
Activation of p53-regulated pro-apoptotic signaling pathways in PrP-mediated myopathy
Upload
independentCategory
view
0download
0
Nebulin expression in patients with nemaline myopathy
Juliana Gurgel-Giannettia, Umbertina Reeda, Marie-Louise Bangb, Katarina Pelinc, Kati Donnerc,Sueli K. Mariea, Mary Carvalhoa, Moacir A.T. Firemand, Edmar Zanotelid, Acary S.B. Oliveirad,
Mayana Zatze, Carina Wallgren-Petterssonc, Siegfried Labeitb, Mariz Vainzofa,e,*
aDepartment of Neurology, LIM 15, School of Medicine, University of SaÄo Paulo, SaÄo Paulo, SP, BrazilbEMBL, Meyerhofstrasse 1, 69012 Heidelberg, Germany
cThe FolkhaÈlsan Institute of Medical Genetics and Department of Medical Genetics, University of Helsinki, Helsinki, FinlanddDepartment of Neurology, UNIFESP-EPM, SaÄo Paulo, SP, Brazil
eCentro de Estudos do Genoma Humano, IB ± University of SaÄo Paulo, R. do MataÄo, 277 SaÄo Paulo, SP-CEP 05508-900, Brazil
Received 28 March 2000; received in revised form 3 July 2000; accepted 24 July 2000
Abstract
Nemaline myopathy is a structural congenital myopathy which may show both autosomal dominant and autosomal recessive inheritance
patterns. Mutations in three different genes have been identi®ed as the cause of nemaline myopathy: the gene for slow a-tropomyosin 3
(TPM3) at 1q22±23, the nebulin gene (NEB) at 2q21.1±q22, and the actin gene (ACTA1) at 1q42. The typical autosomal recessive form
appears to be the most common one and is caused by mutations in the nebulin gene. We have studied the pattern of nebulin labeling, in
patients with the typical congenital form (ten patients), the severe congenital form (two patients) or the mild, childhood-onset form (one
patient), using antibodies against three different domains of nebulin. A qualitative and quantitative nebulin analysis in muscle tissue showed
the presence of nebulin in myo®bers from all patients. Some differences relating to the rod structure were observed. The majority of the
largest subsarcolemmal rods were not labeled with the N2 nebulin antibody (I-band epitope) and showed an indistinct pattern with the two
antibodies directed to the Z-band portion of nebulin (epitopes M176±181 and serine-rich domain). Diffuse rods were not revealed using the
three antibodies. A discordant pattern of nebulin N2 epitope labeling was found in two affected sisters with a mutation in the nebulin gene,
suggesting that modi®cations in nebulin distribution inside the rods might occur with the progression of the disease. Western blot analysis
showed no direct correlation with immuno¯uorescence data. In nine patients, the band had a molecular weight comparable to the normal
control, while in one patient, it was detected with a higher molecular weight. Our results suggest that presence/absence of speci®c nebulin Z-
band epitopes in rod structures is variable and could depend on the degree of rod organization. q 2001 Elsevier Science B.V. All rights
reserved.
Keywords: Nemaline myopathy; Nebulin; Congenital myopathy
1. Introduction
Nemaline myopathy (NM) is a non-progressive muscular
disorder associated with the presence of rod-like structures
(nemaline bodies) inside the muscle ®bers and with an esti-
mated incidence of 2 per 100 000 live births [1].
The presence of `rods' or nemaline bodies in the muscle
®bers is the pathological hallmark of this disorder. Within
the ®bers, the rods can be present as compact sub-sarco-
meric forms, as ®ne diffuse structures, or both [1±5].
Usually, the rods are present in the type I ®bers, that are
often predominant. But the proportion of ®bers containing
rods can also vary among individuals and among different
types of muscles [1,4±9].
Clinically, NM is characterized by the presence of hypo-
tonia as well as proximal and facial weakness associated
with skeletal deformities. According to the degree of muscle
weakness, severity and age at onset, and based on correla-
tions from the international database on nemaline myopa-
thy, the ENMC Nemaline Consortium at its workshop in
June 1999 [10] suggested the following classi®cation: (1)
severe congenital form, with contractures and no sponta-
neous movements or no respiration at birth; (2) the typical
form, with onset in early childhood with weakness espe-
cially pronounced in the facial, bulbar and respiratory
muscles and in the neck ¯exors; proximal more than distal
weakness, milestones delayed but reached, slow or non-
progressive course; (3) intermediate congenital form, with
Neuromuscular Disorders 11 (2001) 154±162
0960-8966/01/$ - see front matter q 2001 Elsevier Science B.V. All rights reserved.
PII: S0960-8966(00)00177-2
www.elsevier.com/locate/nmd
* Corresponding author. Tel.: 155-11-3818-7563; fax: 155-11-3818-
7419.
E-mail address: [email protected] (M. Vainzof).

infantile onset, breathing and moving at birth, but later in
childhood unable to achieve respiratory independence,
sitting or walking, or use of wheelchair before the age of
11 years; (4) mild childhood or juvenile-onset, with no
facial weakness and no foot drop; (5) adult form with
onset of symptoms in adult age; (6) other forms, associated
with cardiomyopathy, ophthalmoplegia, unusual distribu-
tion of weakness or intranuclear nemaline bodies [1,4±21].
Genetically, NM may show both autosomal dominant and
autosomal recessive inheritance patterns [1,5,6,10,21±23].
The dominant and recessive forms are clinically and histo-
logically similar.
