
MOLECULAR EFFECTS of lithium
51
Kurs “ Allgemeine und systematische Pharmakologie und Toxikologie” Sommersemester 2011 Seminarthema II: Psychopharmaka Der Inhalt bzw. die Gliederung der Referate ist frühzeitig mit der/dem zuständigen Dozentin/en abzusprechen. Alle Referate sollten max. 15 Minuten dauern und den Einsatz von Hilfsmitteln (Folien) umfassen. Bei Wiederverwendung von Overhead-Folien von Kolleginnen/en vorangegangener Seminare werden keine Jokerpunkte (siehe Link "Creditsystem") vergeben. Dr. Barbara Möpps N 26-5204 500-65505/65515 Referat I: Therapie der endogenen Depression Belmaker, R.H. and G. Agam (2008): Major depressive disorder. N. Eng. J. Med 358: 55-68. Bschor, T. and M. Adli (2008): Treatment of depressive disorders. Dtsch. Ärztebl. Int. 105: 782-792. Lee, S., Jeong, J., Kwak, Y., and Park, S.K. (2010): Depression research: where are we now? Mol. Brain 3: 8. Ihr Referat sollte folgende Punkte umfassen: - Aminhypothese, Therapie der unipolaren Depression: trizyklische und heterozyklische Antidepressiva, MAO- und reuptake-Inhibitoren - Therapeutische Anwendung und Nebenwirkungen - neue Erkenntnisse zu den Mechanismen der Entstehung von Depressionen Referat II: Therapie der bipolaren Affekterkrankung Quiroz, J.A., Gould, T.D., and Manji, H.K. (2004): Molecular effects of lithium. Mol. Interv. 4: 259-272. Beaulieu, J.M. and Caron, M.G. (2008): Looking at lithium. Mol. Interv. 8: 230-241. Schloesser, R.J., Huang, J., Klein, P.S., and Manji, H.K. (2008): Cellular plasticity cascades in the pathophysiology and treatment of bipolar disorder. Neuropharmacol. 33: 110-133. Ihr Referat sollte folgende Punkte umfassen: - Definition und Therapie der bipolaren Depression - Molekulare und zelluläre Mechanismen der Wirkung von Lithiumionen sowie Pharmakokinetik und Pharmakodynamik von Lithium Referat III: Therapie der Schizophrenie Burlon, M. (2007): Pharmakotherapie der Schizophrenie-“state of the art”. NeuroTransmitter 5, 59-70. Tajima, K., H. Fernandez, J.J. Lopez-Ibor, J.L. Carrasco, and M. Diaz-Marsa (2009): Schizophrenia treatment. Critical review on the drugs and mechanisms of action of antipsychotics. Actas Esp. Psiquatr. 37: 330-342. Tost, H., Alam, T., and Meyer-Lindenberg (2010): Dopamine and psychosis: theory, pathomechanisms and intermediate phenotypes. Neurosci. Biobehav. Rev. 34: 689- 700. Ihr Referat sollte folgende Punkte umfassen: - Pathogenese der Schizophrenie, Dopaminhypothese, neue Ansätze in der Therapie - Therapie: niederpotente versus hochpotente Antipsychotika typische (first) versus atypischen (second) Antipsychotika, NW
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of MOLECULAR EFFECTS of lithium

Kurs “ Allgemeine und systematische Pharmakologie und Toxikologie” Sommersemester 2011
Seminarthema II: Psychopharmaka Der Inhalt bzw. die Gliederung der Referate ist frühzeitig mit der/dem zuständigen Dozentin/en abzusprechen. Alle Referate sollten max. 15 Minuten dauern und den Einsatz von Hilfsmitteln (Folien) umfassen. Bei Wiederverwendung von Overhead-Folien von Kolleginnen/en vorangegangener Seminare werden keine Jokerpunkte (siehe Link "Creditsystem") vergeben.
Dr. Barbara Möpps N 26-5204 500-65505/65515
Referat I: Therapie der endogenen Depression Belmaker, R.H. and G. Agam (2008): Major depressive disorder. N. Eng. J. Med 358: 55-68. Bschor, T. and M. Adli (2008): Treatment of depressive disorders. Dtsch. Ärztebl. Int. 105: 782-792. Lee, S., Jeong, J., Kwak, Y., and Park, S.K. (2010): Depression research: where are we now? Mol. Brain 3: 8.
Ihr Referat sollte folgende Punkte umfassen: - Aminhypothese, Therapie der unipolaren Depression: trizyklische und
heterozyklische Antidepressiva, MAO- und reuptake-Inhibitoren - Therapeutische Anwendung und Nebenwirkungen - neue Erkenntnisse zu den Mechanismen der Entstehung von Depressionen
Referat II: Therapie der bipolaren Affekterkrankung
Quiroz, J.A., Gould, T.D., and Manji, H.K. (2004): Molecular effects of lithium. Mol. Interv. 4: 259-272. Beaulieu, J.M. and Caron, M.G. (2008): Looking at lithium. Mol. Interv. 8: 230-241. Schloesser, R.J., Huang, J., Klein, P.S., and Manji, H.K. (2008): Cellular plasticity cascades in the pathophysiology and treatment of bipolar disorder. Neuropharmacol. 33: 110-133.
Ihr Referat sollte folgende Punkte umfassen: - Definition und Therapie der bipolaren Depression - Molekulare und zelluläre Mechanismen der Wirkung von Lithiumionen
sowie Pharmakokinetik und Pharmakodynamik von Lithium
Referat III: Therapie der Schizophrenie Burlon, M. (2007): Pharmakotherapie der Schizophrenie-“state of the art”. NeuroTransmitter 5, 59-70. Tajima, K., H. Fernandez, J.J. Lopez-Ibor, J.L. Carrasco, and M. Diaz-Marsa (2009): Schizophrenia treatment. Critical review on the drugs and mechanisms of action of antipsychotics. Actas Esp. Psiquatr. 37: 330-342. Tost, H., Alam, T., and Meyer-Lindenberg (2010): Dopamine and psychosis: theory, pathomechanisms and intermediate phenotypes. Neurosci. Biobehav. Rev. 34: 689-700.
Ihr Referat sollte folgende Punkte umfassen: - Pathogenese der Schizophrenie, Dopaminhypothese, neue Ansätze in der Therapie
- Therapie: niederpotente versus hochpotente Antipsychotika typische (first) versus atypischen (second) Antipsychotika, NW

October 2004Volume 4, Issue 5 259
Jorge A. Quiroz, Todd D. Gould, and Husseini K. Manji
Laboratory of Molecular Pathophysiology, Mood and Anxiety Disorders Program, National
Institute of Mental Health, NIH, Bethesda, Maryland 20892
ipolar affective disorder is a common, severe, chronic, and often life-threatening illness, associated
with other medical and psychiatric conditions (i.e., co-morbidity). The treatment of this devastating
disorder was revolutionized by the discovery of lithium’s antimanic effects over fifty years ago. Recent
molecular and cellular biological studies have identified a number of unexpected targets for this mon-
ovalent cation, notably glycogen synthase kinase-3 and neurotrophic signaling cascades. These find-
ings are leading to a reconceptualization of the biological underpinnings of bipolar disorder and are
resulting in considerable interest in utilizing lithium for the treatment of certain neurodegenerative
disorders. We review recent insights into lithium’s actions including its direct inhibitory actions on ino-
sitol monophosphatase, inositol polyphosphate 1-phosphatase, glycogen synthase kinase-3, fructose
1,6-bisphosphatase, bisphosphate nucleotidase, and phosphoglucomutase enzymes. We also discuss
lithium’s intracellular downstream targets including adenylate cyclase, the phosphoinositol cascade
(and its effect on protein kinase C), arachidonic acid metabolism, and effects on neurotrophic cascades.
Many of the new insights of lithium’s actions may lead to the strategic development of improved thera-
peutics for the treatment of bipolar disorder.
MOLECULAR EFFECTS of lithium

260
Review
Introduction
Bipolar disorder is a devastating and relatively common disease, with an overall lifetime incidence of about 1% in the general pop-ulation. A number of studies show that for a high percentage of patients the outcome is poor, with a high rate of chronicity, resid-ual symptoms, relapse, subsyndromes, cognitive and functional impairment, and psychosocial disability (1, 2). The costs associ-ated with disability and premature death represent an economic burden of tens of billions of dollars annually in the United States alone; not surprisingly, the Global Burden of Disease Study has identified bipolar disorder and mood disorders among the lead-ing causes of disability worldwide, with increasing disability likely in the coming years (3). In addition to the tremendous economic cost, suicide is estimated to be the cause of death in 10-20% of the individuals with bipolar disorder, and increasingly, mood dis-orders are associated with many other health-related consequences (4, 5).
The discovery of lithium’s efficacy as an antimanic agent over fifty years ago revolutionized the treatment of patients with bipolar disorder. The remarkable efficacy of lithium has served to spark a revolution that has, over time, reshaped not only medical and scientific but also popular concepts of severe mental illnesses. Indeed, the efforts to understand how a simple monovalent cation like lithium can exert such profound beneficial effects has led investigators to examine the signal transduction pathways involved in bipolar disorder. After nearly fifty years, lithi-um continues to be one of the mainstays of treatment for this disorder, both for the acute manic phase and as prophy-laxis for recurrent manic and depressive episodes. Adequate lithium treatment, particularly in the context of a lithium clinic (an outpatient clinic dedicated to the treatment of bipolar patients and psycho-pharmacological management of lithium), also reportedly reduces the excessive mortal-ity observed in the illness (6, 7). In the last decade, there has been an explosion in the number of options available for the treatment of recur-rent mood disorders with a parallel and unprecedented increase in the interest in the treatment of bipolar disorder
by pharmaceutical companies, clinicians, researchers, and indeed the general public. Despite the introduction of a number of new anticonvulsants and antipsychotics into the pharmacopeia, the last three years have seen a resurgence of interest not only in lithium’s utility in the long-term treatment of bipolar disorder, but possibly also for neurodegenerative disorders. This renewed interest in lithium has come about largely due to converging evidence from biochemical studies that have identified critical signaling and neu-rotrophic molecules as targets for lithium’s actions.
In spite of lithium’s past success and future promises, how-ever, it remains far from the perfect drug. Increasing evidence suggests that a significant number of patients do not respond ade-quately or cannot tolerate its side effects, or both. Similarly, other mood stabilizers such as valproate (VPA) and carbamazepine are ineffective or intolerable for a significant proportion of patients. The recognition of the significant morbidity and mortality of patients with severe mood disorders as well as the growing appre-ciation that a significant percentage of patients respond poorly to existing treatments have made the task of discovering new thera-peutic agents that are both efficacious and have few side effects increasingly more important. We discuss these recent insights into lithium’s actions and discuss their implications not only for chang-ing our existing concepts of the pathophysiology of severe mood disorders, but also for the strategic development of therapeutics that possess better tolerability, fewer side effects, and better toxic-
Enzyme Function
Inositol monophosphatase (IMPase) Rate limiting enzyme in inositol recycling; lithium’s inhibition
of IMPase led to the inositol depletion hypothesis of lithium’s
actions
Inositol polyphosphate 1-phosphatase
(IPPase)
Enzyme involved in inositol recylcing in phosphoinositol signal-
ing; acts prior to IMPase
Bisphosphate nucleotidase (BPNase) Removes phosphate from 3’-phosphoadenosine 5’-phosphate
(PAP) to form adenosine 5’phosphate (AMP); an increase in
PAP inhibits sulphotransferases, which transfer sulfur to bio-
logical molecules
Fructose 1,6-bisphosphastase (FBPase) Key enzyme in glyconeogenesis; catalyzes the removal of the
1-phosphate from fructose 1, 6-bisphosphatase to form fruc-
tose 6-phosphate
Phosphoglucomutase (PGM) Key enzyme in glycogenolysis and glycogenesis; catalyzes the
formation of glucose 1-phosphate from glucose 6-phosphate
during glycogenolysis (and the reverse during glycogenesis)
Glycogen synthase kinase-3 (GSK-3) Normally active kinase that is inhibited by the activity of many
signaling pathways; inhibiting GSK-3 has been linked to neu-
rotrophic support, neuroprotection, and possible modulation of
circadian rhythms
Reproduced with permission from Gould et al., Mol. Psychiatry (2004)
Table 1. Direct Targets of Lithium
October 2004Volume 4, Issue 5
Molecular Effects of Lithium
ity profiles and pharmacodynamic characteristics than lithium, which may improve the treatment of bipolar disorder. It is hoped that by understanding lithium’s true therapeutic target we will be able using a hypothesis driven approach, to attempt to treat patients with novel drugs with lithium-mimetic properties. We begin with an overview of lithium’s direct targets and follow with a discussion of adaptive changes, which are observed with chronic lithium administration in a therapeutically relevant time frame.
Direct Targets of Lithium
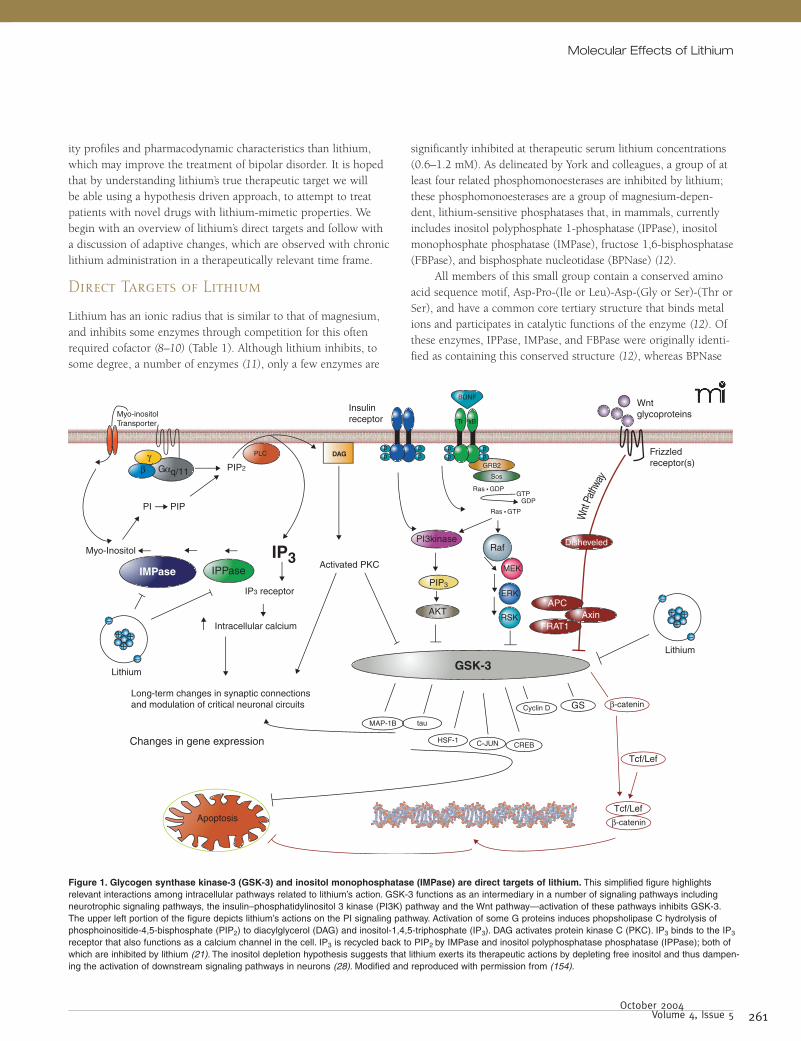
Lithium has an ionic radius that is similar to that of magnesium, and inhibits some enzymes through competition for this often required cofactor (8–10) (Table 1). Although lithium inhibits, to some degree, a number of enzymes (11), only a few enzymes are
significantly inhibited at therapeutic serum lithium concentrations (0.6–1.2 mM). As delineated by York and colleagues, a group of at least four related phosphomonoesterases are inhibited by lithium; these phosphomonoesterases are a group of magnesium-depen-dent, lithium-sensitive phosphatases that, in mammals, currently includes inositol polyphosphate 1-phosphatase (IPPase), inositol monophosphate phosphatase (IMPase), fructose 1,6-bisphosphatase (FBPase), and bisphosphate nucleotidase (BPNase) (12).
All members of this small group contain a conserved amino acid sequence motif, Asp-Pro-(Ile or Leu)-Asp-(Gly or Ser)-(Thr or Ser), and have a common core tertiary structure that binds metal ions and participates in catalytic functions of the enzyme (12). Of these enzymes, IPPase, IMPase, and FBPase were originally identi-fied as containing this conserved structure (12), whereas BPNase
261
Figure 1. Glycogen synthase kinase-3 (GSK-3) and inositol monophosphatase (IMPase) are direct targets of lithium. This simplified figure highlights relevant interactions among intracellular pathways related to lithium’s action. GSK-3 functions as an intermediary in a number of signaling pathways including neurotrophic signaling pathways, the insulin–phosphatidylinositol 3 kinase (PI3K) pathway and the Wnt pathway—activation of these pathways inhibits GSK-3. The upper left portion of the figure depicts lithium’s actions on the PI signaling pathway. Activation of some G proteins induces phopsholipase C hydrolysis of phosphoinositide-4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3). DAG activates protein kinase C (PKC). IP3 binds to the IP3 receptor that also functions as a calcium channel in the cell. IP3 is recycled back to PIP2 by IMPase and inositol polyphosphatase phosphatase (IPPase); both of which are inhibited by lithium (21). The inositol depletion hypothesis suggests that lithium exerts its therapeutic actions by depleting free inositol and thus dampen-ing the activation of downstream signaling pathways in neurons (28). Modified and reproduced with permission from (154).

