
Neural Stem Cell Isolation and Characterization
22
Chapter 11 Neural Stem Cell Isolation, Characterization and Transplantation Jasodhara Ray and Fred H. Gage ■ Introduction During development, nerve cells in the mammalian central nervous system (CNS) are generated by the proliferation of multipotent stem or progenitor cells that migrate, find their site of final destination and ultimately terminally differentiate. However, the mechanisms for development of diversified cell types in the CNS and the environmental stimuli involved in these processes are not well understood. In analogy with the hemat- opoietic system, it has been proposed that during CNS development, a self-renewing population of stem cells gives rise to a more restrictive population of progenitor cells. However, the existence of these elusive progenitor cells has not been proven due to the unavailability of specific phenotypic markers. In the adult brain only a small number of stem cells exist and they differentiate into neurons at specific neurogenic sites at a slow rate (Morshead et al., 1994; Kuhn et al., 1996). Attempts have been made for decades to isolate and culture stem or progenitor cells (will be referred to as stem cells in this chapter) that give rise to neuroblasts and gliob- lasts, which upon differentiation, generate mature neurons, astrocytes and oli- godendrocytes, i.e., the cells that are the major building blocks of the central nervous system (CNS). To obtain long-term proliferative cultures, cells from embryonic brains were immortalized by using oncogenic transgenes like v-myc or SV40 large T antigen (Cepko, 1988a; Lendahl and McKay, 1990; Whittemore and Snyder, 1996). The immor- talization process arrests cells at specific stages of development and halts their terminal differentiation (Cepko, 1988a; Lendahl and McKay, 1990). Clonal cultures of cells repre- senting a specific stage in development were isolated and used for in vitro and in vivo studies (Gage et al., 1995a; Whittmore and Snyder; 1996; Ray et al., 1997; Fisher, 1997). Although immortalized cells offer a number of advantages, they do not always represent their primary counterparts. Isolation and long-term culturing of primary stem/progen- itor cells have been advanced by the findings that the mitogenic growth factors epider- mal growth factor (EGF) and basic fibroblast growth factor (FGF-2) have proliferative effects on these cells (Weiss et al., 1996a; Ray et al., 1997; McKay, 1997). Isolated and cultured stem cells not only provide an important source of cells for in vitro studies to address issues related to fate choice and differentiation, but they are also an important source of CNS cells that can be used in transplantation studies (Ray et al., 1997, 1998). To explore the possible use of these cells for therapy, embryonic or adult brain-derived stem cells have been grafted in normal CNS to determine their in vivo survival and potential for fate choice, differentiation and integration (Hammang et al., 1994; Gage et al., 1995b; Suhonen et al., 1996; Ray et al., 1997). Adult rat hippocampus- derived FGF-2 responsive cells grafted to homo- or heterotypic neurogenic sites (hip- Correspondence to: Jasodhara Ray, Salk Institute for Biological Studies, Laboratory of Genetics, 10010N. Torrey Pines Road, La Jolla, CA, 92037, USA (phone: +01-858-453-4100 ext. 1006; fax: +01 858- 597-0824; e-mail: [email protected])
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Neural Stem Cell Isolation and Characterization

Chapter 11
Neural Stem Cell Isolation, Characterization and Transplantation
Jasodhara Ray and Fred H. Gage
■ Introduction
During development, nerve cells in the mammalian central nervous system (CNS) aregenerated by the proliferation of multipotent stem or progenitor cells that migrate, findtheir site of final destination and ultimately terminally differentiate. However, themechanisms for development of diversified cell types in the CNS and the environmentalstimuli involved in these processes are not well understood. In analogy with the hemat-opoietic system, it has been proposed that during CNS development, a self-renewingpopulation of stem cells gives rise to a more restrictive population of progenitor cells.However, the existence of these elusive progenitor cells has not been proven due to theunavailability of specific phenotypic markers. In the adult brain only a small number ofstem cells exist and they differentiate into neurons at specific neurogenic sites at a slowrate (Morshead et al., 1994; Kuhn et al., 1996).
Attempts have been made for decades to isolate and culture stem or progenitor cells(will be referred to as stem cells in this chapter) that give rise to neuroblasts and gliob-lasts, which upon differentiation, generate mature neurons, astrocytes and oli-godendrocytes, i.e., the cells that are the major building blocks of the central nervoussystem (CNS). To obtain long-term proliferative cultures, cells from embryonic brainswere immortalized by using oncogenic transgenes like v-myc or SV40 large T antigen(Cepko, 1988a; Lendahl and McKay, 1990; Whittemore and Snyder, 1996). The immor-talization process arrests cells at specific stages of development and halts their terminaldifferentiation (Cepko, 1988a; Lendahl and McKay, 1990). Clonal cultures of cells repre-senting a specific stage in development were isolated and used for in vitro and in vivostudies (Gage et al., 1995a; Whittmore and Snyder; 1996; Ray et al., 1997; Fisher, 1997).Although immortalized cells offer a number of advantages, they do not always representtheir primary counterparts. Isolation and long-term culturing of primary stem/progen-itor cells have been advanced by the findings that the mitogenic growth factors epider-mal growth factor (EGF) and basic fibroblast growth factor (FGF-2) have proliferativeeffects on these cells (Weiss et al., 1996a; Ray et al., 1997; McKay, 1997).
Isolated and cultured stem cells not only provide an important source of cells for invitro studies to address issues related to fate choice and differentiation, but they are alsoan important source of CNS cells that can be used in transplantation studies (Ray et al.,1997, 1998). To explore the possible use of these cells for therapy, embryonic or adultbrain-derived stem cells have been grafted in normal CNS to determine their in vivosurvival and potential for fate choice, differentiation and integration (Hammang et al.,1994; Gage et al., 1995b; Suhonen et al., 1996; Ray et al., 1997). Adult rat hippocampus-derived FGF-2 responsive cells grafted to homo- or heterotypic neurogenic sites (hip-
Correspondence to: Jasodhara Ray, Salk Institute for Biological Studies, Laboratory of Genetics,10010N. Torrey Pines Road, La Jolla, CA, 92037, USA (phone: +01-858-453-4100 ext. 1006; fax: +01 858-597-0824; e-mail: [email protected])