Up to now, mutations in three different genes have been
identi®ed as the cause of NM: The ®rst, NEM1 form,
mapped to 1q22±23, was identi®ed by Laing et al. [22]. A
missense point mutation (Met9Arg) in codon 9 of the slow
a-tropomyosin 3 (TPM3) was found in a large Australian
autosomal dominant kindred with late childhood onset [24].
Subsequently, a homozygous missense mutation in the same
gene was found in a patient with presumed recessive nema-
line myopathy and a severe phenotype [25].
The second, NEM2 form, was identi®ed by Wallgren-
Pettersson et al. through linkage analysis, at 2q21.1±q22,
in seven European families [23,26,27]. The candidate
gene in this region was the nebulin gene, and recently,
pathogenic mutations were found [28]. To date, 11 different
mutations have been identi®ed in 11 families of different
ethnic origins. The patients are compound heterozygous for
the mutations in six of the families, and homozygous in ®ve.
The majority of the mutations are small deletions or inser-
tions causing frameshifts, or base substitutions causing stop
codons. The clinical spectrum in these patients showed that
nine had the typical form, three had the severe form, two had
the mild form and one the intermediate form [10].
Very recently, mutations in a third gene, the actin gene at
1q42, were found in patients with either congenital myopa-
thy with excess of thin ®laments (n � 3), severe NM
(n � 11) or mild NM (n � 4). Both autosomal dominant
and autosomal recessive inheritance were described for
this form [29].
The direct correlation between the different mutations
and the expression of the proteins in the muscle is still
under investigation for these three genes.
The main objective of this study was to analyze the loca-
lization and quantity of nebulin in muscle biopsies from
patients with NM, using antibodies for three different
domains of nebulin, and to correlate both with the clinical
variability.
2. Patients and methods
A total of 13 patients (from 12 unrelated families) with a
diagnosis of NM were included in this investigation.
The diagnosis was based on clinical examination, course
of the disease (using the protocol of the ENMC International
Nemaline Myopathy Consortium [10], family history,
serum creatine-kinase levels, electromyography and muscle
biopsy.
Muscle samples were obtained from biceps or deltoid
biopsies, frozen in liquid nitrogen immediately after
removal and stored at 2708C until use. Routine histological
and histochemical procedures were done, with staining for
HE, modi®ed Gomori trichrome, NADH, ATPase 9.4, 4.3
and acid and alkaline phosphatase [9].
The ®ber typing was determined by counting 1000 ®bers
from each patient in ATPase 9.4 and 4.3 reactions, and by
calculating the percentage of type I and type II ®bers. We
considered a predominance of type I ®bers to be present
when there were more than 75% of ®ber type I.
For the analysis of the proportion of ®bers with rods, 800±
1000 ®bers of each patient were analyzed and the percen-
tage of ®bers with and without rods was calculated. In addi-
tion, a classi®cation of the rods and their pattern of
distribution was done as described below: (a) subsarcolem-
mal, when they were localized in a compact manner close to
the ®ber membrane; (b) diffuse, when several small rods
were distributed across the whole ®ber.
The subsarcolemmal rods were classi®ed in large and
small, based on the measurement and calculation of their
relative size inside the ®bers: The diameter and area of 100
®bers and the diameter of their respective rods were
measured, using a speci®c software (KS300, Zeiss). We
also calculated the proportional area that the rods occupy
inside the ®ber and the number and proportion of ®bers with
a large rod (when it occupied more than 11% of the ®ber)
and a small rod (less than 10% of the ®ber) were calculated.
In addition, the average of rod size for each patient was
calculated (Table 2).
Immunohistochemical staining of frozen sections were
done through single and double labeling reactions [30],
using a rabbit polyclonal antibody for a-actinin 2 (kindly
provided by Dr A. Beggs) diluted 1/100, as a marker for rod
structure. Three different antibodies for nebulin were used: a
mouse monoclonal antibody directed to an I-band epitope
near the N2 line region, diluted 1/200 (Sigma [31]). In addi-
tion, two rabbit polyclonal antibodies were used which had
been raised to the expressed serine-rich domain, and to the
M176±181 domains. Both polyclonal antibodies were
diluted 1/10 [32,33]. As second antibodies, anti-rabbit and
anti-mouse IgG antibodies both FITC and CY3 conjugated
were used.
For the analysis of nebulin distribution in muscle ®bers,
the following data were considered: the pattern of labeling
in the entire ®ber and the pattern of labeling of the rod,
identi®ed through labeling with an antibody for ACTN2.
According to the intensity of labeling inside the rods, they
were considered positive (strong labeling), equal (indistin-
guishable from the remaining ®ber cross-sectional area), or
negative (no labeling at all).
Western blot was done with 6% sodium dodecyl sulfate±
polyacrylamide gel electrophoresis gels, and transfer at 150
J. Gurgel-Giannetti et al. / Neuromuscular Disorders 11 (2001) 154±162 155

V for 1 h [32]. The blots were incubated and revealed ®rst
with antibody for nebulin (Sigma), and subsequently, with
antibody against ACTN2. The incubations with primary
antibodies were done overnight, and the detection was
done using alkaline phosphatase-conjugated secondary anti-
body.
To determine the relative protein content of the samples,
a relative quantitative densitometric analysis was done for
each patient, comparing nebulin quantity to ACTN2 band
concentration. In addition, this proportion was assessed in
normal control muscle, at different concentrations of muscle
extract (100, 50, 25%).
