
Immunofluorescent visualization of mouse interneuron subtypes

The FASEB Journal • Research Communication
Neuronal selenoprotein expression is required forinterneuron development and prevents seizures andneurodegeneration
Eva K. Wirth,* Marcus Conrad,§ Jochen Winterer,† Christian Wozny,†
Bradley A. Carlson,� Stephan Roth,* Dietmar Schmitz,† Georg W. Bornkamm,§
Vincenzo Coppola,¶ Lino Tessarollo,¶ Lutz Schomburg,‡ Josef Kohrle,‡
Dolph L. Hatfield,� and Ulrich Schweizer*,‡,1
*Neurobiology of Selenium and †Cellular and Molecular Neurobiology, Neuroscience ResearchCenter, and ‡Institute for Experimental Endocrinology, Charite-Universitatsmedizin Berlin, Berlin,Germany; §Institute of Clinical Molecular Biology and Tumour Genetics, Helmholtz ZentrumMunchen, Munich, Germany; �Molecular Biology of Selenium Section, Laboratory of CancerPrevention, Center for Cancer Research, National Cancer Institute, National Institutes of Health,Bethesda, MD, USA; and ¶Neural Development Group, Mouse Cancer Genetics Program, NationalCancer Institute, Frederick, Maryland, USA
ABSTRACT Cerebral selenium (Se) deficiency is as-sociated with neurological phenotypes including sei-zures and ataxia. We wanted to define whether neuronsrequire selenoprotein expression and which selenopro-teins are most important, and explore the possiblepathomechanism. Therefore, we abrogated the expres-sion of all selenoproteins in neurons by genetic inacti-vation of the tRNA[Ser]Sec gene. Cerebral expressionof selenoproteins was significantly diminished in themutants, and histological analysis revealed progres-sive neurodegeneration. Developing interneuronsfailed to specifically express parvalbumin (PV) in themutants. Electrophysiological recordings, beforeovert cell death, showed normal excitatory transmis-sion, but revealed spontaneous epileptiform activityconsistent with seizures in the mutants. In developingcortical neuron cultures, the number of PV� neuronswas reduced on combined Se and vitamin E depriva-tion, while other markers, such as calretinin (CR) andGAD67, remained unaffected. Because of the syner-gism between Se and vitamin E, we analyzed micelacking neuronal expression of the Se-dependentenzyme glutathione peroxidase 4 (GPx4). Althoughthe number of CR� interneurons remained normal inGpx4-mutant mice, the number of PV� interneuronswas reduced. Since these mice similarly exhibit sei-zures and ataxia, we conclude that GPx4 is a sel-enoenzyme modulating interneuron function and PVexpression. Cerebral SE deficiency may thus act viareduced GPx4 expression.—Wirth, E. K., Conrad, M.,Winterer, J., Wozny, C., Carlson, B. A., Roth, S., Schmitz,D., Bornkamm, G. W., Coppola, V., Tessarollo, L.,Schomburg, L., Kohrle, J., Hatfield, D. L., Schweizer, U.Neuronal selenoprotein expression is required for inter-neuron development and prevents seizures and neurode-generation. FASEB J. 24, 844–852 (2010). www.fasebj.org
Key Words: selenium � parvalbumin � excitability � schizophre-nia � � oscillation
Selenocysteine (Sec) is the 21st proteinogenicamino acid in mammals. It had initially been over-looked when the genetic code was elucidated, becauseit is encoded by the stop codon UGA and is present inonly a small number of proteins (1). This rare aminoacid contains a selenium (Se) atom in place of thesulfur atom of cysteine. The human and mouse genomescontain 25 and 24 genes encoding selenoproteins, respec-tively (2). Since the discovery of Se as an essential traceelement in rats (3), Se deficiency has been related tonumerous disorders in humans (4). Glutathione peroxi-dases (GPxs), thioredoxin reductases (Txnrds), and iodo-thyronine deiodinases (Dios) were the first mammalianselenoenzymes to be identified; hence, virtually all biolog-ical actions of Se were initially explained by their actionson peroxide degradation and thyroid hormone metabo-lism, respectively. Recently, selenoproteins have beenimplicated in protein folding, degradation of misfoldedmembrane proteins, and control of cellular calcium ho-meostasis, all processes known to be deregulated in neu-rodegenerative diseases (5). Dietary Se restriction in ani-mals does not lead to spontaneous neurological damage,because the brain retains its Se during dietary Se shortage(6, 7). However, targeted inactivation of the main plasmaSe transport protein, selenoprotein P (SePP), demon-strated that SePP is epistatic to brain Se content and brainselenoenzyme expression (8). Depending on dietary Sesupply, Sepp�/� mice die prematurely before weaning and
1 Correspondence: Institute for Experimental Endocrinol-ogy, Charite-Universitatsmedizin Berlin, Augustenburger Platz1, 13353 Berlin, Germany. E-mail: [email protected]
doi: 10.1096/fj.09-143974
844 0892-6638/10/0024-0844 © FASEB

display seizures and ataxia (9). Recently, the lipoproteinreceptor ApoER2 was identified as a SePP receptor inbrain, and ApoER2�/� mice suffer neurodegenera-tion similar to Sepp-deficient mice when fed anSe-deficient diet (10, 11). In aggregate, these studiesdemonstrate that SePP/ApoER2-mediated Se uptakeinto brain is essential for normal brain function inmammals.
It is still unclear which selenoproteins mediate Sefunction in the brain. Moreover, the brain is composedof different cell types, such as neurons, astrocytes,oligodendrocytes, microglia, and endothelium, anddysfunction of any of these cell types may lead toneurological impairment or neurodegeneration. Toaddress the role of selenoproteins in brain function,we have abrogated neuronal selenoprotein expres-sion by Cre recombinase-mediated deletion of thegene encoding tRNA[Ser]Sec (gene symbol Trsp).Because of elimination of tRNA[Ser]Sec, biosynthesisof all selenoproteins is simultaneously disrupted intargeted cells. We show that neuron-specific ablation ofselenoprotein expression causes a neurodevelopmentaland -degenerative phenotype affecting the cerebralcortex and hippocampus, specifically the parvalbumin(PV)-positive interneuron population. Given the selec-tive loss of PV neurons in certain neuropsychiatricdiseases, the class of selenoproteins should be evalu-ated for its possible role in human disease.
MATERIALS AND METHODS
Animals
Mice were maintained according to local regulations as de-scribed previously for the Sepp-deficient mice generated in ourlaboratory (12). All animal experiments were approved by thelocal authorities in Berlin and Munich. Conditional Trsp-knock-out mice (Trspfl/fl) have been described previously (13). Thesemice were crossed with transgenic mice expressing Cre recom-binase under control of the tubulin�1 promotor (14) or undercontrol of the CamKII� promotor (15), yielding mice deficientin neuronal selenoprotein biosynthesis, T�1-Cre/Trspfl/fl, andCamK-Cre/Trspfl/fl, respectively. Conditional Gpx4 mice havebeen described previously (16). Mutant mice and littermatecontrols were analyzed between postnatal day (P)3 and P15.Electrophysiological studies were performed on P10.
