
Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway
Upload
independentCategory
view
1download
0
NCLX is an essential component of mitochondrialNa+/Ca2+ exchangeRaz Paltya, William F. Silvermanb, Michal Hershfinkelb, Teresa Caporalec, Stefano L. Sensic,d, Julia Parnise,Christiane Noltee, Daniel Fishmanb, Varda Shoshan-Barmatzf, Sharon Herrmanna, Daniel Khananshvilig,and Israel Seklera,1
Departments of aPhysiology, bMorphology, and cLife Sciences, and the Zlotowski Center for Neuroscience, Ben-Gurion University of the Negev, Beer-Sheva84105, Israel; cDepartment of Basic and Applied Medical Science, Molecular Neurology Unit, CESI–Center for Research on Aging, University G. d’Annunzio,Chieti 66013, Italy; dDepartment of Neurology, University of California, Irvine, CA, 92697; eDepartment of Cellular Neuroscience, Max Delbrueck Center forMolecular Medicine (MDC), D-13122 Berlin-Buch, Germany; Departments of fPhysiology and gPharmacology, Sackler School of Medicine, Tel Aviv University,Ramat-Aviv 69978, Israel
Edited by Harald Reuter, University of Bern, Hinterkappelen, Switzerland, and approved November 10, 2009 (received for review July 22, 2009)
Mitochondrial Ca2+ efflux is linked to numerous cellular activitiesand pathophysiological processes. Although it is established thatan Na+-dependent mechanism mediates mitochondrial Ca2+ efflux,the molecular identity of this transporter has remained elusive.Here we show that the Na+/Ca2+ exchanger NCLX is enriched inmitochondria, where it is localized to the cristae. Employing Ca2+
and Na+ fluorescent imaging, we demonstrate that mitochondrialNa+-dependent Ca2+ efflux is enhanced upon overexpression ofNCLX, is reduced by silencing of NCLX expression by siRNA, andis fully rescued by the concomitant expression of heterologousNCLX. NCLX-mediated mitochondrial Ca2+ transport was inhibited,moreover, by CGP-37157 and exhibited Li+ dependence, both hall-marks of mitochondrial Na+-dependent Ca2+ efflux. Finally, NCLX-mediated mitochondrial Ca2+ exchange is blocked in cells express-ing a catalytically inactive NCLX mutant. Taken together, our re-sults converge to the conclusion that NCLX is the long-soughtmitochondrial Na+/Ca2+ exchanger.
mitochondrial calcium exchanger | mitochondrial calcium homeostasis |sodium calcium exchanger | CGP-37157
Apart from their metabolic role, mitochondria are a major hubof cellular Ca2+ homeostasis (1). Powered by the steep mi-
tochondrial membrane potential, Ca2+ enters into this organellevia a mitochondrial uniporter and is extruded by either an H+- oranNa+-coupledmitochondrial exchanger.Activity of theNa+/Ca2+ exchanger is ubiquitously found in most cell types andtissues studied so far and is particularly robust in excitable cells,whereas the activity of the H+/Ca2+ exchanger is pri-marily found in nonexcitable cells. Recently a mitochondrial H+/Ca2+ exchanger termed Letm1 has been identified, but its role inCa2+ extrusion is not clear (2). By catalyzingNa+-dependent Ca2+
efflux, the putative mitochondrial Na+/Ca2+ exchanger plays afundamental role in regulating mitochondrial Ca2+ homeostasis(3), oxidative phosphorylation (4), and Ca2+ crosstalk amongmitochondria, cytoplasm, and the endoplasmic reticulum (ER)(5). Although the activity of this transporter was documentedmore than 30 years ago (6), its molecular identity remained un-known. We and others previously identified and characterizedNCLX, a novel Na+/Ca2+ exchanger, and the single mammalianmember of a phylogenetically ancestral branch of the Na+/Ca2+
exchanger superfamily (7, 8). Intriguingly, NCLX catalyzes Na+-or Li+-dependent Ca2+ transport at similar rates, a feature sharedonly with the unidentified mitochondrial exchanger. Although theactivity of ectopically expressed humanNCLXwas first monitoredat the plasma membrane, the unique phylogenetic and functionalproperties of NCLX prompted us to investigate whether NCLX islinked to the mitochondrial exchanger.
ResultsWe initially sought to determine if NCLX is found in the mitochon-dria. Comparison of NCLX levels in total versus mitochondrial-
enriched fractions from mouse heart and brain showed that the 50-and 70-kDa forms ofNCLX(7) and an additional 100 kDa formwereenriched in the mitochondrial fraction (Fig. 1A). Augmentation ofNCLX expression was also observed in the mitochondrial fractionfrom HEK-293 cells heterologously or endogenously expressingNCLX (Fig. 1B andD and Figs. S1A and S2C–F). Based on the welldocumented ability of NCX and other membrane proteins to formSDS-resistantdimers,we reasoned that the100-kDaformis related tosuch an NCLX dimer. This was confirmed by the simultaneous re-duction in expression of the 50-kDa and 100-kDa forms of NCLX incells transfected with siNCLX (Fig. 1C), and by coimmunoprecipi-tation of NCLX monomers (see Fig. S1C). Also, dissociation ofthe 100-kDa form to the 50-kDa formwas observed following lengthyincubation in denaturing buffer (Fig. S1B). Intriguingly, the mi-tochondrial NCLX is remarkably similar in size to molecularly un-identified mitochondrial polypeptides that, when purified andreconstituted, exhibited Na+/Ca2+ exchange activity (10, 11).We next determined the subcellular distribution of NCLX by
analyzing endogenous NCLX expression in plasma membrane,ER, and mitochondrial fractions from HEK-293 cells or rat heart(Fig. 1 D and E) and found that endogenous NCLX was enrichedprimarily in the mitochondrial fraction, but not in the ER or theplasma membrane, as previously suggested (7, 9). Altogether,these results indicate that the mitochondria are the major cel-lular site of NCLX localization.Additional support for the mitochondrial localization of
NCLX came from immunoelectron microscopy analysis of brainsections and CHO cells. Intense labeling was observed in mito-chondria, but not in nuclear, sarcoplasmic reticulum, or plasmamembrane structures or in preparations reacted with preimmuneserum (Fig. 2 A–C). Finally, to define the localization of NCLXwithin mitochondria, a postembedding cryo-EM procedure fea-turing protein A gold–labeled, anti-NCLX antibodies was em-ployed (Fig. 2 D–E and Fig. S1D). Discrete gold particles wereobserved within mitochondria, particularly within the cristae, ofcontrol or NCLX-overexpressing SHSY-5Y cells. Individual goldparticles unrelated to mitochondria were occasionally observed(see Materials and methods), showed no organelle or compart-ment preference, and apparently represent nonspecific back-ground staining. Comparison of the number of gold particlesobserved on ultrathin mitochondrial profiles from control and
Author contributions: R.P., W.F.S., D.K., and I.S. designed research; R.P., W.F.S., T.C., S.L.S.,J.P., C.N., D.F., S.H., and D.K. performed research; R.P., W.F.S., M.H., S.L.S., C.N., D.F., V.S.-B., S.H., D.K., and I.S. analyzed data; and R.P., W.F.S., M.H., S.L.S., C.N., V.S.-B., and I.S.wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1To whom correspondence should may be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0908099107/DCSupplemental.
436–441 | PNAS | January 5, 2010 | vol. 107 | no. 1 www.pnas.org/cgi/doi/10.1073/pnas.0908099107

NCLX-overexpressing SHSY-5Y cells revealed that mitochon-dria from the latter contained more particles than did controlmitochondria (35 vs. 14; P < 0.01; Fig. 2F). This result indicatesthat the observed labeling is related to NCLX and that bothoverexpressed and endogenous NCLX, like the mitochondrialexchanger, are localized to the mitochondrial cristae.If NCLX is to play a functional role in mitochondria, it should be
linked tomitochondrialCa2+efflux.MitochondrialCa2+ signalsweredetermined by using the mitochondrial-targeted calcium-sensingproteinPericam [RP-mt (12), also seeFig. S3B].RP-mtwas excitedatthe pH-insensitive 430-nm wavelength and not at the 485-/430-nmratiometric mode because of the pH sensitivity of Pericam to ex-citation at the 485-nm wavelength and the potential interference bymitochondrial pH changes we and others have observed (see Fig. S3E–G and ref. 13). In this set of experiments, we have expressed themurine isoform of NCLX that, consistent with previous studies (9),did not reach the plasma membrane and did not exhibit plasmamembraneNa+/Ca2+ exchange activity (seeFig. S2B andF). PlasmamembraneNa+/Ca2+ exchange activity was also absent in the controlnontransfected cells (seeFig. S2B).These results indicate thatneitherthe endogenous nor the ectopically expressed murine NCLX con-tribute to plasma membrane Ca2+ fluxes. We then asked if over-expression of NCLXwill modulate mitochondrial Ca2+ efflux. Ratesof mitochondrial Ca2+ efflux in control cells were taken as the ref-erence point (100%) and changes in activity following NCLX over-
expression, silencing, or inhibition were presented as percentage ofcontrol. Application of ATP (40 μM) was followed by mitochondrialCa2+ uptake and a subsequent slower Ca2+ efflux phase (Fig. 3A),corresponding to the established activity of the mitochondrial Ca2+
uniporter and the Na+/Ca2+ exchanger, respectively. MitochondrialCa2+ uptake was similar in NCLX-expressing or control cells but therate of Ca2+ efflux was higher in the NCLX-expressing cells (194 ±23% of control; Fig. 3 A and C), indicating that NCLX expression islinked to enhanced mitochondrial Ca2+ efflux. Application of ATPtriggered a similar increase in cytoplasmic Ca2+ (Fig. S4E) in theNCLX-overexpressing cells and control. Furthermore, steady-statemitochondrial Ca2+ levels and membrane potential were similar inNCLX-expressing and control cells (Fig. S3A and Fig. S4 A–C), ar-guing against an indirect effect of NCLX activity on the cytoplasmicCa2+ response or mitochondrial Ca2+ uptake. However, when ATPwas applied for longer intervals in the presence of Ca2+-containingRinger solution, thereby imposing a stronger mitochondrial Ca2+
load, the cytoplasmic Ca2+ levels were elevated in cells over-expressing NCLX (Fig. S4 F and G). This is consistent with thephysiological role of themitochondrial exchanger in also modulatingcytoplasmic Ca2+ responses by enhancingmitochondrial Ca2+ effluxand augmenting the store-operated calcium entry into cells (13).Because of the dimeric nature ofNCLX(Fig. S1B andC and ref.