340 Jasodhara Ray and Fred H. Gage
pocampus or olfactory system) differentiated into neurons (Gage et al., 1995b; Suhonenet al., 1996), whereas when they were grafted to non-neurogenic site (cerebellum) noneuronal differentiation was observed (Suhonen et al., 1996). In contrast, EGF-respon-sive embryonic mouse stem cells grafted in intact neonatal mouse cortex and spinalcord showed poor survival and no neuronal differentiation (Hammang et al., 1994).Similar results were obtained with EGF-responsive stem cells from embryonic rats en-grafted into lesioned adult rat brains (Svendsen et al., 1996). The usefulness of stem cellsas a vehicle to deliver trophic factors in the brain has been explored in two recent stud-ies. EGF-responsive stem cells were cultured from transgenic mice in which GFAP pro-moter drives the expression of human nerve growth factor (NGF). Cells grafted in adultrat striatum survived up to 3 weeks (latest time point examined), did not migrate fromthe graft site and differentiated into astrocytes (Carpenter et al., 1997; Kordower et al.,1997). The secreted bioactive NGF induced hypertrophy and sprouting of the endog-enous cholinergic neurons within the intact rodent brain (Carpenter et al., 1997) andprevented the degeneration of striatal neurons in a rodent model of Huntington’s dis-ease (Kordower et al., 1997). These studies show promise that neural stem cells can beexploited for cell replacement and gene therapy purposes to repair CNS injuries or dis-orders.
Neural tissues are composed of both neuronal and nonneuronal cells as well as con-nective and vascular tissues. When removed from the brain and put in culture, the cellslose the physiological connections, anchorages and the humoral environments. Strate-gies to isolate and culture stem cells from CNS involved: i) isolation of cells from an in-tertwined network of thousands of adhesive contacts without causing damage to thecells; ii) separating stem cells from other brain cells and connective tissue debris; andiii) providing the appropriate environmental conditions, including specific nutrientsand growth factors, required for the survival and the proliferation of stem cells. Neuralcultures are generally maintained at pH 7.2–7.6 and at the appropriate osmolarity. Themost commonly used methods for the isolation and culture of stem cells use serum-freeculture medium supplemented with various hormones and nutrients (Bottenstein andSato, 1979) and mitogenic growth factors EGF or FGF-2. EGF has been used to culturesubependymal/forebrain stem cells as neurospheres from embryonic and adult mouse(Reynolds et al., 1992; Reynolds and Weiss, 1992). However, EGF failed to induce prolif-eration of stem cells isolated from adult mouse spinal cord which required a combina-tion of EGF and FGF-2 for proliferation (Weiss et al., 1996b). In contrast, FGF-2 alonehas been successfully used to establish monolayer cultures of stem cells from differentembryonic and adult brain regions and the spinal cord (Ray et al., 1993, 1994; Gage etal., 1995b; Palmer et al., 1995; Minger et al., 1996; Shihabuddin et al., 1997) and to gen-erate neurospheres from lateral ventricle/forebrain of adult mouse (Gritti et al., 1996).
The choice of a culture method depends on the specific issues being addressed by theresearchers. Methods for culturing neural stem cells have been described previously(Ray et al., 1995). This chapter describes the strategies and the latest techniques for cul-turing stem cells by the two most commonly used methods, and discusses the genera-tion of clonal populations and the characterization of the cells and their grafting in adultrat brain.
■ Outline
Dissect out embryonic or adult brain regions, digest the tissues by enzymatic digestionto release cells from connective tissues, partially purify stem cells from debris and othercells by percoll density gradient centrifugation (optional), and plate cells (Fig.1).
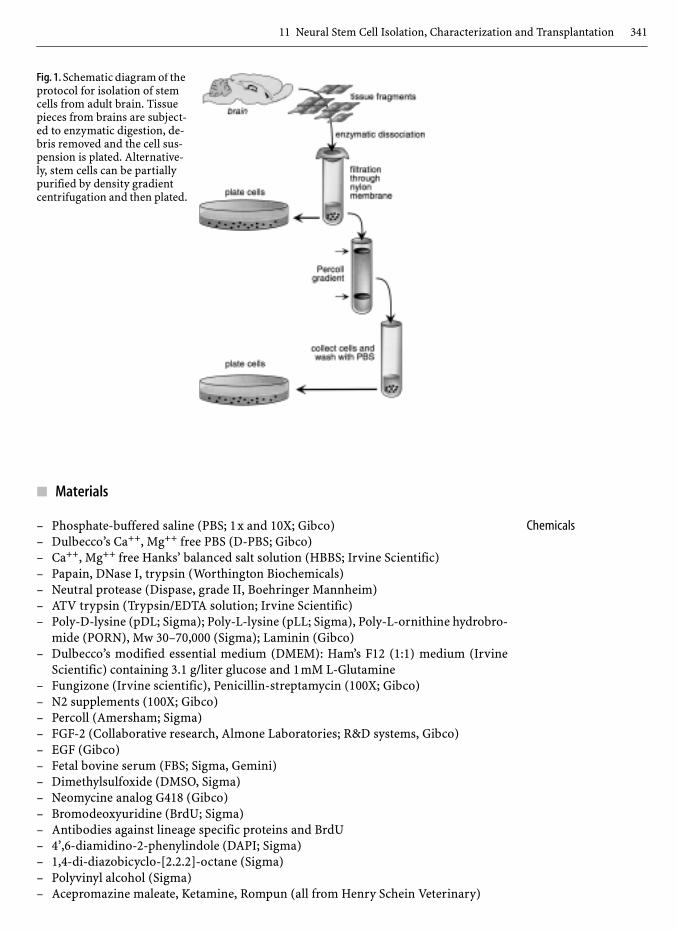
11 Neural Stem Cell Isolation, Characterization and Transplantation 341
■ Materials
Chemicals– Phosphate-buffered saline (PBS; 1x and 10X; Gibco)– Dulbecco’s Ca++, Mg++ free PBS (D-PBS; Gibco)– Ca++, Mg++ free Hanks’ balanced salt solution (HBBS; Irvine Scientific)– Papain, DNase I, trypsin (Worthington Biochemicals)– Neutral protease (Dispase, grade II, Boehringer Mannheim)– ATV trypsin (Trypsin/EDTA solution; Irvine Scientific)– Poly-D-lysine (pDL; Sigma); Poly-L-lysine (pLL; Sigma), Poly-L-ornithine hydrobro-
mide (PORN), Mw 30–70,000 (Sigma); Laminin (Gibco)– Dulbecco’s modified essential medium (DMEM): Ham’s F12 (1:1) medium (Irvine
Scientific) containing 3.1 g/liter glucose and 1mM L-Glutamine– Fungizone (Irvine scientific), Penicillin-streptamycin (100X; Gibco)– N2 supplements (100X; Gibco)– Percoll (Amersham; Sigma)– FGF-2 (Collaborative research, Almone Laboratories; R&D systems, Gibco)– EGF (Gibco)– Fetal bovine serum (FBS; Sigma, Gemini)– Dimethylsulfoxide (DMSO, Sigma)– Neomycine analog G418 (Gibco)– Bromodeoxyuridine (BrdU; Sigma)– Antibodies against lineage specific proteins and BrdU– 4’,6-diamidino-2-phenylindole (DAPI; Sigma)– 1,4-di-diazobicyclo-[2.2.2]-octane (Sigma)– Polyvinyl alcohol (Sigma)– Acepromazine maleate, Ketamine, Rompun (all from Henry Schein Veterinary)
Fig. 1. Schematic diagram of the protocol for isolation of stem cells from adult brain. Tissue pieces from brains are subject-ed to enzymatic digestion, de-bris removed and the cell sus-pension is plated. Alternative-ly, stem cells can be partially purified by density gradient centrifugation and then plated.

342 Jasodhara Ray and Fred H. Gage
Supply – Tissue culture dishes and Labtek or Nunc Chamber slides (Fisher)– Cryovials (Nalgene)– Sterile filters (0.45µm and 0.22µm; Nalgene)– Betadine (J. A. Webster, Inc.)– Nylon mesh (pore size 15µm, Nitex, TETKO, Inc.)
Equipment – Laminar flow hood; CO2 Incubator (Forma Scientific)– Freezing chambers (Nalgene) and liquid nitrogen tank
Solutions and Medium 1. Anesthetic solution– 7.5ml Ketamine (10 mg/ml)– 0.75ml Acepromazine maleate (100 mg/ml)– 1.9ml Rompun (20 mg/ml)– 9.85ml Saline– Inject 0.25 ml/160g rat
2. Artificial cerebrospinal fluid (aCSF)– 124mM NaCl– 5mM KCl– 1.3mM MgCl2– 2mM CaCl2– 26mM NaHCO3– 10mM D-glucose– 1x penicillin-streptomycin– pH 7.35, Total osmolality ~280 mOsm
3. Papain-Protease-DNase (PPD) solution– HBBS supplemented with 12.4mM MgSO4– 0.01% papain– 0.1% neutral protease– 0.01% DNase I– Filter sterilize through 0.22µm filter– Store aliquots at −20°C
4. Trypsin-hyaluronidase-kynurenic acid solution– 124mM NaCl– 5mM KCl– 3.2mM MgCl2– 0.1mM CaCl2– 26mM NaHCO3– 10mM D-glucose– 1x penicillin-streptomycin– 1.33 mg/ml trypsin– 0.67 mg/ml hyaluronidase– 0.2 mg/ml kynurenic acid– pH 7.35, Total osmolality ~280 mOsm
5. N2 supplement culture medium (N2 medium)– DMEM:F12– 2.5µg/ml Fungizone, 1x penicillin-streptomycin– N2 supplement (1x)– Store at 4 °C for <1 month– Prewarm to 37 °C before use

11 Neural Stem Cell Isolation, Characterization and Transplantation 343
6. 4% Paraformaldehyde Solution– Distilled water at 50–60°C– 4% paraformaldehyde– 3–5 pellets of NaOH– 0.2M NaPO4, pH 7.2– Dissolve, cool, filter
7. TCS (Tissue Collecting Solution)/Cryoprotectant– 25% Glycerin– 30% Ethylene Glycol– 0.1M NaPO4, pH 7.2– Store at room temperature for <3 months.
8. PAV-DABCO solution– 25% glycerol– 10% polyvinyl alcohol– 2.5% 1,4-diazobicyclo-[2.2.2]-octane– 100mM Tris HCl, pH 8.5– Store in aliquots at −20°C
■ Procedure
Dissection of Embryonic and Adult CNS Regions
The methods for tissue dissection from embryonic and adult hippocampus and spinal cordof rat are briefly described here. A more detailed protocol can be found in Chapter 14.
Embryonic CNS1. Deeply anesthetize timed pregnant rats by interperitoneal injection of the anesthesiacocktail: ketamine (44 mg/kg), acepromazine (4.0 mg/kg) and rompun (0.75 mg/kg).
2. Remove uterine horns by cesarean section, place on ice, take out embryos from theirindividual sacs and place them in cold D-PBS.
3. Measure the crown-rump length to verify the embryonic age.
Hippocampus1. Remove brain from each embryo and place in D-PBS. Using forceps to stabilize thebrain, peel back the cortex on one side from the midline and lay out flat. Hold the tis-sue pieces with a sharp forceps and cut out the hippocampus, lying just underneaththe cortex, with a pair of erridectomy scissors.
2. Place brains under microscope. Transfer the tissue to D-PBS.3. Take out the hippocampus from the contralateral side. Remove any meningeal mem-
branes or blood vessels still attached to the tissue with sharp forceps. Pool hippo-campi of 15–20 embryos.
Spinal Cord1. Place the embryo on its side in sterile D-PBS under a microscope and make an initialcut lateral to the spinal canal with a pair of erridectomy scissors. Make subsequentcuts on the opposite side, and at the level of the cervical and lower lumbar regions.
2. Remove spinal cord from the embryo and remove residual connective tissue. Poolspinal cords from 10–15 embryos.
Adult CNS Hippocampus1. Anesthetize adult female Fischer 344 rats (3–6 months old), kill by decapitation, re-move brain and place them in cold D-PBS in a petri dish.
2. Place the brain under a microscope, separate the right and the left cortex above tha-lamus by bisecting the corpus collosum and rolling back the posterior margin of each

