
Multiple Viral Genetic Analyses Detect Low-Level Human Immunodeficiency Virus Type 1 Replication...
11
10.1128/JVI.77.10.5721-5730.2003. 2003, 77(10):5721. DOI: J. Virol. Tobin and James I. Mullins Madhumita Mahalanabis, Wilscott E. Naugler, Nicole H. Melvin, Paul F. Lewis, Laura M. Heath, Ingrid A. Beck, Holte, Shannon M. De Vange, Diane M. Pawluk, Ann J. McKernan, Giovanina M. Ellis, Kathleen M. Mohan, Sarah E. Lisa M. Frenkel, Yang Wang, Gerald H. Learn, Jennifer L. Active Antiretroviral Therapy Type 1 Replication during Effective Highly Low-Level Human Immunodeficiency Virus Multiple Viral Genetic Analyses Detect http://jvi.asm.org/content/77/10/5721 Updated information and services can be found at: These include: REFERENCES http://jvi.asm.org/content/77/10/5721#ref-list-1 at: This article cites 58 articles, 23 of which can be accessed free CONTENT ALERTS more» articles cite this article), Receive: RSS Feeds, eTOCs, free email alerts (when new http://journals.asm.org/site/misc/reprints.xhtml Information about commercial reprint orders: http://journals.asm.org/site/subscriptions/ To subscribe to to another ASM Journal go to: on January 6, 2013 by guest http://jvi.asm.org/ Downloaded from
-
Upload
washington -
Category
Documents
-
view
3 -
download
0
Transcript of Multiple Viral Genetic Analyses Detect Low-Level Human Immunodeficiency Virus Type 1 Replication...

10.1128/JVI.77.10.5721-5730.2003.
2003, 77(10):5721. DOI:J. Virol. Tobin and James I. MullinsMadhumita Mahalanabis, Wilscott E. Naugler, Nicole H.Melvin, Paul F. Lewis, Laura M. Heath, Ingrid A. Beck, Holte, Shannon M. De Vange, Diane M. Pawluk, Ann J.McKernan, Giovanina M. Ellis, Kathleen M. Mohan, Sarah E. Lisa M. Frenkel, Yang Wang, Gerald H. Learn, Jennifer L. Active Antiretroviral TherapyType 1 Replication during Effective HighlyLow-Level Human Immunodeficiency Virus Multiple Viral Genetic Analyses Detect
http://jvi.asm.org/content/77/10/5721Updated information and services can be found at:
These include:
REFERENCEShttp://jvi.asm.org/content/77/10/5721#ref-list-1at:
This article cites 58 articles, 23 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on January 6, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from

JOURNAL OF VIROLOGY, May 2003, p. 5721–5730 Vol. 77, No. 100022-538X/03/$08.00�0 DOI: 10.1128/JVI.77.10.5721–5730.2003Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Multiple Viral Genetic Analyses Detect Low-Level HumanImmunodeficiency Virus Type 1 Replication during Effective Highly
Active Antiretroviral TherapyLisa M. Frenkel,1,2* Yang Wang,3 Gerald H. Learn,3 Jennifer L. McKernan,1 Giovanina M. Ellis,1
Kathleen M. Mohan,1 Sarah E. Holte,4 Shannon M. De Vange,1 Diane M. Pawluk,1Ann J. Melvin,1 Paul F. Lewis,5 Laura M. Heath,3 Ingrid A. Beck,1
Madhumita Mahalanabis,1† Wilscott E. Naugler,1‡Nicole H. Tobin,1 and James I. Mullins2,3,6
Departments of Pediatrics,1 Laboratory Medicine,2 Microbiology,3 Biostatistics,4 and Medicine,6 University ofWashington, Seattle, Washington, and Department of Pediatrics, Oregon Health and Science University,
Portland, Oregon5
Received 2 December 2002/Accepted 14 February 2003
To evaluate human immunodeficiency virus type 1 (HIV-1) replication and selection of drug-resistant virusesduring seemingly effective highly active antiretroviral therapy (HAART), multiple HIV-1 env and pol sequenceswere analyzed and viral DNA levels were quantified from nucleoside analog-experienced children prior to andduring a median of 5.1 (range, 1.8 to 6.4) years of HAART. Viral replication was detected at different rates, withapparently increasing sensitivity: 1 of 10 by phylogenetic analysis; 2 of 10 by viral evolution with increasinggenetic distances from the most recent common ancestor (MRCA) of infection; 3 of 10 by selection ofdrug-resistant mutants; and 6 of 10 by maintenance of genetic distances from the MRCA. When four- orfive-drug antiretroviral regimens were given to these children, persistent plasma viral rebound did not occurdespite the accumulation of highly drug-resistant genotypes. Among the four children without genetic evidenceof viral replication, a statistically significant decrease in the genetic distance to the MRCA was detected inthree, indicating the persistence of a greater number of early compared to recent viruses, and their HIV-1 DNAdecreased by >0.9 log10, resulting in lower absolute DNA levels (P � 0.007). This study demonstrates thevariable rates of viral replication when HAART has suppressed plasma HIV-1 RNA for years to a median of<50 copies/ml and that combinations of four or five antiretroviral drugs suppress viral replication even aftershort-term virologic failure of three-drug HAART and despite ongoing accumulation of drug-resistant mu-tants. Furthermore, the decrease of cellular HIV-1 DNA to low absolute levels in those without genetic evidenceof viral replication suggests that monitoring viral DNA during HAART may gauge low-level replication.
Administration of highly active antiretroviral therapy(HAART) has been associated with improved immunologicfunction and survival in human immunodeficiency virus type 1(HIV-1)-infected individuals (22, 35). However, only 40 to65% of subjects have continued suppression of HIV-1 replica-tion to below the limits of detection after 6 months or more oftreatment (22, 29, 38, 50, 51, 59). While partial suppression ofviral replication offers immunologic benefits (6), the durationof these benefits may be limited to several years (7). Multiplefactors have been associated with foreshortening the antiviralactivity of HAART, including the selection of drug-resistantmutants (12, 26, 39, 41, 49). Among individuals in whomHAART appears effective, as defined by plasma HIV-1 levelsconsistently below the limit of detection of currently licensedassays (50 copies of RNA/ml of plasma), viral replication nev-ertheless may persist at low levels (5, 11, 16, 19, 21, 25, 32, 36,43, 48, 61, 63). The extent and effects of low-level replica-
tion on the efficacy of HAART have not been fully character-ized.
To gain insight into the emergence of drug resistant HIV-1during apparently effective therapy, the viral population in theperipheral blood mononuclear cells (PBMC) of children wascharacterized when plasma HIV-1 RNA levels were below thelimit of detection over a median of 5.1 years of HAART. Ofparticular interest were whether the evolution of resistant viruscould be detected within PBMC prior to a rebound of plasmaHIV-1 RNA to detectable levels, whether mutations accumulatedover time with the eventual loss of suppression of viral replication,and whether simple identifiers were associated with low-level viralreplication during apparently effective HAART.
MATERIALS AND METHODS
Study design. Children with plasma HIV-1 RNA levels of �50 copies/ml after1 to 2 years of HAART were enrolled into the study, with consent obtained fromeach child’s guardian by one of the investigators in accordance with InstitutionalReview Board guidelines. Plasma HIV-1 RNA copy numbers were determinedevery 1 to 6 months by using bDNA (Chiron Corp, Emeryville, Calif.), AmplicorHIV-1 Monitor 1.0 (Roche Diagnostics, Montclair, N.J.), or Ultra SensitiveMonitor 1.0 (Roche) assays, with lower limits of detection of 500, 400, and 50copies/ml, respectively. Lymphocyte subsets were determined by fluorescence-activated cell sorter analysis (34). Adherence to prescribed HAART was evalu-ated by a formal questionnaire or parental report of the number of doses missedduring the most recent 2 days and during the entire time interval preceding each
* Corresponding author. Mailing address: 4800 Sand Point Way,NE, 8G-1, Seattle, WA 98105. Phone: (206) 987-5140. Fax: (206)987-3890. E-mail: [email protected].
† Present address: Department of Microbiology, University ofWashington, Seattle, Wash.
