
“on water” wittig reaction laboratory experiment
110
“ON WATER” WITTIG REACTION LABORATORY EXPERIMENT AND THE DEVELOPMENT OF AN “ON WATER” CATALYTIC WITTIG REACTION _______________ A Thesis Presented to the Faculty of San Diego State University _______________ In Partial Fulfillment of the Requirements for the Degree Master of Science in Chemistry _______________ by Lucas Brett Fallot Spring 2014
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of “on water” wittig reaction laboratory experiment

“ON WATER” WITTIG REACTION LABORATORY EXPERIMENT
AND THE DEVELOPMENT OF AN “ON WATER” CATALYTIC
WITTIG REACTION
_______________
A Thesis
Presented to the
Faculty of
San Diego State University
_______________
In Partial Fulfillment
of the Requirements for the Degree
Master of Science
in
Chemistry
_______________
by
Lucas Brett Fallot
Spring 2014


iii
Copyright © 2014
by
Lucas Brett Fallot
All Rights Reserved

iv
DEDICATION
This thesis is dedicated to my beloved family; Laura, Autumn and Preston. You
follow me to the ends of the earth and unconditionally support me through whatever I do.
Thank you for believing in me.

v
ABSTRACT OF THE THESIS
“On Water” Wittig Reaction Laboratory Experiment and the Development of an “On Water” Catalytic Wittig Reaction
by Lucas Brett Fallot
Master of Science in Chemistry San Diego State University, 2014
I. “On Water” Wittig Reaction Laboratory Experiment The aqueous Wittig reaction is a suitable undergraduate experiment which allows for instructors to effectively and very quickly demonstrate and promote a greener alternative of an alkene synthesis in the organic chemistry teaching laboratory. There is an opportunity for students to compare previously reported Wittig reaction approaches and evaluate those with the green “on water” type Wittig reaction methodology. II. Development of an “On Water” Catalytic Wittig Reaction The achievement of an “on water” catalytic Wittig reaction is the next big step in making the Wittig reaction greener. Employing the normal phosphine oxide by-product of the Wittig reaction as the catalyst will improve the reactions overall atom economy. The development of an “on water” catalytic Wittig reaction using triphenylphosphine oxide and 3-methyl-1-phenylphospholane oxide is reported. 3-Methyl-1-phenylphospholane oxide compared to triphenylphosphine oxide is a more promising catalyst to achieve an “on water” catalytic Wittig reaction because it reduces more easily.

vi
TABLE OF CONTENTS
PAGE
ABSTRACT ...............................................................................................................................v
LIST OF TABLES ................................................................................................................... ix
LIST OF FIGURES ...................................................................................................................x
LIST OF ABBREVIATIONS ................................................................................................ xiii
ACKNOWLEDGMENTS ..................................................................................................... xiv
CHAPTER
1 “ON WATER” WITTIG REACTION LABORATORY EXPERIMENT ....................1
1.1 Introduction ........................................................................................................1
1.2 Previous Wittig Reaction Protocols ...................................................................3
1.2.1 Hazardous Reagent Wittig Reaction Protocols .........................................3
1.2.2 Phase Transfer Catalysis ...........................................................................5
1.2.3 “Instant Ylid” ............................................................................................5
1.2.4 Solvent-Free Wittig Reactions ..................................................................6
1.2.4.1 Ball Milling ......................................................................................6
1.2.4.2 Mortar and Pestle .............................................................................8
1.2.5 Microwave Assisted Experiments .............................................................9
1.2.5.1 Household Microwave .....................................................................9
1.2.5.2 Laboratory Grade Microwave Reactor ..........................................10
1.2.6 Stabilized Ylide Wittig Reaction ............................................................11
1.3 “On Water” Wittig Reaction Laboratory Experiment .....................................11
1.3.1 Experimental Procedure ..........................................................................12
1.3.2 Results and Discussion ...........................................................................15
1.3.3 Hazards of the Experiment ......................................................................18
1.4 Laboratory Experiment Comparison ................................................................18
1.5 Conclusion .......................................................................................................20
2 THE ATTEMPTED DEVELOPMENT OF AN “ON WATER” CATALYTIC WITTIG REACTION ..................................................................................................21

vii
2.1 Introduction ......................................................................................................21
2.2 Use of Triphenylphosphine Oxide ...................................................................21
2.2.1 Standard Wittig Reaction Using Ph3P in Organic Solvent .....................23
2.2.2 Reduction Protocols for Ph3PO ...............................................................24
2.2.2.1 Metal Hydrides...............................................................................24
2.2.2.2 Hydrosilanes and a Titanium Catalyst ...........................................24
2.2.2.3 Phosphoric Acid Derivative with a Silane .....................................25
2.2.2.4 Benzoic Acid Derivative with a Silane ..........................................26
2.2.3 Work Towards a Catalytic Wittig Reaction in Organic Solvent .............29
2.2.3.1 Wittig Reaction and Beller Reduction Protocol .............................29
2.2.3.2 O’Brien Catalytic Wittig Reaction in Organic Solvents ................32
2.2.3.3 Catalytic Wittig Reaction Attempt Using O’Brien’s Protocol with Diphenylsilane and Sodium Carbonate ...................33
2.2.4 “On Water” Reduction Protocols for Ph3PO ..........................................34
2.2.5 “On Water” Catalytic Wittig Reaction Attempt Using Ph3PO ...............37
2.3 Use of 3-methyl-1-phenylphospholane Oxide .................................................38
2.3.1 Standard “On Water” Wittig Reaction Using 3-methyl-1-phenylphospholane .................................................................................39
2.3.2 “On Water” Reduction of 3-methyl-1-phenylphospholane oxide with Ph2SiH2 ...........................................................................................39
2.3.3 “On Water” Catalytic Wittig Reaction Attempts Using 3-methyl-1-phenylphospholane oxide ....................................................................40
2.4 Future Work for the Development of an “On Water” Catalytic Wittig Reaction ...........................................................................................................43
2.5 Conclusion .......................................................................................................46
3 THE EXPERIMENTAL PART ...................................................................................47
3.1 General .............................................................................................................47
3.2 “On Water” Wittig Reaction Experiment ........................................................47
3.3 Attempted Catalytic Wittig Reaction Experiments Using Ph3P and Ph3PO ...............................................................................................................48
3.3.1 Standard Wittig Reaction Using Ph3P in Organic Solvent .....................48
3.3.2 Reduction of Ph3PO in Organic Solvent .................................................48
3.3.3 Compatibility Test of Standard Wittig Reaction and Beller Reduction Protocol ..................................................................................49

viii
3.3.4 Experiments to Develop a Catalytic Wittig Reaction in Organic Solvent ....................................................................................................49
3.3.5 Attempted Catalytic Wittig Reaction Experiment Using Ph3PO in Organic Solvents Without NEt3 ..............................................................50
3.3.6 Standard Wittig Reaction Experiment Using Diethoxymethylsilane and Ph3P ..............................................................50
3.3.7 Attempted Catalytic Wittig Reaction Using O’Brien’s Protocol, Sodium Carbonate, and Ph3PO ...............................................................51
3.3.8 “On Water” Reduction of Ph3PO ............................................................51
3.3.9 Reduction of Ph3PO in 15% DMSO and Water ......................................52
3.3.10 Attempted Catalytic Wittig Reaction in Water Using 5% DMSO, NaCO3, Ph3PO, and Ph2SiH2 .....................................................52
3.4 Experimental Work Towards an “On Water” Catalytic Wittig Reaction Using 3-methyl-1-phenylphospholane oxide ...................................................53
3.4.1 Optimized O’Brien Catalytic Wittig Reaction in Toluene .....................53
3.4.2 “On Water” Wittig Reaction Using 3-methyl-1-phenylphospholane .................................................................................53
3.4.3 “On Water” Reduction of 3-methyl-1-phenylphospholane oxide ..........54
3.4.4 Attempted “On Water” Catalytic Wittig Reaction using Optimized O’Brien Catalytic Wittig Reaction Protocol .........................54
3.4.5 Attempted “On Water” Catalytic Wittig Reactions Using 3-methyl-1-phenylphospholane oxide and Na2CO3/NaHCO3 ....................55
3.4.6 Attempted “On Water” Catalytic Wittig Reactions Using O’Brien’s Protocol and Tertiary Nitrogen Bases ....................................55
3.4.7 Initial Optimization Attempts of Experiment 2.31 .................................56
3.4.8 Optimization Attempts of Experiment 2.31 with Greater Equivalents of Methyl Bromoacetate ......................................................56
REFERENCES ........................................................................................................................58
APPENDIX
A JOURNAL OF CHEMICAL EDUCATION “ON WATER” WITTIG REACTION LABORATORY EXPERIMENT SUBMISSION .................................62
B 1H NMR, AND 31P NMR OF REACTIONS ...............................................................68

ix
LIST OF TABLES
PAGE
Table 1.1. Ball-Milling Wittig Reactions ..................................................................................7
Table 1.2. Equipment for “On Water” Wittig Reaction Experiment .......................................13
Table 1.3. Cost, Yield and Reaction Time Comparison of Wittig Reaction Experiments .................................................................................................................19
Table 2.1. Reduction of Ph3PO with TMDS and Ti(OiPr)4 .....................................................25
Table 2.2. Beller Reduction Protocol .......................................................................................26
Table 2.3. O’Brien Reduction Protocol ...................................................................................28
Table 2.4. O’Brien’s Catalytic Wittig Reactions in Organic Solvents ....................................33
Table 2.5. Silane in the Presence of Water ..............................................................................35
Table 2.6. Microwave Reduction of Ph3PO in Water ..............................................................36

x
LIST OF FIGURES
PAGE
Figure 1.1. Twelve principles of green chemistry. ....................................................................2
Figure 1.2. Wittig reaction using a lachrymator and a pyrophoric base. ...................................3
Figure 1.3. Wittig reaction using a pyrophoric starting material. ..............................................4
Figure 1.4. Wittig reaction using sodium hydride. ....................................................................4
Figure 1.5. Phase transfer catalysis using a 50% sodium hydroxide solution. ..........................5
Figure 1.6. “Instant Ylid” Wittig reaction. ................................................................................6
Figure 1.7. Ball-milling Wittig reactions. ..................................................................................7
Figure 1.8. Ball-milling one-pot Wittig reaction. ......................................................................7
Figure 1.9. Mortar and pestle Wittig reaction. ...........................................................................8
Figure 1.10. Domestic household microwave Wittig reaction. .................................................9
Figure 1.11. Microwave reactor Wittig reaction. .....................................................................10
Figure 1.12. Stabilized ylide Wittig reaction. ..........................................................................11
Figure 1.13. “On water” Wittig reaction experiment. ..............................................................11
Figure 1.14. Experiment set-up. ...............................................................................................14
Figure 1.15. Product 1H NMR of (E/Z)-ethyl cinnamate. ........................................................16
Figure 1.16. Starting material 1H NMR of triphenylphosphine. ..............................................17
Figure 1.17. Starting material 1H NMR of ethyl bromoacetate. ..............................................17
Figure 1.18. Starting material 1H NMR of benzaldehyde. .......................................................17
Figure 2.1. Four steps of a catalytic Wittig reaction. ...............................................................22
Figure 2.2. Catalytic Wittig reaction with tributylarsenic. ......................................................22
Figure 2.3. Catalytic Wittig reaction using a tellurium compound. .........................................23
Figure 2.4. Standard Wittig using Ph3P in organic solvent. .....................................................23
Figure 2.5. Reduction of Ph3PO with TMDS and Ti(OiPr)4. ...................................................24
Figure 2.6. Beller reduction protocol. ......................................................................................26
Figure 2.7. Proposed Beller reduction mechanism of Ph3PO. .................................................27
Figure 2.8. O’Brien reduction protocol ....................................................................................27
Figure 2.9. Reduction study of Ph3PO using Ph2SiH2 and 4-nitrobenzoic acid. .....................29

xi
Figure 2.10. Compatibility test of standard Wittig reaction and Beller reduction protocol. ..............................................................................................................30
Figure 2.11. Catalytic Wittig reaction using Ph3PO in toluene. ..............................................30
Figure 2.12. Catalytic Wittig using diphenylsilane and diphenylphosphate. ..........................31
Figure 2.13. Catalytic Wittig reaction using 50 mol % of diphenylphosphate. .......................31
Figure 2.14. Catalytic Wittig reaction without the use of NEt3. ..............................................31
Figure 2.15. Standard Wittig reaction with Ph3P and (EtO)2MeSiH. ......................................32
Figure 2.16. O’Brien’s catalytic Wittig reactions in organic solvents. ....................................33
Figure 2.17. Catalytic Wittig reaction with Ph3PO in toluene. ................................................34
Figure 2.18. Greaney’s one-pot silyl-Reformatsky olefination. ..............................................35
Figure 2.19. Microwave reduction of Ph3PO in water. ............................................................36
Figure 2.20. Ph3PO reduction in 15% DMSO and water. ........................................................37
Figure 2.21. Attempted “on water” catalytic Wittig reaction with DMSO. ............................37
Figure 2.22. O’Brien et al. catalytic Wittig reaction in toluene. ..............................................38
Figure 2.23. Optimized O’Brien catalytic Wittig reaction in toluene. .....................................39
Figure 2.24. “On water” Wittig reaction using 3-methyl-1-phenylphospholane. ....................39
Figure 2.25. “On water” reduction of 3-methyl-1-phenylphospholane oxide. ........................40
Figure 2.26. Catalytic “on-water” Wittig with 3-methyl-1-phenylphospholane oxide. ..........40
Figure 2.27. Attempted “on water” catalytic Wittig experiments with Na2CO3 and NaHCO3. .............................................................................................................41
Figure 2.28. Attempted “on water” catalytic Wittig reactions with tertiary nitrogen bases. ...................................................................................................................42
Figure 2.29. Initial optimization attempts of reaction 2.31. .....................................................42
Figure 2.30. Reaction 2.31 with greater equivalents of methyl bromoacetate. .......................43
Figure 2.31. Side reaction of methyl bromoacetate with base and water. ...............................44
Figure 2.32. Reduction side reaction of methyl bromoacetate with diphenylsilane and water. ...................................................................................................................45
Figure B.1. 1H NMR of reaction 1.1. .......................................................................................69
Figure B.2. 1H NMR of reaction 2.1. .......................................................................................70
Figure B.3. 31P NMR of reaction 2.5. ......................................................................................71
Figure B.4. 1H NMR of reaction 2.5. .......................................................................................72
Figure B.5. 1H NMR of reaction 2.6. .......................................................................................73
Figure B.6. 1H NMR of reaction 2.7. .......................................................................................74

xii
Figure B.7. 1H NMR of reaction 2.8. .......................................................................................75
Figure B.8. 1H NMR of reaction 2.9. .......................................................................................76
Figure B.9. 1H NMR of reaction 2.10. .....................................................................................77
Figure B.10. 1H NMR of reaction 2.11. ...................................................................................78
Figure B.11. 1H NMR of reaction 2.12. ...................................................................................79
Figure B.12. 31P NMR of reaction 2.21. ..................................................................................80
Figure B.13. 1H NMR of reaction 2.21. ...................................................................................81
Figure B.14. 1H NMR of reaction 2.22. ...................................................................................82
Figure B.15. 1H NMR of reaction 2.23. ...................................................................................83
Figure B.16. 1H NMR of reaction 2.24. ...................................................................................84
Figure B.17. 31P NMR of 3-methyl-1-phenylphospholane oxide. ...........................................85
Figure B.18. 31P NMR of reaction 2.25. ..................................................................................86
Figure B.19. 1H NMR of reaction 2.26. ...................................................................................87
Figure B.20. 1H NMR of reaction 2.27. ...................................................................................88
Figure B.21. 1H NMR of reaction 2.28. ...................................................................................89
Figure B.22. 1H NMR of reaction 2.29. ...................................................................................90
Figure B.23. 1H NMR of reaction 2.30. ...................................................................................91
Figure B.24. 1H NMR of reaction 2.31. ...................................................................................92
Figure B.25. 1H NMR of reaction 2.32. ...................................................................................93
Figure B.26. 1H NMR of reaction 2.33. ...................................................................................94
Figure B.27. 1H NMR of reaction 2.34. ...................................................................................95
Figure B.28. 1H NMR of reaction 2.35. ...................................................................................96

xiii
LIST OF ABBREVIATIONS
DCM Dichloromethane
DIPEA Diisopropylethylamine
DMAP Dimethylaminopyridine
DMF Dimethylformamide
DMSO Dimethylsulfoxide
NMR Nuclear Magnetic Resonance
PMHS Polymethylhydrosiloxane
RT Room Temperature
THF Tetrahydrofuran
TMDS Tetramethyldisiloxane

xiv
ACKNOWLEDGMENTS
First and foremost, I would like to thank God for giving my family and I the strength
to endure all of the obstacles we had to face during our time in San Diego. I would like to say
thank you to Dr. Bergdahl for the opportunity to work in his lab and for providing me with an
exceptional research idea for my thesis. I am very proud to say I received my practical
experience of working in an organic chemistry lab from the Bergdahl group! Thank you to all
of the members of the Bergdahl crowd; your patience, wisdom and kindness never ceased to
amaze me. You all contributed greatly to my education and I am forever grateful to have
worked with you and more importantly, call you friends. I would also like to thank all of the
chemistry department staff and faculty at San Diego State University for providing a top-
notch chemistry curriculum. I feel as though I am leaving school with a well-rounded
knowledge of chemistry that will help me to instruct cadets at my following assignment at
West Point. Thank you to Dr. LeRoy Lafferty for teaching me how to operate the NMR
equipment, for aiding me when I needed help and for his countless efforts to maintain the
NMR equipment. Lastly, I would like to make a special thank you to Michael Kelly for
taking me under his wing and helping me every step of the way; even to the emergency
room. His selflessness and goodwill to all whom he encounters is unparalleled. I am forever
grateful for the time and effort he dedicated to help me in the lab.

1
CHAPTER 1
“ON WATER” WITTIG REACTION
LABORATORY EXPERIMENT
1.1 INTRODUCTION
There is ample evidence that the scale and very nature of industrial activity has
permanently changed the chemical dynamics of “stable” systems in nature.1 Environmental
issues that arise due to industrial activity include climate change, stratospheric ozone
depletion, photochemical ozone formation, acidification, and eutrophication.2 Population
growth along with an increased demand of industrial materials has led to an increase of
requirements for natural and synthetic materials. The scale and accelerating rate of change
results in activity and waste streams that disrupt and degrade natural systems worldwide—the
very systems that provide critical services on which society and the economy depend on like
healthy air, clean energy, productive soil and safe food.1
Sustainability is a global concept to achieve a balance between environmental,
economic and social realms of society in order to ensure the development of the present does
not impede the development of future generations.2 Though there are many ways to prevent
pollution, green chemistry is an approach that chemists can take to change chemical
processes so they are of less risk to human health and to improve the balance of
environmental sustainability.3 The concepts of sustainability and green chemistry must be
heavily integrated into the scientific community so that there may be a paradigm shift in
industry to move away from dangerous pollute-then-clean up practices.4
Although, sustainability and environmental protection are two critical issues in our
society, greener practices have only been slowly adopted in the organic chemistry teaching
laboratory. Often, especially at the undergraduate level, many laboratory procedures have
minor changes from those of fifty years ago. In order to increase awareness and adoption of
practical greener chemistry in the future, it is essential to positively inform students early in
their education about the alternatives possible for more environmentally-friendly chemistry.4

2
Educators can use the Wittig reaction along with the Twelve Principles of Green Chemistry
as a way to simplify the concept of green chemistry and use it as a model to practically
demonstrate it (Figure 1.13).
Figure 1.1. Twelve principles of green chemistry. Source: Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice; Oxford University Press: New York, 1998; p 30.
The Wittig reaction is a fundamental introductory organic chemistry reaction used to
produce an alkene from the reaction of a phosphonium ylide with a carbonyl compound.5 The
Wittig reaction can be used to promote practical greener chemistry by way of replacing
organic solvents with water. Water is by its very nature nontoxic, cost-effective and less-
harmful to the environment than the organic solvents traditionally used when conducting the
Wittig reaction. It also represents the outside-the-box thinking that will be necessary for a
more-sustainable approach to common chemical problems without diminishing the larger
lesson: the fundamental utility of the Witting reaction. The positive influence of the Twelve

3
Principles of Green Chemistry along with the Wittig reaction will help convince students
early-on in their education to buy-in to green chemistry, and later in their professional
careers, to help positively influence industry to achieve green sustainability goals and in turn,
protect our world’s natural systems for future generations.
1.2 PREVIOUS WITTIG REACTION PROTOCOLS
In order to best demonstrate the necessary thinking for the implementation of greener
solutions to common chemical problems, it is helpful to first examine and compare some
traditional Wittig reaction protocols from literature. Variations of the Wittig reaction can be
found in many laboratory textbooks as well as in the Journal of Chemical Education.
Among these sources, none demonstrate that a simple Wittig reaction can be done in water in
a way that can be easily and practically conducted by undergraduate students in an organic
chemistry teaching laboratory.
1.2.1 Hazardous Reagent Wittig Reaction Protocols
In some of these early reported experiments, pyrophoric reagents like butyllithium
(Figure 1.2), acutely toxic and flammable reagents like triethylphosphite (Figure 1.3) and
lacrymators, for instance benzyl chloride, are used in order to make an unstable ylide.6,7 One
case in particular reports the use of sodium hydride (Figure 1.4), which appears unsuitable as
a reagent in an undergraduate teaching laboratory because of its pyrophoric properties.8 Even
more startling, in a commonly used textbook today, one Wittig reaction protocol calls for the
use of sodium metal to make the strong base sodium ethoxide.9
Figure 1.2. Wittig reaction using a lachrymator and a pyrophoric base.

4
Figure 1.3. Wittig reaction using a pyrophoric starting material.
Figure 1.4. Wittig reaction using sodium hydride.
In accordance with The Twelve Principles of Green Chemistry, the use of harmful
substances like toxic solvents, pyrophoric reagents and lachrymators should be minimized or
replaced where possible and practical. In addition, for obvious safety concerns, the use of
many of these reagents is wholly unnecessary, and requires laboratories to be equipped with
inert gas support and special waste bins, both factors which can increase the cost and reduce
the practicality of the execution of the experiments in a teaching laboratory. The use of
dangerous substances can require precious lab time for special student training. For example,
in using these harmful reagents, more work is placed on the undergraduate students to rinse
the syringe with concentrated ammonium hydroxide after using a lachrymator and rinse the
syringe with hexanes after using a pyrophoric base. In terms of special training, students have
to be instructed the importance of setting up a reaction vessel that eliminates moisture from
the reaction. Avoiding such reagents will simplify the reaction procedure for students. Lastly,
in terms of energy efficiency, these reactions utilize organic solvents refluxing at elevated
temperatures of around 100 oC. It is greener for teaching laboratories to use protocols carried
out at ambient temperature.

