
Monitoring and quantifying dynamic physiological processes in live neurons using fluorescence...
10
*School of Life Sciences, University of Sussex, Brighton, UK †Department of Physiology and Cell Biology, Faculty of Health Sciences, Zlotowski Center for Neuroscience, Ben-Gurion University of the Negev, Beer-Sheva, Israel Abstract The direct visualization of subcellular dynamic processes is often hampered by limitations in the resolving power achiev- able with conventional microscopy techniques. Fluorescence recovery after photobleaching has emerged as a highly informative approach to address this challenge, permitting the quantitative measurement of the movement of small organelles and proteins in living functioning cells, and offering detailed insights into fundamental cellular phenomena of physiological importance. In recent years, its implementation has benefited from the increasing availability of confocal microscopy systems and of powerful labeling techniques based on genetically encoded fluorescent proteins or other chemical markers. In this review, we present fluorescence recovery after photobleaching and related techniques in the context of contemporary neurobiological research and discuss quantitative and semi-quantitative approaches to their inter- pretation. Keywords: fluorescence, FRAP, GFP, microscopy, synapse, vesicle. J. Neurochem. (2013) 126, 213–222. Movement is typically gauged by observing the displacement of individual elements. However, this approach becomes problematic when the objects are either too small or too intermingled to be resolved, as is true for most molecules of biological interest. In this case, a biological sample with a large amount of macromolecular movement would still appear as a motionless continuum. One informative strategy to reveal mobility is to create a disturbance in the sample and monitor the subsequent dynamics. For example, a drop of colorless water added to the center of an unstirred dye solution reveals the intrinsic mobility of the dye molecules over time as they invade the colorless region. Fluorescence recovery after photobleaching (FRAP), a methodology used to measure the mobility of fluorescently labeled molecules, proteins or small organelles (Reits and Neefjes 2001), draws on these same basic principles. A subregion of a biological sample containing mobile fluorescent elements is rapidly and irreversibly photobleached by targeted irradiation with intense excitation light. This produces a transient distinction between bleached molecules within the target region and intact fluorescent molecules outside it. Subsequently, fluo- rescent moieties move back into the bleached region (influx) and bleached moieties move out (efflux). This can be directly observed as macroscopic recovery of fluorescence within the photobleached area (Fig. 1a–c). Quantitative or semi-quan- titative measurement of the kinetics and extent of recovery reveals the nature of the mobility of the moiety of interest. FRAP encompasses a group of related methodologies that differ in the time scale of the examined phenomena, the initial distribution of the moiety of interest, and the quantitative accuracy of the answer being sought. A com- plementary and inverse approach involves the use of photoactivatable (Patterson and Lippincott-Schwartz 2002; Tsuriel et al. 2006) or photoswitchable fluorescent proteins (Chudakov et al. 2007), or of caged fluorescent compounds (Santamaria et al. 2006). In these, fluorescent moieties are formed by focal irradiation and their dispersal is monitored and quantitatively analyzed. Received January 24, 2013; revised manuscript received March 12, 2013; accepted March 13, 2013. AddresscorrespondenceandreprintrequeststoDanielGitler,Department of Physiology and Cell Biology, Faculty of Health Sciences, Ben-Gurion UniversityoftheNegev,Beer-Sheva84105,Israel.E-mail:[email protected] Abbreviations used: FRAP, fluorescence recovery after photobleach- ing. © 2013 International Society for Neurochemistry, J. Neurochem. (2013) 126, 213--222 213 JOURNAL OF NEUROCHEMISTRY | 2013 | 126 | 213–222 doi: 10.1111/jnc.12240
Transcript of Monitoring and quantifying dynamic physiological processes in live neurons using fluorescence...

*School of Life Sciences, University of Sussex, Brighton, UK
†Department of Physiology and Cell Biology, Faculty of Health Sciences, Zlotowski Center for
Neuroscience, Ben-Gurion University of the Negev, Beer-Sheva, Israel
AbstractThe direct visualization of subcellular dynamic processes isoften hampered by limitations in the resolving power achiev-able with conventional microscopy techniques. Fluorescencerecovery after photobleaching has emerged as a highlyinformative approach to address this challenge, permittingthe quantitative measurement of the movement of smallorganelles and proteins in living functioning cells, and offeringdetailed insights into fundamental cellular phenomena ofphysiological importance. In recent years, its implementationhas benefited from the increasing availability of confocal
microscopy systems and of powerful labeling techniquesbased on genetically encoded fluorescent proteins or otherchemical markers. In this review, we present fluorescencerecovery after photobleaching and related techniques in thecontext of contemporary neurobiological research and discussquantitative and semi-quantitative approaches to their inter-pretation.Keywords: fluorescence, FRAP, GFP, microscopy, synapse,vesicle.J. Neurochem. (2013) 126, 213–222.
Movement is typically gauged by observing the displacementof individual elements. However, this approach becomesproblematic when the objects are either too small or toointermingled to be resolved, as is true for most molecules ofbiological interest. In this case, a biological sample with alarge amount of macromolecular movement would stillappear as a motionless continuum. One informative strategyto reveal mobility is to create a disturbance in the sample andmonitor the subsequent dynamics. For example, a drop ofcolorless water added to the center of an unstirred dyesolution reveals the intrinsic mobility of the dye moleculesover time as they invade the colorless region. Fluorescencerecovery after photobleaching (FRAP), a methodology usedto measure the mobility of fluorescently labeled molecules,proteins or small organelles (Reits and Neefjes 2001), drawson these same basic principles. A subregion of a biologicalsample containing mobile fluorescent elements is rapidly andirreversibly photobleached by targeted irradiation withintense excitation light. This produces a transient distinctionbetween bleached molecules within the target region andintact fluorescent molecules outside it. Subsequently, fluo-rescent moieties move back into the bleached region (influx)and bleached moieties move out (efflux). This can be directly
observed as macroscopic recovery of fluorescence within thephotobleached area (Fig. 1a–c). Quantitative or semi-quan-titative measurement of the kinetics and extent of recoveryreveals the nature of the mobility of the moiety of interest.FRAP encompasses a group of related methodologies thatdiffer in the time scale of the examined phenomena, theinitial distribution of the moiety of interest, and thequantitative accuracy of the answer being sought. A com-plementary and inverse approach involves the use ofphotoactivatable (Patterson and Lippincott-Schwartz 2002;Tsuriel et al. 2006) or photoswitchable fluorescent proteins(Chudakov et al. 2007), or of caged fluorescent compounds(Santamaria et al. 2006). In these, fluorescent moieties areformed by focal irradiation and their dispersal is monitoredand quantitatively analyzed.
Received January 24, 2013; revised manuscript received March 12,2013; accepted March 13, 2013.AddresscorrespondenceandreprintrequeststoDanielGitler,Department
of Physiology and Cell Biology, Faculty of Health Sciences, Ben-GurionUniversityoftheNegev,Beer-Sheva84105,Israel.E-mail:[email protected] used: FRAP, fluorescence recovery after photobleach-
ing.