Enzyme assays
Forebrains were freshly dissected from postnatal mice andimmediately frozen on dry ice. Brain tissue was powderedunder liquid nitrogen using a dismembrator (Braun Melsun-gen, Melsungen, Germany) and enzymatic activities assessedas described previously (12).
Western blot
Equal amounts of protein from tissue homogenates (50 �g)were applied to SDS-PAGE and electroblotted on PVDFmembrane by standard techniques. Even transfer of proteinswas verified by Ponceau staining of the blotted membranesand by immunostaining for Txnrd2 (ATLAS; rabbit poly-
clonal 1:5000). Antibodies against selenoprotein M (SePM,mouse monoclonal 1:5000) and GPx1 and GPx4 (rabbitpolyclonal 1:1000) were from Abcam (Cambridge, MA,USA) or were made in our laboratories as describedpreviously for selenoproteins (R, H, and T). Immunoreac-tive bands were visualized on X-ray film by chemilumines-cence.
Primary neuron culture and neuronal survival
For in vitro cultures of cortical neurons, brains were dis-sected on embryonic day 15, and isolated neurons werecultured in neurobasal medium on poly-l-lysine and colla-gen-coated 12-well plates. Because the commercial supple-ment (B27; Invitrogen, Carlsbad, CA, USA) contains anunspecified amount of vitamin E and selenite, a self-madesupplement based on earlier formulations (17) was pre-pared. Accordingly, the Se� medium contained 83 nMsodium selenite, a concentration sufficient to allow maxi-mal expression of cellular GPx activity (unpublished re-sults). Based on the same report, we added to Vit E�medium 1 �g/ml each of �-tocopherol and �-tocopherol-acetate, similar to the content of 10% calf serum condi-tions. Synthetic media with Se and vitamin E not added areexpected to contain at best trace impurities of these agents.In vitro differentiation of cortical interneurons in cultureproceeded for 17 d and was assessed by double immuno-fluorescent labeling for GAD67 and PV or calretinin (CR).Photomicrographs were evaluated for the fraction of PV�
cells among the GAD67� cells according to previous work(18); in addition, the density of CR� and GAD67� cells permm2 was quantified. All experiments were replicated withat least 3 independent neuron cultures and means calcu-lated. Cell death was assessed by lactic acid dehydrogenaseactivity in the medium and compared with a commericalcalibrator solution (Greiner). MTT oxidation by metabol-ically active cells was assessed as an alternative measure forcellular survival. Several animals of each genotype werealways cultured in parallel as indicated in the figurelegends.
Electrophysiology
Hippocampal slices were prepared from P10 wild-type andknockout mice as described previously (19). Field potentialrecordings were performed with low-resistance patch pipettesfilled with external solution placed in stratum radiatum ofhippocampal area CA1. Schaffer collaterals were extracellu-larly stimulated with low-resistance patch-pipettes filled withexternal solution placed in stratum radiatum of area CA1.Data were digitized at 5 kHz, recorded, and analyzed withcustom-made software in IGOR Pro (WaveMetrics Inc., LakeOswego, OR, USA). Local field potential recordings wereperformed at 32 � 1°C in an interface-type recording cham-ber. Glass microelectrodes with tip diameter of �5 �m werefilled with ACSF before use. Extracellular signal was amplified�1000 and filtered at 1 Hz to 2 or 5 kHz.
Immunohistochemistry
Brains from mouse pups were immediately fixed after dissectionin 4% paraformaldehyde in 0.1 M phosphate buffer (PB), pH7.4, as described previously (20). Free-floating sections werestained with the indicated antibodies at dilutions of 1:1000–1:5000 at 4°C overnight. Polyclonal rabbit �-PV and rabbit �-CRantibodies were from Swant (Bellinzona, Switzerland), mousemonoclonal �-GFAP antibodies were from Sigma (St. Louis,MO, USA), and �-NPY, �-SOM, and �-NeuN antibodies were
845INACTIVATION OF SELENOPROTEIN EXPRESSION IN NEURONS
from Millipore (Billerica, MA, USA). Horseradish peroxidaseand diaminobezidine substrate were used in conjunction withthe Vectastain ABC kit (Vector, Burlingame, CA, USA). Pho-tomicrographs were taken at a Zeiss Axioskop II equipped withAxioCam MRc and operated with Axiovision software (CarlZeiss, Oberkochen, Germany). The area comprising the somato-sensory cortex was determined using Axiovision software (2–4mm2), and neuronal profiles within were counted in severalsections and both hemispheres (up to n106/area for CR� andup to n474/area for PV�).
Statistical analysis
Student’s t test was used to compare differences between 2groups. Multiple comparisons were assessed using ANOVAfollowed by Dunnett’s post hoc test. Calculations were doneusing Excel (Microsoft, Redmond, WA, USA) or GraphPadPrism software (GraphPad, San Diego, CA, USA). Data areexpressed as means � se. Statistical significance was definedas P 0.05, P 0.01, or P 0.001, as indicated.