5
1.2.2 Phase Transfer Catalysis
The use of phase transfer catalysis is another common protocol for conducting Wittig
chemistry.10-16 These reactions are also carried out in two steps—first, in-situ preparation of
the ylide followed by the Wittig reaction itself. These reactions take advantage of
lachrymators in the first step in the formation of the phosphonium salt. The phosphonium salt
is the catalyst that brings the hydroxide ion across into the organic phase to remove a proton
from the phosphonium salt to produce the phosphorous ylide. In contrast to the previous
example using a pyrophoric base, a 50% aqueous sodium hydroxide solution is used to make
the ylide, which is a step forward towards a greener and safer approach in organic chemistry
(Figure 1.5).
Figure 1.5. Phase transfer catalysis using a 50% sodium hydroxide solution.
The phase transfer catalysis reactions are also beneficial to conduct in an organic
chemistry teaching laboratory because they are done at ambient temperature. Despite the
ambient temperature, 50% aqueous sodium hydroxide might prove to be dangerous to
students because of the severe burns it can cause upon exposure to skin.
An additional J. Chem. Ed. paper17 illustrates the one phase transfer catalysis Wittig
experiment, using a 50-fold excess of formaldehyde. This protocol would not be good to use
in an undergraduate teaching laboratory since formaldehyde is a well known carcinogen.
1.2.3 “Instant Ylid”
Taking advantage of an “instant ylid” is an example of a protocol that greatly
simplifies the Wittig reaction,18 but it is unfortunately potentially harmful in a teaching

6
laboratory setting for undergraduates because of the use of alkyl triphenylphosphonium
bromide and sodium amide in anhydrous THF (Figure 1.6).
Figure 1.6. “Instant Ylid” Wittig reaction.
The reaction calls for the protective use of a calcium chloride drying agent in addition
to freshly distilled THF. These precautions must be made because the reactive properties of
sodium amide against water. Although this experiment is seemingly simple to conduct, it
should be avoided in the undergraduate organic chemistry teaching lab because of the
potential for a severe accident occurring if water accidently is added to the sodium amide. In
addition to this, the reaction also calls for the use of ligroin, a flammable petroleum fraction,
which is also a suspected human carcinogen. It is also a skin absorbent and should not be
used around any materials that build up static electricity.
1.2.4 Solvent-Free Wittig Reactions
There are also published experiments which describe more safer and greener
strategies in performing Wittig reactions in the undergraduate teaching laboratory.19,20
Examples of these include solvent-free reaction that takes advantage of either a ball-milling
apparatus or a mortar and pestle to replace the use of organic solvents during the reaction.
1.2.4.1 BALL MILLING
The ball-milling protocol (Figure 1.7, Table 1.1) involves innovative Wittig reactions
with potassium carbonate as the base, but unfortunately, these reactions take an extensive
amount of time (7 to 14 hours) to conduct.

7
Figure 1.7. Ball-milling Wittig reactions.
Table 1.1. Ball-Milling Wittig Reactions
Compound Milling Time, hr Yield, % E/Z Ratio
3a 7 85 1.6:1
3b 8 92 2:1
3c 14 70 3.4:1
3d 12 73 -
Reactions conducted in an undergraduate organic teaching laboratory need to be done
in a reasonable time frame so students can be able to conduct necessary workups after the
reaction is complete. In addition, the reaction uses a ball-milling apparatus, a piece of
laboratory equipment not typically found in undergraduate organic teaching laboratories. It
would be a costly expenditure for a chemistry department to purchase ball-milling
apparatus’.
The ball-milling experiments are greener than previously discussed Wittig reaction
experiments because they do not use an organic solvent and they use a safer base compared
to previously mentioned Wittig protocols. This is one of the few undergraduate organic
chemistry teaching Wittig reactions that describes a one-pot reaction with the generation of
an in-situ ylide at ambient temperature (Figure 1.8).
Figure 1.8. Ball-milling one-pot Wittig reaction.

8
As green as this reaction is, the reaction time is quite long (8 hours). Therefore, it
might not be as suitable for undergraduates to conduct as a laboratory experiment. Typical
undergraduate laboratory experiments at San Diego State University are conducted for 3
hours, hence the reaction time needs to be shorter so students can have enough time to set-up
the reaction, conduct the experiment, perform work-up procedures and then isolate the
product.
1.2.4.2 MORTAR AND PESTLE
The use of a mortar and pestle is a wonderful example of a green Wittig reaction
experiment (Figure 1.9).
Figure 1.9. Mortar and pestle Wittig reaction.
This reaction consists of a commercially purchased phosphonium salt, benzyl
triphenylphosphonium chloride, 4-bromobenzaldehyde, and potassium phosphate to make
(E)-and (Z)-1-(4-bromophenyl)-2-phenylethene. This reaction is very green and needs no
solvent system. The reaction is conducted at ambient temperature and there is no use of
highly toxic chemicals. The most attractive point with this experiment is the actual isolation
of the E-isomer product—the initial crude is washed with water, followed by recrystallization
in ethanol. Similar to the ball-milling solvent-free Wittig reaction, this reaction uses a solid
base, potassium phosphate to generate the ylide during the reaction. In addition to the energy
efficiency of this reaction being done at ambient temperature, all is done by mechanical
movements of a student grinding all the reaction components together with a mortar and
pestle. This is an attractive alternative in a teaching laboratory since the mortar and pestle are
common and very inexpensive laboratory equipment.
Despite the green advantages, there are two issues with this reaction. The first issue is
reproducibility becomes a real problem because of the variables involved in physically
grinding the reactants for 20 minutes. Students not only move the pestle within the mortar
differently, but they also differ with how long they can physically grind the reaction. The

9
variable nature of the students’ grinding motion and endurance might provide different yields
for all the reactions done in the lab.
The second issue with this experiment is that it calls for an already premade
phosphonium salt. The goal for a greener Wittig reaction is for it to be one-pot so that in the
future it may be a catalytic reaction with the regeneration of the phosphine used in the
reaction. In addition to this, because the reaction requires no solvent system, the likelihood of
the development of a catalytic Wittig reaction using a solvent-free reaction methodology is
less likely to occur than a Wittig reaction using a solvent system. It is for these reasons that
even though the mortar and pestle reaction is greener than most Wittig reactions, educators
need to utilize a Wittig reaction that uses reagents that can be a one-pot Wittig reaction with
future potential towards being a catalytic Wittig reaction.
1.2.5 Microwave Assisted Experiments
The microwave accelerated Wittig reaction is another alternative way to introduce
greener chemistry into the teaching laboratory.21,22 One procedure utilizes the common
household microwave, and the other reports the use of a microwave reactor specifically
designed for organic reactions.
1.2.5.1 HOUSEHOLD MICROWAVE
The reported common household microwave experiment does not take the use of a
solvent system—it utilizes potassium carbonate to generate the specific ylide, which is an
attractive opportunity to introduce greener chemistry in the teaching laboratory (Figure 1.10).
Figure 1.10. Domestic household microwave Wittig reaction.
In addition to the benefit of not using a solvent system, the reaction takes around 10
minutes to conduct, adding to the amount of time students have to do any workup procedures
and analysis of results. Another advantage of this procedure is that the common household
microwave is relatively inexpensive. A fourth advantage of this reaction is that it uses

10
common lab equipment found in the organic chemistry teaching laboratory, so there are no
costly expenditures for educators to conduct this experiment.
Although the common household microwave is a relatively inexpensive instrument to
use in a teaching laboratory, it might pose as a potential danger and possibly a liability. It
does not have the built-in safety features found in microwave reactors specifically made for
conducting organic reactions; for example, a reinforced steel reaction chamber, a special
door-locking mechanism in the event of a reaction vessel rupture, fiber-optic thermocouple
temperature monitoring and an automatic temperature and pressure shutoff capability. Thus,
due to the possibility of an accidental overheating and explosion of a reaction, the use of a
household type microwave is not recommended in the undergraduate teaching laboratory.
Though they can be used in a teaching laboratory for the quick and routine heating of non-
harmful aqueous solutions, they are unsafe for complicated synthetic procedures.23
1.2.5.2 LABORATORY GRADE MICROWAVE
REACTOR
In regards to the laboratory experiment utilizing a reactor specifically designed for
organic reactions, the Wittig reaction is conducted in three separate phases and the laboratory
grade type microwave reactor is used only in the first phase to produce high yields of the
phosphonium salt in 6.5 minutes (Figure 1.11).
Figure 1.11. Microwave reactor Wittig reaction.
Although the reaction time, safety features of the microwave reactor, and increased
phosphonium salt yield utilizing the reactor make this reaction appealing, the experiment
calls for the use of xylenes, a chemical that permeates the skin and is potentially unsafe to
use in an undergraduate teaching laboratory. Moreover, the microwave reactor is very
expensive, so to make use of this technology with a great number of laboratory students, the
laboratory would require at least three microwave reactors. This could potentially be a very
costly purchase because a typical teaching laboratory microwave instrument costs between

11
twenty and thirty thousand dollars.23 Lastly, the life cycle replacement of the reaction vessels
and probes used in the reactor would also be expensive to maintain.
1.2.6 Stabilized Ylide Wittig Reaction
The stabilized ylide Wittig reaction is a very simple reaction of a premade stabilized
ylide, (carbethoxymethylene)triphenylphosphorane, with benzaldehyde in an organic solvent
of DCM (Figure 1.12).24
Figure 1.12. Stabilized ylide Wittig reaction.
The experiment is well suited for the teaching laboratory and offers several important
advantages. The synthesis, workup and characterization of the product may be accomplished
in a single laboratory session with commonly available laboratory equipment and glassware.
Furthermore, the product is obtained at high yield and excellent purity, and the reaction does
not require special precautions of pre-dried glassware, solvents, and reagents. This reaction is
an improvement to other Wittig reactions like the ones that use pyrophoric bases, but it uses
dichloromethane as the solvent, a known carcinogen. Although the reaction is very simple, it
can be improved upon by using water as the reaction medium. The reaction can also be made
simpler if the one-pot reaction creates the in-situ ylide rather than employing the pre-made
ylide.
1.3 “ON WATER” WITTIG REACTION LABORATORY EXPERIMENT
A water based Wittig reaction is a complement to the previous protocols mentioned in
that it maximize the exposure of greener chemistry and at the same time, maintains a high
level of safety in the organic chemistry teaching laboratory (Figure 1.13).
Figure 1.13. “On water” Wittig reaction experiment.

12
The adaptation of a simple Wittig reaction can be conducted in aqueous sodium
bicarbonate media—an experiment suitable for the organic teaching laboratory. The reaction
is conducted for 2 hours at ambient temperature and is a “one-pot” Wittig reaction of
triphenylphosphine, benzaldehyde and ethyl bromoacetate with aqueous saturated sodium
bicarbonate as the base. This organic reaction allows educators to provide students with a
safer, simpler, less expensive and more effective means to conduct a greener Wittig reaction
in an undergraduate organic chemistry laboratory setting (Appendix A). Water is not
considered a “solvent” in this reaction, but rather a “medium” since the reagents are quite
hydrophobic and appear insoluble. Aqueous diluted sodium bicarbonate is a very weak
suitable base for a teaching laboratory, used here to generate the in-situ stabilized ylide
needed for the Wittig reaction to produce the alkene product. Though ethyl bromoacetate is
classified as a lachrymator, it is used in small quantities within a ventilated setting. Adding to
the simplicity of the experiment, the reaction is done at ambient temperature utilizing all
common laboratory equipment (Table 1.2).
In consideration of cost, water is obviously much less costly than organic solvents.
Water is also environmentally benign and should always be considered as the medium in
future organic reactions. Also, the proposed experiment does not use any pyrophoric reagents
requiring an inert atmosphere. Not only does that make this experiment much safer, but it
reduces the cost to carry the experiment as well. Rather than purchasing the ylide, it is
proposed herein to be generated in-situ using inexpensive starting materials.
Water is a remarkably efficient medium for the Wittig reaction and can be used with a
wide range of stabilized ylides and aldehydes. Even more astonishing is that that reaction rate
for the Wittig reaction is faster in water than in organic solvents even though the reactions
seem heterogeneous in water.25 Wittig reactions in water work best when large hydrophobic
entities like aromatic functional groups are present. Despite the reactants’ poor solubility,
aqueous Wittig reactions have very high yields with high precedence for trans-(E)-
selectivities.26
1.3.1 Experimental Procedure
The “on water” Wittig lab experiment can be done in one 3 hour lab setting and
utilizes common, inexpensive lab equipment. The procedure is very simple and it can be

13
Tab
le 1
.2. E
qu
ipm
ent
for
“On
Wat
er”
Wit
tig
Rea
ctio
n E
xper
imen
t
Mat
eria
l Q
uan
tity
C
AS
(if
ap
pli
cab
le)
Mat
eria
l Q
uan
tity
C
AS
(if
ap
pli
cab
le)
trip
heny
lpho
sphi
ne
0.36
7 g
603-
35-0
se
para
tion
funn
el, 1
25 m
L
1 -
sodi
um b
icar
bona
te (
aq)
5 m
L
144-
55-8
er
lenm
eyer
fla
sk, 2
50 m
L
2 -
ethy
l bro
moa
ceta
te
0.2
mL
10
5-36
-2
erle
nmey
er f
lask
, 100
mL
1
-
benz
alde
hyde
0.
102
mL
10
0-52
-7
Buc
hner
fun
nel
2 -
ethy
l ace
tate
25
mL
14
1-78
-6
vacu
um f
lask
, 250
mL
1
-
sodi
um c
hlor
ide
(aq)
5
mL
76
47-1
4-5
roun
d bo
ttom
fla
sk, 2
50 m
L
1 -
hexa
ne
95 m
L
110-
54-3
gr
adua
ted
cylin
der,
20
mL
1
-
sodi
um b
isul
fite
(aq
) 10
mL
76
31-9
0-5
reag
ent d
igge
r sp
atul
a 1
-
anhy
drou
s so
dium
sul
fate
2
g 77
57-8
2-6
spat
ula
1 -
silic
a 20
0 m
L
7631
-86-
9 sy
ring
e, 1
mL
2
-
20 m
L s
cint
illat
ion
vial
2
- sy
ring
e, 2
0 m
L
1 -
flat
mag
netic
stir
bar
, 10
mm
1
- va
cuum
hos
e 1
-
mag
netic
stir
rer,
20
mm
1
- ex
tra
long
sta
inle
ss s
teel
luer
lo
ck s
yrin
ge n
eedl
es
2 -

14
conducted individually by all students. In general, this experiment can be recommended as an
initial laboratory procedure to be conducted by unfamiliar undergraduate students enrolled in
an organic chemistry laboratory course. Students are to write down observations from
beginning to end during the experiment in order for them to fully understand the reaction
takes place despite the hydrophobic entities present in the water.
To carry out the reaction, students are to add 0.367 g of triphenylphosphine to a 20
mL scintillation vial with a flat magnetic stir bar (Figure 1.14).
Figure 1.14. Experiment set-up.
The use of a flat magnetic stir bar on a flat vial surface ensures all reagents properly
mix in the reaction vessel. Students are to add the remaining reagents in the following order:
5 mL of aqueous sodium bicarbonate, 0.2 mL of ethyl bromoacetate, and 0.102 mL of
benzaldehyde. It is important for students to stir the heterogeneous reaction mixture
vigorously for two hours at room temperature.
Because the reaction time is quite long (2 hours) for a 3 hour laboratory period, it is
necessary for students to also use the two hour reaction time to prepare their workspace to
conduct the following work-up and purification processes. After the reaction stirs for two
hours the students will add 5 mL of ethyl acetate to the reaction vessel. This will separate the
organic phase from the aqueous phase in preparation for the work-up procedures. During the
work-up, students will separate the organic phase from the aqueous phase using a small 125
mL separation funnel. They will then wash the aqueous phase with small portions of ethyl
acetate (2 x 5 mL) to extract any remaining product from the aqueous phase. The organic
phases will then be combined and subsequently washed with 10% aqueous sodium bisulfite

15
(1 x 10 mL). The organic phase is then washed with saturated sodium chloride (1 x 5 mL)
and dried over anhydrous sodium sulfate. After filtering the dried organic phase under
vacuum using a Buchner funnel, the ethyl acetate was removed under vacuum and the crude
organic residue was the left to air dry.
After the residue is air dried, students will then perform the purification of their crude
product. The pure product has an Rf value of 0.5 using 10% ethyl acetate/hexane as an eluent.
Dissolve the residue in minimum 10% ethyl acetate and hexane and purify on a short silica
plug (5 x 2 cm). After removal of the organic solvent the students then determine their yields
(%) of the product after letting it air dry.
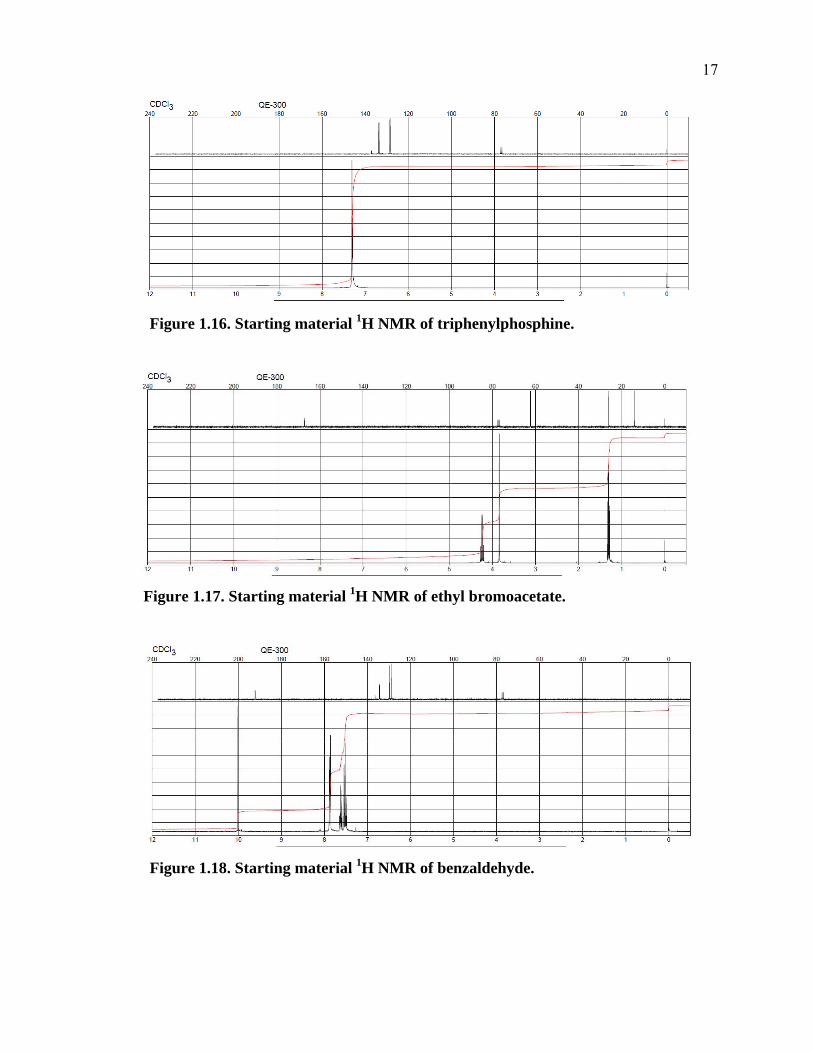
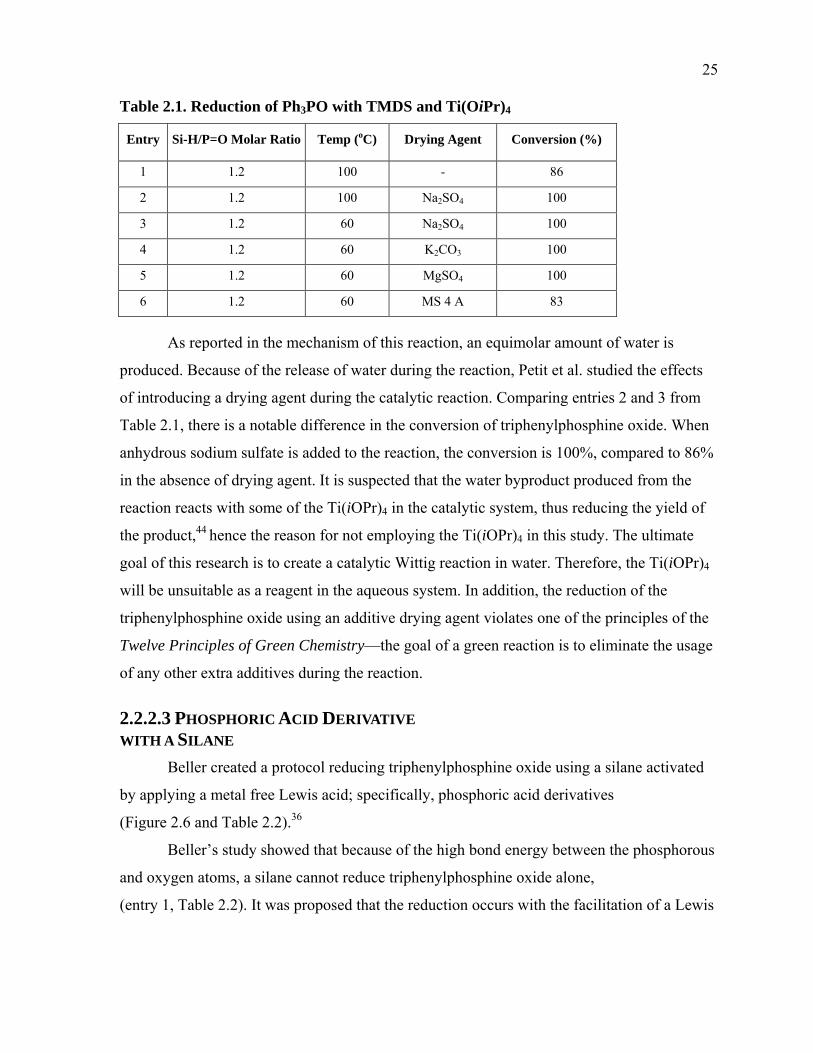
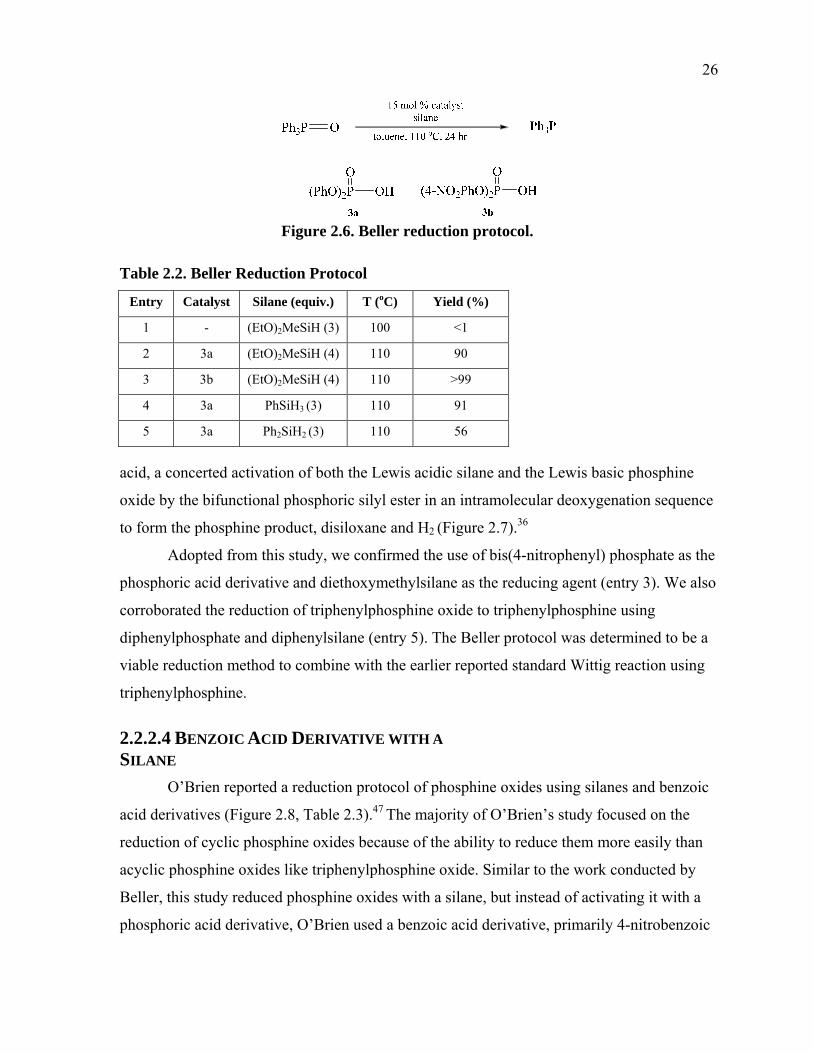
1.3.2 Results and Discussion
The product composition can then be analyzed by 1H NMR spectroscopy
(Figure B.1 in Appendix B). Here the students are able to compare their product 1H NMR
(Figure 1.15) spectrum with 1H NMR (Figures 1.16, 1.17, and 1.18) spectra of the starting
materials. 27-29 Simplicity of the 1H NMRs of the starting materials and the product is an
additional reason this experiment is beneficial to students in an undergraduate organic
chemistry teaching laboratory. The knowledge of the mechanism of the Wittig reaction in
conjunction with the starting material 1H NMRs will help them to conceptually predict what
their product 1H NMRs will look like.
The students will also be able to determine the trans/cis (E/Z) ratio of their alkene
products in their 1H NMR spectrum. In the event of unreacted aldehyde present after the
reaction is complete, it is removed by washing with sodium bisulfite. The triphenylphosphine
oxide formed during the Wittig reaction is removed on a short silica plug. Unreacted
triphenylphosphine remains when diluting the organic phase in 10% ethyl acetate and hexane
because triphenylphosphine is insoluble in hexane. Triphenylphosphine, if remaining, will
make up very little (<1%) of the product mass and has a distinguishable 1H NMR signal just
downfield from the chloroform signal. Unreacted ethyl bromoacetate may make up 6% of the
product mass. A typical student product yield was found to be on average 93% with an E/Z
ratio of circa 92/8.