© 2013 International Society for Neurochemistry, J. Neurochem. (2013) 126, 213--222 213
JOURNAL OF NEUROCHEMISTRY | 2013 | 126 | 213–222 doi: 10.1111/jnc.12240

FRAP has been used extensively in cell biologicalresearch, for example to study the mobility of cytosolicand membrane proteins (Peters et al. 1974; Wang et al.2008; Berkovich et al. 2011), to examine the structure ofmembranes (Teissie et al. 1978), to probe the structure anddirectionality of the cytoskeleton (Iliev and Wouters 2007;Fridman et al. 2009), to probe transcription of mRNA andits processing (Aizer et al. 2008), and to view themovement of various organelles (Mitra and Lippincott-Schwartz 2010; Nadezhdina et al. 2010). Likewise, FRAPand homologous techniques have been widely exploited inneurobiological research, for example to examine mobilityof neurotransmitter receptors or transporters (Axelrod et al.1976; Eriksen et al. 2009; Jaskolski and Henley 2009),polarity of the axonal skeleton (Edson et al. 1993; Gauthi-er-Kemper et al. 2012; Koskinen et al. 2012), diffusional
properties of calcium buffers (Schmidt et al. 2003, 2005,2007), axonal transport and mobility of vesicles (Gaffieldet al. 2006; Kamin et al. 2010; Staras et al. 2010; Forna-siero et al. 2012; Orenbuch et al. 2012; Scott and Roy2012), and the stability of synaptic structure (Tsuriel et al.2006, 2009).This review presents an outline of FRAP approaches and
similar related techniques, with specific reference toneurobiological applications. It includes a general descrip-tion of instrumentation, details on the assumptions made inFRAP experiments, approaches to their analysis, and anexplanation of the type of information that can be readilydeduced.
The FRAP experiment
The initial step in a FRAP experiment is the introduction of adye, or the fluorescent labeling of the structure/protein ofinterest in live cells (Fig. 1a). Exogenous probes can bemicro-injected into cells as free dye or pre-conjugated to aprotein of interest (Schmidt et al. 2005, 2007; Santamariaet al. 2006). Alternatively, they may be introduced asacetoxymethylester dyes (Blatter and Wier 1990) or throughphysiological processes. An example of the latter is the useof styryl dyes, a family of amphipathic proteins usedextensively for labeling recycling synaptic vesicles (Betzand Bewick 1992; Gaffield et al. 2006). Genetically encodedfluorescent proteins [such as the Enhanced Green FluorescentProtein (EGFP)] are widely used for FRAP experiments,although due consideration should be given to the fact thattheir large size means they could affect the mobility of the
(a)
(b)
(c)
(d) (e)
Fig. 1 Outline of a Fluorescence Recovery After Photobleachingexperiment. (a) The cytoplasm of a cell is filled with a mobile
fluorescent dye (green). (b) Enlargement of the area demarcated bya dashed square in (a) reveals that the dye is composed of mobilemolecules moving randomly throughout the imaged area (left, green
circles). The molecules within the dashed circle (corresponding toblack spot in a) are bleached by intense light (center, black circles).Recovery (right) ensues as a result of the continuous movement of
fluorescent molecules into the spot, and of bleached molecules out ofit. Depicted here is an intermediate and not yet complete state ofrecovery. (c) Graphical representation of fluorescence intensity withinthe bleaching spot. The sudden drop in intensity refers to the rapid
bleaching of the fluorophores; it is followed by recovery. The rate ofrecovery depends on the instantaneous rate of movement of themolecules, as well as on the size and shape of the bleached area. A
black diamond marks the timing of the right frame in (b). If themolecules are freely mobile, the intensity recorded after recovery (theasymptote) reflects the remaining fluorescence within the whole cell. If
the total volume is very large in comparison to the bleaching spot, theoverall loss of fluorescence is negligible. (d) Depiction of the recoveryphase when a subset of the molecules is immobilized (red outline). (e)In the case an immobile subset exists, the asymptote of the recovery
curve does not approach the pre-bleach fluorescence, but ratherreflects just the mobile fraction.
© 2013 International Society for Neurochemistry, J. Neurochem. (2013) 126, 213--222
214 K. Staras et al.

target, and are thus less applicable to the direct determinationof protein diffusion. Finally, it is important to select afluorophore which can be readily photobleached by strongillumination yet is sufficiently resistant to bleaching by theweaker illumination required to subsequently monitor itsdistribution.The FRAP experiment itself is divided into three separate
phases (Fig. 1b and c): (i) the pre-bleach phase, (ii)bleaching, and (iii) post-bleach recovery. The pre-bleachimaging phase is intended to provide readout of the initialfluorescence within the observation volume, and to establishthat the biological and experimental systems are stable. Thebleach phase uses intense excitation light to bring aboutlocalized photobleaching within a defined and usually smallcompartment. In the post-bleach phase, fluorescence recov-ery within the bleached region is monitored. While thekinetics of recovery provides a measure of the instantaneousspeed of the mobile moieties that can help to establish thetype of movement executed by the fluorescent moiety, theextent of recovery also provides information about thepresence of immobile moieties (Fig. 1d–e). More detailedinformation concerning the analysis of FRAP experiments isprovided below.
Assumptions and requisites
To quantitatively analyze FRAP experiments the followingconditions should be fulfilled:
(i) For quantitative analysis, the precise shape of thebleaching spot should be known. The simpler thegeometry of the spot and its boundaries, the simpler theformula that describes the recovery phase (Soumpasis1983; Brown et al. 1999; Tsibidis 2009).
(ii) Bleaching should be irreversible, but does not need tobe complete [i.e., not all fluorescence in the spot needsto be bleached (Brown et al. 1999)]. See however(Sinnecker et al. 2005).
(iii) The bleaching process should not in itself influence themobility of the moiety of interest and should not harmthe biological processes within the sample (Bancaudet al. 2010). Bleaching may form free radicals andlocally heat the sample, representing a real concern forany FRAP experiment. Appropriate tests to ensure thatnormal physiological processes are not compromisedshould be performed.
(iv) The bleaching step should be brief and significantlyfaster than the recovery process to minimize substantialrecovery during the bleaching step itself.
(v) Imaging-related bleaching outside of the bleachingphase should be minimal, or at least directly quantifi-able to allow for correction.
(vi) It is imperative that the bleached area remain in focusfor the duration of the experiment.
