
Role of adipokines and peroxisome proliferator-activated ...
Upload
independentCategory
view
3download
0
Peroxisome Proliferator-Activated Receptor � GeneVariants Influence Progression of Coronary Atherosclerosis
and Risk of Coronary Artery DiseaseDavid M. Flavell, PhD; Yalda Jamshidi, PhD; Emma Hawe, MSc; Inés Pineda Torra, PhD;
Marja-Riitta Taskinen, MD; M. Heikki Frick, MD; Markku S. Nieminen, MD; Y. Antero Kesäniemi, MD;Amos Pasternack, MD; Bart Staels, PhD; George Miller, MD; Steve E. Humphries, PhD, FRCPath, MRCP;
Philippa J. Talmud, PhD, DSc, FRCPath; Mikko Syvänne, MD
Background—Peroxisome proliferator-activated receptor � (PPAR�) regulates the expression of genes involved in lipidmetabolism and inflammation, making it a candidate gene for atherosclerosis and ischemic heart disease (IHD).
Methods and Results—We investigated the association between the leucine 162 to valine (L162V) polymorphism and aG to C transversion in intron 7 of the PPAR� gene and progression of atherosclerosis in the Lopid CoronaryAngiography Trial (LOCAT), a trial examining the effect of gemfibrozil treatment on progression of atherosclerosisafter bypass surgery and on risk of IHD in the second Northwick Park Heart Study (NPHS2), a prospective study ofhealthy middle-aged men in the United Kingdom. There was no association with plasma lipid concentrations in eitherstudy. Both polymorphisms influenced progression of atherosclerosis and risk of IHD. V162 allele carriers had lessprogression of diffuse atherosclerosis than did L162 allele homozygotes with a similar trend for focal atherosclerosis.Intron 7 C allele carriers had greater progression of atherosclerosis than did G allele homozygotes. The V162 alleleattenuated the proatherosclerotic effect of the intron 7 C allele. Homozygotes for the intron 7 C allele had increased riskof IHD, an effect modulated by the L162V polymorphism
Conclusions—The PPAR� gene affects progression of atherosclerosis and risk of IHD. Absence of association with plasmalipid concentrations suggests that PPAR� affects atherosclerotic progression directly in the vessel wall. (Circulation.2002;105:1440-1445.)
Key Words: atherosclerosis � genetics � coronary disease
Atherogenesis is a complex process influenced by numer-ous factors, including dyslipidemia, endothelial dys-
function, oxidative stress, and inflammation, and involves theinteraction of monocyte/macrophages, endothelial cells, andsmooth muscle cells.1 Peroxisome proliferator-activated re-ceptor � (PPAR�) is a member of the nuclear hormonereceptor superfamily of ligand-activated transcription fac-tors.2 Ligands for PPAR� include fatty acids, eicosanoids,and the fibrate class of hypolipidemic drugs.3 PPAR� regu-lates the expression of genes involved in lipid metabolism,and fibrate treatment causes a dramatic decrease in triglycer-ides, a lesser decrease in LDL cholesterol, and an increase inHDL cholesterol, producing a less atherogenic lipid pheno-type.4 PPAR� is expressed at high levels in liver, heart,skeletal muscle, and kidney,5 tissues that catabolize fattyacids.
PPAR� also regulates proatherosclerotic processes in thevessel wall. PPAR� is expressed in endothelial cells6 andsmooth muscle cells7 and in monocyte/macrophages in thecirculation8 and in atherosclerotic plaques.9 In these cell types,PPAR� regulates the expression of genes influencing monocyterecruitment (monocyte chemotactic protein-1,10 endothelin11),adhesion (vascular cell adhesion model-1 [VCAM-1]12,13) andtransmigration (matrix metalloproteinase-9 [MMP-9]14), foamcell formation (SR-BI9), reverse cholesterol transport (liver Xreceptor � [LXR�], ATP binding cassette transporter 1[ABCA1]15), and thrombogenicity (tissue factor16,17).
PPAR� also may influence atherosclerosis through antiin-flammatory properties. PPAR� inhibits the activator protein(AP-1)11,18 and nuclear factor-�B7,11,18,19 inflammatory cyto-kine signaling pathways. Fenofibrate lowers plasma concen-trations of proinflammatory cytokines.7,20 PPAR� knockout
Received November 20, 2001; revision received January 18, 2002; accepted January 18, 2002.From the Centre for Cardiovascular Genetics, Department of Medicine, Royal Free and University College of London Medical School, London, UK
(D.M.F., Y.J., E.H., S.E.H., P.J.T.); U.325 INSERM, Département d’Athérosclérose, Institut Pasteur, Lille, France (I.P.T., B.S.); the Department ofMedicine, Helsinki University Central Hospital, Helsinki, Finland (M.-R.T., M.H.F., M.S.N., M.S.); the Department of Medicine, University of Oulu,Oulu, Finland (Y.A.K.); the Department of Medicine, Tampere University Hospital, Tampere, Finland (A.P.); and Medical Research CouncilEpidemiology and Medical Care Unit, Wolfson Institute of Preventive Medicine, The Medical College of St Bartholomew’s Hospital, London, UK(G.M.).
