
MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating...
15
Molecular Immunology 43 (2006) 1129–1143 MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors Klaus Brischwein a , Bernd Schlereth a , Benjamin Guller a , Carola Steiger a , Andreas Wolf a , Ralf Lutterbuese a , Sonja Offner a , Mathias Locher a , Thomas Urbig a , Tobias Raum a , Petra Kleindienst a , Pauline Wimberger c , Rainer Kimmig c , Iduna Fichtner b , Peter Kufer a , Robert Hofmeister a , Antonio J. da Silva a,1 , Patrick A. Baeuerle a,∗ a Micromet AG, Staffelseestr. 2, 81477 Munich, Germany b Experimental Pharmacology and Oncology GmbH, Berlin-Buch, 13122 Berlin, Germany c Department of Gynecology and Obstetrics, University of Essen, 45122 Essen, Germany Received 18 May 2005 Available online 1 September 2005 Abstract We have developed a novel single-chain Ep-CAM-/CD3-bispecific single-chain antibody construct designated MT110. MT110 redirected unstimulated human peripheral T cells to induce the specific lysis of every Ep-CAM-expressing tumor cell line tested. MT110 induced a costimulation independent polyclonal activation of CD4- and CD8-positive T cells as seen by de novo expression of CD69 and CD25, and secretion of interferon gamma, tumor necrosis factor alpha, and interleukins 2, 4 and 10. CD8-positive T cells made the major contribution to redirected tumor cell lysis by MT110. With a delay, CD4-positive cells could also contribute presumably as consequence of a dramatic upregulation of granzyme B expression. MT110 was highly efficacious in a NOD/SCID mouse model with subcutaneously growing SW480 human colon cancer cells. Five daily doses of 1 g MT110 on days 0–4 completely prevented tumor outgrowth in all mice treated. The bispecific antibody construct also led to a durable eradication of established tumors in all mice treated with 1 g doses of MT110 on days 8–12 after tumor inoculation. Finally, MT110 could eradicate patient-derived metastatic ovarian cancer tissue growing under the skin of NOD/SCID mice. MT110 appears as an attractive bispecific antibody candidate for treatment of human Ep-CAM-overexpressing carcinomas. © 2005 Elsevier Ltd. All rights reserved. Keywords: Ep-CAM; CD3; BiTE; Single-chain antibody; Xenograft model; Tumor 1. Introduction The successful treatment of late-stage cancer is one of the greatest challenges in modern medicine. This is reflected by the still unchanged average mortality rate in cancer during the past decades in industrialized countries (Antunes et al., 2003; Hoel et al., 1992; Howe et al., 2001). The invasion of organs by metastasizing tumor cells, the establishment of large tumor masses that are barely accessible to therapeutics, the genetic heterogeneity of tumor cells and the evasion ∗ Corresponding author. Tel.: +49 89 895277601; fax: +49 89 895277205. E-mail address: [email protected] (P.A. Baeuerle). 1 Present address: Direvo Biotech AG, 50829 Cologne, Germany. of tumor cells from immune recognition by a multitude of escape mechanisms are major reasons why surgical, radiation and cytotoxic therapies in most cases are not curative but at best can retard disease progression. Because tumor cells share all genes with normal cells, only subtle differences to normal cells will manifest which can be exploited for targeted therapies designed to very selectively eliminate or control tumor cells. Enhanced proliferation, altered metabolism, aberrant protein expression and covalent modification, mutations and altered splicing may change tumor cell surface immunogenicity, which can potentially provide useful target structures for antibody-based therapies. However, there is always the limitation that not all tumor cells show the targeted phenotype, and not all tumor cells are 0161-5890/$ – see front matter © 2005 Elsevier Ltd. All rights reserved. doi:10.1016/j.molimm.2005.07.034
-
Upload
virginiogomez -
Category
Documents
-
view
1 -
download
0
Transcript of MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating...

Molecular Immunology 43 (2006) 1129–1143
MT110: A novel bispecific single-chain antibody construct with highefficacy in eradicating established tumors
Klaus Brischweina, Bernd Schleretha, Benjamin Gullera, Carola Steigera, Andreas Wolfa,Ralf Lutterbuesea, Sonja Offnera, Mathias Lochera, Thomas Urbiga, Tobias Rauma,
Petra Kleindiensta, Pauline Wimbergerc, Rainer Kimmigc, Iduna Fichtnerb, Peter Kufera,Robert Hofmeistera, Antonio J. da Silvaa,1, Patrick A. Baeuerlea,∗
a Micromet AG, Staffelseestr. 2, 81477 Munich, Germanyb Experimental Pharmacology and Oncology GmbH, Berlin-Buch, 13122 Berlin, Germanyc Department of Gynecology and Obstetrics, University of Essen, 45122 Essen, Germany
Received 18 May 2005Available online 1 September 2005
Abstract
redirectedu nduced ac D25, ands ontributiont dramaticu g SW480h . Theb 12a OD/SCIDm .©
K
1
gtt2olt
degical,
not
onlybe
ivelyion,alentnge
allypies.or
ls are
0d
We have developed a novel single-chain Ep-CAM-/CD3-bispecific single-chain antibody construct designated MT110. MT110nstimulated human peripheral T cells to induce the specific lysis of every Ep-CAM-expressing tumor cell line tested. MT110 iostimulation independent polyclonal activation of CD4- and CD8-positive T cells as seen by de novo expression of CD69 and Cecretion of interferon gamma, tumor necrosis factor alpha, and interleukins 2, 4 and 10. CD8-positive T cells made the major co redirected tumor cell lysis by MT110. With a delay, CD4-positive cells could also contribute presumably as consequence of apregulation of granzyme B expression. MT110 was highly efficacious in a NOD/SCID mouse model with subcutaneously growinuman colon cancer cells. Five daily doses of 1�g MT110 on days 0–4 completely prevented tumor outgrowth in all mice treatedispecific antibody construct also led to a durable eradication of established tumors in all mice treated with 1�g doses of MT110 on days 8–fter tumor inoculation. Finally, MT110 could eradicate patient-derived metastatic ovarian cancer tissue growing under the skin of Nice. MT110 appears as an attractive bispecific antibody candidate for treatment of human Ep-CAM-overexpressing carcinomas2005 Elsevier Ltd. All rights reserved.
eywords: Ep-CAM; CD3; BiTE; Single-chain antibody; Xenograft model; Tumor
. Introduction
The successful treatment of late-stage cancer is one of thereatest challenges in modern medicine. This is reflected by
he still unchanged average mortality rate in cancer duringhe past decades in industrialized countries (Antunes et al.,003; Hoel et al., 1992; Howe et al., 2001). The invasionf organs by metastasizing tumor cells, the establishment of
arge tumor masses that are barely accessible to therapeutics,he genetic heterogeneity of tumor cells and the evasion
∗ Corresponding author. Tel.: +49 89 895277601; fax: +49 89 895277205.E-mail address: [email protected] (P.A. Baeuerle).
1 Present address: Direvo Biotech AG, 50829 Cologne, Germany.
of tumor cells from immune recognition by a multituof escape mechanisms are major reasons why surradiation and cytotoxic therapies in most cases arecurative but at best can retard disease progression.
Because tumor cells share all genes with normal cells,subtle differences to normal cells will manifest which canexploited for targeted therapies designed to very selecteliminate or control tumor cells. Enhanced proliferataltered metabolism, aberrant protein expression and covmodification, mutations and altered splicing may chatumor cell surface immunogenicity, which can potentiprovide useful target structures for antibody-based theraHowever, there is always the limitation that not all tumcells show the targeted phenotype, and not all tumor cel
161-5890/$ – see front matter © 2005 Elsevier Ltd. All rights reserved.oi:10.1016/j.molimm.2005.07.034

1130 K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143
physically accessible to the targeted therapy. Recent yearshave shown that targeted tumor therapy by monoclonal anti-bodies of the human IgG1 isotype can lead in certain patientsto significant anti-tumor responses that frequently profitfrom a co-therapy with standard chemotherapy or radiation(Davis et al., 2004; Hurwitz et al., 2004; Matar et al., 2004;Pegram et al., 2004). Examples are antibodies recognizingoverexpressed epidermal growth factor receptors EGFR(cetuximab) (Khalil et al., 2003) and HER-2 (trastuzumab)(Vogel et al., 2001) used for treatment of solid tumours, anddifferentiation antigens CD20 (rituximab) (Grillo-Lopez etal., 1999; Smith, 2003), CD52 (alemtuzumab) (Faulkner etal., 2004; Hale et al., 1998; Kottaridis et al., 2000) and CD22(epratuzumab) (Furman et al., 2004; Leonard et al., 2004)used for blood-bourne cancers. Many studies have suggestedthat antibody-dependent cellular cytotoxicity (ADCC) is amajor contributor to the therapeutic efficacy of monoclonalantibody therapies (Clynes et al., 2000; Gennari et al., 2004;Maloney, 2001; Nagler et al., 1997).
ADCC is mediated via a bispecific function of IgG1.While the two antigen binding arms fix the antibody to thetumor-associated antigen, the Fc part of IgG1 allows to tran-siently tether and simultaneously activate cytotoxic immunecells bearing Fc� receptors. Most important seem Fc�RIII/CD16-expressing natural killer (NK) cells, as is evidentfrom the impact of genetic polymorphisms in CD16 on ther-a oe ntw ayb andi witht stroym
thepa t al.,2 diesb f Tc r cellsi unec tt aveb 4w esee on Tc m toa anti-b nsea poly-c lar Tc cificT gensa Onec rtiesa ucts,a E)
(Baeuerle et al., 2003; Kufer et al., 2004). A CD19/CD3-bispecific BiTE has been characterized in great detail andcurrently is in phase I clinical trials (Dreier et al., 2003, 2002;Loffler et al., 2000). Here, we report on the construction andcharacterization of a novel BiTE molecule called MT110designed for the treatment of human carcinomas.
As tumor-associated antigen for recognition by MT110,we have selected the epithelial cell adhesion molecule(Ep-CAM), which is among the best characterized targetsfor immunotherapeutic approaches including monoclonalantibody therapies and vaccination strategies (Balzar et al.,1999; Mosolits et al., 2004; Naundorf et al., 2002; Pranget al., 2005). A particular advantage of Ep-CAM is itsfrequent overexpression on almost all human carcinomas(Moldenhauer et al., 1987; Momburg et al., 1987) andlimited accessibility on normal epithelial tissues (Balzar etal., 1999). By targeting Ep-CAM, MT110 has the potentialto be effective in a wide range of human carcinomas.A functional role of Ep-CAM overexpression in tumorproliferation, invasion and migration was recently shownfor breast cancer cells (Munz et al., 2004; Osta et al., 2004),which may relate to a functional antagonism of Ep-CAMwith E-cadherin (Winter et al., 2003). The finding explainswhy breast cancer patients with tumors overexpressingEp-CAM have a poor overall survival (Spizzo et al., 2002,2004).
andh rop-e ule,b l.,2 ivoe lyu rgetr n ofC redt ighlya herr dentT elyp uta-n anc ncert
2
2E
r theg ctsw hagedV es.A ts
peutic efficacy of rituximab (Cartron et al., 2002; Dall’Ozzt al., 2004; Weng and Levy, 2003). The continuous treatmeith IgG1 for months at fairly high serum trough levels me necessary for tumor penetration of both antibody
mmune cells, a permanent decoration of tumor cellshe antibody and for the time-consuming seek-and-deechanism performed by cytotoxic immune cells.Another important class of immune cells with
otential to eradicate even large tumors are T cells (Dudleynd Rosenberg, 2003; Dudley et al., 2005; Glimcher e004), which cannot be recruited by monoclonal antiboecause they lack Fc� receptors. The increasing number oell escape mechanisms discovered in late-stage tumompressively underscores the threat this class of immells is posing to tumor cells (Foss, 2002). During the paswo decades, bispecific antibodies recruiting T cells heen developed (Baeuerle et al., 2003; Kufer et al., 200),hich may have the potential to circumvent some of thscape mechanisms. By binding with one arm to a commell signaling protein, such as CD3 and with the other artumor-associated antigen on the target cell, bispecificodies should theoretically elicit a polyclonal T cell respogainst antibody target-expressing tumor cells. Such alonal response should be largely independent of reguell recognition molecules, such as MHC class I, a specell receptor, costimulatory proteins and peptide anti
long with their processing and transport molecules.lass of bispecific antibodies with very suitable propere bispecific CD3-specific single-chain antibody constrlso referred to as ‘bispecific T cell engager’ (BiT
The present study shows that MT110 is a stableomogenous protein that shares all particular BiTE prties with another previously described BiTE molecscCD19× CD3 (Dreier et al., 2003, 2002; Loffler et a000). These properties include high in vitro and in vfficacy, and induction of lytic activity with previousnstimulated peripheral T cells at low effector-to-taatios. Here, we also demonstrate polyclonal activatioD8- and CD4-positive T cells by MT110, which requi
he presence of target-expressing cells. MT110 was hctive in two NOD/SCID mouse efficacy models. By eitedirecting co-administered human T cells or tumor-resi
cells, low micrograms doses of MT110 completrevented tumor outgrowth or led to eradication of subceously growing solid tumors derived from a SW480 humancer line and from human metastatic ovarian caissue.
