
PIXE and EDXRF studies on banded iron formations from eastern India
10
PIXE and EDXRF studies on banded iron formations from eastern India P.K. Nayak a, * , D. Das b , V. Vijayan c , P. Singh d , V. Chakravortty a,1 a Department of Chemistry, Utkal University, Vani Vihar, Bhubaneswar, Orissa 751 004, India b M€ ossbauer Division, IUC-DAEF, Calcutta Centre, Kolkata 700 091, India c Institute of Physics, Sachivalaya Marg, Bhubaneswar 751 005, India d Department of Geology, Utkal University, Bhubaneswar 751 004, India Received 2 January 2003; received in revised form 8 May 2003 Abstract A selected number of eastern Indian banded iron formations (BIFs) have been studied by using complementary non- destructive X-ray techniques like proton-induced X-ray emission and energy dispersive X-ray fluorescence. Nineteen trace elements including Co, Ni, Zr, Cr, Ti and Y were quantified in these BIFs, which have been used to interpret the various physical–chemical conditions of the depositional basin. The study advocates the use of X-ray techniques complementarily in quantifying trace elements in high-grade ores in order to reveal geochemistry successfully. Ó 2003 Published by Elsevier B.V. Keywords: PIXE; EDXRF; Complementary use of PIXE and EDXRF; Banded iron formation; Trace element studies; Geochemical application 1. Introduction The deposition of banded iron formations (BIFs) occurred between early Archean to late Proterozoic period (3.5–1.8 billion years B.P.) [1,2], and are considered to have formed as chemical precipitates in relatively shallow primi- tive seas [2,3]. These are mostly alternate bands of iron and silica expected to contain lower trace metal concentration. Although a considerable current interest is under progress for understand- ing their trace element geochemistry [4,5] and un- folding the primary–secondary processes those have produced [5], the origin of these BIFs are highly controversial [6–8]. Until recently, there have been few systematic geochemical studies carried out on BIFs, particu- larly of their trace element geochemistry. Now a days various multielemental techniques like atomic absorption spectroscopy (AAS), electron probe micro-analysis (EPMA), inductively cou- pled plasma mass spectroscopy (ICP-MS) and laser-ablation combined with inductively coupled plasma mass spectroscopy (LA-ICP-MS) are be- ing used for quantification of trace elements in geological materials. Among them, ICP-MS and AAS are becoming more routine, but destructive * Corresponding author. Tel.: +91-674-2540466. E-mail address: [email protected] (P.K. Nayak). 1 Posthumously. 0168-583X/$ - see front matter Ó 2003 Published by Elsevier B.V. doi:10.1016/j.nimb.2003.07.006 Nuclear Instruments and Methods in Physics Research B 215 (2004) 252–261 www.elsevier.com/locate/nimb
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of PIXE and EDXRF studies on banded iron formations from eastern India

Nuclear Instruments and Methods in Physics Research B 215 (2004) 252–261
www.elsevier.com/locate/nimb
PIXE and EDXRF studies on banded iron formationsfrom eastern India
P.K. Nayak a,*, D. Das b, V. Vijayan c, P. Singh d, V. Chakravortty a,1
a Department of Chemistry, Utkal University, Vani Vihar, Bhubaneswar, Orissa 751 004, Indiab M€ossbauer Division, IUC-DAEF, Calcutta Centre, Kolkata 700 091, India
c Institute of Physics, Sachivalaya Marg, Bhubaneswar 751 005, Indiad Department of Geology, Utkal University, Bhubaneswar 751 004, India
Received 2 January 2003; received in revised form 8 May 2003
Abstract
A selected number of eastern Indian banded iron formations (BIFs) have been studied by using complementary non-
destructive X-ray techniques like proton-induced X-ray emission and energy dispersive X-ray fluorescence. Nineteen
trace elements including Co, Ni, Zr, Cr, Ti and Y were quantified in these BIFs, which have been used to interpret the
various physical–chemical conditions of the depositional basin. The study advocates the use of X-ray techniques
complementarily in quantifying trace elements in high-grade ores in order to reveal geochemistry successfully.
� 2003 Published by Elsevier B.V.
Keywords: PIXE; EDXRF; Complementary use of PIXE and EDXRF; Banded iron formation; Trace element studies; Geochemical
application
1. Introduction
The deposition of banded iron formations
(BIFs) occurred between early Archean to lateProterozoic period (3.5–1.8 billion years B.P.)
[1,2], and are considered to have formed as
chemical precipitates in relatively shallow primi-
tive seas [2,3]. These are mostly alternate bands of
iron and silica expected to contain lower trace
metal concentration. Although a considerable
current interest is under progress for understand-
* Corresponding author. Tel.: +91-674-2540466.
E-mail address: [email protected] (P.K. Nayak).1 Posthumously.
0168-583X/$ - see front matter � 2003 Published by Elsevier B.V.
