
Non-diffusive spatial segregation of surface reactants in corrosion simulations
12
Non-diffusive spatial segregation of surface reactants in corrosion simulations P. C ordoba-Torres a , K. Bar-Eli b , V. Fair en a, * a Departamento de F ısica Matem atica y Fluidos, Universidad Nacional de Educaci on a Distancia (UNED), Apartado 60141, 28080 Madrid, Spain b School of Chemistry, University of Tel-Aviv, Tel-Aviv, Israel Received 20 January 2004; received in revised form 5 May 2004; accepted 6 May 2004 Available online 2 July 2004 Abstract A metal corrosion mechanism involves several steps with their associated intermediate reactants and rates. The mechanism determines the time evolution of a metal atom through a sequence of states, beginning as soon as it is exposed to the electrolyte and ending when it dissolves, and to which we can associate the idea of a time ordering. On a perfectly smooth or flat surface, this time ordering does not generate heterogeneity in the distribution of reactants on the surface automatically. However, the random nature of individual dissolution events precludes smooth corroding surfaces, leading instead to the development of a certain degree of roughness. We show in this paper that when this is the case, a heterogeneous distribution of surface reactants does indeed emerge as the result of the interplay between the kinetics, which control the time ordering of the species, and morphology, which projects this differential aging process on a spatial dimension. This is done with the help of a 1 + 1-dimensional cellular automaton model of the dissolving metal. We analyze several simulation settings and show that a heterogeneous distribution of reactants evolves along the incisions, predominantly in the direction of their penetration within the metal. The peculiar feature of this surface segregation process is that neither diffusion nor site-dependent reactivity are involved. It is also shown that the segregation phenomenon can be well predicted from the standard macroscopic theory if its results are appropriately interpreted. Ó 2004 Elsevier B.V. All rights reserved. Keywords: Cellular automata simulation; Corrosion; Spatial segregation; Interface roughness; Kinetic model 1. Introduction Surfaces display several specific features that differ from those of the bulk and that affect, among others, such properties as catalysis, corrosion or adsorption [1,2]. One of these features is surface segregation, which is usually understood either in terms of an enrichment of one element at the surface in binary alloys [3] – coseg- regation of two or more in multialloys [4] – or in terms of the formation of adsorbate structures [5]. Both alterna- tives involve mobile particles and are closely related through the adsorption-induced surface enrichment in alloys [6]. Also, surface segregation has been found to affect rough morphologies of interfaces [7]. In two previous papers [8,9], a cellular automaton model (CA) was developed to simulate the dissolution process occurring when a metallic surface dissolves un- der zero current conditions. In spite of the fact that the electrolyte composition was assumed to be perfectly uniform and that the overall process was uniquely controlled by surface reactions – surface and bulk dif- fusion or lateral exchange were not considered – the evidence pointed to an unexpected chemical heteroge- neous distribution coevolving with the emergence of a rough morphology on the metal surface. Inasmuch as particles stay immobile, at least until they dissolve into the electrolyte, we are concerned with a different type of segregation from those mentioned above; it is purely reactive in origin. As the dissolution progresses the surface becomes more corrugated and clusters of metal (designated below NDC – non-dissolved cells) are de- tached from the main bulk. Reactants average coverage * Corresponding author. Tel.: +34-91-3987124; fax: +34-91-3987628/ 6697. E-mail address: [email protected] (V. Fair en). 0022-0728/$ - see front matter Ó 2004 Elsevier B.V. All rights reserved. doi:10.1016/j.jelechem.2004.05.009 Journal of Electroanalytical Chemistry 571 (2004) 189–200 www.elsevier.com/locate/jelechem Journal of Electroanalytical Chemistry
Transcript of Non-diffusive spatial segregation of surface reactants in corrosion simulations

Journal ofElectroanalytical
Chemistry
Journal of Electroanalytical Chemistry 571 (2004) 189–200
www.elsevier.com/locate/jelechem
Non-diffusive spatial segregation of surface reactants incorrosion simulations
P. C�ordoba-Torres a, K. Bar-Eli b, V. Fair�en a,*
a Departamento de F�ısica Matem�atica y Fluidos, Universidad Nacional de Educaci�on a Distancia (UNED), Apartado 60141, 28080 Madrid, Spainb School of Chemistry, University of Tel-Aviv, Tel-Aviv, Israel
Received 20 January 2004; received in revised form 5 May 2004; accepted 6 May 2004
Available online 2 July 2004
Abstract
A metal corrosion mechanism involves several steps with their associated intermediate reactants and rates. The mechanism
determines the time evolution of a metal atom through a sequence of states, beginning as soon as it is exposed to the electrolyte and
ending when it dissolves, and to which we can associate the idea of a time ordering. On a perfectly smooth or flat surface, this time
ordering does not generate heterogeneity in the distribution of reactants on the surface automatically. However, the random nature
of individual dissolution events precludes smooth corroding surfaces, leading instead to the development of a certain degree of
roughness. We show in this paper that when this is the case, a heterogeneous distribution of surface reactants does indeed emerge as
the result of the interplay between the kinetics, which control the time ordering of the species, and morphology, which projects this
differential aging process on a spatial dimension. This is done with the help of a 1+ 1-dimensional cellular automaton model of the
dissolving metal. We analyze several simulation settings and show that a heterogeneous distribution of reactants evolves along the
incisions, predominantly in the direction of their penetration within the metal. The peculiar feature of this surface segregation
process is that neither diffusion nor site-dependent reactivity are involved. It is also shown that the segregation phenomenon can be
well predicted from the standard macroscopic theory if its results are appropriately interpreted.
� 2004 Elsevier B.V. All rights reserved.
Keywords: Cellular automata simulation; Corrosion; Spatial segregation; Interface roughness; Kinetic model
1. Introduction
Surfaces display several specific features that differ
from those of the bulk and that affect, among others,
such properties as catalysis, corrosion or adsorption
[1,2]. One of these features is surface segregation, which
is usually understood either in terms of an enrichment of
one element at the surface in binary alloys [3] – coseg-
regation of two or more in multialloys [4] – or in terms ofthe formation of adsorbate structures [5]. Both alterna-
tives involve mobile particles and are closely related
through the adsorption-induced surface enrichment in
alloys [6]. Also, surface segregation has been found to
affect rough morphologies of interfaces [7].
* Corresponding author. Tel.: +34-91-3987124; fax: +34-91-3987628/
6697.
E-mail address: [email protected] (V. Fair�en).
0022-0728/$ - see front matter � 2004 Elsevier B.V. All rights reserved.
doi:10.1016/j.jelechem.2004.05.009
In two previous papers [8,9], a cellular automatonmodel (CA) was developed to simulate the dissolution
process occurring when a metallic surface dissolves un-
der zero current conditions. In spite of the fact that the
electrolyte composition was assumed to be perfectly
uniform and that the overall process was uniquely
controlled by surface reactions – surface and bulk dif-
fusion or lateral exchange were not considered – the
evidence pointed to an unexpected chemical heteroge-neous distribution coevolving with the emergence of a
rough morphology on the metal surface. Inasmuch as
particles stay immobile, at least until they dissolve into
the electrolyte, we are concerned with a different type of
segregation from those mentioned above; it is purely
reactive in origin. As the dissolution progresses the
surface becomes more corrugated and clusters of metal
(designated below NDC – non-dissolved cells) are de-tached from the main bulk. Reactants average coverage

190 P. C�ordoba-Torres et al. / Journal of Electroanalytical Chemistry 571 (2004) 189–200
fractions measured on these clusters differ from those
obtained at the surface, supporting the conclusion of an
inhomogeneous interface in which segregation of species
occurs as a result of the interplay of kinetics and mor-
phology. Results show that, depending on the reactionkinetic parameters of the model, in some cases one of the
species may concentrate at the rear parts of the corro-
sion front while in other cases that happens for some
other species. This species heterogeneity was found to be
directly related to surface roughness.