. Materials and methods
.1. Selection of MT110 from a panel ofp-CAM-specific BiTE constructs
EpCAM-specific single-chain antibodies used here foeneration of a panel of Ep-CAM-specific BiTE construere either isolated as single-chain antibodies from pisplay libraries (Raum et al., 2001), or constructed fromH and VL domains cloned from various hybridoma lintotal of 21 different Ep-CAM-specific BiTE construc

K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143 1131
were made, expressed, purified and compared for a set ofspecifications deemed important for development of a highlyactive biological drug (see Section3).
All candidates shared the same CD3�-specific single-chain antibody derived from mouse monoclonal antibodyL2K, which is distinct from monoclonal antibodies TR66and OKT3. L2K was subjected to a procedure that identifieshuman T helper cell epitopes and removes by point mutationsMHC class II anchor amino acids within potential epitopes(Jones et al., 2004). This resulted in “de-immunized” L2K(diL2K), which was used to construct MT110 and the major-ity of other BiTEs tested.
2.2. Expression and purification of MT110
MT110 and the other single-chain bispecific antibodieswere constructed by standard DNA technologies. The con-structs were cloned into the expression vector pEF-DHFR.Chinese hamster ovary (CHO) cells were transfected withthe expression vectors and stable transfectants selected aspreviously described (Loffler et al., 2000). For production,transfected cells were grown for 7 days in roller bottles withmodified DMEM medium (HyQ, Perbio Science, Bonn, Ger-many), removed by centrifugation and the supernatant storedat −20◦C. Secreted proteins were enriched from culturesupernatants by immobilized metal affinity chromatography( n( fac-t fferA ures mL)w hedw nts.B ufferB e).E ona Ger-m ny).T rmi-n WG de-c E)a GE,t asr o-g ngec am,F ffera iento 60c cingc en,K roto-c arkp col-l ere
determined using protein assay dye (MicroBCA, Perbio Sci-ence, Bonn Germany) and IgG (Biorad, Munich, Germany)as standard protein. All chemicals were of research grade andpurchased from Sigma (Taufkirchen, Germany) or Merck(Darmstadt, Germany).Akta FPLC System and UnicornSoftware (Amersham, Freiburg, Germany) were used forchromatography.
2.3. Cell lines and culture
The Kato III human gastric carcinoma cell line wasobtained from the European Collection of Cell Cultures(ECACC, Salisbury, UK). The MCF-7, MDA-MB-453 andSK-BR-3 breast cancer cell lines were obtained from theGerman Collection of Microorganisms and Cell Cultures(DSMZ, Braunschweig, Germany). CHO dhfr- and NALM-6cells were also obtained from that source. CHO lines stablyexpressing human Ep-CAM or CD19 were generated bytransfecting cells with plasmids containing the respectivecDNAs. Selection of clones and amplification of expressionwas performed in the presence of methotrexate. Cellswere cultured in RPMI medium supplemented with 10%fetal bovine serum (FBS) at 37◦C in a 5% CO2 chamber.The medium for transfected cell lines was additionallysupplemented with 100 nM methotrexate.
2
t tod facea ofb IT( ro-v sity.B hed.5 e indfs ndi onl ed in1 anI at2 andr ofc liburi Then theb is ofe rismV
2
owc cells
IMAC). IMAC was performed using a Fractogel columMerck, Darmstadt, Germany) according to the manuurers protocol. The column was equilibrated with bu2 (20 mM NaPP pH 7.5, 0.4 M NaCl) and the cell cultupernatant (500 mL) was applied to the column (10ith a flow rate of 3 mL/min. The column was then wasith buffer A2 to remove unbound protein contaminaound protein was eluted using a two-step gradient of b2 (20 mM NaPP, pH 7.5, 0.4 M NaCl, 0.5 M imidazolluted protein fractions were then pooled for gel filtrationSephadex S200 HiPrep column (Amersham, Freiburg,any) equilibrated with PBS (Gibco, Karlsruhe, Germahe column was calibrated for molecular weight deteation using a molecular weight marker kit (Sigma MF-200). Column fractions were subjected to lithium do
yl sulfate (LDS) polyacrylamide gel electrophoresis (PAGnd Western blot for detection. As estimated by LDS-PA
he purity of MT110 preparations after gel filtration woutinely >95%. Final purification of the protein to homeneity was achieved by high-resolution cation exchahromatography using a MiniS column (Amershreiburg, Germany), equilibrated with 20 mM MES but pH 5.5. Bound protein was eluted with a shallow gradf equilibration buffer containing 0–30% of 1 M NaCl inolumn volumes. LDS-PAGE was performed under reduonditions with pre-cast 4–12% Bis-Tris gels (Invitrogarlsruhe, Germany) according to the manufacturer’s pol. The molecular weight was determined with MultiMrotein standard (Invitrogen). Gels were stained with
oidal Coomassie (Invitrogen). Protein concentrations w
.4. Saturation binding assays
Saturation binding experiments were carried ouetermine equilibrium binding constants and cell surntigen density. Quantification of absolute numberinding sites per cell made use of the DAKO QIFIKDakoCytomation GmbH, Hamburg, Germany) which pides calibration beads for determination of antigen denriefly, subconfluent cells were trypsinized and was× 104 cells per well were then added to a 96-well platuplicate. After centrifugation of culture plates at 300× g
or 5 min, cells were re-suspended in 50�L of a three-folderial dilution of primary antibody in FACS buffer ancubated at 2–8◦C for 45 min. To quantify target expressievels, cells and calibration beads were re-suspend00�L of the appropriate secondary antibody (anti-hum
gG FITC or anti-mouse IgG FITC). After incubation–8◦C for 45 min, samples were washed three timese-suspended in 200�L of FACS buffer. Surface stainingells was analyzed by flow cytometry using a FacsCanstrument (Becton Dickinson, Heidelberg, Germany).umber of surface bindings sites was determined fromead calibration curve. Non-linear regression analysquilibrium binding curves was done with GraphPad Persion 3.0 (GraphPad Software, San Diego, CA).
.5. Cytotoxicity assays and immunostaining
Redirected cellular cytotoxicity was assayed by flytometry using human peripheral blood mononuclear

1132 K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143
(PBMCs) as effector cells and various Ep-CAM-positivehuman carcinoma cell lines or transfected CHO cells as tar-gets. PBMC were isolated from healthy donors by ficolldensity gradient centrifugation using standard procedures.Briefly, after centrifugation, cells were incubated in 10 mLof erythrocyte lysis buffer at room temperature for 10 min,washed with PBS, resuspended in RPMI-1640 completemedium and the cell number adjusted to 4× 106 cells/mL.Target cells were stained with PKH26 fluorescent mem-brane dye according to the manufacturer’s instructions(Sigma–Aldrich GmbH, Taufkirchen, Germany). Alterna-tively, Ep-CAM-positive target cells were identified byimmunostaining with a human Ep-CAM-specific mousemonoclonal antibody (EBA-1; BD Bioscience, Heidelberg,Germany) immediately prior to analysis. Cell number wasadjusted to 8× 105 cells/mL in RPMI complete medium.Equal volumes of target and effector cell suspension werethen mixed and 50�L of this suspension transferred to eachof a 96-well round bottom plate. Fifty microlitres of a MT110serial dilution or of RPMI complete medium as a negativecontrol were then added. Unless otherwise indicated, plateswere incubated for 16–20 h at 37◦C in a 5% CO2 humidi-fied incubator. Staining of cell surface markers was carriedout using directly conjugated mAbs against Ep-CAM (EBA-1), CD4 (RPA-T4), CD8 (RPA-T8), CD69 (FN50), CD25(M-A251) and CD3 (UCHT-1) (all obtained from BD Bio-s ce inF t4 toa edb tonD ter-m . Alli is ofc etern sion3
2ip
e-p manC an-u y ofp nal-y cellsw ceH anti-g bH,H wasd tru-m inea per-f ead
Array (CBA) kit following the manufacturer’s instructions(BD Bioscience Heidelberg, Germany). T cell proliferationduring incubation of PBMC with MT110 and target cells wasdetermined as increase of the respective cell count relative toa fixed number of beads (CaliBrite3, BD Bioscience, Heidel-berg, Germany) added to each sample immediately prior toFACS analysis.
2.7. SW480 human colon carcinoma xenograft models
All animal experiments in human xenograft models wereperformed in NOD/SCID mice characterized by T-, B-,NK-cell deficiency and lack of macrophage function (Jack-son Lab., Bar Harbor, USA). The mice were maintainedunder sterile and standardized environmental conditions(20± 1◦C room temperature, 50± 10% relative humidity,12 h light:12 h dark-rhythm). They received autoclaved foodand bedding (ssniff, Soest, Germany) and acidified (pH 4.0)drinking water ad libitum. They were tested for leakiness andonly mice with IgG levels below 100 ng/mL were used. Allexperiments were performed according to the German Ani-mal Protection Law with permission from the responsiblelocal authorities.
Human effector cells were isolated from whole bloodsamples of healthy donors. Peripheral blood mononuclearcells (PBMC) were prepared by Ficoll density centrifugation.E cytele ytesra er ofC asd D4,C erg,Gc ofaa ndS rightfl ni-m spe-c thet l, sixa gle-c BS)1 seso at 4c umorm t day0 hens meso utived with ac mesc ore
cience, Heidelberg, Germany). Cells were washed onACS buffer (PBS, 1% FCS, 0.02% NaN3) and incubated a◦C in 50�L for 30 min. Propidium iodide (PI) was addedfinal concentration of 1�g/mL and samples were analyzy flow cytometry on a FACSCalibur instrument (Becickinson, Heidelberg, Germany). Target cell lysis was deined as the percentage of cells staining positive by PI
ncubations were performed in duplicate. Data analysytotoxicity assays was performed using a four paramon-linear fit model integrated into GraphPad Prism Ver.0 (GraphPad Software, San Diego, CA).
