Regulation of NAMPT in Human Gingival Fibroblasts and Biopsies
Upload
independentCategory
view
1download
0
Molecular Genetics and Metabolism 112 (2014) 294–301
Contents lists available at ScienceDirect
Molecular Genetics and Metabolism
j ourna l homepage: www.e lsev ie r .com/ locate /ymgme
Pharmacological chaperones increase residual β-galactocerebrosidaseactivity in fibroblasts from Krabbe patients
Anna Sara Berardi a,1, Giovanna Pannuzzo b,1, Adriana Graziano b, Elvira Costantino-Ceccarini a,Paola Piomboni a, Alice Luddi a,⁎a Department of Molecular and Developmental Medicine, University of Siena, Siena, Italyb Department of Bio-Medical Sciences, Section of Physiology, University of Catania, Catania, Italy
Abbreviations: GALC, β-galactocerebrosidase; GLD, glenzyme replacement therapy; CNS, central nervous systemtem; LSDs, lysosomal storage diseases.⁎ Corresponding author at: Department of Molecular
Policlinico Le Scotte, viale Bracci, 53100 Siena, Italy.E-mail address: [email protected] (A. Luddi).
1 These authors contributed equally to this work.
http://dx.doi.org/10.1016/j.ymgme.2014.05.0091096-7192/© 2014 Elsevier Inc. All rights reserved.
a b s t r a c t
a r t i c l e i n f oArticle history:Received 25 February 2014Received in revised form 15 May 2014Accepted 15 May 2014Available online 23 May 2014
Keywords:Krabbe diseasePharmacological chaperonesMissense mutationsβ-GalactocerebrosidaseLysosomal storage disease
Krabbe disease or globoid cell leukodystrophy is a degenerative, lysosomal storage disease resulting from the de-ficiency of β-galactocerebrosidase activity. This enzyme catalyzes the lysosomal hydrolysis of galactocerebrosideand psychosine. Krabbe disease is inherited as an autosomal recessive trait, and many of the 70 disease-causingmutations identified in theGALC gene are associatedwith proteinmisfolding. Recent studies have shown that en-zyme inhibitors can sometimes translocate misfolded polypeptides to their appropriate target organellebypassing the normal cellular quality controlmachinery and resulting in enhanced activity. In search for pharma-cological chaperones that could rescue the β-galactocerebrosidase activity, we investigated the effect of α-Lobeline or 3′,4′,7-trihydroxyisoflavone on several patient-derived fibroblast cell lines carrying missense muta-tions, rather than on transduced cell lines. Incubation of these cell lines with α-lobeline or 3′,4′,7-trihydroxyisoflavone leads to an increase of β-galacocerebrosidase activity in p.G553R + p.G553R, in p.E130K +p.N295T and in p.G57S + p.G57S mutant forms over the critical threshold. The low but sustained expression ofβ-galactocerebrosidase induced by these compounds is a promising result; in fact, it is known that residual enzymeactivity of only 15–20% is sufficient for clinical efficacy. The molecular interaction of the two chaperones with β-galactocerebrosidase is also supported by in silico analysis. Collectively, our combined in silico–in vitro approach in-dicateα-lobeline and 3′,4′,7-trihydroxyisoflavone as two potential pharmacological chaperones for the treatmentor improvement of quality of life in selected Krabbe disease patients.
© 2014 Elsevier Inc. All rights reserved.
1. Introduction
Globoid cell leukodystrophy (GLD), also known as Krabbe disease, isa monogenic lysosomal storage disorder (LSD) inherited as an autoso-mal recessive trait [1,2]. GLD is characterized by a deficiency ingalactocerebrosidase (GALC), a lysosomal enzyme essential for normalcatabolism of galactolipids, including a major myelin component,galactocerebroside and psychosine [1]. The characteristic biochemicalfeature of Krabbe disease is the lack of accumulation of the undegradedgalactocerebroside in brain, explained by the early degeneration of themyelin forming cells and the block in the synthesis of galactocerebroside[3,4]. However, GALC deficiency results in abnormal accumulation ofpsychosine, a toxic metabolite which has been demonstrated to induceapoptotic death in oligodendrocytes and Schwann cells throughout
oboid cell leukodistrophy; ERT,; PNS, peripheral nervous sys-
and Developmental Medicine,
respectively the central nervous system (CNS) and peripheral nervoussystem (PNS) [5–7]. Loss of thesemyelin-forming cells causes demyelin-ation in both the CNS and PNS during early developmental stages [8,9].
The biochemical disturbances in GLD aremanifested by different de-grees of brain demyelination, resulting in a broadneurological spectrumof the disease. The rapidly progressive early infantile form is the mostcommon phenotype, which manifests within the first 6 months of lifeand is characterized by severe neuro-developmental delay, progressivemotor dysfunction, and early death [10]. Late onset forms of GLD repre-sent approximately 10% of cases and include the juvenile and adult on-sets that predominantly present with motor weakness, sensory andmotor neuropathy, cognitive impairment, and psychiatric/behavior dis-turbances [5,10].
More than 140 mutations have been identified in patients with allclinical types of GLDs, many of which occur in compound heterozygotepatterns in patients [2,11–16]. As with many other lysosomal storagediseases, late-onset Krabbe patients with similar or identical genotypescan have varied clinical presentations and course of their disease [13,17]. In fact, while some mutations clearly result in the infantile form ifhomozygous or heterozygous with another severe mutation, for mostof the mutations it is difficult to establish a genotype–phenotype

295A.S. Berardi et al. / Molecular Genetics and Metabolism 112 (2014) 294–301
correlation. A more detailed understanding of individual GALC muta-tions must be established to develop a tailored therapy for Krabbe dis-ease, an approach never taken into consideration for this pathology.
The only available therapy for Krabbe disease is hematopoieticcell transplant using bone marrow or umbilical cord blood cells fromhealthy donors. This treatment, if done in pre-symptomatic patients,can prevent the rapid neurological course of the disease and the long-term outcome of transplanted infants [18].
Direct intracerebral injection of vectors or transplantation ofenzyme-producing cells in the mouse model of the disease has beendemonstrated to be effective in enzyme deficiency correction by pro-moting donor-to-recipient cross correction via enzyme secretion-recapture [19–21]. However, in humans, safety concerns may representa limit for this approach.