The influence of segregation on morphology can be
understood qualitatively by resorting to the concept of
cell-lifetime [9], defined for any state as the average timeelapsing for one cell in that state until it is no longer part
of the interface. Then, in a propagating interface as the
dissolution goes on, if cells located at the rear of the
front are in states with cell-lifetimes smaller than those
of cells at the forefront, they are more prone to disap-
pear earlier and this has a smoothing effect on the
morphology; on the contrary, if cells at the back edge of
the interface have associated larger cell-lifetimes thansites at the forward line, the latter tend to be very active
sites that make notches in the metal bulk, enhancing
roughness. In the first case, the process will not depart
very much from a layer-by-layer dissolution (no
roughness), while the second case leads to high levels of
irregularity. If the surface is homogeneous, all cells have
similar cell-lifetimes and dissolution proceeds in a purely
random way, generating a surface with a very charac-teristic degree of roughness. This idea can be formalized
by introducing the differential aging factor – a lumped
parameter in terms of cell lifetimes – shown to be cor-
related to interface roughness [9]. Moreover, an ap-
proximation to this differential aging factor can be
retrieved from the knowledge of the kinetic rate con-
stants alone, which means that roughness can be in-
ferred from purely macroscopic data. However, detailsof the reactants’ segregation and its relation to kinetics,
which are determinant concepts in a comprehensive
understanding of the emergence of surface roughness,
have not been duly formalized and are worth attention.
Our aim in this paper is to investigate in depth the
segregation of surface reactants along the advancing
direction of the corrosion front, and to relate this seg-
regation to the dissolution mechanism. We shall con-clude that this species heterogeneity is the consequence
of two processes working simultaneously: kinetics traps
interface metallic cells in determinate states remaining
for a long time in that state while surface evolution
leaves interface sites at the rear of the front as they age.
The paper is constructed as follows: a brief review of
the electrochemical mechanism and computational
model is given in Section 2; in Section 3, the simulationresults are shown and the preferential location of surface
reactants are described. It is shown that differentiation in
space is due to a differentiation in time. It is also shown
that segregation exerts a feedback effect on morphology,
leading to profiles with different degrees of roughness.
Finally, in Section 4, we develop a mathematical model
that predicts segregation from kinetics constants.
2. Electrochemical mechanism and simulations
The mechanism and the resulting CA will be de-
scribed briefly. For further details and arguments the
reader is referred to [8,9].
The dissolution mechanism considers a bivalent un-
identified metal and a generic cation, both leading to thepresence of two intermediate adsorbed species on the
metallic surface. The reaction scheme is given by:
M ¢k1
k�1
MðIÞads þ e� ð1Þ
MðIÞads!k2MðIIÞsol þ e� ð2Þ
Cþ þ e�!k�3Cads ð3Þ
Cþ þ Cads þ e�!k�4C2;sol ð4Þ
The CA derived from this kinetic scheme involvesfour possible occupation states: empty [/] (indexed as
0), metal [M] (indexed as 3), intermediate adsorbate
[MðIÞads] (indexed as 1) and cation adsorbate [Cads]
(indexed as 2). Transitions between states obey a set of
probabilistic rules that mimic the above electrochemical
mechanism – a complete list of transition rules can be
found in [8]. Each significant allowed transition is given
a probability of success:
R1 ¼ Probability of ½M� ! ½MðIÞads� ð5Þcorresponding to reaction (1) forward;
R2 ¼ Probability of ½MðIÞads� ! ½/� ð6Þcorresponding to reaction (2);
R3 ¼ Probability of ½MðIÞads� ! ½M� ð7Þcorresponding to reaction (1) backward;
R4 ¼ Probability of ½M� ! ½Cads� ð8Þcorresponding to reaction (3). And, finally,
R5 ¼ Probability of Cads½ � ! ½M� ð9Þ
corresponding to reaction (4).
The process is reaction-limited, diffusion from or into
the electrolyte is considered instantaneous and reactionproducts MðIIÞsol and C2;sol are immediately removed
from the interface during the simulation.
The set of rules can be summarized in the following
scheme:
Cads ¢R5
R4
M ¢R1
R3
MðIÞads!R2MðIIÞsol ð10Þ

P. C�ordoba-Torres et al. / Journal of Electroanalytical Chemistry 571 (2004) 189–200 191
Arguments that justify this scheme are cited exten-
sively in [8,9] and will not be repeated here.
Given a set of reaction probabilities, fRig, as de-
fined in (5)–(9), the corresponding simulation starts
from an initial rectangular grid-lattice of widthL0 ¼ 1000 in which all cells, except those at the top
row, are metal – in state [M]. Those in the top row
are initially in the ‘‘empty’’ state – state [/] – and
model the initial electrolyte, which is always consid-
ered structureless – empty cells – for our purpose. As
the simulation proceeds metal cells dissolve progres-
sively from top to bottom of the grid and the other
species, namely, Cads and MðIÞads, also cover thesurface. When empty rows are formed at the top,
more metal bulk rows are added at the bottom, so
the lattice depth can be considered as infinite. The
stochastic nature of the elementary transition pro-
cesses roughens the initially smooth metal surface
until, after some relaxation time T [8], the whole
process progresses in a stationary regime, character-
ized by fluctuating coverage fractions of surface re-actants around well defined mean values we call
steady-state values. From time to time, the junction
of dissolving inlets isolates clusters of non-dissolved
cells from the metal bulk. They were designated NDC
in [8] and must consistently be accounted for in the
rate equations governing the process.
Writing down the mass and charge balance equations
for our system at the steady state t > T , we obtain:
R1H3 � R2ð þ R3ÞH1 � �n1 ¼ 0; ð11Þ
R4H3 � R5H2 � �n2 ¼ 0; ð12Þ
�vR2H1 þ R5H2 þ R3H1 � R1ð þ R4ÞH3 � �n3 ¼ 0; ð13Þ
�v�
� 1�¼P3
i¼1�ni
R2H1
; ð14Þ
J ¼ R1ð � R4ÞH3 þ R2ð � R3ÞH1 � R5H2; ð15Þ
where H1, H2, H3 ¼ 1�H1 �H2, stand for the average
steady-state coverage ratios of MðIÞads, Cads and M, re-
spectively; R1 through R5 denote the reaction probabil-
ities defined above; J is the dimensionless net currentdensity, which is considered null at open circuit condi-
tions; �n1, �n2 and �n3 stand for the averaged NDC loss rate
of MðIÞads, Cads and M, respectively. Finally, �v is the
mean number of newly exposed M sites that, as a result
of a given cell dissolution, become part of the interface –
see [8] for a thorough interpretation of this descriptor.
Besides the standard electrochemical terms, (11)–(15)
include terms that result from the rough topography ofthe surface: the terms �n1 through �n3 and �vR2H1, would
not exist in a smooth metal surface.
From the above equations, one obtains the steady-
state coverage fractions of MðIÞads and Cads as follows:
H1¼R1R5
R4þR5ð Þ R2þR3ð ÞþR1R5
þ�n2R1��n1 R4þR5ð Þ
R4þR5ð Þ R2þR3ð ÞþR1R5
;
ð16Þ
H2¼R4 R2þR3ð Þ
R4þR5ð Þ R2þR3ð ÞþR1R5
þ�n1R4��n2 R1þR2þR3ð ÞR4þR5ð Þ R2þR3ð ÞþR1R5
:
ð17ÞIt is interesting to note that the first terms on the
right-hand side of Eqs. (16) and (17) are precisely those
expressions for the steady-state values derived from thestandard macroscopic rate equations for reactions (1)–
(4), provided we make a one to one correspondence
between R1, R2, R3, R4, R5 and k1, k2, k�1, k�3 and k�4,
respectively. The �n-dependent contributions to (16) and
(17) are purely mesoscopic – having no macroscopic
counterpart – and are related to NDC production.
3. Results
As described in the previous section, the computa-
tions start with a smooth metallic-electrolyte surface anda given set of probabilities fRig, and proceed according
to the cited rules. The initially flat interface moves in the
downward direction as metal cells dissolve, becoming
irregular in a typical succession of indented valleys al-
ternating with summits. After some time T the system
arrives at a steady state as far as average surface frac-
tions are concerned [8]. The steady-state values are given
by (16) and (17). On the other hand, in an infinite lattice(L0 ¼ 1), the height of the corrugated front in the
propagation direction, hmaxðL0; tÞ, given by the distance
in lattice units between the bottom and the top of the
profile, would grow indefinitely. Actually, due to
the lattice finiteness and after some further time, T � > T ,the front height reaches a steady-state value [10],
hssmaxðL0Þ, that depends on the particular set fRig. Fig. 1shows how the interface height grows from the initialflat surface until it saturates to a constant value and
fluctuates around it.
