
Nicotine Blocks Angiotensin II Inhibition of LTP in the Dentate Gyrus
7
Peptides,Vol. 17, No, 7, pp. 1127-1133, 1996 Copyright01996 ElsevierScienceInc. Printedin theUSA. All rightsreserved 0196-9781/96 $15.00 + ,00 PII S0196-9781(96)00179-9 Nicotine Blocks Angiotensin II Inhibition of LTP in the Dentate Gyrus MATTHEW J. WAYNER,’ DEBORAH L. ARMSTRONG AND CLYDE F. PHELIX Division of Lije Sciences, The University of Texas at San Antonio, San Antonio, TX 78249-0662, USA Received 3 May 1996 WAYNER, M. J., D. L. ARMSTRONG AND C. F. PHELIX. Nicotine blocks angiotensinII inhibition of LTP in the dentate gyrus. PEPTIDES 17(7) 1127–1 133, 1996.—Angiotensin (ANG)-containing axons, terminals, and receptors have been found in the hippocampus. When angiotensin II (ANG II) is administered to the dentate gyms, long-term potentiation (LTP) induction, in response to medial perforant path stimulation, is inhibited and it can be blocked by losartan, an ANG II AT1receptor antagonist. ANG II has been shown to mediate impairment of the retention of an inhibitory shock avoidance response and to be involved in ethanol and diazepam inhibition of dentate gyms LTP, all of which can be blocked by losartan. Nicotine acetylcholine receptors are found in the hippocampus and nicotine is involved in the enhancement of complex and important psychological functions that are mediated by the hippocampus; therefore, the possibility that nicotine prevents the ANG 11inhibition of dentate granule cell LTP was examined. Nicotine pretreatment reduced ANG II inhibition of LTP induction in a dose-dependent manner. Mecamyl- amine blocked the nicotine antagonism of ANG II-induced LTP inhibition and normal LTP occurred, whereas hexamethonium was ineffective in blocking these central effects of nicotine. Nicotine by itself did not affect normal LTP under these conditions. Nicotinic blocking of the ANG II inhibition of a frequency dependent type of synaptic plasticity provides a function for central nicotinic receptors and a possible mechanism of action a) to explain the enhancement of learning and memory by nicotine, b) an explanation for tobacco smoking while drinking alcohol, and c) a possible basis for the excessive use of tobacco in depression and schizophrenia that supports a possible therapeutic use of nicotine in some mental disorders. Copyright(!21996 Elsevier Science Inc. Angiotensin II Nicotine Long-term potentiation Hippocampus Memory Mecamylamine Hexamethonium Cytisine Aging Alzheimer’s disease Cognition BECAUSE of the potentially beneficial uses of nicotine (43,44) and related compounds, nicotine has been a topic of considerable research interest. Nicotine appears to be involved in many com- plex and important psychological functions: alertness, arousal, and attention ( 17,20,28,34), learning and memory (23,24), anx- iety ( 10,18,37), schizophrenia (2,3,21), and as an analgesic ( 12,33), all closely associated with the frontal cortex and the limbic system that are richly endowed with a relatively high den- sity of nicotinic receptors (4,30,32). Many reviews have focused on the molecular pharmacology and the diversity of the nicotinic binding protein subunits and tend to agree that there is an obvious lack of data in support of physiological mechanisms and a site of action at the cellular level (38). Recently, the review by McGehee and Role (26) substantiates these points clearly. Our discovery that ANG II inhibits LTP induction in dentate granule cell–medial perforant path synapses ( 14), that the inhibition is mediated by the ANG II AT, receptor because it can be blocked by the specific antagonist losartan (45), and our recent physio- logical data and behavioral results provide strong evidence for a meaningful physiological relationship between nicotine and ANG II in the hippocampus related to learning and the memory process. Ethanol- and diazepam-induced anterograde amnesia as well as the inhibition of LTP by both drugs appem to be mediated by ANG II because the inhibition of LTP can be prevented by pretreatment with losartan (46,47). Additional intoxicating ef- fects of ethanol, reduced activity in an open field (48), and im- pairment of the aerial righting reflex (42) cart be drastically re- duced by pretreatment with losartan. When ANG II is injected directly into the dentate gyms, 24-h retention of an inhibitory shock avoidance response is impaired in a dose-dependent man- ner and the impairment can be prevented by pretreatment with losartan (22). The time required for ANG II to inhibit LTP induction in vivo is long, approximately 1.5 h, and the inhibition is reversible (49). In cultured chromaffin cells internalization, degradation, and re- cycling of ANG II is also relatively long and significant amounts of ANG II are present intracellulm-ly in an active form for at least 40 min ( 13). With the discovery of an intracellular binding pro- tein (39) and widespread intracellular localization of ANG II, including the nucleus ( 15), it seems reasonable that ANG II is playing an important role in one or more intracellular signal transduction mechanisms. Nicotine and the use of tobacco is a powerful reinforcing agent in human behavior. Not only is nicotine involved in the 1Requests for reprints should be addressed to Matthew J. Wayner. Division of Life Sciences, The University of Texas at San Antonio, San Antonio, TX 78249-0662. 1127
Transcript of Nicotine Blocks Angiotensin II Inhibition of LTP in the Dentate Gyrus

Peptides,Vol. 17, No, 7, pp. 1127-1133, 1996Copyright01996 ElsevierScienceInc.Printedin theUSA. All rightsreserved
0196-9781/96 $15.00 + ,00
PII S0196-9781(96)00179-9
Nicotine Blocks Angiotensin II Inhibition of LTPin the Dentate Gyrus
MATTHEW J. WAYNER,’ DEBORAH L. ARMSTRONG AND CLYDE F. PHELIX
Division of Lije Sciences, The University of Texas at San Antonio, San Antonio, TX 78249-0662, USA
Received 3 May 1996
WAYNER, M. J., D. L. ARMSTRONG AND C. F. PHELIX. Nicotine blocks angiotensinII inhibitionof LTP in the dentategyrus. PEPTIDES 17(7) 1127–1 133, 1996.—Angiotensin (ANG)-containing axons, terminals, and receptors have been foundin the hippocampus. When angiotensin II (ANG II) is administered to the dentate gyms, long-term potentiation (LTP) induction,in response to medial perforant path stimulation, is inhibited and it can be blocked by losartan, an ANG II AT1 receptor antagonist.ANG II has been shown to mediate impairment of the retention of an inhibitory shock avoidance response and to be involved inethanol and diazepam inhibition of dentate gyms LTP, all of which can be blocked by losartan. Nicotine acetylcholine receptorsare found in the hippocampus and nicotine is involved in the enhancement of complex and important psychological functions thatare mediated by the hippocampus; therefore, the possibility that nicotine prevents the ANG 11inhibition of dentate granule cellLTP was examined. Nicotine pretreatment reduced ANG II inhibition of LTP induction in a dose-dependent manner. Mecamyl-amine blocked the nicotine antagonism of ANG II-induced LTP inhibition and normal LTP occurred, whereas hexamethoniumwas ineffective in blocking these central effects of nicotine. Nicotine by itself did not affect normal LTP under these conditions.Nicotinic blocking of the ANG II inhibition of a frequency dependent type of synaptic plasticity provides a function for centralnicotinic receptors and a possible mechanism of action a) to explain the enhancement of learning and memory by nicotine, b) anexplanation for tobacco smoking while drinking alcohol, and c) a possible basis for the excessive use of tobacco in depressionand schizophrenia that supports a possible therapeutic use of nicotine in some mental disorders. Copyright(!21996ElsevierScienceInc.