The goal of our study was to assess the therapeutic potential of apharmacological chaperone therapy approach, a novel and emergingtherapeutic strategy using small molecules as drugs for the treatmentof Krabbe disease. This therapeutic strategy has the potential to treatdiseases caused by missense mutations that result in the synthesis ofimproperly folded lysosomal enzymes. Indeed, pharmacological chap-erones are low-molecular-weight molecules designed to selectivelywork as a folding template for the conformational mutant enzymes,thereby facilitating proper folding and increasing rescuing of misfoldedmutant proteins from the endoplasmic reticulum-associated degrada-tion. Due to their small size, pharmacological chaperones have the po-tential to be orally available with broad biodistribution, including theCNS.
The use of small pharmaceutical chaperones for the treatment ofKrabbe disease is based not only on the safety problems and unsatisfac-tory results obtained with gene therapy, but it is also suggested byin vitro studies done on primary oligodendrocytes demonstrating thatover expression of the GALC enzyme is detrimental for oligodendro-cytes [19]. It is known that GALC, as other lysosomal enzymes, is consti-tutively expressed at low levels in all tissues. Our attempt to restore lowbut sustained expression of the enzyme activity is a promising ap-proach, since it is known that in other lysosomal storage diseases a re-sidual enzyme activity of only 15–20% is sufficient for clinical efficacy[22,23].
In the present study, we screened several pharmacological chaper-ones and tested themon severalfibroblast cell lines from infantile, juve-nile and adult Krabbe patients bearing diverse missense mutations. Wehave identified two potential candidates:α-lobeline already known as aweak inhibitor of GALC [24] and 3′,4′,7-trihydroxyisoflavone a novelchaperone, never previously tested on GALC. The approach used foridentification and characterization of the two chaperones was basedon biochemical studies in cellular systems and on in silico analysis onthe interaction between the compounds and the protein. Our studiesclearly show that both compounds selectively increase the residual ac-tivity in diverse forms of the defective protein. We hope that this ap-proach will be further explored for the development of noveltherapeutic treatment of Krabbe patients with responsive mutations,particularly those with severe brain damage.
2. Experimental procedures
2.1. Materials
Fibroblasts from patients with Krabbe disease (p.E130K + p.N295T,p.D187V+ p.G323R, p.G286D+ p.P318R)were kindly provided by thegenetic bio-bank of the Gaslini Institute (Genova, Italy). The fibroblastcell lines carrying the p.G553R + p.G553R and the p.G57S + p.G57Smutation were gift of Prof Balestri (University of Siena) and ProfFederico (University of Siena) respectively. The above GALC mutationsare numbered according to HGVS nomenclature recommendations,numbering from the first methionine of the complete 42-residue signalsequence. Since the traditional nomenclature uses p.M17 as the first
residue, to switching from new to original nomenclature is required tosubtract 16 from their numbers.
The α-lobeline and 3′,4′,7-trihydroxyisoflavone were purchasedfrom Santa Cruz Biotechnology. Stock solutions were prepared inDMSO (Sigma Aldrich) at a concentration of 10 mM, filtered sterileand stored at 20 °C. Fluorogenic substrates were purchased fromMoscerdam Substrates or from Sigma Aldrich.
2.2. GALC enzymatic activity assay
Enzyme activity of cerebroside-ß-galactosidase was measured usingthe fluorogenic substrate 6-hexadecanoylamino-4-methylumbelliferyl-ß-D-galactoside (HMU-ßGal) as originally described [25]. Preliminarystudies comparing natural and HMU-ßGal substrate were carried outin order to test the reliability of the water-soluble substrate for thisstudy. No significant differences in the results were obtained; we havetherefore decided to use the water-soluble substrate.
Duplicates from each sample, containing 10 μg of total protein fromnormal control fibroblasts and 20 μl of substrate solution, were mixedon a 200 μl PCR tube and incubated at 37 °C for 17 h. After incubation,200 μl of stop solution (0.5 M NaHCO3/0.5MNa2CO3 buffer pH 10.7 +0.25% TritonX-100) was added and mixed. The absorbance of thesupernatant was measured (excitation at 404 nm and emission at460 nm) with an F-4500 fluorescence spectrometer (Hitachi, Tokyo,Japan).
2.3. In vitro inhibition assay on GALC
The describedGALC enzymatic activity assaywas used as the screen-ing method to identify GALC inhibitors. Tested drugs or vehicle DMSO(4% v/v) were added to the mixture solution containing 10 μg of totalprotein from normal control fibroblasts and 20 μl of substrate solution.The final concentration of the two compounds in the sample was0.4 mM and each sample was run in triplicate.
2.4. In vitro inhibition assay for other lysosomal enzymes
Measurements of β-glucosidase, β-galactosidase,α-glucosidase andα-galactosidase enzymatic activity were performed using cultured celllysates from primary fibroblasts as previously described [26–29].
2.5. Compound toxicity in cell culture
Normal control fibroblast cells were grown in Dulbecco's modifiedEagle medium media (Sigma Aldrich) containing 4.5 g/l glucose, 10%fetal calf serum, stable glutamine and 1% antibiotics and maintained at37 °C in a humidified CO2 incubator. Cells seeded at 2 × 103 cells/wellin a 96-well plate were treated with of α-lobeline or 3′,4′,7-trihydroxyisoflavone at increasing concentrations (10 to 600 μM) for72 h. To determine the cell toxicity of the two compounds cell viabilitywas assessed using XTT assay kit (Sigma Aldrich). This assay is based onthe reduction of tetrazolium salts (XTT) to formazan by the succinate-tetrazolium reductase system in the mitochondria of metabolically ac-tive cells. The assay kit was used according to the manufacturer's in-structions. The cells were incubated with the tetrazolium salts for 4 hand absorbancewasmeasured at 450-nm using a 550 Ultramarkmicro-titer plate reader (Biorad, Milan, Italy). The experiments were run intriplicates and the results refer to themean of the three measurements.
2.6. Cell-based chaperone assay for restoring GALC activity
Fibroblast cell lines frompatientswere grown inDulbecco'smodifiedEagle's medium (Sigma Aldrich) and 10% fetal bovine serum (Lonza,Basel, Switzerland) supplemented with 1% penicillin/streptomycin(Lonz,a Basel, Switzerland). Cell lines were cultured at 37 °C in a humid-ified incubator with 5% CO2. Prior to treatment, cells were cultured for at

296 A.S. Berardi et al. / Molecular Genetics and Metabolism 112 (2014) 294–301
least 12 h to reach exponential growth phase. α-Lobeline and 3′,4′,7-trihydroxyisoflavone were dissolved in DMSO and added to the culturemedium at various concentrations; control cells were treated with thesame amount of DMSO for the same time periods and treated in quadru-plicates. After treatment, cells were washed, and then harvested by useof trypsin (SigmaAldrich) and the cell lysatewas assayed forGALC activ-ity as described earlier.