doi:10.1016/j.nimb.2003.07.006
ing their trace element geochemistry [4,5] and un-
folding the primary–secondary processes those
have produced [5], the origin of these BIFs are
highly controversial [6–8].Until recently, there have been few systematic
geochemical studies carried out on BIFs, particu-
larly of their trace element geochemistry. Now
a days various multielemental techniques like
atomic absorption spectroscopy (AAS), electron
probe micro-analysis (EPMA), inductively cou-
pled plasma mass spectroscopy (ICP-MS) and
laser-ablation combined with inductively coupledplasma mass spectroscopy (LA-ICP-MS) are be-
ing used for quantification of trace elements in
geological materials. Among them, ICP-MS and
AAS are becoming more routine, but destructive
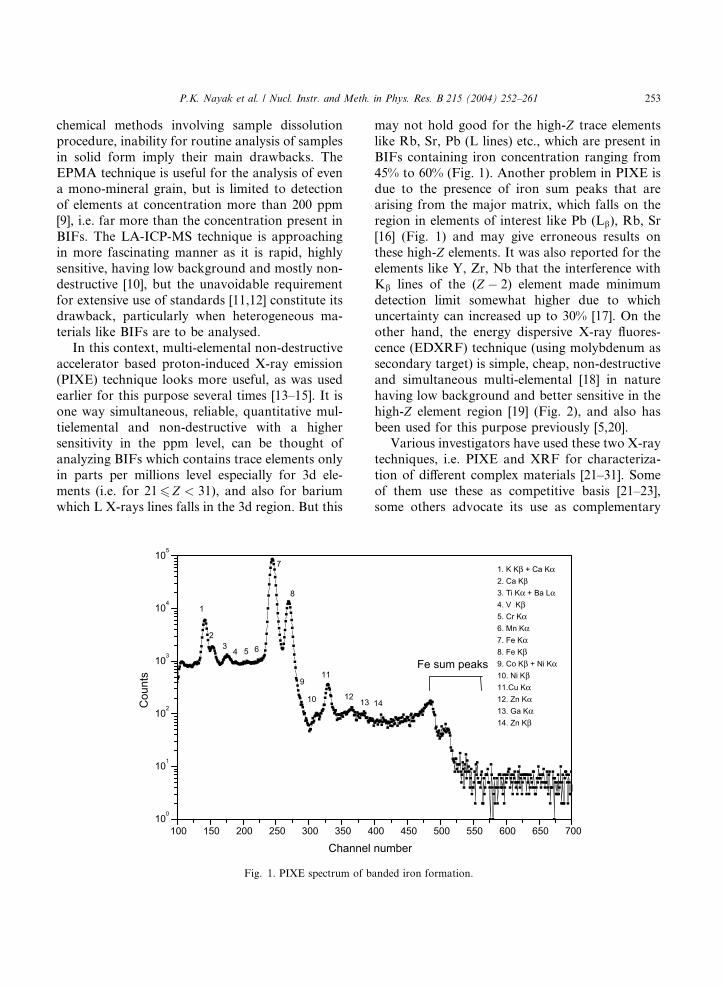
P.K. Nayak et al. / Nucl. Instr. and Meth. in Phys. Res. B 215 (2004) 252–261 253
chemical methods involving sample dissolution
procedure, inability for routine analysis of samples
in solid form imply their main drawbacks. The
EPMA technique is useful for the analysis of evena mono-mineral grain, but is limited to detection
of elements at concentration more than 200 ppm
[9], i.e. far more than the concentration present in
BIFs. The LA-ICP-MS technique is approaching
in more fascinating manner as it is rapid, highly
sensitive, having low background and mostly non-
destructive [10], but the unavoidable requirement
for extensive use of standards [11,12] constitute itsdrawback, particularly when heterogeneous ma-
terials like BIFs are to be analysed.
In this context, multi-elemental non-destructive
accelerator based proton-induced X-ray emission
(PIXE) technique looks more useful, as was used
earlier for this purpose several times [13–15]. It is
one way simultaneous, reliable, quantitative mul-
tielemental and non-destructive with a highersensitivity in the ppm level, can be thought of
analyzing BIFs which contains trace elements only
in parts per millions level especially for 3d ele-
ments (i.e. for 216Z < 31), and also for barium
which L X-rays lines falls in the 3d region. But this
100 150 200 250 300 350 4100
101
102
103
104
105
1312
11
10
9
8
7
6543
2
1
Cou
nts
Channel
Fig. 1. PIXE spectrum of b
may not hold good for the high-Z trace elements
like Rb, Sr, Pb (L lines) etc., which are present in
BIFs containing iron concentration ranging from
45% to 60% (Fig. 1). Another problem in PIXE isdue to the presence of iron sum peaks that are
arising from the major matrix, which falls on the
region in elements of interest like Pb (Lb), Rb, Sr
[16] (Fig. 1) and may give erroneous results on
these high-Z elements. It was also reported for the
elements like Y, Zr, Nb that the interference with
Kb lines of the (Z � 2) element made minimum
detection limit somewhat higher due to whichuncertainty can increased up to 30% [17]. On the
other hand, the energy dispersive X-ray fluores-
cence (EDXRF) technique (using molybdenum as
secondary target) is simple, cheap, non-destructive
and simultaneous multi-elemental [18] in nature
having low background and better sensitive in the
high-Z element region [19] (Fig. 2), and also has
been used for this purpose previously [5,20].Various investigators have used these two X-ray
techniques, i.e. PIXE and XRF for characteriza-
tion of different complex materials [21–31]. Some
of them use these as competitive basis [21–23],
some others advocate its use as complementary
00 450 500 550 600 650 700
Fe sum peaks
1. K Kβ + Ca Kα2. Ca Kβ3. Ti Kα + Ba Lα4. V Kβ 5. Cr Kα6. Mn Kα7. Fe Kα8. Fe Kβ9. Co Kβ + Ni Kα10. Ni Kβ11.Cu Kα12. Zn Kα13. Ga Kα14. Zn Kβ
14
number
anded iron formation.