‡ Present address: Department of Medicine, Oregon Health andScience University, Portland, OR 97239.
5721
on January 6, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from

monthly clinic visit. Multiple HIV-1 pol and env sequences were obtained fromendpoint dilutions of enrollees’ PBMC collected prior to HAART and every 6 to12 months during 1.8 to 6.5 years of effective HAART and, when viremia wasdetected, from endpoint dilutions of plasma.
Sampling of HIV-1 populations by limiting dilution PCR of PBMC DNA andquantification of DNA copy number in CD4� cells. DNA for PCR was obtainedfrom a known quantity of PBMC by lysis with a detergent-proteinase K solution(62) or by extraction using the IsoQuick nucleic acid extraction kit (Orca Re-search Inc, Bothell, Wash.). HIV-1 DNA was quantified by endpoint-dilutionPCR (44). The viral DNA load per 106 CD4� cells was then calculated. Speci-mens were diluted so that no more than 30% of the reactions were positive afternested PCR, providing 70 to 80% probability that a positive reaction was from asingle viral template.
Nucleic acids encoding protease (PR), reverse transcriptase (RT), and the C2-V5region of Env were coamplified in first-round PCRs using either two or three outerprimer sets in the same reaction. The PR- and RT-encoding regions were amplifiedeither individually, with PRL (5�-GGGACCAGCGGCTACACTAGAAGAAATGATGACAGCATGTCAGG-3�) and PR2 (5�-GGAGTATTGTATGGATTTTCAGGCC-3�) plus RT1 and RT2 (18), or amplified as one fragment with PRL andRT2. In either case, env was simultaneously amplified using ED31 and BH2 (17).Second-round PCRs were conducted separately, using pro primers PRC (5�-CTCCCCCTCAGAAGCAGGAGCCGATAGACAAGGAACTGTATCC-3�) and PR4(5�-GGGCCATCCATTCCTGGC-3�), rt primers RT4 and RT3 (18) (or pol primersPRC and RT3), and env primers ES7 and ES8 (9). Samples that did not amplify withthe forward PR primers PRL and PRC were amplified using either pol-forwardouter and pol-forward inner (19), PRF1 (5�-GAGCCAAGTAACAAATTCAGC-3�) and PRF2 (5�-CACCAGAAGAGAGCTTCAGGT-3�), or PRA (5�-CCTAGGAAAAAGGGCTGTTGGAAATGTGG-3�) and PRB (5�-ACTGAGAGACAGGCTAATTTTTTAGGGA-3�). PCR and direct DNA sequencing of PCR productswere performed as previously described (45). The resulting sequences with evidenceof polymorphic residues, thus indicating the presence of multiple templates, wereexcluded from our analyses.
Sequence analysis. Sequences were assembled and error checked using Se-quencher 3.0 (Gene Codes, Ann Arbor, Mich.). Those with substantial G3A mu-tational bias (as determined using HYPERMUT [28]), suggesting hypermutation(24, 57), were omitted from further analyses. Sequence alignments were obtainedusing ClustalW 1.7 (55) and manually edited as necessary. Regions of ambiguousalignment were removed from subsequent evolutionary analyses. Neighbor-joiningphylogenetic trees of each subject’s Env, PR, and RT coding sequences were con-structed using PAUP* version 4.0b4 (54) with evolutionary models selected using theAkaike information criterion (1) under Modeltest 3.0 (42). Consequently, most envtrees used the general time-reversible model with gamma-distributed heterogeneityof substitution rates (31). Model parameters are available from the authors uponrequest. Phylogenetic trees of the nucleotide sequences encoding HIV-1 Env, PR,and RT from each patient were constructed separately and rooted using HIV-1sequences from his or her mother, when available, or several closely related HIV-1sequences from GenBank. The reliability of clustering in phylograms was assessed bybootstrapping analyses (15). More complete information related to the phylogeneticanalyses of virus from all subjects can be found at the website http://ubik.microbiol.washington.edu/HIV/Frenkel-2/index.html.
The most recent common ancestor (MRCA) sequence of a given infection wasinferred as the sequence at the node that included all of the sequences for a givengene region (PR, RT, or env C2-V5) for a given patient. This sequence was obtainedusing maximum likelihood estimation using PAUP*, and the divergence rates fromthe MRCA were calculated using linear regression over time (30).
Statistical analyses. Logistic regression analysis was applied to the prevalenceof genetic mutants over time, with tests to determine whether the coefficient fortime was significantly different from zero. When analysis was conducted on morethan one individual, generalized estimating equations were used to account forthe correlations resulting from repeated measures from the same individual (10).The log10-transformed HIV-1 DNA levels before and during potent HAARTwere compared using Student’s two-tailed t test.
Nucleotide sequence accession numbers. The GenBank accession numbers ofthe HIV-1 pol and env sequences derived in this study are AY075701 to AY077450.
RESULTS
Patient population, antiretroviral history, and HIV-1 RNAand DNA levels. Ten children were studied before and duringa median and mean of 5.1 years (range, 1.8 to 6.4 years) ofprotease inhibitor (PI)-containing HAART. All 10 were naïve
to PI therapy when HAART was initiated; however, all hadreceived other antiretrovirals (Fig. 1). The initial HAARTregimens included three or four drugs, including one to twonucleoside reverse transcriptase inhibitors (NRTI) and one totwo PI, with or without one non-nucleoside reverse transcrip-tase inhibitor (NNRTI), except for one child (L1) who wastreated with only stavudine and ritonavir as part of a clinicaltrial. “Potent HAART,” defined as combination therapy withthree classes of drugs (NRTI, NNRTI, and PI) or with twoNRTI and two PI, was prescribed to 9 of the 10 participantsduring the course of this study.
Adherence to prescribed therapy was judged to be �95%(missing zero to three doses per month) in 7 of the 10 children: inG2, H3, L1, R1, and B1 from the time therapy was initiated; in G1after resolution of pneumonia that developed during the monthhe began three-drug HAART; and in H1 after the placement ofa gastrostomy tube prior to starting a second potent HAARTregimen. H2 had �95% adherence except during a brief timeduring his fourth year of therapy. In contrast, subjects M2 and S1reported missing one to two doses per week. In addition, M2missed multiple doses during 1 month in her fifth year of HAARTdue to problems within her family.
Plasma HIV-1 RNA levels were determined a median of sixtimes per year (range, two to nine) after falling below the limit ofdetection (Table 1; see also Fig. 1, 2, 4, and 5). Four children hada rebound of viral plasma RNA to �500 copies/ml during theirinitial two-class, three-drug HAART regimens (two NRTI plusone PI). Two of these, G2 (see Fig. 5) and R1 (see Fig. 1),reported complete adherence to HAART during this period;however, their regimens included nucleoside analogs that theyhad taken previously. Viral rebound in the other two was associ-ated with difficulties in adherence, due to illness (G1 [see Fig.4A]) and the disagreeable taste of ritonavir (H1 [see Fig. 1]).HAART potency was increased by the addition and switching ofantiretroviral agents in these four and in a fifth child (L1 [see Fig.1]) who did not have plasma viral rebound but began HAARTwith only two drugs. After these modifications in therapy, 9 of 10children were receiving potent HAART. During potent HAART,plasma HIV-1 RNA levels were sustained at a median �50 cop-ies/ml for a combined total of 40 patient years. Rebounds of �250copies/ml occurred in only two during potent HAART, in H2 andM2, with 589 and 1,215 copies/ml, respectively, both during peri-ods of nonadherence.
PBMC viral DNA load decreased in four children by �0.9log10 between 1 and 2 years of potent HAART (Table 2). Thepre-HAART viral DNA load in these four children was similar(P � 0.731) to that in those who had a �0.9 log10 decrease;however, their post-HAART loads were significantly lower (P� 0.007; Student’s t test).
