
Occurrence of an anomalous endocytic compartment in fibroblasts from Sandhoff disease patients
10
Occurrence of an anomalous endocytic compartment in fibroblasts from Sandhoff disease patients Brunella Tancini • Alessandro Magini • Loredana Latterini • Lorena Urbanelli • Virginia Ciccarone • Fausto Elisei • Carla Emiliani Received: 12 June 2009 / Accepted: 16 September 2009 / Published online: 2 October 2009 Ó Springer Science+Business Media, LLC. 2009 Abstract Sandhoff disease (SD) is a lysosomal storage disorder due to mutations in the gene encoding for the b-subunit of b-hexosaminidase, that result in b-hexosa- minidase A (ab) and b-hexosaminidase B (bb) deficiency. This leads to the storage of GM2 ganglioside in endosomes and lysosomes, which ends in a progressive neurodegen- eration. Currently, very little is known about the bio- chemical pathways leading from GM2 ganglioside accumulation to pathogenesis. Defects in transport and sorting by the endosomal–lysosomal system have been described for several lysosomal storage disorders. Here, we have investigated the endosomal–lysosomal compartment in fibroblasts from SD patients and observed that both late endosomes and lysosomes, but not early endosomes, have a higher density in comparison with normal fibroblasts. Moreover, Sandhoff fibroblasts have an intracellular dis- tribution of terminal endocytic organelles that differs from the characteristic perinuclear punctate pattern observed in normal fibroblasts and endocytic vesicles also appear lar- ger. These findings reveal the occurrence of an alteration in the terminal endocytic organelles of Sandhoff fibroblasts, suggesting an involvement of this compartment in the disruption of cell metabolic and signalling pathways and in the onset of the pathological state. Keywords Lysosomal storage disease Sandhoff disease Endocytic compartment GM2 storage Introduction The endosomal–lysosomal system plays important roles in cellular physiology. Beyond the well known function as terminal degradative compartment necessary to maintain the health of the cell, lysosomes are critical for many other cellular processes, such as termination of signaling medi- ated by cell-surface receptors [1] and degradation of internalized peptides in antigen presenting cells [2]. Moreover, the intracellular membrane trafficking related to the endosomal–lysosomal system plays pivotal roles in diverse physiological and pathological processes, such as secretion, plasma membrane repair, and endocytosis [3–5]. Mutations in the genes encoding for lysosomal hydro- lases cause a group of severe diseases named lysosomal storage disorders (LSD). These diseases are characterized by accumulation within the lysosomes of partially unde- graded catabolic products [6]. Currently, very little is known about biochemical mechanisms leading from the accumulation of these substrates to the disruption of cell metabolic and signaling pathways, ending with the onset of the pathology. Many recent studies have shown that several LSD share pathological features with age-related neuro- degenerative diseases such as Alzheimer’s and Parkinson diseases, namely atypical central nervous system inflam- mation [7], altered calcium homeostasis [8], and impaired endosomal–lysosomal degradative capacity [9]. In addi- tion, there is evidence that lysosomes in patients affected by some LSD are characterized by remarkable changes in terms of their chemical–physical and physiological prop- erties [10–13]. B. Tancini A. Magini L. Urbanelli V. Ciccarone C. Emiliani (&) Department of Experimental Medicine and Biochemical Sciences, University of Perugia, via del Giochetto, 06126 Perugia, Italy e-mail: [email protected] L. Latterini F. Elisei Department of Chemistry, University of Perugia, via Elce di Sotto, 06125 Perugia, Italy 123 Mol Cell Biochem (2010) 335:273–282 DOI 10.1007/s11010-009-0277-0
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Occurrence of an anomalous endocytic compartment in fibroblasts from Sandhoff disease patients

Occurrence of an anomalous endocytic compartmentin fibroblasts from Sandhoff disease patients
Brunella Tancini • Alessandro Magini •
Loredana Latterini • Lorena Urbanelli •
Virginia Ciccarone • Fausto Elisei • Carla Emiliani
Received: 12 June 2009 / Accepted: 16 September 2009 / Published online: 2 October 2009
� Springer Science+Business Media, LLC. 2009
Abstract Sandhoff disease (SD) is a lysosomal storage
disorder due to mutations in the gene encoding for the
b-subunit of b-hexosaminidase, that result in b-hexosa-
minidase A (ab) and b-hexosaminidase B (bb) deficiency.
This leads to the storage of GM2 ganglioside in endosomes
and lysosomes, which ends in a progressive neurodegen-
eration. Currently, very little is known about the bio-
chemical pathways leading from GM2 ganglioside
accumulation to pathogenesis. Defects in transport and
sorting by the endosomal–lysosomal system have been
described for several lysosomal storage disorders. Here, we
have investigated the endosomal–lysosomal compartment
in fibroblasts from SD patients and observed that both late
endosomes and lysosomes, but not early endosomes, have a
higher density in comparison with normal fibroblasts.
Moreover, Sandhoff fibroblasts have an intracellular dis-
tribution of terminal endocytic organelles that differs from
the characteristic perinuclear punctate pattern observed in
normal fibroblasts and endocytic vesicles also appear lar-
ger. These findings reveal the occurrence of an alteration in
the terminal endocytic organelles of Sandhoff fibroblasts,
suggesting an involvement of this compartment in the
disruption of cell metabolic and signalling pathways and in
the onset of the pathological state.
Keywords Lysosomal storage disease � Sandhoff
disease � Endocytic compartment � GM2 storage
Introduction
The endosomal–lysosomal system plays important roles in
cellular physiology. Beyond the well known function as
terminal degradative compartment necessary to maintain
the health of the cell, lysosomes are critical for many other
cellular processes, such as termination of signaling medi-
ated by cell-surface receptors [1] and degradation of
internalized peptides in antigen presenting cells [2].
Moreover, the intracellular membrane trafficking related to
the endosomal–lysosomal system plays pivotal roles in
diverse physiological and pathological processes, such as
secretion, plasma membrane repair, and endocytosis [3–5].
