
Occurrence and patterns of nutritional traits and polycyclic ...

©2005 FASEB The FASEB Journal express article 10.1096/fj.04-2269fje. Published online June 6, 2005. Polycyclic aromatic hydrocarbons induce an inflammatory atherosclerotic plaque phenotype irrespective of their DNA binding properties Daniëlle M. J. Curfs,*,† Ad M. Knaapen,* Daniëlle M. F. A. Pachen,* Marion J. J. Gijbels,†,‡ Esther Lutgens,‡ Marjan L. F. Smook,¶ Mark M. Kockx,§ Mat J. A. P. Daemen,‡ and Frederik J. van Schooten* Departments of *Health Risk Analysis and Toxicology, †Molecular Genetics, ‡Pathology, and ¶Immunology, University of Maastricht, Maastricht, The Netherlands; §Department of Pathology, University of Antwerp, Antwerp, Belgium Corresponding author: Frederik J. van Schooten, Department of Health Risk Analysis and Toxicology, University of Maastricht, Maastricht, The Netherlands. E-mail: [email protected]
ABSTRACT
Although it has been demonstrated that carcinogenic environmental polycyclic aromatic hydrocarbons (PAHs) cause progression of atherosclerosis, the underlying mechanism remains unclear. In the present study, we aimed to investigate whether DNA binding events are critically involved in the progression of PAH-mediated atherogenesis. Apolipoprotein E knockout mice were orally (24 wk, once/wk) exposed to 5 mg/kg benzo[a]pyrene (B[a]P), or its nonmutagenic, noncarcinogenic structural isoform benzo[e]pyrene (B[e]P). 32P-postlabeling of lung tissue confirmed the presence of promutagenic PAH-DNA adducts in B[a]P-exposed animals, whereas in B[e]P-exposed and vehicle control animals, these adducts were undetectable. Morphometrical analysis showed that both B[a]P and B[e]P caused an increase in plaque size, whereas location or number of plaques was unaffected. Immunohistochemistry revealed no differences in oxidative DNA damage (8-OHdG) or apoptosis in the plaques. Also plasma lipoprotein levels remained unchanged after PAH-exposure. However, T lymphocytes were increased ≥2-fold in the plaques of B[a]P- and B[e]P-exposed animals. Additionally, B[a]P and to a lesser extent B[e]P exposure resulted in increased TGFβ protein levels in the plaques, that was mainly localized in the plaque macrophages. In vitro studies using the murine macrophage like RAW264.7 cells showed that inhibition of TGFβ resulted in decreased tumor necrosis factor (TNF) α release, suggesting that enhanced TGFβ expression in the plaque macrophages contributes to the proinflammatory effects in the vessel wall. In general, this inflammatory reaction in the plaques appeared to be a local response since peripheral blood cell composition (T cells, B cells, granulocytes, and macrophages) was not changed upon PAH exposure. In conclusion, we showed that both B[a]P and B[e]P cause progression of atherosclerosis, irrespective of their DNA binding properties. Moreover, our data revealed a possible novel mechanism of PAH-mediated atherogenesis, which likely involves a TGFβ-mediated local inflammatory reaction in the vessel wall.
Page 1 of 18(page number not for citation purposes)

Key words: atherosclerosis • chemical carcinogens • DNA adduct formation • inflammation • apoE-KO mice
s the most important clinical manifestation of cardiovascular disease (CVD), atherosclerosis has been the subject of extensive study during the past several decades. This disease of the large arteries starts early in childhood, but often does not show its
first symptoms until middle life (1, 2). Atherosclerosis is a complex disorder that is characterized by an accumulation of lipids and extracellular matrix in the intima of arteries (3). The main idea is that atherosclerosis is an inflammatory response to injury of the large blood vessels (4). One of the causes of this injury could be a toxic insult to the vessel wall (5). Among the risk factors for atherosclerosis is the exposure to environmental carcinogens like polycyclic aromatic hydrocarbons (PAHs) (6–10).
PAHs account for a large group of structurally related, widespread environmental pollutants that are formed during incomplete combustion processes. Paradoxically, PAHs are biologically inert, but upon entering the organism, detoxification processes can lead to the formation of reactive intermediates that can interact with the DNA (11). These so-called PAH-DNA adducts are considered to play a crucial role in PAH-mediated carcinogenesis. Since PAH exposure is associated with high levels of DNA adducts in the aorta, and because bulky DNA adducts correlate with the severity of coronary artery disease (7–10), a mutual role for PAH-DNA adduct formation in the development of both cancer and atherosclerosis has been suggested (11). More specifically, it is hypothesized that due to the chemical-induced arterial DNA damage, a vascular cell mutates and transforms into a proliferative clone, similar to the most widely held theory of carcinogenesis (12). However, although animal and human studies have indeed demonstrated an association between the formation of PAH-DNA adducts and atherogenesis (10, 13), there is still no conclusive evidence on the causal role of DNA adduct formation in the PAH-mediated atherogenic process.
In previous studies, we have already shown that benzo[a]pyrene (B[a]P), a known carcinogenic PAH, is indeed capable of inducing high levels of DNA damage in the aorta of apolipoprotein E knockout (apoE-KO) mice, and that this was accompanied by a progression of atherosclerosis (14, 15). In the current study, we aimed to investigate whether DNA binding events in the vessel wall are critically involved in this progression of B[a]P-mediated atherosclerosis, thereby further answering the question of whether the mechanism of PAH-related atherogenesis mimics that of PAH-induced carcinogenesis. To this end, we performed chronic dosing experiments in the atherogenic apoE-KO mice using either the well-known DNA binding carcinogen B[a]P or its nonbinding, nonmutagenic structural isomer benzo[e]pyrene (B[e]P). Effects of the PAHs on progression of atherosclerosis were evaluated using morphometrical analysis of plaque number and area. Moreover, immunohistochemistry was applied to assess the contribution of DNA damage-related processes (e.g., apoptosis) but also to further elucidate the possible involvement of PAH-related inflammatory processes.
A
Page 2 of 18(page number not for citation purposes)