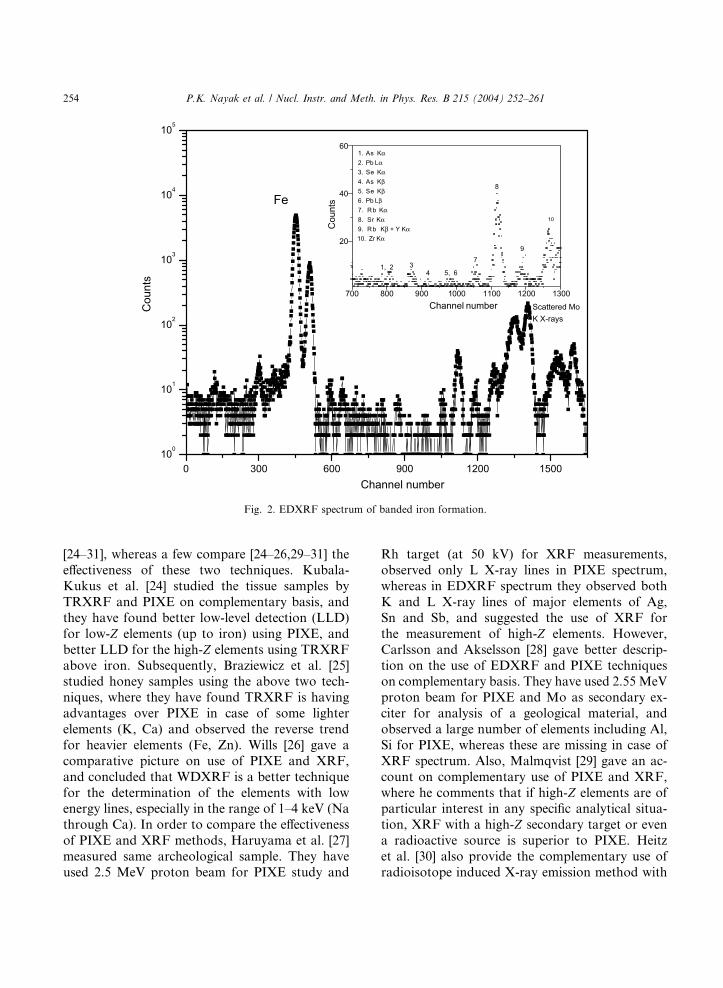
0 300 600 900 1200 1500100
101
102
103
104
105
700 800 900 1000 1100 1200 1300
20
40
60
10
9
8
7
5, 6431, 2
1. As Kα 2. Pb Lα 3. Se Kα 4. As Kβ5. Se Kβ6. Pb Lβ 7. Rb Kα 8. Sr Kα9. Rb Kβ + Y Kα10. Zr Kα
Cou
nts
Channel number Scattered MoK X-rays
Fe
Cou
nts
Channel number
Fig. 2. EDXRF spectrum of banded iron formation.
254 P.K. Nayak et al. / Nucl. Instr. and Meth. in Phys. Res. B 215 (2004) 252–261
[24–31], whereas a few compare [24–26,29–31] the
effectiveness of these two techniques. Kubala-
Kukus et al. [24] studied the tissue samples by
TRXRF and PIXE on complementary basis, and
they have found better low-level detection (LLD)
for low-Z elements (up to iron) using PIXE, and
better LLD for the high-Z elements using TRXRF
above iron. Subsequently, Braziewicz et al. [25]studied honey samples using the above two tech-
niques, where they have found TRXRF is having
advantages over PIXE in case of some lighter
elements (K, Ca) and observed the reverse trend
for heavier elements (Fe, Zn). Wills [26] gave a
comparative picture on use of PIXE and XRF,
and concluded that WDXRF is a better technique
for the determination of the elements with lowenergy lines, especially in the range of 1–4 keV (Na
through Ca). In order to compare the effectiveness
of PIXE and XRF methods, Haruyama et al. [27]
measured same archeological sample. They have
used 2.5 MeV proton beam for PIXE study and
Rh target (at 50 kV) for XRF measurements,
observed only L X-ray lines in PIXE spectrum,
whereas in EDXRF spectrum they observed both
K and L X-ray lines of major elements of Ag,
Sn and Sb, and suggested the use of XRF for
the measurement of high-Z elements. However,
Carlsson and Akselsson [28] gave better descrip-
tion on the use of EDXRF and PIXE techniqueson complementary basis. They have used 2.55MeV
proton beam for PIXE and Mo as secondary ex-
citer for analysis of a geological material, and
observed a large number of elements including Al,
Si for PIXE, whereas these are missing in case of
XRF spectrum. Also, Malmqvist [29] gave an ac-
count on complementary use of PIXE and XRF,
where he comments that if high-Z elements are ofparticular interest in any specific analytical situa-
tion, XRF with a high-Z secondary target or even
a radioactive source is superior to PIXE. Heitz
et al. [30] also provide the complementary use of
radioisotope induced X-ray emission method with

P.K. Nayak et al. / Nucl. Instr. and Meth. in Phys. Res. B 215 (2004) 252–261 255
PIXE on clays and ceramics. After irradiating
a thick clay pellet (3 MeV protons in PIXE and
59.6 keV radiation from 241Am for XRF), they
observed the high sensitivity of XRF for the hea-vier elements.
Benyaich et al. [31] gave fairly detailed com-
parative picture of the use of PIXE and EDXRF
as complementary basis. They have presented
PIXE and XRF spectra for the same sediment
sample, which was a typical silicate rock having
low trace elements content. On the PIXE spectrum
(2 MeV proton beam, 4 h acquisition time), themain peaks originate from Al, Si, K, Ca, Ti, Mn
and Fe while heavier elements such as Rb, Sr, Y
and Zr appear as trace elements with very low
peak to background ratios, and in the energy re-
gion of 10–14 keV, pile-up peaks are present
(similar to Fig. 1). In case of XRF spectrum (Ag
secondary target, 241Am source), it is the heavy
elements Rb, Y, Sr, Zr and Nb that have higherpeak to background ratios, compared to light
elements (Si, Ca, K) whose signals are almost
rubbed out (similar to Fig. 2). In the same report,
Benyaich et al. [31] further plotted the variations
of the intrinsic and effective sensitive ratios
ðSPIXE=SXRFÞ, versus the atomic number of the
emitting target element; computed for K lines for
Z6 42 and for L lines for Z > 42. They inferredthat PIXE is much more sensitive than XRF for
the low energy X-ray lines due to the higher ioni-
sation cross-section of protons, which balances the
strong self-absorption occurring at low energies;
while XRF is more sensitive at high X-ray energies
due to the combination of both high ionisation
cross-section and low self-attenuation. They con-
firm the complimentarily of the two techniques,that PIXE is more sensitive than XRF for low
energy X-ray lines, with MDL values falling down
to few atomic ppm; while at high X-ray energy,
XRF is the most sensitive with MDL values
ranging between 0.6 and 4 atomic ppm. Their
study advocated that in the adopted experimental
conditions (i.e. 2 MeV protons for PIXE, and
22–25 keV Ag K lines for XRF), the two tech-niques PIXE and XRF are complementary, the
former being very sensitive for light elements (low
energy X-ray emitters) and the later for heavier
elements.