Correspondence to David M. Flavell, Centre for Cardiovascular Genetics, Department of Medicine, Royal Free and University College of LondonMedical School, Rayne Building, 5 University St, London WC1E 6JJ, UK. E-mail [email protected]
© 2002 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/01.CIR.0000012145.80593.25
1440

mice have a greater18,21 inflammatory response. Oxidativestress also influences atherosclerosis.22 PPAR� activatorsreduce oxidative stress,23 and PPAR� knockout mice havehigher levels of proinflammatory cytokines and lipidperoxides.19
Fibrates reduced the progression of coronary atherosclero-sis in the Bezafibrate Coronary Atherosclerosis Trial(BECAIT) and the Lopid Coronary Angiography Trial(LOCAT) and reduced the incidence of coronary arterydisease in the Helsinki Heart Study and Veterans Adminis-tration HDL-cholesterol Intervention Trial (VA-HIT).24 Thus,PPAR� is expressed in cell types involved in atherosclerosis,and PPAR� activators have pleiotropic beneficial effects onproatherosclerotic factors. To investigate the role of PPAR�in human atherosclerosis, we examined the association be-tween two polymorphisms in the PPAR� gene, the functionalleucine 162 to valine (L162V) variant25,26 and a novelpolymorphism in intron 7, and progression of atherosclerosisin LOCAT, a trial examining the effect of gemfibroziltreatment on progression of atherosclerosis in Finnish pa-tients with CABG and low HDL27 and in the second North-wick Park Heart Study (NPHS2), a prospective study exam-ining the risk of heart disease in healthy middle-aged men inthe United Kingdom.28
MethodsStudy SamplesA cohort of 395 Finnish men �70 years old with HDL cholesterol�1.1 mmol/L, LDL cholesterol �4.5 mmol/L, and triglycerides�4.0 mmol/L entered the study (baseline characteristics are shownin Table 1). Current smokers and subjects with severe hypertension,obesity, history of diabetes, or fasting glucose concentration�7.8 mmol/L were excluded. Patients had undergone coronaryartery bypass surgery and were treated with placebo or gemfibrozil(1200 mg/d) (Parke Davis) for 2.5 years.27 Coronary angiographywas performed at baseline and on average at 32 months after therapycommencement. Cholesterol and triglyceride concentrations weredetermined in LDL and HDL subfractions.29 Angiographic imageswere analyzed with a computer-assisted quantitative method.30
Diffuse atherosclerosis is defined as a change in average diameter ofcoronary segments (�ADS) from baseline to follow-up angiogram.Focal atherosclerosis is a change in minimum luminal diameter(�MLD) of stenoses. Of subjects with L162V genotype, 302 had�ADS data and 297 subjects had �MLD data. For those with intron7 genotype, 283 had �ADS and 278 had �MLD data.
The Second Northwick Park Heart Study (NPHS2) is a prospec-tive study of ischemic heart disease (IHD) in 3012 middle-aged (50to 61 years) healthy men in the United Kingdom over a period of 7years (see Table 1 for baseline characteristics). Exclusion criteriaincluded history of type 2 diabetes, unstable angina, or previousmyocardial infarction or stroke.28 Plasma concentrations of choles-terol, HDL cholesterol, triglycerides, apolipoprotein (apo)AI, andapoB were determined. “Ischemic events” is a measure of acutemyocardial infarction (n�136), silent myocardial infarction (n�21),or coronary surgery (n�34).