In 11 patients, the TPM3 gene was analyzed for presence
or absence of the Met9Arg mutation (cases 1, 3, 4±13)
according to the methodology described by Laing et al.
[24]. Screening for mutations in the nebulin gene is ongoing
in patients 2, 7 and 8 [28], and 26 exons, starting from the 3 0
end encoding the Z-disc part of this enormous protein, have
been analyzed to date.
3. Results
3.1. Clinical evaluation
Data on clinical assessment are shown in Table 1.
Among the 13 patients (from 12 unrelated families), two
showed the severe congenital form (patients 3 and 4), ten the
typical form (patients 1, 2, 5±12) and one the mild child-
hood or juvenile-onset form (patient 13). The age at biopsy
varied between 10 months and 28 years. The patients with
the severe congenital form have respiratory insuf®ciency
and still require mechanical ventilation. Patient 3 is unable
to walk and patient 4 walks with assistance.
Among the patients classi®ed with the typical form, all
are ambulant. Three (1, 2, 5) suffered some complications
during the ®rst year of life. Patient 1 had swallowing dif®-
culties, necessitating a gastrostomy. Patients 2 and 5
presented with recurrent pneumonia. Their muscle power
improved with time, and they became able to walk with
assistance or independently. Bone deformities (such as
kyphoscoliosis, high arched palate, pes cavus and thoracic
deformities) were observed in nine patients, and facial
weakness was present in eleven patients. Patient 12 died
suddenly from an unknown cause.
Family history suggested an autosomal recessive inheri-
tance in three patients (from two pedigrees), while the
remaining patients were sporadic cases.
3.2. DNA analysis
DNA analysis did not reveal the Met9Arg mutation in the
11 patients in whom the study was done.
Screening for mutations in about 14% of the nebulin gene
in patients 2, 7 and 8 detected a mutation in one allele in
both affected sisters: a 2 bp deletion in exon 173, which
causes a stop at codon 6154, and was not detected in 284
control chromosomes.
3.3. Histological and histochemical analysis
Predominance of type I ®bers was observed in almost all
patients and varied between 100% (in eight patients) and
60% (patient 7) (Table 2).
All patients showed rods in muscle ®bers, with a propor-
tion between 100 and 41% of ®bers (Table 2).
The distribution of the rods inside the ®bers showed the
following patterns: it was predominantly diffuse in two
patients (patients 5 and 12) and sub-sarcolemmal in 11
patients. The subsarcolemmal rods were classi®ed as
being predominantly large in seven patients (1, 2, 4, 7±9,
11) and small in four patients (3, 6, 10, 13). In seven of these
J. Gurgel-Giannetti et al. / Neuromuscular Disorders 11 (2001) 154±162156
Table 1
Clinical data
Patient no. Age of onset Age at biopsy Clinical
forma
Respiratory
insuf®ciency
Gastrostomy Facial
dysmorphism
Bone
deformities
Inheritanceb Maximal motor
ability
1 Birth 14 years TF No Yes Yes S Walks assisted
2 Birth 28 years TF No No Yes Yes S Walks assisted
3 Birth 2 years SCF Yes No Yes Yes S Non-ambulant
4 Birth 8 years SCF Yes Yes Yes Yes S Walks assisted
5 Birth 6 years TF No No Yes Yes S Walks
6 Birth 3 years TF No No Yes Yes S Walks
7c Birth 10 months TF No No Yes No AR Walks
8c Birth 3 years TF No No Yes No AR Walks
9 7±8 months 13 years TF No No Yes Yes S Walks
10 Birth 5 years TF No No Yes AR? Walks
11 4 months 4 years TF No No No Yes S Walks
12 Birth 9 years TF No No Yes Yes S Walks
13 ? 25 years MCF No No No No S Walks
a SCF, severe congenital form; TF, typical form; MCF, mild childhood form.b S, sporadic; AR, autosomal recessive.c Patients 7 and 8 are siblings.

J. Gurgel-Giannetti et al. / Neuromuscular Disorders 11 (2001) 154±162 157
Tab
le2
His
toch
emic
alan
dIm
mu
no
his
toch
emic
alan
aly
sisa
Pat
ien
tn
o.
%T
yp
eI/
II
®b
ers
%O
f®
ber
s
wit
hro
ds
Pre
dom
inan
tro
dpat
tern
Rod
size
pro
port
ion
of
®ber
s
(%occ
upie
din
the
®ber
)
Anti
body
N2:
pat
tern
of
rod
label
ing
on
neb
uli
nst
ain
Anti
body
M101-1
02
Anti
body
M176±181
0±10%
.11%
Mea
nsi
ze
(mm
)
11
�2
WB
mol.
wt.