Mutations in the genes encoding for lysosomal hydro-
lases cause a group of severe diseases named lysosomal
storage disorders (LSD). These diseases are characterized
by accumulation within the lysosomes of partially unde-
graded catabolic products [6]. Currently, very little is
known about biochemical mechanisms leading from the
accumulation of these substrates to the disruption of cell
metabolic and signaling pathways, ending with the onset of
the pathology. Many recent studies have shown that several
LSD share pathological features with age-related neuro-
degenerative diseases such as Alzheimer’s and Parkinson
diseases, namely atypical central nervous system inflam-
mation [7], altered calcium homeostasis [8], and impaired
endosomal–lysosomal degradative capacity [9]. In addi-
tion, there is evidence that lysosomes in patients affected
by some LSD are characterized by remarkable changes in
terms of their chemical–physical and physiological prop-
erties [10–13].
B. Tancini � A. Magini � L. Urbanelli � V. Ciccarone �C. Emiliani (&)
Department of Experimental Medicine and Biochemical
Sciences, University of Perugia, via del Giochetto,
06126 Perugia, Italy
e-mail: [email protected]
L. Latterini � F. Elisei
Department of Chemistry, University of Perugia, via Elce di
Sotto, 06125 Perugia, Italy
123
Mol Cell Biochem (2010) 335:273–282
DOI 10.1007/s11010-009-0277-0

Glycosphingolipid (GSL) storage disorders are a sub-
group of LSD caused by mutations in either hydrolase or
activator protein genes involved in catabolism of GSL. The
defective hydrolysis of these compounds leads to their
accumulation, mainly in neuronal endosomes and lyso-
somes. Fibroblasts from patients affected by a broad group
of GSL storage disorders have been reported to show
defects in lipid sorting and transport along the endocytic
pathway [14, 15]. GSL storage has been suggested to alter
membrane microdomain sphingolipid composition, thus
provoking the mistargeting and disrupted function of
membrane microdomain associated proteins that are
involved in signaling and transport processes [16]. The
alteration of the endocytic pathway has also implications
for therapeutic approaches toward GSL storage disorders.
In fact, enzyme replacement therapy and gene therapy
might fail because of abnormalities in enzyme targeting
within the cell, due to an impairment of the receptor-
mediated endocytosis pathway [17].
Sandhoff disease (SD) is a GM2 gangliosidosis due to
mutations in the gene encoding for the b-subunit of the acidic
glycohydrolase b-hexosaminidase (Hex, EC 3.2.1.52)
[18–20]. This results in the deficit of both b-hexosaminidase
A (ab, Hex A) and b-hexosaminidase B (bb, Hex B) enzy-
matic activities. Lack of Hex A, that is the only isoform that
hydrolyzes GM2 ganglioside, elicits the intralysosomal
accumulation of this substrate mainly in the central nervous
system, leading to neurodegeneration and eventually to
death [18]. In order to gain insight into endosomal–lyso-
somal compartment features in SD we have characterized
some chemical–physical and functional aspects of the
endocytic compartment in a peripheral cell model of the
disease, i.e., fibroblasts from SD patients. Fibroblasts are a
consolidated and easily accessible source to study the path-
ogenic mechanisms that are responsible for neurological
disorders and have revealed a wealth of information on how
such disorders occur [21, 22]. In addition, fibroblasts are
usually used to set up gene transfer strategies and enzyme
replacement protocols for therapy. Here, we report that
Sandhoff fibroblasts show alterations of the endocytic
compartment. These abnormalities have been observed in
late endosomes and lysosomes, but not in early endosomes,
and may be involved in the pathological cascade of events
leading to the onset of the disease.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), Fetal Bovine
Serum, Trypsin, and Penicillin/Streptomycin were from
Seromed, Biospa. 2,20-Azino-bis(3-ethylbenzothiazoline)-6-
sulfonic acid (ABTS), acridine orange, bovine serum albumin,
fluorescein isothiocyanate (FITC)-dextran (PM 10000),
Imidazole, 4-methylumbelliferyl-b-galactoside (MUGal),
4-methylumbelliferone, monoclonal horseradish peroxidase
(HRP)-conjugated anti-rabbit and HRP-conjugated goat anti-
mouse secondary antibodies, HRP Type II, protease inhibitor
cocktail for mammalian cell extracts and sucrose were from
Sigma-Aldrich Fine Chemicals Co. Mouse monoclonal anti-
lysosomal associated membrane protein-2 (LAMP-2)(H4B4),
rabbit polyclonal anti-Rab7 (H-50), goat polyclonal anti-Rab7
(C-19), and rabbit polyclonal anti-Rab5A (S-19) antibodies
were from Santa Cruz Biotechnology, Inc. Rabbit polyclonal
antibody to anti-early endosome antigen 1 (EEA1) was from
Abcam. Alexa Fluor 488-conjugated donkey anti-rabbit,
Alexa Fluor 546-conjugated goat anti-mouse, and Alexa Fluor
546-conjugated donkey anti-goat secondary antibodies were
from Molecular Probes. Hybond-C Extra nitrocellulose and
Enhanced Chemilumiscence (ECL) Western Blotting detec-
tion reagents were from Amersham Biosciences. Protein assay
reagent and chemicals for SDS-PAGE were from Bio-Rad
Laboratories. All other reagents were of analytical grade.
Cell cultures
Fibroblasts from SD patients and healthy subjects matched
for sex and age were kindly provided by Dr. Mirella
Filocamo (Laboratorio di Diagnosi Pre/Post-natale Malattie
Metaboliche, Genova, Italy). All experiments were per-
formed using three different fibroblast samples from both
SD patients and healthy controls. All SD patients were
affected by the early onset form of the disease, character-
ized by neurological and mental deterioration with
regression of psychic and motor functions. The mutations
carried by the three genotypes, [c.299G [ T (r = 0)] ?
[c.512-1G[T (p.G171-L172del)], [c.300-2A[G (p.R101_
S148delfsX12)] ? [c.300-2A[G (p.R101_S148delfsX12)],
[c.850C [ T(p.R284X)] ? [c.1613 ? 2T [ G (p.E538ins
4X5)], have been previously characterized [23]. The activ-
ity of Hex A and Hex B isoenzymes in each cell sample
was measured after their separation by ion-exchange
chromatography and in SD fibroblasts it was less than 0.3%
as compared to control fibroblasts. Cells were cultured in
DMEM supplemented with 10% (v/v) heat-inactivated
fetal bovine serum, 2 mM glutamine, 100 IU/ml penicillin,
100 lg/ml streptomycin in a 5% CO2 incubator at 37�C.