MATERIALS AND METHODS
Animal treatment
Seventy male apoE-KO mice (purchased from IFFA CREDO S.A., a Charles River Company, Lyon, France), 5 wk of age (18.9±0.3 g), were randomly divided into three exposure groups: B[a]P (n=30), B[e]P (n=20), and the control group (n=20). Once per week, all animals were exposed to 5 mg/kg body weight B[a]P, B[e]P, or tricaprylin by oral gavage. This procedure was repeated for 24 consecutive weeks. Mice were housed under standard conditions at the animal facility of the University of Maastricht. Water and standard mouse chow were provided ad libitum.
B[a]P and B[e]P (purity >99.8%) (B1760 and B8382, Sigma, St. Louis, MO) were dissolved in acetone and mixed with tricaprylin (103104, ICN, Irvine, CA) (Fig. 1). A homogenous solution of 0.5 mg B[a]P or B[e]P/ml tricaprylin was created by evaporation of the acetone for 5 h under nitrogen.
Flow cytometry
One week before the end of the exposure protocol, 50 µl of heparinized blood was collected by tail bleeding. Blood was processed immediately, and the erythrocytes were removed by hypotonic lysis with NH4Cl. For the detection of T and B lymphocytes, cells were stained with CD3-FITC (17A2) and CD45RB-PE (RA3-6B2), respectively. Granulocytes and macrophages were detected using Ly-6G and Ly-6C FITC-labeled (RB6-8C5) and CD11b/Mac-1 PE-labeled (M1/70), respectively. All antibodies were supplied by BD PharMingen (San Diego, CA). Flow cytometry was performed on a FACS Calibur, and the results were analyzed using CellQuest software (BD Science, San Diego, CA). Calculations of T and B cells were based on 2 × 104 viable positively stained cells. Percentages of granulocytes and macrophages were calculated based on 5 × 103 viable positively stained cells.
Lipid profile
Plasma cholesterol levels were determined by using standard enzymatic kits (Sigma and Hoffman-La Roche, Basel, Switzerland), and the colorimetric analyses were automated on the Cobas Fara centrifugal analyzer (Hoffmann-La Roche). Standardized serum (Precipath) was used as an internal standard. LDL was calculated using the formula: LDL cholesterol (mmol/l) = total cholesterol – (triglycerides/2.2) – HDL cholesterol.
Tissue processing
For euthanasia, animals were anesthetized by i.p. injection of 0.5 ml/kg Nembutal followed by exsanguination via the portal vein. Tissue processing was performed as described previously (15). In short, the arterial tree was perfused in situ with 0.9% NaCl/20% nitroprusside (3 min) and 1% phosphate-buffered paraformaldehyde (3 min) through a catheter in the apex of the heart. Subsequently, the complete arterial tree was excised, fixed overnight in 1% phosphate-buffered paraformaldehyde, and embedded in paraffin. Lung and liver were taken out and used for histological examination. Lung was further used for DNA isolation and subsequent DNA adduct measurement.
Page 3 of 18(page number not for citation purposes)