16
Figure 1.15. Product 1H NMR of (E/Z)-ethyl cinnamate.
17
Figure 1.16. Starting material 1H NMR of triphenylphosphine.
Figure 1.17. Starting material 1H NMR of ethyl bromoacetate.
Figure 1.18. Starting material 1H NMR of benzaldehyde.

18
1.3.3 Hazards of the Experiment
Before students carry out the laboratory experiment, educators are to brief the hazards
of the reaction to the students. Even though the reaction is greener, all reactions still have
their inherent dangers, for instance, ethyl bromoacetate is a lachrymator and should be used
with caution in a ventilated hood. Even though it is employed herein, it is used in a quite
small quantity. Benzaldehyde, and triphenylphosphine are both irritants and should be used
with caution—they are also used in small quantities. Silica gel in its dust form is probably
one of the more hazardous of materials in this experiment. Long time exposure and
inhalation can cause silicosis. This is a form of occupational lung disease caused by
inhalation of crystalline silica dust, and is marked by inflammation and scarring in the form
of nodular lesions in the upper lobes of the lungs.30 Inhalation of silica dust can be mitigated
easily by students working with it in a well ventilated fume hood. Lastly, the prepared
product ethyl cinnamate is a known irritant and is hazardous only if swallowed.
1.4 LABORATORY EXPERIMENT COMPARISON
It is in every educator’s goal to be able to demonstrate valuable lessons through lab
experiments using the most effective and less costly means possible. The “on water” Wittig
reaction is the least expensive reaction to carry out when comparing the starting materials
used from all of the previous lab experiments discussed (Table 1.3). It costs only $0.23 per
student to carry out. For instance the mortar and pestle and phase transfer catalysis reaction,
previously mentioned herein, are fairly close in cost, $0.29 and $0.37 per student,
respectively. The low cost of this experiment is very attractive, especially when
undergraduate laboratory sizes generally range from 24 to 30 students, and not uncommon,
chemistry departments have multiple laboratory sections to conduct organic chemistry
experiments.
Of the green reactions, the “on water” Wittig reaction is the most well rounded when
looking at the evaluation criteria of cost, yield, and time to carry out the reaction. The green
mortar and pestle reaction is just as inexpensive and takes less time to carry out, but it only
has a yield of 70% compared to the 93% yield of the “on water” reaction. The ball-milling
one-pot also has a high yield, but is quite expensive and takes much longer to carry out
compared to the “on water” Wittig reaction. Comparing the stabilized ylide reaction with the

19
Tab
le 1
.3. C
ost,
Yie
ld a
nd
Rea
ctio
n T
ime
Com
par
ison
of
Wit
tig
Rea
ctio
n E
xper
imen
ts
$/g
Am
ount
Use
d (g
) $/
stud
ent
Pro
duct
T
otal
$/s
tude
nt
Yie
ld
Rea
ctio
n T
ime
$0.6
5 0.
2 $0
.13
tran
s-9-
(2-p
heny
leth
enyl
)ant
hrac
ene
$0.3
7 50
-70%
30
min
$2
.06
0.11
5 $0
.24
$19.
74
0.75
$1
4.81
1,
2-m
ethy
lene
diox
y-4-
(2-m
ethy
lpro
peny
l)-b
enze
ne
$14.
85
60%
30
min
$0.3
0 0.
15
$0.0
5
$0.6
5 0.
9 $0
.59
1-(p
heny
l)-2
-(2-
naph
thyl
)eth
ylen
e $0
.59
85%
7
hr
$0.8
4 3
$2.5
2 1-
(4-b
rom
ophe
nyl)
-2-p
heny
leth
ylen
e $2
.52
92%
8
hr
$2.7
6 0.
7 $1
.93
1-(4
-bro
mop
heny
l)-1
-met
hyl-
2-ph
enyl
ethy
lene
$1
.93
70%
14
hr
$0.6
3 0.
9 $0
.57
2-et
heny
lnap
htha
lene
$0
.57
73%
20
hr
$4.1
6 0.
42
$1.7
5
(E)-
and
(Z
)-1-
(4-b
rom
ophe
nyl)
-2-(
2-na
phth
yl)-
ethy
lene
s $2
.60
93%
8
hr
$1.7
1 0.
36
$0.6
2
$0.4
8 0.
5 $0
.24
$0.6
5 0.
2 $0
.13
(E)-
1-(4
-bro
mop
heny
l)-2
-phe
nyle
then
e $0
.29
70%
20
min
$1
.71
0.09
5 $0
.16
$0.7
0 6.
69
$4.6
8 4-
met
hoxy
-1-(
1-pr
open
yl)b
enze
ne
$5.5
5 42
%
11 m
in
$0.4
0 2.
17
$0.8
7
$0.4
8 2.
2 $1
.06
(E)-
and
(Z
)-1,
4-di
phen
yl-1
,3-b
utad
iene
$1
.27
a 91%
1
hr
$0.1
1 1.
65
$0.1
8
$0.0
7 0.
42
$0.0
3
$2.2
7 2
$4.5
4 (E
)-et
hyl c
inna
mat
e $4
.57
b high
yie
ld
30 m
in
$0.0
5 0.
5 $0
.03
$0.4
8 0.
367
$0.1
7
(E)-
and
(Z
)-et
hyl c
inna
mat
e $0
.23
93%
2
hr
$0.0
5 0.
106
$0.0
1
$0.1
8 0.
3 $0
.05
Not
e: a
) 91
% is
onl
y th
e pe
rcen
t yie
ld o
f th
e ph
osph
oniu
m s
alt f
rom
the
mic
row
ave
reac
tor
reac
tion
. N
o yi
eld
of f
inal
pro
duct
pro
vide
d. b
) no
num
eria
l yie
ld is
pro
vide
d, ju
st "
high
yie
ld".

20
“on water” reaction, even though it reports a “high yield”, and only a 1 hour reaction time, it
is too expensive compared to the “on water” Wittig reaction. If an undergraduate program
has 6 lab sections and 21 students per lab section, the stabilized Wittig reaction would cost
$576 compared to the cost of the “on water” Wittig reaction, $29. Not only is there a
significant difference in cost to conduct an experiment where the same lessons can be
achieved and learned, but the “on water” Wittig reaction is significantly safer as well. The
“on water” Wittig reaction substantially complements all of the prior Wittig reactions.
1.5 CONCLUSION
Water is herein illustrated as a very safe and efficient medium for the Wittig
olefination reaction employing an in-situ prepared stabilized ylide and benzaldehyde in the
teaching laboratory. The separation of triphenylphosphine oxide from the alkene product
without using an organic solvent work-up, however, remains a classic problem. Regardless,
this experiment demonstrates that solubility of the reagents and substrates is not of a
paramount nature in the Wittig reaction, even though pronounced hydrophobic entities are
present.
At the end of this laboratory experiment, with the knowledge of previous Wittig
reaction protocols along with the awareness of the “on water" Wittig reaction and the Twelve
Principles of Green Chemistry, organic chemistry students will have a good initial
understanding of how they can better design green chemistry experiments and hopefully
influence industry practices throughout their future careers.

21
CHAPTER 2
THE ATTEMPTED DEVELOPMENT OF AN “ON
WATER” CATALYTIC WITTIG REACTION
2.1 INTRODUCTION
Switching the solvent from an organic solvent to a water media was an exceptional
leap for making the Wittig reaction a greener alternative. The ultimate goal for making the
Wittig reaction greener is for it to take advantage of a catalytic amount of triphenylphosphine
in water media. During the Wittig reaction, a great deal of phosphine oxide by-product is
formed affecting the Wittig reaction to have poor atom economy; once the phosphine oxide is
produced from a stoichiometric Wittig reaction, it remains a useless by-product in the
reaction.31 In accordance with the Twelve Principles of Green Chemistry, a catalytic Wittig
reaction will make it greener because the use of a catalytic amount of one reagent is far
superior than the use of a stoichiometric amount of the same reagent. Also, the atom
economy will improve because the by-product made in the reaction will be used to continue
the reaction. A catalytic Wittig reaction is illustrated in Figure 2.1.32
There are four steps in a catalytic Wittig reaction. The first step is to form the
phosphonium salt with a phosphine and an alkyl halide. The second step is to generate the in-
situ ylide with a base by removing the α-hydrogen from the phosphonium salt. The third step
in the catalytic Wittig reaction is the actual Wittig transformation and the creation of the
olefin and the phosphine oxide by-product. The fourth–and most critical step–in making the
catalytic Wittig reaction occur is the reduction of the phosphine oxide back to the phosphine
employing a suitable reducing agent compatible with rest of the reagents in this reaction.32
This chapter discusses a study conducted to develop an “on-water” catalytic Wittig reaction.
2.2 USE OF TRIPHENYLPHOSPHINE OXIDE
The development of the catalytic Wittig reaction relies on the reduction of the
phosphine oxide by-product to the phosphine. The P=O bond strength of Ph3PO is 544
kJ/mol, and because of its relative stability, one would expect this reduction to be relatively

22
Figure 2.1. Four steps of a catalytic Wittig reaction.
slow in a catalytic Wittig reaction.32 In comparison to the bond strength of P=O, As=O has a
bond strength much less, 389 kJ/mol. The first catalytic Wittig-type reaction was reported
with the use of tributylarsenic as the catalyst and triphenylphosphite as the reducing agent
(Figure 2.2).33
Figure 2.2. Catalytic Wittig reaction with tributylarsenic.
There is also a report of the use of a tellurium oxide compound as a catalyst34 because
of the Te-O bond strength32 of 376 kJ/mol, a bond strength significantly less than the P=O
bond strength (Figure 2.3).

23
Figure 2.3. Catalytic Wittig reaction using a tellurium compound.
Despite the advantage of arsenic and tellurium oxide compounds being easily reduced
compared to the phosphine oxide, it is important to note that a phosphine oxide compound is
environmentally more complimentary to use for a catalytic olefination reaction. The
corresponding arsenic and tellurium oxides are more toxic and obviously less greener. If
catalytic Wittig reactions are to be conducted in industry at a large scale, Wittig reactions
catalyzed by greener phosphine oxides should be more accepted and employed.35
2.2.1 Standard Wittig Reaction Using Ph3P in Organic Solvent
The development of an “on-water” catalytic Wittig reaction started with the
determination of a successful one-pot Wittig reaction in an organic solvent. The results of
this reaction will be used to compare the catalytic Wittig reactions using triphenylphosphine
oxide hereafter. The standard Wittig reaction consisted of o-anisaldehyde,
triphenylphosphine and methyl bromoacetate in toluene refluxing under argon for 24 hours
(Figure 2.4).
Figure 2.4. Standard Wittig using Ph3P in organic solvent.
The reaction was refluxed for 24 hours in toluene. The reaction condition is similar to
the condition reported for where triphenylphosphine oxide is reduced to triphenylphosphine
in toluene.36 A tertiary amine will be a suitable base since they do not provide the
corresponding imines or enamines with aldehydes. The standard reaction protocol provided a
67% yield with an E/Z ratio of 96/4 (Figure B.2 in Appendix B).

24
2.2.2 Reduction Protocols for Ph3PO
In order to create a catalytic Wittig reaction, it will be important to use a reduction
protocol that selectively reduces the phosphine oxide over other reducible functional groups
like double bonds, carbonyls and halogens. To that end, protocols were sought from multiple
sources for the reduction of phosphine oxides in organic solvents and in aqueous media.
2.2.2.1 METAL HYDRIDES
There have been many reported reduction protocols of phosphine oxides using metal
hydrides like lithium aluminum hydride, diisobutylaluminum hydride, and sodium
borohydride.37-42 Lithium aluminum hydride and diisobutylaluminum hydride would not be
considered as reducing agents for the “on water” catalytic Wittig reaction because they are
too pyrophoric. Sodium borohydride, however, can be used in water, but was not considered
in this study because it will reduce the aldehyde function needed for the Wittig reaction,
which is also the case for lithium aluminum hydride and diisobutylaluminum hydride. In
accordance with the Twelve Principles of Green Chemistry, less toxic and less hazardous
compounds should be used to reduce phosphine oxides.
2.2.2.2 HYDROSILANES AND A TITANIUM CATALYST
There are also phosphine oxide reduction protocols utilizing a combination of
titanium tetraisopropoxide (Ti(OiPr)4) and hydrosilanes.43-46 One in particular uses TMDS
and 10 mol % of Ti(OiPr)4 to reduce triphenylphosphine oxide to triphenylphosphine
(Figure 2.5 and Table 2.1).44
Figure 2.5. Reduction of Ph3PO with TMDS and Ti(OiPr)4.
25
Table 2.1. Reduction of Ph3PO with TMDS and Ti(OiPr)4
Entry Si-H/P=O Molar Ratio Temp (oC) Drying Agent Conversion (%)
1 1.2 100 - 86
2 1.2 100 Na2SO4 100
3 1.2 60 Na2SO4 100
4 1.2 60 K2CO3 100
5 1.2 60 MgSO4 100
6 1.2 60 MS 4 A 83
As reported in the mechanism of this reaction, an equimolar amount of water is
produced. Because of the release of water during the reaction, Petit et al. studied the effects
of introducing a drying agent during the catalytic reaction. Comparing entries 2 and 3 from
Table 2.1, there is a notable difference in the conversion of triphenylphosphine oxide. When
anhydrous sodium sulfate is added to the reaction, the conversion is 100%, compared to 86%
in the absence of drying agent. It is suspected that the water byproduct produced from the
reaction reacts with some of the Ti(iOPr)4 in the catalytic system, thus reducing the yield of
the product,44 hence the reason for not employing the Ti(iOPr)4 in this study. The ultimate
goal of this research is to create a catalytic Wittig reaction in water. Therefore, the Ti(iOPr)4
will be unsuitable as a reagent in the aqueous system. In addition, the reduction of the
triphenylphosphine oxide using an additive drying agent violates one of the principles of the
Twelve Principles of Green Chemistry—the goal of a green reaction is to eliminate the usage
of any other extra additives during the reaction.
2.2.2.3 PHOSPHORIC ACID DERIVATIVE
WITH A SILANE
Beller created a protocol reducing triphenylphosphine oxide using a silane activated
by applying a metal free Lewis acid; specifically, phosphoric acid derivatives
(Figure 2.6 and Table 2.2).36
Beller’s study showed that because of the high bond energy between the phosphorous
and oxygen atoms, a silane cannot reduce triphenylphosphine oxide alone,
(entry 1, Table 2.2). It was proposed that the reduction occurs with the facilitation of a Lewis
26
Figure 2.6. Beller reduction protocol.
Table 2.2. Beller Reduction Protocol
Entry Catalyst Silane (equiv.) T (oC) Yield (%)
1 - (EtO)2MeSiH (3) 100 <1
2 3a (EtO)2MeSiH (4) 110 90
3 3b (EtO)2MeSiH (4) 110 >99
4 3a PhSiH3 (3) 110 91
5 3a Ph2SiH2 (3) 110 56
acid, a concerted activation of both the Lewis acidic silane and the Lewis basic phosphine
oxide by the bifunctional phosphoric silyl ester in an intramolecular deoxygenation sequence
to form the phosphine product, disiloxane and H2 (Figure 2.7).36
Adopted from this study, we confirmed the use of bis(4-nitrophenyl) phosphate as the
phosphoric acid derivative and diethoxymethylsilane as the reducing agent (entry 3). We also
corroborated the reduction of triphenylphosphine oxide to triphenylphosphine using
diphenylphosphate and diphenylsilane (entry 5). The Beller protocol was determined to be a
viable reduction method to combine with the earlier reported standard Wittig reaction using
triphenylphosphine.
2.2.2.4 BENZOIC ACID DERIVATIVE WITH A
SILANE
O’Brien reported a reduction protocol of phosphine oxides using silanes and benzoic
acid derivatives (Figure 2.8, Table 2.3).47 The majority of O’Brien’s study focused on the
reduction of cyclic phosphine oxides because of the ability to reduce them more easily than
acyclic phosphine oxides like triphenylphosphine oxide. Similar to the work conducted by
Beller, this study reduced phosphine oxides with a silane, but instead of activating it with a
phosphoric acid derivative, O’Brien used a benzoic acid derivative, primarily 4-nitrobenzoic
27
Figure 2.7. Proposed Beller reduction mechanism of Ph3PO.
Figure 2.8. O’Brien reduction protocol
acid. In response to Beller’s reduction protocol of phosphine oxides using phosphoric acid
derivatives,36 O’Brien conducted two catalytic Wittig reactions to prove 4-nitrobenzoic acid
is more effective in activating phosphine oxides than bis(4-nitrophenyl)phosphate. The
catalytic Wittig reaction using 4-nitrobenzoic acid achieved a 76% yield compared to the
34% yield of the catalytic Wittig reaction using bis(4-nitrophenyl)phosphate.
28
Table 2.3. O’Brien Reduction Protocol
Entry R3PO Silane Solvent R1 T (oC) t (min) Yield (%)
1 1 PhSiH3 THF H RT 60 94
2 1 Ph2SiH2 THF NO2 RT 60 61
3 1 PhSiH3 THF NO2 RT 2 74
4 2 PhSiH3 Toluene NO2 100 10 50
5 2 4-CF3PhSiH3 Toluene NO2 100 10 81
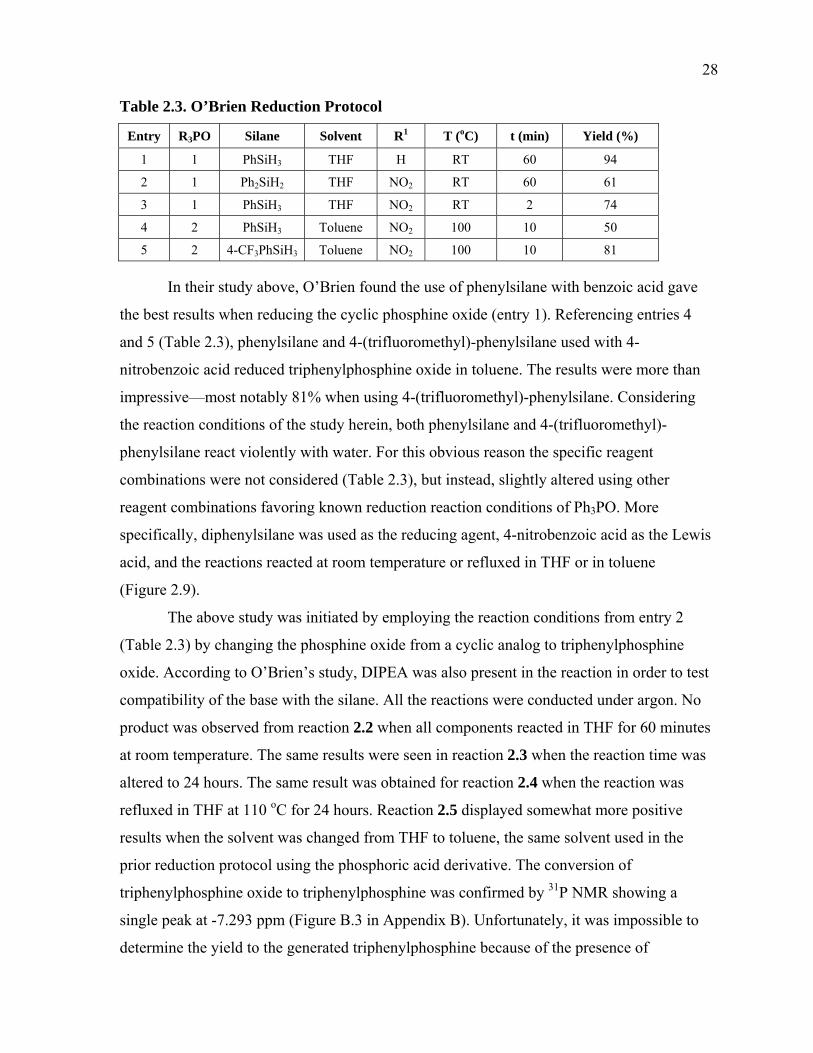
In their study above, O’Brien found the use of phenylsilane with benzoic acid gave
the best results when reducing the cyclic phosphine oxide (entry 1). Referencing entries 4
and 5 (Table 2.3), phenylsilane and 4-(trifluoromethyl)-phenylsilane used with 4-
nitrobenzoic acid reduced triphenylphosphine oxide in toluene. The results were more than
impressive—most notably 81% when using 4-(trifluoromethyl)-phenylsilane. Considering
the reaction conditions of the study herein, both phenylsilane and 4-(trifluoromethyl)-
phenylsilane react violently with water. For this obvious reason the specific reagent
combinations were not considered (Table 2.3), but instead, slightly altered using other
reagent combinations favoring known reduction reaction conditions of Ph3PO. More
specifically, diphenylsilane was used as the reducing agent, 4-nitrobenzoic acid as the Lewis
acid, and the reactions reacted at room temperature or refluxed in THF or in toluene
(Figure 2.9).
The above study was initiated by employing the reaction conditions from entry 2
(Table 2.3) by changing the phosphine oxide from a cyclic analog to triphenylphosphine
oxide. According to O’Brien’s study, DIPEA was also present in the reaction in order to test
compatibility of the base with the silane. All the reactions were conducted under argon. No
product was observed from reaction 2.2 when all components reacted in THF for 60 minutes
at room temperature. The same results were seen in reaction 2.3 when the reaction time was
altered to 24 hours. The same result was obtained for reaction 2.4 when the reaction was
refluxed in THF at 110 oC for 24 hours. Reaction 2.5 displayed somewhat more positive
results when the solvent was changed from THF to toluene, the same solvent used in the
prior reduction protocol using the phosphoric acid derivative. The conversion of
triphenylphosphine oxide to triphenylphosphine was confirmed by 31P NMR showing a
single peak at -7.293 ppm (Figure B.3 in Appendix B). Unfortunately, it was impossible to
determine the yield to the generated triphenylphosphine because of the presence of

29
Ph3P O Ph3POH
OO2N
Ph3P O Ph3POH
OO2N
Ph3P O Ph3POH
OO2N
Ph3P O Ph3POH
OO2N
THF, 110oC, 24 hr
1 equiv.14 equiv. of iPr2NEt
14 equiv. Ph2SiH2
No product formed
1 equiv.
toluene, 110oC, 24 hr
1 equiv.14 equiv. of iPr2NEt
14 equiv. Ph2SiH2
Little product formed
1 equiv.
THF, RT, 24 hr
1 equiv.14 equiv. of iPr2NEt
14 equiv. Ph2SiH2
No product formed
1 equiv.
THF, RT, 60 min
1 equiv.14 equiv. of iPr2NEt
14 equiv. Ph2SiH2
No product formed
1 equiv.
a.
b.
c.
d.
2.2
2.3
2.4
2.5
Figure 2.9. Reduction study of Ph3PO using Ph2SiH2 and 4-nitrobenzoic acid.
by-products in the crude, possibly the presence of hydroxydiphenylsilanes seen in the 1H
NMR (Figure B.4 in Appendix B) spectrum. Moreover, the crude mass was too small and the
triphenylphosphine present would have been little compared to the expected yield.
2.2.3 Work Towards a Catalytic Wittig Reaction in Organic Solvent
This phase of the research sought to develop a catalytic Wittig reaction using Ph3PO
in organic solvents by combining the standard Wittig reaction and the Lewis acid and silane
reduction protocols.
2.2.3.1 WITTIG REACTION AND BELLER
REDUCTION PROTOCOL
This portion of the research started out to test the compatibility of the standard Wittig
reaction using triphenylphosphine and the phosphoric acid derivative protocol in toluene
(Figure 2.10).