Effect of sample geometry
The geometry of the sample can have a significant impact onthe ready interpretation of results. Flat two-dimensionalobjects are especially suitable for FRAP experiments, andwere the first to be examined (Peters et al. 1974). Eventhough the point-spread function of an optical system is acomplex three-dimensional volume, a two-dimensional sam-ple structure permits sharp-edged bleaching at the focal planeof the objective (Brown et al. 1999). Furthermore, the wholebleached area can be conveniently imaged at once. Finally,the dimensionality of diffusion/transport equations can bereduced (Teissie et al. 1978). While FRAP within three-dimensional volumes is possible, a two-photon microscope isneeded to achieve a spot-like bleaching volume (Brown et al.1999; Schmidt et al. 2007). This is based on the fact that thediffraction-delimited oval excitation volume produced bytwo-photon systems are qualitatively different than hourglassbleaching volumes produced by conventional lasers, or thelarger bleaching volumes achievable with arc lamps. Finally,if photobleaching is performed within thin elongatedprocesses, such as axons and dendrites, often the analysisof diffusion or transport can be reduced to one dimension(Schmidt et al. 2007), assuming that axial mobility can beneglected (Zador and Koch 1994; Gabso et al. 1997). Whileonly simple geometries were discussed above, it should beborne in mind that complex geometries can have strongeffects on diffusion. For example, it was shown that thepresence of spines significantly retards diffusion withindendrites (Santamaria et al. 2006), contributing to compart-mentalization.Another factor to note is the influence of the initial
distribution of the fluorophore. The fluorophore may bedistributed homogeneously throughout the cytoplasm of acell (Schmidt et al. 2007), or it may be concentrated indiscrete areas, such as the presynaptic terminal (Orenbuchet al. 2012). In the former case, bleaching is performed onlywithin a subregion of the sample. In the latter, bleaching of asubregion of a terminal is applicable (Gaffield et al. 2006),but so too is bleaching of entire presynaptic terminals. In thiscase, recovery will reflect the movement of fluorophores overlong distances from outside the boundaries of the targetterminal (Darcy et al. 2006a; Orenbuch et al. 2012) (Fig. 2).
Instrumentation
The expected rate of fluorescence recovery and its decom-position into different time scales profoundly affects theplanning and execution of FRAP experiments. The bleachingphase needs to be shorter than the faster kinetic componentsof recovery, whereas the duration of the post-bleaching phaseshould be long enough to reliably determine both the slowerkinetic components and the final extent of recovery. Notsurprisingly, these requirements dictate the properties of the
© 2013 International Society for Neurochemistry, J. Neurochem. (2013) 126, 213--222
Monitoring dynamic physiological processes by FRAP 215
instrumentation. We illustrate this with two contrastingexamples where very fast versus very slow processes wereassessed.
When fast recovery is expected, bleaching is performed byan intense and temporally discrete burst of excitation light.Moreover, sampling of emitted fluorescence is performed at avery high-frequency. Because the recording period is quitebrief, focal stability is usually not a concern. This approach isillustrated by the experimental setup that was used tomeasure FRAP of fluorescent parvalbumin in Purkinjeneurons, an event that occurs in substantially less than 1-s(Schmidt et al. 2007). To produce an intense diffraction-delimited bleaching spot lasting 1–6 ms with a transitiontime of 1 ls, a Pockels cell was placed in the excitation pathof a two-photon microscope. This optical device attenuatesthe intensity of polarized light by electronically rotating thebeam prior to passage through a polarizer. The oval shapeand the size of the bleaching volume were defined by thetwo-photon effect. To permit fast acquisition, the mirrors inthe scan head were held in a parked position. Imaging at afrequency of 250 kHz continued for 1.5 s after the bleachingpulse, sufficient time for completion of recovery.A contrasting example is the case of FRAP experiments
used to look at the exchange rate of fluorescently labeledstructural synaptic proteins, involving recovery over manyhours (Tsuriel et al. 2006, 2009). In this case, the prolongednature of recovery means that it proved acceptable to have ableaching step lasting for several seconds. In these experi-ments, regions of interest were used to define a number oftarget synapses and an acousto-optic tunable filter was usedto specifically subject the regions of interests to intenseillumination during several iterations of frame scanning. Thebleaching volume was determined by the intersection of theaxonal volume and the waist of the hourglass shape of thesingle-photon laser beam. Acquisition was performed fromoptical planes whose depth was determined by the confocalpinhole. Sampling of recovery was performed at a very slowrate, every 5, 10 or 60 min. As a result of the length of therecovery period concerns about focal drift had to beaddressed. To minimize the potential for drift, the temper-ature of the sample and the environment were stabilized, andfocus drift was corrected automatically before acquiring eachtime point. Furthermore, z-stacks spanning a range of focalplanes were acquired at each time-point, so that in-focusframes could be used for analysis. A different approach forfocal stability involves the detection of the reflective surfaceof the coverslip with infrared light, using it as a referencepoint for focus correction (such as the Nikon Perfect Focussystem, Nikon Instruments, Tokyo, Japan). This approach issuitable only in applications where such surfaces are present,for example when imaging cultured cells.Both aforementioned examples utilized the diffraction-
limited spot of a laser-scanning confocal microscope both forbleaching and acquisition. A different approach is to use anepi-fluorescence arc lamp in a wide-field system, wheretargeted bleaching is achieved by restricting the area ofillumination, for example by narrowing the field diaphragm
(a)
(b)
(c)
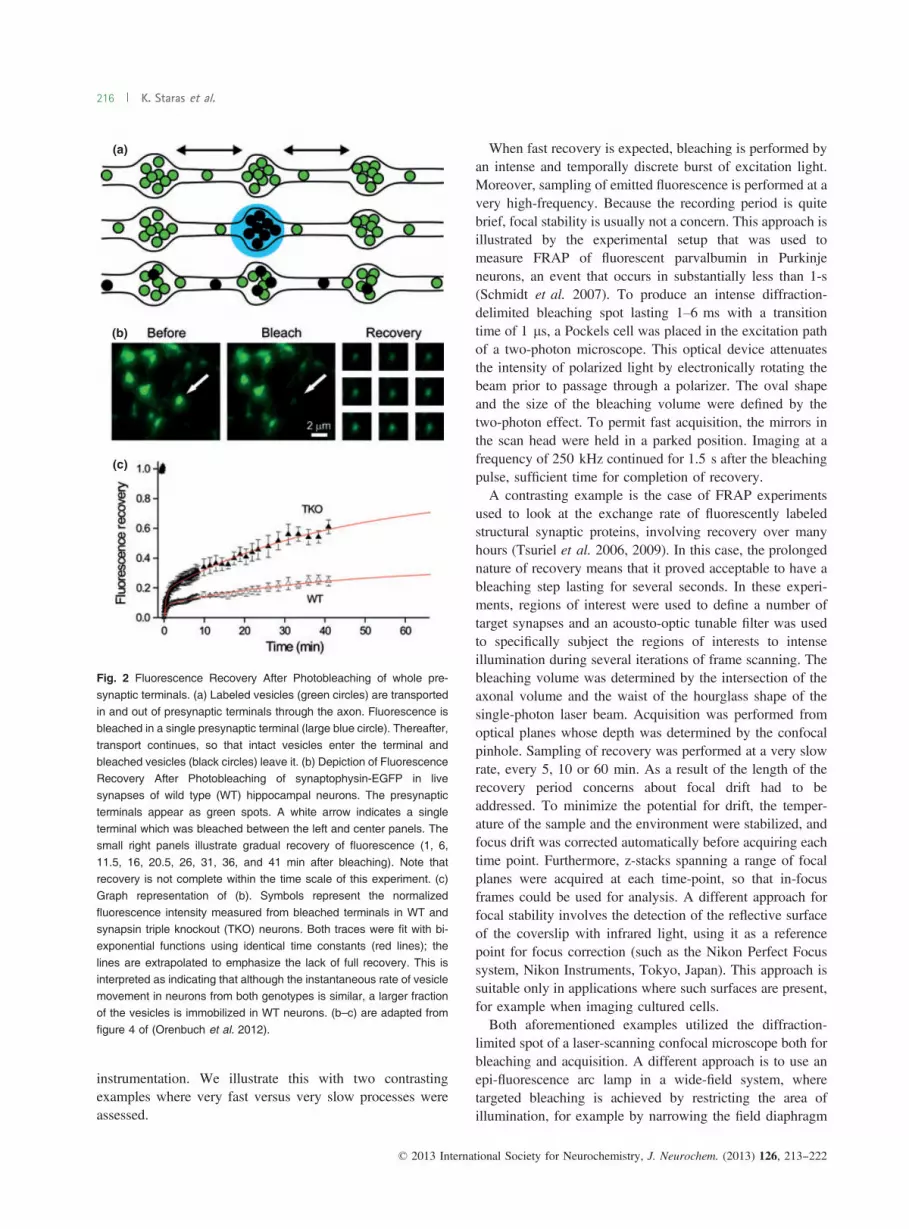
Fig. 2 Fluorescence Recovery After Photobleaching of whole pre-
synaptic terminals. (a) Labeled vesicles (green circles) are transportedin and out of presynaptic terminals through the axon. Fluorescence isbleached in a single presynaptic terminal (large blue circle). Thereafter,
transport continues, so that intact vesicles enter the terminal andbleached vesicles (black circles) leave it. (b) Depiction of FluorescenceRecovery After Photobleaching of synaptophysin-EGFP in livesynapses of wild type (WT) hippocampal neurons. The presynaptic
