
nghiên cứu dược & thông tin thuốc 2020, tập 11, số 6
49
NGHIÊN CỨU DƯỢC & THÔNG TIN THUỐC 2020, TẬP 11, SỐ 6 Journal of Pharmaceutical Research and Drug information 2020, Vol 11. N o 6 MỤC LỤC Số 6, 2020 CONTENTS N o 6, 2020 BÀI NGHIÊN CỨU RESEARCH 2 Nghiên cu chế tạo hệ phân tán rắn telmisartan, ng dụng vào bào chế viên nén Bùi Thị Lan Phương, Lê Thiện Giáp, Phạm Thị Thu Hương, Nguyễn Ngọc Chiến Formulation of solid dispersion of telmisartan, application to tablet preparation Bui Thi Lan Phuong, Le Thien Giap, Pham Thi Thu Huong, Nguyen Ngoc Chien 9 Phân tích tính phù hợp của việc sử dụng kháng sinh carbapenem trong điều trị viêm phổi bệnh viện tại Bệnh viện Đa khoa Hà Đông Nguyễn Thị Thu Thủy, Quách Thị Thu Hà, Lê Vân Anh, Phạm Thị Thúy Vân Appropriateness of carbapenems use in the treatment of hospital acquired pneumonia in Ha Dong General Hospital Nguyen Thi Thu Thuy, Quach Thi Thu Ha, Le Van Anh, Pham Thi Thuy Van 16 Tổng hợp acid (E)-4-dimethylaminobut-2- enoic hydroclorid làm chất trung gian quan trọng cho điều chế afatinib dimaleat Nguyễn Thị Ngọc, Nguyễn Hòa Bình, Bùi Thị Thanh Châm, Trần Hoàng Vũ, Trần Anh Phương, Đào Nguyệt Sương Huyền, Nguyễn Văn Giang, Nguyễn Đình Luyện, Nguyễn Văn Hải Synthesis of (E)-4-(dimethylamino)but-2- enoic acid hydrochloride as an important intermediate for the preparation of afatinib dimaleate Nguyen Thi Ngoc, Nguyen Hoa Binh, Bui Thi Thanh Cham, Tran Hoang Vu, Tran Anh Phuong, Dao Nguyet Suong Huyen, Nguyen Van Giang, Nguyen Dinh Luyen, Nguyen Van Hai 23 Nghiên cu quy trình bào chế viên nang mềm cha hệ nano tự nhũ hóa rosuvastatin Vũ Thị Thu Giang, Phan Thị Nghĩa, Nguyễn Thị Thúy Nga, Nguyễn Thị Linh, Trần Thị Hải Yến, Phạm Bảo Tùng, Nguyễn Đăng Hòa Development of preparing procedure of soft gelatin capsule containing rosuvastatin self- nano emulsifying drug delivery system Vu Thi Thu Giang, Phan Thi Nghia, Nguyen Thi Thuy Nga, Nguyen Thi Linh, Tran Thi Hai Yen, Pham Bao Tung, Nguyen Dang Hoa 31 Tổng hợp và đánh giá tác dụng c chế tế bào ung thư của một số acid hydroxamic mang dị vòng 1,3-thiazol Dương Tiến Anh, Đào Thị Kim Oanh, Nguyễn Hải Nam Synthesis and Evaluation of Novel 1,3- Thiazole-Based Hydroxamic Acids as Histone Deacetylase Inhibitors and Antitumor Agents Duong Tien Anh, Dao Thi Kim Oanh, Nguyen Hai Nam 37 Tổng hợp và thử hoạt tính sinh học một số dẫn chất kết hợp curcumin với diacid Phạm Thị Hiền, Phạm Ngọc Văn, Ngô Quang Trung, Nguyễn Thị Ngọc Bích, Đào Nguyệt Sương Huyền, Nguyễn Văn Giang, Nguyễn Đình Luyện, Nguyễn Văn Hải Synthesis and bioevaluation of some curcumin derivatives conjugated with diacid Pham Thi Hien, Pham Ngoc Van, Ngo Quang Trung, Nguyen Thi Ngoc Bich, Dao Nguyet Suong Huyen, Nguyen Van Giang, Nguyen Dinh Luyen, Nguyen Van Hai 45 ĐIỂM TIN THÔNG TIN THUỐC – CẢNH GIÁC DƯỢC DRUG INFORMATION & PHARMACOVIGILANCE HIGHLIGHTS
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of nghiên cứu dược & thông tin thuốc 2020, tập 11, số 6

NGHIÊN CỨU DƯỢC & THÔNG TIN THUỐC 2020, TẬP 11, SỐ 6
Journal of Pharmaceutical Research and Drug information 2020, Vol 11. No6
MỤC LỤC
Số 6, 2020
CONTENTS
No6, 2020
BÀI NGHIÊN CỨU RESEARCH
2 Nghiên cưu chế tạo hệ phân tán rắn
telmisartan, ưng dụng vào bào chế viên nén
Bùi Thị Lan Phương, Lê Thiện Giáp,
Phạm Thị Thu Hương, Nguyễn Ngọc Chiến
Formulation of solid dispersion of
telmisartan, application to tablet preparation
Bui Thi Lan Phuong, Le Thien Giap,
Pham Thi Thu Huong, Nguyen Ngoc Chien
9 Phân tích tính phù hợp của việc sử dụng
kháng sinh carbapenem trong điều trị viêm
phổi bệnh viện tại Bệnh viện Đa khoa Hà
Đông
Nguyễn Thị Thu Thủy, Quách Thị Thu Hà,
Lê Vân Anh, Phạm Thị Thúy Vân
Appropriateness of carbapenems use in the
treatment of hospital acquired pneumonia in
Ha Dong General Hospital
Nguyen Thi Thu Thuy, Quach Thi Thu Ha,
Le Van Anh, Pham Thi Thuy Van
16 Tổng hợp acid (E)-4-dimethylaminobut-2-
enoic hydroclorid làm chất trung gian quan
trọng cho điều chế afatinib dimaleat
Nguyễn Thị Ngọc, Nguyễn Hòa Bình,
Bùi Thị Thanh Châm, Trần Hoàng Vũ,
Trần Anh Phương, Đào Nguyệt Sương Huyền,
Nguyễn Văn Giang, Nguyễn Đình Luyện,
Nguyễn Văn Hải
Synthesis of (E)-4-(dimethylamino)but-2-
enoic acid hydrochloride as an important
intermediate for the preparation of afatinib
dimaleate
Nguyen Thi Ngoc, Nguyen Hoa Binh,
Bui Thi Thanh Cham, Tran Hoang Vu,
Tran Anh Phuong, Dao Nguyet Suong Huyen,
Nguyen Van Giang, Nguyen Dinh Luyen,
Nguyen Van Hai
23 Nghiên cưu quy trình bào chế viên nang
mềm chưa hệ nano tự nhũ hóa rosuvastatin
Vũ Thị Thu Giang, Phan Thị Nghĩa,
Nguyễn Thị Thúy Nga, Nguyễn Thị Linh,
Trần Thị Hải Yến, Phạm Bảo Tùng,
Nguyễn Đăng Hòa
Development of preparing procedure of soft
gelatin capsule containing rosuvastatin self-
nano emulsifying drug delivery system
Vu Thi Thu Giang, Phan Thi Nghia,
Nguyen Thi Thuy Nga, Nguyen Thi Linh,
Tran Thi Hai Yen, Pham Bao Tung,
Nguyen Dang Hoa
31 Tổng hợp và đánh giá tác dụng ưc chế tế bào
ung thư của một số acid hydroxamic mang
dị vòng 1,3-thiazol
Dương Tiến Anh, Đào Thị Kim Oanh,
Nguyễn Hải Nam
Synthesis and Evaluation of Novel 1,3-
Thiazole-Based Hydroxamic Acids as Histone
Deacetylase Inhibitors and Antitumor Agents
Duong Tien Anh, Dao Thi Kim Oanh,
Nguyen Hai Nam
37 Tổng hợp và thử hoạt tính sinh học một số
dẫn chất kết hợp curcumin với diacid
Phạm Thị Hiền, Phạm Ngọc Văn,
Ngô Quang Trung, Nguyễn Thị Ngọc Bích,
Đào Nguyệt Sương Huyền, Nguyễn Văn Giang,
Nguyễn Đình Luyện, Nguyễn Văn Hải
Synthesis and bioevaluation of some curcumin
derivatives conjugated with diacid
Pham Thi Hien, Pham Ngoc Van,
Ngo Quang Trung, Nguyen Thi Ngoc Bich,
Dao Nguyet Suong Huyen, Nguyen Van Giang,
Nguyen Dinh Luyen, Nguyen Van Hai
45 ĐIỂM TIN THÔNG TIN THUỐC –
CẢNH GIÁC DƯỢC
DRUG INFORMATION &
PHARMACOVIGILANCE HIGHLIGHTS

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 2-8
2
Nghiên cứu chế tạo hệ phân tán rắn telmisartan,
ứng dụng vào bào chế viên nén Bùi Thị Lan Phương, Lê Thiện Giáp, Phạm Thị Thu Hương, Nguyễn Ngọc Chiến*
Trương Đại học Dược Hà Nội
*Tác giả liên hệ: [email protected]
(Ngày gửi đăng: 17/3/2020 – Ngày duyệt đăng: 16/11/2020)
SUMMARY Telmisartan (TEL) is an Angiotensin II Receptor Antagonist, which has been used in
the prevention and treatment of hypertension. However, it is a water-insoluble substance
which causes low dissolution, and poor bioavailability after oral administration (~42 %).
Thus, the objective of this study was to formulate a solid dispersion of telmisartan and
alkaline excipients to improve dissolution of telmisartan in pH 1.2 and 6.8 buffer media. The
solid dispersions containing telmisartan and alkaline excipients were prepared by a solvent
method using PVP K30 as a carrier. The results indicated that a solid dispersion formulation
containing telmisartan, sodium hydroxide and PVP-K30 with a PVP-K30:telmisartan ratio of
1:1 (w:w), dried by a vacuum technique released approximately 90 % of TEL after 60 minutes
in both pH 6.8 and pH 1.2 media. The solid dispersion was stable for one month under a
room condition (15-35 oC). Drug release from the tablets containing the solid dispersion was
approximately 90 % in both pH 1.2 and 6.8 media.
Từ khoá: Telmisartan, hệ phân tán rắn, tá dược kiềm, bốc hơi dung môi.
Đặt vấn đề
Sinh khả dụng của nhiều thuốc dùng theo đường uống bị giới hạn bởi độ tan và tính
thấm kém, do vậy việc làm tăng độ tan dược chất là một trong các biện pháp cải thiện sinh
khả dụng của chúng. Telmisartan là một thuốc điều trị tăng huyết áp hiệu quả cao, tuy nhiên
rất ít tan trong khoảng pH 3-9, nên khả năng giải phóng từ viên nén và sinh khả dụng đường
uống của thuốc thấp và kém ổn định [1], [4], [6]. Nhằm nâng cao sinh khả dụng của
telmisartan chúng tôi thực hiện đề tài: “Nghiên cưu bào chế hệ phân tán rắn chưa hỗn hợp
telmisartan và tá dược kiềm, ưng dụng vào viên nén”.
Nguyên liệu và phương pháp nghiên cứu
Nguyên liệu
Telmisartan (USP 38, Ấn Độ), Avicel PH 102 (USP 38, Đài Loan), polyvinyl
pyrolidon K30 (PVP K30, USP 38), manitol (USP 38), tinh bột (DĐVN V), natri
croscarmelose (EP 8.0), magnesi stearat (USP 38, Trung Quốc), Aerosil (USP 38, Bỉ). Các
hoá chất khác: đạt tiêu chuẩn tinh khiết hoá học.
Thiết bị nghiên cứu:
Máy thử độ hoà tan Pharma-test (Đưc), máy đo quang phổ UV-VIS Optima SP-3000
nano (Nhật), máy đo quang phổ hồng ngoại JASCO FT-IR (Nhật Bản), máy phân tích nhiệt vi
sai METTLER TOLEDO DSC 1 (Mỹ), máy dập viên quay tròn 8 chày SHAKTI (Ấn Độ),
máy phun sấy Buchi Mini Spray Dryer B 191, máy đông khô Christ Alpha 1-LD (Đưc), tủ sấy
chân không LabTech (Hàn Quốc).
Phương pháp nghiên cứu
Bào chế hỗn hợp telmisartan và tá dược kiềm (HH-TEL): Hỗn hợp TEL với các tá
dược kiềm được bào chế như sau: Hòa tan tá dược kiềm vào nước, sau đó thêm TEL theo
công thưc như bảng 1, rồi đun nóng hỗn dịch này ở 60 oC/30 phút trong nồi cách thủy, bốc
hơi dung môi bằng sấy chân không ở 40 oC, áp suất 0,1 atm, 24 giờ thu được hỗn hợp TEL và
tá dược kiềm.
Bào chế hệ phân tán rắn (HPTR) chứa HH-TEL: Hòa tan HH-TEL trong nước ở nhiệt
độ 60 °C, thêm PVP K30 khuấy đến khi tan hoàn toàn, bay hơi dung môi bằng một trong các
phương pháp: sấy chân không (tủ LabTech: 40 oC, áp suất 0,1 atm, 24 giờ), hoặc phun sấy

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 2-8
3
(máy phun sấy Buchi Mini Spray Dryer B 191: tốc độ gió 10-14m3/giờ, tkhí vào = 65 oC, tsản
phẩm = 40 – 41 oC, áp suất súng phun 1,2 – 1,3 bar, tốc độ bơm dịch 1,0ml/phút), hoặc đông
khô (máy đông khô Christ Alpha 1-LD, điều kiện: tiền đông ở -70 oC/12 giờ, sấy khô sơ cấp ở
- 48 ± 2 oC/0,12 ± 0,02 mbar/24 giờ).
Bào chế viên nén TEL 40 mg: Nghiền, rây HPTR chưa HH-TEL qua rây 0,25 mm,
trộn đồng lượng với tá dược độn, tá dược siêu rã, tá dược trơn. Dập viên chày đường kính 9
mm, độ cưng 4-6 kp, mỗi mẻ nghiên cưu 100 viên.
Các phương pháp đánh giá
Định lượng hàm lượng TEL trong HPTR và trong viên nén: Bằng phương pháp sắc kí
lỏng hiệu năng cao (HPLC): Pha động (tỷ lệ 45:55) acetonitril:đệm phosphat 0,02M pH 3,0;
cột sắc ký Zorbax Eclipse XBD C18 250 x 4,6 mm x 5 µm. Thể tích tiêm 20 µl, tốc độ dòng
1,4 ml/phút, dectector UV 230 nm. Chuẩn bị mẫu: Nghiền mịn mẫu HPTR hoặc mẫu viên.
Cân một lượng chính xác mẫu nghiền tương đương 40 mg TEL vào bình định mưc 100 ml,
thêm 1ml NaOH 0,1N và lắc đều. Thêm tiếp 50 ml methanol lắc kỹ và bổ sung đến vạch bằng
methanol. Lọc qua màng cellulose acetat kích thước lỗ lọc 0,45 µm, pha loãng 50 lần với
nước cất. So sánh với dung dịch chuẩn TEL (nguyên liệu) nồng độ 8 µg/ml.
Phương pháp đo phổ hồng ngoại (FT-IR): Các mẫu bột được nghiền mịn, sau đó được
trộn với bột kali bromid. Hỗn hợp sau khi trộn được ép thành viên mỏng rồi được quét phổ
FT-IR với dải bước sóng 4000-400 cm-1, độ phân dải 0,4 cm-1.
Đánh giá độ hoà tan: Máy cánh khuấy, tốc độ khuấy: 75 ± 1 vòng/phút. Môi trường:
thử 1 - 900 ml dung dịch HCl 0,1N pH 1,2; thử 2 – 900 ml dung dịch đệm phosphat pH 6,8.
Nhiệt độ: 37 ± 0,5 oC. Mẫu thử là viên hoặc HPTR chưa tương đương với 40 mg TEL. Sau
các khoảng thời gian nhất định, hút mẫu, lọc, pha loãng tới nồng độ thích hợp và đo mật độ
quang tại bước sóng 291 nm (môi trường HCl 0,1N pH 1,2) hoặc 296 nm (môi trường đệm
phosphat pH 6,8). So sánh với mẫu chuẩn (nguyên liệu). Kết quả trình bày trung bình ± SD
(n=3).
Phương pháp đo phổ phân tích nhiệt vi sai (DSC): Nghiền mịn mẫu phân tích và cho
vào đĩa đo phân tích nhiệt vi sai, đo ở dải nhiệt độ 0 – 300 oC, tốc độ gia nhiệt 10oC/phút, thổi
khí nitrogen ở tốc độ 50 ml/phút.
Đánh giá độ ổn định hỗn hợp: Bảo quản HPTR trong 1 tháng ở điều kiện phòng (15-
35 oC) và điều kiện lão hoá cấp tốc trong tủ vi khí hậu (40 oC ± 2, 75 %± 5) trong lọ thủy tinh
nút kín không màu. Sau thời gian bảo quản, xác định lại hàm lượng TEL bằng phương pháp
HPLC và độ hoà tan của TEL theo phương pháp đo mật quang trên.
Phương pháp xử lí thống kê: Kiểm định hai mẫu độc lập để so sánh phần trăm dược
chất hòa tan ở mỗi thời điểm (* P <0,05 được coi là có ý nghĩa thống kê).
Kết quả nghiên cứu
Kết quả bào chế hỗn hợp telmisartan với các tá dược kiềm
Bảng 1. Công thức bào chế HH-TEL (Đơn vị: mg)
CT
TEL Natri
hydroxyd
Natri
carbonat
Natri
hydrocarbonat
Natri
dihydro
phosphat
Dinatri
hydrophosphat
Natri
phosphat Nước
1 40 3,11 - - - - - 1
2 40 - 8,24 - - - - 1
3 40 - - 6,53 - - - 1
4 40 - - - 9,33 - - 1
5 40 - - - - 11,03 - 1
6 40 - - - - - 12,74 1
Hình 1 cho thấy độ hòa tan của TEL trong môi trường pH 6,8 có sự thay đổi mạnh
giưa các HH-TEL bào chế theo các công thưc khác nhau. Cụ thể, độ hòa tan TEL tại thời

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 2-8
4
điểm 60 phút từ hỗn hợp bào chế theo CT4 (2,2 ± 0,5), CT5 (4,4 ± 0,2), CT6 (0,7 ± 0,2)
không thay đổi nhiều so với nguyên liệu (0,8 ± 0,1), tuy nhiên độ hòa tan TEL từ hỗn hợp bào
chế theo CT1 (NaOH), CT2 (Na2CO3), CT3 (NaHCO3) tăng mạnh (độ hòa tan tại thời điểm
60 phút của CT1 là 73,3 ± 1,6; CT2 là 54,6 ± 2,8 và CT3 là 36,9 ± 1,1; p < 0,01 so với nguyên
liệu). Đặc biệt, khi sử dụng NaOH, độ hòa tan của TEL từ hỗn hợp tăng lên 92 lần sau 60 phút
so với nguyên liệu.
Hình 1. Đồ thị hoà tan của HH-TEL ở môi trương pH 1,2 và pH 6,8 (**p<0,01)
Tuy nhiên, tại môi trường pH 1,2, so với nguyên liệu độ hòa tan của TEL từ hỗn hợp
bào chế theo công thưc CT4 (NaH2PO4), CT5 (Na2HPO4 ), CT6 (Na3PO4) không thay đổi,
trong khi độ hòa tan của các công thưc còn lại đều giảm, nhưng vẫn trên 60 % sau 60 phút.
Trong tất cả hỗn hợp dược chất và tá dược kiềm, công thưc chưa NaOH khả quan nhất. Trước
khi tiến hành thí nghiệm tiếp theo, tiến hành đo phổ hồng ngoại của các mẫu TEL nguyên liệu
và HH-TEL nhằm đánh giá tương tác TEL và các tá dược kiềm, kết quả trình bày ở hình 2.
Hình 2. Phổ hồng ngoại của TEL và hỗn hợp TEL – tá dược kiềm
Từ kết quả phổ hồng ngoại của TEL và các hỗn hợp nhận thấy không có sự dịch
chuyển số sóng của nhóm hydroxyl (O – H) giưa các các mẫu. Tuy nhiên, xét về nhóm
carbonyl (C = O) thì có khác biệt về số sóng và hình dạng pic so với phổ TEL. Phổ của nhóm
hỗn hợp của TEL và Na2HPO4, NaH2PO4, Na3PO4, vẫn cho thấy hai dải hấp thụ riêng biệt tại
1696 cm-1 và 3056 cm-1. Phổ của nhóm hỗn hợp TEL và NaOH, Na2CO3 cho thấy sự thay đổi
trong liên kết C = O, tần số của C = O được dịch chuyển từ 1695 cm-1 đến 1580 cm-1. Sự giảm
tần số dao động của đỉnh carbonyl cho thấy có sự hình thành của anion dạng cacboxylat,
chưng tỏ đã có hiện tượng tạo thành muối của TEL với 2 tá dược kiềm này.
Kết quả bào chế HPTR chứa hỗn hợp TEL và tá dược kiềm
Ảnh hưởng phương pháp bốc hơi dung môi

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 2-8
5
Kết quả trên cho thấy, HH-TEL sử dụng NaOH đã cải thiện đáng kể độ hòa tan của
TEL ở môi trường pH 6,8 và có độ hòa tan cao ở môi trường pH 1,2 (57,2 % sau 10 phút). Do
đó lựa chọn NaOH trong các thí nghiệm tiếp theo ở bảng 2.
Tiến hành bào chế HPTR chưa TEL-NaOH theo phương pháp phun sấy, sấy chân
không và đông khô (CT7, 8, 9). Kết quả thử độ hoà tan được trình bày ở hình 3.
Bảng 2. Công thức bào chế HPTR chứa TEL và natri hydroxyd
Thành phần CT 7 CT 8 CT 9 CT 10 CT 11 CT 12
TEL (mg) 40 40 40 40 40 40
Natri hydroxyd (mg) 3,2 3,2 3,2 3,2 3,2 3,2
Nước (ml) 1 1 1 1 1 1
PVP K30 (mg) 40 40 40 20 120 200
Tỉ lệ TEL:PVP K30 (kl:kl) 1:1 1:1 1:1 2:1 1:3 1:5
PP làm khô Phun sấy SCK Đông khô Sấy chân không (SCK)
Hình 3. Đồ thị hoà tan của HPTR chứa hỗn hợp TEL-NaOH ở môi trương pH 1,2 (a) và
pH 6,8 (b)
Hình 3 cho thấy, tốc độ và mưc độ hòa tan của TEL từ HPTR chưa hỗn hợp TEL-
NaOH trong cả hai môi trường đều tăng lên có ý nghĩa so với hỗn hợp TEL-NaOH và có sự
khác nhau giưa các phương pháp, giảm theo thư tự: đông khô (CT9) > sấy chân không (CT8)
> phun sấy (CT7) (Độ hòa tan ở pH 6,8 tại thời điểm 60 phút của CT9 là 95,2 ± 3,2; CT8 là
89,0 ± 2,6 và CT7 là 88,4 ± 2,1). Phương pháp sấy chân không cho kết quả về độ hòa tan của
sản phẩm ở các môi trường kém phương pháp đông khô, nhưng tốt hơn phương pháp phun
sấy. Sản phẩm sấy chân không có chất lượng khô tơi, ít hút ẩm, độ ổn định tốt hơn sản phẩm
đông khô. Vì vậy, phương pháp sấy chân không được lựa chọn để sử dụng trong các nghiên
cưu tiếp sau.
Ảnh hưởng của tỷ lệ chất mang
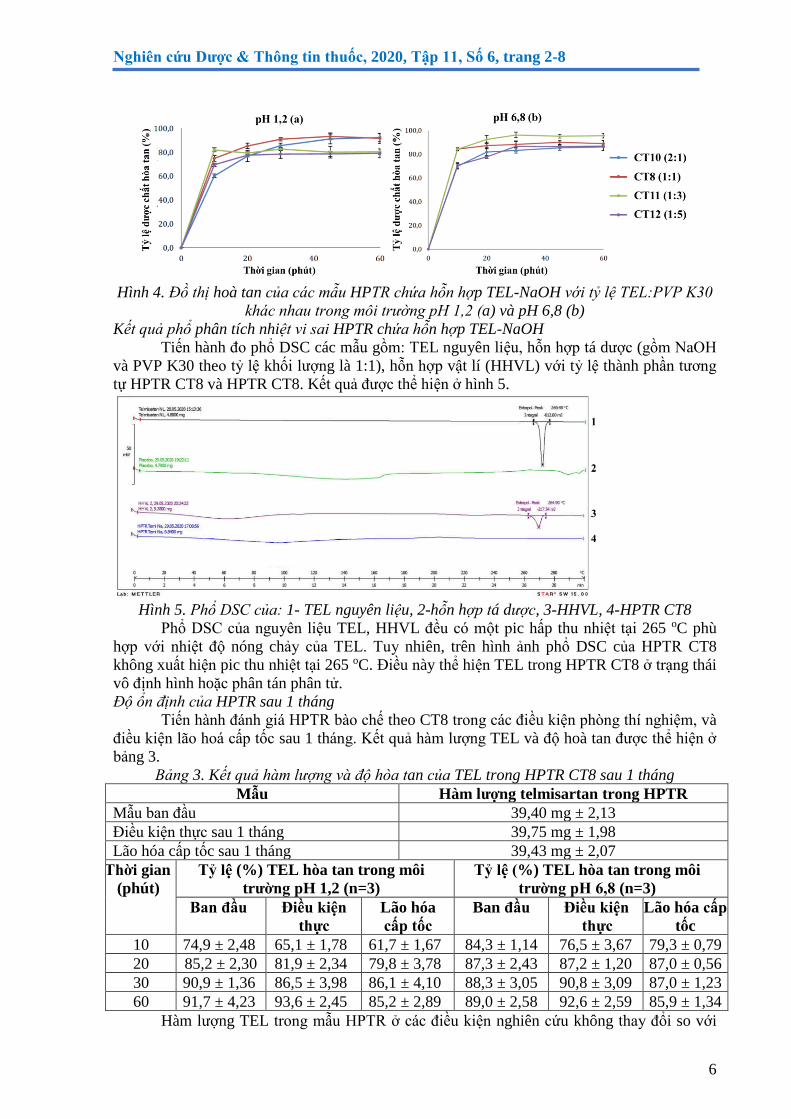
Tiến hành bào chế HPTR chưa hỗn hợp TEL-NaOH theo tỷ lệ TEL:PVP K30 lần lượt
là: 2:1 (CT 10), 1:1 (CT8), 1:3 (CT11), 1:5 (CT12). Kết quả thử độ hoà tan trong các môi
trường của hỗn hợp được trình bày ở hình 4.
Ở môi trường pH 1,2, mưc độ hòa tan của TEL thấp nhất từ hỗn hợp bào chế theo CT
11 (1:3) và CT12 (1:5). Ở môi trường pH 6,8, mưc độ hòa tan của TEL thấp nhất từ hỗn hợp
bào chế theo CT CT12 (1:5) và CT10 (2:1). Sau 60 phút, CT8 (1:1) có độ hòa tan cao nhất
trong môi trường pH 1,2, trong môi trường pH 6,8 tương đối cao. Như vậy HPTR chưa hỗn
hợp TEL-NaOH với tỷ lệ TEL:PVP là 1:1 cho độ hòa tan của TEL ở hai môi trường cao, tốt
hơn hỗn hợp TEL-NaOH ở pH 1,2. Do đó, CT8 với tỷ lệ TEL:PVP K30 là 1:1 được lựa chọn
để tiếp tục nghiên cưu.
Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 2-8
6
Hình 4. Đồ thị hoà tan của các mẫu HPTR chứa hỗn hợp TEL-NaOH với tỷ lệ TEL:PVP K30
khác nhau trong môi trương pH 1,2 (a) và pH 6,8 (b)
Kết quả phổ phân tích nhiệt vi sai HPTR chứa hỗn hợp TEL-NaOH
Tiến hành đo phổ DSC các mẫu gồm: TEL nguyên liệu, hỗn hợp tá dược (gồm NaOH
và PVP K30 theo tỷ lệ khối lượng là 1:1), hỗn hợp vật lí (HHVL) với tỷ lệ thành phần tương
tự HPTR CT8 và HPTR CT8. Kết quả được thể hiện ở hình 5.
Hình 5. Phổ DSC của: 1- TEL nguyên liệu, 2-hỗn hợp tá dược, 3-HHVL, 4-HPTR CT8
Phổ DSC của nguyên liệu TEL, HHVL đều có một pic hấp thu nhiệt tại 265 oC phù
hợp với nhiệt độ nóng chảy của TEL. Tuy nhiên, trên hình ảnh phổ DSC của HPTR CT8
không xuất hiện pic thu nhiệt tại 265 oC. Điều này thể hiện TEL trong HPTR CT8 ở trạng thái
vô định hình hoặc phân tán phân tử.
Độ ổn định của HPTR sau 1 tháng
Tiến hành đánh giá HPTR bào chế theo CT8 trong các điều kiện phòng thí nghiệm, và
điều kiện lão hoá cấp tốc sau 1 tháng. Kết quả hàm lượng TEL và độ hoà tan được thể hiện ở
bảng 3.
Bảng 3. Kết quả hàm lượng và độ hòa tan của TEL trong HPTR CT8 sau 1 tháng
Mâu Hàm lượng telmisartan trong HPTR
Mẫu ban đầu 39,40 mg ± 2,13
Điều kiện thực sau 1 tháng 39,75 mg ± 1,98
Lão hóa cấp tốc sau 1 tháng 39,43 mg ± 2,07
Thời gian
(phút)
Tỷ lệ (%) TEL hòa tan trong môi
trường pH 1,2 (n=3)
Tỷ lệ (%) TEL hòa tan trong môi
trường pH 6,8 (n=3)
Ban đầu Điều kiện
thực
Lão hóa
cấp tốc
Ban đầu Điều kiện
thực
Lão hóa cấp
tốc
10 74,9 ± 2,48 65,1 ± 1,78 61,7 ± 1,67 84,3 ± 1,14 76,5 ± 3,67 79,3 ± 0,79
20 85,2 ± 2,30 81,9 ± 2,34 79,8 ± 3,78 87,3 ± 2,43 87,2 ± 1,20 87,0 ± 0,56
30 90,9 ± 1,36 86,5 ± 3,98 86,1 ± 4,10 88,3 ± 3,05 90,8 ± 3,09 87,0 ± 1,23
60 91,7 ± 4,23 93,6 ± 2,45 85,2 ± 2,89 89,0 ± 2,58 92,6 ± 2,59 85,9 ± 1,34
Hàm lượng TEL trong mẫu HPTR ở các điều kiện nghiên cưu không thay đổi so với