.6. Purification of immune cell subpopulations,ntracellular staining and assessment of T cellroliferation
CD8+ and CD4+ T cells were enriched from freshly prared human PBMC by immunodepletion using the HuD4 and CD8 Subset Column Kit as specified by the mfacturer (R&D Systems, Wiesbaden, Germany). Puritreparations was typically >80% as confirmed by FACS asis. For analysis of granzyme B cytoplasmic expressionere permeabilized (Cytofix/Cytoperm Kit, BD Bioscieneidelberg, Germany) and stained with PE-conjugatedranzyme B antibody (GB12; Caltag Laboratories Gmamburg, Germany). Phenotyping of immune cellsetermined by flow cytometry using a FACSCalibur insent (Becton Dickinson, Heidelberg, Germany). Cytoknalysis of supernatants from cytotoxicity assay was
ormed using the Human Th1/Th2 Cytokine Cytometric B
rythrocytes were removed by incubation with erythroysis buffer (155 mM NH4Cl, 10 mM KHCO3 and 100�Mthylenediamine tetraacetic acid; EDTA) and thrombocemoved via centrifugation of PBMC at 100× g for 10 minnd aspiration of the supernatant. The relative numbD3-, CD4- and CD8-positive T cells per total PBMC wetermined by FACS analysis using anti-human CD3, CD8 and CD45 antibodies (Becton Dickinson, Heidelbermany). 5× 105 PBMC were mixed with 5× 105 SW480
olon carcinoma cells resulting in effective E:T ratiospproximately 1:2 for CD3+:SW480, 1:3 for CD4+:SW480nd 1:7 for CD8+:SW480, respectively. The PBMC aW480 mixture was subcutaneously injected into theank in a final volume of 0.2 mL/mouse. One group of aals was injected with SW480 cells only to evaluate un
ific effects induced by the PBMC effector cells. Forreatment groups in the minimal residual disease modenimals per group were intravenously treated with sinhain bispecific antibodies (MT110) or the vehicle (Ph after SW480/PBMC inoculation at the indicated dof 0.01, 0.1, 1�g/mouse and treatment was repeatedonsecutive days (data not shown). In the established todel, eight animals per group were treated starting a(positive control) and day 8 after SW480 inoculation wubcutaneously growing tumor had reached tumor voluf 50–200 mm3 and treatment was repeated at 4 consecays. Tumor sizes were measured at the indicated daysaliper in two perpendicular dimensions and tumor volualculated according to (width2 × length)/2 as a correlate ffficacy.

K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143 1133
Establishment of the SW480 NOD/SCID mouse modeland the effect of control treatments were recently reported(Schlereth et al., 2005).
2.8. Human ovarian metastases xenograft model
Following surgical resection of peritoneal metastasis ofhistologically proven ovarian cancer patients, primary tumorspecimens were cut into approximately 50 mm3 cubes andsubcutaneously implanted into the right flank of NOD/SCIDmice. Eight animals per group were intravenously treatedwith 5�g MT110/injection or non-specific BiTE control for5 consecutive days at days 5, 12 and 19 after tumor implan-tation. Tumor sizes were measured on the indicated dayswith a caliper in two perpendicular dimensions and tumorvolumes calculated according to the formula: tumor vol-ume = [(width2 × length)/2].
At least four representative pieces of the individual ovar-ian tumor specimens were subjected in parallel to immuno-histochemistry analysis to determine the level of Ep-CAMexpression and presence of T cells. In brief, cryo slides of theprimary ovarian carcinomas were fixed with glutaraldehyde.Endogenous peroxidase activity was quenched with freshlyprepared 3% hydrogen peroxide for 5 min at room tempera-ture. Non-specific antibody binding was blocked with bovineserum albumin for 30 min at room temperature. Sectionsw AM( bod-i for3 strep-t theL y).S een2 eres ivelys
3
3
entE ticsc ndi-d cifics bis-p anC ede nti-b playl 3-s ssedi andc ysis( ncy,
three Ep-CAM-specific SCAs were selected for further opti-mization. A total of 14 recombinant BiTEs with differentdomain arrangements, linkers and SCA orientation were thenconstructed from three different human Ep-CAM-specificSCAs. In this second round of construction and selection,anti-Ep-CAM SCAs were combined with a de-immunizedversion of the human CD3 epsilon-specific murine SCA.The identification and removal of potential human T helpercell epitopes from framework, complementarity determiningregions and linker sequences of the anti-CD3 SCA wasperformed as described elsewhere (Jones et al., 2004).
Expression vectors encoding the various constructs weretransfected into CHO cells and recombinant proteins affinity-purified from cell culture supernatants. The majority ofconstructs were expressed and properly secreted (data notshown). Affinity-purified proteins were then tested for bio-logical activity in cytotoxicity assays. BiTEs were furthercharacterized for stability of the cytotoxic activity followingincubation in human serum and for productivity, percentageof monomer and charge homogeneity.
Six BiTEs showed in the cytotoxicity assays EC50 val-ues below 1 ng/mL, reached comparable levels of overall celllysis during the assay period and had essentially unchangedbiological activity after a 24 h incubation in human serumat 37◦C. Final BiTE selection from this set of proteins wastherefore based on initial productivity data. MT110 consis-t 0%m TEsh e ofm
x-a tideo cyls E)( lec-u uldb -i fiedm ponh iningc mn( ne-i idn n inh lef
anE at-u rci-n anP m-e s as ve aneb7 hips
ere then incubated with the primary anti human Ep-CBerEP4/FITC, Dako, Germany) and human CD8a anties (clone RPA-T8, Bioscience, Heidelberg, Germany)0 min at room temperature. Secondary antibodies and
avidin biotin detection was performed according toSAB kit (DakoCytomation GmbH, Hamburg, Germanections were counterstained with haematoxyline. Betw00 and 400 cells in at least four microscopic views wemi-quantitatively evaluated. The percentage of posittained cells was assessed.
. Results
.1. Generation and characterization of MT110
MT110 was selected from a panel of 21 differp-CAM-specific BiTE molecules based on characterisonsidered to be important for a clinical development caate. In a first round, a total of seven human Ep-CAM-speingle-chain antibodies (SCAs) were used to constructecific T cell engager (BiTEs) using the same humD3�-specific SCA. Ep-CAM-specific SCAs were derivither from VH and VL domains of murine monoclonal aodies of hybridomas or isolated as SCAs from phage dis
ibraries, and positioned in BiTEs N-terminally of the CDpecific SCA. All seven constructs were properly expren CHO cells, affinity-purified from culture supernatantsoncentrations for half maximal redirected target cell lEC50) determined by a cytotoxicity assay. Based on pote
ently showed the highest initial productivity and close to 9onomer content in cell culture supernatant. All other Biad a lower productivity or showed a higher percentagultimers.Affinity purification of MT110 using its C-terminal he
histidine tag yielded a single non-glycosylated polypepf 55 kDa apparent molecular weight upon lithium dodeulfate (LDS) polyacrylamide gel electrophoresis (PAGFig. 1A). MT110 monomer and dimer had the same molar weight (Fig. 1A, compare lanes 2 and 3). They coe separated to baseline by gel filtration (Fig. 1B), ensur
ng isolation of homogenous MT110 monomer. Purionomer of MT110 (>99%) showed a discrete peak uigh-resolution cation exchange chromatography contalose to 90% of the biological activity applied to the coluFig. 1C), indicating a high degree of charge homogety. As determined by gel filtration, monomeric MT110 dot detectably multimerize or aggregate upon incubatiouman serum at 37◦C, prolonged storage or after multip
reeze/thaw cycles (data not shown).The binding affinities of monomeric MT110 to hum
p-CAM and CD3�, respectively, were determined by sration binding assays using the Kato III gastric caoma cell line, and T cells in freshly isolated humBMC. Cell-bound MT110 was detected by flow cytotry using a FITC-conjugated anti-mouse antibody aecondary reagent. The mean of six experiments gaquilibrium dissociation constant (KD) of MT110 for cell-ound Ep-CAM of 1.6± 1.2× 10−8 M and for CD3 of.7± 3.9× 10−8 M. Plasmon resonance analysis using c
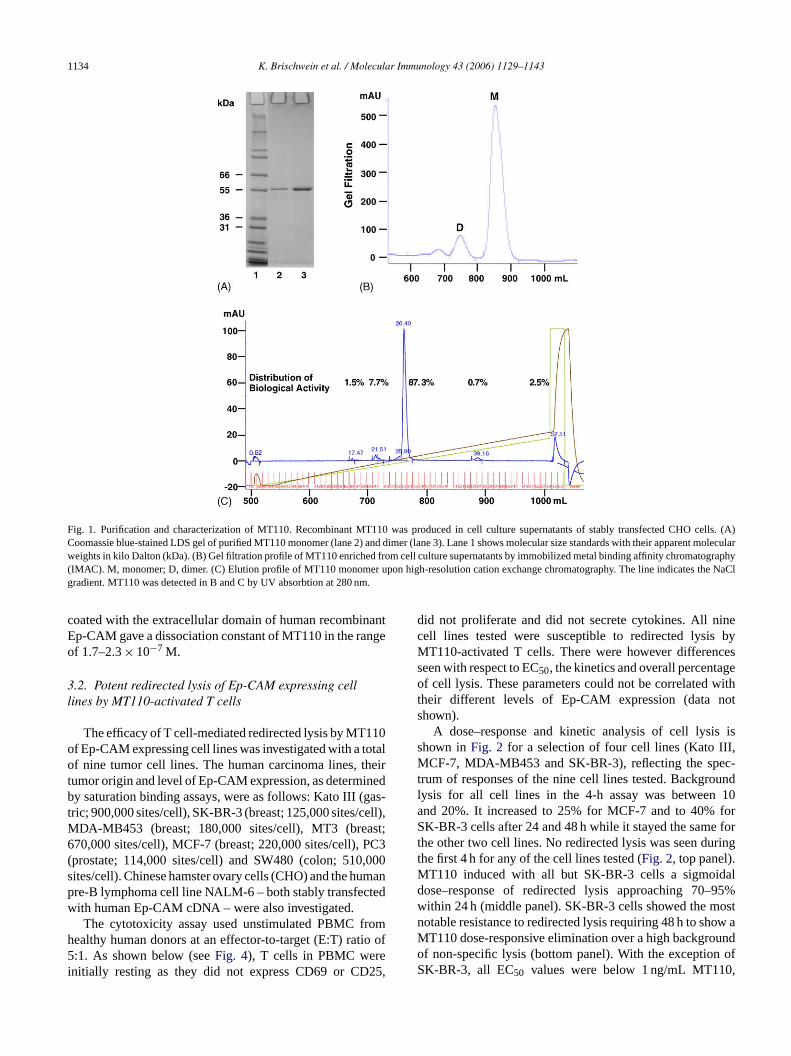
1134 K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143
Fig. 1. Purification and characterization of MT110. Recombinant MT110 was produced in cell culture supernatants of stably transfected CHO cells. (A)Coomassie blue-stained LDS gel of purified MT110 monomer (lane 2) and dimer (lane 3). Lane 1 shows molecular size standards with their apparent molecularweights in kilo Dalton (kDa). (B) Gel filtration profile of MT110 enriched from cell culture supernatants by immobilized metal binding affinity chromatography(IMAC). M, monomer; D, dimer. (C) Elution profile of MT110 monomer upon high-resolution cation exchange chromatography. The line indicates the NaClgradient. MT110 was detected in B and C by UV absorbtion at 280 nm.
coated with the extracellular domain of human recombinantEp-CAM gave a dissociation constant of MT110 in the rangeof 1.7–2.3× 10−7 M.
3.2. Potent redirected lysis of Ep-CAM expressing celllines by MT110-activated T cells
The efficacy of T cell-mediated redirected lysis by MT110of Ep-CAM expressing cell lines was investigated with a totalof nine tumor cell lines. The human carcinoma lines, theirtumor origin and level of Ep-CAM expression, as determinedby saturation binding assays, were as follows: Kato III (gas-tric; 900,000 sites/cell), SK-BR-3 (breast; 125,000 sites/cell),MDA-MB453 (breast; 180,000 sites/cell), MT3 (breast;670,000 sites/cell), MCF-7 (breast; 220,000 sites/cell), PC3(prostate; 114,000 sites/cell) and SW480 (colon; 510,000sites/cell). Chinese hamster ovary cells (CHO) and the humanpre-B lymphoma cell line NALM-6 – both stably transfectedwith human Ep-CAM cDNA – were also investigated.
The cytotoxicity assay used unstimulated PBMC fromhealthy human donors at an effector-to-target (E:T) ratio of5:1. As shown below (seeFig. 4), T cells in PBMC wereinitially resting as they did not express CD69 or CD25,
did not proliferate and did not secrete cytokines. All ninecell lines tested were susceptible to redirected lysis byMT110-activated T cells. There were however differencesseen with respect to EC50, the kinetics and overall percentageof cell lysis. These parameters could not be correlated withtheir different levels of Ep-CAM expression (data notshown).