Angiotensin II Nicotine Long-term potentiation Hippocampus Memory MecamylamineHexamethonium Cytisine Aging Alzheimer’s disease Cognition
BECAUSEof the potentially beneficial uses of nicotine (43,44)and related compounds, nicotine has been a topic of considerableresearch interest. Nicotine appears to be involved in many com-plex and important psychological functions: alertness, arousal,and attention ( 17,20,28,34), learning and memory (23,24), anx-iety ( 10,18,37), schizophrenia (2,3,21), and as an analgesic( 12,33), all closely associated with the frontal cortex and thelimbic system that are richly endowed with a relatively high den-sity of nicotinic receptors (4,30,32). Many reviews have focusedon the molecular pharmacology and the diversity of the nicotinicbinding protein subunits and tend to agree that there is an obviouslack of data in support of physiological mechanisms and a siteof action at the cellular level (38). Recently, the review byMcGehee and Role (26) substantiates these points clearly. Ourdiscovery that ANG II inhibits LTP induction in dentate granulecell–medial perforant path synapses ( 14), that the inhibition ismediated by the ANG II AT, receptor because it can be blockedby the specific antagonist losartan (45), and our recent physio-logical data and behavioral results provide strong evidence for ameaningful physiological relationship between nicotine andANG II in the hippocampus related to learning and the memoryprocess. Ethanol- and diazepam-induced anterograde amnesia as
well as the inhibition of LTP by both drugs appem to be mediatedby ANG II because the inhibition of LTP can be prevented bypretreatment with losartan (46,47). Additional intoxicating ef-fects of ethanol, reduced activity in an open field (48), and im-pairment of the aerial righting reflex (42) cart be drastically re-duced by pretreatment with losartan. When ANG II is injecteddirectly into the dentate gyms, 24-h retention of an inhibitoryshock avoidance response is impaired in a dose-dependent man-ner and the impairment can be prevented by pretreatment withlosartan (22).
The time required for ANG II to inhibit LTP induction in vivois long, approximately 1.5 h, and the inhibition is reversible (49).In cultured chromaffin cells internalization, degradation, and re-cycling of ANG II is also relatively long and significant amountsof ANG II are present intracellulm-ly in an active form for at least40 min ( 13). With the discovery of an intracellular binding pro-tein (39) and widespread intracellular localization of ANG II,including the nucleus ( 15), it seems reasonable that ANG II isplaying an important role in one or more intracellular signaltransduction mechanisms.
Nicotine and the use of tobacco is a powerful reinforcingagent in human behavior. Not only is nicotine involved in the
1Requests for reprints should be addressed to Matthew J. Wayner. Division of Life Sciences, The University of Texas at San Antonio, San Antonio,TX 78249-0662.
1127

1128 WAYNER AND ARMSTRONG
many aforementioned complex psychological functions, it is wellknown that smoking and alcohol drinking usually occur together,and also smoking and depression (9) and stress are related factors(29). Now that ANG II has been identified as a neuromediatorspecifically involved with the AT I receptor in anterograde am-nesia, and possibly other effects produced by ethanol and diaz-epam, and because nicotine enhances some of the same physio-logical functions impaired by ANG II, does nicotine antagonizeANG II inhibition of LTP induction? The purpose of the presentseries of experiments was to examine the effects of various dosesof nicotine on dentate granule cell LTP in response to medialperforant path stimulation. Results demonstrate clearly that pre-treatment with nicotine: a) significantly reduces LTP inhibitionby ANG II; b) the antagonism by nicotine can be prevented bypretreatment with mecamylamine; c) both effects are dose re-lated; d) the nicotine effect is not blocked by hexamethonium;and e) nicotine in the same doses did not affect normal LTP underthese experimental conditions.
METHOD
Animals
Harlan male Sprague–Dawley rats weighing 275-used. Animal rooms were maintained at 21 312L:12D cycle, lights on at 0600 h. The rats weri
375 g were“C with ahoused in
plastic cages containing corn cob chips and covered with venti-lator tops. Commercial Tekland rat chow and water were avail-able ad lib. All rats were allowed a minimum of 2-weeks accli-mation before being used. The rats were food deprived 24 hbefore surgery.
Surgery
Following a brief period of mild metophane inhalation, ani-mals were anesthetized using a dose of 1.4 glkg 25Y0 urethane(Sigma), administered 1P. The rats were then placed in a Nari-shige stereotaxic instrument with the head fixed such that thePaxinos and Watson’s (31) coordinate system could be used. Atthe beginning of all surgeries xylocaine (USP, Schein Pharma-ceutical) was applied in small quantities to the incision area. Thesurface of the skull was exposed, two holes were made with adental drill, and the dura matter was incised. Surgical procedureswere completed within 1 h. To prevent drying 0.970 saline wasperiodically applied to the exposed skull and brain. Throughoutthe surgery and experiment, core body temperatures were mon-itored and maintained at 353 1°Cwith a feedback control systemand a heated pad. Body temperature was monitored visually atl-rein intervals.
