
Novel well-defined glycopolymers synthesized via the reversible addition fragmentation chain...
14
Novel Well-Defined Glycopolymers Synthesized via the Reversible Addition Fragmentation Chain Transfer Process in Aqueous Media ZHICHENG DENG, MARYA AHMED, RAVIN NARAIN Department of Chemistry and Biochemistry, Biomolecular Sciences Program, Laurentian University, 935, Ramsey Lake Rd, Sudbury, Ontario, P3E 2C6, Canada Received 26 August 2008; accepted 3 November 2008 DOI: 10.1002/pola.23187 Published online in Wiley InterScience (www.interscience.wiley.com). ABSTRACT: We describe here the direct synthesis of novel gluconamidoalkyl metha- crylamides by reacting D-gluconolactone with aminoalkyl methacrylamides. The gly- comonomers were then successfully polymerized via the reversible addition-fragmen- tation chain transfer process (RAFT) using 4-cyanopentanoic acid dithiobenzoate (CTP) as chain transfer agent and 4,4 0 -azobis(4-cyanovaleric acid) (ACVA) as the ini- tiator in aqueous media. Well-defined polymers were obtained as revealed by gel per- meation chromatography. Diblock copolymers were then synthesized by the macro- CTA approach. The cationic glycopolymers were subsequently used in the formation of nanostructures via the complexation with plasmid DNA. As noted by dynamic light scattering, monodisperse nanoparticles were obtained via the electrostatic inter- action of the cationic glycopolymer with DNA. The sizes of the nanoparticles formed were found to be stable and independent of pH. In vitro cell viability studies of the glycopolymers were carried out using HELA cell lines. The RAFT synthesized glycopoly- mers and cationic glyco-copolymers revealed to be nontoxic. V V C 2008 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 614–627, 2009 Keywords: biomaterials; block copolymers; cytoxicity; DNA complexation; glycopolymers; reversible addition fragmentation chain transfer INTRODUCTION In recent years, synthetic polymers with pendant saccharides moieties, so-called glycopolymers have received an enormous attention in the bio- chemical and biomedical fields. Glycopolymers are hydrophilic, water-soluble, biocompatible, and potentially bioactive toward specific biomolecules depending on the nature of the pendent saccha- ride moieties. As such, glycopolymers are often designed to mimic glycoproteins in the biological systems. In the biomedical fields, glycopolymers have been applied in molecular recognition processes, 1–9 drug and gene delivery systems, 10–15 surface modifiers, 16–18 responsive hydrogels, 19,20 stationary phases, 21–24 and treatment of dis- eases. 25–30 Glycopolymers at the cell surface can involve in numerous biological functions, adhe- sion, cell growth regulation, cancer cell metasta- sis, and inflammation. For extended applications in the biomedical field, proper control on the molecular weight and molecular weight distribution of the glycopoly- mers have to be achieved. Several synthetic Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 47, 614–627 (2009) V V C 2008 Wiley Periodicals, Inc. Additional Supporting Information may be found in the online version of this article. Correspondence to: R. Narain (E-mail: rnarain@ laurentian.ca) 614
Transcript of Novel well-defined glycopolymers synthesized via the reversible addition fragmentation chain...

Novel Well-Defined Glycopolymers Synthesized viathe Reversible Addition Fragmentation Chain TransferProcess in Aqueous Media
ZHICHENG DENG, MARYA AHMED, RAVIN NARAIN
Department of Chemistry and Biochemistry, Biomolecular Sciences Program, Laurentian University, 935,Ramsey Lake Rd, Sudbury, Ontario, P3E 2C6, Canada
Received 26 August 2008; accepted 3 November 2008DOI: 10.1002/pola.23187Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: We describe here the direct synthesis of novel gluconamidoalkyl metha-crylamides by reacting D-gluconolactone with aminoalkyl methacrylamides. The gly-comonomers were then successfully polymerized via the reversible addition-fragmen-tation chain transfer process (RAFT) using 4-cyanopentanoic acid dithiobenzoate(CTP) as chain transfer agent and 4,40-azobis(4-cyanovaleric acid) (ACVA) as the ini-tiator in aqueous media. Well-defined polymers were obtained as revealed by gel per-meation chromatography. Diblock copolymers were then synthesized by the macro-CTA approach. The cationic glycopolymers were subsequently used in the formationof nanostructures via the complexation with plasmid DNA. As noted by dynamiclight scattering, monodisperse nanoparticles were obtained via the electrostatic inter-action of the cationic glycopolymer with DNA. The sizes of the nanoparticles formedwere found to be stable and independent of pH. In vitro cell viability studies of theglycopolymers were carried out using HELA cell lines. The RAFTsynthesized glycopoly-mers and cationic glyco-copolymers revealed to be nontoxic. VVC 2008 Wiley Periodicals, Inc.
J Polym Sci Part A: Polym Chem 47: 614–627, 2009
Keywords: biomaterials; block copolymers; cytoxicity; DNA complexation;glycopolymers; reversible addition fragmentation chain transfer
INTRODUCTION
In recent years, synthetic polymers with pendantsaccharides moieties, so-called glycopolymershave received an enormous attention in the bio-chemical and biomedical fields. Glycopolymers arehydrophilic, water-soluble, biocompatible, andpotentially bioactive toward specific biomoleculesdepending on the nature of the pendent saccha-
ride moieties. As such, glycopolymers are oftendesigned to mimic glycoproteins in the biologicalsystems. In the biomedical fields, glycopolymershave been applied in molecular recognitionprocesses,1–9 drug and gene delivery systems,10–15
surface modifiers,16–18 responsive hydrogels,19,20
stationary phases,21–24 and treatment of dis-eases.25–30 Glycopolymers at the cell surface caninvolve in numerous biological functions, adhe-sion, cell growth regulation, cancer cell metasta-sis, and inflammation.
For extended applications in the biomedicalfield, proper control on the molecular weight andmolecular weight distribution of the glycopoly-mers have to be achieved. Several synthetic
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 47, 614–627 (2009)VVC 2008 Wiley Periodicals, Inc.
Additional Supporting Information may be found in theonline version of this article.