Selection of drug resistance mutations and viral reboundrelated to the potency of HAART. Despite plasma HIV-1 RNAbelow the limits of detection, new drug-resistant mutants orincreases in the population of existing drug-resistant mutantswere detected (Fig. 2 and 3). New mutants were observed inthree of six children during two-class, three-drug HAART (forPR, N88S in G1 and V82A, L90M in G2 [Fig. 4C and 5C,respectively]); (for RT, M184V in G2 and H1 [data notshown]), and in one of nine during potent HAART (in G2, PRmutants L10I, I54V, A71V/T, V82F, and I84V [Fig. 5C]; RT
5722 FRENKEL ET AL. J. VIROL.
on January 6, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from

FIG. 1. HIV-1 RNA and CD4� cell levels and antiretroviral treatments for eight subjects (data for subjects G1 and G2 are provided below inFig. 4 and 5, respectively). The plasma HIV-1 RNA and CD4� cell levels and antiretroviral drug regimens are shown for nucleoside analog-experienced children whose viral genetics were studied during a median of 5.1 years of HAART. Plasma HIV-1 RNA rebounded in G1, G2, H1,and R1 shortly after starting HAART. All subjects except H3 were ultimately treated with potent HAART, defined as combination therapy withthree classes of drugs (NRTI, NNRTI, and PI) or with two NRTI and two PI, during which the median plasma viral RNA level was �50 copies/ml.
5723
on January 6, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from

mutants V106A, Y181C, and a hypermutated sequence with aT69SEA insertion [data not shown]).
Three of the four episodes of viral rebound during nonpo-tent HAART were coincident with, or followed, detection ofdrug-resistant mutants not previously found in the subjects.
The V82A and L90M in PR and M184V in RT were observedin 10% of subject G2’s PBMC 3.5 months before rebound;however, no specimens were available from the months pre-ceding rebound in the other two with novel mutants detectedat viral rebound (G1 and H1). No novel mutations were asso-ciated with rebound in R1, who reported complete adherenceto three-drug HAART that had been formulated by the addi-tion of ritonavir to ongoing lamivudine and zidovudine. Inaddition, during potent HAART no novel mutations were de-tected with the isolated plasma viral rebounds associated withnonadherence to antiretrovirals by H2 and M2.
Although no sustained viral rebound was detected amongsubjects during potent HAART (Table 1 and Fig. 1 and 2),episodes of transient viremia were detected in subject G2 withplasma viral RNA levels between 50 and 101 copies/ml on 9 of19 occasions during 3.8 years of three-class, five-drug HAART(Fig. 5). The prevalence of PI-resistant mutants increased inhis PBMC (0% in 1996, 30% in 2000, and 55% in 2001; P �0.0005) and in his plasma (58% in late 1999 to 100% in 2000).Notably, mutations conferring high-level resistance to all threeclasses of antiretrovirals predominated in his plasma andPBMC virus load (RT had T69SEA, K103N, V106A, Y181C,M184V, T215F/Y, and K219Q; PR had L10I, I54V, A71V/T,V82A/F, I84V, and L90M), yet viral rebound was not sus-tained. Subsequent to the substitution of lopinavir-ritonavir(Kaletra) for saquinavir and ritonavir, his plasma HIV-1 RNAwas �50 copies/ml in all nine assays over 19 months and hisviral DNA load decreased by �1 log10/106 CD4� cells to 1.8log10 copies/106 CD4� PBMC.
Drug-resistant mutants detected in PBMC prior to the ini-tiation of HAART generally increased in prevalence whenselected for by three-drug and potent HAART regimens (P �0.0001) (Fig. 3A), although in one subject, G1, the frequencyof mutants (V75T) decreased over several years of HAART.Simultaneously, mutants selected by drugs in previous but notthe current regimens decreased in prevalence (P � 0.0013)(Fig. 3B). An absence of relevant drug-resistant mutants pre-vented the evaluation of all subjects for these trends.
Phylogenetic and genetic distance analysis. Three viral phy-logenetic patterns, forward, no, and regressive evolution, weredetermined by statistical analysis of genetic distances from the
TABLE 1. Summary of subjects’ characteristics and HIV-1 evolution
SubjectAge at startof HAART
(years)
Years of HAART(dates)
Distance from MRCA duringHAART (P value of distance over
time compared to zero)a
Episodes of rebound plasma HIV-1 RNAby year of HAARTb
Env RT PR 1 2 3 4 5 6 7
B1 5.9 4.8 (11/97–9/02) 0.009 0.800 0.070 0/7 0/6 0/6 0/6 0/5G1 9.1 6.4 (4/96–9/02) 0.020 �0.001 0.030 1/6 0/6 0/5 0/5 1/8 0/6 0/2G2 11.2 6.4 (4/96–9/02) 0.001 0.001 <0.001 0/4 3/7 3/6 3/5 3/7 0/6 0/2H1 1.7 3.5 (6/97–12/00) 0.444 0.860 0.090 3/9 1/7 0/6 0/5H2 0.2 5.0 (9/97–9/02) 0.290 0.222 0.385 0/7 2/5 0/7 1/5 0/4H3 14.7 3.9 (5/96–3/00) 0.110 0.350 0.990 0/4 0/3 0/2 0/3L1 6.2 5.5 (2/97–9/02) 0.040 0.640 0.120 0/5 1/4 0/5 0/5 1/6 0/3M2 5.5 5.2 (7/97–9/02) 0.850 0.129 0.056 0/9 1/7 1/7 0/6 1/9 0/1R1 2.3 6.1 (8/96–9/02) 0.055 0.720 0.490 2/7 0/6 0/5 1/5 0/5 0/4S1 3.0 1.8 (7/97–3/99) 0.690 0.440 0.740 0/9 1/4
a P values shown in boldface indicate increasing distance.b Number of episodes per number of measurements (per year). Values in bold indicate rebound to �500 copies/ml. Underlined values indicate that subjects were
receiving only two classes of drugs; otherwise, subjects were receiving potent HAART.
TABLE 2. Marked decline and low absolute HIV-1 DNA loadafter 1 to 2 years of HAART in subjects without
genetic evidence for viral replicationa
Subject Viral divergenceduring HAART
Log10 HIV-1 DNA copies/106 CD4� cells
Pre-HAART Post-HAART
With �0.9 log10decreaseH3 Stable 2.8 2.3M2 Trend to increase 2.6 2.6R1 Stable 3.5 2.9S1 Stable 3.2 2.9G2 Increased 2.6 3.6H1 Stable 3.7 3.7
Mean (95% CI)b 3.08 (2.58, 3.57) 2.99 (2.41, 3.58)
With �0.9 log10decreaseG1 Decreased 2.7 1.8L1 Decreased 2.8 1.6B1 Decreased 3.2 2.1H2 Stable 3.3 2.0
Mean (95% CI) 2.98 (2.49, 3.46) 1.90 (1.54, 2.27)P 0.731 0.007
a Subjects were sorted by the magnitude of HIV-1 DNA load change (�0.9log10 or �0.9 log10 decrease in HIV-1 DNA copies per 106 CD4� cells) afterpotent HAART (and three-drug HAART in H3). The absolute pre-HAARTHIV-1 DNA log10-transformed values were similar between the two groups,while the post-HAART values were significantly lower among those with de-creases of �0.9 log10 in HIV-1 DNA copies/106 CD4� cells, Interestingly, thesubjects with a �0.9 log10 decrease had less viral genetic evidence for viralreplication during HAART, with either no viral diversification after startingpotent HAART during primary infection (H2) or decreasing viral divergencefrom the MRCA of infection (see Table 1). In contrast, the viral populations ofthose with a �0.9 log10 or no decrease in HIV-1 DNA load maintained orincreased in divergence from the MRCA during HAART (see Table 1), indi-cating a greater degree of viral replication. Viral replication was indeed detectedin H3, G2, and H1 by selection of drug-resistant mutants (Fig. 3A) and was likelyin M2, R1, and S1 due to the persistence of a genetically diverse viral population.These observations suggest that a �0.9 log10 decrease in HIV-1 DNA copies/106
CD4� cells or a low absolute HIV-1 DNA level indicates profound suppressionof viral replication.
b CI, confidence interval.