3.1. Segregation: preliminary considerations
The fractional coverage of the various species as a
function of the height is defined by
HiðhÞ ¼ niðhÞX3i¼1
niðhÞ, +*
t>T �
; i ¼ 1; 2; 3; ð18Þ
where h is the elevation, measured in number of lattice
rows, with the origin at the bottom floor of the deepest
valley of the interface profile, and niðhÞ denotes the
number of interface cells occupied by species i at a
height h. The symbol h�it>T � indicates that HiðhÞ is the
result of a time average on a simulation, for t > T �. For

0,0 2,0x105
4,0x105
6,0x105
8,0x105
1,0x106
0
20
40
60
80
100
h max
(L0, t
) /l
atti
ce r
ows
t /number of steps
Fig. 1. Time evolution of interface height, hmaxðL0; tÞ, in lattice units –
L0 ¼ 1000. The horizontal continuous line corresponds to the steady-
state value hssmaxðL0Þ ¼ 73 reached after time T �. This simulation was
carried with the following set of reaction probabilities:
R1 ¼ 1:62� 10�2, R2 ¼ 1:54� 10�2, R3 ¼ 0:0, R4 ¼ 2:0� 10�2,
R5 ¼ 1:02� 10�3.
0,0
0,1
0,2~
Θ1(t
CA(h))
0,0
0,2
0,4
0,6
0,8
~ Θ2(t
CA(h))
0 10 20 30 40 50 60 70
0,0
0,2
0,4
0,6
0,8~ Θ
3(t
CA(h))
Θ3(h)
Θ2(h)
Θ1(h)
h /lattice rows
Fig. 2. Height dependence of surface coverage of MðIÞads, Cads and M,
calculated from (18) at the steady-state regime shown in Fig. 1 (solid
points). The corresponding mean interface coverage fractions obtained
from the simulation, H1 ¼ 7:41� 10�2, H2 ¼ 8:54� 10�1 and
H3 ¼ 7:17� 10�2, are indicated with horizontal broken lines. The
continuous lines stand for the macroscopic model developed in Section
4, Eq. (27) for the same set of Rs of Fig. 1.
0 10 20 30 40 50 60 700,000
0,005
0,010
0,015
0,020
0,025
0,030
Ni(h )
h /lattice rows
N1
N2
N3
Fig. 3. Normalized number of MðIÞads cells (solid circles), Cads cells
(open circles) and M cells (triangles) vs. front height, h, corresponding
to the same simulation of Figs. 1 and 2.
192 P. C�ordoba-Torres et al. / Journal of Electroanalytical Chemistry 571 (2004) 189–200
the particular simulation displayed in Fig. 2, we realize
that the coverage is heterogeneous. MðIÞads has higher
than average (shown by the dotted horizontal line in the
figure) surface concentration near valley bottoms,
H1 ðlow hÞ > H1, and lower than the average around
summits, H1 ðhigh hÞ < H1. The spatial distribution is
reversed for the Cads: H2 ðhigh hÞ > H2 andH2 ðlow hÞ < H2. A similar pattern to that shown by the
MðIÞads is observed for the metal M sites.
In order to see this more clearly we show in Fig. 3 the
number (normalized) of cells occupied by the different
species as a function of the height, for the same set of Rs
of Fig. 2, evaluated according to the following equation:
NiðhÞ ¼ niðhÞXhmax
h¼0
niðhÞ !, +*
t>T �
; i ¼ 1; 2; 3: ð19Þ
It is seen that the Cads are preferentially located at
high elevations of the moving interface while both the M
and the MðIÞads cells are more prone to pack together at
low elevations. We will term this situation Cads – seg-
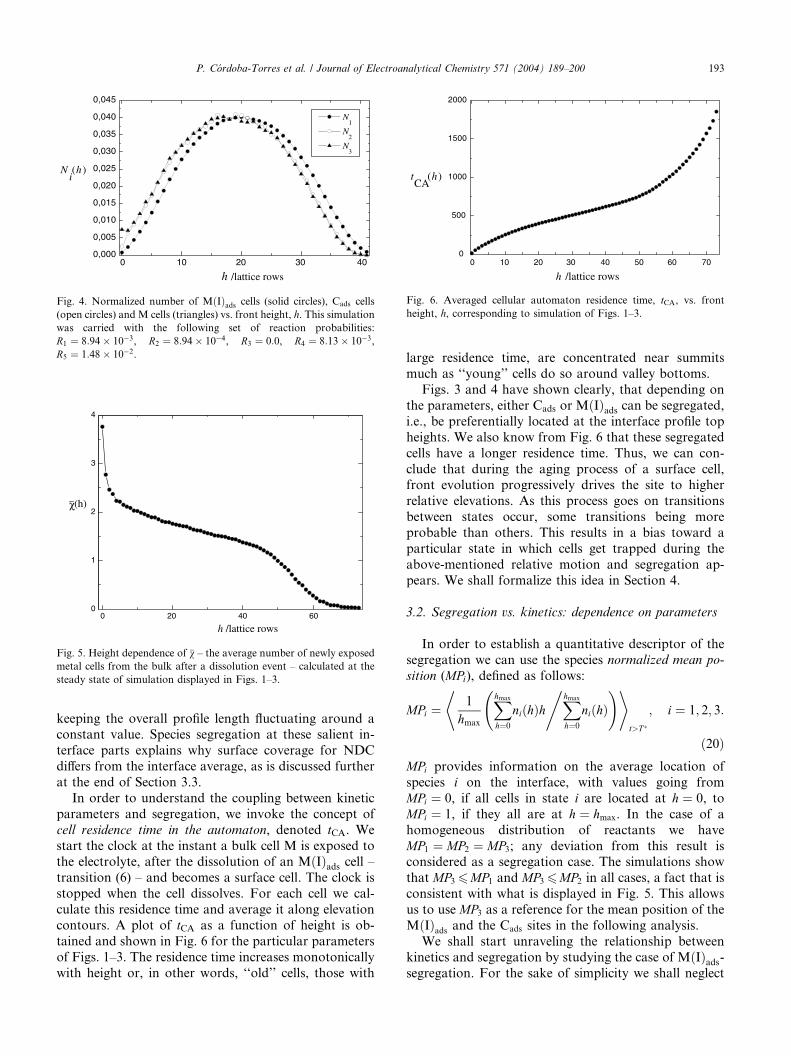
regation in contrast to the case shown in Fig. 4 that
shows the MðIÞads – segregation case, with this species
shifting preferential occupation to higher elevations.
Figs. 3 and 4 are particular examples of Cads orMðIÞads segregation, respectively. Different degrees of
segregation – including the possibility of no segregation,
i.e., homogeneous surface – are possible, depending on
the set of parameters values fRig. Metal cells (M cells)
constitute a distinct case for they always tend to accu-
mulate, on average, at low heights. It happens that at
low elevations an individual dissolution event uncovers,
on average, more fresh new M sites than it does at highelevations, as shown in Fig. 5. �v, the average number of
newly exposed metal cells from the bulk after a disso-
lution event [8], decreases from bottom to top – the
decrease of �v with height is typical for the system and
occurs with all sets of parameters. This mechanism of
enlargement of the interface length is balanced by anNDC-stripping at interface protrusions, thus helping in
0 10 20 30 400,000
0,005
0,010
0,015
0,020
0,025
0,030
0,035
0,040
0,045
Ni(h )
h /lattice rows
N1
N2
N3
Fig. 4. Normalized number of MðIÞads cells (solid circles), Cads cells
(open circles) and M cells (triangles) vs. front height, h. This simulation
was carried with the following set of reaction probabilities:
R1 ¼ 8:94� 10�3, R2 ¼ 8:94� 10�4, R3 ¼ 0:0, R4 ¼ 8:13� 10�3,
R5 ¼ 1:48� 10�2.
0
h /lattice rows20 40 60
0
1
2
3
4
(h)χ
Fig. 5. Height dependence of �v – the average number of newly exposed
metal cells from the bulk after a dissolution event – calculated at the
steady state of simulation displayed in Figs. 1–3.