30
Figure 2.10. Compatibility test of standard Wittig reaction and Beller reduction protocol.
Supported by the results of reaction 2.6, the two protocols are possibly compatible
with each other because a 66% yield of product was attained (Figure B.5 in Appendix B), a
yield close to the 67% yield of the standard Witting reaction. Following this lead, a similar
reaction was conducted, but instead of using triphenylphosphine, triphenylphosphine oxide
was used as a proof of concept to prove the reduction of triphenylphosphine oxide worked if
now the product was derived from the generation of an in-situ ylide (Figure 2.11).
Figure 2.11. Catalytic Wittig reaction using Ph3PO in toluene.
Unfortunately, the product yield of reaction 2.7 was 6% (Figure B.6 in Appendix B).
This result concludes the reduction of the phosphine oxide occurred (or at least partly
occurred) because of the generation of some product, but it is unknown the exact reason of
the observed low yield. It is difficult to ascertain a specific reason because of the many
reagents involved in this one-pot catalytic Wittig reaction. Despite this result, the study was
continued to find a one-pot catalytic Wittig reaction in toluene by changing reaction
conditions. This was done by first replacing the bis(4-nitrophenyl) phosphate to
diphenylphosphate in reaction 2.8 (Figure 2.12), but no product formation was observed.
There was only aldehyde present according to the 1H NMR spectrum
(Figure B.7 in Appendix B).

31
Figure 2.12. Catalytic Wittig using diphenylsilane and diphenylphosphate.
In the original reduction protocol, 15 mol % of the phosphoric acid derivative was
employed. The next change in the portion of this study was to increase the amount of
phosphoric acid derivative used to 50 mol % (Figure 2.13).
Figure 2.13. Catalytic Wittig reaction using 50 mol % of diphenylphosphate.
The increase of the phosphoric acid derivative did not yield any product, only the
aldehyde was present in the 1H NMR (Figure B.8 in Appendix B). Attention was then turned
to the use of triethylamine as the base and thus a reaction was conducted in the absence of
triethylamine (Figure 2.14). This was conducted in order to determine if the base was needed
in the first place and secondly if it was negatively interfering with the reaction.
Figure 2.14. Catalytic Wittig reaction without the use of NEt3.
In the absence of triethylamine of reaction 2.10 there was mostly unreacted aldehyde
according to the 1H NMR spectrum (Figure B.9 in Appendix B). The result obtained from the
reaction in the absence of base seems similar to the result of the reaction in the presence of
triethylamine. Thus, it appears that the presence of a tertiary amine is unneccesary.
Interesting is also which base in reaction 2.14 is responsible for creating the crucial ylide to

32
be formed in order for the Wittig reaction to occur. The assumption is that not only does the
diethoxymethylsilane take part in the reduction of triphenylphosphine oxide, but it also
appears to remove the acidic proton from the phosphonium salt to produce the in-situ ylide.
With this assumption in mind, the standard one-pot Wittig reaction was conducted with
triphenylphosphine, but instead of using triethylamine as the base, the impact of
diethoxymethylsilane was studied (Figure 2.15).
Figure 2.15. Standard Wittig reaction with Ph3P and (EtO)2MeSiH.
Even though only 16% of product was produced from this experiment, it is still
enough generated product to assume that diethoxymethylsilane can play a role in removing
the acidic proton from the phosphonium salt in order to produce the in-situ ylide. There was
mostly unreacted aldehyde. The absence of any methyl bromoacetate according to the 1H
NMR spectrum shows that most likely the ester has reacted with the silane
(Figure B.10 in Appendix B).
2.2.3.2 O’BRIEN CATALYTIC WITTIG
REACTION IN ORGANIC SOLVENTS
Following the start of this research, O’Brien published two articles illustrating his
accomplishment of a one-pot catalytic Wittig reaction in organic solvents
(Figure 2.16, Table 2.4).47,48
O’Brien reported successful catalytic Wittig reactions employing both cyclic and
acyclic phosphine oxides in organic solvents. In this study, more focus was placed on
developing a catalytic Wittig reaction in water media using triphenylphosphine oxide.
Although O’Brien’s group successfully accomplished a catalytic Wittig reaction using
triphenylphosphine oxide as their catalyst, they reported the successful use with the 4-
(trifluoromethyl)-phenylsilane as the alternative reducing agent. However, this specific silane
reacts violently with water; hence it is not useful for a one-pot catalytic Wittig reaction

33
Figure 2.16. O’Brien’s catalytic Wittig reactions in organic solvents.
Table 2.4. O’Brien’s Catalytic Wittig Reactions in Organic Solvents
Entry R3PO Silane Solvent T (oC) Conversion % E/Z
1 1 PhSiH3 THF RT 91 75/25
2 2 4-(trifluoromethyl)-phenylsilane Toluene 100 89 90/10
3 3 Ph3SiH Toluene 100 trace n.d.
4 3 Ph2SiH2 Toluene 100 75 >95/5
5 3 PhSiH3 Toluene 100 46 >95/5
6 3 (MeO)3SiH Toluene 100 61 70/30
in water, which is also the case for phenylsilane. Since O’Brien’s group did not have success
with triphenylsilane, it was also not considered as a silane suitable for aqueous Wittig
chemistry. Although O’Brien was successful in employing trimethoxysilane, this silane was
not considered for this study because of its hazardous nature. In addition, it could very likely
lose its hydride in water like diethoxymethylsilane. Despite O’Brien’s successes for
generating a catalytic Wittig reaction using triphenylphosphine oxide in organic solvents, a
study was still conducted to develop an “on water” catalytic Wittig reaction.
2.2.3.3 CATALYTIC WITTIG REACTION
ATTEMPT USING O’BRIEN’S PROTOCOL
WITH DIPHENYLSILANE AND SODIUM
CARBONATE
Due to the success reported by O’Brien and the goal to find a successful catalytic
one-pot reaction in organic solvent with the potential to be transferred to water, a catalytic

34
one-pot Wittig reaction in toluene was conducted using diphenylsilane, sodium carbonate and
triphenylphosphine oxide. It was determined from O’Brien’s work that sodium carbonate was
an acceptable alternative to DIPEA. This tertiary amine was replaced with sodium carbonate
in this study because of its better solubility in water. Rather than employing o-anisaldehyde
as the standard aldehyde, benzaldehyde was used since it was the preferred aldehyde used in
the work published by O’Brien (Figure 2.17).
Figure 2.17. Catalytic Wittig reaction with Ph3PO in toluene.
O’Brien’s group did most of their work with a cyclic phospholane oxide because of
the ease of reduction of the cyclic phospholane oxide compared to the reduction of
triphenylphosphine oxide, probably due to relieving of the ring strain of the cyclic
phospholane oxides.35 A key catalytic Wittig reaction criteria for the O’Brien group was that
it would be done in 24 hours. In order for this to occur, the pre-catalyst loading of 10 mol %
would have to cycle every 2.4 hours. They felt that triphenylphosphine oxide would not
fulfill this requirement. In light of this information, reaction 2.12 had to be conducted for 96
hours in order for the triphenylphosphine oxide catalyst to be reduced.
The 96 hour reaction time did not matter for the reduction of the triphenylphosphine
oxide–only trace amounts of the product were formed. A greater amount of unreacted
benzaldehyde remained in the reaction crude compared to the product from the results of the 1H NMR (Figure B.11 in Appendix B). The results of this reaction leave us very uncertain as
to why this reaction is not working. The stability of the triphenylphosphine oxide is the most
logical reason as to why this reaction is not functioning catalytically and producing a high
yield of the product.
2.2.4 “On Water” Reduction Protocols for Ph3PO
Reduction protocols “on water” media have been reported, but most are for
hydrogenation, dehalogenation and reduction of carbonyl compounds with water soluble

35
metal catalysts that would not be compatible with the rest of the functional groups from the
catalytic Wittig reaction components.49 Therefore, this study continued to use silanes as the
reducing agent. Before “on water” reduction experiments of triphenylphosphine oxide were
conducted, experiments were first conducted to determine which silane compounds did not
readily lose their hydrides when mixed with water at room temperature for an hour
(Table 2.5).
Table 2.5. Silane in the Presence of Water
Silane In Presence of Water
diphenylsilane Retains hydride
polymethylhydrosiloxane Retains hydride
tetramethyldisiloxane Retains hydride
triisopropylsilane Retains hydride
diethoxymethylsilane Loses hydride
trichlorosilane Reacts violently with water
phenylsilane Reacts violently with water
4-(trifluoromethyl)-phenylsilane Reacts violently with water
Diphenylsilane, PMHS, TMDS, and triisopropylsilane all retain their hydride(s) and
overall structure in the presence of water. Surprisingly, diethoxymethylsilane, the silane with
the best reduction results of triphenylphosphine oxide, does not retain its hydride when
mixed with water. O’Brien reported catalytic Wittig reactions using phenylsilane and 4-
(trifluoromethyl)-phenylsilane, but those silanes react violently with water. Likewise,
trichlorosilane, a silane used by Greaney50 in a catalytic one-pot silyl-Reformatsky
olefination reaction using triphenylphosphine oxide, and triethylamine in a DCM solvent,
also reacts violently in water (Figure 2.18). Therefore these reagents cannot be used in this
study to achieve an aqueous one-pot catalytic Wittig reaction.
Figure 2.18. Greaney’s one-pot silyl-Reformatsky olefination.

36
There is another reduction protocol reported for the reduction of acyclic phosphine
oxides to phosphines using hexachlorodisilane in chloroform at room temperature or
refluxing in benzene.51 Unfortunately, similar to trichlorosilane, the hexachlorodisilane also
reacts violently with water and cannot be employed in this aqueous study.
Following the experiments to determine what silane compounds can be used in water,
”on water” reduction experiments were conducted utilizing the known reduction protocols to
reduce triphenylphosphine oxide in organic solvents. To expedite this process, a laboratory
grade microwave was used to mimic the reaction conditions needed to carry out the
following protocols. To ensure this would work, triphenylphosphine oxide was reduced using
diethoxymethylsilane, and bis(4-nitrophenyl)phosphoric acid in toluene. The reaction yield
was greater than 99%. Multiple “on water” reductions of triphenylphosphine oxide were
attempted using different combinations of silanes and either a phosphoric acid derivative or a
benzoic acid derivative (Figure 2.19 and Table 2.6).
Figure 2.19. Microwave reduction of Ph3PO in water.
Table 2.6. Microwave Reduction of Ph3PO in Water
Silane
Phosphoric or Carboxylic Acid Derivative
Diphenylphosphate bis(4-nitrophenyl) phosphate 4-nitrobenzoic acid
diphenylsilane No Reduction No Reduction No Reduction
triisopropylsilane No Reduction No Reduction No Reduction
PMHS No Reduction - -
TMDS No Reduction - -
No reaction combinations of any silane and Lewis acid resulted in the reduction of
triphenylphosphine oxide. This was confirmed by 1H NMR and especially by 31P NMR
spectroscopy. Despite these results, an effort was made to make the triphenylphosphine oxide
more soluble in water in order to have better reaction results. This was achieved by solvating

37
the triphenylphosphine oxide in DMSO before introducing it to the reaction vessel filled with
water, silane and Lewis acid, in this case diphenylphosphate (Figure 2.20).
Figure 2.20. Ph3PO reduction in 15% DMSO and water.
In reaction 2.21, solvating the triphenylphosphine oxide in 15% DMSO in water
proved to help produce triphenylphosphine, verified by 31P NMR and 1H NMR
(Figure B.12 and B.13 in Appendix B). The triphenylphosphine produced was not able to be
isolated in order to accurately determine the yield for the reaction.
2.2.5 “On Water” Catalytic Wittig Reaction Attempt Using Ph3PO
Due to the earlier mentioned reports of O’Brien developing a catalytic Wittig reaction
in organic solvents, this study continued to work towards an “on water” catalytic Wittig
reaction. Sodium carbonate was first used as the base because it is highly soluble in water
(Figure 2.21). Also, O’Brien estimated the pKa value of the ylide-forming proton of the
phosphonium salt to be approximately 9, hence the pKa of the conjugate acid of the base
needs to be between 9 and 12.48 The pKa of the conjugate acid of sodium carbonate is 10.33.
DMSO was used to better solvate the triphenylphosphine oxide so it is more dispersed
throughout the water media.
H
O
BrO
O
1.5 eq. Na2CO3(aq), 100 oC, 96 hr
5% DMSO
O
O
1 equiv. 1.2 equiv.
O=PPh3 - 10 mol %Ph2SiH2 - 1.1 equiv.
No product formed
2.22
Figure 2.21. Attempted “on water” catalytic Wittig reaction with DMSO.
Unlike the catalytic Wittig reaction 2.12 in toluene, no product was formed in
reaction 2.22, which was confirmed from the 1H NMR spectrum obtained of the crude
product mixture (Figure B.14 in Appendix B); the aldehyde was present and appears not to
react with the other reagents present. However, quite puzzling, there was no methyl

38
bromoacetate remaining according to the 1H NMR spectrum. Due to the uncertainty of the
root cause of the catalytic Wittig reaction not occurring when employing triphenylphosphine
oxide, the focus of this study was turned to the use of 3-methyl-1-phenylphospholane as the
active phosphine with hopes of removing the hardships of reducing the phosphine oxide
during the catalytic process.
2.3 USE OF 3-METHYL-1-PHENYLPHOSPHOLANE OXIDE
Due to the unsuccessful results from the use of triphenylphosphine oxide as the
catalyst in this study and the success reported by O’Brien when developing a catalytic Wittig
reaction in organic solvents with 3-methyl-1-phenylphospholane oxide, this study was
continued with 3-methyl-1-phenylphospholane oxide as the catalyst. O’Brien reported the
cyclic phospholane oxides in his experiments because of the ease of reduction compared to
the reduction of triphenylphosphine oxide. This is most likely due to the need of relieving the
ring strain of the cyclic phospholane oxide.35 Decreasing the hardship of reducing the
phosphine oxide will further aid in the discovery of an “on water” catalytic Wittig reaction.
In addition to this change, instead of using o-anisaldehyde as the aldehyde, this study
changed to the use of benzaldehyde to best mimic the successful work reported by O’Brien.
Below is yet another successful catalytic Wittig reaction reported by O’Brien. In this
example they used the cyclic phospholane oxide, diphenylsilane, benzaldehyde, methyl
bromoacetate, and DIPEA as the base (Figure 2.22).48
Figure 2.22. O’Brien et al. catalytic Wittig reaction in toluene.
In an attempt to recreate and verify this experiment, only a 38% yield of product was
obtained. This is much less than the yield reported by O’Brien (80%). A similar reaction was
conducted, but stoichiometric amounts of the silane and the base were increased as well as
the catalytic amount of the cyclic phospholane oxide (Figure 2.23).

39
Figure 2.23. Optimized O’Brien catalytic Wittig reaction in toluene.
The yield in reaction 2.23 was significantly greater (Figure B.15 in Appendix B)
compared to the yield of the reaction to verify O’Brien catalytic protocol. The protocol used
in Figure 2.23 was used to conduct similar “on water” experiments; though, before
conducting “on water” catalytic experiments, this study first set out to achieve a standard “on
water” Wittig reaction using 3-methyl-1-phenylphospholane and reduce 3-methyl-1-
phenylphospholane oxide “on water”.
2.3.1 Standard “On Water” Wittig Reaction Using 3-methyl-1-phenylphospholane
The standard Wittig reaction in aqueous media consisted of benzaldehyde, 3-methyl-
1-phenylphospholane, methyl bromoacetate and DIPEA refluxing “on water” for 24 hours
(Figure 2.24).
Figure 2.24. “On water” Wittig reaction using 3-methyl-1-phenylphospholane.
Reaction 2.24 provided a 54% product yield with an E/Z ratio of 70/30
(Figure B.16 in Appendix B).
2.3.2 “On Water” Reduction of 3-methyl-1-phenylphospholane oxide with Ph2SiH2
3-Methyl-1-phenylphospholane oxide was reduced “on water” using diphenylsilane
as the reducing agent and refluxing at 100 oC for 24 hours (Figure 2.25).

40
Figure 2.25. “On water” reduction of 3-methyl-1-phenylphospholane oxide.
Reaction 2.25 provided a product yield of 93%. The formation of 3-methyl-1-
phenylphospholane was verified using 31P NMR. The cyclic phosphine oxide has a 95:5
mixture of diastereomers at 59.80 ppm and 59.43 ppm (Figure B.17 in Appendix B) and the
product 31P NMR of reaction 2.25 showed peaks at -13.44 ppm and -13.88 ppm
(Figure B.18 in Appendix B). The reduction of the cyclic phosphine oxide “on water” and the
“on water” Wittig reaction using the cyclic phospholane were great steps forward in
developing an “on water” catalytic Wittig reaction.
2.3.3 “On Water” Catalytic Wittig Reaction Attempts Using 3-methyl-1-phenylphospholane oxide
Due to the promising results from reactions 2.23, 2.24 and 2.25, it was then decided
to combine the “on water” one-pot Wittig reaction protocol using 3-methyl-1-
phenylphospholane and the “on water” reduction protocol (Figure 2.26).
Figure 2.26. Catalytic “on-water” Wittig with 3-methyl-1-phenylphospholane oxide.
The 1H NMR spectrum of this crude reaction showed no product, mostly aldehyde
(Figure B.19 in Appendix B). It is quite surprising that this reaction did not show any product
formation because of the success conducting the same reaction in toluene with 68% yield.
This information gives question to whether or not the water plays a role in hampering this
reaction through a side reaction with another component of the Wittig reaction, like the
methyl bromoacetate.

41
In light of this result, other bases were used with this protocol (Figure 2.27). In
O’Brien’s work,48 they describe the use of sodium carbonate as the base comparable to the
use of DIPEA. They switched from sodium carbonate to DIPEA because it was more soluble
in organic solvents than sodium carbonate. Similar experiments to reaction 2.26 were
conducted, but with the use of sodium carbonate or sodium bicarbonate as the bases. These
two bases are ideal to use to develop an “on water” catalytic Wittig reaction because they are
very green and non-toxic bases.
Figure 2.27. Attempted “on water” catalytic Wittig experiments with Na2CO3 and NaHCO3.
The crude 1H NMR spectra of both reactions, 2.27 and 2.28, showed little olefin
peaks for the product compared to the chemical shift peaks of impurities and the aldehyde
(Figures B.20 and B.21 in Appendix B). The products from both reactions were too small to
be isolated. The purification however was quite difficult to conduct due to the white solid by-
product formed. These experiments were repeated, but in the presence of tertiary nitrogen
bases: DMAP, triethylamine and N,N-dimethylaniline (Figure 2.28). The use of sodium
carbonate and sodium bicarbonate are much greener options compared to the use of DMAP,
triethylamine and N,N-dimethylaniline, but isolating the product proved to be troublesome.
In reaction 2.29, no product was formed when DMAP was used
(Figure B.22 in Appendix B), the same with reaction 2.30 with triethylamine as the base
(Figure B.23 in Appendix B), but there was a less than 1% product yield in reaction 2.31
when N,N-dimethylaniline was used as the base (Figure B.24 in Appendix B). The reaction
work-up and isolation was much easier for reaction 2.31 compared to the work-up and

42
Figure 2.28. Attempted “on water” catalytic Wittig reactions with tertiary nitrogen bases.
isolation attempts when sodium carbonate and sodium bicarbonate were used as the bases.
After achieving a product yield when using N,N-dimethylaniline as the base, optimization
attempts were conducted by changing different variables (Figure 2.29).
Figure 2.29. Initial optimization attempts of reaction 2.31.
Unfortunately, the reactions to optimize the less than 1% yield of reaction 2.31 were
not successful in producing better product yields. No product was produced when a greater
equivalent of methyl bromoacetate was used in reaction 2.32. Reaction 2.33 also had a less

43
than 1% yield of product. Evident in all of the 1H NMR spectra for these reactions is the fact
that there is no methyl bromoacetate left in the reactions (Figures B.25 and B.26 in
Appendix B). Because of this, two more experiments were conducted where the equivalents
of methyl bromoacetate were drastically increased to see if the same results can be achieved
(Figure 2.30).
PPh O
H
O
BrO
O
1 equiv. 3 equiv.
+ +
20 mol %
O
O3 equiv. N,N-dimethylaniline3 equiv. Ph2SiH2
PPh O
H
O
BrO
O
1 equiv. 5 equiv.
+ +
20 mol %
O
O
< 1 % product yield
3 equiv. N,N-dimethylaniline3 equiv. Ph2SiH2
2.34
2.35
3 mL H2O, 100 oC, 24 hr
3 mL H2O, 100 oC, 24 hr
< 1 % product yield
Figure 2.30. Reaction 2.31 with greater equivalents of methyl bromoacetate.
Both reactions 2.34 and 2.35 had less than 1% product yield. Though again, very little
product was formed, aldehyde remained and there was no methyl bromoacetate left
remaining according to the crude reaction NMR spectra (Figures B.27 and B.28 in
Appendix B). It is believed methyl bromoacetate must be consumed via a side reaction and
therefore, is not available long enough to be able to form the in-situ ylides with the reduced
cyclic phospholane oxide.
2.4 FUTURE WORK FOR THE DEVELOPMENT OF AN “ON
WATER” CATALYTIC WITTIG REACTION
Future work to develop an “on water” catalytic Wittig reaction should continue with
the use of 3-methyl-1-phenylphospholane oxide as the catalyst because of the reported
successes of an “on water” Wittig reaction with 3-methyl-1-phenylphospholane and an “on
water” reduction of 3-methyl-1-phenylphospholane oxide.
Due to the possibility of methyl bromoacetate undergoing side reactions quickly and
not being present for further Wittig reaction cycles in the previously attempted “on water”
catalytic Wittig reactions, future work can be done to conduct experiments that incrementally