2.7. Structure determination
In the present study the BLAST program (Basic Local AlignmentSearch Tool) [30] was used to model human GALC. Mouse GALC (PDBaccession code 3zr5A) (residues 40–684) matched to the sequenceand predicted fold of the human GALC with the highest percentage ofsequence identity (83%) [31].
The structure of galactose was downloaded from the complex withmouse GALC from online Protein Data Bank (3zr6A), the protein wassubsequently removed and the structure was subjected to energy min-imization. The structures of α-lobeline and trihydroxyflavone werebuilt as pdb files by means of the server PRODRG2 (http://davapc1.bioch.dundee.ac.uk/prodrg/) [32]. The energies of the protein and themolecules were minimized by the steepest descent algorithm. Simula-tions were performed by using the GROMACS simulation package(4.5.5) [33] and GROMOS 53A6 [34] force field was used to describe at-omistically receptor and ligands respectively.
2.8. Structure analysis
All docking calculations were carried out using software AutoDockVina [35]. For GALC preparation, polar hydrogens were added, andthen Kollman united atom charges and atomic solvation parameterswere assigned. For ligand preparation, Gasteiger partial charges wereadded, non-polar hydrogen atoms were merged and rotatable bondswere defined. The grid points and spacing were computed using theAutogrid program [36]. The grid must surround the region of interestin the macromolecule. The spacing between grid points was 1 Å forblind docking and 0.37 Å for the active site, respectively. A preliminaryblind docking study was performed in order to discriminate the prefer-ential binding sites of the ligand to the receptor. To this aim, for the firstsimulation, the grid size was properly set up in order to contain the en-tire receptor structure up (70, 70, and 78 Å along x, y and z, respectively,space between grid points of 1 Å). Visual Molecular Dynamics (VMD)software was used to visualize assay and post-docking analysis.
2.9. Statistical analysis
All data are presented asmean values ± SE of at least triplicate sam-ples from representative experiments. Each experiment was indepen-dently repeated at least three times.
3. Results
3.1. Selection of pharmacological chaperones
Many efforts are being directed towards exploring the efficacy ofpharmacological chaperones as therapeutic compounds in lysosomaldiseases. In this study we report the results of a pilot enzymatic inhibi-tion assay in which we screened several small molecules that could bepotentially useful for the treatment of Krabbe disease. Among severalcompounds, various iminosugar analogues were initially screened;however, significant inhibitory activity could not be observed for anyof them at the concentration used (400 μM). We then focused on twotest compounds: α-lobeline, previously reported to be active in restor-ing GALC on the D528N mutation despite its weak inhibitory activityon GALC [24], and 3′,4′,7-trihydroxyisoflavone, used for the first time,in this study. Both compounds, tested at the concentration of 400 μM
showed a weak inhibitory activity on GALC but were considered suit-able for further investigation as pharmacological chaperones.
To test the inhibitory activity of α-lobeline and 3′,4′,7-trihydroxyisoflavone on GALC, cell extracts from human control fi-broblasts were exposed to increasing concentration (from 0,1 μMto 1 mM) of the two compounds. The results indicated an IC50 of200 μM for α-lobeline (Fig. 1), confirming the weak inhibitory activityobserved in our pilot screening. We were able to detect only a verysmall inflection for 3′,4′,7-trihydroxyisoflavone in the concentration–response at the highest concentration used (Fig. 1) and we were there-fore unable to estimate an IC50 for this compound.
3.2. Enzyme specificity
To examine the specificity of α-lobeline and 3′,4′,7-trihydroxyisoflavone, the inhibitory activity on normal controlfibroblasts was tested for other lysosomal enzymes hydrolyzing differ-ent glycosidic linkages and/or sugar moieties, namely β-glucosidase,β-galactosidase, α-glucosidase and α-galactosidase. The concentrationused was 400 μM for both compounds in all different enzyme assays.All tested enzymes were not inhibited by α-lobeline, confirming thespecificity of this compound for GALC. The 3′,4′,7-trihydroxyisoflavonehad no inhibitory activity on β-glucosidase and α-glucosidase, butshowed very low inhibitory activity (15%) on α-galactosidase and, asexpected, higher inhibitory activity (40%) on β-galactosidase; in fact,it is indicated as an inhibitor of β galactosidase [37].
3.3. Cell toxicity assay
To assess cellular toxicity of the two molecules, we next studied theeffect of test chaperones on the viability of normal control fibroblasts.The cells were incubated with increasing concentrations of each com-pound. In all cases, the cells well tolerated concentration of the com-pounds from 10 to 200 μM, while at higher concentrations (400 and600 μM) cell viability was significantly reduced (Fig. 2). These datashow that these compounds have low cell toxicity, suggesting thatthey may have a distinct advantage with regard to safety of administra-tion, whichmakes them good candidates for further drug development.
3.4. In silico analysis of molecular interaction between GALC andα-lobelineor 3′,4′,7-trihydroxyisoflavone
The molecular docking analysis of the ligands to their macromo-lecular receptor (GALC) indicated that α-lobeline and 3′,4′,7-trihydroxyisoflavone can bind to GALC in 9 different sites with an en-ergy of binding (ΔG) ranging from −7.3 to −6.8 for α-lobeline andfrom −7.8 to −7.0 for 3′,4′,7-trihydroxyisoflavone, while the ΔGof the binding for the natural substrate is −6.4. Interestingly, only onepose of all potential binding sites for both α-lobeline and 3′,4′,7-trihydroxyisoflavone overlaps to the binding site of the natural sub-strate (Fig. 3A–B), but this pose has lower ΔG, so it is disadvantagedin relation to others, as inferred by the weak inhibitory activity mea-sured in the enzyme assay. The bonding interaction between GALCand galactose is shown in panel C (Fig. 3). Our in silico analysis suggeststhat both compounds interact with GALC and can be, therefore, consid-ered good potential candidates as chaperones.
3.5. Pharmacological chaperoning activity on fibroblast cell cultures
Previous studies have used, to test potential chaperones, COS-1 cellsover-expressing GALC bearing different human mutations [24]. In thisstudy, being the chaperone activity mutation specific, we aimed at iden-tifying new missense mutant forms of GALC responsive to α-lobelineand/or to 3′,4′,7-trihydroxyisoflavone. The residual enzyme activity wasdetermined in fibroblast cell lines, carrying differentmissensemutations.