Correspondence to: R. Narain (E-mail: [email protected])
614

methods have been described in the designed oftailor-made glycopolmyers such as living ionic po-lymerization,31–33 ring-opening metathesis poly-merization (ROMP),6,34,35 nitroxide-mediated po-lymerization (NMP),36–44 and atom transfer radi-cal polymerization (ATRP).45–48 However, mostsynthetic method requires protecting group chem-istry and multiple steps reaction/purificationpathways. Narain and Armes49–51 were the firstto report a direct strategy to synthesize glycopoly-mers via ATRP without protection of the carbohy-drate hydroxyl groups. Heavy metal catalyst resi-due in the product restricts the use in biomedicalapplication. Recently, the progress in reversibleaddition-fragmentation chain transfer (RAFT)52,53
offers an opportunity to synthesize well-definedglycopolymers without using any metal catalyst.Because of the ideal tolerance to a wide range ofreaction conditions and monomers, the RAFT pro-cess earns an increasing attention in the synthe-sis of glycopolymers. Lowe et al.54 first reportedthe direct polymerization of 2-methacryloxyethylglucoside via RAFT in aqueous phase employing4-cyanopentanoic acid dithiobenzoate (CTP) asRAFT chain transfer agent (CTA). Pseudo firstorder kinetics and a near linear relationshipbetween molar masses and conversion wereobtained. Block copolymer with 3-sulfopropylmethacrylate (SPMA) was achieved with no de-tectable low–molecular-weight peak in the sizeexclusion chromatography. However, polydisper-sities increased and some high-molecular-weightimpurities were found. Later, methyl 6-O-metha-cryloyl-a-D-glucoside, 2-hydroxyethyl methacry-late diblock copolymer and subsequently methyl6-O-methacryloyl-a-D-mannoside, 2-methacrylox-yethyl glucoside diblock copolymer55 and methyl6-O-methacryloyl-a-D-glucoside56 were success-fully synthesized by Albertin et al. via RAFTusing the same CTA. The CTA was also success-fully employed to polymerize 6-O-methacryloylmannose.3 In 2007, Ramiah et al.57 polymerized3-O-methacryloyl-1,2:5,6-di-O-isopropylidene-D-glucofuranose via RAFT using cumylphenyldithioacetate as CTA and subsequently copoly-merized with styrene and methyl acrylate tointroduce amphiphilic property. Lowe andWang58 reported well-defined AB diblock copoly-mers of 3-O-methacryloyl-1,2:3,4-di-O-isopropyli-dene-D-galactopyranose with 2-(dimethylami-no)ethyl methacrylate. Recently, Housni et al.59
reported the RAFT polymerization of D-glucona-midoethyl methacrylate (GAMA) without usingprotecting group chemistry and the glycopoly-
mers were subsequently used to stabilize goldnanoparticles. RAFT synthesized glycopolymerswere also reported by Spain et al. to preparefunctionalized gold nanoparticles.60 All of theabove glycomonomers/glycopolymers involveester bond which can hydrolytically degrade sothat it restricts their applications. Several glyco-monomers/glycopolymers with amide linkagewere reported. Stenzel et al.61,62 reported theRAFT polymerization of N-acryloyl glucosaminecopolymerized with N-isopropylacrylamide(NIPAM) to introduce thermal responsive prop-erty employing 3-benzylsulfanylthiocarbonylsul-fanylpropionic acid as CTA and star architec-tures thermal responsive glyco-copolymer wasfurther synthesized. Thermally responsive glyco-copolymers were also synthesized via RAFTemploying protecting group chemistry byOzyurek et al.63 To introduce multi-functionproperties to glycopolymers, copolymerizationwith variety monomers64 and modification ofCTA were reported.65 On the other hand, modifi-cations of the CTA usually involve multiple syn-thesis and/or purification steps. To the best of ourknowledge, only one report exists in the litera-ture regarding the introduction of highly activefunctional group, such as an aldehyde into glyco-polymer.66 However, in that case, they hadrecourse to protecting group chemistry.
Herein, we report the facile synthesis of novelglycomonomers containing stable amide linkageswithout using any protecting group chemistry.Well-defined glycopolymers were prepared by thereversible addition fragmentation chain transferprocess using CTP as CTA and 4,40-azobis(4-cya-novaleric acid) (ACVA) as initiator in aqueousmedia. Furthermore, well-defined cationic glyco-copolymers were subsequently prepared and theirelectrostatic interaction with DNA to generatenanostructures was studied. Cell viability studieswere also carried using HELA cell lines to assessthe toxicity of the glycopolymers and the cationicdiblock glyco-copolymers.
EXPERIMENTAL
Materials
3-aminopropyl methacrylamide hydrochloride(APMA) and 2-aminoethyl methacrylamide hydro-chloride (AEMA) were synthesized according tothe previous work.67 4,40-azobis(4-cyanovalericacid) (ACVA, 97%) was purchased from AcrosOrganics and used as received. The CTA, CTP,
GLYCOPOLYMERS SYNTHESIS VIA RAFT 615
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

was synthesized as previously described.68 HPLCgrade methanol and reagent grade N,N0-dimethyl-formamide (DMF) were purchased from CaledonChemicals. The doubly distilled de-ionized waterwas used in all the experiments.
Synthesis of 3-GluconamidopropylMethacrylamide (GAPMA)
GAPMA monomer was prepared according to themethod reported by Narain and Armes.49–51 D-Glu-conolactone (16 g, 89.8 mmol) was first dissolved in600 mL methanol. A methanolic solution (100 mL)of APMA (20 g, 112.3 mmol) and hydroquinonewere added, followed by the addition of triethyl-amine (16 mL, 114.8 mmol). The mixture wasstirred overnight at room temperature. The result-ing mixture was then concentrated under vacuumand precipitated out in methanol at �10 �C. Thecrude product was washed by 2-propanol threetimes and followed by acetone. To remove any re-sidual inhibitor, GAPMA monomer was first dis-solved in DMF and recovered by a large amountexcess of diethyl ether. Characterization of GAPMAby 1H and 13C NMR (Fig. 1) and mass spectrometer(see Supp. Inform., spectrometer operating in ioni-zation mode; calculated mass for the M � 1 parention was 319.1 found 319.0) confirmed its high pu-rity. The final yield after purification was�70%.
Synthesis of 2-Gluconamidoethyl Methacrylamide(GAEMA)
A similar procedure was carried out for the syn-thesis of 2-gluconamidoethyl methacrylamide(GAEMA) monomer. D-Gluconolactone (16 g, 89.8mmol) was first dissolved in methanol (600 mL).A methanolic solution (100 mL) of AEMA (20 g,121.6 mmol) was subsequently added, followed bythe addition of triethylamine (20.4 mL, 146.4mmol). The mixture was stirred overnight atroom temperature. The crude product was thenfiltered out and washed three times with 2-propa-nol and followed by acetone. Characterization ofGAEMA by 1H and 13C NMR and mass spectros-copy (see Supp. Inform., spectrometer operatingin ionization mode; calculated mass for the M � 1parent ion was 305.1, found 305.0) confirmed itshigh purity. The final yield after purification wasfound to be �80%.
RAFT Polymerization of GAPMA
Prior to polymerization, GAPMA monomer wasfurther purified by precipitating in cold methanol.
The polymerization was achieved at 70 �C,employing ACVA as the radical initiator and CTPas the RAFT CTA. In a 10-mL Schlenk tube,GAPMA (1 g, 3.12 mmol) was dissolved in doubledistilled de-ionized water (5 mL) before the addi-tion of 1 mL CTP (9 mg, 0. 032 mmol, target DPn
¼ 75) and ACVA (4 mg, 0.0097 mmol) N,N0-dime-thylformamide (DMF) stock solution. Afterdegassing via four freeze thaw cycles, the flaskwas placed in a preheated oil bath for 11 h at70 �C. The conversion was determined by 1HNMR using D2O by comparing the vinyl signal atd 5.3–5.8 to the sugar pendant group signal of thepolymer at d 3.0–4.4. The molecular weight andmolecular weight distribution were obtained fromaqueous GPC after the polymer precipitated inacetone.
RAFT Polymerization of GAEMA
The RAFT polymerization of GAEMA was con-ducted via a similar procedure as described abovefor the polymerization of GAPMA. A typical syn-thesis is described below. The polymerization wasachieved at 70 �C, employing ACVA as the radicalinitiator and CTP as the RAFT CTA. In a 10-mL
Figure 1. Assigned (a) 1H and (b) 13C-NMR spectra(D2O) for 3-gluconamidopropyl methacrylamide(GAPMA) monomer.