Recordingand StimulatingElectrodes
Micropipette recording electrodes were prepared from single-barrel borosilicate glass tubing 1.2 mm o.d. X 0.6 mm id. pulledon a Narishige vertical puller and filled with 3 M NaC1. Resis-tance ranged from 1 to 3 Mfl. At the beginning of an experiment,the recording electrodes were positioned approximately 3.5 mmposterior to bregma and 2.0 mm lateral to the midline. The den-tate gyms was identified by single-unit activity characteristic ofgranule cells as well as by an electrode depth of 3.0–3.5 mmbelow the brain surface. The position of the recording electrodewas adjusted to produce an optimal population excitatory post-synaptic potential (pEPSP) in the granule cells.
Frederick Haer #17-90-l stainless steel concentric electrodeswere used for stimulation. The stereotaxic coordinates were ad-justed for variation in rat ages and to maximize the monosynapticresponses of the positive-going pEPSPs produced by granule
cells in response to stimulation of the dorsomedial perforant path.Average stimulating coordinates were 8.5 mm posterior tobregma and 4.2 mm lateral to the midline. At the end of theexperiment, placement in the dorsomedial perforant path wasverified with 1OO-HZtetanic stimulation (27).
Electrophysiological Recording
Stimulation consisted of 50-ps duration monophasic constantcurrent pulses delivered once per minute. Input–output curveswere obtained over the range of 25–460 PA before baseline andat the end of each experiment. Fourteen increasing stimuli wereused. In these experiments, actual baseline intensities selectedranged from 50 to 251 KA. These stimulus intensities producedpEPSP amplitudes of 5–7 mV that were at or slightly belowthreshold for evoking a negative-going waveform, populationspike (PS ), on the declining phase of the pEPSP. RepresentativepEPSPs before and after tetanization and input–output curve datahave been published previously (50). The slope of an individualpEPSP was measured over the first millisecond of the approxi-mately linear rising phase. The slope of each trace was measuredby adjusting the cursors in the on-line display and calculating thedifference in mV. These differences constitute the raw data andare referred to as the slopes. Once determined, stimulus currentremained constant throughout the experiment. After recording 30min of baseline responses, four sets of tetanic stimulation wereadministered to induce LTP, separated by intervals of 10 min at30, 40, 50, and 60 min as illustrated in Figs. 1–4. Each set con-tained five trains, 10 pulses per train at 400 Hz, delivered at arate of one train per second for 5 s. The pulse width in the trainswere 50, 100, 150, and 200 KS,respectively. The posttetanic po-tentiation due to the individual tentanii appear as prominent in-creasing vertical displacements from baseline in all the figures.The pEPSPs were recorded every minute for 130 min. PercentpEPSP was calculated as follows: slope at each minute minus themean baseline slope and then divided by the mean baseline slopeand then X 100. Baseline was the mean slope of the first 30 min.
The granule cell pEPSPs were amplified by a conventionalamplifier at a frequency band of 1.6 Hz to 2 kHz. All potentialswere monitored on an oscilloscope, digitized at a sampling in-terval of 20 Ks, for on-line computer display (RC Electronics).The first millisecond of the pEPSP slopes was used in the anal-ysis. All data were stored on floppy disks.
DrugAdministration
Nicotine. ( – )-Nicotine (Sigma #N3876) was dissolved indouble distilled water and doses were calculated as the base. Nic-otine was administered SC 120 min before the tetanii were ap-plied during the electrophysiological recording period. When theeffects of nicotine alone were determined, 30 min later artificialcerebral spinal fluid (ACSF) was injected above the hippocam-pus in place of the ANG II. The following doses were used: 0.05,0.1,0.2,0.3, and 0.4 mg/kg.
Angioterzsin 11.One microliter of 4.78 PM ANG II, 4.78 pmolpeptide (Sigma #A9525) in ACSF was injected stereotaxicallyabove the hippocampus on the same side of the brain from whichthe recordings were obtained. The stereotaxic coordinates were3.5 mm posterior to bregma, 2.0 mm lateral to the midline, anda depth of 1.8 mm below the brain surface. The l.O-pl volumewas injected in equal amounts of 200 nlhnin. ANG II was ad-ministered 90 min before the first tetanus was applied.
Mecamylamine.Mecamylamine, a neuronal nicotinic cholin-ergic channel blocker, crosses the blood–brain barrier and bindsto brain nicotinic receptors ( 12). Three doses of mecamylamine

ANGIOTENSIN AND NICOTINE INTERACTIONS
in double distilled water, 1, 5, and 10 mg/kg, were injected 1P30 min before the nicotine was administered.
Hexamethonium. Hexamethonium is as effective as meca-mylamine in vitro in competing for nicotinic binding sites bothperipherally and centrally in rat but does not readily cross theblood–brain barrier. In rat brain membrane preparations bothcompounds are equally effective in binding to nicotinic receptors( 1). A single dose of 20 mg/kg in double distilled water wasinjected 1P 30 min before the nicotine was administered.
Cytisine. Cytisine is one of the most effective nicotinic ago-nists ( 19) and more potent than nicotine itself. A single dose of0.4 mg/kg in double distilled water was injected SC 30 min be-fore the 4.78 pkf ANG II was administered.
Experimental Design and Statistical Analysis
Data were expressed as percent change in slope of the pEPSPfrom baseline and are illustrated as means 3 SEM as a functionof time at minute intervals during the experimental protocol. Aone-way ANOVA for independent samples and Student–New-man–Keuls multiple-range post hoc determinations were utilizedto compare the means of the last 10 min of the various groupssubjected to the different experimental treatments. There were 23different groups. There were five animals in each of the groups,with one exception: the 0.1 mg/kg nicotine + 4.78 pkf ANG IIgroup (n = 10). Data were analyzed utilizing SigmaStat and thesignificance level for the post hoc comparisons was set at p <0.05. Because these experiments were conducted over a relativelylong period of time and different shipments of animals vary, evenfrom the same supplier, three different control groups were uti-lized and correspond to three time periods over which these ex-periments were conducted. The same control group was used inthe statistical analysis of the data in Figs. 1 and 2 and two dif-ferent control groups were used in Figs. 3 and 4. Even thoughan ANOVA of the differences between the means of the threegroups was not significant, F’(2, 12) = 1.68, p <0.227, individ-ual treatment comparisons were made with the appropriate con-trol group data for that time period.