Cell viability was determined by the Trypan blue method.
Continuous sucrose gradient
Microsome preparation and subcellular fractionation on
continuous sucrose gradient were performed as reported in
Yeyeodu et al. [24] with minor modifications. Briefly,
274 Mol Cell Biochem (2010) 335:273–282
123

Sandhoff and normal fibroblasts were scraped and resus-
pended on ice-cold 3 mM imidazole buffer, pH 7.4, con-
taining 0.25 M sucrose, 0.5 mM PMSF and 1 mM EDTA.
After an incubation on ice for 15 min, cells were homog-
enized with 25 strokes of a Dounce homogenizer (pestle A)
and centrifuged at 3,000g for 15 min. The supernatant was
recovered and centrifuged again on a Beckman Optima
Max Ultracentrifuge at 100,000g (TLA 100.3 rotor) for 1 h
at 4�C. Finally, the microsome pellet was resuspended,
layered on the top of a continuous sucrose gradient pre-
pared using 0.5 and 1.6 M sucrose (2.4 ml ? 2.4 ml) in
20 mM imidazole buffer, pH 7.4, and centrifuged at
170,000g for 70 min at 4�C on a Beckman MLS-50 rotor.
Fractions (0.260 ml) were collected from the top of the
gradient and aliquots used for Western blots and enzyme
assays.
Western blotting analysis
Samples were subjected to 12% SDS-PAGE under reduc-
ing conditions according to Laemmli [25]. Proteins were
transferred to nitrocellulose and the membrane probed with
primary antibody at the appropriate concentration and then
incubated with HRP-conjugated secondary antibody. Blots
were finally revealed using the ECL detection system.
Densitometric analysis was performed using the Image-
Master2D platinum software (Amersham Bioscience).
Enzyme assay
Triton X-100 (0.1%) was added to an aliquot of each frac-
tion gradient to solubilize cellular membranes. The activity
of the lysosomal enzyme b-galactosidase was determined
using the fluorogenic substrate MUGal 1.5 mM in 0.1 M
citric acid/0.2 M disodium phosphate buffer, pH 4.5 [26].
Fluorescence of the 4-methylumbelliferone product was
measured on a Perkin-Elmer LS B50 fluorimeter (excitation,
360 nm; emission, 446 nm). One enzymatic unit (U) is the
amount of enzyme that hydrolyzes 1 lmol of substrate/min
at 37�C.
Protein concentration was determined by the method of
Bradford [27] using bovine serum albumin as standard.
Fluorescence and confocal microscopy
Cells grown on glass coverslips were washed with phosphate-
buffered saline (PBS), fixed with 3.7% paraformaldehyde in
PBS for 10 min at room temperature, washed again with PBS
and then permeabilized and blocked in 0.2% Triton X-100/
PBS containing 3% bovine serum albumin (blocking solution)
for 1 h. Primary antibodies were diluted at the appropriate
concentration in blocking solution, added to cells and incu-
bated for 1 h. After washings, cells were incubated for 1 h
with the appropriate Alexa Fluor-conjugated secondary anti-
bodies diluted 1:2,000 in blocking solution. All incubation
steps were carried out at room temperature. Coverslips were
then washed with PBS containing 0.2% Triton X-100, rinsed
with water and mounted with Vectashield. For dextran loading
experiments, cells grown on glass coverslips were incubated
for 18 h in culture medium containing 2 mg/ml FITC-dex-
tran, rinsed with PBS, chased for 2 or 4 h in medium devoid of
the fluid-phase marker and then fixed and mounted as above.
For acridine orange staining, cells were grown on glass cov-
erslips and incubated for 10 min at 37�C in the presence of
2.5 lg/ml of acridine orange in the culture medium. After
washing with fresh DMEM, living cells were immediately
viewed by fluorescence microscopy. Cells were visualized
using a fluorescence light microscope (Nikon) through a 609
oil immersion objective and images were acquired with a
Sony camera. Confocal microscopy analysis of the samples
was carried out through a laser scanning confocal microscope
(Nikon, PCM2000). In this case, fluorescence images were
recorded using Ar- or He–Ne-lasers (kexc = 488 and 543 nm,
respectively) as light source and two acquisition channels
which differ for the spectral region selected by band-pass
filters, then visualized in different colors (false colors). Images
were obtained with a 609, 1.4 N.A. oil immersion objective
(512 9 512 pixels). Image processing was performed using
Adobe Photoshop software. For the quantification of the
colocalization all red- and green-colored pixels that were
found at identical positions in a confocal section were com-
pared to those red- and green-colored pixels that could not be
located to the same position. This analysis was performed for
at least 15 cells for each cell sample (45 cells for each
genotype).
Measurement of lysosomal pH
Sandhoff and normal fibroblasts were incubated for 18 h at
37�C in culture medium containing 2 mg/ml FITC-dextran
(mol wt 10 kDa), rinsed with PBS and then chased for
90 min in fresh medium devoid of the fluid-phase marker.
Cells were scraped, resuspended in PBS and analyzed by
spectrofluorimeter to determine the lysosomal pH according
to Ohkuma et al. [28] with minor modifications. Briefly, cell
samples were sequentially excited at 457 and 488 nm and
the emitted fluorescence was measured in each case at
515 nm. The pH of the terminal endocytic organelles was
estimated comparing the ratio of the two fluorescence
intensities with a standard curve generated with FITC-
dextran-loaded microsomes at defined pH. Labeled
microsomes were prepared by loading of fibroblasts with
FITC-dextran as described above and subsequent centrifu-
gation of the post-nuclear supernatant at 100,000g for 1 h.
Aliquots of pelleted microsomes were resuspended in
Mol Cell Biochem (2010) 335:273–282 275
123

buffers of defined pH in the presence of 1 lM nigericin to
dissipate a potential pH gradient.
Fluid-phase endocytosis
Sandhoff and normal fibroblasts were incubated with pre-
warmed 3 mg/ml horseradish peroxidase (HRP) in DMEM
for 2 h at 37�C followed by extensive washing with PBS.