5724 FRENKEL ET AL. J. VIROL.
on January 6, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from

MRCA (Table 1). “Forward evolution” (a statistically signifi-cant increase in distance from the MRCA) occurred in G2(Fig. 5B) and to a lesser degree in M2 (PR distance [P �0.056]) during potent HAART (Table 1). “No evolution” wasdetected in H1, H2, H3, R1, and S1. Nonetheless, low levels ofreplication (Fig. 3A) were indicated in H1 and H3 by increasesin the prevalence of certain drug-resistant mutants during po-tent and nonpotent HAART, respectively, and were suggestedin R1 and S1 by persistence of genetic diversity duringHAART (data not shown). However, there was no diversifica-tion of the viral population of H2, who began HAART duringprimary infection shortly after a course of peripartum zidovu-dine (data not shown). “Regressive evolution,” with viral se-quences becoming more similar to the MRCA of infection(i.e., to early virus) during HAART compared to pre-HAARTsequences, was pronounced in three subjects, B1, G1, and L1,during potent HAART (Table 1). Notably, the three subjectswith regressive evolution and H2 had pronounced decreases inviral DNA load (Table 2).
Another important observation in the phylogenetic analysiswas that plasma viral RNA detected intermittently at low levels(50 to 101 copies/ml in G2 [Fig. 5C] and R1 [data not shown])included genotypes typical of those found shortly after infec-tion, indicating that viral genomes similar to those in long-livedcells were replication competent.
DISCUSSION
Various analyses of viral gene sequences from 10 children dur-ing HAART with median plasma HIV-1 RNA levels of �50copies/ml over a median of 5.1 years detected replication in 1 to6 subjects of 10, depending on the method employed. Whetherviral replication remained at low levels, manifest only by selection
of drug-resistant mutants, or was marked and resulted in resump-tion of detectable plasma viremia appeared to be related to thepotency of the child’s HAART regimen. Children without evi-dence of viral replication by any method had marked decreasesand low absolute levels of HIV-1 DNA per 106 CD4� cells.
Viral replication during HAART when plasma viral RNAwas below the level of detection was indicated in six children byseveral different analyses of viral genotypes. Phylogenetic anal-ysis, as used in previous studies (60, 63), revealed viral repli-cation. However, this was the least sensitive measure of repli-cation, detecting replication in 1 of the 10 children studied.Ranking the methods utilized in this study according to in-creasingly more sensitive measurements of viral replication,after phylogenetic analysis, was as follows: increasing geneticdistances from the MRCA; the selection of novel drug-resis-tant mutants; increases in the frequency of drug-resistantmutants antedating HAART; and stable genetic distances,or divergence, from the MRCA. The four children withoutevidence of replication by any of the aforementioned tests,including no viral diversification following primary infection orwith genetic distances that approached the MRCA, were as-sumed to have insignificant viral replication.
HIV-1 DNA levels normalized to CD4� PBMC appeared tobe a sensitive indicator of residual viral replication in thechildren we studied. While HIV-1 DNA levels before HAARTwere similar among children that did and did not have evi-dence of replication during HAART, those without replicationhad a �0.9 log10 decrease in their HIV-1 DNA and signifi-cantly lower absolute HIV-1 DNA concentration in CD4�
PBMC after 1 to 2 years of HAART, compared to no changein levels among children with replication. These data suggestthat HIV-1 DNA levels might be useful in gauging low-level
FIG. 2. Summary of virologic studies. Subjects, identified by a letter and number, are grouped by intensity of therapy. “HAART” was treatmentwith NRTI and one PI, except for L1 who received a two-drug regimen of stavudine and ritonavir. “Potent HAART” was treatment with threeclasses of drugs, except for G1 who was prescribed two NRTI and two PI. The ranges of plasma HIV-1 RNA levels are indicated by vertical bars,as is the lower limit of detection of the assay used (shaded areas); a horizontal bar (—) indicates that all values were below the limit of detection.A decrease of �0.9 log10 in HIV-1 DNA in CD4� cells within 1 to 2 years of potent HAART is indicated by an asterisk. Findings from sequenceanalyses are indicated in the boxes as follows: light gray, genetic distances of env from MRCA were stable or increased during HAART; dark gray,selection of new and/or increases in established HIV-1 drug-resistant mutations; black, evolution of HIV-1 in phylogram confirmed by distancesfrom MRCA; white, decay of drug-resistant mutant prevalence or no phylogenetic evidence of viral evolution. na, not applicable due to nopre-HAART drug-resistant mutants, or not assessed. Viral replication and associated viral evolution were diminished in association with potentHAART.
VOL. 77, 2003 HIV-1 GENETIC ANALYSES DURING HAART 5725
on January 6, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from
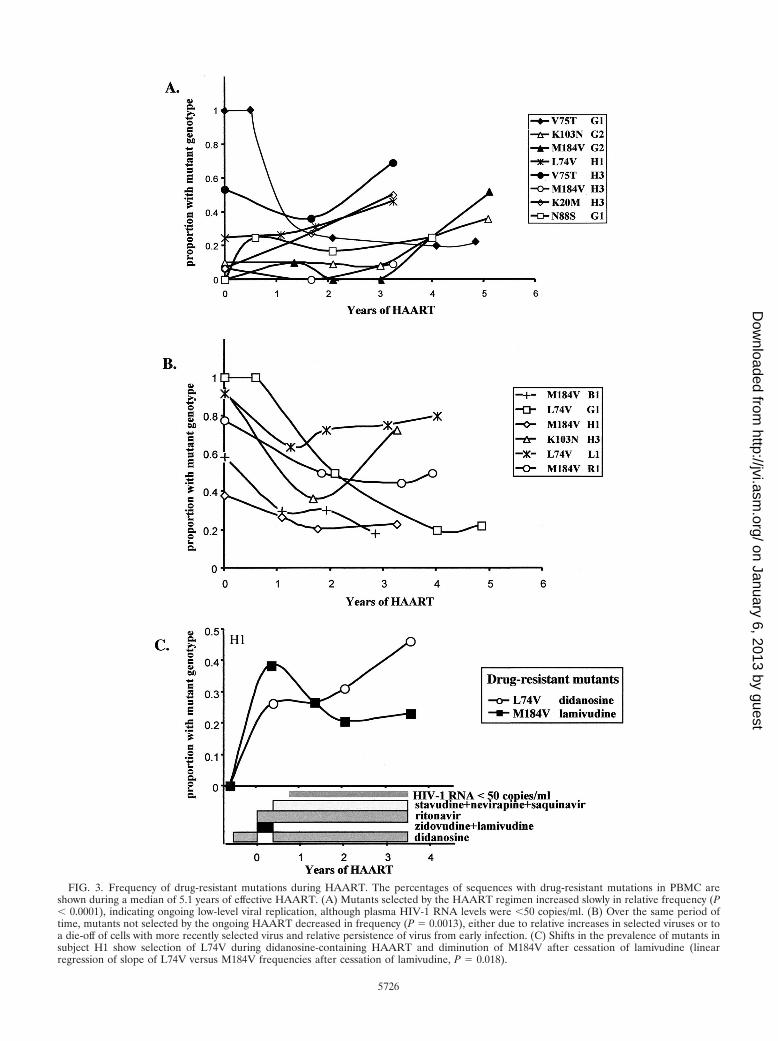
FIG. 3. Frequency of drug-resistant mutations during HAART. The percentages of sequences with drug-resistant mutations in PBMC areshown during a median of 5.1 years of effective HAART. (A) Mutants selected by the HAART regimen increased slowly in relative frequency (P� 0.0001), indicating ongoing low-level viral replication, although plasma HIV-1 RNA levels were �50 copies/ml. (B) Over the same period oftime, mutants not selected by the ongoing HAART decreased in frequency (P � 0.0013), either due to relative increases in selected viruses or toa die-off of cells with more recently selected virus and relative persistence of virus from early infection. (C) Shifts in the prevalence of mutants insubject H1 show selection of L74V during didanosine-containing HAART and diminution of M184V after cessation of lamivudine (linearregression of slope of L74V versus M184V frequencies after cessation of lamivudine, P � 0.018).