14), we hypothesized that a catalytically inactive mutant of NCLXwould exert a dominant-negative effect on mitochondrial Ca2+
efflux, thereby providing an additional and highly specific tool foranalyzing the mitochondrial role of NCLX. Accordingly, we gen-erated a mutant in which threonine 468, which is catalytically es-sential for the activity of NCLX and other members of NCXfamily, was replaced by a serine (NCLX-S468T) (14). Like thenative protein, NCLX-S468T was targeted to the mitochondria(see Fig. S2E). Applying the same paradigm as described in Fig.3A, we found that expression of NCLX-S468T was followed byprofound inhibition of endogenousmitochondrial Ca2+ efflux ratein SHSY-5Y (20 ± 15% of control; Fig. 3 A and C) or HEK-293cells (26± 5% of control; Fig. S5A–B). These results indicate thatNCLX is catalytically essential for mitochondrial Ca2+ efflux.The benzothiazepine compound CGP-37157 is an established
inhibitor of the mitochondrial Na+/Ca2+ exchanger (15). Wereasoned that, if NCLX is this exchanger, its activity should beblocked by this compound. Application of CGP-37157 (10 μM) toSHSY-5Y cells heterologously expressing NCLX or to controlcells resulted in profound inhibition of mitochondrial Ca2+ effluxin both (Fig. 3 B and C). In addition, dose-dependence analysis ofthe effect of CGP-37157 on Ca2+ efflux in NCLX-overexpressingSHSY-5Y cells showed approximately 50% inhibition using 5 μMand complete inhibition of mitochondrial Ca2+ efflux at 7.5 μM(Fig. 3D). This analysis argues against any heterogeneous effect ofCGP-37157 on distinct targeting sites and thus indicates that en-dogenous and overexpressed NCLX share similar sensitivity toGCP-37157.Moreover, the concentration of CGP-37157 requiredfor inhibition of Ca2+ efflux in NCLX-overexpressing SHSY-5Ycells was similar to that reported for inhibition of the mitochon-drial exchanger in intact cells (16), and is considerably lower thanthat reported for partial inhibition of other members of the NCXfamily (17, 18). Hence, the results of this set of experiments pointto NCLX as being the molecular moiety catalytically linked tomitochondrial Ca2+ efflux, and its sensitivity to CGP-37157 pro-vides additional evidence that NCLX is the mitochondrial Na+/Ca2+ exchanger.If NCLX mediates mitochondrial exchange activity, then
reducing its endogenous expression should attenuate mitochon-drial Ca2+ efflux. Transfection of cells with NCLX siRNA(siNCLX) loweredNCLXexpression (Fig. 1C). These cells exhibitsimilar resting mitochondrial Ca2+ levels as untreated cells, theirmitochondria being slightly hyperpolarized (Figs. S3A and S4C).Then the same ATP-induced mitochondrial Ca2+ efflux assaydescribed in Fig. 3A was applied. Whereas mitochondrial Ca2+
Fig. 1. NCLX is localized tomitochondria. (A) Immunoblot analysis of NCLX intotal tissue lysate (Total) and mitochondrial fractions (Mitochondria) purifiedfrom mouse heart and brain (15 μg). (B) Immunoblot analysis of total cellularand mitochondrial fractions purified from HEK-293-T cells overexpressingmurine NCLX (10 μg). (Lower) Immunoblots of ANT or VDAC serving asmarkers. (C) Immunoblot analysis of NCLX in mitochondrial fractions purifiedfrom HEK-293 cells transfected with siNCLX or scrambled siRNA (siControl).Note that siNCLXdiminishes expression levels of both the 50-kDa and 100-kDaforms of NCLX. (D and E) Expression of NCLX in cellular and tissue fractions ofthe indicated components purified from HEK-293 cells (20 μg) (D) or rat heart(10 μg) (E). (Lower) Immunoblots of ANT (mitochondrial marker), Na+/K+ AT-Pase, or N-cadherin (plasma membrane, PM, marker) and Calnexin or Sec-62(ER, marker). Note that the mitochondria are the major site of NCLX local-ization (the slight NCLX signal in cardiac sarcoplasmic reticulum is presumablyrelated to cross-contamination with mitochondria; see ANT staining).
Palty et al. PNAS | January 5, 2010 | vol. 107 | no. 1 | 437
PHYS
IOLO
GY

uptake was unaffected, in cells in which NCLX expression hadbeen silenced, the Ca2+ efflux rate was reduced by approximately75% of the rate measured in control cells (22 ± 6.5% of control;Fig. 4A).A similar decrease inmitochondrial Ca2+ efflux followingsilencing of NCLX expression was also observed in CHO cells(Fig. S5 D–F). We further reasoned that, if NCLX directly par-ticipates in endogenous mitochondrial exchange activity, ex-pression of a heterologous species of NCLX should rescue theactivity reduced by the silencing. To address this hypothesis,SHSY-5Y cells were transfected with a plasmid encoding NCLXshRNA (see Materials and Methods), thereby generating a se-quence that specifically targets aUTR region in the humanNCLXmRNA sequence (i.e., shNCLX) but not in the murine homo-logue. Human SHSY-5Y cells transfected with the shNCLX ex-hibited a 56± 6.7% reduction inmitochondrial Ca2+ efflux activitycompared with control cells (Fig. 4B). In contrast, coexpression ofmurine NCLX with the shNCLX plasmid induced a fourfoldincrease in the rate of Ca2+ efflux versus cells transfected withshNCLX only. Hence, Ca2+ efflux activity had been fully restored(Fig. 4B). Taken together, the results from this set of experimentssupport the concept that NCLX is an essential mediator ofendogenous mitochondrial Ca2+ efflux and that heterologousexpression of NCLX is sufficient to functionally complement theloss of the endogenous activity.We next examined whether NCLX activity is linked to mitochon-
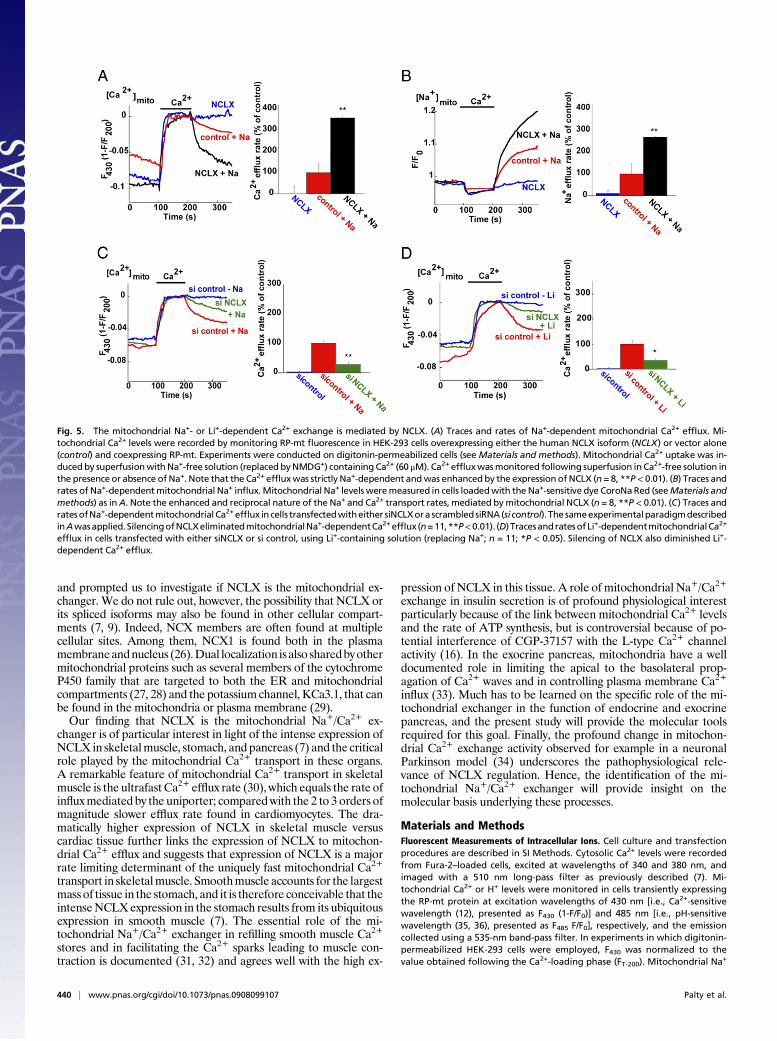
drial Na+/Ca2+ exchange by assessingwhether it displays a strict Na+
dependence in mediating Ca2+ efflux. The Na+ dependence of mi-tochondrial NCLXwas assayed in digitonin-permeabilized cells, thusallowing equilibration of the intracellular and extracellular com-partments and control of the extramitochondrial ionic environmentwhile eliminating any possible interference by plasma membranetransporters. To ascertain that the cells were permeabilized using thisprotocol, they were loaded with carboxyfluorescein diacetate(CFDA) succinimidyl ester dye that was trapped within the cells.Subsequent addition of digitonin triggered a strong decrease ofCFDA fluorescence, suggesting that the entrapped dye was removedoutof thecells andhence that thecellswerepermeabilized (Fig.S3C).Permeabilized HEK-293 cells transiently expressing RP-mt alone ortogetherwithNCLXwere s\perfusedwithaCa2+-containing (60μM)Na+-free (replaced with NMDG+, see Materials and Methods) sol-ution that triggered a similar increase inmitochondrial Ca2+ levels incontrol and NCLX-transfected cells (Fig. S3D). Strong Ca2+ effluxwas monitored only after 20 mM Na+ (Fig. 5A) were reintroduced,
indicating that theCa2+effluxmediatedbyNa+-dependent exchangeis the major mitochondrial Ca2+ extrusion pathway in these cells. Amarked increase (259±45%) inmitochondrialNa+-dependentCa2+
efflux was monitored in NCLX-overexpressing versus control cells(Fig. 5A). To directly determine if Na+ was the counter ion trans-ported into mitochondria by NCLX, the same paradigm was appliedto cells overexpressing NCLX loaded with the mitochondrial Na+-sensitive dyeCoroNaRed.As shown inFig. 5B, applicationof 20mMNa+ followingmitochondrialCa2+ loading triggereda rise inCoroNaRed fluorescence. The rate of Na+ influx was increased (170± 47%)inNCLX-overexpressing cells comparedwith controls, butnot in cellssuperfused with Na+-free solution. indicating that the fluorescenceincrease resulted from Na+ influx into the mitochondria. This ap-parent coupling between mitochondrial Na+ influx and Ca2+ effluxwith elevated expression levels of NCLX is consistent with mi-tochondrial Na+/Ca2+ exchange activity being mediated by thisprotein. Further substantiating the link between NCLX and mi-tochondrial exchanger activity, the observed Na+-dependent Ca2+
effluxwas profoundly inhibited (to 28±8.7%of control) inHEK-293cells transfected with the siNCLX construct (Fig. 5C). Importantly,Ca2+ efflux was virtually eliminated in NCLX-overexpressing (Fig. 5AandB) or control (Fig. 5C) cells thatwere superfusedwithNa+-freesolution. indicating that Ca2+ efflux mediated by the mitoch-ondrial exchanger and mitochondrial NCLX share the same strictNa+ dependence.Finally, Li+/Ca2+ exchange activity is a unique property of both
NCLX (7) and the mitochondrial exchanger (19). We thereforeasked if NCLX conducts mitochondrial Li+-dependent Ca2+
transport. Superfusion of permeabilized HEK-293 control cellswith 30 mM Li+ in a Na+-free solution facilitated robust Ca2+
efflux from the mitochondria, consistent with the unique ability ofthe mitochondrial exchanger to mediate Li+-dependent Ca2+
exchange (Fig. 5D). Importantly, silencing of NCLX expressionwas followed by a strong reduction in the rate of Li+-dependentCa2+ efflux, (35 ± 8% of control; Fig. 5D). Similar attenuation ofLi+-dependent Ca2+ efflux activity upon silencing of NCLX ex-pression was also recorded in CHO cells (see Fig. S5G–I). Theseresults indicate that NCLX is the molecular determinant essentialfor mitochondrial Na+/Ca2+ and Li+/Ca2+ exchange activity.