11
00
/09
8S
ub
sarc
ole
mm
alla
rge
32
68
14.2
02%
98%
N�
V�
V
21
00
/09
7S
ub
sarc
ole
mm
alla
rge
1dif
fuse
b43
57
12.6
2%
±98%
"�
�V
38
0/2
09
0S
ub
sarc
ole
mm
alsm
all
1dif
fuse
b53
47
10.9
Pre
sentc
pre
sent
N�
Mosa
ic
41
00
/01
00
Su
bsa
rco
lem
mal
larg
e8
92
20.3
030%
70%
N�
�5
10
0/0
10
0D
iffu
seN
oR
evea
lfa
ult
yN
��
61
00
/09
2S
ub
sarc
ole
mm
alsm
all
1dif
fuse
b71
29
8.9
42%
30%
28%
N�
�7
60
/40
98
Su
bsa
rco
lem
mal
larg
e1
dif
fuse
b5
95
27.9
Pre
sentc
Pre
sent
N�
�M
osa
ic
89
0/1
01
00
Su
bsa
rco
lem
mal
larg
e1
dif
fuse
b9.5
90.5
20.9
020%
80%
N�
�V
91
00
/07
6S
ub
sarc
ole
mm
alsm
all
and
larg
e46
54
20.7
2%
fault
y51%
fault
y47%
fault
yN
�F
ault
y�
Fau
lty
10
95
/54
4S
ub
sarc
ole
mm
alsm
all
94
65.8
77%
23%
0N
��
11
10
0/0
99
.5S
ub
sarc
ole
mm
alla
rge
1dif
fuse
b42
58
13.3
4%
57%
fault
y39%
N�
�1
27
0/3
04
0.6
Dif
fuse
No
Rev
eal
N�
�M
osa
ic
13
10
09
5S
ub
sarc
ole
mm
alsm
all
1dif
fuse
b95
55.0
0100%
0N
��
aW
B,
Wes
tern
blo
t;N
,as
no
rmal
con
tro
l;",
hig
her
than
norm
alco
ntr
ol;
V,
rods
not
posi
tive
but
stru
ctura
lly
vis
ible
;fa
ult
y,
smal
lar
eas
wit
hno
label
ing
insi
de
the
®ber
s.b
Mo
reth
an3
0%
of
the
®b
ers
con
tain
ing
dif
fuse
rod
sal
so.
cA
nal
ysi
sim
pai
red
du
eto
the
smal
lsi
zeo
fth
e®
ber
.

11 patients, diffuse rods were also present in association
with the subsarcolemmal rods.
3.4. Protein studies
3.4.1. Immunohistochemical analysis in ®ber cross sections
In control muscles, all the three nebulin antibodies
showed the typical cross-striation pattern of sarcomeric
proteins throughout the ®bers. Antibody M176±181 also
showed the mosaic pattern of type I/II ®bers, that was not
observed with the antibodies directed to the N2-line and Z-
line epitopes, with slow type I ®bers more intensely labeled
than fast type II ®bers.
None of the patients showed a complete absence of nebu-
lin in muscle ®bers with the three antibodies for nebulin. In
three patients (5, 9, 10), a faulty pattern was observed, with
small areas with no labeling inside the ®bers. It was more
intense in patient 9, using the N2 antibody (Fig. 1). In
patient 9 only, this faulty pattern was also observed with
the other two nebulin antibodies.
A ®ber-typing mosaic pattern with antibody M176±181
was observed in three patients, which was compatible with
the proportion of type I/II on ATPase staining.
3.4.2. Rod labeling pattern
The proportion of subsarcolemmal rods that were
revealed using antibodies for the ACTN2 and N2 domains
of nebulin was similar in all patients, taking into account
both negative and positive images of these structures.
Immunohistochemical analysis of the rod structure, using
the N2 nebulin antibody, showed that a positive labeling
pattern was predominant in the small rods, while a negative
staining of nebulin was observed in the majority of the
largest sub-sarcolemmal rods. (Table 2 and Fig. 1). With
antibodies from the serine-rich domain (M101±102) and
M176±181, the rods were equally labeled, although the
structure of the rods was visible in some patients (Fig. 2).
The diffuse rods were not revealed by the nebulin reaction
with any of the three antibodies.
3.5. Western blot analysis
Western blot (WB) analysis was possible only with anti-
body N2. In three patients there was not enough protein in
J. Gurgel-Giannetti et al. / Neuromuscular Disorders 11 (2001) 154±162158
Fig. 1. Examples of double immuno¯uorescence patterns for nebulin (N2
antibody) and ACTN2 staining of the rods, showing a positive nebulin
pattern (1), a negative pattern (2), a pattern indistinguishable from that
seen in the areas of the ®ber not occupied by rods (�) and a faulty pattern in
the cross-sectional area of the ®ber.
Fig. 2. Schematic structure of the nebulin gene and immuno¯uorescence analysis of muscle biopsy from patient 1, showing negatively labeled rods with
antibody from the N2 line (left), and indistinguishable labeling with antibodies from the M176±181 (middle) and serine-rich domain (right) antibodies.
Asterisks denote identifying rods.

the muscle extract for analysis (patients 3, 4, 13) while in the
remaining ten patients the presence of nebulin was
con®rmed. In nine of these 10 patients, the band had a
molecular weight comparable to that of the normal control,
while in one patient (patient 2), a band with a higher mole-
cular weight was detected (Fig. 3). The relative proportion
of nebulin to ACTN2 in normal muscle varied between 37
and 52%, and did not depend on the different muscle protein
concentrations. In the 13 patients, nebulin quanti®cation on
blots was compromised in 7 patients due to muscle extract
degradation. In the remaining six, there was apparently no
correlation with the immunohistochemical data: patients
with undetectable nebulin labeling of diffuse rods (patients
5 and 12) as well as those with negative large subsarcolem-
mal rod labeling on IF (patient 8; Table 2 and Fig. 3) showed
strong bands on WB.
3.6. Intrafamilial correlation
Histochemical study in the two sisters showed a higher
proportion of type 2 ®bers in the younger one (40%) when
compared with the older sister (10%). Both of them,
however, showed the same predominant pattern of large
subsarcolemmal rods.