DNA isolation and bulky DNA adduct analysis
DNA was isolated from lung tissue by overnight lysis with SET/SDS containing 50 µg/ml proteinase K at 55°C. DNA was isolated using a three-step standard phenol extraction (16). The DNA was precipitated with 3 M sodiumacetate and cold ethanol, and dissolved in 2 mM Tris (pH 7.4) until a final concentration of 2 mg/ml.
The 32P-postlabeling assay was used to quantify bulky DNA adduct levels, as described previously (16). In short, 10 µg DNA was digested into deoxyribonucleoside 3′-monophosphates using micrococcal endonuclease (0.25 U/µl) and spleen phosphodiesterase (2 µg/µl). Subsequently, nuclease P1 (2.5 µg/µl) adduct enrichment was performed (40 min at 37°C). Nucleotides were labeled with [γ-32P]-ATP (50 µCi/sample) in the presence of T4-polynucleotide kinase (10 U/µl). Radiolabeled adduct nucleotide biphosphates were separated by thin layer chromatography on polyethyleneimine (PEI) cellulose sheets (Magerey Nagel, Düren, Germany). For quantification, 2 standards of [3H]BPDE-modified DNA (1 adduct per 107 and 1 adduct per 108 nucleotides, respectively) were included in each experiment. Quantification toward the standards was performed by means of phosphor-imaging technology (Molecular Dynamics, Sunnyvale, CA) with a detection limit of ~1 adduct per 109 nucleotides.
Morphometry and immunohistochemistry
Four hematoxylin and eosin-stained sections (each 20 µm apart) of the aortic arch as well as the thoracic aorta were used to classify lesions as either initial or advanced according to the guidelines given by the American Heart Association (AHA) (2). Plaque area was measured using the Leica 570 Quantimet morphometry system.
Sections were immunolabeled for the detection of plaque total inflammatory cells (CD45, 1:50, PharMingen), plaque T-lymphocytes (CD3, 1:80, Labvision, Montreal, Canada), TGFβ1 (TGFβ1, 1:30 R&D Systems, Minneapolis, MN), macrophages (Mac-3, 1:30, BP PharMingen), and oxidative DNA damage (8-hydroxydeoxyguanosine, 8-OHdG) (N45.1, 1:150, Japan Institute for the Control of Aging, Fukuroi, Japan). Apoptotic cell death was detected by TUNEL staining (ApopTag kit, Oncor, Gaithersburg, MD) and was performed in the lab of Dr. M. Kockx (Antwerp, Belgium). Total inflammatory cell content, T cell content, 8-OHdG, and apoptotic cell content were quantified by counting the number of CD45, CD3, 8-OHdG, and TUNEL positively stained cells, respectively, divided by the total plaque area. Macrophage content was measured by dividing the MAC3-positive cell area by the total plaque area. Finally, based on the TGFβ1 protein staining intensity of the plaque macrophages, animals were semiquantitatively classified as having low or high TGFβ1 protein expression.
Cell culture and enzyme-linked immunosorbent assay (ELISA)
Murine monocytes/macrophages like RAW264.7 cells were cultured in RPMI supplemented with 10% fetal calf serum (FCS), 1% penicillin/streptomycin, and 1% L-glutamine, and passaged twice per week. To specifically investigate the significance of increased TGFβ1 protein expression in macrophages, we plated cells in a 24-well plate at a density of 5 × 105 cells/well, and incubated with 0.1 µg/ml anti-TGFβ1 antibody (R&D Systems) for 24 h before the exposure
Page 4 of 18(page number not for citation purposes)

protocol. At t = 0, cells were again supplemented with 0.1 µg/ml anti-TGFβ1 antibody and subsequently exposed to 100 ng/ml lipopolysaccharide (LPS), or PAHs (0–5 µM) for 8 h to activate the macrophages. After the exposure protocol, the proinflammatory and proatherogenic cytokine tumor necrosis factor (TNF)-α was assayed in the supernatants by ELISA (Biosource, Etten-Leur, The Netherlands) according to the manufacturer’s instructions. All in vitro experiments were performed in quadruplicate.
Statistical analysis
The results obtained from the in vivo experiments are presented as mean ± SE. Results from the in vitro experiments are presented as mean ± SD. Data were analyzed using the nonparametric Mann-Whitney U test. A Chi-square test was performed to evaluate differences in plaque TGFβ1 protein expression. The level of statistical significance was set at P < 0.05. All analyses were performed by two independent observers who were blinded for the exposure protocol. Both inter- and intraobserver variation was <10%.
RESULTS
General
During the trial, 9 animals died; 3 out of 20 animals of the B[e]P group (15%), 4 out of 30 animals of the B[a]P group (13.3%), and 2 out of 20 animals of the control group (10%). At the end of the trial, the body weight of both B[e]P- and B[a]P-exposed animals was lower than the body weight of the control animals (P<0.05, Table 1). Pathology of lung and liver showed no major abnormalities like tumor formation or excessive inflammation. Plasma lipid levels at the end of the trial were not significantly modulated by the PAH exposure (Table 1).
Since oral B[a]P exposure leads to a general and rather homogeneous distribution of DNA adducts over multiple organs, including lung and aorta (14), we used lung tissue of the animals to control for the efficiency of the different PAHs to produce DNA adducts. The results confirmed that, in contrast with B[a]P, B[e]P did not form any detectable reactive metabolites bound to the DNA (Table 1).
Blood flow cytometry analysis
The effect of PAH exposure on circulating inflammatory cells was measured by means of flow cytometry of blood T-lymphocytes, B-lymphocytes, granulocytes and macrophages. B[a]P and B[e]P exposure did not influence the levels of circulating inflammatory cells compared with the control group (Table 2). Although blood granulocyte levels were decreased in the B[e]P-exposed animals compared with the B[a]P-exposed animals, in comparison with the control group no significant differences could be observed.
Plaque parameters
Plaque burden
In total, 274 lesions were analyzed in the aortic arches of 61 mice: 74 lesions in the B[e]P-exposed animals (4.4 plaques per arch, n=17), 116 lesions in the B[a]P (4.5 plaques per arch,
Page 5 of 18(page number not for citation purposes)