616 DENG, AHMED, AND NARAIN
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

schlenk tube, GAEMA (1 g, 3.27 mmol) was addedto 7 mL double distilled water before the additionof 1 mL CTP (18.3 mg, 0.065 mmol, target DPn
¼ 37) N,N0-dimethylformamide (DMF) stock solu-tion. After degassing via four freeze thaw cycles,the mixture was heated until completely dissolvedand cooled down to room temperature. Fiftymicroliter degassed ACVA (9.2 mg, 0.022 mmol)N,N0-dimethylformamide (DMF) stock solutionwas injected. The flask was subsequently placedin a preheated oil bath for 10 h at 70 �C. The con-version was determined by 1H NMR using D2O bycomparing the vinyl signal at d 5.3–5.8 ppm to thesugar pendant group signal of the polymer at d3.0–4.4 ppm. The molecular weight and molecularweight distribution were obtained from aqueousGPC after the polymer precipitated in acetone.
RAFT Diblock Copolymerizationof GAPMA with APMA
The homopolymerization of GAPMA was carriedout at 70 �C, employing ACVA as the initiator andCTP as the CTA. The poly(GAPMA) homopolymerwas precipitated out in acetone and the residualGAPMA monomer was removed by washing withan excess of methanol. Subsequently, the poly(GAPMA) homopolymer (Mn ¼ 6100 g/mol; Mw/Mn
¼ 1.20) was used as macro chain transfer agent(macro-CTA). A typical protocol is described belowfor the copolymerization of GAPMA with APMA.In a 10-mL schlenk tube, APMA (0.21 g, 1.18mmol), poly (GAPMA) (0.2 g), and ACVA (5 mg,0.0178 mmol, dissolved in 0.1 mL DMF) weremixed in 3 mL doubly distilled de-ionized water.After degassing via four freeze thaw cycles, theflask was immersed in an oil bath for 20 h at 80�C. The reaction was then quenched by cooling thereaction vessel in an ice bath and exposure to air.The molecular weight and molecular weight distri-bution were obtained from aqueous GPC after thepolymer was precipitated in acetone. (Residualmonomerwas removed bywashingwithmethanol).
RAFT Diblock Copolymerizationof GAPMA with AEMA
A similar procedure was employed to synthesizepoly(GAPMA-b-AEMA) diblock copolymer. Thepoly(GAPMA) homopolymer (Mn ¼ 6100 g/mol;Mw/Mn ¼ 1.20) was used as macro-CTA. In a 10-mL Schlenk tube, AEMA (0.25 g, 1.52 mmol), poly(GAPMA) (0.2 g), and ACVA (2.5 mg, 0.0089mmol, dissolved in 0.1 mL DMF) were mixed in
doubly distilled de-ionized water (3 mL). Afterdegassing via four freeze thaw cycles, the flask wasimmersed in an oil bath for 20 h at 80 �C. The reac-tionwas then quenched by cooling the reaction ves-sel in an ice bath and exposure to air. The molecu-lar weight and molecular weight distribution wereobtained from aqueous GPC after the polymer wasprecipitated in acetone (Residual monomer wasremoved bywashingwithmethanol).
RAFT Diblock Copolymerizationof GAEMA with AEMA
The homopolymerization of GAEMA was carriedout at 70 �C according to the process describedabove. ACVA was used as the initiator and CTPwas used as the CTA. The poly(GAEMA) homo-polymer was precipitated out in acetone and theresidual GAEMA monomer was removed bywashing with DMF. Subsequently, the poly(GAEMA) homopolymer (Mn ¼ 9500 g/mol; Mw/Mn ¼ 1.19) was used as macro-CTA. A typical pro-tocol is described below for the copolymerizationof GAEMA with AEMA: In a 10-mL schlenk tube,AEMA (0.09 g, 0.55 mmol), poly(GAEMA) (0.1 g),and ACVA (3 mg, 0.01 mmol, dissolved in 0.1 mLDMF) were mixed in doubly distilled de-ionizedwater (2 mL). After degassing via four freezethaw cycles, the flask was immersed in an oil bathfor 20 h at 80 �C. The reaction was then quenchedby cooling the reaction vessel in an ice bath and ex-posure to air. The molecular weight and molecularweight distribution were obtained from aqueousGPC after the polymer was precipitated in acetone(Residual monomer was removed by washing withmethanol).
RAFT Diblock Copolymerization of GAEMA withMethacryloyloxyethyl Phosphorylcholine (MPC)
The poly(GAEMA) homopolymer (Mn ¼ 9500 g/mol; Mw/Mn ¼ 1.19) was also used as macro-CTAto synthesize poly(GAEMA-b-MPC) diblock co-polymer. A typical protocol is described below: Ina 10-mL schlenk tube, MPC (0.15 g, 0.51 mmol),poly(GAEMA) (0.1 g), and ACVA (2 mg, 0.0071mmol, dissolved in 0.1 mL DMF) were mixed in2 mL acidified doubly distilled de-ionized water(3% v/v acetic acid, pH �4). After degassing viafour freeze thaw cycles, the flask was immersed inan oil bath for 20 h at 80 �C. The reaction wasthen quenched by cooling the reaction vessel in anice bath and exposure to air. The molecularweight and molecular weight distribution were
GLYCOPOLYMERS SYNTHESIS VIA RAFT 617
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

obtained from aqueous GPC after the polymerwas precipitated in acetone.
Complexation of Cationic Glycopolymer[poly(APMA-b-GAPMA)] to Plasmid DNA
A stock solutions of 1 � 10�4 M poly(APMA29-b-GAPMA46) were prepared in appropriate buffers.One hundred microliter of pGlo plasmid (0.08 mg/mL) was incubated with 400 lL of polymer stocksolutions for 20 min. Mixtures were vortexed andfurther incubated for 10 min. Dynamic light scat-tering measurements were performed at roomtemperature using a Viscotek DLS instrumenthaving He-Ne laser at a wavelength of 632 nmand pelltier temperature controller.
Cell Viability/Cytotoxicity of Polymers
Preparation of polymer stock solutions: All poly-mers were dissolved into Dulbecco’s phosphatebuffer at the final concentration of 0.6 mM. Thesamples were autoclaved and different volumes ofprepared solution were added into different vol-umes of DMEMmedium to obtain the final concen-tration of 2, 4, and 6 lMof polymer in themedium.
Cytotoxicity was characterized using MTTassay. HELA cells were plated in a 96-well plateat the density of 15,000 cells/well in a 100 lL ofgrowth medium consisting DMEM with 5% fetal
bovine serum and antibiotics (penicillin 100 U/mLand streptomycin 100 lg/mL). Cells were incu-bated for 24 h at 37 �C and 5% CO2, after whichthe growth medium was replaced with DMEMmedium containing different concentrations ofpolymers, prepared above [poly(APMA), poly(AEMA), poly(GAPMA), poly(GAEMA), and poly(APMA-b-GAPMA)]. The cells were further incu-bated for 24 h under the same conditions. Afterwhich 25 lL of MTT dye was added to each welland plate was incubated for 2 h. Next, 100 lL ofMTT lysis solution was added to each well andcells were incubated overnight. Absorbance wasmeasured at 570 nm (Power wave X plate reader).Survival percentage was calculated by compari-son to blank cells (100% survival).
Characterization
1H and 13C NMR spectra of the monomers andpolymers were recorded on a Varian 200 MHzinstrument.