terminals appear as green spots. A white arrow indicates a singleterminal which was bleached between the left and center panels. Thesmall right panels illustrate gradual recovery of fluorescence (1, 6,
11.5, 16, 20.5, 26, 31, 36, and 41 min after bleaching). Note thatrecovery is not complete within the time scale of this experiment. (c)Graph representation of (b). Symbols represent the normalized
fluorescence intensity measured from bleached terminals in WT andsynapsin triple knockout (TKO) neurons. Both traces were fit with bi-exponential functions using identical time constants (red lines); the
lines are extrapolated to emphasize the lack of full recovery. This isinterpreted as indicating that although the instantaneous rate of vesiclemovement in neurons from both genotypes is similar, a larger fractionof the vesicles is immobilized in WT neurons. (b–c) are adapted from
figure 4 of (Orenbuch et al. 2012).
© 2013 International Society for Neurochemistry, J. Neurochem. (2013) 126, 213--222
216 K. Staras et al.

(Axelrod et al. 1976). Alternatively, an external light sourcemay be used (Roy et al. 2011). These approaches benefitfrom the advantage of simplicity, but typically at the price ofa larger bleaching area which exhibits diffuse edges, unlikethe gaussian edges of the confocal spot (Berkovich et al.2011).
Controls, corrections, and analysis
Controls
Control experiments are necessary to assess whetherassumptions made in planning the FRAP experiment arevalid. Here, we discuss two types of controls; first, thoseaimed at establishing that recovery does indeed stem frommovement, and second, those designed to test for possibleunintended effects on the biological system.In many instances it is possible to compare FRAP in live
and chemically fixed cells, with the assumption that fixationimmobilizes the fluorescent marker. In this way, it is possibleto assess whether a decrease in fluorescence, which isinterpreted as bleaching, is indeed irreversible. This type ofcontrol can also address the possibility of other dynamicchanges in fluorescence properties; for example, photoacti-vation or entry into dark states (Malkani and Schmid 2011).Of course, it is also important to consider that the fixationsteps themselves may have confounding effects that makeinterpretation of the results difficult; for example, thegeneration of an autofluorescence signal (Clancy and Cauller1998). Another type of control experiment is recommendedto establish that the physiological function of the biologicalpreparation is not compromised by the bleaching process.This type of control needs to be separately developed foreach experiment, and depends on the availability of a suitableassay that can determine whether cellular functions areunaffected by bleaching. For example, in experimentsinvolving mitosis, the timing of the cell cycle can becalibrated in control cells to verify that photoperturbationdoes not inhibit cell-cycle progression (Bancaud et al. 2010).In another example, it was shown that bleaching of styryldyes contained within synaptic vesicles in hippocampalsynapses did not deleteriously affect synaptic function bydetermining that recycling of fresh styryl dye was unchangedin synapses that had been previously subjected to bleaching(Darcy et al. 2006a).
Corrections
In some instances technical limitations prevent the fulfill-ment of some of the aforementioned assumptions for aFRAP experiment. In such cases, certain corrections can beimplemented. For example, if it is not possible to keep thebleaching phase sufficiently brief, substantial recovery mayoccur within this period, thus distorting the time course ofrecovery; compensations for this eventuality have beenproposed (Kang et al. 2009; Yang et al. 2010). Likewise, if
sampling during the prebleach and recovery phases resultsin significant bleaching, its extent can usually be quantifiedand can be corrected during offline analysis (Endress et al.2005; Tsuriel et al. 2006; Bancaud et al. 2010; Wu et al.2012).
Analysis
The rigor of analysis is ultimately derived from the overallobjective of the experiment. In some cases, a semi-quanti-tative approach may be deemed sufficient. For example,when the aim is to compare the overall mobility of amolecule/structure of interest under different conditions,direct comparison of the recovery traces or of raw timeconstants may suffice. However, when the aim is tospecifically determine a diffusion coefficient (Schmidt et al.2007), or to establish the type of movement being executedby the moiety of interest [e.g. isotropic, stick-and-diffuse,caged, anomalous or directional (Yeung et al. 2007; Spragueand McNally 2005; Berkovich et al. 2011)], then quantita-tive analysis is called for. Much information can be extractedfrom fitting of the recovery trace with various mobilitymodels (for examples see Bancaud et al. 2010; Spragueet al. 2004), taking into account the geometry of both thebleach spot and the sample. For example, binding of dye-bearing molecules with organelles or the plasma membrane(Berkovich et al. 2011) or the presence of obstructions thatconstrain diffusion (Santamaria et al. 2006) can result insignificantly slower apparent diffusion, but their presence canbe inferred by careful analysis. More importantly, the natureof the constraints (Sprague et al. 2004) and the binding andunbinding constants (Berkovich et al. 2011) can beextracted. A useful reference in this type of study is thedetermination of the diffusion of free dye (Sprague et al.2004). Furthermore, determination of the asymptote of therecovery trace can reveal whether a fraction of the labeledmoieties is immobile (Schmidt et al. 2005; Bancaud et al.2010; Orenbuch et al. 2012), after correcting for thedecrement in the total quantity of fluorescent material inthe sample that is directly attributable to the bleaching step.The rationale for this statement is that bleached immobilemoieties prevent incoming mobile fluorescent ones fromassociating with a limited number of binding sites, effec-tively and proportionally reducing the extent of fluorescencerecovery (Fig. 1d and e). In some cases, redistribution of themoiety of interest may involve active transport, a fact thatcan (although not always) result in directional movement(Iliev and Wouters 2007; Scott et al. 2011), which is notexpected in the case of diffusion. Another handle on thiseventuality is that manipulating the cytoskeleton may arresttransport (Darcy et al. 2006a), but will usually not retardisotropic diffusion.For the purpose of presentation, FRAP traces are typically
normalized by the level of fluorescence measured prior tobleaching, after subtracting the background fluorescence
© 2013 International Society for Neurochemistry, J. Neurochem. (2013) 126, 213--222
Monitoring dynamic physiological processes by FRAP 217

(Bancaud et al. 2010) (Eqn 1); this facilitates visual andstatistical comparison of related experiments.