n=26), and 84 lesions in the control group (4.7 plaques per arch, n=18). The location (inner curve of the arch, brachiocephalic trunk, left subclavian artery, and left common carotid artery) and the number of plaques per arch did not differ between the three groups. Most lesions were advanced lesions (236 out of 274). Whereas the total area of initial plaques per arch did not differ between the three groups, the total area of advanced plaques per arch was significantly increased in both B[a]P- and B[e]P-exposed mice (Fig. 2A).
Segments of the complete thoracic aorta were analyzed to measure the effect of PAH exposure on plaque initiation. The lesions in the thoracic aorta were predominantly initial lesions. There was no difference in the number of lesions per thoracic aorta segment between the three groups: control, 2.1 ± 0.5 (n=9); B[e]P, 2.9 ± 0.3 (n=17); and B[a]P, 2.2 ± 0.3 (n=16). Also the size of the lesions did not differ between the groups (Fig. 2B).
Plaque phenotype
Staining for oxidative DNA damage (8-OHdG) in the nuclei of cells showed that the immunoreactivity for 8-OHdG was mainly localized in the plaque macrophages and the endothelial cell layer covering the plaques. Quantification of the positively stained cells in the plaques showed no difference in the levels of 8-OHdG between the B[e]P, B[a]P, and control animals: 10.2 ± 1.7 × 10−4 cells/µm2 (n=17), 10.5 ± 1.6 × 10−4 cells/µm2 (n=16), and 20.0 ± 1.1 × 10−4 cells/µm2 (n=9), respectively. The media underlying the plaques as well as the nondiseased vessel wall were almost completely negative. TUNEL staining revealed no differences in apoptosis between the B[e]P, B[a]P, and control mice: 24 ± 3.2 × 10−6 cells/µm2, 26 ± 3.5 × 10−6 cells/µm2, and 27 ± 10 ×10−6 cells/µm2, respectively.
Based on initial results from our previous study (15), we further investigated the effects of both PAHs on inflammatory plaque parameters in the aortic arch. Plaque macrophage content was not significantly different between the B[e]P, B[a]P, and control group: 39.4 ± 2.9% (n=17), 44.6 ± 3.1% (n=16), and 37.8 ± 4.1% (n=9), respectively. However, in both B[e]P- and B[a]P-exposed animals, plaque total inflammatory cells (CD45-positive cells) were significantly enhanced compared with the control group (P<0.05, Fig. 2C). No differences could be observed between B[e]P and B[a]P. More specifically, immunohistochemistry showed a clear increase in plaque T lymphocytes (CD3-positive cells) in both B[e]P- and B[a]P-exposed animals, as compared with the control animals (P<0.05, Fig. 2D and Fig. 3A–C).
Expression of TGFβ1 was mainly observed in plaque macrophages (Fig. 3D–F) Based on staining intensity, sections of the aortic arches were classified having low or high protein expression. The control animals exhibited predominantly low staining for TGFβ1, whereas the plaques of the B[a]P and to a lesser extent the B[e]P group were mainly classified as high (B[a]P: χ2=7.0, P<0.05. Fig. 2E).
In vitro experiments using the murine RAW264.7 monocyte/macrophage cells
To investigate the significance of the up-regulation of TGFβ in plaque macrophages in vivo, we performed further in vitro experiments using the murine monocyte/macrophage RAW264.7 cell line. The results showed that neutralization of TGFβ1 in activated RAW264.7 cells significantly reduced the release of the potent atherogenic cytokine TNF-α (P<0.05, Fig. 4). This was
Page 6 of 18(page number not for citation purposes)

paralleled by an inhibiting effect of TGFβ antibodies on LPS-induced expression of TNF-α mRNA (data not shown). No direct effects of B[e]P or B[a]P on release of TNF-α or expression of TGFβ by RAW264.7 cells could be observed (data not shown).
DISCUSSION
The aim of the present study was to investigate whether DNA binding events in the vessel wall are causally involved in the progression of chemical atherogenesis. The major outcome of our study is that both the DNA binding PAH B[a]P and its nonbinding structural isomer B[e]P had comparable effects on development of atherosclerosis in apoE-KO mice, suggesting that there is no functional role for DNA adduct formation in PAH-mediated murine atherogenesis. This finding is highly relevant for the field of (chemical) atherosclerosis research, as it implicates that PAH-mediated atherogenesis cannot be explained simply by the basic mechanisms underlying chemical carcinogenesis. Moreover, further analysis revealed that this PAH-induced atherogenic effect was accompanied by the induction of a local inflammatory response, which is possibly mediated by the induction of TGFβ.
Atherosclerosis is a multicellular disorder in which lipids and extracellular matrix accumulate in the intima of arteries. Due to the activation of endothelial cells, smooth muscle cells, macrophages, and T lymphocytes, a local inflammatory response is elicited (17, 18). In the present study, the specific role of PAH-DNA adduct formation on atherosclerosis in apoE-KO mice was further elucidated by comparing the atherogenic capacity of the adduct-forming carcinogen B[a]P with the chemically almost identical non-adduct-forming, noncarcinogen B[e]P. In contrast to B[a]P, B[e]P has proven to be very weak or noncarcinogenic, most likely because it does not interact with the Ah receptor (19), which is a necessary step to activate the drug metabolizing enzyme system cytochrome P450. Furthermore, even in the presence of metabolizing enzymes, the symmetric structure of B[e]P (Fig. 1) causes steric hinderence, hampering the formation of reactive metabolites. As a result, no or only very low levels of reactive intermediates and thus DNA adducts are formed (20–22). However, despite the dramatic difference in DNA binding, we showed that both PAHs caused a significant and comparable increase in atherosclerotic plaque progression. Because no DNA adducts could be found in B[e]P-treated animals, these data suggest that DNA adduct formation is a rather irrelevant process with respect to the atherogenic effects of both B[a]P and B[e]P. Moreover, we showed that PAH-induced DNA adduct formation did not parallel related processes such as apoptosis, cell proliferation, expression of p53 protein, and oxidative DNA damage (8-OHdG) in atherosclerotic plaques (15).
To find a possible explanation for the atherogenic effects of both PAHs, we extended previous analyses (15) and found that both B[a]P and B[e]P significantly affected the plaque phenotype by causing a profound influx of inflammatory cells into the plaques. For example, exposure to both PAHs significantly increased the T lymphocyte number in the plaques. In general, the capacity of PAHs to modulate immune cell function has also been well documented (23–25), and studies from Johnson et al. (26) suggested that modification of T cell activation could be a specific target in PAH-induced atherogenesis. It has been reported by several groups that B[a]P is able to suppress B lymphocyte responses to T cell-dependent and -independent antigens (27–29). Furthermore, it is also reported that low doses of B[a]P (3 mg/kg day) resulted in an increase in T lymphocytes, whereas high doses (90 mg/kg day) resulted in a decrease of both T and B
Page 7 of 18(page number not for citation purposes)