5726
on January 6, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from

replication among individuals with plasma HIV-1 RNA belowthe limits of detection. Data from others’ studies similarlysuggest that PBMC HIV-1 DNA loads correlate with markersof viral nucleic acid synthesis (2, 4, 20, 27, 37, 61), as haveintracellular viral RNA levels (4) and the ratio of unspliced tomultiply spliced HIV-1 mRNA (19). Decreases in total HIV-1levels associated with viral replication well-suppressed byHAART appear primarily due to decreases in unintegratedHIV-1 DNA (27). HIV-1 DNA levels have also correlatedclosely with the level of plasma RNA, grouped as �3, 3 to 50,and 50 to 200 copies/ml (61), and a strong correlation wasobserved between viral DNA and intracellular viral RNA lev-els (P � 0.005; r � 0.69) among 18 subjects with plasma HIV-1RNA levels of �200 copies/ml, including 15 with �20 cop-ies/ml (4). In contrast to our study, these data correlatingHIV-1 RNA and DNA levels do not prove that full cycles ofviral replication with infection of new cells were ongoing. Viral
particles and mRNA can be derived from provirus withoutresulting in the infection of additional cells. Thus, it wouldfollow that individuals with higher levels of provirus wouldproduce more viral RNA. Our study, by demonstrating in-creases in the proportion of viral DNA with drug resistancemutations, indicates full cycles of viral replication were ongo-ing, with HAART failing to completely suppress the infectionof additional cells.
Genetic bottlenecks affecting viral replication capacity (8,33; T. Wrin, A. Gamarnik, N. Whitehurst, J. Beauchaine, J. M.Whitcomb, N. S. Hellman, and C. J. Petropoulos, abstract fromthe 5th International Workshop on HIV Drug Resistance andTreatment Strategies 2001, Antivir. Ther. 6:20, 2001) and phar-macologic barriers (12, 40) imposed by HAART appearedcritical to limiting viral replication among the children westudied. These effects were demonstrated by the suppression ofviral replication by potent four- or five-drug HAART after
FIG. 4. Clinical history and viral sequence analyses of a subject with regressive viral evolution during potent HAART. (A) Clinical course andlaboratory values, including drug history, plasma HIV-1 RNA levels, and CD4� and CD8� cell numbers. (B) The genetic distances of thesesequences from the inferred MRCA for pol encoding PR are shown. (C) The neighbor-joining phylogenetic analysis for HIV-1 encoding PR isshown, with multiple iterations showing the virus detected at the indicated time points as filled circles and viruses from past time points as opencircles. Pre-HAART specimens demonstrated time-ordered evolution, and within 5 months of beginning three-drug HAART in 1996 a drug-resistant mutant (N88S, within box) was selected. Subsequently, and in association with the intensification of HAART, there was a gradualdiminution of viral sequences that grouped with more recent time points, except for a few drug-resistant variants (N88S) that persisted but did notevolve or increase in prevalence. After 5 years of HAART, the detection of PBMC sequences that grouped with those from earlier time pointssuggested die-off of more recently infected cells, also documented by the decrease in distance from the MRCA.
VOL. 77, 2003 HIV-1 GENETIC ANALYSES DURING HAART 5727
on January 6, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from
virologic failure of three-drug regimens. The selection of in-creasingly drug-resistant but implicitly poorly fit viruses in onesubject during nearly 4 years of potent HAART demonstratedthat the genetic barrier posed by a therapeutic regimen couldpersist for a sustained period of time. The importance of phar-macologic barriers was observed in this same subject whenepisodes of transient viremias ceased following the substitutionof lopinavir-ritonavir for saquinavir and ritonavir in his five-drug regimen. Furthermore, strengthening of the pharmaco-logic barrier by lopinavir-ritonavir was associated with a de-crease in HIV-1 DNA in his CD4� cells to an absolute level inthe range of those in the children without viral genetic evi-dence of replication. While we suspect that the replicationcapacity of his drug-resistant mutants was impaired, this wasnot assessed in vitro (52; Wrin et al., Antivir. Ther. 6:20, 2001),
and thus we did not estimate the relative contribution of thegenetic and pharmacokinetic barriers of his HAART on thesuppression of viral replication.
In the one case evaluated, new drug-resistant mutants weredetected in PBMC DNA prior to rebound of plasma viral RNA.This finding suggests that monitoring virus in PBMC by sensitiveassays (3, 13, 47, 53, 56, 58) may have clinical utility in forecastingviral rebound.
Among our subjects with 5 to 6 years of profound suppres-sion of viral replication, the persisting PBMC viral genotypeswere similar to virus detected early in the course of HIV-1infection. The abundance of early genotypes in the persistingviral population most likely results from a relatively greaternumber of long-lived cells becoming infected during the periodof acute infection compared to similar time intervals during
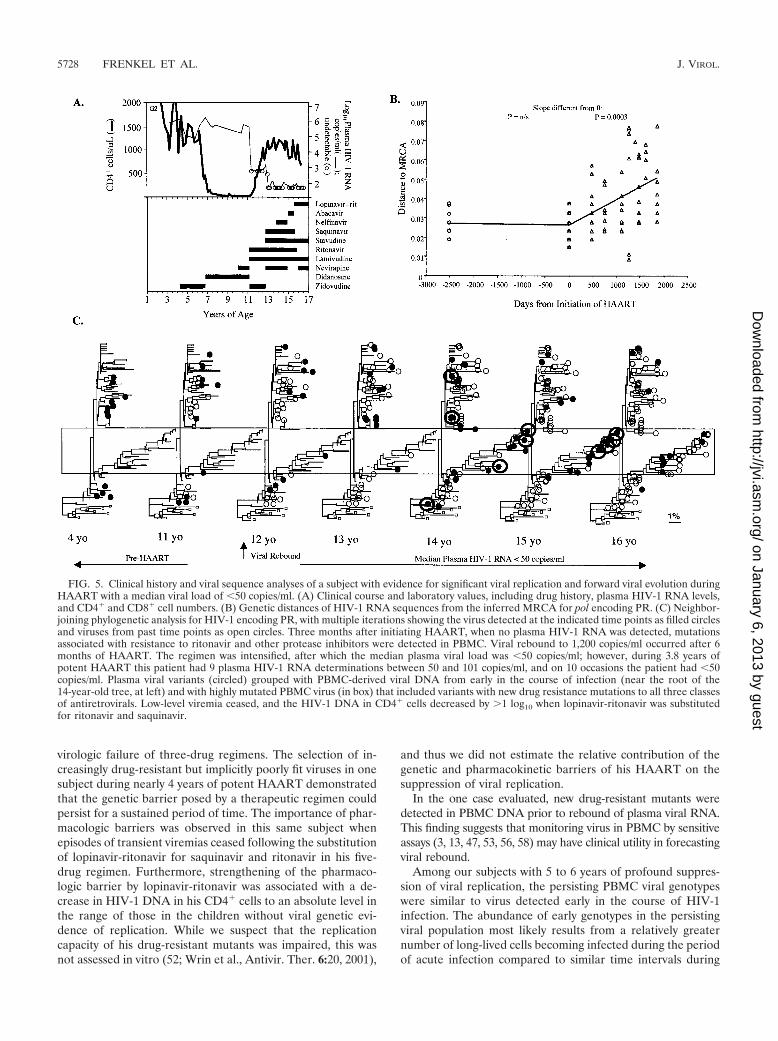
FIG. 5. Clinical history and viral sequence analyses of a subject with evidence for significant viral replication and forward viral evolution duringHAART with a median viral load of �50 copies/ml. (A) Clinical course and laboratory values, including drug history, plasma HIV-1 RNA levels,and CD4� and CD8� cell numbers. (B) Genetic distances of HIV-1 RNA sequences from the inferred MRCA for pol encoding PR. (C) Neighbor-joining phylogenetic analysis for HIV-1 encoding PR, with multiple iterations showing the virus detected at the indicated time points as filled circlesand viruses from past time points as open circles. Three months after initiating HAART, when no plasma HIV-1 RNA was detected, mutationsassociated with resistance to ritonavir and other protease inhibitors were detected in PBMC. Viral rebound to 1,200 copies/ml occurred after 6months of HAART. The regimen was intensified, after which the median plasma viral load was �50 copies/ml; however, during 3.8 years ofpotent HAART this patient had 9 plasma HIV-1 RNA determinations between 50 and 101 copies/ml, and on 10 occasions the patient had �50copies/ml. Plasma viral variants (circled) grouped with PBMC-derived viral DNA from early in the course of infection (near the root of the14-year-old tree, at left) and with highly mutated PBMC virus (in box) that included variants with new drug resistance mutations to all three classesof antiretrovirals. Low-level viremia ceased, and the HIV-1 DNA in CD4� cells decreased by �1 log10 when lopinavir-ritonavir was substitutedfor ritonavir and saquinavir.