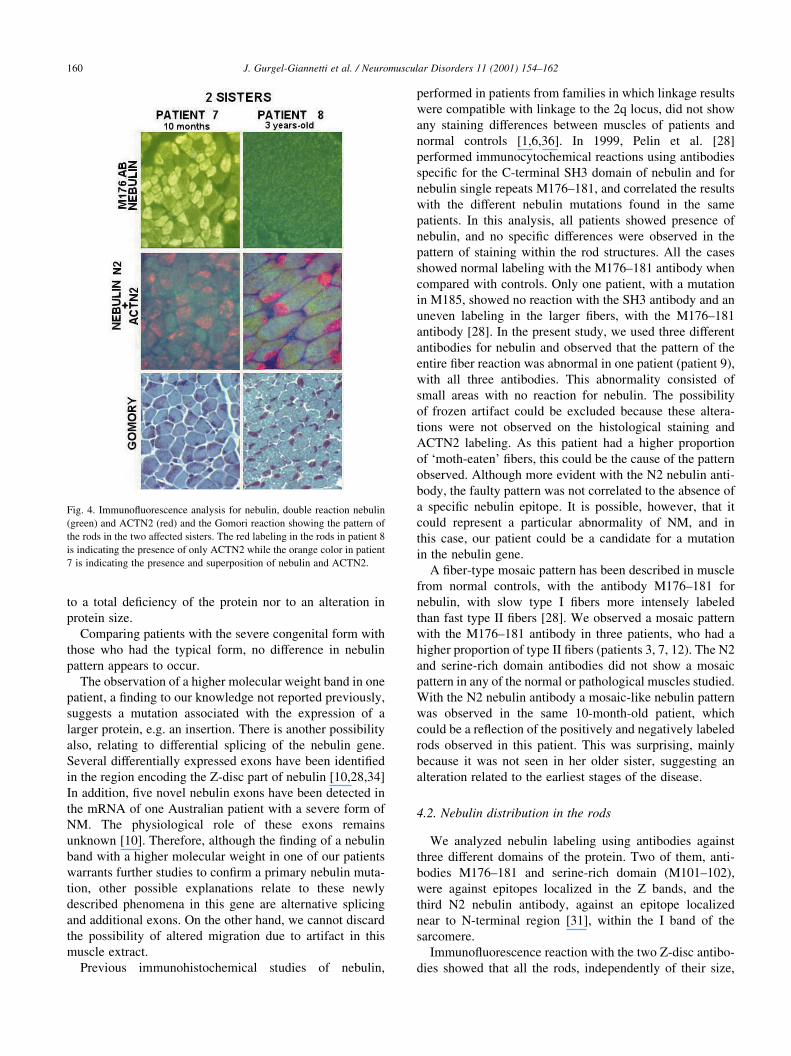
Immuno¯uorescence (IF) study with the N2 nebulin anti-
body showed different patterns of labeling: the younger, 10-
month-old girl (patient 7) showed some large sub-sarcolem-
mal nebulin-de®cient rods, other positively-labeled rods and
a global mosaic-like nebulin pattern on muscle ®bers. Her
older sister (aged 3 years) (patient 8) showed a homoge-
neous pattern of labeling of ®bers, with predominance of
large sub-sarcolemmal rods that were de®cient for nebulin
(Fig. 4). With the two other nebulin antibodies, the rod
structures were indistinct inside the ®ber. A mosaic of
®ber typing was observed with the M176±181 antibody in
the younger sister (Fig. 4).
4. Discussion
Nebulin is a giant protein (600±800 kDa) found in skele-
tal muscles, which accounts for 3±4% of the total myo®-
brillar proteins [33]. The C-terminal region of nebulin is
located at the Z lines, whereas its N-terminal end is at the
pointed end of the thin ®lament [31,33±35]. Nebulin acts as
a molecular ruler for the thin ®lament and is important for
the assembly and integration of Z discs with the sarcomere
[28]. Mutations in the nebulin gene were recently found in
patients affected by the typical congenital form of NM [28].
However, the correlation between the mutations and the
expression of the protein in the muscle is still under inves-
tigation.
According to the clinical course, ten patients from the
present study could be candidates for the nebulin-related
typical NEM2 form [1,6,10]. All presented with clinical
features compatible with the typical congenital form
although not all had facial weakness or skeletal deformities.
At muscle biopsy, in addition to the presence of rods, a
predominance of type I ®bers was observed. Analysis of
the proportion between type I and type II ®bers in the two
affected sisters showed a more preserved mosaic pattern
(60/40 type I/II) in the younger 10-month-old girl when
compared with her 3-year-old sister (90/10 type I/II).
These data are in accordance with previous pathological
studies which suggested a progressive transformation of
type II to type I ®bers [4,8].
4.1. Nebulin in muscle ®bers
The IF analysis for nebulin using three antibodies against
different domains of this protein showed a positive pattern
in the muscle ®bers of all patients. In addition, the presence
of a normal molecular weight nebulin band, using the N2
nebulin antibody, in nine out of ten patients, suggests that
the primary defect in these patients would neither be related
J. Gurgel-Giannetti et al. / Neuromuscular Disorders 11 (2001) 154±162 159
Fig. 3. Double Western blot analysis for nebulin and ACTN2 showing a normal control lane and those of the two sets of NM patients. Myosin content is each
muscle extract is shown in the Ponceau pre-stained blot. Observe the higher molecular weight nebulin band in patient 2, detected in two independent
experiments.
to a total de®ciency of the protein nor to an alteration in
protein size.