The monomers were analyzed by an AgilentHPLC 1100 interfaced with an Electro-Spray Ioni-zation Agilent Mass Spectrometer Model 6120with a Chemstation data system LC-MSDB.03.01.
Molecular weight and molecular weight distri-butions were determined by conventional gel
Scheme 1. Structure of monomers.
618 DENG, AHMED, AND NARAIN
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

permeation chromatography (GPC) system usingaqueous eluents. The following two GPC protocolswere used in this work.
Aqueous GPC at Neutral pH
This protocol was used for the GAPMA andGAEMA homopolymers. Chromatograms wererecorded from a conventional Viscotek GPC sys-tem using a 0.1 M sodium nitrate as eluent, twoWaters WAT011545 columns at room temperatureand a flow rate of 1.0 mL/min. Seven near-mono-disperse Pullulan (polysaccharides) standards,obtained from PolyAnalytik Company, (Mw
¼ 5900–404,000 gmol�1) were used for calibration.
Aqueous GPC at Low pH
Chromatograms for the GAPMA-APMA, GAPMA-AEMA, GAEMA-AEMA, and GAEMA-MPC
diblock copolymers were obtained from a conven-tional Viscotek GPC system using a 0.5 M sodiumacetate/0.5 M acetic acid buffer as eluent, twoWaters WAT011545 columns at room temperatureand a flow rate of 1.0 mL/min. Seven near-mono-disperse Pullulan standards (Mw ¼ 5900–404,000g mol�1) were used for calibration.
Dynamic light scattering measurements wereperformed at room temperature using a ViscotekDLS instrument having He-Ne laser at a wave-length of 632 nm and pelltier temperature con-troller.
RESULTS AND DISCUSSION
Glycomonomer Syntheses
Our motivation in this work is first to synthesizestable vinyl sugar monomers via a one-step chem-ical process without using any protecting group
Scheme 3. RAFT Polymerization of 3-gluconamidopropyl methacrylamide (GAPMA),using CTP as chain transfer agent and ACVA as initiator.
Scheme 2. Synthesis of 2-gluconamidoethyl methacrylamide (GAEMA) and 3-gluco-namidopropyl methacrylamide (GAPMA) monomers.
GLYCOPOLYMERS SYNTHESIS VIA RAFT 619
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

chemistry. Therefore, we have selected the rawmaterial, D-gluconolactone, which is a cheap andabundant raw material and is obtained from theoxidation of D-glucose. The ring opening of sugar
lactones has previously been reported by Narainand Armes.49–51 In this work, we report for thefirst time the direct synthesis of polymerizablevinyl sugars such as GAPMA and GAEMA whichhave stable amide linkages (Scheme 1). The glyco-monomers are well characterized by a range oftechniques. Typical 1H and 13C NMR spectra forGAPMA is illustrated in Figure 1. GAPMA andGAEMA monomers were readily prepared by thereaction of APMA (Scheme 2) or AEMA with D-gluconolactone at room temperature. The mono-mers APMA and AEMA were prepared accordingto the procedure reported by Deng et al.67 In con-trast to the previously reported synthesis of gluco-namidoethyl methacrylate (GAMA),49–51 only aslight excess of the aminoalkyl methacrylamideswere required because of the higher stability ofAPMA and AEMA once deprotected. The deproto-nation of APMA.HCl and AEMA.HCl was carriedwith triethylamine. As previously reported, theaminoalkyl methacrylamides are significantlymuch more stable as compared with the commer-cially available 2-aminoethyl methacrylate.67
Furthermore, the reaction of the aminoalkylmethacrylamides with D-gluconolactone was con-ducted both in the presence and absence of inhib-itor. No autopolymerization was observed in theabsence of the inhibitor and therefore eliminatesany additional purification process required toremove inhibitor before conducting the RAFTpolymerization.
Figure 2. Kinetic plots for the RAFT polymerizationof 3-gluconamidopropyl methacrylamide (GAPMA) fora target degree of polymerization of 75. (a) AqueousGPC curves indicating the evolution of Mn with elu-tion time; (b) Semilogarithmic and conversion plotversus time; (c) Evolution of Mn and Mw/Mn with con-version.
Figure 3. Aqueous GPC curves indicating the evolu-tion of Mn with elution time for the RAFT polymeriza-tion of 3-gluconamidopropyl methacrylamide (GAPMA)with different target degrees of polymerization.
620 DENG, AHMED, AND NARAIN
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

Homopolymerization
GAPMA and GAEMA were homopolymerized viathe reversible addition fragmentation chain trans-fer process at 70 �C employing CTP as CTA and4,40-azobis-(4-cyanovaleric acid) (ACVA) as initia-tor in aqueous media (Scheme 3). Although theCTA is only slightly soluble in water, the highlyhydrophilic nature of the glycomonomers andhence the glycopolymers rendered the polymeriza-tion impossible to be carried out in either purewater or pure organic solvent. Considering thedifference in solubility of CTP, initiator, sugarmonomer, and glycopolymer, a mixture of waterand N,N0-dimethylformamide (DMF) was chosenas the appropriate polymerization solvent for ahomogeneous mixture. In the polymerization ofGAEMA (12% w/v), the ratio of water to DMF isminimally set 7:1 which is necessary for the com-plete dissolution of the sugar monomer. Further-more, the dissolution of GAEMA in the water/DMF mixture was facilitated by warming theSchlenk tube. In contrast to GAEMA, GAPMAmonomer is much more soluble and a ratio of 5:1of water/DMF is used to dissolve the vinyl sugarmonomer at a concentration of 17% w/v. At thesemonomer concentrations and polymerization sol-vent, a homogeneous mixture was noted through-out the RAFT polymerization. To compare therate of the polymerization of GAEMA andGAPMA, the glycomonomers were polymerizedunder similar conditions (same concentrations,similar solvent system (water:DMF ¼ 7:1) andsame degree of polymerization). We found thatthe rate of homopolymerization of GAPMA wasslower than that of GAEMA for similar degree of