RESULTS
Nicotine
Mean i SEM percent potentiation measured in terms of therelative change in pEPSP slope compared to baseline is presentedin Fig. 1 as a function of time in minutes for each of six groups:normal control group (which received distilled water SC) andfive groups corresponding to five doses of nicotine SC: 0.05,0.1,0.2, 0.3, and 0.4 mg/kg. There were five animals in each group.A one-way ANOVA of the means for the final 10 min for all sixgroups was not significant, F(5, 24) = 2.41, p <0.067. There-fore, the nicotine doses used in this study did not have an effecton normal LTP under these experimental conditions.
Nicotine andAngiotensinII
Mean ? SEM percent change in potentiation as a function oftime in minutes is presented in Fig. 2 for each of six groupscorresponding to the same six doses of nicotine SC, as in thepreceding experiment, except that the nicotine was injected 30min prior to the administration of the ANG II. The normal con-trols in Fig. 2(A) are the same control animals as in Fig. 1(A).Also, there are 10 animals in the 0.1 mg/kg nicotine and 4.78pkf ANG II group [Fig. 2(C)]. There are 10 animals in thisgroup because four of the animals showed ANG II inhibition andthe other six were similar to the LTP observed in the 0.2 mg/kgnicotine + 4.78 pkf ANG II group [Fig. 2(D)]. This is what one
+ I50
25
0~P
0 20 40 60 60100120140
‘:=50
L
ii
25 s qklmgk
o
I I I I I I0 20 40 60 60100120140
100E. 0.3wI!wNICOTINE
“.5
I50
25
0w
11,1 ’1’l’l’l’i0 20 40 60 60100120140
o-t- 1
I I I I I 10 20 40 60 80100120140
100D. 0,2m9fkgNICOTINE
“.5
75
50
25
0LJ$i0 20 40 60 60100120140
‘:-
50
25
u
e
o
0 20 40 60 60100120140
TIME (MIN)
1. Mean f SEM percent potentiation, measured in terms of a rel-ative change in pEPSP slopes compared to baseline, presented as a func-tion of time in minutes. The occurrence of the tetanii and resultant post-tetanic potentiation can be seen as prominent vertical displacements at10-rnin intervals following baseline. Normal LTP and pretreatment withfive different doses of nicotine, SC, illustrate that nicotine has no effecton LTP induction.
might expect for a threshold dose; therefore, the additional ani-mals were added to this group to establish that about 50% of theanimals would display inhibition and 509Z0would not. There werefive animals in each of the other five groups. A one-way ANOVAof the means for the final 10 min for all six groups was signifi-cant, F(5, 29) = 6.26, p < 0.0005. Post hoc Newman-Keulscomparisons between the means revealed that only the 0.05 mg/kg dose of nicotine did not block the inhibitory effect of 4.78plf ANG II. This inhibitory effect was normal and did not differsignificantly from the inhibitory effect of ANG II alone [Fig.4(B)]. Therefore, the other four doses significantly blocked theinhibito~ effect of ANG II on LTP induction. The only othersignificant difference occurred between the 0.1 and 0.4 mg/kggroups. The threshold dose was 0.1 mg/kg for blocking the ANGII inhibition of LTP under these conditions. The 0.2 mg/kg wasselected for the other experiments in this study because it wasthe lowest dose that clearly blocked the ANG II inhibition ofLTP induction.
A4ecamylamine, Nicotine, and Angiotensin II
Mecamylamine is a well known ganglionic blocking agentthat readily passes the blood–brain barrier and is also an effectivenoncompetitive nicotinic antagonist in the hippocampus (4). The
1130 WAYNER AND ARMSTRONG
50
25
0~P
“krm—m—d0 20 40 60 80100120140
‘:r25
0
E0 20 40 60 80100120140
100E. 03nwltg NlC0mNE&
4.7sIIMAII
‘:m1111111,10 20 40 60 80100120140‘:rt, I , I ‘ I ‘ I ‘ I ‘ I ‘
0 20 40 80 80100120140
I I I I I I I
0 20 40 60 80100120140
I I I I I I I
0 20 40 60 60100120140
TIME (MIN)
FIG. 2. Same as Fig. 1 exceut that 4.78 uA4ANG II was administeredabove the dorsal hippocamp~s 30 min following administration of thesame doses of nicotine as in Fig. 1. Normaf control group are the sameanimals as in the control group in Fig. 1. The other five groups demon-strate a dose-related decrease in ANG II inhibition of LTP. In (B), abelow threshold dose of nicotine has no effect and normal inhibition ofLTP by ANG II occurs.