lymphocytes (30). In our study, using low doses of B[a]P (5 mg/kg wk), an increase of immune cells was found in the plaques in the absence of an increase in circulating macrophages, neutrophils, and B and T lymphocytes. This further supports the idea that the observed inflammatory plaque phenotype after PAH exposure is due to a local response of the damaged vessel wall. This is also illustrated by the lack of PAH-related changes in liver weight or pathology, such as observed in rats exposed to higher doses of B[a]P (30).
To get more insight in the PAH-induced inflammatory processes in the plaques, we focused on TGFβ since it is considered to play a crucial role in the development of atherosclerosis (12, 31, 32) and is shown to be modulated by PAHs (33). In the present study, we found that TGFβ1 protein expression was increased in the plaque macrophages of B[a]P and, although to a lesser extent, also in plaque macrophages of B[e]P-exposed animals. Importantly, these findings could possibly explain the observed influx of T lymphocytes into the plaques since it has been reported that TGFβ induces the migration of T lymphocytes (34). Our in vitro studies, using RAW264.7, showed that the induction of TGFβ could not be explained by a direct effect of the PAHs on the plaque macrophages, suggesting that the TGFβ expression is merely a consequence of the ongoing PAH-mediated inflammatory changes in the plaques. In general, there is still debate on the role of TGFβ1 in vascular disease. Several studies have associated TGFβ expression with the development of arterial disease (35) whereas others suggest that TGFβ prevents arterial lesion formation (31, 32). We showed that inhibition of TGFβ in activated macrophages was associated with a decreased release of the proatherogenic cytokine TNF-α (36). These findings suggests that the expression of TGFβ in plaque macrophages further increases the local proinflammatory effects in the vessel wall and thereby contributes to the enhanced progression of atherosclerosis in the PAH-exposed ApoE-KO mice.
In conclusion, the results obtained in this study are in contrast with earlier studies that have proposed a link between PAH-DNA adduct formation and atherogenesis. We have shown that the non-adduct-forming B[e]P elicits an inflammatory plaque phenotype in apoE-KO mice that is comparable to the phenotype induced by the DNA adduct-forming B[a]P. These observations suggest that PAH-DNA adduct formation is not causally involved in PAH-modulated atherogenesis. In contrast, our data indicate that PAHs such as B[a]P and B[e]P might exert their atherogenic effect via the stimulation of a general, TGFβ-mediated, local inflammatory process, involving an increased influx of proinflammatory cells into the plaques (Fig. 5).
ACKNOWLEDGMENTS
A.M. Knaapen was supported by a postdoctoral fellowship from the Netherlands Organization for Scientific Research (NWO, grant 916.46.092). E. Lutgens is a postdoctoral research fellow of the Dr. E. Dekker program of the Dutch Heart Foundation (2000T041).
REFERENCES
1. Stary, H. C. (1989) Evolution and progression of atherosclerotic lesions in coronary arteries of children and young adults. Arteriosclerosis 9, I19–I32
2. Stary, H., Blankenhorn, D., Chandler, A., Glagov, S., Insull, W., Richardson, M., Rosenfeld, M., Schaffer, S., Schwartz, C., Wagner, W., et al. (1995) A definition of advanced types of
Page 8 of 18(page number not for citation purposes)