polymerization which is consistent with rateof the homopolymerization of APMA to that ofAEMA (see supporting information). It should benoted that an induction period is observed in thehomopolymerization of both GAEMA and GAPMAmonomers. The evolution of Mn linear with con-version is a clear indication of the successfulRAFT polymerization. Furthermore, polydisper-sities remained relatively narrow throughout thepolymerization and GPC analyses indicatedmonomodal chromatograms in all cases. Pseudofirst order kinetics indicates a constant concentra-tions of radicals, close values of experimental Mn
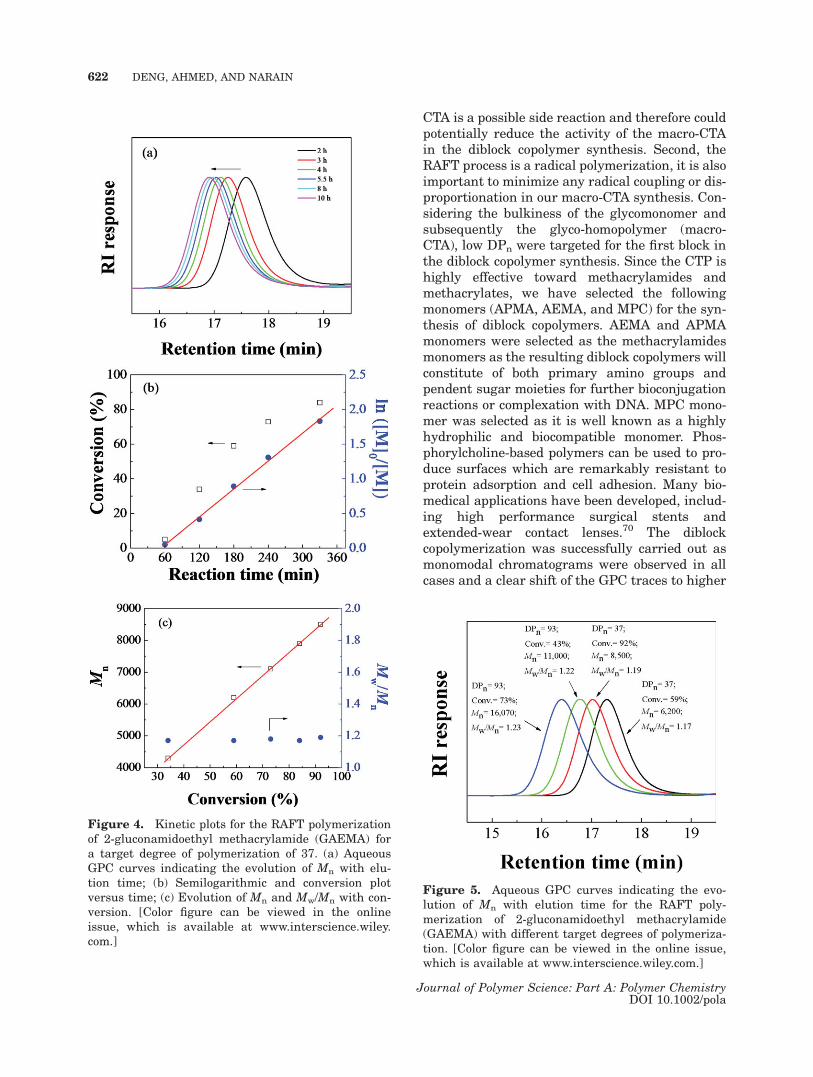
to theoretical values showed that the polymeriza-tions of both GAPMA (Fig. 2, Table 1) andGAEMA (Fig. 4, Table 2) were seen to proceed ina controlled manner. Different target degrees ofpolymerization (DPn) were also studied and rela-tively low polydispersity was obtained (Figs. 3and 5).
RAFT Copolymerization
After establishing the appropriate experimentalconditions for the homopolymerization of GAEMAand GAPMA, diblock copolymers were preparedby the RAFT process via the macro-CTA tech-nique. GAEMA and GAPMA were first homopoly-merized by the RAFT process using the CTP asCTA and ACVA as initiator. The polymerizationwas allowed to proceed to a conversion of 60–75%for the following reasons to ensure a higher effi-ciency of the macro-CTA in the RAFT copolymer-ization. First, since the homopolymerization isconducted in aqueous media, hydrolysis68,69 of
Table 1. Summary of Synthetic Parameters for the RAFT Homopolymerization ofGAPMA at 70 �C Using ACVA as Initiator and CTP as the Chain Transfer Agent fora Target Degree of Polymerization of 75
EntryPolymerizationTime (min)
Conv. (%)1H NMR Mn th
a Mnb (g/mol) Mw/Mn
b
ZD199-1 120 13 3400 3600 1.19ZD199-2 180 30 7400 7600 1.18ZD199-3 240 42 10,300 10,700 1.18ZD199-4 330 56 13,600 13,500 1.19ZD199-5 480 72 17,500 16,300 1.18ZD199-6 630 78 18,900 18,200 1.20
aMn th ¼ [GAPMA]0/([CTP]0 þ [ACVA]0) � 320 � conversion þ MWCTP.b The evolution of the molecular weight with time for the polymerizations were studied with
a conventional Viscotek GPC system using a 0.1 M sodium nitrate as eluent, two WatersWAT011545 columns at room temperature and a flow rate of 1.0 mL/min. Seven near-monodis-perse Pullulan standards (Mw ¼ 5900–404,000 g mol�1) were used for calibration.
GLYCOPOLYMERS SYNTHESIS VIA RAFT 621
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
CTA is a possible side reaction and therefore couldpotentially reduce the activity of the macro-CTAin the diblock copolymer synthesis. Second, theRAFT process is a radical polymerization, it is alsoimportant to minimize any radical coupling or dis-proportionation in our macro-CTA synthesis. Con-sidering the bulkiness of the glycomonomer andsubsequently the glyco-homopolymer (macro-CTA), low DPn were targeted for the first block inthe diblock copolymer synthesis. Since the CTP ishighly effective toward methacrylamides andmethacrylates, we have selected the followingmonomers (APMA, AEMA, and MPC) for the syn-thesis of diblock copolymers. AEMA and APMAmonomers were selected as the methacrylamidesmonomers as the resulting diblock copolymers willconstitute of both primary amino groups andpendent sugar moieties for further bioconjugationreactions or complexation with DNA. MPC mono-mer was selected as it is well known as a highlyhydrophilic and biocompatible monomer. Phos-phorylcholine-based polymers can be used to pro-duce surfaces which are remarkably resistant toprotein adsorption and cell adhesion. Many bio-medical applications have been developed, includ-ing high performance surgical stents andextended-wear contact lenses.70 The diblockcopolymerization was successfully carried out asmonomodal chromatograms were observed in allcases and a clear shift of the GPC traces to higher
Figure 4. Kinetic plots for the RAFT polymerizationof 2-gluconamidoethyl methacrylamide (GAEMA) fora target degree of polymerization of 37. (a) AqueousGPC curves indicating the evolution of Mn with elu-tion time; (b) Semilogarithmic and conversion plotversus time; (c) Evolution of Mn and Mw/Mn with con-version. [Color figure can be viewed in the onlineissue, which is available at www.interscience.wiley.com.]
Figure 5. Aqueous GPC curves indicating the evo-lution of Mn with elution time for the RAFT poly-merization of 2-gluconamidoethyl methacrylamide(GAEMA) with different target degrees of polymeriza-tion. [Color figure can be viewed in the online issue,which is available at www.interscience.wiley.com.]