344 Jasodhara Ray and Fred H. Gage
cortical hemisphere. Separate the fornix and subiculum along the internal and exter-nal edge of each hippocampal formation and remove hippocampus.
Adult CNS Spinal Cord 1. Place anesthetized rat on its side and make a cut lateral to the spinal canal. Make cutson the opposite side and gently remove the spinal cord from the animal. Remove anyconnective tissues attached to the cord. Microdissect sacral, thoracic, lumber andcervical areas.
Establishment of Primary Cultures from Embryonic CNS Regions
Culture of Stem Rat Cells as Monolayers with FGF-2 (Ray et al., 1993)1. Transfer the dissected tissues to a 15ml centrifuge tube containing ~5ml D-PBS, re-
suspend the tissues by gently tapping the tube and wash 3–4 times by centrifugationat 1000g for 3min.
2. Resuspend tissue in 1ml DMEM:F12 medium, break the tissue pieces by titurationwith a fire-polished Pasteur pipet and then make a single cell suspension by repeatedtituration with a large to medium bore (1.5–1.0mm) Pasteur pipet (about 20x). Avoidforming bubbles by not blowing out the last bit of the medium.
3. Wash tissues 1–2x with DMEM:F12 medium by centrifugation to remove debris andmake a final single cell suspension by tituration with a large to medium bore Pasteurpipet (5–10x).
4. Make an appropriate dilution of an aliquot of the cell suspension and count thenumber of cells using a hemocytometer. Adjust the cell density of the suspension andplate 1–2X104 cells/cm2 onto PORN or PORN/laminin coated dishes.
5. Change medium every 3–4 days or longer depending on the confluency. If the celldensity is low, replace half of the medium with fresh medium but increase FGF-2 con-centration to make it 20 ng/ml.
Culture of Stem Mouse Cells with EGF (Reynolds et al., 1992; Reynolds and Weiss, 1996)1. Wash dissected tissues in DMEME:F12 medium, mechanically dissociate the tissue
with a medium bore Pasteur pipet as described before.2. Make a cell suspension, count cells and plate onto PORN coated glass cover slips in
24 well culture dishes at a density of 2500 cells/cm2. Culture cells in N2 medium con-taining 20 ng/ml EGF.
3. After 10–14 days, replace medium with fresh medium and repeat the medium changeevery 2–4 days.
Establishment of Stem Cell Cultures from Adult CNS Regions
Enzymatic Digestion The two most commonly used methods for the digestion of connective tissues to releasethe cells are described below.
Papain-protease-DNase (PPD) Digestion (Ray et al., 1995 )1. Transfer tissue pieces to a 10cm petri dish and cut them into small pieces (1–2 mm3).2. Wash dissected tissues with 5ml HBSS or D-PBS 3x and remove the final wash by as-
piration.3. Resuspend tissue pieces in 5ml PPD solution and incubate for 15min in a 37 °C
waterbath with occasional gentle shaking to keep the tissue pieces resuspended. Tit-urate with a 5ml pipet to break up the large chunk of tissues and incubate for addi-tional 15min in the 37 °C waterbath with occasional shaking. Titurate with a 5ml pi-pet until the cell suspension is free of visible tissue pieces (suspension will turnmilky).

11 Neural Stem Cell Isolation, Characterization and Transplantation 345
4. Remove the remaining tissue pieces by filtration through a nylon mesh. Centrifugethe filtered cell suspension at 1000g for 3min and remove PPD solution by gentle as-piration. Cell suspension is not tightly packed, so do not disturb the cell pellet.
5. Resuspend cells in DMEM:F12 containing 10% FBS (10 ml/g of starting tissue weight)and wash the cell pellet two to five times by centrifugation. Aspirate the medium andresuspend cells in N2 medium containing 10% FBS (1–2ml). The cell suspension stillcontains small tissue pieces, myelin and red blood cells. Cells can be plated withoutfurther purification or can be separated from contaminating cells and debris by per-coll density gradient centrifugation (see subprotocol).
Trypsin-Hyaluronidase-Kynurenic Acid Digestion (Gritti et al., 1996; Weiss et al., 1996b)1. Place the brain/spinal cord of adult mice in 95% O2/5% CO2 oxygenated artificial cer-
ebrospinal fluid (aCSF). Cut the tissues into small pieces (~1–2 mm3) and transfer tospinner flasks (Bellco Glass) containing the enzyme mixture (trypsin, hyaluronidaseand kynurenic acid).
2. Aerate the tissue suspension with 5% O2/5% CO2 and incubate at 32–35 °C for 90minwith constant stirring.
3. Transfer the tissue to DMEM:F12 medium containing 0.7 mg/ml ovamucoid and dis-sociate cells by mechanical tituration with a fire-polished Pasteur pipet. Centrifugedissociated cell suspension at 1000g for 5min, wash pellet once in the same bufferand then plate cells without further purification or separate them from contaminat-ing cells and debris by Percoll density gradient centrifugation (see subprotocol).
Culturing of Rat Cells as Monolayers (Ray et al., 1995; Gage et al., 1995b).1. Resuspend cells in N2 medium containing 10% FBS and plate at least 1x104 cells/cm2
in 10cm uncoated tissue culture plates.2. On the next day, change medium with serum-free N2 medium containing 20 ng/ml
FGF-2.3. Feed cultures every 3–4 days and if the cell density is low, exchange half the medium
with fresh medium. Add double amounts of FGF-2 to increase the concentration to20 ng/ml.
Culturing of Mouse Cells as Neurospheres (Reynolds and Weiss, 1992; Gritti et al., 1996;Weiss et al., 1996b)1. Plate cells (25–1000 cells/cm2) in uncoated 6 well plates in serum-free N2 medium
containing EGF, FGF-2 or both at concentrations of 20 ng/ml. Cells can be maintainedand passaged as neurospheres or can be grown as monolayer cultures (J. Ray, unpub-lished results).
2. To grow mouse stem cells as monolayers, allow spheres to grow until they are big andattach to the substratum.
3. Passage monolayer cultures by trypsinization onto uncoated tissue culture dishes(see subprotocol).
Isolation of Clonal Cultures
The main reason for isolating clonal cultures is to determine whether the stem cells aremultipotent and can give rise to both neurons and glia. Clonal cultures can be generatedin two ways:
Limiting Dilution1. Plate cells from bulk cultures at clonal density (1–2 cells/well; 96 well plate or 1 cell/7cm2 in a 35mm petri dish) onto PORN/laminin coated dishes in serum-free N2 me-dium containing appropriate growth factor.

346 Jasodhara Ray and Fred H. Gage
2. To monitor a particular cell, mark its position on the dish by scratching the bottomof the plate. Stem cells migrate in the dish, so always make sure that the same cell ismonitored.
3. Feed cells every 4–5 days with medium containing EGF or FGF-2 (20 ng/ml) and sup-plemented with 50% conditioned medium collected from a high density stem cellculture (see subprotocol). Since cells present at low density will not be effectively ableto condition their own medium, factors present in the conditioned medium collectedfrom high density culture will support the survival/proliferation of stem cells platedat clonal density.
4. Monitor the cells until the density of the clones reaches a critical mass (>100cells/colony), then either passage cells and expand or characterize the clonal popula-tions by immunocytochemistry.
Genetic Marking ofCells to Establish Clonal
Cultures
1. Infect a bulk population of stem cells with the limiting dilutions of the retroviral vec-tor of choice expressing a marker gene like green fluorescent protein (GFP), E. coliLacZ gene or alkaline phosphatase (Suhonen et al., 1997; see comments).
2. Plate 1% of the cells in the presence of the minimum amount of G418 needed to selectfor stable transfectants. Usually start with 40µg/ml G418 and increase the concentra-tion slowly to 100 µg/ml. To increase cell survival during the selection process, in-crease the concentration of G418 gradually and use medium supplemented with 50%conditioned medium collected from a high density stem cell culture.
3. Feed cells every 3–4 days until clusters of proliferative cells appear. Selection can bestopped when stably transfected cultures are established but select cells periodicallyto remove cells that have lost the selectable marker gene.
4. Passage individual clones with agarose/trypsin (see subprotocol).5. Determine the clonality of cultures by Southern blot analysis (Sambrook et al., 1989).
Briefly, prepare genomic DNA by lysis of cells and digest with appropriate restrictionenzymes that cut once within the vector or once in each viral long terminal repeat.Resolve digested DNA on agarose gels, transfer to nylon membranes and probe with32P-labeled neo or transgene-specific probes.
Immunocytochemical Analysis of Stem Cells (Peterson et al., 1996)
All staining procedures are carried out at room temperature except where indicated andwashing steps are done for 10min each.1. Grow rat or mouse stem cells in PORN/laminin coated glass chamber slides until 50–
70% confluent.2. Fix cells for 10min in 4% paraformaldehyde and wash twice in 100mM Tris-buffered
saline (TBS). Fixed cells can be stored in TBS at 4 °C for about a week or can be pro-cessed immediately for immunocytochemistry.
3. Preincubate cells for at least 1h in TBS containing 10% donkey serum and 0.25% Tri-ton X-100 (blocking buffer).
4. Incubate with pooled primary antibody (polyclonal and monoclonal) diluted in TBScontaining 0.25% Triton X-100 (TBS+). If the antibody recognizes cell surface mole-cules then exclude Triton from the incubation buffer.
5. After 24–48h at 4 °C, wash cells 3x with the blocking buffer and incubate for 2h in thedark with species-specific secondary antibody conjugated to the desired fluorophoreslike fluorescein isothiocyanate (FITC), Texas Red Cy-5 or Cy-3 diluted in TBS+.
6. Wash cells twice in TBS, incubate in TBS containing DAPI (10 ng/ml) for 1min andthen coverslip in PAV-DABCO solution. Analyze cell phenotype by confocal or fluo-rescence microscopy.