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 2-8
7
ban đầu. Độ hòa tan của mẫu sau 1 tháng bảo quản ở điều kiện thực tương tự so với mẫu khi
mới bào chế, tuy nhiên độ hòa tan ở điều kiện lão hóa có xu hướng giảm. HPTR chưa hỗn hợp
TEL-NaOH được lựa chọn để ưng dụng vào bào chế viên nén.
Ứng dụng vào bào chế viên nén
Tiến hành bào chế viên nén chưa HPTR của hỗn hợp TEL-NaOH bằng phương pháp
dập thẳng theo các công thưc ở bảng 4.
Bảng 4. Công thức bào chế viên nén chứa 40mg TEL (mg)
Thành phần CT13 CT14 CT15 CT 16 CT 17 CT 18
HPTR chưa 40mg
TEL
83,2 83,2 83,2 83,2 83,2 83,2
Avicel PH 102 139,28 - - 145,28 133,28 127,28
Manitol - 139,28 - - - -
Tinh bọt - - 139,28 - - -
Croscarmelose 12 12 (5%) 12 6 (2,5%) 18 (7,5%) 24(10%)
Aerosil 0,72 0,72 0,72 0,72 0,72 0,72
Magnesi stearat 4,8 4,8 4,8 4,8 4,8 4,8
Tông 240 240 240 240 240 240
Kết quả thử hòa tan về ảnh hưởng của tá dược độn theo các CT13, CT14, CT15 cho
thấy không có sự khác biệt về mưc độ và tốc độ hòa tan giưa các mẫu trong môi trường đệm
pH 6,8 nhưng trong môi trường pH 1,2, độ hòa tan của các mẫu viên nén giảm dần theo thư tự
avicel (CT13, 79,8 ± 4,56 %) > manitol (CT14, 71,6 ± 1,34 %)> tinh bột (CT15, 62,1 ± 3,10
%) sau 30 phút. Avicel được lựa chọn làm tá dược độn.
Hình 6. Ảnh hưởng tá dược độn và siêu rã đến độ hoà tan của các mẫu viên nén TEL trong
môi trương pH 1,2 (6a) và pH 6,8 (6b)
Kết quả thử hòa tan về ảnh hưởng của tỷ lệ tá dược rã được khảo sát theo các tỷ lệ
tăng dần 2,5 % (CT16), 5 % (CT14), 7,5 % (CT17) và 10 % (CT18) được thể hiện ở hình 6.
Hình 6 cho thấy ở pH 1,2, tăng dần lượng tá dược siêu rã từ 2,5 – 7,5 % thì mưc độ hòa tan
tăng nhưng khi tá dược siêu rã tăng từ 7,5 % lên 10 %, thì mưc độ hòa tan không tăng thêm.
Ở pH 6,8 CT14 (5 %) và CT17 (7,5 %) cho kết quả mưc độ hòa tan cao nhất. Do vậy công
thưc CT17 (7,5 % Croscarmellose) được lựa chọn là công thưc cuối.
Bàn luận
Telmisartan là một acid yếu, tan tốt trong kiềm mạnh, rất ít tan trong nước (độ tan 1,02
μg/ml). Do vậy, các tá dược kiềm thường được sử dụng với vai trò làm vi môi trường hoặc tạo
muối làm tăng độ tan cho TEL [3], [4], [7]. Trong nghiên cưu này, 6 tá dược kiềm có độ base
từ mạnh đến yếu gồm natri hydroxyd, natri phosphat, natri carbonat, dinatri hydrophosphat,
natri hydrocarbonat, natri dihydrophosphat được đưa vào khảo sát với tỷ lệ mol TEL:tá dược
kiềm = 1:1. Kết quả tương tự như các nghiên cưu khác, độ base càng mạnh, độ hòa tan ở pH
6,8 của TEL từ hỗn hợp càng cao [7], [8], tốt nhất là hỗn hợp TEL-NaOH. Điều này có thể
giải thích do ở điều kiện tạo hỗn hợp TEL:tá dược kiềm, có thể tạo ra một phần muối TEL với
các tá dược kiềm mạnh và do vi môi trường kiềm tạo ra. Ở pH 1,2 độ hòa tan của TEL từ hỗn

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 2-8
8
hợp kiềm mạnh (ví dụ TEL-NaOH) lại bị giảm so với nguyên liệu, điều này được giải thích
trong môi trường acid, muối này bị thủy phân ngược và chậm giải phóng TEL.
Phương pháp làm khô ảnh hưởng đến độ hòa tan dược chất từ HPTR. Khi sử dụng
phương pháp đông khô, hệ có cấu trúc bột xốp do đó dễ dàng hút nước phân tán và giải phóng
dược chất nhanh, nên mưc độ hòa tan là cao nhất. Tuy nhiên, phương pháp đông khô lại có
nhiều hạn chế như trang thiết bị đặc biệt, sản phẩm có độ ổn định kém do dễ hút ẩm, quy trình
đông khô thường kéo dài [5]. Phương pháp sấy chân không cho kết quả về độ hòa tan của sản
phẩm ở các môi trường kém phương pháp đông khô, nhưng tốt hơn phương pháp phun sấy.
Phương pháp phun sấy có thời gian thu sản phẩm nhanh nhất, tuy nhiên sản phẩm phun sấy dễ
hút ẩm và đóng vón, ngoài ra phương pháp phun sấy có nhược điểm là hiệu suất thấp, hao phí
lớn [2]. Thực tế, sản phẩm sấy chân không khô, tơi, ít hút ẩm và ổn định về mặt thể chất hơn
sản phẩm thu được từ hai phương pháp còn lại.
Khi đưa hỗn hợp TEL-NaOH vào HPTR với chất mang PVP K30 đã làm tăng độ hòa
tan của TEL ở cả hai môi trường so với hỗn hợp TEL-NaOH. Độ hòa tan của TEL sau 60 phút
ở pH 6,8 cao hơn nhiều so với nguyên liệu và hỗn hợp TEL-NaOH. Tuy nhiên, độ hòa tan của
TEL sau 60 phút ở pH 1,2 thấp hơn nguyên liệu nhưng vẫn trên 85% và cao hơn so với hỗn
hợp TEL-NaOH. Công thưc HPTR có tỷ lệ TEL:PVP = 1:1 cho kết quả độ hòa tan cao ở pH
1,2 và 6,8. Phổ DSC xác nhận TEL trong HPTR CT8 ở trạng thái vô định hình hoặc phân tán
phân tử. Viên nén chưa HPTR trên được bào chế bằng phương pháp dập thẳng vẫn đảm bảo
độ hòa tan khi phối hợp với các tá dược độn rã phù hợp.
Kết luận
Trong nghiên cưu này, độ hòa tan của TEL đã được cải thiện được bằng cách chế tạo
HPTR của TEL với chất mang là PVP K30 và tá dược làm tăng độ tan là NaOH. Công thưc
HPTR tốt nhất được ưng dụng để bào chế viên nén. Viên nén bào chế theo công thưc lựa chọn
có độ hòa tan TEL đạt xấp xỉ 90 % trong cả hai môi trường pH 1,2 và 6,8.
TÀI LIỆU THAM KHẢO
1. Bhise Sucheta D. et al., (2012), “Isolation and Evaluation of Natural Superdisintegrant
from Plantago Ovata in MDTs of Telmisartan”, IJPI’s Journal of Pharmaceutics and
Cosmetology, 2(10), pp. 9-21.
2. Broadhead J., Edmond Rouan S., Rhodes C. (1992), "The spray drying of
pharmaceuticals", Drug development and industrial pharmacy, 18(11-12), pp. 1169-
1206.
3. Cao Y. et al. (2016), "In-Vitro Characterization and Oral Bioavailability of Organic
Solvent-free Solid Dispersions Containing Telmisartan", Iranian journal of
pharmaceutical research: IJPR, 15(2), pp. 385.
4. Gaja JB, Sayyad FJ (2013), "Enhancement of solubility and dissolution rate of
telmisartan by spray drying technique", Indo Am. J. Pharm. Res, 3, pp. 1732- 1745.
5. Nireesha G., Divya L., Sowmya C., et al. (2013), "Lyophilization/freeze drying-an
review", International journal of novel trends in pharmaceutical sciences, 3(4), pp.
87-98.
6. Pandya Vatsal A., Chaudhari Shilpa P. (2012), "Optimization and evaluation of a
formulation containing low soluble antihypertensive agent", Int j curr pharm res, 4(2),
pp. 37-41.
7. Tran Phuong Ha Lien et al. (2008), "Modulation of microenvironmental pH and
crystallinity of ionizable telmisartan using alkalizers in solid dispersions for controlled
release", Journal of Controlled Release, 129(1), pp. 59-65.
8. Zhong L. et al. (2014), "Influence of alkalizers on dissolution properties of telmisartan
in solid dispersions prepared by cogrinding", Drug development and industrial
pharmacy, 40(12), pp. 1660-1669.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 9-15
9
Phân tích tính phù hợp của việc sử dụng kháng sinh carbapenem trong điều
trị viêm phổi bệnh viện tại Bệnh viện Đa khoa Hà Đông Nguyễn Thị Thu Thủy1, Quách Thị Thu Hà1,2, Lê Vân Anh3, Phạm Thị Thúy Vân1*
1Trường Đại học Dược Hà Nội 2Bệnh viện đa khoa Hà Đông
3Bệnh viện Hữu Nghị
*Tác giả liên hệ: [email protected]
(Ngày gửi đăng: 25/8/2020 – Ngày duyệt đăng: 16/11/2020)
SUMMARY
A retrospective, descriptive study was conducted to analyze the appropriateness of
carbapenem use in treatment of hospital acquired pneumonia (HAP). Totally, 136 medical
records in Ha Dong General Hospital in 2018 were analyzed. Carbapenem was mostly
prescribed at Intensive Care Unit (93.4 %). 57.5 % of patients had at least one risk factor for
HAP. Only 25.0 % of patients were indicated microbiological testing before using antibiotics.
The rate of positive results was 58.2 %. Acinetobacter baumannii (33.7 %), Pseudomonas
aeruginosa (26.5 %) and Klebsiella pneumoniae (14.5 %) were three most common isolates.
Less than 50 % of isolates were sensitive to carbapenem. The rates of patients using
carbapenem in initial empirical, alternative empirical and definitive regimen were 58.8 %,
17.6% and 10.3 %, respectively. The rates of appropriateness of indications were 5.0 %, 75.0
% and 21.4 %, respectively. Rational carbapenem dosage was recognized in 43.7 % of dose
regimens. The majority of patients was infused carbapenem over 61 - 120 minutes (73.7 %).
Thus, there are carbapenem - related problems such as rational choice of antibiotic regimen,
dosage and administration that need to be improved in clinical practice.
Từ khóa: sử dụng carbapenem, tuân thủ hướng dẫn, viêm phổi bệnh viện
Đặt vấn đề
Carbapenem là nhóm kháng sinh có hoạt lực mạnh và phổ kháng khuẩn rộng, có tác
dụng trên nhiều chủng vi khuẩn đã đề kháng với nhiều nhóm kháng sinh. Do vậy, đây là
nhóm kháng sinh dự trữ, giữ vai trò quan trọng trong điều trị nhiễm khuẩn nặng, đe dọa tính
mạng và cần được quản lý sử dụng trên lâm sàng. Trong Hướng dẫn thực hiện quản lý sử
dụng kháng sinh của Bộ Y tế ban hành theo Quyết định 772/QĐ-BYT năm 2016, carbapenem
thuộc danh mục kháng sinh cần phê duyệt trước khi sử dụng [1].
Viêm phổi bệnh viện (VPBV) là một trong những bệnh lý nhiễm trùng nặng, phổ biến
với tỷ lệ tử vong cao trong các nhiễm khuẩn bệnh viện. Nhằm giảm tử vong khi điều trị,
phương phap tiếp cận hiện nay là điều trị xuống thang, trong đó carbapenem là nhóm kháng
sinh quan trọng của cac phac đồ điều trị VPBV ở những bệnh nhân có nguy cơ cao mắc vi
khuẩn đa khang hoặc có tình trạng bệnh lý nặng [3].
Bệnh viện đa khoa Hà Đông là bệnh viện hạng I trực thuộc Sở Y tế Hà Nội. Tại đây đã
triển khai một số các biện pháp nhằm hạn chế sử dụng kháng sinh nhóm carbapenem. Tuy
nhiên, dữ liệu khảo sát cho thấy mức độ tiêu thụ carbapenem có xu hướng ngày càng gia tăng
kể từ khi thuốc bắt đầu được đưa vào sử dụng từ năm 2013, trong đó viêm phổi bệnh viện là
chỉ định phổ biến nhất. Bên cạnh đó, dữ liệu vi sinh của bệnh viện cũng ghi nhận xu hướng
giảm nhạy cảm với kháng sinh trong nhóm carbapenem của các vi khuẩn phân lập được. Do
vậy, nghiên cứu này được thực hiện nhằm phân tích tính hợp lý dựa trên cac hướng dẫn hiện
hành của việc sử dụng khang sinh nhóm carbapenem trong điều trị viêm phổi bệnh viện, nhằm
cung cấp thêm thông tin về chất lượng sử dụng kháng sinh, từ đó xây dựng các biện pháp
quản lý kháng sinh tại bệnh viện đa khoa Hà Đông.
Đối tượng và phương pháp nghiên cứu
Đối tượng nghiên cứu: Toàn bộ hồ sơ bệnh an (136 bệnh an) của bệnh nhân người lớn điều
trị nội trú tại bệnh viện đa khoa Hà Đông từ 01/01/2018 đến 31/12/2018 được chẩn đoan viêm
phổi bệnh viện có sử dụng meropenem hoặc imipenem.
Phương pháp nghiên cứu:

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 9-15
10
Thiết kế nghiên cứu: Hồi cứu mô tả dựa trên hồ sơ bệnh án
Chỉ tiêu nghiên cứu: Cac chỉ tiêu nghiên cứu bao gồm:
- Đặc điểm lâm sàng của bệnh nhân: Tuổi, khoa điều trị, yếu tố nguy cơ trong VPBV,
chức năng thận.
- Đặc điểm về vi sinh học: Tỷ lệ bệnh nhân được xét nghiệm vi sinh, các chủng vi
khuẩn phân lập được, độ nhạy cảm với kháng sinh của vi khuẩn phân lập được.
- Đặc điểm về sử dụng phac đồ carbapenem: Vị trí của carbapenem trong điều trị, các
loại phac đồ, tính phù hợp của phac đồ sử dụng so với hướng dẫn, lý do không phù hợp của
cac phac đồ sử dụng.
- Đặc điểm liều dùng, cách dùng của kháng sinh carbapenem: Các chế độ liều, tỷ lệ
liều dùng phù hợp, các chế độ tiêm truyền của kháng sinh carbapenem.
- Đặc điểm số ngày sử dụng kháng sinh, kết quả ra viện.
Qui ước nghiên cứu về đánh giá tính phù hợp
- Đối với phac đồ kháng sinh kinh nghiệm (KSKN) ban đầu có carbapenem: Đánh giá
tính phù hợp dựa trên Hướng dẫn chẩn đoan và điều trị viêm phổi bệnh viện và viêm phổi thở
máy của IDSA/ATS 2016 [9] và của Hội hô hấp – Hồi sức cấp cứu và Chống độc Việt Nam
2017 [3], có điều chỉnh phù hợp với danh mục kháng sinh sử dụng tại bệnh viện (bổ sung
cefoperazon, cefoperazon/sulbactam vào nhóm betalactam có phổ trên P.aeruginosa và
neltimicin vào nhóm aminosid).
- Đối với phac đồ KSKN thay thế có carbapenem: Đánh giá là phù hợp khi bệnh nhân
đã dùng một trong các kháng sinh khác gồm: Piperacilin/tazobactam, cefepim, ceftazidim,
cefoperazon, cefoperazon/sulbactam, levofloxacin, ciprofloxacin nhưng tình trạng lâm sàng
không cải thiện hoặc xấu đi.
- Đối với phac đồ điều trị theo đích vi khuẩn: Đánh giá là phù hợp khi vi khuẩn không
nhạy cảm với piperacilin/tazobactam, cefoperazon/sulbactam, cefepim, ceftazidim,
cefoperazon, levofloxacin, ciprofloxacin và nhạy với ít nhất 1 kháng sinh trong phac đồ sử
dụng.
- Đanh gia tính hợp lý về liều dựa trên liều khuyến cáo cho viêm phổi bệnh viện theo
tờ Hướng dẫn sử dụng của thuốc Tienam®, Meronem® [7], [8].
Xử lý số liệu: Sử dụng phần mềm Exel 2007 trong quản lý, thống kê và phân tích số liệu. Các
biến liên tục có phân phối chuẩn được mô tả bằng giá trị trung bình ± độ lệch chuẩn. Các biến
liên tục có phân phối không chuẩn được mô tả bằng trung vị, khoảng tứ phân vị. Các biến
định tính được mô tả theo số lượng và tỷ lệ %.
Kết quả nghiên cứu
Đặc điểm lâm sàng
Tỷ lệ bệnh nhân điều trị tại khoa Hồi sức tích cực và chống độc là 93,4 %. Tuổi trung
bình là 65,2 ± 18,7 (năm) trong đó 63,2 % bệnh nhân ≥ 60 tuổi. Nghiên cứu ghi nhận 57,4 %
bệnh nhân có ít nhất 1 yếu tố nguy cơ của tử vong hoặc mắc vi khuẩn đa khang trong viêm
phổi bệnh viện (Bảng 1). Các yếu tố nguy cơ phổ biến bao gồm: Suy hô hấp cần thở máy
(35,3 %), dùng khang sinh tĩnh mạch trong 90 ngày trước (41,9 %).
Tỷ lệ lượt bệnh nhân sử dụng meropenem và imipenem/cilastatin cần chỉnh liều theo
chức năng thận (Clcr≤50 ml/phút với meropenem và Clcr≤ 70 ml/phút với imipenem/cilastatin)
lần lượt là 48,2 % và 64,1 %.
Bảng 1. Các yếu tố nguy cơ (YTNC) trong viêm phổi bệnh viện
Đặc điểm Kết quả (n, %)
Tỷ lệ bệnh nhân có ít nhất 1 YTNC 78 (57,4%)
YTNC tử vong
Suy hô hấp cần thở máy 48 (35,3%)
Sốc nhiễm khuẩn 4 (2,9%)
Suy hô hấp và sốc nhiễm khuẩn 1 (0,7%)
YTNC mắc vi khuẩn Gram âm
và Gram dương đa khang
Dùng khang sinh tĩnh mạch trong
90 ngày trước
57 (41,9%)
Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 9-15
11
YTNC mắc vi khuẩn Gram âm
đa kháng
Bệnh cấu trúc phổi 4 (2,9%)
Đặc điểm vi sinh
Số lượt bệnh nhân được chỉ định xét nghiệm vi sinh trước và trong khi điều trị kháng
sinh lần lượt là 34 (25,0 %) và 77 (56,6 %), với tổng số 158 mẫu bệnh phẩm hô hấp. Số lượng
mẫu bệnh phẩm dương tính là 92 (58,2 %), từ đó định danh được 98 chủng vi khuẩn. Loại vi
khuẩn phân lập được nhiều nhất là các vi khuẩn Gram âm bao gồm: Acinetobacter baumannii
(33,7 %), Pseudomonas aeruginosa (26,5 %), Klebsiella pneumoniae (14,5 %), Enterobacter
spp. (6,1 %), Escherichia coli (4,1 %), Proteus mirabilis (3,1 %). Ngoài ra, vi khuẩn Gram
dương Staphylococcus aureus được phân lập ở 11,2 % mẫu bệnh phẩm.
Kết quả khang sinh đồ cho thấy, với hai kháng sinh meropenem và imipenem, tỷ lệ các
chủng vi khuẩn còn nhạy cảm đều dưới 50 %, lần lượt là 25,0 % và 47,4 %. Tuy nhiên, các vi
khuẩn còn nhạy cảm với piperacillin/tazobactam khá cao (77,8 %) (Hình 1). Ngoài ra, 45,7 %
vi khuẩn còn nhạy với amikacin. Độ nhạy cảm với cefoperazon/sulbactam chỉ được xac định
ở 1 chủng phân lập, kết quả cho thấy chủng phân lập được còn nhạy với kháng sinh này.
Hình 1. Tỷ lệ nhạy cảm của vi khuẩn Gram âm phân lập với kháng sinh
Đặc điểm phác đồ carbapenem
Trong mẫu nghiên cứu, 18/136 bệnh nhân đã được sử dụng carbapenem trước khi có
chẩn đoan VPBV. Trên 118 bệnh nhân được sử dụng khi đã có chẩn đoan VPBV, số lượt sử
dụng carbapenem với vai trò là kháng sinh trong phác đồ kinh nghiệm (PĐKN) ban đầu,
PĐKN thay thế và PĐ theo đích vi khuẩn lần lượt là 80 (58,8 %), 24 (17,6 %) và 14 (10,3 %).
Với phác đồ carbapenem sau khi có phân lập vi khuẩn, tổng cộng 16 vi khuẩn đích ghi nhận
được bao gồm: Acinetobacter baumannii (n=3), Pseudomonas aeruginosa (n=6), Klebsiella
pneumoniae (n=4), Enterobacter spp. (n=1), Escherichia coli (n=1) và Staphylococcus aureus
(n=1). Hầu hết bệnh nhân được chỉ định phac đồ phối hợp (98,1 %), với tỷ lệ dùng phối hợp
carbapenem với 1 kháng sinh (KS), 2 KS và 3 KS lần lượt là 64,4 %; 32,7 % và 1,0 % (Bảng
2). Fluoroquinolon và aminoglycosid là hai nhóm được phối hợp chủ yếu.
Bảng 2. Các phác đồ kháng sinh sử dụng
Phác đồ n (%) Tổng
Đơn độc carbapenem 2 (1,9%) 2 (1,9%)
Phối hợp với 1 KS
Quinolon 63 (60,6%) 67 (64,4%)
Kháca 4 (3,9%)
Phối hợp với 2 KS
Quinolon + aminosid 25 (24,0%)
34 (32,7%) Quinolon + macrolid 3 (2,9%)
Quinolon + metronidazol 3 (2,9%)
Khácb 3 (2,9%)
Phối hợp với 3 KS Quinolon + aminosid + TMP/SFX 1 (1,0%) 1 (1,0%)
Ghi chú: a gồm: aminosid, betalactam, macrolid
b gồm: aminosid + macrolid, quinolon + vancomycin