Detection of the Intron 7 and L162V PolymorphismThe intron 7 polymorphism was reported as a TaqI restrictionfragment length polymorphism.31 Human PPAR� genomic sequence(Genbank AL078611) indicated that the polymorphism was situatedin a TaqI fragment spanning exon 7. TaqI digestion of polymerasechain reaction (PCR) products revealed a polymorphic site, andsequencing revealed a G to C transversion at nt 2498 of intron 7 (nt16669 of complementary strand of Genbank AL078611), 396 ntfrom the 3� end of intron 7. Intron 7 PCR primers were forwardACAATCACTCCTTAAATATGGTGG, reverse AAGTAGGGA-CAGACAGGACCAGTA, generating a fragment of 266bp digestedby TaqI to 216bp and 50bp. PCR reactions were performed in 10 �Lcontaining 1� buffer (16 mmol/L [NH4]2SO4/67 mmol/L Tris, pH8.4/0.01% Tween 20/0.02 mmol/L each dNTP), 2 mmol/L MgCl2, 8pmol each primer, 0.1 U Taq polymerase. TaqI digestion wasperformed by adding 3 U of TaqI (Helena), 1.5 �L restrictionenzyme buffer in a volume of 5 �L, and incubation for 4 hours at65°C. L162V polymorphism genotyping has been previouslydescribed.25
Statistical AnalysisThe maximum number of genotypes available for each parameterwas used in analysis. Polymorphisms were checked for deviationfrom Hardy-Weinberg equilibrium by means of the �2 test. Allelicassociation was estimated by means of the Estimate Haplotypeprogram. In LOCAT, the relation between baseline lipid levels,change in lipid levels on treatment, and PPAR� genotype wasexamined by t test or ANOVA. Triglycerides were log-transformed.Multivariate ANOVA was used to control for baseline levels of ADSor MLD, time between the baseline and follow-up angiograms, andrandomized therapy. Results are reported as unadjusted and adjustedleast-squares mean�SEM.
In NPHS2, statistical analysis was conducted with intercooledSTATA version 6.0. Body mass index (BMI), systolic blood pres-sure, triglycerides, and fibrinogen were log-transformed. One-wayANOVA was used to assess the effect of genotype on baselinecharacteristics. Survival analysis was carried out with Cox propor-tional hazards model, with significance assessed by the likelihoodratio test. Adjustment was performed for age, BMI, smoking status,systolic blood pressure, fibrinogen, and cholesterol. Because of thelow numbers of individuals with L162V VV genotype, genotypesLV and VV were combined.
ResultsGenotypes for the L162V and intron 7 polymorphisms weredetermined for 317 and 298 participants, respectively, in theLOCAT study, and for 2620 and 2684 participants, respec-tively, in the Second Northwick Park Heart Study. InLOCAT, allele frequencies were 0.028 (95% CI, 0.015 to0.041) for the V162 allele and 0.134 (95% CI, 0.107 to 0.162)for the intron 7 C allele. In NPHS2, allele frequencies were0.063 (95% CI, 0.056 to 0.069) for the V162 allele and 0.174(95% CI, 0.164 to 0.184) for the intron 7 C allele. Frequenciesof the L162V (P�0.0006) and intron 7 (P�0.01) poly-morphisms were significantly lower in the LOCAT study.Polymorphisms were in Hardy-Weinberg equilibrium, withthe exception of the intron 7 polymorphism in NPHS2
TABLE 1. Baseline Characteristics of Subjects in LOCATand NPHS2
LOCAT(n�297)
NPHS2
Controls(n�2560)
Cases(n�176)
Age, y 58.90�6.86 56.02�3.42 56.72�3.63
BMI, kg/m2 26.44�2.22 26.19�3.37 26.89�3.46
Smokers, % 0 27.5 38.6
Systolic blood pressure, mm Hg 134.9�18.1 136.7�18.6 142.8�20.4
Cholesterol, mmol/L 5.17�0.71 5.71�1.01 6.09�1.05
Triglycerides, mmol/L 1.59�0.74 1.78�0.93 2.08�1.11
Fibrinogen, g/L N/A 270.0�51.4 283.7�51.0
LOCAT data are for the 297 subjects with both genotype data available. Dataare mean�SD.
Flavell et al PPAR� Gene Variants and Heart Disease 1441

(P�0.04), due to regional variation in allele frequency. TheV162 and the intron 7 C allele were in allelic association inLOCAT (��0.32, P�0.01) and NPHS2 (��0.34,P�0.0001). Estimated haplotype frequencies are shown inTable 2.
The association between the PPAR� gene polymorphismsand plasma lipid concentrations at baseline and change inplasma lipid concentrations after gemfibrozil treatment wasexamined. There were no significant differences in plasmalipid concentrations in either study (Table 3) or in the changein plasma lipids in response to gemfibrozil in LOCAT (notshown) by PPAR� genotype.
Association between PPAR� polymorphisms and progres-sion of atherosclerosis was examined. Compared with L162homozygotes, V162 allele carriers had significantly reducedprogression of diffuse atherosclerosis in native coronaryarteries (LL, 0.026�0.006 mm, n�284; LV, 0.032�0.025 mm, n�18; P�0.022) (Figure 1). In multivariateANOVA, adjusting for baseline ADS, time between angio-grams, and treatment, the effect remained significant(P�0.049). The effect was similar in the placebo and treatedgroups, with no evidence of interaction between genotype andtreatment. Moreover, the LV genotype showed a protectiveinfluence against progression of focal atherosclerosis incoronary arteries (�MLD) (LL, 0.073�0.010 mm, n�279,versus LV, 0.006�0.041 mm, n�18; P�0.064) (Figure 1A),which was again independent of gemfibrozil treatment. Thus,carriers of the V162 allele were protected from progression ofatherosclerosis.
