, distribution, anchorage, positioning). These properties can change both in space and in time within cells.
Human mitochondrial DNA: roles of inherited and somatic mutationsEric A. Schon1,2, Salvatore DiMauro1 and Michio Hirano1
Abstract | Mutations in the human mitochondrial genome are known to cause an array of diverse disorders, most of which are maternally inherited, and all of which are associated with defects in oxidative energy metabolism. It is now emerging that somatic mutations in mitochondrial DNA (mtDNA) are also linked to other complex traits, including neurodegenerative diseases, ageing and cancer. Here we discuss insights into the roles of mtDNA mutations in a wide variety of diseases, highlighting the interesting genetic characteristics of the mitochondrial genome and challenges in studying its contribution to pathogenesis.
D I S E A S E M E C H A N I S M S
R E V I E W S
878 | DECEMBER 2012 | VOLUME 13 www.nature.com/reviews/genetics
© 2012 Macmillan Publishers Limited. All rights reserved

Primary pathogenic mtDNA mutationsMutations that can compromise OXPHOS function and cause disease. They arise in mtDNA through processes such as random errors during normal mtDNA replication.
Mitochondrial function and dysfunctionThe human mitochondrial genome is a tiny 16.6 kb circle containing only 37 genes: 13 of these genes encode pro-teins and the remaining 24 — consisting of 2 ribosomal RNAs (rRNAs) and 22 tRNAs — are used for transla-tion of those 13 polypeptides8 (FIG. 1a). Remarkably, all 13 polypeptides are components of a single metabolic entity: the OXPHOS system (FIG. 1b). Pathogenic mutations
resulting in respiratory chain deficiency can arise from mutations in either genome9. In addition to these struc-tural subunits, there are more than 50 nDNA-encoded ancillary gene products required for the proper assem-bly and functioning of the OXPHOS system (FIG. 1b). Although mitochondrial OXPHOS disorders are quite varied, they typically display a number of ‘canonical’ biochemical and morphological features (BOX 3).
Box 1 | Primary mitochondrial DNA-related diseases
Leber’s hereditary optic neuropathy (LHON). This is the most common mitochondrial DNA (mtDNA)-related disorder, causing subacute loss of central vision in young adults, predominantly men. LHON is usually due to homoplasmic mutations in one of three genes encoding complex I subunits (m.11778G→A in NADH dehydrogenase 4 (ND4), m.3460G→A in ND1 and m.14484T→C in ND6). Although mutations are predominantly homoplasmic, the retinal ganglion cells are affected selectively. The marked predominance of LHON in men, which is not accounted for by differences in mitochondrial genetics between the sexes, has been attributed to the relative abundance of oestrogen receptors120 and may be the first example of the role of epigenetics in the expression of mtDNA-related diseases.
Leigh’s syndrome. This disease is more commonly due to nuclear DNA (nDNA) mutations than to mtDNA mutations. The mutations are most frequently in subunits of complex I or in assembly factors of complex IV. Despite the striking variety of specific aetiologies, Leigh’s syndrome has common clinical features (developmental delay or regression, respiratory abnormalities, recurrent vomiting, nystagmus, ataxia, dystonia and early death) and characteristic radiological changes (bilateral symmetrical lesions affecting mostly basal ganglia and the brainstem), a reflection of the devastating effects of energy shortage on the developing nervous system.
Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS). This is a multisystem disorder in which brain, muscle and the endocrine system are predominantly involved and is often fatal in childhood or in young adulthood121. Unique to MELAS are the transient eponymous stroke-like episodes, which are most often due to infarcts in the temporal and occipital lobes and are associated with hemiplegia and cortical blindness. Although we still do not understand the cause, their occurrence highlights the fact that MELAS is also an angiopathy, making it almost unique among the mitochondrial diseases122. Although the most common causal mutation is m.3243A→G in tRNALeu(UUR), more than a dozen other mutations have been associated with MELAS, affecting both tRNA and protein-coding genes.
Myoclonus epilepsy and ragged red fibres (MERRF). For unknown reasons, MERRF is due almost exclusively to mutations in tRNALys. It is associated with a rather different set of signs and symptoms from the other diseases, often including cervical lipomas as well as myoclonus and epilepsy123. As in MELAS, the pathogenic cause seems to be a post-transcriptional failure to modify the tRNAs with taurine, thereby affecting translational efficiency124. Moreover, in MERRF and MELAS, there is mitochondrial proliferation not only in muscle (that is, the ragged red fibres (RRFs)) but also in blood vessels (strongly SDH-positive vessels (SSVs))125. Although blood vessel involvement can be found in patients with both disorders, angiopathy is less prevalent in MERRF. Interestingly, both the RRFs and SSVs in MERRF are negative for cytochrome c oxidase (COX) histochemical activity, whereas those in MELAS are typically COX-positive125,126 (BOX 3). Herein lies a paradox: why is it that MERRF, which has COX-negative SSVs, is not primarily an angiopathy, whereas MELAS, which has ostensibly ‘more normal’ COX-positive SSVs, is an angiopathy? One possible explanation is that it is the COX positivity that triggers the stroke-like episodes in MELAS, because the absolute amount of COX in these mitochondria-laden SSVs is far greater than normal126. As COX is known to bind nitric oxide (NO)127,128,
which is a key signalling molecule for vasoconstriction (via the conversion of l‑arginine to NO by nitric oxide synthase and the subsequent binding of NO to soluble guanylate cyclase129), it could be that in MELAS (but not in MERRF), NO is titrated out by supernormal levels of COX after a NO-mediated signal to dilate blood vessels. This could thereby prevent vasodilation and precipitate the strokes126. If this possibility is true, agents that increase compensatory NO levels to allow more normal vasodilation might be of therapeutic value. Indeed, treatment of patients with MELAS with l‑arginine has been reported to mitigate the frequency and severity of strokes in this disorder130.
Neuropathy, ataxia and retinitis pigmentosa, and maternally inherited Leigh’s syndrome (NARP and MILS, respectively). These are two possible clinical outcomes of mutations at the same nucleotide (m.8993T→G or, less frequently, m.8993T→C). In the family in which these were originally identified, three adults had one or more of the acronymic features, whereas a maternally related child had severe developmental delay, pyramidal signs, retinitis pigmentosa, ataxia and cerebellar and brainstem atrophy, features that are characteristic of Leigh’s syndrome. We now know that onset and severity depend on the mutation load: patients with NARP harbour ~70% mutant mtDNAs, whereas those with MILS harbour >90%.
Reversible respiratory chain deficiency. Although it is rare, mutation in another tRNA — a homoplasmic m.14674T→C mutation in the discriminator base of tRNAGlu — deserves to be mentioned. This mutation causes a highly unusual disorder, namely a ‘reversible’ form of OXPHOS deficiency in which infants are severely ill but, if sustained vigorously during the perinatal period, recover full mitochondrial function spontaneously within 2 years131. Interestingly, the disease can also be caused by mutations in TRMU132, a nucleus-encoded mitochondrial protein that catalyses the 2-thiolation of uridine at the wobble position of three mt-tRNAs133, including tRNAGlu. These mutations may also modify the expression of a deafness-associated mutation at nucleotide 1,555 in 12S ribosomal RNA134.
Sporadic deletions of mtDNA. Kearns–Sayre syndrome (KSS)25 is defined by the onset before age 20 of ophthalmoplegia (that is, paralysis of the muscles that move the eyeballs), ptosis (that is, droopy eyelids), pigmentary retinopathy and at least one of the following: complete heart block, cerebrospinal fluid protein levels above 100 mg dL–1 and cerebellar ataxia. In this multisystemic disorder, partially deleted mtDNAs (Δ-mtDNAs) are present in all examined tissues (implying that the deletion event occurred in the germ line or soon after fertilization). In chronic progressive external ophthalmoplegia (CPEO)26 — a myopathy with the defining features of ophthalmoplegia and ptosis — Δ-mtDNAs are found only in muscle (implying that the deletion event occurred after fertilization in the muscle lineage of mesoderm). In Pearson’s syndrome27, which is characterized by sideroblastic anaemia and exocrine pancreas dysfunction, Δ-mtDNAs are initially abundant in haematopoietic cells. Fascinatingly, patients with Pearson’s syndrome who survive their anaemia often develop KSS as teenagers: as the Δ-mtDNA load declines in blood (a rapidly dividing tissue), the Δ-mtDNA load increases in more terminally differentiated tissues (for example, in the muscles or brain). This phenomenon demonstrates two other features of mitochondrial genetics: namely, the different thresholds for dysfunction in various tissues and the autonomy of mitochondrial division (and mtDNA replication) even within terminally differentiated cells.
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 13 | DECEMBER 2012 | 879
© 2012 Macmillan Publishers Limited. All rights reserved
NystagmusAn involuntary, quick, rhythmic movement of the eyeball, which can be horizontal, vertical or rotary.
HemiplegiaParalysis of the left or right side of the body owing to a brain lesion affecting motor pathways.
Strongly SDH-positive vessels(SSVs). Abnormal, massive accumulation of mitochondria in blood vessels, as visualized by enzyme histochemistry to detect the activity of succinate dehydrogenase (SDH; complex II of the respiratory chain).
Pathogenic mtDNA mutations: distribution and threshold effects. Primary mtDNA-related diseases may be defined as those disorders that arise directly from a mutation in mtDNA — either a point mutation or a DNA rearrangement (that is, a deletion, duplication or inversion) — that directly compromises OXPHOS function. Since 1988, more than 270 point mutations
have been described, affecting every mtDNA gene (examples are provided in TABLE 1). Remarkably, more than half of these mutations are located in tRNA genes, even though tRNAs comprise only about 10% of the total coding capacity of the genome. Conversely, the polypeptide-coding genes, comprising almost 70% of the genome, account for only about 40% of the
Box 2 | Cybrids
Because there is currently no technology available for introducing exogenous DNA into mammalian mitochondria stably and heritably, the pathogenicity of many primary mtDNA mutations discussed in this Review has been established using cytoplasmic hybrid, or cybrid, technology. In this approach, patient mitochondria and mitochondrial DNAs (mtDNAs) are transferred to a host ρ0 cell containing mitochondria that are devoid of endogenous mtDNAs135 (see the figure). Thus, cybrids can be assayed for donor mitochondrial function in a ‘standard’ nuclear background93.
In the most common approach for making ρ0 cells, a transformed cell line that is deficient in thymidine kinase activity (TK–) is exposed to ethidium bromide (EtBr), which is an inhibitor of mtDNA replication (mitochondria have a high membrane potential and take up EtBr, as it is ionizable). The ρ0 line is auxotrophic for uridine and pyruvate (U–, P–). The ρ0 cells are repopulated by forming hybrids with cytoplasts (cells that lack nuclei but still contain mitochondria) from a patient cell line. After fusion, cells are plated in media containing bromodeoxyuridine (+BrdU) and lacking either pyruvate or uridine (–U or –P), which permits only the growth of ρ0 cells that have fused with cytoplasts containing functional mitochondria (TK+ donor cells are not able to grow in the presence of BrdU). If heteroplasmic cells are used as donors, it is possible to isolate cybrid clones that harbour varying proportions of mutated mtDNAs, ranging from 0% mutant (that is, 100% wild type) to 100% mutant (that is, 0% wild type). Both the parental ρ0 line and homoplasmic cybrid lines harbouring nonsense- or frameshift-mutated mtDNAs must be grown in media supplemented with uridine (+U; ~200 μM) and pyruvate (+P; ~1 mM)136.
Nature Reviews | Genetics
Heteroplasmic
Repopulate individual cellular clones
Homoplasmic normal Homoplasmic mutantρ0
TK– TK– TK–
TK–
Cybrid
U+, P+
U+, P+
TK–TK+
Enucleate EtBr
Cytoplast fusion:–U or –P, +BrdU
TK–
TK–
U+, P+
U–, P–
U+, P+
U+, P+ U+, P+U–, P– U–, P–
ρ0 cellCytoplast
ρ+ cellPatient cell
R E V I E W S
880 | DECEMBER 2012 | VOLUME 13 www.nature.com/reviews/genetics
© 2012 Macmillan Publishers Limited. All rights reserved

Figure 1 | Mitochondrial DNA and the human respiratory chain/oxidative phosphorylation system. a | The human mitochondrial genome. The 37 mitochondrial DNA (mtDNA)- encoded genes include seven subunits of complex I (ND1, 2, 3, 4, 4L, 5 and 6), one subunit of complex III (cytochrome b (Cyt b)), three subunits of complex IV (Cyt c oxidase (COX) I, II and III), two subunits of complex V (A6 and A8), two rRNAs (12S and 16S) and 22 tRNAs (one-letter code). Also shown are the origins of replication of the heavy strand (O
H) and the
light strand (OL), and the promoters of transcription of
the heavy strand (HSP) and light strand (LSP). b | Oxidative phosphorylation (OXPHOS) complexes. The system is comprised of complex I (NADH dehydrogenase-CoQ reductase), complex II (succinate dehydrogenase), complex III (ubiquinone-Cyt c oxidoreductase), complex IV (COX) and complex V (ATP synthase; F
0 and F
1 denote the
proton-transporting and catalytic subcomplexes, respectively), plus two electron carriers, coenzyme Q (CoQ; also called ubiquinone) and Cyt c. Complexes I–IV pump NADH- and FADH
2-derived protons (produced by the
tricarboxylic acid (TCA) cycle and the β-oxidation ‘spiral’) from the matrix across the mitochondrial inner membrane to the intermembrane space to generate a proton gradient while at the same time transferring electrons to molecular oxygen to produce water. The proton gradient, which makes up most of the mitochondrial transmembrane potential (Δψ
m), is used to do work by being dissipated
across the inner membrane in the opposite direction through the fifth complex (ATP synthase), thereby generating ATP from ADP and free phosphate. Complexes I, III, IV and V contain both mtDNA- and nuclear DNA (nDNA)-encoded subunits, whereas complex II, which is also part of the TCA cycle, has only nDNA-encoded subunits. Note that CoQ also receives electrons from dihydroorotate dehydrogenase (DHOD), an enzyme of pyrimidine synthesis. Polypeptides encoded by nDNA are in blue (except for DHOD, which is in pink); those encoded by mtDNA are in colours corresponding to the colours of the polypeptide-coding genes (shown in a). The ‘assembly’ proteins are all nDNA-encoded. IMS, intermembrane space; MIM, mitochondrial inner membrane. Part a is modified from REF. 149 © (2006) Wiley.
H+
A8A6
Nature Reviews | Genetics
COX ICOX II
COX III
Cyt b
CoQ
Cyt c
ND4
ND3
ND5
ND6
ND4L
ND2ND1
DHOD
Complex IPolypeptides Complex IIIComplex II Complex VComplex IV
7mtDNA-encodedsubunits 10 23
~39nDNA-encodedsubunits 104 ~1410
~11Assemblyproteins ~9~2 ~3~30
Succinate Fumarate
O2 2H2O
e–
e–
e– e–
e–e–
ADP ATP
F1
F0
H+H+
b
a
H+Dihydroorotate Orotate
ND5
ND4
ND4L
ND3
ND6E
P
Q
ANCY
S1
S2H
R
GK
D
W
M
L1
VF
OHHSP
I
A8 A6
L1
OL
LSP
T
Cyt b
D loop
COX I
COX II
COX III
16S
12S
MIM
Matrix
IMS
ND2
ND1
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 13 | DECEMBER 2012 | 881
© 2012 Macmillan Publishers Limited. All rights reserved

HeteroplasmicA term that describes the presence of two or more mtDNA genotypes within a cell, tissue or organism.
mutations, and the two rRNA genes (15% of coding capacity) account for only about 2%.
This unexpected distribution is likely to be due to the fact that most patients with mtDNA mutations are heteroplasmic, which means that they harbour a mixture
of normal and mutated mtDNAs. Moreover, with one exception10, all of these heteroplasmic mtDNA mutations are ‘recessive’: an extremely high amount of mutation is required before a clinical phenotype evinces itself. For unknown reasons, this ‘threshold effect’ varies among mutation types, and in skeletal muscle the muta-tion load for any particular tRNA (~90%)11 is typically higher than that for large-scale partial deletions of mtDNA (~70–80%)12. The threshold for mutation load in polypeptide-coding genes can be similarly broad, with low levels of mutation causing one type of clini-cal presentation and higher levels causing another. An excellent example of this behaviour is the m.8993T→G mutation in the ATP synthase 6 (ATP6) gene: at muta-tion loads above 90%, patients present with maternally inherited Leigh’s syndrome (MILS), which is a fatal encephalopathy13
(BOX 1), whereas at mutation loads in the range of 70–90% patients present with neuropathy, ataxia and retinitis pigmentosa (NARP)14, which is a late-onset, debilitating but non-fatal disorder. By contrast, a patient with 70% mutation in a tRNA will rarely display overt disease. Thus, tRNA mutations are ‘tolerated’ over a broader range of mutant load than are mutations in polypeptide-coding genes. Of course, after the mutation load has risen above 90–95%, no matter what the type of mutation is, OXPHOS deficiency occurs, usually with devastating results.
These high thresholds for pathogenicity also mask the actual frequency of potentially pathogenic mtDNA mutations in the population. Although the prevalence of mtDNA-related disease is about 1 in 5,000, the popu-lation frequency of the ten most common pathogenic mtDNA mutations is much higher — approximately 1 in 200 — suggesting that many normal individuals carry subthreshold levels of potentially harmful mtDNA mutations15.
The skewed distribution of mutations towards tRNA errors initially implies that maternally inherited muta-tions in structural subunits of the respiratory chain are selected against, presumably during embryogenesis, whereas mutations in tRNAs are somehow less ‘severe’ during embryogenesis. Indeed, maternally inherited mutations in tRNAs might be under less-stringent puri-fying selection through the germ line than are muta-tions in polypeptide-coding genes16. However, it is easy to imagine that the reverse might also hold true: namely, that mutations in rRNA and tRNA genes that disrupt global protein synthesis would have a greater deleteri-ous effect than mutations in single respiratory chain sub-units. The paucity of mutations in rRNA genes supports this alternative view: there is only one known mutation in 16S rRNA associated with a primary mitochondrial disease, and none of the five known mutations in the 12S rRNA gene is lethal, but they do cause non-syndromic deafness17 for reasons related to an effect on 12S rRNA methylation and subsequent nuclear E2F1-mediated transcription18 rather than to the role that 12S rRNA has in protein synthesis. Thus, in addition to differen-tial degrees of purifying selection, the predilection of mutations for tRNA genes may be due to other, currently unknown reasons.
Box 3 | Biochemical and morphological features of mitochondrial disease
Although mitochondrial oxidative phosphorylation (OXPHOS) disorders are quite varied, they typically display a number of ‘canonical’ biochemical and morphological features. An obvious feature is respiratory chain deficiency, which typically manifests itself as reduced enzymatic function in one or more respiratory chain complexes, with a concomitant reduction in cellular oxygen consumption and ATP synthesis. In addition, patients often have increased resting lactic acid levels in the blood and cerebrospinal fluid9. Under normal conditions, pyruvate — the end product of anaerobic glycolysis — is transported into mitochondria for further oxidation in the tricarboxylic acid (TCA) cycle to produce the reducing equivalents (namely, NADH and FADH
2) that are required
for proton pumping by the respiratory chain. However, if the respiratory chain is compromised, NADH and pyruvate accumulate in the cytosol. Excess cytosolic pyruvate is converted to lactate by lactate dehydrogenase, thereby accounting for the increased lactic acid found in many mitochondrial diseases and especially in those affecting infants or young children9.
A prominent morphological feature of OXPHOS disease is the ragged red fibre (RRF), which reflects a massive proliferation of OXPHOS-defective mitochondria29 in muscle; this can be visualized with the modified Gomori trichrome stain137. The accumulation can also be observed using nitro-blue tetrazolium to detect the activity of succinate dehydrogenase (SDH; that is, complex II)138, which contains only nuclear DNA (nDNA)-encoded subunits. In this case, the RRFs appear dark blue (see the figure). The figure shows RRFs and strongly SDH-positive vessels (SSVs) in serial sections of skeletal muscle from patients with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) and myoclonus epilepsy and ragged red fibres (MERRF) visualized by histochemistry to detect the activities of SDH (left) and cytochrome c oxidase (COX; right). Note that RRFs and SSVs are COX-negative in MERRF, whereas they are COX-positive in MELAS. RRFs are a sufficient but not necessary feature for determining mitochondrial disease (as is increased lactate in the blood or cerebrospinal fluid).
The vast majority of OXPHOS mutations impair the cell’s ability to produce ATP in amounts that are sufficient for maintaining viability, but sometimes OXPHOS problems can leave ATP synthesis intact and yet can be deleterious by, for example, stimulating the overproduction of reactive oxygen species (ROS)139 or inducing an autoimmune response140.
Photographs are provided courtesy of Eduardo Bonilla and Kurenai Tanji, Columbia University, New York, USA.
SDH COX
SSV
SSV
SSV
SSV
RRF
RRF
RRF
RRF
MEL
AS
MER
RF
Nature Reviews | Genetics
R E V I E W S
882 | DECEMBER 2012 | VOLUME 13 www.nature.com/reviews/genetics
© 2012 Macmillan Publishers Limited. All rights reserved