RESULTS
Loss of selenoprotein expression in neuron-specificTrsp-knockout brain leads to neurodegeneration
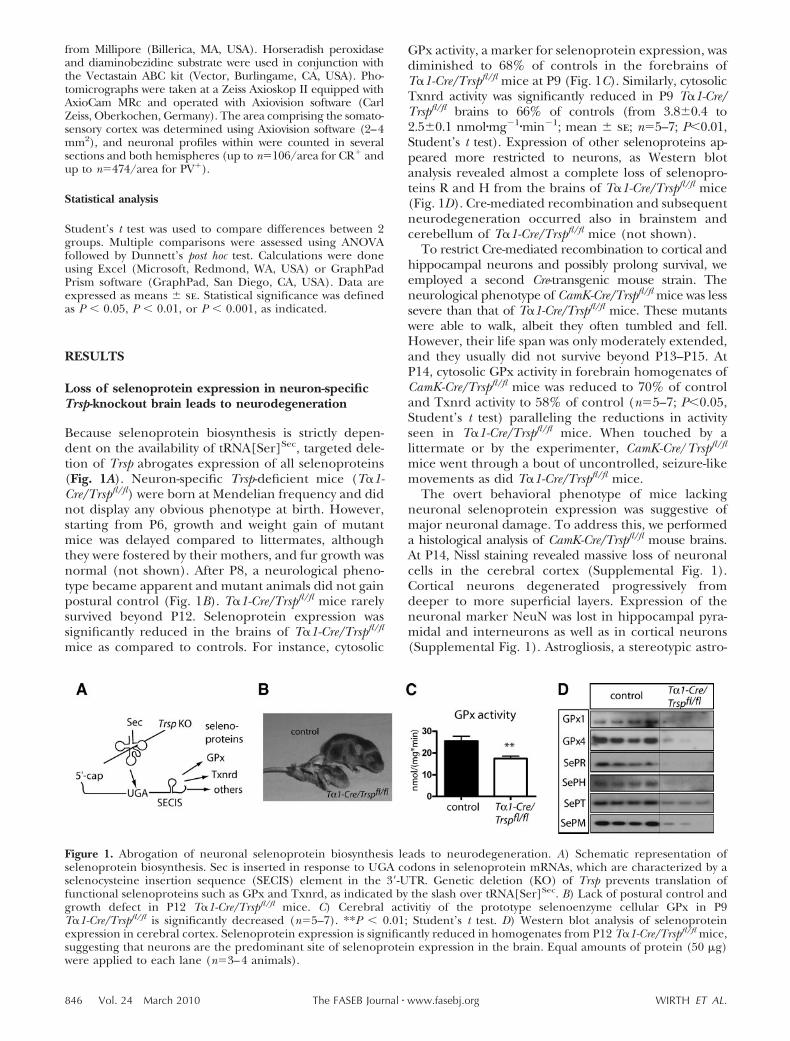
Because selenoprotein biosynthesis is strictly depen-dent on the availability of tRNA[Ser]Sec, targeted dele-tion of Trsp abrogates expression of all selenoproteins(Fig. 1A). Neuron-specific Trsp-deficient mice (T�1-Cre/Trspfl/fl) were born at Mendelian frequency and didnot display any obvious phenotype at birth. However,starting from P6, growth and weight gain of mutantmice was delayed compared to littermates, althoughthey were fostered by their mothers, and fur growth wasnormal (not shown). After P8, a neurological pheno-type became apparent and mutant animals did not gainpostural control (Fig. 1B). T�1-Cre/Trspfl/fl mice rarelysurvived beyond P12. Selenoprotein expression wassignificantly reduced in the brains of T�1-Cre/Trspfl/fl
mice as compared to controls. For instance, cytosolic
GPx activity, a marker for selenoprotein expression, wasdiminished to 68% of controls in the forebrains ofT�1-Cre/Trspfl/fl mice at P9 (Fig. 1C). Similarly, cytosolicTxnrd activity was significantly reduced in P9 T�1-Cre/Trspfl/fl brains to 66% of controls (from 3.8�0.4 to2.5�0.1 nmol�mg�1�min�1; mean � se; n5–7; P0.01,Student’s t test). Expression of other selenoproteins ap-peared more restricted to neurons, as Western blotanalysis revealed almost a complete loss of selenopro-teins R and H from the brains of T�1-Cre/Trspfl/fl mice(Fig. 1D). Cre-mediated recombination and subsequentneurodegeneration occurred also in brainstem andcerebellum of T�1-Cre/Trspfl/fl mice (not shown).
To restrict Cre-mediated recombination to cortical andhippocampal neurons and possibly prolong survival, weemployed a second Cre-transgenic mouse strain. Theneurological phenotype of CamK-Cre/Trspfl/fl mice was lesssevere than that of T�1-Cre/Trspfl/fl mice. These mutantswere able to walk, albeit they often tumbled and fell.However, their life span was only moderately extended,and they usually did not survive beyond P13–P15. AtP14, cytosolic GPx activity in forebrain homogenates ofCamK-Cre/Trspfl/fl mice was reduced to 70% of controland Txnrd activity to 58% of control (n5–7; P0.05,Student’s t test) paralleling the reductions in activityseen in T�1-Cre/Trspfl/fl mice. When touched by alittermate or by the experimenter, CamK-Cre/Trspfl/fl
mice went through a bout of uncontrolled, seizure-likemovements as did T�1-Cre/Trspfl/fl mice.
The overt behavioral phenotype of mice lackingneuronal selenoprotein expression was suggestive ofmajor neuronal damage. To address this, we performeda histological analysis of CamK-Cre/Trspfl/fl mouse brains.At P14, Nissl staining revealed massive loss of neuronalcells in the cerebral cortex (Supplemental Fig. 1).Cortical neurons degenerated progressively fromdeeper to more superficial layers. Expression of theneuronal marker NeuN was lost in hippocampal pyra-midal and interneurons as well as in cortical neurons(Supplemental Fig. 1). Astrogliosis, a stereotypic astro-
Figure 1. Abrogation of neuronal selenoprotein biosynthesis leads to neurodegeneration. A) Schematic representation ofselenoprotein biosynthesis. Sec is inserted in response to UGA codons in selenoprotein mRNAs, which are characterized by aselenocysteine insertion sequence (SECIS) element in the 3�-UTR. Genetic deletion (KO) of Trsp prevents translation offunctional selenoproteins such as GPx and Txnrd, as indicated by the slash over tRNA[Ser]Sec. B) Lack of postural control andgrowth defect in P12 T�1-Cre/Trspfl/fl mice. C) Cerebral activitiy of the prototype selenoenzyme cellular GPx in P9T�1-Cre/Trspfl/fl is significantly decreased (n5–7). **P 0.01; Student’s t test. D) Western blot analysis of selenoproteinexpression in cerebral cortex. Selenoprotein expression is significantly reduced in homogenates from P12 T�1-Cre/Trspfl/fl mice,suggesting that neurons are the predominant site of selenoprotein expression in the brain. Equal amounts of protein (50 �g)were applied to each lane (n3–4 animals).
846 Vol. 24 March 2010 WIRTH ET AL.The FASEB Journal � www.fasebj.org

cytic reaction to neurodegeneration, followed the pat-tern of neuronal loss as judged by the analysis of GFAPimmunoreactivity (Supplemental Fig. 1).
Impaired cortical interneuron development
A histological study using a number of neuronal mark-ers revealed the absence of PV� interneurons in cortexand hippocampus of Trsp mutant mice (Fig. 2 and notshown). We thus asked whether PV� interneurons hadalready degenerated or whether the development ofthese interneurons was specifically impaired. There-fore, the development of major classes of interneuronswas studied in the cerebral cortex from P8 to P15. Incontrol mice, the first cortical PV� cells were observedat P8, and their density increased from P11 to P15. InCamK-Cre/Trspfl/fl mice, expression of PV was not de-tectable at any time point investigated (Fig. 2A, E). Thisindicates that cortical PV� interneuron developmentand/or maturation depends on selenoprotein expres-sion. Interneurons expressing somatostatin-14 (SOM)or neuropeptide Y (NPY) are developmentally related
to PV� neurons and also arise in the medial ganglioniceminence. Their numbers, however, were similar incontrol and Trsp mutant cortex (Fig. 2B, C). Anotherclass of GABAergic interneurons, CR� cells, originatesin the caudal ganglionic eminence and follows a differ-ent specification program. These cells were present innormal number and distribution in the cortex of Trspmutants at P8, but they progressively degeneratedthereafter (Fig. 2D, F). Similar results were found in thehippocampus, and PV� neurons in the globus pallidusmaintained strong PV expression at all time pointsinvestigated (not shown). We thus concluded thatdifferentiation of PV� interneurons is specifically im-paired in the absence of selenoprotein expression.