The intron 7 polymorphism also influenced the rate ofprogression of atherosclerosis. C allele homozygotes (n�7)
were combined with C allele carriers. Carriers of the C alleleshowed a nonsignificant trend toward greater progression ofdiffuse atherosclerosis (�ADS) than G allele homozygotes(GG, �0.017�0.007 mm, n�213; GC CC, �0.041� 0.015mm, n�70; P�0.095) (Figure 1B). Intron 7 genotype hadsimilar effects in both placebo and treated groups. Intron 7genotype significantly influenced progression of focal athero-sclerosis (�MLD). Carriers of the C allele had a significantlygreater decrease in MLD than G allele homozygotes (GG,
TABLE 2. Estimated Haplotype Frequencies in LOCATand NPHS2
L162V Intron 7 LOCAT
NPHS2
Total Controls Cases
L162 G 0.860 0.804 0.806 0.776
L162 C 0.113 0.132 0.129 0.178
V162 G 0.006 0.021 0.021 0.014
V162 C 0.021 0.041 0.043 0.032
TABLE 3. Association Between PPAR� Polymorphisms and Baseline Plasma Lipid Concentrations
LL LV VV P GG GC CC P
LOCAT (n�284) (n�18) (n�0) (n�213) (n�63) (n�7)
Triglycerides 1.62�0.73 1.52�0.71 � � � 0.55 1.61�0.74 1.72�0.69 1.17�0.27 0.12
Total cholesterol 5.17�0.71 5.17�0.74 � � � 0.99 5.18�0.74 5.19�0.60 5.40�0.82 0.74
LDL 3.42�0.60 3.41�0.54 � � � 0.94 3.44�0.61 3.38�0.49 3.71�0.16 0.34
HDL total 1.02�0.17 1.05�0.17 � � � 0.55 1.03�0.18 1.01�0.16 1.11�0.17 0.31
NPHS2 (n�2302) (n�307) (n�11) (n�1847) (n�740) (n�97)
Triglycerides 1.80�0.96 1.79�0.91 1.80�0.79 0.95 1.78�0.93 1.84�0.98 1.74�0.85 0.35
Total cholesterol 5.74�1.02 5.71�1.02 6.01�1.34 0.61 5.74�1.01 5.73�1.02 5.69�1.17 0.89
ApoB 0.88�0.26 0.84�0.26 0.94�0.32 0.11 0.88�0.26 0.88�0.26 0.87�0.29 0.77
HDL 0.80�0.24 0.80�0.25 0.69�0.14 0.35 0.80�0.25 0.79�0.24 0.82�0.22 0.48
ApoAI 1.61�0.34 1.60�0.34 1.56�0.24 0.88 1.62�0.34 1.59�0.34 1.61�0.34 0.29
Values are mmol/L, mean�SD.
Figure 1. Effect of PPAR� genotype on progression of athero-sclerosis in LOCAT. A, L162V genotype. B, Intron 7 genotype.Carriers and homozygotes for the intron 7 C allele are com-bined. Values are mean�SEM. Values in brackets indicate num-ber of subjects. C, Regression model for PPAR� L162V andintron 7 variants. Values are �-coefficients.
1442 Circulation March 26, 2002
�0.050�0.011 mm, n�209; GCCC, �0.115�0.019 mm,n�69; P�0.003) (Figure 1B), an effect again observed inboth placebo and treated groups. Thus intron 7 C allelecarriers show increased progression of atherosclerosis.
The L162V and intron 7 polymorphisms are in positive allelicassociation yet have opposing effects on progression of atheroscle-rosis. The estimated frequency of the V162-intron 7 C haplotype inLOCAT was 0.021, whereas the haplotype frequency of theV162-G haplotype was 0.006; that is, 78% of V162 alleles are onthe same haplotype as the intron 7 C allele (Table 2). In a regressionmodel, both polymorphisms showed significant effects on progres-sion of diffuse (�ADS: constant, �0.018�0.007 mm, P�0.014;V162 allele, 0.082�0.029 mm, P�0.005; intron 7 C allele,�0.038�0.015 mm, P�0.013) and focal (�MLD: constant,�0.054�0.011 mm, P�0.001; V162 allele, 0.132�0.045 mm,P�0.003; intron 7 C allele, �0.086�0.024 mm, P�0.001) athero-sclerosis (Figure 1C). In a multivariate regression model includingbaseline measures, time between angiogram, both PPAR� geno-types and examining for interaction between the PPAR� variants, asignificant interaction was observed between PPAR� genotypes fordiffuse atherosclerosis (�ADS, P�0.05), with a similar trendobserved for focal atherosclerosis (�MLD P�0.07).