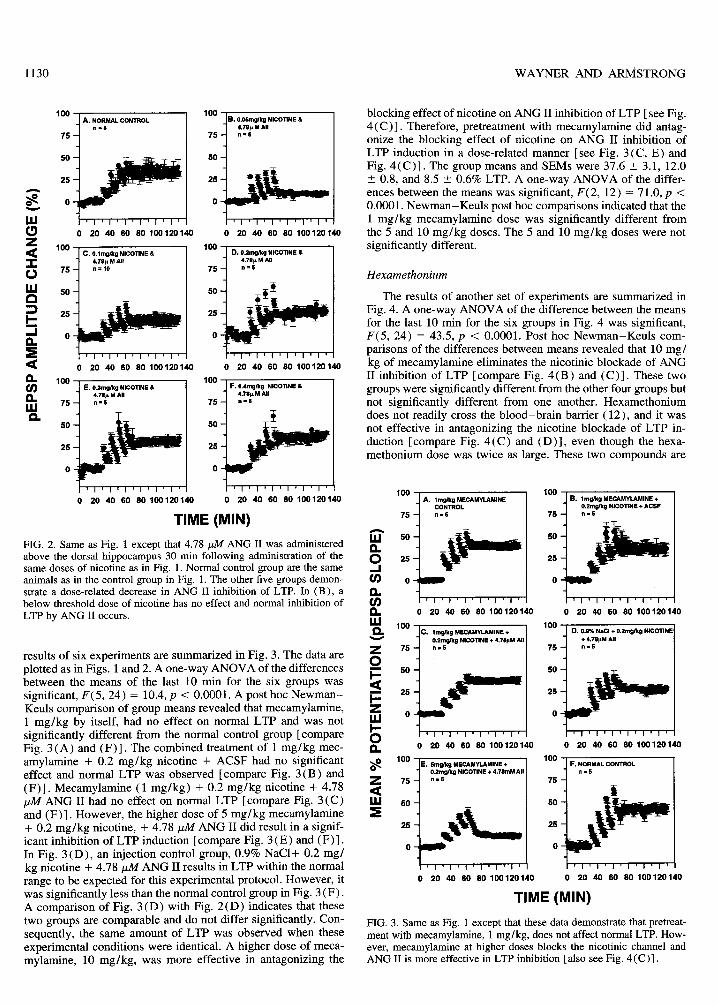
results of six experiments are summarized in Fig. 3. The data areplotted as in Figs. 1 and 2. A one-way ANOVA of the differencesbetween the means of the last 10 tnin for the six groups wassignificant, F(5, 24) = 10.4, p <0.0001. A post hoc Newman–Keuls comparison of group means reverded that mecarnylamine,1 mg/kg by itself, had no effect on normal LTP and was notsignificantly different from the normal control group [compareFig. 3(A) and (F)]. The combined treatment of 1 mg/kg mec-arnylaruine + 0.2 mg/kg nicotine + ACSF had no significanteffect and normal LTP was observed [compare Fig. 3(B) and(F)]. Mecamylamine (1 mg/kg) + 0.2 mg/kg nicotine + 4.78jull ANG II had no effect on normal LTP [compare Fig. 3(C)and (F)]. However, the higher dose of 5 mg/kg mecamylamine+ 0.2 mg/kg nicotine, + 4.78 pll ANG II did result in a signif-icant inhibition of LTP induction [compare Fig. 3(E) and (F)].In Fig. 3(D), an injection control group, 0.9% NaCl+ 0.2 mg/kg nicotine + 4.78 @4 ANG II results in LTP within the normalrange to be expected for this experimental protocol. However, itwas significantly less than the normal control group in Fig. 3(F).A comparison of Fig. 3(D) with Fig. 2(D) indicates that thesetwo groups are comparable and do not differ significantly. Con-sequently, the same amount of LTP was observed when theseexperimental conditions were identical. A higher dose of meca-mylamine, 10 mg/kg, was more effective in antagonizing the
blocking effect of nicotine on ANG II inhibition of LTP [see Fig.4(C)]. Therefore, pretreatment with mecamylamine did antag-onize the blocking effect of nicotine on ANG II inhibition ofLTP induction in a dose-related manner [see Fig. 3(C, E) andFig. 4(C)]. The group means and SEMS were 37.6 t 3.1, 12.0t 0.8, and 8.5 3 0.6Y0LTP. A one-way ANOVA of the differ-ences between the means was significant, F(2, 12) = 71.0, p <0.0001. Newman–Keuls post hoc comparisons indicated that the1 mg/kg mecamylamine dose was significantly different fromthe 5 and 10 mg/kg doses. The 5 and 10 mg/kg doses were notsignificantly different.
Hexamethonium
The results of another set of experiments are summarized inFig. 4. A one-way ANOVA of the difference between the meansfor the last 10 min for the six groups in Fig. 4 was significant,F(5, 24) = 43.5, p <0.0001. Post hoc Newman–Keuls com-parisons of the differences between means revealed that 10 mg/kg of mecarnylamine eliminates the nicotinic blockade of ANGII inhibition of LTP [compare Fig. 4(B) and (C)]. These twogroups were significantly different from the other four groups butnot significantly different from one another. Hexamethoniumdoes not readily cross the blood–brain barrier ( 12), and it wasnot effective in antagonizing the nicotine blockade of LTP in-duction [compare Fig. 4(C) and (D)], even though the hexa-methonium dose was twice as large. These two compounds are
100A. 1m@ig MECAMVIAMIME
CONTROL
b+-75 n=5
50
25
0t- 1Ir,,trl’i’li,l0 20 40 60 60100120140
+ I
I I I I I I I
0 20 40 60 60100120140
100
r
B. 1m@kgMECAMYLAMINE+0,2mgJkgNlC0nNE+ACSF
75 . -s
50
25 *Ill
o
1’l’l’l’l’l ’1’0 20 40 60 80100120140
100
n
D. 0.W4N.CI +o,2mglkg Nlc0nNE+4781LMA11
75 n-s
: k
o
1,1,1’1’lrl’110 20 40 60 80100120140
100F. ::~WL.CONIROL
75
I,,,,i’i’ilrl0 20 40 60 80100120140
t,, , ,,1’1 ‘1’1 ,10 20 40 60 60100120140
TIME (MIN)
FIG. 3. Same as Fig. 1 except that these data demonstrate that pretreat-ment with mecarnylamine, 1 mg/kg, does not affect normal LTP. How-ever, mecamylamine at higher doses blocks the nicotinic channel andANG II is more effective in LTP inhibition [also see Fig. 4(C)].