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 9-15
12
Khi so sánh với quy ước nghiên cứu, tỷ lệ sử dụng phù hợp ở nhóm sử dụng
carbapenem trong PĐKN ban đầu, PĐKN thay thế và PĐ theo đích vi khuẩn lần lượt là 5,0 %,
75,0 % và 21,4 % (Hình 2). Ở nhóm sử dụng PĐKN ban đầu, các lý do không phù hợp bao
gồm: phac đồ thiếu KS có phổ trên tụ cầu vàng kháng methicilin (MRSA) ở bệnh nhân có yếu
tố nguy cơ tử vong (n=47), phac đồ thiếu 1 KS có phổ trên P.aeruginosa ở bệnh nhân có yếu
tố nguy cơ mắc vi khuẩn đa khang (n=6), phac đồ thừa 1 KS có phổ trên P.aeruginosa ở bệnh
nhân không có yếu tố nguy cơ (n=28) và ở bệnh nhân có yếu tố nguy cơ mắc vi khuẩn đa
kháng (n=9).
Hình 2. Tỷ lệ phù hợp của các phác đồ sử dụng so với quy ước nghiên cứu
Đặc điểm chế độ liều dùng carbapenem
Trong mẫu nghiên cứu, có 83 lượt bệnh nhân dùng meropenem và 78 lượt bệnh nhân
dùng imipenem/cilastatin, (tổng cộng 161 lượt) với tương ứng 167 chế độ liều được sử dụng.
So sánh với quy ước nghiên cứu về liều dùng phù hợp, số lượt phù hợp với meropenem và
imipenem và cả nhóm carbapenem lần lượt là 45 (52,3 %), 28 (34,6 %) và 73 (43,7 %). Các
chế độ liều của meropenem 1 g/8 giờ, 1 g/12 giờ và imipenem 1g/8 giờ có tỷ lệ sử dụng phù
hợp lần lượt là 41,8 %, 60,0 % và 35,3 % (Hình 3). Tỷ lệ không phù hợp liên quan đến khía
cạnh liều 1 lần, số lần đưa thuốc trong ngày hoặc cả hai khía cạnh trên lần lượt là: 19 (20,2
%), 23 (24,5 %) và 52 (55,3 %).
Hình 3. Tình phù hợp của các chế độ liều carbapenem sử dụng trên bệnh nhân
Đặc điểm đường dùng, cách dùng carbapenem
100 % bệnh nhân trong mẫu nghiên cứu được sử dụng carbapenem theo đường truyền
tĩnh mạch, với tổng số lượt chế độ truyền là 171. Trong số đó, chủ yếu bệnh nhân được truyền
từ 61 – 120 phút, chiếm 73,7 %; sau đó là 31- 60 phút, chiếm 19,9 %. Chỉ 4,1 % bệnh nhân
được truyền từ 3 giờ trở lên.
Đặc điểm về số ngày dùng kháng sinh và hiệu quả điều trị
Số ngày sử dụng carbapenem trung bình là 11,7 ± 8,1 (ngày), số ngày sử dụng kháng
sinh trung bình là 19,6 ± 13,8 (ngày). Sau thời gian nằm viện trung bình là 23,0 ± 14,9 (ngày),
kết quả ra viện ghi nhận tỷ lệ đỡ/giảm chiếm 47,1 %, tỷ lệ bệnh nhân ra viện trong tình trạng
nặng và không thay đổi đều bằng 25,7 % và 1,5 % bệnh nhân tử vong khi ra viện.
Bàn luận

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 9-15
13
Về đặc điểm lâm sàng
Đa phần bệnh nhân trong mẫu nghiên cứu có tuổi trung bình cao (63,2 % bệnh nhân ≥
60 tuổi). 93,4 % bệnh nhân điều trị tại Khoa Hồi sức tích cực và Chống độc, phù hợp với mô
hình Khoa Hồi sức thường tiếp nhận điều trị cac nhiễm khuẩn nặng kém đap ứng với cac
khang sinh khac hoặc nhiễm khuẩn do vi khuẩn đa khang thuốc, đặc biệt nhiễm khuẩn hô hấp
có kèm biến chứng suy hô hấp cần thở may hoặc sốc nhiễm khuẩn. Đa số bệnh nhân (57,4 %)
có ít nhất một yếu tố nguy cơ tử vong và/hoặc mắc cac chủng đa khang thuốc trong viêm phổi
bệnh viện. Cac yếu tố nguy cơ mắc cac chủng đa khang bao gồm: Sử dụng khang sinh đường
tĩnh mạch trong 90 ngày trước đó (41,9 %), mắc kèm bệnh cấu trúc phổi (2,9 %). Do vậy cần
lưu ý phân tầng nguy cơ để lựa chọn phac đồ khang sinh kinh nghiệm phù hợp, từ đó tăng khả
năng đạt đap ứng lâm sàng sớm trong điều trị.
Về đặc điểm vi sinh
Các Hướng dẫn điều trị hiện nay đều khuyến cao cấy mẫu vi sinh trước khi sử dụng
khang sinh trên tất cả bệnh nhân viêm phổi bệnh viện. Tuy nhiên, tỷ lệ này ghi nhận trong
nghiên cứu rất thấp, chỉ chiếm 34 lượt (25,0 %). Ngược lại, tỷ lệ được lấy mẫu trong qua trình
điều trị tương đối cao, chiếm 77 (56,6 %) bệnh nhân; cho thấy bac sĩ thường chỉ định xét
nghiệm vi sinh khi diễn biến bệnh trên bệnh nhân chưa cải thiện, làm căn cứ để điều chỉnh
phac đồ khang sinh. Chúng tôi đề xuất cần tăng tỷ lệ xét nghiệm vi sinh trước khi dùng kháng
sinh. Kết quả vi sinh sớm cùng cac đanh gia lâm sàng sau 48 – 72 giờ điều trị sẽ giúp điều
chỉnh phac đồ kịp thời trên bệnh nhân.
Nghiên cứu ghi nhận ba chủng phân lập được phổ biến nhất bao gồm Acinetobacter
baumannii (33,7 %), Pseudomonas aeruginosa (26,5 %) và Klebsiella pneumoniae (14,5 %),
phù hợp với mô hình vi sinh tại Việt Nam [5], [6], [10]. Căn nguyên tụ cầu chỉ ghi nhận ở
11,2 % mẫu bệnh phẩm, trong khi tại cac quốc gia khac như Mỹ, tụ cầu vàng là căn nguyên
gây bệnh phổ biến nhất [2], [3], [9]. Sự khac biệt về mô hình vi sinh dẫn tới việc cần lưu ý khi
ap dụng phac đồ khang sinh kinh nghiệm trong Hướng dẫn điều trị viêm phổi bệnh viện của
IDSA/ATS 2016 vào bối cảnh tại Việt Nam nói chung và tại Bệnh viện đa khoa Hà Đông nói
riêng [2].
Mặc dù cỡ mẫu nhỏ, tỷ lệ được xét nghiệm vi sinh chưa cao, nghiên cứu đã bước đầu
ghi nhận độ nhạy cảm của cac chủng phân lập được. Piperacilin/tazobactam là kháng sinh có
độ nhạy cảm tương đối khả quan (77,8 %), do vậy có thể cân nhắc được sử dụng trong phac
đồ khang sinh kinh nghiệm tại Bệnh viện đa khoa Hà Đông để tăng tỷ lệ bao phủ trên cac
chủng gây bệnh khang thuốc trong khi đợi kết quả vi sinh. Ngoài ra, amikacin có tỷ lệ vi
khuẩn còn nhạy tới 45,7 %, do vậy có thể cân nhắc trong cac phac đồ kinh nghiệm phối hợp
cùng kháng sinh betalactam. Đối với hai khang sinh carbapenem là meropenem và imipenem,
tỷ lệ cac chủng phân lập được còn nhạy dưới 50%. Điều này gợi ý cần thực hiện mạnh mẽ
hơn nữa cac giải phap hướng đến bảo tồn nhóm khang sinh quan trọng này.
Về phác đồ carbapenem được sử dụng
Tỷ lệ carbapenem giữ vai trò trong phac đồ kinh nghiệm khởi đầu chiếm tới 58,8 %, lý
do có thể xuất phát từ lo ngại về mức độ nghiêm trọng của VPBV và xu hướng gia tăng đề
kháng của các vi khuẩn Gram âm. Tuy nhiên, tỷ lệ này chưa tương xứng với tỷ lệ làm vi sinh
trước khi dùng kháng sinh còn thấp (25,0 %), gợi ý cần có thêm các giải pháp quản lý sử dụng
kháng sinh trong thời gian tới về khía cạnh xét nghiệm vi sinh. Tỷ lệ PĐKN ban đầu phù hợp
chỉ đạt ở 5 % bệnh nhân. Kết quả này thấp hơn rất nhiều so với nghiên cứu của Nguyễn Bửu
Huy khi cùng đanh gia tính phù hợp của phac đồ kinh nghiệm với hướng dẫn của IDSA/ATS
2016, tuy nhiên cũng có thể giải thích do nghiên cứu này coi phac đồ là phù hợp khi ít nhất
một khang sinh được dùng phù hợp với khuyến cáo [4]. Các lý do không phù hợp ghi nhận
trong mẫu nghiên cứu của chúng tôi bao gồm: Thiếu KS phổ MRSA ở bệnh nhân có yếu tố
nguy cơ tử vong, thiếu 1 KS có phổ trên P.aeruginosa ở bệnh nhân có yếu tố nguy cơ mắc vi
khuẩn đa khang, phac đồ thừa 1 hoặc 2 KS có phổ trên P.aeruginosa ở bệnh nhân không có
yếu tố nguy cơ và ở bệnh nhân có yếu tố nguy cơ mắc vi khuẩn đa khang. Hiện nay, các
Hướng dẫn điều trị đều khuyến nghị cần phân tầng nguy cơ bệnh nhân dựa theo yếu tố nguy

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 9-15
14
cơ tử vong và/hoặc yếu tố nguy cơ mắc vi khuẩn đa khang để tiếp cận lựa chọn PĐKN.
Chúng tôi gợi ý cần tăng cường phổ biến thông tin tới bac sĩ về cac phac đồ điều trị cập nhật
và dữ liệu vi sinh tại bệnh viện để bac sĩ có căn cứ lựa chọn PĐKN khởi đầu phù hợp.
Ngoài ra, 24 lượt bệnh nhân (17,6 %) được chỉ định carbapenem trong phac đồ kinh
nghiệm để thay thế cho phac đồ khác với tỷ lệ phù hợp tới 75,0 %. Như vậy, vẫn còn 25,0 %
bệnh nhân có thể còn tối ưu hóa được các phac đồ khac trước khi dùng đến carbapenem.
Có 14 bệnh nhân (10,3 %) sử dụng carbapenem trong phac đồ điều trị theo đích vi
khuẩn. Tỷ lệ sử dụng phù hợp và không phù hợp lần lượt là là 21,4 % và 42,9 %. Đang chú ý,
nghiên cứu vẫn ghi nhận nhiều trường hợp không chọn kháng sinh phù hợp với diễn tiến lâm
sàng và kết quả kháng sinh đồ. Kết quả khảo sát cho thấy, một số trường hợp vi khuẩn vẫn
còn nhạy với khang sinh khac được khuyến cáo trong điều trị VPBV nhưng chưa được sử
dụng; ngược lại, trong một số trường hợp, carbapenem được lựa chọn, mặc dù khang sinh đồ
thể hiện vi khuẩn đã đề kháng mà vẫn nhạy cảm với kháng sinh khác. Cần có thêm các chiến
lược tăng cường áp dụng hiệu quả kết quả khang sinh đồ trong sử dụng kháng sinh hợp lý.
Trên nhóm được chỉ định carbapenem trong phac đồ kinh nghiệm, gần 50 % bệnh
nhân có thay đổi phac đồ. Khoảng 74 % bệnh nhân được đổi sang phac đồ giữ nguyên số
lượng kháng sinh hoặc thậm chí tăng số lượng khang sinh. Điều này có thể do tỷ lệ phac đồ
kinh nghiệm ban đầu chưa phù hợp tương đối cao do vậy bác sỹ tiếp tục điều chỉnh phac đồ
theo diễn biến lâm sàng hoặc theo kết quả khang sinh đồ trên bệnh nhân.
Về chế độ liều và cách dùng
Meropenem và imipenem là hai kháng sinh với tỷ lệ thải trừ qua thận ở dạng còn hoạt
tính tương đối cao, lần lượt là 70 % và khoảng 70 – 80 %. Do vậy, bên cạnh lựa chọn liều phù
hợp với chẩn đoan viêm phổi bệnh viện, chế độ liều của thuốc cần được hiệu chỉnh phù hợp,
đặc biệt khi bệnh nhân có suy giảm chức năng thận. Mẫu nghiên cứu ghi nhận tỷ lệ lượt bệnh
nhân dùng liều dùng phù hợp là 43,7 %, với riêng imipenem là 34,6 % và meropenem là 52,3
%. Hơn 50 % bệnh nhân sử dụng liều không phù hợp về cả khía cạnh liều dùng 1 lần và số lần
đưa thuốc trong ngày.
Tất cả bệnh nhân đều được truyền tĩnh mạch carbapenem. Hiện nay trong điều trị
VPBV, cac Hướng dẫn đều khuyến cáo truyền kéo dài 3 giờ để tối ưu hóa hiệu quả điều trị
thông qua tăng chỉ số T > MIC. Tuy nhiên, trong mẫu nghiên cứu, chủ yếu bệnh nhân được
truyền từ > 60 – 120 phút. Chỉ có 4,1 % bệnh nhân được truyền từ 3 giờ trở lên. Do vậy chế
độ liều và cách dùng của kháng sinh carbapenem cần được lưu ý cải tiến tốt hơn.
Về số ngày dùng kháng sinh và hiệu quả ra viện
Nhìn chung, số ngày sử dụng carbapenem nói riêng và kháng sinh nói chung tương đối
dài, trung bình là 11,7 và 19,6 ngày; so với liệu trình thông thường là 7 ngày. Kết quả ra viện
ghi nhận tỷ lệ bệnh nhân đỡ/giảm đạt dưới 50 %. Điều này có thể liên quan đến tình trạng
bệnh lý nặng, việc sử dụng phac đồ KSKN ban đầu chưa phù hợp, dẫn tới việc bệnh nhân cần
chuyển phac đồ nhiều lần trong qua trình điều trị. Trong nghiên cứu này, một số bệnh nhân
phải thay đổi tới 7 - 8 lần phac đồ khang sinh. Việc thay đổi phac đồ theo diễn biến lâm sàng
chiếm đến 79,4 %, trong khi đó theo khang sinh đồ chỉ là 12,4 %. Điều này có thể liên quan
đến thực trạng tỷ lệ cấy mẫu vi sinh thấp, dẫn tới thiếu căn cứ hướng dẫn thay đổi phac đồ.
Cac kết quả này càng nhấn mạnh tầm quan trọng của việc sử dụng phac đồ kinh nghiệm phù
hợp và tăng cường lấy mẫu vi sinh để kịp thời điều chỉnh phac đồ khang sinh, hướng tới giảm
số ngày dùng thuốc, giảm nguy cơ phat sinh cac chủng đa khang, tối ưu hóa hiệu quả điều trị.
Bên cạnh đó, đảm bảo sử dụng liều tối ưu, cach truyền tối ưu cũng có thể góp phần vào hiệu
quả tổng thể ở bệnh nhân.
Kết luận
Nghiên cứu đã phân tích được tính phù hợp trong việc sử dụng carbapenem ở 136
bệnh nhân mắc viêm phổi bệnh viện tại bệnh viện Đa khoa Hà Đông. Những điểm nổi bật
nhất ghi nhận được là tỷ lệ bệnh nhân được làm xét nghiệm vi sinh trước khi dùng kháng sinh
còn thấp (25,0 %); tỷ lệ sử dụng carbapenem trong phac đồ kinh nghiệm cao (76,4 %); tỷ lệ
phù hợp của phac đồ kinh nghiệm ban đầu thấp (5,0 %); dưới 50,0 % bệnh nhân dùng chế độ

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 9-15
15
liều phù hợp; 95,9 % bệnh nhân chưa được truyền kéo dài carbapenem và thời gian sử dụng
carbapenem và sử dụng kháng sinh trung bình tương đối dài so với thời gian điều trị kháng
sinh thông thường. Thực trạng này sẽ là căn cứ quan trọng cho việc xây dựng các chiến lược
trong chương trình quản lý kháng sinh, góp phần nâng cao chất lượng sử dụng thuốc tại bệnh
viện, bao gồm: tăng cường chỉ định hợp lý carbapenem kinh nghiệm, tăng cường lấy mẫu vi
sinh trước khi dùng kháng sinh, các chiến lược nhằm tối ưu hóa chế độ liều và thời gian
truyền thuốc.
TÀI LIỆU THAM KHẢO
1. Bộ Y Tế (2016), "Quyết định về việc ban hành tài liệu “Hướng dẫn thực hiện quản lý
sử dụng kháng sinh trong bệnh viện”"
2. Bộ Y tế (2015), Hướng dẫn sử dụng kháng sinh (ban hành kèm theo Quyết định số
708/QĐ-BYT ngày 02/3/2015)
3. Hội hô hấp Việt Nam – Hội hồi sức cấp cứu và chống độc Việt Nam (2017), Khuyến
cáo chẩn đoán và điều trị viêm phổi bệnh viện và viêm phổi thở máy, Nhà xuất bản Y
học
4. Nguyễn Bửu Huy (2018), Phân tích tình hình sử dụng kháng sinh trên bệnh nhân viêm
phổi bệnh viện/viêm phổi thở máy điều trị tại Khoa Hồi sức tích cực, Bệnh viện Đa
khoa Thành phố Cần Thơ, Trường đại học Dược Hà Nội, Hà Nội
5. Nguyễn Thanh Hiền (2012), Đánh giá việc sử dụng kháng sinh nhóm Carbapenem
trên bệnh nhân điều trị tại phòng hồi sức tích cực Bệnh viện Việt Đức, Trường Đại
học Dược Hà Nội, Hà Nội
6. Balkhy HH, Al Othman A, et al. (2015), "Consumption of carbapenems in different
intensive care units in a Saudi tertiary care hospital", Antimicrobial resistance and
infection control, 4(1), pp. P181.
7. EMA, "Thông tin sản phẩm Meronem", Retrieved, from
https://www.ema.europa.eu/en/documents/referral/meronem-article-30-referral-annex-
i-ii-iii_en.pdf.
8. EMA, "Thông tin sản phẩm Tienam", Retrieved, from
https://www.ema.europa.eu/en/documents/referral/tienam-article-30-referral-annex-
iii_en.pdf.
9. Kalil A. C., Metersky M. L., et al. (2016), "Management of Adults With Hospital-
acquired and Ventilator-associated Pneumonia: 2016 Clinical Practice Guidelines by
the Infectious Diseases Society of America and the American Thoracic Society", Clin
Infect Dis, 63(5), pp. e61-e111.
10. Rhodes A., Evans L. E., et al. (2017), "Surviving Sepsis Campaign: International
Guidelines for Management of Sepsis and Septic Shock: 2016", Crit Care Med, 45(3),
pp. 486-552.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 16-22
16
Tổng hợp acid (E)-4-dimethylaminobut-2-enoic hydroclorid làm chất trung
gian quan trọng cho điều chế afatinib dimaleat Nguyễn Thị Ngọc1,2, Nguyễn Hòa Bình1, Bùi Thị Thanh Châm2, Trần Hoàng Vũ,
Trần Anh Phương1, Đào Nguyệt Sương Huyền1, Nguyễn Văn Giang1,
Nguyễn Đình Luyện1, Nguyễn Văn Hải1* 1Trường Đại học Dược Hà Nội
2Trường Đại học Y Dược Thái Nguyên
*Tác giả liên hệ: [email protected]
(Ngày gửi đăng: 02/10/2020 – Ngày duyệt đăng: 09/12/2020 )
SUMMARY
Afatinib dimaleate is an effective second-generation EGFR tyrosine kinase inhibitors
(TKIs) approved for the first-line treatment of EGFR mutation-positive non-small cell
lung cancer (NSCLC). The current paper reports for the first time in Vietnam an efficient
and convenient synthesis of (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (1) as
one of the important intermediates in the synthetic routes of this drug, starting from the
commercially available crotonic acid (2). The overall yield has been improved by our
synthetic process up to 60.5 % which is higher than that of the published literature. Some
conditions for synthesis and purification, including: radical formation catalyst, molar
ratio of reactants, additive base and crystallization solvents have been improved. These
conditions have retained the original E-alken configuration. This simple synthetic
procedure with mild reaction conditions is useful for further optimizing and developing
synthesis process of afatinib.
Từ khóa: afatinib, acid (E)-4-bromobut-2-enoic, acid crotonic, acid (E)-4-
(dimethylamino)but-2-enoic
Đặt vấn đề
Afatinib dimaleat là thuốc chống ung thư tác dụng hướng đích nhóm ức chế tyrosin-
kinase (TKIs) [1], [2]. Thuốc có hiệu quả điều trị tốt, được chỉ định như là lựa chọn đầu
cho ung thư phổi tế bào không nhỏ có gen đột biến [3]. Tuy nhiên, giá thành của thuốc
này ở Việt Nam còn khá cao đối với đa số bệnh nhân (24,3 triệu đồng/20 gam nguyên liệu
afatinib dimaleat 99,0 % theo báo giá của Công ty thiết bị y tế và vật tư khoa học kỹ thuật
Nam Thành, từ 700 nghìn đồng/viên nén 40 mg [4]). Do đó, việc nghiên cứu phát triển
quy trình, từng bước tạo được nguyên liệu afatinib với giá thành hợp lý và công nghệ chủ
động trong nước là điều cần thiết. Cấu trúc afatinib có thể chia thành 4 hợp phần: nhân
chính quinazolin, 3 mạch nhánh ở các vị trí C-4, C-6 và C-7. Đáng chú ý là mạch nhánh
tại C-7 có cấu trúc alcol bất đối (S)-(tetrahydrofuran-3-yl)oxy và tại C-4 có cấu hình E-
alken 4-(dimethylamino)but-2-enamido. Chính vì thế, quy trình tổng hợp afatinib thường
khá dài (~ 9 bước) và việc xây dựng các mạch nhánh nói trên cũng phải được tiến hành
theo phương pháp phù hợp để tránh tạo tạp đồng phân [5], [6], [7], [8], [9]. Ở bài báo
trước, chúng tôi đã triển khai thành công tổng hợp (S)-3-hydroxytetrahydrofuran từ nguồn
nguyên liệu sẵn có acid L-malic với điều kiện phản ứng nhẹ nhàng, dễ thực hiện và sản
phẩm đạt yêu cầu ( = +15,15°, c = 3,33 trong methanol) để xây dựng hợp phần tại C-
7 trong phân tử afatinib [10]. Để xây dựng hợp phần (E)-4-(dimethylamino)but-2-enamido
tại C-4, nhiều tài liệu chỉ ra có 2 cách tiếp cận chính là: (a) sử dụng mạch E-alken có sẵn
sau đó tiến hành N-acyl hóa nhân quinazolin với tác nhân acid (i) hoặc clorid acid (ii)
hoặc ester [7], [11], [12], [13], [14]; (b) dùng phản ứng Horner-Wadsworth-Emmons của
carbanion phosphonat với aldehyd (iii) hoặc với acid α-hydroxysulfonic (iv) để tạo E-
alken một cách chọn lọc [14], [15] (xem Hình 1). Theo cách (a), quy trình bắt buộc phải
sử dụng mắt xích acid (E)-4-dimethylaminobut-2-enoic (dạng muối HCl) để ngưng tụ trực
tiếp tạo amid, hoặc để chuyển thành dạng clorid tương ứng (với thionyl clorid hoặc oxalyl
clorid). Tại Việt Nam, chưa có nghiên cứu nào về tổng hợp chất acid (E)-4-
dimethylaminobut-2-enoic. Bài báo này được thực hiện với mục tiêu tổng hợp được dạng

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 16-22
17
muối hydroclorid của acid (E)-4-dimethylaminobut-2-enoic (1) từ acid crotonic (2) ở quy
mô phòng thí nghiệm.
Hình 1. Các phương pháp xây dựng hợp phần (E)-4-(dimethylamino)but-2-enamido trong
cấu trúc afatinib
Nguyên liệu và phương pháp
Nguyên liệu
Acid acetic (99,5 %), acid crotonic (99,0 %), benzoyl peroxid (99,0 %), N-
bromosuccinimid (NBS, 99,0 %), carbon tetraclorid (99,5 %), dicloromethan (DCM, 99,5
%), dimethylamin (Me2NH, dung dịch nước 33 %), 4-dimethylaminopyridin (DMAP,
99,5%), ethanol (99,9%), ether dầu hỏa 70-90 (95 %), ethyl acetat (99,5 %), n-hexan (97
%), n-pentan (98 %), natri hydrocarbonat (99,5 %), natri sulfat khan (99,5 %), pyridin
(99,0 %), tetrahydrofuran (THF, 99,9 %), toluen (99,9 %), triethylamin (TEA, 99,0 %)
được mua từ hãng Xilong (Trung Quốc). Các hóa chất được sử dụng trực tiếp, không qua
tinh chế.
Phương pháp nghiên cứu
Tổng hợp các dẫn chất được thực hiện tại Phòng Tổng hợp hóa dược - Bộ môn Công
nghiệp Dược - Trường Đại học Dược Hà Nội qua 2 giai đoạn (3 phản ứng): bromo hóa
acid crotonic (2) với NBS thu được acid 4-bromocrotonic (acid (E)-4-bromobut-2-enoic,
3), N-alkyl hóa dimethylamin với 3 thu được acid (E)-4-dimethylaminobut-2-enoic, sau đó
tạo muối hydroclorid 1 (Hình 2).
Hình 2. Sơ đồ tổng hợp acid (E)-4-dimethylaminobut-2-enoic hydroclorid (1)
Theo dõi phản ứng bằng phương pháp sắc ký lớp mỏng (SKLM) sử dụng bản mỏng
silica gel 60 F254 (Merck) với hệ dung môi khai triển phù hợp. Quan sát vết sắc ký dưới
đèn UV 254 nm. Đo nhiệt độ nóng chảy (t°nc) bằng máy EZ-Melt (Mỹ). Sử dụng phương
pháp cất, chiết, kết tinh, lọc, rửa để tinh chế sản phẩm.
Xác định cấu trúc của sản phẩm tổng hợp bằng các phương pháp phổ: phổ khối lượng
(MS), phổ hồng ngoại (IR), phổ cộng hưởng từ hạt nhân (1H-NMR, 13C-NMR). Phổ MS
được ghi bằng máy Agilent 1100 LC-MSD Trap. Phổ IR được ghi bằng máy GX-Perkin-
Elmer với kỹ thuật viên nén KBr trong vùng 4000-400 cm-1. Phổ NMR được ghi trên máy
Bruker 500 MHz Ascend. Các phổ được đo tại Viện Hóa học - Viện Hàn lâm Khoa học và
Công nghệ Việt Nam và tại Khoa Hóa học - Trường Đại học Khoa học Tự nhiên, Đại học
Quốc gia Hà Nội.
Thực nghiệm và kết quả
Tổng hợp acid (E)-4-bromobut-2-enoic (3):
Hòa tan 2,00 g (23,2 mmol; 1 eq) acid crotonic (2) vào 10 mL carbon tetraclorid (CCl4)
trong bình cầu 2 cổ dung tích 100 mL có sinh hàn. Tiến hành khuấy từ và đun nóng đến
~77 °C. Bổ sung 6,20 g (34,8 mmol; 1,5 eq) NBS và 0,05 g (0,2 mmol; 0,01 eq) benzoyl
peroxid vào bình cầu. Duy trì đun hồi lưu hỗn hợp phản ứng trong 4 giờ. Kết thúc phản

