Combined Natural Killer T-Cell Based Immunotherapy Eradicates Established Tumors in Mice
11
Combined Natural Killer T-Cell–Based Immunotherapy Eradicates Established Tumors in Mice Michele W.L. Teng, 1 Jennifer A. Westwood, 1 Phillip K. Darcy, 1,7 Janelle Sharkey, 1 Moriya Tsuji, 2 Richard W. Franck, 3 Steven A. Porcelli, 4 Gurdyal S. Besra, 5 Kazuyoshi Takeda, 1 Hideo Yagita, 6 Michael H. Kershaw, 1,7 and Mark J. Smyth 1,7 1 Cancer Immunology Program, Peter MacCallum Cancer Centre, East Melbourne, Victoria, Australia; 2 HIV and Malaria Vaccine Program, Aaron Diamond AIDS Research Center, The Rockefeller University; 3 Hunter College, City University of New York, New York, New York; 4 Department of Microbiology and Immunology, Albert Einstein College of Medicine, Bronx, New York; 5 School of Biosciences, University of Birmingham, Birmingham, United Kingdom; 6 Department of Immunology, Juntendo University School of Medicine, Tokyo, Japan; and 7 Department of Pathology, University of Melbourne, Parkville, Victoria, Australia Abstract A rational monoclonal antibody (mAb)-based antitumor therapy approach has previously been shown to eradicate various established experimental and carcinogen-induced tumors in a majority of mice. This therapy comprised an agonistic mAb reactive with tumor necrosis factor–related apoptosis-inducing ligand receptor (DR5), expressed by tumor cells, an agonistic anti-CD40 mAb to mature dendritic cells, and an agonistic anti-4-1BB mAb to costimulate CD8 + T cells. Because agonists of CD40 have been toxic in patients, we were interested in substituting anti-CD40 mAb with other dendritic cell–maturing agents, such as glycolipid ligands recognized by invariant natural killer T (iNKT) cells. Here, we show that CD1d-restricted glycolipid ligands for iNKT cells effectively substitute for anti-CD40 mAb and reject established experi- mental mouse breast and renal tumors when used in combination with anti-DR5 and anti-4-1BB mAbs (termed ‘‘NKTMab’’ therapy). NKTMab therapy–induced tumor rejec- tion was dependent on CD4 + and CD8 + T cells, NKT cells, and the cytokine IFN-;. NKTMab therapy containing either A-galactosylceramide (A-GC) or A-C-galactosylceramide (A-c-GC) at high concentrations induced similar rates of tumor rejection in mice; however, toxicity was observed at the highest doses of A-GC (>250 ng/injection), limiting the use of this glycolipid. By contrast, even very low doses of A-c-GC (25 ng/injection) retained considerable antitumor activity when used in combination with anti-DR5/anti-4-1BB, and thus, A-c-GC showed a considerably greater therapeutic index. In summary, sequential tumor cell apoptosis and amplifica- tion of dendritic cell function by NKT cell agonists represents an exciting and novel approach for cancer treatment. [Cancer Res 2007;67(15):7495–504] Introduction Immunotherapy is now gaining recognition as a valid approach for the treatment of cancer. The promise of immunotherapy is based on graft versus leukemia activity following hematopoietic stem cell transplantation, the use of monoclonal antibodies (mAb) to target lymphoma (Rituximab; ref. 1), breast (Trastuzumab; ref. 2), and colorectal (bevacizumab and cetuximab; ref. 3) cancer, and the use of vaccines (4) or adoptive T-cell transfer (5) in melanoma. However, response rates using these therapies are generally low and are often not complete and tumors can relapse. Hence, it is our contention that, although mAbs have been one of the most successful and safe forms of immunotherapy to date, no single mAb against a tumor antigen is going to provide the ‘‘magic bullet’’ that eliminates established cancer. Tumor cell apoptosis is the basis of many cancer therapies, and several apoptosis-inducing receptors have been targeted with mAb or ligand with the aim of triggering tumor cell death. Recently, it has been shown that tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) preferentially induces apoptosis in tumor cells, but not normal cells, and that it plays a critical role in tumor immune surveillance (6–8). Hence, the TRAIL receptor is considered an attractive target for mAb-mediated tumor therapy. Indeed, experimental tumor models have shown the potent tumoricidal activities of recombinant soluble forms of TRAIL or agonistic mAb specific for cell death–inducing human TRAIL receptors (DR4 and DR5; ref. 7) and humanized versions have reached phase II clinical trials. We now also appreciate that tumor- specific CTL induction plays a critical role in the successful antitumor effects of some mAb-based therapies (7, 9, 10), and indeed, induction of CTL reactive with tumors has been a goal of most immunotherapies. We have shown that anti-mouse DR5 mAb generated adaptive tumor-specific immunity, and we hypothesized that additionally maturing dendritic cells and costimulating CTL might potentially result in an even more effective cancer therapy than anti-DR5 mAb alone. Indeed, we have now combined the tumor apoptosis mediated by anti-DR5 mAb with agonistic mAbs against CD40 and 4-1BB that mature dendritic cells and costimulate T cells, respectively. Surprisingly, this combined therapy that we have termed ‘‘TriMab’’ resulted in the eradication of several different preestablished tumors and multiorgan metas- tases in mice (11). Notably, however, the reported toxicity observed in experimental animal models and patients receiving agonists of CD40 (12–15), and a recent study suggesting that triggering of CD40 on endothelial cells might actually promote tumor growth (16), has given us the impetus to investigate substituting anti-CD40 mAb in the combination therapy with other agents that effectively mature dendritic cells. Natural killer T (NKT) cells are unique population of T cells capable of regulating a broad range of immune responses, including autoimmunity, allergy, infection, and tumor rejection (17, 18). NKT cells recognize glycolipids presented in the context Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/). Requests for reprints: Mark J. Smyth, Cancer Immunology Program, Peter MacCallum Cancer Centre, Locked Bag 1, A’Beckett Street, Melbourne, Victoria 8006, Australia. Phone: 61-3-9656-3728; Fax: 61-3-9656-1411; E-mail: [email protected]. I2007 American Association for Cancer Research. doi:10.1158/0008-5472.CAN-07-0941 www.aacrjournals.org 7495 Cancer Res 2007; 67: (15). August 1, 2007 Research Article Research. on February 16, 2016. © 2007 American Association for Cancer cancerres.aacrjournals.org Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Combined Natural Killer T-Cell Based Immunotherapy Eradicates Established Tumors in Mice

Combined Natural Killer T-Cell–Based Immunotherapy
Eradicates Established Tumors in Mice
Michele W.L. Teng,1Jennifer A. Westwood,
1Phillip K. Darcy,
1,7Janelle Sharkey,
1
Moriya Tsuji,2Richard W. Franck,
3Steven A. Porcelli,
4Gurdyal S. Besra,
5
Kazuyoshi Takeda,1Hideo Yagita,
6Michael H. Kershaw,
1,7and Mark J. Smyth
1,7
1Cancer Immunology Program, Peter MacCallum Cancer Centre, East Melbourne, Victoria, Australia; 2HIV and Malaria VaccineProgram, Aaron Diamond AIDS Research Center, The Rockefeller University; 3Hunter College, City University of New York, New York,New York; 4Department of Microbiology and Immunology, Albert Einstein College of Medicine, Bronx, New York; 5School of Biosciences,University of Birmingham, Birmingham, United Kingdom; 6Department of Immunology, Juntendo University School of Medicine, Tokyo,Japan; and 7Department of Pathology, University of Melbourne, Parkville, Victoria, Australia
Abstract
A rational monoclonal antibody (mAb)-based antitumortherapy approach has previously been shown to eradicatevarious established experimental and carcinogen-inducedtumors in a majority of mice. This therapy comprised anagonistic mAb reactive with tumor necrosis factor–relatedapoptosis-inducing ligand receptor (DR5), expressed by tumorcells, an agonistic anti-CD40 mAb to mature dendritic cells,and an agonistic anti-4-1BB mAb to costimulate CD8+ T cells.Because agonists of CD40 have been toxic in patients, we wereinterested in substituting anti-CD40 mAb with other dendriticcell–maturing agents, such as glycolipid ligands recognized byinvariant natural killer T (iNKT) cells. Here, we show thatCD1d-restricted glycolipid ligands for iNKT cells effectivelysubstitute for anti-CD40 mAb and reject established experi-mental mouse breast and renal tumors when used incombination with anti-DR5 and anti-4-1BB mAbs (termed‘‘NKTMab’’ therapy). NKTMab therapy–induced tumor rejec-tion was dependent on CD4+ and CD8+ T cells, NKT cells, andthe cytokine IFN-;. NKTMab therapy containing eitherA-galactosylceramide (A-GC) or A-C-galactosylceramide(A-c-GC) at high concentrations induced similar rates oftumor rejection in mice; however, toxicity was observed at thehighest doses of A-GC (>250 ng/injection), limiting the use ofthis glycolipid. By contrast, even very low doses of A-c-GC(25 ng/injection) retained considerable antitumor activitywhen used in combination with anti-DR5/anti-4-1BB, andthus, A-c-GC showed a considerably greater therapeutic index.In summary, sequential tumor cell apoptosis and amplifica-tion of dendritic cell function by NKT cell agonists representsan exciting and novel approach for cancer treatment. [CancerRes 2007;67(15):7495–504]