262
Review
was identified subsequently based upon commonly shared amino acid sequences (13). Newer technology utilizing computer-assisted molecular modeling may allow for more extensive structural char-acterization of the properties of this binding site, with the poten-tial to discover novel enzymes inhibited by lithium that do not contain this specific motif.
Lithium also inhibits the metabolic enzymes phosphogluco-mutase (PGM) (14–17) and glycogen synthase kinase-3 (GSK-3), a serine–threonine kinase that functions as an intermediary in numerous intracellular signaling pathways (18, 19) (Table 1). Significant research effort has focused on IMPase and GSK-3 as possible therapeutically relevant targets of lithium inhibition (Figure 1) based predominantly on the roles these enzymes play in CNS functions (20). Pharmaceutical companies have focused on both of these lithium targets, and it is very likely that a future pharmaceutical inhibitor of either IMPase or GSK-3 may have lithium-mimetic properties in the treatment of bipolar disorder.
IMPase and IPPase
IMPase and IPPase are enzymes involved in recycling and de novo synthesis of inositol, which is a necessary component of the phos-phoinositol (PI) signaling pathway. Many extracellular receptors [such as the serotonin (5-HT)2, α1, and muscarinic (M)1, 3, and 5 receptors] are coupled to the G protein Gq/11, which, through activation of phospholipase C (PLC) mediates the hydrolysis of a cellular membrane phospholipid, phosphoinositide 4,5-bisphos-phate (PIP2), to form the second messengers diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3) (21, 22). DAG and IP3 subse-quently modulate the activity of a multitude of intracellular events (see below).
A number of inositol phosphate phosphatase (IPPase) enzymes are involved in the dephosphorylation (recycling) of IP3 to inositol, a precursor of membrane PIP2 (21). This recycling is necessary to maintain PI-mediated signaling in cell types where inositol is not freely available. The enzyme IMPase catalyzes the final (and rate-limiting) step in the conversion of IP3 into inositol. IPPase removes a phosphate from inositol-1,4-bisphosphate, at the point just prior to where IMPase participates. Both appear to be critical steps in the maintenance of inositol levels and continua-tion of PI-mediated signaling (23).
Lithium’s direct effect on IMPase (24, 25) and secondarily on IPPase (26, 27) led to the inositol depletion hypothesis of lithium’s action (28, 29) (Figure 1). The inositol depletion hypothesis sug-gests that lithium exerts its mood stabilizing effect by inhibiting IMPase, decreasing inositol concentrations and thus the amount of PIP2 available for signaling cascades that rely upon this pathway, including but not limited to neurotrophin signaling pathways, receptor tyrosine kinase pathways, and some G protein–medi-ated signaling (28). It is hypothesized that the brain is particularly sensitive to lithium because of inositol’s relatively poor penetra-tion across the blood-brain barrier (28) or to a reduced ability of
specific neuronal populations to transport inositol across their cell membranes (23). Furthermore, based on the noncompetitve inhi-bition profile of lithium, more active cells and brain regions may be affected to a greater degree (30); however, a recent study sug-gests that depletion of inositol may not have major effects on PI-mediated signaling. Specifically, Berry and colleagues found that the reduction of intracellular inositol in the brain sodium–myo-inositol transporter (SMIT1) knockout in mice has no effect on PI levels (31).
Although the data are not entirely consistent, lithium does decrease free inositol levels in brain sections and in the brains of rodents treated with lithium (32, 33). Lithium treatment also decreases myoinositol (another form of inositol) in human sub-jects (34). Thus, it is our contention that, although it was first proposed more than a decade ago (28, 29), the inositol depletion hypothesis remains a viable one for the mechanism of action of lithium. However, no clinically-approved inhibitors of either IPPase or IMPase are available, and therefore it remains difficult to test the inositol depletion hypothesis in patients with bipolar disorder. Past pharmaceutical industry efforts have attempted to develop a brain-penetrating IMPase inhibitor by altering the primary substrate of IMPase—inositol monophosphate (35). Compounds with sufficient inhibition properties were developed, but have thus far failed to advance through clinical trials because they are too highly charged (36), or extremely lipophilic (37), both properties that limit bioavailability in the brain (35). The pub-lished crystal structure and modeling studies of IMPase may help to develop novel inhibitors (38, 39). As we discuss below, down-stream molecules [notably protein kinase C (PKC)] of IMPase sig-naling and the PI pathway may also be relevant targets.
GSK-3
GSK-3 is a serine–threonine kinase that is normally highly active in cells, and is deactivated by signals originating from numerous signaling pathways [for example the Wnt pathway, PI-3` kinase (PI3K) pathway, protein kinase A, protein kinase C, among many others]. It is found in two isoforms, α and β, that have similar, but not always identical, biological functions. Cellular targets of GSK-3 are numerous and often depend on the signaling pathway that is acting upon it (due to cellular localization and regional sequestration). For example, Wnt pathway-mediated inhibition of GSK-3 activates the transcription factor β-catenin, whereas in the insulin–PI3K signaling pathway, inhibition of GSK-3 results in activation of the enzyme glycogen synthase. Targets of GSK-3 include, among others, transcription factors [β-catenin, cyclic AMP response element binding protein (CREB), c-Jun], proteins bound to microtubules [Tau, microtubule-associated protein (MAP)-1B, kinesin light chain], cell cycle mediators (cyclin D, human ninein), and regulators of metabolism (glycogen synthase, pyruvate dehydrogenase) (20, 40) (Figure 1).
As a component of many signaling pathways, with mul-tiple cellular targets to choose from, GSK-3 is able to regulate a

263 October 2004
Volume 4, Issue 5
Molecular Effects of Lithium
diverse array of cellular processes such as glycogen synthesis, gene transcription, events related to synaptic plasticity, apoptosis (cell death), and the circadian cycle (41–44). Although many of these functions are likely critically important to both cellular and organ-ism functioning, GSK-3 is currently receiving the most interest as a regulator of apoptosis and cellular resilience (Figure 1). Generally, increased activity of GSK-3 has pro-apoptotic effects, whereas inhibiting GSK-3 attenuates or prevents apoptosis (42, 44).
Evidence suggests an association between mood disorders and impairments of neuroplasticity and cellular resilience—with both in vivo and postmortem studies suggesting neuron and/or glial cell loss or atrophy in circumscribed brain areas (45, 46). Importantly, lithium likely has neuroprotective effects, both clini-cally and in rodent and cell-based models (45, 47). Lithium may exert these neuroprotective effects at least partly by inhibiting GSK-3 (42, 44).
In 1996, Klein and Melton noted that lithium administration to developing Xenopus embryos had the same effect—duplication of the dorsal axis (48)—as did down-regulation of GSK-3 activ-ity (49). These parallel observations led them to study the direct effects of lithium on GSK-3 (18). Lithium was initially found to inhibit GSK-3 with an enzyme inhibition constant (Ki) of 1–2 mM (serum therapeutic range 0.6 to 1.2 mM) (18, 19); however, evidence showing that lithium inhibits GSK-3 by competing with magnesium (9, 50) suggests that the original studies using higher than physiological levels of magnesium may have underestimated the degree of inhibition.
Early studies suggested that peripheral administration of lithium inhibited brain GSK-3 in the 7-day-old rat brain (51). More recently, studies suggest that this enzyme is significantly inhibited in the rodent brain in the presence of therapeutic serum lithium concentrations during long-term treatment. For example, it was demonstrated that nine days of lithium treatment (at a mean serum concentration of 0.8 mM) of rats increased cytosolic protein levels of β-catenin, a transcription factor regulated directly by GSK-3 (52). This protein level increase was accompanied by a small but significant decrease in β-catenin mRNA levels (reflecting cellular compensation), further suggesting that lithium exerted its actions posttranslationally by inhibiting GSK-3 (52). Confirmatory findings reporting that chronic lithium indeed activates β-catenin- dependent transcription in the mouse brain has been recently published (53). Furthermore, Phiel and colleagues found that three weeks of lithium treatment (at serum levels of 0.8–1.2 mM) decreased the amount of amyloid-β peptide in the brains of AP-Swedish/Tg2576 mice (used in modeling familial Alzheimer Disease), a finding that is likely due to inhibition of GSK-3 (54) given lithium’s effect on accumulation of amyloid-β in cell culture (54–56). These preclinical (i.e., animal or cellular) studies clearly suggest that therapeutic serum concentrations of lithium produce a biologically significant inhibition of GSK-3 in the mammalian brain.
Although GSK-3 was identified in 1996 as the target of lithium responsible for the developmental effects in Xenopus
embryos (18), only recently has further evidence been obtained substantially supporting the claim that GSK-3 represents a thera-peutic target of lithium. As discussed, GSK-3 represents a strong candidate as a mediator of lithium’s neuroprotective effects most likely because GSK-3 in the brain is significantly inhibited by therapeutic lithium concentrations. Additionally, recent evidence suggests that the behavioral effects of lithium, at least in rodent models, may also be due to inhibition of GSK-3. Three groups have found that administration of GSK-3 inhibitors results in anti-depressant-like effects in the forced swim test paradigm following either intracerebral ventricle injections in mice (57), peripheral administration to rats (58), or lithium administration to mice (53). Furthermore, O’Brien and colleagues have recently examined the behavioral effects of knocking out a single copy of the GSK-3β gene, observing in these animals the same antidepressant-like behavior induced by alternate pharmacological inhibition and by lithium administration (i.e., increased mobility in the forced swim test) (53). Further supporting the hypothesis that the effects of antidepressants may be mediated in a GSK-3-dependent manner, Li and colleagues reported that inhibitory phosphorylation (on Ser9) of GSK-3 is acutely increased by increasing the concentra-tions of 5-HT in the brain through a variety of pharmacological mechanisms (59). Thus, GSK-3 inhibition may represent a thera-peutically relevant downstream consequence of antidepressant drugs that initially target serotonin levels.
Amphetamine-induced hyperactivity is the most established rodent model for mania. This behavior is reproducibly attenuated by a number of mood stabilizers including lithium, anticonvul-sants, and antipsychotics. Beaulieu et al. recently reported that dopamine-dependent activity increases in mice are mediated in large part via a GSK-3-dependent mechanism (60). They report that both lithium and alternative GSK-3 inhibitors attenuate the hyperactivity in mice lacking the dopamine transporter. They also found that amphetamine administration to wild-type mice results in a decrease in the inhibitory phosphorylation of GSK-3, and that mice heterozygous for GSK-3 have an attenuated response to amphetamine administration. Accordingly, peripheral administra-tion of a GSK-3 inhibitor decreases amphetamine-induced hyper-activity in rats (58). In toto, these data support the possibility that inhibition of GSK-3 may represent lithium’s antimanic as well as its antidepressant target. It will be critical to future understanding of mood disorder etiology to determine which GSK-3 target(s) are responsible for behavior in models of both mania and depression.
In addition to its possible usefulness in the treatment of bipolar disorder (20), inactivation of GSK-3 has been suggested as a potential therapy for a number of diseases, with diabetes and Alzheimer disease receiving the most attention. Diabetes has drawn interest because GSK-3 phosphorylates and deactivates glycogen synthase (61). Alzheimer disease is a target of interest because GSK-3 participates in both the phosphorylation of tau (62, 63) and in the assembly of amyloid-β (54, 55, 64), both of which are thought to be significantly involved in the neurobiol-ogy of Alzheimer disease. Specifically, hyperphosphorylation of