Examples of point mutations in mtDNA. Among the point mutations (TABLE 1), the most common are: an A→G transition at position 3,243 in the tRNALeu(UUR) gene, which causes mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS); an A→G transition at position 8,344 in tRNALys, which causes myoclonus epilepsy and ragged red fibres (MERRF) syn-drome; and the aforementioned T→G transversion at position 8,993 in ATP6, which causes NARP and MILS (BOX 1). These three disorders highlight the diversity of biochemical, morphological and clinical presentations that are a hallmark of mitochondrial disorders and that not only challenge the diagnostic capabilities of clini-cians but also pose fundamental questions regarding basic mitochondrial biology.
NARP and MILS highlight additional aspects of mito-chondrial disease and behaviour. These disorders are due to mutations in the membrane-spanning (so-called F0) portion of ATP synthase (FIG. 1b) that compromise ATP synthesis. Whereas all of the mutations described above affect respiratory chain integrity, thereby reducing the mitochondrial transmembrane potential (Δψm)19,20, ATP synthase mutations typically leave the respiratory chain intact but decouple proton flow from ATP generation21. As such, Δψm in NARP or MILS is either unchanged or is actually higher than normal21. This crucial difference in transmembrane potential will render cells from patients with NARP or MILS refractory to treatment strategies that are predicated on selectively eliminating mitochondria with a low Δψm (REFS 19,20).
Examples of large-scale deletions of mtDNA. The most easily observable mtDNA mutations are the large-scale partial deletions: mtDNAs with these deletions are des-ignated Δ-mtDNAs. Discovered in 1988 (REFS 22,23), these mutations remove ~2–10 kb of mtDNA, and the deletion breakpoints are often flanked by short (5–13 bp) direct repeats24. The deletions are typically sporadic (that is, only the proband has the mutation; mothers and siblings do not), and although they vary among patients, each patient harbours only a single species of deletion. These facts imply that the deletion event occurred either in the germ line of the mother or in early embryogenesis of the proband; the deletion event arose randomly in a single mtDNA molecule, and this single molecule of Δ-mtDNA was massively ampli-fied during pre- and postnatal development through clonal expansion. The deletions can be located almost anywhere in the mitochondrial genome24, but they all cause one of three pathogenically similar disorders, as distinguished by their tissue proclivities9: Kearns–Sayre syndrome (KSS)25; chronic progressive external ophthalmoplegia (CPEO, or simply PEO)26; or Pearson’s syndrome27 (BOX 1).
In all three syndromes, the deleted region encom-passes at least one tRNA gene. Indeed, this is the basis for the underlying pathogenesis of these syndromes and demonstrates two additional aspects of mitochon-drial behaviour. First, the loss of a single tRNA gene is sufficient to compromise the translation of all 13 mtDNA-encoded polypeptides28. Second, this failure
to translate mt-mRNAs occurs even in heteroplasmic cells but only if the fraction of Δ-mtDNA within each cell (or muscle segment) is above ~80% and the amount of normal mtDNAs is insufficient to complement the defect by providing the ‘missing’ tRNAs20,29,30. If the mutation load is below this threshold, complemen-tation will occur20, as normal mRNAs and polypep-tides can move fairly freely, not only within individual organelles but also as a result of the constant fusion and fission of mitochondria. By contrast, the mtDNAs themselves are more ‘confined’, as they are bound in mtDNA–protein complexes called nucleoids that are attached to the inner face of the inner membrane and operate as semi-autonomous genetic elements20.
Secondary mtDNA defects: multiple deletions and depletion of mtDNA. Secondary mtDNA-related dis-orders are those in which mtDNA integrity is compro-mised but not via a ‘direct hit’ on the mitochondrial genome. Rather, mtDNA mutations in these disorders (secondary mtDNA mutations) are caused ‘indirectly’ as a consequence of a Mendelian-inherited mutation in a nuclear gene that encodes a protein required for the proper replication and/or maintenance of the mito-chondrial genome, ultimately resulting in mtDNA instability. This instability can manifest itself through the generation of multiple point mutations or large-scale Δ-mtDNAs that arise over the lifetime of the patient31; loss of the entire mitochondrial genome (that is, mtDNA depletion32) can also occur. Often, all three types of error coexist within tissues of the same patient33. Strictly speaking, these syndromes are not considered to be primary mtDNA diseases. However, because at a basic level these diseases are defects in the dialogue between the nucleus and the mitochondria, they share many of the features of the primary disor-ders, including their population-genetic behaviour (for example, heteroplasmy and threshold effects).
There are various mutations that can cause mtDNA instability, including those in nDNA-encoded proteins required for maintenance of mitochondrial nucleo-tide pools34, nucleotide transport35, DNA replica-tion36 and RNA transcription37. For example, PEO can occur with Mendelian inheritance owing to mutations in a nucleus-encoded mitochondrial DNA helicase called Twinkle37; these mutations result in the genera-tion and accumulation in muscle of multiple large- scale Δ-mtDNAs that are structurally similar to the ‘clonal’ Δ-mtDNAs observed in patients with sporadic PEO and KSS. Depending on the loads of depleted and mutated mtDNAs and their tissue distributions, secondary mtDNA-related disorders can have various presentations. Examples of syndromes include mito-chondrial neurogastrointestinal encephalomyopathy (MNGIE)34, Alpers’ sydrome38 and PEO in isolation or as a part of syndromes that include PEO plus other features.
Ageing and neurodegenerationAccumulation of mtDNA mutations in normal ageing. The mutation load of the clonal Δ-mtDNAs found in
Lactic acidosisThe abnormal accumulation of lactic acid causing lower pH in blood in a resting individual (that is, not during or immediately after exercise), which is seen in many patients with mitochondrial disease.
MyoclonusInvoluntary jerky movement of an area of the body (usually a limb).
Mitochondrial transmembrane potential(Δψm). This is the electrical potential generated across the mitochondrial inner membrane due to differences in the distribution of ions (for example, H+, Ca2+, Na+, K+ and ionized ATP species) between the matrix and the intermembrane space.
Secondary mtDNA mutationsMutations that can compromise OXPHOS function and cause disease and that arise in mtDNA owing to a mutation in the nuclear genome that compromises mtDNA integrity (for example, a mutation in mitochondrial DNA polymerase-γ causing systemic errors in mtDNA replication).
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 13 | DECEMBER 2012 | 883
© 2012 Macmillan Publishers Limited. All rights reserved

sporadic KSS, PEO and Pearson’s syndrome is extremely high (>80% in affected issues)39. Interestingly, these mutations also accumulate during the lifetime of normal people, albeit at extremely low levels that are detectable only by PCR40 and with different amounts in different tissues41. It appears that these Δ-mtDNAs accumulate during our lifetimes, especially in postmitotic tissues such as the brain, heart and muscle42–44. Although they arise spontaneously as the result of somatic muta-tion, each individual species of Δ-mtDNA has the capacity to expand clonally45; this expansion is particu-larly noticeable in muscle from aged normal individuals46 (and in patients with Mendelian PEO)47. Although each individual species of Δ-mtDNA may represent less than 0.1% of total mtDNA43,44, sufficient numbers of different Δ-mtDNA species accumulate so that, depending on their tissue distribution and concentration48, they can have deleterious consequences49.
Although Δ-mtDNAs clearly accumulate in ageing, the evidence that the same holds for point mutations is weaker43. There are many ongoing studies aimed at detecting somatic mtDNA mutations, but it still remains uncertain whether these mutations cause overt disease or whether they have a major role in normal ageing. Perhaps studies of experimental models that age more rapidly than humans (for example, flies, worms and even yeast) will shed light on this question.
mtDNA mutations in late-onset neurodegenerative dis-ease. There is a growing interest in the potential role of mitochondrial dysfunction in general, and of mtDNA mutations in particular, in late-onset neurodegen-erative diseases, such as Alzheimer’s, Parkinson’s and Huntington’s diseases, amyotrophic lateral sclerosis and hereditary spastic paraplegias50. Although there seems to be little doubt of a mitochondrial connection, it seems to relate more to problems of mitochondrial dynamics (for example, fission, fusion, distribution and anchorage) or quality control (for example, autophagy) than to mutations in mtDNA50. However, there is one neurodegenerative disorder in which mtDNA mutations may have a role (albeit probably a secondary one), and that is Parkinson’s disease. Studies of proteins mutated in familial Parkinson’s disease — for example, PINK1, a mitochondrially targeted kinase51, and parkin, a cyto-solic ubiquitin ligase that translocates to low-Δψm dys-functional mitochondria52 — have implicated altered mitochondrial quality control as an underlying element in the pathogenesis of both the familial and sporadic forms of the disease. However, it has also been found that the substantia nigra — the brain region that is most affected in Parkinson’s disease (and the main site of dopaminergic neuron degeneration in patients with Parkinson’s disease) — harbours substantial levels of the types of Δ-mtDNAs found in KSS and in normal age-ing53,54. This finding has led to the hypothesis that these somatically produced Δ-mtDNAs may eventually cause bioenergetic deficit, leading to the disease. This hypoth-esis has received indirect support from studies of mice in which mtDNA deficiency was induced specifically in the substantia nigra and that displayed many of the
Table 1 | mtDNA mutations in human primary respiratory chain disorders
Gene product Number of mutations Main disorder
rRNAs
12S rRNA 5 Deafness
16S rRNA 1 Atypical MELAS
tRNAs
tRNAAla 3 Myopathy
tRNAArg 2 Various
tRNAAsn 5 Myopathy
tRNAAsp 2 Various
tRNACys 3 Various
tRNAGln 3 Various
tRNAGlu 7 Reversible respiratory chain deficiency
tRNAGly 3 Various
tRNAHis 4 Various
tRNAIle 14 PEO
tRNALeu(CUN) 8 Myopathy
tRNALeu(UUR) 23 MELAS
tRNALys 14 MERRF
tRNAMet 2 Various
tRNAPhe 14 Myopathy
tRNAPro 5 Multisystem
tRNASer(AGY) 4 Myopathy
tRNASer(UCN) 12 Myopathy; deafness
tRNAThr 2 Various
tRNATrp 12 Encephalomyopathy
tRNATyr 4 Myopathy
tRNAVal 6 Multisystem
Polypeptides
ATP synthase 6 13 NARP or MILS
ATP synthase 8 2 Various
COX I 10 Various
COX II 8 Various
COX III 6 Myopathy
Cytochrome b 21 Sporadic myopathy
ND1 16 MELAS; LHON
ND2 3 Various
ND3 5 Leigh’s syndrome
ND4 5 LHON
ND4L 1 LHON
ND5 12 MELAS
ND6 11 LHON
This is a ‘minimal’ list that was compiled by the authors. A more comprehensive list (>300 mutations) is available at MITOMAP, which contains confirmed pathogenic mutations (including those selected for this table), those that are possibly associated with pathology and those that appear to confer a higher risk of developing disease or are modifiers of disease presentation. COX, cytochrome c oxidase; LHON, Leber’s hereditary optic neuropathy; MELAS, mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes; MERRF, myoclonus epilepsy and ragged red fibres; MILS, maternally inherited Leigh’s syndrome; mtDNA, mitochondrial DNA; NARP, neuropathy, ataxia and retinitis pigmentosa; ND, NADH-ubiquinone oxidoreductase; PEO, progressive external ophthalmoplegia; rRNA, ribosomal RNA.
R E V I E W S
884 | DECEMBER 2012 | VOLUME 13 www.nature.com/reviews/genetics
© 2012 Macmillan Publishers Limited. All rights reserved

HomoplasmicA term that describes the presence of a single mtDNA genotype within a cell, tissue, or organism.
Aerobic glycolysisThe conversion of glucose to lactate (and the production of ATP by substrate level phosphorylation) even in the presence of oxygen, especially in tumours.
pathological features of Parkinson’s disease55. We note, however, that patients with KSS who have high levels of Δ-mtDNAs do not, as a rule, have features of Parkinson’s disease and that only one mutation in mtDNA has been associated with such features56. Further analysis in model systems, including mice harbouring clonal57 or multi-ple58 Δ-mtDNAs, or analysis of PINK1- or PARK2 (also known as parkin)-mutated mice for the presence of Δ-mtDNAs, may help to resolve this issue.
Inherited mtDNA variants in complex traitsIn addition to the potential roles of somatic mtDNA mutations in ageing and neurodegenerative diseases, are there mtDNA haplotypes or specific polymorphisms that are associated with predisposion to neurodegenerative diseases or that are associated with other complex traits? Different ethnic groups have distinct mitochondrial hap-lotypes that have resulted from the accumulation of vari-ations from the mtDNA of our ancestral ‘mitochondrial Eve’59. Some of these haplotypes may confer vulnerabil-ity to, or protection from, various common diseases. In addition, the genetic variation might be responsible for subtle biochemical differences among haplogroups. For example, it has been postulated that mtDNA hap-lotypes that favour a looser coupling between oxidation and phosphorylation — thereby generating a surplus of heat at the expense of ATP — may have favoured the set-tling of human populations in colder climates60; however, this hypothesis is controversial61,62.
Mitochondrial haplotypes as modifiers. It is clear that bona fide pathogenic mtDNA mutations can cause common disorders such as diabetes mellitus63, hearing loss64,65 or exercise intolerance66, but such mutations account for less than 2% of patients with these complex traits. What is less clear is the role of mtDNA haplo-type variants in such common disorders. Theoretically, an mtDNA variant with mild functional consequences could interact with other polymorphisms in mtDNA or nDNA to produce complex traits synergistically67. A well-documented example is the effect of mtDNA haplotype on the expressivity of Leber’s hereditary optic neuropathy (LHON) mutations. The variable penetrance of these mutations cannot be accounted for by hetero-plasmy, because the mtDNA defect is homoplasmic in most patients. Haplotype analyses of LHON mutations have revealed an increased risk of vision loss in the set-ting of haplotypes J1, J2 and K68,69. Analyses in vitro of cybrids (BOX 2) and fibroblast cells that harbour either the m.11778G→A or m.14484T→C mutations confirmed that haplogroup J conferred sensitivity to the neurotoxin n-hexane70, an environmental toxin that, like smoke and pesticides, has been implicated as an environmental trigger of optic neuropathy in patients with LHON70.
Susceptibility to complex disorders. Evidence for mtDNA susceptibility polymorphisms and/or haplo-groups in complex disorders is highly controversial. There is evidence for and against such links in a num-ber of diseases, including age-related macular degen-eration, Alzheimer’s disease, amyotrophic lateral
sclerosis, frontotemporal dementia, hereditary spastic paraplegia, Huntington’s disease, ischaemic stroke, metabolic syndrome, myocardial infarction, psychi-atric disorders, Parkinson’s disease, sarcopaenia and type 2 diabetes, among others71–83. Although it has been argued that those studies in which associations were not observed typically had a low statistical power owing to small sample sizes and the genotypic complexity of mtDNA lineages and sublineages, the fact remains that very few ‘primary’ mtDNA mutations (which have been found on diverse mtDNA haplotypic backgrounds) are associated with these disorders, and they are far fewer in number than are nuclear mutations found in these conditions50. Moreover, most of these studies were either correlative, descriptive or both and were also sus-ceptible to technical difficulties that were unique to the analysis of mtDNA polymorphisms84. These included problems in determining the functional consequences of one haplotype as compared to another85 and in the misattribution of mutations to mtDNA resulting from the co-amplification of authentic mtDNA with nucleus-embedded mtDNA pseudogenes (described below). Taken together, we feel that although there may be important contributions of mtDNA haplotypes to complex traits and disorders, the evidence to date is too weak to support such a conclusion.
mtDNA mutations in cancerPerhaps nowhere is the role of mtDNA mutations in pathology more contentious than in cancer. Moreover, the discovery of mtDNA mutations in tumours has also contributed to the recent resurgence of inter-est in the Warburg hypothesis of cancer, which states that tumours rely on ‘aerobic glycolysis’ rather than on OXPHOS in normoxic conditions to survive and even to thrive86.
In 1998, Bert Vogelstein’s group reported that colo-rectal cancers harbour numerous mtDNA mutations (typically, point mutations, not rearrangements, with many in the homoplasmic state); adjacent non-cancer-ous tissues had only normal mtDNAs87. Since then, this result has been replicated numerous times88 but, given our understanding of mitochondrial behaviour, espe-cially in primary mitochondrial disease, these findings raise many questions. Why are there so many mutations, and why are many of the mutations in cis (that is, on the same mtDNA circle)? How do they arise rapidly? Why do some tumours have nonsense mutations, whereas others have synonymous or missense mutations, many of which are apparently functionally neutral? Why is there no apparent difference in tumour biology between those tumours that harbour homoplasmic mutations and those that harbour heteroplasmic mutations (some-times below the threshold level typical of primary mito-chondrial disease)? Fundamentally, these questions are really asking the same thing: what is the relationship of mtDNA mutations and OXPHOS function to cancer biology (BOX 4)?
Although there are dozens of studies document-ing mtDNA mutations in cancer, almost none of them has asked whether a specific mtDNA genotype affects
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 13 | DECEMBER 2012 | 885
© 2012 Macmillan Publishers Limited. All rights reserved

mitochondrial function and whether such dysfunc-tion has a role in tumorigenesis. There have been a few reports of tumours that are, for example, cytochrome c oxidase (COX)-deficient5,89, but it is not clear whether the deficiency is due to a mutation in an mtDNA-encoded COX gene, or whether this is the result of a general downregulation of respiratory chain function or even the result of reduced mtDNA copy number. Moreover, it is noteworthy that of the known patho-genic point mutations causing primary mitochondrial disease (TABLE 1), none is unequivocally associated with cancer. Thus, mtDNA mutations seem more likely to be a marker of cancer and/or to arise as a secondary effect of altered tumour biology than to be a cause of cancer.
An alternative idea is that mtDNA mutations result in downregulation of OXPHOS function, and this pro-cess somehow promotes tumour survival. One idea in support of this view is that defects in OXPHOS lead to activation of the AKT cell survival pathway, including downregulation of apoptosis via a mechanism mediated by cellular NADH, which is increased when mitochon-drial respiration is blocked90. However, defective bioen-ergetics in tumours, either through mtDNA mutation or reduction in mtDNA copy number91 (but the issue of copy number is contentious92), raises a conceptual problem: mitochondrial respiratory chain function is indispensable for the synthesis of the pyrimidines that cells — especially highly proliferating tumour cells — need to divide, and in the absence of a functioning respiratory chain, cells eventually die93. This is because a key reaction in de novo pyrimidine synthesis — the conversion of dihydroorotate to orotate (a precursor of uridine monophosphate (UMP)) — is catalysed by the mitochondrial enzyme dihydroorotate dehydroge-nase, which depends on an intact respiratory chain for its function94; more specifically, the electron acceptor for this redox reaction is reduced ubiquinone (that is, CoQ) and, through complexes III and IV, molecular oxy-gen (FIG. 1b). If a similar effect operates in tumours that contain homoplasmic loss-of-function mtDNA muta-tions, it would mitigate against, not for, the generation of mtDNA mutations. We note in this regard that cells that are completely devoid of mtDNA (called ρ0 cells (BOX 2)) do not form tumours when they are injected into mice subcutaneously95,96, but they are tumorigenic when injected intramuscularly96. This is perhaps because the subcutaneous site is less vascularized than the muscle and cannot take up uridine from the circulation (uridine is ~5 μM in plasma97).
Are tumour mtDNA mutations artefacts? If some base-line level of normal OXPHOS function is required for tumours to be viable, why have so many mtDNA muta-tions been detected? One possibility is that at least some of these could be technical artefacts. If a low-stringency BLAST of mtDNA is run against the human genome, it is possible to detect ~1,000 nucleus-embedded mito-chondrial sequences (NUMTs), ranging in size from ~50 bp to >15 kb (that is, almost full-length)98,99. These NUMTs have been relocalized from the mitochondria to the nucleus in evolutionary time98 and have subse-quently been subjected to genetic drift, with individual NUMTs picking up polymorphic mutations in differ-ent human lineages100,101 (that is, they are essentially mtDNA-derived pseudogenes). What is less appreci-ated is that NUMTs can also enter the nucleus during the lifetime of an individual, especially under conditions of stress. This rapid relocalization of mtDNA sequences has been documented in plants102, yeast103, rodents104 and humans105–107. Of course, in these cases, the sequences of authentic mtDNA and of recently relocalized NUMTs will be extremely similar or possibly identical.
Chromosomal instability in tumours has the poten-tial to increase the NUMT copy number in several ways: the NUMTs could be located in unstable regions that
Box 4 | Mitochondrial function in cancer
Mitochondrial function is altered in numerous ways in tumours, allowing them to survive and to circumvent cell-death pathways141. Some of these changes are summarized below.
Loss of p53 function. Among its numerous functions, the tumour suppressor p53 regulates respiratory chain function and glycolysis via positive transcriptional regulation of synthesis of cytochrome oxidase 2 (SCO2), which encodes a complex IV assembly protein, and negative regulation of TP53‑induced glycolysis regulator (TIGAR; also known as C12orf5). When TP53 (the gene that encodes p53) is mutated or lost in tumours, glycolysis (and the mitochondrial tricarboxylic acid (TCA) cycle) is stimulated142 and the respiratory chain is depressed. This phenomenon is known as the Crabtree effect.
Evasion of hypoxia-mediated cell death. Poorly vascularized, rapidly dividing tumour cells become hypoxic, stimulating cell-death pathways mediated by the transcription factor hypoxia-inducible factor 1 (HIF1). Among other effects, this change promotes vascularization, cell survival and invasion. HIF1 subunit-α is normally degraded by the proteasome after hydroxylation by prolyl hydroxylase and subsequent ubiquitylation by the von Hippel–Lindau E3 ubiquitin ligase, but increased levels of TCA-derived succinate in tumours inhibit the hydroxylase, thus stabilizing HIF1. Notably, mutations in nuclear genes encoding two TCA cycle enzymes — namely, succinate dehydrogenase (SDH) and fumarase, both of which result in increased cytosolic succinate levels — can give rise to cancer. These cancers are pheochromocytomas and paragangliomas in the case of SDH143 and leiomyomas in the case of fumarase144. These observations lend support to the idea that HIF1α has a role in tumorigenesis and to the conceptual underpinnings of the Warburg hypothesis145.
Evasion of apoptosis. Cell death by apoptosis is regulated by a balance between interacting pro-apoptotic factors (for example, BAX and the truncated form of BH3-interacting domain death agonist (tBID)) and anti-apoptotic factors (for example, BCL-2). In cancer, this balance is shifted towards anti-apoptotic factors. For example, the glycolytic enzyme hexokinase, which binds to voltage-dependent anion-selective channel (VDAC1) on the mitochondrial outer membrane, is upregulated in tumours; this prevents the interaction of VDAC1 with tBID146,147.
Role in DNA replication. In addition to the requirement of a respiratory chain to support pyrimidine synthesis (see text), it was recently shown that purine biosynthesis in tumours is also mediated by mitochondria via pathways requiring glycine. In mitochondria, glycine is converted to tetrahydrofolate (by the glycine cleavage system), which provides one-carbon units for incorporation into the purine ring. Although glycine is made both in the cytosol (de novo from carbon dioxide and ammonia and salvaged by serine and tetrahydrofolate) and in mitochondria (also from serine and tetrahydrofolate), tumours (but not rapidly dividing normal cells) consume glycine that is metabolized specifically by the mitochondrial pathway148. Thus, DNA replication using both purine and pyrimidine precursors — the sine qua non of tumour growth — rely on mitochondrial function.
This brief overview vastly understates the complicated interplay between mitochondrial and tumour biology141 but gives some sense of the potential effects of a possible role that mitochondria and mitochondrial DNA mutations might have in cancer.
R E V I E W S
886 | DECEMBER 2012 | VOLUME 13 www.nature.com/reviews/genetics
© 2012 Macmillan Publishers Limited. All rights reserved