A dose–response and kinetic analysis of cell lysis isshown inFig. 2 for a selection of four cell lines (Kato III,MCF-7, MDA-MB453 and SK-BR-3), reflecting the spec-trum of responses of the nine cell lines tested. Backgroundlysis for all cell lines in the 4-h assay was between 10and 20%. It increased to 25% for MCF-7 and to 40% forSK-BR-3 cells after 24 and 48 h while it stayed the same forthe other two cell lines. No redirected lysis was seen duringthe first 4 h for any of the cell lines tested (Fig. 2, top panel).MT110 induced with all but SK-BR-3 cells a sigmoidaldose–response of redirected lysis approaching 70–95%within 24 h (middle panel). SK-BR-3 cells showed the mostnotable resistance to redirected lysis requiring 48 h to show aMT110 dose-responsive elimination over a high backgroundof non-specific lysis (bottom panel). With the exception ofSK-BR-3, all EC50 values were below 1 ng/mL MT110,

K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143 1135
Fig. 2. Kinetics and dose–response of redirected lysis by MT110 of fourhuman carcinoma target cell lines. The gastric carcinoma cell line Kato III(�) and the breast carcinoma cell lines MCF-7 (�), MDA-MB-453 (�) andSK-BR-3 (�) were incubated in the presence of freshly isolated PBMC at aratio of 1:5 and increasing concentrations of MT110. Target cell lysis wasdetermined by flow cytometry as the percentage of target cells becomingpropidium iodide-positive after 4 h (A), 24 h (B) and 48 h (C).
i.e., below 18 pM. Consistent with previous experiments(Dreier et al., 2002; Schlereth et al., 2005), we have notobserved that target-negative cells were detectably lysed inthe presence of high amounts of BiTE and T cells (data notshown).
Lysis of Kato III cells by MT110-activated T cells wasstudied in more detail. EC50 values were determined withPBMC from a total of 24 healthy donors. The range of EC50values was from 20 to 1000 pg/mL with a mean of 397 pg/mLand a standard deviation of±357 pg/mL.
Fig. 3. Specificity of target cell lysis by MT110 and a CD19-specific BiTE.CHO cells stably transfected with either human Ep-CAM (CHO-Ep-CAM)or human CD19 (CHO-CD19) were used as target cells in a cytotoxicityassay in the presence of human PBMC at a ratio of 1:5 and increasingconcentrations of either MT110 or bscCD19× CD3 BiTE (MT103); (�)CHO-Ep-CAM cells treated with MT110; (�) CHO-Ep-CAM cells treatedwith MT103; (�) CHO-CD19 cells treated with MT110; (�) CHO-CD19cells treated with MT103. Target cell lysis was determined by flow cytom-etry as the percentage of target cells becoming propidium iodide-positiveafter 20 h of incubation.
3.3. Specificity of redirected lysis by MT110 and aCD19-specific BiTE
The specificity of redirected lysis by BiTEs was ear-lier demonstrated in detail for a CD19-specific BiTE calledbscCD19× CD3 (Dreier et al., 2002). Here, we wanted tocompare the activity and specificity of MT110 with that ofbscCD19× CD3 in the same cellular background. RodentCHO cell lines were established that stably expressed trans-fected cDNAs encoding either human Ep-CAM or humanCD19. The CHO-Ep-CAM line expressed 600,000 Ep-CAMbinding sites/cell and the CHO-CD19 line 250,000 CD19binding sites/cell. CHO-Ep-CAM cells were potently elimi-nated by MT110-activated T cells in a 16-h assay at a PBMCto target ratio of 5:1 (Fig. 3). Likewise, CHO-CD19 cellswere eliminated by bscCD19× CD3-activated T cells. Bothcell lines however were fully resistant to lysis in the pres-ence of T cells (i.e., PBMC) if the respective other BiTEwas added up to concentrations of 100 or 200 ng/mL, respec-tively. Because MT110 and bscCD19× CD3 share the sameCD3-specific SCA, binding of the BiTE to only T cells was,therefore, insufficient to trigger redirected target cell lysis.Non-transfected rodent CHO cells were fully resistant to redi-rected lysis in the presence of MT110 and human PBMC (datanot shown).
3
ulara te, Tc ativec E
.4. Target cell-dependent T cell activation by MT110
We next explored whether MT110 could induce a regctivation of T cells in the presence of target cells. Of noells incubated with MT110 in the presence of target-negells did not elicit any lytic activity even at very high BiT
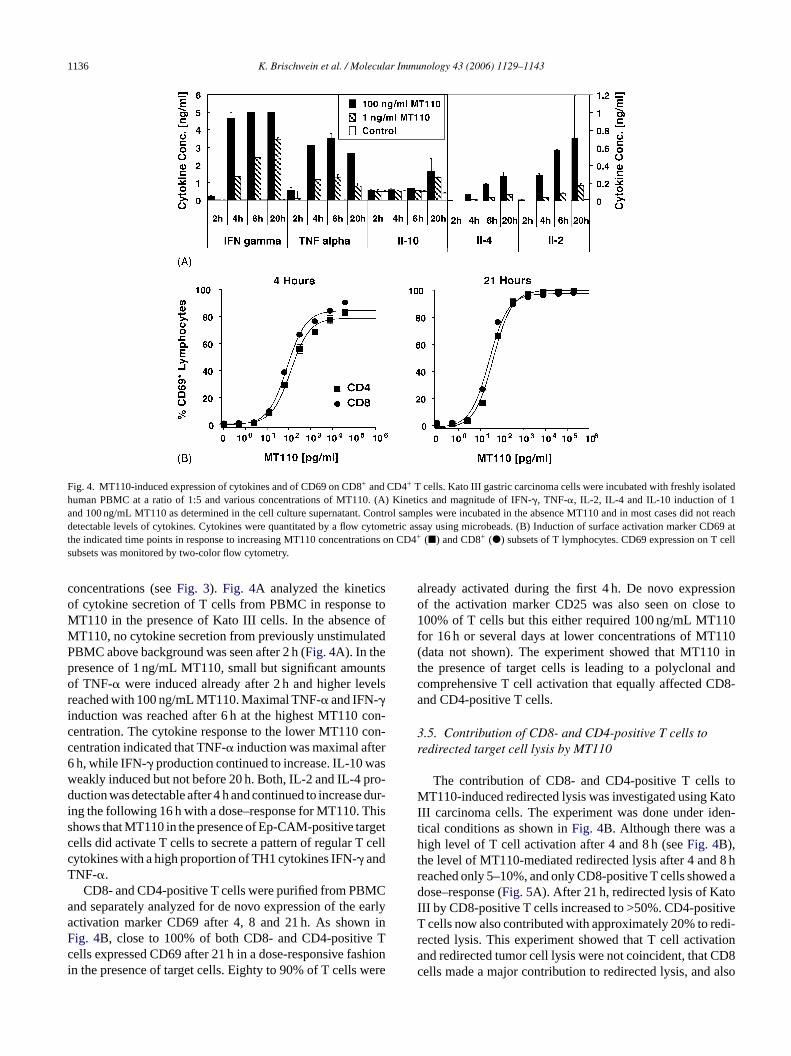
1136 K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143
Fig. 4. MT110-induced expression of cytokines and of CD69 on CD8+ and CD4+ T cells. Kato III gastric carcinoma cells were incubated with freshly isolatedhuman PBMC at a ratio of 1:5 and various concentrations of MT110. (A) Kinetics and magnitude of IFN-�, TNF-�, IL-2, IL-4 and IL-10 induction of 1and 100 ng/mL MT110 as determined in the cell culture supernatant. Control samples were incubated in the absence MT110 and in most cases did not reachdetectable levels of cytokines. Cytokines were quantitated by a flow cytometric assay using microbeads. (B) Induction of surface activation marker CD69 atthe indicated time points in response to increasing MT110 concentrations on CD4+ (�) and CD8+ (�) subsets of T lymphocytes. CD69 expression on T cellsubsets was monitored by two-color flow cytometry.
concentrations (seeFig. 3). Fig. 4A analyzed the kineticsof cytokine secretion of T cells from PBMC in response toMT110 in the presence of Kato III cells. In the absence ofMT110, no cytokine secretion from previously unstimulatedPBMC above background was seen after 2 h (Fig. 4A). In thepresence of 1 ng/mL MT110, small but significant amountsof TNF-� were induced already after 2 h and higher levelsreached with 100 ng/mL MT110. Maximal TNF-� and IFN-�induction was reached after 6 h at the highest MT110 con-centration. The cytokine response to the lower MT110 con-centration indicated that TNF-� induction was maximal after6 h, while IFN-� production continued to increase. IL-10 wasweakly induced but not before 20 h. Both, IL-2 and IL-4 pro-duction was detectable after 4 h and continued to increase dur-ing the following 16 h with a dose–response for MT110. Thisshows that MT110 in the presence of Ep-CAM-positive targetcells did activate T cells to secrete a pattern of regular T cellcytokines with a high proportion of TH1 cytokines IFN-� andTNF-�.
CD8- and CD4-positive T cells were purified from PBMCand separately analyzed for de novo expression of the earlyactivation marker CD69 after 4, 8 and 21 h. As shown inFig. 4B, close to 100% of both CD8- and CD4-positive Tcells expressed CD69 after 21 h in a dose-responsive fashionin the presence of target cells. Eighty to 90% of T cells were
already activated during the first 4 h. De novo expressionof the activation marker CD25 was also seen on close to100% of T cells but this either required 100 ng/mL MT110for 16 h or several days at lower concentrations of MT110(data not shown). The experiment showed that MT110 inthe presence of target cells is leading to a polyclonal andcomprehensive T cell activation that equally affected CD8-and CD4-positive T cells.