5728 FRENKEL ET AL. J. VIROL.
on January 6, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from

later stages of disease. The persisting early viral populationappeared to include replication-competent virus, since HIV-1genotypes typical of early infection were detected in theplasma of two children we studied and by others studyinglatent cellular and low-level virus in the blood (23, 46). Impor-tantly, PBMC containing drug-resistant mutants, while de-creasing in relative frequency during HAART, persisted at lowlevels in all individuals for years following the cessation of theselecting drug, as others have observed (23, 46). The relativeloss of more recently infected PBMC and the persistence ofvirus from early infection may explain why plasma viremiaduring effective HAART is often genetically similar to archivalvirus (23) and why the viral phenotype has been observed torevert from X4/syncytium- to R5/non-syncytium-inducing virusduring HAART (14). However, immune reconstitution andreturn to a more healthful cytokine milieu could also limit thereplication of X4/syncytium-inducing viruses.
In summary, analysis of viral population genetics over amedian of 5.1 years of effective HAART detected ongoingreplication in a significant subset of children, with the mostsensitive indicators being a selection of new or an increasingfrequency of preexisting drug-resistant mutants in PBMC andmaintenance of the mean genetic distance from the MRCA ofinfection. Among children with ongoing selection of drug-re-sistant mutants documented, viral rebound appeared to belimited by potent four- and five-drug HAART regimens, pre-sumably by selection of viruses with impaired replication ca-pacity. Furthermore, subjects without genetic evidence of viralreplication had a greater persistence of viral genotypes typicalof early infection compared to recently evolved viral se-quences. In addition, their HIV-1 DNA levels decreased mark-edly with HAART and to lower absolute levels compared tosubjects with ongoing viral replication, providing a rationalefor studies evaluating whether HIV-1 DNA levels predict sub-sequent virologic failure among subjects with plasma HIV-1RNA levels of �50 copies/ml.
ACKNOWLEDGMENTS
This project was supported by U.S. Public Health Service grants, theUniversity of Washington Center for AIDS Research, and the FosterFoundation.
REFERENCES
1. Akaike, H. 1974. A new look at statistical model identification. IEEE Trans.Autom. Contr. 19:716–723.
2. Andreoni, M., L. Sarmati, L. Ercoli, E. Nicastri, G. Giannini, C. Galluzzo,M. F. Pirillo, and S. Vella. 1997. Correlation between changes in plasma HIVRNA levels and in plasma infectivity in response to antiretroviral therapy.AIDS Res. Hum. Retrovir. 13:555–561.
3. Beck, I. A., M. Mahalanabis, G. Pepper, A. Wright, S. Hamilton, E. Lang-ston, and L. M. Frenkel. 2002. A rapid and sensitive oligonucleotide ligationassay for the detection of mutations in human immunodeficiency virus type1 associated with high-level resistance to protease inhibitors. J. Clin. Micro-biol. 40:1413–1419.
4. Burgard, M., J. Izopet, B. Dumon, C. Tamalet, D. Descamps, A. Ruffault, A.Vabret, G. Bargues, M. Mouroux, I. Pellegrin, S. Ivanoff, O. Guisthau, V.Calvez, J. M. Seigneurin, and C. Rouzioux. 2000. HIV RNA and HIV DNAin peripheral blood mononuclear cells are consistent markers for estimatingviral load in patients undergoing long-term potent treatment. AIDS Res.Hum. Retrovir. 16:1939–1947.
5. Chun, T. W., L. Stuyver, S. B. Mizell, L. A. Ehler, J. A. Mican, M. Baseler,A. L. Lloyd, M. A. Nowak, and A. S. Fauci. 1997. Presence of an inducibleHIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl.Acad. Sci. USA 94:13193–13197.
6. Deeks, S. G. 2001. Durable HIV treatment benefit despite low-level viremia:reassessing definitions of success or failure. JAMA 286:224–226.
7. Deeks, S. G., J. D. Barbour, R. M. Grant, and J. N. Martin. 2002. Durationand predictors of CD4 T-cell gains in patients who continue combinationtherapy despite detectable plasma viremia. AIDS 16:201–207.
8. Deeks, S. G., T. Wrin, T. Liegler, R. Hoh, M. Hayden, J. D. Barbour, N. S.Hellman, C. J. Petropoulos, J. M. McCune, M. Hellerstein, and R. M. Grant.2001. Virologic and immunologic consequences of discontinuing combina-tion antiretroviral drug therapy in HIV-infected patients with detectableviremia. N. Engl. J. Med. 344:472–480.
9. Delwart, E. L., E. G. Shpaer, F. E. McCutchan, J. Louwagie, M. Grez, H.Rubsamen-Waigmann, and J. I. Mullins. 1993. Genetic relationships deter-mined by a DNA heteroduplex mobility assay: analysis of HIV-1 env genes.Science 262:1257–1261.
10. Diggle, P. J., K. Y. Liang, and S. L. Zeger. 1994. Analysis of longitudinaldata. Clarendon Press, Oxford, United Kingdom.
11. Dornadula, G., H. Zhang, B. VanUitert, J. Stern, L. Livornese, Jr., M. J.Ingerman, J. Witek, R. J. Kedanis, J. Natkin, J. DeSimone, and R. J.Pomerantz. 1999. Residual HIV-1 RNA in blood plasma of patients takingsuppressive highly active antiretroviral therapy. JAMA 282:1627–1632.
12. Durant, J., P. Clevenbergh, R. Garraffo, P. Halfon, S. Icard, P. Del Giudice,N. Montagne, J. M. Schapiro, and P. Dellamonica. 2000. Importance ofprotease inhibitor plasma levels in HIV-infected patients treated with geno-typic-guided therapy: pharmacological data from the Viradapt Study. AIDS14:1333–1339.
13. Edelstein, R. E., D. A. Nickerson, V. O. Tobe, L. A. Manns-Arcuino, andL. M. Frenkel. 1998. Oligonucleotide ligation assay for detecting mutationsin the human immunodeficiency virus type 1 pol gene that are associated withresistance to zidovudine, didanosine, and lamivudine. J. Clin. Microbiol.36:569–572.
14. Equils, O., E. Garratty, L. S. Wei, S. Plaeger, M. Tapia, J. Deville, P.Krogstad, M. S. Sim, K. Nielsen, and Y. J. Bryson. 2000. Recovery ofreplication-competent virus from CD4 T cell reservoirs and change in core-ceptor use in human immunodeficiency virus type 1-infected children re-sponding to highly active antiretroviral therapy. J. Infect. Dis. 182:751–757.
15. Felsenstein, J. 1985. Confidence limits on phylogenies: an approach usingthe bootstrap. Evolution 39:783–791.
16. Finzi, D., M. Hermankova, T. Pierson, L. M. Carruth, C. Buck, R. E.Chaisson, T. C. Quinn, K. Chadwick, J. Margolick, R. Brookmeyer, J. Gal-lant, M. Markowitz, D. D. Ho, D. D. Richman, and R. F. Siliciano. 1997.Identification of a reservoir for HIV-1 in patients on highly active antiret-roviral therapy. Science 278:1295–1300.
17. Frenkel, L. M., J. I. Mullins, G. H. Learn, L. Manns-Arcuino, B. L. Herring,M. L. Kalish, R. W. Steketee, D. M. Thea, J. E. Nichols, S.-L. Liu, A.Harmache, X. He, D. Muthui, A. Madan, L. Hood, A. T. Haase, M. Zupancic,K. Staskus, S. M. Wolinsky, P. Krogstad, J. Zhao, I. Chen, R. Koup, D. D.Ho, B. T. Korber, R. J. Apple, R. W. Coombs, S. Pahwa, and N. J. J. Roberts.1998. Genetic evaluation of suspected cases of transient HIV-1 infection ofinfants. Science 280:1073–1077.
18. Frenkel, L. M., L. E. Wagner II, S. M. Atwood, T. J. Cummins, and S.Dewhurst. 1995. Specific, sensitive, and rapid assay for human immunode-ficiency virus type 1 pol mutations associated with resistance to zidovudineand didanosine. J. Clin. Microbiol. 33:342–347.