11 Neural Stem Cell Isolation, Characterization and Transplantation 347
7. If necessary, the signal for specific antigens can be amplified by using biotin-strepta-vidin amplification. After the primary antibody and wash step, incubate cells in bi-otinylated donkey anti-species antibody diluted in TBS+ for 2h, wash twice in thesame buffer and then incubate in streptavidin conjugated to the desired fluorophore.
8. To detect both cell surface and nuclear or cytoplasmic antigens in the same cell, pre-incubate cells in TBS containing 10% donkey serum and then incubate with antibo-dies against cell surface antigens for a minimum of 2h at room temperature or over-night at 4 °C. Wash cells three times in TBS, postfix in 4% paraformaldehyde for5min, wash three times in TBS and then preincubate in blocking buffer. Proceed withthe antibody staining protocol as described before.
Differentiation of Stem Cells
1. Plate cells in PORN/laminin coated glass chamber slides at a density of 1X105/cm2
(high density) or 2.5–3X103/cm2 (low density) and grow for 24h in serum-free N2medium containing FGF-2.
2. Replace the medium with fresh medium containing differentiating agents like serum(0.5, 2 or 10%), retinoic acid (1µM), forskolin (5µM), brain-derived neurotrophicfactor (BDNF; 20 ng/ml), neurotrophin 3 (NT-3; 40 ng/ml), ciliary neurotrophic fac-tor (CNTF; 10–20 ng/ml), leukemia inhibitory factor (LIF; 10 ng/ml) and thyroid hor-mone T3 (3 ng/ml). To density arrest, plate cells in the absence or the presence of alow concentration of FGF (1 ng/ml) (Vicario-Abejon et al., 1995; Johe et al., 1996;Palmer et al., 1997).
3. Change medium every 2 days and allow the cells to differentiate for 6 days. Fix cellswith paraformaldehyde and analyze by immunocytochemistry.
Stereotaxic Implantation of Stem Cells into the Adult Brain
The procedure for the preparation of animals for the implantation of stem cells is simi-lar to that described for the implantation of fetal cells in Chapter 14. In addition, de-tailed protocols related to the grafting procedures, perfusion of the animals, sectioningof the brain tissues and their characterization by using histochemical and immunocy-tochemical methods can be found in Suhonen et al. (1997). Methods are briefly de-scribed below.
Preparation of Cells for Grafting
1. Passage stem cell cultures 3–5 days prior to grafting from a 70–80% confluent cultureat 1:1 or 1:2 split ratio.
2. Cultures should be 50–60% confluent by the start of BrdU treatment. Two days priorto grafting change medium with fresh medium containing appropriate growth fac-tor(s) and 5µM BrdU (stock 5 mM in water).
3. Repeat the process the next day.4. Detach cells from the flasks by using ATV trypsin, transfer to a 15ml centrifuge tube
with D-PBS. Centrifuge at 1000g for 3min, wash cells with D-PBS twice and resus-pend in D-PBS by using medium-to-small bore Pasteur pipets.
5. Count the cell number in a hemocytometer.6. Remove the appropriate number of cells to a 0.5ml Eppendorf tube, spin for 1min in
a microcentrifuge and resuspend cells at a concentration of 5X104-1X105 cells/µl inD-PBS containing 20 ng/ml of FGF-2 for rat or EGF or both the factors and heparinfor mouse.

348 Jasodhara Ray and Fred H. Gage
Grafting of Stem Cells inAdult Rat Brain
1. Determine the coordinates of the injection sites from either the adult rat brain atlas(Paxinos and Watson, 1986) or by injecting a small amount of dye at a particular co-ordinate to determine its location (should be done as a separate experiment priorto grafting experiments. See Suhonen et al., 1997).
2. Anesthetize the recipient animals with an intramuscular injection of anesthesia so-lution.
3. Shave the skull, sterilize the skin with Betadine and place the anesthetized rat in thestereotaxic frame.
4. Make a single incision in the skin from a point midline between the eyes to a pointbetween the ears using a No. 10 scalpel blade. Separate the skin flaps, clean and drythe skull surface of blood and connective tissue with swabs.
5. Identify Bregma. Drill a 1.0mm wide hole over the desired point into the cranium.Cut the dura with the point of 26 gauge needle.
6. Gently resuspend the cells by tapping the tube containing the cell suspension. Drawup the required volume (1–3µl; 5X104-1X105 cells/µl) of cell suspension into the sy-ringe mounted to the electrode manipulator on the stereotaxic frame, avoiding airbubbles.
7. Lower the syringe to the specific vertical distance below dura assuming a stereotax-ic frame of reference measured from Bregma for the anterior-posterior and medial-lateral coordinates, and from dura for the vertical coordinates. Slowly inject re-quired volume of solution at a rate of 1µl per min or slower (2–3µl/min).
8. When the injection is completed, raise the needle 1mm and then leave the syringeneedle in place for an additional 2min to minimize cell diffusion up the needle trackand then gently withdraw over a 1–2min period.
9. If there is more than one injection site, repeat injection steps at the new sites.10. Remove the rat from the stereotaxic frame, clean the skull, sprinkle antibiotic pow-
der, put the skin flaps together, close the skin incision with wound clips and transferthe animal to a recovery cage.
Perfusion of Adult Rat
A detailed protocol can be found in Suhonen et al. (1997).1. At appropriate time points after surgery anesthetize the rats.2. Perfuse the animal transcardially with ice-cold 0.9% saline (50 ml/rat) using a per-
fusion pump at a flow rate of ~1000 ml/h followed by fixation with 4% paraformal-dehyde (250 ml/rat or 150ml for head perfusion only). Add 0.1% glutaraldehyde ifelectron microscopy is planned or if certain antibodies require glutaraldehyde fixa-tion. In our experience, the presence of a low concentration of glutaraldehyde doesnot interfere with the antigenicity of the tissue.
3. Remove brains, postfix overnight at 4 °C on a shaker table. Transfer the brains to 30%sucrose (in 0.1M NaPO4, pH 7.2), and keep at 4 °C for at least 3 days or until thebrains sink before sectioning. If fresh tissue is used for the experiments then omit thesucrose step.
Sectioning of the Rat Brains
1. To cut brain sections in a freezing sliding microtome, trim the brain to the minimumsize containing the desired target areas.
2. Mount the trimmed-down brain on the chuck of a freezing sledge microtome con-taining OTC compound and then freeze with powdered dry ice for 15minutes. OTCcompound facilitates the sectioning by holding the tissue firmly to the chuck.