0
2
4
6
8
10
12
14
16
18
untreated 0.1 µM 1 µM 10 µM 100 µM 200 µM 500 µM 1 mM
GA
LC
act
ivity
Fig. 1. In vitro inhibition of the normal control human GALC. IC50 values were determined by increasing the inhibitor concentrations. Assays were performed with cell extracts in 0.1 Mcitrate buffer (pH 5.2), using 6-hexadecanoylamino-4-methylumbelliferyl-ß-D-galactoside as substrate. Black bar: untreated; gray bars, α-lobeline treated; open bars: 3′,4′,7-trihydroxyisoflavone treated.
297A.S. Berardi et al. / Molecular Genetics and Metabolism 112 (2014) 294–301
Themutations analyzed are the following: p.G553R, p.E130K+ p.N295T,p.G57S, p.D187V + p.G323R and p.G286D + p.P318R.
3.5.1. p.G553R + p.G553RThe fibroblast cell line we have analyzed was derived from a patient
affected by the infantile form of Krabbe disease bearing p.G553R muta-tion in homozygosis; the residual enzymatic activity reported in associ-ation with this mutation is very low [38]. In silico analysis indicates thatthismutation induces severemisfolding of GALC [39]. To assess if the se-lected chaperones could rescue GALC activity, fibroblasts were treatedwith α-lobeline and 3′,4′,7-trihydroxyisoflavone at the concentrationof 50, 100 and 200 μM for 72 h.
The treatmentwithα-lobeline at lower concentrations (50–100 μM)significantly raised (two folds) the enzyme activity, while at the highestconcentration used the enzyme activity returned to the level of untreat-ed cells (Fig. 4). In the 3′,4′,7-trihydroxyisoflavone treated cells, a steadyincrease in the GALC activity was observed with increasing concen-tration of the test compound (Fig. 4), with the highest activityreached (6 nmol/17 h/mg protein) at 200 μM. This value, corre-sponding to about 30% of the mean value of normal fibroblasts (con-trol range 16–42, n=17), is well over the 15–20% needed to preventaccumulation of storage material [22]. These data clearly show theefficacy of 3′,4′,7-trihydroxyisoflavone in restoring GALC activity inthis mutation.
0
20
40
60
80
100
120
140
160
untreated α-Lobeline
Cel
l via
bilit
y (%
of
surv
ival
)
10 µM 100 µM 200 µM 400 µM 600 µM
Fig. 2. Viability of normal control fibroblast cell-line in the presence of vehicle,α-lobeline or 3′the mean of three reactions +SD.
3.5.2. p.E130K + p.N295TTreatment with α-lobeline or whith 3′,4′,7-trihydroxyisoflavone
was also successful in increasing GALC activity in a fibroblast line bear-ing themissensemutations p.E130K+ p.N295T.When combined thesemutations cause the infantile form of Krabbe disease. As for the p.G553Rmutation, the p.E130K induces severemisfolding, while the p.N295T in-duces loss of stabilizing hydrogen bonds [39]; both mutant formschange the structural conformation of GALC. In these fibroblasts thetreatment with α-lobeline or 3′,4′,7-trihydroxyisoflavone induced anincrease of GALC activity to similar levels (Fig. 5). The concentrationrange needed to increase by four folds the enzyme activitywas between50 μMand 100 μMof both compounds; when the concentration of bothcompounds was increased to 200 μM the specific activity decreased sig-nificantly, although it remained higher than that of untreated cells. It isnot possible, at this time, to determine if one of the twomutations is re-sponsive to both inhibitors or which of the twomutant forms is respon-sive to which compound.
3.5.3. p.G57S + p.G57SWehave focused our efforts at testing the effectiveness of the select-
ed chaperones toward the p.G57S + p.G57S mutation causing a lateonset form of Krabbe disease. This mutation is present with high inci-dence in a restricted geographical area of southern Italy [15]. Findinga pharmacological chaperone active in restoring GALC activity inthese patients can be considered an important step forward the
0
20
40
60
80
100
120
140
160
10 µM 100 µM 200 µM 400 µM 600 µM
untreated 3’,4’,7-Trihydroxyisoflavone
Cel
l via
bilit
y (%
of
surv
ival
)
,4′,7-trihydroxyisoflavone for 72 h. Cell viability was assessed by XTT assay. The values are
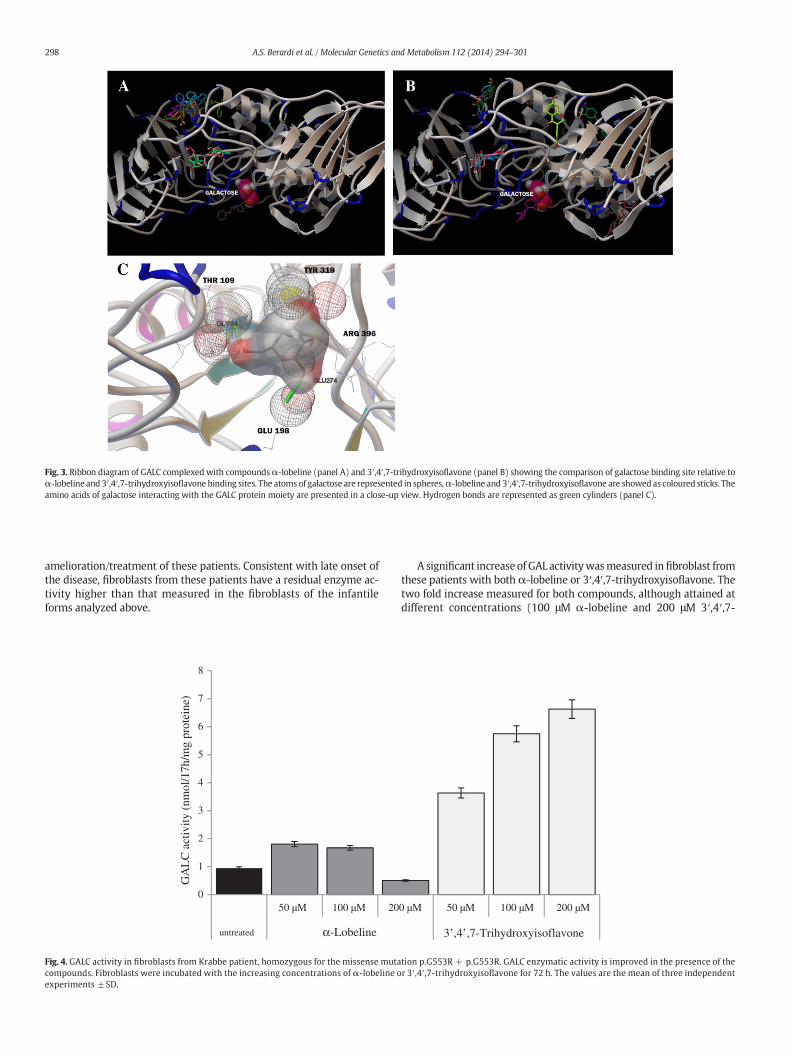
Fig. 3. Ribbon diagram of GALC complexed with compoundsα-lobeline (panel A) and 3′,4′,7-trihydroxyisoflavone (panel B) showing the comparison of galactose binding site relative toα-lobeline and 3′,4′,7-trihydroxyisoflavone binding sites. The atomsof galactose are represented in spheres,α-lobeline and 3′,4′,7-trihydroxyisoflavone are showed as coloured sticks. Theamino acids of galactose interacting with the GALC protein moiety are presented in a close-up view. Hydrogen bonds are represented as green cylinders (panel C).