0 10 20 30 40 50 60 700
500
1000
1500
2000
tCA
(h)
h /lattice rows
Fig. 6. Averaged cellular automaton residence time, tCA, vs. front
height, h, corresponding to simulation of Figs. 1–3.
P. C�ordoba-Torres et al. / Journal of Electroanalytical Chemistry 571 (2004) 189–200 193
keeping the overall profile length fluctuating around aconstant value. Species segregation at these salient in-
terface parts explains why surface coverage for NDC
differs from the interface average, as is discussed further
at the end of Section 3.3.
In order to understand the coupling between kinetic
parameters and segregation, we invoke the concept of
cell residence time in the automaton, denoted tCA. We
start the clock at the instant a bulk cell M is exposed tothe electrolyte, after the dissolution of an MðIÞads cell –transition (6) – and becomes a surface cell. The clock is
stopped when the cell dissolves. For each cell we cal-
culate this residence time and average it along elevation
contours. A plot of tCA as a function of height is ob-
tained and shown in Fig. 6 for the particular parameters
of Figs. 1–3. The residence time increases monotonically
with height or, in other words, ‘‘old’’ cells, those with
large residence time, are concentrated near summits
much as ‘‘young’’ cells do so around valley bottoms.
Figs. 3 and 4 have shown clearly, that depending on
the parameters, either Cads or MðIÞads can be segregated,
i.e., be preferentially located at the interface profile top
heights. We also know from Fig. 6 that these segregated
cells have a longer residence time. Thus, we can con-
clude that during the aging process of a surface cell,front evolution progressively drives the site to higher
relative elevations. As this process goes on transitions
between states occur, some transitions being more
probable than others. This results in a bias toward a
particular state in which cells get trapped during the
above-mentioned relative motion and segregation ap-
pears. We shall formalize this idea in Section 4.
3.2. Segregation vs. kinetics: dependence on parameters
In order to establish a quantitative descriptor of the
segregation we can use the species normalized mean po-
sition (MPi), defined as follows:
MPi ¼1
hmax
Xhmax
h¼0
niðhÞhXhmax
h¼0
niðhÞ, ! +*
t>T �
; i ¼ 1; 2; 3:
ð20ÞMPi provides information on the average location of
species i on the interface, with values going from
MPi ¼ 0, if all cells in state i are located at h ¼ 0, to
MPi ¼ 1, if they all are at h ¼ hmax. In the case of a
homogeneous distribution of reactants we have
MP1 ¼ MP2 ¼ MP3; any deviation from this result isconsidered as a segregation case. The simulations show
that MP3 6MP1 and MP3 6MP2 in all cases, a fact that is
consistent with what is displayed in Fig. 5. This allows
us to use MP3 as a reference for the mean position of the
MðIÞads and the Cads sites in the following analysis.
We shall start unraveling the relationship between
kinetics and segregation by studying the case of MðIÞads-segregation. For the sake of simplicity we shall neglect
5 10 15 20
1,05
1,10
1,15
1,20
(a)
MP
2/ MP
3
5 10 15 20
1,02
1,03
1,04
1,05
1,06
R3/R
2=0
R3/R
2=20
MP
2/ MP
3
(b)
R3/R
2=0
R3/R
2=20
0 5 10 15 201,010
1,015
1,020
1,025
1,030
1,035
1,040
1,045
R3/R
2=0
R3/R
2=20
MP
2/ MP
3
(c)
R4/R
5
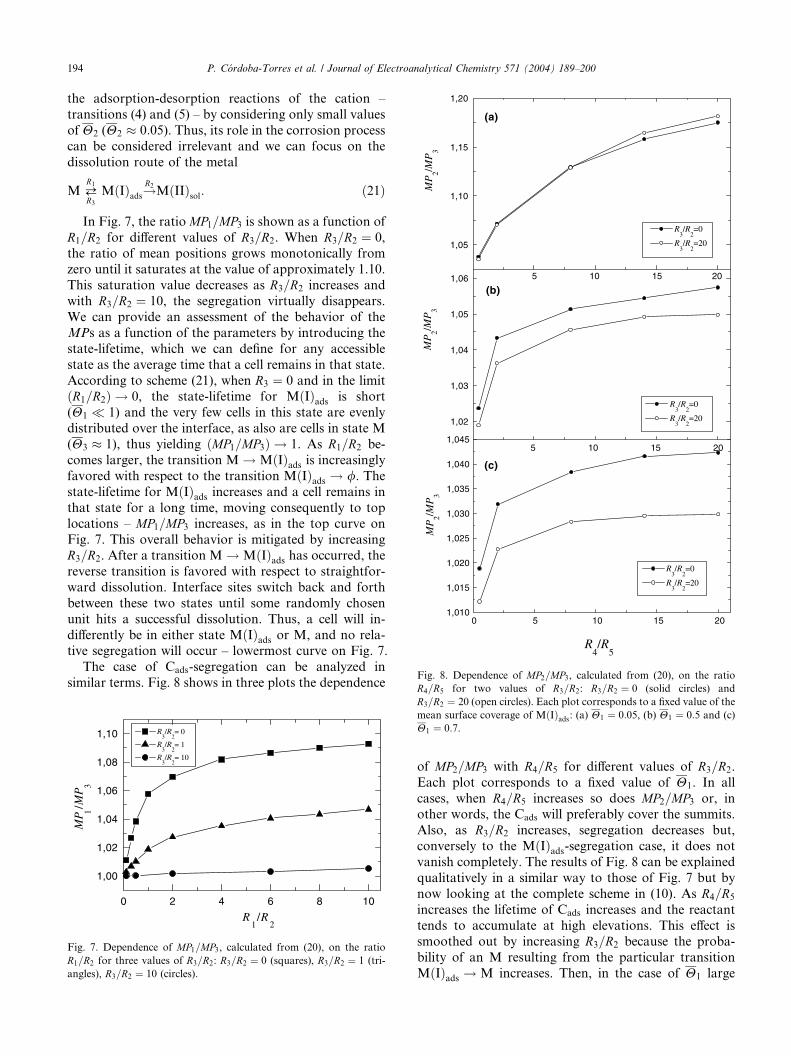
Fig. 8. Dependence of MP2=MP3, calculated from (20), on the ratio
R4=R5 for two values of R3=R2: R3=R2 ¼ 0 (solid circles) and
194 P. C�ordoba-Torres et al. / Journal of Electroanalytical Chemistry 571 (2004) 189–200
the adsorption-desorption reactions of the cation –
transitions (4) and (5) – by considering only small values
of H2 (H2 � 0:05). Thus, its role in the corrosion process
can be considered irrelevant and we can focus on the
dissolution route of the metal
M ¢R1
R3
MðIÞads!R2MðIIÞsol: ð21Þ
In Fig. 7, the ratio MP1=MP3 is shown as a function of
R1=R2 for different values of R3=R2. When R3=R2 ¼ 0,
the ratio of mean positions grows monotonically fromzero until it saturates at the value of approximately 1.10.
This saturation value decreases as R3=R2 increases and
with R3=R2 ¼ 10, the segregation virtually disappears.
We can provide an assessment of the behavior of the
MPs as a function of the parameters by introducing the
state-lifetime, which we can define for any accessible
state as the average time that a cell remains in that state.
According to scheme (21), when R3 ¼ 0 and in the limitðR1=R2Þ ! 0, the state-lifetime for MðIÞads is short
(H1 � 1) and the very few cells in this state are evenly
distributed over the interface, as also are cells in state M
(H3 � 1), thus yielding ðMP1=MP3Þ ! 1. As R1=R2 be-
comes larger, the transition M ! MðIÞads is increasinglyfavored with respect to the transition MðIÞads ! /. Thestate-lifetime for MðIÞads increases and a cell remains in
that state for a long time, moving consequently to toplocations – MP1=MP3 increases, as in the top curve on
Fig. 7. This overall behavior is mitigated by increasing
R3=R2. After a transition M ! MðIÞads has occurred, thereverse transition is favored with respect to straightfor-
ward dissolution. Interface sites switch back and forth
between these two states until some randomly chosen
unit hits a successful dissolution. Thus, a cell will in-
differently be in either state MðIÞads or M, and no rela-tive segregation will occur – lowermost curve on Fig. 7.