FðtÞ � BGFðPBÞ � BG
ð1Þ
where F(t) is the fluorescence at time t, F(PB) is thefluorescence prior to bleaching and BG is the backgroundvalue. Note that F(0), the fluorescence just after bleaching,is not necessarily 0.If the extent of bleaching does not influence the time
course of recovery [but see (Brown et al. 1999)], it is helpfulto subtract the fluorescence remaining just after bleaching(Eqn 2), thus rescaling the calculated value of recovery to bewithin a range of 0–1.
FðtÞ � Fð0ÞFðPBÞ � Fð0Þ ð2Þ
When the kinetics of recovery, but not its extent, areunchanged by an experimental perturbation (Campbell andKnight 2007), normalization by the end-point (Eqn 3) canillustrate that even though the proportion of mobile moietiesdiffers, their instantaneous rate of movement does not(Orenbuch et al. 2012).
FðtÞ � Fð0ÞFð1Þ � Fð0Þ ð3Þ
where F(∞) is the fluorescence at the endpoint (Saxton et al.1984).We stress that quantitative analysis of a FRAP experiment
depends strongly on the specific implementation of FRAP.Consequently, we do not strive to provide here a fullmathematical derivation of FRAP analysis. Readers inter-ested in this subject are referred to excellent publications thatprovide examples which can be further tailored to the specificrequirements of novel experimental conditions (Brown et al.1999; Sprague et al. 2004; Weiss 2004; Sprague andMcNally 2005; Santamaria et al. 2006; Jonsson et al.2008; Berkovich et al. 2011). These examples address thegeometry of the bleach spot, the sharpness of its edges, thedimensionality of the diffusion model, interactions withcellular structures, and obstruction to free diffusion.
Alternatives to FRAP
Several viable alternatives to FRAP exist, which addressmobility using different principles; some of these aredescribed briefly below.
Photo activation
This method relies on photoactivatable fluorescent proteins,which in their initial state do not have a fluorescence emission(Patterson and Lippincott-Schwartz 2002; Subach et al.2009). Brief irradiation at an appropriate wavelength causes
a chemical change in the fluorophore, so that it becomesstrongly fluorescent. Activation is performed in a confinedarea and the time-dependent dispersion of fluorescence isrecorded (Tsuriel et al. 2009; Roy et al. 2011). A similarapproach involves the use of photoswitchable proteins, whichirreversibly change their emission properties after a ‘switch-ing’ illumination step (Chudakov et al. 2007; Staras et al.2010). While photoactivation is essentially a mirror-image ofFRAP, some differences between them should be noted. Toachieve photoactivation, typically near-ultraviolet light isbriefly applied. Although irradiation at this wavelength can bemore biologically damaging, this fact is usually offset by thefact that the intensity of irradiation needed to achievephotoactivation is significantly weaker than that typicallyused for bleaching. Another difference refers to the signal tonoise ratio. In FRAP, the initial signal is weak, and increaseswith time, while in photo activation the inverse is true,resulting in an inversion in the signal to noise ratio betweenthe two methods (Tsuriel et al. 2006). This should beconsidered when deciding on the specific methodology to use.
Super-resolution and single-molecule localizationIn many cases, it is feasible to use live-cell super-resolutiontechniques to directly view the mobility of individualorganelles or of protein complexes. For example, stimulatedemission depletion microscopy was used to measure themobility of styryl-dye loaded vesicles (Westphal et al. 2008;Kamin et al. 2010). Similarly, three-dimensional tracking ofsingle vesicles loaded with quantum dots was achieved at aresolution of approximately 20 nm to examine activity-dependent changes in vesicle mobility (Park et al. 2012).Another important example is the direct visualization of themobility of glutamate receptors into and out of post-synapticcontacts by nanoscopic single-molecule tracking and local-ization (Cognet et al. 2006).
Fluorescence correlation spectroscopy
In a fluorescence correlation spectroscopy experiment, asmall volume is illuminated within a labeled sample.Labeling needs to be sufficiently sparse to ensure that atany given time, very few fluorophores enter or exit theillumination volume. By analyzing the fluctuation in thefluorescence readout, much can be deduced concerning themobility of the labeled moieties (Gennerich and Schild 2000,2002). This method has been employed, for example, toexamine mobility of synaptic vesicles within the presynapticterminal (Yeung et al. 2007). A related technique, Fluores-cence fluctuation spectroscopy has been employed in asimilar capacity (Jordan et al. 2005).
Applications in neuroscience
FRAP and related techniques have been used to studynumerous questions of fundamental neurobiological interest.
© 2013 International Society for Neurochemistry, J. Neurochem. (2013) 126, 213--222
218 K. Staras et al.

In this section, we explore some of these. The sectionconcludes with a discussion of a particularly powerfulapplication of FRAP for evaluating inter-synaptic vesiclemovement in small central synapses.
Mobility of neurotransmitter receptors and transportersNeurotransmitter receptors are known to be highly concen-trated in the post-synaptic specializations formed oppositethe active zone. Early work revealed that acetylcholinereceptors found on the surface of developing muscle are veryrestricted in their movement (Axelrod et al. 1976). Later,FRAP and other techniques expanded this view by demon-strating that receptors, as well as transporters, move on thesurface of the neurons in and out of synaptic areas, inaddition to being internalized by endocytotic mechanisms(Jacob et al. 2005; Rasse et al. 2005; Bats et al. 2007; Linand Huganir 2007; Eriksen et al. 2009).
The cytoskeleton and axonal transport
The cytoskeleton of axons and dendrites has been exten-sively investigated with FRAP. Its longitudinal organizationand dynamic nature is especially amenable to examination bythis technique. Moreover, FRAP was instrumental inexploring the differences in cytoskeletal stability in differentlocales and under various physiological conditions (Edsonet al. 1993; Takeda et al. 1995; Star et al. 2002; Iliev andWouters 2007). Another related subject, which has beenaddressed exquisitely with FRAP and photoactivationapproaches, is the nature of axonal transport mechanismsof various classes of neuronal proteins (Roy et al. 2011;Tang et al. 2012).
Mobility of calcium and its buffers
Because of its involvement in a myriad of processes, thedynamics of calcium ions within neurons is of intenseinterest (Augustine et al. 2003). Research on this subject hasrevealed that the properties of endogenous calcium buffersare critical determinants of these dynamics, since theystrongly influence the concentration and distribution ofcalcium ions (Gabso et al. 1997). The diffusive propertiesof two of these buffers, parvalbumin and calbindin D28k,have been studied by FRAP (Schmidt et al. 2003, 2005,2007). A quantitative approach revealed that parvalbumin isfreely diffusible, while the mobility of calbindin D28k isrestricted by intermolecular interactions.
Synapse tenacity
Although it has long been known that the synapse is plastic,the extent of exchange of its components had not been well-characterized. This topic was addressed directly in a series ofFRAP studies (Tsuriel et al. 2006, 2009). It was shown thatdifferent structural components of the synapse (post-synapticdensity proteins, active zone proteins, vesicle-associatedproteins etc.) are replaced at vastly different rates. Moreover,
while some of these rates of movements were influenced byneuronal activity patterns, others were not.