11 Neural Stem Cell Isolation, Characterization and Transplantation 349
3. Cut the sections and transfer the slices from the knife into tissue culture wells (24 or96 wells) containing TCS. Sections can be stored in a −20 °C freezer indefinitely.
Coating of Tissue Culture Plates
All procedures should be done under sterile conditions (in a laminar flow hood) usingsterile solutions. For cell attachment and growth as a monolayer, cultures are grown ontissue culture dishes coated with PORN or PORN/laminin, pDL or pLL, PORN orPORN/laminin (Ray et al., 1995)1. Make a 10 mg/ml stock solution of PORN in sterile water and filter sterilize by pass-
ing through 0.22µm filter. Store aliquots at −20 °C.2. Add enough PORN (10µg/ml in water) to entirely cover the surface of the dish and
incubate at room temperature for 24h.3. Wash 2–3 times with sterile water, store plates in water in sealed plastic bags at −20 °C
for future use.4. Alternatively, air dry PORN coated plates at room temperature in a laminar flow
hood. Before plating the cells, wash plates 2–3 times with water followed by a washwith the culture medium. If coating with laminin, use PBS instead of water for wash-ing.
5. Make a stock solution of laminin (5 mg/ml mouse or rat laminin) in PBS. Store insmall aliquots at −80 °C. Do not freeze-thaw laminin more than once or twice.
6. Add enough laminin (5µg/ml in PBS) to PORN coated plates to cover the surface andincubate at 37 °C for 24h.
7. Store the plates sealed in plastic bags at −20 °C. The plates can be stored for 1–2months. Wash once in PBS or medium before plating cells.
pDL or pLL (Juurlink, 1992)
1. Make a stock solution of pDL or pLL (1.0 mg/ml) in water or in 0.1M boric acid-NaOH buffer, pH 8.4, and filter sterilize. Store in small aliquots at −20 °C.
2. Dilute the stock in water or appropriate buffer to make a solution of desired concen-tration (10–50µg/ml). Cover the surface of the plates with enough pDL or pLL solu-tion, incubate at 37 °C for 2–24h.
3. Wash plates with sterile water 3–4 times and air dry. Plates can be used for severalweeks.
Percoll Gradient Purification of Stem Cells
Stem cells can be partially purified from contaminating debris and other cell types byusing Percoll density gradients.1. Dilute stock Percoll solution 9:1 (vol/vol) with 10x PBS.2. For discontinuous density gradient, form layers containing 50, 40, 30, 20 and 10%
Percoll. Layer cell suspension (obtained after enzymatic digestion and filtration)over step gradient (Maric et al., 1997).
3. Centrifuge at 400g for 20–30min at room temperature.4. Collect the layer between 40–50% gradient.5. Dilute 2–5 fold in cold PBS (containing antibiotics and fungizone) and wash cells at
least 3 times by centrifugation at 1000g for 3min. The cell pellet is very small; to avoidlosing the cell pellet leave behind ~1ml wash liquid in between aspiration and wash-es.
6. Resuspend the cell pellet in 1ml of the appropriate plating medium, count cells andthen plate ~1.3–4X104 cells/cm2 (1–3X106 cells/75cm2 flask). If a smaller number ofcells is obtained, plate in 35 or 60mm plates.

350 Jasodhara Ray and Fred H. Gage
7. Cells can also be separated from debris by continuous density gradient centrifuga-tion. Mix cells with Percoll (1:1), centrifuge and then collect the liquid in betweenmyelin layer at the top and the red blood cell layer at the bottom. Proceed from step 5.
Passaging and Re-culturing of Neural Stem Cells
Monolayer Cultures 1. Add 1.0–1.5ml ATV trypsin/10cm plate or T 75 flask (add less for smaller dishes) pre-warmed to 37 °C. Swirl plate/flask to distribute the liquid evenly.
2. Let sit for 1min. Hit the side of the plate gently to dislodge cells.3. Transfer cells to a 15ml sterile centrifuge tube by using PBS. Wash the plate once with
PBS and transfer to the same tube. Centrifuge at 1000g for 3min.4. Remove supernatant slowly so as not to disturb the cell pellet. Resuspend cells in 1
ml serum-free N2 medium, titurate with a medium bore fire-polished Pasteur pipet.5. Plate portions of the cells (split-ratio will depend on initial cell density and the
growth rate of cells) on PORN/laminin coated (rat cells) or uncoated (mouse cells)plates in the same medium containing FGF-2 (rat cells) or EGF, FGF-2 and heparin(mouse cells).
6. If necessary freeze cells in liquid nitrogen for long-term storage.
Passaging ofNeurospheres
1. Collect the culture medium containing the spheres in 15ml sterile centrifuge tubesand centrifuge at 1000g for 3min. Remove supernatant slowly without disturbing thecell pellet.
2. Resuspend cells in 1ml serum-free N2 medium containing EGF and make a single cellsuspension by tituration (10–20x) with a medium bore Pasteur pipet.
3. Plate cells onto uncoated plates or freeze cells in liquid nitrogen for long-term stor-age.
Freezing of Cells 1. Suspend cells in serum-free N2 medium containing 10% DMSO and appropriategrowth factors.
2. Aliquot 1ml in each freezing vial.3. Put the vials in freezing chambers and place the chamber in −70 °C freezer to allow
the cells to freeze slowly.4. On the next day transfer the vials to a box kept in liquid nitrogen.
Re-culturing ofFrozen Cells
1. Remove vials from liquid nitrogen and thaw cells quickly by constant shaking of thetube in 37°C waterbath.
2. Transfer cells to a sterile 15ml centrifuge tube with DMEM:F12 medium, centrifugeat 1000 g for 3min. Remove supernatant.
3. Wash cells once in the same medium. Resuspend cells in 1ml N2 medium, make a sin-gle cell suspension by titurating with a medium bore Pasteur pipet.
4. Plate on PORN/laminin coated (for rat cells) or uncoated (for mouse cells) plates inserum-free N2 medium containing appropriate growth factors.
Agarose/trypsin Methodfor Passaging Cells
1. Pick clones from plates that have relatively big (>100 cells/clone) and well-separatedcolonies. Mark a clone by marking it on the back of the dish.
2. Melt 3% agarose solution (made in PBS) in a microwave oven, cool to ~45–50 °C andmix 1 ml agarose with 2ml ATV trypsin (warmed to 37 °C).
3. Remove culture medium and immediately add agarose/trypsin mixture to the dishcontaining the colonies, swirl the plate to gently spread over the cells and allow to so-lidify for 2–3min.
4. With a sterile Pasteur pipet gently cut around the clones and lift the agarose plugs(with cells attached to them) and transfer them to individual wells of a 24 well plate

11 Neural Stem Cell Isolation, Characterization and Transplantation 351
containing serum-free N2 medium supplemented with 50% conditioned mediumand G418. Gently wash the area of the plug twice with the medium (~100µl) andtransfer it to the well containing the cells.
5. Change medium every 3–4 days and allow the cells to grow until desired confluencyis reached and cells are ready to be passaged.
Preparation of Condition Medium for Use in Culturing
1. Collect the conditioned medium from a high density stem cell culture after at least 24h incubation.
2. Centrifuge at 1000g for 5min and freeze aliquots. The conditioned medium can alsobe filter sterilized to prevent accidental contamination with residual cells.
Analyses of Brain Sections
The fate and the nature of the grafted stem cells in vivo are characterized by immuno-cytochemistry, in situ hybridization and electron microscopy. Only the immunocyto-chemistry method is described here.1. To detect BrdU, pretreat free-floating CNS sections for 2h in 50% formamide/2XSSC
at 65°C followed by a 30-min incubation in 2M HCl at 37 °C.2. Analyze the pretreated sections by immunocytochemistry (as described above) for
the expression of specific marker proteins followed by confocal or fluorescence mi-croscopy. Co-localization of BrdU and a specific marker protein will identify the invivo fate of the grafted cells.
Cell numbers in brain sections are quantified by using unbiased stereology. The methodhas been described in detail previously (Sterio, 1984; Peterson et al., 1994; West and Slo-mianka, 1998) and only an outline is given here.1. Sample a systematic series of tissue sections completely containing the structure to
be quantified in a uniform, random manner using the optical dissector principle sothat all cells within the tissue have equal probability of being sampled.
2. Count cells directly within the three-dimensional unbiased counting frame accord-ing to the sampling criteria for the optical dissector.
3. Calculate results either directly from the known subsample of the total structure (theoptical fractionator procedure) or by combining the numerical density achievedfrom the optical dissector counts with the estimation of total structure volume deter-mined using the Cavalieri procedure (the NV-VRef procedure).
■ Results
Growth Properties and Morphology of Rat and Mouse CNS-derived Stem Cells
When rat stem cells are plated as monolayers at a high density (2–5X104 cells/cm2), pro-liferating cells can be seen within 3–5 days (depending on the initial plating density;Fig.2a). At least half the cells in culture have a stem cell morphology of small phasebright cell bodies and two or more long processes (Fig.2 b, c). The cultures also containcells of different morphologies, including some flat and some long phase dark elongatedcells (Fig.2 b, c). Stem cells seem to be generated from flat cells (Fig.2 c). The flat cellsdo not express any stem/progenitor cell marker proteins (nestin, vimentin, O4 andA2B5) and may represent true stem cell populations (J. Ray, unpublished observations).