298 A.S. Berardi et al. / Molecular Genetics and Metabolism 112 (2014) 294–301
amelioration/treatment of these patients. Consistent with late onset ofthe disease, fibroblasts from these patients have a residual enzyme ac-tivity higher than that measured in the fibroblasts of the infantileforms analyzed above.
0
1
2
3
4
5
6
7
8
untreated α-Lobeline
50 µM 100 µM 20
GA
LC
act
ivity
(nm
ol/1
7h/m
g pr
otei
ne)
Fig. 4. GALC activity in fibroblasts from Krabbe patient, homozygous for the missense mutacompounds. Fibroblasts were incubated with the increasing concentrations of α-lobeline oexperiments ±SD.
A significant increase of GAL activitywasmeasured infibroblast fromthese patients with both α-lobeline or 3′,4′,7-trihydroxyisoflavone. Thetwo fold increase measured for both compounds, although attained atdifferent concentrations (100 μM α-lobeline and 200 μM 3′,4′,7-
0 µM 50 µM 100 µM 200 µM
3’,4’,7-Trihydroxyisoflavone
tion p.G553R + p.G553R. GALC enzymatic activity is improved in the presence of ther 3′,4′,7-trihydroxyisoflavone for 72 h. The values are the mean of three independent

0.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
untreated
50 µM 100 µM 200 µM 50 µM 100 µM 200 µM
α-Lobeline 3’,4’,7-Trihydroxyisoflavone
GA
LC
act
ivity
(nm
ol/1
7h/m
g pr
otei
ne)
Fig. 5.GALC activity infibroblasts fromKrabbe patient, bearingmissensemutations p.E130K+ p.N295T. Fibroblastswere incubatedwith increasing concentrations ofα-lobeline or 3′,4′,7-trihydroxyisoflavone for 72 h. GALC enzymatic activity is improved in the presence of both compounds at the lowest concentration used. At the highest concentration of both compoundsthe specific activity decreased significantly but remained higher than untreated cells. The values are the mean of three independent experiments ±SD.
299A.S. Berardi et al. / Molecular Genetics and Metabolism 112 (2014) 294–301
trihydroxyisoflavone) is equal to 15% of normal control values (range16–42, n=17) (Fig. 6) and in the range expected to prevent accumula-tion of storage material [22].
3.5.4. p.D187V + p.G323R and p.G286D + p.P318RWe also investigated the effect of α-lobeline and 3′,4′,7-
trihydroxyisoflavone on two fibroblast cell lines from heterozygotecompound patients; these mutations do not affect the folding of theprotein [39]. The G286D + p.P318R mutation is associated with theadult onset; G286D introduces an acidic side chain, while the P318R in-troduces a hydrophobic side chain near the substrate binding site. TheD187V+ p.G323R is associated to the late infantile form of Krabbe dis-ease. It has been reported that D187V mutation causes the substitutionof an acidic side chain to a smaller hydrophobic side chain, determininga change of the local surface charge. For the G323R no in silico hypothe-sis has been published. When the fibroblast of these patients were
0
0.5
1
1.5
2
2.5
3
3.5
50 µM 100 µM 20
untreated α-Lobeline
GA
LC
act
ivity
(nm
ol/1
7h/m
g pr
otei
ne)
Fig. 6. GALC activity in fibroblasts from Krabbe patient homozygous for the missense mutatiolobeline or 3′,4′,7-trihydroxyisoflavone for 72 h. Both compounds significantly increase the GA
treated with α-lobeline or 3′,4′,7-trihydroxyisoflavone, no changes inGALC specific activity were measured at all concentration used.
4. Discussion
The search for new strategies to optimize therapies and to target thedifferent cellular and phenotypic aspects is becoming an important goalof current research due to the limitations of the therapeutic approachesavailable to treat LSDs. The several attempts and strategies,made to cor-rect the enzyme activity in brain of neuronopathic LSDs, are still a majortherapeutic goal. More than 70 GALCmutations causing Krabbe diseasehave been reported, and many of them cause amino acid substitutions(HumanGeneMutation Database; http://www.hgmd.org). Aswith sev-eral other LSDs, Krabbe patients with similar or identical genotypes canhave different clinical presentations and course of their disease [17];thus, the development of a tailored therapy should represent a success-ful strategy for the treatment of this patients. Recent studies indicated
0 µM 50 µM 100 µM 200 µM
3’,4’,7-Trihydroxyisoflavone
n p.G57S + p.G57S. Fibroblasts were incubated with the increasing concentrations of α-LC activity. The values are the mean of three independent experiments ±SD.

300 A.S. Berardi et al. / Molecular Genetics and Metabolism 112 (2014) 294–301
the pharmacological or molecular chaperone therapy as a potentialtargeted therapeutic approach for LSDs caused by missense mutations[40].
The study presented shows the identification of two compoundsthat selectively rescue GALC activity in fibroblasts derived from patientbearing missense mutations causing infantile, late infantile or adultforms of Krabbe disease. In this study only fibroblast from Krabbe pa-tientswere used rather than transduced cell lines as reported in thema-jority of the studies on chaperones [24,41]. This approach is preferablewhen measuring the functional activity of lysosomal enzymes involvedin lipid degradation because these cell extracts contain all of the neces-sary co-factors, i.e. saposins, for enzyme activation in the natural envi-ronment. Our results show that α-lobeline has a good chaperoningactivity on GALC mutations other than those previously reported [24].Moreover, the 3′,4′,7-trihydroxyisoflavone has been tested for the firsttime for chaperoning activity and our data demonstrated that this com-pound can efficiently rescue GALC activity in 4 differentmissensemuta-tions. In silico analysis suggests that the best-ranked poses ofα-lobelineand 3′,4′,7-trihydroxyisoflavone interact with GALC with a binding en-ergy higher than that of the natural substrate. The modeling data aresupported by the in vitro results showing that α-lobeline is a weak in-hibitor and that 3′,4′,7-trihydroxyisoflavone affects GALC activity onlyat very high concentrations.