264
tau is associated with the formation of neurofibrillary tangles, and accumulation of amyloid-β leads to amyloid plaques. GSK-3 inhibitors may also be useful for the treatment of cardiac ischemic injury (65), baldness and alopecia [the Wnt pathway is involved in hair growth (66)], other neurodegenerative disorders (45, 47) and stroke and other neurotraumatic injuries (47, 67, 68).
FBPase, BPNase, and PGM
Lithium inhibits FBPase, BPNase, and PGM at therapeutic con-centrations (10). Fructose-1, 6-bisphosphate (a regulator of gluconeogenesis), removes the 1-phosphate from FBPase to form fructose 6-phosphate. Lithium’s inhibition of FBPase was originally described a number of years ago (14, 69, 70), and more recent studies support these findings (71, 72). Lithium-dependent inhibition of FBPase has not received much attention, however, probably because dysfunction of glyconeogenesis is not a primary theory of bipolar disorder pathophysiology. Inhibitors of FBPase are under development as possible treatments for diabetes (73).
Mammalian BPNase acts on bisphosphorylated nucleotides such as 3´-phosphoadenosine 5´-phosphate (PAP), where it removes the 3´ phosphate to form adenosine 5´-phosphate (AMP) (13, 74, 75); hence, BPNase is also referred to as PAP phosphatase. Sulfotransferases are enzymes that transfer a sulfate group to vari-ous biomolecules, using 3´-phosphoadenosine 5´-phosphosulfate (PAPS) as a sulfate donor. PAP is produced following the removal of the sulfate group from PAPS, and acts as an inhibitor of sulfo-transferases. Therefore, inhibition of BPNase (and the subsequent buildup of PAP) would be expected to inhibit sulfotransferases. Although studies in mammalian systems are lacking, biochemical reactions potentially modulated by BPNase and/or PAP accumula-tion include RNA processing metabolism, sodium homeostasis, and sulfation.
The development of nephrogenic diabetes insipidus in patients undergoing lithium therapy might arise from the inhibi-tion of BPNase (13). BPNase, similar to IPPase, hydrolyzes ino-sitol-1,4-bisphosphate, and lithium prevents BPNase-mediated hydrolysis of both substrates (13, 74, 75). Thus, lithium inhibition of BPNase would be expected to have important effects on inosi-tol recycling, similar to inhibiting IMPase or IPPase. The recently described crystal structure of BPNase should help promote the development of novel inhibitors (76), and a recent review has noted some of the possible roles of BPNase in bipolar disorder (77).
PGM catalyzes the formation of glucose 1-phosphate from glucose 6-phosphate during glycogenolysis (and the reverse dur-ing glycogenesis). Lithium was originally identified to inhibit the rabbit and rat PGM enzyme (14–16), and more recently has been found to inhibit human and yeast PGM (17). The role of PGM as a therapeutic target in bipolar disorder treatment has been mostly overlooked due to limited evidence that metabolism of glycogen is involved in this disorder.
Downstream Targets of Lithium
Several signaling pathways exist that are regulated by a number of mood stabilizers (10); those signaling pathways where lithium plays an important role, i.e., the adenylate cyclase (AC), phos-phoinositide (PI), arachidonic acid (AA) and neurotrophic-related signaling pathways, are further discussed in detail here.
Cyclic AMP-mediated Signal Transduction
Significant lithium-dependent modulation of cyclic adenosine monophosphate (cAMP)-mediated signaling has been reported. G proteins modulate intracellular cAMP levels by mediating the effect of neurotransmitters (via extracellular receptors) on AC, an integral membrane protein of which there exist numerous subtypes. AC catalyzes the conversion of adenosine triphosphate (ATP) to cAMP. Stimulation of the G proteins Gαs and Gαolf increases AC activity, whereas stimulation of Gαi results in a decrease in AC activity. The physiologic effects of cAMP appear to be mediated primarily by activation of protein kinase A (PKA), an enzyme that phosphorylates and regulates many proteins includ-ing ion channels, cytoskeletal elements, transcription factors, and other enzymes. One direct target in the central nervous system (CNS) for the actions of PKA is the transcription factor CREB, which plays a major role in long-term neuroplasticity, and is a downstream target of antidepressants. It is noteworthy that tran-scription of the CREB gene increases following long-term treat-ment of rodents with a variety of anti-depressants (22, 78).
One of the genes activated by CREB is brain-derived neuro-trophic factor (BDNF), a protein implicated in neuronal survival and synaptic plasticity. There is a growing body of data suggesting that agents that directly modulate the cAMP–PKA–CREB–BDNF signaling cascade may be useful in the treatment of depression (79). In addition to antidepressant effects on cAMP-mediated sig-naling, mood stabilizers also appear to regulate this pathway. Both lithium and VPA increase BDNF levels in the brains of rats treated chronically with these drugs (80–82). Thus, it is useful to keep in mind that multiple interactions between signaling pathways, e.g., CREB activity and BDNF expression, are regulated by multiple signaling pathways including neurotrophic signaling pathways (as discussed later in this review), and that the cAMP signaling path-way does much more than simply regulate CREB activity.
Lithium appears to have complex effects on cAMP-medi-ated signaling, with the preponderance of the data demonstrating an elevation of basal AC activity, but also a reduction of recep-tor-stimulated responses in both preclinical and clinical studies [see (83) for an excellent and thorough review of these data]. Thus, a number of independent research laboratories have found in preclinical models that the ability of the receptor-mediated signal to be propagated via AC is decreased after lithium treat-ment (22, 83). These extensive cellular findings are consistent with an animal model wherein cholera toxin (a stimulator of the
Review

October 2004Volume 4, Issue 5
G proteins Gs and Golf) induces hyperactivity when injected into the nucleus accumbens of rats. Cholera toxin-induced hyperac-tivity was decreased by lithium administration (84), consistent with decreased Gs and/or Golf activity during lithium treatment. But whereas stimulated levels are decreased, there is evidence to suggest an increase in basal cAMP activity (83). These complex, potentially regional specific effects on basal activity and stimulated AC activity may arise from lithium’s effects on G proteins, AC sub-types, and their relative abundance in different brain regions (83).
Postmortem and peripheral cell studies are also consistent with a role of cAMP in mood disorders. Postmortem brain stud-ies of patients who had bipolar disorder reveal increased levels of Gαs and post-receptor stimulated AC activity (85, 86). Generally, the experiments measuring AC activity in unipolar depression find both reduced immediate and long-term effects (87). Thus, although an oversimplification, the majority of the evidence reports increased activity of the AC system in bipolar disorder and a decrease in activity in unipolar depression.
Caution is warranted when attempting to correlate these pre-clinical and postmortem studies with human disease; however, the available evidence is noteworthy. There are numerous compounds that inhibit AC activity. Particularly, a good deal of specificity has been observed with analogs of the nucleoside adenosine, also called P-site inhibitors (88, 89). Ideally, novel compounds would be isoform-selective in order to avoid peripheral side effects due to the widespread distribution of multiple AC isoforms in different organs in the body. The development of these compounds sug-gests the eventual possibility of trials with these medications in the treatment of bipolar disorder. Stimulators of AC (e.g., forskolin) may be useful for challenge studies.
PI-Mediated Signaling
Inositol phospholipids play a major role in receptor-mediated signal-transduction pathways, involved in a diverse range of responses such as cell division, secretion, neuronal excitability, and responsiveness. The PI pathway is initiated by the activation of G protein–coupled receptors. M1, M2, M3, α1, and 5-HT2 receptors coupled to Gαq/11 induce PLC hydrolysis of the mem-brane component PIP2. Hydrolysis of PIP2 by PLC results in the formation of the intracellular second messengers IP3 and DAG, an endogenous activator of PKC. IP3 binds to the IP3 receptor facili-tating the release of calcium from intracellular stores, in particular the endoplasmic reticulum (22).
Among other proteins, the Ca2+-receptor protein calmodulin (CaM) stimulates calmodulin-dependent protein kinases (CaMKs) that regulate the activity of diverse proteins, including ion chan-nels, signaling molecules, proteins that regulate apoptosis, scaf-folding proteins, and transcription factors (90). As described ear-lier, IPPase and IMPase (enzymes that are involved in recycling of IP3 back to PIP2) are directly inhibited by lithium (21) (Figure 1). Lithium’s inhibition of these enzymes led to the inositol depletion
hypothesis of lithium’s action, which suggests that lithium, via inhibition of IMPase, decreases the availability of myoinositol, and thus the amount of PIP2 available for G protein–mediated signal-ing events that rely upon this pathway (28).
The inositol depletion hypothesis led to a number of studies, both in cultured cells and in animal models, to determine if the PI pathway may be involved in the pathophysiology or treatment of bipolar disorder (91). Interestingly, a number of studies have sug-gested the possibility that multiple distinct mood stabilizers may regulate the PI signaling pathway. These include studies of SMIT1, a high affinity myoinositol transport system that has been charac-terized in various cell types, including those of neural origin (92). The activity and expression of SMIT mRNA in cultured astrocytes is downregulated after chronic treatment with therapeutic con-centrations of lithium (92, 93). Decreased expression of SMIT was also observed after VPA or carbamazepine treatment (92, 93). If replicated in vivo, these findings suggest that SMIT may represent a novel target for the development of new drugs.
Another finding implicating PI signaling in the actions of mood stabilizers comes from Williams and colleagues, who used a tissue-culture assay that measures sensory neuron growth-cone stability to conclude that the depletion of neuronal IP3 may be a common mechanism of action of mood stabilizers (94). These investigators demonstrated that lithium, VPA, and carbamaze-pine all inhibit the collapse of sensory neuron growth cones and increase growth-cone area, effects which were reversed by inositol. The authors then used Dictyostelium, a soil-living organism that relies on IP3 for its development, to identify mutants that confer resistance to the drugs: null mutations of prolyl oligopeptidase confer lithium resistance and elevate intracellular levels of IP3. The authors established a link between lithium and IP3 by showing that prolyl oligopeptidase inhibitors abolished the effects of lithi-um, carbamazepine, and VPA on growth-cone collapse and area in their tissue-culture assay (94).
PKC and Myristoylated Alanine-Rich C Kinase Substrate (MARCKS)PKC is a primary target of DAG (Figure 1), and as such, has been an object of intense research in regard to the actions of lithium and other mood stabilizers on the PI pathway. PKC is a ubiquitous enzyme, highly enriched in the brain, where it plays a significant role in regulating both pre- and postsynaptic aspects of neuro-transmission (95). Recent studies have suggested that PKC activa-tion may facilitate neurotransmitter release via a variety of mecha-nisms, including: 1) modulation of several ionic conductances regulating Ca2+ influx; 2) upstream steps regulating release of Ca2+ from intracellular stores; 3) recruitment of neurotransmitter-con-taining vesicles to at least two distinct vesicle pools; and 4) the Ca2+ sensitivity of the release process itself. PKC is active in many other cellular processes, including stimulating transmembrane glu-cose transport, secretion, exocytosis, smooth muscle contraction, gene expression, modulation of ion conductance, cell prolifera-
Molecular Effects of Lithium
265

266
tion, and desensitization of extracellular receptors (95). PKC and PKC signaling appear to be a target of both lithium
and VPA (91). Chronic lithium treatment decreases the level of PKC isozymes α and ε (96–98) in cell culture and in treated rodents. The precise mechanisms by which lithium exerts these isozyme-selective actions is unknown, but there is evidence that it is partly due to lithium’s inhibition of IMPase (91, 96). Further evi-dence supporting the effects of lithium on PKC are data showing that lithium decreases the levels and phosphorylation of a major PKC substrate, myristoylated alanine-rich C kinase substrate (MARCKS), following chronic treatment in rats (99). In cultured cells, this lithium-mediated effect appears to be dependent on low concentrations of inositol in the media, thus implicating lithium’s inhibition of IMPase and/or IPPase as a causative factor (91, 100).
PKC Signaling in Animal Models of Mood DisordersCurrent animal models of mania that have been used in the study of mood disorders include kindling, behavioral/amphetamine sensitization, and glucocorticoid administration (43, 101, 102), and PKC activity is implicated in all of these models. Kindling is an animal model for epilepsy that has been proposed to have similarities with pathophysiological aspects of bipolar disorder, in which repeated administration of an electrical stimulus (that is, subthreshold to produce seizures) results in a convulsion and a permanent state of hyperexcitability to the stimulus. These studies on rats have consistently shown hippocampal kindling leads to increased PKC activity and protein concentration (103–108), find-ings that also were demonstrated to be valid in other brain struc-tures such as the amygdala (109, 110) and neocortex (111, 112).
Studies have also implicated alterations in PKC activity as mediators of long-term alterations in neuronal excitability in the brain following chronic stimulant use. Several independent labo-ratories have demonstrated that both acute and chronic amphet-amine produce an alteration in PKC activity, its relative cytosol-to-membrane distribution, as well as the phosphorylation of a major PKC substrate, growth-associated protein (GAP)-43, which has been implicated in long-term alterations of neurotransmitter release (113–117). Furthermore, PKC inhibitors have been shown to block the acute responses (as assessed by both behavioral and in vivo microdialysis studies) to both amphetamine (118) and cocaine as well as cocaine-induced sensitization (119, 120). To further explore the possibility that the PKC signaling may play a role in mood stabilization, a series of studies were undertaken to investigate the behavioral sequelae of PKC inhibition by testing the effects of tamoxifen on three psychostimulant-induced behav-iors, representing different validated animal models of mania. Although not a selective agent (better known for its antiestrogenic effects), tamoxifen represents the only CNS-penetrant PKC inhibi-tor currently available for human use. Tamoxifen significantly reduced acute or chronic amphetamine-induced hyperactivity in a large open field without affecting spontaneous activity levels. However, the same treatment normalized amphetamine-induced
increase in visits to the center of an open field (representing risk-taking behavior) and reduced hedonic-like amphetamine-induced conditioned place preference (Einat et al., unpublished data). Additionally, recent nonhuman primate studies investigating cog-nitive deficits similar to those observed in mania have also dem-onstrated the efficacy of a selective PKC inhibitor (Birnbaum et al., unpublished data).
Thus, although considerable caution needs to be employed when extrapolating from rodent brain and animal behavioral mod-els, the fact that the various animal models of mania are associated with opposite effects on PKC signaling to those observed with chronic lithium or VPA is compelling. In toto, the preclinical data supports further exploration of PKC inhibition as a possible target for new medications. Indeed, CNS-penetrant PKC inhibitors may not only have considerable utility in the treatment of acute mania, but may also exert their effects much more rapidly than existing medications. Such a contention is supported by the findings of a pilot study demonstrating the antimanic effects of tamoxifen (121); large scale clinical trials of PKC inhibitors are clearly warranted.
Neurotrophic Signaling Cascades
Neurotrophins are a family of regulatory factors that mediate the differentiation and survival of neurons, as well as the modulation of synaptic transmission and synaptic plasticity. The neurotrophin family now includes, among others, nerve growth factor (NGF), BDNF, neurotrophin (NT)-3, NT-4, NT-5, and NT-6. BDNF and other neurotrophic factors are necessary for the survival and func-tion of neurons, implying that a sustained reduction of these fac-tors could affect neuronal viability. BDNF also has a number of much more acute effects on synaptic plasticity and neurotransmit-ter release and facilitates the release of glutamate, γ-aminobutyric acid (GABA), dopamine, and serotonin (122).
BDNF is best known for its long-term neurotrophic and neuroprotective effects, which may be very important for its puta-tive role in the pathophysiology and treatment of mood disorders. Although endogenous neurotrophic factors have traditionally been viewed as increasing cell survival by providing necessary tro-phic support, it is now clear that their survival-promoting effects are mediated in large part by inhibiting cell death (apoptosis) cascades (122). Increasing evidence suggests that neurotrophic factors inhibit cell death cascades by activating the extracellular-regulated kinase (ERK) signaling pathway, the PLC-γ cascade, and the PI3K–Akt pathway. Chronic stress (21 days of foot-shock, an animal model of depression) in rats induced a pronounced and persistent ERK1/2 hyper-phosphorylation in dendrites of the higher prefrontal cortical layers of rat brains, whereas phospho-CREB was reduced in several cortical regions including the frontal cortex (123). Because CREB is phosphorylated and activated by phospho-ERK1/2 directly, this phospho-CREB reduction indicates that chronic stress could downregulate CREB phosphorylation indirectly, and subsequently downregulate the transcription of some neurotrophic genes such as bcl-2 and BDNF.
Review