Introduction
Immunotherapy is now gaining recognition as a valid approachfor the treatment of cancer. The promise of immunotherapy isbased on graft versus leukemia activity following hematopoieticstem cell transplantation, the use of monoclonal antibodies (mAb)
to target lymphoma (Rituximab; ref. 1), breast (Trastuzumab; ref. 2),and colorectal (bevacizumab and cetuximab; ref. 3) cancer, and theuse of vaccines (4) or adoptive T-cell transfer (5) in melanoma.However, response rates using these therapies are generally lowand are often not complete and tumors can relapse. Hence, it is ourcontention that, although mAbs have been one of the mostsuccessful and safe forms of immunotherapy to date, no singlemAb against a tumor antigen is going to provide the ‘‘magic bullet’’that eliminates established cancer.Tumor cell apoptosis is the basis of many cancer therapies, and
several apoptosis-inducing receptors have been targeted with mAbor ligand with the aim of triggering tumor cell death. Recently, ithas been shown that tumor necrosis factor (TNF)-relatedapoptosis-inducing ligand (TRAIL) preferentially induces apoptosisin tumor cells, but not normal cells, and that it plays a critical rolein tumor immune surveillance (6–8). Hence, the TRAIL receptor isconsidered an attractive target for mAb-mediated tumor therapy.Indeed, experimental tumor models have shown the potenttumoricidal activities of recombinant soluble forms of TRAIL oragonistic mAb specific for cell death–inducing human TRAILreceptors (DR4 and DR5; ref. 7) and humanized versions havereached phase II clinical trials. We now also appreciate that tumor-specific CTL induction plays a critical role in the successfulantitumor effects of some mAb-based therapies (7, 9, 10), andindeed, induction of CTL reactive with tumors has been a goal ofmost immunotherapies. We have shown that anti-mouse DR5 mAbgenerated adaptive tumor-specific immunity, and we hypothesizedthat additionally maturing dendritic cells and costimulating CTLmight potentially result in an even more effective cancer therapythan anti-DR5 mAb alone. Indeed, we have now combined thetumor apoptosis mediated by anti-DR5 mAb with agonistic mAbsagainst CD40 and 4-1BB that mature dendritic cells andcostimulate T cells, respectively. Surprisingly, this combinedtherapy that we have termed ‘‘TriMab’’ resulted in the eradicationof several different preestablished tumors and multiorgan metas-tases in mice (11). Notably, however, the reported toxicity observedin experimental animal models and patients receiving agonists ofCD40 (12–15), and a recent study suggesting that triggering ofCD40 on endothelial cells might actually promote tumor growth(16), has given us the impetus to investigate substituting anti-CD40mAb in the combination therapy with other agents that effectivelymature dendritic cells.Natural killer T (NKT) cells are unique population of T cells
capable of regulating a broad range of immune responses,including autoimmunity, allergy, infection, and tumor rejection(17, 18). NKT cells recognize glycolipids presented in the context
Note: Supplementary data for this article are available at Cancer Research Online(http://cancerres.aacrjournals.org/).
Requests for reprints:Mark J. Smyth, Cancer Immunology Program, PeterMacCallumCancer Centre, Locked Bag 1, A’Beckett Street, Melbourne, Victoria 8006, Australia. Phone:61-3-9656-3728; Fax: 61-3-9656-1411; E-mail: [email protected].
I2007 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-07-0941
www.aacrjournals.org 7495 Cancer Res 2007; 67: (15). August 1, 2007
Research Article
Research. on February 16, 2016. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
of MHC-1–like molecule, CD1d, and in mice the invariant NKT(iNKT) cell subpopulation expresses a biased T-cell receptor(TCR) repertoire characterized by an invariant Va14-Ja18 chaincoupled with either Vh8.2, 7, or 2, whereas human iNKT cellsexpress the homologous TCR gene Va24-Ja18 in conjunction withVb11 (17). The most commonly used glycolipid ligand for thestudy of NKT cell activation is a-galactosylceramide (a-GC). a-GC is presented by CD1d-expressing antigen-presenting cells(APC) and potently activates iNKT cells to rapidly produce both Thelper (Th) 1 and Th2 cytokines, such as IFN-g and interleukin(IL)-4 (17). Importantly, activation of iNKT cells with a-GC leadsto potent downstream activation of CD8+ T cells, natural killer(NK) cells, and APC, such as dendritic cells and B cells (19–21).Bystander activation of these cells is crucial to the protectiveantitumor and microbial immunity mediated by a-GC. Further-more, given the potent immunoregulatory role and antitumorfunctions of iNKT cells in mice, a-GC has been used in clinicaltrials as an antitumor agent (22–26). a-GC seems to have limitedantitumor activity when used alone, but it does cause down-stream activation of T cells (26). Thus, the ability of a-GC–activated iNKT cells to cross-talk with mature dendritic cellsmakes a-GC and some related analogues attractive replacementsfor agonistic anti-CD40 mAbs.
In this study, we have shown that substituting glycolipid ligandsfor anti-CD40 mAb in combination with anti-DR5 and anti-CD137mAbs (termed ‘‘NKTMab’’ therapy) is similarly effective to TriMabtherapy in rejecting established experimental tumors. These dataillustrate for the first time the utility of some CD1d-binding iNKTcell ligands when used in combination therapies that both causetumor cell apoptosis directly and costimulate resultant tumor-specific T-cell immunity.
Materials and Methods
MiceInbred BALB/c mice were purchased from The Walter and Eliza Hall
Institute of Medical Research. BALB/c IFN-g�/�, BALB/c IL-4�/�, BALB/cIL-13�/�, and BALB/c Ja18-deficient mice (Ja18�/�) were bred and
maintained at the Peter MacCallum Cancer Centre (PMCC). All gene-targeted mice were backcrossed onto the BALB/c background for at least 10
generations and genotyped by either PCR or flow cytometry. Mice >6 weeks
old were used in all experiments. Female mice were used for all experimentswith the 4T1 tumor, and all experiments were done in accordance with
guidelines set out by the PMCC animal experimentation ethics committee.
Tumor Cell LinesBALB/c-derived TRAIL-sensitive 4T1 mammary carcinoma (27, 28),
TRAIL-sensitive Renca renal carcinoma (29), TRAIL-sensitive Renca variant
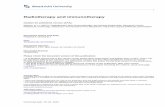
Figure 1. Comparable eradication of established R331 tumors by NKTMab or TriMab therapy. Groups of BALB/c mice (n = 5) were inoculated s.c. with therenal carcinoma cell line R331 (5 � 105). On days 7, 11, and 15 after tumor inoculation, mice were i.p. treated with either NKTMab (n) or TriMab (.) therapy orwith anti-DR5/a-GC (D), anti-4-1BB/a-GC (E), or control Ig (cIg ; 5). A to D, decreasing doses of a-GC (500, 250, 100, and 50 ng) were used in theNKTMab therapy or in combination with anti-4-1BB or anti-DR5 mAb. Points, mean of tumor sizes; bars, SE. Parentheses, tumor rejection rates. Statistical differencesin tumor size between control Ig/vehicle-treated and NKTMab-treated mice were determined by Mann-Whitney rank sum test. *, P < 0.05. Similar results wereobtained in two other independent experiments.
Cancer Research
Cancer Res 2007; 67: (15). August 1, 2007 7496 www.aacrjournals.org
Research. on February 16, 2016. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
R331, and TRAIL-resistant variant R331-FLIP (27, 29) were maintained asdescribed previously.
Antibodies and GlycolipidsWe prepared and purified agonistic mAb to mouse DR5 (MD5-1),
agonistic mAb to mouse CD40 (FGK45; provided by A. Rolink, University ofBasel, Basel, Switzerland; ref. 30), agonistic mAb to mouse 4-1BB (CD137;
3H3; provided by R. Mittler, Emory University, Atlanta, GA; ref. 31), mAb to
mouse CD4 (GK1.5), and mAb to mouse CD8 (53-6.7) as described
previously (7). We purchased anti-asialo-GM1 (ASGM1) from Wako PureChemicals. a-GC was provided by the Pharmaceutical Research Laborato-
ries, Kirin Brewery, and prepared as described (32). a-C-galactosylceramide
(a-c-GC) and a-GC C20:2 (C20:2) were synthesized as described (33, 34).
Glycolipids and the control vehicle were resuspended in saline supple-mented with 0.5% polysorbate-20 (w/v).