Most of the chaperones so far identified for the treatment of LSDs areactive site-directed molecules and are reversible competitive inhibitorsof the target enzymes. This issue has raised concerns and stimulated theidentification of a new generation of chaperones able to protect the en-zymes from degradation without interfering with its activity [42–44].
We believe that the compounds we have identified are related tothis new generation of chaperonesmaking them suitable for further ex-ploration as pharmacological chaperones.
In addition, our data show that, at the concentration used, thesecompounds are relatively not toxic and do not inhibit other lysosomalenzymes with closely related activities, confirming that these com-pounds could have a role in the therapy of Krabbe patients. Moreovertheir potential therapeutic role for diseases with CNS involvement isalso supported by the demonstrated capacity of α-lobeline and 3′,4′,7-trihydroxyisoflavone to cross the blood brain barrier [45–47], and itcan be hypothesized that they penetrate cell membranes and achievetherapeutic concentrations in specific cell compartments.
The efficacy of α-lobeline and 3′,4′,7-trihydroxyisoflavone in res-cuing GALC activity has been demonstrated in our in vitro studies. Infibroblasts carrying the p.G553R mutations both compounds werehelpful, but the 3′,4′,7-trihydroxyisoflavone was found to be moreeffective than α-lobeline in increasing GALC activity. This cell linewas previously used in a different study for the selection of smallmolecules from a library of pharmacological active compounds, butnone of the examined compounds was active in increasing the resid-ual GALC activity [41].
We were particularly interested in analyzing the efficacy of α-lobeline and 3′,4′,7-trihydroxyisoflavone on p.G57S because of thehigh incidence of this mutation in a restricted geographical area ofSouthern Italy [15]. The development of a tailored chaperone therapycould benefit the patients carrying thismutation. In this case both chap-eroneswere able to rescue GALC activity to the same extent (15% of nor-mal values); however, lower concentrations of α-lobeline were neededto reach this value. It has been demonstrated that a threshold activity ofapproximately 10% is sufficient to prevent storage in LSDs [23]. Thus, theincrease to 15% of normal activity induced by α-lobeline and 3′,4′,7-trihydroxyisoflavone is likely to have an impact on disease pathologyand to be beneficial for these Krabbe patients. The increase of theGALC activity over the critical threshold is significant from a therapeuticaspect, and in fact, is sufficient to deplete and prevent storage of the sub-strate. However, even if the increase in enzyme activity remains belowthe critical threshold, the rescued activity could still be sufficient to in-duce clinical benefit and ameliorate the patients' quality of life.
5. Conclusion
In summary, our combined in silico–in vitro approach allowed for thesuccessful identification of two pharmacological chaperones,α-lobelineand 3′,4′,7-trihydroxyisoflavone, for the treatment of Krabbe disease.Thus, further studies will have to evaluate whether these compoundsact similarly on other mutations, and preclinical studies will indicate ifthese compounds are amenable for development of an individualizedtherapy for Krabbe disease.
Conflict of interest
The authors declare that the research was conducted in the absenceof any commercial or financial relationships that could be construed as apotential conflict of interest.
Acknowledgments
This study was supported by funds to Alice Luddi from Tuscany Re-gion “Bando salute 2009” n°158. Three of the cell lines used from pa-tients affected by Krabbe disease were obtained from the G. GasliniInstitute–Telethon Genetic Biobank Network (project GTB07001). Oneof the cell lines used from patients affected by Krabbe disease was ob-tained from Prof Paolo Balestri, University of Siena. One of the celllines used from patients affected by Krabbe disease was obtained fromProf Antonio Federico, University of Siena. We are also thankful toVera Cardile for the kind collaboration.
References
[1] K. Suzuki, Y. Suzuki, Globoid cell leucodystrophy (Krabbe's disease): deficiency ofgalactocerebroside beta-galactosidase, Proc. Natl. Acad. Sci. U. S. A. 66 (1970)302–309.
[2] D.A. Wenger, M.A. Rafi, P. Luzi, Molecular genetics of Krabbe disease (globoid cell leu-kodystrophy): diagnostic and clinical implications, Hum. Mutat. 10 (1997) 268–279,http://dx.doi.org/10.1002/(SICI)1098-1004(1997)10:4b268::AID-HUMU2N3.0.CO;2-D([pii] 1002/(SICI)1098–1004(1997)10:4b268::AID-HUMU2N3.0.CO;2-D).
[3] M.T. Vanier, L. Svennerholm, Chemical pathology of Krabbe's disease. III. Ceramide-hexosides and gangliosides of brain, Acta Paediatr. Scand. 64 (1975) 641–648.
[4] L. Svennerholm, M.T. Vanier, J.E. Mansson, Krabbe disease: a galactosylsphingosine(psychosine) lipidosis, J. Lipid Res. 21 (1980) 53–64.
[5] D.J. Costello, A.F. Eichler, F.S. Eichler, Leukodystrophies: classification, diagnosis, andtreatment, Neurologist 15 (2009) 319–328, http://dx.doi.org/10.1097/NRL.0b013e3181b287c8 (00127893-200911000-00004 [pii]).
[6] H. Nagara, H. Ogawa, Y. Sato, T. Kobayashi, K. Suzuki, The twitcher mouse: degener-ation of oligodendrocytes in vitro, Brain Res. 391 (1986) 79–84.
[7] T. Taketomi, K. Nishimura, Physiological activity of psychosine, Jpn. J. Exp. Med. 34(1964) 255–265.
[8] F. Seitelberger, Demyelination and leukodystrophy at an early age, Bol. Estud. Med.Biol. 31 (1981) 373–382.
[9] T. Kobayashi, I. Goto, T. Yamanaka, Y. Suzuki, T. Nakano, K. Suzuki, Infantileand fetal globoid cell leukodystrophy: analysis of galactosylceramide andgalactosylsphingosine, Ann. Neurol. 24 (1988) 517–522, http://dx.doi.org/10.1002/ana.410240407.