Cells were then trypsinized, pelleted and resuspended in
40 mM potassium phosphate buffer containing 0.5% (v/v)
Triton X-100, pH 6.8. After 30 min of incubation on ice,
cell lysates were centrifuged at 16,000g for 10 min and the
supernatants were used for HRP activity determination
using 2,20-Azino-bis(3-ethylbenzothiazoline)-6-sulfonic
acid (ABTS) as substrate [29, 30]. Briefly, 2–4 ll of
diluted extract were added to 1 ml of 0.7 mM ABTS in
100 mM potassium phosphate buffer, pH 5.0 at 25�C; then
30 ll of fresh prepared 0.3% H2O2 were added, samples
were mixed by inversion and the increase in absorbance at
405 nm was immediately recorded for 4 min. DA405 nm/min
was calculated using the maximum linear rate. The same
procedure was performed with untreated cells as negative
control. One unit (U) is the amount of enzyme that oxidizes
1 lmol of substrate/min at pH 5.0 at 25�C.
Results
Subcellular fractionation on continuous sucrose
gradient of Sandhoff fibroblast endocytic organelles
Continuous density gradients in the range of 0.3–1.6 M
sucrose were optimized to determine the buoyant density of
fibroblast endocytic organelles. Microsomal fractions from
both Sandhoff and normal fibroblasts were layered on the
top of the gradient and subjected to ultracentrifugation, as
described in the ‘‘Materials and methods’’ section. The
endocytic organelle buoyant density of Sandhoff and nor-
mal fibroblasts was compared by Western blotting analysis
of gradient fractions with organelle specific markers. Pro-
teins detected were Rab5 as early endosomal marker [31],
Rab7 as late endosomal marker [32], and LAMP-2 as
lysosomal marker [33]. Among density gradient systems
tested, the best separation profile was achieved using a
0.5–1.6 M sucrose gradient, although a very similar dis-
tribution pattern was also observed with a 0.7–1.5 M
sucrose gradient (data not shown). Results are reported in
Fig. 1. The comparison of normal and pathological orga-
nelle distribution profiles reveals differences between
Sandhoff and normal fibroblasts related to terminal
Fig. 1 Distribution of endocytic organelles in continuous sucrose
gradient. Normal (NF) and Sandhoff (SF) fibroblast microsomes were
resolved on a 0.5–1.6 M sucrose density gradient, and 0.260 ml
fractions were collected from the top. Proteins were separated by
SDS–PAGE and visualized by immunoblotting. Representative
Western blots (a) and densitometric analysis (b) showing the
distribution of LAMP-2, Rab7, and Rab5 proteins in the gradient
fractions are reported. The results are expressed as percentages of the
respective band volume in each fraction to the sum of all fractions.
Three different fibroblast samples from both SD patients and healthy
controls were used and three independent experiments for each cell
sample were performed. Very similar results were obtained for each
genotype. Here, representative results of one experiment with SD and
normal fibroblasts are reported
276 Mol Cell Biochem (2010) 335:273–282
123

endocytic vesicles distribution. In fact, although lysosomes
were found to be typically distributed in two major peaks in
both cell types as already described [24], their buoyant
density profiles were dissimilar: light lysosomes appeared
mainly localized in the gradient fractions 3–4 in both
Sandhoff and normal fibroblasts, while dense lysosomes
appeared mainly localized in the gradient fractions 11–12
in control fibroblasts and 16–17 in Sandhoff fibroblasts
(Fig. 1-LAMP-2). Moreover, late endosomes detected by
the Rab7 marker protein resulted mainly distributed in the
gradient fractions 4–5 and 9–10 in control fibroblasts and
in the fractions 6–7 and 10–11 in Sandhoff fibroblasts
(Fig. 1-Rab7). The buoyant property of early endosomes
was also assessed and in this case the distribution of the
early endosome marker Rab5 resulted very similar in
Sandhoff and normal fibroblasts, as determined by Western
blot analysis of gradient fractions. In fact, as shown by
densitometry profiles, early endosomes were broadly dis-
tributed in the fractions ranging from 4 to 11 in both cell
types (Fig. 1-Rab5).
As the observed difference could be due to abnormali-
ties in the delivery of the protein marker LAMP-2, a
membrane protein that is targeted to lysosomes by clathrin-
coated pits, the gradient distribution of lysosomes was also
assessed by evaluation of the enzymatic activity of
b-galactosidase, a soluble lysosomal glycohydrolase that is
targeted to lysosomes by a mannose 6-phosphate receptor-
dependent pathway [34]. The density gradient pattern
obtained with b-galactosidase assays overlapped that
observed with anti-LAMP-2 antibody, thus confirming that
in Sandhoff fibroblasts the population of dense lysosomes
is denser than in normal fibroblasts (Fig. 2).
Analysis of endocytic compartment
by immunofluorescence
Immunofluorescence microscopy was used to specifically
label endocytic organelles of Sandhoff and normal fibro-
blasts. Early endosomes were labeled using EEA1 (early
endosome antigen 1) as marker [35] and fluorescence
microscopy analysis (Fig. 3-EEA1) showed homogeneously
dispersed granules in the cytoplasm of both normal and
pathological fibroblasts. The lysosomal compartment
stained with anti-LAMP-2 antibodies evidenced the char-
acteristic perinuclear punctate pattern in normal fibroblasts,
whereas Sandhoff fibroblast lysosomes appeared as aggre-
gate structures mainly localized in a restricted region of the
cytoplasm (Fig. 3-LAMP-2).
Confocal microscopy analysis was then used to further
characterize the late endocytic compartment in normal and
pathological cells. It was previously reported that in normal
fibroblasts the lysosomal membrane protein LAMP-2 par-
tially colocalized with the late endosome marker Rab7 [36].
Sandhoff and normal fibroblasts were stained for LAMP-2
and Rab-7 and representative images are shown in Fig. 4,
panel A. We confirmed that in normal fibroblasts LAMP-2
partially colocalize with Rab7. However, in Sandhoff
fibroblasts the extent of Rab7 and LAMP-2 signals over-
lapping was less consistent. The quantification of the colo-
calization degree (see ‘‘Materials and methods’’ for details)
Fig. 2 Distribution of b-galactosidase in continuous sucrose gradi-
ent. Normal (NF) and Sandhoff (SF) fibroblast microsomes were
resolved on a 0.5–1.6 M sucrose density gradient and 0.260 ml
fractions were collected from the top. b-galactosidase distribution in
the gradient fractions was determined by enzyme activity assay. The
results are expressed as percentages of the respective enzymatic
activity in each fraction to the total activity. Three different fibroblast
samples from both SD patients and healthy controls were used and
three independent experiments for each cell sample were performed.