DiscussionNumerous studies have reported that the Na+-dependent mi-tochondrial exchanger, similar to other members of the NCX
Fig. 2. NCLX is found on the inner membrane of mi-tochondria. (A–C) Electron micrographs of rat cortical slices(A) or CHO cells (B) stained with anti-NCLX antibodies orNCLX preimmune serum (C). N, ER, and PM denote the nu-cleus, endoplasmic reticulum, and plasma membrane struc-tures, respectively. Positive immunolabeling, defined as thepresence of dense DAB precipitate, is observed primarily inthe mitochondria. (Scale bar, 0.5 μm.) (D and E) Immunogoldlabeling of SHSY-5Y cells overexpressing (D) or endogenouslyexpressing (E) NCLX. Note that NCLX labeling is found pri-marily in the cristae of the mitochondria. (Scale bar, 0.2 μm.)(F) The distribution of numbers of gold particles per mi-tochondrion is shown. Quantitative analysis of mitochondrialgold particle distribution shows that the overall number ofparticles in NCLX-overexpressing cells was 35 versus 14 in thecontrol cells (number of mitochondria examined, n = 10 foreach group; **P < 0.01).
438 | www.pnas.org/cgi/doi/10.1073/pnas.0908099107 Palty et al.

family, mediates Na+/Ca2+ exchange, albeit with unique functionalproperties (15, 19), including altered selectivity to monovalentcations and distinct sensitivity to inhibitors. In our studies, we havefocused on NCLX because, although it is a member of the NCXfamily, it is phylogenetically and functionally distinct from otherknown NCX or NCKX family members (20). Support for NCLXbeing the mitochondrial Na+/Ca2+ exchanger come from the fol-lowing: (i) NCLX is expressed inmitochondria, where it is localizedto the innermembraneof this organelle, and has amolecularweightsimilar to that of the purified mitochondrial exchanger (21). (ii)NCLX expression accelerates mitochondrial Ca2+ efflux activity,whereas silencing of NCLX expression attenuates this process.Importantly, Ca2+ efflux was fully rescued in NCLX-silenced cellsby concomitant overexpression of NCLX, indicating that the ex-pression of NCLX is essential and sufficient to functionally com-plement mitochondrial Na+/Ca2+ exchange. Despite the profoundeffect of NCLX expression onmitochondrial Ca2+ efflux, it did notaffect the steady-state resting mitochondrial Ca2+ level. The rela-tively lowaffinityof themitochondrial exchanger toCa2+ (10) or theeffective phosphate-based mitochondrial Ca2+ buffering systemthat is capable of keeping steady-state Ca2+ concentration constantover a wide range by a reversible formation or dissociation of in-soluble Ca2+-phosphate precipitates may account for this effect
(22). Hence, NCLX may be playing a similar role to plasma mem-brane members of the NCX family that primarily mediate and re-spond to rapid and robust Ca2+ changes rather than fine tuning ofthe steady-state Ca2+ level (23). (iii) Mitochondrial Ca2+ effluxmediated by NCLX is strictly Na+-dependent and coupled to ac-celerated mitochondrial Na+ influx. The strong endogenous Na+
dependence of Ca2+ efflux that we have assayed in permeabilizedcells (Fig. 5) indicates that NCLX is the dominant Ca2+ extrudingtransporter in these cells. Further studies comparing the activity ofNCLX and the H+/Ca2+ exchanger are required to address theirspecific roles in distinct cell types and tissues. (iv) Replacement of asingle residue in the putative catalytic α-domain of NCLX wassufficient to fully block mitochondrial Ca2+ efflux. The role of theα-domains as the catalytic core of NCX members is firmly estab-lished and hence this finding will provide the basis and rational forfurther studies on the unique and common structural/functionalproperties of the mitochondrial Na+/Ca2+ exchanger. (v) NCLX isfully inhibited by the mitochondrial Na+/Ca2+ exchange inhibitorCGP-37157 and (vi) NCLX mediates Li+/Ca2+ exchange, a func-tional property that, amongNCXproteins, is shared exclusivelywiththe mitochondrial exchanger. Thus, the mitochondrial localization,the link betweenNCLX expression/inactivation withmitochondrialexchange activity, and its functional properties all indicate thatNCLX is the mitochondrial exchanger.Our results show that endogenous expression of NCLX in HEK
293 cells, as well as in other cells studied, is most prominent inmitochondria (Fig. 1); this is also seen in cells overexpressing themurineNCLXisoform.Thus, theenhancedplasmamembraneCa2+
transport activity that was previously found in HEK-293 cells ec-topically expressing the human isoform of NCLX (7) is probablylinked to a “spillover” of NCLX to the plasma membrane, a phe-nomenon commonly linked to strong overexpression in HEK-293and other cell types (24, 25). Importantly, this spillover of the humanNCLX protein to the plasma membrane fortuitously provided uswith the seminal finding that NCLX mediates Li+/Ca2+ exchange
Fig. 4. Silencing the expression of endogenous NCLX decreases mitochondrialCa2+ efflux that can be rescued by expression of recombinant murine NCLX. (A)Mitochondrial Ca2+ responses following application ofATP (40 μM, as in Fig. 3A)was measured in HEK-293 cells cotransfected with either the siNCLX or ascrambled siRNA construct (si control) and the RP-mt–expressing plasmid.AveragedratesofmitochondrialCa2+effluxare shown(Right) (n=9,**P<0.01).(B) The same experiment as in A was performed on SHSY-5Y cells transfectedwith NCLX shRNA plasmid alone or together with the murine NCLX-encodingplasmid (which is insensitive to the NCLX shRNA construct). Averaged rates ofmitochondrial Ca2+ efflux are shown (Right) (n = 11, *P < 0.05).
Fig. 3. Expression of NCLX enhancesmitochondrial Ca2+ efflux that is blockedby mutation in the catalytic site of NCLX and by the mitochondrial exchangerinhibitor CGP-37157. (A) SHSY-5Y cells transfected with plasmids encoding themouse NCLX, NCLX-S468Tmutant, or the vector (control) were cotransfected toexpress RP-mt. Cells were superfused with ATP-containing Ringer solution (40μM at the indicated time) while monitoring mitochondrial Ca2+ fluorescence.Note that enhancement ofNCLX levels increasedmitochondrial Ca2+ efflux, andthemutantwasnotonly inactivebutalsoexertedadominant-negativeeffectonendogenous activity. (B) Illustration of the same experimental paradigm as inArepeated in control or NCLX-expressing cells in the presence of the mitochon-drial exchanger inhibitor CGP-37157 (10 μM). Similar inhibition of Ca2+ efflux isseen in both. (C) Averaged mitochondrial Ca2+ efflux rates (n = 9; **P < 0.01).(Inset) Experimental paradigm for Ca2+ efflux rate measurement based on de-termination of the initial rate of the mitochondrial Ca2+ efflux phase. (D) Dose-dependence analysis of the effect of CGP-37157 onmitochondrial Ca2+ efflux incells expressing NCLX. Mitochondrial Ca2+ efflux rate was measured in thepresence of the indicated concentrations of CGP-37157 and presented as per-centageof the ratemeasured in its absence. Thedashed line is afit of thedata tothe equation I = Io / [1
+([CGP] / IC50)].