622 DENG, AHMED, AND NARAIN
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

molar masses was noted (Fig. 6). The results forthe copolymerization are summarized in Table 3.The molar mass distribution of the resultingdiblock was found to be slightly higher as com-pared with the homopolymers due to a tailing atthe low-molecular weight region. This behaviorwas not observed in the block copolymerization ofAPMAwith AEMA. Therefore, the following threepossibilities may have caused the broadening ofthe GPC peak in the chromatogram: (1) A smallportion of the macro-CTA is inactive (no RAFTagent at polymer chain end) to allow chain exten-sion; (2) The macro-CTA may be sterically hin-dered in the diblock copolymer synthesis; (3) Theconventional GPC protocol used to analyze thehomopolymers may not be suitable in the analysisof the resulting ionic diblock copolymers obtained.Therefore, a self-chain extension experiment ofpoly(GAPMA) was conducted to better understandthe broadening of the molar mass distributionchromatograms that have occurred in the diblockcopolymerizations. GPC analysis of poly(GAPMA)after chain extension via the macro-CTA approachrevealed that the presence of a tail in the chromat-ogram at the low-molecular weight regions. There-fore, we can conclude that the tailoring in theGPC traces of the ionic diblock copolymers is aresult of an inefficiency of the macro-CTA (prob-ably due to hydrolysis) in the polymerization ofthe second monomer.71 However, the compositionsof the final diblock copolymers were found to beconsistent with the initial feed ratio of the mono-mers. (See Supp. Inform., Fig. S7)
Complexation of Cationic Glycopolymer[poly(APMA-b-GAPMA)] to Plasmid DNA
Cationic polymers can easily condense DNA andfuse and destabilize cell membranes and henceenhance gene delivery. The cationic glycopolymer,poly(APMA29-b-GAPMA46), of molar mass wasselected to study the formation of nanostructuresvia the complexation with plasmid DNA. Anexcess of the cationic glycopolymer was used toelectrostatically interact with the plasmid DNA.The formation of nanoparticles was monitoredextensively by dynamic light scattering (DLS) asa function of pH. At acidic and neutral pH, theDLS data of the pure cationic glycopolymer indi-cate a size of 1.8 nm. Incubation of plasmid DNAwith an excess of the cationic glycopolymerresulted in the formation of nanoparticles of sizes30–35 nm. Furthermore, the complete disappear-ance of the DLS peak at 1.8 nm indicates the com-plete complexation of the cationic glycopolymerwith DNA. It should be noted that the size of thenanoparticles is independent of the pH of the so-lution (Table 4). This result shows that the cati-onic glycopolymer retains its ability to complex toplasmid DNA at the physiological pH of 7.4. Fur-thermore, it is also noted that the DLS size ofplasmid DNA (34 nm) is slightly larger than thesize of nanoparticles produced with the cationicglycopolymer at pH 7.4. It may be due to thestrong electrostatic interactions between the posi-tively charged glycopolymer and the negativelycharged DNA that resulted in compact nanostruc-tures and slight decrease in the size of plasmid.
Table 2. Summary of Synthetic Parameters for the RAFT Homopolymerization of2-Gluconamidoethyl Methacrylamide (GAEMA) at 70 �C Using ACVA as Initiatorand CTP as the Chain Transfer Agent for a Target Degree of Polymerization of 37
EntryPolymerizationTime (min)
Conv. (%)1H NMR Mn th
a Mnb (g/mol) Mw/Mn
b
ZD201-1 0 0 0 – –ZD201-2 60 5 850 – –ZD201-3 120 34 4200 4300 1.17ZD201-4 180 59 7000 6200 1.17ZD201-5 240 73 8600 7100 1.18ZD201-6 330 84 9900 7900 1.17ZD201-7 480 92 10,800 8500 1.19ZD201-8 600 93 10,900 8900 1.18
aMn th ¼ [GAEMA]0/([CTP]0 þ [ACVA]0) � 306 � conversion þ MWCTP.b The evolution of the molecular weight with time for the polymerizations were studied with
a conventional Viscotek GPC system using a 0.1 M sodium nitrate as eluent, two WatersWAT011545 columns at room temperature and a flow rate of 1.0 mL/min. Seven near-monodis-perse Pullulan standards (Mw ¼ 5900–404,000 g mol�1) were used for calibration.
GLYCOPOLYMERS SYNTHESIS VIA RAFT 623
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

This behavior has previously been reported byTan et al.72 where they incubated PEO-b-PDEApolycation (PDEA—poly(diethylaminoethyl meth-acrylate)) with hrGFP plasmid of 3.7 kilobasepair(kbp) and the size of plasmid decreases from 88.5to 49.7 nm. The decrease in nanoparticle size isproportional to the net amount of cationic polymerin the mixture. They have also proved that lowerconcentration of cationic polymer strongly bindswith the plasmid to produce compact nanopar-
ticles, but when the concentration of cationic poly-mer increases in the mixture, negative charge ofDNA counteracts the positive charge of the poly-mer and size of nanoparticles increases.
In Vitro Cell Viability/Cytotoxicity Studies
The in vitro cytotoxicity of the polymers isobtained using MTT assay on HELA cell line. Theassay determines the number of viable cells basedon the mitochondrial activity of the cells. TheMTT dye produces formazan crystals in the wellsbased on the mitochondrial activity of cells in thewell. MTT lysis buffer dissolves the formazancrystals produced and the absorbance can be readat 570 nm, giving the number of viable cells ineach well. Figure 7 shows the percent cell viabil-ity as a function of concentration of different poly-mers. The cationic polymer poly(APMA) (Mn
¼ 21,700 g/mol) is found to be toxic at relativelylow concentration (2 lM) as the cell viability isonly about 20%. The RAFT synthesized block poly(APMA29-b-GAPMA46) (Mn ¼ 19,100 g/mol) showsexcellent biocompatibility as the cell viabilityincreases from 20 to 80%. This is an ideal exampleof the structure-property relationship where theglycopolymer component in the copolymer playsan important role in reducing significantly thetoxicity of poly(APMA) component. It is also clearfrom the graph that although the toxicity of thepolymer is concentration dependent, only a slightincrease in toxicity of the copolymer is observed
Figure 6. GPC traces for the blocking experimentsof poly(GAPMA) and poly(GAEMA) via RAFT at80 �C. (a) GPC traces showing the homopolymer poly(GAPMA) and the resulting diblock copolymer poly(GAPMA19-b-APMA36) and poly(GAPMA19-b-AEMA44);(b) GPC traces showing the homopolymer poly(GAEMA) and the resulting diblock copolymer poly(GAEMA31-b-AEMA59) and poly(GAEMA31-b-MPC52).[Color figure can be viewed in the online issue, which isavailable at www.interscience.wiley.com.]
Figure 7. Percent cell viability/cytotoxicity as afunction of concentration of different polymers. [Colorfigure can be viewed in the online issue, which isavailable at www.interscience.wiley.com.]
624 DENG, AHMED, AND NARAIN
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

with the increase in polymer concentration. Thetoxicity of block copolymer is 89% for up to 6 lMconcentration of the polymer. This data is verysimilar to the one reported by Wang and Hsiue,73
where lactose sugar has been used to decrease thetoxicity of cationic polyethyleneimine (PEI) poly-mer. The percentage cell viability reported is upto 80%. The synthesis of biocompatible cationicblock copolymers containing sugar moieties isvery important for the gene delivery purposes.These block copolymer exhibit PEG like biocom-patible characteristics and can prove to be moreefficient for gene delivery purposes. Overall, sugarbased polymer are completely nontoxic, poly(GAPMA) (Mn ¼ 11,900 g/mol) seems to enhancethe cell proliferation, while poly(GAEMA) (Mn
¼ 16,100 g/mol) yields 100% cell viability.
CONCLUSIONS
Two novel glycomonomers with amide linkagewere successfully synthesized by the direct reac-tion of D-gluconolactone with aminoalkyl metha-
crylamides. The reversible addition fragmentationchain transfer process of the GAPMA andGAEMA were successfully carried out using CTPas CTA and 4-40-azobis(4-cyanovaleric acid)(ACVA) as initiator to generate a range of welldefined glycopolymers of controlled dimensions.The glycopolymers were subsequently used asmacro-CTA agent for the synthesis of a range ofwell-defined diblock glyco-copolymers. The com-plexation of the cationic glyco-copolymers withplasmid DNA revealed the formation of welldefined nanostructures. Furthermore, cell viabil-ity studies were also carried using HELA celllines to assess the toxicity of the glycopolymersand the cationic diblock glyco-copolymers. Theglycopolymers and glyco-copolymers revealed tobe nontoxic at a concentration range of 2–6 lM.