Spontaneous epileptiform activity in mice lackingneuronal selenoproteins
Lack of PV� interneurons or their dysfunction mightbe responsible for neurological findings such as unco-ordinated seizure-like movements in CamK-Cre/Trspfl/fl
mice. To investigate changes in neuronal excitability or
Figure 2. Development of cortical interneurons isdisrupted in Trsp-deficient brain. Expression ofinterneuron markers in somatosensory cortex inCamK-Cre/Trspfl/fl and control mice at P8, P11, andP15. A–D) Parvalbumin expression is not detectedat any time point in Trsp-deficient mice (A),whereas other interneuron markers, such as soma-tostatin 14 (B), neuropeptide Y (C), and calretinin(D) appear normal. E, F) Quantitification of PV�
(E) and CR� (F) neuronal profiles in somatosen-sory cortex, respectively. Solid bars, control;
shaded bars, CamK-Cre/Trspfl/fl. **P 0.01, ***P 0.001; 2-sided Student’s t test. Scale bars 100 �m (A–C); 200 �m (D).
847INACTIVATION OF SELENOPROTEIN EXPRESSION IN NEURONS

network function, we performed electrophysiologicalrecordings on hippocampal slices from P10 animals. Atthis age, CamK-Cre/Trspfl/fl mice already displayed aclearly visible neurological phenotype, although theydid not exhibit tissue damage as determined by Nisslstaining. To assess the strength of synaptic transmis-sion, we compared the size of the presynaptic fibervolley (input) with the slope of the EPSP (output) instratum radiatum of hippocampal area CA1. We foundthat synaptic transmission was not altered in CamK-Cre/Trspfl/fl mice (n6) in comparison to littermate controls(n9; Fig. 3A), confirming that tissue integrity in themutant was still preserved at this age.
We next addressed whether disruption of Trsp leadsto alterations of hippocampal network oscillations. Inuntreated acute hippocampal slices of P10 animals,profound differences in network synchronization be-tween CamK-Cre/Trspfl/fl and control mice were ob-served (Fig. 3B). In slices from mutant animals (n10;n3), we detected spontaneous interictal discharges(epileptiform activity) in 40% of the slices tested. Incontrast, we did not observe any spontaneous epilepti-form activity in control slices (n10; n3). Further-more, we investigated the effects of cholinergic activa-tion on the network rhythms in the hippocampus,which receives a major cholinergic input from themedial septum/diagonal band (21). Bath applicationof carbachol (5–20 �M) induced epileptiform activityin 66% of the slices of the CamK-Cre/Trspfl/fl mice,whereas in the control animals just one single sliceshowed interictal discharges on application of 20 �Mcarbachol (Fig. 3C). These electrophysiological find-
ings suggest that deficiency of neuronal selenoproteinsresults in pathological network synchronization, compat-ible with compromised PV� interneuron development.
Maturation of PV interneurons in vitro depends on Seand vitamin E
Given the influence of selenoproteins on PV expressionin vivo, we wanted to investigate whether interneurondevelopment in vitro is also modulated by selenoproteins.Because Trsp-deficient neurons do not survive for a suffi-ciently long period of time in culture (see SupplementalFig. 2), we cultured wild-type neurons in the presence orabsence of Se and vitamin E. After 17 d in vitro (DIV),cultures were stained for PV, GAD67, and CR (Fig. 4A).Withdrawal of Se from the medium alone did not signif-icantly reduce the fraction of PV� interneurons in thepresence of high vitamin E concentrations; however,deprivation of Se and vitamin E significantly reduced thenumber of PV-expressing GAD67� cells (Fig. 4B). Be-cause the absolute density of GAD67� cells was notinfluenced by Se and vitamin E, cell death cannot accountfor the reduced PV� expression among interneurons(Fig. 4C). As a further control, we quantified the densityof CR� cells, some of which are GAD67� interneurons,but most comprise cholinergic interneurons. Again, thedensity of CR� cells was not changed by Se or vitamin E inthe culture medium (Fig. 4D). We thus concluded thatthe loss of PV� interneurons may not represent cell death,but reflect a failure to properly differentiate into themature PV� phenotype. The interaction of Se and vita-min E pointed to a possible role of the selenoenzyme
Figure 3. Spontaneous epileptiform activity inacute hippocampal slices in vitro. A) Unalteredinput/output function of the CA1-fEPSP in Trspmutant mice. Top panel: representative fEPSPtraces recorded from 2 individual control andCamK-Cre/Trspfl/fl slices showing similarly in-creased responses to higher stimulation inten-sity applied in the stratum radiatum. Bottompanel; summary graph showing fEPSP slope vs. FVamplitude. B) Interictal discharges in hippocam-pal slices of CamK-Cre/Trspfl/fl mice. Representa-tive traces of local field potentials in slices ofcontrol and Trsp mutant animals, recorded instratum pyramidale of hippocampal CA1 in 10 �Mcarbachol (CCh). Bottom panel: magnification of
representative traces. C) Summary plot of untreated and CCh treated slices from wild-type (n10/group) and Trsp mutantanimals (untreated, n10; CCh treated, n6).
848 Vol. 24 March 2010 WIRTH ET AL.The FASEB Journal � www.fasebj.org

phospholipid hydroperoxide GPx4 in the maturation ofPV� cells.