Palty et al. PNAS | January 5, 2010 | vol. 107 | no. 1 | 439
PHYS
IOLO
GY
and prompted us to investigate if NCLX is the mitochondrial ex-changer. We do not rule out, however, the possibility that NCLX orits spliced isoforms may also be found in other cellular compart-ments (7, 9). Indeed, NCX members are often found at multiplecellular sites. Among them, NCX1 is found both in the plasmamembraneandnucleus (26).Dual localization isalso sharedbyothermitochondrial proteins such as several members of the cytochromeP450 family that are targeted to both the ER and mitochondrialcompartments (27, 28) and the potassium channel, KCa3.1, that canbe found in the mitochondria or plasma membrane (29).Our finding that NCLX is the mitochondrial Na+/Ca2+ ex-
changer is of particular interest in light of the intense expression ofNCLX in skeletalmuscle, stomach, and pancreas (7) and the criticalrole played by the mitochondrial Ca2+ transport in these organs.A remarkable feature of mitochondrial Ca2+ transport in skeletalmuscle is the ultrafast Ca2+ efflux rate (30), which equals the rate ofinfluxmediated by the uniporter; comparedwith the 2 to 3 orders ofmagnitude slower efflux rate found in cardiomyocytes. The dra-matically higher expression of NCLX in skeletal muscle versuscardiac tissue further links the expression of NCLX to mitochon-drial Ca2+ efflux and suggests that expression of NCLX is a majorrate limiting determinant of the uniquely fast mitochondrial Ca2+
transport in skeletalmuscle. Smoothmuscle accounts for the largestmass of tissue in the stomach, and it is therefore conceivable that theintenseNCLXexpression in the stomach results from its ubiquitousexpression in smooth muscle (7). The essential role of the mi-tochondrial Na+/Ca2+ exchanger in refilling smooth muscle Ca2+
stores and in facilitating the Ca2+ sparks leading to muscle con-traction is documented (31, 32) and agrees well with the high ex-
pression of NCLX in this tissue. A role of mitochondrial Na+/Ca2+
exchange in insulin secretion is of profound physiological interestparticularly because of the link between mitochondrial Ca2+ levelsand the rate of ATP synthesis, but is controversial because of po-tential interference of CGP-37157 with the L-type Ca2+ channelactivity (16). In the exocrine pancreas, mitochondria have a welldocumented role in limiting the apical to the basolateral prop-agation of Ca2+ waves and in controlling plasma membrane Ca2+
influx (33). Much has to be learned on the specific role of the mi-tochondrial exchanger in the function of endocrine and exocrinepancreas, and the present study will provide the molecular toolsrequired for this goal. Finally, the profound change in mitochon-drial Ca2+ exchange activity observed for example in a neuronalParkinson model (34) underscores the pathophysiological rele-vance of NCLX regulation. Hence, the identification of the mi-tochondrial Na+/Ca2+ exchanger will provide insight on themolecular basis underlying these processes.
Materials and MethodsFluorescent Measurements of Intracellular Ions. Cell culture and transfectionprocedures are described in SI Methods. Cytosolic Ca2+ levels were recordedfrom Fura-2–loaded cells, excited at wavelengths of 340 and 380 nm, andimaged with a 510 nm long-pass filter as previously described (7). Mi-tochondrial Ca2+ or H+ levels were monitored in cells transiently expressingthe RP-mt protein at excitation wavelengths of 430 nm [i.e., Ca2+-sensitivewavelength (12), presented as F430 (1-F/F0)] and 485 nm [i.e., pH-sensitivewavelength (35, 36), presented as F485 F/F0], respectively, and the emissioncollected using a 535-nm band-pass filter. In experiments in which digitonin-permeabilized HEK-293 cells were employed, F430 was normalized to thevalue obtained following the Ca2+-loading phase (FT-200). Mitochondrial Na+
Fig. 5. The mitochondrial Na+- or Li+-dependent Ca2+ exchange is mediated by NCLX. (A) Traces and rates of Na+-dependent mitochondrial Ca2+ efflux. Mi-tochondrial Ca2+ levels were recorded by monitoring RP-mt fluorescence in HEK-293 cells overexpressing either the human NCLX isoform (NCLX) or vector alone(control) and coexpressing RP-mt. Experiments were conducted on digitonin-permeabilized cells (seeMaterials and methods). Mitochondrial Ca2+ uptake was in-ducedby superfusionwithNa+-free solution (replaced byNMDG+) containing Ca2+ (60 μM). Ca2+ effluxwasmonitored following superfusion in Ca2+-free solution inthe presence or absence of Na+. Note that the Ca2+ effluxwas strictly Na+-dependent andwas enhanced by the expression of NCLX (n = 8, **P< 0.01). (B) Traces andrates of Na+-dependentmitochondrial Na+ influx.Mitochondrial Na+ levels weremeasured in cells loadedwith theNa+-sensitive dye CoroNaRed (seeMaterials andmethods) as inA. Note the enhanced and reciprocal nature of the Na+ and Ca2+ transport rates, mediated bymitochondrial NCLX (n = 8, **P < 0.01). (C) Traces andratesofNa+-dependentmitochondrial Ca2+efflux in cells transfectedwitheither siNCLXora scrambledsiRNA(si control). The sameexperimentalparadigmdescribedinAwasapplied.SilencingofNCLXeliminatedmitochondrialNa+-dependentCa2+efflux (n=11,**P<0.01). (D) TracesandratesofLi+-dependentmitochondrialCa2+
efflux in cells transfected with either siNCLX or si control, using Li+-containing solution (replacing Na+; n = 11; *P < 0.05). Silencing of NCLX also diminished Li+-dependent Ca2+ efflux.
440 | www.pnas.org/cgi/doi/10.1073/pnas.0908099107 Palty et al.

levels were monitored in CoroNa-Red (Molecular Probes)–loaded cells ex-cited at 545 nm and imaged using a 630-nm long-pass filter. Unless statedotherwise, all experiments in nonpermeabilized cells were conducted usingRinger solutions containing 130 mM NaCl or NMDG (Na+-free), 20 mMHepes, 15 mM glucose, 5 mM KCl, and 0.8 mM MgCl2, with the pH adjustedto 7.4. Ringer solution was supplemented with 0.5 or 2 mM CaCl2 (for onlyexperiments described in SI Materials and Methods; Figs. S2 and S4F) or in0.5 mM EDTA and 40 μM ATP as indicated. Mitochondrial Ca2+ or Na+ indigitonin-permeabilized (0.007% digitonin) cells were determined as pre-viously described (37) in a buffer containing 220 mM sucrose, 10 mM Hepes,5 mM succinate, 2.5 mM KH2PO4, 0.4 mM EGTA, and 1 μM cyclosporine A,with the pH adjusted to 7.4 with KOH. Following the addition of 60 μM Ca2+,and Ca2+ loading of the mitochondria, 1 μM ruthenium red was added toeliminate a leak of Ca2+ via this pathway. Then 20 mM NaCl, NMDG Cl, or 30mM LiCl were added as indicated.
For all single-cell imaging experiments, traces of averaged responses,recorded from 5 to 20 cells in each experiment, were analyzed and plottedusing KaleidaGraph. The rate of ion transportwas calculated from each graph(summarizing an individual experiment) by a linear fit of the change in thefluorescence (ΔF) over a period of 30 s following initiation of apparent ef-flux/influx (ΔF/dt). Rates from n experiments (as mentioned in legends to thefigures) were averaged and displayed in bar graph format. Analysis with thet test was performed to determine significance when relevant.
Immunoelectron Microscopy. Brain sections were processed and imaged asdescribed in SI Materials and Methods. Analysis of the distribution of NCLXin control and NCLX-overexpressing cells was performed blindly using thegold-labeled preparations for the EM. Each mitochondrion containing atleast one gold particle was digitally-captured at ×25,000 magnification. Onlyone mitochondrion was imaged per cell section, with 20 cells overall (i.e., 20mitochondria): 10 overexpressing and 10 controls. Only mitochondria thatexhibited a clear outer membrane and cristae were selected for the analysis.Each cell was counted as an event, and we then compared the gold particlenumber for each event. Similar numbers of extramitochondrial gold particles(n = 1–2) were observed in approximately 40% of the sections obtainedeither from control or NCLX-overexpressing cells.
ACKNOWLEDGMENTS. The authors thank Dr. Ronit Ben-Romano, SalahAbu-Hamad, Rina Jaeger, Elizabeth Weinfeld, and Nichole Kahn for theirtechnical help; Dr. Jonathan Lytton for the mouse NCLX isoform and NCKX4plasmids; Dr. Atsushi Miyawaki for the RP-mt–encoding plasmid; and Dr.Claude Brodski for his critical reading of the manuscript. This work waspartially supported by Israel Science Foundation (ISF) Grant 985/05 andGerman Israeli Foundation Grant 917-119.1/2006 (to I.S. and C.N.); ISF Grant450/03 (to M.H.), and FIRB 2003 and PRIN 2006 grants from the Ministryof Research and Education of Italy (S.L.S.). R.P. was supported by the AdamsFellowship Program of the Israel Academy of Sciences and Humanities.
1. Szabadkai G, Duchen MR (2008) Mitochondria: the hub of cellular Ca2+ signaling.Physiology (Bethesda) 23:84–94.
2. Jiang D, Zhao L, Clapham DE (2009) Genome-wide RNAi screen identifies Letm1 as amitochondrial Ca2+/H+ antiporter. Science 326:144–147.
3. Gunter TE, Buntinas L, Sparagna G, Eliseev R, Gunter K (2000) Mitochondrial calciumtransport: mechanisms and functions. Cell Calcium 28:285–296.
4. Cox DA, Matlib MA (1993) A role for the mitochondrial Na(+)-Ca2+ exchanger in theregulation of oxidative phosphorylation in isolated heart mitochondria. J Biol Chem268:938–947.
5. Szabadkai G, et al. (2006) Mitochondrial dynamics and Ca2+ signaling. BiochimBiophys Acta 1763:442–449.
6. Carafoli E, Tiozzo R, Lugli G, Crovetti F, Kratzing C (1974) The release of calcium fromheart mitochondria by sodium. J Mol Cell Cardiol 6:361–371.
7. Palty R, et al. (2004) Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J Biol Chem 279:25234–25240.
8. Lytton J (2007) Na+/Ca2+ exchangers: three mammalian gene families control Ca2+transport. Biochem J 406:365–382.
9. Cai X, Lytton J (2004) Molecular cloning of a sixth member of the K+-dependentNa+/Ca2+ exchanger gene family, NCKX6. J Biol Chem 279:5867–5876.
10. Paucek P, Jabůrek M (2004) Kinetics and ion specificity of Na(+)/Ca(2+) exchangemediated by the reconstituted beef heart mitochondrial Na(+)/Ca(2+) antiporter.Biochim Biophys Acta 1659:83–91.
11. Li W, et al. (1992) Reconstitution, identification, purification, and immunologicalcharacterization of the 110-kDa Na+/Ca2+ antiporter from beef heart mitochondria. JBiol Chem 267:17983–17989.
12. Nagai T, Sawano A, Park ES, Miyawaki A (2001) Circularly permuted green fluorescentproteins engineered to sense Ca2+. Proc Natl Acad Sci USA 98:3197–3202.