atherosclerotic lesions and a histological classification of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 15, 1513–1531
3. Lusis, A. J. (2000) Atherosclerosis. Nature 407, 233–241
4. Ross, R. (1999) Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 340, 115–126
5. Benditt, E. P. (1974) Evidence for a monoclonal origin of human atherosclerotic plaques and some implications. Circulation 50, 650–652
6. Izzotti, A., Cartiglia, C., Lewtas, J., and De Flora, S. (2001) Increased DNA alterations in atherosclerotic lesions of individuals lacking the GSTM1 genotype. FASEB J. 15, 752–757
7. Van Schooten, F. J., Hirvonen, A., Maas, L. M., De Mol, B. A., Kleinjans, J. C., Bell, D. A., and Durrer, J. D. (1998) Putative susceptibility markers of coronary artery disease: association between VDR genotype, smoking, and aromatic DNA adduct levels in human right atrial tissue. FASEB J. 12, 1409–1417
8. Binkova, B., Strejc, P., Boubelik, O., Stavkova, Z., Chvatalova, I., and Sram, R. J. (2001) DNA adducts and human atherosclerotic lesions. Int. J. Hyg. Environ. Health 204, 49–54
9. De Flora, S., Izzotti, A., Randerath, K., Randerath, E., Bartsch, H., Nair, J., Balansky, R., van Schooten, F., Degan, P., Fronza, G., et al. (1996) DNA adducts and chronic degenerative disease. Pathogenetic relevance and implications in preventive medicine. Mutat. Res. 366, 197–238
10. De Flora, S., Izzotti, A., Walsh, D., Degan, P., Petrilli, G. L., and Lewtas, J. (1997) Molecular epidemiology of atherosclerosis. FASEB J. 11, 1021–1031
11. International Agency for Research on Cancer (1983) Polynuclear aromatic compounds. Part 1. Chemical, environmental and experimental data. In IARC Monograph on the Evaluation of the Carcinogenic Risk of Chemicals on Humans, no. 32, pp. 1–453. IARC, Lyon, France
12. Ross, J. S., Stagliano, N. E., Donovan, M. J., Breitbart, R. E., and Ginsburg, G. S. (2001) Atherosclerosis and cancer: common molecular pathways of disease development and progression. Ann. N.Y. Acad. Sci. 947, 271–292, discussion 292–293
13. Wakabayashi, K. (1990) International Commission for Protection Against Environmental Mutagens and Carcinogens. ICPEMC Working Paper 7/1/3. Animal studies suggesting involvement of mutagen/carcinogen exposure in atherosclerosis. Mutat. Res. 239, 181–187
14. Curfs, D. M., Beckers, L., Godschalk, R. W., Gijbels, M. J., and van Schooten, F. J. (2003) Modulation of plasma lipid levels affects benzo[a]pyrene-induced DNA damage in tissues of two hyperlipidemic mouse models. Environ. Mol. Mutagen. 42, 243–249
15. Curfs, D. M., Lutgens, E., Gijbels, M. J., Kockx, M. M., Daemen, M. J., and van Schooten, F. J. (2004) Chronic exposure to the carcinogenic compound benzo[a]pyrene induces larger
Page 9 of 18(page number not for citation purposes)

and phenotypically different atherosclerotic plaques in ApoE-knockout mice. Am. J. Pathol. 164, 101–108
16. Godschalk, R. W., Maas, L. M., Van Zandwijk, N., van't Veer, L. J., Breedijk, A., Borm, P. J., Verhaert, J., Kleinjans, J. C., and van Schooten, F. J. (1998) Differences in aromatic-DNA adduct levels between alveolar macrophages and subpopulations of white blood cells from smokers. Carcinogenesis 19, 819–825
17. Hansson, G. K. (2001) Immune mechanisms in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 21, 1876–1890
18. Ross, R. (1993) The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362, 801–809
19. Hankinson, O. (1995) The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 35, 307–340
20. Wood, A. W., Chang, R. L., Huang, M. T., Levin, W., Lehr, R. E., Kumar, S., Thakker, D. R., Yagi, H., Jerina, D. M., and Conney, A. H. (1980) Mutagenicity of benzo(e)pyrene and triphenylene tetrahydroepoxides and diol-epoxides in bacterial and mammalian cells. Cancer Res. 40, 1985–1989
21. Thakker, D. R., Levin, W., Buening, M., Yagi, H., Lehr, R. E., Wood, A. W., Conney, A. H., and Jerina, D. M. (1981) Species-specific enhancement by 7,8-benzoflavone of hepatic microsomal metabolism of benzo[e]pyrene 9,10-dihydrodiol to bay-region diol epoxides. Cancer Res. 41, 1389–1396
22. Wood, A. W., Levin, W., Thakker, D. R., Yagi, H., Chang, R. L., Ryan, D. E., Thomas, P. E., Dansette, P. M., Whittaker, N., Turujman, S., et al. (1979) Biological activity of benzo[e]pyrene. An assessment based on mutagenic activities and metabolic profiles of the polycyclic hydrocarbon and its derivatives. J. Biol. Chem. 254, 4408–4415
23. Burchiel, S. W., Hadley, W. M., Barton, S. L., Fincher, R. H., Lauer, L. D., and Dean, J. H. (1988) Persistent suppression of humoral immunity produced by 7,12-dimethylbenz(A)anthracene (DMBA) in B6C3F1 mice: correlation with changes in spleen cell surface markers detected by flow cytometry. Int. J. Immunopharmacol. 10, 369–376
24. Ladics, G. S., Kawabata, T. T., and White, K. L., Jr. (1991) Suppression of the in vitro humoral immune response of mouse splenocytes by 7,12-dimethylbenz[a]anthracene metabolites and inhibition of immunosuppression by alpha-naphthoflavone. Toxicol. Appl. Pharmacol. 110, 31–44
25. Holladay, S. D., and Smith, B. J. (1995) Benzo[a]pyrene-induced alterations in total immune cell number and cell-surface antigen expression in the thymus, spleen and bone marrow of B6C3F1 mice. Vet. Hum. Toxicol. 37, 99–104
26. Johnson, C. D., Balagurunathan, Y., Lu, K. P., Tadesse, M., Falahatpisheh, M. H., Carroll, R. J., Dougherty, E. R., Afshari, C. A., and Ramos, K. S. (2003) Genomic profiles and
Page 10 of 18(page number not for citation purposes)