Considering all these, we come to a reasonable
conclusion that at this particular situation PIXE
can give better low level detection at lower X-ray
energies for quantification of 3d elements (alongwith barium) whereas EDXRF (molybdenum as
secondary target) has privilege for higher X-ray
energies having better low level detection, and
both could be considered for quantification com-
plementarily. In this present investigation, an at-
tempt was made by the authors to quantify trace
elements in banded iron formations from eastern
Indian geological belt using PIXE and EDXRF,and the results were used in interpreting the envi-
ronmental condition of the depositional basin.
2. Experimental
2.1. Sample description and preparation
Banded iron formation (BIF) samples were
collected from Precambrian iron ore supergroup,
which is found in three major distinct geological
settings [32–34], known as BIF1, BIF2 and BIF3.
A total of 12 representative samples have been
selected for the present investigation. The geo-
graphical locations and local name of the deposits
(sampling stations) are provided in Table 1. Threesamples coded as IFA1, IFA2 and IFA3 are from
the mines of BIF1 region, four samples IFB1,
IFB2, IFB3 and IFB4 are from BIF2 region,
whereas five samples coded as IFC1, IFC2, IFC3,
IFC4 and IFC5 are selected from BIF 3 region.
As the original samples contain the alternative
bands of iron and silica, the required powdered
samples have been prepared only from the ironband. XRD and 57Fe M€ossbauer spectroscopic
studies of these samples reveal the presence of
hematite, magnetite and goethite as chief iron
bearing phases, also provided in Table 1 for in-
formation, and have been discussed elaborately
elsewhere [5,33,34]. For PIXE measurements, the
powdered samples were heated at 105 �C for 12 h
in an oven and allowed to cool slowly [15,22].After this, these samples are thoroughly mixed
with extra pure graphite powder (Merck) in the
ratio 1:1 by mass (200 mg each) [22]. Thick targets
are prepared by pressing the above mixture with a

Table 1
Sample location, their longitude and latitude and iron phases present therein
Region [32] Sample code Location Longitude Latitude Iron phases present
[5,33,34]
BIF 1 IFA1 Badampahar 22�040 86�050 Hematite, Goethite
IFA2 Gorumahisani 22�010 85�530 Hematite, Goethite
IFA3 Sulaipat 22�090 86�150 Hematite
BIF 2 IFB1 Daitary I 21�030 85�450 Hematite
IFB2 Daitary II 21�030 85�580 Hematite, Magnetite
IFB3 Tomaka I 21�120 85�490 Hematite, Goethite
IFB4 Tomaka II 21�120 85�530 Hematite
BIF 3 IFC1 Khondadhar 21�470 85�070 Hematite
IFC2 Malangtoli 21�480 85�190 Hematite
IFC3 Sakardihi 21�490 85�230 Hematite
IFC4 Mahaparbat 21�520 85�260 Hematite
IFC5 Palsa 21�550 85�250 Hematite
256 P.K. Nayak et al. / Nucl. Instr. and Meth. in Phys. Res. B 215 (2004) 252–261
KBr press. Pellets of NIST coal fly ash standard
1632b and USGS nodule standard Nod-A-1 are
also prepared by the same procedure for calibra-
ting the PIXE set up. For EDXRF experiments,
powdered samples are mixed with cellulose (as
binder) in 1:1 ratios by mass and thick targets have
been prepared [5,38].
2.2. Instrumental measurements
The 3 MeV collimated proton beam, obtained
from the 3MV Tandem pelletron accelerator at
Institute of Physics (Bhubaneswar, India) was used
to irradiate the sample in vacuum (10�6 Torr) in-
side a PIXE chamber [33]. The targets were held at
45� to the beam direction on a sample holder [35].Measurements were carried out with a maximum
beam current of 3 nA. A Si (Li) detector (Can-
berra, active area 30 mm2, beryllium window
thickness of 8 m) with a resolution of 170 eV at
5.9 keV placed at 90� to the beam direction was
used to detect characteristic X-rays emitted from
the targets. X-rays exit the PIXE chamber through
a 50 lm Mylar window before entering the de-tector [36]. Spectra were recorded by using a
Canberra series S-100 MCA calibrated with 241Am
X-ray source [37].
The EDXRF system used for the analysis con-
sists of a low power (50 W) tungsten anode; air-
cooled X-ray tube with tri-axial geometry and a
multi-channel analyser [5,38]. Molybdenum is used
as a secondary target and the characteristic K
X-rays were used to irradiate the pellets. X-rays
from the samples are detected by a Si(Li) detector
(the same detector as used for PIXE). The signals
from the detector were amplified, fed to PC based
MCA and were recorded [22].
2.3. Data analysis
The PIXE spectral analyses were performed
using GUPIX-2000 software [39]. This provides a
non-linear least squares fitting of the spectrum,
together with subsequent conversion of the fitted
X-ray peak intensities into elemental concentra-
tions, utilizing the fundamental parameter method
(FPM) for quantitative analysis. It requires pa-rameters of experimental geometry and Si(Li) de-
tector (H -parameter), chamber window thickness,
energy of the incident particle, the net charge
collected, the target thickness along with the major
matrix, and the invisible element oxygen [33,
36,39,40]. The concentrations of the low-Z ele-
ments were estimated from the intensities of K
X-ray lines while that of barium was from theintensities of L X-ray lines, and the results of dif-
ferent trace elements thus obtained for various
banded iron formations are provided in Table 2.