13. Malli R, et al. (2003) Sustained Ca2+ transfer across mitochondria is Essential formitochondrial Ca2+ buffering, sore-operated Ca2+ entry, and Ca2+ store refilling. JBiol Chem 278:44769–44779.
14. Palty R, et al. (2006) Single alpha-domain constructs of the Na+/Ca2+ exchanger,NCLX, oligomerize to form a functional exchanger. Biochemistry 45:11856–11866.
15. Cox DA, Conforti L, Sperelakis N, Matlib MA (1993) Selectivity of inhibition ofNa(+)-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. JCardiovasc Pharmacol 21:595–599.
16. Luciani DS, Ao P, Hu X,Warnock GL, Johnson JD (2007) Voltage-gated Ca(2+) influx andinsulin secretion in human and mouse beta-cells are impaired by the mitochondrialNa(+)/Ca(2+) exchange inhibitor CGP-37157. Eur J Pharmacol 576:18–25.
17. Czyz A, Kiedrowski L (2003) Inhibition of plasmalemmal Na(+)/Ca(2+) exchange bymitochondrial Na(+)/Ca(2+) exchange inhibitor 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP-37157) in cerebellar granule cells.Biochem Pharmacol 66:2409–2411.
18. Omelchenko A, et al. (2003) Inhibition of canine (NCX1.1) and Drosophila (CALX1.1)Na(+)-Ca(2+) exchangers by 7-chloro-3,5-dihydro-5-phenyl-1H-4,1-benzothiazepine-2-one (CGP-37157). J Pharmacol Exp Ther 306:1050–1057.
19. Crompton M, Künzi M, Carafoli E (1977) The calcium-induced and sodium-inducedeffluxes of calcium from heart mitochondria. Evidence for a sodium-calcium carrier.Eur J Biochem 79:549–558.
20. Cai X, Lytton J (2004) The cation/Ca(2+) exchanger superfamily: phylogenetic analysisand structural implications. Mol Biol Evol 21:1692–1703.
21. Kar P, Chakraborti T, Samanta K, Chakraborti S (2008) mu-Calpain mediated cleavageof the Na+/Ca2+ exchanger in isolated mitochondria under A23187 induced Ca2+stimulation. Arch Biochem Biophys .
22. Nicholls DG, Chalmers S (2004) The integration of mitochondrial calcium transportand storage. J Bioenerg Biomembr 36:277–281.
23. Hilgemann DW, Yaradanakul A, Wang Y, Fuster D (2006) Molecular control of cardiacsodium homeostasis in health and disease. J Cardiovasc Electrophysiol 17 (Suppl 1):S47–S56.
24. Ginger RS, et al. (2008) SLC24A5 encodes a trans-Golgi network protein withpotassium-dependent sodium-calcium exchange activity that regulates humanepidermal melanogenesis. J Biol Chem 283:5486–5495.
25. Schultz BD, Frizzell RA, Bridges RJ (1999) Rescue of dysfunctional deltaF508-CFTRchloride channel activity by IBMX. J Membr Biol 170:51–66.
26. Xie X, Wu G, Lu ZH, Rohowsky-Kochan C, Ledeen RW (2004) Presence of sodium-calcium exchanger/GM1 complex in the nuclear envelope of non-neural cells: natureof exchanger-GM1 interaction. Neurochem Res 29:2135–2146.
27. Addya S, et al. (1997) Targeting of NH2-terminal-processed microsomal protein tomitochondria: a novel pathway for the biogenesis of hepatic mitochondrial P450MT2.J Cell Biol 139:589–599.
28. Robin MA, et al. (2000) Vesicular transport of newly synthesized cytochromes P4501Ato the outside of rat hepatocyte plasma membranes. J Pharmacol Exp Ther 294:1063–1069.
29. De Marchi U, et al. (2009) Intermediate conductance Ca2+-activated potassiumchannel (KCa3.1) in the inner mitochondrial membrane of human colon cancer cells.Cell Calcium 45:509–516.
30. Rudolf R, Mongillo M, Magalhães PJ, Pozzan T (2004) In vivo monitoring of Ca(2+)uptake into mitochondria of mouse skeletal muscle during contraction. J Cell Biol 166:527–536.
31. Balemba OB, Bartoo AC, Nelson MT, Mawe GM (2008) Role of mitochondria inspontaneous rhythmic activity and intracellular calcium waves in the guinea piggallbladder smooth muscle. Am J Physiol Gastrointest Liver Physiol 294:G467–G476.
32. Poburko D, Liao CH, van Breemen C, Demaurex N (2009) Mitochondrial regulationof sarcoplasmic reticulum Ca2+ content in vascular smooth muscle cells. Circ Res104:104–112.
33. Tinel H, et al. (1999) Active mitochondria surrounding the pancreatic acinar granuleregion prevent spreading of inositol trisphosphate-evoked local cytosolic Ca(2+)signals. EMBO J 18:4999–5008.
34. Gandhi S, et al. (2009) PINK1-associated Parkinson's disease is caused by neuronalvulnerability to calcium-induced cell death. Mol Cell 33:627–638.
35. Filippin L, Magalhães PJ, Di Benedetto G, Colella M, Pozzan T (2003) Stableinteractions between mitochondria and endoplasmic reticulum allow rapidaccumulation of calcium in a subpopulation of mitochondria. J Biol Chem 278:39224–39234.
36. Frieden M, et al. (2004) Ca(2+) homeostasis during mitochondrial fragmentation andperinuclear clustering induced by hFis1. J Biol Chem 279:22704–22714.
37. Lee B, et al. (2003) Inhibition of mitochondrial Na+-Ca2+ exchanger increasesmitochondrial metabolism and potentiates glucose-stimulated insulin secretion in ratpancreatic islets. Diabetes 52:965–973.
Palty et al. PNAS | January 5, 2010 | vol. 107 | no. 1 | 441
PHYS
IOLO
GY

Supporting InformationPalty et al. 10.1073/pnas.0908099107
SI Materials and MethodsCell Culture and Transfection Procedures. HEK-293-T, HEK-293,SHSY-5Y, and CHO cells were cultured in DMEM as previouslydescribed (1). Transfection of HEK-293 or SHSY-5Y cells wasperformed using Ca2PO4 as described previously (1). CHO cellswere transfected using Lipofectamine 2000 (Invitrogen) orTransfect-it CHO reagent (Mirus) according to themanufacturer'sprotocol. Fluorescent ion measurements or cell harvesting wereconducted 30 to 72 h after transfection.
Plasmid and siRNA Preparation. Preparation of the human NCLX-encoding plasmid was described previously (1). NCLXS468T- andNCLX-6His-encoding plasmids were generated by replacing a NotIfragment from the α2 S273T or WT α2-6His plasmid (2), re-spectively, with the corresponding fragment of theNCLX-encodingplasmid. The murine NCLX variant and the NCKX4-encodingplasmids were provided by Jonathan Lytton (Calgary, AB, Canada)(3). The RP-mt plasmid was provided by AtsushiMiyawaki (Wako,Japan) (4). Double-stranded siRNAs used to silence NCLX ex-pression was obtained from Ambion (Applied Biosystems). Thesequence of 21 nucleotides corresponding to the sense strands usedfor the NCLX siRNAwas AACGGCCACUCAACUGUCUtt andthat for the control siRNA was AACGCGCAUCCAACUGU-CUtt. The humanNCLXshRNAplasmidwas obtained fromSigma(Mission TRC shRNA Target Set TRCN-5045).
Fluorescent Measurements of Intracellular Ions and MitochondrialMembrane Potential. Changes in inner mitochondrial membranepotential (ΔΨm) were monitored by detecting microfluorimetricchanges in fluorescence (ΔF) of the membrane potential–sensitiveprobes TMRE (5) (for HEK293 cells). HEK293 cells loaded with0.05 μM TMRE were washed in a static bath of Ringer solutioncontaining the probe and then exposed to 10 μM of CCCP toobtain maximal loss of ΔΨm.. TMRE-loaded cells were excitedwith a 530-nm (±30) band-pass filter and emission monitored atgreater than 590 nm. TMRE ΔF was calculated as fluorescencevalues at baseline and after exposure to CCCP.
Flow Cytometry Analysis of ΔΨm. ToevaluateΔΨminSHSY-5Ycells,5 × 105 cells were incubated with 500 nM of 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidazolyl carbocyanine iodide (JC1; Molec-ular Probes) in the growthmedium for 10min at 37°C in the dark andimmediately analyzed by flow cytometry using the FACSCalibur in-strument (BD Bioscience). Data were processed with FlowJo soft-wareandpresentedas the ratiobetweenmeanfluorescence intensitiesmeasured with the FL1 (green) and FL2 (red) photomultipliers.
Generation of Anti-NCLX Antibodies. The generation of the antibodyused in Figs. 1 and 2 in the main text was previously described (1).Additional antibodies used in theexperiment described inFig. S1wasraised against peptide CSRSHTEVKLEDPDGL (CSR antibody),located close to theC terminus ofNCLX, as previously described (1).