predictive biological networks in oxidant-induced atherogenesis. Physiol. Genomics 13, 263–275
27. Dean, J. H., Luster, M. I., Boorman, G. A., Lauer, L. D., Leubke, R. W., and Lawson, L. (1983) Selective immunosuppression resulting from exposure to the carcinogenic congener of benzopyrene in B6C3F1 mice. Clin. Exp. Immunol. 52, 199–206
28. White, K. L., Jr., and Holsapple, M. P. (1984) Direct suppression of in vitro antibody production by mouse spleen cells by the carcinogen benzo(a)pyrene but not by the noncarcinogenic congener benzo(e)pyrene. Cancer Res. 44, 3388–3393
29. White, K. L., Jr., Lysy, H. H., and Holsapple, M. P. (1985) Immunosuppression by polycyclic aromatic hydrocarbons: a structure-activity relationship in B6C3F1 and DBA/2 mice. Immunopharmacology 9, 155–164
30. De Jong, W. H., Kroese, E. D., Vos, J. G., and Van Loveren, H. (1999) Detection of immunotoxicity of benzo[a]pyrene in a subacute toxicity study after oral exposure in rats. Toxicol. Sci. 50, 214–220
31. Lutgens, E., Gijbels, M., Smook, M., Heeringa, P., Gotwals, P., Koteliansky, V. E., and Daemen, M. J. (2002) Transforming growth factor-beta mediates balance between inflammation and fibrosis during plaque progression. Arterioscler. Thromb. Vasc. Biol. 22, 975–982
32. Mallat, Z., Gojova, A., Marchiol-Fournigault, C., Esposito, B., Kamate, C., Merval, R., Fradelizi, D., and Tedgui, A. (2001) Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ. Res. 89, 930–934
33. Sherman, J. H., Miller, M. L., Albert, R. E., and Baxter, C. S. (1993) Dose- and time-dependent expression of transforming growth factor-beta 1 mRNA and protein in mouse epidermis and papillomas after repeated topical application of benzo[a]pyrene. Mol. Carcinog. 8, 264–271
34. Adams, D. H., Hathaway, M., Shaw, J., Burnett, D., Elias, E., and Strain, A. J. (1991) Transforming growth factor-beta induces human T lymphocyte migration in vitro. J. Immunol. 147, 609–612
35. Schulick, A. H., Taylor, A. J., Zuo, W., Qiu, C. B., Dong, G., Woodward, R. N., Agah, R., Roberts, A. B., Virmani, R., and Dichek, D. A. (1998) Overexpression of transforming growth factor beta1 in arterial endothelium causes hyperplasia, apoptosis, and cartilaginous metaplasia. Proc. Natl. Acad. Sci. USA 95, 6983–6988
36. Branen, L., Hovgaard, L., Nitulescu, M., Bengtsson, E., Nilsson, J., and Jovinge, S. (2004) Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 24, 2137–2142
Received November 2, 2004; accepted April 8, 2005.
Page 11 of 18(page number not for citation purposes)

Table 1
Growth, DNA adducts, and lipoprotein levelsa Control (n=18) B[e]P (n=17) B[a]P (n=26)
Weight gain (g)
12.4 ± 0.4 10.8 ± 0.3b
11.2 ± 0.2b
DNA adducts (per 108 nucl.)
Lung
<0.1 <0.1 14.3 ± 0.9c
Lipoprotein levels (mmol/l) Control (n=15) B[e]P (n=6) B[a]P (n=15) Total cholesterol
11.4 ± 0.6
11.9 ± 2.2
11.8 ± 0.8
Total glycerol 1.2 ± 0.1 1.2 ± 0.2 1.4 ± 0.1 HDL cholesterol 0.3 ± 0.03 0.5 ± 0.03 0.3 ± 0.04 Free glycerol 0.3 ± 0.05 0.2 ± 0.02 0.2 ± 0.03 LDL cholesterol 10.7 ± 0.6 10.9 ± 2.1 11.1 ± 0.8
aValues are mean ± se. bP < 0.05 vs. controls. cP < 0.01 vs. B[e]P and controls.
Page 12 of 18(page number not for citation purposes)

Table 2 Blood flow cytometrya Control
(n=5) B[e]P (n=16)
B[a]P (n=14)
T-lymphocytes 18.8 ± 1.7 21.9 ± 1.2 21.8 ± 1.3 B-lymphocytes 43.0 ± 1.4 45.8 ± 1.7 43.5 ± 1.5 Granulocytes 19.3 ± 2.4 15.6 ± 0.8b 20.5 ± 1.8 Macrophages 15.3 ± 1.1 15.0 ± 0.6 13.4 ± 0.8 aValues are mean ± se. bP < 0.05 vs. B[a]P.
Page 13 of 18(page number not for citation purposes)