We would like to mention here that the Co Kb line
has been fitted to the spectra in order to determine
its concentration as suggested in GUPIX especially
for iron-rich materials [39]. Furthermore, it must

Table 2
PIXE and EDXRF data of banded iron-formation samples (concentrations are in ppm)
Elements IFA1 IFA2 IFA3 IFB1 IFB2 IFB3 IFB4 IFC1 IFC2 IFC3 IFC4 IFC5 Average
Caa 1555.8 ± 38.7 1456.8 ± 30.4 1854.9 ± 46.5 1102.8 ± 25.6 842.7 ± 18.5 1457.2 ± 33.6 705.4 ± 17.5 2607.4 ± 57.2 1305.4 ± 30.0 1277.5± 28.4 1375.6 ± 31.0 1345.4 ± 31.0 1407.2
Sca bdl 22.4± 1.2 15.3 ± 0.9 bdl bdl bdl bdl bdl 17.5 ± 1.0 bdl bdl bdl 4.6
Tia 165.5 ± 10.2 135.4 ± 8.2 266.7 ± 15.2 19.9 ± 1.5 56.4 ± 3.6 26.5 ± 1.6 47.1± 2.8 25.2 ± 1.5 40.0 ± 2.4 27.0± 1.6 31.7 ± 1.8 75.0± 4.4 76.4
Va 18.0 ± 1.8 130.3 ± 11.9 28.8 ± 3.1 23.0 ± 2.4 18.6 ± 2.0 12.3 ± 1.1 16.1± 1.5 42.2 ± 3.9 24.3 ± 2.2 24.7± 2.2 19.3 ± 1.8 32.2± 2.9 32.4
Cra 11.6 ± 0.9 17.4± 1.4 13.6 ± 1.0 39.6 ± 3.1 bdl 46.5 ± 4.6 16.4± 1.3 16.0 ± 1.3 33.1 ± 2.8 29.5± 2.2 37.8 ± 3.2 45.8± 4.1 25.6
Mna 98.5 ± 7.8 178.5 ± 15.9 181.7 ± 16.0 140.5 ± 13.7 434.4 ± 28.7 157.8 ± 13.5 70.5± 5.4 510.9 ± 37.2 280.5 ± 20.1 335.4 ± 23.5 303.7 ± 21.5 202.5 ± 14.2 241.2
Coa 19.3 ± 3.4 32.8± 5.6 12.8 ± 2.1 26.7 ± 4.2 24.6 ± 4.3 12.3 ± 2.4 11.8± 2.0 31.5 ± 4.8 18.5 ± 3.0 21.3± 3.5 16.4 ± 2.8 12.6± 2.2 20.0
Nia 36.4 ± 4.6 66.4± 8.4 44.7 ± 7.5 42.4 ± 5.6 65.2 ± 8.0 27.4 ± 3.8 19.5± 2.6 69.4 ± 8.6 62.4 ± 8.1 62.7± 7.9 67.5 ± 7.9 45.8± 6.0 50.8
Cua 99.1 ± 5.9 128.8 ± 7.7 113.7 ± 7.1 80.7 ± 4.1 165.6 ± 9.1 106.6 ± 6.0 60.5± 3.9 215.6 ± 13.0 123.4 ± 6.9 81.4± 4.8 161.5 ± 9.1 82.9± 5.2 118.3
Zna 47.4 ± 2.9 62.6± 3.6 52.6 ± 3.1 19.5 ± 1.3 65.2 ± 3.9 29.5 ± 1.9 28.7± 1.7 68.3 ± 4.3 27.4 ± 1.8 25.8± 1.7 31.3 ± 2.1 57.5± 3.7 43.0
Gaa 5.4 ± 0.2 14.1± 0.6 7.8 ± 0.3 6.4 ± 0.3 4.4 ± 0.2 8.4 ± 0.4 4.5 ± 0.2 6.0 ± 0.3 7.6 ± 0.3 3.8 ± 0.2 5.3 ± 0.2 11.7± 0.5 7.1
Asb 12.7 ± 0.6 bdl 16.7 ± 0.7 15.5 ± 0.6 48.3 ± 1.9 17.4 ± 0.7 16.6± 0.7 92.4 ± 3.1 21.0 ± 0.9 13.2± 0.5 17.3 ± 0.7 22.4± 1.0 24.4
Seb 7.8 ± 0.6 2.2 ± 0.2 14.2 ± 1.2 7.8 ± 0.6 5.6 ± 0.5 15.6 ± 1.3 5.4 ± 0.4 5.3 ± 0.4 14.3 ± 1.0 5.5 ± 0.5 9.4 ± 0.8 16.3± 1.1 9.1
Rbb 6.8 ± 0.4 4.2 ± 0.3 5.7 ± 0.3 11.2 ± 0.7 15.3 ± 1.2 74.5 ± 4.0 26.2± 1.3 9.3 ± 0.3 28.2 ± 1.4 20.8± 1.1 24.5 ± 1.3 14.2± 0.5 126.8
Srb 86.9 ± 6.4 64.2± 4.3 75.1 ± 5.1 66.3 ± 4.4 60.7 ± 4.2 64.6 ± 4.3 39.4± 2.4 35.6 ± 1.9 79.3 ± 5.3 47.7± 2.7 55.3 ± 3.8 22.6± 1.2 58.1
Yb 9.6 ± 0.6 12.6± 0.7 9.8 ± 0.6 7.3 ± 0.5 15.3 ± 0.8 14.2 ± 0.7 18.3± 0.9 12.6 ± 0.6 10.7 ± 0.5 16.2± 0.8 13.0 ± 0.7 17.6± 0.8 13.1
Zrb 21.2 ± 0.6 45.8± 1.2 27.5 ± 0.8 25.0 ± 0.7 41.9 ± 1.7 41.0 ± 1.7 52.6± 2.4 36.3 ± 1.6 32.8 ± 1.5 49.4± 2.3 41.8 ± 2.0 47.4± 2.2 38.5
Baa 140.0 ± 7.2 115.7 ± 5.5 152.1 ± 7.4 107.8 ± 5.1 98.8 ± 4.9 124.4 ± 6.1 76.1± 3.8 268.7 ± 12.5 142.4 ± 6.9 94.0± 4.8 98.3 ± 4.7 102.9 ± 5.1 20.7
Pbb 23.3 ± 1.2 16.7± 0.9 13.3 ± 6.8 19.7 ± 1.0 28.7 ± 1.5 bdl 30.7± 1.6 19.3 ± 1.2 14.3 ± 0.9 19.0± 1.2 23.6 ± 1.4 10.6± 0.6 18.3
bdl¼ below detection limit.aQuantification by PIXE.bQuantification by EDXRF.