[10] J. Aicardi, The inherited leukodystrophies: a clinical overview, J. Inherit. Metab. Dis.16 (1993) 733–743.
[11] R. De Gasperi, M.A. Gama Sosa, E.L. Sartorato, S. Battistini, H. MacFarlane, J.F. Gusella,W. Krivit, E.H. Kolodny, Molecular heterogeneity of late-onset forms of globoid-cellleukodystrophy, Am. J. Hum. Genet. 59 (1996) 1233–1242.
[12] H. Furuya, Y. Kukita, S. Nagano, Y. Sakai, Y. Yamashita, H. Fukuyama, Y. Inatomi, Y.Saito, R. Koike, S. Tsuji, et al., Adult onset globoid cell leukodystrophy (Krabbe dis-ease): analysis of galactosylceramidase cDNA from four Japanese patients, Hum.Genet. 100 (1997) 450–456.
[13] L. Fu, K. Inui, T. Nishigaki, N. Tatsumi, H. Tsukamoto, C. Kokubu, T. Muramatsu, S.Okada, Molecular heterogeneity of Krabbe disease, J. Inherit. Metab. Dis. 22 (1999)155–162.
[14] C. Xu, N. Sakai, M. Taniike, K. Inui, K. Ozono, Six novel mutations detected in theGALC gene in 17 Japanese patients with Krabbe disease, and new genotype-phenotype correlation, J. Hum. Genet. 51 (2006) 548–554, http://dx.doi.org/10.1007/s10038-006-0396-3.
[15] W. Lissens, A. Arena, S. Seneca, M. Rafi, G. Sorge, I. Liebaers, D.Wenger, A. Fiumara, Asingle mutation in the GALC gene is responsible for themajority of late onset Krabbedisease patients in the Catania (Sicily, Italy) region, Hum. Mutat. 28 (2007) 742,http://dx.doi.org/10.1002/humu.9500.
[16] D.A. Wenger, M.L. Escolar, P. Luzi, M.A. Rafi, Scriver's The Online Metabolic and Mo-lecular Bases of Inherited Disease (OMMBID). Chapter 147 Krabbe Disease (GloboidCell Leukodystrophy), Available at: http://www.ommbid.com/OMMBID/the_

301A.S. Berardi et al. / Molecular Genetics and Metabolism 112 (2014) 294–301
online_metabolic_and_molecular_bases_of_inherited_disease/b/abstract/Part16/ch147 2013 ([accessed 28 Oct 2013]).
[17] A. Fiumara, R. Barone, A. Arena, M. Filocamo, W. Lissens, L. Pavone, G. Sorge, Krabbeleukodystrophy in a selected population with high rate of late onset forms: longersurvival linked to c.121GNA (p.Gly41Ser) mutation, Clin. Genet. 80 (2011)452–458, http://dx.doi.org/10.1111/j.1399-0004.2010.01572.x.
[18] P.K. Duffner, V.S. Caviness Jr., R.W. Erbe, M.C. Patterson, K.R. Schultz, D.A. Wenger, C.Whitley, The long-term outcomes of presymptomatic infants transplanted forKrabbe disease: report of the workshop held on July 11 and 12, 2008, Holiday Valley,New York, Genet. Med. 11 (2009) 450–454, http://dx.doi.org/10.1097/GIM.0b013e3181a16e04.
[19] E. Costantino-Ceccarini, A. Luddi, M. Volterrani, M. Strazza, M.A. Rafi, D.A. Wenger,Transduction of cultured oligodendrocytes from normal and twitcher mice by a ret-roviral vector containing human galactocerebrosidase (GALC) cDNA, Neurochem.Res. 24 (1999) 287–293.
[20] A. Luddi, M. Volterrani, M. Strazza, A. Smorlesi, M.A. Rafi, J. Datto, D.A. Wenger, E.Costantino-Ceccarini, Retrovirus-mediated gene transfer and galactocerebrosidaseuptake into twitcher glial cells results in appropriate localization and phenotypecorrection, Neurobiol. Dis. 8 (2001) 600–610, http://dx.doi.org/10.1006/nbdi.2001.0407 (S0969-9961(01)90407-3 [pii]).
[21] M.A. Rafi, H.Z. Rao, P. Luzi, M.T. Curtis, D.A. Wenger, Extended normal life afterAAVrh10-mediated gene therapy in the mouse model of Krabbe disease, Mol.Ther. 20 (2012) 2031–2042, http://dx.doi.org/10.1038/mt.2012.153.
[22] P. Leinekugel, S. Michel, E. Conzelmann, K. Sandhoff, Quantitative correlation be-tween the residual activity of beta-hexosaminidase A and arylsulfatase A and the se-verity of the resulting lysosomal storage disease, Hum. Genet. 88 (1992) 513–523.
[23] U.H. Schueler, T. Kolter, C.R. Kaneski, G.C. Zirzow, K. Sandhoff, R.O. Brady, Correlationbetween enzyme activity and substrate storage in a cell culture model system forGaucher disease, J. Inherit. Metab. Dis. 27 (2004) 649–658.
[24] W.C. Lee, D. Kang, E. Causevic, A.R. Herdt, E.A. Eckman, C.B. Eckman, Molecular char-acterization of mutations that cause globoid cell leukodystrophy and pharmacolog-ical rescue using small molecule chemical chaperones, J. Neurosci. 30 (2010)5489–5497, http://dx.doi.org/10.1523/JNEUROSCI.6383-09.2010 (30/16/5489 [pii]).
[25] G.Wiederschain, S. Raghavan, E. Kolodny, Characterization of 6-hexadecanoylamino-4-methylumbelliferyl-beta-D- galactopyranoside as fluorogenic substrate ofgalactocerebrosidase for the diagnosis of Krabbe disease, Clin. Chim. Acta 205(1992) 87–96.
[26] D. Robinson, The fluorimetric determination of beta-glucosidase: its occurrence inthe tissues of animals, including insects, Biochem. J. 63 (1956) 39–44.
[27] R. Kwapiszewski, J. Szczudlowska, K. Kwapiszewska,M. Chudy, Z. Brzozka, Determina-tion of acid beta-galactosidase activity: methodology and perspectives, Indian J. Clin.Biochem. 29 (2014) 57–62, http://dx.doi.org/10.1007/s12291-013-0318-z (318 [pii]).
[28] K. Umapathysivam, J.J. Hopwood, P.J. Meikle, Determination of acid alpha-glucosidase activity in blood spots as a diagnostic test for Pompe disease, Clin.Chem. 47 (2001) 1378–1383.