The authors thank the Natural Sciences and Engineer-ing Research Council of Canada for financial support ofthis work.
REFERENCES AND NOTES
1. Dai, X.; Dong, C.; Yan, D. J Phys Chem B 2008,112, 3644–3652.
2. Huang, M.; Shen, Z.; Zhang, Y.; Zeng, X.; Peng,G.; Wang, B. Med Chem Lett 2007, 17, 5379–5383.
3. Granville, A. M.; Quemener, D.; Davis, T. P.;Barner-Kowollik, C.; Stenzel, M. H. MacromolSymp 2007, 255, 81–89.
4. (a) Sharon, N.; Lis, H. Sci Am 1993, 268, 82–89;(b) Ambrosi, M.; Cameron, N. R.; Davis, B. G. OrgBiomol Chem 2005, 3, 1593–1608.
5. Gabius, H. J.; Siebert, H. C.; Andre, S.; Jimenez-Barbero, J.; Rudiger, H. Chembiochem 2004, 5,741–764.
Table 3. Summary of Synthetic Parameters for the RAFT Copolymerization of GAPMA and GAEMA at 80 �C
Copolymer Type
Homopolymer Copolymer
Mna Mw/Mn
a Mn, th Mna Mw/Mn
a
poly(GAPMA19-b-APMA36) 6100 1.20 12,500 18,400 1.40poly(GAPMA19-b-AEMA44) 6100 1.20 13,700 21,300 1.43poly(GAEMA31-b-AEMA59) 9500 1.19 18,400 15,300 1.44poly(GAEMA31-b-MPC52) 9500 1.19 23,800 25,040 1.39poly(APMA29-b-GAPMA46) 5200b 1.23b 15,100 19,100 1.39
aThe evolution of the molecular weight with time for the polymerizations were studied with a conventional Viscotek GPCsystem using a 0.5 M sodium acetate/0.5 M acetic acid buffer as eluent, two Waters WAT011545 columns at room temperatureand a flow rate of 1.0 mL/min. Seven near-monodisperse Pullulan standards (Mw ¼ 5900–404,000 g mol�1) were used for cali-bration.
b Six near-monodisperse PEO standards (Mp ¼ 1010–101,200 g mol�1) were used for calibration.
Table 4. Complexation of the Cationic Glycopolymer[poly(APMA29-b-GAPMA46)] to Plasmid DNA
Copolymer Sample Buffer
DLSSize(nm)
DLSSize
Distribution
poly(APMA29-b-GAPMA46) 7.4 1.8 0.04poly(APMA29-b-GAPMA46) 5 1.8 0.05poly(APMA29-b-GAPMA46)þ plasmid
7.4 29.7 0.04
poly(APMA29-b-GAPMA46)þ plasmid
5 34.2 0.11
plasmid 7.4 33.7 0.16
GLYCOPOLYMERS SYNTHESIS VIA RAFT 625
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

6. Mortell, K. H.; Gingras, M.; Kiessling, L. L. J AmChem Soc 1994, 116, 12053–12054.
7. Kanai, M.; Mortell, K. H.; Kiessling, L. L. J AmChem Soc 1997, 119, 9931–9932.
8. Kiessling, L. L.; Gestwicki, J. E.; Strong, L. E.Curr Opin Chem Biol 2000, 4, 696–703.
9. Cairo, C. W.; Gestwicki, J. E.; Kanai, M.; Kies-sling, L. L. J Am Chem Soc 2002, 124, 1615–1619.
10. Palomino, E. Adv Drug Deliv Rev 1994, 13, 311–323.
11. Chen, X. M.; Dordick, J. S.; Rethwisch, D. G.Macromolecules 1995, 28, 6014–6019.
12. Li, J.; Zacharek, S.; Chen, X.; Wang, J.; Zhang,W.; Janczuk, A.; Wang, P. G. Bioorg Med Chem1999, 7, 1549–1558.
13. Garcia-Martin, M. G.; Jimenez-Hidalgo, C.; Al-Kass, S. S. J.; Caraballo, I.; de Paz, M. V.; Galbis,J. A. Polymer 2000, 41, 821–826.
14. Roche, A. C.; Fajac, I.; Grosse, S.; Frison, N.; Ron-danino, C.; Mayer, R.; Monsigny, M. Cell Mol LifeSci 2003, 60, 288–297.
15. Yun, Y. H.; Goetz, D. J.; Yellen, P.; Chen, W. Bio-materials 2004, 25, 147–157.
16. Wulff, G.; Zhu, L.; Schmidt, H. Macromolecules1997, 30, 4533–4539.
17. Wulff, G.; Schmidt, H.; Zhu, L. Macromol ChemPhys 1999, 200, 774–782.
18. Klein, J.; Kunz, M.; Kowalczyk, J. MacromolChem 1990, 191, 517–528.
19. Novick, S. J.; Dordick, J. S. Chem Mater 1998,10, 955–958.
20. Miyata, T.; Uragami, T.; Nakamae, K. Adv DrugDeliv Rev 2002, 54, 79–98.
21. Karamuk, E.; Mayer, J.; Wintermantel, E.;Akaike, T. Artif Organs 1999, 23, 881–887.
22. Kim, S.-H.; Kim, J.-H.; Akaike, T. FEBS Lett2003, 553, 433–439.
23. Taguchi, T.; Kishida, A.; Sakamoto, N.; Akashi,M. J Biomed Mater Res 1998, 41, 386–391.
24. Liu, X.-C.; Dordick, J. S. J Polym Sci Part A:Polym Chem 1999, 37, 1665–1671.
25. Choi, S.-K.; Mammen, M.; Whitesides, G. M.J Am Chem Soc 1997, 119, 4103–4111.
26. Gordon, E. J.; Strong, L. E.; Kiessling, L. L. Bio-org Med Chem 1998, 6, 1293–1299.
27. Yoshida, T.; Akasaka, T.; Choi, Y.; Hattori, K.; Yu,B.; Mimura, T.; Kaneko, Y.; Nakashima, H.; Ara-gaki, E.; Premanathan, M.; Yamamoto, N.; Uryu, T.J PolymSci Part A: PolymChem 1999, 37, 789–800.
28. Roy, R.; Baek, M.-G. Rev Mol Biotechnol 2002, 90,291–309.
29. Fleming, C.; Maldjian, A.; Costa, D. D.; Rullay, A.K.; Haddleton, D. M.; St. John, J.; Penny, P.;Noble, R. C.; Cameron, N. R.; Davis, B. G. NatChem Biol 2005, 1, 270–274.
30. Petronia, M. G.; Mansi, A.; Gallinelli, C.; Pisani,S.; Seganti, L.; Chaiarini, F. Chemotherapy 1997,43, 211–217.
31. Yamada, K.; Minoda, M.; Miyamoto, T. Macromo-lecules 1999, 32, 3553–3558.
32. Labeau, M.; Cramail, H.; Deffieux, A. MacromolChem Phys 1998, 199, 335–342.
33. (a) Yamada, K.; Minoda, M.; Miyamoto, T. JPolym Sci Part A: Polym Chem 1997, 35, 751–757; (b) Hirao, A.; Kato, K.; Nakahama, S. Macro-molecules 1992, 25, 535–540.