Intra- and inter-synaptic vesicle mobility
Close examination of the presynaptic terminal reveals withinit a large collection of vesicles that are packed at a highdensity in the vicinity of the active zone (Palay 1956). Incentral synapses and many peripheral junctions, this vesicleclustering is consistent with the idea that vesicles areimmobilized, probably by interactions with cytoskeletalelements and by cross-interactions between the vesiclesthemselves through vesicle-associated proteins such as thesynapsins (Gitler et al. 2004b; Denker and Rizzoli 2010;Pechstein and Shupliakov 2010). Nonetheless, vesicles alsohave some intra-terminal mobility, as revealed by FRAP,fluorescence correlation spectroscopy and fluorescence fluc-tuation spectroscopy (Li and Murthy 2001; Jordan et al.2005; Gaffield et al. 2006; Yeung et al. 2007), perhapsreflecting the mobilization of vesicles from the cluster to theactive zone upon demand (Kamin et al. 2010). Conversely,in some specialized terminals, such as lizard cone photore-ceptors, FRAP experiments reveal that their vesicles arehighly mobile with movement dynamics approaching the rateof free diffusion (Rea et al. 2004). Recent work in hippo-campal neurons, using a combination of FRAP, photocon-version of styryl dyes, and electron microscopy (Darcy et al.2006a,b; Staras et al. 2010) has observed that a significantnumber of functional releasable vesicles are also transportedactively between distinct terminals (Staras and Branco 2010).This observation significantly extends our view of thepresynaptic terminal, by adding the synapse-spanning vesicle‘superpool’ (Westphal et al. 2008; Staras et al. 2010) to theknown list of functional vesicle pools (Denker and Rizzoli2010; Alabi and Tsien 2012). The discovery of the superpoolalso raised the question of the regulation of inter-synaptictransport of vesicles (Fig. 2). Because deletion of thesynapsins is known to cause a pronounced decrease in thevesicle population within presynaptic terminals of hippocam-pal neurons (Gitler et al. 2004a; Siksou et al. 2007), it washypothesized that in the absence of synapsins more vesiclesare mobile, and that consequently, they should redistributeinto the axon. Indeed, FRAP of the entire vesicle population ofpresynaptic terminals revealed that vesicle mobility wasenhanced by the deletion of the synapsins, and in agreement,3D electron-microscopic reconstruction of the terminals andadjacent axonal segments found more vesicles in the axons ofneurons devoid of synapsins (Fornasiero et al. 2012; Oren-buch et al. 2012). Moreover, extrapolation of the FRAP tracesillustrated that deletion of the synapsins enlarged the fractionof mobile vesicles (Orenbuch et al. 2012) (Fig. 2b–c).Finally, synapsins were found to specifically affect themobility of resting pool vesicles, by comparing FRAPexperiments using the styryl dye FM1-43, which labelsrecycling-pool vesicles, and of synaptophysin I-EGFP, which
© 2013 International Society for Neurochemistry, J. Neurochem. (2013) 126, 213--222
Monitoring dynamic physiological processes by FRAP 219

labels all synaptic vesicles (Orenbuch et al. 2012). Interest-ingly, the list of proteins participating in the regulation ofinter-synaptic vesicle traffic was recently expanded to includea-synuclein, a protein associated with Parkinson’s disease(Scott and Roy 2012).
Concluding remarks
FRAP is a group of related techniques useful for probingmobility of sub-resolution particles, be they proteins ororganelles, in living functional cells. The ability to geneti-cally manipulate cells to introduce a host of differentfluorescent labels into a myriad of structures and proteinshas substantially enhanced the utility of FRAP. Advances ininstrumentation, such as the introduction of secondaryscanners and of ultrafast and sensitive super-resolutionacquisition, will no doubt amplify the applicability of FRAPin traditionally quantitative research fields like neuroscience.
Acknowledgements
This work was supported by grant 1427/12 of the Israel ScienceFoundation (DG). The authors do not report any conflict of interest.
References
Aizer A., Brody Y., Ler L. W., Sonenberg N., Singer R. H. and Shav-Tal Y. (2008) The dynamics of mammalian P body transport,assembly, and disassembly in vivo. Mol. Biol. Cell 19, 4154–4166.
Alabi A. A. and Tsien R. W. (2012) Synaptic vesicle pools anddynamics. Cold Spring Harb. Perspect. Biol. 4, a013680.
Augustine G. J., Santamaria F. and Tanaka K. (2003) Local calciumsignaling in neurons. Neuron 40, 331–346.
Axelrod D., Ravdin P., Koppel D. E., Schlessinger J., WebbW.W., ElsonE. L. and Podleski T. R. (1976) Lateral motion of fluorescentlylabeled acetylcholine receptors in membranes of developing musclefibers. Proc. Natl Acad. Sci. USA 73, 4594–4598.
Bancaud A., Huet S., Rabut G. and Ellenberg J. (2010) Fluorescenceperturbation techniques to study mobility and molecular dynamicsof proteins in live cells: FRAP, photoactivation, photoconversion,and FLIP. Cold Spring Harb. Protoc. 2010; doi:10.1101/pdb.top90.
Bats C., Groc L. and Choquet D. (2007) The interaction betweenStargazin and PSD-95 regulates AMPA receptor surfacetrafficking. Neuron 53, 719–734.
Berkovich R., Wolfenson H., Eisenberg S., Ehrlich M., Weiss M.,Klafter J., Henis Y. I. and Urbakh M. (2011) Accuratequantification of diffusion and binding kinetics of non-integralmembrane proteins by FRAP. Traffic 12, 1648–1657.
Betz W. J. and Bewick G. S. (1992) Optical analysis of synaptic vesiclerecycling at the frog neuromuscular junction. Science 255, 200–203.
Blatter L. A. and Wier W. G. (1990) Intracellular diffusion, binding, andcompartmentalization of the fluorescent calcium indicators indo-1and fura-2. Biophys. J. 58, 1491–1499.
Brown E. B., Wu E. S., Zipfel W. and Webb W. W. (1999) Measurementof molecular diffusion in solution by multiphoton fluorescencephotobleaching recovery. Biophys. J. 77, 2837–2849.
Campbell J. J. and Knight M. M. (2007) An improved confocal FRAPtechnique for the measurement of long-term actin dynamics inindividual stress fibers. Microsc. Res. Tech. 70, 1034–1040.
Chudakov D. M., Lukyanov S. and Lukyanov K. A. (2007) Trackingintracellular protein movements using photoswitchable fluorescentproteins PS-CFP2 and Dendra2. Nat. Protoc. 2, 2024–2032.
Clancy B. and Cauller L. J. (1998) Reduction of backgroundautofluorescence in brain sections following immersion insodium borohydride. J. Neurosci. Methods 83, 97–102.
Cognet L., Groc L., Lounis B. and Choquet D. (2006) Multiple routes forglutamate receptor trafficking: surface diffusion and membranetraffic cooperate to bring receptors to synapses. Sci. STKE 327, pe13.