P.K.Nayaket
al./Nucl.
Instr.
andMeth
.in
Phys.Res.
B215(2004)252–261
257

258 P.K. Nayak et al. / Nucl. Instr. and Meth. in Phys. Res. B 215 (2004) 252–261
be noted here that in case of some other elements,
the K X-ray component of one element ðZÞ over-laps with the Ka component of higher element (i.e.
Z þ 1), which were taken care by GUPIX itself,where there is a provision to correct for this
overlap, and thus giving the correct intensities of
the elements [39].
The EDXRF spectra were analysed using the
software AXIL [41]. The instrument was cali-
brated and checked using various certified refer-
ence materials before the analyses of these BIFs,
and was also verified with earlier published data[5].
3. Results and discussion
The measured elemental concentrations are
provided in Table 2. It is worth mentioning here
that the concentrations of seven elements such asAs, Se, Rb, Sr, Y, Zr and Pb were estimated from
EDXRF spectra (triplicate measurements), while
the rest of the elements have been quantified from
PIXE spectra (duplicate measurements).
The concentration of titanium has been con-
sidered by various investigators in order to differ-
entiate iron ores formed at high temperature
magmatic process from low temperature sedi-
Table 3
Comparative average trace elemental concentration of present study w
beltsa
Elements Present study
[32,33]
Olary Domain,
South Australia [8]
Kushtag
belt, Ind
Ti 76.4 719.2 N.R.
V 32.4 115 N.R.
Cr 25.6 24 109.9
Mn 241.2 309.8 N.R.
Co 20.0 35 1.6
Ni 50.8 14 25.3
Cu 118.3 42 N.R.
Zn 43.0 17 30.8
Rb 20.7 21 0.8
Sr 58.1 161 13.2
Y 13.1 10 7.1
Zr 38.5 24 10.1
Ba 126.8 6714 N.R.
Co/Ni ratio 0.394 2.5 0.06
N.R.¼ not reported.a Concentrations are in ppm.
mentary process. It may be noted here that the
Clarke value of lithospheric titanium for iron ores
is 1000 ppm [22]. In these iron-formation samples
the concentration of Ti is well below that of theClarke value suggesting the source of the materials
to be other than of igneous origin, and this favours
the theory of sedimentary deposition. Vanadium,
an element of industrial interest, is usually rich in
titanium-bearing minerals, and it is evidenced
from our earlier studies [22] that this is also an
important geochemical indicator. The abundance
of vanadium in these ores appears to be high, ingeneral, as compared to ores of Lake Superior type
of USA [42], but is rather low in comparison with
Algoma type of Canada [42] and South Australia
[8] (Table 3).
Frietsch [43] reported that Cr is present below
the lithospheric content in iron ores of N-Sweden,
which are non-magmatic in origin. Very low av-
erage chromium (25.6 ppm) content of the pres-ently studied BIFs, which are well below the
Clarke value of sediments (110 ppm) [44], suggests
the initial formation condition to be of sedimen-
tary environment. Hagemann and Albrecht [45]
reported fairly low concentrations of manganese,
generally up to 150 ppm in hematite from typical
sedimentary ores. In contrast, the Mn content in
magnetites from similar deposits is generally of the
ith that of different world deposits along with two major Indian
i schist
ia [50]
Sandur schist
belt, India [4]
Algoma facies,
Canada [42]
Lake-Superior
facies, USA [42]
300 860 160
11.3 97 30
55.7 78 122
N.R. 1400 4600
33.7 38 27
10.2 83 32
12.5 96 10
11.2 N.R. N.R.
2.7 N.R. N.R.
3.8 42 98
1.8 N.R. N.R.
4.0 56 84
14.5 170 180
3.3 0.457 0.843

50 100 150 20050
100
150
200
250
300
Con
entra
tion
of b
ariu
m (p
pm)
Concentration of copper (ppm)
Fig. 3. Scatter plot between Cu and Ba.
P.K. Nayak et al. / Nucl. Instr. and Meth. in Phys. Res. B 215 (2004) 252–261 259
order of Clarke value for the upper lithosphere, i.e.
about 1000 ppm [43]. In this light, the observed
low levels of Mn in these iron formations appar-
ently offer strong support to the contention thatthe bulk of the iron was precipitated by sedimen-
tary process.
According to R€ossler and Lange [46], the av-
erage values of nickel in igneous rocks are 80–200
ppm. In these samples, nickel concentration is well
below (average 50.8 ppm, Table 2) the R€ossler andLange�s value for rocks of igneous origin. How-
ever, in some specific cases (samples IFC1, IFC4,IFA2, IFB2 and IFC4) the value is quite close to
the Clarke value (80 ppm), which is probably due
to the comparable radii of Ni2þ and Fe2þ, and this
may result in increase in nickel concentration.
Generally, the element strontium follows calcium
and barium in natural conditions [15] as it belongs
to the same group in the Moseley�s Periodic Table.The low content of strontium in these BIFs (bar-ium and calcium poor ores) seems to be geo-
chemically appropriate. Furthermore, the low
concentration of calcium (average 1407.2 ppm)
supports the non-volcanogenic origin of these
BIFs [47].
Another important geochemical parameter zir-
conium has been used as an effective indicator of
provenance, scarcely prone to weathering, and isbelieved not to be affected by transportation dur-
ing weathering or by any other specific later geo-
chemical events. It is immobile, highly stable and
rarely decomposes in nature [22]. So the concen-
tration of zirconium in ores represents the con-
stituent of source rock. In these samples the
content of zirconium is rather low (of an average
of 38.5 ppm) in comparison to other specific worlddeposits [8,42], leading to the situation that the
source solution had concentration of lower zirco-
nium-containing materials. Furthermore, barium
and zirconium are adsorbed in clay structure due to
their larger ionic radii and lower ionic potentials
[48]. The lower concentrations of these two ele-
ments (of an average of 126.8 and 38.5 ppm, re-
spectively) indicate the source material wasdeficient in clay minerals.