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 16-22
18
ứng, làm lạnh khối phản ứng xuống 0-5 °C, lọc tách tủa, thu lấy dịch CCl4. Rửa tủa bằng 5
mL CCl4, gộp dịch lọc và dịch rửa, sau đó cất loại CCl4 thu được dịch đặc màu vàng.
Chiết dịch đặc 4 lần với n-hexan ấm (10 mL/lần, 50 °C). Gộp dịch chiết n-hexan, lọc
trong, sau đó cất loại dung môi còn khoảng ½ theo thể tích. Để kết tinh lạnh (0-5 °C) qua
đêm. Lọc thu lấy tinh thể thô màu trắng hơi vàng. Kết tinh lại sản phẩm trong khoảng 15
mL n-hexan. Sấy khô ở 45 °C thu được sản phẩm 3 là chất rắn kết tinh hình kim, màu
trắng có khối lượng 2,86 g (hiệu suất 74,5 %). t°nc 69,0-70,8 °C. Rf = 0,42 (DCM : AcOH,
50/1) và Rf = 0,69 (EtOAc : n-hexan, 1/1). ESI-MS (MeOH), m/z: 162,5 và 164,6 [M-H]-
(CTPT: C4H5BrO2; M = 164,99 đvC). IR (KBr), νmax (cm-1): 2920 (C-H no); 1704 (C=O);
1652 (C=C); 1291 (C-O); 974 (C-H trong trans-CH=CH-C=O); 689 (C-Br). 1H-NMR
(500 MHz, DMSO-d6), δ (ppm): 6,88-6,81 (1H, td, J1 = 7,5 Hz, J2 = 15,5 Hz, H-3); 6,05
(1H, dd, J1 = 6,0 Hz, J2 = 15,5 Hz, H-2); 4,26-4,23 (2H, m, H-4). 13C-NMR (125 MHz,
DMSO-d6), δ (ppm): 166,4 (C-1); 142,1 (C-3); 125,0 (C-2); 30,7 (C-4).
Tổng hợp acid (E)-4-dimethylaminobut-2-enoic (1):
Cho 2,00 g (12,1 mmol; 1 eq) chất 3 vào bình cầu 1 cổ 100 mL, thêm 15 mL THF và
4,5 mL (330,0 mmol; 27,5 eq) TEA. Sau đó, làm lạnh khối phản ứng về 0-5 °C, nhỏ từ từ
2,0 mL (14,7 mmol; 1,25 eq) dung dịch dimethylamin 33 % (trong nước). Đưa phản ứng
về nhiệt độ phòng và khuấy qua đêm (18 giờ). Kết thúc phản ứng, thêm 5 mL NaHCO3
bão hòa (đến pH ~ 8) rồi tiến hành cất loại dung môi ở 70 °C thu được chất rắn. Thêm tiếp
10 mL THF vào bình cầu và cất loại dung môi để loại bỏ hoàn toàn amin tồn dư (TEA và
dimethylamin). Hòa tan cắn thu được trong 5 mL nước cất, acid hóa với dung dịch HCl
2M đến pH = 2-3. Rửa pha nước với ethyl acetat (3 lần x 10 mL). Cất quay dịch nước ở
70 °C đến khi còn khoảng 5 mL, thêm 20 mL THF vào để kết tủa sản phẩm. Lọc lấy chất
rắn. Kết tinh lại sản phẩm bằng cách hòa tan chất rắn trong 5 mL nước cất, thêm dần THF
đến tạo mầm tinh thể, để kết tinh qua đêm. Lọc lấy tinh thể, sấy khô ở nhiệt độ 40 °C
trong tủ sấy chân không. Sản phẩm 1 thu được là chất rắn màu trắng có khối lượng 1,63 g
(hiệu suất 81,2 %). t°nc 159,0-160,3 °C. Rf = 0,41 (DCM : EtOH, 5/1). IR (KBr), νmax (cm-
1): 2947 (C-H no); 2668 và 2481 (NH+, dạng muối của amin bậc ba); 1719 (C = O); 1668
(C = C); 977 (C-H trong hệ trans-CH=CH-C=O). ESI-MS (MeOH), m/z: 130,03 [M-Cl]+
(CTPT: C6H11O2N.HCl; M = 165,62 đvC). 1H-NMR (500 MHz, DMSO-d6), δ (ppm):
6,81 (1H, td, J1 = 7,0 Hz, J2 = 16,0 Hz, H-3); 6,18 (1H, d, J = 15,5 Hz, H-2); 3,89 (2H, dd,
J1 = 1,0 Hz, J2 = 6,0 Hz, H-4); 2,50 (6H, s, H-5, H-6). 13C-NMR (125 MHz, DMSO-d6), δ
(ppm): 165,9 (C-1); 135,7 (C-3); 129,5 (C-2); 56,2 (C-4); 41,7 (C-5, C-6).
Các kết quả đã khảo sát
Sau phân tích các tài liệu, chúng tôi lựa chọn dung môi phản ứng tổng hợp 3 từ 2 là
carbon tetraclorid, nhiệt độ phản ứng là hồi lưu (77 °C) theo tài liệu [25], thay tác nhân
AIBN bằng benzoyl peroxid, khảo sát ảnh hưởng tỷ lệ mol và dung môi tinh chế, đánh giá
hiệu suất phản ứng và nhiệt độ nóng chảy. Kết quả khảo sát được thể hiện trong bảng 1 và
2.
Bảng 1. Kết quả khảo sát ảnh hưởng của tỷ lệ mol NBS : 2
TT Tỷ lệ mol NBS : 2 m sản phẩm 3 (g) Hiệu suất (%) t°nc (°C)
1 1,1 : 1 1,79 46,7 68,9-70,4
2 1,3 : 1 2,40 62,5 68,8-70,2
3 1,5 : 1 2,86 74,5 69,0-70,8
4 1,7 : 1 2,85 74,2 68,4-71,9
Nhận xét: Khi tăng tỷ lệ mol NBS : 2, từ 1,1 : 1 đến 1,5 : 1, hiệu suất phản ứng tăng,
sản phẩm có màu trắng, tất cả đều đạt khoảng t°nc. Tuy nhiên, khi vượt quá tỷ lệ mol 1,5 :
1 thì hiệu suất phản ứng không tăng nhiều, đồng thời trên SKLM quan sát thấy nhiều tạp
hơn, sản phẩm có cảm quan màu trắng hơi vàng. Tỷ lệ mol NBS : 2 = 1,5 : 1 cho kết quả
tốt nhất.
Bảng 2. Kết quả khảo sát dung môi tinh chế chất 3

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 16-22
19
TT Dung môi tinh chế m sản phẩm 3 (g) Hiệu suất
(%) t°nc (°C)
1 Ether dầu hỏa 2,69 70,1 68,0-70,9
2 n-Pentan 2,38 62,0 68,8-70,6
3 n-Hexan 2,86 74,5 69,0-70,8
4 DCM 2,10 54,7 65,2-69,7
5 Toluen 2,84 74,0 69,3-70,5
Nhận xét: Sử dụng n-hexan cho hiệu suất tốt nhất (74,5 %), tinh khiết theo t°nc. Sử
dụng toluen theo tài liệu [25] cho kết quả tương đương (74 %) so với n-hexan, song khó
cất loại hơn. So với các dung môi, DCM hòa tan sản phẩm kém chọn lọc, nên cho hiệu
suất thấp (54,7 %) và sản phẩm chứa nhiều tạp hơn.
Kết quả khảo sát đối với phản ứng tổng hợp 1 từ 3 được ghi trong bảng 3 và 4.
Bảng 3. Kết quả khảo sát ảnh hưởng của base bổ trợ
TT Base m sản phẩm 1 (g) Hiệu suất (%) t°nc (°C)
1 NaOH 10% (pH 9) -*
2 TEA 1,63 81,2 159,0-160,3
3 DMAP 1,58 79,7 154,1-158,1
4 Pyridin 1,30 64,8 153,0-156,6
*: không thu được sản phẩm sau xử lý.
Nhận xét: Sử dụng NaOH làm khối phản ứng biến màu nhanh và khó xử lý, không thu
được sản phẩm dạng bột. Sử dụng pyridin và DMAP cho hiệu suất thấp hơn so với TEA,
sản phẩm cũng kém tinh khiết hơn theo SKLM và t°nc.
Bảng 4. Kết quả khảo sát ảnh hưởng của tỷ lệ mol dimethylamin : 3
TT Tỷ lệ mol Me2NH : 3 m sản phẩm 1 (g) Hiệu suất (%) t°nc (°C)
1 1,0 : 1 1,45 72,4 159,1-160,5
2 1,25 : 1 1,63 81,2 159,0-160,3
3 1,5 : 1 1,58 78,9 158,2-160,1
4 1,75 : 1 1,57 78,3 158,1-160,4
Nhận xét: Tỷ lệ mol Me2NH : 3 = 1,25 : 1 cho hiệu suất phản ứng tốt nhất. Khi sử dụng
dimethylamin quá dư, xuất hiện vết tạp màu do oxi hóa amin, dẫn đến kéo dài thời gian xử
lí loại bỏ amin, hiệu suất phản ứng có xu hướng giảm.
Bàn luận
Về lựa chọn phương pháp tổng hợp chất 1
Phân tích các tài liệu tham khảo, chúng tôi nhận thấy rằng có 5 con đường (pp1-5) tổng
hợp chất 1, đi từ 4 nguyên liệu là: acid crotonic (2), ethyl 4-chloro-3-hydroxybutanoat (4),
natri dimethylamino-1-hydroxy-ethansulfonat (5) và alkyn 6 (xem hình 3) [6], [16], [17],
[18], [19], [20], [21], [22], [23], [24].

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 16-22
20
Hình 3. Các phương pháp tổng hợp 1 theo tài liệu tham khảo
Phương pháp pp5 đi từ alkyn 6 khó cho sản phẩm chọn lọc E-alken, mà chủ yếu cho
đồng phân Z-alken [24]. Quy trình pp4 đi từ nguyên liệu 5 tuy cho hiệu suất tốt, sản phẩm
tương đối tinh khiết, song sử dụng tác nhân phosphonat (phản ứng Horner-Wadsworth-
Emmons) có giá thành cao, và khó mua tại Việt Nam (theo báo giá của các công ty hóa
chất trong nước) [23]. Phương pháp pp3 từ nguyên liệu 4 khó nâng cấp do có nhiều giai
đoạn, đòi hỏi tinh chế trung gian ethyl 4-bromocrotonat phức tạp để đạt được sản phẩm
cuối tinh khiết [6], [19]. Từ nguyên liệu acid crotonic (2), có 2 con đường tổng hợp, trong
đó con đường pp2 sử dụng bromo hóa sau khi đã ester hóa acid crotonic (bằng các nhóm
methyl, ethyl, tert-butyl, trimethylsilyl) giúp cải tiến được hiệu suất, song số bước tổng
hợp là 4, tinh chế phức tạp nên hiệu suất toàn phần cũng chưa cao [6], [16], [17], [18],
[19], [20]. Con đường bromo hóa trực tiếp acid crotonic (2), sau đó N-alkyl hóa tạo sản
phẩm 1 (con đường pp1) có ưu điểm là: nguyên liệu 2 có sẵn trên thị trường, giá thành
thấp, dễ mua, dễ bảo quản, quy trình tổng hợp chỉ gồm 2 giai đoạn [25]. Tuy nhiên, tài
liệu công bố hiệu suất toàn phần mới chỉ đạt 20,2 % [25]. Trên cở sở đó, chúng tôi đã lựa
chọn phương pháp này để tiến hành các khảo sát thực nghiệm, từ đó phân tích, lựa chọn
thông số phù hợp nhằm nâng cao hiệu suất sản phẩm.
Về phản ứng tổng hợp acid (E)-4-bromobut-2-enoic (3)
Phản ứng tổng hợp chất 3 là phản ứng brom hóa acid crotonic với tác nhân NBS theo
cơ chế gốc tự do với sự có mặt của xúc tác tạo gốc trong dung môi carbon tetraclorid.
Phản ứng thế xảy ra tương đối chọn lọc tại vị trí C-4 do C-4 thuộc cấu trúc allyl. Tài liệu
[25] sử dụng azobis(isobutyronitril) (AIBN) làm xúc tác tạo gốc cho hiệu suất 56 %, tuy
nhiên tác giả không đưa ra đủ dữ liệu khẳng định cấu hình trans. Tuy AIBN có khả năng
xúc tác mạnh, cho thời gian phản ứng tương đối ngắn (5 giờ), song AIBN có giá thành khá
cao, khó bảo quản và sinh tạp tetramethylsuccinonitril với độc tính cao. Vì vậy, trong quá
trình nghiên cứu, chúng tôi đã lựa chọn xúc tác là benzoyl peroxid có giá thành thấp hơn
và ít độc hơn. Những quan sát bước đầu của chúng tôi khi làm phản ứng này cho thấy, tốc
độ phản ứng chậm do acid crotonic tan kém trong dung môi CCl4 và NBS bị phân hủy
theo thời gian. Để tốc độ phản ứng đạt tối đa, chúng tôi tiến hành ở nhiệt độ sôi của hỗn
hợp (~ 77 °C) [25] và khảo sát tăng tỷ lệ mol NBS so với acid crotonic. Khi tăng tỷ lệ mol
NBS : 2 hiệu suất phản ứng tăng, tuy nhiên khi vượt quá tỷ lệ 1,5 : 1 thì hiệu suất phản
ứng chỉ tăng không đáng kể, đồng thời quan sát thấy nhiều tạp phân hủy của NBS
(succinimid, brom tự do), từ đó làm giảm hiệu suất tinh chế sản phẩm (bảng 1). Do đó,
chúng tôi đã lựa chọn tỷ lệ mol NBS : 2 tốt nhất là 1,5 : 1. Ở tỷ lệ này, thời gian phản ứng
kéo dài (hơn 4 giờ) sẽ dẫn đến nhiều tạp (xuất hiện trên SKLM), bao gồm succinimid, tạp
màu (do brom hòa tan) và tạp do sự kết hợp của các gốc tự do ở giai đoạn tắt mạch theo
cơ chế gốc. Đáng chú ý là, chúng tôi đã tìm được dung môi n-hexan ở nhiệt độ 50 °C có

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 16-22
21
khả năng hòa tan sản phẩm 3, trong khi đó, ở nhiệt độ lạnh (0-5 °C) sản phẩm 3 lại kém
tan, hầu hết các tạp nói trên tan tốt. Các dung môi còn lại cho kết quả kém hơn khi thử ở
điều kiện tương tự (chiết nóng sau đó để lạnh). Vì vậy, n-hexan được chọn làm dung môi
chiết nóng cũng như kết tinh sản phẩm ở nhiệt độ lạnh (bảng 2). Qua đó, chúng tôi đã thu
được sản phẩm có độ sạch và hiệu suất đạt 74,5 %, cao hơn so với tài liệu đã công bố
bằng con đường tương tự (pp1; 20,2 %) [25].
Về tổng hợp acid (E)-4-dimethylaminobut-2-enoic (1)
Phản ứng N-alkyl hóa dimethylamin bằng tác nhân 3 sinh ra sản phẩm phụ là acid
mạnh HBr, có khả năng tạo muối với nguyên liệu amin ban đầu, làm giảm khả năng ái
nhân của dimethylamin. Vì vậy, trong quy trình, chúng tôi đã khảo sát một số base làm
chất bổ trợ (additive reagent) để đảm bảo nguyên liệu dimethylamin ở dạng tự do, nâng
khả năng phản ứng của nguyên liệu, từ đó đã lựa chọn được TEA (bảng 3). Ngoài ra, việc
dùng TEA còn có ưu điểm là dễ dàng cất loại hơn so với pyridin (nhiệt độ sôi 115 °C) và
DMAP (dạng rắn). Khi xử lý, chúng tôi dùng NaHCO3 nhằm mục đích chuyển hết các
muối amin về dạng amin tự do, qua đó các amin (TEA/pyridin và dimethylamin dư) được
cất loại triệt để dưới dạng bay hơi cùng dung môi THF, làm cho quá trình kết tinh sản
phẩm về sau được dễ dàng hơn. Bên cạnh đó, cần chú ý sử dụng hợp lý tỷ lệ mol
dimethylamin : 3 là 1,25 : 1 (bảng 4). Dùng quá thừa amin gây kéo dài thời gian cất loại,
tốn kém nguyên liệu và tăng tạo tạp màu (oxy hóa amin) khi xử lý phản ứng. Giá trị pH 2-
3 để tạo muối với HCl 2M được lựa chọn dựa trên tài liệu [25]. Các tạp được loại đi nhờ
quá trình rửa với ethyl acetat, kế tiếp kết tinh trong THF/nước cũng là hệ dung môi được
chúng tôi khảo sát và lựa chọn mà chưa có trong tài liệu công bố. Sản phẩm thu được đạt
tinh khiết SKLM và t°nc. Hiệu suất đạt 81,2 % cao hơn so với tài liệu đã công bố.
Như vậy, hiệu suất toàn quy trình tổng hợp 1 từ acid crotonic đạt 60,5 %, cao hơn so
với tài liệu công bố ở trên. Trong đó, đã lựa chọn được một số điều kiện cho giai đoạn
bromo hóa (xúc tác tạo gốc là benzoyl peroxid; tỷ lệ mol NBS : 2 là 1,5 : 1; dung môi kết
tinh là n-hexan); cho giai đoạn N-alkyl hóa (tỷ lệ mol dimethylamin : 3 là 1,25 : 1; sử
dụng base bổ trợ là TEA; base trung hòa là NaHCO3), lựa chọn dung môi kết tinh sản
phẩm cuối là hỗn hợp THF/nước. Thao tác tiến hành trong quy trình khá đơn giản, điều
kiện phản ứng nhẹ nhàng. Các dữ liệu phổ IR, MS, NMR đã khẳng định đúng cấu trúc của
các chất trung gian và sản phẩm cuối, tương tự như các tài liệu đã công bố [17],[18], [25].
Trong đó, giá trị hằng số tương tác J của proton H-2 và H-3 trong sản phẩm là 15,5-16,0
Hz, không có sự xuất hiện của tạp đồng phân trên NMR. Điều này chỉ ra, các điều kiện
của quy trình đã bảo toàn được cấu hình E-alken ban đầu. Đây là những tiền đề để tiếp tục
tối ưu hóa, nâng cấp quy trình điều chế afatinib đưa vào ứng dụng trong nước.
Kết luận
Chúng tôi đã tổng hợp được sản phẩm acid (E)-4-dimethylaminobut-2-enoic hydrolorid
từ nguồn nguyên liệu acid crotonic ở quy mô phòng thí nghiệm qua 2 giai đoạn với hiệu
suất toàn phần đạt 60,5 %, cao hơn so với các tài liệu đã công bố. Trong đó, đã lựa chọn
được một số điều kiện phù hợp về tổng hợp và tinh chế, bao gồm: xúc tác tạo gốc, tỷ lệ
mol các chất phản ứng, base bổ trợ và dung môi kết tinh. Cấu trúc các chất được khẳng
định bằng các phân tích phổ IR, MS, 1H-NMR và 13C-NMR. Các điều kiện của quy trình
đã bảo toàn được cấu hình E-alken ban đầu.
TÀI LIỆU THAM KHẢO 1. Joint Formulary Committee (2020), “Afatinib”, British National Formulary (BNF) 79,
London: BMJ Group and Pharmaceutical Press, pp. 998-999.
2. Shah R. and Lester J.F. (2020), “Tyrosine kinase inhibitors for the treatment of EGFR
mutation-positive non-small-cell lung cancer: A clash of the generations”, Clin. Lung Cancer,
21(3), pp. e216-e228.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 16-22
22
3. Holleman M.S., van Tinteren H., Groen H.J. et al. (2019), “First-line tyrosine kinase
inhibitors in EGFR mutation-positive non-small-cell lung cancer: a network meta-analysis”,
Onco. Targets Ther., 12, pp. 1413-1421.
4. https://drugbank.vn/.
5. Himmelsbach F., Langkopf E., Blech S. et al. (2002), “Quinazoline derivatives,
medicaments containing said compounds, their utilization and method for the production
thereof”, PCT Patent, WO200250043A1.
6. Choi Y.-H., Chang W.-T. (2010), “Method for producing an alkyl amine derivative”,
PCT Patent, WO2010131921.
7. Schroeder J., Dziewas G., Fachinger T. et al. (2012), “Process for preparing
aminocrotonylamino-substituted quinazoline derivatives”. U.S. patent, US8188274B2.
8. Xu X. (2013), “Afatinib preparation method”, CN Patent, CN103242303A.
9. Kovacevic T., Mesic A., Avdagic A. et al. (2018), “An alternative synthesis of the
non-small cell lung carcinoma drug afatinib”, Tetrahedron Letters, 59(47), pp. 4180-4182.
10. Nguyễn Thị Ngọc, Chu Hà Phương, Nguyễn Văn Giang, Nguyễn Đình Luyện, Nguyễn
Văn Hải (2019), “Bước đầu nghiên cứu tổng hợp (S)-3-hydroxytetrahydrofuran”, Tạp chí Hóa
học, 57(6E1,2), tr. 136-139.
11. Zhang Q., Zhu H. (2011), “Novel quinazoline derivatives”, PCT Patent,
WO201184796A2.
12. Slobbe P., Poot A. J., Van Dongen A. A. M. S. et al. (2015), “Radiolabeled
quinazoline derivatives”, U.S. Patent, US20150368230A1.
13. Liu Y., Liu X., Liu L. (2019), “A kind of Afatinib highly finished product synthetic
method”, CN Patent, CN109776514A.
14. Tuskar M., Ratkaj M., Zegarac M. (2015), “Crystalline forms of afatinib dimaleate”,
PCT Patent, WO2015103456A1.
15. Guo Z. (2016), “A kind of method that high selectivity prepares maleic acid afatinib”,
CN Patent, CN106243092B.
16. Yang C., Rao Z., Bai C. et al. (2018), “A kind of felodipine class compound and its
application”, CN Patent, CN107556289A.
17. Hong J., Xu X., Yue X. (2016), “Pyrazolopyrimidine derivative, preparation method,
pharmaceutical composition and purposes”, CN Patent, CN106146511A.
18. Cha M.Y., Kim M.R., Kang S.J., et al. (2011), “Novel pyrimidine derivative for
inhibiting the growth of cancer cells”, PCT Patent, WO2011099764A2.
19. Zheng J., Deng D., Guanghua L.V. et al. (2015), “Process for the manufacture of (E)-
4-N,N-dialkylamino crotonic acid in HX salt form and use thereof for synthesis of EGFR
tyrosine kinase inhibitors”, U.S. patent, US20150183764A1.
20. Considine J. L., Daigneault S., Chew W. et al. (2006), “Synthesis of 4-(amino)-2-
butenoyl chlorides and their use in the preparation of 3-cyano quinolines”, U.S. patent,
US7126025B2.
21. Verma S. S., Singh S. K., Singh K. et al. (2015), “Process for the preparation of 4-
dimethylaminocrotonic acid”, PCT patent, WO2015186065A1.
22. Liu Z., Feng Q., Zou X. et al. (2015), “Method for preparing highly pure trans-4-
dimethylamino crotonic acid hydrochloride through one-pot technology”, CN Patent,
CN105669479A.
23. Tian D., He L. (2016), “Trans-4-dimethylaminocrotonic acid hydrochloride
preparation method”, CN Patent, CN105439879A.
24. Lee K.-O., Cha M.Y., Kim M.R., et al. (2008), “Novel amide derivative for inhibiting
the growth of cancer cells”, PCT Patent, WO2008150118A2.
25. Tao J. (2017), “Chemical synthesis method of Maihuatinib (EGFR/HER2 high-
efficient dual inhibitor)”, CN Patent, CN106432105A.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 23-30
23
Nghiên cứu quy trình bào chế viên nang mềm chứa hệ nano
tự nhũ hóa rosuvastatin Vũ Thị Thu Giang1, Phan Thị Nghĩa1,2, Nguyễn Thị Thúy Nga1,3,
Nguyễn Thị Linh1, Trần Thị Hải Yến1, Phạm Bảo Tùng1, Nguyễn Đăng Hòa1*
1 Trường Đại học Dược Hà Nội 2 Viện Kiểm nghiệm thuốc Trung ương
3 Trường Đại học Điều dưỡng
*Tác giả liên hệ: [email protected]
(Ngày gửi đăng: 06/10/2020 – Ngày duyệt đăng: 09/12/2020)
SUMMARY The influence of technical parameters in the preparing procedure of capsule shell
fluid, capsule shell, and soft gelatin capsules containing rosuvastatin self-nano
emulsifying drug delivery system (SNEDDS) was investigated. The studied technical
parameters included swelling time, temperature, and dissolving time of gelatin;
incubation condition; the temperature of congelation and sealing; drying time. The
obtained results showed that the selected parameters ensured the preparation procedure's
stability and the quality standards of capsule shell solution, capsule shell, and soft gelatin
capsules containing rosuvastatin SNEDDS. The selected technical parameters were as
following: swelling time, temperature, and dissolving time of gelatin were was 30 – 45
minutes, 60 – 70 oC and 1.5 – 2.0 hours respectively; incubation condition was at 55 – 60 oC and in 12 – 16 hours; the temperature of congelation and sealing were 18 – 20 oC and
40 – 41 oC respectively; drying time was 16 – 20 hours.
Từ khóa: nang mềm, gelatin, rosuvastatin, hệ nano tự nhũ hóa (SNEDDS).
Đặt vấn đề
Trong nghiên cứu trước đây, công thức bào chế vỏ nang mềm chứa hệ nano tự nhũ
hóa (SNEDDS) rosuvastatin đã được xây dựng dựa trên kết quả khảo sát ảnh hưởng của
các thành phần như: tỷ lệ và loại gelatin, tỷ lệ và loại chất hóa dẻo, tương tác giữa màng
vỏ nang và dịch thuốc đóng nang, đánh giá các đặc tính của dịch vỏ nang, màng vỏ nang.
Từ kết quả nghiên cứu thu được cũng như một số nghiên cứu đã công bố khác cho thấy có
nguy cơ xảy ra tương tác giữa vỏ nang mềm với SNEDDS rosuvastatin đóng trong nang
dẫn đến kéo dài thời gian hòa tan màng vỏ nang, ảnh hưởng đến quá trình giải phóng dược
chất [1], [5]. Nghiên cứu đã chọn được tỷ lệ và loại gelatin, tỷ lệ và loại tá dược hóa dẻo
thích hợp có khả năng hạn chế được tương tác giữa vỏ nang và dịch thuốc đóng nang. Vì
vậy, nghiên cứu này được tiếp tục thực hiện với mục tiêu xây dựng được quy trình bào
chế viên nang mềm SNEDDS rosuvastatin 10 mg.
Nguyên liệu, thiết bị và phương pháp nghiên cứu
Nguyên liệu
Calci rosuvastatin (Ros) (TCNSX – Enaltec, Ấn độ); Capryol 90 (EP – Gattefossé,
Pháp); Cremophor RH 40 và polyoxyethylen glycol 400 (PEG 400) (EP – BASF, Đức);
gelatin 150 BL (loại B, TCNSX - NITTA gelatin InC, Nhật); dung dịch sorbitol 70 %
(EP/USP – Roquette, Pháp); glycerin, methyl paraben và propyl paraben (TCNSX, Trung
Quốc); acetonitril và methanol (HPLC – Fisher, Mỹ). Các thuốc thử khác đều đạt tiêu
chuẩn tinh khiết phân tích, mua từ Beijing, Trung Quốc).
Thiết bị nghiên cứu
Thiết bị Texture Analyzer CT3 1500 (Mỹ), Máy đo độ nhớt DVE Viscometer
Brookfield (Mỹ), Bể điều nhiệt Memmert (Đức), Máy khuấy từ IKA (Đức), Tủ làm mát
Kangaroo (Việt Nam), thiết bị đo độ dày màng (chính xác đến 0,01 mm, Thượng Hải),
Máy hút chân không (Trung Quốc), khuôn nhúng tạo vỏ nang (0,4 ml), thiết bị sấy nang
mềm Drymax temperature and RH control unit DMTH (Canada), thiết bị hàn nang thủ
công, thiết bị cán tạo vỏ nang thủ công, …