19. Furtado, M. R., D. S. Callaway, J. P. Phair, K. J. Kunstman, J. L. Stanton,C. A. Macken, A. S. Perelson, and S. M. Wolinsky. 1999. Persistence ofHIV-1 transcription in peripheral-blood mononuclear cells in patients re-ceiving potent antiretroviral therapy. N. Engl. J. Med. 340:1614–1622.
20. Garrigue, I., I. Pellegrin, B. Hoen, B. Dumon, M. Harzic, M. H. Schrive, D.Sereni, and H. Fleury. 2000. Cell-associated HIV-1-DNA quantitation afterhighly active antiretroviral therapy-treated primary infection in patients withpersistently undetectable plasma HIV-1 RNA. AIDS 14:2851–2855.
21. Gunthard, H. F., S. D. Frost, A. J. Leigh-Brown, C. C. Ignacio, K. Kee, A. S.Perelson, C. A. Spina, D. V. Havlir, M. Hezareh, D. J. Looney, D. D. Rich-man, and J. K. Wong. 1999. Evolution of envelope sequences of humanimmunodeficiency virus type 1 in cellular reservoirs in the setting of potentantiviral therapy. J. Virol. 73:9404–9412.
22. Hammer, S. M., K. E. Squires, M. D. Hughes, J. M. Grimes, L. M. Demeter,J. S. Currier, J. J. Eron, Jr., J. E. Feinberg, H. H. Balfour, Jr., L. R. Deyton,J. A. Chodakewitz, and M. A. Fischl. 1997. A controlled trial of two nucle-oside analogues plus indinavir in persons with human immunodeficiencyvirus infection and CD4 cell counts of 200 per cubic millimeter or less.N. Engl. J. Med. 337:725–733.
23. Hermankova, M., S. C. Ray, C. Ruff, M. Powell-Davis, R. Ingersoll, R. T.D’Aquila, T. C. Quinn, J. D. Siliciano, R. F. Siliciano, and D. Persaud. 2001.HIV-1 drug resistance profiles in children and adults with viral load of �50copies/ml receiving combination therapy. JAMA 286:196–207.
24. Hirsch, V. M. 1987. Structure of the simian immunodeficiency virus and itsrelationship to human immunodeficiency viruses. Ph.D. dissertation. Har-vard University, Cambridge, Mass.
25. Hockett, R. D., J. M. Kilby, C. A. Derdeyn, M. S. Saag, M. Sillers, K. Squires,S. Chiz, M. A. Nowak, G. M. Shaw, and R. P. Bucy. 1999. Constant meanviral copy number per infected cell in tissues regardless of high, low, orundetectable plasma HIV RNA. J. Exp. Med. 189:1545–1554.
26. Hogg, R. S., B. Yip, K. J. Chan, E. Wood, K. J. Craib, M. V. O’Shaughnessy, and
VOL. 77, 2003 HIV-1 GENETIC ANALYSES DURING HAART 5729
on January 6, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from

J. S. Montaner. 2001. Rates of disease progression by baseline CD4 cell countand viral load after initiating triple-drug therapy. JAMA 286:2568–2577.
27. Izopet, J., M. Cazabat, C. Pasquier, K. Sandres-Saune, E. Bonnet, B. Mar-chou, P. Massip, and J. Puel. 2002. Evolution of total and integrated HIV-1DNA and change in DNA sequences in patients with sustained plasma virussuppression. Virology 302:393–404.
28. Korber, B. T., and P. Rose. 1999. HYPERMUT. Los Alamos NationalLaboratory, Los Alamos, N.Mex.
29. Krogstad, P., S. Lee, G. Johnson, K. Stanley, J. McNamara, J. Moye, J. B.Jackson, R. Aguayo, A. Dieudonne, M. Khoury, H. Mendez, S. Nachman,and A. Wiznia. 2002. Nucleoside-analogue reverse-transcriptase inhibitorsplus nevirapine, nelfinavir, or ritonavir for pretreated children infected withhuman immunodeficiency virus type 1. Clin. Infect. Dis. 34:991–1001.
30. Learn, G. H., D. Muthui, S. J. Brodie, T. Zhu, K. Diem, J. I. Mullins, and L.Corey. 2002. Virus population homogenization following acute human im-munodeficiency virus type 1 infection. J. Virol. 76:11953–11959.
31. Leitner, T., S. Kumar, and J. Albert. 1997. Tempo and mode of nucleotidesubstitutions in gag and env gene fragments in human immunodeficiency virustype 1 populations with a known transmission history. J. Virol. 71:4761–4770.
32. Martinez-Picado, J., M. P. DePasquale, N. Kartsonis, G. J. Hanna, J. Wong,D. Finzi, E. Rosenberg, H. F. Gunthard, L. Sutton, A. Savara, C. J. Petro-poulos, N. Hellmann, B. D. Walker, D. D. Richman, R. Siliciano, and R. T.D’Aquila. 2000. Antiretroviral resistance during successful therapy of HIVtype 1 infection. Proc. Natl. Acad. Sci. USA 97:10948–10953.
33. Martinez-Picado, J., A. V. Savara, L. Sutton, and R. T. D’Aquila. 1999.Replicative fitness of protease inhibitor-resistant mutants of human immu-nodeficiency virus type 1. J. Virol. 73:3744–3752.
34. Melvin, A. J., K. M. Mohan, L. A. Arcuino, R. E. Edelstein, and L. M.Frenkel. 1997. Clinical, virologic and immunologic responses of children withadvanced human immunodeficiency virus type 1 disease treated with pro-tease inhibitors. Pediatr. Infect. Dis. J. 16:968–974.
35. Montaner, J. S., P. Reiss, D. Cooper, S. Vella, M. Harris, B. Conway, M. A.Wainberg, D. Smith, P. Robinson, D. Hall, M. Myers, and J. M. Lange. 1998.A randomized, double-blind trial comparing combinations of nevirapine,didanosine, and zidovudine for HIV-infected patients: the INCAS trial, Italy,The Netherlands, Canada and Australia Study. JAMA 279:930–937.
36. Natarajan, V., M. Bosche, J. A. Metcalf, D. J. Ward, H. C. Lane, and J. A.Kovacs. 1999. HIV-1 replication in patients with undetectable plasma virusreceiving HAART, highly active antiretroviral therapy. Lancet 353:119–120.
37. Ngo-Giang-Huong, N., C. Deveau, I. Da Silva, I. Pellegrin, A. Venet, M.Harzic, M. Sinet, J. F. Delfraissy, L. Meyer, C. Goujard, and C. Rouzioux.2001. Proviral HIV-1 DNA in subjects followed since primary HIV-1 infec-tion who suppress plasma viral load after one year of highly active antiret-roviral therapy. AIDS 15:665–673.
38. Paediatric European Network for Treatment of AIDS. 2002. Comparison ofdual nucleoside-analogue reverse-transcriptase inhibitor regimens with andwithout nelfinavir in children with HIV-1 who have not previously beentreated: the PENTA 5 randomised trial. Lancet 359:733–740.
39. Paterson, D. L., S. Swindells, J. Mohr, M. Brester, E. N. Vergis, C. Squier,M. M. Wagener, and N. Singh. 2000. Adherence to protease inhibitor therapyand outcomes in patients with HIV infection. Ann. Intern. Med. 133:21–30.
40. Perno, C. F., F. M. Newcomb, D. A. Davis, S. Aquaro, R. W. Humphrey, R.Calio, and R. Yarchoan. 1998. Relative potency of protease inhibitors inmonocytes/macrophages acutely and chronically infected with human immu-nodeficiency virus. J. Infect. Dis. 178:413–422.
41. Phillips, A. N., S. Staszewski, R. Weber, O. Kirk, P. Francioli, V. Miller, P.Vernazza, J. D. Lundgren, and B. Ledergerber. 2001. HIV viral load re-sponse to antiretroviral therapy according to the baseline CD4 cell count andviral load. JAMA 286:2560–2567.