Double minute DNAs(dmDNAs). Tiny fragments of extrachromosomal nuclear DNA found in many tumours, derived from the amplification of small regions of chromosomal DNA.
lead to aneuploidy; they could be located in homo-geneously staining regions (HSRs), which are tandemly repeated regions of the genome with increased copy number; and they can be selectively amplified within small regions of the chromosome as double-minute DNAs (dmDNAs)108. In the cases of HSRs and dmDNAs, the copy number in the repeated regions can vary, rang-ing from ~3 to ~30 repeats, with a median value of ~10 repeats108,109. Because the typical dmDNA is about 1–3 Mb in size, it will contain on average one NUMT (on the basis of there being ~1,000 NUMTs in a human genome) and will be amplified selectively by approxi-mately tenfold over its pre-cancerous level110,111. We therefore speculate that NUMT targets can increase in tumours in ways that vary from individual to individual and from tumour to tumour.
When using PCR of total cellular DNA to analyse mtDNA in normal tissues, the number of authen-tic mtDNA molecules far exceeds the number of NUMTs detectable by any particular pair of PCR prim-ers, and the PCR preferentially amplifies the normal mtDNA. However, in tumours, these PCR reactions might amplify one or more NUMTs along with (or even in preference to) authentic mtDNA owing to a reduc-tion in the mtDNA/NUMT target DNA ratio. There are at least three possible reasons for this: a reduction in mtDNA copy number91; the presence of large num-bers of potentially highly polymorphic and diverged NUMTs in tumours; and the potential to amplify specific NUMTs selectively in dmDNAs. Therefore, depending on the choice of primers (FIG. 2), mutations in tumour NUMT DNA may be misattributed to those in authentic tumour mtDNA112. We note that when
mitochondria were purified before PCR amplification, no pathogenic mtDNA mutations were found in mouse brain tumours113, and the number of mtDNA mutations in human colorectal tumours was actually lower than that in the surrounding tissue114.
These caveats are speculative, and for that reason they should not be allowed to obscure the very real possibility that authentic mtDNAs are indeed mutated in tumours (especially as the rapid cellular division in cancers may allow for the accumulation and fixation of spontaneous mtDNA mutations115). Moreover, even if the mutated DNAs are artefacts derived from amplified NUMTs116 and are less biologically informative, they are nevertheless still extremely useful as early markers of cancer117,118, and thus they may have clinical use.
Concluding remarksThe notion that mutations in mtDNA, and the subse-quent alterations in mitochondrial oxidative energy metabolism, are confined to a small group of pathologies no longer holds true. Together, primary mtDNA-related diseases, which were previously deemed to be extremely rare, have now been shown to be as frequent as some Mendelian disorders, such as Duchenne’s muscular dystrophy or cystic fibrosis. Moreover, mtDNA muta-tions — either partial deletions or point mutations — that are present at high levels in primary mitochondrial disorders have now been identified at low levels in infant cord blood15, at somewhat higher levels in the ageing population49,54 and at fairly high levels within target tis-sues in some of the most common pathologies that afflict humankind53. Although we have made some progress in using the basic concepts of mitochondrial biology and
Figure 2 | Example of the potential to amplify inadvertently a nucleus-embedded mitochondrial sequence. Comparison of a region of the 16S rRNA gene within authentic mitochondrial DNA (mtDNA) (top lines; numbering according to the standard ‘Cambridge’ mtDNA sequence8) to that of a nucleus-embedded mitochondrial sequence (NUMT) located on chromosome 17p11.2 (bottom lines; genome numbering according to the February 2009 release of the human genome (GRCh37/hg19)). The primer pairs used (Forward and Reverse 1, and Forward and Reverse 2)112 have the potential to amplify both mtDNA and the NUMT. Sequence differences between mtDNA (black) and NUMT DNA (red) are shown in bold.
Nature Reviews | Genetics
Forward
Reverse 1
Reverse 2
mtDNA: NUMT:
mtDNA:NUMT:
mtDNA: NUMT:
mtDNA: NUMT:
mtDNA:NUMT:
22023970
324722024002
3215
220239203166
311622023871
306622023821
tgagtTCAGACCGGAGTAATCCAGGttggtttctatctattctacatttc
ctgccccatagatgatattatctcagcattttactgtacccacacccacc
caagaacagggtTTGTTAAGATGGCAGAGCCC
ctccctgtacgaaaggacaagagaaataaggcctacttcacaaagcgcctttcca-gtacgaaaggataagagaaatggggcccacttcataaagtgccc
-tcccccgtaaatgatatcatctcaacttagtattatacccacacccacc
CAGCCGCTATTAAAGGTTCGtttgttcaacgattaaagtcctacgtgatcCAGCCGCTATTAAAGGTTCGtttgttcaacgattagagtcctatgtgatc
tgagtTCAGACCGGAGTAATCCAGGtcggtttctatctaccttcaaattc
caagaacagggtTTGTTAAGATGGCAGACCCC
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 13 | DECEMBER 2012 | 887
© 2012 Macmillan Publishers Limited. All rights reserved

genetics to deduce the pathogenesis of primary mito-chondrial OXPHOS disease, we are only now begin-ning to identify and to understand the role of secondary mtDNA mutations in ageing, in Parkinson’s disease and perhaps in other disorders as well.
Two technological issues will determine the pace of further progress in the field. With respect to under-standing the relationship of underlying mtDNA hap-lotypes to complex traits, the application of recent technological advances — such as whole-exome sequencing, whole-genome sequencing, high-through-put whole-transcriptome shotgun sequencing and various ‘omics’ approaches (especially proteomics, lipidomics and metabolomics) — to this problem may enable us to understand more clearly the relationship of genotype to phenotype. However, there is currently
no proven technology that will allow us to introduce exogenous mtDNA into mammalian mitochondria in a stable and heritable way. Thus, we cannot make tar-geted mtDNA mutations to study basic issues in mito-chondrial biogenesis (for example, dissecting mtDNA elements controlling replication, transcription or trans-lation) nor can we introduce targeted mutations into mitochondria to generate cellular or animal models of mtDNA-based disorders, be they primary, secondary or complex. However, after this major technical hurdle has been overcome, the field will be poised to enter a revolutionary new phase in our understanding of mito-chondrial biology and disease. To the question posed in 2000 regarding the phenotypic consequences of mtDNA mutations — ‘are we scraping the bottom of the barrel?’119 — the answer is a definite ‘no’.
1. Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 14, 255–274 (1967).
2. Garcia-Rodriguez, L. J. Appendix 1. Basic properties of mitochondria. Methods Cell Biol. 80, 809–812 (2007).
3. Calvo, S. E. et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 4, 118ra10 (2012).This was one of the first applications of sequencing the ‘mitochondrial exome’ (that is, ~1,500 nuclear genes encoding mitochondrial-targeted proteins) to identify pathogenic mutations causing mitochondrial disease.
4. Schaefer, A. M. et al. Prevalence of mitochondrial DNA disease in adults. Ann. Neurol. 63, 35–39 (2008).
5. Greaves, L. C., Reeve, A. K., Taylor, R. W. & Turnbull, D. M. Mitochondrial DNA and disease. J. Pathol. 226, 274–286 (2012).
6. Koopman, W. J., Willems, P. H. & Smeitink, J. A. Monogenic mitochondrial disorders. N. Engl. J. Med. 366, 1132–1141 (2012).
7. Ylikallio, E. & Suomalainen, A. Mechanisms of mitochondrial diseases. Ann. Med. 44, 41–59 (2012).This is a comprehensive overview of mitochondrial respiratory chain disorders.
8. Anderson, S. et al. Sequence and organization of the human mitochondrial genome. Nature 290, 457–465 (1981).
9. DiMauro, S. & Schon, E. A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 348, 2656–2668 (2003).
10. Sacconi, S. et al. A functionally dominant mitochondrial DNA mutation. Hum. Mol. Genet. 17, 1814–1820 (2008).
11. Yoneda, M., Miyatake, T. & Attardi, G. Heteroplasmic mitochondrial tRNALys mutation and its complementation in MERRF patient-derived mitochondrial transformants. Muscle Nerve 3, S95–S101 (1995).
12. Sciacco, M., Bonilla, E., Schon, E. A., DiMauro, S. & Moraes, C. T. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum. Mol. Genet. 3, 13–19 (1994).
13. Santorelli, F. M., Shanske, S., Macaya, A., DeVivo, D. C. & DiMauro, S. The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh’s syndrome. Ann. Neurol. 34, 827–834 (1993).
14. Holt, I. J., Harding, A. E., Petty, R. K. & Morgan-Hughes, J. A. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am. J. Hum. Genet. 46, 428–433 (1990).
15. Elliott, H. R., Samuels, D. C., Eden, J. A., Relton, C. L. & Chinnery, P. F. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 83, 254–260 (2008).This was the first large-scale epidemiological survey of the prevalence of common pathological mtDNA mutations in the normal population.
16. Stewart, J. B. et al. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 6, e10 (2008).
17. Prezant, T. R. et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nature Genet. 4, 289–294 (1993).
18. Raimundo, N. et al. Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell 148, 716–726 (2012).
19. Gilkerson, R. W. et al. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 21, 978–990 (2012).
20. Gilkerson, R. W., Schon, E. A., Hernandez, E. & Davidson, M. M. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J. Cell Biol. 181, 1117–1128 (2008).
21. Sgarbi, G. et al. Inefficient coupling between proton transport and ATP synthesis may be the pathogenic mechanism for NARP and Leigh syndrome resulting from the T8993G mutation in mtDNA. Biochem. J. 395, 493–500 (2006).
22. Holt, I. J., Harding, A. E. & Morgan-Hughes, J. A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331, 717–719 (1988).
23. Lestienne, P. & Ponsot, G. Kearns-Sayre syndrome with muscle mitochondrial DNA deletion. Lancet 331, 885–886 (1988).
24. Damas, J. et al. Mitochondrial DNA deletions are associated with non-B DNA conformations. Nucleic Acids Res. 40, 7606–7621 (2012).
25. Kearns, T. P. & Sayre, G. P. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: unusual syndrome with histologic study in one of two cases. AMA Arch. Ophthalmol. 60, 280–289 (1958).
26. Moraes, C. T. et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns–Sayre syndrome. N. Engl. J. Med. 320, 1293–1299 (1989).
27. Pearson, H. A. et al. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 95, 976–984 (1979).
28. Nakase, H. et al. Transcription and translation of deleted mitochondrial genomes in Kearns–Sayre syndrome: implications for pathogenesis. Am. J. Hum. Genet. 46, 418–427 (1990).This was one of the first studies providing insight into the pathomechanism of mtDNA deletion disorders.
29. Mita, S., Schmidt, B., Schon, E. A., DiMauro, S. & Bonilla, E. Detection of “deleted” mitochondrial genomes in cytochrome-c oxidase-deficient muscle fibers of a patient with Kearns-Sayre syndrome. Proc. Natl Acad. Sci. USA 86, 9509–9513 (1989).
30. Moraes, C. T. et al. Molecular analysis of the muscle pathology associated with mitochondrial DNA deletions. Nature Genet. 1, 359–367 (1992).
31. Ashley, N. et al. Defects in maintenance of mitochondrial DNA are associated with intramitochondrial nucleotide imbalances. Hum. Mol. Genet. 16, 1400–1411 (2007).
32. Moraes, C. T. et al. Mitochondrial DNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am. J. Hum. Genet. 48, 492–501 (1991).This was the first description of a quantitative defect in mtDNA copy number causing disease.
33. Nishigaki, Y., Marti, R., Copeland, W. C. & Hirano, M. Site-specific somatic mitochondrial DNA point mutations in patients with thymidine phosphorylase deficiency. J. Clin. Invest. 111, 1913–1921 (2003).This was the first description of a secondary mitochondrial disorder causing mtDNA point mutations.
34. Nishino, I., Spinazzola, A. & Hirano, M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 283, 689–692 (1999).
35. Kaukonen, J. et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 289, 782–785 (2000).
36. Van Goethem, G., Dermaut, B., Lofgren, A., Martin, J. J. & Van Broeckhoven, C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nature Genet. 28, 211–212 (2001).
37. Spelbrink, J. N. et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nature Genet. 28, 223–231 (2001).
38. Naviaux, R. K. & Nguyen, K. V. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann. Neurol. 55, 706–712 (2004).
39. Moraes, C. T. et al. Phenotype-genotype correlations in skeletal muscle of patients with mtDNA deletions. Muscle Nerve 3, S150–S153 (1995).
40. Cortopassi, G. A., Shibata, D., Soong, N. W. & Arnheim, N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl Acad. Sci. USA 89, 7370–7374 (1992).This was among the first reports that called attention to the accumulation of somatic mtDNA deletions in normal ageing.
41. Corral-Debrinski, M. et al. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nature Genet. 2, 324–329 (1992).
42. Meissner, C. et al. The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: a useful biomarker or more? Exp. Gerontol. 43, 645–652 (2008).
43. Pallotti, F., Chen, X., Bonilla, E. & Schon, E. A. Evidence that specific mtDNA point mutations may not accumulate in skeletal muscle during normal human aging. Am. J. Hum. Genet. 59, 591–602 (1996).
44. Simonetti, S., Chen, X., DiMauro, S. & Schon, E. A. Accumulation of deletions in human mitochondrial DNA during normal aging: analysis by quantitative PCR. Biochim. Biophys. Acta 1180, 113–122 (1992).
45. Oldfors, A. et al. Mitochondrial DNA deletions and cytochrome c oxidase deficiency in muscle fibres. J. Neurol. Sci. 110, 169–177 (1992).
R E V I E W S
888 | DECEMBER 2012 | VOLUME 13 www.nature.com/reviews/genetics
© 2012 Macmillan Publishers Limited. All rights reserved

46. Bodyak, N. D., Nekhaeva, E., Wei, J. Y. & Khrapko, K. Quantification and sequencing of somatic deleted mtDNA in single cells: evidence for partially duplicated mtDNA in aged human tissues. Hum. Mol. Genet. 10, 17–24 (2001).
47. Vu, T. H. et al. Analysis of mtDNA deletions in muscle by in situ hybridization. Muscle Nerve 23, 80–85 (2000).
48. Petruzzella, V. et al. Extremely high levels of mutant mtDNAs co-localize with cytochrome c oxidase-negative ragged-red fibers in patients harboring a point mutation at nt-3243. Hum. Mol. Genet. 3, 449–454 (1994).
49. Bua, E. et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 79, 469–480 (2006).
50. Schon, E. A. & Przedborski, S. Mitochondria: the next (neurode)generation. Neuron 70, 1033–1053 (2011).
51. Vives-Bauza, C. et al. Control of mitochondrial integrity in Parkinson’s disease. Prog. Brain Res. 183, 99–113 (2010).
52. Narendra, D. P. & Youle, R. J. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal. 14, 1929–1938 (2011).
53. Bender, A. et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature Genet. 38, 515–517 (2006).This was the first report of correlating a neurodegenerative disease with the accumulation of mtDNA mutations in the clinically relevant target tissue.
54. Kraytsberg, Y. et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nature Genet. 38, 518–520 (2006).
55. Ekstrand, M. I. et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl Acad. Sci. USA 104, 1325–1330 (2007).
56. De Coo, I. F. et al. A 4-base pair deletion in the mitochondrial cytochrome b gene associated with parkinsonism/MELAS overlap syndrome. Ann. Neurol. 45, 130–133 (1999).
57. Inoue, K. et al. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nature Genet. 26, 176–181 (2000).
58. Tyynismaa, H. et al. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc. Natl Acad. Sci. USA 102, 17687–17692 (2005).
59. Cann, R. L., Stoneking, M. & Wilson, A. C. Mitochondrial DNA and human evolution. Nature 325, 31–36 (1987).
60. Ruiz-Pesini, E., Mishmar, D., Brandon, M., Procaccio, V. & Wallace, D. C. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 303, 223–226 (2004).
61. Amo, T. & Brand, M. D. Were inefficient mitochondrial haplogroups selected during migrations of modern humans? A test using modular kinetic analysis of coupling in mitochondria from cybrid cell lines. Biochem. J. 404, 345–351 (2007).
62. Saxena, R. et al. Comprehensive association testing of common mitochondrial DNA variation in metabolic disease. Am. J. Hum. Genet. 79, 54–61 (2006).
63. Kadowaki, T. et al. A subtype of diabetes mellitus associated with a mutation of mitochondrial DNA. N. Engl. J. Med. 330, 962–968 (1994).This was an important survey that demonstrated the importance of a specific mtDNA mutation as a cause of diabetes mellitus.
64. Bitner-Glindzicz, M. et al. Prevalence of mitochondrial 1555A→G mutation in European children. N. Engl. J. Med. 360, 640–642 (2009).
65. Vandebona, H. et al. Prevalence of mitochondrial 1555A→G mutation in adults of European descent. N. Engl. J. Med. 360, 642–644 (2009).
66. Andreu, A. L. et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N. Engl. J. Med. 341, 1037–1044 (1999).
67. Elson, J. L., Majamaa, K., Howell, N. & Chinnery, P. F. Associating mitochondrial DNA variation with complex traits. Am. J. Hum. Genet. 80, 378–382; author reply 382–383 (2007).
68. Carelli, V. et al. Haplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of Leber hereditary optic neuropathy pedigrees. Am. J. Hum. Genet. 78, 564–574 (2006).This paper provieds a good example of the ‘synergistic’ relationship between specific mtDNA haplotypes and the severity of pathogenic mtDNA point mutations.
69. Hudson, G. et al. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am. J. Hum. Genet. 81, 228–233 (2007).
70. Ghelli, A. et al. The background of mitochondrial DNA haplogroup J increases the sensitivity of Leber’s hereditary optic neuropathy cells to 2,5-hexanedione toxicity. PLoS ONE 4, e7922 (2009).
71. Anderson, C. D. et al. Common mitochondrial sequence variants in ischemic stroke. Ann. Neurol. 69, 471–480 (2011).
72. Arning, L. et al. Mitochondrial haplogroup H correlates with ATP levels and age at onset in Huntington disease. J. Mol. Med. 88, 431–436 (2010).
73. Ingram, C. J. et al. Analysis of European case-control studies suggests that common inherited variation in mitochondrial DNA is not involved in susceptibility to amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 13, 341–346 (2012).
74. Hiona, A. & Leeuwenburgh, C. The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 43, 24–33 (2008).
75. Khusnutdinova, E. et al. A mitochondrial etiology of neurodegenerative diseases: evidence from Parkinson’s disease. Ann. NY Acad. Sci.1147, 1–20 (2008).
76. Mancuso, M., Filosto, M., Orsucci, D. & Siciliano, G. Mitochondrial DNA sequence variation and neurodegeneration. Hum. Genom. 3, 71–78 (2008).
77. Nishigaki, Y., Fuku, N. & Tanaka, M. Mitochondrial haplogroups associated with lifestyle-related diseases and longevity in the Japanese population. Geriatr. Gerontol. Int. 10, S221–S235 (2010).
78. Rose, G. et al. No evidence of association between frontotemporal dementia and major European mtDNA haplogroups. Eur. J. Neurol. 15, 1006–1008 (2008).
79. SanGiovanni, J. P. et al. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS ONE 4, e5508 (2009).
80. Sanchez-Ferrero, E. et al. Mitochondrial DNA polymorphisms/haplogroups in hereditary spastic paraplegia. J. Neurol. 259, 246–250.
81. Santoro, A. et al. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer’s disease. PLoS ONE 5, e12037 (2012).
82. Sequeira, A. et al. Mitochondrial mutations and polymorphisms in psychiatric disorders. Front. Genet. 3, 103 (2012).
83. Tranah, G. J. Mitochondrial-nuclear epistasis: implications for human aging and longevity. Ageing Res. Rev. 10, 238–252 (2012).
84. McRae, A. F., Byrne, E. M., Zhao, Z. Z., Montgomery, G. W. & Visscher, P. M. Power and SNP tagging in whole mitochondrial genome association studies. Genome Res. 18, 911–917 (2008).
85. Battersby, B. J. & Shoubridge, E. A. Selection of a mtDNA sequence variant in hepatocytes of heteroplasmic mice is not due to differences in respiratory chain function or efficiency of replication. Hum. Mol. Genet. 10, 2469–2479 (2001).
86. Warburg, O., Wind, F. & Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 (1927).
87. Polyak, K. et al. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nature Genet. 20, 291–293 (1998).
88. Yu, M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 89, 65–71 (2011).
89. Melnick, P. J. Enzyme patterns of tumors demonstrated histochemically in cryostat sections. Ann. NY Acad. Sci.125, 689–715 (1965).
90. Pelicano, H. et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 175, 913–923 (2006).
91. Yu, M. et al. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life 59, 450–457 (2007).
92. Bai, R. K. et al. Mitochondrial DNA content varies with pathological characteristics of breast cancer. J. Oncol. 2011, 496189 (2011).
93. King, M. P., Koga, Y., Davidson, M. & Schon, E. A. Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNALeu(UUR) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol. Cell. Biol. 12, 480–490 (1992).
94. Miller, R. W. Dihydroorotate-quinone reductase of Neurospora crassa mitochondria. Arch. Biochem. Biophys. 146, 256–270 (1971).
95. Hayashi, J., Takemitsu, M. & Nonaka, I. Recovery of the missing tumorigenicity in mitochondrial DNA-less HeLa cells by introduction of mitochondrial DNA from normal human cells. Somat. Cell. Mol. Genet. 18, 123–129 (1992).
96. Morais, R. et al. Tumor-forming ability in athymic nude mice of human cell lines devoid of mitochondrial DNA. Cancer Res. 54, 3889–3896 (1994).
97. Peters, G. J. et al. In vivo inhibition of the pyrimidine de novo enzyme dihydroorotic acid dehydrogenase by brequinar sodium (DUP-785; NSC 368390) in mice and patients. Cancer Res. 50, 4644–4649 (1990).
98. Mourier, T., Hansen, A. J., Willerslev, E. & Arctander, P. The Human Genome Project reveals a continuous transfer of large mitochondrial fragments to the nucleus. Mol. Biol. Evol. 18, 1833–1837 (2001).
99. Ramos, A. et al. Nuclear insertions of mitochondrial origin: database updating and usefulness in cancer studies. Mitochondrion 11, 946–953 (2011).
100. Hazkani-Covo, E., Zeller, R. M. & Martin, W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 6, e1000834 (2010).
101. Lang, M. et al. Polymorphic Numts trace human population relationships. Hum. Genet. 131, 757–771 (2012).
102. Wang, D., Lloyd, A. H. & Timmis, J. N. Environmental stress increases the entry of cytoplasmic organellar DNA into the nucleus in plants. Proc. Natl Acad. Sci. USA 109, 2444–2448 (2012).
103. Cheng, X. & Ivessa, A. S. The migration of mitochondrial DNA fragments to the nucleus affects the chronological aging process of Saccharomyces cerevisiae. Aging Cell 9, 919–923 (2010).This is a fascinating example of how mtDNA fragments can be transferred to the nucleus in ‘real time’ (that is, during the lifetime of an individual).
104. Caro, P. et al. Mitochondrial DNA sequences are present inside nuclear DNA in rat tissues and increase with age. Mitochondrion 10, 479–486 (2010).
105. Goldin, E. et al. Transfer of a mitochondrial DNA fragment to MCOLN1 causes an inherited case of mucolipidosis IV. Hum. Mutat. 24, 460–465 (2004).
106. Turner, C. et al. Human genetic disease caused by de novo mitochondrial-nuclear DNA transfer. Hum. Genet. 112, 303–309 (2003).
107. Willett-Brozick, J. E., Savul, S. A., Richey, L. E. & Baysal, B. E. Germ line insertion of mtDNA at the breakpoint junction of a reciprocal constitutional translocation. Hum. Genet. 109, 216–223 (2001).
108. Storlazzi, C. T. et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome Res. 20, 1198–1206 (2010).
109. Valsesia, A. et al. Network-guided analysis of genes with altered somatic copy number and gene expression reveals pathways commonly perturbed in metastatic melanoma. PLoS ONE 6, e18369 (2011).
110. Hirano, T. et al. Co-localization of mitochondrial and double minute DNA in the nuclei of HL-60 cells but not normal cells. Mutat. Res. 425, 195–204 (1999).
111. Lee, J. H., Ryu, T. Y., Cho, C. H. & Kim, D. K. Different characteristics of mitochondrial microsatellite instability between uterine leiomyomas and leiomyosarcomas. Pathol. Oncol. Res. 17, 201–205 (2011).
112. Ellinger, J. et al. Circulating mitochondrial DNA in serum: A universal diagnostic biomarker for patients with urological malignancies. Urol. Oncol. 30, 509–515 (2012).
113. Kiebish, M. A. & Seyfried, T. N. Absence of pathogenic mitochondrial DNA mutations in mouse brain tumors. BMC Cancer 5, 102 (2005).
114. Ericson, N. G. et al. Decreased mitochondrial DNA mutagenesis in human colorectal cancer. PLoS Genet. 8, e1002689 (2012).
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 13 | DECEMBER 2012 | 889
© 2012 Macmillan Publishers Limited. All rights reserved