352 Jasodhara Ray and Fred H. Gage
Mouse stem cell proliferation and neurosphere formation are detected 3–5 days afterplating (Fig.3 a) and they increase in size with time in culture (Fig.3 b). When some ofthe neurospheres reach a critical mass, they attach to the substratum and start to gen-erate streams of cells. The cells grow out of the spheres (Fig.3 c), and on subsequent pas-saging cells will grow as monolayers (Fig.3 d). Some spheres do not attach and grow asspheres even after passaging. Although it is not possible to determine the morphologyof cells within the neurospheres, the morphology of cells grown as monolayers is differ-ent from rat cells. They have bigger, more elongated cell bodies and smaller processesthan rat stem cells.
■ Troubleshoot
– Purity of the cultures to a large extent depends on the clean dissection of tissues.Contaminating connective tissues can increase the non-neuronal cell population thatwill eventually overtake the cultures.
– Enzymatic digestion of tissues with PPD should not be done for more than 40min.Longer enzymatic digestion will lower the yield of stem cells.
– Embryonic rat cells plated and cultured in uncoated tissue culture dishes in serum-free medium containing EGF generated neurospheres, but they grew poorly and
Fig. 2. Morphology of adult rat neural stem cells cultured in serum-free N2 medium containing FGF-2. (a) Proliferating cellscan be seen by 3–5 days in vitro (DIV). Stem cells have small phase bright cell bodies and two or more long processes (smallarrows in a, d). (b) By 14 DIV, a large number of stem cells are present. (c) The cultures also contain flat cells (indicated by longarrows) which do not stain for any stem or precursor cell markers. Small phase bright cells seem to generate on top of theseflat cells. (d) Mostly stem cells are present in the passaged cultures. Scale bar: 100µm

11 Neural Stem Cell Isolation, Characterization and Transplantation 353
could not be expanded for more than 4 weeks (Svendsen et al., 1997; J. Ray, unpub-lished observation).
– Survival of rat stem cells grown as monolayer cultures is density dependent, and whenplated at <1000 cells/cm2 they do not survive even in the presence of FGF-2J. Ray, un-published observation). However, when mouse stem cells are grown as neurospheres,where cells remain in close contact, they grow even when plated at a low density, albeitat a slow rate. For example, adult mouse striatal stem cells plated in EGF or FGF-2 at200 cells/cm2 began to divide by 5 days in vitro (DIV) and generated spherical clustersof cells by 21 DIV (Gritti et al., 1996). No significant difference was found between thenumber of spheres per plate generated in response to EGF or FGF-2.
– The culturing conditions and requirements of factors for the growth of rat, mouse,monkey and human CNS-derived stem cells are different (Ray et al., 1995; Svendsenet al., 1997; Sah et al., 1997; J. Ray, unpublished observation). The best condition togrow adult rat stem cells, irrespective of CNS regions, is to culture them as monolay-ers in serum-free N2 medium containing FGF-2 (20 ng/ml) (Gage et al., 1995b). Con-ditions for the establishment of stem cell cultures from mouse CNS regions are quitedifferent from those for rat. The plating conditions used for rat cells do not generatestem cell populations from mouse CNS (J. Ray, unpublished observation)
– Do not trypsinize cells for more than 2min. Longer incubation will result in extensivecell death. Stem cells detach easily and shorter trypsin treatment will enrich pas-
Fig. 3. Morphology of adult mouse neural stem cells cultured in serum-free N2 medium containing EGF, FGF-2 and heparin. (a)Neurospheres are visible by 5 DIV. With time in culture neurospheres increase in size (b) and some of the spheres attach to thesubstratum (c). Cells stream out of the spheres and grow as monolayer. (d) Upon passage attached cells grow as monolayers.Mouse stem cells have more elongated cell bodies (arrows in c, d) and smaller processes than rat stem cells. Scale bar: 100µm

354 Jasodhara Ray and Fred H. Gage
saged cultures with stem-like cells while leaving behind flat cells and more differen-tiated glia and neurons.
– For fixing cells, use fresh or frozen (−20 °C) aliquots of paraformaldehyde. For per-fusion, prepare paraformaldehyde just before use, keep on ice or in the cold room un-til use.
– To achieve maximum cell survival after grafting, prepare cells just before grafting,keep them at room temperature during the grafting process and resuspend once eve-ry 30–40minutes.– Graft cells within 3h after the preparation of the cell suspension. If grafting a large
number of animals, prepare the cells in batches to minimize the time they are keptin suspension before grafting.
– The optimal concentration of cell suspension is important as a large number ofcells grafted at one site produce a dense graft and prevent the nutrients necessaryfor graft survival from reaching the center of the graft. As a result cells start to dieand yellow necrotic centers are formed. A concentration of 5X104–1X105 cells/µland a deposit of 1–2µl are used in the authors’ laboratory. If it is necessary to im-plant more cells, make multiple deposits.
– Use 26 gauge or smaller size needles (that are not too narrow) to avoid shearingcells as this will result in poor survival of the grafted cells. To avoid clogging, reg-ularly rinse the syringe and the needle with ethanol and saline solution.
– To check the viability of the cells used for grafting, replate the extra cells and ana-lyze how many cells survive after 1–2 days.
– For immunostaining it is best to use 40–50µm free-floating brain sections althoughthinner sections can result in better resolution. Depending on the desired thicknesscut the sections on a freezing sledge microtome (30–100µm) or on the cryostat (1–50µm).
■ Comments
Some of the parameters important to consider for in vitro culturing of stem cells andtheir analyses are discussed below.
Age and Brain Regions
Although stem cells can be cultured from both embryonic and adult brain from differentspecies, it is easier to establish cultures from embryonic brains. Development of brainregions and the cascade of expression of various receptors for growth factors that areessential for their survival and proliferation take place during discrete developmentalperiods. For culturing EGF or FGF-2-responsive stem cells choose the optimum embry-onic age at which the receptors for these mitogenic growth factors are expressed abun-dantly. Use freshly dissected tissues whenever possible as this will generate better yieldof stem cells.
Proteolytic Digestion
No comparison has been made of the two most commonly used enzymatic digestionmethods described here to extricate live cells from the three-dimensional adult CNSnetwork and it is not possible to say which is the better method. In the authors’ labora-

11 Neural Stem Cell Isolation, Characterization and Transplantation 355
tory the PPD digestion method is routinely used to culture stem cells from various adultbrain regions as well as from different species and has yielded large numbers of viablecells. A number of other proteases alone or in combination have also been successfullyused to isolate stem cells from adult brain (Reynolds and Weiss, 1992; Weiss et al., 1996).Comparison of the viability of cells isolated from embryonic brains by various protocolsinvolving mechanical or enzymatic (collagenase, trypsin or papain) methods showedthat papain optimizes cell viability compared to other methods (Maric et al., 1997). Astudy that compared the ability of different proteases (trypsin/proteinase K, proteaseXXIII, papain, collagenase and dispase) to isolate viable neurons from 6-week-old ratsshowed that, although almost equal numbers of cells could be isolated with different en-zymes, the viability of cells isolated with papain alone was better (Brewer, 1997).
Medium and Supplements
Most methods described for the culture of neural stem cells use DMEM or DMEM:F12medium, N2 supplement and EGF, FGF-2 or both. Neurobasal medium (Gibco) contain-ing B27 supplements and EGF has also been used to culture embryonic (E18) rat brainstem cells (Svendsen et al., 1995) and adult rat hippocampal neurons (Brewer, 1997). B27supplement includes a range of hormones, anti-oxidants and retinal acetate (Brewer etal., 1993) in addition to the basic formulation of N2 (Bottenstein and Sato, 1979). How-ever, the survival rather than the proliferation of stem cells following their initial platingis strongly increased in B27 medium containing EGF (Svendsen et al., 1995). To generateproliferating stem cells, cultures established in B27 medium can be transferred to N2medium containing EGF without any detrimental effect.
Substratum
Composition of the substratum is important for the adhesion, survival, proliferationand differentiation of cells. To grow cells as monolayer cultures they are plated on un-coated plastics in the presence of serum for 2–24h. Factors in serum provide compo-nents for cell attachment. Alternatively, plates can be coated with agents like polymersof basic amino acids PORN or pLL or pDL and cells will attach on the basis of charge.Laminin, a cell adhesion molecule, can be used in addition to PORN as substratum.However, neural stem cells can also be cultured in uncoated tissue culture dishes wherethey form neurospheres and remain as suspension cultures. Some of the neurospheresloosely attach to the plates and grow as monolayers. Substratum has also been reportedto influence the fate choice of stem cells (Stemple and Anderson, 1992). For example,subclones of neural crest cells generate only astrocytes when plated on fibronectin butneurons when plated on fibronectin/pDL.
Choice of EGF vs. FGF-2
Although both FGF-2, EGF or their combinations have been successfully used to culturestem cells from mouse and rat, the cells from different species respond differently tothese growth factors. Direct comparison of growth rates and survival of stem cell neuro-spheres isolated from embryonic rat or mouse striatum and cultured in EGF showedthat, although mouse cells can be expanded over 50 days, rat cells died between days 21and 28 (Svendsen et al., 1997). FGF-2 alone did not lead to an expansion of rat striatalcells under the conditions used in the experiments. Although combinations of FGF-2