Measuring Serum TransaminasesGroups of BALB/c wild-type (WT) mice were inoculated s.c. with 2 �
105 4T1 tumor cells on day 0. Seven days after tumor inoculation, groupsof mice were either left untreated or treated i.p. with the following:
NKTMab therapy (100 Ag each of anti-DR5 and anti-4-1BB mAbs and the
indicated dose of a-GC or a-c-GC), TriMab therapy (100 Ag each of anti-
DR5, anti-4-1BB, and anti-CD40 mAbs), and anti-4-1BB or anti-DR5 mAbalone or in combination or a-GC alone at the indicated dose at day 7
(treatment 1) and day 11 (treatment 2). Mice were eye bled the day after
treatment 1 (day 8), before treatment 2 (day 11), and a day after treatment
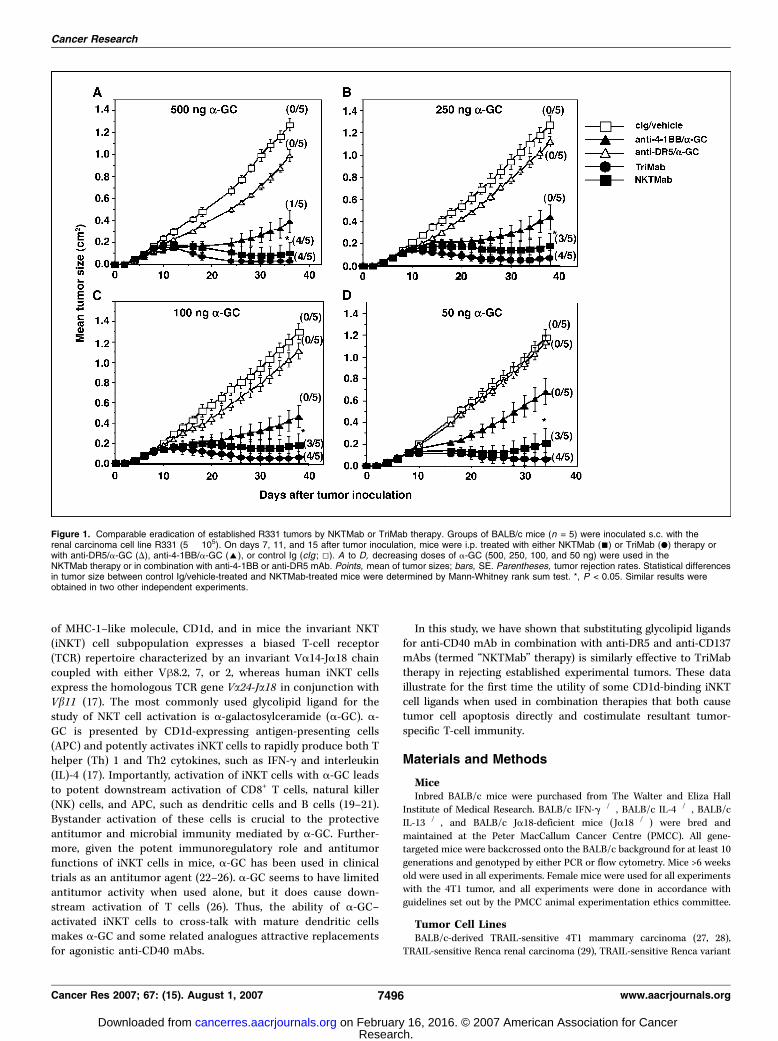
Figure 2. Serum ALT levels of tumor-bearing mice following NKTMab therapy.Groups of BALB/c mice (n = 3–7) wereinoculated s.c. with the mammarycarcinoma cell line 4T1 (2 � 105). Sevendays after tumor inoculation, mice wereeither left untreated (C) or injected i.p.with mAbs and/or a-GC (T) at days 7(treatment 1) and 11 (treatment 2). Micewere eye bled after treatment 1 (Post Txt 1),before treatment 2 (Pre Txt 2), aftertreatment 2 (Post Txt 2), and 4 d aftertreatment 2 (4 days post Txt 2), and theirsera were assayed for the presence of ALT.A, ALT levels of tumor-bearing mice afterNKTMab (containing 500 ng a-GC) therapy.B, ALT levels of tumor-bearing mice eitherafter single-agent therapy [a-GC (500 ng),anti-DR5 mAb (100 Ag), and anti-4-1BBmAb (100 Ag)], dual therapy [a-GC(500 ng)/anti-DR5 mAb (100 Ag) and a-GC(500 ng)/anti-4-1BB mAb (100 Ag)], orTriMab therapy. C, ALT levels of tumor-bearing mice after NKTMab therapycontaining decreasing doses of a-GC(25–500 ng). B and C, only ALT levels aftertreatment 1 and after treatment 2 are shown.Each point represents ALT level of a singlemouse and where shown the mean of thegroup is represented by the cross bar .
NKT Cell–Based Immunotherapy
www.aacrjournals.org 7497 Cancer Res 2007; 67: (15). August 1, 2007
Research. on February 16, 2016. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
2 (day 12). Briefly, blood collected from retro-orbital puncture was spun at
13,000 rpm for 15 min at 11jC. Sera were then harvested and either usedimmediately for analysis or stored at �20jC. Before analysis, sera were
thawed and diluted 1 in 5 or 1 in 10 in sterile saline and serum
transaminase [alanine aminotransferase (ALT) and aspartate aminotrans-
ferase (AST)] levels measured on the Advia 1200 Chemistry System (BayerHealthCare).
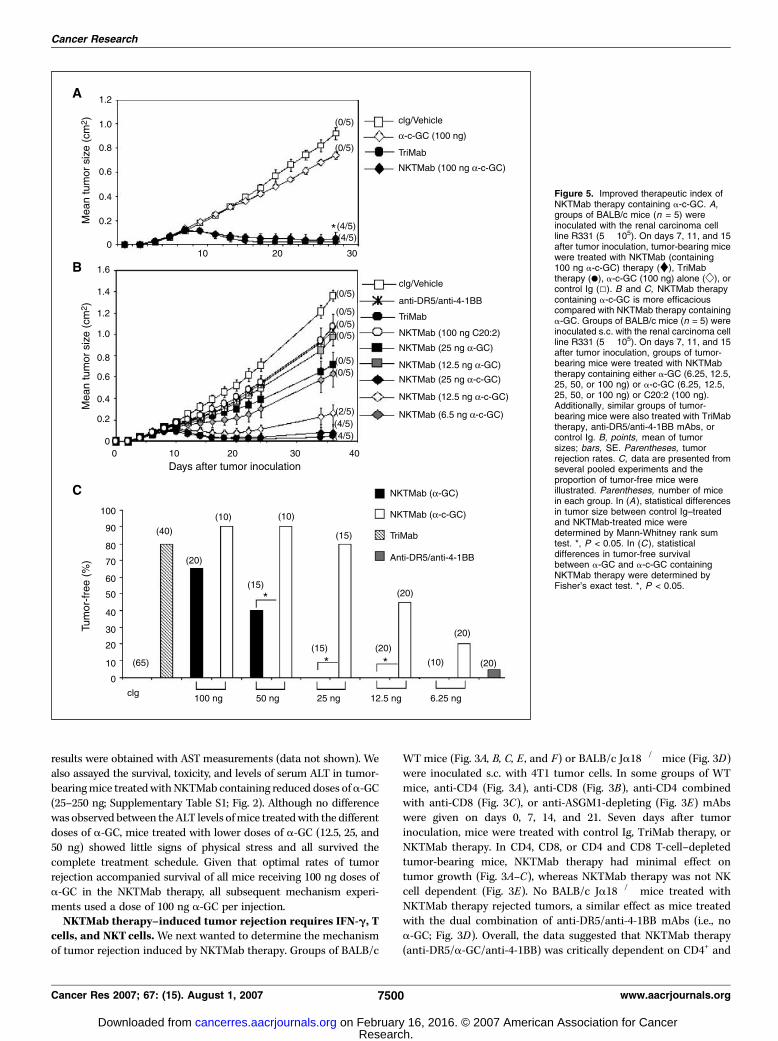
Therapy of Transplanted TumorsR331 renal tumor growth. Groups of five BALB/c WT mice were
inoculated s.c. with 5 � 105 R331 tumor cells on day 0. Seven days aftertumor inoculation, groups of mice were treated i.p. with the following:
control Ig (300 Ag), TriMab therapy (100 Ag each of anti-DR5, anti-4-1BB,
and anti-CD40 mAbs), NKTMab therapy (100 Ag each of anti-DR5, anti-4-1BB mAbs, and the indicated dose of a-GC, a-c-GC, or C20:2), a
combination of anti-4-1BB or anti-DR5 mAbs with the indicated dose ofa-GC, and a combination of anti-4-1BB and anti-DR5 mAbs or theindicated dose of a-c-GC or C20:2. Tumor size was measured with acaliper and recorded as the product of two perpendicular diameters(cm2).
4T1 mammary tumor growth. Groups of five BALB/c WT, BALB/cIFN-g�/�, BALB/c IL-4�/�, or BALB/c Ja18�/�, and BALB/c IL-13�/� mice wereinoculated s.c. with 2 � 105 4T1 tumor cells on day 0. Seven days after tumorinoculation, groups of mice were treated i.p. with the following: control Ig (100Ag), NKTMab therapy (100 Ag each of anti-DR5 and anti-4-1BB mAbs and 100 nga-GC), or a combination of anti-4-1BB and anti-DR5 mAbs (100 Ag each). Insome experiments, mice were depleted of CD4+ (GK1.5), CD8+ (53.6.72), acombination of CD4+ and CD8+ T cells, or NK cells on days 0, 7, 14, and 21.Tumor size was measured using a caliper and recorded as the product of twoperpendicular diameters (cm2).