We investigated whether PPAR� variants influence risk ofIHD in the prospective second Northwick Park Heart Study.L162V genotype did not influence risk of events in a Coxproportional hazards model adjusted for age, BMI, cholester-ol, fibrinogen, smoking, and systolic blood pressure. Carriersand homozygotes for the V162 allele combined had a hazardratio of 0.75 (95% CI, 0.45 to 1.26, P�0.28). Intron 7 C allelehomozygotes showed a nonsignificant trend for greater riskof IHD, with a hazard ratio of 1.83 (95% CI, 0.96 to 3.51,
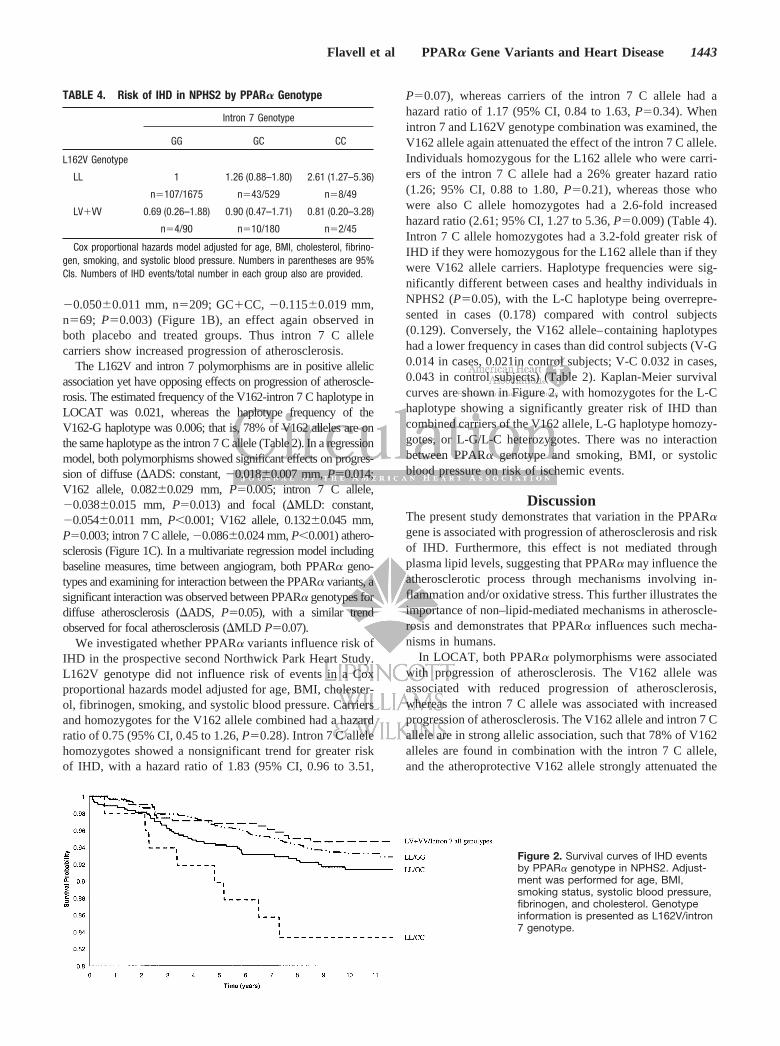
P�0.07), whereas carriers of the intron 7 C allele had ahazard ratio of 1.17 (95% CI, 0.84 to 1.63, P�0.34). Whenintron 7 and L162V genotype combination was examined, theV162 allele again attenuated the effect of the intron 7 C allele.Individuals homozygous for the L162 allele who were carri-ers of the intron 7 C allele had a 26% greater hazard ratio(1.26; 95% CI, 0.88 to 1.80, P�0.21), whereas those whowere also C allele homozygotes had a 2.6-fold increasedhazard ratio (2.61; 95% CI, 1.27 to 5.36, P�0.009) (Table 4).Intron 7 C allele homozygotes had a 3.2-fold greater risk ofIHD if they were homozygous for the L162 allele than if theywere V162 allele carriers. Haplotype frequencies were sig-nificantly different between cases and healthy individuals inNPHS2 (P�0.05), with the L-C haplotype being overrepre-sented in cases (0.178) compared with control subjects(0.129). Conversely, the V162 allele–containing haplotypeshad a lower frequency in cases than did control subjects (V-G0.014 in cases, 0.021in control subjects; V-C 0.032 in cases,0.043 in control subjects) (Table 2). Kaplan-Meier survivalcurves are shown in Figure 2, with homozygotes for the L-Chaplotype showing a significantly greater risk of IHD thancombined carriers of the V162 allele, L-G haplotype homozy-gotes, or L-G/L-C heterozygotes. There was no interactionbetween PPAR� genotype and smoking, BMI, or systolicblood pressure on risk of ischemic events.