Comparing patients with the severe congenital form with
those who had the typical form, no difference in nebulin
pattern appears to occur.
The observation of a higher molecular weight band in one
patient, a ®nding to our knowledge not reported previously,
suggests a mutation associated with the expression of a
larger protein, e.g. an insertion. There is another possibility
also, relating to differential splicing of the nebulin gene.
Several differentially expressed exons have been identi®ed
in the region encoding the Z-disc part of nebulin [10,28,34]
In addition, ®ve novel nebulin exons have been detected in
the mRNA of one Australian patient with a severe form of
NM. The physiological role of these exons remains
unknown [10]. Therefore, although the ®nding of a nebulin
band with a higher molecular weight in one of our patients
warrants further studies to con®rm a primary nebulin muta-
tion, other possible explanations relate to these newly
described phenomena in this gene are alternative splicing
and additional exons. On the other hand, we cannot discard
the possibility of altered migration due to artifact in this
muscle extract.
Previous immunohistochemical studies of nebulin,
performed in patients from families in which linkage results
were compatible with linkage to the 2q locus, did not show
any staining differences between muscles of patients and
normal controls [1,6,36]. In 1999, Pelin et al. [28]
performed immunocytochemical reactions using antibodies
speci®c for the C-terminal SH3 domain of nebulin and for
nebulin single repeats M176±181, and correlated the results
with the different nebulin mutations found in the same
patients. In this analysis, all patients showed presence of
nebulin, and no speci®c differences were observed in the
pattern of staining within the rod structures. All the cases
showed normal labeling with the M176±181 antibody when
compared with controls. Only one patient, with a mutation
in M185, showed no reaction with the SH3 antibody and an
uneven labeling in the larger ®bers, with the M176±181
antibody [28]. In the present study, we used three different
antibodies for nebulin and observed that the pattern of the
entire ®ber reaction was abnormal in one patient (patient 9),
with all three antibodies. This abnormality consisted of
small areas with no reaction for nebulin. The possibility
of frozen artifact could be excluded because these altera-
tions were not observed on the histological staining and
ACTN2 labeling. As this patient had a higher proportion
of `moth-eaten' ®bers, this could be the cause of the pattern
observed. Although more evident with the N2 nebulin anti-
body, the faulty pattern was not correlated to the absence of
a speci®c nebulin epitope. It is possible, however, that it
could represent a particular abnormality of NM, and in
this case, our patient could be a candidate for a mutation
in the nebulin gene.
A ®ber-type mosaic pattern has been described in muscle
from normal controls, with the antibody M176±181 for
nebulin, with slow type I ®bers more intensely labeled
than fast type II ®bers [28]. We observed a mosaic pattern
with the M176±181 antibody in three patients, who had a
higher proportion of type II ®bers (patients 3, 7, 12). The N2
and serine-rich domain antibodies did not show a mosaic
pattern in any of the normal or pathological muscles studied.
With the N2 nebulin antibody a mosaic-like nebulin pattern
was observed in the same 10-month-old patient, which
could be a re¯ection of the positively and negatively labeled
rods observed in this patient. This was surprising, mainly
because it was not seen in her older sister, suggesting an
alteration related to the earliest stages of the disease.
4.2. Nebulin distribution in the rods
We analyzed nebulin labeling using antibodies against
three different domains of the protein. Two of them, anti-
bodies M176±181 and serine-rich domain (M101±102),
were against epitopes localized in the Z bands, and the
third N2 nebulin antibody, against an epitope localized
near to N-terminal region [31], within the I band of the
sarcomere.
Immuno¯uorescence reaction with the two Z-disc antibo-
dies showed that all the rods, independently of their size,
J. Gurgel-Giannetti et al. / Neuromuscular Disorders 11 (2001) 154±162160
Fig. 4. Immuno¯uorescence analysis for nebulin, double reaction nebulin
(green) and ACTN2 (red) and the Gomori reaction showing the pattern of
the rods in the two affected sisters. The red labeling in the rods in patient 8
is indicating the presence of only ACTN2 while the orange color in patient
7 is indicating the presence and superposition of nebulin and ACTN2.

were labeled with the same intensity as the remaining ®ber.
The presence of some of the largest, dense subsarcolemmal
rods could be identi®ed because of differences in the stria-
tion pattern. However, with the nebulin antibody from the I
band, it was observed that many of the largest subsarcolem-
mal rods were predominantly negative, while small sarco-
lemmal rods were mostly positive. Additionally, the diffuse
rods were not detectable by nebulin antibody labeling.
Therefore, the presence of speci®c epitopes of nebulin in
these structures may be related to the size and the degree of
organization of the rods. It is tempting to suggest that the
large subsarcolemmal rod would present a demarcated
structure, with a compaction of proteins predominantly
from the Z disc, while the diffuse and small rods, positively
labeled with all antibodies, would be formed by a wider
mixture of proteins.
Nebulin analysis in the rods from the two sisters gives
further support to the hypothesis that as the disease
progresses, modi®cations in nebulin presence and/or distri-
bution inside these structures could occur.
The mechanism of rod formation is still unknown. A
primary defect in the nebulin gene could lead to abnormal-
ities in the nebulin structure causing an altered connection
of this protein at the Z disc and, as a consequence, sarco-
mere instability and rod formation. Perhaps through similar
mechanisms, mutations in other genes for the proteins of the
thin ®lament and Z disc, such as the tropomyosin and actin
genes, also lead to rod formation. Our results suggest that
the presence/absence of the nebulin in rod structures is vari-
able and could depend on the degree organization and
compaction of rods. Alternatively, the immunohistochem-
ical visualization of nebulin at rod structures could be
impaired by masking of the epitopes by other sarcomeric
proteins related to the Z disc, as already suggested for other
proteins [37,38].