The case of Cads-segregation can be analyzed in
similar terms. Fig. 8 shows in three plots the dependence
0 2 4 6 8 10
1,00
1,02
1,04
1,06
1,08
1,10
MP 1
/ MP 3
R1/R
2
R3/R
2= 0
R3/R
2= 1
R3/R
2= 10
Fig. 7. Dependence of MP1=MP3, calculated from (20), on the ratio
R1=R2 for three values of R3=R2: R3=R2 ¼ 0 (squares), R3=R2 ¼ 1 (tri-
angles), R3=R2 ¼ 10 (circles).
R3=R2 ¼ 20 (open circles). Each plot corresponds to a fixed value of the
mean surface coverage of MðIÞads: (a) H1 ¼ 0:05, (b) H1 ¼ 0:5 and (c)
H1 ¼ 0:7.
of MP2=MP3 with R4=R5 for different values of R3=R2.
Each plot corresponds to a fixed value of H1. In all
cases, when R4=R5 increases so does MP2=MP3 or, inother words, the Cads will preferably cover the summits.
Also, as R3=R2 increases, segregation decreases but,
conversely to the MðIÞads-segregation case, it does not
vanish completely. The results of Fig. 8 can be explained
qualitatively in a similar way to those of Fig. 7 but by
now looking at the complete scheme in (10). As R4=R5
increases the lifetime of Cads increases and the reactant
tends to accumulate at high elevations. This effect issmoothed out by increasing R3=R2 because the proba-
bility of an M resulting from the particular transition
MðIÞads ! M increases. Then, in the case of H1 large
1,05
1,10
1,15
MP
/MP 1
P. C�ordoba-Torres et al. / Journal of Electroanalytical Chemistry 571 (2004) 189–200 195
(Fig. 8(c)) there are many MðIÞads cells distributed all
over the front height, meaning that more M cells will be
located at higher h. This is why MP2=MP3 decreases withrespect to the R3=R2 ¼ 0 case. The above effect resulting
from the MðIÞads ! M transition loses intensity whenH1 decreases (Fig. 8(a) and (b)) and so does the sepa-
ration between the curves.
0,95
1,00
2 4 6 8 10 12 14
L / L0
MP
/MP 2
MP
/MP 3
0,96
0,98
1,00
1,02
1,04
1,06
1,08
2 4 6 8 10 12 14
1,00
1,05
1,10
1,15
2 4 6 8 10 12 14
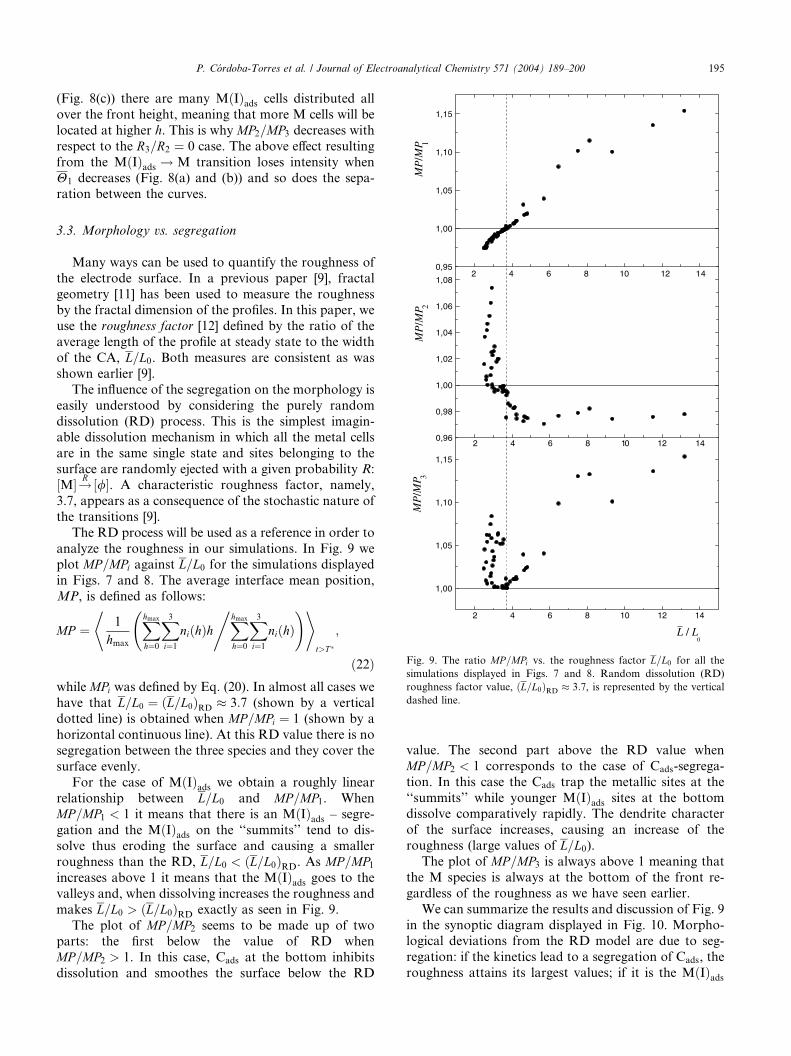
Fig. 9. The ratio MP=MPi vs. the roughness factor L=L0 for all the
simulations displayed in Figs. 7 and 8. Random dissolution (RD)
roughness factor value, ðL=L0ÞRD � 3:7, is represented by the vertical
dashed line.
3.3. Morphology vs. segregation
Many ways can be used to quantify the roughness of
the electrode surface. In a previous paper [9], fractal
geometry [11] has been used to measure the roughnessby the fractal dimension of the profiles. In this paper, we
use the roughness factor [12] defined by the ratio of the
average length of the profile at steady state to the width
of the CA, L=L0. Both measures are consistent as was
shown earlier [9].
The influence of the segregation on the morphology is
easily understood by considering the purely random
dissolution (RD) process. This is the simplest imagin-able dissolution mechanism in which all the metal cells
are in the same single state and sites belonging to the
surface are randomly ejected with a given probability R:
½M�!R ½/�. A characteristic roughness factor, namely,
3.7, appears as a consequence of the stochastic nature of
the transitions [9].
The RD process will be used as a reference in order to
analyze the roughness in our simulations. In Fig. 9 weplot MP=MPi against L=L0 for the simulations displayed
in Figs. 7 and 8. The average interface mean position,
MP, is defined as follows:
MP ¼ 1
hmax
Xhmax
h¼0
X3i¼1
niðhÞhXhmax
h¼0
X3i¼1
niðhÞ, ! +*
t>T �
;
ð22Þwhile MPi was defined by Eq. (20). In almost all cases we
have that L=L0 ¼ ðL=L0ÞRD � 3:7 (shown by a vertical
dotted line) is obtained when MP=MPi ¼ 1 (shown by a
horizontal continuous line). At this RD value there is no
segregation between the three species and they cover the
surface evenly.
For the case of MðIÞads we obtain a roughly linear
relationship between L=L0 and MP=MP1. WhenMP=MP1 < 1 it means that there is an MðIÞads – segre-
gation and the MðIÞads on the ‘‘summits’’ tend to dis-
solve thus eroding the surface and causing a smaller
roughness than the RD, L=L0 < ðL=L0ÞRD. As MP=MP1increases above 1 it means that the MðIÞads goes to the
valleys and, when dissolving increases the roughness and
makes L=L0 > ðL=L0ÞRD exactly as seen in Fig. 9.
The plot of MP=MP2 seems to be made up of twoparts: the first below the value of RD when
MP=MP2 > 1. In this case, Cads at the bottom inhibits
dissolution and smoothes the surface below the RD
value. The second part above the RD value when
MP=MP2 < 1 corresponds to the case of Cads-segrega-
tion. In this case the Cads trap the metallic sites at the
‘‘summits’’ while younger MðIÞads sites at the bottomdissolve comparatively rapidly. The dendrite character
of the surface increases, causing an increase of the
roughness (large values of L=L0).The plot of MP=MP3 is always above 1 meaning that
the M species is always at the bottom of the front re-
gardless of the roughness as we have seen earlier.