Fig. 1
Figure 1. Chemical structure of the DNA-binding mutagenic and carcinogenic PAH benzo[a]pyrene and its non-DNA binding and noncarcinogenic isomer benzo[e]pyrene.
Page 14 of 18(page number not for citation purposes)
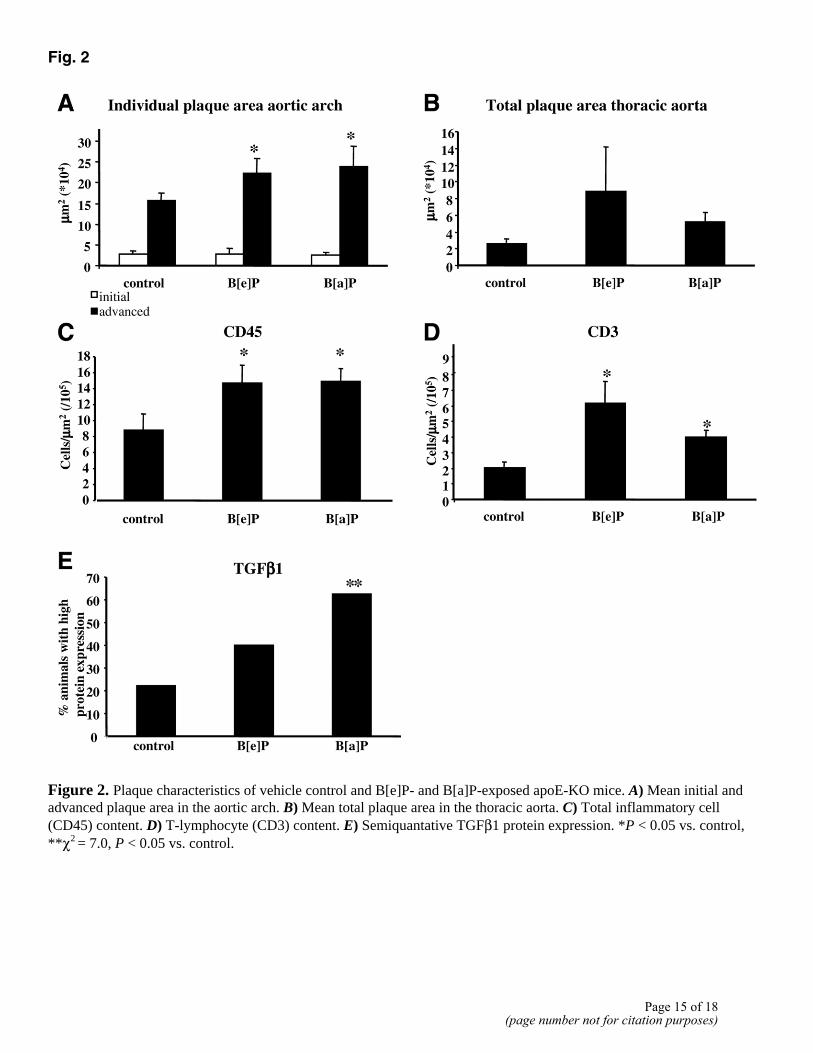
Fig. 2
Figure 2. Plaque characteristics of vehicle control and B[e]P- and B[a]P-exposed apoE-KO mice. A) Mean initial and advanced plaque area in the aortic arch. B) Mean total plaque area in the thoracic aorta. C) Total inflammatory cell (CD45) content. D) T-lymphocyte (CD3) content. E) Semiquantative TGFβ1 protein expression. *P < 0.05 vs. control, **χ2 = 7.0, P < 0.05 vs. control.
Page 15 of 18(page number not for citation purposes)

Fig. 3
Figure 3. Representative plaque characteristics of apoE-KO mice exposed to B[a]P, B[e]P, or vehicle (control). CD3 staining of plaques in the aortic arch of a control (A), B[e]P-exposed (B), and B[a]P-exposed (C) apoE-KO mouse, ×40. TGFβ1 staining in plaques of a control (D), B[e]P-exposed (E), and B[a]P-exposed (F) apoE-KO mouse, ×20.
Page 16 of 18(page number not for citation purposes)

Fig. 4
Figure 4. Effect of inhibition of TGFβ on TNF-α release by RAW264.7 cells exposed to LPS (100 ng/ml, 8 h). (TGFβ1-AB = antibodies against TGFβ1, 100 ng/ml, 24 h) *P < 0.05 vs. negative control, **P < 0.05 vs. LPS control.
Page 17 of 18(page number not for citation purposes)

Fig. 5
Figure 5. General overview of the proposed mechanism of PAH-mediated atherogenesis in apoE-KO mice. A cross (×) indicates the exclusion of a possible route of action. PAHs are suggested to cause progression of atherosclerosis by inducing a local inflammatory reaction in the vessel wall. Our data further indicate a mediating role of TGFβ by enhancing the inflammatory processes in the vessel wall.
Page 18 of 18(page number not for citation purposes)
Copyright © 2022 FDOKUMEN




