44
add the methyl bromoacetate to the reaction vessel over the 24 hour reaction period.
Figure 2.31 is a possible side reaction where N,N-dimethylaniline and water react with
methyl bromoacetate and convert it to methyl hydroxyacetate, thus removing methyl
bromoacetate from reacting with more cycles of the reduced cyclic phospholane oxide.
Figure 2.31. Side reaction of methyl bromoacetate with base and water.
Future work can be done using a different α-halo ester like methyl chloroacetate.
Chlorine is a more electronegative atom compared to bromine, so it is more strongly bonded
to the α-carbon of the α-halo ester. Also, a chlorine atom is smaller than a bromine atom, so
its bond overlap with the α-carbon of the methyl acetate will be stronger. Due to these two
physical properties of the chlorine atom, chlorine is a worse leaving group compared to
bromine and the methyl chloroacetate would possibly not undergo side reactions before
reacting with the phosphine to make the phosphonium salt. Along with the same idea of
changing the bromine atom to a chlorine atom of the α-halo ester, a tosylate leaving group
can be used instead of a halide. The steric bulk of that group might help deter side reactions
from occurring.
In organic non-polar solvents like toluene, O’Brien was successful in achieving a
catalytic Wittig reaction using methyl bromoacetate in the presence of diphenylsilane. Due to
the success of these reactions, methyl bromoacetate was not reduced by the diphenylsilane.
Although there are reports of esters undergoing reduction by transition metal catalysts and
hydrosilanes like diphenylsilane,52 the methyl bromoacetate in O’Brien’s work did not go
through a reduction side reaction; their product yields are too high to suggest a side reaction
occurred. This is not the case for the attempted “on water” catalytic Wittig reaction
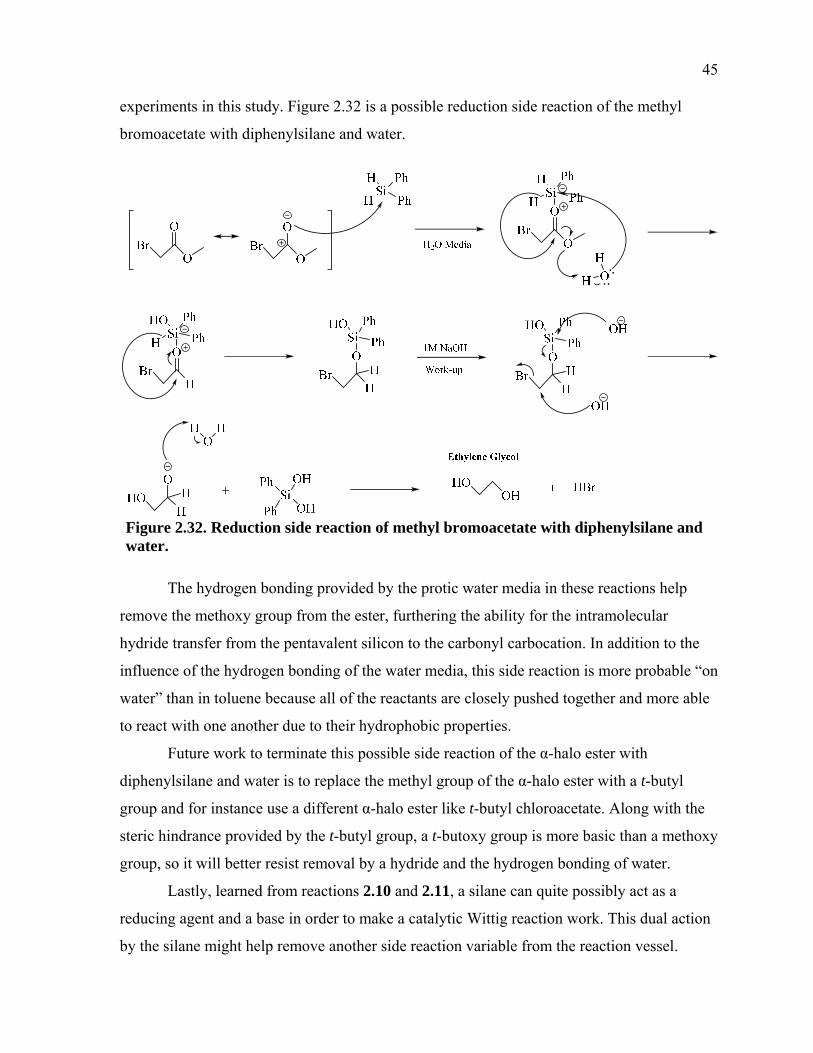
45
experiments in this study. Figure 2.32 is a possible reduction side reaction of the methyl
bromoacetate with diphenylsilane and water.
Figure 2.32. Reduction side reaction of methyl bromoacetate with diphenylsilane and water.
The hydrogen bonding provided by the protic water media in these reactions help
remove the methoxy group from the ester, furthering the ability for the intramolecular
hydride transfer from the pentavalent silicon to the carbonyl carbocation. In addition to the
influence of the hydrogen bonding of the water media, this side reaction is more probable “on
water” than in toluene because all of the reactants are closely pushed together and more able
to react with one another due to their hydrophobic properties.
Future work to terminate this possible side reaction of the α-halo ester with
diphenylsilane and water is to replace the methyl group of the α-halo ester with a t-butyl
group and for instance use a different α-halo ester like t-butyl chloroacetate. Along with the
steric hindrance provided by the t-butyl group, a t-butoxy group is more basic than a methoxy
group, so it will better resist removal by a hydride and the hydrogen bonding of water.
Lastly, learned from reactions 2.10 and 2.11, a silane can quite possibly act as a
reducing agent and a base in order to make a catalytic Wittig reaction work. This dual action
by the silane might help remove another side reaction variable from the reaction vessel.

46
Future work can be done to see if an “on water” catalytic Wittig reaction using 3-methyl-1-
phenyl-phospholane oxide can be achieved with just a silane and no other base present.
2.5 CONCLUSION
Standard “on water” Wittig reactions can be achieved using triphenylphosphine and
3-methyl-1-phenylphospolane; though, only 3-methyl-1-phenylphospholane oxide can easily
be reduced “on water”. The cyclic phospholane oxide is a much more feasible catalyst for the
discovery of an “on water” catalytic Wittig reaction compared to the use of
triphenylphosphine oxide. Though there was little success in producing product when N,N-
dimethylaniline was used as the base for the “on water” catalytic Wittig reaction, it is less
favored to use than greener bases like sodium carbonate and sodium bicarbonate.

47
CHAPTER 3
THE EXPERIMENTAL PART
3.1 GENERAL
All reagents used were commercially purchased. Flash chromatography was
conducted using silica gel (60 Å, 230-400 mesh). Flash chromatography was performed on
Biotage SP1 using 25 mL columns. 1H NMR and 31P NMR spectra were recorded on a 400-
MHz instrument at 25 oC, with chemical shift values reported in ppm relative to TMS as an
internal standard (δ=ppm). Coupling patterns are abbreviated as: s (singlet), d (doublet), t
(triplet), q (quartet), and m (multiplet). Reactions done using a microwave reactor were done in
2 mL capped vials using a Biotage Initiator Classic 2.0.
3.2 “ON WATER” WITTIG REACTION EXPERIMENT
(E,Z)-Ethyl cinnamate (1.1). Add 0.367 g of triphenylphosphine to a 20 mL
scintillation vial with a flat magnetic stir bar. The use of a flat magnetic stir bar on a flat vial
surface ensures all reagents properly mix in the reaction vessel. Next, add 5 mL of saturated
aqueous sodium bicarbonate, 0.2 mL of ethyl bromoacetate and 0.102 mL of benzaldehyde to
the reaction vessel. Stir the heterogeneous reaction mixture vigorously for 2 hours at RT and
then add 5 mL of ethyl acetate to the reaction vessel. Separate the organic phase using a
small separation funnel. Wash the aqueous phase with small portions of ethyl acetate (2 x 5
mL). Combine and wash the organic phases once with 10% aqueous sodium bisulfite (1 x 10
mL). Wash the organic phase with saturated sodium chloride (1 x 5 mL). Dry the organic
phase over anhydrous sodium sulfate. Filter the dry organic phase under vacuum using a
Buchner funnel. Evaporate the ethyl acetate and let the crude organic residue air dry. The
pure product has an Rf value of 0.5 using 10% ethyl acetate/hexane as an eluent. Dissolve the
residue in minimum 10% ethyl acetate and hexane and purify on a short silica plug (5 x 2
cm). After evaporating the organic solvent determine the yield (%) of the product after letting
it air dry. 1H NMR (400 MHz, CDCl3): δ 1.24 (t, 3H), 1.34 (t, 3H), 4.17 (q, 2H), 4.27 (q,
2H), 5.95 (d, 1H), 6.44 (d, 1H), 6.94 (d, 1H), 7.3-7.6 (m, 8H), 7.7 (d, 1H).

48
3.3 ATTEMPTED CATALYTIC WITTIG REACTION
EXPERIMENTS USING PH3P AND PH3PO
The following experiments are those done using Ph3P and Ph3PO in development
towards an “on water” catalytic Wittig reaction.
3.3.1 Standard Wittig Reaction Using Ph3P in Organic Solvent
3-(2'-Methoxyphenyl)- (E,Z)-propenoic acid methyl ester (2.1). The reaction was
done in a 50 mL round bottom with magnetic stir bar, refluxing in toluene at 110 oC, under
argon for 24 hours. Add 0.290 g of triphenylphosphine (1.1 mmol), and then 5 mL of
toluene. Put reaction vessel under argon. Add the remaining reagents in the following order:
0.11 mL (0.9 mmol) of o-anisaldehyde, 0.11 mL (1.1 mmol) of methyl bromoacetate, and
0.56 mL (4 mmol) of triethylamine. After the reaction runs for 24 hours, let cool to RT and
then add 5 mL of 1M HCl to quench the triethylamine. Add 5 mL of DCM to the reaction
vessel and then wash the aqueous phase once with DCM. Combine organic phases and dry
over anhydrous MgSO4. Product separated using flash column with 20% ethyl acetate in
hexanes as the eluent. 1H NMR (400 MHz, CDCl3): δ 3.66 (s, 3H), 3.80 (s, 3H), 3.83 (s, 3H),
3.89 (s, 3H), 5.98 (d, 1H), 6.53 (d, 1H), 7.18 (d, 1H), 7.3-7.85 (m, 6H), 7.993 (d, 1H).
3.3.2 Reduction of Ph3PO in Organic Solvent
Triphenylphosphine (2.2 thru 2.5). Reactions 2.2 and 2.3 were done at RT, under
argon, with THF as the solvent in a 20 mL scintillation vial with a flat magnetic stir bar.
Reaction 2.2 reacted for 60 minutes and then reaction 2.3 reacted for 24 hours. Reactions 2.4
and 2.5 were done in a 25 mL bulb with stir bar, under argon, 2.4 in THF and 2.5 in toluene,
under argon refluxing for 24 hours. Add 0.0278 g (0.1 mmol) of triphenylphosphine oxide
and charge reaction vessel with argon. Add the remainder reagents in the following order: 3
mL of solvent, 0.017 g (0.1 mmol) of 4-nitrobenzoic acid, 0.26 mL (1.4 mmol) of
diphenylsilane, and then 0.24 mL (1.4 mmol) of DIPEA. Crude products dried under vacuum
and analyzed using 1H NMR and 31P NMR. 1H NMR (400 MHz, CDCl3): δ 7.33 (m, 15H). 31P NMR (400 MHz, CDCl3): δ -7.29 (s, 1P).

49
3.3.3 Compatibility Test of Standard Wittig Reaction and Beller Reduction Protocol
3-(2'-Methoxyphenyl)-(E,Z)-propenoic acid methyl ester (2.6). Reaction done in a 25
mL round bottom with stir bar, under argon, refluxing in toluene for 24 hours. Add 0.289 g
(1.1 mmol) of triphenylphosphine and 0.056 g (15 mol %) of bis(4-nitrophenyl)phosphate to
the reaction vessel and then charge it with argon. Add remaining reagents in the following
order: 10 mL of toluene, 0.109 mL (0.9 mmol) of o-anisaldehyde, 0.104 mL of methyl
bromoacetate (1.1 mmol), 0.66 mL (4 mmol) of diethoxymethylsilane and 0.56 mL (4 mmol)
of triethylamine. After reacting for 24 hours, cool to 0 oC and then add 5 mL of 1M KOH in
methanol. After 3 hours of mixing, add 3 mL of H2O. Extract organic layer with ethyl acetate
(3 x 20 mL). Wash organic phase with 1 M HCl (3 x 5 mL) and wash the organic layer with
1M NaHCO3 (3 x 5 mL). Dry organic layer with anhydrous NaSO4 and purify with a silica
plug using 20% ethyl acetate in hexanes. 1H NMR (400 MHz, CDCl3): δ 3.66 (s, 3H), 3.80
(s, 3H), 3.83 (s, 3H), 3.89 (s, 3H), 5.98 (d, 1H), 6.53 (d, 1H), 7.18 (d, 1H), 7.3-7.85 (m, 6H),
7.993 (d, 1H).
3.3.4 Experiments to Develop a Catalytic Wittig Reaction in Organic Solvent
3-(2'-Methoxyphenyl)-(E,Z)-propenoic acid methyl ester (2.7 thru 2.9). All reactions
were done in a 50 mL round bottom flask with stir bar, in toluene, refluxing for 24 hours
under argon. Add 0.306 g (1.1 mmol) of triphenylphophine oxide to reaction 2.7 and add
0.145 (0.5 mmol) to reactions 2.8 and 2.9. Add 0.151 g of bis(4-nitrophenyl)phosphate (15
mol % of Ph3PO) to 2.7, add 0.02 g (15 mol % of Ph3PO) of diphenylphosphate to 2.8 and
0.07 g of diphenylphosphate (50 mol % of Ph3PO) to 2.9. Charge reaction vessels with argon
and add the following remaining reagents to the reaction vessels: Add 10 mL of toluene to
each reaction vessel; add 0.109 mL (0.9 mmol) of o-anisaldehyde to 2.7, 0.063 mL (0.5
mmol) of o-anisaldehyde to 2.8 and 2.9; add 0.104 mL (1.1 mmol) of methyl bromoacetate to
2.7 and add 0.05 mL (0.5 mmol) of methyl bromo acetate to 2.8 and 2.9.; add 0.64 mL (4
mmol) of diethoxymethylsilane to 2.7 and add 0.33 mL (2 mmol) of diethoxymethylsilane to
2.8 and 2.9; add 0.56 mL (4 mmol) triethylamine to 2.7 and add 0.3 mL (2 mmol) of
triethylamine to 2.8 and 2.9. After reacting for 24 hours, cool to 0 oC and then add 5 mL of
1M KOH in methanol. After 3 hours of mixing, add 3 mL of H2O. Extract organic layer with

50
ethyl acetate (3 x 20 mL). Wash organic phase with 1M HCl (3 x 5 mL) and wash the organic
layer with 1M NaHCO3 (3 x 5 mL). Dry organic layer with anhydrous NaSO4 and purify with
a silica plug using 20% ethyl acetate in hexanes. 1H NMR (400 MHz, CDCl3): δ 3.66 (s, 3H),
3.80 (s, 3H), 3.83 (s, 3H), 3.89 (s, 3H), 5.98 (d, 1H), 6.53 (d, 1H), 7.18 (d, 1H), 7.3-7.85 (m,
6H), 7.993 (d, 1H).
3.3.5 Attempted Catalytic Wittig Reaction Experiment Using Ph3PO in Organic
Solvents Without NEt3
3-(2'-Methoxyphenyl)-(E,Z)-propenoic acid methyl ester (2.10). The reaction was
done in a 50 mL round bottom flask with stir bar, in toluene, refluxing for 24 hours under
argon. Add 0.145 (0.5 mmol) triphenylphosphine oxide and 0.07 g (50 mol % of Ph3PO) to
the reaction vessel and then charge it with argon. Add the remaining reagents in the
following order: 10 mL of tolune, 0.06 mL (0.5 mmol) of o-anisaldehyde, 0.05 mL (0.5
mmol) of methyl bromoacetate, and 0.33 mL (2 mmol) of diethoxymethylsilane. After
reacting for 24 hours, cool to 0 oC and then add 5 mL of 1M KOH in methanol. After 3 hours
of mixing, add 3 mL of H2O. Extract organic layer with ethyl acetate (3 x 20 mL). Wash
organic phase with 1M HCl (3 x 5 mL) and wash the organic layer with 1M NaHCO3 (3 x 5
mL). Dry organic layer with anhydrous NaSO4 and purify with a silica plug using 20% ethyl
acetate in hexanes. 1H NMR (400 MHz, CDCl3) δ: 3.66 (s, 3H), 3.80 (s, 3H), 3.83 (s, 3H),
3.89 (s, 3H), 5.98 (d, 1H), 6.53 (d, 1H), 7.18 (d, 1H), 7.3-7.85 (m, 6H), 7.993 (d, 1H).
3.3.6 Standard Wittig Reaction Experiment Using Diethoxymethylsilane and Ph3P
3-(2'-Methoxyphenyl)-(E,Z)-propenoic acid methyl ester (2.11). This reaction was
carried out in a 25 mL round bottom with stir bar, under argon, and refluxing in toluene for
24 hours. Add 0.306 g (1.1 mmol) of triphenylphosphine, and charge reaction vessel with
argon. Add the following remaining reagents in the following order: 10 mL of toluene, 0.121
mL (1 mmol) of o-anisaldehyde, 0.104 mL (1.1 mmol) methyl bromoacetate, and 0.50 mL (3
mmol) of diethoxymethylsilane. After the reaction goes for 24 hours, let cool to RT and add
5 mL of 1M HCl. Add 3 mL of DCM to reaction vessel and separate organic phase using
DCM. Wash the aqueous layer with DCM (3 x 5 mL). Dry the combined layers using

51
anhydrous MgSO4. Run crude through a dry silica plug using 20% ethyl acetate in hexanes. 1H NMR (400 MHz, CDCl3): δ 3.66 (s, 3H), 3.80 (s, 3H), 3.83 (s, 3H), 3.89 (s, 3H), 5.98 (d,
1H), 6.53 (d, 1H), 7.18 (d, 1H), 7.3-7.85 (m, 6H), 7.993 (d, 1H).
3.3.7 Attempted Catalytic Wittig Reaction Using O’Brien’s Protocol, Sodium Carbonate, and Ph3PO
(E,Z)-Methyl cinnamate (2.12). The reaction was conducted in a 50 mL round bottom
flask with stir bar, under argon and refluxing in toluene for 96 hours. First add 0.160 g (1.5
mmol) of sodium carbonate and 28 mg (10 mol %) of triphenylphosphine oxide to the
reaction vessel and put the reaction vessel under argon. Add the remaining reagents in the
following order: 10 mL of toluene, 0.102 mL (1 mmol) of benzaldehyde, 0.123 mL (1.3
mmol) methyl bromoacetate, and 0.21 mL (1.1 mmol) of diphenylsilane. Let the reaction
cool to RT after the 96 hour reaction time. Add 5 mL of ethyl acetate to the reaction vessel.
Wash the organic phase with H2O (3 x 10 mL). Wash the aqueous phase with ethyl acetate (1
x 10 mL). Dry organic layer with anhydrous NaSO4 and purify with a silica plug using 20%
ethyl acetate in hexanes. 1H NMR (400 MHz, CDCl3): δ 3.71 (s, 3H), 3.809 (s, 3H), 5.95 (d,
1H), 6.44 (d, 1H), 6.98 (d, 1H), 7.4-7.6 (m, 8H), 7.69 (d, 1H).
3.3.8 “On Water” Reduction of Ph3PO
Triphenylphosphine (2.13 thru 2.20). Reactions 2.13 thru 2.25 were done using a
microwave reactor. Reaction conditions were determined based off of a conversion chart. To
simulate water refluxing at 100 oC for 24 hours, the reactions ran for 13 minutes at 160 oC in
the microwave reactor. All the reactions were done in a 10 mL vial with a v-stir bar and stop
cap filled with 2 mL of H2O. For reactions 2.13 to 2.15, diphenylsilane is used as the silane.
For reactions 2.16 to 2.18, triisopropylsilane is used as the silane. For reaction 2.19, PMHS
was used and for reaction 2.20, TMDS was used. For reaction 2.13, add 0.07 (0.25 mmol) of
triphenylphosphine oxide, 0.19 mL (1 mmol) of diphenylsilane and 0.009 g (15 mol %) of
diphenylsilane. For reaction 2.14, add 0.07 g (0.25 mmol) of triphenylphosphine oxide, 0.19
mL (1 mmol) of diphenylsilane and 0.013 g (15 mol %) of bis(4-nitrophenyl)phosphate. For
reaction 2.15, add 0.021 g of triphenylphosphine oxide (0.075 mmol), 0.013 g (0.075 mmol)
of 4-nitrobenzoic acid, 0.18 mL (1.05 mmol) of DIPEA, and 0.020 mL (1.05 mmol) of
diphenylsilane. For reaction 2.16, add 0.07 g (0.25 mmol) of triphenylphosphine oxide, 0.013

52
g (15 mol %) of bis(4-nitrophenyl)phosphate, and 0.205 mL (1 mmol) of triisopropylsilane).
For reaction 2.17, add 0.07 g (0.25 mmol) of triphenylphosphine oxide, 0.009 g (15 mol %)
of diphenylphosphate, and 0.205 mL (1 mmol) of triisopropylsilane). For reaction 2.18, add
0.021 g (0.075 mmol) of triphenylphosphine oxide, 0.013 g (0.075 mmol) of 4-nitrobenzoic
acid, 0.22mL (1.05 mmol) of triisopropylsilane, and 0.18 mL (1.05 mmol) of DIPEA. For
reaction 2.19, add 0.13 mL (0.052 mmol) of PMHS, 0.0145 g (0.052mmol) of
triphenylphosphine oxide, and 0.002 g (15mol %) of diphenylphosphate. For reaction 2.20,
add 0.03 g (0.1 mmol) of triphenylphosphine oxide, 0.05 mL (0.1 mmol) of TMDS, and
0.004 g (15 mol %) of diphenylphosphate. Separate organic phase with DCM. Wash aqueous
phase with DCM (1 x 5 mL). Dry combined organic phases with anhydrous NaSO4. 1H NMR
(400 MHz, CDCl3): δ 7.33 (m, 15H). 31P NMR (400 MHz, CDCl3): δ -7.29 (s, 1P).
3.3.9 Reduction of Ph3PO in 15% DMSO and Water
Triphenylphosphine (2.21). Reaction was conducted in a 50 mL round bottom with
stir bar refluxing at 100 oC for 24 hours. First, add 0.07 g (0.025 mmol) of
triphenylphosphine oxide and then dilute in 0.7 mL (15% of 4.7 mL) of DMSO. Add stir bar
and then add the remaining reagents in the following order: 4 mL of H2O, 0.14 mL (0.75
mmol) of diphenylsilane, and 0.009 g (15 mol %) of diphenylsilane. Reaction cooled to RT
after 24 hours. Organic separated with 5 mL of ethyl acetate. Aqueous layer washed with
ethyl acetate (1 x 5 mL). Organic layers combined and dried using anhydrous NaSO4. 1H
NMR (400 MHz, CDCl3): δ 7.33 (m, 15H). 31P NMR (400 MHz, CDCl3): δ -7.29 (s, 1P).
3.3.10 Attempted Catalytic Wittig Reaction in Water Using 5% DMSO, NaCO3, Ph3PO, and Ph2SiH2
(E,Z)-Methyl cinnamate (2.22). This reaction was done in a 50 mL round bottom with
stir bar in 10 mL of H2O for 96 hours. First, add 0.028 (10 mol %) triphenylphosphine oxide
and then add 0.5 mL (5% of 10.5 mL) DMSO. Stir until triphenylphosphine oxide is
homogenous throughout mixture. Add remaining reagents in the following order: 0.102 mL
(1 mmol) of benzaldehyde, 0.123 mL (1.3 mmol) of methyl bromoacetate, 0.123 mL (1.1
mmol) of diphenylsilane and 0.160 g (1.5 mmol) of sodium carbonate. Let reaction run for 96
hours and then let cool to RT. Add 5 mL of ethyl acetate and separate organic phase from
aqueous phase. Wash aqueous phase once with ethyl acetate (1 x 5 mL). Dry combined