We can summarize the results and discussion of Fig. 9
in the synoptic diagram displayed in Fig. 10. Morpho-logical deviations from the RD model are due to seg-
regation: if the kinetics lead to a segregation of Cads, the
roughness attains its largest values; if it is the MðIÞads

M(I)ads
segregation
Cads
segregation
RD ModelRou
ghne
ss
Fig. 10. Scheme describing the relationship between the morphology of
the interface and the type of species segregation.
0,96 0,98 1,00 1,02 1,04 1,06 1,08 1,10 1,12 1,14 1,160,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
MP/MP2
Θ1N
DC /
Θ1
MP/MP1
0,96 0,98 1,00 1,02 1,04 1,06 1,08
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
Θ3N
DC /
Θ3
Θ2N
DC /
Θ2
0,98 1,00 1,02 1,04 1,06 1,08 1,10 1,12 1,14 1,16
0,0
0,2
0,4
0,6
0,8
1,0
MP/MP3
Fig. 11. Plots of the surface coverage of the species in the NDC, HNDC
i ,
relative to that on the interface, Hi, as a function of the species relative
mean position, MP=MPi, for all the simulations displayed in Figs. 7
and 8.
196 P. C�ordoba-Torres et al. / Journal of Electroanalytical Chemistry 571 (2004) 189–200
that is segregated, the profiles display a lower degree ofirregularity.
Segregation, however, does not always lead to a de-
parture from the RD model. There is a singular case of
segregation showing the same morphology as in the RD
model: the limit H1 ! 1. It is obtained when
R1=R2 ! 1 and R3=R2 ¼ 0. The interface is practically
covered by MðIÞads and MP=MP1 ! 1, but there is in-
stead a great segregation between MðIÞads and both Mand Cads (Fig. 7). We have thus MP=MP2 > 1 and
MP=MP3 > 1. The case to which we refer would corre-
spond to the limit point in the prolongation of the left
branch of the plots MP=MP2 and MP=MP3 in Fig. 9: as
R1=R2 ! 1 while R3=R2 ¼ 0, MP=MP2 and MP=MP3increase and L=L0 ! ðL=L0ÞRD.
We shall conclude the section by focusing on NDC
surface composition. As described in the introduction,the corrugated surface, formed by the dissolution pro-
cess, may break from time to time and detached clusters
of metal, designated non-dissolved cells – NDC, are
formed. It is reasonable to expect that the NDC will be
formed preferentially from the summits of the corru-
gated surface. Indeed, the plots of Fig. 11 are in agree-
ment with this expectation. The figure shows HNDC
i =Hi
as a function of MP=MPi, i.e., the average species cov-erage on the NDC, as compared to that of the interface
coverage, as a function of the species relative mean
position. The effect of the MðIÞads segregation is dis-
played in the first plot of Fig. 11: when the MðIÞadsspecies is located preferentially at the summits
(MP=MP1 < 1), the contours of NDC have more MðIÞadsthan expected, H
NDC
1 =H1 > 1. The Cads segregation case
behaves in a similar way: when MP=MP2 < 1 we observe
that HNDC
2 =H2 > 1, i.e., the NDC are covered prefer-
entially with Cads; again, our anticipation that the NDC
originate at the top is confirmed. Finally, in the last plot
of Fig. 9 we saw that MP=MP3 > 1 in all cases, showing
that the metal atoms are always located at the bottom.Therefore, one expects the NDC surface to be depleted
of the metal atoms; Fig. 11 shows that in all cases
0,25
0,30
P. C�ordoba-Torres et al. / Journal of Electroanalytical Chemistry 571 (2004) 189–200 197
HNDC3 =H3 < 1, i.e., there are less M cells on the surface
of NDC than on the bulk surface.
0,00
0,05
0,10
0,15
0,20
0,0
0,2
0,4
0,6
0,8
0 100 200 300 400 500 600 700
0,0
0,2
0,4
0,6
0,8
~
~
~
Θ3(t)
Θ2(t)
Θ1(t)
t
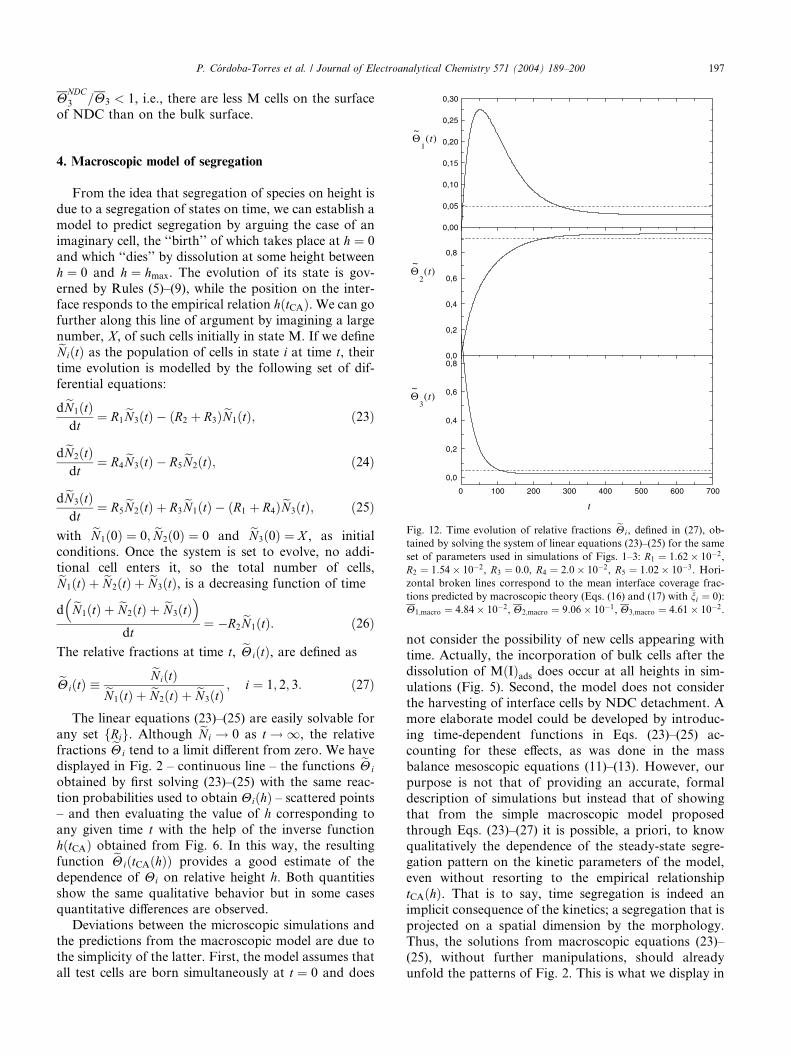
Fig. 12. Time evolution of relative fractions eHi, defined in (27), ob-
tained by solving the system of linear equations (23)–(25) for the same
set of parameters used in simulations of Figs. 1–3: R1 ¼ 1:62� 10�2,
R2 ¼ 1:54� 10�2, R3 ¼ 0:0, R4 ¼ 2:0� 10�2, R5 ¼ 1:02� 10�3. Hori-
zontal broken lines correspond to the mean interface coverage frac-
tions predicted by macroscopic theory (Eqs. (16) and (17) with �ni ¼ 0):
H1;macro ¼ 4:84� 10�2, H2;macro ¼ 9:06� 10�1, H3;macro ¼ 4:61� 10�2.