Darcy K. J., Staras K., Collinson L. M. and Goda Y. (2006a)Constitutive sharing of recycling synaptic vesicles betweenpresynaptic boutons. Nat. Neurosci. 9, 315–321.
Darcy K. J., Staras K., Collinson L. M. and Goda Y. (2006b) Anultrastructural readout of fluorescence recovery afterphotobleaching using correlative light and electron microscopy.Nat. Protoc. 1, 988–994.
Denker A. and Rizzoli S. O. (2010) Synaptic vesicle pools: an update.Front. Synaptic. Neurosci. 2, 135.
Edson K. J., Lim S. S., Borisy G. G. and Letourneau P. C. (1993) FRAPanalysis of the stability of the microtubule population alongthe neurites of chick sensory neurons. Cell Motil. Cytoskeleton 25,59–72.
Endress E., Weigelt S., Reents G. and Bayerl T. M. (2005) Derivation ofa closed form analytical expression for fluorescence recovery afterphoto bleaching in the case of continuous bleaching during readout. Eur. Phys. J. E. Soft Matter 16, 81–87.
Eriksen J., Rasmussen S. G., Rasmussen T. N., Vaegter C. B., Cha J. H.,Zou M. F., Newman A. H. and Gether U. (2009) Visualization ofdopamine transporter trafficking in live neurons by use offluorescent cocaine analogs. J. Neurosci. 29, 6794–6808.
Fornasiero E. F., Raimondi A., Guarnieri F. C., Orlando M., Fesce R.,Benfenati F. and Valtorta F. (2012) Synapsins contribute to thedynamic spatial organization of synaptic vesicles in an activity-dependent manner. J. Neurosci. 32, 12214–12227.
Fridman V., Gerson-Gurwitz A., Movshovich N., Kupiec M. and GheberL. (2009) Midzone organization restricts interpolar microtubuleplus-end dynamics during spindle elongation. EMBO Rep. 10,387–393.
Gabso M., Neher E. and Spira M. E. (1997) Low mobility of the Ca2+buffers in axons of cultured Aplysia neurons. Neuron 18, 473–481.
Gaffield M. A., Rizzoli S. O. and Betz W. J. (2006) Mobility of synapticvesicles in different pools in resting and stimulated frog motornerve terminals. Neuron 51, 317–325.
Gauthier-Kemper A., Weissmann C., Reyher H. J. and Brandt R. (2012)Monitoring cytoskeletal dynamics in living neurons usingfluorescence photoactivation. Methods Enzymol. 505, 3–21.
Gennerich A. and Schild D. (2000) Fluorescence correlationspectroscopy in small cytosolic compartments depends criticallyon the diffusion model used. Biophys. J. 79, 3294–3306.
Gennerich A. and Schild D. (2002) Anisotropic diffusion in mitral celldendrites revealed by fluorescence correlation spectroscopy.Biophys. J. 83, 510–522.
Gitler D., Takagishi Y., Feng J., Ren Y., Rodriguiz R. M., Wetsel W. C.,Greengard P. and Augustine G. J. (2004a) Different presynapticroles of synapsins at excitatory and inhibitory synapses.J. Neurosci. 24, 11368–11380.
Gitler D., Xu Y., Kao H.-T., Lin D., Lim S., Feng J., Greengard P. andAugustine G. J. (2004b) Molecular determinants of synapsintargeting to presynaptic terminals. J. Neurosci. 24, 3711–3720.
Iliev A. I. and Wouters F. S. (2007) Application of simplephotobleaching microscopy techniques for the determination of
© 2013 International Society for Neurochemistry, J. Neurochem. (2013) 126, 213--222
220 K. Staras et al.

the balance between anterograde and retrograde axonal transport.J. Neurosci. Methods 161, 39–46.
Jacob T. C., Bogdanov Y. D., Magnus C., Saliba R. S., Kittler J. T.,Haydon P. G. and Moss S. J. (2005) Gephyrin regulates the cellsurface dynamics of synaptic GABAA receptors. J. Neurosci. 25,10469–10478.
Jaskolski F. and Henley J. M. (2009) Synaptic receptor trafficking: thelateral point of view. Neuroscience 158, 19–24.
Jonsson P., Jonsson M. P., Tegenfeldt J. O. and Hook F. (2008) Amethod improving the accuracy of fluorescence recovery afterphotobleaching analysis. Biophys. J. 95, 5334–5348.
Jordan R., Lemke E. A. and Klingauf J. (2005) Visualization of synapticvesicle movement in intact synaptic boutons using fluorescencefluctuation spectroscopy. Biophys. J. 89, 2091–2102.
Kamin D., Lauterbach M. A., Westphal V., Keller J., Schonle A., Hell S.W. and Rizzoli S. O. (2010) High- and low-mobility stages in thesynaptic vesicle cycle. Biophys. J. 99, 675–684.
Kang M., Day C. A., Drake K., Kenworthy A. K. and DiBenedetto E.(2009) A generalization of theory for two-dimensional fluorescencerecovery after photobleaching applicable to confocal laser scanningmicroscopes. Biophys. J. 97, 1501–1511.
Koskinen M., Bertling E. and Hotulainen P. (2012) Methods to measureactin treadmilling rate in dendritic spines. Methods Enzymol. 505,47–58.
Li Z. and Murthy V. N. (2001) Visualizing postendocytic trafficof synaptic vesicles at hippocampal synapses. Neuron 31, 593–605.
Lin D. T. and Huganir R. L. (2007) PICK1 and phosphorylation of theglutamate receptor 2 (GluR2) AMPA receptor subunit regulatesGluR2 recycling after NMDA receptor-induced internalization.J. Neurosci. 27, 13903–13908.
Malkani N. and Schmid J. A. (2011) Some secrets of fluorescentproteins: distinct bleaching in various mounting fluids andphotoactivation of cyan fluorescent proteins at YFP-excitation.PLoS ONE 6, e18586.
Mitra K. and Lippincott-Schwartz J. (2010) Analysis of mitochondrialdynamics and functions using imaging approaches. Curr. Protoc.Cell Biol. 46, 4.25.1–4.25.21.
Nadezhdina E. S., Lomakin A. J., Shpilman A. A., Chudinova E. M. andIvanov P. A. (2010) Microtubules govern stress granule mobilityand dynamics. Biochim. Biophys. Acta 1803, 361–371.
Orenbuch A., Shalev L., Marra V., Sinai I., Lavy Y., Kahn J., Burden J.J., Staras K. and Gitler D. (2012) Synapsin selectively controls themobility of resting pool vesicles at hippocampal terminals.J. Neurosci. 32, 3969–3980.
Palay S. (1956) Synapses in the central nervous system. J. Biophys.Biochem. Cytol. 2, 193–202.
Park H., Li Y. and Tsien R. W. (2012) Influence of synaptic vesicleposition on release probability and exocytotic fusion mode. Science335, 1362–1366.
Patterson G. H. and Lippincott-Schwartz J. (2002) A photoactivatableGFP for selective photolabeling of proteins and cells. Science 297,1873–1877.
Pechstein A. and Shupliakov O. (2010) Taking a back seat: synapticvesicle clustering in presynaptic terminals. Front. Synaptic.Neurosci. 2, 143.