53
organic phases with anhydrous NaSO4. Dry organic layer with NaSO4 and purify with a silica
plug using 10% ethyl acetate in hexanes. 1H NMR (400 MHz, CDCl3): δ 3.71 (s, 3H), 3.809
(s, 3H), 5.95 (d, 1H), 6.44 (d, 1H), 6.98 (d, 1H), 7.4-7.6 (m, 8H), 7.69 (d, 1H).
3.4 EXPERIMENTAL WORK TOWARDS AN “ON WATER”
CATALYTIC WITTIG REACTION USING 3-METHYL-1-PHENYLPHOSPHOLANE OXIDE
The following experiments are those done with 3-methyl-1-phenylphospholane and 3-
methyl-1-phenylphospholane oxide in development of an “on water” catalytic Wittig
reaction.
3.4.1 Optimized O’Brien Catalytic Wittig Reaction in Toluene
(E,Z)-Methyl cinnamate (2.23). This reaction took place in a 50 mL round bottom
with magnetic stir bar refluxing in H2O for 24 hours. Add the reagents in the following order:
19 mg (20 mol %) 3-methyl-1-phenylphospholane oxide, 0.051 mL (0.5 mmol)
benzaldehyde, 0.051 mL methyl bromoacetate (0.55 mmol), 0.28 mL (1.5 mmol)
diphenylsilane, and 0.26 (1.5 mmol) DIPEA. Add 10 mL of ethyl acetate to reaction after
cooling reaction to RT. Separate the organic phase from aqueous phase using ethyl acetate.
Wash organic phase with 1 M HCl (2 x 10 mL). Wash combined organic phases with 10%
NaOH (aq) (2 x 10 mL). Use brine when needed. Dry combined organic phase with
anhydrous NaSO4. Run a flash column using 10% ethyl acetate in hexanes. Product has an Rf
value of 0.5 in 10% ethyl acetate in hexanes. Next, run a second column using DCM as the
solvent. The isolated product has an Rf value of 0.8 in DCM. 1H NMR (400 MHz, CDCl3): δ
3.71 (s, 3H), 3.809 (s, 3H), 5.95 (d, 1H), 6.44 (d, 1H), 6.98 (d, 1H), 7.4-7.6 (m, 8H), 7.69 (d,
1H).
3.4.2 “On Water” Wittig Reaction Using 3-methyl-1-phenylphospholane
(E,Z)-Methyl cinnamate (2.24). The reaction was done in a 50 mL round bottom with
magnetic stir bar refluxing for 24 hours. The reagents were added in the following order:
0.05 mL (0.5 mmol) benzaldehyde, 0.05 mL (0.55 mmol) methyl bromoacetate, 0.26 mL (1.5
mmol) DIPEA, and 0.098 g (0.55 mmol) of 3-methyl-1-phenylphospholane. After 24 hours,

54
reaction was cooled to RT. Add 5 mL of 1M HCl. Add 5 mL of ethyl acetate and separate
organic phase using ethyl acetate. Wash aqueous phase once with 5 mL ethyl acetate. Dry
combined organic phases using anhydrous NaSO4 and purify with a silica plug using 10%
ethyl acetate in hexanes. 1H NMR (400 MHz, CDCl3): δ 3.71 (s, 3H), 3.809 (s, 3H), 5.95 (d,
1H), 6.44 (d, 1H), 6.98 (d, 1H), 7.4-7.6 (m, 8H), 7.69 (d, 1H).
3.4.3 “On Water” Reduction of 3-methyl-1-phenylphospholane oxide
3-Methyl-1-phenylphospholane (2.25). Reaction was done in a 25 mL round bottom
with a stir bar in water refluxing for 24 hours. The reagents were added in the following
order: 0.097 g (0.5 mmol) 3-methyl-1-phenylphospholane oxide, and 0.28 mL (1.5 mmol) of
diphenylsilane. Cool reaction to RT. Separate organic phase from the aqueous phase using
ethyl acetate. Wash aqueous phase once with ethyl acetate. Dry combined organic phase
under anhydrous NaSO4. Wash organic phase multiple times with hexanes to further purify
and separate from white solid. Analyze using 31P NMR. 31P NMR (500 MHz, CDCl3): δ -
13.58 (s, 1P), -13.98 (s, 1P).
3.4.4 Attempted “On Water” Catalytic Wittig Reaction using Optimized O’Brien Catalytic
Wittig Reaction Protocol
(E,Z)-Methyl cinnamate (2.26). This reaction took place in a 50 mL round bottom
with magnetic stir bar refluxing in H2O for 24 hours. Add the reagents in the following order:
19 mg (20 mol %) 3-methyl-1-phenylphospholane oxide, 0.051 mL (0.5 mmol)
benzaldehyde, 0.051 mL methyl bromoacetate (0.55 mmol), 0.28 mL (1.5 mmol)
diphenylsilane, and 0.26 (1.5 mmol) DIPEA. Cool reaction to RT after running for 24 hours.
Adjust the pH to 5.5 using 1M H2SO4. Add 5 mL of ethyl acetate to the reaction and separate
the organic phase from the aqueous phase using ethyl acetate. Wash the aqueous phase once
with ethyl acetate. Dry combined organic phase with anhydrous NaSO4. 1H NMR (400 MHz,
CDCl3): δ 3.71 (s, 3H), 3.809 (s, 3H), 5.95 (d, 1H), 6.44 (d, 1H), 6.98 (d, 1H), 7.4-7.6 (m,
8H), 7.69 (d, 1H).

55
3.4.5 Attempted “On Water” Catalytic Wittig Reactions Using 3-methyl-1-phenylphospholane oxide
and Na2CO3/NaHCO3
(E,Z)-Methyl cinnamate (2.27 and 2.28). These reactions took place in a 50 mL
round bottom with magnetic stir bar refluxing in H2O for 24 hours. For reaction 2.27, add the
reagents in the following order: 19 mg (20 mol %) 3-methyl-1-phenylphospholane oxide,
0.051 mL (0.5 mmol) benzaldehyde, 0.051 mL methyl bromoacetate (0.55 mmol), 0.28 mL
(1.5 mmol) diphenylsilane, and 0.158 g (1.5 mmol) sodium carbonate. For reaction 2.28, add
the reagents in the following order: 19 mg (20 mol %) 3-methyl-1-phenylphospholane oxide,
0.051 mL (0.5 mmol) benzaldehyde, 0.051 mL methyl bromoacetate (0.55 mmol), 0.28 mL
(1.5 mmol) diphenylsilane, and 0.126 g (1.5 mmol) sodium bicarbonate. Add 5 mL of ethyl
acetate to the reaction and separate the organic phase from the aqueous phase using ethyl
acetate. Wash the aqueous phase once with ethyl acetate. Dry combined organic phase with
anhydrous NaSO4. Run crude through a silica plug using 10% ethyl acetate in hexanes. Wash
organic phase multiple times with hexanes to remove a white solid. 1H NMR (400 MHz,
CDCl3): δ 3.71 (s, 3H), 3.809 (s, 3H), 5.95 (d, 1H), 6.44 (d, 1H), 6.98 (d, 1H), 7.4-7.6 (m,
8H), 7.69 (d, 1H).
3.4.6 Attempted “On Water” Catalytic Wittig Reactions Using O’Brien’s Protocol and Tertiary
Nitrogen Bases
(E,Z)-Methyl cinnamate (2.29 to 2.31). These reactions took place in a 50 mL round
bottom with magnetic stir bar refluxing in H2O for 24 hours. For reaction 2.29, add the
reagents in the following order: 19 mg (20 mol %) 3-methyl-1-phenylphospholane oxide,
0.051 mL (0.5 mmol) benzaldehyde, 0.051 mL methyl bromoacetate (0.55 mmol), 0.28 mL
(1.5 mmol) diphenylsilane, and 0.29 g (1.5 mmol) DMAP. For reaction 2.30, add the
reagents in the following order: 19 mg (20 mol %) 3-methyl-1-phenylphospholane oxide,
0.051 mL (0.5 mmol) benzaldehyde, 0.051 mL methyl bromoacetate (0.55 mmol), 0.28 mL
(1.5 mmol) diphenylsilane, and 0.21 mL (1.5 mmol) triethylamine. For reaction 2.31, add the
reagents in the following order: 19 mg (20 mol %) 3-methyl-1-phenylphospholane oxide,
0.051 mL (0.5 mmol) benzaldehyde, 0.051 mL methyl bromoacetate (0.55 mmol), 0.28 mL
(1.5 mmol) diphenylsilane, and 0.19 mL (1.5 mmol) N,N-dimethylaniline. Add 5 mL of ethyl

56
acetate to the reaction and separate the organic phase from the aqueous phase using ethyl
acetate. Wash the aqueous phase once with ethyl acetate. Dry combined organic phase with
anhydrous NaSO4. Run crude through a flash column using 10% ethyl acetate in hexanes.
Next, run a second column using DCM as the solvent. The isolated product has an Rf value of
0.8 in DCM. Analyze using 1H NMR. 1H NMR (400 MHz, CDCl3): δ 3.71 (s, 3H), 3.809 (s,
3H), 5.95 (d, 1H), 6.44 (d, 1H), 6.98 (d, 1H), 7.4-7.6 (m, 8H), 7.69 (d, 1H).
3.4.7 Initial Optimization Attempts of Experiment 2.31
(E,Z)-Methyl cinnamate (2.32 and 2.33). These reactions took place in a 50 mL
round bottom with magnetic stir bar refluxing in H2O for 24 hours. For reaction 2.32, add the
reagents in the following order: 19 mg (20 mol %) 3-methyl-1-phenylphospholane oxide,
0.051 mL (0.5 mmol) benzaldehyde, 0.09 mL methyl bromoacetate (0.9 mmol), 0.28 mL (1.5
mmol) diphenylsilane, and 0.19 mL (1.5 mmol) N,N-dimethylaniline. For reaction 2.33, add
the reagents in the following order: 19 mg (20 mol %) 3-methyl-1-phenylphospholane oxide,
0.051 mL (0.5 mmol) benzaldehyde, 0.09 mL methyl bromoacetate (0.9 mmol), 0.28 mL (1.5
mmol) diphenylsilane, and 0.152 mL (0.6 mmol) N,N-dimethylaniline. Add 5 mL of ethyl
acetate to the reaction and separate the organic phase from the aqueous phase using ethyl
acetate. Wash the aqueous phase once with ethyl acetate. Dry combined organic phase with
anhydrous NaSO4. Run crude through a flash column using 10% ethyl acetate in hexanes.
Next, run a second column using DCM as the solvent. The isolated product has an Rf value of
0.8 in DCM. 1H NMR (400 MHz, CDCl3): δ 3.71 (s, 3H), 3.809 (s, 3H), 5.95 (d, 1H), 6.44
(d, 1H), 6.98 (d, 1H), 7.4-7.6 (m, 8H), 7.69 (d, 1H).
3.4.8 Optimization Attempts of Experiment 2.31 with Greater Equivalents of Methyl Bromoacetate
(E,Z)-Methyl cinnamate (2.34 and 2.35). These reactions took place in a 50 mL
round bottom with magnetic stir bar refluxing in H2O for 24 hours. For reaction 2.33, add the
reagents in the following order: 19 mg (20 mol %) 3-methyl-1-phenylphospholane oxide,
0.051 mL (0.5 mmol) benzaldehyde, 0.151 mL methyl bromoacetate (1.58 mmol), 0.28 mL
(1.5 mmol) diphenylsilane, and 0.19 mL (1.5 mmol) N,N-dimethylaniline. For reaction 2.34,
add the reagents in the following order: 19 mg (20 mol %) 3-methyl-1-phenylphospholane

57
oxide, 0.051 mL (0.5 mmol) benzaldehyde, 0.24 mL methyl bromoacetate (2.5 mmol), 0.28
mL (1.5 mmol) diphenylsilane, and 0.152 mL (0.6 mmol) N,N-dimethylaniline. Add 5 mL of
ethyl acetate to the reaction and separate the organic phase from the aqueous phase using
ethyl acetate. Wash the aqueous phase once with ethyl acetate. Dry combined organic phase
with anhydrous NaSO4. Run crude through a flash column using 10% ethyl acetate in
hexanes. Next, run a second column using DCM as the solvent. The isolated product has an
Rf value of 0.8 in DCM. 1H NMR (400 MHz, CDCl3): δ 3.71 (s, 3H), 3.809 (s, 3H), 5.95 (d,
1H), 6.44 (d, 1H), 6.98 (d, 1H), 7.4-7.6 (m, 8H), 7.69 (d, 1H).

58
REFERENCES
1. National Research Council. Sustainability in the Chemical Industry; The National Academies Press: Washington, D.C., 2006; pp 68-69.
2. Jimenez-Gonzalez, C.; Constable, D. Green Chemistry and Engineering: A Practical Design Approach; John Wiley & Sons, Inc.: New Jersey, 2011; pp 42-51.
3. Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice; Oxford University Press: New York, 1998; p 30.
4. Hjeresen, D. L.; Schutt, D. L.; Boese, J. M. Green Chemistry and Education. J. Chem. Educ. 2000, 77, 1543-1547.
5. Kolodiazhnyi, O. Phosphorous Ylides: Chemistry and Application in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 1999; p 359.
6. Roberts, M. R.; Gilbert, J. C.; Rodewald, L. B.; Wingrove, A. S. An Introduction to Modern Experimental Organic Chemistry; Holt, Rinehart and Winston, Inc.: New York, 1974; pp 271-279.
7. Garner, C. M. Techniques and Experiments for Advanced Organic Laboratory; John Wiley & Sons, Inc.: New York, 1997; pp 97-100.
8. Breton, G. W. The Wittig Reaction: Generation, Observation and Reactivity of a Phosphorous Ylide Intermediate. An Experiment for the Advanced Organic Chemistry Laboratory Course. J. Chem. Educ. 1997, 74, 114-115.
9. Pavia, D.; Lampman, G.; Kriz, G.; Engel, R. Introduction to Organic Laboratory Techniques A Small Scale Approach, 2nd ed.; Thompson Brooks Cole: Belmont, CA, 2005; pp 341-347.
10. Lampman, G. M.; Koops, R. W.; Olden, C. C. Phosphorus and sulfur ylide formation: Preparation of 1-benzoyl-2-phenylcyclopropane and 1,4-diphenyl-1,3-butadiene by phase transfer catalysis. J. Chem. Educ. 1985, 62, 267-268.
11. Gillois, J.; Guillerm, G.; Savignac, M.; Stephan, E.; Vo-Quanq, L. Diphenylbutadienes syntheses by means of the Wittig reaction: Experimental introduction to the use of phase transfer catalysis. J. Chem. Educ. 1980, 57, 161-162.
12. Warner, J. C.; Anastas, P.T.; Anselme, J.-P. The Wittig Reaction in the Undergraduate Organic Laboratory. J. Chem. Educ. 1985, 62, 346.
13. Silversmith, E. F. A Wittig reaction that gives only one stereoisomer. J. Chem. Educ. 1986, 63, 645.
14. Breuer, S. W. The Wittig synthesis of alkenes by phase-transfer catalysis: The syntheses of 4,4'-dichlorostilbenes and of E, E-1,4 –diphenylbutadiene. J. Chem. Educ. 1991, 68, A58-A60.

59
15. Wilcox, C. F.; Wilcox, M. F. Experimental Organic Chemistry: A Small-Scale Approach, 2nd ed.; Prentice Hall: Upper Saddle River, NJ, 1995; pp 369-370.
16. Jaworek, C.; Iacobucci, S. Wittig Reaction: The Synthesis of trans-9-(2-Phenylethenyl)anthracene Revisited. J. Chem. Educ. 2002, 79, 111.
17. Broos, R.; Tavernier, D.; Anteunis, M. p-Carboxystyrene. A Wittig procedure in aqueous medium. J. Chem. Educ. 1978, 55, 813.
18. Pike, R. M.; Mayo, D. W.; Butcher, S. S.; Butcher, D. J.; Hinkle, R. J. Microscale organic laboratory: IV: A simple and rapid procedure for carrying out Wittig reactions. J. Chem. Educ. 1986, 63, 917-918.
19. Balema, V. P.; Wiench, J. W.; Pruski, M.; Pecharsky, V. K. Mechanically Induced Solid-State Generation of Phosphorus Ylides and the Solvent-Free Wittig Reaction. J. Am. Chem. Soc. 2002, 124, 6244-6245.
20. Leung, S. H.; Angel, S. A. Solvent-Free Wittig Reaction: A Green Organic Chemistry Laboratory Experiment. J. Chem. Educ. 2004, 81, 1492-1493.
21. Martin, E.; Kellen-Yuen, C. Microwave-Assisted Organic Synthesis in the Organic Teaching Lab: A Simple, Greener Wittig Reaction. J. Chem. Educ. 2007, 84, 2004-2006.
22. Baar, M.; Falcone, D.; Gordon, C. Microwave-Enhanced Organic Syntheses for the Undergraduate Laboratory: Diels−Alder Cycloaddition, Wittig Reaction, and Williamson Ether Synthesis. J. Chem. Educ. 2010, 87, 84-86.
23. Zovinka, E.; Stock, A. Microwave Instruments: Green Machines for Green Chemistry? J. Chem. Educ. 2010, 87, 350-352.
24. Speed, Traci J.; McIntyre, Jean P.; Thamattoor, Dasan M. Wittig Reaction Using a Stabilized Phosphorus Ylid: An Efficient and Stereoselective Synthesis of Ethyl trans-Cinnamate. J. Chem. Educ. 2004, 81, 1355-1356.
25. Narayan, S.; Muldoon, J.; Finn, M.; Fokin, V.; Kolb, H.; Sharpless, B. “On Water”: Unique Reactivity of Organic Compounds in Aqueous Suspension. Angew. Chem. Int. Ed. 2005, 44, 3275-3279.
26. El-Batta, A.; Jiang, C.; Zhao, W.; Anness, R.; Cooksy, A. L.; Bergdahl, M. Wittig Reactions in Water Media Employing Stabilized Ylides with Aldehydes. Synthesis of α,β-Unsaturated Esters from Mixing Aldehydes, α-Bromoesters, and Ph3P in Aqueous NaHCO3. J. Org. Chem. 2007, 72, 5244-5259.
27. Sigma Aldrich. Proton NMR of triphenylphosphine. http://www.sigmaaldrich.com/ catalog/product/fluka/93090?lang=en®ion=US (accessed Jan 2014).
28. Sigma Aldrich. Proton NMR of ethyl bromoacetate. http://www.sigmaaldrich.com/ catalog/product/aldrich/133973?lang=en®ion=US (accessed Jan 2014).
29. Sigma Aldrich. Proton NMR of benzaldehyde. http://www.sigmaaldrich.com/ catalog/product/aldrich/418099?lang=en®ion=US (accessed Jan 2014).
30. R. W. Sidley. Silica Dust Material Safety Data Sheet. http://www.rwsidley .com/MSDS/manley.pdf (accessed Jan 2014).

60
31. Van Kalkeren, H.; Blom, A.; Rutjes, F.; Huijbregts, M. On the usefulness of life cycle assessment in early chemical methodology development: the case of organophosphorus-catalyzed Appel and Wittig reactions. Green Chem. 2013, 15, 1255-1263.
32. Fairlamb, Ian. J. S. The Phosphine-Catalyzed Wittig Reaction: A New Vista for Olefin Synthesis? ChemSusChem. 2009, 2, 1021-1024.
33. Shi, L.; Wang, W.; Wang, Y.; Huang, Y. –Z. The first example of a catalytic Wittig-type reaction. Tri-n-butylarsine-catalyzed olefination in the presence of triphenyl phosphite. J. Org. Chem. 1989, 54, 2027-2028.
34. Huang, Z. –Z.; Tong, Y. Unexpected Catalyst for Wittig-Type and Dehalogenation Reactions. J. Org. Chem. 2002, 67, 5320-5326.
35. O’Brien, C. J.; Tellez, J.; Nixon, Z.; Kang, L.; Carter, A.; Kunkel, S.; Przeworski, K.; Chass, G. Recycling the Waste: The Development of a Catalytic Wittig Reaction. Angew. Chem. Int. Ed. 2009, 48, 6836-6839.
36. Li, Y.; Lu, L. –Q.; Das, S. Pisiewicz, S. Junge, K.; Beller, M. Highly Chemoselective Metal-Free Reduction of Phosphine Oxides to Phosphines. J. Am. Chem. Soc. 2012, 134, 18325-18329.
37. Henson, P.; Naumann, K.; Mislow, K. Stereomutation of phosphine oxides by lithium aluminum hydride. J. Am. Chem. Soc. 1969, 91, 5645.
38. Kobayashi, S.; Suzuki, M; Saegusa, T. A New Reduction Method of Poly(phosphine oxide) to Polyphosphine. Preparation of Poly[(P-phenyl)trimethylenephosphine]. Polym. Bull. 1982, 8, 417.
39. Imamoto, T.; Takeyama, T.; Kusumoto, T. Facile Reduction of Organic Halides and Phosphine Oxides with LiAlH4–CeCl3.Chem. Lett. 1985, 10, 1491.
40. Imamoto, T.; Kikuchi, S.; Miura,T.; Wada, Y. Stereospecific Reduction of Phosphine Oxides to Phosphines by the Use of a Methylation Reagent and Lithium Aluminum Hydride. Org. Lett. 2001, 3, 87.
41. Busacca, C.; Raju, R.; Grinberg, N.; Haddad, N.; James-Jones, P. Lee, H.; Lorenz, J.; Saha, A; Senanayake, C. Reduction of Tertiary Phosphine Oxides with DIBAL-H. J. Org. Chem. 2008, 73, 1524.
42. Rajendran, K.; Gilheany, D. Simple unprecedented conversion of phosphine oxides and sulfides to phosphine boranes using sodium borohydride. Chem. Comm. 2012, 48, 817.
43. Bethod, M.; Favre-Reguillon, A.; Mohamad, J.; Mignani, G.; Docherty, G.; Lemaire, M. A Catalytic Method for the Reduction of Secondary and Tertiary Phosphine Oxides. Synlett 2007, 10, 1545.
44. Petit, C.; Favre-Reguillon, A.; Albela, B.; Bonneviot, L.; Mignani, G.; Lemaire, M. Mechanistic Insight into the Reduction of Tertiary Phosphine Oxides by Ti(OiPr)4/TMDS. Organometallics 2009, 28, 6379.