FURTHER INFORMATIONEric Schon’s homepage 1: http://web.neuro.columbia.edu/members/profiles.php?id=207Eric Schon’s homepage 2: http://asp.cumc.columbia.edu/facdb/profile_list.asp?DepAffil=Genetics&uni=eas3Salvatore DiMauro’s homepage: http://web.neuro.columbia.edu/members/profiles.php?id=24Michio Hirano’s homepage: http://web.neuro.columbia.edu/members/profiles.php?id=42
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
115. Chinnery, P. F., Samuels, D. C., Elson, J. & Turnbull, D. M. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet 360, 1323–1325 (2002).
116. Parr, R. L. et al. The pseudo-mitochondrial genome influences mistakes in heteroplasmy interpretation. BMC Genomics 7, 185 (2006).
117. Maki, J. et al. Mitochondrial genome deletion aids in the identification of false- and true-negative prostate needle core biopsy specimens. Am. J. Clin. Pathol. 129, 57–66 (2008).
118. Parr, R. L. et al. Somatic mitochondrial DNA mutations in prostate cancer and normal appearing adjacent glands in comparison to age-matched prostate samples without malignant histology. J. Mol. Diagn. 8, 312–319 (2006).
119. DiMauro, S. & Andreu, A. L. Mutations in mtDNA: are we scraping the bottom of the barrel? Brain Pathol. 10, 431–441 (2000).
120. Giordano, C. et al. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain 134, 220–234 (2011).
121. Kaufmann, P. et al. Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology 77, 1965–1971 (2011).
122. Tay, S. H. et al. Aortic rupture in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes. Arch. Neurol. 63, 281–283 (2006).
123. Chong, P. S., Vucic, S., Hedley-Whyte, E. T., Dreyer, M. & Cros, D. Multiple symmetric lipomatosis (Madelung’s disease) caused by the MERRF (A8344G) mutation: a report of two cases and review of the literature. J. Clin. Neuromuscul. Dis. 5, 1–7 (2003).
124. Suzuki, T., Nagao, A. & Suzuki, T. Human mitochondrial diseases caused by lack of taurine modification in mitochondrial tRNAs. Wiley Interdiscip. Rev. RNA 2, 376–386 (2011).
125. Hasegawa, H., Matsuoka, T., Goto, Y. & Nonaka, I. Cytochrome c oxidase activity is deficient in blood vessels of patients with myoclonus epilepsy with ragged-red fibers. Acta Neuropathol. 85, 280–284 (1993).
126. Naini, A. et al. Hypocitrullinemia in patients with MELAS: an insight into the “MELAS paradox”. J. Neurol. Sci. 229–230, 187–193 (2005).
127. Shiva, S., Brookes, P. S., Patel, R. P., Anderson, P. G. & Darley-Usmar, V. M. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc. Natl Acad. Sci. USA 98, 7212–7217 (2001).
128. Torres, J., Darley-Usmar, V. & Wilson, M. T. Inhibition of cytochrome c oxidase in turnover by nitric oxide: mechanism and implications for control of respiration. Biochem. J. 312, 169–173 (1995).
129. Rodriguez-Juarez, F., Aguirre, E. & Cadenas, S. Relative sensitivity of soluble guanylate cyclase and mitochondrial respiration to endogenous nitric oxide at physiological oxygen concentration. Biochem. J. 405, 223–231 (2007).
130. Koga, Y. et al. l‑arginine improves the symptoms of strokelike episodes in MELAS. Neurology 64, 710–712 (2005).
131. Horvath, R. et al. Molecular basis of infantile reversible cytochrome c oxidase deficiency myopathy. Brain 132, 3165–3174 (2009).An unexpected connection is presented in this paper between an mtDNA mutation and a mitochondrial disorder with an unusual course.
132. Uusimaa, J. et al. Reversible infantile respiratory chain deficiency is a unique, genetically heterogenous mitochondrial disease. J. Med. Genet. 48, 660–668 (2011).
133. Umeda, N. et al. Mitochondria-specific RNA-modifying enzymes responsible for the biosynthesis of the wobble base in mitochondrial tRNAs. Implications for the molecular pathogenesis of human mitochondrial diseases. J. Biol. Chem. 280, 1613–1624 (2005).
134. Yan, Q. et al. Human TRMU encoding the mitochondrial 5-methylaminomethyl-2-thiouridylate-methyltransferase is a putative nuclear modifier gene for the phenotypic expression of the deafness-associated 12S rRNA mutations. Biochem. Biophys. Res. Commun. 342, 1130–1136 (2006).
135. King, M. P. & Attardi, G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503 (1989).This was the first description of the use of cybrid technology in mammalian cells.
136. King, M. P. & Attardi, G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. 264, 304–313 (1996).
137. Engel, W. K. & Cunningham, G. G. Rapid examination of muscle tissue. An improved trichrome method for fresh-frozen biopsy sections. Neurology 13, 919–923 (1963).
138. Bonilla, E. et al. New morphological approaches to the study of mitochondrial encephalomyopathies. Brain Pathol. 2, 113–119 (1992).In this paper, a comprehensive survey is presented of morphological approaches used to diagnose and study mitochondrial diseases.
139. Quinzii, C. M. et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. 24, 3733–3743 (2010).
140. Loveland, B., Wang, C. R., Yonekawa, H., Hermel, E. & Lindahl, K. F. Maternally transmitted histocompatibility antigen of mice: a hydrophobic peptide of a mitochondrially encoded protein. Cell 60, 971–980 (1990).
141. Gogvadze, V., Orrenius, S. & Zhivotovsky, B. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol. 18, 165–173 (2008).
142. Madan, E. et al. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget 2, 948–957 (2011).
143. van Nederveen, F. H. et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 10, 764–771 (2009).
144. Tomlinson, I. P. et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nature Genet. 30, 406–410 (2002).
145. Selak, M. A. et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 7, 77–85 (2005).
146. Lemasters, J. J. & Holmuhamedov, E. Voltage-dependent anion channel (VDAC) as mitochondrial governator — thinking outside the box. Biochim. Biophys. Acta 1762, 181–190 (2006).
147. Majewski, N., Nogueira, V., Robey, R. B. & Hay, N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol. Cell. Biol. 24, 730–740 (2004).
148. Jain, M. et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044 (2012).
149. DiMauro, S., Hirano, M. & Schon, E. A. Approaches to the treatment of mitochondrial diseases. Muscle Nerve 34, 265–283 (2006).
AcknowledgementsThe authors were supported by grants from the US National Institutes of Health (HD32062), the US Department of Defense (W911NF-12-1-0159), the Muscular Dystrophy Association, the Ellison Medical Foundation, the Alzheimer Drug Discovery Foundation and the Marriott Mitochondrial Disorder Clinical Research Fund.
Competing interests statementThe authors declare competing financial interests: see Web version for details.
R E V I E W S
890 | DECEMBER 2012 | VOLUME 13 www.nature.com/reviews/genetics
© 2012 Macmillan Publishers Limited. All rights reserved

Seminar
www.thelancet.com Vol 379 May 12, 2012 1825
Lancet 2012; 379: 1825–34
Published OnlineApril 5, 2012DOI:10.1016/S0140-6736(11)61305-6
Department of Clinical Neurosciences, Institute of Neurology, University College London, London, UK (Prof A H V Schapira MD)
Correspondence to:Prof Anthony H V Schapira, Department of Clinical Neurosciences, Institute of Neurology, London NW3 2PF, [email protected]
Mitochondrial diseasesAnthony H V Schapira
Mitochondria have a crucial role in cellular bioenergetics and apoptosis, and thus are important to support cell function and in determination of cell death pathways. Inherited mitochondrial diseases can be caused by mutations of mitochondrial DNA or of nuclear genes that encode mitochondrial proteins. Although many mitochondrial disorders are multisystemic, some are tissue specifi c—eg, optic neuropathy, sensorineural deafness, and type 2 diabetes mellitus. In the past few years, several disorders have been associated with mutations of nuclear genes responsible for mitochondrial DNA maintenance and function, and the potential contribution of mitochondrial abnormalities to progressive neurodegenerative diseases such as Parkinson’s disease and Alzheimer’s disease has been recognised. The process of mitochondrial fi ssion-fusion has become a focus of attention in human disease. Importantly, the mitochondrion is now a target for therapeutic interventions that encompass small molecules, transcriptional regulation, and genetic manipulation, off ering opportunities to treat a diverse range of diseases.
IntroductionAdvances in genetics and cell biology have provided valuable insights into the function of mitochondria and into the contribution of defects of mitochondrial metabolism to human disease. Almost 200 diff erent mutations of mitochondrial DNA (mtDNA) have been reported in human beings. This Seminar provides an update on the pathogenesis of diseases (particularly neurodegenerative disorders) associated with mitochon-drial dysfunction, and reviews the emerging area of mitochondrial pharmacology. The appendix summarises the basic biology of mitochondria and disease.1–3 Other important areas of interest to mitochondrial biology and pathology that cannot be covered in detail here include neoplasia and anticancer therapies. The relation between neurodegeneration and cancer has been highlighted4 and the potential for somatic mutations of mtDNA to aff ect both the cell biology of neoplasia and the sensitivity or resistance of cells to chemotherapy has been noted.
Defi nitionAbnormalities of mitochondrial function that contribute or lead to human disease can derive from several sources. Mutations of mtDNA are a primary cause and can be due to inherited sequence changes in the mtDNA genome (eg, deletions, rearrangements, point mutations [table 1]), secondary to nuclear gene mutations causing, for example, mtDNA multiple deletions or depletion (table 2), or somatic—eg, as result of free-radical-mediated damage or faulty repair. Since the genome encodes 13 proteins of the respiratory chain, the resulting mutations of mtDNA usually, but not always, induce defects of respiratory chain function.
Primary respiratory chain diseases can be thought of as the result of mutations of mtDNA or nuclear genes encoding respiratory chain subunits (table 3). Mutations of nuclear genes encoding respiratory chain subunits tend to present early in infancy or childhood (although adult onset cases are described) and are usually fatal. Nuclear gene mutations can aff ect not only mtDNA structure (deletions) and content (depletion), but also assembly and maintenance of respiratory chain
complexes, causing secondary respiratory chain defects. Mutations in several nuclear genes encoding other mitochondrial proteins have been described. These mutations aff ect many mitochondrial functions and can have secondary eff ects on oxidative phosphorylation and other processes. Some of these mutations are consistently associated with specifi c phenotypes—eg, parkin and PINK1 with Parkinson’s disease, MFN2 with Charcot-Marie-Tooth disease type 2A, and frataxin with Friedreich’s ataxia (table 4). Thus mitochondrial diseases can be associated with several primary genetic causes aff ecting either genome and conceptually, need not be restricted to defects of the respiratory chain.
Mitochondrial dysfunction has been identifi ed in several other disorders—eg, Alzheimer’s disease, Hunt-ington’s disease, and amyotrophic lateral sclerosis—where either no causative mutation has been noted or the mitochondrial eff ects are secondary to biochemical abnormalities induced by the identifi ed mutation (eg, huntingtin in Huntington’s disease). Whether such mitochondrial abnormalities contribute to the patho-genesis of these diseases remains to be defi ned.
Mitochondrial quality controlMitochondria can divide (fi ssion) or fuse, and these processes are important to mitochondrial transport. Fission-fusion is regulated by several signalling proteins, such as mitofusins 1 and 2, which mediate fusion of mitochondrial outer membranes, and optic atrophy
Search strategy and selection criteria
I searched PubMed and Medline with the search terms “mitochondrial DNA”, “polymerase gamma”, “mitochondria”, “Parkinson’s disease”, “mitochondrial biogenesis” “neurodegeneration”, “diabetes”, “Alzheimer’s disease”, and “Huntington’s disease” for articles published between January, 2005, and July, 2011. Papers were also identifi ed from the references lists of relevant articles and through searches of my fi les. Only papers published in English or with abstracts in English were selected.
See Online for appendix

Seminar
1826 www.thelancet.com Vol 379 May 12, 2012
protein 1, a dynamin-like GTPase involved in fusion of the inner membranes. Dynamin-1-like protein 1 and fi ssion 1 homologue regulate mitochondrial fi ssion.5
An important function of the fi ssion-fusion process is to maintain the eff ectiveness, or quality, of cell mito-chondria.6 Defective mitochondrial function as a con-sequence of mtDNA or nuclear mutations, or as a result of certain external toxins, can reduce effi ciency of ATP synthesis and increase free-radical production. These factors will reduce membrane potential and the threshold for apoptotic cell death. Thus, repair or removal of defective mitochondria helps to maintain cell integrity and function. Part of the signalling system for mito-chondrial quality control involves the activation of the AMP-activated protein kinase, which in turn reduces func tion of the mammalian target of rapamycin complex 1. This complex is an important activator of mitochondrial metabolism and biogenesis, which might turn the balance in favour of mitochondrial fi ssion and destruction via autophagy (mitophagy) rather than fusion and biogenesis
(fi gure).5 Some evidence suggests that inhibition of mitochondrial fi ssion by knockdown of dynamin-1-like protein 1 or fi ssion 1 homologue can lead to an increase in the proportion of mutant mtDNA in cells carrying the mtDNA mutation A3243G, which is associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS).7 Knockout of mitofusin 2 in mice results in a substantial reduction in mtDNA content, which is followed by an increase in mtDNA mutation content and morphological abnormalities in skeletal muscle. These fi ndings further emphasise the importance of fi ssion-fusion to mtDNA mutation load, the potential expression of disease, and the opportunity to manipulate this process in favour of wildtype mtDNA.
Parkin and PTEN-induced putative kinase 1 (PINK1) proteins have an important role in the mitophagy path-way.8 Parkin is a ubiquitin protein ligase and translocates from the cytosol to the mitochondrion in response to a fall in mitochondrial membrane potential.9 This translocation might be preceded by phosphoryl ation of parkin by PINK1.10 Parkin is also involved in the ubiquitination of mitofusins 1 and 2.11 Mutations of the parkin, PINK1, and HTRA2 genes lead to Parkinson’s disease,12 and evidence is accumulating that defects of the mitophagy pathway contribute to the pathogenesis of this disease.13
That mitochondrial function declines with age has been known for some time, with an accumulation of mtDNA deletions (usually multiple), deteriorating res-piratory chain function, and evidence of increased damage mediated by free radicals.14–16 An increase in rates of mtDNA mutation or a reduction in cell antioxidant concentrations accelerates senescence,17 whereas upregulation of antioxidant enzymes can reverse this acceleration.18 Evidence suggests that mitochondrial quality control declines with age,19 which in turn will result in further free-radical-mediated damage from accumulating defective mitochondria. Protein disposal activity by the ubiquitin proteasomal and autophagy systems declines,20 which promotes protein accumula tion and aggregation. Thus the normal
Common genotype
Chronic progressive external ophthalmoplegia Single or multiple deletions
Kearns-Sayre syndrome Single deletions
Pearson marrow-pancreas syndrome Single deletions
Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes tRNA mutation (eg, 3243A→G)
Myoclonic epilepsy with ragged red fi bres tRNA mutation (eg, 8344A→G)
Leber’s hereditary optic neuropathy Complex I gene mutations (eg, 11778G→A, 14484 T→C, 3460 G→A)
Neurogenic ataxia with retinitis pigmentosa Complex V mutations (eg, 8993 T→G/C)
Myopathy Deletions (eg, 14709 T→C, cytochrome b)
Cardiomyopathy tRNA mutations (eg, 3243 A→G, 4269 A→G)
Diabetes or deafness, or both tRNA mutations (eg, 3243 A→G, 12258 C→A, 7445 A→G)
tRNA=transfer ribonucleic acid.
Table 1: Common mitochondrial DNA mutation diseases, by phenotype
mtDNA Phenotype
Polymerase gamma Multiple deletions, depletion CPEO (autosomal dominant or autosomal recessive)
Adenine nucleotide transporter 1 Multiple deletions CPEO
Twinkle Multiple deletions CPEO
Deoxyguanosine kinase Depletion Infantile hepatocerebral failure
Thymidine kinase 2 Depletion Infantile myopathy
Thymidine phosphorylase Multiple deletions, depletion Mitochondrial neurogastrointestinal encephalomyopathy
Ribonucleotide reductase Multiple deletions, depletion Kearns-Sayre syndrome, CPEO, encephalopathy, and tubulopathy
Succinate coenzyme A ligase Depletion Leigh’s syndrome, deafness, and methylmalonic aciduria
MPV17 encoding inner membrane protein
Depletion Infantile hepatocerebral failure
CPEO=chronic progressive external ophthalmoplegia.
Table 2: Non-oxidative phosphorylation nuclear gene defects aff ecting mtDNA structure or function, by protein

Seminar
www.thelancet.com Vol 379 May 12, 2012 1827
process of mito chondrial senescence promotes the same patho genetic pathways that are thought to bring about cell dysfunction in several neurodegenerative diseases.
mtDNA mutations and diseaseThe prevalence of mtDNA mutations is diffi cult to establish with accuracy, especially because of the high asymptomatic carrier rate. However, studies have shown a prevalence for specifi c mutations of 0·14–0·20%.21,22 mtDNA mutations are associated with a very wide range of clinical expression (table 1). Manifestations of mtDNA mutations vary from oligo symptomatic states (eg, isolated sensorineural deafness or type 2 diabetes) to complex multisystem syndromes that might involve neurological, endocrine, cardiological, dermatological, and gastro-enterological features. Many of these disorders have been reviewed extensively, in combination with the ideas of heteroplasmy, clonal expansion, mutation thresholds, and genetic bottleneck.1,2 Most mtDNA mutations function recessively, in that 60–90% mutation load is needed before a biochemical defect is noted. DNA sequence variations (nuclear or mitochondrial) can aff ect the expression of specifi c mtDNA mutations.23,24
mtDNA rearrangements in the form of deletions and a quantitative reduction of mtDNA concentrations (depletion) are important causes of disease.25 Single deletion populations are usually inherited from the germline, whereas multiple deletions and depletion are often the result of primary mutations in nuclear genes that are involved in mtDNA function. Mutations of the mitochondrial DNA polymerase gamma (POLG) gene cause multiple deletions or depletion of mtDNA alone or in combination.26 Patients might present in infancy with myopathy or hepatocerebral disease, in adulthood with ophthalmoplegia and myopathy, or with a more complex phenotype that could include myopathic features, encephalopathy, polyneuropathy, reduced fertility, and parkinsonism. Thus the genotype of mtDNA depletion is associated with a wide range of clinical manifestations and has many diff erent genetic causes (table 2).27
Several models of mitochondrial disease help to explain some of the mechanisms involved in pathogenesis— eg, yeast, transmitochondrial cybrids, and mouse models, which include those that incorporate mutations of nuclear genes that produce mtDNA deletions or depletion. Mouse models of POLG mutations have expressed features of accelerated senescence (progeria) and parkinsonism.17,28 Disruption of mitochondrial transcription factor A in mice also leads to loss of dopaminergic neurons and some elements of a parkinsonian phenotype.29
mtDNA mutations impair respiratory chain function and oxidative phosphorylation if present in suffi cient proportions, and so reduce cellular energy supply. Additionally, there could be an associated increase in free-radical production and potentially in excitotoxic-mediated damage.30 Mitochondria have an important role in mediation of cell signals for the initiation of apoptosis.31
A mutation in the mitochondrial-associated apoptosis initiating factor has been associated in infancy with X-linked mitochondrial encephalomyopathy that is partially responsive to ribofl avin.32 The relation between mtDNA mutations and their eff ect on cell survival and response to apoptotic signals or endoplasmic-reticulum-induced stress is likely to be complex.
Respiratory chain defectsThe fi ve protein complexes of the oxidative phos-phorylation system can be rendered defective by any of the mtDNA mutations outlined in this Seminar, or by mutations of nuclear genes encoding oxidative phos-phorylation proteins. Selective dysfunction of a complex can occur, but combinations of defects are more common (table 3). Pure complex I defects are seen as a result of mutations of mtDNA complex I genes, such as in Leber’s hereditary optic neuropathy, or of nuclear complex I genes, which often cause Leigh’s syndrome. Complex II defects are rare and caused by mutations in the nuclear genome. These defects can be associated with Leigh’s
Gene Common phenotype
Nuclear gene defects aff ecting individual oxidative phosphorylation proteins, by protein complex
Complex I NDUFS 1–4, 6–8, V1, V2, AI, A2, AII, FI, F2
Leigh’s syndrome
Complex II SDHA–SDHD Leigh’s syndrome, tumours
Complex III BCS1L Myopathy, encephalopathy, liver disease
Complex IV SURF1, SCO1, SCO2, COX10, COX15, COX6B1, FASTKD2, ETHE1
Leigh’s syndrome, encephalopathy, liver failure
Complex V ATPAF2, ATP5E Encephalopathy
Combined oxidative phosphorylation defects, by protein
Mitochondrial ribosomal protein S16 MRPS16 Agenesis of the corpus callosum, dysmorphism, and fatal neonatal lactic acidosis
Mitochondrial elongation factor G1 GFM Leigh’s syndrome
Mitochondrial elongation factor Tu TUFM Leigh’s syndrome
Mitochondrial translation elongation factor Ts
TSFM Encephalomyopathy, hypertrophic cardiomyopathy
Table 3: Nuclear gene mutations causing oxidative phosphorylation defects
Function Disease
Mitofusin 2 Dynamin-related protein Charcot-Marie-Tooth disease type 2
Coenzyme Q10 biosynthesis protein
Q10 function Ataxia, Leigh’s syndrome; hepatorenal failure
ABC7 Iron exporter X-linked ataxia/sideroblastic anaemia
Frataxin Iron storage protein Friedreich’s ataxia
Paraplegin Metalloprotease Hereditary spastic paraplegia
Optic atrophy protein 1 Dynamin-related protein Autosomal dominant optic atrophy
DJ1 Oxidant-sensing protein Parkinsonism
PTEN-induced kinase 1 Respiratory chain function, mtDNA maintenance, mitophagy
Parkinsonism
Parkin Mitophagy Juvenile parkinsonism
Table 4: Non-oxidative phosphorylation nuclear-encoded mitochondrial proteins and disease, by protein

Seminar
1828 www.thelancet.com Vol 379 May 12, 2012
syndrome, carotid body paragangliomas, and phaeo-chromocytomas. Complex III defects are rare either alone or in combination, but mutations of the mtDNA gene encoding cytochrome b or the nuclear BCS1L gene account for most described to date. Patients might present with myopathy alone or complex multisystem disorders. Complex IV (cytochrome oxidase) defects are common and can be caused by mutations of the COX mtDNA genes, nuclear genes of cytochrome oxidase subunits, or cytochrome oxidase assembly and main-tenance proteins. Clinical presentation can vary from pure myopathy to Leigh’s syndrome. Mutations of complex V (ATPase) genes in either mitochondrial or nuclear genomes have been described, with clinical eff ects of varying severity.
Management of diseases with mtDNA mutationsGuidelines for the diagnosis of mtDNA diseases have been published.33 Early attempts to treat primary mitochondrial disorders used supplements, vitamins, substrates, or electron carriers—eg, coenzyme Q10.34,35 However, many of these treatments were of little value and no randomised studies with a suffi ciently large number of patients to provide a defi nitive result were done. Coenzyme Q is of clear benefi t to patients with primary coenzyme Q10 defi ciency, and creatine is useful in the treatment of creatine defi ciency.36 Benefi t of L-arginine in MELAS on the basis of its vasodilatory action has been reported anecdotally,37 but further assess-ment is needed. Mitochondrial neurogastrointestinal encephalomyopathy is due to a defect of thymidine phosphorylase and might be the result of mutations in nuclear or mitochondrial genomes.38 The disorder can be treated temporarily with platelet infusions (that contain
thymidine phosphorylase), or more defi nitively with bone marrow transplantation.39
Alternative strategies such as resistance exercise training programmes have improved oxidative phos-phorylation but no change was noted in the mutant to wildtype ratio of deleted mtDNA.40 The changes in oxidative phos phorylation can be aff ected partly by an increase in activation of muscle satellite cells. Endurance training has improved oxidative phosphorylation capacity and exercise tolerance in patients with myopathy,41 but was accompanied by an increase in mutant mtDNA load in one study.42 A range of techniques to manipulate mutant mtDNA and its products are in early development.43
Genetic counselling for mtDNA diseases is a challenge because of the diffi culty with prediction of clinical expression in off spring.1 Importantly, appropriate coun-selling for mtDNA defects relies on an accurate molecular genetic diagnosis. Primary mtDNA point mutations such as those that cause MELAS and myo-clonic epilepsy with ragged red fi bres are inherited through the female line alone and can usually be detected in the blood. Aff ected men do not transmit the disease. Single mtDNA deletions usually arise in the maternal germline, are rarely present in the blood, and although off spring might be aff ected, subsequent transmission is uncommon. Multiple mtDNA deletions are usually secondary to a nuclear gene defect and, together with other mitochondrial diseases due to nuclear gene muta-tions, are subject to mendelian inheritance. Diag nosis by abnormal muscle morphology or defective respiratory chain enzyme function is not suffi cient to provide the basis for accurate counselling as both can be caused by mutations of either genome.
A novel strategy to prevent transmission that combines both counselling and treatment is mitochondrial gene replacement that involves the exchange of maternal mtDNA with that of a healthy donor.44,45 This technique requires in-vitro fertilisation with the parent ovum and sperm, removal of the pronucleus from the resulting zygote, and fusion into an enucleated donor oocyte (cytoplast). The reconstituted zygote can then be implanted into the mother’s uterus for development. Inevitably, this process requires the molecular diagnosis to have been made in the host woman (and excluded in the donor), and is an important potential therapy for female mutation carriers.
Mitochondrial dysfunction and Parkinson’s diseaseMitochondrial dysfunction in Parkinson’s disease was fi rst identifi ed in 1989, and accumulating evidence suggests a primary role in pathogenesis.46–48 An early consideration was whether mtDNA mutations might account for a proportion of Parkinson’s disease cases. However, no large-scale mtDNA rearrangements were recorded in brains of patients with Parkinson’s disease,49
Figure: A simplifi ed representation of the mitochondrial fi ssion-fusion cycleMitofusins 1 and 2 (MFN1 and MFN2) and optic atrophy protein 1 (OPA1) mediate mitochondrial fusion. Defects in mitochondrial function can promote fi ssion mediated through fi ssion 1 homologue (FIS1) and dynamin-1-like protein 1 (DRP1). Repaired daughter mitochondria re-enter the mitochondrial pool by fusion, and remaining defective mitochondria with reduced mitochondrial membrane potential are targeted by PTEN-induced putative kinase 1 (PINK1) and parkin to mediate destruction by autophagy (mitophagy).
Reduced membranepotential
Mitophagy
PINK1, parkin
Defectivemitochondria
FIS1, DRP1 Fission
Fusion
Healthymitochondria
MFN1, MFN2
OPA1