356 Jasodhara Ray and Fred H. Gage
and EGF acted synergistically on the growth of rat cells, they did not prevent their senes-cence and death. In summary, EGF, FGF-2 or their combinations generate neurospheresfrom mouse and rat brains but the presence of FGF-2 limits the expansion capability ofrat cell neurospheres. In contrast, rat stem cells cultured as monolayers in the presenceof FGF-2 can be expanded and maintained in culture for a long period of time (Ray etal., 1993; Ray and Gage, 1994; Gage et al., 1995b; Shihabuddin et al., 1997). This differ-ence may reflect the difference between two culture conditions, or EGF and FGF-2 maybe recruiting cells that differ in their “stemness.” Future studies will address these issues.
Neurospheres vs. Monolayer Cultures
It must be noted that no study at this point has compared the properties of stem cells cul-tured as monolayers or neurospheres with FGF-2 or EGF alone or with a combination ofthe factors. As a result it is not possible to say whether cells isolated and cultured by dif-ferent methods using the two growth factors recruit the same type of cells and expandthem in culture or if they are different cells. It is possible that the same cells are recruitedbut that they exhibit different properties in EGF and FGF-2. For example, EGF-respon-sive stem cells can be subsequently expanded in FGF-2 (Vescovi et al., 1993) but FGF-2-responsive cells cannot be subsequently cultured in EGF (J. Ray, unpublished observa-tion).
There are several disadvantages for culturing cells as neurospheres. The morphologyof cells in the spheres cannot be determined visually and due to lack of penetration ofthe antibodies, they will immunostain cells present on or close to the surface of thespheres. Consequently, the nature of cells present within the spheres cannot be deter-mined. If the spheres are big, cells situated well within the spheres would die due to lackof nutrients needed for their survival and hence cell yield will reduce with time and pas-sage. Neurosphere cultures cannot be used for some biochemical assays like ligandbinding studies to determine the number of specific receptor binding sites present onthe cells, rate of cell division and for proliferation assay by 3H-thymidine incorporationto determine the mitogenic effects of growth factors.
Retroviral Vectors
Retroviral vectors are derived from Moloney murine leukemia virus (MoMLV) and con-tain the gene of interest (transgene) and a selectable marker like neomycin-resistantgene, neo (Verma and Somia, 1997). Retroviral vectors can infect only dividing cells andintegrate randomly in the cellular genome. As a result, all the progeny of a single infect-ed cell will inherit a unique and identifiable integration site (Cepko, 1988b) and theclonality of a population of cell can be determined by restriction enzyme digest fol-lowed by Southern blotting (Sambrook et al., 1989).
Choice of Methods for Sectioning of Brain
Thick cryostat or freezing sliding microtome-cut sections are used for immunohisto-chemical analyses and thin cryostat sections for in situ hybridization. Freezing slidingmicrotome is used to cut thicker (40–50µm) sections from fixed brains and it is difficultto get sections <25µm thick. The thicker sections are well suited for analysis by confocalmicroscopy where the requisition of multiple focal planes produces a data set fromwhich three-dimensional information about the cytoarchitecture can be obtained. An-other advantage of the microtome is that multiple brains can be sectioned simultane-ously. The cryostat can be used to cut thinner sections (10–30µm) from fresh or fixed

11 Neural Stem Cell Isolation, Characterization and Transplantation 357
brains and mounted to glass slides directly after cutting. Thin cryostat sections cut fromfresh brain are used for in situ hybridization. Brain sections cut in the vibratome arethicker (>50µm) and are generally used for electron microscopy. One disadvantage ofvibratome cut sections is that they are susceptible to compression artifacts.
■ Applications
The ability to generate long-term cultures of neural stem cells from normal or transgen-ic animals, to obtain clonal cultures and to transfer transgene into these cells hasopened up the possibility of studying how environmental stimuli influence the fatechoice and differentiation potentials of these cells bothin vivo and in vitro. In addition,stem cells can be genetically modified to express specific growth factors and neuro-transmitters. Stem cells, normal or genetically modified, can be used in animal modelsof neurodegenerative diseases with the ultimate aim of providing an expandable sourceof well-characterized cells for cell replacement therapy or gene therapy in humans.
Acknowledgement: We thank M. L. Gage for her helpful critique of the manuscript. Thework in our laboratory was supported by grants from American Paralysis Association,Hollfelder Foundation, STTR fund from NIA (R42 AG12576–03) and by Contract NO1-NS-6-2348 from NIH. The content of this publication does not necessarily reflect theviews or policies of the Department of Human and Health Services; neither does men-tion of trade names, commercial products, or organizations imply endorsement by theU. S. Government.
■ References
Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993) Optimized survival of hippocampal neurons inB27-supplemented Neurobasal, a new serum-free combination. J Neurosci Res 35:567–765
Brewer GJ (1997) Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods71:143–155
Bottenstein JE, Sato G (1979) Growth of rat neurobalstoma cell line in serum-free supplementedmedium. Proc Natl Acad Sci USA 76:514–517
Carpenter MK, Winkler C, Fricker R, Emerich DF, Wong SC, Greco C, Chen E-Y, Chu Y, KordowerJH, Messing A, Björklund A, Hammang JP (1997) Generation, and transplantation of EGF-re-sponsive neural stem cells derived from GFAP-hNGF transgenic mice. Exp Neurol 148:187–204
Cepko CL (1988a) Immortalization of neural cells via retrovirus-mediated oncogene transduc-tion. Trends Neurosci. 11:6–8.
Cepko CL (1988b) Retroviral vectors and their applications in neurobiology. Neuron 1:345–353Fisher LJ (1997) Neural precursor cells: applications for the study and repair of the central nerv-
ous system. Neurobiol Dis 4:1–22.Gage FH, Ray J, Fisher LJ (1995a) Isolation, characterization and use of stem cells from the CNS.
Ann Rev Neurosci 18:159–192Gage FH, Coates PW, Palmer TD, Kuhn HG, Fisher LJ, Suhonen JO, Peterson DA, Suhr ST, Ray J
(1995b) Survival and differentiation of adult neuronal progenitor cells transplanted to theadult brain. Proc Natl Acad Sci USA 92:11879–11883.
Gritti A, Parati EA, Cova L, Frolichsthal P, Galli R, Wanke E, Faravelli L, Morassutti DJ, Roisen F,Nickel DD, Vescovi AL (1996) Mutipotential stem cells from the adult mouse brain proliferateand self-renew in response to basic fibroblast growth factor. J Neurosci 16:1091–1100.
Hammang JP, Reynolds BA, Weiss S, Messing A, Duncan ID (1994) Transplantation of epidermalgrowth factor-responsive neural stem cell progeny into the murine central nervous system.Methods in Neurosci 21:281–293
Johe KK, Hazel TG, Muller T, Dugich-Djordjevic MM, McKay RD (1996) Single factor direct thedifferentiation of stem cells from the fetal and adult central nervous system. Genes Develop10:3129–40.