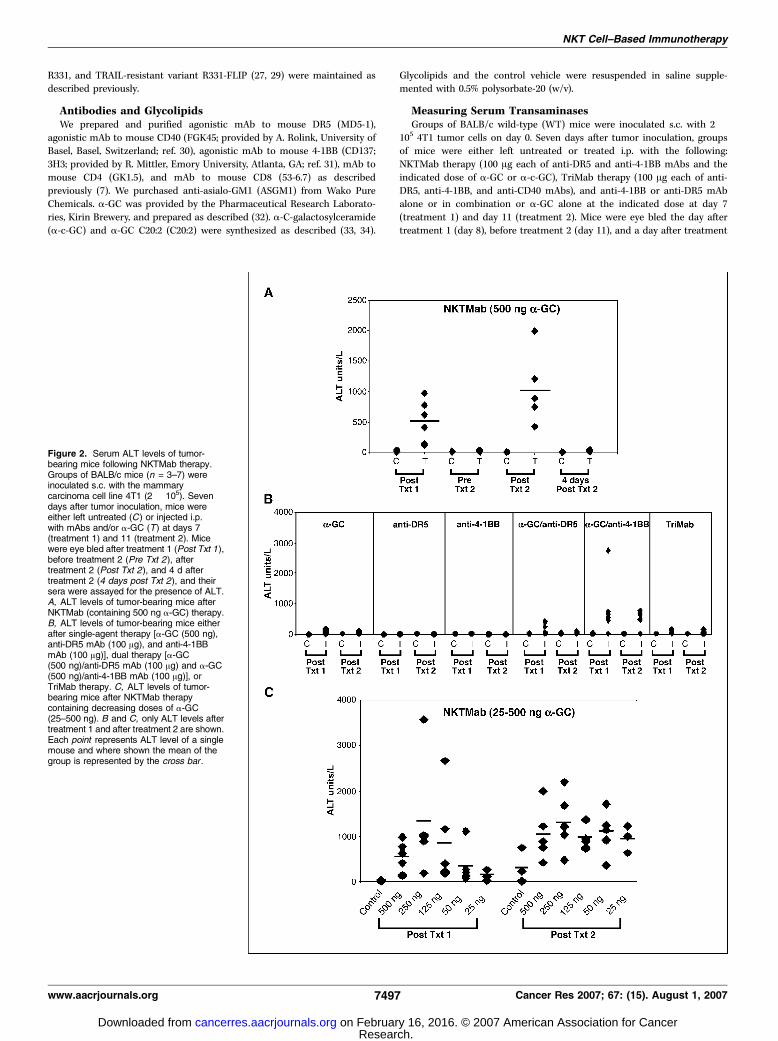
Figure 3. T cells and NKT cells arerequired for NKTMab-induced rejection oftumors. Groups of BALB/c WT (A, B, C, E ,and F ) or BALB/c Ja18�/� (D ) mice (n = 5)were inoculated s.c. with the mammarycarcinoma 4T1 (2 � 105). On days 7, 11,and 15 after tumor inoculation, mice weretreated with either NKTMab therapy(containing 100 ng a-GC; n), TriMabtherapy (.), anti-DR5/anti-4-1BB mAbs(D), or control Ig (5). Additionally, somegroups of mice were also i.p. injected ondays 0, 7, 14, and 21 with either CD4-depleting mAbs (A), CD8-depleting mAbs(B), CD4- and CD8-depleting mAbs (C),anti-ASGM1 antibody (E), or control Ig (F ).Points, mean of tumor sizes; bars, SE.Parentheses, tumor rejection rates.Statistical differences in tumor sizebetween WT mice and antibody-depletedWT mice receiving NKTMab therapy weredetermined by Mann-Whitney ranksum test. *, P < 0.05.
Cancer Research
Cancer Res 2007; 67: (15). August 1, 2007 7498 www.aacrjournals.org
Research. on February 16, 2016. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
Statistical AnalysisStatistical significance of tumor growth or tumor-free survival was assessed
through the use of the Mann-Whitney rank sum test or Fisher’s exact test as
appropriate.
Results
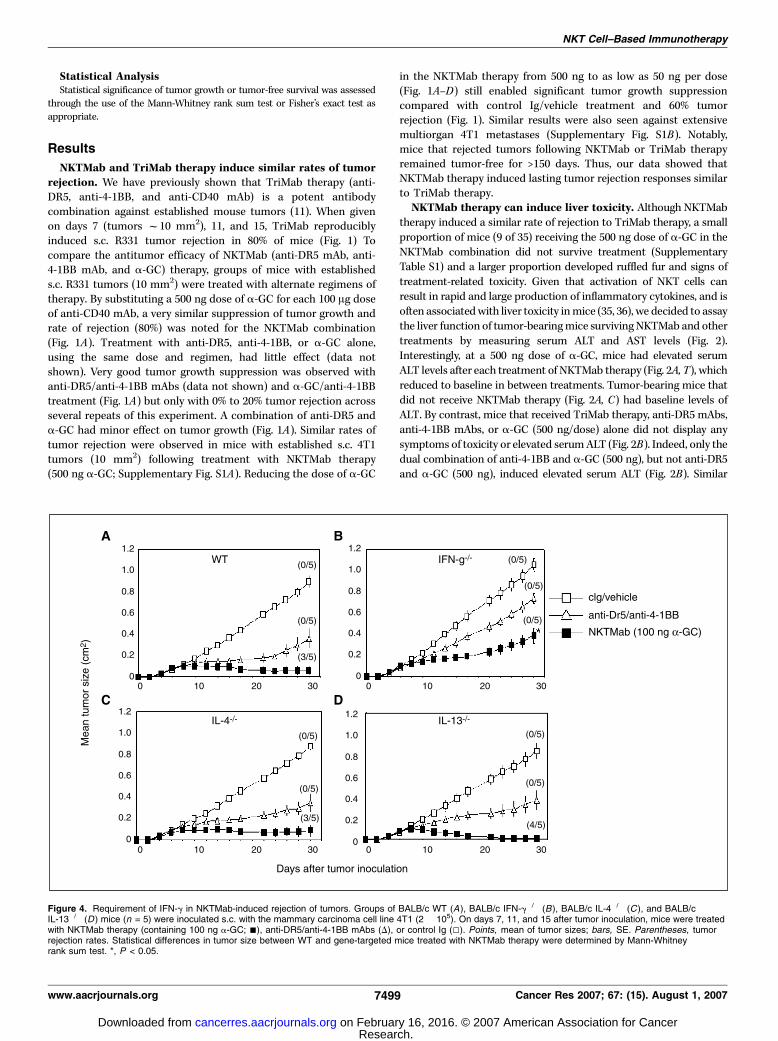
NKTMab and TriMab therapy induce similar rates of tumorrejection. We have previously shown that TriMab therapy (anti-DR5, anti-4-1BB, and anti-CD40 mAb) is a potent antibodycombination against established mouse tumors (11). When givenon days 7 (tumors f10 mm2), 11, and 15, TriMab reproduciblyinduced s.c. R331 tumor rejection in 80% of mice (Fig. 1) Tocompare the antitumor efficacy of NKTMab (anti-DR5 mAb, anti-4-1BB mAb, and a-GC) therapy, groups of mice with establisheds.c. R331 tumors (10 mm2) were treated with alternate regimens oftherapy. By substituting a 500 ng dose of a-GC for each 100 Ag doseof anti-CD40 mAb, a very similar suppression of tumor growth andrate of rejection (80%) was noted for the NKTMab combination(Fig. 1A). Treatment with anti-DR5, anti-4-1BB, or a-GC alone,using the same dose and regimen, had little effect (data notshown). Very good tumor growth suppression was observed withanti-DR5/anti-4-1BB mAbs (data not shown) and a-GC/anti-4-1BBtreatment (Fig. 1A) but only with 0% to 20% tumor rejection acrossseveral repeats of this experiment. A combination of anti-DR5 anda-GC had minor effect on tumor growth (Fig. 1A). Similar rates oftumor rejection were observed in mice with established s.c. 4T1tumors (10 mm2) following treatment with NKTMab therapy(500 ng a-GC; Supplementary Fig. S1A). Reducing the dose of a-GC
in the NKTMab therapy from 500 ng to as low as 50 ng per dose(Fig. 1A–D) still enabled significant tumor growth suppressioncompared with control Ig/vehicle treatment and 60% tumorrejection (Fig. 1). Similar results were also seen against extensivemultiorgan 4T1 metastases (Supplementary Fig. S1B). Notably,mice that rejected tumors following NKTMab or TriMab therapyremained tumor-free for >150 days. Thus, our data showed thatNKTMab therapy induced lasting tumor rejection responses similarto TriMab therapy.NKTMab therapy can induce liver toxicity. Although NKTMab
therapy induced a similar rate of rejection to TriMab therapy, a smallproportion of mice (9 of 35) receiving the 500 ng dose of a-GC in theNKTMab combination did not survive treatment (SupplementaryTable S1) and a larger proportion developed ruffled fur and signs oftreatment-related toxicity. Given that activation of NKT cells canresult in rapid and large production of inflammatory cytokines, and isoften associatedwith liver toxicity inmice (35, 36), we decided to assaythe liver function of tumor-bearingmice survivingNKTMab and othertreatments by measuring serum ALT and AST levels (Fig. 2).Interestingly, at a 500 ng dose of a-GC, mice had elevated serumALT levels after each treatment of NKTMab therapy (Fig. 2A, T), whichreduced to baseline in between treatments. Tumor-bearing mice thatdid not receive NKTMab therapy (Fig. 2A, C) had baseline levels ofALT. By contrast, mice that received TriMab therapy, anti-DR5 mAbs,anti-4-1BB mAbs, or a-GC (500 ng/dose) alone did not display anysymptoms of toxicity or elevated serumALT (Fig. 2B). Indeed, only thedual combination of anti-4-1BB and a-GC (500 ng), but not anti-DR5and a-GC (500 ng), induced elevated serum ALT (Fig. 2B). Similar
Figure 4. Requirement of IFN-g in NKTMab-induced rejection of tumors. Groups of BALB/c WT (A), BALB/c IFN-g�/� (B), BALB/c IL-4�/� (C ), and BALB/cIL-13�/� (D ) mice (n = 5) were inoculated s.c. with the mammary carcinoma cell line 4T1 (2 � 105). On days 7, 11, and 15 after tumor inoculation, mice were treatedwith NKTMab therapy (containing 100 ng a-GC; n), anti-DR5/anti-4-1BB mAbs (D), or control Ig (5). Points, mean of tumor sizes; bars, SE. Parentheses, tumorrejection rates. Statistical differences in tumor size between WT and gene-targeted mice treated with NKTMab therapy were determined by Mann-Whitneyrank sum test. *, P < 0.05.