267 October 2004
Volume 4, Issue 5
In this context, it is noteworthy that severe stress exacer-bates stroke outcome by suppressing bcl-2 expression (124); mice exposed to aggressive social stress expressed approximately 70% less bcl-2 mRNA than unstressed mice following ischemia. Furthermore, stress greatly exacerbated infarct area in control mice, but not in transgenic mice that constitutively express increased neuronal bcl-2. Finally, high corticosterone concentra-tions were significantly correlated with larger infarcts in wild-type mice but not in transgenic mice overexpressing bcl-2. Thus, enhanced bcl-2 expression appears to be capable of offsetting the potentially deleterious consequences of stress-induced neuronal endangerment, and suggests that pharmacologically-induced upregulation of bcl-2 may have considerable utility in the treat-ment of a variety of disorders associated with endogenous or acquired impairments of cellular resilience.
Overall, it is clear that the neurotrophic factor–ERK/MAP kinase–bcl-2 signaling cascade plays a critical role in cell survival in the CNS, and that there is a fine balance maintained between the levels and activities of cell survival and cell death factors. Dysregulation of the BDNF–ERK–CREB coordination may be a key mechanism by which prolonged stress induces atrophy of selective subpopulations of vulnerable neurons and/or distal den-drites. Conceivably, the precise kinetics of ERK and CREB activa-tion will ultimately dictate whether the activated kinases partici-pate in a cell survival– or death-promoting pathway.
Neurotrophic Effects of Lithium in AnimalsHow does the important role of ERK/MAP kinases in mediating long-term neuroplastic events relate to the molecular actions of lithium? Lithium and VPA, at therapeutically relevant concentra-tions, activate the ERK/MAP kinase cascade in human neuroblas-toma SH-SY5Y cells (125) and in critical limbic and limbic-related areas of the rodent brain (80). Neurotrophic factors are now known to promote cell survival by activating MAP kinases to sup-press intrinsic, cellular apoptotic machinery, not only by inducing cell survival pathways (122). Thus, a downstream target of the MAP kinase cascade, ribosomal S6 kinase (Rsk) phosphorylates CREB, leading to the induction of bcl-2 gene expression (Figure 1). Consistent with an activation of neurotrophic signaling cas-cades, chronic treatment of rats with the animal equivalent of therapeutic doses of lithium or VPA produces an increase in the activation of Rsk and CREB, and eventually a doubling of bcl-2 levels in frontal cortex, effects which are primarily due to a marked increase in the number of bcl-2 immunoreactive cells in layers II and III of the frontal cortex (126–128). Interestingly, the importance of neurons in layers II–IV of the frontal cortex in mood disorders has recently been emphasized, because primate studies indicate that these areas are important for providing con-nections with other cortical regions, and that they are targets for subcortical input (129).
Further suggestive evidence that lithium and VPA activate the MAP kinase pathway and/or targets of this pathway comes from
the data showing that chronic administration of lithium or VPA can increase the expression of BDNF in the rodent brain (80, 81).
Consistent with its effects on neurotrophic signaling cascades, lithium is neuroprotective in animal models of ischemia and Huntington disease can promote neurogenesis in the hippocam-pus of rats to increase the regeneration of CNS axons (130) and is neuroprotective in many cell culture models (45, 47). Recent evi-dence suggests that the neuroprotective effect of lithium in cortical neurons requires BDNF expression (131).
Neurotrophic Effects of Lithium in HumansThe body of preclinical data demonstrating neurotrophic and neuroprotective effects of mood stabilizers is striking, yet consid-erable caution must be exercised in extrapolating these data to the clinical situation with humans. In view of lithium’s robust effects on the levels of the cytoprotective protein bcl-2 in the frontal cortex, Drevets and associates reanalyzed older data demonstrat-ing an approximate 40% reduction in subgenual prefrontal cortex volumes in familial mood disorder subjects (132). Consistent with neurotrophic and neuroprotective effects of lithium, they found that the patients treated with chronic lithium or VPA had sub-genual prefrontal cortex volumes that were significantly greater relative to untreated patients, and not significantly different from controls (Wayne Drevets, personal communication). In a more recent study, Drevets and colleagues investigated glial cell densities in mood disordered patients, and although the sample sizes were small, unipolar patients in this study exhibited reduced glial-cell densities, whereas only the bipolar patients who discontinued chronic lithium or VPA exhibited similar reductions (133), sug-gesting a neuroprotective role associated with the utilization of these agents in patients.
Although the results of the studies noted previously suggest that mood stabilizers may have provided neuroprotective effects during naturalistic use, small sample sizes and the cross-sectional nature of the studies warrant caution. To investigate the potential neurotrophic effects of lithium in humans more definitively, a longitudinal clinical study was recently undertaken using proton magnetic resonance spectroscopy (1H MRS) to measure N-acetyl-aspartate (NAA, a putative marker of neuronal viability) levels (134). Four weeks of lithium treatment produced a significant increase in NAA levels, effects that were localized almost exclu-sively to gray matter (135). These findings provide intriguing indirect support for the contention that chronic lithium increases neuronal viability and function in the human brain. Furthermore, a very high correlation (R = 0.97) between lithium-induced NAA increases and regional voxel (i.e., volume pixel, the smallest dis-tinguishable box-shaped part of a three-dimensional image) gray matter content was observed, thereby providing evidence for co-localization with increased expression of bcl-2 in specific regions observed (e.g., gray vs white matter) in the rodent brain cortices. These results suggest that chronic lithium may not only exert robust neuroprotective effects (as has been demonstrated in a vari-
Molecular Effects of Lithium

268
ety of preclinical paradigms), but also exerts neurotrophic effects in humans.
A follow-up volumetric magnetic resonance imaging (MRI) study demonstrated that four weeks of lithium treatment also significantly increased total gray matter content in the human brain (136), suggesting an increase in the volume of the neuropil, the moss-like layer comprised of axonal and dendritic fibers that occupies much of the cortex grey matter volume. A finer grained subregional analysis of this brain imaging data is ongoing, but clearly shows that lithium produces a regionally- selective increase in gray matter, with prominent effects observed in the hippocam-pus and caudate (unpublished observations; G.J. Moore and H.K. Manji). Furthermore, no changes in overall gray matter volume are observed in healthy volunteers treated chronically with lithi-um, suggesting that lithium is truly producing a reversal of illness-related atrophy, rather than non-specific gray matter increases. Recently, cross-sectional studies have corroborated the gray matter findings (137) and NAA findings (138).
Arachidonic Acid (AA) Metabolism
AA functions as an important mediator of second messenger pathways within the brain (139, 140). AA is released from mem-brane phospholipids via receptor–G protein–initiated activation of phospholipase A2 (PLA2) (141). This action results in release of AA from the cellular membrane, and cyclooxygenase (COX)-mediated production of eicosanoid metabolites such as prostaglandins and thromboxanes. These metabolites mediate numerous subsequent intracellular responses and, due to their lipid permeable nature, transynaptic responses.
AA metabolism as a target of mood stabilizers was originally suggested by studies done by Rapoport, Chang, and colleagues in 1996 and 2001, showing that chronic lithium or VPA treatment of rats results in selective reductions in the turnover rate in the brain phospholipids of AA (142–144). In the case of lithium, the reduc-tion of AA turnover was 80%, and subsequently it was shown that lithium decreased the gene expression and protein levels of an AA-specific PLA2 (i.e., cytosolic PLA2 [cPLA2]) (145, 146) and the protein levels of COX-2 (147). VPA also decreased the turnover of AA by 33% (142), had no apparent effect on cPLA2 protein levels (142), but decreased protein levels of COX-1 and COX-2 (148). Most recently it has been observed that carbamazepine downregu-lates PLA2 mediated release of arachidonic acid and its subsequent conversion to prostaglandin E2 by cyclooxygenase (149). These findings suggest that effects of mood stabilizers on cell mem-branes—and specifically AA turnover—might be relevant to the pharmacological action of lithium and VPA (140, 144).
Further general support for the involvement of the AA sig-naling pathway in bipolar disorder comes from other preclinical studies. Recent studies in rats found that administration of non-selective COX inhibitors indomethacin and piroxicam prevented amphetamine-stimulated locomotor activity (150) and blocked
cocaine sensitization (151) ––both rodent models of mania (102). Also, NS-398, a specific COX-2 inhibitor, attenuates restraint stress (a model of depression)–induced oxidative changes (152). The inflammatory hypothesis of bipolar disorder has led to a clini-cal trial addressing the effect of a specific COX-2 inhibitor as an adjunct treatment in bipolar patients (153).
Conclusions
Bipolar disorder affects approximately 1–3 percent of the world’s population. There has been little progress, however, in develop-ing truly novel drugs for the treatment of bipolar disorder. In fact, most recent additions to the pharmacopeia are brain-penetrant drugs developed for the treatment of epilepsy or schizophrenia (e.g., anticonvulsants such as carbamazepine and antipsychotics such as olanzapine). Thus, there exists a critical need to develop novel approaches for the treatment of bipolar disorder. Although the task of developing novel medications is very difficult, recent insights into lithium’s actions have identified a number of promis-ing and unexpected targets. Moreover, the demonstration of robust neurotrophic and neuroprotetive effects of lithium suggests that one of psychiatry’s oldest treatments may have considerable utility in the treatment of neurodegenerative disorders as well (47, 54).
The pharmaceutical industry has yet to develop brain-pen-etrant IMPase inhibitors, but GSK-3 inhibitors are rapidly being developed, and we believe these will be invaluable to discern the role of inhibition of GSK-3 in the treatment of bipolar disorder. Tamoxifen, an antiestrogen agent utilized in the therapy of breast cancer, is currently being investigated as an antimanic agent because of its properties as an inhibtor of protein kinase C; initial results from a single-blind study are encouraging, and these are being followed up by large double-blind studies. Mechanisms to enhance neurotrophic pathways are a major focus for the treat-ment of neurodegenerative disorders, and it is likely that novel medications for this intent may be soon available. We are opti-mistic that recent novel insights into the mechanisms of action of lithium will ultimately lead to improved medications for the treat-ment of those who suffer from bipolar disorder. doi:10.1124/mi.4.5.6
Acknowledgments
We are grateful for the support of the Intramural Research Program of the National Institute of Mental Heath, the National Association for Research on Schizophrenia and Depression (NARSAD; Young Investigators Awards to JAQ and TDG) and the Stanley Medical Research Institute (HKM).
References1. Goodwin, F.K. and Ghaemi, S.N. The course of bipolar disorder and the
nature of agitated depression. Am. J. Psychiatry 160, 2077–2079 (2003).
2. Judd, L.L. and Akiskal, H.S. The prevalence and disability of bipolar spectrum disorders in the US population: Re-analysis of the ECA database taking into account subthreshold cases. J. Affect. Disord. 73,
Review