ANGIOTENSIN AND NICOTINE INTERACTIONS 1131
100 A. N0RL2ALC0NlR0Ln.5
754 I50
25
0
/!JP
I I I I I I I
0 20 40 60 60100120140
100
0.2mIylIgNICOTINE+75 4.78pMAll
n-s
50
25
0
0 20 40 60 80100120140
100
w
E. 0.4mWC7mSlNE+4.78@J 1
76 ..5
50
25
0
1,1’ 1,1, 1’1’1’10 20 40 60 80100120140
100B. 4,78WAIICONTROL
“.575
50
25
0b
I i I I I 1
0 20 40 60 80100120140
100
r
D. 20m@g HEX4MElH0NlUM+02m@QNlC011NE+
75 4,78@2All
iifi@Q-
..5
50
25
0
1,1, ’1,1 ’r ’1”0 20 40 60 80100120140
100F. 0.4m@k0NICOTINE+
4.78*MAU75 n-5
1,, ,,, ,,, ,,,1’1
0 20 40 60 80100120140
TIME (rein)FIG. 4. Same as Fig. 1 except: (B) illustrates the inhibition of LTPinduction by ANG II injected above the dorsal hippocampus, (D) showsthat hexamethonium, which does not cross the blood–brain barrier, doesnot block the effect of nicotine and LTP occurs, and (E) shows thatcytisine, a nicotine agonist, is more effective than nicotine [compare (E)and (F)]
equally potent in terms of binding characteristics in brain neu-ronal membrane preparations ( 1). Although pretreatment withhexamethonium did not result in normal LTP as illustrated bythe normal control LTP in Fig. 4(A), it was almost the same asobtained with 0.4 mg/kg nicotine + 4.78 pif ANG II [see Fig.4(F)]. It is possible that at this high dose some hexamethoniumpasses the blood–brain barrier ( 12). Failure of the hexarrtethon-ium to significantly attenuate the inhibition of LTP induced by0.2 mg/kg nicotine + 4.78 @l ANG II substantiates the centralmediation of the effects reported in this study.
Cytisine
Cytisine is a more potent agonist than nicotine ( 19). A com-parison of the relative effectiveness of 0.4 mg/kg cytisine to 0.4mg/kg of nicotine in antagonizing the ANG II inhibition of LTPcan be made by examining Fig. 4(E) and (F). The differencebetween the two means is significant. However, the differencebetween the 0.4 mg/kg cytisine + 4.78 pkf ANG II and thenormal control data was not significant [compare Fig. 4(A) and(E)], whereas the difference between the 0.4 mg/kg nicotine +4.78 pikf ANG II and the normal control group was significant.Therefore, cytisine was more potent than nicotine under theseexperimental conditions in blocking ANG II inhibition of LTP.
Posttetanic Potentiation
The effects of posttetanic stimulation are very prominent forthe low doses of nicotine in Fig. 1(B, C, and D), more so thanin any of the other experimental groups in the other figures. Nic-otine appears to enhance posttetanic potentiation and it is reducedby mecamylamine at the two lowest doses [see Fig. 3(C, E)].
DISCUSSION
These results demonstrate a clear finding that nicotine canprevent a peptide, ANG II, from inhibiting LTP induction in me-dial perforant path–dentate granule cell synapses and provide aplausible explanation for some of the cognitive and other behav-ior effects of nicotine. Nicotine alone had no effect on LTP; how-ever, this particular experimental protocol produces a maximumlevel of LTP and, at levels closer to threshold, enhancementmight have occurred. The effect of nicotine was blocked by mec-amylamine but not by hexatnethonium. Cytisine, a potent ago-nist, was more effective than nicotine. These data confirm thatblocking the ANG II inhibition of LTP is mediated by the nic-otinic acetylcholine receptor and that the effects are mediatedcentrally.
A mechanism by which ANG II might block LTP is unknown;however, it does involve the AT, receptor because the inhibitionis blocked by losartan. The signal transduction pathway for theATI G-protein linked receptor involves increased phosphoinos-itol hydrolysis, which results in intracellular Ca2+ mobilizationand protein kinase C activation (8,40). Results of patch-clampexperiments that examined ANG II effects on ionic currents haveshown that ANG II inhibits voltage-gated calcium currents inacutely dissociated rat sympathetic neurons (36) and culturednodose neurons from neonatal rats (5) via AT, receptor activa-tion. A possible mechanism by which ANG II inhibits LTP wouldbe the reduction of NMDA glutamate receptor-mediated calciuminflux. Activation of NMDA receptors during tetanic stimulationis necessary for LTP induction in medial perforant path–granulecell synapses ( 11,16). However, the effect of ANG II on NMDAreceptor-mediated responses has yet to be determined.
Under these experimental conditions nicotine alone had noeffect on LTP determined in terms of pEPSP slopes. These resultsare supported by the findings in guinea pig hippocampal sliceswhere, during paired pulse stimulation of the dentate molecularlayer, nicotine did not change the pEPSP slopes; however, theamplitude of the second population spike was increased, and thiseffect was blocked in the presence of bicuculline, indicating thatthe enhancement of somatic excitability was mediated by a re-duction in GABA intemeuron activity (35).
The data on nicotine effects on GABA release are somewhatcomplicated. Micromolar concentrations stimulate the release ofGABA from rat hippocarnpal synaptosomes (51 ) and guinea pigcortical slices (6); however, in vivo studies in freely movinganimals showed a decrease in corticaf GABA outflow (6). There-fore, in the intact animal systemic application of nicotine mightactivate intact neural circuits not functional in the slice andthereby other neurotransrnitter systems that inhibit GABA re-lease.
The effects of nicotine on glutamate release at the medialhabenuku excitatory synapse in the interpeduncular nucleus, animportant limbic system relay, has been described recently (25).Nicotine increased the frequency of spontaneous EPSCS medi-ated by AMPA receptors with no detectable change in postsyn-aptic membrane excitability. The increase in transmitter releasewas linked to rises in presynaptic calcium, presumably mediatedby presynaptic nicotine receptors. These effects were eliminatedby CNQX, an AMPA receptor antagonist. It is possible that the

1132
ability of nicotine to prevent ANG II inhibition of LTP in thedentate is also due to increased glutamate release from medialperforant path axon terminals. If the increased levels of glutamateactivate postsynaptic glutamate metabotropic receptors, a seriesof intracellular events could be set into action that would blocksubsequent ANG II mechanisms. In all of our experiments nic-otine is present for 30 min prior to ANG II application and 90min before the tetanii were applied, providing sufficient time fora relatively small amount of nicotine-induced glutamate releaseto activate a metabotropic glutamate receptor (mGluR) and sig-nal transduction mechanism. Recently, it was reported that me-tabotropic glutamate receptors “. may have a modulatory ef-fect on LTP induction that is most evident for near thresholdpatterns of synaptic stimulation” (41). The basis for this hy-pothesis is a mGluR activated “molecular switch” (7). There-fore, a threshold amount of glutamate is required to “set theswitch” and activate a biochemical process that would proceedwithout additional glutamate stimulation. If we assume that our
WAYNER AND ARMSTRONG
results can be explained in terms of nicotine activating presnyap-tic nicotinic receptors that release glutamate, then it is possiblethat the glutamate activates the molecuku switch and sets in ac-tion a biochemical process that will induce or enhance LTP. Con-sequently, under our experimental conditions when ANG II isinjected into the dentate gyms 30 min later, the molecular switchhas been activated and the ANG II will not be effective. Theseexperiments need to be repeated in presence of mgluR antago-nists such as MCPG to determine if the results presented here aredependent upon metabotropic glutamate receptors.