NKT Cell–Based Immunotherapy
www.aacrjournals.org 7499 Cancer Res 2007; 67: (15). August 1, 2007
Research. on February 16, 2016. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
results were obtained with ASTmeasurements (data not shown). Wealso assayed the survival, toxicity, and levels of serum ALT in tumor-bearingmice treatedwithNKTMab containing reduced doses ofa-GC(25–250 ng; Supplementary Table S1; Fig. 2). Although no differencewas observed between the ALT levels ofmice treatedwith the differentdoses of a-GC, mice treated with lower doses of a-GC (12.5, 25, and50 ng) showed little signs of physical stress and all survived thecomplete treatment schedule. Given that optimal rates of tumorrejection accompanied survival of all mice receiving 100 ng doses ofa-GC in the NKTMab therapy, all subsequent mechanism experi-ments used a dose of 100 ng a-GC per injection.NKTMab therapy–induced tumor rejection requires IFN-;, T
cells, and NKT cells.We next wanted to determine the mechanismof tumor rejection induced by NKTMab therapy. Groups of BALB/c
WTmice (Fig. 3A, B, C, E , and F) or BALB/c Ja18�/� mice (Fig. 3D)were inoculated s.c. with 4T1 tumor cells. In some groups of WTmice, anti-CD4 (Fig. 3A), anti-CD8 (Fig. 3B), anti-CD4 combinedwith anti-CD8 (Fig. 3C), or anti-ASGM1-depleting (Fig. 3E) mAbswere given on days 0, 7, 14, and 21. Seven days after tumorinoculation, mice were treated with control Ig, TriMab therapy, orNKTMab therapy. In CD4, CD8, or CD4 and CD8 T-cell–depletedtumor-bearing mice, NKTMab therapy had minimal effect ontumor growth (Fig. 3A–C), whereas NKTMab therapy was not NKcell dependent (Fig. 3E). No BALB/c Ja18�/� mice treated withNKTMab therapy rejected tumors, a similar effect as mice treatedwith the dual combination of anti-DR5/anti-4-1BB mAbs (i.e., noa-GC; Fig. 3D). Overall, the data suggested that NKTMab therapy(anti-DR5/a-GC/anti-4-1BB) was critically dependent on CD4+ and
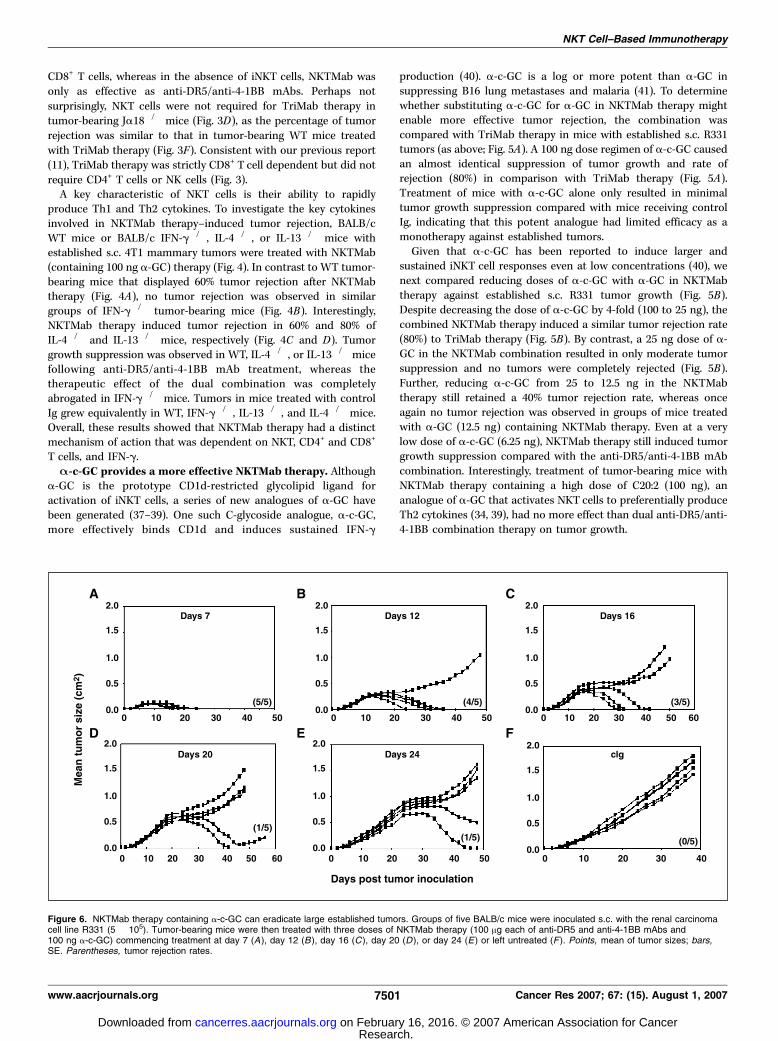
Figure 5. Improved therapeutic index ofNKTMab therapy containing a-c-GC. A,groups of BALB/c mice (n = 5) wereinoculated with the renal carcinoma cellline R331 (5 � 105). On days 7, 11, and 15after tumor inoculation, tumor-bearing micewere treated with NKTMab (containing100 ng a-c-GC) therapy (y), TriMabtherapy (.), a-c-GC (100 ng) alone ( w ), orcontrol Ig (5). B and C, NKTMab therapycontaining a-c-GC is more efficaciouscompared with NKTMab therapy containinga-GC. Groups of BALB/c mice (n = 5) wereinoculated s.c. with the renal carcinoma cellline R331 (5 � 105). On days 7, 11, and 15after tumor inoculation, groups of tumor-bearing mice were treated with NKTMabtherapy containing either a-GC (6.25, 12.5,25, 50, or 100 ng) or a-c-GC (6.25, 12.5,25, 50, or 100 ng) or C20:2 (100 ng).Additionally, similar groups of tumor-bearing mice were also treated with TriMabtherapy, anti-DR5/anti-4-1BB mAbs, orcontrol Ig. B, points, mean of tumorsizes; bars, SE. Parentheses, tumorrejection rates. C, data are presented fromseveral pooled experiments and theproportion of tumor-free mice wereillustrated. Parentheses, number of micein each group. In (A ), statistical differencesin tumor size between control Ig–treatedand NKTMab-treated mice weredetermined by Mann-Whitney rank sumtest. *, P < 0.05. In (C ), statisticaldifferences in tumor-free survivalbetween a-GC and a-c-GC containingNKTMab therapy were determined byFisher’s exact test. *, P < 0.05.
Cancer Research
Cancer Res 2007; 67: (15). August 1, 2007 7500 www.aacrjournals.org
Research. on February 16, 2016. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
CD8+ T cells, whereas in the absence of iNKT cells, NKTMab wasonly as effective as anti-DR5/anti-4-1BB mAbs. Perhaps notsurprisingly, NKT cells were not required for TriMab therapy intumor-bearing Ja18�/� mice (Fig. 3D), as the percentage of tumorrejection was similar to that in tumor-bearing WT mice treatedwith TriMab therapy (Fig. 3F). Consistent with our previous report(11), TriMab therapy was strictly CD8+ T cell dependent but did notrequire CD4+ T cells or NK cells (Fig. 3).A key characteristic of NKT cells is their ability to rapidly
produce Th1 and Th2 cytokines. To investigate the key cytokinesinvolved in NKTMab therapy–induced tumor rejection, BALB/cWT mice or BALB/c IFN-g�/�, IL-4�/�, or IL-13�/� mice withestablished s.c. 4T1 mammary tumors were treated with NKTMab(containing 100 ng a-GC) therapy (Fig. 4). In contrast to WT tumor-bearing mice that displayed 60% tumor rejection after NKTMabtherapy (Fig. 4A), no tumor rejection was observed in similargroups of IFN-g�/� tumor-bearing mice (Fig. 4B). Interestingly,NKTMab therapy induced tumor rejection in 60% and 80% ofIL-4�/� and IL-13�/� mice, respectively (Fig. 4C and D). Tumorgrowth suppression was observed in WT, IL-4�/�, or IL-13�/� micefollowing anti-DR5/anti-4-1BB mAb treatment, whereas thetherapeutic effect of the dual combination was completelyabrogated in IFN-g�/� mice. Tumors in mice treated with controlIg grew equivalently in WT, IFN-g�/�, IL-13�/�, and IL-4�/� mice.Overall, these results showed that NKTMab therapy had a distinctmechanism of action that was dependent on NKT, CD4+ and CD8+
T cells, and IFN-g.A-c-GC provides a more effective NKTMab therapy. Although
a-GC is the prototype CD1d-restricted glycolipid ligand foractivation of iNKT cells, a series of new analogues of a-GC havebeen generated (37–39). One such C-glycoside analogue, a-c-GC,more effectively binds CD1d and induces sustained IFN-g
production (40). a-c-GC is a log or more potent than a-GC insuppressing B16 lung metastases and malaria (41). To determinewhether substituting a-c-GC for a-GC in NKTMab therapy mightenable more effective tumor rejection, the combination wascompared with TriMab therapy in mice with established s.c. R331tumors (as above; Fig. 5A). A 100 ng dose regimen of a-c-GC causedan almost identical suppression of tumor growth and rate ofrejection (80%) in comparison with TriMab therapy (Fig. 5A).Treatment of mice with a-c-GC alone only resulted in minimaltumor growth suppression compared with mice receiving controlIg, indicating that this potent analogue had limited efficacy as amonotherapy against established tumors.Given that a-c-GC has been reported to induce larger and
sustained iNKT cell responses even at low concentrations (40), wenext compared reducing doses of a-c-GC with a-GC in NKTMabtherapy against established s.c. R331 tumor growth (Fig. 5B).Despite decreasing the dose of a-c-GC by 4-fold (100 to 25 ng), thecombined NKTMab therapy induced a similar tumor rejection rate(80%) to TriMab therapy (Fig. 5B). By contrast, a 25 ng dose of a-GC in the NKTMab combination resulted in only moderate tumorsuppression and no tumors were completely rejected (Fig. 5B).Further, reducing a-c-GC from 25 to 12.5 ng in the NKTMabtherapy still retained a 40% tumor rejection rate, whereas onceagain no tumor rejection was observed in groups of mice treatedwith a-GC (12.5 ng) containing NKTMab therapy. Even at a verylow dose of a-c-GC (6.25 ng), NKTMab therapy still induced tumorgrowth suppression compared with the anti-DR5/anti-4-1BB mAbcombination. Interestingly, treatment of tumor-bearing mice withNKTMab therapy containing a high dose of C20:2 (100 ng), ananalogue of a-GC that activates NKT cells to preferentially produceTh2 cytokines (34, 39), had no more effect than dual anti-DR5/anti-4-1BB combination therapy on tumor growth.