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 23-30
24
Phương pháp nghiên cứu
Phương pháp bào chế viên nang mềm
Trên cơ sở các kết quả nghiên cứu đã công bố [1], viên nang mềm SNEDDS
rosuvastatin được bào chế theo công thức như sau:
Bảng 1. Công thức mẻ 300 viên nang mềm SNEDDS rosuvastatin
Vỏ nang Dịch nhân
Thành phần Khối lượng (g) Thành phần Khối lượng (g)
Gelatin 86,00 Rosuvastatin calci 3,12
Glycerin 13,26 Cremophor RH 40 49,09
Sorbitol 26,62 Capryol 90 49,09
Methyl paraben 0,18 PEG 400 18,70
Propyl paraben 0,02
Nước 73,92
- Bào chế dịch vỏ nang:
• Hòa tan các paraben, glycerin và sorbitol trong nước nóng trên 80 °C.
• Ngâm trương nở gelatin trong dung dịch thu được trong thời gian thích hợp rồi
đun cách thủy và khuấy hòa tan.
• Loại bọt khí trong dịch vỏ nang trong điều kiện hút chân không duy trì áp suất – 0,6
đến – 0,4 Mpa
• Ủ dịch dịch gelatin ở nhiệt độ thích hợp đến trong.
- Bào chế vỏ nang:
Nhúng khuôn ngập dịch vỏ nang kết hợp xoay để vỏ nang bám đều lên bề mặt
khuôn, nhấc khuôn ra. Làm đông đặc vỏ nang ở nhiệt độ thích hợp sau đó bóc vỏ nang ra
khỏi khuôn.
- Đóng thuốc vào nang:
Dùng bơm tiêm hút và bơm SNEDDS rosuvastatin vào đầy vỏ nang rồi hàn kín
viên nang bằng máy hàn nang thủ công.
- Làm khô viên nang:
Viên được làm khô ở nhiệt độ 21 - 24 °C, độ ẩm 25 - 30 % đến khi độ ẩm vỏ viên nang
đạt 9 – 14 %.
Các thông số kỹ thuật trong quá trình bào chế viên nang mềm như thời gian ngâm
trương nở, nhiệt độ và thời gian hòa tan gelatin, nhiệt độ và thời gian ủ dịch vỏ nang, nhiệt
độ làm đông đặc và hàn kín vỏ nang, thời gian sấy khô nang được xác định thông qua
nghiên cứu khảo sát ảnh hưởng của các thông số này đến đặc tính của dịch vỏ nang, màng
vỏ nang và viên nang mềm bào chế được.
Phương pháp đánh giá
- Đánh giá tương tác giữa vỏ nang mềm và SNEDDS rosuvastatin đóng nang:
Thiết bị đánh giá: Máy Nicolet Thermo IS50, chế độ đo phổ FTIR. Mẫu đánh giá
là vỏ nang trước khi đóng dịch nhân và vỏ viên nang chứa SNEDDS rosuvastatin sau 2
tháng bảo quản ở điều kiện thường. Phổ FTIR của các mẫu được tiến hành chồng phổ để
phân tích sự tương tác giữa SNEDDS rosuvastatin và vỏ nang.
- Đánh giá SNEDDS rosuvastatin:
SNEDDS rosuvastatin được đánh giá các chỉ tiêu: Hàm lượng dược chất, kích
thước giọt (KTG) và phân bố kích thước (PDI), tỷ lệ rosuvastatin được nano nhũ hóa và
độ ổn định vật lý theo các phương pháp đã công bố trong nghiên cứu bào chế hệ nano tự
nhũ hóa rosuvastatin [2].
- Đánh giá dịch vỏ nang:
Dịch vỏ nang được đánh giá các đặc tính về tính chất, độ nhớt và độ bền gel theo
các phương pháp đã công bố trong nghiên cứu xây dựng công thức vỏ nang mềm chứa hệ
nano tự nhũ hóa rosuvastatin [1].
- Đánh giá màng vỏ nang:

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 23-30
25
Màng vỏ nang được đánh giá các chỉ tiêu độ ẩm, độ dày và độ đồng đều, lực kéo
đứt và khả năng giãn dài, khả năng hòa tan theo các phương pháp đã công bố [1].
- Đánh giá viên nang mềm SNEDDS rosuvastatin:
• Định lượng bằng phương pháp HPLC [1].
• Độ hòa tan [1].
• Độ đồng đều hàm lượng: theo phương pháp 1, phụ lục 11.2 trong DĐVN V.
• Độ rã: tiến hành trong môi trường nước theo phụ lục 11.6 của DĐVN V
Phương pháp xử lý số liệu
Các kết quả thu được sẽ được xử lý thống kê với sự hỗ trợ của phần mềm Excel
2010 và được trình bày dưới dạng TB ± SD hoặc TB ± RSD trong đó TB là giá trị trung
bình, SD là độ lệch chuẩn, RSD là độ lệch chuẩn tương đối.
Kết quả nghiên cứu Quy trình bào chế viên nang mềm SNEDDS rosuvastatin 10 mg được xây dựng
trên cơ sở khảo sát ảnh hưởng của các thông số kỹ thuật trong quá trình bào chế đến đặc
tính của viên nang mềm SNEDDS rosuvastatin để lựa chọn ra các thông số phù hợp nhất.
Kết quả thu được như sau:
Ảnh hưởng của thời gian ngâm trương nở gelatin
Tiến hành ngâm trương nở gelatin trong các khoảng thời gian 15 phút (G15), 30
phút (G30) và 45 phút (G45). Cố định nhiệt độ hòa tan và ủ dịch vỏ nang sau hòa tan là 60 oC. Kết quả đánh giá được trình bày trong bảng 2.
Bảng 2. Ảnh hưởng thời gian ngâm trương nở gelatin đến đặc tính của dịch vỏ và màng vỏ
nang (n=3, TB ± SD)
Tiêu chí Mẫu dịch và màng vỏ tạo thành
G15 G30 G45
Thời gian hòa tan gelatin (giờ) 2,5 2,25 2,25
Thời gian ủ để dịch trong (giờ) 15 14 14
Độ nhớt (cP) 160,0 ± 1,1 152,0 ± 1,1 150 ± 1,1
Độ Bền gel (g) 325,4 ± 0,1 365,0 ± 0,1 361,1 ± 0,1
Độ dày màng (mm) 0,33 ± 0,04 0,36 ± 0,03 0,36 ± 0,03
Thời gian hòa tan màng (phút) 39,7 ± 0,5 40,3 ± 0,5 39,3 ± 0,9
Lực kéo đứt (N) 6,9 ± 0,3 10 ± 0,1 10,2 ± 0,1
Kết quả nghiên cứu cho thấy thời gian ngâm trương nở gelatin có ảnh hưởng tới
đặc tính dịch vỏ nang và màng vỏ nang. Khi tăng thời gian ngâm trương nở gelatin, độ
bền gel của dịch vỏ nang và lực kéo đứt có xu hướng tăng trong khi độ nhớt dịch vỏ
gelatin, thời gian hòa tan gelatin và thời gian để dịch hết bọt lại có xu hướng giảm. Đặc
tính của các mẫu dịch và màng vỏ nang G30 và G45 khác nhau không đáng kể nhưng có
thời gian để dịch trong ngắn hơn, lực kéo đứt lớn hơn so với mẫu G15. Trên cơ sở kết quả
nghiên cứu thu được, thời gian ngâm trương nở gelatin trong khoảng 30 - 45 phút được
lựa chọn.
Ảnh hưởng của nhiệt độ hòa tan gelatin
Tiến hành ngâm trương nở gelatin trong 30 phút. Khảo sát nhiệt độ hòa tan gelatin
là 60 oC (M60), 70 oC (M70), 80 oC (M80). Cố định nhiệt độ ủ dịch vỏ nang sau hòa tan là
60 oC. Kết quả đánh giá một số đặc tính của dịch vỏ nang và màng vỏ nang được trình bày
trong hình 1 và bảng 3.
Kết quả nghiên cứu còn cho thấy khi tăng nhiệt độ hòa tan gelatin từ 60 oC lên
70oC và 80oC, thời gian cần thiết để hòa tan vỏ nang giảm tương ứng từ 2,25 giờ xuống
1,5 và 1,0 giờ; thời gian cần để ủ dịch vỏ nang đến trong cũng giảm tương ứng từ 14 giờ
xuống 12 và 11 giờ.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 23-30
26
Bảng 3. Ảnh hưởng của nhiệt độ hòa tan gelatin tới đặc tính của màng vỏ nang
(n=3, TB ± SD)
Tiêu chí Mẫu dịch và màng vỏ tạo thành
M60 M70 M80 Thời gian hòa tan màng (phút) 40,3 ± 0,5 39,0 ± 0,8 30,3 ± 1,2 Độ dày màng (mm) 0,36 ± 0,01 0,38 ± 0,01 0,37 ± 0,04
Lực kéo đứt (N) 10,0 ± 0,1 11,2 ± 0,2 8,3 ± 0,4
Hình 1. Ảnh hưởng của nhiệt độ hòa tan gelatin tới độ bền gel và độ nhớt
của dịch vỏ nang
Kết quả thể hiện trong hình 1 cho thấy: Khi tăng nhiệt độ hòa tan gelatin lên đến 80 oC, độ nhớt, độ bền gel của dịch vỏ nang giảm xuống. Ngoài ra, nhiệt độ hòa tan gelatin
tăng lên không ảnh hưởng nhiều đến độ dày màng vỏ nang nhưng đều có xu hướng làm
giảm thời gian hòa tan cũng như lực kéo đứt vỏ nang (bảng 3). Sự thay đổi đặc tính của
dịch và màng vỏ nang có thể liên quan đến khả năng gelatin bị thủy phân một phần ở nhiệt
độ cao. Từ kết quả nghiên cứu thu được, nhiệt độ hòa tan gelatin được chọn trong khoảng
60 - 70 oC với thời gian hòa tan tương ứng là 1,5 – 2 giờ.
Ảnh hưởng của nhiệt độ ủ dịch vỏ nang
Tiến hành ngâm trương nở gelatin trong 30 phút. Nhiệt độ hòa tan gelatin 70 oC,
thời gian hòa tan gelatin 1,5 giờ. Khảo sát nhiệt độ ủ dịch vỏ nang sau hòa tan là 55 oC
(G55), 60 oC (G60) và 65 oC (G65). Kết quả đánh giá dịch vỏ nang và màng vỏ nang được
trình bày trong bảng 4.
Bảng 4. Ảnh hưởng của nhiệt độ ủ dịch gelatin tới đặc tính của dịch vỏ nang và màng vỏ
nang (n =3, TB ± SD)
Tiêu chí Mẫu dịch và màng vỏ tạo thành
G55 G60 G65
Thời gian ủ đến dịch trong (giờ) 16 12 9
Độ nhớt (cP) 183,5 ± 1,1 149,6 ± 1,2 138,5 ± 1,5
Độ bền gel (g) 327,0 ± 0,1 322,4 ± 0,2 315,2 ± 0,2
Độ dày màng (mm) 0,39 ± 0,02 0,38 ± 0,01 0,40 ± 0,04
Thời gian hòa tan (phút) 40,3 ± 1,2 39,0 ± 0,8 33,7 ± 2,6
Lực kéo đứt (N) 11,6 ± 0,4 11,2 ± 0,2 6,2 ± 0,4
Nhận thấy nhiệt độ ủ dịch vỏ gelatin có ảnh hưởng tới đặc tính của dịch vỏ nang và
màng vỏ nang. Khi nhiệt độ ủ dịch vỏ tăng, các chỉ tiêu chất lượng của dịch và màng vỏ
nang đều có xu hướng giảm. Mẫu G65 có sự giảm mạnh chỉ tiêu lực kéo đứt và độ nhớt,
đồng thời độ đồng đều bề dày màng giảm đi nhiều so với hai mẫu còn lại. Không có sự
quá khác biệt về độ bền gel dịch vỏ, thời gian hòa tan và lực kéo đứt màng vỏ nang giữa
hai mẫu ủ ở nhiệt độ 55 oC và 60 oC. Trên cơ sở phân tích trên, nhiệt độ ủ dịch vỏ nang
thích hợp là 55 – 60 oC với thời gian ủ dịch gelatin tương ứng là 12 – 16 giờ.
Ảnh hưởng nhiệt độ làm đông đặc vỏ nang và hàn vỏ nang

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 23-30
27
Bào chế viên nang mềm với công thức vỏ nang và thông số kỹ thuật đã lựa chọn.
Khảo sát nhiệt độ làm đông đặc vỏ nang ở nhiệt độ 18 – 20 oC và 2 – 8 oC. Đánh giá khả
năng hàn kín vỏ nang ở các nhiệt độ 40, 41 và 42 oC. Kết quả được trình bày ở bảng 5.
Bảng 5. Độ kín của viên nang được bào chế với nhiệt độ đông đặc vỏ nang
và nhiệt độ hàn khác nhau (n = 3, TB ± SD)
Nhiệt độ
hàn vỏ
Nhiệt độ đông đặc vỏ nang
2 – 8 oC 18 – 20 o C
Ban đầu Ban đầu Sau 2 tuần
40 oC - + Viên kín, không rò dịch
41 oC - + Viên kín, không bị rò dịch.
42 oC ± -
Ghi chú:
(+): Viên được hàn kín, không bị rò rỉ dịch khi bóp nhẹ viên và làm khô
(±): Vỏ nang hàn dính được nhưng bị rò dịch khi bóp nhẹ và làm khô.
(-): Không hàn dính được vỏ nang
Kết quả khảo sát cho thấy:
Mẫu vỏ nang làm đông đặc ở điều kiện 2 – 8 oC: Khi hàn kín vỏ nang ở nhiệt độ 40
– 41 oC vỏ nang không chảy lỏng nên không hàn được viên. Ở nhiệt độ hàn 42 oC, mép
hàn kết dính được vỏ nang với nhau nhưng viên bị rò dịch khi bóp nhẹ và làm khô.
Mẫu vỏ nang làm đông đặc ở điều kiện 18 – 20 oC: Với nhiệt độ hàn vỏ nang 40 oC
có thể hàn kín viên, đảm bảo viên không bị rò dịch sau làm khô hoặc bóp nhẹ nhưng khó
cắt viên khỏi vị trí hàn. Ở nhiệt độ hàn 41 oC, có thể hàn được viên, viên không bị rò dịch
sau làm khô, mép hàn vẫn kín khi bóp nhẹ viên. Ở nhiệt độ hàn 42 oC, vỏ nang chảy lỏng
nhanh nên không hàn kín được.
Từ kết quả nghiên cứu thu được, nhiệt độ đông đặc và nhiệt độ hàn kín vỏ nang
được chọn lần lượt là 18 – 20 oC và 40 – 41 oC.
Thời gian làm khô nang
Bào chế viên nang mềm theo theo các thông số kỹ thuật đã chọn ở trên. Khảo sát
các khoảng thời gian làm khô 16 giờ, 20 giờ, 24 giờ và 28 giờ. Kết quả đánh giá độ ẩm
của viên sau làm khô ở khoảng thời gian khác nhau thể hiện trong bảng 6.
Bảng 6. Độ ẩm của viên nang bào chế sau khi làm khô ở các khoảng thời gian khác nhau (n
= 3, TB ± SD)
Thời gian làm khô nang (giờ) Độ ẩm (%)
16 giờ 14,0 ± 0,4
20 giờ 9,0 ± 0,7
24 giờ 7,3 ± 0,6
28 giờ 5,8 ± 0,2
Kết quả nghiên cứu chứng tỏ để độ ẩm viên nang đạt yêu cầu 9 - 14 %, cần làm khô
trong thời gian 16 – 20 giờ.
Bào chế viên nang mềm SNEDDS rosuvastatin 10 mg
Trên cơ sở kết quả khảo sát và lựa chọn các thông số kỹ thuật trong từng giai đoạn,
tiến hành bào chế viên nang mềm rosuvastatin theo công thức trong bảng 1 và các thông số
kỹ thuật đã chọn. Đánh giá các chỉ tiêu chất lượng của dịch vỏ nang, SNEDDS rosuvastatin
đóng nang và viên nang mềm, trên cơ sở đó đề xuất tiêu chuẩn bán thành phẩm và thành
phẩm. Kết quả thu được thể hiện trong các bảng 7, 8.
Bảng 7. Kết quả đánh giá và đề xuất tiêu chuẩn bán thành phẩm (n = 3, TB ± SD)
Chỉ tiêu Kết quả
Đề xuất Mẻ 1 Mẻ 2 Mẻ 3
Dịch đóng nang (SNEDDS rosuvastatin)
Định lượng (%) 100,92 ± 0,84 100,72 ± 0,90 100,74 ± 0,67 90,0 - 110,0

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 23-30
28
KTG (d.nm) 19,38 ± 0,22 21,08 ± 0,74 20,08 ± 0,13 ≤ 200
PDI 0,180 ± 0,010 0,200 ± 0,010 0,200 ± 0,020 ≤ 0,3
Tỷ lệ dược chất được
nano nhũ hóa (%) 93,33 ± 0,47 92,75 ± 1,57 93,20 ± 0,58 ≥ 90 %
Độ ổn định vật lý Không được tách lớp sau khi ly tâm 5000 vòng/phút trong 30 ph
Dịch vỏ nang
Độ nhớt (cP) 150,3 ± 1,8 149,6 ± 1,7 150,8 ± 1,9 145 - 155
Độ bền gel (g) 321,2 ± 1,0 322,4 ± 0,3 321,0 ± 0,4 310 - 330
Tính chất Dịch sánh nhớt, trong, màu vàng đậm, không có bọt khí
Kết quả nghiên cứu cho thấy các chỉ tiêu chất lượng của dịch đóng nang, dịch vỏ
nang và viên nang mềm bào chế ổn định, không có sự khác biệt đáng kể giữa 03 mẻ bào
chế. Như vậy quy trình bào chế và các thông số kỹ thuật đã lựa chọn là phù hợp.
Bảng 8. Kết quả đánh giá và đề xuất tiêu chuẩn chất lượng viên nang mềm.
Chỉ tiêu Mẻ 1 Mẻ 2 Mẻ 3 Đề xuất yêu cầu
Tính chất Viên nang mềm có hình trứng, màu vàng nhạt, bên trong chứa
dịch thuốc không màu.
Định tính
Trên sắc ký đồ dung dịch thử cho pic chính có thời gian lưu
tương ứng với thời gian lưu của pic calci rosuvastatin thu được
từ sắc ký đồ của dung dịch chuẩn.
Định lượng (%) 100,07 ± 0,03 101,38 ± 0,96 100,94 ± 0,81
90 – 110 % so với
hàm lượng ghi trên
nhãn
Độ hòa tan sau 60 ph (%) 100,85 ± 0,66 100,7 ± 0,65 100,64 ± 0,88 ≥ 80 %
Đồng đều hàm lượng
(%, n=10) 98,83 ± 1,75 99,65 ± 1,59 99,42 ± 2,13
Đạt yêu cầu trong
phụ lục 11.2 –
DĐVN V
Độ rã (phút, n=6) 10,3 ± 0,8 10,4 ± 0,6 10,4 ± 0,8 ≤ 30 phút
Đánh giá tương tác giữa vỏ và dịch đóng nang
Tiến hành quét phổ hồng ngoại FTIR của màng vỏ nang gelatin ở thời điểm ban
đầu và sau 02 tháng đóng thuốc và bào quản ở điều kiện thường theo phương pháp đánh
giá tương tác giữa vỏ nang mềm và SNEDDS rosuvastatin đã mô tả ở trên. Hình ảnh phổ
FTIR vỏ nang ban đầu và sau 2 tháng được thể hiện ở hình 2 và 3.
Phổ FTIR của màng vỏ nang gelatin tại thời điểm ban đầu được quét ở các vị trí
khác nhau, các mặt đo khác nhau. Kết quả mẫu có hệ số tương đồng phổ trên 99 % giữa
các lần đo.
Hình 2. Phổ FTIR của mẫu vỏ nang ban đầu

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 23-30
29
Hình 3. Kết quả chồng phổ của mẫu ban đầu và mẫu sau 2 tháng
Viên nang mềm SNEDDS rosuvastatin đã bảo quản 02 tháng được tiến hành cắt
vỏ, loại dịch đóng nang, lau khô cả mặt trong và ngoài. Tiến hành quét phổ FTIR của vỏ
nang ở nhiều vị trí khác nhau, bên trong viên và bên ngoài viên. Đánh giá mức độ ảnh
hưởng của dịch ruột nang lên vỏ nang bằng cách chồng phổ vỏ nang ban đầu (vỏ nang 02)
và sau 2 tháng bảo quản (vỏ nang T1.5). Kết nghiên cứu cho thấy phổ FTIR của vỏ nang
tại thời điểm ban đầu và sau khi đóng nang 2 tháng có hệ số tương đồng phổ trên 95 %.
Như vậy, kết quả nghiên cứu bước đầu cho thấy ở thời điểm đánh giá không có biểu hiện
tương tác giữa vỏ nang và SNEDDS rosuvastatin đóng nang.
Bàn luận
Việc khảo sát và lựa chọn các thông số kỹ thuật trong quá trình bào chế thuốc là
cần thiết để đảm bảo tính ổn định của qui trình cũng như sự đồng nhất về chất lượng thuốc
bào chế giữa các lô mẻ. Thực tế nghiên cứu cho thấy, các thông số kỹ huật đã khảo sát đều
ảnh hưởng tới các đặc tính của dịch vỏ nang, vỏ nang cũng như viên nang mềm bào chế
được. Độ bền gel và độ nhớt của gel là hai thông số thường được sử dụng để đánh giá
mức độ phân hủy gelatin [9]. Trong đó, độ bền của gel được cho là chỉ số nhạy cảm hơn
của phản ứng phân hủy. Sự thủy phân gelatin (biểu hiện hiện là sự giảm độ bền gel theo
thời gian) đã được chứng minh là tuân theo động học phản ứng bậc nhất, với tốc độ phản
ứng thay đổi theo độ pH và nhiệt độ [6]. Sự thủy phân gelatin cũng dẫn đến giảm độ nhớt
và khả năng tạo gel của dung dịch gelatin trong quá trình bào chế viên nang mềm [6], [8].
Kết quả nghiên cứu cũng cho thấy, nhiệt độ có ảnh hưởng tới độ nhớt, độ bền gel
của dịch vỏ nang. Khi nhiệt độ hòa tan gelatin tăng từ 60 oC đến 80 oC độ nhớt của dung
dịch gelatin giảm tương ứng từ 152 xuống 116 (cP) và độ bền gel của dịch vỏ nang giảm
mạnh từ 365,0 xuống 257,0 (g). Khi nhiệt độ ủ dịch vỏ nang tăng từ 55 oC đến 65 oC, độ
nhớt và độ bền gel của dịch vỏ nang giảm tương ứng từ 183,5 xuống 138,5 (cP) và 327,0
xuống 315,2 (g). Nhiệt độ hòa tan gelatin và nhiệt độ ủ dịch gelatin được chọn lần lượt là
60 – 70 oC và 55 – 60 oC. Kết quả nghiên cứu thu được cũng phù hợp với một số nghiên
cứu đã công bố [3], [4], [6], [7].
Các thông số về điều kiện làm khô nang mềm cần được xác định cụ thể cho từng
công thức vỏ và dịch thuốc đóng nang và cần được kiểm soát chặt chẽ. Việc loại bỏ quá
nhiều nước trong vỏ nang có thể dẫn đến viên bị cứng, giòn, dễ bị nứt. Thời gian sấy ngắn
không đủ làm khô có thể làm cho các viên nang dính với nhau. Ngoài ra còn có thể có sự
di chuyển nước từ vỏ nang vào dịch thuốc hoặc ngược lại. Nếu điều kiện sấy loại bỏ nước
quá nhanh, gây ra bởi nhiệt độ sấy cao, độ ẩm tương đối thấp hoặc luồng khí thổi mạnh có
thể xảy ra hiện tượng bề mặt bên ngoài của viên nang khô nhanh chóng và tạm thời ngăn
chặn nước thoát ra khỏi vỏ, độ ẩm dư thừa từ bên trong vỏ nang di chuyển chậm ra ngoài
vỏ trong quá trình bảo quản khiến cho vỏ nang mềm, dính vào nhau [6]. Độ ẩm của viên
nang có thể thay đổi bởi thành phần vỏ nang và thuốc đóng nang. Với thuốc nang mềm
bào chế trong nghiên cứu này, khi sấy nang đến độ ẩm 6 – 8% thì vỏ nang có dấu hiệu
khô, đanh không còn dẻo dai bình thường nên chúng tôi chọn điều kiện làm khô ở nhiệt
độ 21 – 24 °C, độ ẩm 25 - 30 % và thời gian làm khô từ 16 – 20 giờ đến khi độ ẩm viên