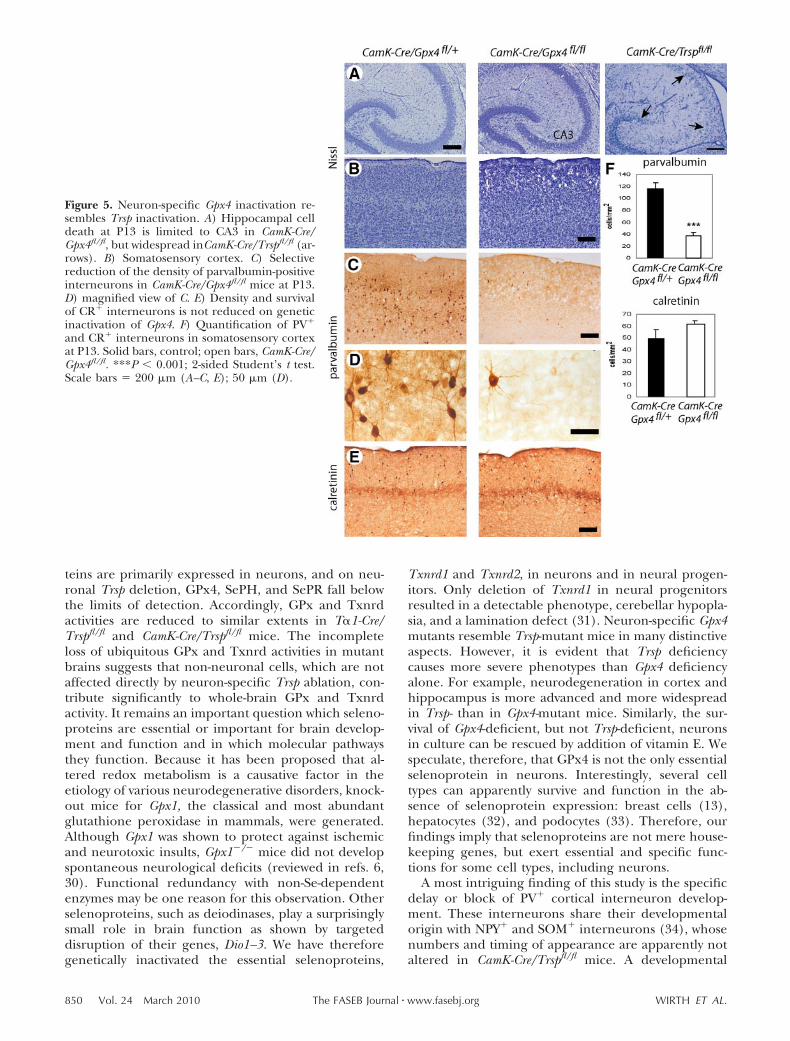
Neuronal Gpx4 mutants partially resembleTrsp-mutant mice
GPx4 is a critical regulator of cellular survival and inter-feres with a lipoxygenase-dependent apoptotic pathway(16). Gpx4-deficient mice do not survive until birth (16,22). To assess the role of Gpx4 in brain development inrelation to a total loss of neuronal selenoproteins, wegenerated CamK-Cre/Gpx4fl/fl mice and compared themwith CamK-Cre/Trspfl/fl mice. Gpx4-deficient mice dis-played the same growth defect as Trsp-mutant mice (notshown). Their neurological phenotype was milder, butCamK-Cre/Gpx4fl/fl mice were still hyperexcitable anddisplayed an awkward gait. Nissl staining at P13 re-vealed that cell death was mostly concentrated in theCA3 region of the hippocampus as opposed to acomplete degeneration of hippocampal neurons asobserved in Trsp-deficient mice (Fig. 5A, B). Neverthe-less, the number of PV� interneurons was significantlyreduced in the somatosensory cortex of CamK-Cre/Gpx4fl/fl mice (Fig. 5C, D, F), higher than in Trsp-mutantmice (Fig. 2). As in Trsp-deficient animals, this devel-opmental defect of prospective PV� interneurons wassubtype specific, because the density of CR� corticalinterneurons was not reduced in Gpx4-mutant mice(Fig. 5E, F). The normal number of CR� cells inGpx4-deficient mice as opposed to Trsp-deficient micefurther demonstrated the greater tissue damage inTrsp-mutant animals. This finding suggested that, inaddition to GPx4, at least one more selenoprotein isessential for neurons. Since we have shown that Gpx4-deficient neurons and fibroblasts can be rescued fromcell death by increased vitamin E in the culture me-dium (16), we tested whether vitamin E is also able torescue Trsp-mutant neurons in culture. Survival of Trspmutant neurons in vitro was severely compromised after9 DIV, but could be rescued by addition of vitamin E
(Supplemental Fig. 2A, B). In contrast, vitamin E wasnot able to sustain Trsp-deficient neurons for 14 DIV(Supplemental Fig. 2C, D). This finding is compatiblewith the notion that GPx4 is not the only essentialselenoprotein in neurons.
DISCUSSION
The aim of this study was to define whether neuronsrequire selenoproteins, try to characterize the patho-mechanism of the neurological phenotype, and possi-bly identify which selenoprotein is crucial for theobserved phenotypes. Here we demonstrate that sel-enoproteins are essential for neuronal function in vitroand in vivo. Beyond a simple role for neuronal survival,we provide evidence that selenoprotein function isparticularly important for the function and differenti-ation of cortical inhibitory PV� interneurons, and weshow that isolated deficiency in Gpx4 alone is sufficientto provoke the interneuron phenotype in vivo.
Because mice deficient in the Se transport protein,SePP, or its receptor ApoER2 develop signs of neuro-degeneration, including ataxia and seizures (9, 11, 12,23–25), it is possible that reduced neuronal GPx4activity may be involved in their neurological pheno-types. SePP is not only a plasma protein, but expressedin the human brain and represents a constituent ofcerebrospinal fluid (26). Moreover, early studies onprimary cortical neuron culture identified SePP as aneurotrophic activity from fetal calf serum (27, 28). Inour hypothetical model, SePP/ApoER2 provides Se forneuronal selenoprotein biosynthesis. If this Se uptakemechanism is impaired, GPx4 biosynthesis may fallunder a pathological threshold.
Recently we have shown at the transcript level thatcerebral cortex, hippocampus, cerebellum, and olfac-tory bulb express the highest numbers of selenopro-teins (29). In the current study, we confirm on theprotein level that, within the brain, several selenopro-
Figure 4. In vitro interneuron differentiationdepends on Se and vitamin E (Vit E) in theculture medium. A) Representative images ofPV�, GAD67�, and CR� interneurons in corti-cal neuron culture at 17 d in culture. B) Frac-tion of PV� cells among GAD67� interneuronsis reduced in cultures deprived of both Se andVit E. C, D) Density of GAD67� interneurons(C) and CR� neurons (D) does not depend onSe and Vit E. Data are means of 3 independentcultures performed in triplicate. �, 83 nMselenite or 1 �g/ml each of �-tocopherol and�-tocopherol-acetate; �, exclusion of seleniteor Vit E. **P 0.01; ANOVA with Dunnett’spost hoc test.