Figure 6. NKTMab therapy containing a-c-GC can eradicate large established tumors. Groups of five BALB/c mice were inoculated s.c. with the renal carcinomacell line R331 (5 � 105). Tumor-bearing mice were then treated with three doses of NKTMab therapy (100 Ag each of anti-DR5 and anti-4-1BB mAbs and100 ng a-c-GC) commencing treatment at day 7 (A ), day 12 (B ), day 16 (C ), day 20 (D ), or day 24 (E) or left untreated (F ). Points, mean of tumor sizes; bars,SE. Parentheses, tumor rejection rates.
NKT Cell–Based Immunotherapy
www.aacrjournals.org 7501 Cancer Res 2007; 67: (15). August 1, 2007
Research. on February 16, 2016. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

The significant improvement in antitumor efficacy when a-c-GC,rather than a-GC, was used in the NKTMab therapy was bestillustrated by a statistical improvement in tumor-free survival inmice with R331 treated with either 50, 25, or 12.5 ng of glycolipid(Fig. 5C). Importantly, NKTMab (containing a-GC or a-c-GC) wasnot as effective against a TRAIL-resistant tumor cell transfectant ofR331 (R331-FLIP; Supplementary Fig. S2), consistent with ourprevious studies that illustrated the resistance of FLIP-expressingtumors to DR5-mediated therapy (7, 11). Very similar efficacy ofa-c-GC containing NKTMab therapy was also observed againstextensive multiorgan 4T1 metastases (Supplementary Fig. S3). Todetermine the potency of a-c-GC containing NKTMab therapy, wehave also examined the efficacy of NKTMab therapy (100 ng a-c-GC) against increasingly large s.c. R331 tumors (Fig. 6). Impres-sively, tumor rejection rates of 60% to 80% were observed whentreatment commenced on day 12 (mean tumor size, 0.26 cm2) orday 16 (mean tumor size, 0.38 cm2; Fig. 6B and C). Remarkably,starting NKTMab (a-c-GC) therapy of mice on day 20 with meantumor size equal to 0.55 cm2 still induced significant tumor growthsuppression and tumor rejection in 20% of treated mice (Fig. 6D).The efficacy of NKTMab (a-c-GC) and TriMab therapy in BALB/cmice bearing established orthotopic Renca tumors was also shown(Supplementary Fig. S4).A-c-GC provides a safer NKTMab therapy. Surprisingly, mice
receivinga-c-GC inNKTMab combination therapy showed no signs oftoxicity. Given that liver toxicity was observed in tumor-bearing micetreatedwith NKTMab therapy containinga-GC, we next assessed seralevels of ALT in tumor-bearing mice following NKTMab therapycontaining the highest dose of a-c-GC (500 ng/dose). Unlike micetreatedwithNKTMab therapy containinga-GC, only very low levels ofALT were induced following NKTMab (a-c-GC) therapy and theselevels were not increased following the second injection of thetreatment schedule (Supplementary Fig. S5). These data indicatedthat NKTMab therapy (comprising anti-DR5, anti-4-1BB, and a-c-GC)not only induced a similar level of tumor rejection to TriMab therapybut did so in the absence of any appreciable toxicity.
Discussion
In this study, we have shown that CD1d-restricted glycolipidligands reactive with iNKT cells effectively substitute for anti-CD40mAbs and can reject established experimental mouse breast andrenal tumors when used in combination with anti-DR5 and anti-4-1BB mAbs. This combination, which we termed NKTMab therapy,induced tumor rejection that required CD4+ and CD8+ T cells, NKTcells, and the cytokine IFN-g. NKTMab therapy containing eithera-GC or a-c-GC at higher concentrations induced similar rates oftumor rejection inmice; however, toxicity was observed at the highestdoses of a-GC (>250 ng/injection). By contrast, very low doses ofa-c-GC (25 ng/injection) retained considerable antitumor activitywhen used in combination with anti-DR5/anti-4-1BB, and thus,a-c-GC showed a considerably greater therapeutic index. Given theshown toxicities of CD40 agonists in humans and mice (13, 16),this study illustrates the alternative of using NKT cell agonists thatin synergy with an anti-DR5/anti-4-1BB combination preferentiallydrive IFN-g production to suppress tumor growth and metastases.Although TriMab (anti-DR5/anti-CD40/anti-4-1BB) has shown
tremendous promise in preclinical mouse models (11), the knowntoxicities of CD40 agonists in humans may limit the clinical utilityof this approach. The effective replacement of anti-CD40 mAbswith glycolipids in our combination therapy approach represents a
feasible alternative of activating and maturing dendritic cells in asimilarly effective tumor immunotherapy. The reciprocal activationof NKT cells and dendritic cells is initiated on the presentation ofglycolipids on CD1d molecules by resting dendritic cells to NKTcells. NKTcells are thus induced to up-regulate CD40L, Th1, and Th2cytokines and chemokines, whereas CD40 cross-linking inducesdendritic cells to up-regulate CD80, CD86, and IL-12, which in turnenhances NKT cell activation and cytokine production (17).Although a-GC has been the prototype CD1d ligand used in the
activation of iNKT cells to exert antitumor effects in experimentalmodels of cancers, several new structurally distinct analogues of a-GC have been synthesized (37–41). Recently, a synthetic isosteric C-glycoside analogue of a-GC (a-c-GC) was synthesized and reportedto exhibit a 1,000-fold more potent antimalaria activity and a 100-fold more potent antimetastatic activity than a-GC (41). Theimproved antitumor efficacy of a-c-GC over a-GC was recentlyshown by Fujii et al. (40) to be due to the ability of a-c-GC to bindmore stably to dendritic cells, resulting in more prolongedproduction of Th1 cytokines, such as IL-12 and IFN-g, but notIL-4 or TNF-a. By substituting a-GC with a-c-GC in our NKTMabtherapy, we showed the improved antitumor efficacy of a-c-GC toinduce tumor rejection at doses considerably lower than a-GC. Inaddition, the importance of using glycolipids that preferentiallyinduce Th1 cytokine production was also illustrated in our study bya complete lack of tumor rejection in mice receiving NKTMabtherapy containing the C20:2 analogue of a-GC. Our findings wereconsistent with the key role of IFN-g in tumor rejection and thatthe C20:2 analogue contains a diunsaturated C20 fatty acid and hasbeen shown to potently induce NKT cell to preferentially secreteTh2 cytokines, with diminished IFN-g production and reducedNKT cell expansion (34, 39). More recently, the synthesis of a newtruncated nonisosteric a-c-GC analogue has been reported (42). Itwas shown to induce cytokine production from NKT cells with thehighest IFN-g to IL-4 and IFN-g to IL-13 ratios compared witha-c-GC and a-GC. This novel mimetic of a-GC might potentiallyfurther promote Th1 immune response over that induced bya-c-GC, and future experiments will be undertaken to directlycompare this glycolipid with a-c-GC in the R331 tumor model.Surprisingly, despite a potent antitumor effect of NKTMab the-
rapy containing a-GC, at the highest dose of a-GC tested, thetreated mice developed ruffled fur, had elevated serum ALTand ASTlevels, and on rare occasions died. By contrast, mice that receiveda-c-GC in NKTMab therapy at all doses tested (6–500 ng/injection)only displayed moderate and transient elevations in ALT at thehighest dose and never severe signs of toxicity. These datahighlighted the far greater therapeutic index of the anti-DR5/a-c-GC/anti-4-1BB combination. Among the various reductions of thiscombination, only anti-4-1BB/a-GC (500 ng/dose) induced signifi-cantly elevated serum ALT levels. We will now determine whichcytokines contribute to elevations in serum ALT/AST using tumor-bearingWTmice and IFN-g, IL-4, IL-13, or other gene-targetedmice.Although liver toxicities were seen in tumor-bearing mice followingNKTMab (a-GC) therapy, the safety of a-GC alone in humans haspreviously been shown in several clinical trials. Administration ofa-GC directly into cancer patients (24, 25) or transfer of a-GC–activated NKTcells (23) or a-GC–pulsed dendritic cells (22) has beenfound to be well tolerated in patients with advanced diseases.Humans contain fewer iNKT cells in the liver compared with mice(43), and it is possible that the current toxicity observed in ourstudies may not manifest in humans. The safety of a-c-GC in theNKTMab therapy has yet to be tested in the clinic.