3.5. Contribution of CD8- and CD4-positive T cells toredirected target cell lysis by MT110
The contribution of CD8- and CD4-positive T cells toMT110-induced redirected lysis was investigated using KatoIII carcinoma cells. The experiment was done under iden-tical conditions as shown inFig. 4B. Although there was ahigh level of T cell activation after 4 and 8 h (seeFig. 4B),the level of MT110-mediated redirected lysis after 4 and 8 hreached only 5–10%, and only CD8-positive T cells showed adose–response (Fig. 5A). After 21 h, redirected lysis of KatoIII by CD8-positive T cells increased to >50%. CD4-positiveT cells now also contributed with approximately 20% to redi-rected lysis. This experiment showed that T cell activationand redirected tumor cell lysis were not coincident, that CD8cells made a major contribution to redirected lysis, and also
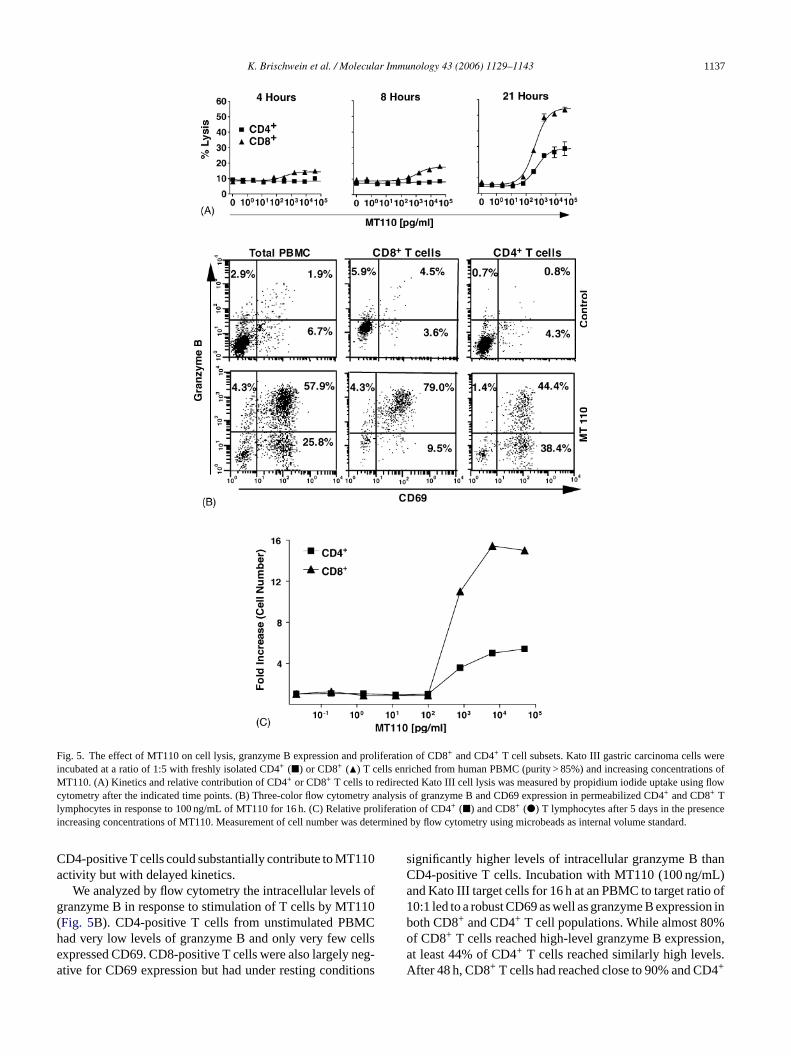
K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143 1137
Fig. 5. The effect of MT110 on cell lysis, granzyme B expression and proliferation of CD8+ and CD4+ T cell subsets. Kato III gastric carcinoma cells wereincubated at a ratio of 1:5 with freshly isolated CD4+ (�) or CD8+ (�) T cells enriched from human PBMC (purity > 85%) and increasing concentrations ofMT110. (A) Kinetics and relative contribution of CD4+ or CD8+ T cells to redirected Kato III cell lysis was measured by propidium iodide uptake using flowcytometry after the indicated time points. (B) Three-color flow cytometry analysis of granzyme B and CD69 expression in permeabilized CD4+ and CD8+ Tlymphocytes in response to 100 ng/mL of MT110 for 16 h. (C) Relative proliferation of CD4+ (�) and CD8+ (�) T lymphocytes after 5 days in the presenceincreasing concentrations of MT110. Measurement of cell number was determined by flow cytometry using microbeads as internal volume standard.
CD4-positive T cells could substantially contribute to MT110activity but with delayed kinetics.
We analyzed by flow cytometry the intracellular levels ofgranzyme B in response to stimulation of T cells by MT110(Fig. 5B). CD4-positive T cells from unstimulated PBMChad very low levels of granzyme B and only very few cellsexpressed CD69. CD8-positive T cells were also largely neg-ative for CD69 expression but had under resting conditions
significantly higher levels of intracellular granzyme B thanCD4-positive T cells. Incubation with MT110 (100 ng/mL)and Kato III target cells for 16 h at an PBMC to target ratio of10:1 led to a robust CD69 as well as granzyme B expression inboth CD8+ and CD4+ T cell populations. While almost 80%of CD8+ T cells reached high-level granzyme B expression,at least 44% of CD4+ T cells reached similarly high levels.After 48 h, CD8+ T cells had reached close to 90% and CD4+
1138 K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143
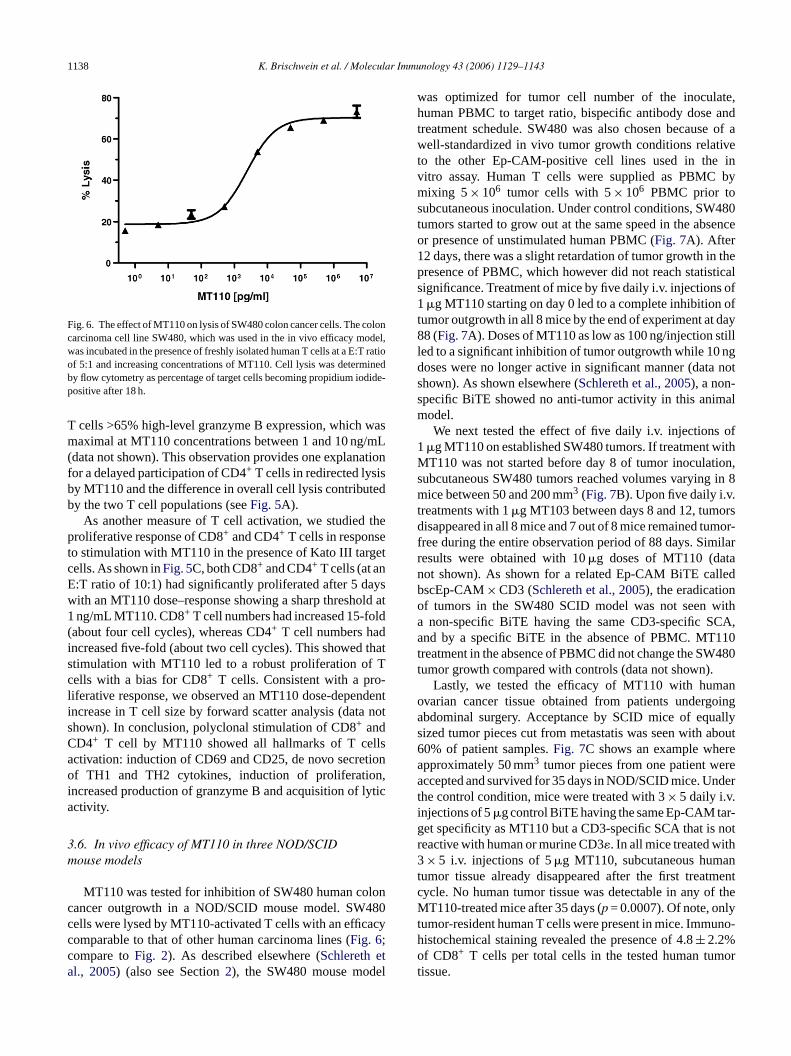
Fig. 6. The effect of MT110 on lysis of SW480 colon cancer cells. The coloncarcinoma cell line SW480, which was used in the in vivo efficacy model,was incubated in the presence of freshly isolated human T cells at a E:T ratioof 5:1 and increasing concentrations of MT110. Cell lysis was determinedby flow cytometry as percentage of target cells becoming propidium iodide-positive after 18 h.
T cells >65% high-level granzyme B expression, which wasmaximal at MT110 concentrations between 1 and 10 ng/mL(data not shown). This observation provides one explanationfor a delayed participation of CD4+ T cells in redirected lysisby MT110 and the difference in overall cell lysis contributedby the two T cell populations (seeFig. 5A).
As another measure of T cell activation, we studied theproliferative response of CD8+ and CD4+ T cells in responseto stimulation with MT110 in the presence of Kato III targetcells. As shown inFig. 5C, both CD8+ and CD4+ T cells (at anE:T ratio of 10:1) had significantly proliferated after 5 dayswith an MT110 dose–response showing a sharp threshold at1 ng/mL MT110. CD8+ T cell numbers had increased 15-fold(about four cell cycles), whereas CD4+ T cell numbers hadincreased five-fold (about two cell cycles). This showed thatstimulation with MT110 led to a robust proliferation of Tcells with a bias for CD8+ T cells. Consistent with a pro-liferative response, we observed an MT110 dose-dependentincrease in T cell size by forward scatter analysis (data notshown). In conclusion, polyclonal stimulation of CD8+ andCD4+ T cell by MT110 showed all hallmarks of T cellsactivation: induction of CD69 and CD25, de novo secretionof TH1 and TH2 cytokines, induction of proliferation,increased production of granzyme B and acquisition of lyticactivity.
3m
lonc 480c acycc ta el
was optimized for tumor cell number of the inoculate,human PBMC to target ratio, bispecific antibody dose andtreatment schedule. SW480 was also chosen because of awell-standardized in vivo tumor growth conditions relativeto the other Ep-CAM-positive cell lines used in the invitro assay. Human T cells were supplied as PBMC bymixing 5× 106 tumor cells with 5× 106 PBMC prior tosubcutaneous inoculation. Under control conditions, SW480tumors started to grow out at the same speed in the absenceor presence of unstimulated human PBMC (Fig. 7A). After12 days, there was a slight retardation of tumor growth in thepresence of PBMC, which however did not reach statisticalsignificance. Treatment of mice by five daily i.v. injections of1�g MT110 starting on day 0 led to a complete inhibition oftumor outgrowth in all 8 mice by the end of experiment at day88 (Fig. 7A). Doses of MT110 as low as 100 ng/injection stillled to a significant inhibition of tumor outgrowth while 10 ngdoses were no longer active in significant manner (data notshown). As shown elsewhere (Schlereth et al., 2005), a non-specific BiTE showed no anti-tumor activity in this animalmodel.
We next tested the effect of five daily i.v. injections of1�g MT110 on established SW480 tumors. If treatment withMT110 was not started before day 8 of tumor inoculation,subcutaneous SW480 tumors reached volumes varying in 8mice between 50 and 200 mm3 (Fig. 7B). Upon five daily i.v.t orsd mor-f ilarr tan ledb no itha CA,a 110t 480t
ano oinga allys bout6 rea rea nderti r-g notr h3 ant mentc f theM yt uno-ho ort
.6. In vivo efficacy of MT110 in three NOD/SCIDouse models
MT110 was tested for inhibition of SW480 human coancer outgrowth in a NOD/SCID mouse model. SWells were lysed by MT110-activated T cells with an efficomparable to that of other human carcinoma lines (Fig. 6;ompare toFig. 2). As described elsewhere (Schlereth el., 2005) (also see Section2), the SW480 mouse mod
reatments with 1�g MT103 between days 8 and 12, tumisappeared in all 8 mice and 7 out of 8 mice remained tu
ree during the entire observation period of 88 days. Simesults were obtained with 10�g doses of MT110 (daot shown). As shown for a related Ep-CAM BiTE calscEp-CAM× CD3 (Schlereth et al., 2005), the eradicatiof tumors in the SW480 SCID model was not seen wnon-specific BiTE having the same CD3-specific S
nd by a specific BiTE in the absence of PBMC. MTreatment in the absence of PBMC did not change the SWumor growth compared with controls (data not shown).
Lastly, we tested the efficacy of MT110 with humvarian cancer tissue obtained from patients undergbdominal surgery. Acceptance by SCID mice of equized tumor pieces cut from metastatis was seen with a0% of patient samples.Fig. 7C shows an example whepproximately 50 mm3 tumor pieces from one patient weccepted and survived for 35 days in NOD/SCID mice. U
he control condition, mice were treated with 3× 5 daily i.v.njections of 5�g control BiTE having the same Ep-CAM taet specificity as MT110 but a CD3-specific SCA that iseactive with human or murine CD3�. In all mice treated wit× 5 i.v. injections of 5�g MT110, subcutaneous hum
umor tissue already disappeared after the first treatycle. No human tumor tissue was detectable in any oT110-treated mice after 35 days (p = 0.0007). Of note, onl
umor-resident human T cells were present in mice. Immistochemical staining revealed the presence of 4.8± 2.2%f CD8+ T cells per total cells in the tested human tum
issue.