Seminar
www.thelancet.com Vol 379 May 12, 2012 1829
although more detailed analysis with laser capture of dopaminergic neurons from parkinsonian brains showed a greater proportion of deleted mtDNA than for age-matched controls.50 An analysis reported no bias towards either maternal inheritance or the mtDNA haplotype 10398G in Parkinson’s disease.51
However, mendelian genetics seem to have a substan-tial role in linkage of mitochondrial dysfunction to Parkinson’s disease.52 Three of the gene products causing autosomal recessive familial Parkinson’s disease have a mitochondrial location. PINK1 protein has been localised to mitochondria in several tissues.53 Its function is not completely understood, but the protein is important in mitochondrial free-radical metabolism, calcium homoeo-stasis, and mtDNA maintenance.54–56 Fibroblasts from patients with Parkinson’s disease with PINK1 mutations and PINK1 gene knockout models show defective oxidative phosphorylation, increased free radical damage, and reduced mitochondrial numbers.57–59
Parkin mutations predominantly cause parkinsonism in patients younger than 30 years. Mitochondrial dysfunction and increased oxidative stress have been described in parkin-defi cient Drosophila, mouse models, and periph-eral tissues in patients with parkin-mutation-positive Parkinson’s disease.60–62 Parkin is a ubiquitin E3 ligase and regulates expression of peroxisome-proliferator-activated receptor γ coactivator 1α (PGC1α) through interaction with parkin interacting substrate protein, which represses expression of PGC1α and nuclear respiratory factor 1.63 Importantly, the mutations of parkin or PINK1 that cause Parkinson’s disease seem to interfere with mitophagy effi ciency and result in accumulation of defective mito-chondria.11 This process can be reversed by upregulation of parkin or PINK1 proteins and the removal of defective mitochondria. The demonstration of abnormal expression of autophagy proteins in the brain of patients with Parkinson’s disease has further drawn attention to the importance of degradation pathways to the pathogenesis of the disease.64 These fi ndings off er intriguing oppor-tunities for the development of future therapies for disorders of impaired mitochondrial function. That up-regulation of the translation inhibitor 4E-BP counteracts the eff ects of PINK1 or parkin mutants in Drosophila should be noted. 65 Rapamycin, a drug that activates 4E-BP and autophagy, is also protective in these mutants.65
Mutations of DJ1 are a rare cause of familial Parkinson’s disease. DJ1-knockout mice had downregulated mito-chondrial uncoupling proteins 4 and 5, impaired calcium-induced uncoupling, and increased oxidant damage.66 DJ1 is thought to have a protective role in reduction of protein misfolding and aggregation that might be a result of oxidative stress and can reduce α-synuclein aggregation.67
Genetic causes of Parkinson’s disease, other than those encoding mitochondrial proteins, can also aff ect mito-chondrial function. α-synuclein is a major component of Lewy bodies and neurites present in the brains of patients
with Parkinson’s disease. Point mutations or multiplications of the α-synuclein gene cause familial Parkinson’s disease. α-synuclein is predominantly cytosolic but a fraction has been identifi ed in mito-chondria,68 and has been noted to interact directly with mitochondrial membranes, including at the neuronal synapse,69 and to inhibit complex I in a dose-dependent manner that shows regional expression of the protein.70,71
In addition to genetic causes, several environmental factors have been associated with both mitochondrial dysfunction and Parkinson’s disease or parkinsonism, including a range of mitochondrial complex I inhibitors that are toxic to dopaminergic neurons.72–74
Mitochondrial dysfunction and Alzheimer’s diseaseApart from age, important risk factors for Alzheimer’s disease include apolipoprotein ε4 status and mutations of amyloid precursor protein or presenilin. Investigators of several studies have reported abnormalities of mito-chondrial structure or function, or mtDNA defects in the brain or other tissues and cells of patients with Alzheimer’s disease, but the presence and relevance of these fi ndings have remained controversial, and the data derived have not always been reproducible.
However, new fi ndings have brought a diff erent perspective to mitochondrial involvement in Alzheimer’s disease pathogenesis. Polymorphism of the TOMM40 gene (2 kilobases away from the APOE 4 gene on chromosome 19) seems to be an important risk factor for Alzheimer’s disease and age of onset.75 TOMM40 is an outer mitochondrial membrane protein that forms part of a pore that serves as the import site for cytoplasmic proteins to enter the mitochondrion. Amyloid precursor protein accumulates in this pore.76 Presenilins 1 and 2, and γ-secretase are associated with the mitochondria-associated membrane,77 a connection site between the endoplasmic reticulum and the mitochondrion that is dependent on mitofusin 2 function.78 The mitochondria-associated membrane plays an important part in lipid metabolism, including the synthesis of phosphatidyl-ethanolamine, which is transported into mitochondria and has a modulatory role in tau phosphorylation. Mitochondria-associated membranes also contain acyl-CoA cholesterol acyltransferase—an important enzyme in cholesterol metabolism that is needed for the formation of amyloid β,79 which in turn can localise to mitochondria.80 Impaired mitochondria-associated membrane function could also aff ect intracellular calcium homoeostasis and be relevant to calcium dysregulation by the endoplasmic reticulum and abnormal neuronal calcium handling detected in Alzheimer’s disease models and patients.81,82 The mitochondria-associated membrane is rich in inositol-1,4,5-trisphosphate receptors that bring about calcium traffi cking.83 Both presenilins 1 and 2 bind to these receptors, which substantially increases intracellular calcium and amyloid β production.83

Seminar
1830 www.thelancet.com Vol 379 May 12, 2012
Triple transgenic mice expressing amyloid and tangle pathology similar to that noted in Alzheimer’s disease had pronounced mitochondrial abnormalities,84 such as reduced oxidative phosphorylation, decreased activities of complexes I and IV, lowered mitochondrial membrane potential, and increased free-radical generation. Thus, the potential contribution of mitochondria to Alzheimer’s disease continues to develop from a primary role in terms of mtDNA mutations or polymorphisms, to a pivotal role in mediation of downstream biochemical events that aff ect intracellular bioenergetics and homoeostasis.
Mitochondrial dysfunction and Huntington’s diseaseHuntington’s disease is caused by a triplet repeat expansion in the huntingtin gene encoding an enlarged polyglutamine sequence in the mature protein. Mito-chondrial defects have been described in patients with Huntington’s disease in vivo, in aff ected brains post mortem, and in cell and animal models of the disease.85–88
The mitochondrial defects in Huntington’s disease are associated with abnormalities of calcium handling, increased susceptibility to calcium-induced opening of the mitochondrial permeability pore, and reduced respiration.89 Evidence shows that mutant huntingtin protein associates with mitochondrial membranes and can impair axonal traffi cking of mitochondria and reduce synaptic ATP concentrations.90 Mutant huntingtin protein interacts with, and increases the sensitivity of, the inositol-1,4,5-trisphosphate receptor at the mitochondria-associated membrane and thus contributes to calcium dysregulation in Huntington’s disease.91
Mutant huntingtin protein also sensitises cells to free-radical-mediated damage, reduces ATP production and mitochondrial fusion, and induces increased fragmen-tation—changes that were prevented by overexpression of mitofusin 2.92 The role of mitochondrial quality control is further supported by the fi nding that the calcineurin-mediated dephosphorylation of dynamin-like protein 1 is increased in cells of patients with Huntington’s disease and leads to increases in mitochondrial translocation, promotion of fragmentation, and an increased cell susceptibility to apoptosis.93 Mutant huntingtin protein has an important role in transcription regulation, and the expression of a range of mitochondrial proteins is modifi ed in Huntington’s disease striatal neurons—such as downregulation of cytochrome oxidase subunit 2, mitofusin 1, mitochondrial transcription factor A, and PGC1α—correlating with disease severity and increased expression of dynamin-like protein 1.94
The regulation of PGC1α by mutant huntingtin protein is of particular interest because of the potential to manipulate the expression of this molecule with drugs. Mutant huntingtin protein binds to the PGC1α promoter and blocks transcription of target genes in Huntington’s disease models and patient muscle.95,96 In view of the important regulatory role for PGC1α in mitochondrial
biogenesis, this process might contribute to the mito-chondrial dysfunction seen in Huntington’s disease, including impaired defence against free radicals.97
Other neurodegenerative diseasesMitochondrial dysfunction has been identifi ed in several other neurodegenerative diseases. Secondary abnor-malities of mitochondrial morphology and function have been recorded in amyotrophic latereral sclerosis,98 whereas in other disorders the causative gene mutation involves a mitochondrial protein (table 4)—eg, Friedreich’s ataxia99 and hereditary spastic paraplegia.100 Mutations in the MFN2 gene are a common cause of autosomal dominant Charcot-Marie-Tooth type 2 disease—an early-onset axonal sensorimotor neuropathy.101 A proportion of patients have additional abnormalities, such as optic atrophy or deafness.102 Mutations of OPA1 cause autosomal dominant optic atrophy, but the phenotype can include peripheral neuropathy, deafness, ataxia, and ophthal-moplegia with multiple mtDNA deletions.103 The part that both mitofusin 2 and optic atrophy protein 1 play in fi ssion-fusion might at least partly explain both the patho-physiology of neuronal–axonal dysfunction and the overlapping phenotypes of mutations aff ecting these proteins,104 although an additional role in mtDNA maintenance cannot be excluded.
Mitochondrial dysfunction and visual failureMitochondrial dysfunction has been a topic of interest to ophthalmological practice since the identifi cation of mtDNA mutations as a cause of Leber’s hereditary optic neuropathy.105 OPA1 mutations are a cause of optic atrophy, and mtDNA mutations associated with enceph-alomyopathies can be associated with ophthalmoplegia or retinal pigmentation. Attention is now focused on the potential involvement of mitochondria in glaucoma. The cause of glaucoma is multifactorial; age and ocular pressure are risk factors, although the disease can occur at normal ocular pressures. Age-related mitochondrial dysfunction and oxidative stress might contribute to risk for the development of glaucoma.106,107 Nucci and colleagues108 reported evidence of excitotoxic-mediated damage to the retina in glaucoma; mitochondrial dysfunction, specifi cally that induced, for example, by the A3243G mtDNA mutation, lowers the threshold for excitotoxic eff ects.30
Mitochondrial dysfunction and diabetes mellitusDiabetes mellitus is a recognised feature of many mitochondrial disorders. It can be the sole expression of mtDNA mutations, but is also frequently recorded as part of an encephalomyopathy (eg, MELAS), and is associated with Friedreich’s ataxia and Huntington’s disease. Mitochondria play a crucial part in glucose signalling for insulin release and mitochondrial defects impair this process, as in turn does persistent hyper-glycaemia, leading to mitochondrial dysfunction and

Seminar
www.thelancet.com Vol 379 May 12, 2012 1831
reduced insulin release.109 Many mechanisms are asso-ciated with the pathogenesis of type 2 diabetes, including impaired mitochondrial capacity or function; altered insulin signalling due to cellular lipid accumulation, proinfl ammatory signals, and endoplasmic reticulum stress; and reduced incretin-dependent and incretin-independent cell insulin secretion.110
Abnormal mitochondrial metabolism with reduced mitochondrial mass has been detected in muscle biopsies, and in vivo in patients with type 2 diabetes and people who are obese.111 Defects specifi c for patients with type 2 diabetes—eg, impaired response to exercise stimulus— have been detected.112 Reduced mitochondrial mass is associated with a 20–30% global reduction in nuclear-encoded subunits of oxidative phosphorylation.113 Several factors probably aff ect the contribution of mitochondrial metabolism to type 2 diabetes, including genetic contri-bution to oxidative phosphorylation enzymes114 and expression of PINK1, which is suppressed in type 2 diabetic muscle.115 Similar mitochondrial abnormalities are noted in liver and adipose tissue. Thus, increasing evidence shows that mitochondrial dysfunction is present in type 2 diabetes and could participate in a complex interacting cycle to promote cell damage and insulin resistance. However, some of the mitochondrial changes could be a response, rather than a cause, of the metabolic abnormalities.116
The decrease in mitochondrial mass and downregulation of oxidative phosphorylation subunits might also be secondary to the reduced expression of PGC1α and PGC1β (appendix), which is detected both in patients with type 2 diabetes and in insulin resistance.117 Manipulation of PGC concentrations has therefore been of interest in the potential treatment of type 2 diabetes.118
Mitochondria as a therapeutic targetSubstrates, carriers, and antioxidantsThe discovery of mitochondrial defects in several common neurodegenerative diseases provided the basis and suffi cient numbers of patients to begin to assess applicability of compounds such as coenzyme Q10 and creatine to treat disorders of mitochondrial function.119 Coenzyme Q10 is an electron carrier and antioxidant. At a dose of 1200 mg per day, coenzyme Q10 seemed to slow progression in a pilot study in early Parkinson’s disease,120 although other studies using similar bio-available doses have not shown benefi t.121 Further studies of coenzyme Q10 in early Parkinson’s disease are underway, but preliminary results suggest that these are negative. Coenzyme Q10 seem ed to slow progression in Huntington’s disease, but this eff ect was not signifi cant.122 In Friedreich’s ataxia, a combination of coenzyme Q10 and vitamin E improved neurological progression compared with 4 year cross-sectional natural progression data.123 A low dose was as eff ective as high-dose therapy in improving function in patients with coexisting coenzyme Q defi ciency.124 Idebenone,
another electron carrier, did not show benefi t in a 6 month controlled trial in Friedreich’s ataxia,125 although other studies have shown improved cardiac function.126 Modifi ed agents have been produced—eg, Mito-Q, which has enhanced mitochondrial penetration and activity in vitro.127
In a small study of patients with Parkinson’s disease, creatine was of no benefi t,128 but it did improve motor scores in a larger randomised futility analysis.129 Some clinical benefi t has been associated with doses of 10 g of creatine daily in Huntington’s disease, 130 and concen-trations of a peripheral marker of oxidative damage were also reduced.131
Transcription regulationAn interesting strategy for reversing the eff ects of mitochondrial dysfunction might be through the regulation of mitochondrial protein transcription and biogenesis. PGC1α is an important regulator of mito-chondrial activity (appendix) and functions together with sirtuin 1 to alter genetic programmes for mitochondrial biogenesis. SIRT1 is one of a family of situins, known to catalyse NAD+-dependent protein deacetylation, yielding nicotinamide and O-acetyl-ADP-ribose; and to regulate longevity, apoptosis, and DNA repair.132 SIRT1 interacts directly with and deacetylates PGC1α and increases its activity, leading to the induction of gene transcription. Both proteins promote adaptation to caloric restriction by regulation of the genetic programmes for gluco-neogenesis and glycolysis.133 Activation of peroxisome-proliferator-activated receptor γ transactivates target genes with the support of PGC1α. Drugs that activate this receptor include rosiglitazone, pioglitazone, and troglitazone, which are used to treat type 2 diabetes.
Resveratrol is a natural polyphenolic compound found in the skin of grapes that substantially increases SIRT1 activity.134 Resveratrol enhances PGC1α activity, increases mitochondrial biogenesis, decreases insulin resistance, and renders animals resistant to diet-induced obesity.135 Resveratrol and bezafi brate, another agonist, have shown protective properties in animal models of Parkinson’s disease,136 Huntington’s disease,137,138 Alzheimer’s disease,139 and other diseases including a mouse model of mito-chondrial myopathy.140
Importantly, the upregulation of mitochondrial bio-genesis through the PGC1α pathway potentially targets all mitochondria (wildtype and mutant) and so could have applications in diseases caused by mtDNA muta-tions and in other disorders associated with mitochondrial dysfunction.Confl icts of interestI declare that I have no confl icts of interest.
References1 Schapira AHV. Mitochondrial disease. Lancet 2006; 368: 70–82.2 Duchen MR, Szabadkai G. Roles of mitochondria in human
disease. Essays Biochem 2010; 47: 115–37.3 McFarland R, Taylor RW, Turnbull DM. A neurological perspective
on mitochondrial disease. Lancet Neurol 2010; 9: 829–40.
For more information about the coenzyme Q10 studies see http://clinicaltrials.gov/ct2/show/CT00740714?term=q10&rank=7

Seminar
1832 www.thelancet.com Vol 379 May 12, 2012
4 Plun-Favreau H, Lewis PA, Hardy J, Martins LM, Wood NW. Cancer and neurodegeneration: between the devil and the deep blue sea. PLoS Genet 2010; 6: e1001257.
5 de Castro I, Martins LM, Tufi R. Mitochondrial quality control and neurological disease: an emerging connection. Expert Rev Mol Med 2010; 12: e12.
6 Dagda RK, Chu CT. Mitochondrial quality control: insights on how Parkinson's disease related genes PINK1, parkin, and Omi/HtrA2 interact to maintain mitochondrial homeostasis. J Bioenerg Biomembr 2009; 41: 473–79.
7 Malena A, Loro E, Di RM, Holt IJ, Vergani L. Inhibition of mitochondrial fi ssion favours mutant over wild-type mitochondrial DNA. Hum Mol Genet 2009; 18: 3407–16.
8 Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA 2008; 105: 1638–43.
9 Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183: 795–803.
10 Kim Y, Park J, Kim S, et al. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun 2008; 377: 975–80.
11 Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 2010; 19: 4861–70.
12 Schapira AH. Etiology of Parkinson’s disease. Neurology 2006; 66 (10 suppl 4): S10–S23.
13 Gegg ME, Schapira AH. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: Implications for Parkinson disease pathogenesis. Autophagy 2011; 7: 243–45.
14 Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem 2010; 79: 683–706.
15 Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen 2010; 51: 440–50.
16 Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 2006; 38: 518–20.
17 Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004; 429: 417–23.
18 Schriner SE, Linford NJ, Martin GM, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005; 308: 1909–11.
19 Luce K, Weil AC, Osiewacz HD. Mitochondrial protein quality control systems in aging and disease. Adv Exp Med Biol 2010; 694: 108–25.
20 Bandyopadhyay U, Cuervo AM. Chaperone-mediated autophagy in aging and neurodegeneration: lessons from alpha-synuclein. Exp Gerontol 2007; 42: 120–28.
21 Bitner-Glindzicz M, Pembrey M, Duncan A, et al. Prevalence of mitochondrial 1555A→G mutation in European children. N Engl J Med 2009; 360: 640–42.
22 Vandebona H, Mitchell P, Manwaring N, et al. Prevalence of mitochondrial 1555A→G mutation in adults of European descent. N Engl J Med 2009; 360: 642–44.
23 Cai W, Fu Q, Zhou X, Qu J, Tong Y, Guan MX. Mitochondrial variants may infl uence the phenotypic manifestation of Leber's hereditary optic neuropathy-associated ND4 G11778A mutation. J Genet Genomics 2008; 35: 649–55.
24 Cock HR, Tabrizi SJ, Cooper JM, Schapira AH. The infl uence of nuclear background on the biochemical expression of 3460 Leber's hereditary optic neuropathy. Ann Neurol 1998; 44: 187–93.
25 Copeland WC. Inherited mitochondrial diseases of DNA replication. Annu Rev Med 2008; 59: 131–46.
26 Taanman JW, Rahman S, Pagnamenta AT, et al. Analysis of mutant DNA polymerase gamma in patients with mitochondrial DNA depletion. Hum Mutat 2009; 30: 248–54.
27 Rahman S, Poulton J. Diagnosis of mitochondrial DNA depletion syndromes. Arch Dis Child 2009; 94: 3–5.
28 Williams SL, Huang J, Edwards YJ, et al. The mtDNA mutation spectrum of the progeroid Polg mutator mouse includes abundant control region multimers. Cell Metab 2010; 12: 675–82.
29 Ekstrand MI, Terzioglu M, Galter D, et al. Progressive parkinsonism in mice with respiratory-chain-defi cient dopamine neurons. Proc Natl Acad Sci USA 2007; 104: 1325–30.
30 DiFrancesco JC, Cooper JM, Lam A, et al. MELAS mitochondrial DNA mutation A3243G reduces glutamate transport in cybrids cell lines. Exp Neurol 2008; 212: 152–56.
31 Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet 2009; 43: 95–118.
32 Ghezzi D, Sevrioukova I, Invernizzi F, et al. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet 2010; 86: 639–49.
33 Finsterer J, Harbo HF, Baets J, et al. EFNS guidelines on the molecular diagnosis of mitochondrial disorders. Eur J Neurol 2009; 16: 1255–64.
34 Morgan-Hughes JA, Sweeney MG, Cooper JM, et al. Mitochondrial DNA (mtDNA) diseases: correlation of genotype to phenotype. Biochim Biophys Acta 1995; 1271: 135–40.
35 Chinnery P, Majamaa K, Turnbull D, Thorburn D. Treatment for mitochondrial disorders. Cochrane Database Syst Rev 2006; 1: CD004426.
36 Montini G, Malaventura C, Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 defi ciency. N Engl J Med 2008; 358: 2849–50.
37 Moutaouakil F, El Otmani H, Fadel H, Sefrioui F, Slassi I. l-arginine effi ciency in MELAS syndrome. A case report. Rev Neurol (Paris) 2009; 165: 482–85 (in French).
38 Taanman JW, Daras M, Albrecht J, et al. Characterization of a novel TYMP splice site mutation associated with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Neuromuscul Disord 2009; 19: 151–54.
39 Halter J, Schupbach WM, Casali C, et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant 2011; 46: 330–37
40 Murphy JL, Blakely EL, Schaefer AM, et al. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain 2008; 131: 2832–40.
41 Jeppesen TD, Duno M, Schwartz M, et al. Short- and long-term eff ects of endurance training in patients with mitochondrial myopathy. Eur J Neurol 2009; 16: 1336–39.
42 Taivassalo T, Shoubridge EA, Chen J, et al. Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic eff ects. Ann Neurol 2001; 50: 133–41.
43 Kyriakouli DS, Boesch P, Taylor RW, Lightowlers RN. Progress and prospects: gene therapy for mitochondrial DNA disease. Gene Ther 2008; 15: 1017–23.
44 Tachibana M, Sparman M, Sritanaudomchai H, et al. Mitochondrial gene replacement in primate off spring and embryonic stem cells. Nature 2009; 461: 367–72.
45 Craven L, Tuppen HA, Greggains GD, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 2010; 465: 82–85.
46 Schapira AHV, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I defi ciency in Parkinson's disease. Lancet 1989; 333: 1269.
47 Schapira AHV, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I defi ciency in Parkinson’s disease. J Neurochem 1990; 54: 823–27.
48 Schapira AHV. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol 2008; 7: 97–109.
49 Mann VM, Cooper JM, Schapira AH. Quantitation of a mitochondrial DNA deletion in Parkinson’s disease. FEBS Lett 1992; 299: 218–22.
50 Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 2006; 38: 515–17.
51 Simon DK, Pankratz N, Kissell DK, et al. Maternal inheritance and mitochondrial DNA variants in familial Parkinson's disease. BMC Med Genet 2010; 11: 53.
52 Schapira AH, Agid Y, Barone P, et al. Perspectives on recent advances in the understanding and treatment of Parkinson's disease. Eur J Neurol 2009; 16: 1090–99.