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 23-30
30
đạt 9 – 14 %. Thực tế nghiên cứu cho thấy viên đạt các yêu cầu chất lượng đề ra khi sấy
đến độ ẩm công bố. Sau 2 tháng bảo quản ở điều kiện phòng thí nghiệm, không thấy hiện
tượng rò dịch đóng nang. Kết quả phân tích phổ FTIR vỏ nang ban đầu và sau 02 tháng
bảo quản đã không quan sát thấy biểu hiện tương tác giữa vỏ nang và SNEDDS
rosuvastatin đóng nang.
Kết luận
Đã khảo sát được một số yếu tố ảnh hưởng tới quy trình bào chế viên nang mềm
SNEDDS rosuvastatin 10mg ở quy mô 300 viên/mẻ. Từ đó đã lựa chọn được các ác thông
số kỹ thuật thích hợp là: thời gian ngâm trương nở gelatin: 30 – 45 phút, nhiệt độ hòa tan
60 – 70 oC và thời gian hòa tan gelatin 1,5 - 2,0 giờ; nhiệt độ và thời gian ủ dịch vỏ nang
lần lượt là 55 – 60 oC và 12 – 16 giờ; nhiệt độ đông đặc vỏ nang 18 – 20 oC; nhiệt độ hàn
viên 40 – 41 oC; điều kiện và thời gian làm khô viên nang: 21 – 24 °C, độ ẩm 25 - 30 %
trong 16 – 20 giờ. Với quy trình đã lựa chọn, dịch vỏ nang, vỏ nang và viên nang mềm
SNEDDS rosuvastatin đều đạt các yêu cầu chất lượng đề ra.
Lời cảm ơn
Nghiên cứu này được tài trợ bởi chương trình “Nghiên cứu ứng dụng và phát triển
công nghệ tiên tiến phục vụ bảo vệ và chăm sóc sức khỏe cộng đồng” trong đề tài
“Nghiên cứu bào chế viên nang SNEDDS rosuvastatin. Mã số KC.10.34/16-20.
Trong quá trình thực hiện, nghiên cứu còn nhật được sự hỗ trợ, giúp đỡ từ công ty
Cổ phần Dược phẩm Fresh Life. ½ lô 25+26+29, cụm công nghiệp An Xá, phường Mỹ
Xá, thành phố Nam Định.
Tài liệu tham khảo
1. Vũ Thị Thu Giang, Nguyễn Thị Thúy Nga và cộng sự (2020), “Xây dựng công thức vỏ
nang mềm chứa hệ nano tự nhũ hóa rosuvstatin”, Tạp chí Nghiên cứu dược & Thông tin
thuốc, tập 11 (1+2), tr. 54 – 60.
2. Vũ Thị Thu Giang, Phan Thị Nghĩa và cộng sự (2020), “Nghiên cứu bào chế hệ nano tự
nhũ hóa rosuvastatin”, Tạp chí Nghiên cứu dược & Thông tin thuốc, tập 11 (4), tr. 2 – 10.
3. Cumper C. Alexander A (1952), "The viscosity and rigidity of gelatin in concentrated
aqueous systems. I. Viscosity", Australian Journal of Chemistry, 5 (1), pp. 146-152.
4. Gullapalli R. P (2010), "Soft gelatin capsules (softgels)", Journal of pharmaceutical
sciences, 99 (10), pp. 4107-4148.
5. Marques M. R. J. A. P (2014), "Enzymes in the dissolution testing of gelatin capsules",
AAPS PharmSciTech. 15 (6), pp. 1410-1416.
6. Podczeck F., Jones B. E. (2004), Pharmaceutical capsules, 2nd Edition, London – Chicago
Pharmaceutical Press, pp. 201 – 210.
7. Reich G. J. P. C., Pharmaceutical Press, London (2004), "Formulation and physical
properties of soft capsules", pp. 201-212.
8. Rivero S, García MA, et al. (2010), "Correlations between structural, barrier, thermal and
mechanical properties of plasticized gelatin films", Innovative Food Science & Emerging
Technologies, 11(2), pp. 369-375.
9. Thomazine M., Carvalho R.A., et al. (2005), "Physical properties of gelatin films
plasticized by blends of glycerol and sorbitol", Journal of Food Science, 70(3), pp. E172-
E176.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 31-36
31
Tổng hợp và đánh giá tác dụng ức chế tế bào ung thư của một số acid
hydroxamic mang dị vòng 1,3-thiazol Dương Tiến Anh, Đào Thị Kim Oanh, Nguyễn Hải Nam*
Trường Đại học Dược Hà Nội
* Tác giả liên hệ: [email protected]
(Ngày gửi đăng: 11/8/2020 – Ngày duyệt đăng: 10/12/2020) SUMMARY
In the process of discovering novel small molecules targeting deacetylase histone,
we have designed and synthesized a series of novel hydroxamic acids bearing 2-
benzamidothiazol (3a-g) scaffold. Biological evaluation showed that these hydroxamic
acids comparably inhibited HDACs with IC50 values in sub-micromolar range. These
compounds also were generally cytotoxic against three human cancer cell lines
(SW620, colon cancer; PC-3, prostate cancer; NCI-H23, lung cancer). Among them,
the derivative 3f displayed the most potential cytotoxicity to cancer cells, especially on
SW620. The synthesized derivatives are expected to reduce the side effects of
traditional hydroxamic acids.
Từ khóa: ức chế histon deacetylase, HDAC, acid hydroxamic, thiazol.
Đặt vấn đề
Nhiều nghiên cứu đã chỉ ra rằng HDAC không chỉ liên quan đến cấu trúc của
chromatin và sự biểu hiện của gen mà còn tác động đến chu trình phát triển của tế bào và
tiến trình ung thư bao gồm sự hình thành của các khối u ác tính [6]. Khởi đầu với SAHA
(Zolinza®) là chất ức chế enzym HDAC đầu tiên được FDA cấp phép lưu hành năm 2006,
và sau đó lần lượt romidepsin (Istodax®), belinostat (Beleodaq®) và panobinostat
(Farydax®) cũng đã được cấp phép sử dụng trong điều trị một số bệnh ung thư [4]. Tiếp
cận với xu thế chung, nhóm nghiên cứu tại bộ môn Hóa Dược, Trường Đại học Dược Hà
Nội cũng đã thiết kế, tổng hợp, thử hoạt tính nhiều dãy chất dẫn xuất acid hydroxamic
hướng ức chế HDAC có hoạt tính kháng tế bào ung thư tốt [1], [3].
Bên cạnh đó, dị vòng 1,3-thiazol có mặt trong nhiều hợp chất có hoạt tính sinh học
như: chống nấm, kháng khuẩn, chống lao... đặc biệt một số dẫn chất ức chế kinase
(Dasatinib) và các hợp chất có tiềm năng chống ung thư như tiazofurin, S3U937 [2]. Do
vậy, chúng tôi tiếp tục triển khai hướng nghiên cứu về các dẫn chất acid hydroxamic là
các dẫn chất benzamid kết hợp với dị vòng 1,3-thiazol. Nội dung dưới đây công bố một
phần các kết quả nghiên cứu này.
Nguyên liệu và phương pháp nghiên cứu
Dung môi, hóa chất và thiết bị
Các hóa chất được sử dụng cho việc tổng hợp các phản ứng hữu cơ được mua và
sử dụng trực tiếp khi nhận từ các hãng Merck (Đức) và Aldrich (Mỹ). Nhiệt độ nóng
chảy được đo bằng máy đo điểm chảy nhiệt điện Smp3. Sắc ký lớp mỏng sử dụng bản
mỏng nhôm tráng sẵn silica gel GF254. Phổ hồng ngoại ghi bằng máy Shimadzu FTIR
Affinity-1S. Phổ cộng hưởng từ hạt nhân ghi bằng máy Bruker AV-500. Phổ khối
lượng ghi bằng máy khối phổ Agilent 6530 Accurate Mass QTOF LC/MS (Agilent
technology, Santa Clara, California, United States).
Các enzym HDAC (chiết từ tế bào Hela) được mua của Enzo Life Sciences Inc.
(Mỹ). Các dòng tế bào thử nghiệm được mua từ Ngân hàng Tế bào Ung thư của Viện
nghiên cứu khoa học sinh học và công nghệ Hàn Quốc (KRIBB). Môi trường nuôi cấy
và các hóa chất để thử tác dụng sinh học khác được mua của GIBCO Co. ltd. (Mỹ).

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 31-36
32
Phương pháp nghiên cứu
Tổng hợp hóa học
Các acid hydroxamic mục tiêu được tổng hợp theo Hình 1.
Hình 1. Sơ đồ tổng hợp các acid hydroxamic mang dị vòng 1,3-thiazol (3a-g)
Xác định cấu trúc
Sử dụng các phương pháp phổ bao gồm: Phổ hồng ngoại (IR); phổ khối (HR-
MS); phổ cộng hưởng từ hạt nhân (1H-NMR, 13C-NMR).
Thử hoạt tính kháng tế bào ung thư in vitro
Thử hoạt tính kháng tế bào ung thư in vitro được thực hiện tại Khoa Dược,
Trường Đại học Chungbuk, Cheongju, Hàn Quốc trên 3 dòng tế bào ung thư người là
SW620 (ung thư đại tràng), PC3 (ung thư tuyến tiền liệt), và NCI-H23 (ung thư phổi).
Độc tính tế bào của các chất nghiên cứu được xác định bằng phương pháp đo màu cải
tiến [5]. Giá trị IC50 được tính theo phương pháp Probits [7] và lấy kết quả trung bình
của 3 lần thực nghiệm độc lập (SD ≤ 10 %).
LogP được xác định dựa trên phần mềm KOWWIN v1.67.
Thử tác dụng ức chế enzym histon deacetylase
Thử tác dụng ức chế enzym HDAC trên hỗn hợp HDAC tổng tách từ tế bào Hela
được thực hiện tại Khoa Dược Trường Đại học Chungbuk, Cheongju, Hàn Quốc và
Viện nghiên cứu sinh học và công nghệ sinh học Hàn Quốc (KRIBB). Định lượng
enzym được thực hiện bằng phương pháp huỳnh quang với bộ Fluorogenic HDAC
Assay Kit (Enzo Life Sciences Inc.) theo hướng dẫn của nhà sản xuất. Tác dụng ức chế
HDAC của các dẫn chất được tính bằng phần mềm GraphPad Prism (Mỹ).
Kết quả nghiên cứu
Tổng hợp hóa học
Hoà tan 0,34 g (2 mmol) ethyl 2-aminothiazol-4-carboxylat bằng 10 mL DCM
trong bình cầu 50 mL. Thêm 0,24 g (2 mmol) DMAP và tiến hành khuấy ở điều kiện
phòng trong 10 phút. Thêm 2,2 mmol các dẫn chất của benzoyl clorid, khuấy hỗn hợp
ở 50 ºC trong 12 giờ. Kết thúc phản ứng, hỗn hợp được cất quay dưới áp suất giảm
loại bỏ dung môi. Thêm 30 mL dung dịch NaHCO3 5 %, tiến hành lọc tủa thu được,
rửa tủa bằng nước lạnh và sấy tủa. Tinh chế sản phẩm thu được bằng sắc ký cột với hệ
pha động DCM: MeOH = 95:5 thu được sản phẩm 2a-g là chất rắn màu trắng với hiệu
suất tinh chế các chất lần lượt là 2a (76 %), 2b (79 %), 2c (82 %), 2d (81 %), 2e
(79 %), 2f (80 %), 2g (67 %).
Phân tán các ester trung gian 2a-g trong 10 mL MeOH. Thêm tiếp 10 đương
lượng NH2OH.HCl vào bình phản ứng. Tiến hành nhỏ từng giọt dung dịch NaOH
50% đã được làm lạnh đến pH = 12. Khuấy hỗn hợp phản ứng ở nhiệt độ -5 ºC trong
khoảng 1-2 giờ đến khi phản ứng kết thúc. Xử lý phản ứng bằng cách thêm vào hỗn
hợp sản phẩm 50 mL nước lạnh, trung hòa đến pH = 7 bằng cách nhỏ từng giọt dung

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 31-36
33
dịch HCl 5 %. Tiến hành lọc tủa thu được, kết tinh lại sản phẩm trong MeOH thu được
các dẫn chất cần tổng hợp 3a-g.
Xác định cấu trúc
Cấu trúc của các sản phẩm cuối cùng được khẳng định thông qua các dữ liệu
phân tích phổ IR, HR-MS, 1H-NMR và 13C-NMR.
2-Benzamido-N-hydroxythiazol-4-carboxamid (3a)
Chất rắn màu trắng; hiệu suất: 68 % (0,35 g). tnc: 189-188 oC. Rf = 0,52 (DCM :
MeOH : AcOH = 90 : 5 : 1). IR (KBr, cm-1): 3339 (NH); 3262, 3111 (OH); 3057, 2994
(CH, aren); 1655 (C=O); 1601, 1545 (C=C). 1H-NMR (500 MHz, DMSO-d6, ppm): δ
12,64 (1H, s, NHOH); 10,73 (1H, s, CO-NH); 9,05 (1H, s, NHOH); 8,01 (2H, d, J =
8,00 Hz, H-2’, H-6’); 7,71 (1H, d, J = 2,50 Hz, H-5); 7,57 (1H, t, J = 7,50 Hz, H-4’);
7,47 (2H, t , J = 7,75 Hz, H-3’, H-5’). 13C NMR (125 MHz, DMSO-d6, ppm): δ165,98;
159,80; 158,99; 143,72; 133,24; 132,29; 129,12; 128,64; 117,50. HR-MS (ESI) m/z:
262,0302 [M+H]+.
2-(2-Clorobenzamido)-N-hydroxythiazol-4-carboxamid (3b)
Chất rắn màu trắng; hiệu suất: 72 % (0,42 g). tnc: 198-199oC. Rf = 0,54 (DCM :
MeOH : AcOH = 90 : 5 : 1). IR (KBr, cm-1): 3337 (NH); 3175, 3117 (OH); 3057, 2967,
2872 (CH, aren); 1680, 1655 (C=O); 1589, 1545 (C=C). 1H-NMR (500 MHz, DMSO-
d6, ppm): δ 12,80 (1H, s, NHOH); 10,75 (1H, s, CO-NH); 9,02 (1H, s, NHOH); 7,75
(1H, s, H-5); 7,58-7,37 (4H, m, H-3’, H-4’, H-5’, H-6’). 13C NMR (125 MHz, DMSO-
d6, ppm): δ 165,80; 159,56; 158,02; 143,84; 134,65; 132,48; 130,70; 130,24; 129,90;
127,72; 117,88. HR-MS (ESI) m/z: 295,9898 [M+H]+.
2-(3-Clorobenzamido)-N-hydroxythiazol-4-carboxamid (3c)
Chất rắn màu trắng; hiệu suất: 74 % (0,44 g). tnc: 201-202 oC. Rf = 0,54 (DCM :
MeOH : AcOH = 90 : 5 : 1). IR (KBr, cm-1): 3393 (NH); 3125 (OH); 3065, 2967 (CH,
aren); 1651 (C=O); 1599, 1549 (C=C). 1H-NMR (500 MHz, DMSO-d6, ppm): δ 8,07
(1H, d, J = 1,50 Hz, H-2’); 7,96 (1H, d, J = 8,00 Hz, H-6’); 7,77 (1H, s, H-5); 7,61
(1H, dd, J = 8,00 Hz, J’ = 1,50 Hz, H-4’); 7,49 (1H, t, J = 7,75 Hz, H-5’). 13C NMR
(125 MHz, DMSO-d6, ppm): δ 164,76; 159,71; 157,59; 143,62; 134,43; 133,90;
132,91; 131,05; 128,48; 127,40; 117,62. HR-MS (ESI) m/z: 295,9903 [M+H]+.
2-(4-Clorobenzamido)-N-hydroxythiazol-4-carboxamid (3d)
Chất rắn màu trắng; hiệu suất: 74 % (0,44 g). tnc: 207-208 oC. Rf = 0,54 (DCM :
MeOH : AcOH = 90 : 5 : 1). IR (KBr, cm-1): 3395, 3332 (NH); 3142 (OH); 3061, 2968,
2803 (CH, aren); 1651 (C=O); 1593, 1547 (C=C). 1H-NMR (500 MHz, DMSO-d6,
ppm): δ 12,75 (1H, s, NHOH) ;10,75 (1H, s, CO-NH); 9,07 (1H, s, NHOH); 8,02 (2H,
d, J = 9,00 Hz, H-2’, H-6’); 7,72 (1H, s, H-5); 7,54 (2H, d, J = 8,50 Hz, H-3’, H-5’). 13C NMR (125 MHz, DMSO-d6, ppm): δ 165,06; 159,74; 158,94; 143,73; 138,11;
131,18; 130,60; 129,22; 117,59. HR-MS (ESI) m/z: 295,9905 [M+H]+.
2-(4-Florobenzamido)-N-hydroxythiazol-4-carboxamid (3e)
Chất rắn màu trắng; hiệu suất: 75 % (0,42 g). tnc: 188-189 oC. Rf = 0,45 (DCM :
MeOH : AcOH = 90 : 5 : 1). IR (KBr, cm-1): 3399, 3321 (NH); 3142 (OH); 3075, 2984,
2820 (CH, aren); 1657 (C=O); 1601, 1547, 1510 (C=C). 1H-NMR (500 MHz, DMSO-
d6, ppm): δ 12,46 (1H, s, NHOH); 10,73 (1H, s, CO-NH); 9,07 (1H, s, NHOH); 8,10
(2H, dd, J = 8,50 Hz, J’ = 5,50 Hz, H-2’, H-6’); 7,73 (1H, s, H-5); 7,31 (2H, t, J =
8,75 Hz, H-3’, H-5’). 13C NMR (125 MHz, DMSO-d6, ppm): δ 166,22; 164,92; 164,22;
159,82; 158,95; 143,69; 131,64; 131,57; 128,82; 128,80; 117,47; 116,25; 116,08. HR-
MS (ESI) m/z: 280,0204 [M+H]+.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 31-36
34
N-hydroxy-2-(4-methylbenzamido)thiazol-4-carboxamid (3f)
Chất rắn màu trắng; hiệu suất: 72 % (0,40 g). tnc: 179-180 oC. Rf = 0,49 (DCM :
MeOH : AcOH = 90 : 5 : 1). IR (KBr, cm-1): 3447 (NH); 3221 (OH); 3107, 2974, 2903
(CH, aren); 1649 (C=O); 1609, 1541 (C=C). 1H-NMR (500 MHz, DMSO-d6, ppm): δ
12,64 (1H, s, NHOH); 10,70 (1H, s, CO-NH); 9,05 (1H, s, NHOH); 7,92 (2H, d, J =
8,50 Hz, H-2’, H-6’); 7,70 (1H, s, H-5); 7,27 (2H, d, J = 8,00 Hz, H-3’, H-5’); 2,30
(3H, s, 4’-CH3). 13C NMR (125 MHz, DMSO-d6, ppm): δ 165,80; 159,88; 159,20;
143,70; 143,55; 129,67; 129,43; 128,69; 117,41; 21,57. HR-MS (ESI) m/z: 276,0454
[M+H]+.
N-hydroxy-2-(4-methoxybenzamido)thiazol-4-carboxamid (3g)
Chất rắn màu trắng; hiệu suất: 76 % (0,45 g). tnc: 197-198 oC. Rf = 0,48 (DCM :
MeOH : AcOH = 90 : 5 : 1). IR (KBr, cm-1): 3337 (NH); 3215, 3179, 3117 (OH); 2992,
2837 (CH, aren); 1661 (C=O); 1605, 1547, 1501 (C=C). 1H-NMR (500 MHz, DMSO-
d6, ppm): δ 12,50 (1H, s, NHOH); 10,67 (1H, s, CO-NH); 9,04 (1H, s, NHOH); 8,02
(2H, d, J = 8,50 Hz, H-2’, H-6’); 7,69 (1H, s, H-5); 7,00 (2H, d, J = 8,50 Hz, H-3’, H-
5’); 3,77 (3H, s, 4’-OCH3). 13C NMR (125 MHz, DMSO-d6, ppm): δ 165,25; 163,31;
159,92; 159,14; 143,66; 130,78; 124,25; 117,26; 114,42; 56,02. HR-MS (ESI) m/z:
294,0539 [M+H]+.
Kết quả thử tác dụng ức chế enzym HDAC và độc tính tế bào
Kết quả thử tác dụng ức chế HDAC và hoạt tính kháng tế bào ung thư của các
chất tổng hợp được trình bày trong bảng 1.
Bảng 1. Kết quả thử tác dụng ức chế HDAC và độc tính tế bào của các chất tổng hợp
được
Chất Ar LogP1
Ức chế
HDAC tổng
(Hela)
(IC50,2 M)
Độc tính tế bào (IC50,2 M) trên
từng dòng tế bào3
SW620 PC3 NCI-H23
3a H 1,07 0,037±0,002 4,22±0,17 8,48±1,11 6,14±0,27
3b 2-Cl 1,72 0,051±0,001 5,99±0,48 3,78±0,53 7,69±0,83
3c 3-Cl 1,72 0,038±0,007 3,02±0,20 5,02±0,82 5,02±0,37
3d 4-Cl 1,72 0,033±0,001 4,42±0,56 6,57±0,50 6,78±0,23
3e 4-F 1,27 0,035±0,002 3,47±0,21 4,64±0,43 6,24±0,02
3f 4-CH3 1,62 0,131±0,052 2,21±0,01 4,34±0,64 3,29±0,51
3g 4-OCH3 1,15 0,073±0,009 6,54±0,01 6,05±0,08 6,41±0,08
SAHA4 264,32 1,44 0,025±0,002 1,12±0,10 1,82±0,09 1,44±0,17 1Tính bằng phần mềm ChemDraw 9.0; 2Nồng độ (M) chất thử gây giảm 50% hoạt tính enzym hoặc
sự phát triển tế bào. Số liệu là trung bình của 3 lần thực nghiệm; 3Dòng tế bào thử: SW620 (ung thư
đại tràng); PC3 (ung thư tiền liệt tuyến); NCI-H23 (ung thư phổi); 4SAHA, acid suberoylanilid
hydroxamic, chất đối chứng dương tính.
Bàn luận
Bàn luận về tổng hợp hóa học
Quá trình tổng hợp các dẫn chất 3a-g trải qua 2 bước được mô tả theo Hình 1.
Phản ứng tổng hợp các chất 2a-g

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 31-36
35
Phản ứng đầu tiên là phản ứng tạo liên kết amid với tác nhân là các dẫn chất của
benzoyl clorid. Đây là một tác nhân acyl hoá mạnh nên phản ứng diễn ra dễ dàng và
có hiệu suất cao. Phản ứng xảy ra trong dung môi DCM có thể dễ dàng loại bỏ bằng
cách cất quay dưới áp suất giảm. DMAP được thêm vào nhằm tạo trạng thái trung
gian, giúp phản ứng acyl xảy ra dễ dàng hơn (Hình 2). Ngoài ra, DMAP giúp trung
hòa HCl sinh ra, tránh việc amin bị proton hóa làm giảm tốc độ phản ứng. Lượng
benzoyl clorid được thêm vào dư có thể dễ dàng được loại bằng NaHCO3.
Hình 2. Vai trò của xúc tác DMAP
Phản ứng tổng hợp các chất 3a-g
Phản ứng thứ hai là phản ứng tạo acid hydroxamic. Tác nhân NH2OH được sử
dụng dưới dạng muối hydroclorid bởi ưu điểm ổn định hơn, bảo quản dễ hơn dạng
base. NaOH được thêm vào dư giúp chuyển dạng muối NH2OH.HCl thành dạng tự do
đồng thời giúp sản phẩm acid hydroxamic tạo dạng muối, hòa tan trong dịch phản ứng.
Tuy nhiên, môi trường kiềm mạnh do NaOH dư tạo ra sẽ gây thủy phân các ester 2a-g
tạo tạp (là các acid carboxylic rất khó loại bỏ) và giảm hiệu suất phản ứng. Vì vậy, cần
tiến hành trong nhiệt độ thấp (khoảng -5 ºC), đồng thời nhỏ từ từ NaOH đã được làm
lạnh vào bình phản ứng để tránh nhiệt độ tăng đột ngột làm mất NH2OH tự do. Xác
định thời điểm phản ứng kết thúc bằng sắc ký lớp mỏng, đồng thời nhận diện sự có
mặt của acid hydroxamic bằng phản ứng tạo phức màu tím với FeCl3. Bàn luận về tác dụng ức chế enzym HDAC và độc tính tế bào
Các dẫn chất tổng hợp sau khi được khẳng định cấu trúc thông qua phổ hồng
ngoại (IR), phổ khối lượng (MS), phổ cộng hưởng từ hạt nhân (1H-NMR, 13C-NMR),
được tiến hành thử tác dụng ức chế HDAC và độc tính tế bào với kết quả được mô tả
ở bảng 1.
Kết quả cho thấy tất cả các dẫn chất được tổng hợp được đều có tác dụng ức chế
HDAC với IC50 cỡ µM. Ảnh hưởng của vị trí nhóm thế đến tác dụng ức chế enzym
này được thể hiện ở 3 dẫn chất có cùng nhóm thế -Cl (3b-d). Dẫn chất 3d với IC50
thấp nhất (IC50, 0,033 ± 0,001 µM) đã cho thấy ưu thế của vị trí thế 4. Bên cạnh đó,
việc dẫn chất 4-Cl (3d) có tác dụng ức chế tốt hơn 4-CH3 và 4-OCH3 (3f-g) cũng
mang dự đoán rằng các nhóm thế hút điện tử có tác dụng ức chế enzym HDAC tốt
hơn. Tuy nhiên nhóm thế 4-F (3e) lại cho kết quả ngược lại (IC50, 3,124 ± 0,262 µM).
Điều này có thể giải thích bởi kích thước phân tử bé của F làm giảm khả năng tạo các
tương tác của bề mặt nhận diện.
Về độc tính tế bào, 7 dẫn chất (3a-g) đều có độc tính trên 3 dòng tế bào thử
nghiệm. So với dẫn chất 3a (IC50: 4,22-8,48 µM), nếu thêm nhóm thế -Cl ở các vị trí 2
hoặc 4 không cải thiện độc tính tế bào trong khi vị trí 3 khiến độc tính tế bào tăng
(IC50: 3,02-5,02 µM). Cùng vị trí thế 4, dẫn chất thế -CH3 (3f) cho độc tính tế bào cao
hơn -Cl và -F (3d, 3e). Dẫn chất này cũng có độc tính trên tế bào cao nhất 7 chất (3a-
g), đặc biệt trên dòng tế bào SW620 (IC50 = 2,21 µM). Từ đó có thể dự đoán rằng các
mật độ điện tử trên bề mặt nhận diện tỉ lệ thuận với tác dụng ức chế tế bào ung thư.
Tuy nhiên dẫn chất thế 4-OCH3 (3g) lại có độc tính tế bào kém nhất trong 4 dẫn chất
(3d-g). Kết quả này có thể được giải thích bằng việc dẫn chất 3g thân nước hơn khiến