It is known that WB studies are unable to detect small
alterations in protein quantity. However, the lack of correla-
tion between WB data and IF pattern of nebulin in the rods
provides further evidences for the hypothesis that the
presence or absence of nebulin in the rod structures is not
a re¯ection of quantitative alterations but a consequence of
reorganization of proteins.
In summary, based on IF and WB analysis, our studies
show the presence of nebulin in muscle ®bers of all the
patients studied with congenital NM. Abnormalities of
nebulin distribution within the rods could be a consequence
of reorganization of sarcomeric proteins.. WB analysis may
in some cases help to reveal a nebulin abnormality.
Acknowledgements
Antibody for ACTN2 was kindly given by Dr Alan
Beggs, to whom we are very grateful. We also wish to
thank Marta Canovas and Fabiana da Silva, for the excellent
technical assistance, Valdir Caldeiras and the Departament
of Biology, IB±USP, for the support on the confocal micro-
scopy and all the physicians who referred affected patients.
M.L.B. was supported by a Marie-Curie Fellowship from
the European Union. K.P. and K.D. were supported by
grants to C.W.-P. from the Association Francaise contre
les Myopathies, the Swedish Cultural Foundation of
Finland, the Finska LaÈkaresaÈllskapet, and the Medicinska
understoÈdsfoÈreningen Liv och HaÈlsa. For organizational and
®nancial support to the ENMC International Consortium on
Nemaline Myopathy, we are indebted to the European
Neuromuscular Centre (ENMC) and its main sponsors:
Association Francaise contre les Myopathies (France),
Italian Telethon Committee (Italy), Muscular Dystrophy
Group of Great Britain and Northern Ireland (UK), Vereni-
ging Spierziekten Nederland (The Netherlands) and
Deutsche Gesellschaft fuÈr Muskelkranke (Germany),
Schweizerische Stiftung fuÈr die Erforschung der Muskelk-
rankheiten (Switzerland), Prinses Beatrix Fonds (The Neth-
erlands), Verein zur Erforschung von Muskelkrankheiten
bei Kindern (Austria) and Muskelsvindfonden (Denmark)
as well as its associate members: Unione Italiana Lotta
alla Distro®a Muscolare and Muscular Dystrophy Associa-
tion of Finland. This work was supported by grants from
FundacËaÄo de Amparo aÁ Pesquisa do Estado de SaÄo Paulo
(FAPESP), FundacËaÄo Faculdade de Medicina, PRONEX,
CNPq and AssociacËaÄo Brasileira de Distro®a Muscular
(ABDIM).
References
[1] North KN, Laing NG, Wallgreen-Pettersson CJ, ENMC International
Consortium on Nemaline Myopathy. Nemaline myopathy: current
concepts. Med Genet 1997;34:705±713.
[2] Shy GM, Engel WK, Somers JE, Wanko T. Nemaline myopathy. A
new congenital myopathy. Brain 1963;86:793±810.
[3] Goebel HH. Congenital myopathies. Acta Paediatr Jpn 1991;33:247±
255.
[4] Wallgreen-Pettersson C, Rapola J, Donner M. Pathology of congeni-
tal nemaline myopathy. A follow-up study. J Neurol Sci
1988;83:243±257.
[5] Wallgren-Pettersson C, Laing NG. 40th ENMC International Work-
shop: Nemaline Myopathy. Neuromusc Disord 1996;5:389±391.
[6] Wallgren-Pettersson C, Beggs AH, Laing NG. 51st ENMC Interna-
tional Workshop: Nemaline Myopathy. Neuromusc Disord
1998;8:53±56.
[7] Shimomura C, Nonaka I. Nemaline myopathy: comparative muscle
histochemistry in the severe neonatal, moderate congenital and adult-
onset forms. Pediatr Neurol 1988;5:25±31.
[8] Nonaka I, Ishiura S, Arahata K, Ishibashi-Ueda H, Maruyama A, Li
K. Progression in nemaline myopathy. Acta Neuropathol
1989;78:484±491.
[9] Dubowitz V. Muscle disorders in childhood, 2nd ed. London: Saun-
ders, 1995. pp. 134±177.
[10] Wallgren-Pettersson C, Laing NG. Report of the 70th ENMC Inter-
national Workshop: Nemaline Myopathy. Neuromusc Disord
2000;313±320.
[11] Schmalbruch H, Kamieniekka Z, Arroe M. Early fatal nemaline
myopathy: case report and review. Dev Med Child Neurol
1987;29:800±804.
J. Gurgel-Giannetti et al. / Neuromuscular Disorders 11 (2001) 154±162 161

[12] McMenamin JB, Curry B, Taylor GP, Becker LE, Murphy EG. Fatal
nemaline myopathy in infancy. Can J Neurol Sci 1984;11:305±309.
[13] Norton P, Ellison P, Sulaiman AR, Harb J. Nemaline myopathy in the
neonate. Neurology 1983;33:351±356.
[14] Oferil CJ, Castilho LJ, Arguelles PP, Torrez VI, Alvarez FE. Severe
and mild forms of nemaline myopathy: a report of three cases. Na Esp
Pediatr 1993;39:517±520.