The Co/Ni ratio has been used by various
workers to differentiate between igneous and sed-
imentary origin of iron ores [43,48]. According to
Frietsch [43], Co/Ni ratio below unity for iron
oxides is indicative of low temperature and sedi-
mentary origin but higher values are obtained in
the iron oxides of igneous origin. In the presentwork this ratio is well below (average value 0.394,
Table 2) to that of Frietsch�s ratio confirming the
clear sedimentary environment of deposition.
However, from the concentrations of different
trace elements (Table 2), it is observed that there
are positive correlations and some similar asso-
ciative behaviour between two groups of elements,
i.e. Cu–Ba (Fig. 3) and Mn–Ni (Fig. 4) althoughwe do not know the exact cause and mechanism. It
may be possible that there is simultaneous pre-
cipitation between these two groups of elements
with that of the source solution. We would further
like to mention here that the presence and high
content of yttrium (average of 13.1 ppm) in these
samples indicates towards the possibility of the
existence of lanthanides in these BIFs [5], andfurther supported by a report by Majumdar et al.
[49].
The average trace elements� concentration in
Table 2 while compared with other world [8,42]
and Indian deposits [4,50] (Table 3), indicates that
these iron formations are neither comparable with
other world deposits nor with other Indian de-
posits, and were deposited at specific environmentcondition. The original source material is having
lower trace elements� content. Hence, on the basis

0 100 200 300 400 500
20
30
40
50
60
70
Con
cent
ratio
n of
Ni (
ppm
)
Concentartion of Mn (ppm)
Fig. 4. Scatter plot between Mn and Ni.
260 P.K. Nayak et al. / Nucl. Instr. and Meth. in Phys. Res. B 215 (2004) 252–261
of PIXE and EDXRF study, one can conclude
that the Eastern Indian iron ore basin signifies
specific and independent deposition, and is notcomparable to any other iron formations either of
Canada, USA and Australia (Table 3). In supple-
mentary, we would like to mention here that the
variation of three large-ion lithophile elements (K,
U and Th) in these BIFs have been reported else-
where [51]. Further investigation is under progress
on the variation and distribution of rare earth el-
ements and isotopic ratios, which may explain thedepositional environment of the present geological
belt more comprehensively.
4. Conclusion
Selected numbers of banded iron formations
have been studied by using PIXE and EDXRF. Atotal of nineteen elements including Ca, Ti, V, Cr,
Mn, Ni, Cu, Zn, Sr, Y, Zr, Ba and Pb were esti-
mated in these ores. The Co/Ni ratio and the con-
tent of trace elements such as Ti, Cr, Ni, Zr have
been used in explaining the environment of the
depositional basin of these banded iron forma-
tions. The large variations in trace elements have
been observed though the positive correlationsbetween two groups of elements, i.e. Cu–Ba and
Mn–Ni have also been observed. The comparison
of the average trace element concentration with
that of other Indian and world banded iron for-
mation deposits indicates the eastern Indian iron
ore basin�s specific and independent deposition.
Acknowledgements
Authors wish to express their posthumous
gratitude to Prof. V. Chakravortty (Utkal Uni-
versity, Vani Vihar, Bhubaneswar). Heartfelt
thanks are due to Prof. S. Jena, Department of
Chemistry, Utkal University, Vani Vihar (Bhu-
baneswar), Prof. R.K. Choudhury and Prof. D.P.Mahapatra of Institute of Physics (Bhubaneswar)
and Prof. S. Acharya, Ex-Vice-Chancellor, Utkal
University, Vani Vihar (Bhubaneswar) for useful
discussions regarding the various aspects of the
present investigation. P.K.N. thanks to Prof. S.N.
Bhattacharyya, Dr. A.K. Sinha and Dr. A. Saha of
IUC-DAEF, Calcutta Center, Kolkata for their
constant inspiration. The scientific staffs andmembers of Ion Beam Lab, Institute of Physics are
thanked for providing accelerator facilities and
various necessary supports. P.K.N. is grateful to
IUC-DAEF, Calcutta Center (Kolkata) for a re-
search Fellowship. Authors acknowledge and are
grateful to an anonymous referee for the signifi-
cant improvement of the manuscript at the revi-
sion stage.
References
[1] S.J. Mojsis, G. Arrhenius, K.D. KcKeegan, T.M. Harri-
son, A.P. Nutman, C.R.L. Friend, Nature 384 (1996) 55.
[2] J.F. Kasting, Science 259 (1993) 920.
[3] I.W. Croudace, J.M. Gilligan, X-ray Spectrom. 19 (1990)
117.
[4] C. Manikyamba, V. Balaram, S.M. Naqvi, Precamb. Res.
61 (1993) 137.
[5] P.K. Nayak, D. Das, V. Vijayan, P. Singh, V. Chakra-
vortty, Nucl. Instr. and Meth. B 184 (2001) 649.
[6] M.M. Kimberley, Ore Geol. Rev. 5 (1989) 13.
[7] E. Dimroth, Geol. Soc. Ind. 28 (1986) 239.
[8] P.M. Ashley, B.G. Lottermoser, J.M. Westaway, Contr.
Miner. Petr. 64 (1998) 187.
[9] S. Murao, S.H. Sie, Resour. Geol. 47 (1) (1997) 21.
[10] J.S. Becker, Spectrochim. Acta B 57 (2002) 1805.
[11] J.D. Robertson, H. Neff, B. Higgins, Nucl. Instr. and
Meth. B 189 (2002) 378.
[12] L. Kempenaers, N.H. Bings, T.E. Jeffries, B. Vakemans, K.
Janssens, J. Anal. At. Spectrom. 16 (2001) 1006.

P.K. Nayak et al. / Nucl. Instr. and Meth. in Phys. Res. B 215 (2004) 252–261 261
[13] I. Brissaud, A. de Chateau-Thierry, J.P. Frontier, G.
Lagarde, J. Radioanal. Nucl. Chem. 102 (1) (1986) 131.
[14] C.G. Ryan, D.R. Cousens, S.H. Sie, W.L. Griffin, G.F.
Suter, E. Clayton, Nucl. Instr. and Meth. B 87 (1990) 55.