269 October 2004
Volume 4, Issue 5
123–131 (2003).
3. Murray, C.J. and Lopez, A.D. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 349, 1498–1504 (1997).
4. Schulz, R., Beach, S.R., Ives, D.G., Martire, L.M., Ariyo, A.A., and Kop, W.J. Association between depression and mortality in older adults: The Cardiovascular Health Study. Arch. Intern. Med. 160, 1761–1768 (2000).
5. Musselman, D.L., Evans, D.L., and Nemeroff, C.B. The relationship of depression to cardiovascular disease: Epidemiology, biology, and treat-ment. Arch. Gen. Psychiatry 55, 580–592 (1998).
6. Goodwin FK, Fireman B, Simon GE, Hunkeler EM, Lee J, Revicki D: Suicide risk in bipolar disorder during treatment with lithium and dival-proex. JAMA 290, 1467–1473 (2003). This article presents long-term follow-up data from 20,638 bipolar patients under mood stabilizers treatment, showing lower risk of suicide attempt and suicide death during treatment with lithium compared to valproate (VPA).
7. Baldessarini, R.J., Tondo, L., and Hennen, J. Lithium treatment and sui-cide risk in major affective disorders: Update and new findings. J. Clin. Psychiatry 64 Suppl. 5, 44–52 (2003).
8. Amari, L., Layden, B., Rong, Q., Geraldes, C.F., and Mota de Freitas, D. Comparison of fluorescence, 31P-NMR, and 7Li-NMR spectroscopic methods for investigating Li+/Mg2+ competition for biomolecules. Anal. Biochem. 272, 1–7 (1999).
9. Ryves, W.J. and Harwood, A.J. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem. Biophys. Res. Commun. 280, 720–725 (2001).
10. Gould, T.D., Chen, G., and Manji, H.K. Mood stabilizer psychopharma-cology. Clin. Neurosci. Res. 24, 193–212 (2002).
11. Davies, S.P., Reddy, H., Caivano, M., and Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105 (2000). Report the specificity of various kinase inhibitors.
12. York, J.D., Ponder, J.W., and Majerus, P.W. Definition of a metal-depen-dent/Li+-inhibited phosphomonoesterase protein family based upon a conserved three-dimensional core structure. Proc. Natl. Acad. Sci. U.S.A. 92, 5149–5153 (1995). This work identified a conserved motif con-served among lithium-sensitive phosphomonoesterases.
13. Spiegelberg, B.D., Xiong, J.P., Smith, J.J., Gu, R.F., and York, J.D. Cloning and characterization of a mammalian lithium-sensitive bispho-sphate 3`-nucleotidase inhibited by inositol 1,4 bisphosphate. J. Biol. Chem. 274, 13619–13628 (1999).
14. Nordenberg, J., Kaplansky, M., Beery, E., Klein, S., and Beitner, R. Effects of lithium on the activities of phosphofructokinase and phospho-glucomutase and on glucose-1,6-diphosphate levels in rat muscles, brain and liver. Biochem. Pharmacol. 31, 1025–1031 (1982).
15. Rhyu, G.I., Ray, W.J., Jr., and Markley, J.L. Enzyme-bound intermedi-ates in the conversion of glucose 1-phosphate to glucose 6-phosphate by phosphoglucomutase. Phosphorus NMR studies. Biochemistry 23, 252–260 (1984).
16. Ray, W.J., Jr., Szymanki, E.S., and Ng, L. The binding of lithium and of anionic metabolites to phosphoglucomutase. Biochim. Biophys. Acta 522, 434–442 (1978).
17. Masuda, C.A., Xavier, M.A., Mattos, K.A., Galina, A., and Montero-Lomeli, M. Phosphoglucomutase is an in vivo lithium target in yeast. J. Biol. Chem. 276, 37794–37801 (2001).
18. Klein, P.S. and Melton, D.A. A molecular mechanism for the effect of lithi-um on development. Proc. Natl. Acad. Sci. U.S.A. 93, 8455–8459 (1996). First article to report that lithium is a direct inhibitor of GSK-3.
19. Stambolic, V., Ruel, L., and Woodgett, J.R. Lithium inhibits glycogen syn-thase kinase-3 activity and mimics wingless signaling in intact cells. Curr. Biol. 6, 1664–1668 (1996).
20. Gould, T.D., Zarate, C.A., and Manji, H.K. Glycogen synthase kinase-3: A target for novel bipolar disorder treatments. J. Clin. Psychiatry 65, 10–21 (2004).
21. Majerus, P.W. Inositol phosphate biochemistry. Annu. Rev. Biochem. 61, 225–250 (1992).
22. Gould, T.D. and Manji, H.K. Signaling networks in the pathophysiology and treatment of mood disorders. J. Psychosom. Res. 53, 687–697 (2002).
23. Gani, D., Downes, C.P., Batty, I., and Bramham, J. Lithium and myo-ino-sitol homeostasis. Biochim. Biophys. Acta 1177, 253–269 (1993).
24. Naccarato, W.F., Ray, R.E., and Wells, W.W. Biosynthesis of myo-inositol
in rat mammary gland. Isolation and properties of the enzymes. Arch. Biochem. Biophys. 164, 194–201 (1974).
25. Hallcher, L.M. and Sherman, W.R. The effects of lithium ion and other agents on the activity of myo-inositol-1-phosphatase from bovine brain. J. Biol. Chem. 255, 10896–10901 (1980).
26. Ragan, C.I., Watling, K.J, Gee, N.S. et al. The dephosphorylation of ino-sitol-1,4-bisphosphate to inositol in liver and brain involves two distinct Li+-sensitive enzymes and proceeds via inositol 4-phosphate. Biochem. J. 249, 143–148 (1988).
27. Inhorn, R.C. and Majerus, P.W. Properties of inositol polyphosphate 1-phosphatase. J. Biol. Chem. 263, 14559–14565 (1988).
28. Berridge, M.J., Downes, C.P., and Hanley, M.R. Neural and developmen-tal actions of lithium: A unifying hypothesis. Cell 59, 411–419 (1989). This early article proposed in detail what has come to be known as the inositol depletion hypothesis for lithium’s action.
29. Berridge, M.J., Downes, C.P., and Hanley, M.R. Lithium amplifies ago-nist-dependent phosphatidylinositol responses in brain and salivary glands. Biochem. J. 206, 587–595 (1982).
30. Nahorski, S.R., Ragan, C.I., and Challiss, R.A. Lithium and the phos-phoinositide cycle: An example of uncompetitive inhibition and its phar-macological consequences. Trends Pharmacol. Sci. 12, 297–303 (1991).
31. Berry, G.T., Buccafusca, R., Greer, J.J., and Eccleston, E. Phosphoinositide deficiency due to inositol depletion is not a mechanism of lithium action in brain. Mol. Genet. Metab. 82, 87–92 (2004).
32. Allison, J.H. and Stewart, M.A. Reduced brain inositol in lithium-treated rats. Nat. New Biol. 233, 267–268 (1971).
33. Atack, J.R. Lithium, phosphatidylinositol signaling, and bipolar disorder. In: Bipolar medications: Mechanism of action. Belmaker, R.H. (ed.) Washington, DC: American Psychiatric Press, Inc. (2000).
34. Moore, G.J., Bebchuk, J.M., Parrish, J.K., Faulk, M.W., Arfken, C.L., Strahl-Bevacqua, J., and Manji, H.K. Temporal dissociation between lithi-um-induced changes in frontal lobe myo-inositol and clinical response in manic–depressive illness. Am. J. Psychiatry 156,1902–1908 (1999).
35. Atack, J.R. Inositol monophosphatase inhibitors––lithium mimetics? Med. Res. Rev. 17, 215–24 (1997). This article provides a thorough review of progress made in the development of novel inositol monophos-phatase inhibitors.
36. Atack, J.R., Cook, S.M., Watt, A.P., Fletcher, S.R., and Ragan, C.I. In vitro and in vivo inhibition of inositol monophosphatase by the bisphos-phonate L-690,330. J. Neurochem. 60, 652–658 (1993).
37. Atack, J.R., Prior, A.M., Fletcher, S.R., Quirk, K., McKernan, R., and Ragan, C.I. Effects of L-690,488, a prodrug of the bisphosphonate inosi-tol monophosphatase inhibitor L-690,330, on phosphatidylinositol cycle markers. J. Pharmacol. Exp. Ther. 270, 70–76 (1994).
38. Pollack, S.J., Atack, J.R., Knowles, M.R., McAllister, G., Ragan, C.I., Baker, R., Fletcher, S.R, Iversen, L.L., and Broughton, H.B. Mechanism of inositol monophosphatase, the putative target of lithium therapy. Proc. Natl. Acad. Sci. U.S.A. 91, 5766–5770 (1994).
39. Bone, R., Springer, J.P., and Atack, J.R. Structure of inositol monophos-phatase, the putative target of lithium therapy. Proc. Natl. Acad. Sci. U.S.A. 89, 10031–10035 (1992).
40. Frame, S. and Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 359, 1–16 (2001).
41. Woodgett, J.R. Judging a protein by more than its name: Gsk-3. Sci. STKE RE12 (2001).
42. Gould, T.D. and Manji, H.K. The wnt signaling pathway in bipolar disor-der. Neuroscientist 8, 497–511 (2002).
43. Lenox, R.H., Gould, T.D., and Manji, H.K. Endophenotypes in bipolar dis-order. Am. J. Med. Genet. 114, 391–406 (2002). [Erratum in: Am. J. Med. Genet. 114, 592 (2002).]
44. Jope, R.S. and Bijur, G.N. Mood stabilizers, glycogen synthase kinase-3beta and cell survival. Mol. Psychiatry 7 Suppl. 1, S35–S45 (2002).
45. Manji, H.K., Moore, G.J., and Chen, G. Lithium at 50: Have the neuro-protective effects of this unique cation been overlooked? Biol Psychiatry 46, 929–40 (1999).
46. Quiroz, J.A., Singh, J., Gould, T.D., Denicoff, K.D., Zarate, C.A., and Manji, H.K. Emerging experimental therapeutics for bipolar disorder: Clues from the molecular pathophysiology. Mol Psychiatry 9, 756–776 (2004). This is a very up-to date review of current strategies to develop novel medications based on the knowledge obtained of the underlying pathophysiology of bipolar disorder.
47. Chuang, D.M., Chen, R., Chalecka-Franaszek, E. et al. Neuroprotective
Molecular Effects of Lithium

270
effects of lithium in cultured cells and animal models of diseases. Bipolar Disord. 4, 129–136 (2002).
48. Kao, K.R., Masui, Y., and Elinson, R.P. Lithium-induced respecification of pattern in Xenopus laevis embryos. Nature 322, 371–373 (1986).
49. He, X., Saint-Jeannet, J.P., Woodgett, J.R., Varmus, H.E., and Dawid, I.B. Glycogen synthase kinase-3 and dorsoventral patterning in Xenopus embryos. Nature 375, 617–622 (1995).
50. Gurvich, N. and Klein, P.S. Lithium and valproic acid: Parallels and con-trasts in diverse signaling contexts. Pharmacol. Ther. 96, 45–66 (2002).
51. Munoz-Montano, J.R., Moreno, F.J., Avila, J., and Diaz-Nido, J. Lithium inhibits Alzheimer’s disease-like tau protein phosphorylation in neurons. FEBS Lett. 411, 183–188 (1997).
52. Gould, T.D., Chen, G., and Manji, H.K. In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology 29, 32–38 (2004).
53. O’Brien, W.T., Harper, A.D., Jove, F., Woodgett, J.R., Maretto, S., Piccolo, S., and Klein, P.S. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 24, 6791–6798 (2004). This article presents compelling evidence to sup-port a central role for GSK-3beta in mediating behavioral responses to lithium.
54. Phiel, C.J., Wilson, C.A., Lee, V.M., and Klein, P.S. GSK-3alpha regu-lates production of Alzheimer’s disease amyloid-beta peptides. Nature 423, 435–439 (2003).
55. Sun, X., Sato, S., Murayama, O., Murayama, M., Park, J.M., Yamaguchi, H., and Takashima, A. Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci. Lett. 321, 61–64 (2002).
56. Su, Y., Ryder, J., Li, B. et al. Lithium, a common drug for Bipolar Disorder treatment, regulates amyloid-β precursor protein processing. Biochemistry 43, 6899–6908 (2004).
57. Kaidanovich-Beilin, O., Milman, A., Weizman, A., Pick, C.G., and Eldar-Finkelman, H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hip-pocampus. Biol. Psychiatry 55, 781–784 (2004). This study found that in vivo inhibition of GSK-3 in mice produces antidepressant-like effects in the forced swim test.
58. Gould, T.D., Einat, H., Bhat, R., and Manji, H.K. AR–A014418, a selec-tive GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int. J. Neuropsychopharmacol. Jul 26, 1–4 (2004). [Epub ahead of print] This study found that in-vivo inhibition of GSK-3 in rats produces antidepressant-like effects in the forced swim test.
59. Li, X., Zhu, W., Roh, M.S., Friedman, A.B., Rosborough, K., and Jope, R.S. In vivo regulation of glycogen synthase kinase-3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacology 29, 1426–1431 (2004).
60. Beaulieu, J.M., Sotnikova, T.D., Yao, W.D., Kockeritz, L., Woodgett, J.R., Gainetdinov, R.R., and Caron, M.G. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 5099–5104 (2004).
61. Kaidanovich, O. and Eldar-Finkelman, H. The role of glycogen synthase kinase-3 in insulin resistance and Type 2 diabetes. Expert Opin. Ther. Targets 6, 555–561 (2002).
62. Bhat, R.V. and Budd, S.L. GSK3beta signalling: Casting a wide net in Alzheimer’s disease. Neurosignals 11, 251–261 (2002).
63. Alvarez, G., Munoz-Montano, J.R., Satrustegui, J., Avila, J., Bogonez, E., and Diaz-Nido, J. Regulation of tau phosphorylation and protection against beta-amyloid–induced neurodegeneration by lithium. Possible implications for Alzheimer’s disease. Bipolar Disord. 4, 153–165 (2002).
64. Su, Y., Ryder, J., Li, B. et al. Lithium, a common drug for bipolar dis-order treatment, regulates amyloid-beta precursor protein processing. Biochemistry 43, 6899–6908 (2004).
65. Tong, H., Imahashi, K., Steenbergen, C., and Murphy, E. Phosphorylation of glycogen synthase kinase-3beta during precondition-ing through a phosphatidylinositol-3-kinase–dependent pathway is car-dioprotective. Circ. Res. 8, 377–379 (2002).
66. Frame, S., and Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 359, 1–16 (2001).
67. Ren, M., Senatorov, V.V., Chen, R.W, and Chuang, D.M. Postinsult treatment with lithium reduces brain damage and facilitates neurologi-cal recovery in a rat ischemia/reperfusion model. Proc. Natl. Acad. Sci.
U.S.A. 100, 6210–6215 (2003).
68. Bhat, R.V., Shanley, J., Correll, M.P., Fieles, W.E., Keith, R.A., Scott, C.W., and Lee, C.M. Regulation and localization of tyrosine216 phos-phorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 97, 11074–11079 (2000).
69. Marcus, F. and Hosey, M.M. Purification and properties of liver fructose 1,6-bisphosphatase from C57BL/KsJ normal and diabetic mice. J. Biol. Chem. 255, 2481–2486 (1980).
70. Nakashima, K. and Tuboi, S. Size-dependent allosteric effects of mon-ovalent cations on rabbit liver fructose-1,6-bisphosphatase. J. Biol. Chem. 251, 4315–4321 (1976).
71. Villeret, V., Huang, S., Fromm, H.J., and Lipscomb, W.N. Crystallographic evidence for the action of potassium, thallium, and lithium ions on fruc-tose-1,6-bisphosphatase. Proc. Natl. Acad. Sci. U.S.A. 92, 8916–8920 (1995).
72. Zhang, R., Villeret, V., Lipscomb, W.N., and Fromm, H. Kinetics and mechanisms of activation and inhibition of porcine liver fructose-1,6-bisphosphatase by monovalent cations. Biochemistry 35, 3038–3043 (1996).
73. Wright, S.W., Carlo, A.A., Danley, D.E. et al. 3-(2-carboxyethyl)-4,6-dichloro-1H-indole-2-carboxylic acid: An allosteric inhibitor of fructose-1,6-bisphosphatase at the AMP site. Bioorg. Med. Chem. Lett. 13, 2055–2058 (2003).
74. Lopez-Coronado, J.M., Belles, J.M., Lesage, F., Serrano, R., and Rodriguez. P.L. A novel mammalian lithium-sensitive enzyme with a dual enzymatic activity, 3`-phosphoadenosine 5`-phosphate phosphatase and inositol-polyphosphate 1-phosphatase. J. Biol. Chem. 274, 16034–16039 (1999).
75. Yenush, L., Belles, J.M., Lopez-Coronado, J.M., Gil–Mascarell, R., Serrano, R., and Rodriguez, P.L. A novel target of lithium therapy. FEBS Lett. 467, 321–325 (2000).
76. Patel, S., Yenush, L., Rodriguez, P.L., Serrano, R., and Blundell, T.L. Crystal structure of an enzyme displaying both inositol-polyphosphate-1-phosphatase and 3`-phosphoadenosine-5`-phosphate phosphatase activities: A novel target of lithium therapy. J. Mol. Biol. 315, 677–685 (2002).
77. Agam, G. and Shaltiel, G. Possible role of 3`(2`)-phosphoadenosine-5`-phosphate phosphatase in the etiology and therapy of bipolar disorder. Prog. Neuropsychopharm. Biol. Psych. 27, 723–727 (2003).
78. Duman, R.S. Synaptic plasticity and mood disorders. Mol. Psychiatry 7 Suppl. 1, S29–S34 (2002).
79. Manji, H.K. and Duman, R.S. Impairments of neuroplasticity and cellular resilience in severe mood disorders: Implications for the development of novel therapeutics. Psychopharmacol. Bull. 35, 5–49 (2001).
80. Einat, H., Yuan, P., Gould, T.D., Li, J., Du, J., Zhang, L., Manji, H.K., and Chen, G. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J. Neurosci. 23, 7311–7316 (2003).
81. Fukumoto, T., Morinobu, S., Okamoto, Y., Kagaya, A., and Yamawaki, S. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology (Berl.) 158, 100–106 (2001).
82. Angelucci, F., Aloe, L., Jimenez-Vasquez, P., and Mathe, A.A. Lithium treatment alters brain concentrations of nerve growth factor, brain-derived neurotrophic factor and glial cell line–derived neurotrophic factor in a rat model of depression. Int. J. Neuropsychopharmacol. 6, 225–231 (2003).
83. Jope, R.S. A bimodal model of the mechanism of action of lithium. Mol. Psychiatry 4, 21–25 (1999).
84. Kofman, O., Li, P.P., and Warsh, J.J. Lithium, but not carbamazepine, potentiates hyperactivity induced by intra-accumbens cholera toxin. Pharmacol. Biochem. Behav. 59, 191–200 (1998).
85. Extein, I., Tallman, J., Smith, C.C., and Goodwin, F.K. Changes in lym-phocyte beta-adrenergic receptors in depression and mania. Psychiatry Res. 191–197 (1979).
86. Ebstein, R.P., Oppenheim, G., Ebstein, B.S., Amiri, Z., and Stessman, J. The cyclic AMP second messenger system in man: The effects of hered-ity, hormones, drugs, aluminum, age and disease on signal amplification. Prog. Neuropsychopharmacol. Biol. Psychiatry 10, 323–353 (1986).
87. Wang. J.-F., Young, L.T., Li, P.P., and Warsh, J.J. Signal transduction abnormalities in bipolar disorder. In: Bipolar disorder: Biological Models
Review