Seminar
www.thelancet.com Vol 379 May 12, 2012 1833
53 Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304: 1158–60.
54 Abramov AY, Gegg M, Grunewald A, Wood NW, Klein C, Schapira AH. Bioenergetic consequences of PINK1 mutations in Parkinson disease. PLoS One 2011; 6: e25622.
55 Gandhi S, Wood-Kaczmar A, Yao Z, et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 2009; 33: 627–38.
56 Gegg ME, Cooper JM, Schapira AH, Taanman JW. Silencing of PINK1 expression aff ects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS One 2009; 4: e4756.
57 Hoepken HH, Gispert S, Morales B, et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol Dis 2007; 25: 401–11.
58 Grunewald A, Gegg ME, Taanman JW, et al. Diff erential eff ects of PINK1 nonsense and missense mutations on mitochondrial function and morphology. Exp Neurol 2009; 219: 266–73.
59 Gispert S, Ricciardi F, Kurz A, et al. Parkinson phenotype in aged PINK1-defi cient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One 2009; 4: e5777.
60 Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 2003; 100: 4078–83.
61 Muftuoglu M, Elibol B, Dalmizrak O, et al. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov Disord 2004; 19: 544–48.
62 Grunewald A, Voges L, Rakovic A, et al. Mutant Parkin impairs mitochondrial function and morphology in human fi broblasts. PLoS One 2010; 5: e12962.
63 Shin JH, Ko HS, Kang H, et al. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell 2011; 144: 689–702.
64 Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 2010; 67: 1464–72.
65 Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci 2009; 12: 1129–35.
66 Guzman JN, Sanchez-Padilla J, Wokosin D, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010; 468: 696–700.
67 Batelli S, Albani D, Rametta R, et al. DJ-1 modulates alpha-synuclein aggregation state in a cellular model of oxidative stress: relevance for Parkinson’s disease and involvement of HSP70. PLoS One 2008; 3: e1884.
68 Li WW, Yang R, Guo JC, et al. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 2007; 18: 1543–46.
69 Nakamura K, Nemani VM, Wallender EK, Kaehlcke K, Ott M, Edwards RH. Optical reporters for the conformation of alpha-synuclein reveal a specifi c interaction with mitochondria. J Neurosci 2008; 28: 12305–17.
70 Liu G, Zhang C, Yin J, et al. alpha-Synuclein is diff erentially expressed in mitochondria from diff erent rat brain regions and dose-dependently down-regulates complex I activity. Neurosci Lett 2009; 454: 187–92.
71 Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 2008; 283: 9089–100.
72 Warner TT, Schapira AH. Genetic and environmental factors in the cause of Parkinson's disease. Ann Neurol 2003; 53 (suppl 3): S16–S23.
73 Schapira AH. Complex I: inhibitors, inhibition and neurodegeneration. Exp Neurol 2010; 224: 331–35.
74 Hollerhage M, Matusch A, Champy P, et al. Natural lipophilic inhibitors of mitochondrial complex I are candidate toxins for sporadic neurodegenerative tau pathologies. Exp Neurol 2009; 220: 133–42.
75 Roses AD, Lutz MW, Amrine-Madsen H, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer's disease. Pharmacogenomics J 2010; 10: 375–84.
76 Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci 2006; 26: 9057–68.
77 Area-Gomez E, de Groof AJ, Boldogh I, et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 2009; 175: 1810–16.
78 de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008; 456: 605–10.
79 Puglielli L, Konopka G, Pack-Chung E, et al. Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat Cell Biol 2001; 3: 905–12.
80 Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 2006; 15: 1437–49.
81 Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem 2010; 285: 12463–68.
82 Supnet C, Bezprozvanny I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 2010; 47: 183–89.
83 Cheung KH, Mei L, Mak DO, et al. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal 2010; 3: ra22.
84 Rhein V, Song X, Wiesner A, et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc Natl Acad Sci USA 2009; 106: 20057–62.
85 Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol 1996; 39: 385–89.
86 Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann Neurol 1999; 45: 25–32.
87 Browne SE, Bowling AC, MacGarvey U, et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 1997; 41: 646–53.
88 Lodi R, Schapira AH, Manners D, et al. Abnormal in vivo skeletal muscle energy metabolism in Huntington’s disease and dentatorubropallidoluysian atrophy. Ann Neurol 2000; 48: 72–76.
89 Lim D, Fedrizzi L, Tartari M, et al. Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J Biol Chem 2008; 283: 5780–89.
90 Orr AL, Li S, Wang CE, et al. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial traffi cking. J Neurosci 2008; 28: 2783–92.
91 Kaltenbach LS, Romero E, Becklin RR, et al. Huntingtin interacting proteins are genetic modifi ers of neurodegeneration. PLoS Genet 2007; 3: e82.
92 Wang H, Lim PJ, Karbowski M, Monteiro MJ. Eff ects of overexpression of huntingtin proteins on mitochondrial integrity. Hum Mol Genet 2009; 18: 737–52.
93 Costa V, Giacomello M, Hudec R, et al. Mitochondrial fi ssion and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol Med 2010; 2: 490–503.
94 Kim J, Moody JP, Edgerly CK, et al. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum Mol Genet 2010; 19: 3919–35.
95 Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006; 127: 59–69.
96 Chaturvedi RK, Adhihetty P, Shukla S, et al. Impaired PGC-1alpha function in muscle in Huntington’s disease. Hum Mol Genet 2009; 18: 3048–65.
97 St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006; 127: 397–408.
98 Martin LJ. Mitochondrial pathobiology in Parkinson’s disease and amyotrophic lateral sclerosis. J Alzheimers Dis 2010; 20 (suppl 2): S335–S356.

Seminar
1834 www.thelancet.com Vol 379 May 12, 2012
99 Santos R, Lefevre S, Sliwa D, Seguin A, Camadro JM, Lesuisse E. Friedreich ataxia: molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid Redox Signal 2010; 13: 651–90.
100 Ferreirinha F, Quattrini A, Pirozzi M, et al. Axonal degeneration in paraplegin-defi cient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest 2004; 113: 231–42.
101 Calvo J, Funalot B, Ouvrier RA, et al. Genotype-phenotype correlations in Charcot-Marie-Tooth disease type 2 caused by mitofusin 2 mutations. Arch Neurol 2009; 66: 1511–16.
102 Del BR, Moggio M, Rango M, et al. Mutated mitofusin 2 presents with intrafamilial variability and brain mitochondrial dysfunction. Neurology 2008; 71: 1959–66.
103 Hudson G, Amati-Bonneau P, Blakely EL, et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain 2008; 131: 329–37.
104 Cho DH, Nakamura T, Lipton SA. Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol Life Sci 2010; 67: 3435–47.
105 Carelli V, La MC, Valentino ML, Barboni P, Ross-Cisneros FN, Sadun AA. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta 2009; 1787: 518–28.
106 Kong GY, Van Bergen NJ, Trounce IA, Crowston JG. Mitochondrial dysfunction and glaucoma. J Glaucoma 2009; 18: 93–100.
107 Lascaratos G, Garway-Heath DF, Willoughby CE, Chau KY, Schapira AH. Mitochondrial dysfunction in glaucoma: understanding genetic infl uences. Mitochondrion 2012; 12: 202–12.
108 Nucci C, Tartaglione R, Rombola L, Morrone LA, Fazzi E, Bagetta G. Neurochemical evidence to implicate elevated glutamate in the mechanisms of high intraocular pressure (IOP)-induced retinal ganglion cell death in rat. Neurotoxicology 2005; 26: 935–41.
109 Jitrapakdee S, Wutthisathapornchai A, Wallace JC, MacDonald MJ. Regulation of insulin secretion: role of mitochondrial signalling. Diabetologia 2010; 53: 1019–32.
110 Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev 2010; 31: 364–95.
111 Ritov VB, Menshikova EV, Azuma K, et al. Defi ciency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am J Physiol Endocrinol Metab 2010; 298: E49–E58.
112 Kacerovsky-Bielesz G, Chmelik M, Ling C, et al. Short-term exercise training does not stimulate skeletal muscle ATP synthesis in relatives of humans with type 2 diabetes. Diabetes 2009; 58: 1333–41.
113 Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 2003; 100: 8466–71.
114 Ronn T, Poulsen P, Tuomi T, et al. Genetic variation in ATP5O is associated with skeletal muscle ATP50 mRNA expression and glucose uptake in young twins. PLoS One 2009; 4: e4793.
115 Scheele C, Nielsen AR, Walden TB, et al. Altered regulation of the PINK1 locus: a link between type 2 diabetes and neurodegeneration? FASEB J 2007; 21: 3653–65.
116 Schiff M, Loublier S, Coulibaly A, Benit P, de Baulny HO, Rustin P. Mitochondria and diabetes mellitus: untangling a confl ictive relationship? J Inherit Metab Dis 2009; 32: 684–98.
117 Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34: 267–73.
118 Lira VA, Benton CR, Yan Z, Bonen A. PGC-1alpha regulation by exercise training and its infl uences on muscle function and insulin sensitivity. Am J Physiol Endocrinol Metab 2010; 299: E145–E161.
119 Kerr DS. Treatment of mitochondrial electron transport chain disorders: a review of clinical trials over the past decade. Mol Genet Metab 2010; 99: 246–55.
120 Shults CW, Oakes D, Kieburtz K, et al. Eff ects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol 2002; 59: 1541–50.
121 Storch A, Jost WH, Vieregge P, et al. Randomized, double-blind, placebo-controlled trial on symptomatic eff ects of coenzyme Q(10) in Parkinson disease. Arch Neurol 2007; 64: 938–44.
122 The Huntington Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington's disease. Neurology 2001; 57: 397–404.
123 Hart PE, Lodi R, Rajagopalan B, et al. Antioxidant treatment of patients with Friedreich ataxia: four-year follow-up. Arch Neurol 2005; 62: 621–26.
124 Cooper JM, Korlipara LV, Hart PE, Bradley JL, Schapira AH. Coenzyme Q10 and vitamin E defi ciency in Friedreich’s ataxia: predictor of effi cacy of vitamin E and coenzyme Q10 therapy. Eur J Neurol 2008; 15: 1371–19.
125 Lynch DR, Perlman SL, Meier T. A phase 3, double-blind, placebo-controlled trial of idebenone in Friedreich ataxia. Arch Neurol 2010; 67: 941–47.
126 Di Prospero NA, Baker A, Jeff ries N, Fischbeck KH. Neurological eff ects of high-dose idebenone in patients with Friedreich’s ataxia: a randomised, placebo-controlled trial. Lancet Neurol 2007; 6: 878–86.
127 Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol 2007; 47: 629–56.
128 Bender A, Koch W, Elstner M, et al. Creatine supplementation in Parkinson disease: a placebo-controlled randomized pilot trial. Neurology 2006; 67: 1262–64.
129 NINDS NET-PD Investigators. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology 2006; 66: 664–71.
130 Tabrizi SJ, Blamire AM, Manners DN, et al. High-dose creatine therapy for Huntington disease: a 2-year clinical and MRS study. Neurology 2005; 64: 1655–56.
131 Hersch SM, Gevorkian S, Marder K, et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2'dG. Neurology 2006; 66: 250–52.
132 Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem 2004; 73: 417–35.
133 Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005; 434: 113–18.
134 Howitz KT, Bitterman KJ, Cohen HY, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003; 425: 191–96.
135 Lagouge M, Argmann C, Gerhart-Hines Z, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006; 127: 1109–22.
136 Khan MM, Ahmad A, Ishrat T, et al. Resveratrol attenuates 6-hydroxydopamine-induced oxidative damage and dopamine depletion in rat model of Parkinson’s disease. Brain Res 2010; 1328: 139–51.
137 Maher P, Dargusch R, Bodai L, Gerard PE, Purcell JM, Marsh JL. ERK activation by the polyphenols fi setin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum Mol Genet 2011; 20: 261–70.
138 Ho DJ, Calingasan NY, Wille E, Dumont M, Beal MF. Resveratrol protects against peripheral defi cits in a mouse model of Huntington’s disease. Exp Neurol 2010; 225: 74–84.
139 Anekonda TS, Reddy PH. Neuronal protection by sirtuins in Alzheimer’s disease. J Neurochem 2006; 96: 305–13.
140 Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic defi cit and eff ectively improves a mitochondrial myopathy phenotype. Cell Metab 2008; 8: 249–56.

MOLECULAR GENETICS OF
MITOCHONDRIAL DISORDERS
Lee-Jun C. Wong*Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas
Mitochondrial respiratory chain (RC) disorders (RCDs) are a groupof genetically and clinically heterogeneous diseases because of the factthat protein components of the RC are encoded by both mitochondrialand nuclear genomes and are essential in all cells. In addition, the bio-genesis, structure, and function of mitochondria, including DNA replica-tion, transcription, and translation, all require nuclear-encoded genes. Inthis review, primary molecular defects in the mitochondrial genome andmajor classes of nuclear genes causing mitochondrial RCDs, includinggenes underlying mitochondrial DNA (mtDNA) depletion syndrome, aswell as genes encoding RC subunits, complex assembly genes, and trans-lation factors, are described. Diagnostic methodologies used to detectcommon point mutations, large deletions, and unknown point mutationsin the mtDNA and to quantify mutation heteroplasmy are also discussed.Finally, the selection of nuclear genes for gold standard sequence analy-sis, application of novel technologies including oligonucleotide array com-parative genomic hybridization, and massive parallel sequencing of targetgenes are reviewed. ' 2010 Wiley-Liss, Inc.Dev Disabil Res Rev 2010;16:154–162.
Key Words: mitochondrial disorder; mtDNA mutation; mtDNA depletion;
mtDNA deletion
INTRODUCTION
The major function of mitochondria is to use molecularoxygen to convert the chemical energy of food intoATP, the cellular energy currency. To carry out this
oxidative phosphorylation (OXPHOS) reaction, the mito-chondria must maintain intact structure and function, whichrequires the participation of �1,500 genes encoded by boththe mitochondrial and nuclear genomes [Calvo et al., 2006].By Krebs cycle and beta-oxidation, carbohydrates and fattyacids are oxidized to form NADH and FADH2, respectively.Electrons from these reducing agents are passed through theelectron transport chain complexes, creating a proton-gradient,which is the driving force for ATP synthesis (Fig. 1). Mostpatients with mitochondrial disorders have molecular defectsaffecting the mitochondrial OXPHOS system, which is madeup of �87 protein subunits forming five multiprotein com-plexes (Complexes I–V) embedded in the inner mitochondrialmembrane. Among these 87 proteins, 13 are encoded by themitochondrial genome and the remainder are encoded by thenuclear genome. To form the OXPHOS complexes, a numberof nuclear-encoded assembly factors are required [Tirantiet al., 1998; Valnot et al., 2000; de Lonlay et al., 2001; Anto-nicka et al., 2003; Tay et al., 2004; Dieteren et al., 2008;
Hoefs et al., 2008; Pagliarini et al., 2008; Willems et al.,2008; Saada et al., 2009].
The human mitochondrial genome is a circular, double-stranded, 16.6-kb DNA encoding 13 protein subunits ofOXPHOS complexes and 2 ribosomal RNAs (rRNAs) and22 transfer RNAs (tRNAs) essential for mitochondrial proteinsynthesis. Mitochondrial DNA (mtDNA) is susceptible to oxi-dative DNA damage because of its close vicinity to where thereactive oxygen species (ROS) are produced, the lack of pro-tective histone proteins, and the limited DNA proof-readingand repair systems in mitochondria. The biogenesis ofmtDNA requires nuclear-encoded genes such as DNA poly-merase gamma (POLG) and DNA helicase (TWINKLE) formtDNA replication and purine/pyrimidine nucleoside kinases,DGUOK and TK2, for the salvage synthesis and maintenanceof deoxynucleoside triphosphate (dNTP) pools in the mito-chondria. Defects in any of these genes will cause mtDNAdepletion [Spinazzola and Zeviani, 2009; Spinazzola et al.,2009] or mtDNA multiple deletions [Milone et al., 2008].
Mitochondrial biogenesis, complex assembly, mtDNAreplication, transcription, and mitochondrial protein biosyn-thesis, all require nuclear DNA-encoded factors [Spinazzolaand Zeviani, 2005; Pagliarini et al., 2008; Koene and Smei-tink, 2009]. Furthermore, the roles of mitochondria inautophagy, apoptosis, and ROS production have recently pro-ven to be indispensable in the pathogenic mechanisms of neu-rodegenerative diseases (Parkinson, Alzheimer, Huntington,etc.) and cancer [Corral-Debrinski et al., 1992; Soong et al.,1992; Bianchi et al., 1995; Horton et al., 1996; Jessie et al.,2001; Tan et al., 2002; Geisler et al., 2010]. It is thus conceiv-able that defects in any of the �1,500 genes targeted to mito-chondria are potentially detrimental to mitochondrial structureand function [Wallace, 1999; Smeitink et al., 2001; DiMauro,2004; Geisler et al., 2010]. Since mitochondria are the essen-tial energy-producing organelles in animal cells, virtually allorgan systems may be affected if there is a mitochondrialdefect. However, the tissues with high-energy demand, such
*Correspondence to: Lee-Jun C. Wong, PhD, Department of Molecular and HumanGenetics, Baylor College of Medicine, One Baylor Plaza, NAB 2015, Houston, TX77030. E-mail: [email protected] 13 June 2010; Accepted 29 June 2010View this article online at wileyonlinelibrary.com.DOI: 10.1002/ddrr.104
DEVELOPMENTAL DISABILITIESRESEARCH REVIEWS 16: 154 – 162 (2010)
' 2010Wiley -Liss, Inc.

as muscle and nerve, are most suscepti-ble. In general, neuromuscular symp-toms are the major clinical features ofmitochondrial diseases, including seiz-ures, skeletal muscle weakness, exerciseintolerance, cardiomyopathy, sensori-neural hearing loss, optic atrophy, reti-nitis pigmentosa, ophthalmoplegia, dia-betes mellitus, hypothyroidism, gastroin-testinal reflux, renal dysfunction, andimmunodeficiency. However, diagnosisof mitochondrial respiratory chain (RC)disorders cannot be solely based onclinical features because of their diverseand often nonspecific nature [Haaset al., 2007, 2008].
Accordingly, mitochondrial disor-ders are a complex dual genome diseasethat can be caused by molecular defectsin both nuclear and mitochondrialgenomes. The disease is genetically het-erogeneous. Depending upon the pri-mary defect, mitochondrial disordersmay be autosomal recessive, autosomaldominant, X-linked, or maternallyinherited. Mitochondrial disorders arealso clinically heterogeneous because ofvariation in tissue distribution of themutation, the degree of heteroplasmy inaffected tissues, and variable thresholdsand penetrance effects for differentmutations [Wallace et al., 1995, 2001].Although there are some recognizablesyndromes, in most cases, diagnosis ischallenging because the same mutationsor different mutations in the same genesmay cause different clinical phenotype,and different mutations or differentgenes may cause the same clinical symp-toms. For example, the most commonmtDNA mutation, m.3243A>G, maycause MELAS (Mitochondrial Ence-phalopathy, Lactic Acidosis and Stroke-like episodes) or diabetes, hearing loss,and retinopathy. Similarly, hearing lossand progressive external ophthalmople-gia may be caused by mtDNA deletionor mtDNA multiple deletions secondaryto defects in nuclear genes such asPOLG, OPA1, and TWINKLE [Miloneet al., 2008, 2009; Van Hove et al.,2009].
A definitive molecular diagnosisof a primary mitochondrial RC disor-der (RCD) is important to administerprompt treatment and appropriatepatient care. In addition, identificationof the disease-causing genes, specificmutations, and the mode of inheritancewill facilitate accurate genetic counsel-ling. Furthermore, upon identificationof mutations, genetic testing of at-riskfamily members will become available.As most of the autosomal recessive mi-tochondrial disorders are severe and
fatal, a definitive molecular diagnosisand confirmed parental carrier statusmay assist in future prenatal and/or pre-implantation genetic diagnosis.
In this article, the molecular genet-ics of mitochondrial disorders, currentmolecular diagnostic approaches, meth-odologies used, and advances toward asingle-step comprehensive moleculardiagnostic approach are described.
PRIMARY DEFECTS IN THEMITOCHONDRIAL GENOME
Each animal cell contains hundredsto thousands of mtDNA molecules. Themutation rate of mtDNA is 10–20 timeshigher than that of nuclear DNA. Dele-terious mutations of mtDNA usuallycoexist with wild-type mtDNA mole-cules in a heteroplasmic state. mtDNAmutations are maternally inherited[Smeitink et al., 2001; Wallace et al.,2001; DiMauro, 2004]. There are recur-rent mtDNA mutations, including themost common m.3243A>G mutation intRNAleu(uur) for MELAS syndrome andthe m.8344A>G mutation the tRNAlys
gene for MERRF (Myoclonic Epilepsy,Ragged Red Fibers) syndrome. Them.3243A>G mutation has much moresevere clinical course than them.8344A>G mutation. Clinical symp-toms may be observed at m.3243A>Gmutation heteroplasmy <20% in blood,while the threshold for m.8344A>G ismuch higher, typically >80%. Them.3243A>G mutation is predominantlydetected in pediatric patients. ThetRNAleu(uur) and tRNAlys genes aremutation hot spots where other recur-rent mutations such as m.3271T>C(MELAS), m.8356T>C (MERRF), andm.8363G>A (peripheral neuropathy andcardiomyopathy) are located.
Mutations in protein-codingregions include m.8993T>G (p.L156R,ATP6) and m.8993T>C (p.L156P, ATP6)for NARP (Neuropathy, Ataxia, RetinitisPigmentosa) syndrome and m.11778G>A(p.R340H, ND4), m.3460G>A (p.A52T,ND1), and m.14484T>C (p.M64V,ND6) for LHON (Leber HereditaryOptic Neuropathy). Depending on thedegree of mutation heteroplasmy, them.8993T>G (p.L156R, ATP6) mutationcan cause a continuous spectrum of syn-dromes from asymptomatic (<60%) toretinopathy (60–75%), NARP (75–90%),and Leigh syndrome (>90%) [Enns et al.,2006]. Mutations causing LHON are usu-ally homoplasmic, and there is a differencein penetrance between male and femalecarriers. Mutations causing LHON pri-marily affect the eyes. Nuclear modifiergenes and tissue-specific factors are specu-
lated to play a role in disease expression ofmatrilineal relatives who are homoplasmicfor the mutations and have variable phe-notypic expression [Wallace et al., 1995,2001; Gropman et al., 2004].
The most well-known point muta-tions in mitochondrial rRNA genes arethe m.1555A>G and the m.1494T>Cmutations in the 12S rRNA gene, whichcause hearing loss upon exposure to oto-toxic aminoglycosides. In addition tocommon mtDNA point mutations, largemtDNA deletions are a frequent cause ofpathology accounting for Pearson syn-drome (PS), Kearns–Sayre syndrome(KSS), progressive external ophthalmo-plegia (PEO), or myopathy [Wong,2001]. PS is characterized by infantilemarrow and pancreas abnormalities withsideroblastic anemia. KSS is characterizedby onset at the second decade of life withPEO, pigmentary retinopathy, and atleast one of the following: cerebellarataxia, cardiac conduction defect, or ele-vated CSF protein.
In addition to the qualitativechanges in mtDNA, which include pointmutations and large deletions, quantita-tive alterations in mtDNA content havealso been reported in patients with mito-chondrial disorders and severely reducedcontent of mtDNA molecules. This phe-nomenon is called mtDNA depletion,where mtDNA molecules do not containdetectable mutations, but there is areduction in the copy number ofmtDNA molecules. Such quantitativechanges in mtDNA content are causedby primary mutations in nuclear-encoded genes responsible for mtDNAbiosynthesis [Spinazzola and Zeviani,2009; Spinazzola et al., 2009].
NUCLEAR GENE DEFECTSTHAT CAUSEMITOCHONDRIAL DISORDERS
An estimated 1,500 nuclear-encoded proteins are targeted to mito-chondria [Calvo et al., 2006] (Table 1).Defects in any of these genes can poten-tially cause mitochondrial disorders.However, mutations have been identifiedin only about 150 nuclear genes (Table2). Except for the analysis of whole mi-tochondrial genome, which is now rou-tinely being sequenced for diagnosticpurposes, the limited nuclear subset ofgenes that are available for clinicalsequence analysis is listed in Table 2.
mtDNA Depletion: Nuclear GeneDefects
The identification of mtDNAdepletion or multiple deletions maysuggest a Mendelian disorder because
Dev Disabil Res Rev � MOLECULAR GENETICS OF MITOCHONDRIAL DISORDERS � WONG 155