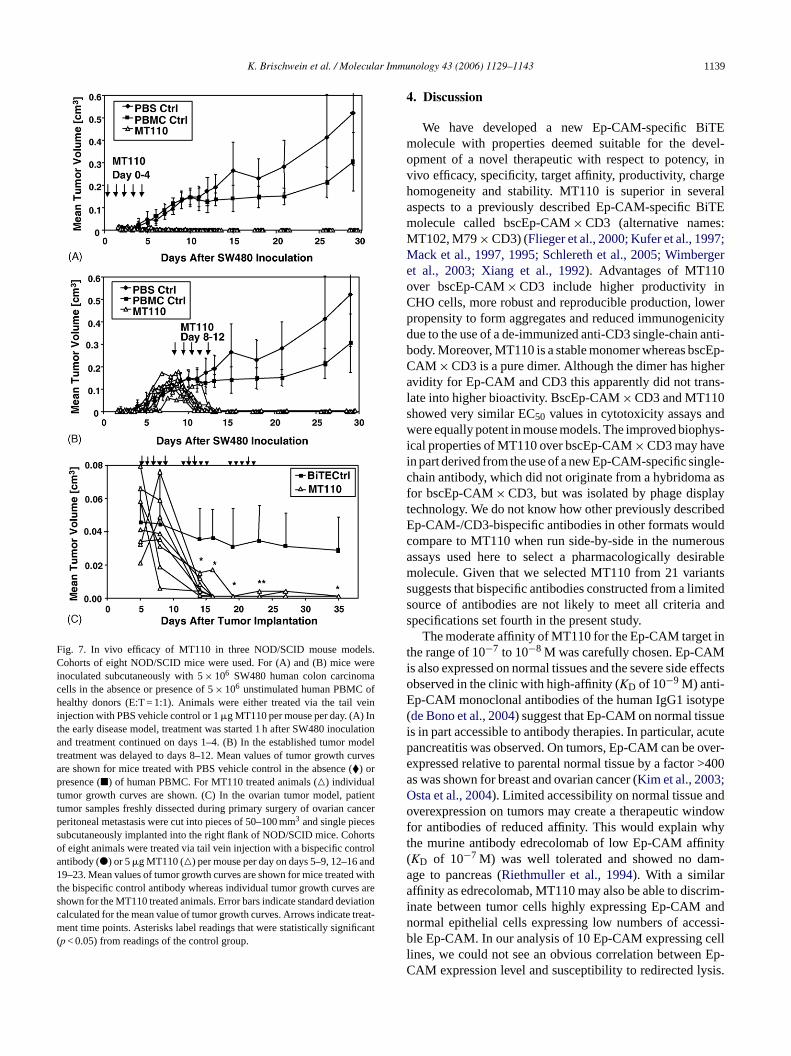
K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143 1139
Fig. 7. In vivo efficacy of MT110 in three NOD/SCID mouse models.Cohorts of eight NOD/SCID mice were used. For (A) and (B) mice wereinoculated subcutaneously with 5× 106 SW480 human colon carcinomacells in the absence or presence of 5× 106 unstimulated human PBMC ofhealthy donors (E:T = 1:1). Animals were either treated via the tail veininjection with PBS vehicle control or 1�g MT110 per mouse per day. (A) Inthe early disease model, treatment was started 1 h after SW480 inoculationand treatment continued on days 1–4. (B) In the established tumor modeltreatment was delayed to days 8–12. Mean values of tumor growth curvesare shown for mice treated with PBS vehicle control in the absence (�) orpresence (�) of human PBMC. For MT110 treated animals (�) individualtumor growth curves are shown. (C) In the ovarian tumor model, patienttumor samples freshly dissected during primary surgery of ovarian cancerperitoneal metastasis were cut into pieces of 50–100 mm3 and single piecessubcutaneously implanted into the right flank of NOD/SCID mice. Cohortsof eight animals were treated via tail vein injection with a bispecific controlantibody (�) or 5�g MT110 (�) per mouse per day on days 5–9, 12–16 and19–23. Mean values of tumor growth curves are shown for mice treated withthe bispecific control antibody whereas individual tumor growth curves areshown for the MT110 treated animals. Error bars indicate standard deviationcalculated for the mean value of tumor growth curves. Arrows indicate treat-ment time points. Asterisks label readings that were statistically significant(p < 0.05) from readings of the control group.
4. Discussion
We have developed a new Ep-CAM-specific BiTEmolecule with properties deemed suitable for the devel-opment of a novel therapeutic with respect to potency, invivo efficacy, specificity, target affinity, productivity, chargehomogeneity and stability. MT110 is superior in severalaspects to a previously described Ep-CAM-specific BiTEmolecule called bscEp-CAM× CD3 (alternative names:MT102, M79× CD3) (Flieger et al., 2000; Kufer et al., 1997;Mack et al., 1997, 1995; Schlereth et al., 2005; Wimbergeret al., 2003; Xiang et al., 1992). Advantages of MT110over bscEp-CAM× CD3 include higher productivity inCHO cells, more robust and reproducible production, lowerpropensity to form aggregates and reduced immunogenicitydue to the use of a de-immunized anti-CD3 single-chain anti-body. Moreover, MT110 is a stable monomer whereas bscEp-CAM × CD3 is a pure dimer. Although the dimer has higheravidity for Ep-CAM and CD3 this apparently did not trans-late into higher bioactivity. BscEp-CAM× CD3 and MT110showed very similar EC50 values in cytotoxicity assays andwere equally potent in mouse models. The improved biophys-ical properties of MT110 over bscEp-CAM× CD3 may havein part derived from the use of a new Ep-CAM-specific single-chain antibody, which did not originate from a hybridoma asfor bscEp-CAM× CD3, but was isolated by phage displayt ibedE uldc rousa irablem antss iteds ands
t int Mi effectsoE ype( uei cutep over-e >400a ;O ndo ndowf hyt ity( m-aa rim-i andn ssi-b elll Ep-C sis.
echnology. We do not know how other previously descrp-CAM-/CD3-bispecific antibodies in other formats woompare to MT110 when run side-by-side in the numessays used here to select a pharmacologically desolecule. Given that we selected MT110 from 21 vari
uggests that bispecific antibodies constructed from a limource of antibodies are not likely to meet all criteriapecifications set fourth in the present study.
The moderate affinity of MT110 for the Ep-CAM targehe range of 10−7 to 10−8 M was carefully chosen. Ep-CAs also expressed on normal tissues and the severe sidebserved in the clinic with high-affinity (KD of 10−9 M) anti-p-CAM monoclonal antibodies of the human IgG1 isot
de Bono et al., 2004) suggest that Ep-CAM on normal tisss in part accessible to antibody therapies. In particular, aancreatitis was observed. On tumors, Ep-CAM can bexpressed relative to parental normal tissue by a factors was shown for breast and ovarian cancer (Kim et al., 2003sta et al., 2004). Limited accessibility on normal tissue averexpression on tumors may create a therapeutic wior antibodies of reduced affinity. This would explain whe murine antibody edrecolomab of low Ep-CAM affinKD of 10−7 M) was well tolerated and showed no dage to pancreas (Riethmuller et al., 1994). With a similarffinity as edrecolomab, MT110 may also be able to disc
nate between tumor cells highly expressing Ep-CAMormal epithelial cells expressing low numbers of accele Ep-CAM. In our analysis of 10 Ep-CAM expressing c
ines, we could not see an obvious correlation betweenAM expression level and susceptibility to redirected ly

1140 K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143
This could be due to the overall high Ep-CAM expression onthese cell lines, which in all cases was in excess of 100,000binding sites/cell. Differences in susceptibility to redirected Tcell lysis observed for some tumor cell lines may thus be dueto other factors. The retarded lytic response seen with targetcell line SK-BR-3 could come, for instance, from an acquiredescape mechanism targeting an early response to granzymeB-mediated target cell lysis, such as the activation of caspase-3 (Adrain et al., 2004). Future studies will be required tounderstand the influence of various T cell escape mechanismsof tumor cells on the kinetics and magnitude of redirected Tcell responses. Lastly, in vivo studies are needed to explorethe therapeutic window of MT110 and to investigate its activ-ity against tumors presenting with various T cell escapemechanisms.
MT110 and other BiTEs show a remarkable specificity.T cells only get activated, i.e., produce cytokines, expressactivation markers, develop lytic activity and start to pro-liferate, when BiTEs are added together with target cells.Neither binding of a BiTE to T cells alone, nor target cells ontheir own are capable of activating T cells. This indicates thatBiTEs need to be clustered on the surface of target cells inorder to stimulate T cells. This is reminiscent of T cell activa-tion by certain monoclonal anti-CD3 antibodies, which canonly potently activate T cells when coated on plastic surfaces.Formation of a polyvalent matrix of anti-CD3 binding armso jux-t apsef iTEsa areb ise,m D3-s cificm -c
xon.A 3w -rT bero tics effi-c rgetc ientfa
cti-v heres ellsi novoe e top D8-p sa hereI ug-g 10
concentrations and had other kinetics as needed to elicitexpression of surface activation markers. Prolonged inter-action between T and target cells or an enlarged interactionsurface between cells may be necessary to initiate and main-tain multiple rounds of cell cycle.
A surprising observation was that MT110 could dramat-ically upregulate granzyme B expression in CD4+ T cells.This may well explain the delayed but substantial contri-bution of CD4+ T cells to overall target cell lysis (Macket al., 1997). It also suggests that a large proportion of CD8+
and CD4+ peripheral T cells can ultimately participate in redi-rected tumor target cell lysis by MT110. The recruitment ofCD4+ T cells and participation of this T cell subpopulationin tumor cell lysis has also been shown with a different kindof bispecific antibody (Porakishvili et al., 2004). The robustproliferation of T cells in response to BiTE and target cellsargues against a significant induction of apoptosis of T cells.Future studies need to investigate how T cells behave afterwithdrawal of MT110 (and other BiTEs), and what the impactof BiTE stimulation is on naıve CD4+ and CD8+ T cells.
As described previously for other BiTEs (Dreier et al.,2003; Dreier et al., 2002), MT110 did not need extra T cellstimuli to activate resting T cells. As investigated with aCD19-specific BiTE (Loffler et al., 2003), stimulation of Tcells with CD28 and IL-2 could augment cytotoxicity butwas likewise not needed to activate resting T cells. This isi tiono nsi ncei ularf eres culard notr
dys rtiesa 19(e inr es,t AMa thes ivityo dentc HCc in thea
es le tot fa p-Cb Dm lowm tha ere,
n target cells by BiTEs may be necessary to very closelyapose T and target cells as a prerequisite for lytic synormation and subsequent T cell activation. Because Bre monovalent for CD3, no significant effects on T cellse triggered via crosslinking of CD3 complexes. Likewonomeric BiTEs are unlikely to lead to redirected C
pecific T cell lysis, as is possible with bivalent CD3-speonoclonal antibodies (Wong et al., 1990) or dimeric bispe
ific antibodies.The extreme potency of BiTEs appears as a parado
lthough the affinity of MT110 for both Ep-CAM and CDas only in the range of 10−7 to 10−8 M, half maximal redi
ected target cell lysis was in the range of 10−11 to 10−12 M.his large difference may indicate that a very low numf MT110 on target (or T cells) is sufficient to trigger lyynapse formation. In this respect, BiTEs appear as anient substitute for MHC class I/peptide complexes on taells where only a 1–2 digit number is thought to be sufficor induction of cell lysis by specific T cell clones (Appel etl., 2000; Offner et al., 2006).
Another remarkable trait of BiTEs is their polyclonal aation of T cells in the presence of target cells. We havehown that essentially all CD3-positive peripheral T cn the assay were activated as determined by their dexpression of CD69 and CD25. However, when it camroliferation and contribution to cell lysis the subset of Cositive T cells was dominant. CD8+ T cell dominance walso reflected by the spectrum of cytokines induced w
FN-� and TNF-� were most prominent. Our data also sest that induction of proliferation required higher MT1
n contrast to other bispecific antibodies where stimulaf resting T cells with anti-CD28, anti-CD3, IL-2 or lecti
s mandatory for potent redirected cell lysis. This differes difficult to explain but appears to relate to the particormat of bispecific single-chain antibody constructs whimultaneous epitope recognition and a particular moleesign may allow for an optimal T/target cell recognitionequiring additional activating signals.
The in-depth characterization of MT110 in this stuhows that the molecule has principally the same propes bscCD19× CD3, a BiTE molecule directed against CDLoffler et al., 2000). Although bscCD19× CD3 showedven lower EC50 values with human B lymphoma linesedirected target cell lysis as MT110 with carcinoma linhe two BiTEs showed comparable activity when Ep-Cnd CD19 targets were expressed to similar levels iname background of a rodent tumor cell line. The actf redirected human T cells against target-positive roells also underscores that BiTEs fully substitute for Mlass I/peptide complexes and do work across speciesbsence of matching co-stimulatory molecules.