Cell Fractionation, Immunoprecipitation, and Immunoblot Analysis.Subcellular fractionation of ER/sarcoplasmic reticulum or mito-chondrial fractions fromHEK293 (6) cells or from rat heart using asmall-scale protocol (7, 8)wereperformed as previously described.Briefly, rat ventricular tissue (approximately 20 g) was homogen-ized 3 times in Polytron, PT3000 (9,000 rpm × 30 s) with ice-coldTMS buffer (0.25M sucrose/20mMTris-Cl, pH 7.4, 1 mMMgCl2,1 mM PMSF, and proteinase inhibitor mixture). The lysates werefurther homogenized with a glassDouncer and then centrifuged at
800 × g for 15 min to remove the contractile proteins and nuclei.The supernatant was centrifuged twice at 8,000 × g for 20 min topellet the mitochondrial fraction. The mitochondrial fraction wasseparately washed whereas the collected supernatants were cen-trifuged at 100,000× g for 60min to pellet themicrosomal fraction.The pellet was suspended in the TMS buffer and the sarcoplasmicreticulum and sarcolemmamembranes were separated by sucrosegradient as previously described (9). The subcellular fractionswere resuspended in 0.25 M sucrose/20 mM Tris-Cl, pH 7.4, andstored at −20°C.Purification of the plasma membrane–enriched fraction was
performed using a cell surface protein isolation kit (Pierce) ac-cording to the manufacturer's instructions. Protein concentrationwas determined by the bicinchoninate assay (Pierce), and 20 μg ofprotein was separated by SDS/PAGE and immunoblotted. In thecase of the plasma membrane fraction, protein samples and BSAstandards were precipitated in 80% acetone, suspended in ddH2O,and then assayed by the bicinchoninate method. Extracted proteins(20 μg) were separated by 10% SDS/PAGE and transferred onto apolyvinylidene difluoride membrane (Amersham). Membraneswere probed using the following antibodies: anti-NCLX [1:500 (2)],anti-ANT (1:100, Santa Cruz Biotechnology), anti-Sec62 (1:5,000;provided by T. Sommer, Berlin, Germany), anti-Na/K ATPase[1:500 (10)], and anti–N-cadherin (1:1,000; BD Biosciences; do-nated byFrankKirchner, Berlin,Germany), all diluted into 5%milkin tris-buffered saline solution with Tween 20.Isolation of mitochondria from transfected HEK293-T cells or
from native mouse tissues was performed as previously described(11), with minor modifications. Briefly, cells were homo-genized in isolation buffer containing (in mM): mannitol 225,sucrose 75, EDTA 0.5, HEPES 10, with the pH being adjusted to7.4 with KOH. The crude homogenate samples were centrifugedat 1,500 × g for 5 min in 4°C, the supernatant was collected, andthe samples were washed with fresh homogenization buffer andrecentrifuged. Pellet was saved (crude membranes) and the su-pernatant collected and recentrifuged for 4 min at 12,000 × g at4°C. Supernatant containing ER fragments and cytosolic pro-teins was saved (cytosolic fraction). The pellet, containing themitochondria, was then washed 3 times with homogenizationbuffer. All samples were analyzed for protein concentration us-ing the Bradford method (Bio-Rad). Equal amounts of proteinwere then resolved by SDS/PAGE and transferred onto nitro-cellulose membranes for Western blot analysis.Immunoprecipitation procedure was performed using the
ExactaCruz kit (Santa Cruz Biotechnology) according to manu-facturer's protocol. Briefly, total cell lysate from HEK-293-Tcells expressing NCLX bearing a c-Myc tag, NCLX bearing a6His-tag, or both were incubated with 20 μg of an antibodytargeted against the 6His epitope (Santa Cruz Biotechnology)overnight at 4°C. Subsequently, 50 μL of immunoprecipitationmatrix was added to the lysate and beads were sedimented andfurther processed according to manufacturer's instructions.Immunoblot analysis was carried out as described previously
(1). Antibodies used in this study included NCLX-antiserum(1:1,000–2,000 dilution), anti–β-actin (1:40,000; Sigma), mono-clonal anti-VDAC antibodies against the N-terminal region of31HL human porin (12) (1:10,000; Calbiochem), anti-ANT(1:100; Santa Cruz Biotechnology), anti-c-Myc (1:500; SantaCruz Biotechnology), and anti-FLAG (1:1,000; VWR).
Immunoelectron Microscopy. Brains sections were processed for pre-embedding immunoelectron microscopy according to a protocol
Palty et al. www.pnas.org/cgi/content/short/0908099107 1 of 7
Supporting Information Corrected / /20100 75 0

described previously (13). TheNCLXantibodywas used at a dilutionof 1:200.The postembedding immunoelectron microscopy protocol was a
modification of the method of Tokuyasu (14). Briefly, control orNCLX-overexpressing SHSY-5Y cells were fixed with 2% paraf-
ormaldehyde, 0.25% glutaraldehyde in 0.1 M phosphate buffer.Cryosections were cut on a Leica UltraCut UCT Ultramicrotomeequipped with a low-temperature sectioning system. Analysis wasperformedon a JEOLJEM-1230 electronmicroscope equippedwitha Tietz TemCam F214 CCD camera (TVIPS, Gauting, Germany).
1. Palty R, et al. (2004) Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J Biol Chem 279:25234–25240.
2. Palty R, et al. (2006) Single alpha-domain constructs of the Na+/Ca2+ exchanger,NCLX, oligomerize to form a functional exchanger. Biochemistry 45:11856–11866.
3. Cai X, Lytton J (2004) Molecular cloning of a sixth member of the K+-dependentNa+/Ca2+ exchanger gene family, NCKX6. J Biol Chem 279:5867–5876.
4. Nagai T, Sawano A, Park ES, Miyawaki A (2001) Circularly permuted green fluorescentproteins engineered to sense Ca2+. Proc Natl Acad Sci USA 98:3197–3202.
5. Farkas DL, Wei MD, Febbroriello P, Carson JH, Loew LM (1989) Simultaneous imagingof cell and mitochondrial membrane potentials. Biophys J 56:1053–1069.
6. Bozidis P, Williamson CD, Colberg-Poley AM (2007) Isolation of endoplasmic reticulum,mitochondria, andmitochondria-associatedmembrane fractions from transfected cellsand fromhuman cytomegalovirus-infected primaryfibroblasts. Curr Protoc Cell Biol 37:3.27.1–3.27.23.
7. Khananshvili D, Shaulov G, Weil-Maslansky E, Baazov D (1995) Positively chargedcyclic hexapeptides, novel blockers for the cardiac sarcolemma Na(+)-Ca2+ exchanger.J Biol Chem 270:16182–16188.
8. Pallotti F, Lenaz G (2007) Mitochondria, eds Pon LA, Schon EA , (Elsevier, New York),Vol 80, pp 3–44.
9. Baazov D, Wang X, Khananshvili D (1999) Time-resolved monitoring of electrogenicNa+–Ca2+ exchange in the isolated cardiac sarcolemma vesicles by using a rapid-response fluorescent probe. Biochemistry 38:1435–1445.
10. Patchornik G, Goldshleger R, Karlish SJ (2000) The complex ATP-Fe(2+) serves as aspecific affinity cleavage reagent in ATP-Mg(2+) sites of Na,K-ATPase: altered ligationof Fe(2+) (Mg(2+)) ions accompanies the E(1)—>E(2) conformational change. ProcNatl Acad Sci USA 97:11954–11959.
11. Vergun O, Reynolds IJ (2005) Distinct characteristics of Ca(2+)-induced depolarizationof isolated brain and liver mitochondria. Biochim Biophys Acta 1709:127–137.
12. Babel D, et al. (1991) Studies on human porin. VI. Production and characterization ofeight monoclonal mouse antibodies against the human VDAC “Porin 31HL” and theirapplication for histotopological studies in human skeletal muscle. Biol Chem HoppeSeyler 372:1027–1034.
13. Elgazar V, et al. (2005) Zinc-regulating proteins, ZnT-1, andmetallothionein I/II are presentin different cell populations in the mouse testis. J Histochem Cytochem 53:905–912.
14. Tokuyasu KT (1986) Application of cryoultramicrotomy to immunocytochemistry. JMicrosc 143:139–149.
15. RenX, Nicoll DA, GalangG, PhilipsonKD (2008) Intermolecular cross-linkingof Na+-Ca2+ exchanger proteins: evidence for dimer formation. Biochemistry 47:6081–6087.
16. Filippin L, Magalhães PJ, Di Benedetto G, Colella M, Pozzan T (2003) Stableinteractions between mitochondria and endoplasmic reticulum allow rapidaccumulation of calcium in a subpopulation of mitochondria. J Biol Chem 278:39224–39234.
17. Frieden M, et al. (2004) Ca(2+) homeostasis during mitochondrial fragmentation andperinuclear clustering induced by hFis1. J Biol Chem 279:22704–22714.
Palty et al. www.pnas.org/cgi/content/short/0908099107 2 of 7
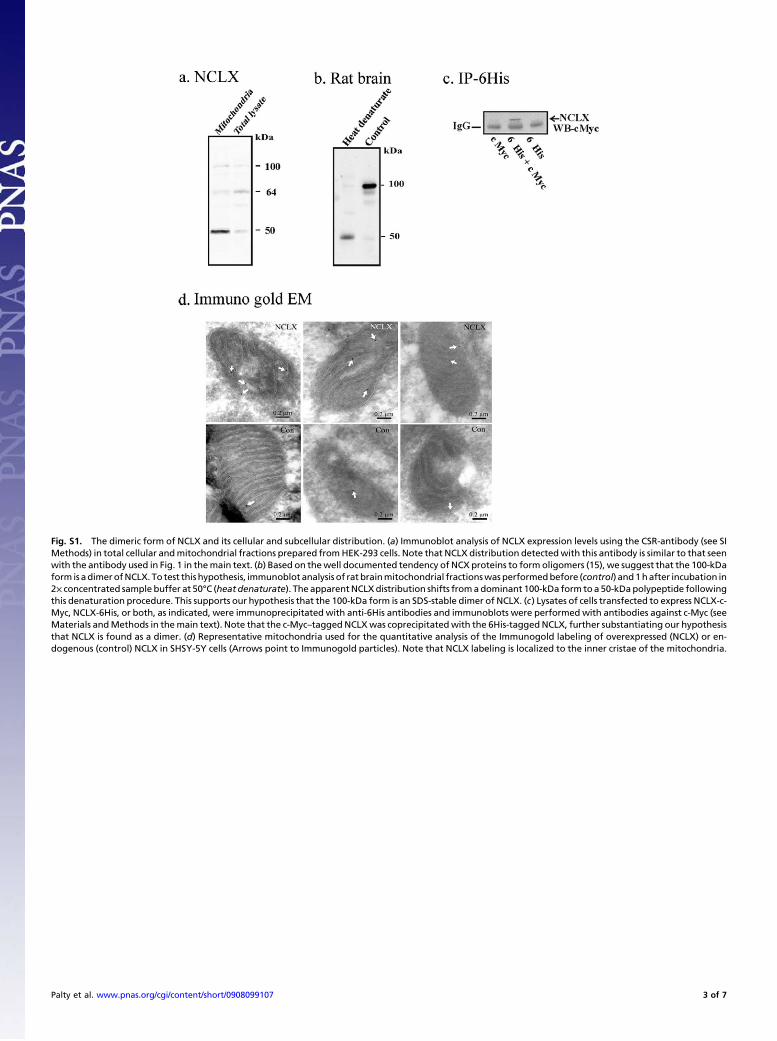
Fig. S1. The dimeric form of NCLX and its cellular and subcellular distribution. (a) Immunoblot analysis of NCLX expression levels using the CSR-antibody (see SIMethods) in total cellular andmitochondrial fractions prepared fromHEK-293 cells. Note that NCLX distribution detectedwith this antibody is similar to that seenwith the antibody used in Fig. 1 in themain text. (b) Based on thewell documented tendency of NCX proteins to form oligomers (15), we suggest that the 100-kDaform is adimerofNCLX. To test this hypothesis, immunoblot analysis of rat brainmitochondrial fractionswasperformedbefore (control) and1hafter incubation in2× concentrated sample buffer at 50°C (heat denaturate). The apparentNCLXdistribution shifts fromadominant 100-kDa form toa 50-kDapolypeptide followingthis denaturation procedure. This supports our hypothesis that the 100-kDa form is an SDS-stable dimer of NCLX. (c) Lysates of cells transfected to express NCLX-c-Myc, NCLX-6His, or both, as indicated, were immunoprecipitated with anti-6His antibodies and immunoblots were performed with antibodies against c-Myc (seeMaterials andMethods in themain text). Note that the c-Myc–taggedNCLXwas coprecipitatedwith the 6His-taggedNCLX, further substantiating our hypothesisthat NCLX is found as a dimer. (d) Representative mitochondria used for the quantitative analysis of the Immunogold labeling of overexpressed (NCLX) or en-dogenous (control) NCLX in SHSY-5Y cells (Arrows point to Immunogold particles). Note that NCLX labeling is localized to the inner cristae of the mitochondria.