358 Jasodhara Ray and Fred H. Gage
Juurlink BH (1992) Chick spinal somatic motoneurons in culture. In: Federoff S and RichardsonA (eds) Protocols for neural cell culture, Humana Press, Totowa, NJ. pp 39–51.
Kordower JH, Chen E-Y, Winkler C, Fricker R, Charles V, Messing A, Mufson EJ, Wong SC, Rosen-stein JM, Björklund A, Emerich DF, Hammang JP, Carpenter MK (1997) Grafts of EGF-respon-sive neural stem cells derived from GFAP-hNGF transgenic mice: trophic and tropic effects ina rodent model of Huntington’s disease. J Comp Neurol 387:96–113
Kuhn HG, Dickinson-Anson H, Gage FH (1996) Neurogenesis in the dentate gyrus of the adult rat:age-related decrease of neuronal progenitor proliferation. J Neurosci 16:2027–2033.
Lendahl U, McKay RDG (1990) The use of cell lines in neurobiology. Trends Neurosci 13:132–137.Maric O, Maric I, Ma W, Lahojuji F, Somogyi R, Wen X, Sieghart W, Fritschy J-M, Barker JL (1997)
Anatomical gradients in proliferation and differentiation of embryonic rat CNS accessed bybuoyant density fractionation: alpha 3, beta 3 and gamma 3 GABAA receptor subunit co-ex-pression by post-mitotic neocortical neurons correlates directly with cell buoyancy. Eur J Neu-rosci 9:507–522.
McKay RD (1997) Stem cells in the central nervous system. Science 276:66–71.Minger SL, Fisher LJ, Ray J, Gage FH (1996) Long-term survival of transplanted basal forebrain neu-
rons following in vitro propagation with basic fibroblast growth factor. Exp Neurol 141:12–24.Morshead CM, Reynolds BA, Craig CG, McBurney MW, Staines WA, Morassutti D, Weiss S, van
der Kooy D (1994) Neural stem cells in the adult mammalian forebrain: A relatively quiescentsubpopulation of subependymal cells. Neuron 13:1071–1082.
Palmer TD, Ray J, Gage FH (1995) FGF-2-Responsive neuronal progenitors reside in proliferativeand quiescent regions of the adult rodent brain. Mol Cell Neurosci 6:474–486
Palmer TD, Takahashi J, Gage FH (1997) The adult rat hippocampus contains primordial neuralstem cells. Mol Cell Neurosci 8:389–404.
Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates. Academic Press, San Diego, CA.Peterson DA, Lucidi-Phillipi CA, Eagle KL, Gage FH (1994) Perforant path damage results in pro-
gressive neuronal death and somal atrophy in layer II of entorhinal cortex and functional im-pairment with increasing postdamage age. J Neurosci 14:6872–6885
Peterson DA, Lucidi-Phillipi CA, Murphy D, Ray J, Gage FH (1996) FGF-2 protects layer II entro-hinal glutamatergic neurons from axotomy-induced death. J Neurosci 16:886–898
Ray J, Peterson DA, Schinstine M, Gage FH (1993) Proliferation, differentiation, and long-termculture of primary hippocampal neurons. Proc Natl Acad Sci USA 90:3602–3606.
Ray J, Gage FH (1994) Spinal cord neuroblasts proliferate in response to basic fibroblast growthfactor. J Neurosci 14:3548–3564.
Ray J, Raymon HK, Gage FH (1995) Generation and culturing of precursor cells and neuroblastsfrom embryonic and adult central nervous system. In: Vogt PK, Verma IM (eds) Oncogenetechniques. Methods in Enzymology, vol 254. Academic Press, San Diego, pp 20–37
Ray J, Palmer TD, Suhonen JO, Takahasi J, Gage FH (1997) Neurogenesis in the adult brain: Les-sons learned from the studies of progenitor cells from embryonic and adult central nervoussystem In: Gage FH and Christen Y (eds) Research and Perspective in Neurosciences. Isolation,characterization and utilization of CNS stem cells. Fondation IPSEN, Springer, Heidelberg, pp129–149
Ray J, Palmer TD, Shihabuddin LS, Gage FH (1998) The use of neural progenitor cells for therapyin the CNS disorders. In: Tuszynski MH, Kordower J H and Bankiewicz K (eds) CNS regener-ation: basic science and clinical applications Academic Press, San Diego. (in press)
Reynolds BA, Tetzlaff W, Weiss S (1992) A multipotent progenitor cell produces neurons and as-trocytes. J Neurosci 12:4565–4574.
Reynolds BA, Weiss S (1992) Generation of neurons and astrocytes from isolated cells of the adultmammalian central nervous system. Science 255:1707–1710.
Reynolds BA, Weiss S (1996) Clonal and population analyses demonstrate that an EGF-responsivemammalian embryonic CNS precursor is a stem cell. Develop Biol 175:1–13.
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold SpringHarbor Laboratory, Cold Spring Harbor, NY.
Sah DWY, Ray J, Gage FH (1997) Bipotent progenitor cell lines from the human CNS respond dif-ferently to external cues. Nature Biotech 15:574–580.
Shihabuddin LS, Ray J, Gage FH (1997) FGF-2 alone is sufficient to isolate progenitors found inthe adult mammalian spinal cord. Exp Neurol 148:577–586
Stemple DL and Anderson DJ (1992) Isolation of a stem cell for neurons and glia from the mam-malian neural crest. Cell 71:973–985
Sterio DC (1984) The unbiased estimation of number and size of arbitrary particles using the dis-sector. J Microsc 134:127–136

11 Neural Stem Cell Isolation, Characterization and Transplantation 359
Suhonen JO, Peterson DA, Ray J, Gage FH (1996) Differentiation of adult-derived hippocampalprogenitor cells into olfactory bulb neurons. Nature 382:624–627
Suhonen JO, Ray J, Blömer U, Gage FH (1997). Ex vivo and in vivo gene delivery to the brain. CurrProt Hum Gene Supplement 11:13.3.1–13.3.24
Svendsen CN, Fawcett JW, Bentlag C, Dunnett SB (1995) Increased survival of rat EGF-generatedCNS precursor cells using B27 supplemented medium. Exp Brain Res 102:407–414.
Svendsen CN, Clarke DJ, Rosser AE, Dunnett SB (1996) Survival and differentiation of rat and hu-man epidermal growth factor-responsive precursor cells following grafting into the lesionedadult ventral nervous system. Exp Neurol 137:376–388
Svendsen CN, Skepper J, Rosser AE, ter Borg MG, Tyres P, Ryken T (1997) Restricted growth po-tential of rat neural precursors as compared to mouse. Dev Brain Res 99:253–258
Verma IM, Somia N (1997) Gene therapy – promises, problems and prospects. Nature 389:239–42.Vescovi AL, Reynolds BA, Fraser DD, Weiss S (1993) bFGF regulates the proliferative fate of uni-
potent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells.Neuron 11:951–966.
Vicario-Abejon C, Johe KK, Hazel TG, Collazo D, McKay RD (1995) Function of basic fibroblastgrowth factor and neurotrophins in the differentiation of hippocampal neurons. Neuron15:105–114
Weiss, S, Reynolds BA, Vescovi AL, Morshead C, Craig CG, van der Kooy D (1996a) Is there a neu-ral stem cell in the mammalian forebrain? TINS 19:387–393
Weiss S, Dunne C, Hewson J, Wohl C, Wheatley M, Peterson A C, Reynold B A. (1996b) Multipo-tent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis.J Neurosci 16:7599–7609
West JM, Slomianka L (1998) Total number of neurons in the layers of the human entorhinal cor-tex. Hippocampus 8:69–82
Whittemore SR, Snyder EY (1996) Physiological relevance and functional potential of centralnervous-system-derived cell lines. Mol Neurobiol 12:13–38.
■ Suppliers
Company: Almone Laboratories, Shatner Center 3P.O. Box 4287, Jerusalem, 91042, Israel(phone: 972–2-652–8002)
Company: Amersham Corp., 2636S. Clearbrook Dr., Arlington Hts., IL, 60005, USA (phone: 800–323–9750)
Company: Boehringer Mannheim, P.O. Box 50414, Indianapolis, IN, 46250, USA (phone: 800–262–1640)
Company: Collaborative Research Inc., Two Oak Pk., Bedford, MA, 01730, USA (phone: 800–343–2035)
Company: Fischer Scientific, 2761 Walnut Ave., Tustin, CA, 92081, USA (phone: 800–766–7000)Company: Forma Scientific, P.O. Box 649, Marietta, OH, 45750, USA (phone: 800–848–3080)Company: Gemini Bioproducts, 5115-M Douglas Fir Road, Calabasas, CA, 91302–1441, USA
(phone: 800–543–6464)Company: Gibco BRL (Life Technologies), P.O. Box 6009, Garthersburg, MD, 20877, USA
(phone: 800–638–8992)Company: Henry Schein Veterinary, 5 Harbor Park Drive, Port Washington, New York, NY, 11050,
USA (phone: 800–872–4346)Company: Irvine Scientific, 2511 Daimler St., Santa Ana, CA, 92905–5588, USA (phone: 714–261–
7800)Company: J. A. Webster, Inc., 86 Leaminister Rd., Sterling, MA, 05164–7911, USA (phone: 800–
225–7911)Company: Nalgene Brand Products, 75 Panorama Creek Dr., P.O. Box 20365, Rochester, NY,
14602–0365, USA (phone: 716–264–3942)Company: R&D Systems, 614 McKinley Place N. E., 6 Minneapolis, MN, 55413, USA (phone: 800–
343–7475)Company: Sigma Chemical Co., P.O. Box 14508, St. Louis, MO, 63178, USA (phone: 800–325–3010)Company: Tetko, 525 Monterey Pass Rd., Monterey Park, CA, 91754, USA (phone: 800–283–8182)Company: Worthington Biochemicals, Halls Mill Rd., Freehold, NJ, 07728, USA (phone: 800–445–
9603)

360 Jasodhara Ray and Fred H. Gage
■ Abbreviations
FGF-2 Basic fibroblast growth factorEGF Epidermal growth factorPORN PolyornithinepDL Poly-D-lysinepLL Poly-L-lysinePBS Phosphate-buffered salineDMEM Dulbecco’s modified essential mediumBrdU BromodeoxyuridineDAPI 4’,6-diamidino-2-phenylindoleDAVCO 1,4-di-diazobicyclo-[2.2.2]-octaneBDNF Brain derived neurotrophic factorNT-3 Neurotrophin 3CNTF Ciliary neurotrophic factorLIF Leukemia inhibitory factor
■ Glossary
Stem cells A self-sustaining population of cells that give rise to all the cells of the CNS. Ret-roviral vectors: Most retroviral vectors are derived from Moloney murine leuke-mia virus (MoMLV) in which the native viral protein sequences are replaced withrecombinant sequences like the gene of interest and selectable marker genes. Thetransgene is expressed from long terminal repeats (LTR) promoter or from an in-ternal promoter.



