the maintenance of mtDNA integrity ishighly dependent on a number of nu-clear-encoded proteins. The mtDNAdepletion syndromes (MDDSs) areautosomal recessive disorders caused bymolecular defects in nuclear genes thatare involved in mtDNA biogenesis andmaintenance of dNTP pools, and whenmutated result in a reduction in cellularmtDNA content. At present, mutationsin at least nine genes (TYMP, POLG,DGUOK, TK2, SUCLA2, MPV17,SUCLG1, RRM2B, and C10orf2) havebeen found to result in mtDNA deple-tion with somewhat tissue-specificinvolvement [Hakonen et al., 2007;Sarzi et al., 2007; Spinazzola andZeviani, 2007; Wong et al., 2007, 2008;Dimmock et al., 2008; Spinazzola et al.,2009].
Mutations in the POLG gene arethe most common nuclear gene defectscausing mitochondrial disorders. Alperssyndrome is the most common autoso-mal recessive form of POLG deficiency,which is characterized by refractoryseizures, psychomotor regression, andliver failure [Naviaux and Nguyen,2004; Nguyen et al., 2005]. However,
the clinical manifestations of patientswith POLG mutations can be extremelyheterogeneous [DiMauro, 2004; Wonget al., 2008b]. It includes the mostsevere form of childhood myocerebro-hepatopathy spectrum disorders, spino-cerebellar ataxia with epilepsy, sensoryataxic neuropathy with dysarthria andophthalmoparesis (SANDO), and auto-somal dominant and recessive forms ofPEO [Van Goethem et al., 2001, 2003;Lamantea et al., 2002].
In addition to Alpers syndrome,the hepatocerebral form of MDDS is acommon cause of severe infantile mito-chondrial disease with liver failure andencephalopathy, which includes molec-ular defects in MPV17, DGUOK, andTWINKLE genes. MPV17 is a mito-chondrial inner membrane proteinwhose function is not clear [Spinazzolaet al., 2006]. Deficiency of MPV17 ischaracterized by infantile hepatic failure.Patients die before neurological deficitsstarted [Wong et al., 2007; El-Hattabet al., 2010]. Deoxyguanosine kinase(DGUOK) is responsible for the salvageof purine nucleosides in the mitochon-dria. Patients with DGUOK deficiency
often have neonatal onset of severe liverdisease with neurological featuresincluding hypotonia, developmentaldelay, nystagmus, neuropathy, encephal-opathy, and early death [Mandel et al.,2001; Dimmock et al., 2008]. In con-trast, patients with POLG deficiencyhave a predominantly encephalopathicpresentation and hepatic failure usuallyoccurs long after the initial encephalop-athy [Horvath et al., 2006; Wong et al.,2008b]. Hepatic failure may be inducedby infection or the antiepileptic drug,valproic acid [Lutz et al., 2009; Sanetoet al., 2010]. The TWINKLE geneencodes a DNA helicase [Spelbrinket al., 2001]. Heterozygous TWINKLEmutations result in mtDNA multipledeletions and cause the autosomal dom-inant form of PEO, while autosomal re-cessive TWINKLE mutations cause ahepatocerebral form of MDDS[Hakonen et al., 2007].
Mutations in TK2 and SUCLA2are responsible for the severe myopathicform of MDDS [Elpeleg et al., 2005;Galbiati et al., 2006; Carrozzo et al.,2007; Ostergaard et al., 2007a,b]. Themitochondrial thymidine kinase (TK2)
Table 1. Categories of Nuclear Genes Targeted to Mitochondria
Category Number Category Number Category Number Category Number
Apoptosis 50 Metabolisma 350a Proteases 25 tRNA synthase 22Chaperone 20 Mito dynamics 10 Nucleases 5 Transportersd 75Cytochromes 15 Replication 10 RNA mod 5 Protein folding and modification 12Cytoskeletal 10 Transcription 30 Translationb 15 Protein import: TIMMs and TOMMs 30DNA repair 12 Regulation 5 MRPLsc 55 RC complex and assembly 150Fe-S cluster 3 Porins 4 MRPSsc 36 RNA modification and processing 10Immune 2 ROS 15 Others 175 Unknown 150
aMetabolic pathways include amino acids, carbohydrates, creatine, iron, ketones, Kreb cycle, lipids, nucleotide, phospholipid, porphyrin, steroid, tetrahydrofolate, tRNA, CoQs, pyruvate, etc.bTranslation: initiation, elongation, termination proteins and factors.cMRP: mitochondrial ribosomal proteins large and small subunits.dTransporters: ATP-binding cassette, iron, nucleotides, solutes, and uncouplers.
Table 2. Nuclear Genes Causing Mitochondrial Disorders That Are Currently Analyzed by ClinicalDiagnostic Laboratories
1. OXPHOS Subunit GenesNDUFA1, NDUFA2, NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS5, NDUFS6, NDUFS7, NDUFS8, NDUFV1, NDUFV2, NDUFA11,COX6B1, SDHA, SDHB, SDHC, SDHD
2. OXPHOS Complex Assembly GenesNDUFAF1, NDUFAF2, SDH5, BCS1L, SCO1, SCO2, SURF1, LRPPRC, COX15, COX10, ATPAF1, COX6B2, TMEM70
3. EnzymesABCB7, APTX, ASS1, BCKDHA, BCKDHB, CABC1, COQ2, COQ9, CYCS, DLD, ETHE1, ETFA, ETFB, ETFDH, FH, HADHA, HADHB,HMGCL, MCCC2, MCCC1, PC, PCK2, PDHA1, PDHB, PDHX, PDSS1, PDSS2, PPM1B, TAZ, C12orf65, C14orf156, C18orf22, CPT1A, CPT1B,CPT2, FASTKD2, HLCS, PDK1, MMAA, MMAB, MMACHC, MMADHC, MRP63, MUT, OGDH, OTC, CPS1, NAGS, OXA1L
4. Mitochondrial Transcription Factors, tRNA Post-Transcription Modification Enzymes, Amino Acyl tRNA Synthetases, TranslationFactors, and Mitochondrial Ribosomal ProteinsTFAM, NRF1, PGC1, GABPA, GABPB1, PUS1, DARS2, RARS2, GFM1, TSFM, TUFM, MRPS16, MRPL33
5. Mitochondrial Membrane Proteins, Carriers, and TransportersDNC, ANT1, TIMM8A, PHC, VDAC1, CACT
6. Mitochondrial DNA Synthesis and Mitochondrial DynamicsDGUOK, MPV17, POLG, POLG2, c10orf2, RRM2B, SUCLA2, SUCLG2, SUCLG1, TK2, TYMP, DNA2, MFN2, OPA1, OPA3, PINK1, SPG7
156 Dev Disabil Res Rev � MOLECULAR GENETICS OF MITOCHONDRIAL DISORDERS � WONG

is an enzyme responsible for the salvageof pyrimidine nucleosides. Patients withTK2 deficiency present with congenitalmuscular dystrophy, elevated CK,ragged red fibers, and COX-deficientfibers. Deficiency of SUCLG1 (SuccinylCoA Ligase GDP-binding protein sub-unit alpha) also causes myopathy inaddition to severe lactic acidosis andencephalopathy [Ostergaard et al.,2007a,b]. MDDS due to defects inSUCLA or SUCLG genes can be dis-tinguished from others by a mild eleva-tion of methylmalonic acid [Elpeleget al., 2005; Carrozzo et al., 2007;Ostergaard et al., 2007a,b].
The GI form of MDDS, MNGIE(mitochondrial neurogastrointestinalencephalomyopathy), is causes by thy-midine phosphorylase (TP) deficiency,which affects primarily the smoothmuscle and the brain [Nishino et al.,1999]. Mutations in p-53-inducibleribonucleotide reductase, RRM2B,cause mtDNA depletion in multipleorgans and a wide clinical spectrumincluding myopathy, Leigh-like, andMNGIE-like syndromes [Bourdonet al., 2007; Shaibani et al., 2009].
Because of the diverse, yet overlap-ping, clinical features of MDDSs and theirmultiple gene etiology, a quick way toscreen for and establish mtDNA depletionbefore sequence analysis of candidate nu-clear genes would be ideal. This can beachieved by quantitative PCR (qPCR)analysis to determine mtDNA copy num-ber in affected tissues [Dimmock et al.,2010; Wong et al., 2010].
mtDNA multiple deletions inmuscle have been found in SANDOpatients with autosomal recessive muta-tions in POLG and in patients withautosomal dominant mutations in genesresponsible for the maintenance ofmtDNA integrity, including TWIN-KLE, ANT1, POLG2, and OPA1[Naimi et al., 2006; Milone et al.,2009]. Mutations in this group of genesmay cause an autosomal dominant formof PEO characterized by muscle weak-ness, ptosis, and ophthalmoplegia.
OXPHOS Complex Subunits andComplex Assembly Genes
Molecular defects in nuclear geneslikely account for the majority of thesevere mitochondrial disorders that arepresent in infants and children. In additionto genes responsible for MDDSs, nuclear-encoded complex subunits (�75 of them)and complex assembly factors are anothergroup of genes that cause autosomal reces-sive mitochondrial RCDs. The mostcommon of these are defects in Complex
IV (cytochrome c oxidase) assembly fac-tors, including SURF1 (Leigh), SCO1(encephalohepatopathy), SCO2 (cardio-myopathy), COX10 (encephalonephrop-athy), and COX15 (myopathy) [Tirantiet al., 1998; Valnot et al., 2000; Antonickaet al., 2003; Tay et al., 2004]. Mutationsin a Complex III assembly gene, BCS1L,have also been reported [de Lonlay et al.,2001]. Recently, molecular defects inTMEM70, an assembly factor for Com-plex V, ATP synthetase, have beenreported in patients with elevated3-methyl glutaconic acid and cardiomy-opathy [Willems et al., 2008; Housteket al., 2009]. Molecular defects inOXPHOS subunit genes or genes essen-tial for complex assembly may cause anisolated complex deficiency depending onthe defective gene [Dieteren et al., 2008;Hoefs et al., 2008; Willems et al., 2008;Saada et al., 2009]. Thus, assay of RCcomplex activities may prioritize genes forsequence analysis.
Other Nuclear Genes Involved inMitochondrial Structure andFunction
Mitochondrial encephalomyopa-thies caused by Coenzyme Q10 (CoQ10)deficiency are now well recognizedbecause of the discovery of mutations ingenes involved in the CoQ10 biosyntheticpathway, including PDSS1, PDSS2,COQ2, CABC1, and COQ9 [Quinziiet al., 2005; Mollet et al., 2007, 2008;Lagier-Tourenne et al., 2008]. SecondaryCoQ10 deficiency was also found to beassociated with ETFDH and APTXmuta-tions [Quinzii et al., 2006; Gempel et al.,2007]. Several groups of genes play crucialroles in the mitochondrial protein transla-tion machinery. These include mitochon-drial ribosomal proteins (MRPs), tRNAposttranscriptional modification enzymes,amino acyl-tRNA synthetases, and mito-chondrial protein translation factors. Pro-tein subunits of mitochondrial large andsmall ribosomes (named MRPL’s andMRPS’s, respectively) play important rolesin maintaining the ribosomal structurenecessary for mitochondrial protein syn-thesis [Kenmochi et al., 2000]. It is verylikely that defects in any of the MRPs willcause mitochondrial disorders, but becauseof the lack of an efficient molecular testingmethod, currently only two such caseshave been reported [Miller et al., 2004].
All 22 mitochondrial tRNAs aresubject to posttranscriptional modifica-tion. Molecular defects in PUS1 (pseu-douridine synthase 1) have been reportedin several patients with MLASA (myopa-thy, lactic acidosis, and sideroblastic ane-mia) [Bykhovskaya et al., 2004, 2007;
Patton et al., 2005; Fernandez-Vizarraet al. 2007]. Mutations have also beenidentified in mitochondrial arginyl- andaspartyl-tRNA synthetase genes (RARS2and DARS2) [Edvardson et al., 2007;Scheper et al., 2007]. Furthermore, mito-chondrial translation elongation factorsare also targets in which mutations causemitochondrial disease [Smeitink et al.,2006; Valente et al., 2009]. Evidently, allmitochondrial tRNA synthetases, tRNAnucleotide modification enzymes, andtranslation factors are potential causes ofmitochondrial disorders. Thus, all thesegenes need to be considered in the molec-ular diagnosis of mitochondrial disease.
MOLECULARMETHODOLOGIES USED FORDIAGNOSIS OFMITOCHONDRIAL DISORDERS
The recurrent common mtDNApoint mutations can be easily screenedby using various mutation detectionmethods, including PCR/restrictionfragment length polymorphism (RFLP),PCR/allele-specific oligonucleotide(ASO) dot blot analysis, TaqMan allelediscrimination assay, allele refractorymutation system (ARMS), or directsequencing. The multiplex PCR/ASOdot blot method appeared to be themost cost effective because it allowed si-multaneous analysis of all commonpoint mutations with a sensitivity ofdetecting heteroplasmy as low as 0.5%[Wong and Senadheera, 1997]. TheARMS qPCR analysis, however, allowsreal-time one-step detection and quan-tification of the mutation heteroplasmyas low as 0.1% [Bai and Wong, 2004].For the detection of mtDNA largedeletions, Southern blot analysis is usu-ally used [Shanske and Wong, 2004].
Since most pathogenic mtDNAmutations are heteroplasmic, quantifica-tion of mutation heteroplasmy is impor-tant to permit correlation with diseaseexpression. Most of the mutation detec-tion methods mentioned earlier do notallow quantification of mutant hetero-plasmy load. Traditionally, the last cyclehot PCR linked to RFLP analysis fol-lowed by phosphoimaging detection hasbeen used for the quantification of het-eroplasmy levels [Shanske and Wong,2004]. However, this method requiresradioactive material and quantification isnot always reliable. A more sophisti-cated method, ARMS qPCR, has beendeveloped for this purpose [Bai andWong, 2004] (see below).
Previous studies demonstrated thatanalysis of a panel of the 12 most com-mon point mutations for MELAS,
Dev Disabil Res Rev � MOLECULAR GENETICS OF MITOCHONDRIAL DISORDERS � WONG 157

MERRF, NARP, Leigh disease, LHON,and large deletions detected mutations inonly about 6% of the patients suspectedof having mitochondrial disorders [Liangand Wong, 1998]. Search for unknownmutations in the entire mitochondrialgenome by sequencing analysis slightlyimproved the mutation detection rate[Smeitink et al., 2001; Spinazzola andZeviani, 2005; Brautbar et al., 2008;Elliott et al., 2008]. This suggests thatthe vast majority of the molecular defectscausing mitochondrial RC deficiency re-side in nuclear genes.
Identification of molecular defectsin nuclear genes is usually performed bydirect Sanger sequencing analysis of thecandidate genes following PCR amplifi-cation of the coding exons [Wonget al., 2007, 2008; Dimmock et al.,
2008; El-Hattab et al., 2010]. Theselection of candidate genes forsequencing analysis is based on individ-ual clinical presentation and biochemicalevaluation. For recognizable syndromes,a group of candidate genes may besequenced. However, because of geneticand clinical heterogeneity underlyingmitochondrial disorders, it is often diffi-cult to determine the exact ‘‘syndrome’’or genes to be sequenced. Thus, molec-ular or biochemical screening methodsthat can quickly determine mtDNAcopy number or RC enzyme complexactivities may be helpful in the rationalselection of genes to be sequenced[Dimmock et al., 2010; Wong et al.,2010]. For example, Southern blot anal-ysis with radioactive probes for bothmtDNA and nuclear DNA (for exam-
ple, the 18S rRNA gene), followed bydensitometric analysis of the autoradio-gram or phosphoimage [Shanske andWong, 2004], has been used to deter-mine mtDNA content. However, thismethod is tedious and requires a largeamount of DNA. Recently, real-timeqPCR (RT qPCR) analysis has beendeveloped for this purpose [Dimmocket al., 2010]. Using the qPCR method,mtDNA content in affected tissues, suchas muscle or liver, can be rapidly deter-mined. If there is an indication ofmtDNA depletion, the relevant nucleargenes can then be selected based onclinical presentation for sequence analy-sis. Increased mtDNA content couldsuggest a compensatory amplification ofmtDNA because of mitochondrial dys-function. Increased mtDNA content hasbeen observed in patients with pointmutations in mitochondrial tRNAgenes or with mtDNA deletions.
Enzymatic assays of RC complexactivities can be used to determine thedefective complex and whether the defi-ciency is in an isolated complex or affectsmultiple complexes. For example, if themolecular defects reside in a complexsubunit or assembly gene, isolated defi-ciency of the specific complex will beobserved [Zhu et al., 1998]. In contrast,mtDNA depletion will cause a reductionin activities of all complexes containingmtDNA-encoded protein subunits [Wonget al., 2007; Dimmock et al., 2008].Combined deficiencies of Complexes I þIII and II þ III are suggestive of defectsin the biosynthetic pathways of CoQ10[Quinzii et al., 2005; Mollet et al., 2007,2008; Lagier-Tourenne et al., 2008].
CURRENT DIAGNOSTICAPPROACHES
Current practice in the moleculardiagnosis of mitochondrial disordersbegins with a thorough clinical evalua-tion followed by metabolic screeningand imaging studies [Haas et al., 2007,2008; Wong et al., 2010]. Based on theresults of clinical, biochemical, andimaging evaluations, if a mitochondrialdisorder is indicated, screening for themost common mtDNA mutations,including m.3243A>G, m.8344A>T,m.8993T>G, and m.8993T>C pointmutations, as well as single mtDNAdeletions (for multisystem disorderswith variable clinical presentation) isusually the first step to promptly con-firm the diagnosis. Since, to date,POLG1 is the most frequently mutatednuclear gene that causes highly variableclinical phenotypes, DNA sequenceanalysis of POLG1 gene is usually rec-
Fig. 1. Major function of mitochondria: producing energy. [Color figure can be viewed in theonline issue, which is available at wileyonlinelibrary.com.]
Fig. 2. Recognizable mitochondrial syndromes and causative genes. [Color figure can beviewed in the online issue, which is available at wileyonlinelibrary.com.]
158 Dev Disabil Res Rev � MOLECULAR GENETICS OF MITOCHONDRIAL DISORDERS � WONG

ommended following negative commonmtDNA mutation screening [Wonget al., 2008b, 2010]. If only a bloodsample is available, screening analysis formtDNA deletions and point mutationsis negative, and the patient presents witha recognizable clinical syndrome (Fig. 2);specific nuclear gene(s) or genes can beselected for DNA sequence analysis. Ifthere is clear indication of maternal in-heritance, sequence analysis of the entiremitochondrial genome is warranted,which is preferably performed on DNAextracted from affected tissues such asmuscle or liver. As mentioned earlier, ifmuscle or liver tissue is available for test-ing, results from qPCR analysis ofmtDNA content and electron transportchain activities in these tissues may helpwith the diagnosis and the selection ofgenes for sequence analysis.
Currently, the gold standard of con-firming a suspected diagnosis of mito-chondrial disease is to perform sequenceanalysis of the candidate gene(s) to identifythe disease-causing pathogenic mutation(s).Large deletions that cannot be detected bysequencing may be detected by traditionalSouthern blot analysis or targeted oligonu-cleotide array comparative genomichybridization (aCGH) [Lacbawan et al.,2000; Wong et al., 2008a, 2010; Chinaultet al., 2009; Lee et al., 2009; Mardis,2009; Shchelochkov et al., 2009]. To cor-relate pathogenic mtDNA mutations withclinical phenotype, it is important todetermine the degree of mutation hetero-plasmy in various tissues by last cycle hotPCR/RFLP or ARMS qPCR [Bai andWong, 2004; Shanske and Wong, 2004;Enns et al., 2006]. The degree of hetero-plasmy for mtDNA deletion and contentof mtDNA molecules may be determinedby qPCR or targeted aCGH [Chinaultet al., 2009; Dimmock et al., 2010].
QUANTITATIVE SCREENINGFOR MDDS
MDDS is by far the most severeand common mitochondrial disordercaused by mutations in a group of nu-clear genes responsible for the biogen-esis of mtDNA and the maintenanceof mtDNA integrity. RT qPCR anal-ysis has shown that the mtDNA con-tent increases with age in muscle tis-sue, but decreases with age in bloodsamples [Dimmock et al., 2010]. Table3 shows that in 158 samples (122blood, 21 muscle, and 15 liver) frompatients with a molecularly provenMDDS, severe mtDNA depletion wasdetected in liver and muscle tissues[Dimmock et al., 2010]. The mtDNAcontent in blood samples of patients
Table 3. mtDNA Depletion in Blood, Muscle, and LiverSpecimens
Mutationof Genes
Blood Specimens Muscle Specimens Liver Specimens
Number ofPatients % Mean 6 SD
Number ofPatients % Mean 6 SD
Number ofPatients % Mean 6 SD
POLG 83 78 6 33 8 35 6 20 2 14 6 17DGUOK 12 52 6 21 2 35 6 20 4 5 6 1.5MPV17 8 50 6 15 4 28 6 27 8 10 6 9TK2 5 79 6 13 3 25 6 2 1 N.A.TP 5 70 6 20SUCLG1 3 72 6 23 1 N.A.RRM2B 4 39 6 13 2 13 6 8SUCLA2 2 86 6 34 1 N.A.
Blood specimens: total number of patients 5 122, average % mean 6 SD 5 72 6 31; muscle specimens: total number of patients 521, average % mean 6 SD 5 33 6 21; liver specimens: total number of patients 5 15, average % mean 6 SD 5 8 6 7.The averagepercentage of mtDNA content in patients with a molecularly proven mtDNA depletion syndrome compared to mtDNA content inage- and tissue-matched pooled controls is given. The mtDNA copy number of each affected patient was measured by real-time qPCRand compared to the mtDNA copy number of age- and tissue-matched mean to obtain the percentage. mtDNA contents (in referenceto matched control mean) of patients with molecular defects in the same gene were then averaged and tabulated.
Fig. 3. Targeted oligonucleotide aCGH: simultaneous detection of nuclear gene copy numberchange and mtDNA depletion. A: Detection of a 1.8-kb heterozygous deletion encompassing Exon4 of DGUOK gene in the affected son. Mother carried the deletion. Father carried a point mutationthat was detected by sequence analysis. B: The black dots illustrate the approximate position of theoligonucleotide probes that are lost. The coordinates indicated the deletion breakpoints subse-quently determined by PCR/sequencing. C: The mtDNA profile in the MitoMet array. The mtDNAcontent in the affected liver specimen is severely depleted to about 6% of tissue-matched control.The mtDNA content in blood is also reduced (50% of age- and tissue- matched control). ThemtDNA content in the blood specimen of the carrier mother and father is within the normal range.[Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Dev Disabil Res Rev � MOLECULAR GENETICS OF MITOCHONDRIAL DISORDERS � WONG 159