271 October 2004
Volume 4, Issue 5
and Their Clinical Application. Joffe, R.T. (ed.) New York: Dekker, 41–79 (1997).
88. Dessauer, C.W., Tesmer, J.J., Sprang, S.R., and Gilman, A.G. The inter-actions of adenylate cyclases with P-site inhibitors. Trends Pharmacol. Sci. 20, 205–210 (1999).
89. Iwatsubo, K., Tsunematsu, T., and Ishikawa, Y. Isoform-specific regula-tion of adenylyl cyclase: A potential target in future pharmacotherapy. Expert Opin. Ther. Targets 7, 441–451 (2003).
90. Soderling, T.R. CaM-kinases: Modulators of synaptic plasticity. Curr. Opin. Neurobiol. 10, 375–380 (2000).
91. Manji, H.K. and Lenox, R.H. Ziskind–Somerfeld Research Award. Protein kinase C signaling in the brain: Molecular transduction of mood stabili-zation in the treatment of manic-depressive illness. Biol. Psychiatry 46, 1328–1351 (1999).
92. van Calker, D. and Belmaker, R.H. The high affinity inositol transport system––implications for the pathophysiology and treatment of bipolar disorder. Bipolar Disord. 2, 102–107 (2000).
93. Lubrich, B. and van Calker, D. Inhibition of the high affinity myo-inositol transport system: A common mechanism of action of antibipolar drugs? Neuropsychopharmacology 21, 519–529 (1999).
94. Williams, R.S., Cheng, L., Mudge, A.W., and Harwood, A.J. A com-mon mechanism of action for three mood-stabilizing drugs. Nature 417, 292–295 (2002).
95. Nishizuka, Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 258, 607–614 (1992).
96. Manji, H.K., Bersudsky, Y., Chen, G., Belmaker, R.H., and Potter, W.Z. Modulation of protein kinase C isozymes and substrates by lithium: The role of myo-inositol. Neuropsychopharmacology 15, 370–381 (1996).
97. Manji, H.K., Etcheberrigaray, R., Chen, G., and Olds, J.L. Lithium decreases membrane-associated protein kinase C in hippocampus: Selectivity for the alpha isozyme. J. Neurochem. 61, 2303–2310 (1993).
98. Li, X. and Jope, R.S. Selective inhibition of the expression of signal transduction proteins by lithium in nerve growth factor–differentiated PC12 cells. J. Neurochem. 65, 2500–2508 (1995).
99. Lenox, R.H., Watson, D.G., Patel, J., and Ellis, J. Chronic lithium admin-istration alters a prominent PKC substrate in rat hippocampus. Brain Res. 570, 333–340 (1992).
100. Watson, D.G., and Lenox, R.H. Chronic lithium-induced down-regulation of MARCKS in immortalized hippocampal cells: Potentiation by musca-rinic receptor activation. J. Neurochem. 67, 767–777 (1996).
101. Post, R.M., Susan, R., and Weiss, B. Sensitization, kindling, and car-bamazepine: An update on their implications for the course of affective illness, in Pharmacopsychiatry 25, 41–43 (1992).
102. Einat, H., Manji, H.K., and Belmaker, R.H. New approaches to modeling bipolar disorder. Psychopharm. Bull. 37, 47–63 (2003).
103. Daigen, A., Akiyama, K., Itoh, T., Kohira, I., Sora, I., Morimoto, K., and Otsuki, S. Long-lasting enhancement of the membrane-associated pro-tein kinase C activity in the hippocampal-kindled rat. Jpn. J. Psychiatry Neurol. 45, 297–301 (1991).
104. Kohira, I., Akiyama, K., Daigen, A., Itoh, T., Ono, M., Morimoto, K., Sora, I., and Otsuki, S. Permanent increase in membrane-associated protein kinase C activity in the hippocampal-kindled rat. Jpn. J. Psychiatry Neurol. 46, 510–512 (1992).
105. Osonoe, K., Ogata, S., Iwata, Y., and Mori, N. Kindled amygdaloid sei-zures in rats cause immediate and transient increase in protein kinase C activity followed by transient suppression of the activity. Epilepsia 35, 850–854 (1994).
106. Ono, M., Akiyama, K., Tsutsui, K., and Kuroda, S. Differential changes in the activities of multiple protein kinase C subspecies in the hippocampal-kindled rat. Brain Res. 660, 27–33 (1994).
107. Akiyama, K., Ono, M., Kohira, I., Daigen, A., Ishihara, T., and Kuroda, S. Long-lasting increase in protein kinase C activity in the hippocampus of amygdala-kindled rat. Brain Res. 679, 212–220 (1995).
108. Kamphuis, W., Hendriksen, E., and Lopes da Silva, F.H. Isozyme-spe-cific changes in the expression of protein kinase C isozyme (alpha-zeta) genes in the hippocampus of rats induced by kindling epileptogenesis. Brain Res. 702, 94–100 (1995).
109. Beldhuis, H.J., Everts, H.G, Van der Zee, E.A., Luiten, P.G., and Bohus, B. Amygdala kindling-induced seizures selectively impair spatial memory. 1. Behavioral characteristics and effects on hippocampal neuronal pro-tein kinase C isoforms. Hippocampus 2, 397–409 (1992).
110. Chen, S.J., Desai, M.A., Klann, E., Winder, D.G., Sweatt, J.D., and Conn, P.J. Amygdala kindling alters protein kinase C activity in dentate gyrus. J. Neurochem. 59, 1761–1769 (1992).
111. Buzsaki, G., Hsu, M., Horvath, Z., Horsburgh, K., Sundsmo, M., Masliah, E., and Saitoh, T. Kindling-induced changes of protein kinase C levels in hippocampus and neocortex. Epilepsy Res. Suppl. 9, 279–283 (1992).
112. Beldhuis, H.J., De Ruiter, A.J., Maes, F.W., Suzuki, T., and Bohus, B. Long-term increase in protein kinase C–gamma and muscarinic acetyl-choline receptor expression in the cerebral cortex of amygdala-kindled rats––a quantitative immunocytochemical study. Neuroscience 55, 965–973 (1993).
113. Giambalvo, C.T. Protein kinase C and dopamine transport–2. Effects of amphetamine in vitro. Neuropharmacology 31, 1211–1222 (1992).
114. Giambalvo, C.T. Protein kinase C and dopamine transport–1. Effects of amphetamine in vivo. Neuropharmacology, 31, 1201–1210 (1992).
115. Gnegy, M.E., Hong, P., and Ferrell, S.T. Phosphorylation of neuromodulin in rat striatum after acute and repeated, intermittent amphetamine. Brain Res. Mol. Brain Res. 20, 289–298 (1993).
116. Iwata, S.I., Hewlett, G.H., Ferrell, S.T., Kantor, L., and Gnegy, M.E. Enhanced dopamine release and phosphorylation of synapsin I and neuromodulin in striatal synaptosomes after repeated amphetamine. J. Pharmacol. Exp. Ther. 283, 1445–1452 (1997).
117. Iwata, S., Hewlett, G.H., and Gnegy, M.E. Amphetamine increases the phosphorylation of neuromodulin and synapsin I in rat striatal synapto-somes. Synapse 26, 281–291 (1997).
118. Kantor, L. and Gnegy, M.E. Protein kinase C inhibitors block amphet-amine-mediated dopamine release in rat striatal slices. J. Pharmacol. Exp. Ther. 284, 592–598 (1998).
119. Steketee, J.D. Injection of the protein kinase inhibitor H7 into the A10 dopamine region blocks the acute responses to cocaine: Behavioral and in vivo microdialysis studies. Neuropharmacology 32, 1289–1297 (1993).
120. Steketee, J.D. Intra-A10 injection of H7 blocks the development of sensi-tization to cocaine. Neuroreport 6, 69–72 (1994).
121. Bebchuk, J.M., Arfken, C.L., Dolan-Manji, S., Murphy, J., Hasanat, K., and Manji, H.K. A preliminary investigation of a protein kinase C inhibitor in the treatment of acute mania. Arch. Gen. Psychiatry 57, 95–97 (2000). This study suggested that the protein kinase C inhibitor tamoxifen may have efficacy in the treatment of bipolar mania.
122. Du, J., Gould, T.D., and Manji, H.K. Neurotrophic signaling in mood dis-orders. In: Signal Transduction and Human Disease. Gutkind, J.S. (ed.) Hoboken, NJ: John Wiley & Sons, Inc., 411–446 (2003).
123. Trentani, A., Kuipers, S.D., Ter Horst, G.J., and Den Boer, J.A. Selective chronic stress–induced in vivo ERK1/2 hyperphosphorylation in medial prefrontocortical dendrites: Implications for stress-related cortical pathol-ogy? Eur. J. Neurosci. 15, 1681–1691 (2002).
124. DeVries, A.C., Joh, H.D., Bernard, O., Hattori, K., Hurn, P.D., Traystman, R.J., and Alkayed, N.J. Social stress exacerbates stroke outcome by suppressing Bcl-2 expression. Proc. Natl. Acad. Sci. U.S.A. 98, 11824–11828 (2001).
125. Yuan, P.X., Huang, L.D., Jiang, Y.M., Gutkind, J.S., Manji, H.K., and Chen, G. The mood stabilizer valproic acid activates mitogen-activated protein kinases and promotes neurite growth. J. Biol. Chem. 276, 31674–31683 (2001).
126. Chen, G., Zeng, W.Z., Yuan, P.X., Huang, L.D., Jiang, Y.M., Zhao, Z.H., and Manji, H.K. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J. Neurochem. 72, 879–882 (1999). This article reports that lithium and valproate (VPA) increase CNS bcl-2 levels in rodents.
127. Manji, H.K. and Chen, G. PKC, MAP kinases and the bcl-2 family of proteins as long-term targets for mood stabilizers. Mol. Psychiatry 7 Suppl.1, S46–S56 (2002).
128. Chen, G., Huang, L.D., Zeng, W.Z., and Manji, H.K. Mood stabiliz-ers regulate cytoprotective and mRNA-binding proteins in the brain: Long–term effects on cell survival and transcript stability. Int. J. Neuropsychopharmacol. 4, 47–64 (2001).
129. Rajkowska, G. Histopathology of the prefrontal cortex in major depres-sion: What does it tell us about dysfunctional monoaminergic circuits? Prog. Brain Res. 126, 397–412 (2000).
130. Huang, X., Wu, D.Y., Chen, G., Manji, H., and Chen, D.F. Support of retinal ganglion cell survival and axon regeneration by lithium through a Bcl-2-dependent mechanism. Invest. Ophthalmol. Vis. Sci. 44, 347–354
Molecular Effects of Lithium