Peters R., Peters J., Tews K. H. and Bahr W. (1974) A microfluorimetricstudy of translational diffusion in erythrocyte membranes. Biochim.Biophys. Acta 367, 282–294.
Rasse T. M., Fouquet W., Schmid A. et al. (2005) Glutamate receptordynamics organizing synapse formation in vivo. Nat. Neurosci. 8,898–905.
Rea R., Li J., Dharia A., Levitan E. S., Sterling P. and Kramer R. H.(2004) Streamlined synaptic vesicle cycle in cone photoreceptorterminals. Neuron 41, 755–766.
Reits E. A. and Neefjes J. J. (2001) From fixed to FRAP: measuringprotein mobility and activity in living cells. Nat. Cell Biol. 3,E145–E147.
Roy S., Yang G., Tang Y. and Scott D. A. (2011) A simplephotoactivation and image analysis module for visualizing andanalyzing axonal transport with high temporal resolution. Nat.Protoc. 7, 62–68.
Santamaria F., Wils S., De Schutter E. and Augustine G. J. (2006)Anomalous diffusion in Purkinje cell dendrites caused by spines.Neuron 52, 635–648.
Saxton W. M., Stemple D. L., Leslie R. J., Salmon E. D., Zavortink M.and McIntosh J. R. (1984) Tubulin dynamics in culturedmammalian cells. J. Cell Biol. 99, 2175–2186.
Schmidt H., Brown E. B., Schwaller B. and Eilers J. (2003) Diffusionalmobility of parvalbumin in spiny dendrites of cerebellar Purkinjeneurons quantified by fluorescence recovery after photobleaching.Biophys. J. 84, 2599–2608.
SchmidtH., Schwaller B. andEilers J. (2005)CalbindinD28k targetsmyo-inositol monophosphatase in spines and dendrites of cerebellarPurkinje neurons. Proc. Natl Acad. Sci. USA 102, 5850–5855.
Schmidt H., Arendt O., Brown E. B., Schwaller B. and Eilers J. (2007)Parvalbumin is freely mobile in axons, somata and nuclei ofcerebellar Purkinje neurones. J. Neurochem. 100, 727–735.
Scott D. and Roy S. (2012) Alpha-synuclein inhibits intersynapticvesicle mobility and maintains recycling-pool homeostasis.J. Neurosci. 32, 10129–10135.
Scott D. A., Das U., Tang Y. and Roy S. (2011) Mechanistic logicunderlying the axonal transport of cytosolic proteins. Neuron 70,441–454.
Siksou L., Rostaing P., Lechaire J. P. et al. (2007) Three-dimensionalarchitecture of presynaptic terminal cytomatrix. J. Neurosci. 27,6868–6877.
Sinnecker D., Voigt P., Hellwig N. and Schaefer M. (2005) Reversiblephotobleaching of enhanced green fluorescent proteins.Biochemistry 44, 7085–7094.
Soumpasis D. M. (1983) Theoretical analysis of fluorescencephotobleaching recovery experiments. Biophys. J. 41, 95–97.
Sprague B. L. and McNally J. G. (2005) FRAP analysis of binding:proper and fitting. Trends Cell Biol. 15, 84–91.
Sprague B. L., Pego R. L., Stavreva D. A. and McNally J. G. (2004)Analysis of binding reactions by fluorescence recovery afterphotobleaching. Biophys. J. 86, 3473–3495.
Star E. N., Kwiatkowski D. J. and Murthy V. N. (2002) Rapid turnoverof actin in dendritic spines and its regulation by activity. Nat.Neurosci. 5, 239–246.
Staras K. and Branco T. (2010) Sharing vesicles between centralpresynaptic terminals: implications for synaptic function. Front.Synaptic Neurosci. 2, 20.
Staras K., Branco T., Burden J. J., Pozo K., Darcy K., Marra V.,Ratnayaka A. and Goda Y. (2010) A vesicle superpool spansmultiple presynaptic terminals in hippocampal neurons. Neuron 66,37–44.
Subach F. V., Patterson G. H., Manley S., Gillette J. M., Lippincott-Schwartz J. and Verkhusha V. V. (2009) Photoactivatable mCherryfor high-resolution two-color fluorescence microscopy. Nat.Methods 6, 153–159.
Takeda S., Funakoshi T. and Hirokawa N. (1995) Tubulin dynamics inneuronal axons of living zebrafish embryos. Neuron 14, 1257–1264.
Tang Y., Das U., Scott D. A. and Roy S. (2012) The slow axonaltransport of alpha-synuclein–mechanistic commonalities amongstdiverse cytosolic cargoes. Cytoskeleton (Hoboken) 69, 506–513.
Teissie J., Tocanne J. F. and Baudras A. (1978) A fluorescence approachof the determination of translational diffusion coefficients of lipids
© 2013 International Society for Neurochemistry, J. Neurochem. (2013) 126, 213--222
Monitoring dynamic physiological processes by FRAP 221

in phospholipid monolayer at the air-water interface. Eur. J.Biochem. 83, 77–85.
Tsibidis G. D. (2009) Quantitative interpretation of binding reactions ofrapidly diffusing species using fluorescence recovery afterphotobleaching. J. Microsc. 233, 384–390.
Tsuriel S., Geva R., Zamorano P., Dresbach T., Boeckers T.,Gundelfinger E. D., Garner C. C. and Ziv N. E. (2006) Localsharing as a predominant determinant of synaptic matrix moleculardynamics. PLoS Biol. 4, e271.
Tsuriel S., Fisher A., Wittenmayer N., Dresbach T., Garner C. C. and ZivN. E. (2009) Exchange and redistribution dynamics of thecytoskeleton of the active zone molecule bassoon. J. Neurosci.29, 351–358.
Wang Q., Zhang X., Zhang L., He F., Zhang G., Jamrich M. and WenselT. G. (2008) Activation-dependent hindrance of photoreceptor Gprotein diffusion by lipid microdomains. J. Biol. Chem. 283,30015–30024.
Weiss M. (2004) Challenges and artifacts in quantitative photobleachingexperiments. Traffic 5, 662–671.
Westphal V., Rizzoli S. O., Lauterbach M. A., Kamin D., Jahn R. andHell S. W. (2008) Video-rate far-field optical nanoscopy dissectssynaptic vesicle movement. Science 320, 246–249.
Wu J., Shekhar N., Lele P. P. and Lele T. P. (2012) FRAP analysis:accounting for bleaching during image capture. PLoS ONE 7,e42854.
Yang J., Kohler K., Davis D. M. and Burroughs N. J. (2010) An improvedstrip FRAP method for estimating diffusion coefficients: correctingfor the degree of photobleaching. J. Microsc. 238, 240–253.
Yeung C., Shtrahman M. and Wu X. L. (2007) Stick-and-diffuse andcaged diffusion: a comparison of two models of synaptic vesicledynamics. Biophys. J. 92, 2271–2280.
Zador A. and Koch C. (1994) Linearized models of calcium dynamics:formal equivalence to the cable equation. J. Neurosci. 14,4705–4715.
© 2013 International Society for Neurochemistry, J. Neurochem. (2013) 126, 213--222
222 K. Staras et al.




