DiscussionThe present study demonstrates that variation in the PPAR�gene is associated with progression of atherosclerosis and riskof IHD. Furthermore, this effect is not mediated throughplasma lipid levels, suggesting that PPAR� may influence theatherosclerotic process through mechanisms involving in-flammation and/or oxidative stress. This further illustrates theimportance of non–lipid-mediated mechanisms in atheroscle-rosis and demonstrates that PPAR� influences such mecha-nisms in humans.
In LOCAT, both PPAR� polymorphisms were associatedwith progression of atherosclerosis. The V162 allele wasassociated with reduced progression of atherosclerosis,whereas the intron 7 C allele was associated with increasedprogression of atherosclerosis. The V162 allele and intron 7 Callele are in strong allelic association, such that 78% of V162alleles are found in combination with the intron 7 C allele,and the atheroprotective V162 allele strongly attenuated the
TABLE 4. Risk of IHD in NPHS2 by PPAR� Genotype
Intron 7 Genotype
GG GC CC
L162V Genotype
LL 1 1.26 (0.88–1.80) 2.61 (1.27–5.36)
n�107/1675 n�43/529 n�8/49
LVVV 0.69 (0.26–1.88) 0.90 (0.47–1.71) 0.81 (0.20–3.28)
n�4/90 n�10/180 n�2/45
Cox proportional hazards model adjusted for age, BMI, cholesterol, fibrino-gen, smoking, and systolic blood pressure. Numbers in parentheses are 95%CIs. Numbers of IHD events/total number in each group also are provided.
Figure 2. Survival curves of IHD eventsby PPAR� genotype in NPHS2. Adjust-ment was performed for age, BMI,smoking status, systolic blood pressure,fibrinogen, and cholesterol. Genotypeinformation is presented as L162V/intron7 genotype.
Flavell et al PPAR� Gene Variants and Heart Disease 1443

proatherosclerotic effect of the intron 7 C allele. The PPAR�
intron 7 polymorphism was also associated with risk of IHDin NPHS2, and although there was no significant associationwith the L162V polymorphism and risk, the V162 allele againreduced the effect of the intron 7 polymorphism. The risk-associated L-C haplotype is overrepresented in subjects withIHD, whereas the V-G and V-C haplotypes are found at alower frequency in cases than in control subjects.
Surprisingly, given the well-documented effects of fibratetreatment on plasma lipid levels, the PPAR� gene poly-morphisms had no effect on plasma lipid concentrations ineither study, confirming our previous findings.25 It has beenreported that apoB and LDL cholesterol concentrations arehigher in carriers of the V162 allele,32,33 though we did notobserve such a finding, possibly because of differences in thesubject selection criteria. In the LOCAT study, a similareffect of PPAR� genotype on progression of atherosclerosiswas observed in both placebo and treated groups, suggestingthat these variants in the PPAR� gene do not dramaticallyinfluence response to fibrate treatment, as previously ob-served.25,34 The absence of an association between PPAR�
polymorphisms and plasma lipid levels suggests that theeffect of the PPAR� polymorphisms on IHD is not plasmalipid–mediated unless subtle alterations occur in plasma lipidprofiles. Additionally, the risk of IHD was unchanged whenplasma cholesterol levels were included in the Cox propor-tional hazards model.
PPAR� is also expressed in vessel wall cell types andregulates the expression of genes that influence proathero-sclerotic processes in these cells. Inflammation plays a majorrole in both the initiation and progression of atherosclerosis,1
and PPAR� activation has antiinflammatory actions.35
PPAR� also influences oxidative stress.19,23 Thus, PPAR�
polymorphisms may affect progression of atherosclerosisthrough inflammatory and/or oxidative stress mechanisms.
The mechanism by which the intron 7 variant influencesthe atherosclerotic process remains unclear. PPAR�-V162has higher transcriptional activation in vitro.25,26 No othercommon missense variants have been identified in thePPAR� gene coding region.25,26,33 Given the opposing effectsof the V162 allele and the intron 7 C allele and the beneficialeffects of stimulation of PPAR� activity with fibrates, wehypothesize that the intron 7 C allele is associated with lowerlevels of PPAR�. It is unlikely that the intron 7 variant isitself functional, due to its position in an intron. However, wesuggest that it is in allelic association with a functionalvariant in a promoter or enhancer element of the PPAR� genethat results in reduced PPAR� gene expression. Surprisingly,PPAR� knockout mice crossed with the apoE knockoutmouse on an atherogenic diet show reduced aortic atheroscle-rosis compared with apoE knockout control mice.36
Thus, PPAR� affects factors that contribute to the athero-sclerotic process, and common variation in the humanPPAR� gene influences these factors. These data demonstratethe important role of PPAR� in human atherogenesis andprovide genetic evidence that PPAR� has antiatheroscleroticeffects in humans in vivo and so influences risk of IHD.