Palty et al. www.pnas.org/cgi/content/short/0908099107 3 of 7
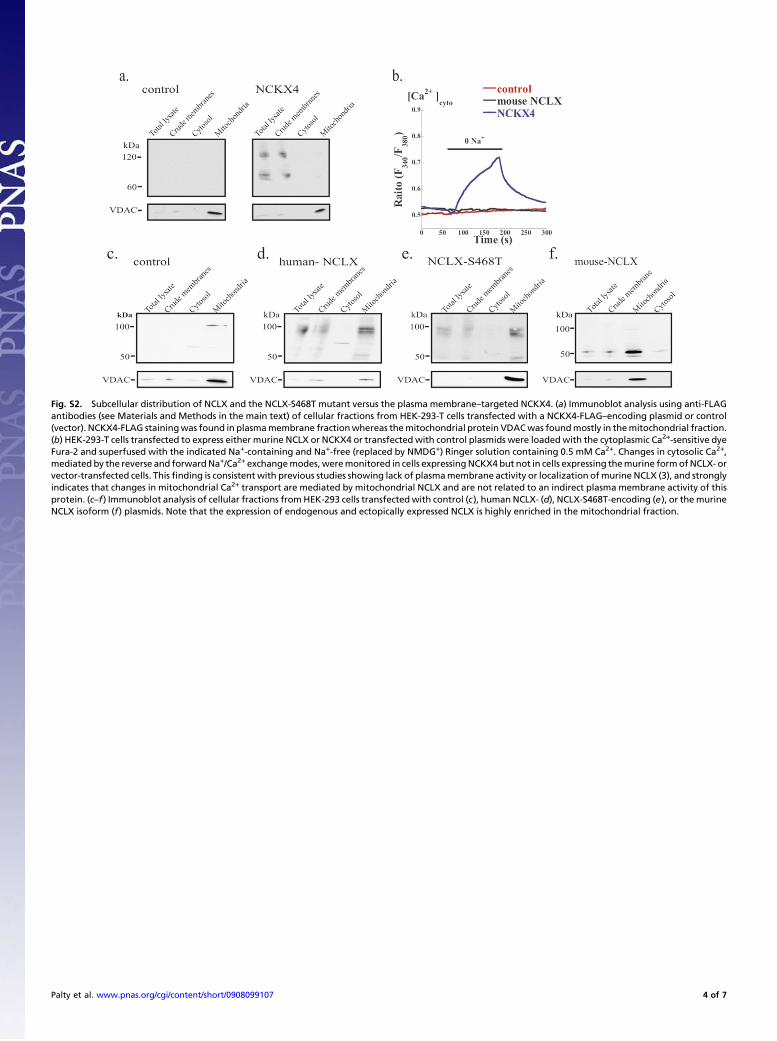
Fig. S2. Subcellular distribution of NCLX and the NCLX-S468T mutant versus the plasma membrane–targeted NCKX4. (a) Immunoblot analysis using anti-FLAGantibodies (see Materials and Methods in the main text) of cellular fractions from HEK-293-T cells transfected with a NCKX4-FLAG–encoding plasmid or control(vector). NCKX4-FLAG stainingwas found in plasmamembrane fractionwhereas themitochondrial protein VDACwas foundmostly in themitochondrial fraction.(b) HEK-293-T cells transfected to express either murine NCLX or NCKX4 or transfected with control plasmids were loaded with the cytoplasmic Ca2+-sensitive dyeFura-2 and superfused with the indicated Na+-containing and Na+-free (replaced by NMDG+) Ringer solution containing 0.5 mM Ca2+. Changes in cytosolic Ca2+,mediated by the reverse and forwardNa+/Ca2+ exchangemodes,weremonitored in cells expressingNCKX4 but not in cells expressing themurine formofNCLX- orvector-transfected cells. Thisfinding is consistent with previous studies showing lack of plasmamembrane activity or localization ofmurine NCLX (3), and stronglyindicates that changes in mitochondrial Ca2+ transport are mediated by mitochondrial NCLX and are not related to an indirect plasma membrane activity of thisprotein. (c–f) Immunoblot analysis of cellular fractions fromHEK-293 cells transfectedwith control (c), human NCLX- (d), NCLX-S468T-encoding (e), or themurineNCLX isoform (f) plasmids. Note that the expression of endogenous and ectopically expressed NCLX is highly enriched in the mitochondrial fraction.
Palty et al. www.pnas.org/cgi/content/short/0908099107 4 of 7
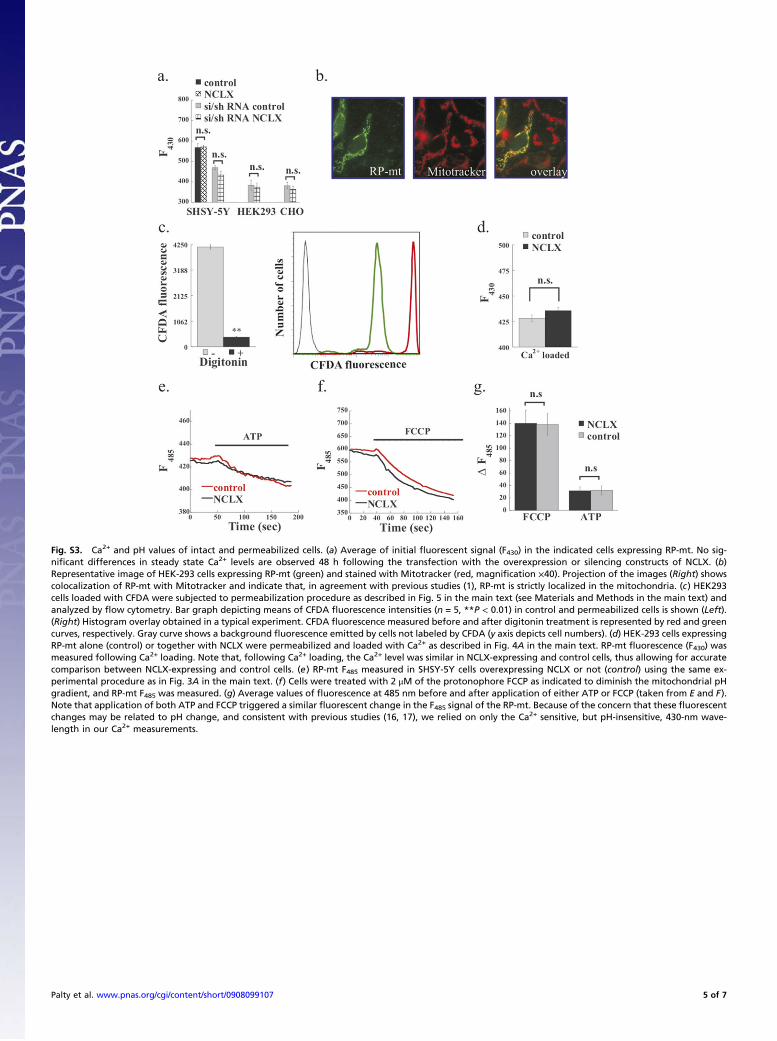
Fig. S3. Ca2+ and pH values of intact and permeabilized cells. (a) Average of initial fluorescent signal (F430) in the indicated cells expressing RP-mt. No sig-nificant differences in steady state Ca2+ levels are observed 48 h following the transfection with the overexpression or silencing constructs of NCLX. (b)Representative image of HEK-293 cells expressing RP-mt (green) and stained with Mitotracker (red, magnification ×40). Projection of the images (Right) showscolocalization of RP-mt with Mitotracker and indicate that, in agreement with previous studies (1), RP-mt is strictly localized in the mitochondria. (c) HEK293cells loaded with CFDA were subjected to permeabilization procedure as described in Fig. 5 in the main text (see Materials and Methods in the main text) andanalyzed by flow cytometry. Bar graph depicting means of CFDA fluorescence intensities (n = 5, **P < 0.01) in control and permeabilized cells is shown (Left).(Right) Histogram overlay obtained in a typical experiment. CFDA fluorescence measured before and after digitonin treatment is represented by red and greencurves, respectively. Gray curve shows a background fluorescence emitted by cells not labeled by CFDA (y axis depicts cell numbers). (d) HEK-293 cells expressingRP-mt alone (control) or together with NCLX were permeabilized and loaded with Ca2+ as described in Fig. 4A in the main text. RP-mt fluorescence (F430) wasmeasured following Ca2+ loading. Note that, following Ca2+ loading, the Ca2+ level was similar in NCLX-expressing and control cells, thus allowing for accuratecomparison between NCLX-expressing and control cells. (e) RP-mt F485 measured in SHSY-5Y cells overexpressing NCLX or not (control) using the same ex-perimental procedure as in Fig. 3A in the main text. (f) Cells were treated with 2 μM of the protonophore FCCP as indicated to diminish the mitochondrial pHgradient, and RP-mt F485 was measured. (g) Average values of fluorescence at 485 nm before and after application of either ATP or FCCP (taken from E and F).Note that application of both ATP and FCCP triggered a similar fluorescent change in the F485 signal of the RP-mt. Because of the concern that these fluorescentchanges may be related to pH change, and consistent with previous studies (16, 17), we relied on only the Ca2+ sensitive, but pH-insensitive, 430-nm wave-length in our Ca2+ measurements.