Very similar results were obtained for each genotype. The results
shown the means ± SD of all the experiments performed with each
genotype
Fig. 3 Intracellular distribution of early endosomes and lysosomes in
normal and Sandhoff fibroblasts. Normal (NF) and Sandhoff (SF)
fibroblasts were fixed, permeabilized and immunostained with
antibodies to anti-EEA1 and anti-LAMP-2 to label early endosomes
and lysosomes, respectively. The labeled compartments were then
visualized by fluorescence microscopy. The images show represen-
tative cells, based on experiments with three independent cell samples
of both SD patient and human normal fibroblasts
Mol Cell Biochem (2010) 335:273–282 277
123

revealed that about 82% of Rab7 could be colocalized with
LAMP-2 in normal, while this percentage decreased to 49%
in Sandhoff fibroblasts. Very poor colocalization was
observed between the early endosome marker EEA1 and
the late endosome marker Rab 7 in both cell types (Fig. 4,
panel B), thus confirming that the early endosomes com-
partment did not differ between pathological and normal
conditions.
Uptake of FITC-dextran
Sandhoff and normal fibroblasts were incubated with FITC-
dextran for 24 h (pulse), then the fluid-phase marker was
chased for 2 or 4 h. The longest period of chase was carried
out to ensure that internalized FITC-dextran molecules had
reached the final destination. After the treatment, cells were
analyzed by fluorescence microscopy and representative
results are reported in Fig. 5. It clearly appeared (panel A)
that pathological cells, as well as their normal counterpart,
were able to internalize FITC-dextran molecules. However,
the intracellular distribution of fluorescent granules was
different in Sandhoff fibroblasts with respect to normal cells,
as perinuclear punctate structures were observed in normal
fibroblasts, while in Sandhoff fibroblasts FITC-dextran loa-
ded vesicles appeared as aggregates mainly localized in a
restricted region of the cytoplasm. Furthermore, the endo-
cytic vesicles of Sandhoff fibroblasts appeared brighter than
those of normal cells, suggesting that a larger amount of
FITC-dextran had been loaded. No differences were
observed between 2 and 4 h of chase (data not shown).
Confocal analysis revealed that endocytic vesicles
reached by dextran were lysosomes in normal fibroblasts as
well as in Sandhoff fibroblasts. In fact, in both cell types
the fluorescent fluid-phase marker showed a high degree of
colocalization with the lysosomal marker LAMP-2 (Fig. 5,
panel B). Again, Sandhoff fibroblast vesicles loaded with
FITC-dextran appeared more fluorescent compared to
normal fibroblast vesicles.
As it is known that the FITC-dextran fluorescence
properties are dependent on the pH, the lumenal pH of the
Fig. 4 Immunofluorescence
microscopy colocalization of
endocytic organelles in normal
and Sandhoff fibroblasts. Normal
(NF) and Sandhoff (SF)
fibroblasts were fixed,
permeabilized and
immunostained with: antibodies
to anti-LAMP-2 and anti-Rab7 to
label lysosomes and late
endosomes, respectively,
(panel A) or antibodies to anti-
Rab7 and anti-EEA1 to label late
endosomes and early
endosomes, respectively
(panel B). Cells were visualized
by confocal microscopy. Right
columns display the merged
images. The images show
representative cells, based on
experiments with three
independent cell samples of both
SD patient and human normal
fibroblasts
278 Mol Cell Biochem (2010) 335:273–282
123

terminal endocytic compartment of normal and pathologi-
cal fibroblasts was measured to verify if the observed dif-
ferences in vesicle fluorescence brightness could arise from
a difference in the pH of the acidic environment of these
organelles. The apparent lysosomal pH, estimated by a
fluorimetric method as described in the ‘‘Materials and
methods’’ section, resulted to be 5.13 (SD ± 0.01) in
normal fibroblasts and 5.15 (SD ± 0.03) in Sandhoff
fibroblasts, thus indicating that pathological cells have a
normal lysosomal pH value, in good agreement with data
available for other mammalian cell types (Fig. 6) [12].
Acridine orange is a lysosomotropic fluorescent probe
that shifts from green to orange light emission upon pro-
tonation at low pH. Staining with acridine orange con-
firmed both the normal acidic pH and the perinuclear
aggregation of terminal endocytic vesicles observed by
FITC-dextran uptake in Sandhoff cells (Fig. 7). It is worth
underlying that the aggregate appearance of pathological
fibroblast organelles appeared to be consistent with their
higher density with respect to the control, as resulted by
sucrose density gradient experiments.