[15] J.E. Martin, R. Garcia-Tenorio, M.A. Respaldiza, M.A.
Ontalba, J.E. Boliver, M.F. Da Silva, Appl. Radiat. Isot.
50 (1999) 445.
[16] J.L. Campbell, in: S.A.E. Johansson, K.G. Malmqvist
(Eds.), Particle Induced X-ray Emission Spectrometry:
Chemical Analysis Series, Chapter 6, Vol. 133, John Wiley
and Sons Inc., New York, 1995, p. 319.
[17] A.P. Santo, A. Paccerillo, P. Del Carmine, F. Lucarelli,
J.D. Macarthur, P.A. Mando, Nucl. Instr. and Meth. B 64
(1992) 517.
[18] A.C. Mandal, M. Sarkar, D. Bhattacharya, Eur. Phys. J.
AP 17 (2002) 81.
[19] J. Parus, W. Raab, D.L. Donohue, X-ray Spectrom. 30
(2001) 296.
[20] A.G. Revenko, X-ray Spectrom. 31 (2002) 264.
[21] F. Arauzo, T. Pinheiro, L.C. Alves, P. Valerio, F. Gaspar,
J. Alves, Nucl. Instr. and Meth. B 136–137 (1998) 1005.
[22] P.K. Nayak, D. Das, S.N. Chintalapudi, P. Singh, S.
Acharya, V. Vijayan, V. Chakravortty, J. Radioanal. Nucl.
Chem. 254 (2) (2002) 351.
[23] V. Vijayan, S.N. Behera, V.S. Ramamurthy, S. Puri, J.S.
Shahi, N. Singh, X-ray Spectrom. 26 (1997) 65.
[24] A. Kubala-Kukus, J. Braziewicz, D. Banas, U. Majewska,
S. Gozdz, A. Urbaniak, Nucl. Instr. and Meth. B 150
(1999) 193.
[25] J. Braziewicz, I. Fijal, C. Czyzewski, M. Jaskola, A.
Korman, D. Banas, A. Kubala-Kukus, U. Majewska, L.
Zemlo, Nucl. Instr. and Meth. B 187 (2002) 231.
[26] P. Wills, Nucl. Instr. and Meth. B 35 (1988) 378.
[27] Y. Haruyama, M. Saito, T. Muneda, M. Mitan, R.
Yamamoto, K. Yoshida, Int. J. PIXE 9 (3–4) (1999) 181.
[28] L.-E. Carlsson, K.R. Akselsson, Adv. X-ray Anal. 24
(1981) 313.
[29] K.G. Malmqvist, Nucl. Instr. and Meth. B 14 (1986) 86.
[30] C. Heitz, G. Lagarde, A. Pape, T. Tenorio, C. Zarate, M.
Menu, L. Scotee, A. Jaidar, R. Acosta, R. Alviso, D.
Gonzalez, V. Gonzalez, Nucl. Instr. and Meth. B 14 (1986)
93.
[31] F. Benyaich, A. Makhtari, L. Torrisi, G. Foti, Nucl. Instr.
and Meth. B 132 (1997) 481.
[32] S. Acharya, Ind. J. Earth Sci., SEISM seminar volume,
1984, p. 19.
[33] P.K. Nayak, Characterization of Iron Ores of Orissa by
PIXE, EDXRF and M€ossbauer spectroscopy, Ph.D. The-
sis, Utkal University, Bhubaneswar, India, 2003.
[34] P.K. Nayak, D. Das, P. Singh, V. Chakravortty, Commu-
nicated to J. Radioanal. Nucl. Chem., 2004, to be
published.
[35] R.K. Dutta, M. Sudarshan, S.N. Bhattacharyya, V.
Vijayan, S. Ghosh, V. Chakravortty, S.N. Chintalapudi,
J. Radioanal. Nucl. Chem. 221 (1997) 193.
[36] V. Vijayan, P.K. Nayak, V. Chakravortty, Ind. J. Phys. A
76 (2002) 477.
[37] P.K. Nayak, V. Chakravortty, V. Vijayan, P. Singh, Int. J.
PIXE, accepted.
[38] M. Ashok, T.R. Rautray, P.K. Nayak, V. Vijayan, V.
Jayanthi, S. Narayana Kalkura, J. Radioanal. Nucl. Chem.
257 (2) (2003) 333.
[39] J.L. Campbell, T.L. Hopman, J.A. Maxwell, Z. Nezedely,
Nucl. Instr. and Meth. B 170 (2000) 193.
[40] M. Sudarshan, R.K. Dutta, V. Vijayan, S.N. Chintalapudi,
Nucl. Instr. and Meth. B 168 (2000) 553.
[41] R.E. Van Griken, A.A. Markowictz, Handbook of X-ray
Spectrometry, Marcel Dekker, New York, 1993.
[42] G.A. Gross, C.R. Mcleod, Canadian Miner. 181 (1980)
223.
[43] R. Frietsch, Sveriges Geol. Unders Arsbok 64 (1970) 1.
[44] S. Landergren, Sveriges geol Unders Ser C 42 (5) (1948) 1.
[45] F. Hagemann, F. Albrecht, Chemie de Erde 17 (1954) 81.
[46] H.J. R€ossler, H. Lange, Geochemical Tables, Elsevier
publication, Amsterdam, 1972.
[47] T. Majumdar, Bull. Geol. Survey Finland 331 (1985) 201.
[48] T. Majumdar, K.L. Chakraborty, A. Bhattacharya, Miner.
Deposita 17 (1982) 107.
[49] T. Majumdar, J.E. Whitley, K.L. Chakraborty, Chem.
Geol. 45 (1984) 203.
[50] R.M.K. Khan, S. Das Sharma, D.J. Patil, S.M. Naqvi,
Geochim. Cosmochim. Acta 60 (1996) 3285.
[51] P.K. Nayak, V. Vijayan, V. Chakravortty, D. Bhatta, P.
Singh, Ind. J. Phys. A 77 (2003) 503.




