ACKNOWLEDGEMENTS
This research was supported by a grant from The Council for TobaccoResearch-U. S.A., Inc., #4038. We appreciate the help of Marianne VanWagner in typing the manuscript and the technical assistance of BarbaraSmith, Sibylle Lang, and John Chiu in data collection and statisticalanalysis.
REFERENCES
1. Abood, L. G.; Reynolds, D. T.; Booth, H.; Bidlack, J. M. Sites andmechanisms for nicotine’s action in the brain. Neurosci. Biobehav.Rev. 5:479–486; 1981.
2. Alder, L. E.; Hoffer, L. D.; Griffith, J.; Waldo, M. C.; Freedman, R.Normalization by nicotine of deficient auditory sensory gating in therelatives of schizophrenics. Biol. Psychiatry 32:607–616; 1992.
3. Adler, L. E.; Hoffer, L. D.; Wiser, A.; Freedman, R. Norrnafizationof auditory physiology by cigarette smoking in schizophrenic pa-tients. Am. J. Psychiatry 150( 12):1856–1861; 1993.
4. Alkondon, M.; Albuquerque, E. Diversity of nicotinic acetylcholinereceptors in rat hippocampal neurons. I. Pharrnacologicat and func-tional evidence for distinct structural subtypes. J. Pharrnacol. Exp.Ther. 265:1455-1473; 1993.
5. Bacal, K.; Kunze, D. L. Dual effects of angiotensin II on calciumcurrents in neonatal rat nodose neurons. J. Neurosci. 14:7159–7167;1994.
6. Beani, L.; Bianchi, C.; Ferraro, L.; Nilsson, L.; Nordberg, A.; Ro-manelli, L.; Spalluto, P.; Sundwall, A.; Tanganelli, S. Effect of nic-otine on the release of acetylcholine and amino acids in the brain.Prog. Brain Res. 79:149–155; 1989.
7. Bortolotto, Z. A.; Bashir, Z. I.; Davies, C. H. A molecular switchactivated by metabotropic glutamate receptors regulates induction oflong-term potentiation. Nature 368(6473):740–743; 1994.
8. Bottari, S. P.; de Gasparo, M.; Muscha Steckelings, U.; Levens, N.R. Angiotensin II receptor subtypes: Characterization, signalingmechanisms, and possible physiological implications. Front. Neu-roendocrinol. 14:123– 171; 1993.
9. Breslau, N.; Kilbey, M. M.; Andreski, P. DSM-11 1-R nicotine de-pendence in young adults: Prevalence, correlates and associated psy-chiatric disorders. Addiction 89(6) :743–754; 1994.
10. Burkholder, T.; Wilkins, L.; Collins, A. C. A genetic comparison ofbehavioral actions of ethanol and nicotine in the mirrored chamber.Pharmacol. Biochem. Behav. 45(4):803–809; 1993.
11. Collingridge, G. L. The mechanism of induction of NMDA receptor-dependent long-term potentiation in the hippocampus. Exp. Physiol.77:771–797; 1993.
12. Damaj, M. I.; Creasy, K. R.; Grove, A. D.; Rosecrans, J. A.; Martin,B. R. Pharmacological effects of epibatidine optical enatiomers.Brain Res. 664:34–40; 1994.
13. De Potter, W. P.; Wang, J. M. Binding, internalization, degradationand recycling of angiotensin II in primary cultured bovine chromti-fin cells. Soc. Neurosci. Abstr. 19:726; 1993.
14. Denny, J. B.; Polan–Curtain, J.; Wayner, M. J.; Armstrong, D. L.Angiotensin II blocks hippocampal long-term potentiation. BrainRes. 567:321–324; 1991.
15. Erdmann, B.; Fuxe, K.; Ganten, D. Subcellular localization of an-giotensin II imnmnoreactivity in the rat cerebella cortex. Hyperten-sion (in press).
16. Errington, M. L.; Lynch, M. A.; Bliss, T. V. P. Long-term potentia-tion in the dentate gyros: Induction and increased release are blockedby D( – )arninophosphonovalerate. Neuroscience 20:279–284;1987.
17. Foulds, J.; Ghodse, A. H. The role of nicotine in tobacco smoking:Implications for tobacco control policy. J. R. Soc. Health 115(4):225–30; 1995.
18. Gilbert, D. G.; Gilbefi, B. O. Personality, psychopathology, and nic-otine response as mediators of the genetics of smoking. Behav. Ge-net. 251 (2): 133– 147; 1995.
19. Houghtling, R. A.; Davila–Garcia, M. I.; Kellar, K. J. Characteri-zation of ( t)-[3H]epibatidine binding to nicotinic cholinergic re-ceptors in rat and human brain. Mol. Pharrnacol. 48:280–287; 1995.
20. Jones, G. M. M.; Sohakiou, B. J.; Levy, R.; Warburton, D. M.; Gray,J. A. Effects of acute subcutaneous nicotine on attention, informationprc)cessing and short term memory in Alzheimer’s disease. Psycho-pharmacology (Berlin) 108:485–494; 1992.
21. Kirch, D. G.; Wyatt, R. J. Nicotine: Associations with schizophreniaand tardive dyskinesia. ACNP Abstr. 28th Annual Meeting 28:39;1989.
22. Lee, E. H. Y.; Ma, Y. L.; Wayner, M. J.; Armstrong, D. L. Impairedretention by angiotensin II mediated by the AT, receptor. Peptides16:1069–1071; 1995.