Fluid-phase endocytosis
The pH measurements excluded the occurrence of an
alteration in the acidic environment of the endocytic
organelles as possible cause of Sandhoff fibroblast vesicles
higher fluorescence. Therefore, to explain the nature of the
more intense fluorescence of pathological fibroblasts with
respect to controls, the function of the endocytic pathway
was assessed by determining the uptake of the fluid-phase
marker horseradish peroxidase (HRP) into both normal and
Sandhoff cells. Cells were incubated with 3 mg/ml of HRP
for 2 h and then the amount of accumulated HRP in the
cells was quantified by enzymatic assay. As reported in
Fig. 8, the intracellular accumulation of HRP was found to
be about twice in Sandhoff fibroblasts as compared to
Fig. 5 Fluorescence microscopy characterization of endocytic vesi-
cles in normal and Sandhoff fibroblasts. Normal (NF) and Sandhoff
(SF) fibroblasts were loaded for 18 h with FITC-dextran followed by
2 h chase in the absence of the fluid-phase marker. The cells were
then fixed and the labeled vesicles were visualized by fluorescence
microscopy (panel A). For confocal microscopy measurements (panel
B), FITC-dextran loaded cells were fixed, permealized and immuno-
stained with antibodies to anti-LAMP-2 to label lysosomes. The right
columns display the merged images. The images show representative
cells, based on experiments with three independent cell samples of
both SD patient and human normal fibroblasts
Fig. 6 Apparent lysosomal pH measurement. Normal (NF) and
Sandhoff (SF) fibroblasts were loaded with FITC-dextran as described
in ‘‘Materials and methods’’ section. The fluorescence intensity ratio
of cell samples were compared with a standard curve generated with
FITC-dextran-loaded microsomes at defined pH. Three independent
cell samples of both SD patient and human normal fibroblasts were
used and three independent measurements for each standard curve
point were performed
Fig. 7 Lysosome acridine orange staining. Normal (NF) and Sand-
hoff (SF) fibroblasts were fixed, permeabilized and stained with the
fluorescent day acridine orange to visualize the lysosomes by
fluorescence microscopy. The images show representative cells,
based on experiments with three independent cell samples of both SD
patient and human normal fibroblasts
Mol Cell Biochem (2010) 335:273–282 279
123
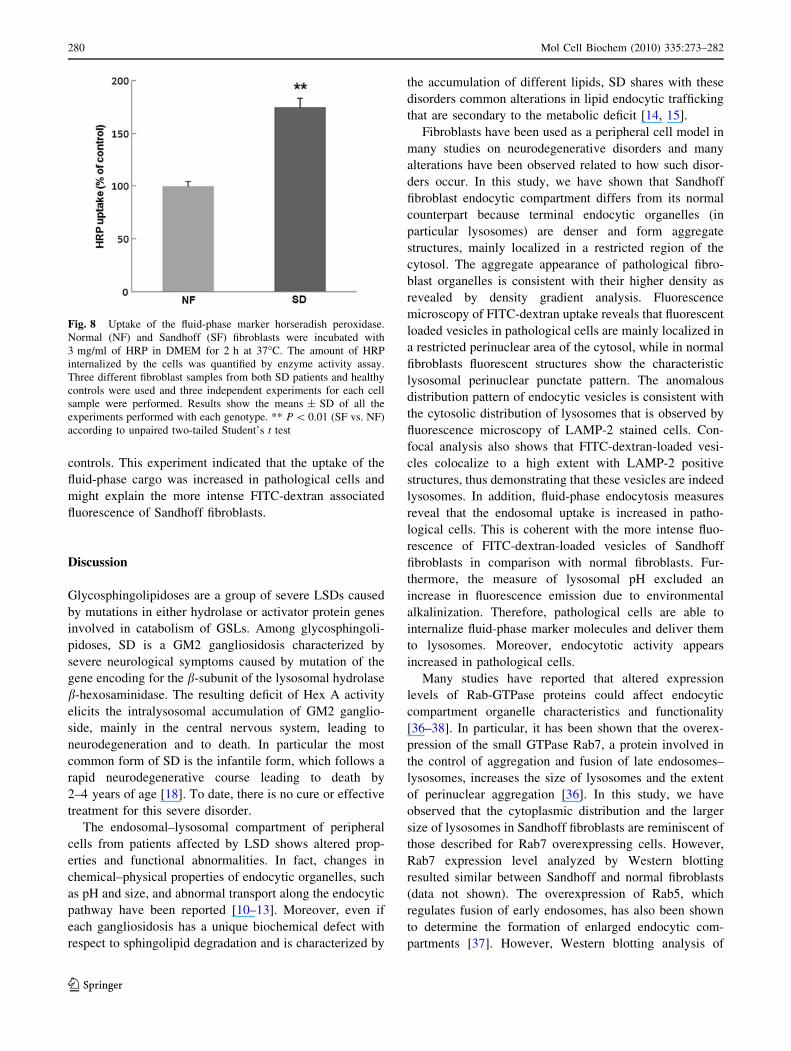
controls. This experiment indicated that the uptake of the
fluid-phase cargo was increased in pathological cells and
might explain the more intense FITC-dextran associated
fluorescence of Sandhoff fibroblasts.
Discussion
Glycosphingolipidoses are a group of severe LSDs caused
by mutations in either hydrolase or activator protein genes
involved in catabolism of GSLs. Among glycosphingoli-
pidoses, SD is a GM2 gangliosidosis characterized by
severe neurological symptoms caused by mutation of the
gene encoding for the b-subunit of the lysosomal hydrolase
b-hexosaminidase. The resulting deficit of Hex A activity
elicits the intralysosomal accumulation of GM2 ganglio-
side, mainly in the central nervous system, leading to
neurodegeneration and to death. In particular the most
common form of SD is the infantile form, which follows a
rapid neurodegenerative course leading to death by
2–4 years of age [18]. To date, there is no cure or effective
treatment for this severe disorder.
The endosomal–lysosomal compartment of peripheral
cells from patients affected by LSD shows altered prop-
erties and functional abnormalities. In fact, changes in
chemical–physical properties of endocytic organelles, such
as pH and size, and abnormal transport along the endocytic
pathway have been reported [10–13]. Moreover, even if
each gangliosidosis has a unique biochemical defect with
respect to sphingolipid degradation and is characterized by
the accumulation of different lipids, SD shares with these
disorders common alterations in lipid endocytic trafficking
that are secondary to the metabolic deficit [14, 15].
Fibroblasts have been used as a peripheral cell model in
many studies on neurodegenerative disorders and many
alterations have been observed related to how such disor-
ders occur. In this study, we have shown that Sandhoff
fibroblast endocytic compartment differs from its normal
counterpart because terminal endocytic organelles (in
particular lysosomes) are denser and form aggregate
structures, mainly localized in a restricted region of the
cytosol. The aggregate appearance of pathological fibro-
blast organelles is consistent with their higher density as
revealed by density gradient analysis. Fluorescence
microscopy of FITC-dextran uptake reveals that fluorescent
loaded vesicles in pathological cells are mainly localized in
a restricted perinuclear area of the cytosol, while in normal
fibroblasts fluorescent structures show the characteristic
lysosomal perinuclear punctate pattern. The anomalous
distribution pattern of endocytic vesicles is consistent with
the cytosolic distribution of lysosomes that is observed by
fluorescence microscopy of LAMP-2 stained cells. Con-
focal analysis also shows that FITC-dextran-loaded vesi-
cles colocalize to a high extent with LAMP-2 positive
structures, thus demonstrating that these vesicles are indeed
lysosomes. In addition, fluid-phase endocytosis measures
reveal that the endosomal uptake is increased in patho-
logical cells. This is coherent with the more intense fluo-
rescence of FITC-dextran-loaded vesicles of Sandhoff
fibroblasts in comparison with normal fibroblasts. Fur-
thermore, the measure of lysosomal pH excluded an
increase in fluorescence emission due to environmental
alkalinization. Therefore, pathological cells are able to
internalize fluid-phase marker molecules and deliver them
to lysosomes. Moreover, endocytotic activity appears
increased in pathological cells.