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 31-36
36
tính thấm của qua màng sinh học giảm. Bên cạnh đó, cấu trúc phân tử cồng kềnh cũng
là một bất lợi cho việc tiếp xúc với enzym, khiến khả năng ức chế tế bào không được
phát huy.
Mặc dù khả năng gây độc tế bào của các benzamid mang khung 1,3-thiazol
không mạnh bằng SAHA song các dẫn chất acid hydroxamic mạch ngắn thường có ưu
điểm là tác dụng chọn lọc hơn trên HDAC6, là một isozym chỉ có trong bào tương,
không có trong nhân. Vì vậy, các dẫn chất benzamid chứa khung 1,3-thiazol này được
kỳ vọng là sẽ có ít độc tính, ít tác dụng phụ hơn các acid hydroxamic truyền thống.
Kết luận
Chúng tôi đã tổng hợp được 7 dẫn chất acid hydroxamic mang khung 1,3-thiazol
(3a-g) có tiềm năng kháng 3 dòng thế bào ung thư thử nghiệm. Tất cả các dẫn chất
được tổng hợp đều có tác dụng ức chế HDAC và độc tính tế bào với IC50 cỡ µM, đặc
biệt dẫn chất 3f có khả năng ức chế tế bào ung thư tốt hơn cả, nhất là trên dòng tế bào
SW620.
Lời cảm ơn
Nghiên cứu được tài trợ bởi NAFOSTED (đề tài 104.01-2020.19) và một phần
bởi chính phủ Hàn Quốc (NRF, Grant number 2017R1A5A2015541).
Tài liệu tham khảo
1. Anh D. T., Thuan N. T., Huong L.-T.-T., Yen N. T., et al, (2019), "Design,
Synthesis and Evaluation of Novel 3/4-((Substituted benzamidophenoxy)
methyl)-N-hydroxybenzamides/Propenamides as Histone Deacetylase
Inhibitors and Antitumor Agents", Anti-Cancer Agents in Medicinal Chemistry,
19 (4), pp. 546-556.
2. He H., Wang X., Shi L., Yin W., et al, (2016), "Synthesis, antitumor activity and
mechanism of action of novel 1, 3-thiazole derivatives containing hydrazide–
hydrazone and carboxamide moiety", Bioorganic & Medicinal Chemistry
Letters, 26 (14), pp. 3263-3270.
3. Hieu D. T., Anh D. T., Tuan N. M., Hai P.-T., et al, (2018), "Design, synthesis and
evaluation of novel N-hydroxybenzamides/N-hydroxypropenamides
incorporating quinazolin-4 (3H)-ones as histone deacetylase inhibitors and
antitumor agents", Bioorganic chemistry, 76 pp. 258-267.
4. Peng X., Liao G., Sun P., Yu Z., et al, (2019), "An overview of HDAC inhibitors
and their synthetic routes", Current Topics In Medicinal Chemistry, 19 (12), pp.
1005-1040.
5. Skehan P., Storeng R., Scudiero D., Monks A., et al, (1990), "New colorimetric
cytotoxicity assay for anticancer-drug screening", J Natl Cancer Inst, 82 (13),
pp. 1107-1112.
6. Witt O. et al, (2009), "HDAC family: What are the cancer relevant targets?",
Cancer letters, 277 (1), pp. 8-21.
7. Wu L., Smythe A. M., Stinson S. F., Mullendore L. A., et al, (1992), "Multidrug-
resistant phenotype of disease-oriented panels of human tumor cell lines used
for anticancer drug screening", Cancer Res, 52 (11), pp. 3029-3034.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 37-44
37
Tổng hợp và thử hoạt tính sinh học một số dẫn chất kết hợp
curcumin với diacid Phạm Thị Hiền1*, Phạm Ngọc Văn2, Ngô Quang Trung1,
Nguyễn Thị Ngọc Bích2, Đào Nguyệt Sương Huyền2,
Nguyễn Văn Giang2, Nguyễn Đình Luyện2, Nguyễn Văn Hải2 1Viện Công nghệ Dược phẩm Quốc gia - Trường Đại học Dược Hà Nội,
2Bộ môn Công nghiệp Dược - Trường Đại học Dược Hà Nội
*Tác giả liên hệ: [email protected]
(Ngày gửi đăng: 16/10/2020 – Ngày duyệt đăng: 18/12/2020) Summary Two compounds 2,2’-(curcumin-di-O-yl) diethyl disuccinate (LH-12020) and 2,2’-
(curcumin-di-O-yl) diethyl diglutarate (LH-22020) were synthesized from curcumin in
two steps: O-alkylation and acylation. The chemical structure of the compounds was
confirmed by MS-, IR- and NMR-spectroscopies. The results of bioactivity evaluation
showed that, both LH-12020 and LH-22020 have weaker antioxidant activity (DPPH)
than curcumin but exhibited anti-inflammatory activity through inhibition of NO
production with the IC50 of LH-12020 and LH-22020: 11.92 ± 0,86 and 12.06 ± 0.42
µM, respectively, that is stronger than curcumin (17.89 ± 1.98µM). LH-22020 and
LH-12020 exhibited toxicity against Hela, HL-60, MCF-7 and HepG2 cell lines. LH-
22020 showed better activity than curcumin at 1,2-2.24 times, while LH-12020
exhibited inhibitory activity 2 times better than that of curcumin against MCF-7 cell
lines.
Từ khóa: Chống ung thư, chống viêm, chống oxy hóa, curcumin, diacid.
Đặt vấn đề Curcumin là một trong số polyphenol có tác dụng dược lý phong phú, được sử
dụng dưới nhiều dạng bào chế trên thị trường dược phẩm và thực phẩm bảo vệ sức
khỏe với quy mô lớn ở Việt Nam và các nước trên thế giới [1], [2], [3], [4], [5], [6].
Mặt khác, hoạt chất này cũng đang được thử nghiệm trong nhiều nghiên cứu lâm sàng
để điều trị các bệnh khác nhau về thần kinh, tim mạch, hô hấp, tiêu hóa, xương khớp,
mắt; bệnh liên quan đến chuyển hóa và ung thư [1], [2], [3], [4], [5], [6]. Tuy nhiên,
curcumin có nhược điểm là sinh khả dụng đường uống thấp (dưới 1 %) do hấp thu
kém, chuyển hóa và đào thải nhanh khỏi cơ thể, trong đó yếu tố hóa-lý quan trọng ảnh
hưởng đến điều này là tính tan kém trong nước của curcumin (<0,1 µg/mL, 25 °C) [7].
Để giải quyết vấn đề này, nhiều kỹ thuật bào chế đã được tiến hành như: chế tạo đồng
tinh thể (co-crystal), nhũ tương nano, vi nhũ tương, liposome, exosome, micell,
dendrimer, hệ phân tán rắn, dạng phức với các cyclodextrin, phospholipid... [8], [1],
[2], [3]. Song song với đó, cách tiếp cận biến đổi hóa học phân tử curcumin cũng được
nhiều nhà nghiên cứu thực hiện với các phương pháp kinh điển là liên hợp khung
curcumin với các nhóm thân nước (phân tử đường, acid amin, polymer tan trong nước,
dẫn chất của glucuronic....). Những nghiên cứu này đã đạt được một số thành công
nhất định trong việc mở rộng/tăng cường hoạt tính sinh học, sinh khả dụng và độ ổn
định của curcumin [9], [10]. Năm 2010, C. Changtam và cộng sự đã tổng hợp được
dẫn chất di-O-(2-hydroxylethyl)-curcumin thể hiện khả năng chống ký sinh trùng tốt
hơn trên một số loài thuộc chi Trypanosoma và Leishmaina [11]. Năm 2011, W.
Wichitnithad và các cộng sự đã thực hiện tổng hợp một số dẫn chất succinat của
curcumin, trong đó dẫn chất curcumin diethyl disuccinat thể hiện hoạt tính kháng tế
bào ung thư biểu mô đại trực tràng ở người (Caco-2) in vitro tốt hơn so với curcumin

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 37-44
38
[12]. Đồng thời, dẫn chất này có khả năng giảm đau tốt trên chuột với độ ổn định hóa
học cao hơn curcumin trong dung dịch đệm phosphat 0,1 M (pH = 7,4) [13]. Tuy
nhiên, chất này tan trong nước kém do ở dạng diethyl ester. Tiếp tục theo hướng tạo
dẫn chất diacid, năm 2018 C. Muangnoi và các cộng sự đã tiến hành nghiên cứu tổng
hợp và thử tác dụng sinh học của acid curcumin di-glutaric giúp cải thiện được độ tan
và tăng khả năng chống nhiễm trùng trên chuột [14]. Trong phát triển thuốc, kết hợp
hoạt chất với diacid succinic đã có những ví dụ kinh điển và được ứng dụng rộng trong
lâm sàng như cloramphenicol succinat, artesunat, α-tocopherol succinat,
methylprednisolon hemisuccinat, estriol succinat... [15]. Cùng với acid succinic, các
diacid khác như acid maleic, glutaric cũng được nhiều nhà nghiên cứu phát triển thuốc
chú ý nhờ các ưu điểm: cải thiện được độ tan, dược động học, độ ổn định, an toàn sinh
dược học và có khả năng tạo dạng muối phù hợp cho bào chế [14]. Những phân tích
trên cho thấy, kết hợp curcumin với các diacid nhằm phát huy những tác dụng tốt của
phân tử này là một hướng đi hấp dẫn, có ý nghĩa thực tiễn. Hiện nay, chưa có tài liệu
nào công bố về kết hợp curcumin với diacid qua cầu nối ethylenoxy. Nghiên cứu này
được chúng tôi tiến hành với hai mục tiêu: (i) Tổng hợp được hai dẫn chất mới ester
glutarat và ester succinat của curcumin; (ii) Đánh giá được hoạt tính chống oxy hóa,
chống viêm và kháng tế bào ung thư của hai dẫn chất này.
Nguyên liệu và phương pháp
Nguyên liệu
Curcumin đạt hàm lượng > 96 % (HPLC); các hóa chất: 2-bromoethanol (95 %,
Acros) và aceton, anhydrid glutaric, anhydrid succinic, clorofom, dicloromethan
(DCM), isopropanol (IPA), kali carbonat, methanol (MeOH), n-hexan, natri hydroxid,
natri sulfat khan, triethylamin (TEA) đạt tiêu chuẩn tinh khiết tổng hợp (AR, Trung
Quốc).
Phương pháp nghiên cứu
Tổng hợp các dẫn chất được tiến hành tại phòng Tổng hợp Hóa dược - Bộ môn
Công nghiệp Dược - Trường Đại học Dược Hà Nội, sử dụng các phản ứng O-alkyl
hóa, acyl hóa theo sơ đồ Hình 1.
Theo dõi tiến triển của phản ứng bằng phương pháp sắc ký lớp mỏng (SKLM) sử
dụng bản mỏng silica gel 60 F254 (Merck) với hệ dung môi khai triển phù hợp. Vết sắc
ký được quan sát nhờ ánh sáng thường và dưới đèn UV bước sóng 254 nm. Đo nhiệt
độ nóng chảy bằng máy EZ-Melt (Mỹ). Sử dụng phương pháp cất, chiết lỏng-lỏng, kết
tủa/kết tinh, lọc, rửa để tinh chế sản phẩm.
Xác định cấu trúc của sản phẩm tổng hợp bằng các phương pháp phổ: MS được
ghi trên máy Agilent 1100 LC-MSD-Trap-SL, sử dụng dung môi MeOH. IR được ghi
trên máy Perkin Elmer, sử dụng kỹ thuật viên nén kali bromid trong vùng 4000-400
cm-1. NMR được ghi trên máy Bruker 500 MHz, sử dụng dung môi DMSO-d6, chất
chuẩn nội là tetramethyl silan. Thực hiện đo các phổ tại Viện Hóa học - Viện Hàn lâm
Khoa học và Công nghệ Việt Nam.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 37-44
39
Hình 1. Sơ đồ tổng hợp hai dẫn chất ester của di-O-(2-hydroxyethyl)-curcumin
Thử hoạt tính sinh học được thực hiện tại Viện Công nghệ sinh học - Viện Hàn
lâm Khoa học và Công nghệ Việt Nam.
Hoạt tính chống oxy hoa được thực hiện theo phương pháp DPPH (1,1-diphenyl-2-
picrylhydrazyl), máy đọc ELISA 96 giếng (Bio-rad), chất đối chứng acid ascorbic.
Nồng độ mẫu thử: 200, 100, 50, 25, 12,5 g/mL. Đo các giá trị mật độ quang OD
(optical density) hấp thụ tại các giếng, tính khả năng trung hòa gốc oxy hóa tự do SA
(scavenging activities) sinh ra từ DPPH theo công thức: % SA = (ODđối chứng - ODmẫu
thử)*100/ODđối chứng (%).
Hoạt tính ức chế sinh NO được thử trên dòng đại thực bào RAW264.7, sử dụng máy
đọc ELISA 96 giếng (Bio-rad), chất tham khảo NG-methyl-L-arginin acetat (L-
NMMA), chứng âm: lipopolysaccharid (LPS). Xác định khả năng ức chế sinh NO của
mẫu theo công thức: % ức chế = 100 % - [hàm lượng NOmẫu thử/hàm lượng
NOLPS]*100. Ngoài ra, đánh giá tác động của mẫu nghiên cứu đến sự sống sót của tế
bào RAW264.7 nhờ phép thử MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoli
bromid).
Hoạt tính gây độc tế bào ung thư được thử trên các dòng HepG2 (ung thư gan ở
người, human hepatocellular carcinoma), MCF7 (ung thư vú ở người, human breast
carcinoma), HL-60 (ung thư bạch cầu cấp ở người) và Hela (ung thư tử cung ở người,
human cervical carcinoma). Trong đó, đối với các dòng tế bào HepG2, MCF7 và
Hela sử dụng sulforhodamin B (SRB) để nhuộm protein tế bào, qua đó xác định mật
độ quang OD [5]. Còn với dòng tế bào HL-60 sử dụng phương pháp MTT để đánh giá
[4]. Các phép thử được lặp lại 3 lần. Giá trị SC50 (nồng độ trung hòa được 50 % gốc tự
do của DPPH) và các giá trị IC50 (nồng độ ức chế 50 % sự hình thành NO, hoặc 50 %
sự phát triển tế bào) được xác định nhờ phần mềm máy tính TableCurve 2D v4.
Thực nghiệm và kết quả Tổng hợp hóa học
Tổng hợp di-O-(2-hydroxyethyl)-curcumin (LH-22017)
Cho hỗn hợp gồm 2,0 g (5,4 mmol; 1 eq) curcumin; 3,38 g (24,5 mmol; 4,5 eq)
kali carbonat khan; 160 mL aceton khan và 3,8 mL (53,6 mmol; 10 eq) 2-bromoethanol
vào bình cầu 500 mL có sinh hàn. Khuấy và đun hồi lưu hỗn hợp trong 10 giờ (theo dõi
phản ứng bằng SKLM với hệ dung môi DCM : MeOH = 9 : 1). Kết thúc phản ứng, lọc
loại kali carbonat, cất loại aceton. Hòa tan cắn còn lại với 200 mL DCM trong bình

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 37-44
40
chiết 500 mL. Rửa pha hữu cơ 1 lần với 50 mL nước cất. Tiếp tục thêm 150 mL nước
cất vào pha hữu cơ trong bình chiết, kiềm hóa bằng dung dịch NaOH 1M đến pH 12,
lắc kỹ bình chiết, tách lấy pha hữu cơ. Rửa lại pha hữu cơ 3 lần với nước cất đến pH
trung tính, làm khan bằng natri sulfat. Lọc loại chất làm khan, cất loại bớt dung môi
đến thể tích còn khoảng 30 mL. Để kết tinh qua đêm thu được tinh thể hình kim màu
đỏ cam. Sấy khô sản phẩm ở 60 °C trong 2 giờ, thu được LH-22017. Khối lượng 1,22
g (hiệu suất 29,3 %). t°nc 176,5-178,1 °C. Rf = 0,54 (DCM : MeOH = 9 : 1). ESI-MS
(MeOH), m/z: 457,0 [M+H]+; 455,1 [M-H]- (CTPT C25H28O8, M = 456,1). IR (KBr),
ῡmax (cm-1): 3526, 3328 (O-Halcol); 3001 (C-Hkhông no); 2925, 2866 (C-Hno); 1625
(C=Oβ-diceton); 1598, 1582, 1511 (C=C); 1262, 1228, 1133, 1081 (C-O). 1H-NMR (500
MHz, DMSO-d6), δ (ppm): 7,58 (2H, d, J = 16,0 Hz, H-1, H-7); 7,35 (2H, d, J = 1,0
Hz, H-2’, H-2”); 7,24 (2H, dd, J1 = 1,5 Hz, J2 = 8,5 Hz, H-6’, H-6”); 7,01 (2H, d, J =
8,0 Hz, H-5’, H-5”); 6,82 (2H, d, J = 15,5 Hz, H-2, H-6); 6,10 (1H, s, H-4, CH enol);
4,87 (1H, br.s, H-4, OH enol); 4,03 (4H, t, J = 5,0 Hz, H-8’, H-8”); 3,84 (6H, s, J = 10
Hz H-7’, H-7”); 3,73 (4H, pseudo singlet, H-8’, H-8”). 13C-NMR (125 MHz, DMSO-
d6), δ (ppm): 183,2 (C-3, C-5); 150,4 (C-4’, C-4’’); 149,1 (C-3’, C-3”); 140,4 (C-1, C-
7); 127,5 (C-1’, C-1’’); 122,8 (C-2, C-6); 122,0 (C-6’, C-6’’); 112,7 (C-5’, C-5’’);
110,7 (C-2’, C-2’’); 101,0 (C-4); 70,2 (C-7’, C-7’’); 59,4 (C-8’, C8’’); 55,5 (C-9’, C-
9’’).
Tổng hợp dẫn chất 2,2’-(curcumin-di-O-yl) diethyl disuccinat (LH-12020)
Tinh chế dung môi: Lắc 200 mL dung môi cloroform với H2SO4 đặc 3 lần, mỗi
lần 10 mL, sau đó rửa pha hữu cơ với nước cất 3 lần, mỗi lần 40 mL. Làm khan với
Na2SO4, lọc loại bỏ chất làm khan. Cất bằng máy cất quay ở 61 °C thu lấy cloroform.
Hòa tan 0,20 g (0,44 mmol; 1 eq) LH-22017 vào 40 mL cloroform trong bình
cầu 2 cổ dung tích 250 mL. Cho vào bình 0,30 mL (2,2 mmol; 5 eq) TEA, khuấy đều.
Sau đó, tiếp tục cho 0,22 g (2,2 mmol; 5 eq) anhydrid succinic vào bình. Sục khí N2
vào khối phản ứng trong ~ 5 phút. Khuấy và đun hồi lưu hỗn hợp trong 2 giờ. Sau đó,
chuyển hỗn hợp phản ứng vào bình chiết 250 mL. Rửa pha hữu cơ bằng HCl 1M lạnh
3 lần, mỗi lần 10 mL để loại TEA. Rửa tiếp pha hữu cơ với nước cất lạnh 4 lần, đến
pH 4. Làm khan pha hữu cơ bằng natri sulfat khan. Lọc loại chất làm khan. Cất thu hồi
dung môi dưới áp suất giảm để loại cloroform đến khi thu được cắn khô. Cắn khô
được rửa với n-hexan 3 lần thu được sản phẩm thô. Hòa tan sản phẩm thô trong 5 mL
IPA, sau đó để kết tinh qua đêm ở nhiệt độ 0 °C. Lọc thu tinh thể, rửa 3 lần bằng IPA
lạnh. Sấy khô sản phầm dưới đèn hồng ngoại trong 60 phút. Cân thu được sản phẩm
LH-12020 là bột vô định hình màu vàng. Khối lượng 0,22 g (hiệu suất 76,6 %). t°nc
119,2-121,5 °C. Rf = 0,52 (DCM : MeOH,7/1). ESI-MS (MeOH), m/z: 657,0 [M+H]+;
655,1 [M-H]- (CTPT C33H36O14, M = 656,12). IR (KBr), ῡmax (cm-1): 3449 (-OH);
2924 (C-Hno); 1735, 1695 (C=Oester, acid); 1626 (C=Oβ-diceton); 1589, 1566, 1508 (C=C);
1263, 1232, 1162, 1140 (C-O). 1H-NMR (500 MHz, DMSO-d6), δ (ppm): 12,19 (2H,
br.s, OH acid); 7,59 (2H, d, J = 15,5 Hz, H-1, H-7); 7,37 (2H, br.s, H-2’,H-2’’); 7,25
(2H, br.d, J = 7,5 Hz, H-6’, H-6’’); 7,04 (2H, d, J = 8,5 Hz, H-5’, H-5’’); 6,84 (2H, d,
J = 16,0 Hz, H-2, H-6); 6,12 (1H, s, H-4, CH enol); 4,35 (4H, t, J = 4,5 Hz, H-9’, H-
9’’); 4,23 (4H, t, J = 4,5 Hz, H-8’, H-8’’); 3,84 (6H, s, H-7’, H-7’’); 2,54 (4H, t, J =
6,0 Hz, H-11’, H-11’’); 2,48 (4H, t, J = 6,5 Hz, H-12’, H-12’’). 13C-NMR (125 MHz,
DMSO-d6), δ (ppm): 183,1 (C-13’, C-13’’); 173,3 (C-10’, C-10’’); 172,1 (C-3, C-5);
149,8 (C-3’, C-3’’); 149,2 (C-4’, C-4’’); 140,3 (C-1, C-7); 128,1 (C-2, C-6); 122,7 (C-
1’, C-1’’); 122,3 (C-6’, C-6’’); 113,3 (C-5’, C-5’’); 111,1 (C-2’, C-2’’); 101,1 (C-4);

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 37-44
41
66,6 (C-8’, C-8’’); 62,6 (C-9’, C-9’’); 55,7 (C-7’, C-7’’); 28,6 (C-11’, C-11’’, C-12’,
C-12’’).
Tổng hợp dẫn chất 2,2’-(curcumin-di-O-yl) diethyl diglutarat (LH-22020):
Hòa tan 0,20 g (0,44 mmol; 1 eq) LH-22017 vào 40 mL cloroform đã tinh chế
trong bình cầu 2 cổ dung tích 250 mL. Cho vào bình 0,36 mL (2,64 mmol; 6 eq) TEA,
khuấy đều. Tiếp tục cho 0,30 g (2,64 mmol; 6 eq) anhydrid glutaric vào bình. Sục khí
N2 vào khối phản ứng trong khoảng 5 phút. Khuấy và đun hồi lưu hỗn hợp phản ứng
trong 7 giờ. Theo dõi bằng SKLM với hệ dung môi DCM: MeOH (7 : 1). Sau khi phản
ứng kết thúc, chuyển hỗn hợp phản ứng vào bình chiết 250 mL. Rửa pha hữu cơ bằng
HCl 1M lạnh 3 lần, mỗi lần 10 mL để loại TEA. Rửa tiếp pha hữu cơ với nước cất lạnh
4 lần đến pH 4. Làm khan pha hữu cơ bằng natri sulfat khan. Lọc loại chất làm khan.
Cất chân không thu hồi dung môi đến khi thu được cắn khô. Rửa cắn 3 lần với n-hexan
để kết tủa sản phẩm. Cho 5 mL IPA vào cắn, sau đó để kết tinh qua đêm ở nhiệt độ 0
°C. Lọc thu tinh thể, rửa 3 lần với IPA lạnh. Sấy khô sản phẩm dưới đèn hồng ngoại
trong 60 phút. Cân thu được sản phẩm LH-22020 là bột vô định hình màu vàng cam.
Khối lượng 0,16 g (hiệu suất 53,4 %). t°nc 113,5 - 116,9 °C. Rf = 0,51 (DCM : MeOH,
9/1). ESI-MS (MeOH), m/z: 685,1 [M+H]+, 683,1 [M-H]- (CTPT C35H40O14, M =
684,12). IR (KBr), ῡmax (cm-1): 3428 (-OH); 2945 (C-Hno); 1728, 1703 (C=Oester, acid);
1620 (C=Oβ-diceton); 1580, 1511 (C=C); 1257, 1176, 1132 (C-O). 1H-NMR (500 MHz,
DMSO-d6), δ (ppm): 12,06 (2H, br.s, OH acid); 7,58 (2H, d, J = 15,5 Hz, H-1, H-7);
7,36 (2H, br.s, H-2’, H-2’’); 7,25 (2H, br.d, J = 8,0 Hz, H-6’, H-6’’); 7,03 (2H, d, J =
8,0 Hz, H-5’, H-5’’); 6,83 (2H, d, J = 16,0 Hz, H-2, H-6); 6,11 (1H, s, H-4, CH enol);
4,36 (4H, pseudo singlet, H-9’, H-9’’); 4,23 (4H, pseudo singlet, H-8’, H-8’’); 3,83
(6H, s, J = 8,0 Hz, H-7’, H-7’’); 2,37 (4H, t, J =7 ,5 Hz, H-13’, H-13’’); 2,26 (4H, t, J
= 7,0 Hz, H-11’, H-11’’); 1,75 (4H, quintet, J = 7,5 Hz, H-12’, H-12’’);. 13C-NMR
(125 MHz, DMSO-d6), δ (ppm): 183,2 (C-14’, C-14’’) ; 174,0 (C-10’, C-10’’); 172,5
(C-3, C-5); 149,8 (C-3’, C-3’’); 149,3 (C-4’, C-4’’); 140,3 (C-1, C-7); 128,1 (C-2, C-
6); 122,7 (C-1’, C-1’’); 122,3 (C-6’, C-6’’); 113,3 (C-5’, C-5’’); 111,0 (C-2’, C-2’’);
101,1 (C-4); 66,6 (C-8’, C-8’’); 62,3 (C-9’, C-9’’); 55,7 (C-7’, C-7’’); 32,5 (C-11’, C-
11’’, C-13’, C-13’’); 19,9 (C-12’, C-12’’).
Thử tác dụng sinh học
Các kết quả đánh giá hoạt tính sinh học được trình bày trong các bảng 1, 2 và 3.
Bảng 1. Kết quả thử hoạt tính chống oxy hóa hệ DPPH* Nồng độ LH-12020 LH-22020 Curcumin acid ascorbic
SC50 (µg/mL) 196,20 ± 6,25 210,05 ± 15,52 9,29 ± 1,23 7,66 ± 0,29
Bảng 2. Khả năng ức chế sinh NO trên dòng tế bào RAW264.7* Nồng độ LH-12020 LH-22020 Curcumin L-NMMA
IC50 (µM) 11,92 ± 0,86 12,06 ± 0,42 17,89 ± 1,98 38,83 ± 1,7
Bảng 3. Kết quả thử hoạt tính gây độc tế bào ung thư*
Hợp chất IC50 (µM)
Hep-G2 MCF-7 HL-60 Hela
LH-12020 69,69±1,50 46,13±5,03 62,13 ± 3,36 73,67±2,36
LH-22020 30,36±2,66 36,19±3,80 32,56 ± 2,56 33,18±3,67
Curcumin 43,00±3,69 80,54±1,64 74,19±7,03
Ellipticin (chứng dương) 1,38 ± 0,08 1,58 ± 0,20 1,34 ± 0,12 1,46 ± 0,16
Ghi chú: * Các kết quả chính xác với r2≥ 0,95.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 37-44
42
Bàn luận Về các phản ứng tổng hợp
Phản ứng tạo dẫn chất di-O-(2-hydroxyethyl)-curcumin (LH-22017) đã được
tác giả C. Changtam đề cập trong tài liệu [11]. Khi alkyl hóa curcumin với tác nhân 2-
bromoethanol, phản ứng không cho sản phẩm chọn lọc, mà thường có các tạp chất là
sản phẩm của quá trình mono-alkyl hóa, tri-alkyl hóa (xảy ra trên OH phenol, nhóm
CH2 linh động và OH enol của dạng hỗ biến) và chính curcumin dư. Tài liệu [11] sử
dụng sắc ký cột để tinh chế, cho hiệu suất thu LH-22017 đạt 35 %. Nghiên cứu của
chúng tôi không sử dụng đến sắc ký cột để tinh chế. Điều này đạt được là nhờ kiểm
soát các điều kiện phản ứng và thay đổi quá trình tinh chế. Dựa theo SKLM, chúng tôi
xác định thời điểm dừng phản ứng là khi bắt đầu xuất hiện tạp tri-alkyl hóa. Khi đó,
tạp chủ yếu của phản ứng chỉ là curcumin và dẫn chất mono-alkyl hóa (mono-O-(2-
hydroxyethyl)-curcumin). Hai tạp này đều có nhóm OH phenol tự do, tan tốt trong
kiềm, nên đã được chúng tôi loại đi khi rửa với NaOH ở pH 12 ở tỷ lệ thể tích pha hữu
cơ/ pha nước kiềm phù hợp (~ 1/1).
Các điều kiện của phản ứng tổng hợp LH-12017 và LH-22017 ban đầu được
dựa theo tài liệu [14], trong đó sử dụng xúc tác TEA, dung môi là DCM và thời gian
hồi lưu ở 40 °C là 2 giờ. Tuy nhiên, khi thực hiện theo tài liệu [14], chúng tôi nhận
thấy phản ứng xảy ra chậm, còn dư nhiều nguyên liệu trên SKLM (ngay cả khi kéo dài
thời gian đến 24 giờ). Sử dụng cloroform để thay thế cho DCM giúp nâng cao nhiệt độ
phản ứng (đến ~ 61 °C) và đẩy nhanh tốc độ phản ứng. Khi đó, các tạp (bao gồm
nguyên liệu LH-22017, sản phẩm phụ acyl hóa trên OH phenol, trên OH enol của cấu
trúc hỗ biến) trên sắc ký lớp mỏng có xuất hiện nhưng không đáng kể. Tác nhân
anhydrid glutaric có khả năng phản ứng kém hơn so với andhydrid succinic, nên lượng
sử dụng cần nhiều hơn và thời gian phản ứng cũng lâu hơn. Việc dùng cloroform mới
tinh chế cũng giúp hạn chế tạp và làm phản ứng xảy ra nhanh hơn. Xử lý phản ứng với
HCl 1M lạnh nhằm mục đích tạo muối với TEA, chuyển sản phẩm về dạng acid
RCOOH và đảm bảo hạn chế tối đa thủy phân liên kết ester. Giai đoạn rửa với nước
lạnh giúp loại bỏ các tạp tan trong nước trong đó có diacid (succinic và glutaric). Cần
chú ý thực hiện cất quay loại dung môi ở nhiệt độ thấp (~ 35 °C) để tránh thủy phân
liên kết ester dạng hemi-carboxylat. Các tạp liên quan chứa khung diaryl heptadien
(nguyên liệu LH-22017, dẫn chất monoacyl và triacyl hóa) được loại hoàn toàn khi xử
lý với n-hexan và kết tinh lạnh trong IPA.
Về cấu trúc các chất
Phổ MS của cả 3 chất LH-22017, LH-12020, LH-22020 đều cho pic m/z đúng
với khối lượng phân tử tương ứng của các chất, không xuất hiện các pic tạp liên quan
của mono-alkyl hóa, mono-acyl hóa hay tri-acyl hóa. Phổ IR cho đủ bộ đỉnh hấp thụ
đặc trưng của các nhóm chức trong khung curucmin, trong đó dải hấp thụ của nhóm β-
diceton xuất hiện với số sóng giảm mạnh (1620-1626 cm-1) so với số sóng của nhóm
ceton thông thường (có ῡmax = 1715 cm-1). Điều này chỉ ra, cả 3 dẫn chất đều tồn tại ở
dạng hỗ biến ceto-enol với liên kết hydro nội phân tử. Bên cạnh đó, trên phổ IR của
LH-12020 và LH-22020 còn xuất hiện thêm đỉnh hấp thụ đặc trưng của nhóm chức
C=O ester (1735, 1728 cm-1) và C=O acid (1703, 1695 cm-1). Trên phổ 1H-NMR của
cả 3 dẫn chất xuất hiện đủ bộ tín hiệu proton theo dự kiến. Trong đó, bộ tín hiệu H-2,
H-6, H-1, H-7 của khung heptadien xuất hiện dưới dạng doublet với hằng số J 15,5-
16,0 Hz của cấu hình 1E, 6E giống như cấu hình khung curcumin thiên nhiên ban đầu.
Quan sát thấy rõ proton của OH acid và các nhóm -CH2- trong mạch nhánh