34. Descotes, G.; Ramza, J.; Basset, J.-M.; Pagano, S.Tetrahedron Lett 1994, 35, 7379–7382.
35. Fraser, C.; Grubbs, R. H. Macromolecules 1995,28, 7248–7255.
36. Georges, M. K.; Veregin, R. P. N.; Kazmaier, P.M.; Hamer, G. K. Macromolecules 1993, 26, 2987–2988.
37. Ohno, K.; Fukuda, T.; Kitano, H. Macromol ChemPhys 1998, 199, 2193–2197.
38. Ohno, K.; Izu, Y.; Yamamoto, S.; Miyamoto, T.;Fukuda, T. Macromol Chem Phys 1999, 200,1619–1625.
39. Chen, Y.; Wulff, G. Macromol Chem Phys 2001,202, 3426–3431.
40. Chen, Y.; Wulff, G. Macromol Chem Phys 2001,202, 3273–3278.
41. Narumi, A.; Matsuda, T.; Kaga, H.; Satoh, T.;Kakuchi, T. Polym J (Tokyo) 2001, 33, 939–945.
42. Narumi, A.; Matsuda, T.; Kaga, H.; Satoh, T.;Kakuchi, T. Polymer 2002, 43, 4835–4840.
43. Narumi, A.; Satoh, T.; Kaga, H.; Kakuchi, T. Mac-romolecules 2002, 35, 699–705.
44. Gotz, H.; Harth, E.; Schiller, S. M.; Frank, C. W.;Knoll, W.; Hawker, C. J. J Polym Sci Part A:Polym Chem 2002, 40, 3379–3391.
45. Ohno, K.; Tsujii, Y.; Fukuda, T. J Polym Sci PartA: Polym Chem 1998, 36, 2473–2481.
46. (a) Chen, Y. M.; Wulff, G. Macromol Chem Phys2001, 202, 3273–3278; (b) Chen, Y. M.; Wulff, G.Macromol Chem Phys 2001, 202, 3426–3431.
47. Chen, Y. M.; Wulff, G. Macromol Rapid Commun2002, 23, 59–63.
48. Ejaz, M.; Ohno, K.; Tsujii, Y.; Fukuda, T. Macro-molecules 2000, 33, 2870–2874.
49. Narain, R.; Armes, S. P. Chem Commun 2002, 23,2776–2777.
50. Narain, R.; Armes, S. P. Macromolecules 2003, 36,4675–4678.
51. Narain, R.; Armes, S. P. Biomacromolecules 2003,4, 1746–1758.
52. Chiefari, J.; Chong, Y. K.; Ercole, F.; Krstina, J.;Jeffery, J.; Le, T. P. T.; Mayadunne, R. T. A.;Meijs, G. F.; Moad, C. L.; Moad, G.; Rizzardo, E.;Thang, S. H. Macromolecules 1998, 31, 5559–5562.
53. (a) Lowe, A. B.; McCormick, C. L. Prog Polym Sci2007, 32, 283–351; (b) Favier, A. Charreyre, M.-T.Macromol Rapid Commun 2006, 27, 653–692; (c)Mael, B.; Franck, D.; Roger, S.; Charreyre, M.-T.;Thierry, D. J Am Chem Soc 2006, 128, 2546–2547.
626 DENG, AHMED, AND NARAIN
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

54. Lowe, A. B.; Sumerlin, B. S.; McCormick, C. L.Polymer 2003, 44, 6761–6765.
55. (a) Albertin, L.; Stenzel, M.; Barner-Kowollik, C.;John, L.; Foster, R.; Davis, T. P. Macromolecules2004, 37, 7530–7537; (b) Albertin, L.; Stenzel, M.H.; Barner-Kowollik, C.; John, L.; Foster, R.;Davis, T. P. Macromolecules 2005, 38, 9075–9084.
56. Albertin, L.; Cameron, N. R. Macromolecules2007, 40, 6082–6093.
57. Ramiah, V.; Matahwa, H.; Weber, W.; McLeary, J.B.; Sanderson, R. D. Macromol Symp 2007, 255,70–80.
58. Lowe, A. B.; Wang, R. Polymer 2007, 48, 2221–2230.59. (a) Housni, A.; Cai, H.-J.; Liu, S.-Y.; Pun, S. H.;
Narain, R. Langmuir 2007, 23, 5056–5061; (b)Narain, R.; Housni, A.; Gody, G.; Boullanger, P.;Charreyre, M.-T.; Delair, T. Langmuir 2007, 23,12835–12841.
60. (a) Spain, S. G.; Albertin, L.; Cameron, N. R.Chem Commun 2006, 40, 4198–4200; (b) Spain, S.G.; Gibson, M. I.; Cameron, N. R. J Polym SciPart A: Polym Chem 2007, 45, 2059–2349.
61. Stenzel, M.; Zhang, L.; Huck, Wilhelm T. S. Mac-romol Rapid Commun 2006, 27, 1121–1126.
62. Bernard, J.; Hao, X.-J.; Davis, T. P.; Barner-Kowollik, C.; Stenzel, M. H. Biomacromolecules2006, 7, 232–238.
63. Ozyurek, Z.; Komber, H.; Gramm, S.; Schmaljo-hann, D.; Muller, Axel H. E.; Voit, B. MacromolChem Phys 2007, 208, 1035–1049.
64. Zhang, L.; Bernard, J.; Davis, T. P.; Barner-Kowollik, C.; Stenzel, M. H. Macromol RapidCommun 2008, 29, 123–129.
65. Gody, G.; Boullanger, P.; Ladaviere, C.; Charreyre,M.-T.; Delair, T. Macromol Rapid Commun 2008,29, 511–519.
66. Xiao, N.-Y.; Li, A.-L.; Liang, H.; Lu, J. Macromole-cules 2008, 41, 2374–2380.
67. Deng, Z.-C.; Bouchekif, H.; Babooram, K.; Housni,A.; Choytun, N.; Narain, R. J. Polym Sci Part A:Polym Chem 2008, 46, 4984–4996.
68. Benaglia, M.; Rizzardo, E.; Alberti, A.; Guerra, M.Macromolecules 2005, 38, 3129–3140.
69. Thomas, D. B.; Convertine, A. J.; Hester, R. D.;Lowe, A. B.; McCormick, C. L. Macromolecules2004, 37, 1735–1741.
70. (a) Lewis, A. L. Colloids Surf B 2000, 18, 261–275; (b) Lewis, A. L.; Hughes, P. D.; Kirkwood,L. C.; Leppard, S. W.; Redman, R. P.; Tolhurst,L. A.; Stratford, P. W. Biomaterials 2000, 21,1847–1859.
71. (a) Suzuki, S.; Whittaker, M. R.; Grøndahl, L.;Monteiro, M. J.; Wentrup-Byrne, E. Biomacromo-lecules 2006, 7, 3178–3187; (b) Plummer, R.; Goh,Y.-K.; Whittaker, A. K.; Monteiro, M. J. Macromo-lecules 2005, 38, 5352–5355.
72. Tan, J. F.; Too, P. H.; Hatton, A. T.; Tam, K. C.Langmuir 2006, 22, 3744–3750.
73. Wang, C.-H.; Hsiue, G.-H. Bioconjug Chem 2005,16, 391–396.
GLYCOPOLYMERS SYNTHESIS VIA RAFT 627
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola




