[15] Hefferman LP, Rewcastle NB, Humphy MD. The spectrum of myopa-
thies. Arch Neurol 1968;18:529±542.
[16] Kuitunen P, Rapola J, Noponen AL, Donner M. Nemaline myopathy.
Report of four cases and review of the literature. Acta Paediatr Scand
1972;61:353±361.
[17] Martinez BA, Lane BD. Childhood nemaline myopathy: a review of
clinical presentation in relation to prognosis. Dev Med Child Neurol
1987;29:815±820.
[18] Meier C, Voellny W, Gertsch M, Zimmermann A, Geissbuthler J.
Nemaline myopathy appearing in adults as cardiomyopathy. A clin-
icopathologic study. Arch Neurol 1984;41:443±445.
[19] Palmucci L, Dorigiuzzi C, Morgini T, Chiado-Piat L. Adult onset
nemaline myopathy: a distinct nosology entity. Clin Neuropathol
1993;12:153±155.
[20] Shahar E, Tervo RC, Murphy EG. Heterogeneity of nemaline myopa-
thy. A follow-up study of 13 cases. Pediatr Neurosci 1988;14:236±
240.
[21] Wallgren-Pettersson C, KaÈaÈriaÈinen H, Rapola J, Salmi T, JaÈaÈskelaÈi-
nen J, Donner M. Genetics of congenital nemaline myopathy: a study
of ten families. J Med Genet 1990;27:480±487.
[22] Laing NG, Majda BT, Akkari PA, et al. Assignment of a gene
(NEM1) for autosomal dominant nemaline myopathy to chromosome
1. Am J Hum Genet 1992;50:576583.
[23] Wallgren-Pettersson C, Avela K, Marchand S, et al. A gene for auto-
somal recessive nemaline myopathy assigned to chromosome 2q by
linkage analyses. Neuromusc Disord 1995;5:441±443.
[24] Laing NG, Wilton SD, Akkari PA, et al. A mutation in the alpha
tropomyosin gene TPM3 associated with autosomal dominant nema-
line myopathy. Nat Genet 1995;9:75±79.
[25] Tan P, Briner J, Boltshauser E, et al. Homozygosity for a nonsense
mutation in the a-tropomyosin gene TPM3 in a patient with severe
nemaline myopathy. Neuromusc Disord 1999;9:573±579.
[26] Wallgren-Pettersson C, Pelin K, HilpelaÈ P, et al. Clinical and genetic
heterogeneity in autosomal recessive nemaline myopathy. Neuromusc
Disord 1999;9:564±572.
[27] Wallgren-Pettersson C. Genetics of nemaline myopathies and
myotubular myopathies. Neuromusc Disord 1998;8:401±404.
[28] Pelin K, HilpelaÈ P, Donner K, et al. Mutations in the nebulin gene
associated with autosomal recessive nemaline myopathy. Proc Natl
Acad Sci USA 1999;96:2305±2310.
[29] Nowak KJ, Wattanasirichaigoon D, Goebel NH, et al. Mutations in
the skeletal muscle alpha actin gene in patients with actin myopathy
and nemaline myopathy. Nat Genet 1999;23:208±212.
[30] Vainzof M, Zubrzycka-Gaarn EE, Rapaport D, et al. Immuno¯uores-
cence dystrophin study in Duchenne dystrophy through the concomi-
tant use of two antibodies directed against the carboxy-terminal and
the amino- terminal region of the protein. J Neurol Sci 1991;101:141±
147.
[31] FuÈrst DO, Osborn M, Nave R, Weber K. The organization of titin
®laments in the half-sarcomere revealed by monoclonal antibodies I.
Immunoelectron microscopy: a map of ten nonrepetitive epitopes
starting at Z line extends close to the M line. J Cell Biol
1988;106:1563±1572.
[32] Ho-Kim M-A, Bedard A, Vincent M, Rogers PA. Dystrophin: a sensi-
tive and reliable immunochemical assay in tissue and cell culture
homogenates. Biochem Biophys Res Commun 1991;181:1164±
1172.
[33] Wang K, Wright J. Architecture of the sarcomere matrix of skeletal
muscle: immunoelectron microscopic evidence that suggests a set of
parallel inextensible nebulin ®laments anchored at Z line. J Cell Biol
1988;6:2199±2212.
[34] Millevoi S, Trombitas K, Lolmerer B, et al. Characterization of nebul-
ette and nebulin and emerging concepts of their roles for Z-discs. J
Mol Biol 1998;282:111±123.
[35] Labeit S, Lolmerer B. The complete primary structure of human
nebulin and its correlation to muscle structure. J Mol Biol
1995;248:308±315.
[36] Sewry CA. The role of immunocytochemistry in congenital myopa-
thies. Neuromusc Disord 1998;8:394±400.
[37] Jockush BM, Veldman H, Grif®ths GW, Van Oost BA, Jennekens
FGI. Immuno¯uorescence microscopy of a myopathy. a-Actinin is
a major constituent of nemaline rods. Exp Cell Res 1980;127:409±
420.
[38] Yamagushi M, Robson RM, Stromer MH, Dahl DS, Oda T. Actin
®laments form the backbone of nemaline myopathy rods. Nature
1978;271(5642):265±267.
J. Gurgel-Giannetti et al. / Neuromuscular Disorders 11 (2001) 154±162162
Copyright © 2022 FDOKUMEN




