[29] N.A. Chamoles, M. Blanco, D. Gaggioli, Fabry disease: enzymatic diagnosis in driedblood spots on filter paper, Clin. Chim. Acta 308 (2001) 195–196.
[30] S.F. Altschul, W. Gish, W. Miller, E.W. Myers, D.J. Lipman, Basic local alignmentsearch tool, J. Mol. Biol. 215 (1990) 403–410, http://dx.doi.org/10.1016/S0022-2836(05)80360-2 (S0022-2836(05)80360-2 [pii]).
[31] J. Kopp, T. Schwede, The SWISS-MODEL repository of annotated three-dimensionalprotein structure homology models, Nucleic Acids Res. 32 (2004) D230–D234,http://dx.doi.org/10.1093/nar/gkh008 (32/suppl_1/D230 [pii]).
[32] A.W. Schuttelkopf, D.M. van Aalten, PRODRG: a tool for high-throughput crystallog-raphy of protein–ligand complexes, Acta Crystallogr. D Biol. Crystallogr. 60 (2004)1355–1363, http://dx.doi.org/10.1107/S0907444904011679 (S0907444904011679[pii]).
[33] S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M.R. Shirts, J.C. Smith,P.M. Kasson, D. van der Spoel, et al., GROMACS 4.5: a high-throughput and highlyparallel open source molecular simulation toolkit, Bioinformatics 29 (2013)845–854, http://dx.doi.org/10.1093/bioinformatics/btt055 (btt055 [pii]).
[34] C. Oostenbrink, T.A. Soares, N.F. van der Vegt, W.F. van Gunsteren, Validation of the53A6 GROMOS force field, Eur. Biophys. J. 34 (2005) 273–284, http://dx.doi.org/10.1007/s00249-004-0448-6.
[35] O. Trott, A.J. Olson, AutoDock Vina: improving the speed and accuracy of dockingwith a new scoring function, efficient optimization, and multithreading, J. Comput.Chem. 31 (2010) 455–461, http://dx.doi.org/10.1002/jcc.21334.
[36] G.M. Morris, L.G. Green, Z. Radic, P. Taylor, K.B. Sharpless, A.J. Olson, F. Grynszpan,Automated dockingwith protein flexibility in the design of femtomolar “click chem-istry” inhibitors of acetylcholinesterase, J. Chem. Inf. Model. 53 (2013) 898–906,http://dx.doi.org/10.1021/ci300545a.
[37] L. Governini, P. Carrarelli, A.L. Rocha, V.D. Leo, A. Luddi, F. Arcuri, P. Piomboni, C.Chapron, L.M. Bilezikjian, F. Petraglia, FOXL2 in humanendometrium: hyperexpressedin endometriosis, Reprod. Sci. (2014), http://dx.doi.org/10.1177/1933719114522549(1933719114522549 [pii]).
[38] B. Tappino, R. Biancheri,M.Mort, S. Regis, F. Corsolini, A. Rossi,M. Stroppiano, S. Lualdi,A. Fiumara, B. Bembi, et al., Identification and characterization of 15 novel GALC genemutations causing Krabbe disease, Hum. Mutat. 31 (2010) E1894–E1914, http://dx.doi.org/10.1002/humu.21367.
[39] J.E. Deane, S.C. Graham, N.N. Kim, P.E. Stein, R. McNair, M.B. Cachon-Gonzalez, T.M.Cox, R.J. Read, Insights into Krabbe disease from structures of galactocerebrosidase,Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 15169–15173, http://dx.doi.org/10.1073/pnas.1105639108 (1105639108 [pii]).
[40] K.J. Valenzano, R. Khanna, A.C. Powe, R. Boyd, G. Lee, J.J. Flanagan, E.R. Benjamin,Identification and characterization of pharmacological chaperones to correct en-zyme deficiencies in lysosomal storage disorders, Assay Drug Dev. Technol. 9(2011) 213–235, http://dx.doi.org/10.1089/adt.2011.0370.
[41] J. Ribbens, G. Whiteley, H. Furuya, N. Southall, X. Hu, J. Marugan, M. Ferrer, G.H.Maegawa, A high-throughput screening assay using Krabbe disease patient cells,Anal. Biochem. 434 (2013) 15–25, http://dx.doi.org/10.1016/j.ab.2012.10.034(S0003-2697(12)00553-2 [pii]).
[42] G. Parenti, C. Pignata, P. Vajro, M. Salerno, New strategies for the treatment of lyso-somal storage diseases (review), Int. J. Mol. Med. 31 (2013) 11–20, http://dx.doi.org/10.3892/ijmm.2012.1187.
[43] S. Patnaik, W. Zheng, J.H. Choi, O. Motabar, N. Southall, W. Westbroek, W.A. Lea, A.Velayati, E. Goldin, E. Sidransky, et al., Discovery, structure–activity relationship,and biological evaluation of noninhibitory small molecule chaperones ofglucocerebrosidase, J. Med. Chem. 55 (2012) 5734–5748, http://dx.doi.org/10.1021/jm300063b.
[44] J.J. Marugan, W. Zheng, M. Ferrer, O. Motabar, N. Southall, E. Goldin, W. Westbroek,E. Sidransky, Discovery, SAR, and Biological Evaluation of a Non-Inhibitory Chaper-one for Acid Alpha Glucosidase, 2010. (doi: NBK153221 [bookaccession]).
[45] T.H. Tsai, Concurrentmeasurement of unbound genistein in the blood, brain and bileof anesthetized rats using microdialysis and its pharmacokinetic application, J.Chromatogr. A 1073 (2005) 317–322.
[46] M. Malinowska, F.L. Wilkinson, K.J. Langford-Smith, A. Langford-Smith, J.R. Brown, B.E. Crawford, M.T. Vanier, G. Grynkiewicz, R.F. Wynn, J.E. Wraith, et al., Genistein im-proves neuropathology and corrects behaviour in a mouse model of neurodegener-ative metabolic disease, PLoS One 5 (2010) e14192, http://dx.doi.org/10.1371/journal.pone.0014192.
[47] A. Friso, R. Tomanin, M. Salvalaio, M. Scarpa, Genistein reduces glycosaminoglycanlevels in a mouse model of mucopolysaccharidosis type II, Br. J. Pharmacol. 159(2010) 1082–1091, http://dx.doi.org/10.1111/j.1476-5381.2009.00565.x (BPH565[pii]).
Copyright © 2022 FDOKUMEN