4. Macroscopic model of segregation
From the idea that segregation of species on height is
due to a segregation of states on time, we can establish a
model to predict segregation by arguing the case of an
imaginary cell, the ‘‘birth’’ of which takes place at h ¼ 0
and which ‘‘dies’’ by dissolution at some height between
h ¼ 0 and h ¼ hmax. The evolution of its state is gov-
erned by Rules (5)–(9), while the position on the inter-face responds to the empirical relation hðtCAÞ. We can go
further along this line of argument by imagining a large
number, X, of such cells initially in state M. If we defineeNiðtÞ as the population of cells in state i at time t, their
time evolution is modelled by the following set of dif-
ferential equations:
deN1ðtÞdt
¼ R1eN3ðtÞ � R2ð þ R3ÞeN1ðtÞ; ð23Þ
deN2ðtÞdt
¼ R4eN3ðtÞ � R5
eN2ðtÞ; ð24Þ
deN3ðtÞdt
¼ R5eN2ðtÞ þ R3
eN1ðtÞ � R1ð þ R4ÞeN3ðtÞ; ð25Þ
with eN1ð0Þ ¼ 0; eN2ð0Þ ¼ 0 and eN3ð0Þ ¼ X , as initial
conditions. Once the system is set to evolve, no addi-tional cell enters it, so the total number of cells,eN1ðtÞ þ eN2ðtÞ þ eN3ðtÞ, is a decreasing function of time
d eN1ðtÞ þ eN2ðtÞ þ eN3ðtÞ� �
dt¼ �R2
eN1ðtÞ: ð26Þ
The relative fractions at time t, eHiðtÞ, are defined as
eHiðtÞ �eNiðtÞeN1ðtÞ þ eN2ðtÞ þ eN3ðtÞ
; i ¼ 1; 2; 3: ð27Þ
The linear equations (23)–(25) are easily solvable for
any set fRig. Although eNi ! 0 as t ! 1, the relative
fractions eHi tend to a limit different from zero. We have
displayed in Fig. 2 – continuous line – the functions eHi
obtained by first solving (23)–(25) with the same reac-
tion probabilities used to obtain HiðhÞ – scattered points– and then evaluating the value of h corresponding to
any given time t with the help of the inverse function
hðtCAÞ obtained from Fig. 6. In this way, the resulting
function eHiðtCAðhÞÞ provides a good estimate of the
dependence of Hi on relative height h. Both quantities
show the same qualitative behavior but in some cases
quantitative differences are observed.
Deviations between the microscopic simulations andthe predictions from the macroscopic model are due to
the simplicity of the latter. First, the model assumes that
all test cells are born simultaneously at t ¼ 0 and does
not consider the possibility of new cells appearing with
time. Actually, the incorporation of bulk cells after the
dissolution of MðIÞads does occur at all heights in sim-
ulations (Fig. 5). Second, the model does not consider
the harvesting of interface cells by NDC detachment. A
more elaborate model could be developed by introduc-ing time-dependent functions in Eqs. (23)–(25) ac-
counting for these effects, as was done in the mass
balance mesoscopic equations (11)–(13). However, our
purpose is not that of providing an accurate, formal
description of simulations but instead that of showing
that from the simple macroscopic model proposed
through Eqs. (23)–(27) it is possible, a priori, to know
qualitatively the dependence of the steady-state segre-gation pattern on the kinetic parameters of the model,
even without resorting to the empirical relationship
tCAðhÞ. That is to say, time segregation is indeed an
implicit consequence of the kinetics; a segregation that is
projected on a spatial dimension by the morphology.
Thus, the solutions from macroscopic equations (23)–
(25), without further manipulations, should already
unfold the patterns of Fig. 2. This is what we display in
5 10 15 20
5 10 15 20
5 10 15 20
1,3
1,4
1,5
1,6
1,7
1,8
1,9
2,0
~~
t 2/ t 3
~~
t 2/ t 3
(a)
R3/R
2=0
R3/R
2=20
1,2
1,3
1,4
1,5
1,6
1,7
1,8
1,9
2,0
R3/R
2=0
R3/R
2=20
(b)
1,1
1,2
1,3
1,4
1,5
1,6
1,7
1,8
1,9
2,0
R3/R
2=0
R3/R
2=20
~~
t 2/ t 3
(c)
R4/R
5
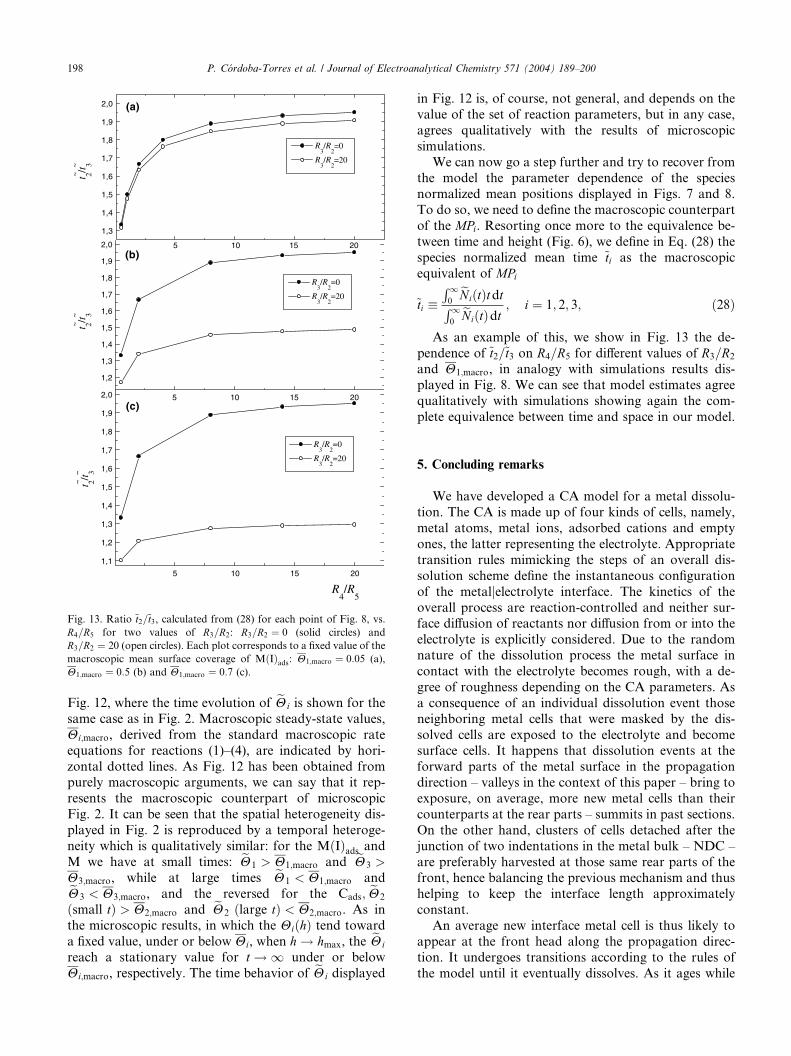
Fig. 13. Ratio ~t2=~t3, calculated from (28) for each point of Fig. 8, vs.
R4=R5 for two values of R3=R2: R3=R2 ¼ 0 (solid circles) and
R3=R2 ¼ 20 (open circles). Each plot corresponds to a fixed value of the
macroscopic mean surface coverage of MðIÞads: H1;macro ¼ 0:05 (a),
H1;macro ¼ 0:5 (b) and H1;macro ¼ 0:7 (c).
198 P. C�ordoba-Torres et al. / Journal of Electroanalytical Chemistry 571 (2004) 189–200
Fig. 12, where the time evolution of eHi is shown for the
same case as in Fig. 2. Macroscopic steady-state values,
Hi;macro, derived from the standard macroscopic rate
equations for reactions (1)–(4), are indicated by hori-
zontal dotted lines. As Fig. 12 has been obtained frompurely macroscopic arguments, we can say that it rep-
resents the macroscopic counterpart of microscopic
Fig. 2. It can be seen that the spatial heterogeneity dis-
played in Fig. 2 is reproduced by a temporal heteroge-
neity which is qualitatively similar: for the MðIÞads andM we have at small times: eH1 > H1;macro and eH3 >H3;macro, while at large times eH1 < H1;macro andeH3 < H3;macro, and the reversed for the Cads; eH2
ðsmall tÞ > H2;macro and eH2 ðlarge tÞ < H2;macro. As in
the microscopic results, in which the HiðhÞ tend toward
a fixed value, under or below Hi, when h ! hmax, the eHi
reach a stationary value for t ! 1 under or below
Hi;macro, respectively. The time behavior of eHi displayed
in Fig. 12 is, of course, not general, and depends on the
value of the set of reaction parameters, but in any case,
agrees qualitatively with the results of microscopic
simulations.
We can now go a step further and try to recover fromthe model the parameter dependence of the species
normalized mean positions displayed in Figs. 7 and 8.