272
(2003).
131. Hashimoto, R., Takei, N., Shimazu, K., Christ, L., Lu, B., and Chuang, D.M. Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: An essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology 43, 1173–1179 (2002).
132. Drevets, W.C., Price, J.L., Simpson, J.R., Jr., Todd, R.D., Reich, T., Vannier, M., and Raichle, M.E. Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386, 824–827 (1997).
133. Bowley, M.P., Drevets, W.C., Ongur, D., and Price, J.L. Low glial num-bers in the amygdala in major depressive disorder. Biol. Psychiatry 52, 404–412 (2002).
134. Tsai, G. and Coyle, J.T. N–acetylaspartate in neuropsychiatric disorders. Prog. Neurobiol. 46, 531–540 (1995).
135. Moore, G.J., Bebchuk, J.M., Hasanat, K. et al. Lithium increases N-ace-tyl-aspartate in the human brain: in vivo evidence in support of bcl-2’s neurotrophic effects? Biol. Psychiatry 48, 1–8 (2000).
136. Moore, G.J., Bebchuk, J.M., Wilds, I.B., Chen, G., and Manji, H.K. Lithium-induced increase in human brain grey matter. Lancet 356, 1241–1242 (2000). This work was first to demonstrate that lithium induced increases in grey matter volume in patients with bipolar disorder.
137. Sassi, R., Nicoletti, M., Brambilla, P., Mallinger, A., Frank, E., Kupfer, D., Keshavan, M., and Soares, J. Increased gray matter volume in lithium-treated bipolar disorder patients. Neurosci. Lett. 329, 243 (2002).
138. Silverstone, P.H., Wu, R.H., O’Donnell, T., Ulrich, M., Asghar, S.J., and Hanstock, C.C. Chronic treatment with lithium, but not sodium valproate, increases cortical N-acetyl-aspartate concentrations in euthymic bipolar patients. Int. Clin. Psychopharmacol. 18, 73–79 (2003).
139. Axelrod, J., Burch, R.M., and Jelsema, C.L. Receptor-mediated activa-tion of phospholipase A2 via GTP-binding proteins: Arachidonic acid and its metabolites as second messengers. Trends Neurosci. 11, 117–123 (1988).
140. Rapoport, S.I. In vivo fatty acid incorporation into brain phosholipids in relation to plasma availability, signal transduction and membrane remod-eling. J. Mol. Neurosci. 16, 243–261 (2001).
141. Axelrod, J. Phospholipase A2 and G proteins. Trends Neurosci. 18, 64–65 (1995).
142. Chang, M.C., Contreras, M.A., Rosenberger, T.A., Rintala, J.J., Bell, J.M., and Rapoport, S.I. Chronic valproate treatment decreases the in vivo turnover of arachidonic acid in brain phospholipids: A possible com-mon effect of mood stabilizers. J. Neurochem. 77, 796–803 (2001).
143. Chang, M.C., Grange, E., Rabin, O., Bell, J.M., Allen, D.D., and Rapoport, S.I. Lithium decreases turnover of arachidonate in several brain phospholipids. Neurosci. Lett. 220, 171–174 (1996). Erratum in: Neurosci. Lett. 222, 141 (1997). This manuscript is the first to report that lithium reduced arachidonic acid turnover.
144. Rapoport, S.I. and Bosetti, F. Do lithium and anticonvulsants target the brain arachidonic acid cascade in bipolar disorder? Arch. Gen. Psychiatry 59, 592–596 (2002).
145. Chang, M.C. and Jones, C.R. Chronic lithium treatment decreases brain phospholipase A2 activity. Neurochem. Res. 23, 887–892 (1998).
146. Rintala, J., Seemann, R., Chandrasekaran, K., Rosenberger, T.A., Chang, L., Contreras, M.A., Rapoport, S.I., and Chang, M.C. 85-kDa cytosolic phospholipase A2 is a target for chronic lithium in rat brain. Neuroreport 10, 3887–3890 (1999).
147. Bosetti, F., Rintala, J., Seemann, R., Rosenberger, T.A., Contreras, M.A., Rapoport, S.I., and Chang, M.C. Chronic lithium downregulates cyclo-oxygenase-2 activity and prostaglandin E2 concentration in rat brain. Mol. Psychiatry 7, 845–850 (2002).
148. Bosetti, F., Weerasinghe, G.R., Rosenberger, T.A., and Rapoport, S.I. Valproic acid down-regulates the conversion of arachidonic acid to eicosanoids via cyclooxygenase-1 and -2 in rat brain. J. Neurochem. 85, 690–696 (2003).
149. Ghelardoni, S., Tomita, Y.A., Bell, J.M., Rapoport, S.I., and Bosetti, F. Chronic carbamazepine selectively downregulates cytosolic phos-pholipase A2 expression and cyclooxygenase activity in rat brain. Biol. Psychiatry, 56, 248–254 (2004).
150. Ross, B.M., Brooks, R.J., Lee, M., Kalasinsky, K.S., Vorce, S.P., Seeman, M., Fletcher, P.J., and Turenne, S.D. Cyclooxygenase inhibi-tor modulation of dopamine-related behaviours. Eur. J. Pharmacol. 450, 141–151 (2002).
151. Reid, M.S., Ho, L.B., Hsu, K., Fox, L., Tolliver, B.K., Adams, J.U., Franco, A., and Berger, S.P. Evidence for the involvement of cyclooxy-genase activity in the development of cocaine sensitization. Pharmacol. Biochem. Behav. 71, 37–54 (2002).
152. Madrigal, J.L., Moro, M.A., Lizasoain, I., Lorenzo, P., Fernandez, A.P., Rodrigo, J., Bosca, L., and Leza, J.C. Induction of cyclooxygenase-2 accounts for restraint stress-induced oxidative status in rat brain. Neuropsychopharmacology 28, 1579–1588 (2003).
153. Soares, J. and Bowden, C. March treatment trial grant. Stanley Medical Institute. www.stanleyresearch.org (2002).
154. Gould, T.D., Quiroz, J.A., Singh, J., Zarate, C.A., and Manji, H.K. Emerging experimental therapeutics for bipolar disorder: Insights from the molecular and cellular actions of current mood stabilizers. Mol. Psychiatry 9, 734–755 (2004).
Review
Jorge A. Quiroz, MD, is Research Fellow at the Laboratory of Molecular Pathophysiology, Mood, and Anxiety Disorders Program of the National Institute of Mental Health, NIH, HHS. His research in translational neurosciences is focused in the investigation of the role of new pharmacological agents that impact
cellular resilience and brain neuroplasticity and its clinical application in major depression and bipolar disorder. In addition, Dr. Quiroz is involved in Magnetic Resonance Spectroscopy research (MRS).
Todd D. Gould, MD, is an NIMH Seymour S. Kety Memorial Research Fellow at the Laboratory of Molecular Pathophysiology, Mood, and Anxiety Disorders Program of the National Institute of Mental Health, NIH, HHS. His research focuses upon understanding the pharmacology of psychotropic medications, with a
particular interest in mood stabilizer targets including glycogen synthase kinase-3.
Husseini K. Manji, MD, is Chief of the Laboratory of Molecular Pathophysiology at the National Institute of Mental Health, NIH, HHS. He is actively involved in research investigating the molecular and cellular mechanisms of action of mood-stabilizing agents and helped to establish a Mood Disorders
Research Unit, which conducts an integrated series of preclinical and clinical studies focusing on signal transduction pathways in unipolar depression and bipolar disorder. Address correspondence to HKM. E-mail [email protected]; fax: 301-480-0123.

230
Lithium and other mood-stabilizing drugs are used for the management of bipolar mood disorders and, to a lesser extent, for augmentation of other psychoactive drugs. Lithium also has neuroprotec-tive properties that may be useful for treatment of neurodegenerative diseases such as Alzheimer’s
disease and amyotrophic lateral sclerosis. Over the years, lithium has been shown to inhibit inositol monophosphatases and glycogen synthase kinase 3, but the relevance of such enzyme inhibition to the therapeutic effects of lithium has remained difficult to assess. Here, we provide an overview of recent advances in the identification of molecular mechanisms involved in the regulation of behavior by lithium. We also highlight recent findings suggesting that lithium could exert some of its behavioral effects by acting on a dopamine receptor–regulated signaling complex composed of Akt, protein phosphatase 2A, and the multifunctional protein scaffold beta-arrestin 2.
Jean-Martin Beaulieu1 and Marc G. Caron2
1Department of Anatomy and Physiology, Université Laval/CRULRG, Québec, Canada G1J 2G3
and 2Department of Cell Biology, Duke University Medical Center, Durham, North Carolina 27710

231 October 2008
Volume 8, Issue 5
Molecular Targets of Lithium
Introduction
Since the discovery of its therapeutic effects on mania (1, 2) lithium has become the prototypical member of a small family of structurally disparate mood stabilizers. These drugs are used pri-marily for the management of bipolar disorders and cyclothymia—two psychiatric ailments, each affecting one to two percent of the US population, characterized by alternating episodes of manic and depressive symptoms. In addition to bipolar disorders, lithium is also used as part of combination therapies with drugs affecting monoaminergic neurotransmission, such as antidepressants and antipsychotics (3, 4). Clinical trials have shown that lithium in combination therapy can be effective for treatment-resistant forms of depression (5). Furthermore, lithium has also been reported to lower suicide rates in individuals with bipolar disorder, major depression, or schizoaffective disorders (6, 7).
Apart from its effects on psychiatric illnesses, recent findings have suggested that lithium could also have, at least in preclinical animal models, beneficial effects in several neurodegenerative con-ditions. Lithium has been shown to reduce the toxicity of mutant Huntington disease protein in flies (8) and improve behavioral and histological phenotypes in a transgenic mouse model of spinocer-ebral ataxia (9). Lithium also reduces the phosphorylation of the microtubule associated protein tau (10)—an important component of neurofibrillary tangles—and the production of beta-amyloid peptides (11) in transgenic mouse models of Alzheimer disease. Finally, lithium is neuroprotective in mice that overexpress a mutant form of superoxide dismutase 1 (SOD1) that is associated with amyotrophic lateral sclerosis (ALS), and it is also neuropro-tective in patients with ALS (12–14).
Despite important clinical applications, the molecular mecha-nisms by which lithium and other mood stabilizers exert their therapeutic effects in humans or preclinical animal models are not well understood. Over the years, lithium has been shown to inhib-it various enzymes, including inositol monophosphatases (IMPAs) (15) and glycogen synthase kinase 3 (GSK3) (16, 17). However, in the absence of clear etiological animal models for bipolar disor-ders, the implications of these targets in the therapeutic actions of lithium have remained less than fully explored.
For a molecule or group of molecules to be established as direct targets of lithium in the regulation of behavior, a number of criteria should be satisfied: 1) The “target” must be directly and specifically affected by lithium in vitro; 2) the target must be affected by lithium in the mammalian brain (at serum concentra-tions of 0.6 to 1.5 mM); 3) activation or inhibition of the target should recapitulate the effects of lithium in vivo with respect to established biochemical markers; 4) pharmacological and genetic modulation of target activity should reflect the effects of lithium in model systems of depression or mania; and 5) elimination of the target should abolish both biochemical and behavioral responsive-ness to lithium in these models.
In this article, we provide an overview of recent advances in the identification of molecular mechanisms involved in the regula-tion of behavior by lithium. We limit our review of targets affected by lithium, in vitro or in vivo, to studies that have addressed behavioral components of the potential targets. We also highlight recent findings suggesting that lithium may exert some of its effects not by directly inhibiting enzymes, but by destabilizing specific protein complexes involved in the regulation of enzyme activity. One such complex, identified in the brain through the activation of dopamine D
2 receptors, is composed of protein
kinase B (Akt), protein phosphatase 2A (PP2A), and the multi-functional protein scaffold β-arrestin 2 (βArr2) (18). This principle may have relevance to the previously proposed targets of lithium.
The Inositol Depletion Hypothesis
One of the first compelling mechanisms proposed for lithium action arose from its characterization as an uncompetitive inhibitor of various phophoinositol phosphatases, with kinetic parameters compatible with therapeutic concentrations (15). Inositol deriva-tives are one of the major second messenger systems in cells (Figure 1). Activation of phosphoinositide-specific phospholipase C (PLC) isoforms by G protein–coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs) leads to the cleavage of membrane phos-phatidylinositol bisphosphate (PIP
2) into dyacylclycerol (DAG) and
inositol-1,4,5 trisphosphate (IP3) (15, 19). DAG activates protein
kinase C (PKC) isoforms, whereas IP3 triggers the release of calcium
from intracellular stores and thus leads to activation of cacium-regulated signaling proteins such as calcineurin (PP2B) and calcium calmodulin regulated kinases (CaMKs). Subsequently, the second messenger IP
3 is rapidly hydrolyzed, by an enzymatic cascade
involving multiple phosphoinositol phosphatases, to terminate the signal and recycle inositol for the crucial resynthesis of PI, PIP, and PIP
2. Inhibition of these lipid phosphatases by lithium has led to the
hypothesis that lithium therapy may deplete cellular inositol (i.e., myoinositol), thus interfering with IP
3-mediated cell signaling (15).
Two isoforms of inositol monophosphatase—IMPA1 and IMPA2—have received considerable attention. Both IMPAs func-tion as homodimers and require magnesium for enzymatic activ-ity (20, 21), and it has been postulated that lithium may inhibit IMPAs by competing with magnesium for binding at the active site (22). Alternatively there are also lines of evidence that displace-ment of magnesium by lithium may prevent the dimerization of IMPAs (32).
Lithium and Inositol Homeostasis: In Vivo Evidence
In Xenopus, lithium interferes with embryonic development, resulting in the duplication of dorsal and anterior structures; in Dictyostelium lithium interferes with the life cycle of this slime