AcknowledgmentsDrs Jamshidi, Humphries, and Torra are supported by British HeartFoundation grants FS/98058 and RG/95007. Dr Talmud is supportedby a European Community grant (ERBFMBICT983214), and BartStaels is supported by grants of the Institut Pasteur de Lille andINSERM. The LOCAT study was supported by Parke-Davis, theFinnish Foundation for Cardiovascular Research, the Aarne KoskeloFoundation, and the Finnish Society of Angiology. NPHS2 wassupported by the Medical Research Council, the US NationalInstitutes of Health (grant NHLBI 33014) and Du Pont Pharma.
References1. Ross R. Atherosclerosis-an inflammatory disease. N Engl J Med. 1999;
340:115–126.2. Issemann I, Green S. Activation of a member of the steroid hormone
receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650.
3. Krey G, Braissant O, L’Horset F, et al. Fatty acids, eicosanoids, andhypolipidemic agents identified as ligands of peroxisome proliferator-ac-tivated receptors by coactivator-dependent receptor ligand assay. MolEndocrinol. 1997;11:779–791.
4. Fruchart J-C, Duriez P, Staels B. Peroxisome proliferator-activatedreceptor-alpha activators regulate genes governing lipoprotein metabo-lism, vascular inflammation and atherosclerosis. Curr Opin Lipidol. 1999;10:245–257.
5. Auboeuf D, Rieusset J, Fajas L, et al. Tissue distribution and quantifi-cation of the expression of mRNAs of peroxisome proliferator-activatedreceptors and liver X receptor-alpha in humans: no alteration in adiposetissue of obese and NIDDM patients. Diabetes. 1997;46:1319–1327.
6. Inoue I, Shino K, Noji S, et al. Expression of peroxisome proliferator-activated receptor alpha (PPAR alpha) in primary cultures of humanvascular endothelial cells. Biochem Biophys Res Commun. 1998;246:370–374.
7. Staels B, Koenig W, Habib A, et al. Activation of human aortic smooth-muscle cells is inhibited by PPAR� but not by PPAR� activators. Nature.1998;393:790–793.
8. Chinetti G, Griglio S, Antonucci M, et al. Activation of proliferator-ac-tivated receptors alpha and gamma induces apoptosis of humanmonocyte-derived macrophages. J Biol Chem. 1998;273:25573–25580.
9. Chinetti G, Gbaguidi FG, Griglio S, et al. CLA-1/SR-BI is expressed inatherosclerotic lesion macrophages and regulated by activators of per-oxisome proliferator activated receptors. Circulation. 2000;101:2411–2417.
10. Zhu L, Bisgaier CL, Aviram M, et al. 9-Cis retinoic acid inducesmonocyte chemoattractant protein-1 secretion in human monocyticTHP-1 cells. Arterioscler Thromb Vasc Biol. 1999;19:2105–2111.
11. Delerive P, Martin Nizard F, Chinetti G, et al. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 pro-duction in human vascular endothelial cells by inhibiting the activatorprotein-1 signaling pathway. Circ Res. 1999;85:394–402.
12. Jackson SM, Parhami F, Xi XP, et al. Peroxisome proliferator-activatedreceptor activators target human endothelial cells to inhibit leukocyte-en-dothelial cell interaction. Arterioscler Thromb Vasc Biol. 1999;19:2094–2104.
13. Marx N, Sukhova GK, Collins T, et al. PPAR� activators inhibit cyto-kine-induced vascular cell adhesion molecule-1 expression in humanendothelial cells. Circulation. 1999;99:3125–3131.
14. Shu H, Wong B, Zhou G, et al. Activation of PPARalpha or gammareduces secretion of matrix metalloproteinase 9 but not interleukin 8 fromhuman monocytic THP-1 cells. Biochem Biophys Res Commun. 2000;267:345–349.
15. Chinetti G, Lestavel S, Bocher V, et al. PPAR-alpha and PPAR-gammaactivators induce cholesterol removal from human macrophage foam cellsthrough stimulation of the ABCA1 pathway. Nat Med. 2001;7:53–58.