849INACTIVATION OF SELENOPROTEIN EXPRESSION IN NEURONS
teins are primarily expressed in neurons, and on neu-ronal Trsp deletion, GPx4, SePH, and SePR fall belowthe limits of detection. Accordingly, GPx and Txnrdactivities are reduced to similar extents in T�1-Cre/Trspfl/fl and CamK-Cre/Trspfl/fl mice. The incompleteloss of ubiquitous GPx and Txnrd activities in mutantbrains suggests that non-neuronal cells, which are notaffected directly by neuron-specific Trsp ablation, con-tribute significantly to whole-brain GPx and Txnrdactivity. It remains an important question which seleno-proteins are essential or important for brain develop-ment and function and in which molecular pathwaysthey function. Because it has been proposed that al-tered redox metabolism is a causative factor in theetiology of various neurodegenerative disorders, knock-out mice for Gpx1, the classical and most abundantglutathione peroxidase in mammals, were generated.Although Gpx1 was shown to protect against ischemicand neurotoxic insults, Gpx1�/� mice did not developspontaneous neurological deficits (reviewed in refs. 6,30). Functional redundancy with non-Se-dependentenzymes may be one reason for this observation. Otherselenoproteins, such as deiodinases, play a surprisinglysmall role in brain function as shown by targeteddisruption of their genes, Dio1–3. We have thereforegenetically inactivated the essential selenoproteins,
Txnrd1 and Txnrd2, in neurons and in neural progen-itors. Only deletion of Txnrd1 in neural progenitorsresulted in a detectable phenotype, cerebellar hypopla-sia, and a lamination defect (31). Neuron-specific Gpx4mutants resemble Trsp-mutant mice in many distinctiveaspects. However, it is evident that Trsp deficiencycauses more severe phenotypes than Gpx4 deficiencyalone. For example, neurodegeneration in cortex andhippocampus is more advanced and more widespreadin Trsp- than in Gpx4-mutant mice. Similarly, the sur-vival of Gpx4-deficient, but not Trsp-deficient, neuronsin culture can be rescued by addition of vitamin E. Wespeculate, therefore, that GPx4 is not the only essentialselenoprotein in neurons. Interestingly, several celltypes can apparently survive and function in the ab-sence of selenoprotein expression: breast cells (13),hepatocytes (32), and podocytes (33). Therefore, ourfindings imply that selenoproteins are not mere house-keeping genes, but exert essential and specific func-tions for some cell types, including neurons.
A most intriguing finding of this study is the specificdelay or block of PV� cortical interneuron develop-ment. These interneurons share their developmentalorigin with NPY� and SOM� interneurons (34), whosenumbers and timing of appearance are apparently notaltered in CamK-Cre/Trspfl/fl mice. A developmental
Figure 5. Neuron-specific Gpx4 inactivation re-sembles Trsp inactivation. A) Hippocampal celldeath at P13 is limited to CA3 in CamK-Cre/Gpx4fl/fl, but widespread inCamK-Cre/Trspfl/fl (ar-rows). B) Somatosensory cortex. C) Selectivereduction of the density of parvalbumin-positiveinterneurons in CamK-Cre/Gpx4fl/fl mice at P13.D) magnified view of C. E) Density and survivalof CR� interneurons is not reduced on geneticinactivation of Gpx4. F) Quantification of PV�
and CR� interneurons in somatosensory cortexat P13. Solid bars, control; open bars, CamK-Cre/Gpx4fl/fl. ***P 0.001; 2-sided Student’s t test.Scale bars 200 �m (A–C, E); 50 �m (D).
850 Vol. 24 March 2010 WIRTH ET AL.The FASEB Journal � www.fasebj.org

block or selective degeneration before maturation ofthe PV� class of GABAergic interneurons is consistentwith the functional impairment of the inhibitory systemand spontaneous epileptiform activity in vitro. Theunchanged slope of EPSP over the amplitude of thefiber volley suggested that the glutamatergic cells re-sponded normally and again pointed to a dysfunctionalGABAergic system. The electrophysiological findingswere supported by the apparent hyperexcitable andjerky phenotype of the mutant mice. Recently condi-tional inactivation of the transcription factor Nkx2.1,which is expressed in cortical interneuron precursors,revealed a selective loss of PV� cortical neurons inassociation with seizures in juvenile mice (35). Hyperex-citability and seizures are also features of Sepp-deficientmice and patients with childhood epilepsy associated withSe deficiency (36, 37). Mice carrying a hypomorphic alleleof Trsp are also hyperexcitable and have reduced numbersof PV� neurons in the cerebral cortex (38).
PV� neurons, in particular, are sensitive to oxidativestress, and induction of oxidative stress with the NMDAreceptor antagonist, ketamine, leads to loss of PVexpression and impaired � oscillations (18, 39). Inschizophrenia, � oscillations are impaired, and thenumber of PV� interneurons is reduced (40). Interest-ingly, the levels of glutathione, the major cofactor ofGPx4 and other glutathione-dependent enzymes, arereduced in the brains of schizophrenic patients, andmutations in enzymes involved in glutathione biosyn-thesis are frequent in schizophrenic patients (41). Ourfinding of a selective developmental deficit of PV�
interneurons in Gpx4-mutant mice is thus consistentwith a specific role of GPx4 for PV expression ininterneurons. Our model, however, cannot rule out aparacrine mechanism in which (GPx4-deficient) corti-cal neurons express less BDNF, a known factor promot-ing PV interneuron maturation (42). To test this hy-pothesis, one would need to inactivate Gpx4 in aninterneuron-specific manner.
The maturation of PV� interneurons is also delayedin hypothyroid rodents (43) and in mice expressing adominant negative thyroid hormone receptor (44).Because all deiodinases are selenoenzymes, one mayargue that Trsp-deficient mice are hypothyroid, andtherefore interneuron maturation is impaired. How-ever, Gpx4 deficiency replicates the cortical interneu-ron phenotype of Trsp-deficient mice, thereby render-ing deiodinase deficiency unlikely as the primary causefor the interneuron phenotype.
How can redox state or lipid peroxidation influ-ence interneuron differentiation? Elucidating theprecise mechanism will require further study, but it isknown that the differentiation of neural precursorcells is modulated by cellular redox state (45). Re-cently it was shown that activity of the redox sensinghistone deacetylase Sirt1 reduces proliferation andpromotes astroglial differentiation of neural progen-itor cells (46). It remains to be shown whether PV�
interneuron differentiation is blocked by Sirt1 actionand whether Sirt1 can sense lipid peroxides. We have
recently reported that GPx4 acts in a 12/15-lipoxygen-ase apoptotic pathway in fibroblasts, and we blockedcell death in the absence of GPx4 with a lipoxygenaseinhibitor (16). However, primary neurons do not toler-ate the inhibitor at effective concentrations, and thuswe were not able to probe the role of this pathway ininterneuron differentiation.
Given the essential roles of selenoproteins in neurons,the question arises whether metabolic disturbances of Seutilization or selenoprotein biosynthesis may contributeto neurodegenerative disease in humans. The epilepticphenotype of Sepp-deficient mice suggests that a moderatereduction of brain Se content suffices to impair brainfunction. We can only speculate whether a slight reduc-tion in brain Se content over decades may precipitateneurodegeneration in humans.
The authors thank Dr. Claudia Iserhot, who performed initialphysiological studies. SiJie Zhang performed some of the neu-ron cultures. CamK-Cre mice were generously provided byGunther Schutz (Deutsches Krebsforschungszentrum, Heidel-berg, Germany). The authors gratefully acknowledge the tech-nical assistance of Vartiter Seher, Anita Kinne, Silke Kappler,Antje Kretschmer, and Heidi Forster. Dorette Freyer (Experi-mental Neurology, Charite Universitatsmedizin Berlin), initiallyhelped with primary neuron culture. Funding for this study wasprovided by Deutsche Forschungsgemeinschaft Scho849/2–2,SFB 665/A7, Ko922/11–1, and Charite Universitatsmedizin Ber-lin and in part by the Intramural Research Program of the U.S.National Institutes of Health, National Cancer Institute, Centerfor Cancer Research.