Many studies have reported that altered expression
levels of Rab-GTPase proteins could affect endocytic
compartment organelle characteristics and functionality
[36–38]. In particular, it has been shown that the overex-
pression of the small GTPase Rab7, a protein involved in
the control of aggregation and fusion of late endosomes–
lysosomes, increases the size of lysosomes and the extent
of perinuclear aggregation [36]. In this study, we have
observed that the cytoplasmic distribution and the larger
size of lysosomes in Sandhoff fibroblasts are reminiscent of
those described for Rab7 overexpressing cells. However,
Rab7 expression level analyzed by Western blotting
resulted similar between Sandhoff and normal fibroblasts
(data not shown). The overexpression of Rab5, which
regulates fusion of early endosomes, has also been shown
to determine the formation of enlarged endocytic com-
partments [37]. However, Western blotting analysis of
Fig. 8 Uptake of the fluid-phase marker horseradish peroxidase.
Normal (NF) and Sandhoff (SF) fibroblasts were incubated with
3 mg/ml of HRP in DMEM for 2 h at 37�C. The amount of HRP
internalized by the cells was quantified by enzyme activity assay.
Three different fibroblast samples from both SD patients and healthy
controls were used and three independent experiments for each cell
sample were performed. Results show the means ± SD of all the
experiments performed with each genotype. ** P \ 0.01 (SF vs. NF)
according to unpaired two-tailed Student’s t test
280 Mol Cell Biochem (2010) 335:273–282
123

Rab5 detected the same expression level between Sandhoff
and normal fibroblasts (data not shown). This demonstrates
that perinuclear aggregation of lysosomes is not due to
alterated expression of these Rab-GTPase proteins.
Furthermore, the normal level of Rab5 is consistent
with the fact that fluorescence microscopy and density
gradient fractionation have not revealed any significant
differences in the early endosome compartment between
pathological and control fibroblasts. Based on these
results we have inferred that early endosomes are normal
in Sandhoff cells and we have excluded that the observed
alterations of the endocytic compartment function might
arise from alteration of these organelles. Confocal anal-
ysis shows that in Sandhoff fibroblasts the degree of
Rab7/LAMP-2 colocalization is lower than in normal
fibroblasts. The density gradient centrifugation analysis
also supports confocal microscopy evidences. As a matter
of fact, densitometric profiles of normal and pathological
cells obtained by Western blotting analysis of gradient
fractions with anti-Rab7 and anti-LAMP-2 antibodies
overlap to a minor extent in Sandhoff than control
fibroblasts.
Provided results underline some peculiar differences
between the terminal but not the early endocytic com-
partment in Sandhoff fibroblasts as compared to normal
fibroblasts. In particular, the aggregate formation, the
anomalous cytosolic distribution of lysosomes and the
reduced extent of Rab7/LAMP-2 colocalization suggest
the occurrence of an alteration in the dynamic properties
of terminal endocytic organelles, as indicated for other
GSL storage diseases such as Niemann-Pick C [15, 16].
Our findings suggest that in Sandhoff cells an abnormal
lipid membrane composition due to the lysosomal GSL
accumulation might alter the distribution of proteins
controlling endosomal membrane dynamics, thus per-
turbing the functionality of the endocytic pathway. As the
integrity of endocytic compartment is important for axo-
nal and dendritic transport, this event might critically
impair cell function at central nervous system level.
Although more experiments are needed to verify if the
observed events occur in cell types other than fibroblasts
such as neurons, which are particularly affected in
Sandhoff disease, our results provide insight into the cell
response to the enzymatic deficiency leading to the GM2
ganglioside lysosomal storage. Fibroblasts are not
strongly compromised by the accumulation of GM2, so
the observed endosomal–lysosomal alteration highlights
an aspect of the cellular dysfunction causing the disease
not directly related to the substrate accumulation. In
addition, the integrity of endocytic compartment may be
critical for the effectiveness of therapeutic approaches
such as enzyme replacement and gene therapy. As a
matter of fact, the observed abnormalities could represent
an obstacle to the efficacy of these therapeutic strategies
[17], as their success requires the effective transport of
the therapeutic enzyme into lysosomes by an endocytosis
mechanism.
Acknowledgments This work was supported by COFIN-PRIN
(Cofinanziamento-Progetto di Ricerca di Interesse Nazionale) and
FIRB (Fondo per gli Investimenti della Ricerca di Base) grants to
C.E. This work was also supported by Fondazione Cassa di Risparmio
di Perugia, Grant 2008.021.375 to C.E. We thank the ‘‘Diagnosi
PrePostnatale Malattie Metaboliche’’ Laboratory (G.Gaslini Institute)
for providing us with specimens from the ‘‘Cell line and DNA bank
from patients affected by Genetic diseases’’ Biobank- Telethon
Genetic Biobank Network (project no. GTB07001A). We thank
Dr. Maria Ragano Caracciolo for the valuable comments and critical
reading of the manuscript.
References
1. Stoscheck CM, Carpenter G (1984) Down regulation of epider-
mal growth factor receptors: direct demonstration of receptor
degradation in human fibroblasts. J Cell Biol 98:1048–1053
2. Trejo J, Hammes SR, Coughlin SR (1998) Termination of sig-
naling by protease-activated receptor-1 is linked to lysosomal
sorting. Proc Natl Acad Sci USA 95:3698–3702
3. Blott EJ, Griffiths GM (2002) Secretory lysosomes. Nat Rev Mol
Cell Biol 3:122–131
4. Reddy A, Caler EV, Andrews NW (2001) Plasma membrane
repair is mediated by Ca2?-regulated exocytosis of lysosomes.