To do so, we need to define the macroscopic counterpart
of the MPi. Resorting once more to the equivalence be-
tween time and height (Fig. 6), we define in Eq. (28) the
species normalized mean time ~ti as the macroscopic
equivalent of MPi
~ti �R10eNiðtÞtdtR1
0eNiðtÞdt
; i ¼ 1; 2; 3; ð28Þ
As an example of this, we show in Fig. 13 the de-
pendence of ~t2=~t3 on R4=R5 for different values of R3=R2
and H1;macro, in analogy with simulations results dis-
played in Fig. 8. We can see that model estimates agree
qualitatively with simulations showing again the com-
plete equivalence between time and space in our model.
5. Concluding remarks
We have developed a CA model for a metal dissolu-
tion. The CA is made up of four kinds of cells, namely,
metal atoms, metal ions, adsorbed cations and empty
ones, the latter representing the electrolyte. Appropriate
transition rules mimicking the steps of an overall dis-
solution scheme define the instantaneous configuration
of the metaljelectrolyte interface. The kinetics of the
overall process are reaction-controlled and neither sur-face diffusion of reactants nor diffusion from or into the
electrolyte is explicitly considered. Due to the random
nature of the dissolution process the metal surface in
contact with the electrolyte becomes rough, with a de-
gree of roughness depending on the CA parameters. As
a consequence of an individual dissolution event those
neighboring metal cells that were masked by the dis-
solved cells are exposed to the electrolyte and becomesurface cells. It happens that dissolution events at the
forward parts of the metal surface in the propagation
direction – valleys in the context of this paper – bring to
exposure, on average, more new metal cells than their
counterparts at the rear parts – summits in past sections.
On the other hand, clusters of cells detached after the
junction of two indentations in the metal bulk – NDC –
are preferably harvested at those same rear parts of thefront, hence balancing the previous mechanism and thus
helping to keep the interface length approximately
constant.
An average new interface metal cell is thus likely to
appear at the front head along the propagation direc-
tion. It undergoes transitions according to the rules of
the model until it eventually dissolves. As it ages while

P. C�ordoba-Torres et al. / Journal of Electroanalytical Chemistry 571 (2004) 189–200 199
the metaljelectrolyte interface keeps propagating, it is
subjected to a displacement, relative to the mean posi-
tion of the front in a direction opposite to that of
propagation. Therefore, the cell positions along the
propagation direction depend monotonically on theirage and thus a space–time relationship can be estab-
lished.
By controlling the CA parameters we can define
which transition or step is slowest. The reactants par-
ticipating in the slowest will have a longer lifetime than
the other intervening species and consequently surface
cells will be trapped in that particular state. As they age
they tend to concentrate at the back of the interfacealong the propagation direction, thus unfolding a het-
erogeneous interface along that same direction. We
speak of a segregation of that particular reactant.
Here, we have dealt with two intermediate reactants:
a metal ion that takes part in the metal dissolution route
and an adsorbed cation. From a morphological point of
view this brings opposite consequences. Metal ion –
MðIÞads – segregation has a leveling effect on the surface,diminishing roughness. On the contrary, by delaying
dissolution the adsorbed cation – Cads – segregation
promotes dendrite formation as large portions of the
interface covered by cells in state Cads remain lagging
behind while dissolution proceeds within deep cuts into
the metal bulk, thus enhancing roughness.
From a kinetic point of view, the importance of the
spatial heterogeneity becomes crucial when bimolecularreactions are included in the reactive scheme. That is the
case of the Tafel reaction: Cads þ Cads ! C2;sol. It has
been shown [13] that, even in a motionless scenario, the
Tafel reaction is well described by fractional order ki-
netics in which the reaction order depends on the aver-
age local coverage of Cads around a Cads, namely, Hlocal
2 .
When this local coverage is equal to the global average,
i.e., Hlocal
2 ¼ H2, the second-order kinetics predicted bythe mean field (MF) theory – which is essentially based
on a perfect mixing of reactants – is then recovered. If,
however, we have Hlocal
2 6¼ H2, then the reaction order
differs from the value of two and the kinetics departs
from the standard macroscopic equations derived from
the MF approach. When the distribution of Cads on the
surface is heterogeneous, as in the Cads-segregation case,
we have Hlocal
2 6¼ H2 and, consequently, the kinetics arecharacterized by a fractional reaction order. If the seg-
regation is very pronounced, we may have Hlocal
2 � 1,
thus giving first-order kinetics for the Tafel reaction.
Finally, a simple macroscopic model that imitates the
CA behavior by describing the evolution of a closed
population of cells was developed and its results com-
pared to those of the CA. As such, this macroscopic
model consists of a set of ordinary differential equationsfor the time evolution of the species coverage fractions.
Once the equations were integrated and the time vari-
able projected into a space variable a good qualitative
agreement was obtained with the CA. This is not sur-
prising inasmuch as the segregation is actually an effect
of the reaction kinetics – diffusion of reactants is not
allowed – and becomes apparent if the appropriate
projection on a spatial scale is conveniently chosen.Consequently, if time and space can be mapped indif-
ferently into each other and the reaction kinetics gov-
erns the ordering in time, then segregation should be
inferable from an integration of the standard macro-
scopic equations without resorting to any spatial map-
ping: that is, reading the time axis as if it was a spatial
coordinate. As shown, this is perfectly possible. The
standard macroscopic equations can give the clue tospatial heterogeneity obtained in the simulations.
Preliminary results obtained from simulations of
more complex reactive schemes, including several routes
of dissolution or considering up to six intermediate
species for the metal, have shown that the conclusions
obtained in the present work are independent of the
model used: states corresponding to reactants partici-
pating in slow reactive steps have longer state-lifetimes,leading to a segregation pattern of those species. This
has a feedback effect on the morphology: if the segre-
gated reactant takes part in the direct dissolution route
of the metal, i.e., it is a metal anion, the resulting mor-
phology has a lower degree of irregularity than the RD
model. If, on the contrary, this is not the case and the
segregated species belongs to an alternative route of
dissolution, it may lead to a high degree of roughness.The macroscopic model developed in Section 4, when it
is properly adapted for the specific reactive scheme used,
has also proved to be very useful in the prediction and
analysis of the segregation results. These results will be
covered in a forthcoming publication.
Acknowledgements
K. Bar-Eli has enjoyed a sabbatical year from The
Ministerio de Educaci�on y Ciencia, Spain. The authors
express their thanks to R.P. Nogueira for his fruitful
discussions.
References
[1] G.A. Somorjai, Introduction to Surface Chemistry and Catalysis,
Wiley, New York, 1994.
[2] C. Stampfl, M.V. Ganduglia-Pirovano, K. Reuter, M. Scheffler,
Surf. Sci. 500 (2002) 368.
[3] M. Polak, L. Rubinovich, Surf. Sci. Rep. 38 (2000) 127.
[4] L.N. Gumen, E.P. Feldman, V.M. Yurchenko, T.N. Mel’nik,
A.A. Krokhin, Surf. Sci. 445 (2000) 526.
[5] V.P. Zhdanov, Surf. Sci. 500 (2002) 966.
[6] E. Christoffersen, P. Stoltze, J.K. Nørskov, Surf. Sci. 505 (2002)
200.
[7] B. Koiller, R.B. Capaz, H. Chacham, Phys. Rev. B 60 (1999) 1787.

200 P. C�ordoba-Torres et al. / Journal of Electroanalytical Chemistry 571 (2004) 189–200
[8] P. C�ordoba-Torres, R.P. Nogueira, L. de Miranda, L. Brenig, J.
Wallenborn, V. Fair�en, Electrochim. Acta 46 (2001) 2975.
[9] P. C�ordoba-Torres, R.P. Nogueira, V. Fair�en, J. Electroanal.
Chem. 529 (2002) 109.
[10] A.L. Barab�asi, H.E. Stanley, Fractal Concepts in Surface Growth,
Cambridge University Press, Cambridge, 1995.
[11] B.B. Mandelbrot, The Fractal Geometry of Nature, Freeman, San
Francisco, CA, 1997.
[12] A.J. Arvia, R.C. Salvarezza, J.M. Vara, Electrochim. Acta 37
(1992) 2155.
[13] P. C�ordoba-Torres, R.P. Nogueira, V. Fair�en, J. Electroanal.
Chem. 560 (2003) 25.




