42. Posada, D., and K. A. Crandall. 1998. MODELTEST: testing the model ofDNA substitution. Bioinformatics 14:817–818.
43. Ramratnam, B., J. E. Mittler, L. Zhang, D. Boden, A. Hurley, F. Fang, C. A.Macken, A. S. Perelson, M. Markowitz, and D. D. Ho. 2000. The decay of thelatent reservoir of replication-competent HIV-1 is inversely correlated withthe extent of residual viral replication during prolonged anti-retroviral ther-apy. Nat. Med. 6:82–85.
44. Rodrigo, A. G., P. C. Goracke, K. Rowhanian, and J. I. Mullins. 1997.Quantitation of target molecules from polymerase chain reaction-based lim-iting dilution assays. AIDS Res. Hum. Retrovir. 13:737–742.
45. Rodrigo, A. G., E. W. Hanley, P. C. Goracke, and G. H. Learn. 2001.Sampling and processing HIV molecular sequences: a computational evolu-tionary biologist’s perspective, p. 1–17. In A. G. Rodrigo and G. H. Learn(ed.), Computational and evolutionary analyses of HIV sequences. KluwerAcademic Publishers, Boston, Mass.
46. Ruff, C. T., S. C. Ray, P. Kwon, R. Zinn, A. Pendleton, N. Hutton, R.Ashworth, S. Gange, T. C. Quinn, R. F. Siliciano, and D. Persaud. 2002.Persistence of wild-type virus and lack of temporal structure in the latent
reservoir for human immunodeficiency virus type 1 in pediatric patients withextensive antiretroviral exposure. J. Virol. 76:9481–9492.
47. Schmit, J. C., L. Ruiz, L. Stuyver, K. Van Laethem, I. Vanderlinden, T. Puig,R. Rossau, J. Desmyter, E. De Clerg, B. Clotet, and A. M. Vandamme. 1998.Comparison of the LiPA HIV-1 RT test, selective PCR and direct solidphase sequencing for the detection of HIV-1 drug resistance mutations.J. Virol. Methods 73:77–82.
48. Sharkey, M. E., I. Teo, T. Greenough, N. Sharova, K. Luzuriaga, J. L.Sullivan, R. P. Bucy, L. G. Kostrikis, A. Haase, C. Veryard, R. E. Davaro,S. H. Cheeseman, J. S. Daly, C. Bova, R. T. Ellison III, B. Mady, K. K. Lai,G. Moyle, M. Nelson, B. Gazzard, S. Shaunak, and M. Stevenson. 2000.Persistence of episomal HIV-1 infection intermediates in patients on highlyactive anti-retroviral therapy. Nat. Med. 6:76–81.
49. Skowron, G., J. C. Street, and E. M. Obee. 2001. Baseline CD4� cell count,not viral load, correlates with virologic suppression induced by potent anti-retroviral therapy. J. Acquir. Immune Defic. Syndr. 28:313–319.
50. Starr, S. E., C. V. Fletcher, S. A. Spector, F. H. Yong, T. Fenton, R. C.Brundage, D. Manion, N. Ruiz, M. Gersten, M. Becker, J. McNamara, L. M.Mofenson, L. Purdue, S. Siminski, B. Graham, D. M. Kornhauser, W. Fiske,C. Vincent, H. W. Lischner, W. M. Dankner, and P. M. Flynn. 1999. Com-bination therapy with efavirenz, nelfinavir, and nucleoside reverse-transcrip-tase inhibitors in children infected with human immunodeficiency virus type1. N. Engl. J. Med. 341:1874–1881.
51. Staszewski, S., J. Morales-Ramirez, K. T. Tashima, A. Rachlis, D. Skiest, J.Stanford, R. Stryker, P. Johnson, D. F. Labriola, D. Farina, D. J. Manion,and N. M. Ruizm. 1999. Efavirenz plus zidovudine and lamivudine, efavirenzplus indinavir, and indinavir plus zidovudine and lamivudine in the treatmentof HIV-1 infection in adults. N. Engl. J. Med. 341:1865–1873.
52. Stoddart, C. A., T. J. Liegler, F. Mammano, V. D. Linquist-Stepps, M. S.Hayden, S. G. Deeks, R. M. Grant, F. Clavel, and J. M. McCune. 2001.Impaired replication of protease inhibitor-resistant HIV-1 in human thymus.Nat. Med. 7:712–718.
53. Stuyver, L., A. Wyseur, A. Rombout, J. Louwagie, T. Scarcez, C. Verhofstede, D.Rimland, R. Schinazi, and R. Rossau. 1997. Line probe assay for rapid detectionof drug-selected mutations in the human immunodeficiency virus type 1 reversetranscriptase gene. Antimicrob. Agents Chemother. 41:284–291.
54. Swofford, D. L. 1999. PAUP 4.0: phylogenetic analysis using parsimony (andother methods), ed. 4.0b2a. Sinauer Associates, Inc., Sunderland, Mass.
55. Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W:improving the sensitivity of progressive multiple sequence alignment throughsequence weighting, position-specific gap penalties and weight matrix choice.Nucleic Acids Res. 22:4673–4680.
56. Van Laethem, K., K. Van Vaerenbergh, J.-C. Schmit, S. Sprecher, P. Her-mans, V. De Vroey, R. Schuurman, T. Harrer, M. Witvrouw, E. Van Wijn-gaerden, L. Stuyver, M. Van Ranst, J. Desmyter, E. De Clercq, and A.-M.Vandamme. 1999. Phenotypic assays and sequencing are less sensitive thanpoint mutation assays for detection of resistance in mixed HIV-1 genotypicpopulations. J. Acquir. Immune Defic. Syndr. 22:107–118.
57. Vartanian, J.-P., A. Meyerhans, B. Asjo, and S. Wain-Hobson. 1991. Selec-tion, recombination, and G3A hypermutation of human immunodeficiencyvirus type I genome. J. Virol. 65:1779–1788.
58. Villahermosa, M. L., I. Beck, L. Perez-Alvarez, G. Contreras, L. M. Frenkel,S. Osmanov, E. Vazquez de Parga, E. Delgado, N. Manjon, M. T. Cuevas,M. M. Thomson, L. Medrano, and R. Najera. 2001. Detection and quanti-fication of multiple drug resistance-associated mutation in HIV-1 reversetranscriptase by an oligonucleotide ligation assay. J. Hum. Virol. 4:238–248.
59. Wiznia, A., K. Stanley, P. Krogstad, G. Johnson, S. Lee, J. McNamara, J.Moye, J. B. Jackson, H. Mendez, R. Aguayo, A. Dieudonne, A. Kovacs, M.Bamji, E. Abrams, S. Rana, J. Sever, and S. Nachman. 2000. Combinationnucleoside analog reverse transcriptase inhibitor(s) plus nevirapine, nelfina-vir, or ritonavir in stable antiretroviral therapy-experienced HIV-infectedchildren: week 24 results of a randomized controlled trial. AIDS Res. Hum.Retrovir. 16:1113–1121.
60. Wong, J. K., M. Hezareh, H. F. Gunthard, D. V. Havlir, C. C. Ignacio, C. A.Spina, and D. D. Richman. 1997. Recovery of replication-competent HIVdespite prolonged suppression of plasma viremia. Science 278:1291–1300.
61. Yerly, S., T. V. Perneger, S. Vora, B. Hirschel, and L. Perrin. 2000. Decay ofcell-associated HIV-1 DNA correlates with residual replication in patientstreated during acute HIV-1 infection. AIDS 14:2805–2812.
62. Zack, J. A., A. M. Haislip, P. Krogstad, and I. S. Chen. 1992. Incompletelyreverse-transcribed human immunodeficiency virus type 1 genomes in qui-escent cells can function as intermediates in the retroviral life cycle. J. Virol.66:1717–1725.
63. Zhang, L., B. Ramratnam, K. Tenner-Racz, Y. He, M. Vesanen, S. Lewin, A.Talal, P. Racz, A. S. Perelson, B. T. Korber, M. Markowitz, and D. D. Ho.1999. Quantifying residual HIV-1 replication in patients receiving combina-tion antiretroviral therapy. N. Engl. J. Med. 340:1605–1613.
5730 FRENKEL ET AL. J. VIROL.
on January 6, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from




