Cell 106:157–169
5. Cataldo AM, Peterhoff CM, Troncoso JC et al (2000) Endocytic
pathway abnormalities precede amyloid beta deposition in spo-
radic Alzheimer’s disease and Down syndrome: differential
effects of APOE genotype and presenilin mutations. Am J Pathol
157:277–286
6. Scriver CR, Beaudet AL, Sly WS, Valle DD (eds) (2001) Lyso-
somal disorders. The metabolic and molecular bases of inherited
disease, 8th edn, vol III. McGraw-Hill, New York, pp 3371–3894
7. Jeyakumar M, Dwek RA, Butters TD, Platt FM (2005) Storage
solutions: treating lysosomal disorders of the brain. Nat Rev
Neurosci 6:713–725
8. Ginzburg L, Kacher Y, Futerman AH (2004) The pathogenesis of
glycosphingolipid storage disorder. Semin Cell Dev Bio 15:
417–431
9. Bahr BA, Bendiske J (2002) The neuropathogenic contributions
of lysosomal dysfunction. J Neurochem 83:481–489
10. Ivleva TS, Ogloblina TA, Litinskaya LL, Wiederschain GY
(1991) Estimation and comparison of lysosomal and cytoplasmic
pH of human fibroblasts from healthy donors and patients with
lysosomal storage diseases. Biomed Sci 2:398–402
11. Bach G, Chen CS, Pagano RE (1999) Elevated lysosomal pH in
Mucolipidosis type IV cells. Clin Chim Acta 280:173–179
12. Schmid JA, Mach L, Paschke E, Glossl J (1999) Accumulation of
sialic acid in endocytic compartments interferes with the forma-
tion of mature lysosomes. J Biol Chem 274:19063–19071
13. Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y et al (2006) TRP-ML1
regulates lysosomal pH and acidic lysosomal lipid hydrolytic
activity. J Biol Chem 281:7294–7301
14. Pagano RE (2003) Endocytic trafficking of glycosphingolipids in
sphingolipid storage diseases. Philos Trans R Soc Lond B Biol
Sci 358:885–891 Review
15. Sillence DJ, Platt FM (2004) Glycosphingolipids in endocytic
membrane transport. Semin Cell Dev Biol 15:409–416
Mol Cell Biochem (2010) 335:273–282 281
123

16. Vruchte D, Lloyd-Evans E, Vldman RJ et al (2004) Accumula-
tion of glycosphingolipids in Niemann-Pick C disease disrupts
endosomal transport. J Biol Chem 279:26167–26175
17. Martino S, Emiliani C, Tancini B et al (2002) Absence of met-
abolic cross-correction in Tay-Sachs cells: implications for gene
therapy. J Biol Chem 277:20177–20184
18. Mahuran DJ (1999) Biochemical consequences of mutations
causing the GM2 gangliosidoses. Biochim Biophys Acta 1455:
105–138
19. Mencarelli S, Cavalieri C, Magini A et al (2005) Identification of
plasma membrane associated b-Hexosaminidase A, active
towards GM2 ganglioside, in human fibroblasts. FEBS Lett 579:
5501–5506
20. Magini A, Mencarelli S, Tancini B et al (2008) Identification and
characterization of mature beta-hexosaminidases associated with
human placenta lysosomal membrane. Biosci Rep 28:229–237
21. Bifsha P, Landry K, Ashmarina L et al (2007) Altered gene
expression in cells from patients with lysosomal storage disorders
suggests impairment of the ubiquitin pathway. Cell Death Differ
14:511–523
22. Connolly GP (1998) Fibroblast models of neurological disorders:
fluorescence measurement studies. Trends Pharmacol Sci 19:
171–177
23. Zampieri S, Filocamo M, Buratti E et al (2009) Molecular and
functional analysis of the HEXB gene in Italian patients affected
with Sandhoff disease: identification of six novel alleles. Neu-
rogenetics 10:49–58
24. Yeyeodu S, Ahn K, Madden V et al (2000) Procathepsin L self-
association as a mechanism for selective secretion. Traffic 1:
724–737
25. Laemmli UK (1970) Cleavage of structural protein during the
assembly of the head of bacteriophage T4. Nature 227:
680–685
26. Emiliani C, Beccari T, Tabilio A et al (1990) An enzyme with
properties similar to those of beta N-acetylhexosaminidase S is
expressed in the promyelocytic cell line HL-60. Biochem J
267:111–117
27. Bradford MM (1976) A rapid and sensitive method for the
quantitation of microgram quantities of protein utilizing the
principle of protein–dye binding. Anal Biochem 72:248–254
28. Ohkuma S, Poole B (1978) Fluorescence probe measurement of
the intralysosomal pH in living cells and the perturbation of pH
by various agents. Proc Natl Acad Sci USA 75:3327–3331
29. Keesey J (ed) (1987) Biochemica information, 1st edn. Boeh-
ringer Mannheim Biochemicals, Indianapolis, pp 19–20
30. Putter J, Becker R (1983) Peroxidase. In: Bergmeyer HW (ed)
Methods of enzymatic analysis, vol III, 3rd edn. Verlag-Chemie,
Weinheim, Germany, pp 286–293
31. Martinez O, Goud B (1998) Rab proteins. Biochim Biophys Acta
1404:101–112 Review
32. Zerial M, McBride H (2001) Rab proteins as membrane orga-
nizers. Nat Rev Mol Cell Biol 2:107–117
33. Eskelinen EL, Tanaka Y, Saftig P (2003) At the acidic edge:
emerging function for lysosomal membrane proteins. Trends Cell
Biol 13:137–145
34. D’Azzo A, Hoogeveen A, Reuser AJ, Robinson D, Galjaard H
(1982) Molecular defect in combined beta-galactosidase and
neuraminidase deficiency in man. Proc Natl Acad Sci USA
79:4535–4539
35. Mu F-T (1995) EEA1, an early endosome-associated protein.
J Biol Chem 270:13503–13511
36. Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B
(2000) Rab7: a key to lysosome biogenesis. Mol Biol Cell
11:467–480
37. Rosenfeld JL, Moore RH, Zimmer KP et al (2001) Lysosome
proteins are redistributed during expression of a GTP-hydrolysis-
defective rab5a. J Cell Sci 114:4499–4508
38. Lebrand C, Corti M, Goodson H et al (2002) Late endosome
motility depends on lipids via the small GTPase Rab7. EMBO J
21:1289–1300
282 Mol Cell Biochem (2010) 335:273–282
123




