Cancer Research
Cancer Res 2007; 67: (15). August 1, 2007 7502 www.aacrjournals.org
Research. on February 16, 2016. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

We have shown that NKTMab therapies (at the highest doses ofa-GC or a-c-GC) can induce similar rates of tumor rejection toTriMab therapy against two different tumor types in s.c. ormetastatic settings. Encouragingly, even larger established tumorswere quite effectively suppressed by the optimal NKTMab therapy.However, to determine the widespread applicability of NKT cell–based therapy, NKTMab treatment of other mouse experimentaltumors from different tissue origins will also be examined.Alternatively, the effectiveness of NKTMab therapy can also beassessed in transgenic models of carcinogenesis, such as BALB/cHER-2/neu mice, which provides valuable insight on diseaseprogression and treatment because they have distinct stagescomparable with those observed in human carcinomas.One of the hallmark features of an effective immunotherapy is its
ability to stimulate lasting tumor-specific immunity. We havepreviously shown that TriMab therapy generates tumor-specificmemory responses detectable by secondary tumor challenge. LikeTriMab therapy, NKTMab therapy–cured mice were also able tomount a tumor-specific response following secondary tumorchallenge (data not shown). Similar to TriMab therapy, IFN-g wasfound to be an important mediator of tumor rejection in NKTMabtherapy. However, unlike TriMab therapy, where CD8+ T cells alonewere enough to induce tumor rejection, our current studies showeda critical requirement for both CD4+ and CD8+ T cells and NKT cellsfor effective NKTMab-induced tumor rejection. However, we cannotdiscount the possibility that antibody depletion of CD4+ cells maydeplete some CD4+ NKT cells. Interestingly, TriMab therapy ofJa18�/� tumor-bearingmice induced a similar rate of tumor rejectionas WT tumor-bearing mice, showing that several ways to activatedendritic cells exist: CD40 ligation or cross-talk with NKT cells.However, it is also possible to activate dendritic cells via CD1d
independently of NKTcells activation. Recently, CD1d ligation alone,in the absence of NKT cells, has been shown to rapidly stimulateproduction of active IL-12p70 by CD1d+ human peripheral bloodmonocytes and immature dendritic cells (44). IFN-g was withouteffect alone but significantly increased CD1d-stimulated IL-12production. In concert with this representing a physiologic response,monocyte differentiation, as evidenced by CD1a and CD40 up-regulation, was also accelerated by CD1d stimulation. This workillustrated an innate immune signaling function for CD1d andprovided amechanism by whichmonocytes and immature dendriticcells could be rapidly activated in the absence of anti-CD40 orglycolipid ligands. Because it is not clear whether concurrentNKT cell activation is necessary to achieve optimal therapeuticantitumor activity, future work will determine whether agonisticanti-mouse CD1d mAbs can substitute for anti-CD40 or a-GC/a-c-GC using the R331 tumor model. It will also be interestingwhether a blocking anti-CD1d mAb might also inhibit type IINKT cell activation and thereby promote downstream tumorimmunity (45).Several factors could potentially limit the antitumor efficacy of
NKT cell–based immunotherapy, and as such, their effect will haveto be duly assessed. It has been documented that frequencies ofhuman NKT cells are more variable between individuals and that
cell numbers are typically lower than that found in mice. Forexample, levels of NKT cells in the thymus are at least 100-foldlower in humans (46), and in the liver of humans, only 1% oflymphocytes are NKT cells compared with 30% in the liver of mice(43), which is also one possible explanation for the low levels oftoxicity observed in clinical trials to date. The lower number ofNKT cells in humans will thus have to be taken into considerationwhen designing any potential trial with a NKTMab-like immuno-therapy. Similar to T cells, immune suppression of NKT cells hasalso been commonly encountered in cancer patients where theyhave either been reported to be decreased compared with healthyhumans or functionally hyporeactive (47). Yanagisawa et al.recently reported that a-GC–induced NKT cell function incancer-bearing mice was impaired. This was due to an increasein CD11b+ Gr-1+ cells, which mediated their suppression on NKTcell function in a nitric oxide–mediated fashion. However, theauthors were able to relieve this suppression through administra-tion of all-trans-retinoic acid, which decreased the CD11b+ Gr-1+
population (48), and a similar strategy might be considered inhumans. It has also been reported that tumors may modulate theirCD1d molecules or shed glycosphingolipids in vitro to inhibitstimulation of NKT cells (49, 50) as another means of avoiding NKTcell activation. Although NKTMab therapy successfully inducedrejection of established tumors in most mice in our studies, it willbe interesting to further deplete NKTMab-treated tumor-bearingmice (that have not rejected tumors) of regulatory T cells or toadditionally give anti-CTLA-4 mAb to observe whether greaternumbers of cures can be achieved. Notably, we have not detectedany signs of autoimmunity in the NKTMab-treated mice, butadditional modifications that potentially break tolerance mecha-nisms will have to be considered with caution.In summary, NKTMab therapy, which uses glycolipid ligands for
NKT cells to mature dendritic cells in combination with antibodiesthat induce tumor cell apoptosis and T-cell activation, can inducerejection of established tumors in several experimental settings.Given that most of the components proposed in our combinationhave been tested for safety in phase I clinical trials in advancedcancer patients, we anticipate that these data bring us closer toimplementing an effective combination of immunotherapies to theclinic for the treatment of cancer.
Acknowledgments
Received 3/13/2007; revised 5/9/2007; accepted 5/24/2007.Grant support: National Health and Medical Research Council of Australia Senior
Principal Research Fellowship, NIH grant R01, Parker Foundation and Susan G. KomenFoundation Grant (M.J. Smyth); Susan G. Komen Foundation Grant and the CancerCouncil of Victoria (M.W.L. Teng); and National Breast Cancer Foundation of Australiaand National Health and Medical Research Council of Australia R.D. WrightPostdoctoral Fellowships (M.H. Kershaw and P.K. Darcy).
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Preethi Guru and Carole van Puyenbroek for technical assistance in theRenca orthotopic kidney tumor model and Shannon Griffiths of the PMCC animalfacilities for maintenance of mice. G.S. Besra acknowledges support in the form of aPersonal Research Chair from Mr. James Bardrick and from the Medical ResearchCouncil (United Kingdom) and The Wellcome Trust.
References
1. Cheson BD. Monoclonal antibody therapy for B-cellmalignancies. Semin Oncol 2006;33:S2–14.
2. Jackisch C. HER-2-positive metastatic breast cancer:optimizing trastuzumab-based therapy. Oncologist 2006;11 Suppl 1:34–41.3. Arsene D, Galais MP, Bouhier-Leporrier K, Reimund JM.
Recent developments in colorectal cancer treatment bymonoclonal antibodies. ExpertOpin Biol Ther 2006;6:1175–92.4. Faries MB, Morton DL. Therapeutic vaccines formelanoma: current status. BioDrugs 2005;19:247–60.
NKT Cell–Based Immunotherapy
www.aacrjournals.org 7503 Cancer Res 2007; 67: (15). August 1, 2007
Research. on February 16, 2016. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from

Cancer Research
Cancer Res 2007; 67: (15). August 1, 2007 7504 www.aacrjournals.org
5. Dudley ME, Rosenberg SA. Adoptive-cell-transfertherapy for the treatment of patients with cancer.Nat Rev Cancer 2003;3:666–75.6. Takeda K, Hayakawa Y, Smyth MJ, et al. Involvement oftumor necrosis factor-related apoptosis-inducing ligandin surveillance of tumor metastasis by liver natural killercells. Nat Med 2001;7:94–100.7. Takeda K, Yamaguchi N, Akiba H, et al. Induction oftumor-specific T cell immunity by anti-DR5 antibodytherapy. J Exp Med 2004;199:437–48.8. Smyth MJ, Cretney E, Takeda K, et al. Tumor necrosisfactor-related apoptosis-inducing ligand (TRAIL) contrib-utes to interferong-dependent natural killer cell protectionfrom tumor metastasis. J Exp Med 2001;193:661–70.9. Selenko N, Maidic O, Draxier S, et al. CD20 antibody(C2B8)-induced apoptosis of lymphoma cells promotesphagocytosis by dendritic cells and cross-priming ofCD8+ cytotoxic T cells. Leukemia 2001;15:1619–26.10. zum Buschenfelde CM, Hermann C, Schmidt B,Peschel C, Bernhard H. Antihuman epidermal growthfactor receptor 2 (HER2) monoclonal antibody trastu-zumab enhances cytolytic activity of class I-restrictedHER2-specific T lymphocytes against HER2-overexpress-ing tumor cells. Cancer Res 2002;62:2244–7.11. Uno T, Takeda K, Kojima Y, et al. Eradication ofestablished tumors in mice by a combination antibody-based therapy. Nat Med 2006;12:693–8.12. Hixon JA, Anver MR, Blazar BR, Panoskaltsis-MortariA, Wiltrout RH, Murphy WJ. Administration of eitheranti-CD40 or interleukin-12 following lethal total bodyirradiation induces acute lethal toxicity affecting thegut. Biol Blood Marrow Transplant 2002;8:316–25.13. Hixon JA, Blazar BR, Anver MR, Wiltrout RH, MurphyWJ. Antibodies to CD40 induce a lethal cytokine cascadeafter syngeneic bone marrow transplantation. BiolBlood Marrow Transplant 2001;7:136–43.14. Vonderheide RH, Dutcher JP, Anderson JE, et al.Phase I study of recombinant human CD40 ligand incancer patients. J Clin Oncol 2001;19:3280–7.15. van Mierlo GJD, den Boer AT, Medema JP, et al. CD40stimulation leads to effective therapy of CD40� tumorsthrough induction of strong systemic cytotoxic Tlymphocyte immunity. Proc Natl Acad Sci U S A 2002;99:5561–6.16. Chiodoni C, Iezzi M, Guiducci C, et al. TriggeringCD40 on endothelial cells contributes to tumor growth.J Exp Med 2006;203:2441–50.17. Godfrey DI, Kronenberg M. Going both ways:immune regulation via CD1d-dependent NKT cells.J Clin Invest 2004;114:1379–88.18. Smyth MJ, Thia KY, Street SE, et al. Differential tumorsurveillance by natural killer (NK) and NKT cells. J ExpMed 2000;191:661–8.19. Hayakawa Y, Godfrey Dale I, Smyth Mark J.a-Galactosylceramide: potential immunomodulatoryactivity and future application general articles. CurrMed Chem 2004;11:241–52.20. Kitamura H, Iwakabe K, Yahata T, et al. The naturalkiller T (NKT) cell ligand a-galactosylceramide demon-strates its immunopotentiating effect by inducinginterleukin (IL)-12 production by dendritic cells andIL-12 receptor expression on NKT cells. J Exp Med 1999;189:1121–8.