The in vivo efficacy of MT110 in NOD/SCID micupplemented with human PBMC was also comparabhat of bscCD19× CD3 (Dreier et al., 2003), and that o
dimeric Ep-CAM-specific BiTE molecule, called bscEAM × CD3 (Schlereth et al., 2005). BscCD19× CD3 andscEp-CAM× CD3 were both highly active in SCID/NOouse xenograft models where sub-nanograms andicrograms doses led to inhibition of tumor outgrownd eradication of established tumors, respectively. H

K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143 1141
we show that also the monomeric Ep-CAM-specific BiTEMT110 had a high potential to inhibit tumor outgrowth as wellas eradicate solid tumors derived either from a human coloncarcinoma cell line or from human ovarian cancer metas-tases. Of note, anti-tumor activity was seen with unstimulatedhuman PBMC, which is in contrast to other animal modelswhere bispecific antibodies required pre-conditioned humanT cells or the presence of anti-CD28 antibodies (Cochloviuset al., 2000). With just low micrograms amounts of anti-body construct inducing complete tumor rejection, MT110 isamong the most potent biologicals known to eradicate tumorsin animal models.
The NOD/SCID mouse model using human metastaticcancer tissue is extremely demanding. Not only do suchtumors present with the full genetic heterogeneity of late-stage human cancer cells and possibly express a variety of Tcell escape mechanisms, but also immune effector T cells insuch tumors are scarce and frequently tolerated (De Visseret al., 2003). In our animal model, no additional T cellsfrom healthy donors were added but MT110 solely reliedon tumor-resident T cells. Nevertheless, we observed that inall mice treated with MT110 already the first course of treat-ment led to a substantial reduction of tumor mass, and norelapse was seen following subsequent treatments. Similarresults were obtained with samples from other patients usingMT110 (data not shown) and bscEp-CAM× CD3 (Schlerethe thep roma peri-m withr ofa gnif-i fromt
char-a anE ftedt ford
A
rst n thea em-i
R
A thezyme
A tterns. 12,
Appel, H., Gauthier, L., Pyrdol, J., Wucherpfennig, K.W., 2000. Kineticsof T-cell receptor binding by bivalent HLA-DR. Peptide complexesthat activate antigen-specific human T-cells. J. Biol. Chem. 275,312–321.
Baeuerle, P.A., Kufer, P., Lutterbuse, R., 2003. Bispecific antibodies forpolyclonal T-cell engagement. Curr. Opin. Mol. Ther. 5, 413–419.
Balzar, M., Winter, M.J., de Boer, C.J., Litvinov, S.V., 1999. The biologyof the 17-1A antigen (Ep-CAM). J. Mol. Med. 77, 699–712.
Cartron, G., Dacheux, L., Salles, G., Solal-Celigny, P., Bardos, P., Colom-bat, P., Watier, H., 2002. Therapeutic activity of humanized anti-CD20monoclonal antibody and polymorphism in IgG Fc receptor Fcgam-maRIIIa gene. Blood 99, 754–758.
Clynes, R.A., Towers, T.L., Presta, L.G., Ravetch, J.V., 2000. InhibitoryFc receptors modulate in vivo cytoxicity against tumor targets. Nat.Med. 6, 443–446.
Cochlovius, B., Kipriyanov, S.M., Stassar, M.J., Christ, O., Schuhmacher,J., Strauss, G., Moldenhauer, G., Little, M., 2000. Treatment of humanB cell lymphoma xenografts with a CD3× CD19 diabody and T cells.J. Immunol. 165, 888–895.
Dall’Ozzo, S., Tartas, S., Paintaud, G., Cartron, G., Colombat, P., Bardos,P., Watier, H., Thibault, G., 2004. Rituximab-dependent cytotoxicityby natural killer cells: influence of FCGR3A polymorphism on theconcentration–effect relationship. Cancer Res. 64, 4664–4669.
Davis, T.A., Kaminski, M.S., Leonard, J.P., Hsu, F.J., Wilkinson, M.,Zelenetz, A., Wahl, R.L., Kroll, S., Coleman, M., Goris, M., Levy,R., Knox, S.J., 2004. The radioisotope contributes significantly tothe activity of radioimmunotherapy. Clin. Cancer Res. 10, 7792–7798.
de Bono, J.S., Tolcher, A.W., Forero, A., Vanhove, G.F., Takimoto, C.,Bauer, R.J., Hammond, L.A., Patnaik, A., White, M.L., Shen, S.,Khazaeli, M.B., Rowinsky, E.K., LoBuglio, A.F., 2004. ING-1, a
ced
D cell
D B.,G.,
t andub-by anol.
D U.,P.A.,cyto-ngle-
D y for675.
D S.L.,kis,, Pel-.M.,sferther-oma.
F A.P.,S.,
M-tation65
F 000.3 ineffi-tes
t al., 2005). There are also a number of limitations ofresent model including: (i) the sample size derived fmetastasis, which restricts the number of control exents possible; (ii) the heterogeneity of patient samples
espect to viability in NOD/SCID mice; (iii) procurementhigh enough sample number to achieve statistically si
cant response rates; (iv) the exclusion of side effectsargeting normal tissue.
In conclusion, the present study shows that the basiccteristics of a CD19-specific BiTE are transferable top-CAM-specific BiTE, and that new BiTEs can be cra
o fulfill a demanding set of specifications as requiredevelopment of a biological therapeutic.
cknowledgements
This work was fully funded by Micromet AG. The authohank Monika Becker for excellent technical assistance inimal experiments and Dr. B. Elbe for immunohistoch
stry.
eferences
drain, C., Murphy, B.M., Martin, S.J., 2004. Molecular ordering ofcaspase activation cascade initiated by the CTL/NK protease granB. J. Biol. Chem. 280, 4663–4673.
ntunes, J.L., Toporcov, T.N., de Andrade, F.P., 2003. Trends and paof cancer mortality in European countries. Eur. J. Cancer Prev367–372.
monoclonal antibody targeting Ep-CAM in patients with advanadenocarcinomas. Clin. Cancer Res. 10, 7555–7565.
e Visser, K.E., Schumacher, T.N., Kruisbeek, A.M., 2003. CD8+ Ttolerance and cancer immunotherapy. J. Immunother. 26, 1–11.
reier, T., Baeuerle, P.A., Fichtner, I., Grun, M., Schlereth,Lorenczewski, G., Kufer, P., Lutterbuse, R., Riethmuller,Gjorstrup, P., Bargou, R.C., 2003. T cell costimulus-independenvery efficacious inhibition of tumor growth in mice bearing scutaneous or leukemic human B cell lymphoma xenograftsCD19-/CD3-bispecific single-chain antibody construct. J. Immu170, 4397–4402.
reier, T., Lorenczewski, G., Brandl, C., Hoffmann, P., Syring,Hanakam, F., Kufer, P., Riethmuller, G., Bargou, R., Baeuerle,2002. Extremely potent, rapid and costimulation-independenttoxic T-cell response against lymphoma cells catalyzed by a sichain bispecific antibody. Int. J. Cancer 100, 690–697.
udley, M.E., Rosenberg, S.A., 2003. Adoptive-cell-transfer therapthe treatment of patients with cancer. Nat. Rev. Cancer 3, 666–
udley, M.E., Wunderlich, J.R., Yang, J.C., Sherry, R.M., Topalian,Restifo, N.P., Royal, R.E., Kammula, U., White, D.E., MavroukaS.A., Rogers, L.J., Gracia, G.J., Jones, S.A., Mangiameli, D.P.letier, M.M., Gea-Banacloche, J., Robinson, M.R., Berman, DFilie, A.C., Abati, A., Rosenberg, S.A., 2005. Adoptive cell trantherapy following non-myeloablative but lymphodepleting chemoapy for the treatment of patients with refractory metastatic melanJ. Clin. Oncol. 23, 2346–2357.
aulkner, R.D., Craddock, C., Byrne, J.L., Mahendra, P., Haynes,Prentice, H.G., Potter, M., Pagliuca, A., Ho, A., Devereux,McQuaker, G., Mufti, G., Yin, J.L., Russell, N.H., 2004. BEAalemtuzumab reduced-intensity allogeneic stem cell transplanfor lymphoproliferative diseases: GVHD, toxicity, and survival inpatients. Blood 103, 428–434.
lieger, D., Kufer, P., Beier, I., Sauerbruch, T., Schmidt-Wolf, I.G., 2A bispecific single-chain antibody directed against EpCAM/CDcombination with the cytokines interferon alpha and interleukin-2ciently retargets T and CD3+CD56+ natural-killer-like T lymphocy

1142 K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143
to EpCAM-expressing tumor cells. Cancer Immunol. Immunother. 49,441–448.
Foss, F.M., 2002. Immunologic mechanisms of antitumor activity. Semin.Oncol. 29, 5–11.
Furman, R.R., Coleman, M., Leonard, J.P., 2004. Epratuzumab in non-Hodgkin’s lymphomas. Curr. Treat Options Oncol. 5, 283–288.
Gennari, R., Menard, S., Fagnoni, F., Ponchio, L., Scelsi, M., Tagliabue,E., Castiglioni, F., Villani, L., Magalotti, C., Gibelli, N., Oliviero, B.,Ballardini, B., Da Prada, G., Zambelli, A., Costa, A., 2004. Pilot studyof the mechanism of action of preoperative trastuzumab in patientswith primary operable breast tumors overexpressing HER2. Clin. Can-cer Res. 10, 5650–5655.
Glimcher, L.H., Townsend, M.J., Sullivan, B.M., Lord, G.M., 2004.Recent developments in the transcriptional regulation of cytolyticeffector cells. Nat. Rev. Immunol. 4, 900–911.
Grillo-Lopez, A.J., White, C.A., Varns, C., Shen, D., Wei, A., McClure,A., Dallaire, B.K., 1999. Overview of the clinical development ofrituximab: first monoclonal antibody approved for the treatment oflymphoma. Semin. Oncol. 26, 66–73.
Hale, G., Zhang, M.J., Bunjes, D., Prentice, H.G., Spence, D., Horowitz,M.M., Barrett, A.J., Waldmann, H., 1998. Improving the outcome ofbone marrow transplantation by using CD52 monoclonal antibodiesto prevent graft-versus-host disease and graft rejection. Blood 92,4581–4590.
Hoel, D.G., Davis, D.L., Miller, A.B., Sondik, E.J., Swerdlow, A.J., 1992.Trends in cancer mortality in 15 industrialized countries, 1969–1986.J. Natl. Cancer Inst. 84, 313–320.
Howe, H.L., Wingo, P.A., Thun, M.J., Ries, L.A., Rosenberg, H.M., Fei-gal, E.G., Edwards, B.K., 2001. Annual report to the nation on thestatus of cancer (1973 through 1998), featuring cancers with recentincreasing trends. J. Natl. Cancer Inst. 93, 824–842.
H rth,ara,umabectal
J ders,.,
of agG1virus
K wthncer.
K .H.,enti-
ients
K K.,ngell,, G.,ch,1Hstem
K cific
K ller,ant
imal193–
L , R.,ener,
elds,man-
ized anti-CD22 antibody, in aggressive non-Hodgkin’s lymphoma:phase I/II clinical trial results. Clin. Cancer Res. 10, 5327–5334.
Loffler, A., Gruen, M., Wuchter, C., Schriever, F., Kufer, P., Dreier, T.,Hanakam, F., Baeuerle, P.A., Bommert, K., Karawajew, L., Dorken,B., Bargou, R.C., 2003. Efficient elimination of chronic lympho-cytic leukaemia B cells by autologous T cells with a bispecificanti-CD19/anti-CD3 single-chain antibody construct. Leukemia 17,900–909.