REFERENCES
1. Hatfield, D. L., and Gladyshev, V. N. (2002) How selenium hasaltered our understanding of the genetic code. Mol. Cell. Biol.22, 3565–3576
2. Kryukov, G. V., Castellano, S., Novoselov, S. V., Lobanov, A. V.,Zehtab, O., Guigo, R., and Gladyshev, V. N. (2003) Character-ization of mammalian selenoproteomes. Science 300, 1439–1443
3. Schwarz, K., and Foltz, C. M. (1957) Selenium as an integral partof factor 3 against dietary necrotic liver degeneration. Nutrition15, 255–264
4. Rayman, M. P. (2000) The importance of selenium to humanhealth. Lancet 356, 233–241
5. Andersen, J. K. (2004) Oxidative stress in neurodegeneration:cause or consequence? Nat. Med. 10(Suppl.), S18–S25
6. Schweizer, U., Schomburg, L., and Savaskan, N. E. (2004) Theneurobiology of selenium: lessons from transgenic mice. J. Nutr.134, 707–710
7. Behne, D., Hilmert, H., Scheid, S., Gessner, H., and Elger, W.(1988) Evidence for specific selenium target tissues and newbiologically important selenoproteins. Biochim. Biophys. Acta 966,12–21
8. Burk, R. F., and Hill, K. E. (2005) Selenoprotein P: an extracel-lular protein with unique physical characteristics and a role inselenium homeostasis. Annu. Rev. Nutr. 25, 215–235
9. Schweizer, U., Michaelis, M., Kohrle, J., and Schomburg, L.(2004) Efficient selenium transfer from mother to offspring inselenoprotein-P-deficient mice enables dose-dependent rescueof phenotypes associated with selenium deficiency. Biochem. J.378, 21–26
10. Olson, G. E., Winfrey, V. P., Nagdas, S. K., Hill, K. E., and Burk,R. F. (2007) Apolipoprotein E receptor-2 (ApoER2) mediatesselenium uptake from selenoprotein P by the mouse testis.J. Biol. Chem. 282, 12290–12297
11. Burk, R. F., Hill, K. E., Olson, G. E., Weeber, E. J., Motley, A. K.,Winfrey, V. P., and Austin, L. M. (2007) Deletion of apolipopro-
851INACTIVATION OF SELENOPROTEIN EXPRESSION IN NEURONS

tein E receptor-2 in mice lowers brain selenium and causessevere neurological dysfunction and death when a low-seleniumdiet is fed. J. Neurosci. 27, 6207–6211
12. Schomburg, L., Schweizer, U., Holtmann, B., Flohe, L., Sendt-ner, M., and Kohrle, J. (2003) Gene disruption discloses role ofselenoprotein P in selenium delivery to target tissues. Biochem. J.370, 397–402
13. Kumaraswamy, E., Carlson, B. A., Morgan, F., Miyoshi, K.,Robinson, G. W., Su, D., Wang, S., Southon, E., Tessarollo, L.,Lee, B. J., Gladyshev, V. N., Hennighausen, L., and Hatfield,D. L. (2003) Selective removal of the selenocysteine tRNA[Ser]Sec gene (Trsp) in mouse mammary epithelium. Mol. Cell.Biol. 23, 1477–1488
14. Coppola, V., Barrick, C. A., Southon, E. A., Celeste, A., Wang, K.,Chen, B., Haddad, E., Yin, J., Nussenzweig, A., Subramaniam,A., and Tessarollo, L. (2004) Ablation of TrkA function in theimmune system causes B cell abnormalities. Development 131,5185–5195
15. Casanova, E., Fehsenfeld, S., Mantamadiotis, T., Lemberger, T.,Greiner, E., Stewart, A. F., and Schutz, G. (2001) A CamKIIalphaiCre BAC allows brain-specific gene inactivation. Genesis 31, 37–42
16. Seiler, A., Schneider, M., Forster, H., Roth, S., Wirth, E. K.,Culmsee, C., Plesnila, N., Kremmer, E., Radmark, O., Wurst, W.,Bornkamm, G. W., Schweizer, U., and Conrad, M. (2008)Glutathione peroxidase 4 senses and translates oxidative stressinto 12/15-lipoxygenase dependent- and AIF-mediated celldeath. Cell. Metab. 8, 237–248
17. Brewer, G. J., and Cotman, C. W. (1989) Survival and growth ofhippocampal neurons in defined medium at low density: advan-tages of a sandwich culture technique or low oxygen. Brain Res.494, 65–74
18. Kinney, J. W., Davis, C. N., Tabarean, I., Conti, B., Bartfai, T.,and Behrens, M. M. (2006) A specific role for NR2A-containingNMDA receptors in the maintenance of parvalbumin andGAD67 immunoreactivity in cultured interneurons. J. Neurosci.26, 1604–1615
19. Breustedt, J., Vogt, K. E., Miller, R. J., Nicoll, R. A., and Schmitz, D.(2003) Alpha1E-containing Ca2� channels are involved in synap-tic plasticity. Proc. Natl. Acad. Sci. U. S. A. 100, 12450–12455
20. Wirth, E. K., Roth, S., Blechschmidt, C., Holter, S. M., Becker,L., Racz, I., Zimmer, A., Klopstock, T., Gailus-Durner, V., Fuchs,H., Wurst, W., Naumann, T., Brauer, A., de Angelis, M. H.,Kohrle, J., Gruters, A., and Schweizer, U. (2009) Neuronal 3�, 3,5-triiodothyronine (T3) uptake and behavioral phenotype ofmice deficient in Mct8, the neuronal T3 transporter mutated inAllan-Herndon-Dudley syndrome. J. Neurosci. 29, 9439–9449
21. Fisahn, A., Pike, F. G., Buhl, E. H., and Paulsen, O. (1998)Cholinergic induction of network oscillations at 40 Hz in thehippocampus in vitro. Nature 394, 186–189
22. Yant, L. J., Ran, Q., Rao, L., Van Remmen, H., Shibatani, T.,Belter, J. G., Motta, L., Richardson, A., and Prolla, T. A. (2003)The selenoprotein GPX4 is essential for mouse developmentand protects from radiation and oxidative damage insults. FreeRadic. Biol. Med. 34, 496–502
23. Hill, K. E., Zhou, J., McMahan, W. J., Motley, A. K., Atkins, J. F.,Gesteland, R. F., and Burk, R. F. (2003) Deletion of selenopro-tein P alters distribution of selenium in the mouse. J. Biol. Chem.278, 13640–13646
24. Hill, K. E., Zhou, J., McMahan, W. J., Motley, A. K., and Burk, R. F.(2004) Neurological dysfunction occurs in mice with targeteddeletion of the selenoprotein p gene. J. Nutr. 134, 157–161