Palty et al. www.pnas.org/cgi/content/short/0908099107 5 of 7
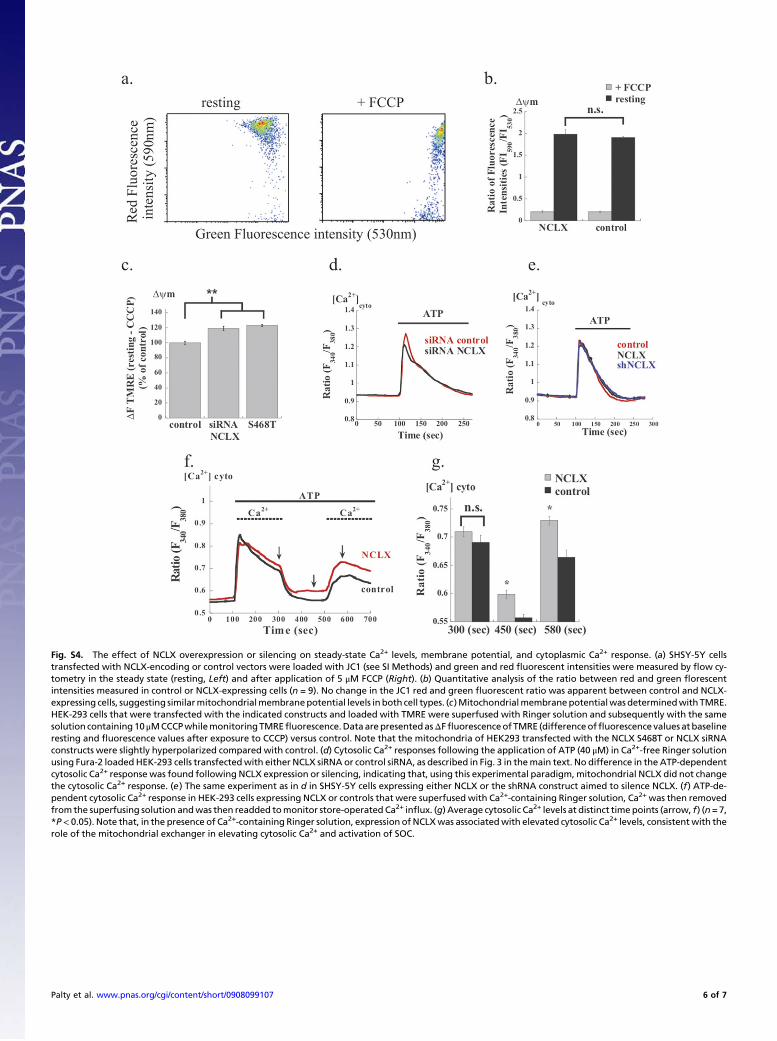
Fig. S4. The effect of NCLX overexpression or silencing on steady-state Ca2+ levels, membrane potential, and cytoplasmic Ca2+ response. (a) SHSY-5Y cellstransfected with NCLX-encoding or control vectors were loaded with JC1 (see SI Methods) and green and red fluorescent intensities were measured by flow cy-tometry in the steady state (resting, Left) and after application of 5 μM FCCP (Right). (b) Quantitative analysis of the ratio between red and green florescentintensities measured in control or NCLX-expressing cells (n = 9). No change in the JC1 red and green fluorescent ratio was apparent between control and NCLX-expressing cells, suggesting similarmitochondrialmembranepotential levels inboth cell types. (c)Mitochondrialmembranepotentialwas determinedwith TMRE.HEK-293 cells that were transfected with the indicated constructs and loaded with TMRE were superfused with Ringer solution and subsequently with the samesolution containing10μMCCCPwhilemonitoringTMREfluorescence.DataarepresentedasΔFfluorescenceof TMRE (differenceoffluorescence values at baselineresting and fluorescence values after exposure to CCCP) versus control. Note that the mitochondria of HEK293 transfected with the NCLX S468T or NCLX siRNAconstructs were slightly hyperpolarized compared with control. (d) Cytosolic Ca2+ responses following the application of ATP (40 μM) in Ca2+-free Ringer solutionusing Fura-2 loaded HEK-293 cells transfectedwith either NCLX siRNA or control siRNA, as described in Fig. 3 in themain text. No difference in the ATP-dependentcytosolic Ca2+ response was found following NCLX expression or silencing, indicating that, using this experimental paradigm, mitochondrial NCLX did not changethe cytosolic Ca2+ response. (e) The same experiment as in d in SHSY-5Y cells expressing either NCLX or the shRNA construct aimed to silence NCLX. (f) ATP-de-pendent cytosolic Ca2+ response in HEK-293 cells expressing NCLX or controls that were superfused with Ca2+-containing Ringer solution, Ca2+ was then removedfrom the superfusing solution andwas then readded tomonitor store-operated Ca2+ influx. (g) Average cytosolic Ca2+ levels at distinct timepoints (arrow, f) (n=7,*P< 0.05). Note that, in the presence of Ca2+-containing Ringer solution, expression of NCLXwas associatedwith elevated cytosolic Ca2+ levels, consistent with therole of the mitochondrial exchanger in elevating cytosolic Ca2+ and activation of SOC.
Palty et al. www.pnas.org/cgi/content/short/0908099107 6 of 7
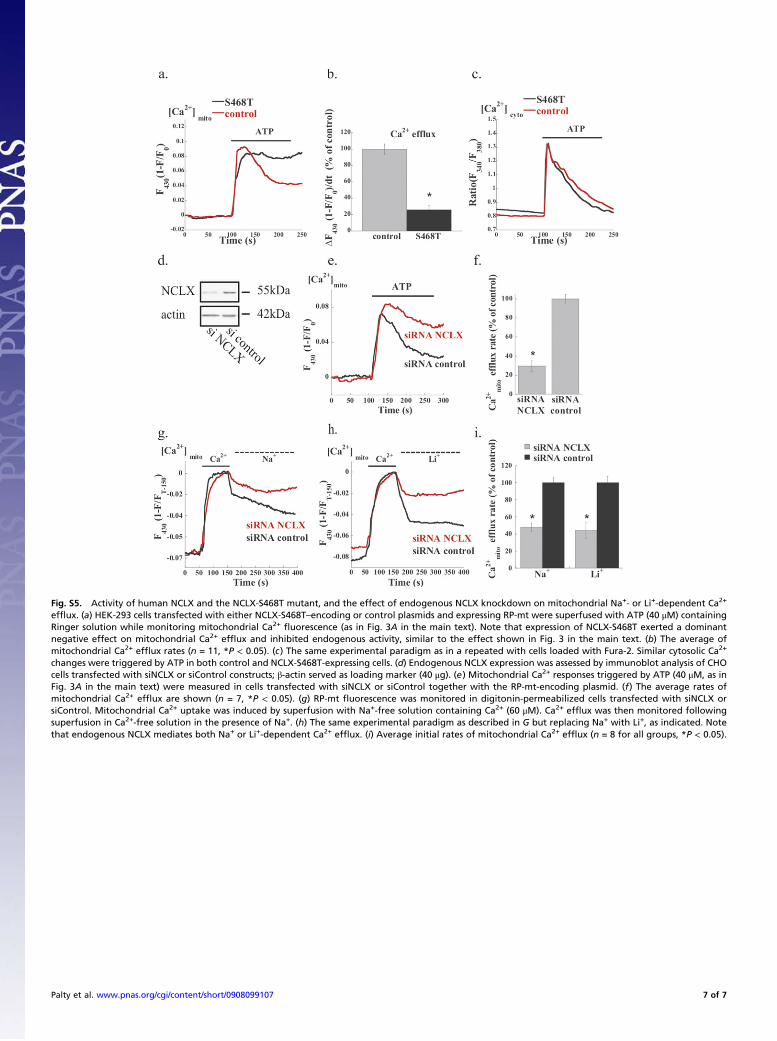
Fig. S5. Activity of human NCLX and the NCLX-S468T mutant, and the effect of endogenous NCLX knockdown on mitochondrial Na+- or Li+-dependent Ca2+
efflux. (a) HEK-293 cells transfected with either NCLX-S468T–encoding or control plasmids and expressing RP-mt were superfused with ATP (40 μM) containingRinger solution while monitoring mitochondrial Ca2+ fluorescence (as in Fig. 3A in the main text). Note that expression of NCLX-S468T exerted a dominantnegative effect on mitochondrial Ca2+ efflux and inhibited endogenous activity, similar to the effect shown in Fig. 3 in the main text. (b) The average ofmitochondrial Ca2+ efflux rates (n = 11, *P < 0.05). (c) The same experimental paradigm as in a repeated with cells loaded with Fura-2. Similar cytosolic Ca2+
changes were triggered by ATP in both control and NCLX-S468T-expressing cells. (d) Endogenous NCLX expression was assessed by immunoblot analysis of CHOcells transfected with siNCLX or siControl constructs; β-actin served as loading marker (40 μg). (e) Mitochondrial Ca2+ responses triggered by ATP (40 μM, as inFig. 3A in the main text) were measured in cells transfected with siNCLX or siControl together with the RP-mt-encoding plasmid. (f) The average rates ofmitochondrial Ca2+ efflux are shown (n = 7, *P < 0.05). (g) RP-mt fluorescence was monitored in digitonin-permeabilized cells transfected with siNCLX orsiControl. Mitochondrial Ca2+ uptake was induced by superfusion with Na+-free solution containing Ca2+ (60 μM). Ca2+ efflux was then monitored followingsuperfusion in Ca2+-free solution in the presence of Na+. (h) The same experimental paradigm as described in G but replacing Na+ with Li+, as indicated. Notethat endogenous NCLX mediates both Na+ or Li+-dependent Ca2+ efflux. (i) Average initial rates of mitochondrial Ca2+ efflux (n = 8 for all groups, *P < 0.05).
Palty et al. www.pnas.org/cgi/content/short/0908099107 7 of 7
Copyright © 2022 FDOKUMEN



