61
45. Petit, C.; Favre-Reguillon, A.; Mignani, G.; Lemaire, M. A straightforward synthesis of unsymmetrical secondary phosphine boranes. Green Chem. 2010, 12, 326.
46. Coumbe, T. Lawrence, N.; Muhammad, F. Titanium (IV) catalysis in the reduction of phosphine oxides. Tetrahedron Lett. 1994, 35, 625–628.
47. O’Brien, C. J.; Lavigne, F.; Coyle, E. E.; Holohan, A. J.; Doonan, B. Breaking the Ring through a Room Temperature Catalytic Wittig Reaction. Chem. Eur. J. 2013, 19, 5854-5858.
48. O’Brien, C. J.; Nixon, Z.; Holohan, A.; Kunkel, S.; Tellez, J.; Doonan, B.; Coyle, E.; Lavigne, F.; Kang, L.; Przeworski, K. Part I: The Development of the Catalytic Wittig Reaction. Chem. Eur. J. 2013, 19, 15281-15289.
49. Fringuelli, F.; Piermatti, O.; Pizzo, F. Oxidation and Reductions in Water. In Organic Synthesis in Water, Grieco, P., Ed.; Springer: Netherlands, 1997; pp. 236-246.
50. Greaney, M.; Smith, J. A one-pot silyl-Reformatsky olefination. Tetrahedron Lett. 2007, 48, 8687-8690.
51. Naumann, K.; Zon, G.; Mislow, K. Use of hexachlorodisilane as a reducing agent. Stereospecific deoxygenation of acyclic phosphine oxides. J. Am. Chem. Soc. 1969, 91, 7012-7023.
52. Addis, D.; Das, S.; Junge, K.; Beller, M. Selective Reduction of Carboxylic Acid Derivatives by Catalytic Hydrosilylation. Angew. Chem. Int. Ed. 2011, 50, 6004-6011.

62
APPENDIX A
JOURNAL OF CHEMICAL EDUCATION “ON
WATER” WITTIG REACTION LABORATORY
EXPERIMENT SUBMISSION

63
The “On Water” type Wittig Reaction as an Essential Experiment in the Undergraduate Organic Chemistry Teaching Laboratory Lucas B. Fallot, Michael Kelly, and B. Mikael Bergdahl* Department of Chemistry and Biochemistry, San Diego State University, San Diego, California 92182-1030, United States
ABSTRACT The aqueous Wittig reaction is a suitable undergraduate experiment which allows for instructors to effectively and very quickly demonstrate and promote a greener alternative of an alkene synthesis in the organic chemistry teaching laboratory. There is an opportunity for students to compare previously reported Wittig reaction approaches and evaluate those with this proposed greener “on-water” type Wittig reaction methodology.
KEYWORDS Green Chemistry, Aqueous, Wittig Reaction, Organic Chemistry, Alkene, Synthesis
Sustainability and environmental protection are two increasingly critical issues in our society, but in many cases greener practices have been only slowly adopted in the chemistry laboratory. Often, especially at the undergraduate level, many laboratory procedures are little changed from those of fifty years ago. In order to increase awareness and adoption of practical greener chemistry in the future, it is essential to positively inform students early in their education about the alternatives possible for more environmentally-friendly chemistry.1 The Wittig reaction is a fundamental introductory organic chemistry reaction that can be used to promote practical greener chemistry by way of replacing organic solvents with water. Water is by its very nature nontoxic, cost-effective and less-harmful to the environment than the organic solvents traditionally used for the Wittig reaction, and it represents the outside-the-box thinking that will be necessary for a more-sustainable approach to common chemical problems without diminishing the larger lesson: the fundamental utility of the Witting reaction.
In order to best demonstrate the thinking necessary for the implementation of greener solutions to common chemical problems, it is helpful to first examine and compare some traditional Wittig reaction protocols from literature. The Wittig reaction is a fundamental method for the synthesis of alkenes and variations of it can be found in many laboratory textbooks as well as in the Journal of Chemical Education, as the samples below show. Among these sources, none discuss green chemistry or demonstrate that a simple Wittig reaction can be done in water in a way that can be easily and practically conducted by undergraduate students in a teaching laboratory.
In accordance with The Twelve Principles of Green Chemistry,2 harmful substances like toxic solvents, pyrophoric reagents and lachrymators should be minimized or replaced where possible and practical. Yet in some of these published protocols intended for undergraduate laboratories, pyrophoric reagents like butyllithium and lachrymators, like benzyl chloride, are used in order to make an unstable ylide.3,4 One case calls for the use of sodium hydride, which would seem unsuitable as a reagent in an undergraduate teaching laboratory.5a Another Wittig protocol in one of the textbooks still commonly used calls for

64
the use of sodium metal to make the strong base sodium ethoxide.5b In addition to obvious safety concerns, the use of many of these reagents is wholly unnecessary, and requires laboratories to be equipped with inert gas support and special waste bins, both factors which can increase the cost and reduce the practicality of the execution of the experiments in a teaching laboratory, and which can require precious lab time for special student training.
The use of phase transfer catalysis is another common protocol for conducting Wittig chemistry.6-12 These reactions are also carried out in two steps: the in situ preparation of the ylide followed by the Wittig reaction itself. These reactions also commonly use lachrymators in the first step to form the phosphonium salt. However, in contrast to the previous examples which used pyrophoric or air-sensitive bases, a 50% aqueous sodium hydroxide solution is used to produce the ylide, which is a step toward a greener and safer approach to the chemistry, yet still holds more hazards than other options. An additional J. Chem. Ed. paper13 illustrates a one-phase transfer catalysis Wittig experiment using a 50-fold excess of formaldehyde, a well-known carcinogen.
In another Wittig protocol, the use of an “instant ylid” greatly simplifies the reaction,14 but is unfortunately potentially harmful in a teaching laboratory setting because of the use of an alkyl triphenylphosphonium bromide and sodium amide, an unnecessarily strong, toxic base, in anhydrous THF. It also reduces the practicality of the experiment because of its water sensitivity.
There are published experiments which describe much safer and greener strategies in performing Wittig reactions in the undergraduate teaching laboratory.15,16 Examples of these include solvent-free Wittig reactions that use either a ball milling apparatus or a mortar and pestle to replace the use of organic solvents during the reaction. The ball milling protocol involves an innovative one-pot Wittig reaction with very safe and inexpensive potassium carbonate as the base, but unfortunately this reaction takes 8 hours to conduct. The use of a mortar and pestle in a solvent-free reaction is another wonderful example of a green innovation in Wittig chemistry, but reproducibility becomes a real problem because of the variables involved in physically grinding the reactants.
The microwave accelerated Wittig reaction is another alternative way to introduce greener chemistry into the teaching laboratory.17,18 One procedure utilizes a common household microwave, and the other reports the use of a microwave reactor specifically designed for organic reactions. The reported common household microwave experiment does not require the use of a solvent, and it utilizes potassium carbonate to generate the ylide, which seems like an attractive greener alternative. Although the common household microwave is a relatively inexpensive instrument to use in a teaching laboratory, it might become a liability. It does not have the built-in safety features found in microwave reactors specifically made for conducting organic reactions; for example, a reinforced steel reaction chamber and a special door-locking mechanism in the event of a reaction vessel rupture. Thus, with the potential for overheating and explosions, the use of a household type microwave is not recommended in the undergraduate teaching laboratory. In regard to the laboratory experiment utilizing a microwave reactor specifically designed for organic reactions, the Wittig reaction is conducted in 3 separate phases and the reactor is used only in the first phase to produce high yields of the phosphonium salt in 6.5 minutes. Although the reaction time, safety features of the microwave reactor, and increased phosphonium salt yield utilizing the reactor make this reaction appealing, the experiment calls for the use of xylenes, a chemical that permeates the skin and is not safe to use with undergraduate students, and the cost of maintaining such a piece of equipment makes this approach less practical.
In order to emphasize the importance and methods of practical environmentally-friendly chemistry and at the same time maintain a high level of safety in the organic chemistry teaching laboratory, we hereby propose the introduction of a water-based Wittig reaction as a complement to previously reported methods.

65
We recommend the adaptation of a simple Wittig reaction protocol that can be conducted in aqueous saturated sodium bicarbonate media using safer reagents where possible – an experiment suitable for the organic teaching laboratory (Scheme 1). This organic reaction allows educators to provide students with a safer, fast, simpler, less expensive and more effective means to conduct a greener Wittig reaction. Water is not considered a “solvent” in this reaction but rather a “medium” since the reagents are quite hydrophobic and appear insoluble. Aqueous sodium bicarbonate is a relatively weak and safe base for students, used here to generate in-situ the stabilized ylide needed for the Wittig reaction to produce the alkene product. Though ethyl bromoacetate is classified as a lachrymator, its use is minimized. The proposed experiment does not use pyrophoric reagents requiring an inert atmosphere or any special training. Adding to the simplicity of the experiment, the reaction is done at ambient temperature utilizing common laboratory equipment. In consideration of cost, water is obviously much less costly than organic solvents. Water is also environmentally benign and has been shown to be effective as a medium in many organic reactions, and therefore can be considered as an option to replace harmful solvents in other organic reactions. Rather than purchasing the ylide, here it is generated in-situ using inexpensive starting materials. The proposed reaction and work-up can be conducted during a three hour organic laboratory period. Students without the experience of conducting the purification on a silica-plug need an additional laboratory period of circa three hours.
Water is a remarkably efficient medium for the Wittig reaction and can be used with a wide range of stabilized ylides and aldehydes. Even more astonishing is the increase in reaction rate that is evident in water as compared to common organic solvents even though the reaction seems heterogeneous in water.19 We have also reported that Wittig reactions in water work best when large hydrophobic entities like aromatic functional groups are present.20 Despite the reactants’ poor solubility, aqueous Wittig reactions have very high yields with high precedence for trans (E)-selectivities.
EXPERIMENTAL PROCEDURE
Add 0.367 g (1.40 mmol) of triphenylphosphine to a 20 mL scintillation vial with a flat magnetic stir bar (Figure 1). The use of a flat magnetic stir bar on a flat vial surface ensures all reagents properly mix in the reaction vessel. Next, add 5 mL of aqueous saturated sodium bicarbonate, 0.20 mL (1.80 mmol) of ethyl bromoacetate and 0.102 mL (1.00 mmol) of benzaldehyde to the reaction vessel. Stir the heterogeneous reaction mixture vigorously for 2 hours at room temperature and then add 5 mL of ethyl acetate to the reaction vessel. Separate the organic phase using a small separation funnel. Wash the aqueous phase with small portions of ethyl acetate (2 x 5 mL). Combine the organic phases and wash once with 10% aqueous sodium bisulfite (10 mL). Following this, wash once with saturated sodium chloride (5 mL). Dry the organic phase over anhydrous sodium sulfate. Filter the dry organic phase under vacuum on a Buchner funnel and evaporate the ethyl acetate. The pure product has an Rf of circa 0.5 using 10% ethyl acetate/hexane as an eluent. Dissolve the residue in minimum 10% ethyl acetate and hexane and purify on a short silica plug (5 x 2 cm). After evaporating the organic solvent determine the yield (%) and analyze the product using 1H-NMR (Figure 2).

66
RESULTS AND DISCUSSION The product composition can be analyzed by 1H-NMR spectroscopy. The students are
able to compare their product 1H-NMR data with 1H-NMR data of the starting material and reagents. The students will also be able to determine the trans/cis (E/Z) ratio of their alkene products in their NMR spectrum. By washing the organic phase with sodium bisulfite it is possible to remove unreacted aldehyde in the crude reaction mixture. The triphenylphosphine oxide formed during the Wittig reaction is removed on a short silica plug. Most of the unreacted triphenylphosphine remains as a residue when dissolving the crude residue in 10% ethyl acetate in hexane. The triphenylphosphine, which remains will make up very little (~<1%) of the product mass has quite a distinguishable 1H-NMR signal just downfield from the chloroform signal. A typical student product yield was found to be on average 93% with an E/Z ratio of circa 92/8.
Water is shown to be a safe and efficient medium for the Wittig olefination reaction employing an in-situ prepared stabilized ylide and benzaldehyde in a teaching laboratory, despite the heterogeneous appearance of the reaction mixture. The separation of the organic materials from the water still requires the use of a small amount of organic solvent, but benign solvents can be used, and their use can be minimized.
HAZARDS
Ethyl bromoacetate is a lachrymator and should be used with caution in a ventilated hood. Benzaldehyde, and triphenylphosphine are irritants and should be used with caution. Do not inhale silica dust, carefully use in a ventilated hood. Consult safety data sheets for all reagents prior to conducting the laboratory experiment.
ASSOCIATED CONTENT
Supporting Information
Laboratory materials, teacher preparation instructions, student handout, and 1H-NMR data are included. This material is available via the Internet at http://pubs.acs.org.
AUTHOR INFORMATION
Corresponding Author *E-mail: [[email protected]]
Notes The authors declare no competing financial interest.
ACKNOWLEDGMENT The authors wish to thank San Diego State University for the “University Grant
Program” in support for the development of this experiment. We also would like to thank the faculty and students at SDSU for their effort in incorporating this laboratory experiment into the undergraduate organic laboratory curriculum.
REFERENCES 1. Hjeresen, D. L.; Schutt, D. L.; Boese, J. M. J. Chem. Educ. 2000, 77, 1543-1547. 2. Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice; Oxford University Press: New
York, 1998; p 30. 3. Roberts, M. R.; Gilbert, J. C.; Rodewald, L. B.; Wingrove, A. S. An Introduction to Modern
Experimental Organic Chemistry; Holt, Rinehart and Winston, Inc.: New York, 1974; pp 271-279. 4. Garner, C. M. Techniques and Experiments For Advanced Organic Laboratory; John Wiley & Sons,
Inc.: New York, 1997; pp 97-100. 5. a) Breton, G. W. J. Chem. Educ. 1997, 74, 114-115.; b) Pavia, D.; Lampman, G.; Kriz, G.; Engel, R.
Introduction to Organic Laboratory Techniques A Small Scale Approach, 2nd ed., Thompson Brooks Cole: Belmont, CA, 2005; pp 341-347.
6. Lampman, G. M.; Koops, R. W.; Olden, C. C. J. Chem. Educ. 1985, 62, 267-268.

67
7. Gillois, J.; Guillerm, G.; Savignac, M.; Stephan, E.; Vo-Quanq, L. J. Chem. Educ. 1980, 57, 161-162.
8. Warner, J. C.; Anastas, P.T.; Anselme, J.-P. J. Chem. Educ. 1985, 62, 346. 9. Silversmith, E. F. J. Chem. Educ. 1986, 63, 645. 10. Breuer, S. W. J. Chem. Educ. 1991, 68, A58-A60. 11. Wilcox, C. F.; Wilcox, M. F. Experimental Organic Chemistry: A Small-Scale Approach, 2nd ed.;
Prentice Hall: Upper Saddle River, NJ, 1995; pp 369-370. 12. Jaworek, C.; Iacobucci, S. J. Chem. Educ. 2002, 79, 111. 13. Broos, R.; Tavernier, D.; Anteunis, M. J. Chem. Educ. 1978, 55, 813. 14. Pike, R. M.; Mayo, D. W.; Butcher, S. S.; Butcher, D. J.; Hinkle, R. J. J. Chem. Educ. 1986, 63,
917-918. 15. Balema, V. P.; Wiench, J. W.; Pruski, M.; Pecharsky, V. K. J. Am. Chem. Soc. 2002, 124, 6244-
6245. 16. Leung, S. H.; Angel, S. A. J. Chem. Educ. 2004, 81, 1492-1493. 17. Martin, E.; Kellen-Yuen, C. J. Chem. Educ. 2007, 84, 2004-2006. 18. Baar, M.; Falcone, D.; Gordon, C. J. Chem. Educ. 2010, 87, 84-86. 19. Narayan, S.; Muldoon, J.; Finn, M.; Fokin, V.; Kolb, H.; Sharpless, B. Angew. Chem. Int. Ed.
2005, 44, 3275-3279. 20. El-Batta, A.; Jiang, C.; Zhao, W.; Anness, R.; Cooksy, A. L.; Bergdahl, M. J. Org. Chem. 2007, 72,
5244-5259.

68
APPENDIX B
1H NMR, AND 31P NMR OF REACTIONS
69
F
igu
re B
.1. 1 H
NM
R o
f re
acti
on 1
.1.

70
F
igu
re B
.2. 1 H
NM
R o
f re
acti
on 2
.1.
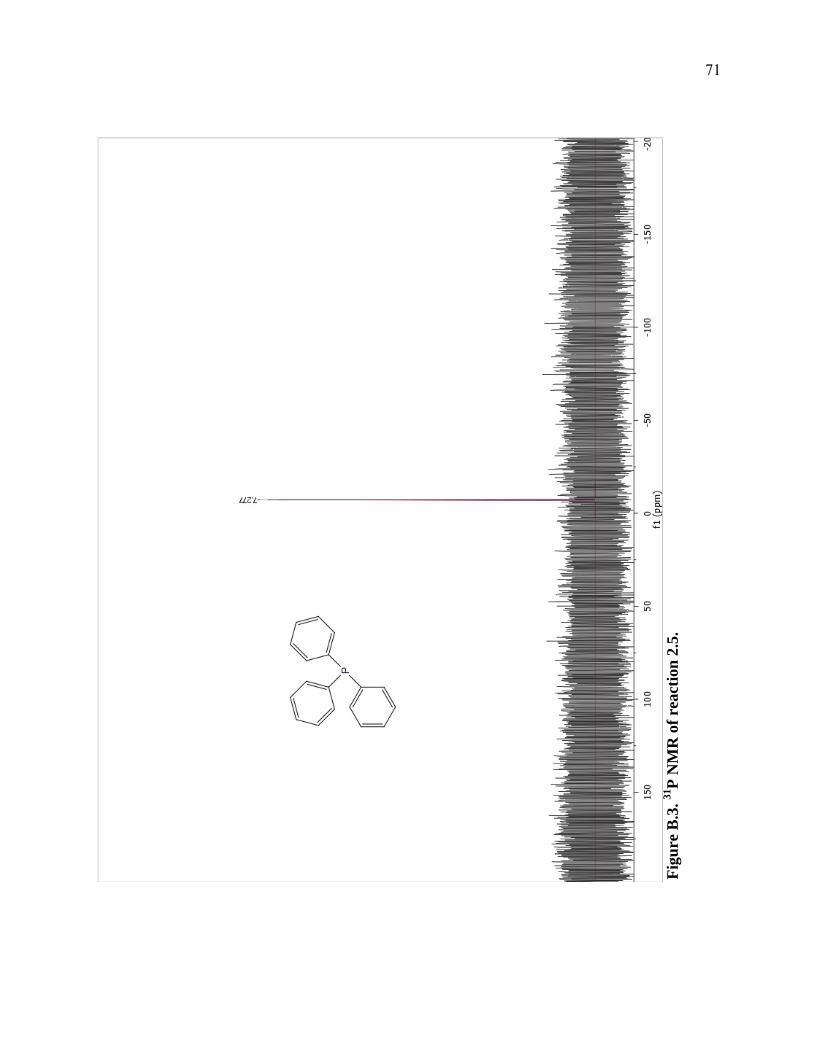
71
F
igu
re B
.3. 31
P N
MR
of
reac
tion
2.5
.

72
F
igu
re B
.4. 1 H
NM
R o
f re
acti
on 2
.5.

73
F
igu
re B
.5. 1 H
NM
R o
f re
acti
on 2
.6.

74
F
igu
re B
.6. 1 H
NM
R o
f re
acti
on 2
.7.

75
F
igu
re B
.7. 1 H
NM
R o
f re
acti
on 2
.8.

76
F
igu
re B
.8. 1 H
NM
R o
f re
acti
on 2
.9.

77
F
igu
re B
.9. 1 H
NM
R o
f re
acti
on 2
.10.

78
F
igu
re B
.10.
1 H N
MR
of
reac
tion
2.1
1.
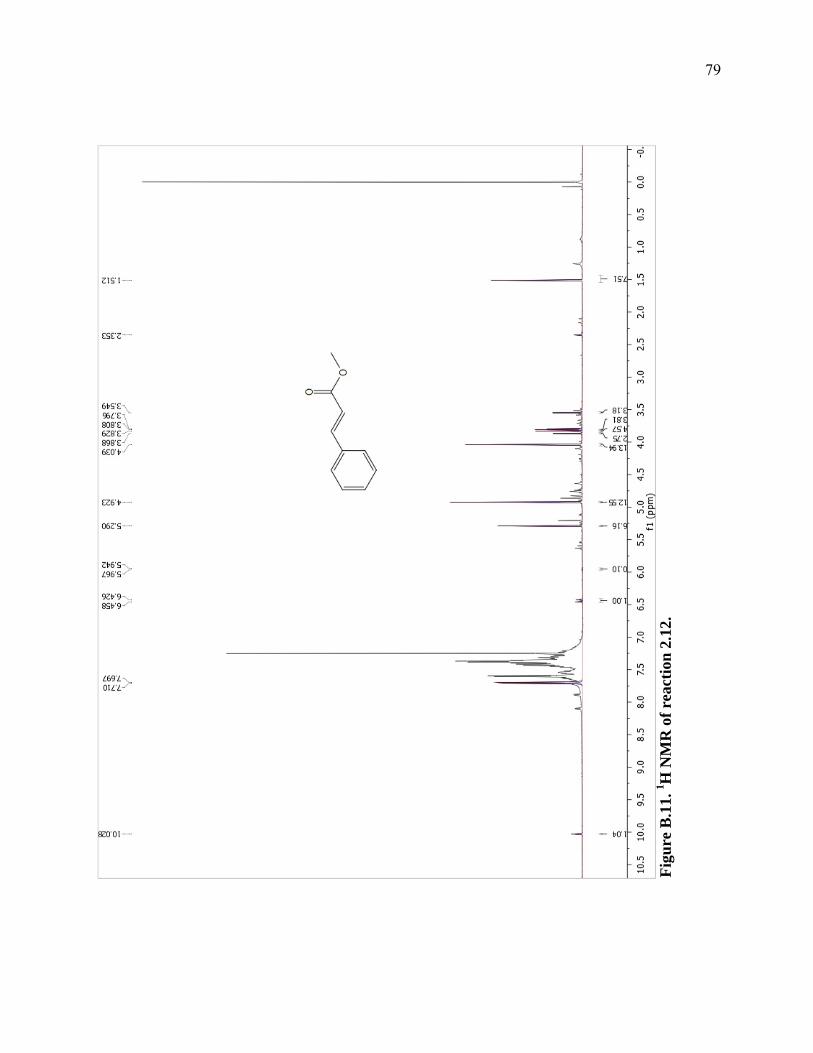
79
F
igu
re B
.11.
1 H N
MR
of
reac
tion
2.1
2.