with MDDS is reduced but not assignificantly as in muscle or liverspecimens [Dimmock et al., 2010].Thus, qPCR can be used to quicklyscreen for mtDNA depletion in mus-cle or liver specimens. Following thisscreening method, mutations in thePOLG, RRM2B, and MPV17 genescan be identified prospectively [Dim-mock et al., 2010].
THE UTILITY OFOLIGONUCLEOTIDE aCGH
Direct sequencing of relevantgenes is currently the primary clinicaltechnique available to identify mutationsin human diseases. However, thisapproach will not detect large intragenicor whole gene deletions. Large dele-tions are conventionally detected bySouthern blot [Shanske and Wong,2004] or multiplex ligation-dependentprobe amplification analysis. These pro-cedures are tedious and do not estimatethe deletion breakpoints. Recently, atargeted custom oligonucleotide-basedmicroarray (MitoMet1) has beendesigned to provide high-density cover-age of a set of 192 nuclear genesinvolved in the pathogenesis of meta-bolic and mitochondrial disorders[Wong et al., 2008a; Chinault et al.,2009]. Using the MitoMet array, intra-genic deletions in nuclear genes(including DGUOK, TK2, MPV17,and POLG1) have been detected andreported [Lee et al., 2009; Zhang et al.,
2010]. The oligonucleotide aCGH hasexcellent utility for the detection ofintragenic large deletions, particularly inautosomal recessive mtDNA depletiondisorders when only one mutant alleleis detected by sequencing despite cleardemonstration of mtDNA depletion bycopy number analysis [Lee et al., 2009;Zhang et al., 2010]. Figure 3 providesan example of a hepatocerebral form ofMDDS of an infant referred for tyrosi-nemia detected on newborn screeningwho subsequently developed liver fail-ure. DNA sequence analysis revealed aheterozygous missense mutation(c.679G>A, p.E227K) in the deoxygua-nosine kinase (DGUOK) gene, whileqPCR analysis of liver DNA showed asevere reduction in mtDNA content.Using oligonucleotide aCGH, an intra-genic heterozygous 1.8-kb deletionencompassing Exon 4 of the DGUOKgene (Figs. 3A and 3B) was detected.The mtDNA profile on the same arrayshowed mtDNA depletion in liver (6%)and in blood (50%; Fig. 3C) [Lee et al.,2009]. Similarly, a heterozygous intra-genic deletion of �5.8 kb was detectedin two deceased siblings with a congen-ital myopathic form of MDDS but whoharbored only one sequence detectableheterozygous point mutation, p.I254N,in the TK2 gene [Zhang et al., 2010].
The current procedures for the di-agnosis of mtDNA deletion syndromesbased on Southern analysis are particularlyinefficient in determining deletion break-
points and the percentage of mtDNA de-letion heteroplasmy. The same custom-designed oligonucleotide MitoMetaCGH, described earlier, that containednot only dense coverage of mitochondrialtargeted nuclear genes but also tiled cover-age of the entire 16.6-kb mitochondrialgenome was used to quickly evaluate formtDNA deletions and depletion. Asshown in Figure 4, the deletion break-points and the percent mtDNA deletionheteroplasmy can be easily deduced fromthe oligonucleotide aCGH [Chinaultet al., 2009]. These studies strongly sug-gest that the targeted oligonucleotide cus-tom array is capable of reliably detectingmtDNA deletions while simultaneouslyelucidating of the deletion breakpointsand percentage of heteroplasmy. In addi-tion, simultaneous detection of copynumber changes in both nuclear and mi-tochondrial genomes makes this dual ge-nome array of tremendous value in thediagnoses of MDDSs [Chinault et al.,2009].
PITFALLS OF CURRENTMOLECULAR DIAGNOSTICAPPROACH FORMITOCHONDRIAL DISORDERS
The current diagnosis of mitochon-drial disorders involves multiple steps andmultiple techniques that include screeningof common mtDNA point mutations andlarge deletions, sequencing of the wholemitochondrial genome, sequentialsequencing of a group of candidate nu-clear genes, specific methods to quantifymtDNA mutation heteroplasmy, mtDNAcopy number, and large mtDNA dele-tions. Sequencing candidate genes one byone is tedious. Further, it is impossible toanalyze all 1,500 nuclear genes, whichproduce proteins that function within mi-tochondria. Rather, sequence analysis isclinically available on only a small fraction(about 100–150) of these genes. Even afterexhaustive sequence analysis, majority ofpatients suspected of having mitochon-drial disorders remain without a definitivediagnosis.
FUTURE APPROACHESTOWARD ONE-STEP,COMPREHENSIVE, DUALGENOME ANALYSIS
To overcome the obstacles inherentin the current diagnosis of mitochondrialdisorders, a comprehensive method isneeded that would allow simultaneousqualitative and quantitative analysis of allgenes (both nuclear- and mitochondrial-encoded) targeted to mitochondria in atime and cost-effective fashion. This canbe achieved by the application of novel,
Fig. 4. mtDNA deletion and depletion illustrated by MitoMet aCGH. Lanes A–C show the ap-proximate deletion breakpoint and deletion heteroplasmy load. A: Pearson syndrome patient,5-kb deletion, 85% heteroplasmy; B: hearing loss/diabetes, 7.5-kb deletion, 20% hetero-plasmy; C: KSS, 4.1-kb deletion, 75% heteroplasmy; and D: mtDNA depletion, to about 50%of control mtDNA copy number levels. [Color figure can be viewed in the online issue, whichis available at wileyonlinelibrary.com.]
160 Dev Disabil Res Rev � MOLECULAR GENETICS OF MITOCHONDRIAL DISORDERS � WONG

high-throughput sequencing technologiesto bi-genomic analysis [Vasta et al., 2009].A large number of target genes may becaptured by custom-designed oligonu-cleotide probes on microarrays or selectedby hybridization to RNA baits in solu-tion. Captured target DNA template isthen massively parallel-sequenced withdeep coverage using one of the next gen-eration sequencing instruments [Vastaet al., 2009]. The characteristics of thecapture design must include the mito-chondrial genome and take into consider-ation that the mitochondrial genome ispresent in 300-fold higher copy numberthan average nuclear genes. To detect largedeletions and estimate mtDNA mutationheteroplasmy, the target sequences mustbe completely captured and sequenced ata reasonable depth of about 100-fold (fornuclear genes) to 1,000-fold (for mito-chondrial genes). Such an approach willallow quick and comprehensive analysessimultaneously in both nuclear and mito-chondrial genomes for point mutations,large deletions, copy number changes, andfor mtDNA, mutation heteroplasmy load.New disease genes are expected to be dis-covered using this approach of compre-hensive target gene capture followed bydeep genome sequencing. n
REFERENCESAntonicka H, Mattman A, Carlson CG, et al.
2003. Mutations in COX15 produce adefect in the mitochondrial heme biosyn-thetic pathway, causing early-onset fatal hy-pertrophic cardiomyopathy. Am J HumGenet 72:101–114.
Bai RK, Wong LJ. 2004. Detection and quantifi-cation of heteroplasmic mutant mitochon-drial DNA by real-time amplification refrac-tory mutation system quantitative PCR anal-ysis: a single-step approach. Clin Chem50:996–1001.
Bianchi MS, Bianchi NO, Bailliet G. 1995. Mito-chondrial DNA mutations in normal and tu-mor tissues from breast cancer patients.Cytogenet Cell Genet 71:99–103.
Bourdon A, Minai L, Serre V, et al. 2007. Muta-tion of RRM2B, encoding p53-controlledribonucleotide reductase (p53R2), causessevere mitochondrial DNA depletion. NatGenet 39:776–780.
Brautbar A, Wang J, Abdenur JE, et al. 2008. Themitochondrial 13513G>A mutation is asso-ciated with Leigh disease phenotypes inde-pendent of complex I deficiency in muscle.Mol Genet Metab 94:485–490.
Bykhovskaya Y, Casas K, Mengesha E, et al. 2004.Missense mutation in pseudouridine synthase1 (PUS1) causes mitochondrial myopathyand sideroblastic anemia (MLASA). Am JHum Genet 74:1303–1308.
Bykhovskaya Y, Mengesha E, Fischel-Ghodsian N.2007. Pleiotropic effects and compensationmechanisms determine tissue specificity in mi-tochondrial myopathy and sideroblastic anemia(MLASA). Mol Genet Metab 91:148–156.
Calvo S, Jain M, Xie X, et al. 2006. Systematicidentification of human mitochondrial dis-
ease genes through integrative genomics.Nat Genet 38:576–582.
Carrozzo R, Dionisi-Vici C, Steuerwald U, et al.2007. SUCLA2 mutations are associatedwith mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deaf-ness. Brain 130:862–874.
Chinault AC, Shaw CA, Brundage EK, et al.2009. Application of dual-genome oligonu-cleotide array-based comparative genomichybridization to the molecular diagnosis ofmitochondrial DNA deletion and depletionsyndromes. Genet Med 11:518–526.
Corral-Debrinski M, Horton T, Lott MT, et al.1992. Mitochondrial DNA deletions inhuman brain: regional variability and increasewith advanced age. Nat Genet 2:324–329.
de Lonlay P, Valnot I, Barrientos A, et al. 2001.A mutant mitochondrial respiratory chainassembly protein causes complex III defi-ciency in patients with tubulopathy, ence-phalopathy and liver failure. Nat Genet29:57–60.
Dieteren CE, Willems PH, Vogel RO, et al. 2008.Subunits of mitochondrial complex I exist aspart of matrix- and membrane-associatedsubcomplexes in living cells. J Biol Chem283:34753–34761.
DiMauro S. 2004. The many faces of mitochon-drial diseases. Mitochondrion 4:799–807.
Dimmock D, Tang LY, Schmitt ES, et al. 2010.Quantitative evaluation of the mitochondrialDNA depletion syndrome. Clin Chem 56:1119–1127.
Dimmock DP, Zhang Q, Dionisi-Vici C, et al.2008. Clinical and molecular features of mi-tochondrial DNA depletion due to muta-tions in deoxyguanosine kinase. Hum Mutat29:330–331.
Edvardson S, Shaag A, Kolesnikova O, et al. 2007.Deleterious mutation in the mitochondrialarginyl-transfer RNA synthetase gene isassociated with pontocerebellar hypoplasia.Am J Hum Genet 81:857–862.
El-Hattab AW, Li FY, Schmitt E, et al. 2010.MPV17-associated hepatocerebral mitochon-drial DNA depletion syndrome: newpatients and novel mutations. Mol GenetMetab 99:300–308.
Elliott HR, Samuels DC, Eden JA, et al. 2008.Pathogenic mitochondrial DNA mutationsare common in the general population. AmJ Hum Genet 83:254–260.
Elpeleg O, Miller C, Hershkovitz E, et al. 2005.Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with ence-phalomyopathy and mitochondrial DNAdepletion. Am J Hum Genet 76:1081–1086.
Enns GM, Bai RK, Beck AE, et al. 2006. Molec-ular–clinical correlations in a family withvariable tissue mitochondrial DNA T8993Gmutant load. Mol Genet Metab 88:364–371.
Fernandez-Vizarra E, Berardinelli A, Valente L, et al.2007. Nonsense mutation in pseudouridylatesynthase 1 (PUS1) in two brothers affected bymyopathy, lactic acidosis and sideroblastic anae-mia (MLASA). J Med Genet 44:173–180.
Galbiati S, Bordoni A, Papadimitriou D, et al.2006. New mutations in TK2 gene associ-ated with mitochondrial DNA depletion.Pediatr Neurol 34:177–185.
Geisler S, Holmstrom KM, Skujat D, et al. 2010.PINK1/Parkin-mediated mitophagy is de-pendent on VDAC1 and p62/SQSTM1.Nat Cell Biol 12:119–131.
Gempel K, Topaloglu H, Talim B, et al. 2007.The myopathic form of coenzyme Q10 defi-ciency is caused by mutations in the elec-
tron-transferring-flavoprotein dehydrogenase(ETFDH) gene. Brain 130:2037–2044.
Gropman A, Chen TJ, Perng CL, et al. 2004.Variable clinical manifestation of homoplas-mic G14459A mitochondrial DNA muta-tion. Am J Med Genet A 124A:377–382.
Haas RH, Parikh S, Falk MJ, et al. 2007. Mitochon-drial disease: a practical approach for primarycare physicians. Pediatrics 120:1326–1333.
Haas RH, Parikh S, Falk MJ, et al. 2008. The in-depth evaluation of suspected mitochondrialdisease. Mol Genet Metab 94:16–37.
Hakonen AH, Isohanni P, Paetau A, et al. 2007.Recessive Twinkle mutations in early onsetencephalopathy with mtDNA depletion.Brain 130:3032–3040.
Hoefs SJ, Dieteren CE, Distelmaier F, et al. 2008.NDUFA2 complex I mutation leads to Leighdisease. Am J Hum Genet 82:1306–1315.
Horton TM, Petros JA, Heddi A, et al. 1996.Novel mitochondrial DNA deletion foundin a renal cell carcinoma. Genes Chromo-somes Cancer 15:95–101.
Horvath R, Hudson G, Ferrari G, et al. 2006.Phenotypic spectrum associated with muta-tions of the mitochondrial polymerasegamma gene. Brain 129:1674–1684.
Houstek J, Kmoch S, Zeman J. 2009. TMEM70protein—a novel ancillary factor of mamma-lian ATP synthase. Biochim Biophys Acta1787:529–532.
Jessie BC, Sun CQ, Irons HR, et al. 2001. Accu-mulation of mitochondrial DNA deletionsin the malignant prostate of patients of dif-ferent ages. Exp Gerontol 37:169–174.
Kenmochi N, Yoshihama M, Higa S, et al. 2000.The human ribosomal protein L6 gene in acritical region for Noonan syndrome. JHum Genet 45:290–293.
Koene S, Smeitink J. 2009. Mitochondrial medi-cine: entering the era of treatment. J InternMed 265:193–209.
Lacbawan F, Tifft CJ, Luban NL, et al. 2000.Clinical heterogeneity in mitochondrialDNA deletion disorders: a diagnostic chal-lenge of Pearson syndrome. Am J MedGenet 95:266–268.
Lagier-Tourenne C, Tazir M, Lopez LC, et al.2008. ADCK3, an ancestral kinase, ismutated in a form of recessive ataxia associ-ated with coenzyme Q10 deficiency. Am JHum Genet 82:661–672.
Lamantea E, Tiranti V, Bordoni A, et al. 2002.Mutations of mitochondrial DNA polymerasegammaA are a frequent cause of autosomaldominant or recessive progressive externalophthalmoplegia. Ann Neurol 52:211–219.
Lee NC, Dimmock D, Hwu WL, et al. 2009. Si-multaneous detection of mitochondrialDNA depletion and single-exon deletion inthe deoxyguanosine gene using array-basedcomparative genomic hybridisation. ArchDis Child 94:55–58.
Liang MH, Wong LJ. 1998. Yield of mtDNAmutation analysis in 2,000 patients. Am JMed Genet 77:395–400.
Lutz RE, Dimmock D, Schmitt ES, et al. 2009.De novo mutations in POLG presentingwith acute liver failure or encephalopathy. JPediatr Gastroenterol Nutr 49:126–129.
Mandel H, Szargel R, Labay V, et al. 2001. Thedeoxyguanosine kinase gene is mutated inindividuals with depleted hepatocerebral mi-tochondrial DNA. Nat Genet 29:337–341.
Mardis ER. 2009. New strategies and emergingtechnologies for massively parallel sequenc-ing: applications in medical research. Ge-nome Med 1:40.
Dev Disabil Res Rev � MOLECULAR GENETICS OF MITOCHONDRIAL DISORDERS � WONG 161

Miller C, Saada A, Shaul N, et al. 2004. Defectivemitochondrial translation caused by a ribo-somal protein (MRPS16) mutation. AnnNeurol 56:734–738.
Milone M, Brunetti-Pierri N, Tang LY, et al.2008. Sensory ataxic neuropathy with oph-thalmoparesis caused by POLG mutations.Neuromuscul Disord 18:626–632.
Milone M, Younge BR, Wang J, et al. 2009. Mito-chondrial disorder with OPA1 mutation lackingoptic atrophy. Mitochondrion 9:279–281.
Mollet J, Delahodde A, Serre V, et al. 2008.CABC1 gene mutations cause ubiquinonedeficiency with cerebellar ataxia and seizures.Am J Hum Genet 82:623–630.
Mollet J, Giurgea I, Schlemmer D, et al. 2007.Prenyldiphosphate synthase, subunit 1(PDSS1) and OH-benzoate polyprenyltrans-ferase (COQ2) mutations in ubiquinonedeficiency and oxidative phosphorylationdisorders. J Clin Invest 117:765–772.
Naimi M, Bannwarth S, Procaccio V, et al. 2006. Mo-lecular analysis of ANT1, TWINKLE andPOLG in patients with multiple deletions ordepletion of mitochondrial DNA by a dHPLC-based assay. Eur J Hum Genet 14:917–922.
Naviaux RK, Nguyen KV. 2004. POLG muta-tions associated with Alpers’ syndrome andmitochondrial DNA depletion. Ann Neurol55:706–712.
Nguyen KV, Ostergaard E, Ravn SH, et al. 2005.POLG mutations in Alpers syndrome. Neu-rology 65:1493–1495.
Nishino I, Spinazzola A, Hirano M. 1999. Thy-midine phosphorylase gene mutations inMNGIE, a human mitochondrial disorder.Science 283:689–692.
Ostergaard E, Christensen E, Kristensen E, et al.2007a. Deficiency of the alpha subunit ofsuccinate-coenzyme A ligase causes fatal in-fantile lactic acidosis with mitochondrialDNA depletion. Am J Hum Genet 81:383–387.
Ostergaard E, Hansen FJ, Sorensen N, et al.2007b. Mitochondrial encephalomyopathywith elevated methylmalonic acid is causedby SUCLA2 mutations. Brain 130:853–861.
Pagliarini DJ, Calvo SE, Chang B, et al. 2008. Amitochondrial protein compendium eluci-dates complex I disease biology. Cell134:112–123.
Patton JR, Bykhovskaya Y, Mengesha E, et al.2005. Mitochondrial myopathy and sidero-blastic anemia (MLASA): missense mutationin the pseudouridine synthase 1 (PUS1)gene is associated with the loss of tRNApseudouridylation. J Biol Chem 280:19823–19828.
Quinzii C, Naini A, Salviati L, et al. 2006. Amutation in para-hydroxybenzoate-poly-prenyl transferase (COQ2) causes primarycoenzyme Q10 deficiency. Am J HumGenet 78:345–349.
Quinzii CM, Kattah AG, Naini A, et al. 2005.Coenzyme Q deficiency and cerebellarataxia associated with an aprataxin mutation.Neurology 64:539–541.
Saada A, Vogel RO, Hoefs SJ, et al. 2009. Muta-tions in NDUFAF3 (C3ORF60), encodingan NDUFAF4 (C6ORF66)-interactingcomplex I assembly protein, cause fatal neo-natal mitochondrial disease. Am J HumGenet 84:718–727.
Saneto RP, Lee IC, Koenig MK, et al. 2010.POLG DNA testing as an emerging standardof care before instituting valproic acid ther-
apy for pediatric seizure disorders. Seizure19:140–146.
Sarzi E, Goffart S, Serre V, et al. 2007. Twinklehelicase (PEO1) gene mutation causes mito-chondrial DNA depletion. Ann Neurol62:579–587.
Scheper GC, van der Klok T, van Andel RJ, et al.2007. Mitochondrial aspartyl-tRNA synthe-tase deficiency causes leukoencephalopathywith brain stem and spinal cord involvementand lactate elevation. Nat Genet 39:534–539.
Shaibani A, Shchelochkov OA, Zhang S, et al.2009. Mitochondrial neurogastrointestinalencephalopathy due to mutations inRRM2B. Arch Neurol 66:1028–1032.
Shanske S, Wong LJ. 2004. Molecular analysis formitochondrial DNA disorders. Mitochond-rion 4:403–415.
Shchelochkov OA, Li FY, Geraghty MT, et al. 2009.High-frequency detection of deletions andvariable rearrangements at the ornithine trans-carbamylase (OTC) locus by oligonucleotidearray CGH. Mol Genet Metab 96:97–105.
Smeitink J, van den Heuvel L, DiMauro S. 2001.The genetics and pathology of oxidativephosphorylation. Nat Rev Genet 2:342–352.
Smeitink JA, Elpeleg O, Antonicka H, et al. 2006.Distinct clinical phenotypes associated with amutation in the mitochondrial translationelongation factor EFTs. Am J Hum Genet79:869–877.
Soong NW, Hinton DR, Cortopassi G, et al.1992. Mosaicism for a specific somatic mito-chondrial DNA mutation in adult humanbrain. Nat Genet 2:318–323.
Spelbrink JN, Li FY, Tiranti V, et al. 2001.Human mitochondrial DNA deletions asso-ciated with mutations in the gene encodingTwinkle, a phage T7 gene 4-like proteinlocalized in mitochondria. Nat Genet28:223–231.
Spinazzola A, Invernizzi F, Carrara F, et al. 2009.Clinical and molecular features of mitochon-drial DNA depletion syndromes. J InheritMetab Dis 32:143–158.
Spinazzola A, Viscomi C, Fernandez-Vizarra E,et al. 2006. MPV17 encodes an inner mito-chondrial membrane protein and is mutatedin infantile hepatic mitochondrial DNAdepletion. Nat Genet 38:570–575.
Spinazzola A, Zeviani M. 2005. Disorders ofnuclear–mitochondrial intergenomic signal-ing. Gene 354:162–168.
Spinazzola A, Zeviani M. 2007. Disorders ofnuclear–mitochondrial intergenomic com-munication. Biosci Rep 27:39–51.
Spinazzola A, Zeviani M. 2009. Disorders from per-turbations of nuclear–mitochondrial intergeno-mic cross-talk. J Intern Med 265:174–192.
Tan DJ, Bai RK, Wong LJ. 2002. Comprehensivescanning of somatic mitochondrial DNAmutations in breast cancer. Cancer Res62:972–976.
Tay SK, Shanske S, Kaplan P, et al. 2004. Associa-tion of mutations in SCO2, a cytochrome coxidase assembly gene, with early fetallethality. Arch Neurol 61:950–952.
Tiranti V, Hoertnagel K, Carrozzo R, et al. 1998.Mutations of SURF-1 in Leigh disease asso-ciated with cytochrome c oxidase deficiency.Am J Hum Genet 63:1609–1621.
Valente L, Shigi N, Suzuki T, et al. 2009. TheR336Q mutation in human mitochondrialEFTu prevents the formation of an activemt-EFTu.GTP.aa-tRNA ternary complex.Biochim Biophys Acta 1792:791–795.
Valnot I, Osmond S, Gigarel N, et al. 2000.Mutations of the SCO1 gene in mitoc-hondrial cytochrome c oxidase deficiencywith neonatal-onset hepatic failure andencephalopathy. Am J Hum Genet 67:1104–1109.
Van Goethem G, Dermaut B, Lofgren A, et al.2001. Mutation of POLG is associated withprogressive external ophthalmoplegia charac-terized by mtDNA deletions. Nat Genet28:211–212.
Van Goethem G, Martin JJ, Dermaut B, et al.2003. Recessive POLG mutations presentingwith sensory and ataxic neuropathy in com-pound heterozygote patients with progressiveexternal ophthalmoplegia. NeuromusculDisord 13:133–142.
Van Hove JL, Cunningham V, Rice C, et al.2009. Finding twinkle in the eyes of a 71-year-old lady: a case report and review ofthe genotypic and phenotypic spectrum ofTWINKLE-related dominant disease. Am JMed Genet A 149A:861–867.
Vasta V, Ng SB, Turner EH, et al. 2009. Nextgeneration sequence analysis for mitochon-drial disorders. Genome Med 1:100.
Wallace DC. 1999. Mitochondrial diseases in manand mouse. Science 283:1482–1488.
Wallace DC, Lott MT, Brown MD, et al. 2001.Mitochondria and neuro-ophthalmologicdiseases. In:Scriver CR, Beaudet AL, SlyWS, et al., editors.The metabolic andmolecular bases of inherited disease, VolII. New York: McGraw-Hill. p2425–2509.
Wallace DC, Shoffner JM, Trounce I, et al. 1995.Mitochondrial DNA mutations in humandegenerative diseases and aging. BiochimBiophys Acta 1271:141–151.
Willems PH, Valsecchi F, Distelmaier F, et al.2008. Mitochondrial Ca2þ homeostasis inhuman NADH:ubiquinone oxidoreductasedeficiency. Cell Calcium 44:123–133.
Wong LJ. 2001. Recognition of mitochondrialDNA deletion syndrome with non-neuro-muscular multisystemic manifestation. GenetMed 3:399–404.
Wong LJ, Brunetti-Pierri N, Zhang Q, et al.2007. Mutations in the MPV17 gene are re-sponsible for rapidly progressive liver failurein infancy. Hepatology 46:1218–1227.
Wong LJ, Dimmock D, Geraghty MT, et al.2008a. Utility of oligonucleotide array-basedcomparative genomic hybridization fordetection of target gene deletions. ClinChem 54:1141–1148.
Wong LJ, Naviaux RK, Brunetti-Pierri N, et al.2008b. Molecular and clinical genetics ofmitochondrial diseases due to POLG muta-tions. Hum Mutat 29:E150–E172.
Wong LJ, Scaglia F, Graham BH, et al. 2010. Cur-rent molecular diagnostic algorithm for mi-tochondrial disorders. Mol Genet Metab100:111–117.
Wong LJ, Senadheera D. 1997. Direct detection ofmultiple point mutations in mitochondrialDNA. Clin Chem 43:1857–1861.
Zhang S, Li FY, Bass HN, et al. 2010. Applicationof oligonucleotide aCGH to the simultane-ous detection of a deletion in the nuclearTK2 gene and mtDNA depletion. MolGenet Metab 99:53–57.
Zhu Z, Yao J, Johns T, et al. 1998. SURF1,encoding a factor involved in the biogenesisof cytochrome c oxidase, is mutated in Leighsyndrome. Nat Genet 20:337–343.
162 Dev Disabil Res Rev � MOLECULAR GENETICS OF MITOCHONDRIAL DISORDERS � WONG
Copyright © 2022 FDOKUMEN




