25. Valentine, W. M., Abel, T. W., Hill, K. E., Austin, L. M., andBurk, R. F. (2008) Neurodegeneration in mice resulting fromloss of functional selenoprotein P or its receptor apolipoproteinE receptor 2. J. Neuropathol. Exp. Neurol. 67, 68–77
26. Scharpf, M., Schweizer, U., Arzberger, T., Roggendorf, W.,Schomburg, L., and Kohrle, J. (2007) Neuronal and ependymalexpression of selenoprotein P in the human brain. J. NeuralTransm. 114, 877–884
27. Yan, J., and Barrett, J. N. (1998) Purification from bovine serumof a survival-promoting factor for cultured central neurons andits identification as selenoprotein-P. J. Neurosci. 18, 8682–8691
28. Kaufman, L. M., and Barrett, J. N. (1983) Serum factor support-ing long-term survival of rat central neurons in culture. Science220, 1394–1396
29. Zhang, Y., Zhou, Y., Schweizer, U., Savaskan, N. E., Hua, D., Kipnis,J., Hatfield, D. L., and Gladyshev, V. N. (2008) Comparative
analysis of selenocysteine machinery and selenoproteome geneexpression in mouse brain identifies neurons as key functional sitesof selenium in mammals. J. Biol. Chem. 283, 2427–2438
30. Schweizer, U., Brauer, A. U., Kohrle, J., Nitsch, R., and Savaskan,N. E. (2004) Selenium and brain function: a poorly recognizedliaison. Brain Res. Brain Res. Rev. 45, 164–178
31. Soerensen, J., Jakupoglu, C., Beck, H., Forster, H., Schmidt, J.,Schmahl, W., Schweizer, U., Conrad, M., and Brielmeier, M.(2008) The role of thioredoxin reductases in brain develop-ment. PLoS ONE 3, e1813
32. Schweizer, U., Streckfuss, F., Pelt, P., Carlson, B. A., Hatfield,D. L., Kohrle, J., and Schomburg, L. (2005) Hepatically derivedselenoprotein P is a key factor for kidney but not for brainselenium supply. Biochem. J. 386, 221–226
33. Blauwkamp, M. N., Yu, J., Schin, M. A., Burke, K. A., Berry, M. J.,Carlson, B. A., Brosius, F. C., III, and Koenig, R. J. (2008) Podocytespecific knock out of selenoproteins does not enhance nephropa-thy in streptozotocin diabetic C57BL/6 mice. BMC Nephrol. 9, 7
34. Wonders, C. P., and Anderson, S. A. (2006) The origin andspecification of cortical interneurons. Nat. Rev. Neurosci. 7,687–696
35. Butt, S. J., Sousa, V. H., Fuccillo, M. V., Hjerling-Leffler, J.,Miyoshi, G., Kimura, S., and Fishell, G. (2008) The requirementof Nkx2–1 in the temporal specification of cortical interneuronsubtypes. Neuron 59, 722–732
36. Weber, G. F., Maertens, P., Meng, X. Z., and Pippenger, C. E.(1991) Glutathione peroxidase deficiency and childhood sei-zures. Lancet 337, 1443–1444
37. Ramaekers, V. T., Calomme, M., Vanden Berghe, D., andMakropoulos, W. (1994) Selenium deficiency triggering intrac-table seizures. Neuropediatrics 25, 217–223
38. Carlson, B. A., Schweizer, U., Perella, C., Shrimali, R. K.,Feigenbaum, L., Shen, L., Speransky, S., Floss, T., Jeong, S. J.,Watts, J., Hoffmann, V., Combs, G. F., Gladyshev, V. N., andHatfield, D. L. (2009) The selenocysteine tRNA STAF-bindingregion is essential for adequate selenocysteine tRNA status,selenoprotein expression and early age survival of mice. Biochem.J. 418, 61–71
39. Behrens, M. M., Ali, S. S., Dao, D. N., Lucero, J., Shekhtman, G.,Quick, K. L., and Dugan, L. L. (2007) Ketamine-induced loss ofphenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 318, 1645–1647
40. Lewis, D. A., Hashimoto, T., and Volk, D. W. (2005) Corticalinhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6,312–324
41. Gysin, R., Kraftsik, R., Sandell, J., Bovet, P., Chappuis, C., Conus,P., Deppen, P., Preisig, M., Ruiz, V., Steullet, P., Tosic, M.,Werge, T., Cuenod, M., and Do, K. Q. (2007) Impaired gluta-thione synthesis in schizophrenia: convergent genetic and func-tional evidence. Proc. Natl. Acad. Sci. U. S. A. 104, 16621–16626
42. Huang, Z. J., Kirkwood, A., Pizzorusso, T., Porciatti, V., Morales,B., Bear, M. F., Maffei, L., and Tonegawa, S. (1999) BDNFregulates the maturation of inhibition and the critical period ofplasticity in mouse visual cortex. Cell 98, 739–755
43. Gilbert, M. E., Sui, L., Walker, M. J., Anderson, W., Thomas, S.,Smoller, S. N., Schon, J. P., Phani, S., and Goodman, J. H.(2007) Thyroid hormone insufficiency during brain develop-ment reduces parvalbumin immunoreactivity and inhibitoryfunction in the hippocampus. Endocrinology 148, 92–102
44. Wallis, K., Sjogren, M., van Hogerlinden, M., Silberberg, G.,Fisahn, A., Nordstrom, K., Larsson, L., Westerblad, H., Morre-ale, D. E., Shupliakov, O., and Vennstrom, B. (2008) Locomotordeficiencies and aberrant development of subtype-specificGABAergic interneurons caused by an unliganded thyroid hor-mone receptor alpha1. J Neurosci. 28, 1904–1915
45. Smith, J., Ladi, E., Mayer-Proschel, M., and Noble, M. (2000)Redox state is a central modulator of the balance betweenself-renewal and differentiation in a dividing glial precursor cell.Proc. Natl. Acad. Sci. U. S. A. 97, 10032–10037
46. Prozorovski, T., Schulze-Topphoff, U., Glumm, R., Baumgart, J.,Schroter, F., Ninnemann, O., Siegert, E., Bendix, I., Brustle, O.,Nitsch, R., Zipp, F., and Aktas, O. (2008) Sirt1 contributescritically to the redox-dependent fate of neural progenitors.Nat. Cell. Biol. 10, 385–394
Received for publication September 2, 2009.Accepted for publication October 15, 2009.
852 Vol. 24 March 2010 WIRTH ET AL.The FASEB Journal � www.fasebj.org


Copyright © 2022 FDOKUMEN




