21. Fujii S-i, Shimizu K, Smith C, Bonifaz L, SteinmanRM. Activation of natural killer T cells by a-galacto-sylceramide rapidly induces the full maturation ofdendritic cells in vivo and thereby acts as an adjuvantfor combined CD4 and CD8 T cell immunity to acoadministered protein. J Exp Med 2003;198:267–79.22. Ishikawa A, Motohashi S, Ishikawa E, et al. A phaseI study of a-galactosylceramide (KRN7000)-pulseddendritic cells in patients with advanced and recur-rent non-small cell lung cancer. Clin Cancer Res 2005;11:1910–7.23. Motohashi S, Ishikawa A, Ishikawa E, et al. A phase Istudy of in vitro expanded natural killer T cells inpatients with advanced and recurrent non-small celllung cancer. Clin Cancer Res 2006;12:6079–86.24. Nieda M, Okai M, Tazbirkova A, et al. Therapeuticactivation of Va24+Vh11+ NKT cells in human subjectsresults in highly coordinated secondary activation ofacquired and innate immunity. Blood 2004;103:383–9.25. Giaccone G, Punt CJA, Ando Y, et al. A phase I studyof the natural killer T-cell ligand a-galactosylceramide(KRN7000) in patients with solid tumors. Clin CancerRes 2002;8:3702–9.26. Chang DH, Osman K, Connolly J, et al. Sustainedexpansion of NKT cells and antigen-specific T cellsafter injection of a-galactosyl-ceramide loaded maturedendritic cells in cancer patients. J Exp Med 2005;201:1503–17.27. Takeda K, Smyth MJ, Cretney E, et al. Critical role fortumor necrosis factor-related apoptosis-inducing ligandin immune surveillance against tumor development.J Exp Med 2002;195:161–9.28. Cretney E, Takeda K, Yagita H, Glaccum M, PeschonJJ, Smyth MJ. Increased susceptibility to tumor initiationand metastasis in TNF-related apoptosis-inducingligand-deficient mice. J Immunol 2002;168:1356–61.29. Seki N, Hayakawa Y, Brooks AD, et al. Tumor necrosisfactor-related apoptosis-inducing ligand-mediated apo-ptosis is an important endogenous mechanism forresistance to liver metastases in murine renal cancer.Cancer Res 2003;63:207–13.30. French RR, Chan HT, Tutt AL, Glennie MJ. CD40antibody evokes a cytotoxic T-cell response thateradicates lymphoma and bypasses T-cell help.Nat Med 1999;5:548–53.31. Melero I, Shuford WW, Newby SA, et al. Monoclo-nal antibodies against the 4-1BB T-cell activationmolecule eradicate established tumors. Nat Med 1997;3:682–5.32. Smyth MJ, Crowe NY, Pellicci DG, et al. Sequentialproduction of interferon-g by NK1.1(+) T cells andnatural killer cells is essential for the antimetastaticeffect of a-galactosylceramide. Blood 2002;99:1259–66.33. Chen G, Chien M, Tsuji M, Franck RW. E andZ a-C-galactosylceramides by Julia-Lythgoe-Kocienskichemistry: a test of the receptor-binding model forglycolipid immunostimulants. Chembiochem 2006;7:1017–22.34. Yu KO, Im JS, Molano A, et al. Modulation of CD1d-restricted NKT cell responses by using N -acyl variants ofa-galactosylceramides. Proc Natl Acad Sci U S A 2005;102:3383–8.35. Biburger M, Tiegs G. a-Galactosylceramide-induced
liver injury in mice is mediated by TNF-a but independentof Kupffer cells. J Immunol 2005;175:1540–50.36. Fujii H, Seki S, Kobayashi S, et al. A murine modelof NKT cell-mediated liver injury induced by a-galactosylceramide/D-galactosamine. Virchows Arch2005;446:663–73.37. Franck RW, Tsuji M. a-C-galactosylceramides: syn-thesis and immunology. Acc Chem Res 2006;39:692–701.38. Miyamoto K, Miyake S, Yamamura T. A syntheticglycolipid prevents autoimmune encephalomyelitis byinducing TH2 bias of natural killer T cells. Nature 2001;413:531–4.39. Forestier C, Takaki T, Molano A, et al. Improvedoutcomes in NOD mice treated with a novel Th2cytokine-biasing NKT cell activator. J Immunol 2007;178:1415–25.40. Fujii S, Shimizu K, Hemmi H, et al. Glycolipid a-C-galactosylceramide is a distinct inducer of dendritic cellfunction during innate and adaptive immune responsesof mice. Proc Natl Acad Sci U S A 2006;103:11252–7.41. Schmieg J, Yang G, Franck RW, Tsuji M. Superiorprotection against malaria and melanoma metastasesby a C-glycoside analogue of the natural killer T cellligand a-galactosylceramide. J Exp Med 2003;198:1631–41.42. Lu X, Song L, Metelitsa LS, Bittman R. Synthesis andevaluation of an a-C-galactosylceramide analogue thatinduces Th1-biased responses in human natural killer Tcells. Chembiochem 2006;7:1750–6.43. Kenna T, Golden-Mason L, Porcelli SA, et al. NKTcells from normal and tumor-bearing human livers arephenotypically and functionally distinct from murineNKT cells. J Immunol 2003;171:1775–9.44. Yue SC, Shaulov A, Wang R, Balk SP, Exley MA. CD1dligation on human monocytes directly signals rapidNF-nB activation and production of bioactive IL-12.Proc Natl Acad Sci U S A 2005;102:11811–6.45. Terabe M, Swann J, Ambrosino E, et al. A nonclassicalnon-Va14Ja18 CD1d-restricted (type II) NKT cell issufficient for down-regulation of tumor immunosurveil-lance. J Exp Med 2005;202:1627–33.46. Baev DV, Peng XH, Song L, et al. Distinct homeostaticrequirements of CD4+ and CD4� subsets of Va24-invariant natural killer T cells in humans. Blood 2004;104:4150–6.47. Dhodapkar MV, Geller MD, Chang DH, et al. Areversible defect in natural killer T cell functioncharacterizes the progression of premalignant tomalignant multiple myeloma. J Exp Med 2003;197:1667–76.48. Yanagisawa K, Exley MA, Jiang X, Ohkochi N,Taniguchi M, Seino K. Hyporesponsiveness to naturalkiller T-cell ligand a-galactosylceramide in cancer-bearing state mediated by CD11b+ Gr-1+ cells producingnitric oxide. Cancer Res 2006;66:11441–6.49. Sriram V, Cho S, Li P, et al. Inhibition of glycolipidshedding rescues recognition of a CD1+ T cell lymphomaby natural killer T (NKT) cells. Proc Natl Acad Sci U S A2002;99:8197–202.50. Fiedler T, Walter W, Reichert TE, Maeurer MJ.Regulation of CD1d expression by murine tumor cells:escape from immunosurveillance or alternate targetmolecules? Int J Cancer 2002;98:389–97.
Research. on February 16, 2016. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from
2007;67:7495-7504. Cancer Res Michele W.L. Teng, Jennifer A. Westwood, Phillip K. Darcy, et al. Eradicates Established Tumors in Mice
Based Immunotherapy−Combined Natural Killer T-Cell
Updated version
http://cancerres.aacrjournals.org/content/67/15/7495
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2007/08/02/67.15.7495.DC1.html
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/67/15/7495.full.html#ref-list-1
This article cites 50 articles, 31 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/67/15/7495.full.html#related-urls
This article has been cited by 11 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on February 16, 2016. © 2007 American Association for Cancercancerres.aacrjournals.org Downloaded from