Loffler, A., Kufer, P., Lutterbuse, R., Zettl, F., Daniel, P.T., Schwenken-becher, J.M., Riethmuller, G., Dorken, B., Bargou, R.C., 2000. Arecombinant bispecific single-chain antibody, CD19× CD3, inducesrapid and high lymphoma-directed cytotoxicity by unstimulated Tlymphocytes. Blood 95, 2098–2103.
Mack, M., Gruber, R., Schmidt, S., Riethmuller, G., Kufer, P., 1997. Bio-logic properties of a bispecific single-chain antibody directed against17-1A (EpCAM) and CD3: tumor cell-dependent T cell stimulationand cytotoxic activity. J. Immunol. 158, 3965–3970.
Mack, M., Riethmuller, G., Kufer, P., 1995. A small bispecific anti-body construct expressed as a functional single-chain molecule withhigh tumor cell cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 92, 7021–7025.
Maloney, D.G., 2001. Mechanism of action of rituximab. AnticancerDrugs 12 (Suppl. 2), S1–S4.
Matar, P., Rojo, F., Cassia, R., Moreno-Bueno, G., Di Cosimo, S.,Tabernero, J., Guzman, M., Rodriguez, S., Arribas, J., Palacios, J.,Baselga, J., 2004. Combined epidermal growth factor receptor tar-geting with the tyrosine kinase inhibitor gefitinib (ZD1839) and themonoclonal antibody cetuximab (IMC-C225): superiority over single-agent receptor targeting. Clin. Cancer Res. 10, 6487–6501.
Moldenhauer, G., Momburg, F., Moller, P., Schwartz, R., Hammerling,G.J., 1987. Epithelium-specific surface glycoprotein of Mr 34,000
r 56,
M 987.man
t tis-
M .G.,Ep-nsectal
M 04.and
N mentcells.
N am,.A.,
an100,
O 006.gle-Mol.
O un,d inerapy.
P , R.,Riva,
.J.,doc-nced
urwitz, H., Fehrenbacher, L., Novotny, W., Cartwright, T., HainswoJ., Heim, W., Berlin, J., Baron, A., Griffing, S., Holmgren, E., FerrN., Fyfe, G., Rogers, B., Ross, R., Kabbinavar, F., 2004. Bevacizplus irinotecan, fluorouracil, and leucovorin for metastatic colorcancer. N. Engl. J. Med. 350, 2335–2342.
ones, T.D., Hanlon, M., Smith, B.J., Heise, C.T., Nayee, P.D., SanD.A., Hamilton, A., Sweet, C., Unitt, E., Alexander, G., Lo, K.MGillies, S.D., Carr, F.J., Baker, M.P., 2004. The developmentmodified human IFN-alpha2b linked to the Fc portion of human Ias a novel potential therapeutic for the treatment of hepatitis Cinfection. J. Interferon Cytokine Res. 24, 560–572.
halil, M.Y., Grandis, J.R., Shin, D.M., 2003. Targeting epidermal grofactor receptor: novel therapeutics in the management of caExpert Rev. Anticancer Ther. 3, 367–380.
im, J.H., Herlyn, D., Wong, K.K., Park, D.C., Schorge, J.O., Lu, KSkates, S.J., Cramer, D.W., Berkowitz, R.S., Mok, S.C., 2003. Idfication of epithelial cell adhesion molecule autoantibody in patwith ovarian cancer. Clin. Cancer Res. 9, 4782–4791.
ottaridis, P.D., Milligan, D.W., Chopra, R., Chakraverty, R.Chakrabarti, S., Robinson, S., Peggs, K., Verfuerth, S., PetteR., Marsh, J.C., Schey, S., Mahendra, P., Morgan, G.J., HaleWaldmann, H., de Elvira, M.C., Williams, C.D., Devereux, S., LinD.C., Goldstone, A.H., Mackinnon, S., 2000. In vivo CAMPATH-prevents graft-versus-host disease following nonmyeloablativecell transplantation. Blood 96, 2419–2425.
ufer, P., Lutterbuse, R., Baeuerle, P.A., 2004. A revival of bispeantibodies. Trends Biotechnol. 22, 238–244.
ufer, P., Mack, M., Gruber, R., Lutterbuse, R., Zettl, F., RiethmuG., 1997. Construction and biological activity of a recombinbispecific single-chain antibody designed for therapy of minresidual colorectal cancer. Cancer Immunol. Immunother. 45,197.
eonard, J.P., Coleman, M., Ketas, J.C., Chadburn, A., FurmanSchuster, M.W., Feldman, E.J., Ashe, M., Schuster, S.J., WegW.A., Hansen, H.J., Ziccardi, H., Eschenberg, M., Gayko, U., FiS.Z., Cesano, A., Goldenberg, D.M., 2004. Epratuzumab, a hu
is a widely distributed human carcinoma marker. Br. J. Cance714–721.
omburg, F., Moldenhauer, G., Hammerling, G.J., Moller, P., 1Immunohistochemical study of the expression of a Mr 34,000 huepithelium-specific surface glycoprotein in normal and malignansues. Cancer Res. 47, 2883–2891.
osolits, S., Markovic, K., Frodin, J.E., Virving, L., Magnusson, CSteinitz, M., Fagerberg, J., Mellstedt, H., 2004. Vaccination withCAM protein or anti-idiotypic antibody induces Th1-biased respoagainst MHC class I- and II-restricted Ep-CAM epitopes in colorecarcinoma patients. Clin. Cancer Res. 10, 5391–5402.
unz, M., Kieu, C., Mack, B., Schmitt, B., Zeidler, R., Gires, O., 20The carcinoma-associated antigen EpCAM upregulates c-mycinduces cell proliferation. Oncogene 23, 5748–5758.
agler, A., Condiotti, R., Lubina, A., Deutsch, V.R., 1997. Enhanceof megakaryocytopoiesis by Campath-1G-treated natural killerBone Marrow Transplant. 20, 525–531.
aundorf, S., Preithner, S., Mayer, P., Lippold, S., Wolf, A., HanakF., Fichtner, I., Kufer, P., Raum, T., Riethmuller, G., Baeuerle, PDreier, T., 2002. In vitro and in vivo activity of MT201, a fully hummonoclonal antibody for pancarcinoma treatment. Int. J. Cancer101–110.
ffner, S., Hofmeister, R., Romaniuk, A., Kufer, P., Baeuerle, P.A., 2Induction of regular cytolytic T cell synapses by bispecific sinchain antibody constructs on MHC class I-negative tumor cells.Immunol. 43, 763–771.
sta, W.A., Chen, Y., Mikhitarian, K., Mitas, M., Salem, M., HannY.A., Cole, D.J., Gillanders, W.E., 2004. EpCAM is overexpressebreast cancer and is a potential target for breast cancer gene thCancer Res. 64, 5818–5824.
egram, M.D., Pienkowski, T., Northfelt, D.W., Eiermann, W., PatelFumoleau, P., Quan, E., Crown, J., Toppmeyer, D., Smylie, M.,A., Blitz, S., Press, M.F., Reese, D., Lindsay, M.A., Slamon, D2004. Results of two open-label, multicenter phase II studies ofetaxel, platinum salts, and trastuzumab in HER2-positive advabreast cancer. J. Natl. Cancer Inst. 96, 759–769.

K. Brischwein et al. / Molecular Immunology 43 (2006) 1129–1143 1143
Porakishvili, N., Kardava, L., Jewell, A.P., Yong, K., Glennie, M.J., Akbar,A., Lydyard, P.M., 2004. Cytotoxic CD4+ T cells in patients with Bcell chronic lymphocytic leukemia kill via a perforin-mediated path-way. Haematologica 89, 435–443.
Prang, M., Preithner, S., Brischwein, K., Goster, P., Woppel, A., Muller,J., Carola Steiger, C., Malte Peters, M., Baeuerle, P., da Silva, A.,2005. Cellular and complement-dependent cytotoxicity of Ep-CAMspecific monoclonal antibody MT201 against breast cancer cell lines.Br. J. Cancer 92, 342–349.
Raum, T., Gruber, R., Riethmuller, G., Kufer, P., 2001. Anti-self anti-bodies selected from a human IgD heavy chain repertoire: a novelapproach to generate therapeutic human antibodies against tumor-associated differentiation antigens. Cancer Immunol. Immunother. 50,141–150.
Riethmuller, G., Schneider-Gadicke, E., Schlimok, G., Schmiegel, W.,Raab, R., Hoffken, K., Gruber, R., Pichlmaier, H., Hirche, H.,Pichlmayr, R., et al., 1994. Randomised trial of monoclonal anti-body for adjuvant therapy of resected Dukes’ C colorectal carcinoma.German Cancer Aid 17-1A Study Group. Lancet 343, 1177–1183.
Schlereth, B., Fichtner, I., Lorenczewski, G., Kleindienst, P., Brischwein,K., da Silva, A., Kufer, P., Lutterbuese, R., Junghahn, I., Kasimir-Bauer, S., Wimberger, P., Kimmig, R., Baeuerle, P.A., 2005. Erad-ication of tumors from a human colon cancer cell line and fromovarian cancer metastases in immunodeficient mice by a single-chainEp-CAM-/CD3-bispecific antibody construct. Cancer Res. 65, 2882–2889.
Smith, M.R., 2003. Rituximab (monoclonal anti-CD20 antibody): mech-anisms of action and resistance. Oncogene 22, 7359–7368.
Spizzo, G., Obrist, P., Ensinger, C., Theurl, I., Dunser, M., Ramoni,A., Gunsilius, E., Eibl, G., Mikuz, G., Gastl, G., 2002. Prognostic
significance of Ep-CAM AND Her-2/neu overexpression in invasivebreast cancer. Int. J. Cancer 98, 883–888.
Spizzo, G., Went, P., Dirnhofer, S., Obrist, P., Simon, R., Spichtin, H.,Maurer, R., Metzger, U., Von Castelberg, B., Bart, R., Stopatschin-skaya, S., Kochli, O.R., Haas, P., Mross, F., Zuber, M., Dietrich, H.,Bischoff, S., Mirlacher, M., Sauter, G., Gastl, G., 2004. High Ep-CAM expression is associated with poor prognosis in node-positivebreast cancer. Breast Cancer Res. Treat. 86, 207–213.
Vogel, C., Cobleigh, M.A., Tripathy, D., Gutheil, J.C., Harris, L.N.,Fehrenbacher, L., Slamon, D.J., Murphy, M., Novotny, W.F., Burch-more, M., Shak, S., Stewart, S.J., 2001. First-line, single-agent Her-ceptin(trastuzumab) in metastatic breast cancer: a preliminary report.Eur. J. Cancer 37 (Suppl 1), S25–S29.
Weng, W.K., Levy, R., 2003. Two immunoglobulin G fragment C recep-tor polymorphisms independently predict response to rituximab inpatients with follicular lymphoma. J. Clin. Oncol. 21, 3940–3947.
Wimberger, P., Xiang, W., Mayr, D., Diebold, J., Dreier, T., Baeuerle,P.A., Kimmig, R., 2003. Efficient tumor cell lysis by autologous,tumor-resident T lymphocytes in primary ovarian cancer samples byan EP-CAM-/CD3-bispecific antibody. Int. J. Cancer 105, 241–248.
Winter, M.J., Nagelkerken, B., Mertens, A.E., Rees-Bakker, H.A., Briaire-de Bruijn, I.H., Litvinov, S.V., 2003. Expression of Ep-CAM shiftsthe state of cadherin-mediated adhesions from strong to weak. Exp.Cell. Res. 285, 50–58.
Wong, J.T., Eylath, A.A., Ghobrial, I., Colvin, R.B., 1990. The mechanismof anti-CD3 monoclonal antibodies. Mediation of cytolysis by inter-Tcell bridging. Transplantation 50, 683–689.
Xiang, J., Pan, Z., Attah-Poku, S., Babiuk, L., Zhang, Y., Liu, E., 1992.Production of hybrid bispecific antibody recognizing human colorectalcarcinoma and CD3 antigen. Mol. Biother. 4, 15–23.




