23. Levin, E. D. Psychopharmacological effects in the radial-arm maze.Neurosci. Biobehav. Rev. 12:169–175; 1988.
24. Levin, E. D. Nicotinic systems and cognitive function. Psychophw-macology (Berlin) 108:417–431; 1992.
25. McGehee, D. S.; Heath, M. J. S.; Gelber, S.; Devay, P.; Role, L. W.Nicotine enhancement of fast excitatory synaptic transmission inCNS by presynaptic receptors. Science 269:1692-1696; 1995.
26. McGehee, D. S.; Role, L. W. Physiological diversity of nicotinicacetylcholine receptors expressed by vertebrate neurons. Annu. Rev.Physiol. 57:521-546; 1995.
27. McNaughton, B. L.; Barnes, C. A. Physiological identification andanalysis of dentate granule cell responses to stimulation of the me-dial and lateral perforant pathways in the rat. J. Comp. Neurol.175:439–454; 1977.
28. Parrott, A. C. Acute pharrnacodynarnic tolerance to subjective ef-fects of cigarette smoking. Psychopharmacology (Berlin) 116:93-97; 1994.
29. Parrott, A. C. Stress modulation over the day in cigarette smokers.Addiction 90(2):233–234; 1995.
30. Paul, B. S.; Schwartz, R. D.; Paul, S. M.; Pert, C. B.; Pert, A. Nic-otinic binding in rat brain: Autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [’251]-a-bungarotoxin. J. Neurosci.5(5):1307-1315; 1985.
31. Paxinos, G.; Watson, C. The rat brain in stereotaxic coordinates.New York: Academic Press; 1982.

ANGIOTENSIN AND NICOTINE INTERACTIONS 1133
32. Perry, E. K.; Court, J. A.; Johnson, M.; Piggott, M. A.; Perry, R. H.Autoradiographic distribution of [3H]nicotine binding in human cor-tex: Relative abundance in subicular complex. J. Chem. Neuroanat.5(5):399-405; 1992.
33. Qian, C.; Li, T.; Shen, T. Y.; Libertine-Garahan, L.; Eckman, J.;Biftu, T.; Ip, S. Epibatidine is a nicotinic analgesic. Eur. J. Phar-macol. 250:R13–R14; 1993.
34. Rusted, J. M.; Warburton, D. M. Facilitation of memory by trialadministration of nicotine: Evidence for an attentional explanation.Psychopharmacology (Berlin) 108:452–455; 1992.
35. Sawada, S.; Ohno–Shosaku, T.; Yamamoto, C. Augmenting actionof nicotine on population spikes in the dentate gyms of the guineapig. Neurosci. Res. 20(4):317-322; 1994.
36. Shapiro, M. S.; Wollmuth, L. P.; Hille, B. Angiotensin II inhibitscalcium and M current channels in rat sympathetic neurons via Gproteins. Neuron 12:1319-1329; 1994.
37. Shervington, D. O. Attitudes and practices of Afkican-Americanwomen regarding cigarette smoking: Implications for interventions.J. Natl. Med. Assoc. 86(5):337–343; 1994.
38. Sivilotti, L.; Colquhoun, D. Acetylcholine receptors: Too manychannels, too few functions. Science 269: 1681– 1682; 1995.
39. Sugiura, N.; Hagiwara, H.; Hirose, S. Molecular cloning of porcinesoluble angiotensin-binding protein. J. Biol. Chem. 267:18067–18072; 1992.
40. Sumners, C.; Raizada, M. K.; Kang, J.; Li, D.; Posner, P. Receptor-mediated effects of angiotensin II on neurons. In: Martini, L.; Gan-ong, W. F., eds. Frontiers in neuroendocrinology, vol. 15. New York:Raven; 1994:1–28.
41. Thomas, M. J.; O’Dell, T. J. The molecular switch hypothesis failsto explain the inconsistent effects of the metabotropic glutamate re-
ceptor antagonist MCPG on long-term potentiation. Brain Res.695( 1):45-52; 1995.
42. Tracy, H, A., Jr.; Wayner, M. J.; Armstrong, D. L. Angiotensin IIantagonists block ethanol effects on the aerial righting reflex. Al-cohol 13:287–289; 1996.
43. Warburton, D. Nicotine: An addictive substance or a therapeuticagent? Prog. Drug Res. 133:9–41; 1989.
44. Warburton, D. Nicotine as a cognitive enhancer. Prog. Neuropsy -chopharmacol. Biol. Psychiatry 16:181–191; 1992.
45. Wayner, M. J.; Armstrong, D. L.; Polan–Curtain, J. L.; Denny, J. B.Role of angiotensin H and AT, receptors in hippocampal LTP. Phar-macol. Biochem. Behav. 45:455–464; 1993.
46. Wayner, M. J.; Armstrong, D. L.; Polan–Curtain, J. L.; Denny,J. B. Ethanol and diazepam inhibition of hippocampal LTP ismediated by angiotensin II and AT1 receptors. Peptides 14:441–444; 1993.
47. Wayner, M. J.; Polan-Curtain, J. L.; Armstrong, D. L. AngiotensinAII AT, receptor mediates ethanol-diazepam inhibition of hippo-campal LTP. Chin. J. Physiol. 37(2) :55–61; 1994.
48. Wayner, M. J.; PolamCurtain, J. L.; Chiu, S.-C.; Armstmng, D. L. b-sartanreduces ethanol intoxicationin the rat. Afcohol 11:343–346;1994.
49. Wayner, M. J.; Polan–Cnrtain, J.; Armstrong, D. L. Dose and timedependency of angiotensin II inhibition of hippocampal long-termpotentiation. Peptides 16:1079–1082; 1995.
50. Wayner, M. J.; Chitwood, R.; Armstrong, D. L.; Phelix, C. Ethanolaffects hypothalamic neurons projecting to the hippocampus and in-hibits dentate granule cell LTP. Alcohol (in press).
51. Wonnacott, S.; Irons, J.; Rapier, C.; Theme, B.; Lunt, G. G. Presy-naptic modulation of transmitter release by nicotinic receptors. Prog.Brain Res. 79:157–163; 1989.




