16. Neve BP, Corseaux D, Chinetti G, et al. PPAR� agonists inhibit tissuefactor expression in human monocytes and macrophages. Circulation.2001;103:207–212.
17. Marx N, Mackman N, Schonbeck U, et al. PPAR� activators inhibittissue factor expression and activity in human monocytes. Circulation.2001;103:213–219.
18. Delerive P, De Bosscher K, Besnard S, et al. Peroxisome proliferator-ac-tivated receptor alpha negatively regulates the vascular inflammatorygene response by negative cross-talk with transcription factorsNF-kappaB and AP-1. J Biol Chem. 1999;274:32048–32054.
1444 Circulation March 26, 2002

19. Poynter ME, Daynes RA. Peroxisome proliferator-activated receptoralpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production inaging. J Biol Chem. 1998;273:32833–32841.
20. Madej A, Okopien B, Kowalski J, et al. Effects of fenofibrate on plasmacytokine concentrations in patients with atherosclerosis and hyperlipo-proteinemia IIb. Int J Clin Pharmacol Ther. 1998;36:345–349.
21. Devchand PR, Keller H, Peters JM, et al. The PPARalpha-leukotriene B4pathway to inflammation control. Nature. 1996;384:39–43.
22. Kunsch C, Medford RM. Oxidative stress as a regulator of geneexpression in the vasculature. Circ Res. 1999;85:753–766.
23. Inoue I, Noji S, Awata T, et al. Bezafibrate has an antioxidant effect:peroxisome proliferator- activated receptor alpha is associated withCu2, Zn2-superoxide dismutase in the liver. Life Sci. 1998;63:135–144.
24. Watts GF, Dimmitt SB. Fibrates, dyslipoproteinaemia and cardiovasculardisease. Curr Opin Lipidol. 1999;10:561–574.
25. Flavell DM, Pineda Torra I, Jamshidi Y, et al. Variation in the PPAR�gene is associated with altered function in vitro and plasma lipid concen-trations in type II diabetic subjects. Diabetologia. 2000;43:673–680.
26. Sapone A, Peters JM, Sakai S, et al. The human peroxisome proliferator-activated receptor � gene: identification and functional characterizationof two natural allelic variants. Pharmacogenetics. 2000;10:321–333.
27. Frick MH, Syvanne M, Nieminen MS, et al. Prevention of the angio-graphic progression of coronary and vein-graft atherosclerosis by gemfi-brozil after coronary bypass surgery in men with low levels of HDLcholesterol: Lopid Coronary Angiography Trial (LOCAT) Study Group.Circulation. 1997;96:2137–2143.
28. Miller GJ, Bauer KA, Barzegar S, et al. Increased activation of thehaemostatic system in men at high risk of fatal coronary heart disease.Thromb Haemost. 1996;75:767–771.
29. Syvanne M, Nieminen MS, Frick MH, et al. Associations betweenlipoproteins and the progression of coronary and vein-graft atherosclero-sis in a controlled trial with gemfibrozil in men with low baseline levelsof HDL cholesterol. Circulation. 1998;98:1993–1999.
30. Syvanne M, Nieminen MS, Frick MH. Accuracy and precision of quan-titative arteriography in the evaluation of coronary artery disease aftercoronary bypass surgery. A validation study. Int J Card Imaging. 1994;10:243–252.
31. Sher T, Yi HF, McBride OW, et al. cDNA cloning, chromosomalmapping, and functional characterization of the human peroxisome pro-liferator activated receptor. Biochemistry. 1993;32:5598–5604.
32. Vohl M-C, Lepage P, Gaudet D, et al. Molecular scanning of the humanPPAR� gene: association of the L162V mutation with hyperapobetali-poproteinemia. J Lipid Res. 2000;41:945–952.
33. Lacquemant C, Lepretre F, Pineda T, et al. Mutation screening of thePPARalpha gene in type 2 diabetes associated with coronary heartdisease. Diabetes Metab. 2000;26:393–401.
34. Jamshidi Y, Flavell DM, Hawe E, et al. Genetic determinants of theresponse to bezafibrate treatment in the Lower Extremity Arterial DiseaseEvent Reduction (LEADER) trial. Atherosclerosis. In press.
35. Delerive P, Fruchart C, Staels B. Peroxisome proliferator-activatedreceptors in inflammation control. J Endocrinol.;2001;169:453–459.
36. Tordjman K, Bernal-Mizrachi C, Zemany L, et al. PPARalpha deficiencyreduces insulin resistance and atherosclerosis in apoE-null mice. J ClinInvest. 2001;107:1025–1034.
Flavell et al PPAR� Gene Variants and Heart Disease 1445
Copyright © 2022 FDOKUMEN




