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 37-44
43
hemisucinyl, hemiglutaryl . Phổ 13C-NMR thể hiện đầy đủ bộ tín hiệu carbon đặc
trưng cho phân tử 3 dẫn chất. Các số liệu phổ NMR thể hiện tính đối xứng của các cấu
trúc (nhân thơm, các mạch nhánh). Các kết quả phân tích trên giúp khẳng định chất
trung gian LH-22017 và 2 sản phẩm diacid LH-12020, LH-22020 có cấu trúc đúng
như dự kiến.
Về hoạt tính sinh học
Kết quả đánh giá hoạt tính cho thấy, LH-12020 và LH-22020 có hoạt tính
chống oxy hóa yếu hơn so với curcumin qua phép thử trung hòa gốc tự do của DPPH
với SC50 (µg/mL) lần lượt là 196,20 ± 6,25 và 210,05 ± 15,52 so với curcumin là 9,29
± 1,23 µg/mL. Khả năng chống oxy hóa của curcumin cao hơn LH-12020 và LH-
22020 có thể do trong phân tử curcumin còn có 2 nhóm -OH phenol tự do có khả năng
dọn dẹp gốc tự do, trong khi hai dẫn chất LH-12020 và LH-22020 cả hai nhóm -OH
phenol tự do đều đã bị thế, dẫn tới tác dụng chống oxy hóa của phân tử giảm mạnh.
Điều này cũng phù hợp với tác giả Michal H. đã phân tích về cơ chế chống oxy hóa
của curcumin [16].
Tuy nhiên, cả hai dẫn chất đều thể hiện hoạt tính chống viêm thông qua ức chế
sinh NO mạnh với IC50 của LH-12020 và LH-22020 lần lượt là: 11,92 ± 0,86 và 12,06
± 0,42 µM, mạnh hơn curcumin (IC50 = 17,89 ± 1,98 µM).
LH-12020 và LH-22020 thể hiện hoạt tính gây độc tế bào trên tất cả các dòng
tế bào ung thư thử nghiệm. Trong đó, LH-12020 thể hiện hoạt tính gây độc tế bào tốt
hơn 2 lần so với curcumin trên dòng tế bào MCF-7; LH-22020 thể hiện hoạt tính trên
các dòng tế bào Hela, MCF-7 và HepG2 tốt hơn so với curcumin, tương ứng gấp 2,24;
2,2 và 1,2 lần.
Kết luận
Đã tổng hợp được hai dẫn chất mới là 2,2’-(curcumin-di-O-yl) diethyl
disuccinat (LH-12020) và 2,2’-(curcumin-di-O-yl) diethyl diglutarat (LH-22020) từ
curcumin qua 2 giai đoạn: di-alkyl hóa tạo ra LH-22017, sau đó acyl hóa LH-22017
bằng các tác nhân anhydrid glutaric và anhydrid succinic trong triethylamin/cloroform.
Kết quả thử hoạt tính sinh học xác định được, LH-12020 và LH-22020 có hoạt tính
chống oxy hóa yếu hơn so với curcumin (SC50 9,29 ± 1,23µg/mL) qua phép thử trung
hòa gốc tự do của DPPH với SC50 lần lượt là 196,20 ± 6,25 và 210,05 ± 15,52 µg/mL.
Cả hai dẫn chất đều thể hiện hoạt tính chống viêm thông qua ức chế sinh NO khá
mạnh với IC50 của LH-12020 và LH-22020 lần lượt là 11,92 ± 0,86 và 12,06 ± 0,42
µM, mạnh hơn curcumin (17,89 ± 1,98 µM). LH-12020 và LH-22020 thể hoạt tính
gây độc tế bào trên tất cả các dòng đã thử nghiệm. Trong đó, LH-12020 thể hiện hoạt
tính gây độc tế bào tốt hơn 2 lần so với curcumin trên dòng tế bào MCF-7; LH-22020
thể hiện hoạt tính trên các dòng tế bào Hela, MCF-7 và HepG2 tốt hơn so với
curcumin, tương ứng gấp 2,24; 2,2 và 1,2 lần. Tài liệu tham khảo
1. Fan X., Zhang C., Liu D.B. et al. (2013), “The clinical applications of curcumin:
current state and the future”, Curr. Pharm. Des., 19(11), pp. 2011-2031.
2. Yang C., Su X., Liu A. et al. (2013), “Advances in clinical study of curcumin”,
Curr. Pharm. Des., 19(11), pp. 1966-1973. 3. Priyadarsini K.I. (2014), “The chemistry of curcumin: from extraction to therapeutic
agent”, Molecules, 19(12), pp. 20091-20112.
4. Prasad S., Gupta S. C., Tyagi A. K. et al. (2014), “Curcumin, a component of
golden spice: From bedside to bench and back”, Biotech. Adv., 32(6), p. 1053-1064.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 37-44
44
5. Sunagawa Y., Katanasaka Y., Hasegawa K. et al. (2015), “Clinical applications of
curcumin”, PharmaNutrition, 3(4), pp. 131-135.
6. Bachmeier B. E., Melchart D. (2019), “Therapeutic effects of curcumin - from
traditional past to present and future clinical applications”, Int. J. Mol. Sci., 20, p.
3757.
7. Salem M., Rohani S., Gillies E.R. (2014), “Curcumin, a promising anti-cancer
therapeutic: a review of its chemical properties, bioactivity and approaches to cancer
cell delivery”, RSC Advances, 4(21), pp. 10815-10829.
8. Ratnatilaka Na Bhuket P., El-Magboub A., Haworth I. S. et al. (2017),
“Enhancement of curcumin bioavailability via the prodrug approach: challenges and
prospects”, Eur. J. Drug Metab. Pharmacokinet., 42(3), pp. 341-353.
9. Parvathy K.S. (2009), “Chemical approaches toward preparation of water-soluble
curcumin derivatives”, Ph.D. Thesis, University of Mysore, India.
10. Ding L., Ma S., Lou H. et al. (2015), “Synthesis and biological evaluation of
curcumin derivatives with water-soluble groups as potential antitumor agents: An in
vitro investigation using tumor cell lines”, Molecules, 20(12), pp. 21501-21514.
11. Changtam C., Hongmanee P., Suksamrarn A. (2010), “Curcuminoid analogs with
potent activity against Trypanosoma and Leishmania species”, Eur. J. Med. Chem.,
45(3), pp. 941-956.
12. Wichitnithad W., Nimmannit U., Wacharasindhu S. et al. (2011), “Synthesis,
characterization and biological evaluation of succinate prodrugs of curcuminoids for
colon cancer treatment”, Molecules, 16(2), pp. 1888-1900.
13. Muangnoi C., Ratnatilaka N. B. P., Jithavech P. et al. (2019), “Scale-up synthesis
and in vivo anti-tumor activity of curcumin diethyl disuccinate, an ester prodrug of
curcumin, in HepG2-Xenograft mice”, Pharmaceutics, 11(8), pii: E373.
14. Muangnoi C., Jithavech P., Ratnatilaka N. B. P. et al. (2018), “A curcumin-
diglutaric acid conjugated prodrug with improved water solubility and
antinociceptive properties compared to curcumin”, Biosci. Biotechnol. Biochem.
82(8), pp. 1301-1308.
15. Jornada D. H. dos Santos Fernandes G. F., Chiba D. E. et al. (2016), “The prodrug
approach: a successful tool for improving drug solubility”, Molecules, 21(1), p. 42.
16. Michal H. et al. (2014), “The Molecular Basis for the Pharmacokinetics and
Pharmacodynamics of Curcumin and Its Metabolites in Relation to Cancer”,
Pharmacol Rev, 66, pp. 222–307.

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 45-49
45
ĐIỂM TIN THÔNG TIN THUỐC – CẢNH GIÁC DƯỢC
Tránh sử dụng NSAIDs khi mang thai từ tuần thứ 20 trở đi: Khuyến cáo của
FDA Hoa Kỳ
Nguồn: https://www.fda.gov/safety/medical-product-safety-information/nonsteroidal-
anti-inflammatory-drugs-nsaids-drug-safety-communication-avoid-use-nsaids-
pregnancy-20
Điểm tin: Nguyễn Phương Thúy
Ngày 15/10/2020, Cơ quan Quản lý Dược phẩm và Thực phẩm Hoa Kỳ (FDA
Hoa Kỳ) khuyến cáo việc sử dụng các thuốc chống viêm không steroid (NSAID) sau
tuần thai thứ 20 trở đi có thể gây ra các bệnh lý thận hiếm gặp và nghiêm trọng ở thai
nhi. Bệnh lý thận có thể làm giảm lượng nước ối xung quanh thai nhi, dẫn tới các biến
chứng thai kỳ khác. Đối với NSAID phải kê đơn, FDA Hoa kỳ yêu cầu các cơ sở đăng
ký thuốc phải cập nhật tờ hướng dẫn sử dụng thuốc để bổ sung thông tin mô tả về nguy
cơ bệnh lý thận dẫn tới thiểu ối trong thai kỳ. Đối với NSAID không kê đơn (OTC)
được sử dụng cho người lớn, FDA Hoa Kỳ cũng có yêu cầu tương tự. Hiện nay, tờ
hướng dẫn sử dụng của các chế phẩm chứa NSAID lưu hành tại Hoa Kỳ mới chỉ có
cảnh báo tránh sử dụng NSAID trong 3 tháng cuối của thai kỳ vì nguy cơ gây ra các
bệnh lý cho thai nhi hoặc các biến chứng trong quá trình chuyển dạ, đồng thời khuyến
cáo phụ nữ mang thai và cho con bú cần trao đổi với cán bộ y tế trước khi sử dụng loại
thuốc này.
Thông tin dành cho người bệnh:
- Người bệnh mang thai từ tuần thứ 20 trở đi không nên sử dụng NSAID trừ khi
được bác sỹ chỉ định vì loại thuốc này có thể gây ra một số bệnh lý cho thai
nhi.
- Nhiều loại thuốc không kê đơn có chứa NSAID, bao gồm cả những loại thuốc
được sử dụng để giảm đau, điều trị triệu chứng của cảm lạnh, cúm và mất ngủ.
Vì vậy, người bệnh cần đọc kỹ tờ hướng dẫn sử dụng thuốc để xác định liệu
thuốc có chứa NSAID hay không.
- Trao đổi với bác sỹ hoặc dược sỹ nếu người bệnh có bất kỳ băn khoăn nào khi
sử dụng NSAID hoặc các loại chế phẩm có chứa NSAID.
- Các loại thuốc khác, ví dụ như paracetamol, là thuốc OTC có thể được sử dụng
để giảm đau và hạ sốt trong thai kỳ, tuy nhiên người bệnh cần trao đổi với
dược sỹ hoặc bác sỹ để được tư vấn và giúp đưa ra quyết định lựa chọn loại
thuốc phù hợp nhất.
Thông tin dành cho cán bộ y tế
- Sử dụng NSAID từ tuần thứ 20 trong thai kỳ trở đi có thể gây rối loạn chức
năng thận của thai nhi dẫn đến thiểu ối và trong một số trường hợp gây suy
thận ở trẻ sơ sinh. Vì vậy, FDA Hoa Kỳ khuyến cáo bác sỹ nên hạn chế kê đơn
NSAID cho phụ nữ mang thai trong khoảng thời gian từ 20 đến 30 tuần của

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 45-49
46
thai kỳ và tránh kê đơn sau tuần thứ 30 của thai kỳ. Nếu điều trị bằng NSAID
được xác định là cần thiết, cần sử dụng liều thấp nhất có hiệu quả trong thời
gian ngắn nhất có thể. Cân nhắc siêu âm theo dõi lượng nước ối nếu điều trị
NSAID kéo dài hơn 48 giờ và ngừng NSAID nếu phát hiện thiểu ối.
- Những nguy cơ trên có thể xảy ra trong vài ngày đến vài tuần sau khi dùng
thuốc. Tuy nhiên, đã ghi nhận trường hợp thiểu ối ngay trong 48 giờ sau khi
bắt đầu dùng NSAID và trường hợp thiểu ối không tự hồi phục khi ngừng sử
dụng thuốc.
- Biến chứng của tình trạng thiểu ối kéo dài có thể bao gồm co cứng các chi và
chậm trưởng thành phổi. Trong một số trường hợp có thể xuất hiện tình trạng
suy giảm chức năng thận ở trẻ sơ sinh sau khi xuất viện, dẫn tới cần phải thực
hiện các thủ thuật xâm lấn như truyền dịch hoặc lọc máu.
- Nếu cần thiết phải điều trị bằng NSAID trong khoảng thời gian từ tuần thứ 20
đến tuần thứ 30 của thai kỳ, hãy sử dụng ở mức liều thấp nhất có hiệu quả và
trong thời gian ngắn nhất có thể. Tránh kê đơn NSAID ở tuần thai thứ 30 trở đi
vì nguy cơ đóng sớm ống động mạch ở thai nhi.
- Các khuyến cáo trên không áp dụng cho aspirin liều thấp (81 mg) được kê đơn
trong một số tình trạng bệnh lý nhất định trong thai kỳ.
- Cân nhắc siêu âm theo dõi lượng nước ối nếu điều trị NSAID kéo dài hơn 48
giờ. Ngừng NSAID nếu xảy ra thiểu ối và theo dõi chặt chẽ các dấu hiệu lâm
sàng sau đó.
Nguy cơ xảy ra sai sót trong sử dụng thuốc khi sử dụng acid tranexamic đường
tiêm: Cảnh báo từ FDA Hoa Kỳ
Nguồn: https://www.fda.gov/drugs/drug-safety-and-availability/fda-alerts-healthcare-
professionals-about-risk-medication-errors-tranexamic-acid-injection-resulting
Điểm tin: Nguyễn Phương Thúy
Ngày 03/12/2020, Cơ quan Quản lý Dược phẩm và Thực phẩm Hoa Kỳ (FDA
Hoa Kỳ) cảnh báo cán bộ y tế nguy cơ vô ý tiêm nhầm acid tranexamic vào tủy sống
có thể dẫn đến những tổn thương nghiêm trọng, thậm chí đe dọa tính mạng như lên
cơn tai biến, loạn nhịp tim, liệt chi dưới, tổn thương thần kinh vĩnh viễn và tử vong.
Trong những báo cáo mà FDA Hoa Kỳ nhận được, bệnh nhân được chỉ định gây tê tủy
sống bằng các thuốc gây tê như bupivacain nhưng lại bị tiêm nhầm thành acid
tranexamic.
Acid tranexamic thường ở dạng ống hoặc lọ đơn liều dung tích 10 mL chứa
1.000 mg acid tranexamic và được tiêm tĩnh mạch. Acid tranexamic, bupivacain và các
thuốc đường tiêm khác sử dụng trong quá trình phẫu thuật có hình thức tương tự nhau
nên dễ gây nhầm lẫn cho cán bộ y tế.
Để hạn chế việc nhầm lẫn đường dùng của thuốc, FDA Hoa Kỳ yêu cầu thay
đổi nhãn thuốc, tờ hướng dẫn sử dụng và bao bì của các chế phẩm chứa acid
tranexamic, đồng thời khuyến cáo:

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 45-49
47
- Cán bộ y tế cần thận trọng khi bảo quản thuốc tiêm acid tranexamic, sắp xếp vị
trí các lọ thuốc acid tranexamic tách biệt với các loại thuốc khác, quay nhãn
thuốc ra ngoài để tránh việc nhận diện thuốc qua màu sắc của nhãn thuốc
- Dán thêm bảng cảnh báo tại nơi đặt các lọ thuốc tiêm acid tranexamic
- Luôn kiểm tra nhãn của lọ thuốc trước khi sử dụng
Atezolizumab và nguy cơ phản ứng có hại trên da nghiêm trọng: Thông tin từ
Medsafe
Nguồn: https://www.medsafe.govt.nz/safety/DHCPLetters/TecentriqNovember2020.pdf
Điểm tin: Nguyễn Phương Thúy
Nhận được sự đồng ý của Cơ quan Quản lý An toàn Dược phẩm và Thiết bị Y
tế New Zealand (Medsafe), Công ty Dược phẩm Roche tại New Zealand, đã gửi thư
cho nhân viên y tế (Dear Healthcare Professional Letter) để cập nhật thông tin an toàn
về thuốc atezolizumab.
Thông tin chính
- Các phản ứng có hại trên da nghiêm trọng (SCAR) liên quan đến việc sử dụng thuốc,
bao gồm cả nhóm thuốc ức chế điểm kiểm soát miễn dịch, tuy hiếm gặp nhưng có khả
năng gây độc cho da và có thể dẫn đến tử vong.
- Phân tích toàn diện dữ liệu hiện có trong chương trình phát triển lâm sàng
atezolizumab đã xác định được nguy cơ xảy ra SCAR sau khi sử dụng atezolizumab.
Tỷ lệ mắc SCAR ở tất cả các mức độ trong nghiên cứu lâm sàng sử dụng phác đồ đơn
độc và phác đồ phối hợp với thuốc khác lần lượt là 0,7% và 0,6%.
- Mục Cảnh báo/Thận trọng trong tờ hướng dẫn sử dụng thuốc atezolizumab lưu hành
ở New Zealand sẽ được bổ sung, chỉnh sửa để cung cấp thông tin và hướng dẫn quản
lý bệnh nhân nghi ngờ gặp SCAR. Mục phản ứng có hại của thuốc cũng được cập nhật
tần suất xuất hiện của phản ứng có hại này.
Thông tin chung
SCAR là một nhóm triệu chứng không đồng nhất của các phản ứng trên da liên
quan đến thuốc bùng phát qua trung gian miễn dịch. Mặc dù hiếm khi xảy ra nhưng
những biến cố này có khả năng gây tử vong và chủ yếu được cấu thành bởi các biến
chứng tổng quát cấp tính như mụn mủ, hội chứng Stevens-Johnson (SJS), hoại tử biểu
bì nhiễm độc (TEN) và phát ban do thuốc cùng với tăng bạch cầu ái toan và các triệu
chứng toàn thân (DRESS). Theo dữ liệu dịch tễ học, tỷ lệ mắc bệnh SJS và TEN trong
dân cư nói chung lần lượt dao động từ 0,8 đến 5,3 và 1,2 đến 6 trên một triệu người-
năm
Một phân tích tích lũy trong cơ sở dữ liệu an toàn của Công ty Dược phẩm
Roche về atezolizumab đã ghi nhận 99 trường hợp, trong đó 36 trường hợp SCAR đã
được xác nhận bằng mô bệnh học hoặc chẩn đoán chuyên khoa. Khoảng 23.654 bệnh
nhân tham gia thử nghiệm lâm sàng và 106.316 bệnh nhân tham gia giám sát hậu mại
đã phơi nhiễm với atezolizumab kể từ ngày 17 tháng 5 năm 2020. Tỷ lệ mắc SCAR ở
tất cả mức độ nghiêm trọng từ đơn trị liệu atezolizumab (N = 3178) hoặc phối hợp
atezolizumab với thuốc khác (N = 4371) trong các nghiên cứu lâm sàng do công ty tài

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 45-49
48
trợ lần lượt là 0,7 % và 0,6 %. Một trường hợp tử vong do TEN đã được báo cáo ở một
bệnh nhân nữ 77 tuổi sử dụng atezolizumab đơn độc.
Do đó, Medsafe và Công ty Dược phẩm Roche New Zealand khuyến cáo:
- Đối với các phản ứng nghi ngờ là SCAR, nên chuyển bệnh nhân đến bác sĩ da
liễu để được chẩn đoán và xử trí.
- Ngừng sử dụng atezolizumab khi có dấu hiệu bị SJS hoặc TEN. Ngừng sử
dụng atezolizumab vĩnh viễn khi có chẩn đoán xác định SJS hoặc TEN.
- Thận trọng khi xem xét việc sử dụng atezolizumab ở bệnh nhân đã có tiền sử
phản ứng có hại trên da nghiêm trọng hoặc đe dọa tính mạng khi điều trị với các chất
chống ung thư kích thích miễn dịch khác.
Rối loạn tâm thần liên quan đến cloroquin và hydroxycloroquin: Thông tin từ
EMA
Nguồn: https://www.ema.europa.eu/en/news/meeting-highlights-pharmacovigilance-
risk-assessment-committee-prac-23-26-november-2020
Điểm tin: Nguyễn Phương Thúy
Ngày 27/11/2020, Ủy ban đánh giá nguy cơ Cảnh giác Dược của Cơ quan Quản
lý Dược phẩm châu Âu (PRAC) đã khuyến cáo cập nhật thông tin của tất cả các chế
phẩm chưa cloroquin hoặc hydroxycloroquin sau khi báo cáo đánh giá tất cả các dữ
liệu hiện có xác nhận mối liên quan giữa việc sử dụng các thuốc này và nguy cơ rối
loạn tâm thần và có hành vị tự tử.
Quá trình đánh giá này được khởi động từ tháng 05/2020 sau khi EMA nhận
được thông tin về 6 ca rối loạn tâm thần ở những bệnh nhân mắc COVID-19 sử dụng
hydroxycloroquin cao hơn liều khuyến cáo từ Cơ quan Quản lý Dược phẩm Tây Ban
Nha (Spanish Medicines Agency – AEMPS). Cloroquin và hydroxycloroquin được
cấp phép trong lãnh thổ EU để điều trị một số bệnh tự miễn nhất định như viêm khớp
dạng thấp và lupus ban đỏ cũng như để dự phòng và điều trị sốt rét. Các thuốc này
không được phê duyệt để điều trị COVID-19 nhưng trong thực tế chúng đã được sử
dụng cho những bệnh nhân nhiễm loại virus này (off-label). Tuy nhiên, trong những
thử nghiệm lâm sàng ngẫu nhiên với cỡ mẫu lớn, cloroquin và hydroxycloroquin chưa
chứng minh bất kỳ lợi ích nào trong điều trị COVID-19.
Khi đánh giá lại việc sử dụng hai thuốc này trong đại dịch COVID-19, EMA đã
có thông báo gửi tới cán bộ y tế về nguy cơ của thuốc vào tháng 4 và tháng 5
năm 2020. Ngay cả khi được sử dụng theo đúng chỉ định và liều dùng đã được phê
duyệt, cloroquin và hydroxycloroquin vẫn có thể gây ra rối loạn tâm thần phổ rộng.
Trong tờ hướng dẫn sử dụng của một số chế phẩm chứa cloroquin và
hydroxycloroquin, các triệu chứng loạn thần và hành vi tự sát hoặc có ý định tự sát
được coi là phản ứng hiếm gặp và chưa xác định rõ tần suất xuất hiện.
Bản đánh giá xác định rối loạn thâm thần có thể xảy ra ở mức độ nghiêm trọng
trên cả bệnh nhân có hoặc không có tiền sử bệnh lý tâm thần. Dựa trên các dữ liệu hiện
có, bản đánh giá nhận định rằng phản ứng này có thể xuất hiện ngay trong tháng đầu

Nghiên cứu Dược & Thông tin thuốc, 2020, Tập 11, Số 6, trang 45-49
49
tiên sử dụng hydroxycloroquin. Đối với cloroquin, hiện chưa có đủ dữ liệu để xác định
thời gian khởi phát.
PRAC khuyến cáo cập nhật tờ hướng dẫn sử dụng của các thuốc này để cảnh
báo cán bộ y tế về nguy cơ tự sát và các rối loạn tâm thần khác. Bệnh nhân sử dụng
cloroquin hoặc hydroxycloroquin nếu thấy xuất hiện các triệu chứng tâm thần (ví dụ:
kích động, lo âu, ảo giác, lú lẫn, trầm cảm, xuất hiện ý nghĩ tự hành hạ bản thân hoặc
tự sát) hoặc những người xung quanh bệnh nhân phát hiện những triệu chứng này, cần
đến gặp bác sĩ để được thăm khám ngay lập tức.




