80
F
igu
re B
.12.
31P
NM
R o
f re
acti
on 2
.21.
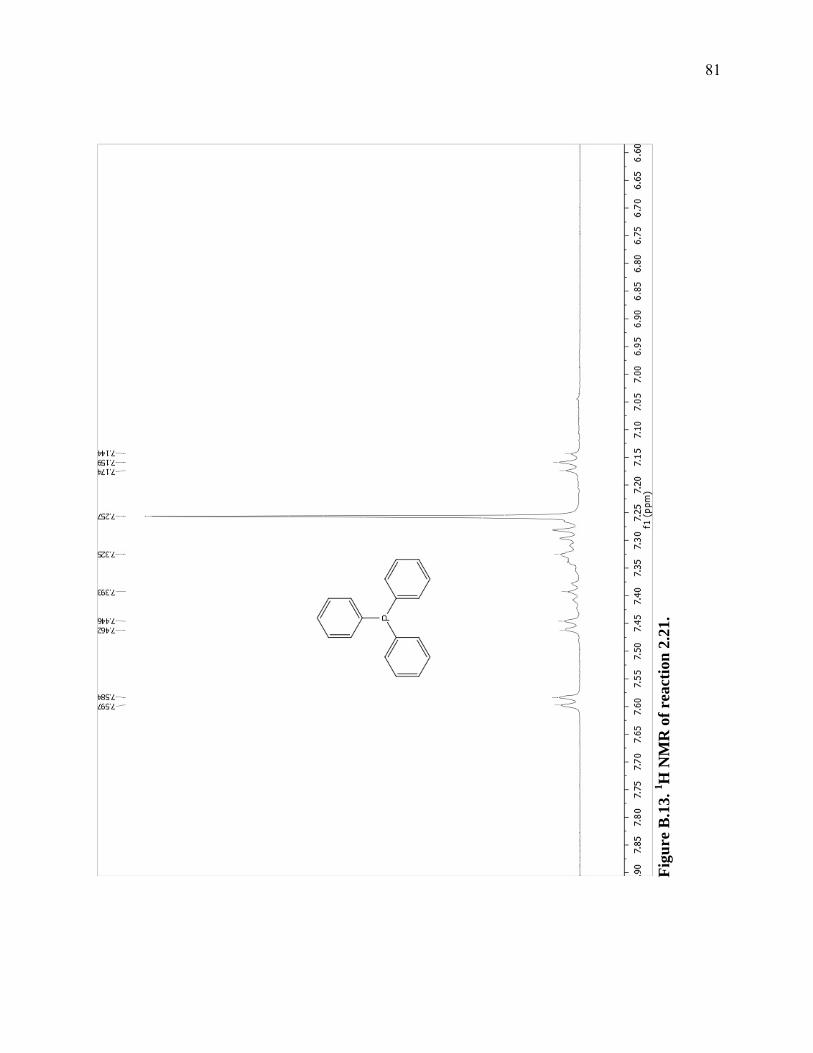
81
F
igu
re B
.13.
1 H N
MR
of
reac
tion
2.2
1.
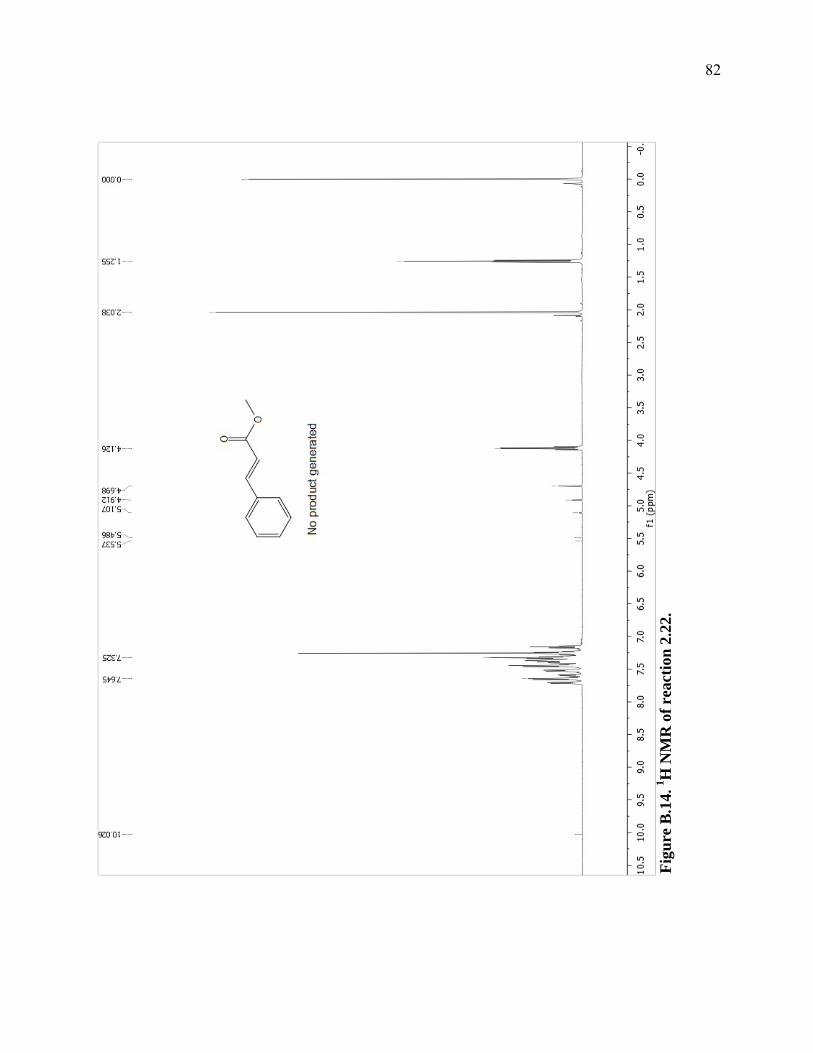
82
F
igu
re B
.14.
1 H N
MR
of
reac
tion
2.2
2.
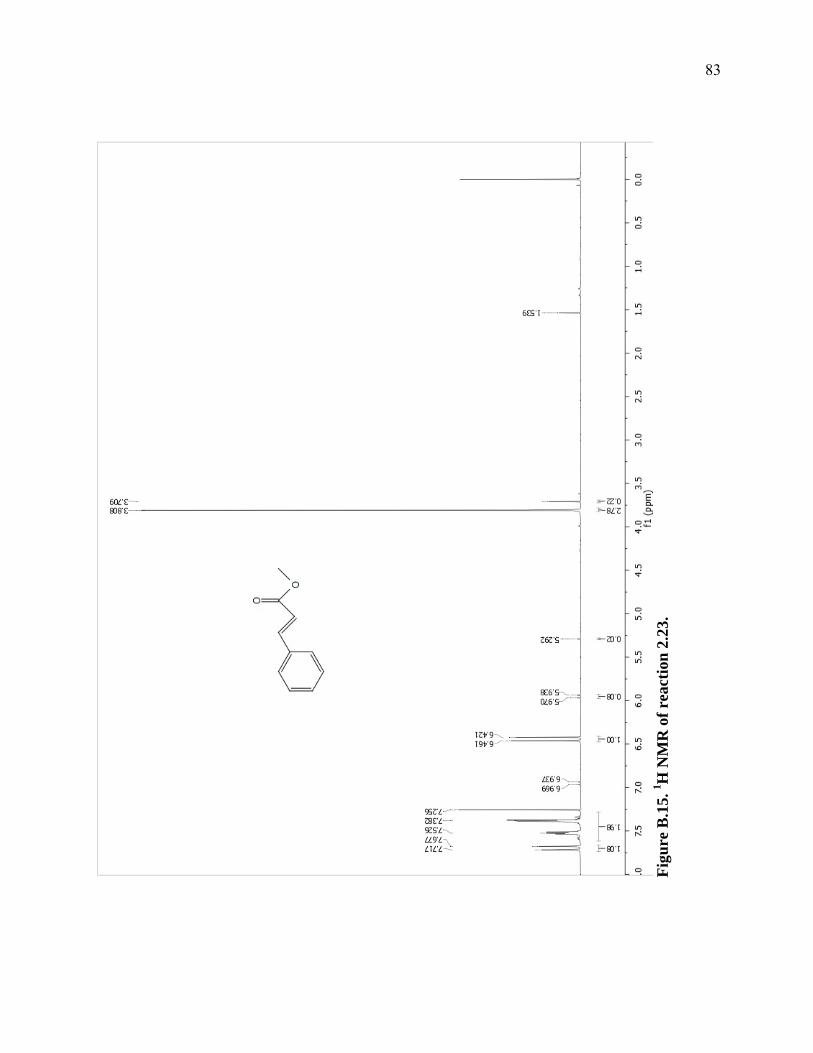
83
F
igu
re B
.15.
1 H N
MR
of
reac
tion
2.2
3.

84
F
igu
re B
.16.
1 H N
MR
of
reac
tion
2.2
4.

85
F
igu
re B
.17.
31P
NM
R o
f 3-
met
hyl
-1-p
hen
ylp
hos
ph
olan
e ox
ide.

86
F
igu
re B
.18.
31P
NM
R o
f re
acti
on 2
.25.
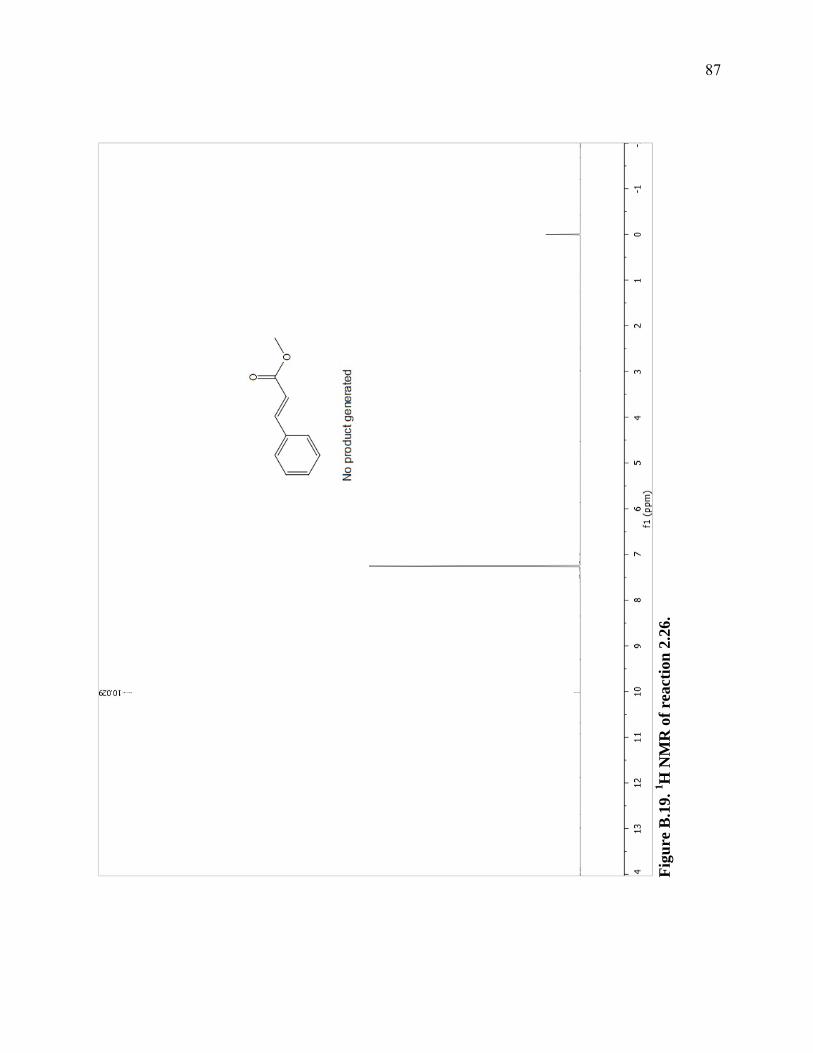
87
F
igu
re B
.19.
1 H N
MR
of
reac
tion
2.2
6.
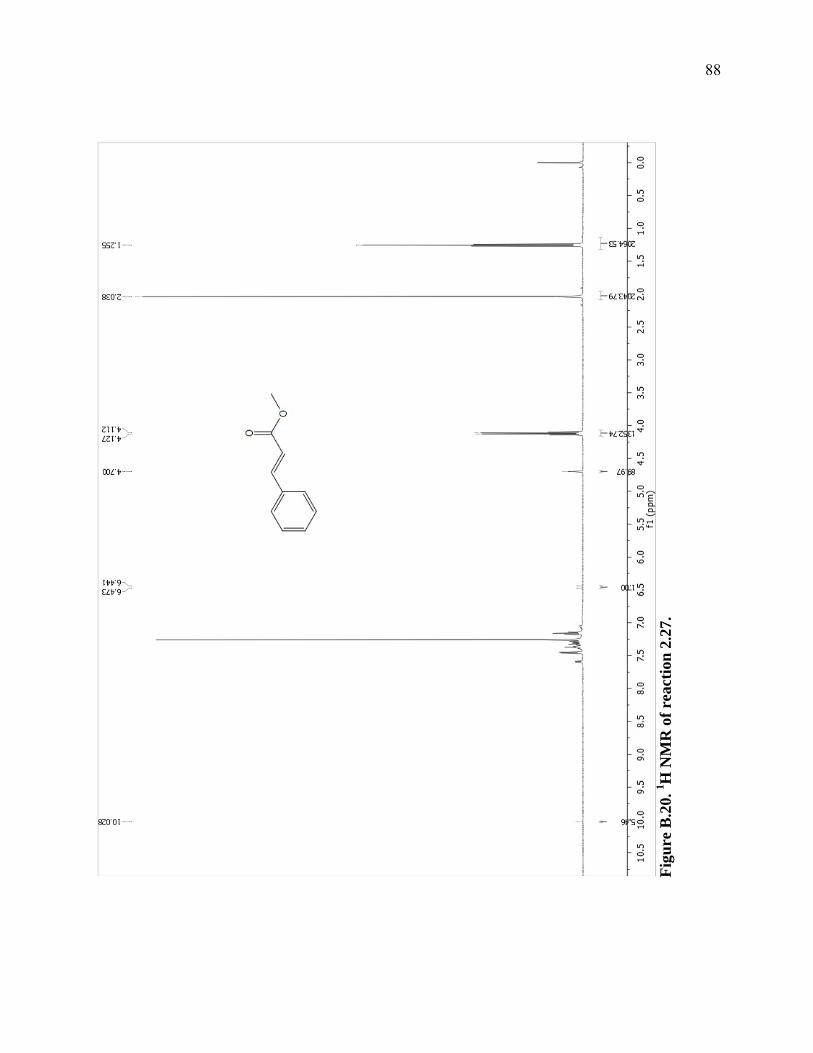
88
F
igu
re B
.20.
1 H N
MR
of
reac
tion
2.2
7.

89
F
igu
re B
.21.
1 H N
MR
of
reac
tion
2.2
8.
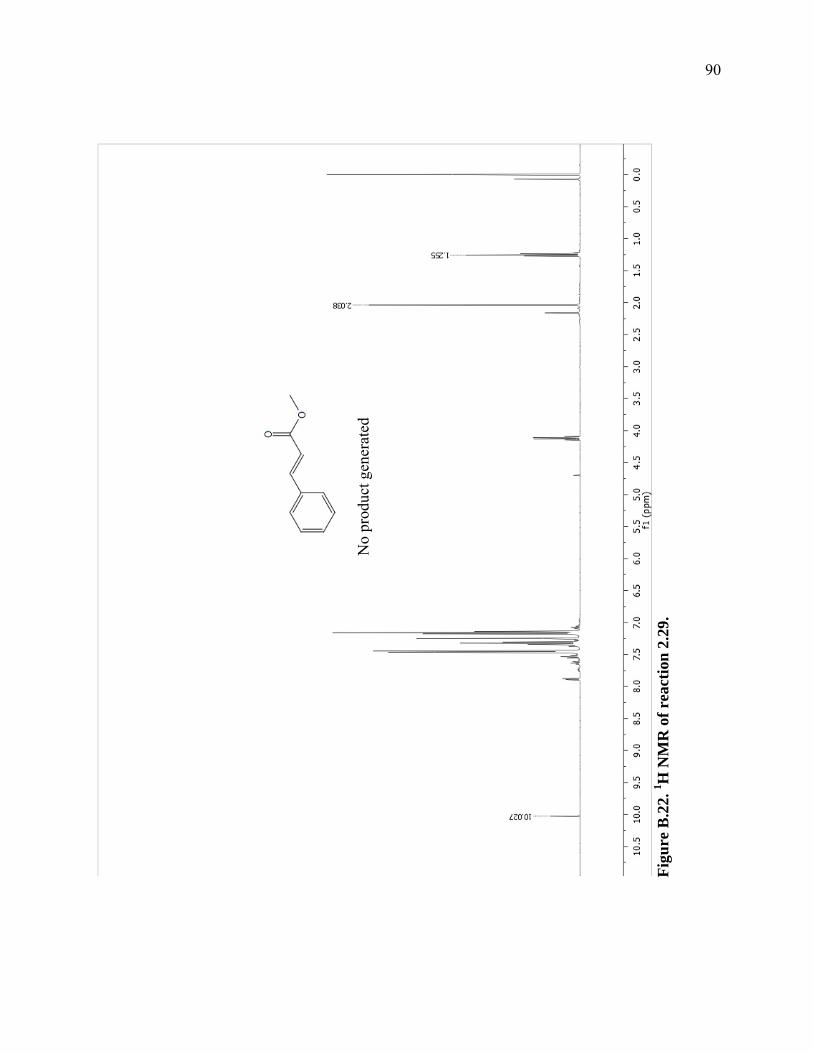
90
F
igu
re B
.22.
1 H N
MR
of
reac
tion
2.2
9.
No
prod
uct g
ener
ated

91
F
igu
re B
.23.
1 H N
MR
of
reac
tion
2.3
0.

92
F
igu
re B
.24.
1 H N
MR
of
reac
tion
2.3
1.
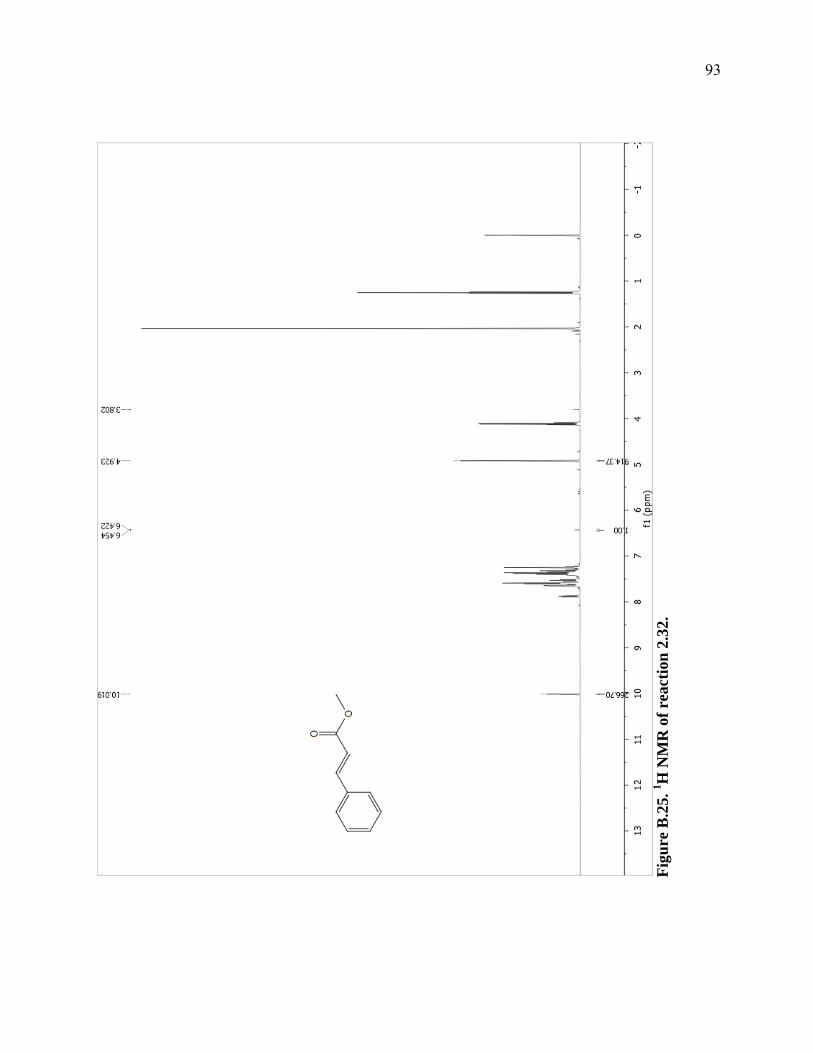
93
F
igu
re B
.25.
1 H N
MR
of
reac
tion
2.3
2.

94
F
igu
re B
.26.
1 H N
MR
of
reac
tion
2.3
3.

95
F
igu
re B
.27.
1 H N
MR
of
reac
tion
2.3
4.
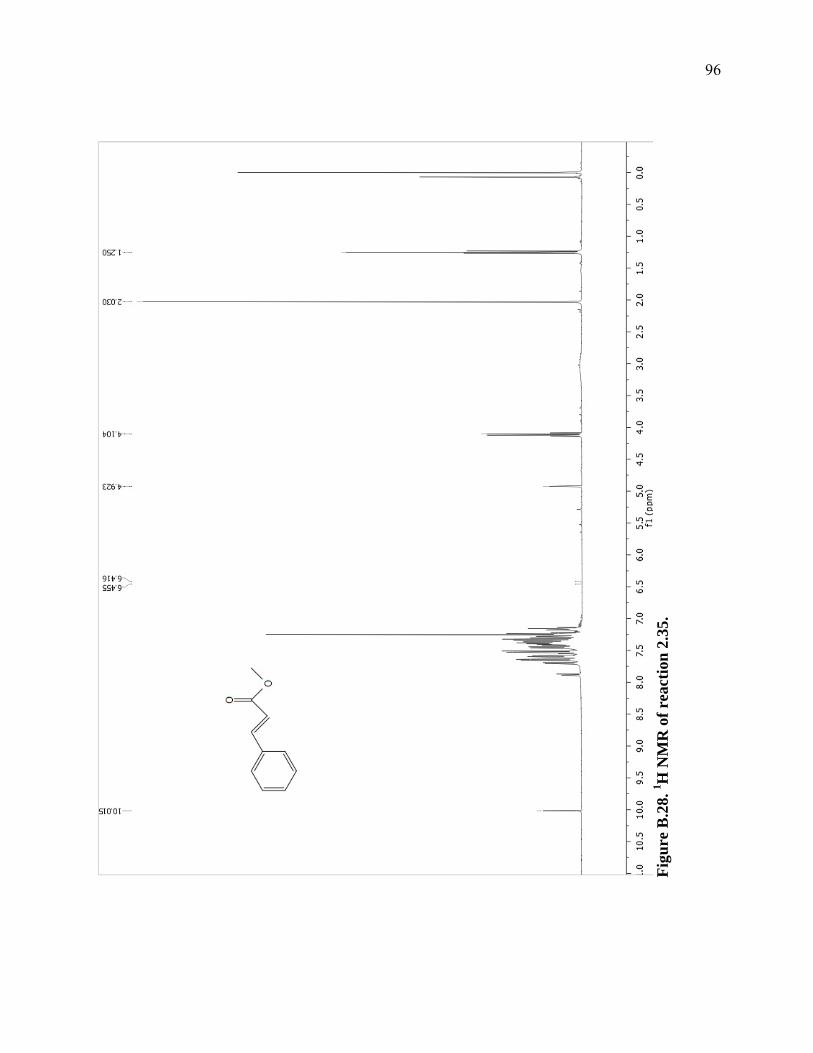
96
F
igu
re B
.28.
1 H N
MR
of
reac
tion
2.3
5.




