232
Review
mold and leads to an expanded stalk cell population. In both organisms, lithium induces inositol depletion, and its metabolic effects are antagonized by addition of exogenous inositol (15, 19). Furthermore, lithium—albeit at concentrations three- to fivefold above therapeutic doses—and other mood stabilizers have been reported to induce inositol depletion and trigger growth cone col-lapse in primary neuronal cultures (24). Finally, chronic lithium treatment in rodents reproducibly reduces the inositol content of various brain tissues by about twenty to twenty-five percent (25, 26). Although these observations point toward a possible involvement of inositol depletion in the effects elicited by lithium, attempts to replicate the behavioral effects of lithium in mammals by inducing brain inositol depletion have been less conclusive.
Lithium and Inositol Depletion: Behavioral Studies
Mutational inactivation of the IMPA1-encoding gene (Impa1) in mice results in an embryonic lethal phenotype that can be rescued by administration of inositol to the mother during pregnancy and lactation (27). In contrast, Impa2-knockout mice develop normally (28). Curiously, deficiency in either Impa1 or Impa2 does not sig-nificantly affect brain inositol content in adult mice (27, 28). When submitted to a battery of tests to evaluate depressive-like (e.g., tail-suspension and forced-swim tests) and manic-like behaviors (e.g., amphetamine-induced locomotor activity), Impa2-knockout mice show no significant behavioral differences relative to their wild-type littermates (28), thus failing to replicate typical antidepressant-
and antimanic-like effects of lithium ( 18, 29, 30). In con-trast, inositol-rescued Impa1-knockout mice display loco-motor hyperactivity, a behavior that is normally antagonized by lithium in rodents (1, 27, 30). Unfortunately, the hyper-active phenotype of the Impa1-knockout mice precludes their utility in addressing antide-pressant agents, as changes in activity can significantly confound such studies (27). Finally, it is of interest that Impa1-knockout mice, like lithium-treated wildtype mice, display enhanced sensitivity to pilocarpine, a seizure-inducing agent (27).
Apart from recycling of phosphoinositol, cellular uptake by the sodium/inositol cotransporter (Smit1) is a major
source of inositol in cells. Smit1-knockout mice die during embryo-genesis but can be rescued into adulthood by inositol supplementa-tion during pregnancy (31). Further investigations conducted in Smit1 hemizygous mice show that Smit1 gene expression correlates to levels of brain inositol; brain inositol levels in hemizygous mice (60–70% of the wild-type level) are slightly superior to those obtained following chronic lithium treatment (75–80%)(25). Despite this similarity of effect on inositol brain levels, Smit1 hemizygotes do not display behavioral responses similar to those of lithium-treated mice in tests of depressive-like behaviors (25). Furthermore, extensive depletion of inositol (> 90%) in the brains of Smit1-knockout mice elicit no dramatic effects on phosphatidylinositol (PtdIns) levels, thus indicating that the effect of inositol depletion on PIP
2- and IP
3-mediated signaling in vivo may be less important
than previously thought (32, 33). Finally, it is of interest that rescued Smit1-knockout mice have enhanced sensitivity to pilocarpine, thereby replicating a phenotype of Impa1-knockout mice (34).
Lithium and the Role of Inositol Depletion: Summary
The inositol depletion model of lithium action represents one of the original attempts to explain, in molecular terms, its therapeu-tic action. Despite its common appeal, the relevance of the model has been questioned by in vivo studies. Inositol depletion that is more drastic than that resulting from treatment with lithium fails to affect predicted behaviors (25). Interestingly, studies conducted in either Impa1- or Smit1-knockout mice suggest that inositol
GPCR SMIT1
myoinositol
IPIP2
PKC PKC[Ca2+]i [Ca2+]i
DAG + IP3 IP3 + DAG
IP2
GqPLC β
RTK
PIP2PIPPIPIPIPPIP2 PIP2PIPPIPIPIPPIP2
PLC γ
IMPA1/2
Figure 1. The phosphatydilinositol cycle in signal transduction. G protein–coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs) mediate the activation of phospholipases C–(PLC)-β and -γ), which cleave phosphoinositide bis-phosphosphate (PIP2) to yield diacylglycerol (DAG) and inositol trisphosphate (IP3). DAG and IP3 activate protein kinase C (PKC) and release calcium [Ca2+]i from intracellular stores, respectively. IP3 is rapidly hydrolyzed to inositol bisphosphate (IP2) and inositol monophosphate (IP). Inositol monophosphatase-(IMAP)-1 and -2 hydrolyze IP to regenerate myoinositol. Inositol gains access to the cellular cytoplasm via uptake by the sodium/inositol cotransporter (SMIT1).

233 October 2008
Volume 8, Issue 5
Molecular Targets of Lithium
depletion contributes to the enhanced sensitivity to pilocarpine that is induced by lithium. Still, the relationship between pilo-carpine sensitization and effects on mood is not clear, and indeed, other mood stabilizers, such as valproic acid, have an anticonvul-sant effect in the pilocarpine paradigm (35). Despite these strong reservations, one must consider that perturbations of inositol homeostasis, such as those evoked by lithium, may exert greater behavioral effects in humans with bipolar disorder than they do in mice. Furthermore, it is possible that even a small depletion of inositol, within the context of other molecular targets in the bipo-lar brain, may still be crucial to the effects of lithium on behavior.
Inhibition of Glycogen Synthase Kinase 3 (GSK3)
A second compelling mechanism for the action of lithium was suggested by the discovery in the mid 1990s that lithium is also an uncompetitive inhibitor of members of the GSK3 family (16, 17). The therapeutic relevance of these findings, however, has remained unclear, given the high K
i (~3.5mM for GSK3a and
~2.0mM for GSK3β) of lithium for GSK3 isoforms (19, 36). GSK3a and GSK3β are closely related serine/threonine kinases originally implicated in the regulation of glycogen synthesis in response to insulin (37, 38). Each is constitutively active and can be inactivated through the phosphorylation of a single serine residue within the N-terminal regulatory domain (Ser 21 of GSK3a; Ser9 of GSK3β) (38). The serine/threonine kinase Akt, also termed protein kinase B (PKB), has been shown to inhibit GSK3a and GSK3β (Figure 2)
in response to multiple hormones and growth factors, including BDNF, IGF, and insulin (38–40). Akt is regulated through phos-phatidylinositol-mediated signaling. Figure 2 shows the activation of Akt according to two steps: 1) its recruitment to the plasma membrane by phosphorylated phosphatidylinositol [Ptdlns(3,4,5)P
3, Ptdlns(3,4)P
2]; and 2) its subsequent phosphorylation at
Thr308 (catalyzed by PDK1) and Ser473 (catalyzed by PDK2) (41–44). GSK3 isoforms have been implicated in multiple physiological functions, including glycogenesis, embryonic development, Wnt signaling, and apoptosis, as well as dopaminergic and serotoniner-gic neurotransmission (30, 38, 45, 46).
The mechanism by which lithium inhibits GSK3 activity is still incompletely understood. One attractive theory is that lithium acts as a competitive inhibitor for the binding of the cofactor magne-sium (36). Competition for the binding of magnesium has also been suggested as a mechanism for the lithium-mediated inhibition of IMPAs (see above). Indeed, magnesium and lithium ions share simi-lar ionic radii (0.065 and 0.060 nm, respectively) (47). The pro-posed competitive mechanism of inhibition by lithium predicts that the in vivo inhibition of GSK3 may be greater than inhibition levels observed in in vitro studies that generally monitor GSK3 activity at optimal concentrations of magnesium ion (36).
In Vivo Evidence of GSK3 Inhibition by Lithium
Multiple independent lines of evidence indicate that both acute and chronic lithium treatment can reduce GSK3 activity in vivo.
Signal
Active Inactive
p110
Akt
PH
AktPDK1 PDK2
PH
Akt
PH
Thr308
Ser473
Ser
P P P
P
P
P
P
PSubstrate
Cellular targets
PIP2 PIP3PIP2 PIP3
Substrate
GSK3
Primingphosphatesite
Activesite
NH2
PI3K
p85
C2
p85
BD
CD
GSK3
Primingphosphatesite
Activesite
Ser
NH2Figure 2. The extracellular signal–dependent Akt regulation of glycogen synthase kinase function. Extracellular signals through RTK and GPCR activate phosphoinositol-3 kinase (PI3K), which converts PIP2 to PIP3 and directs Akt to the plasma membrane via interaction of its PH domain with PIP3. Akt is then activated by phosphorylation by phosphatidylinositol dependent kinase-(PDK)-1 and -2. Akt can then phosphorylate a series of cellular substrates. See text for details.

234
Early studies conducted in cell culture systems have shown that lithium prevents the phosphorylation of proteins that are sub-strates for GSK3, such as the microtubule-associated proteins tau and MAP-1B as well as β-catenin, a transcription factor in the Wnt pathway (16, 17, 19, 48). Furthermore, inhibition of GSK3 in Xenopus and Dictyostelium recapitulates the effects of lithium (see above) (17). More recently, multiple studies of neurodegenerative and neuroinflammatory processes have shown that chronic lithium treatment produces effects that are compatible with the inhibition of GSK3 as observed in the mouse central nervous system (9–11, 49). Finally, lithium treatment increases the expression of β-catenin in the mouse brain (18, 50, 51). Given that the degradation of cyto-plasmic β-catenin is promoted upon its phosphorylation by GSK3 (52), increased β-catenin levels in response to lithium have gener-ally been attributed to reduced GSK3 activity, but a direct role for GSK3 inhibition in therapeutic responses to lithium has not been established. Apart from its action as a GSK3 inhibitor, lithium has also been reported to activate neuronal Akt, a regulatory kinase that phosphorylates and thereby inhibits GSK3 (30, 53–55). In this way, it is conceivable that reduced GSK3 activity, as a component of lithium therapy, could reflect the targeting of either or both kinases, depending on dosage.
Evidence for GSK3 Inhibition in Lithium-Regulated Behavior
Both direct and circumstan-tial lines of evidence support an involvement of GSK3 in the regulation of symptoms of mania and depression by lithium. Research conducted in our laboratories has shown that GSK3 is involved in the regulation of behavior by the monoamine neurotransmit-ters dopamine and serotonin (5-HT), which are important pharmacological targets for the management of depression and psychosis (18, 30, 46, 56–69). Furthermore, various kinase inhibitors, acting directly on GSK3 or indirectly to limit its activity, replicate the effect of lithium in in vivo models of depression and mania (18, 30, 46, 60). Finally, genetic down-
regulation of GSK3 in GSK3β haploinsuficient mice recapitulates many behavioral effects of lithium (30, 51), whereas overexpres-sion of a Ser9 mutant of GSK3β, which results in constitutive GSK3 activity, reproduces behavioral correlates of hyperactivity and mania (61).
Lithium and the Role of GSK3 Inhibition: Summary
Recent literature provides strong evidence for the involvement of GSK3 inhibition in the regulation of behavior by lithium, and there is no doubt that under certain conditions lithium inhibits GSK3. Nevetheless, the relative degrees to which lithium functions as a direct inhibitor of GSK3, as opposed to acting as a regulator of Akt activity, are not clear in a behavioral context. One way out of this conundrum would be to conduct behavioral studies on transgenic mice that lack the N-terminal regulatory serine residues of GSK3a and β (62). Such mice would express GSK3 isoforms that cannot be inhibited by Akt and should thus constitute a good model to sort out the contribution of direct GSK3 inhibition in the behavioral effects of lithium. Another point of caution is that, because mice that lack a functional Gsk3b allele die during devel-opment (63), most behavioral studies of lithium that support the involvement of GSK3 have been conducted in GSK3β haploinsufi-cient mice. These mice are useful for studying the contribution of
Review
A Cβα
PKA
ATP
Dopamine
ATP
ADP
cAMP
PPi
Internalization
� -Arrestin � -Arrestin
GRK
AP2
Clathrin
Akt
β-Arrestin 2 β-Arrestin 2
γ
P
P
P P
P
P P P
PP2A
γα
β
Akt
Canonical G-protein–dependent signaling cAMP–
independentsignaling(D2 receptor)
Figure 3. Dual role of β−arrestins in mediat-ing GPCR desensitization and G protein–independent signaling. Activation of dopamine receptors (DAR) activates G proteins, which leads to activation (Gas) or inhibition (Gai/o) of adenylyl cyclase and modulation of the cAMP-dependent protein kinase PKA (G protein–dependent signaling). This is rapidly followed by receptor phosphorylation by GRKs and the recruitment of β−arrestins, leading to termina-tion of G protein signaling and formation of an internalization complex formed by β−arrestin 1 and/or β−arrestin 2, AP2, clathrin. Recruitment of β−arrestin 2 following activation of D2-class receptors also results in the formation of a sig-naling complex composed at least of β−arrestin 2, PP2A, and Akt, which results in a deactivation of Akt by PP2A and subsequent stimulation of GSK3-mediated signaling.

235 October 2008
Volume 8, Issue 5
GSK3 to some behaviors, but it should be noted that they express normal levels of GSK3a and show only about 25% reduction in total GSK3 activity (30). Therefore, the effects of lithium observed in GSK3β haploinsuficient mice may be significantly influenced by variations in experimental conditions, whereas therapeutic effects of lithium may be predicated on the existence of two GSK3 (30, 46, 59). There are presently no animal models in which the behavioral effects of lithium can be assessed in the total absence of neuronal GSK3. Obviously, animal models in which both GSK3 isoforms could be engineered and experimentally manipulated would be greatly help to elucidate the mechanism of lithium action with respect to GSK3 activity.
A Network of Effects: the Akt·βarr2·PP2A Protein Complex as a Lithium Target
The initial evidence for lithium as an upstream regulator of GSK3 came from cellular studies examining the ability of lithium to protect against neuronal glutamate toxicity (54) and from in vivo paradigms examining signaling responses to mood stabilizers in rodents (30, 53). Direct biochemical evidence has further estab-lished that lithium can regulate Akt and GSK3 in vivo by disrupt-ing the formation of a protein complex involved in GPCR signal-ing (18, 30, 58, 64).
Following stimulation and activation of their cognate G pro-teins, GPCRs are rapidly phosphorylated by members of a family of GPCR kinases (GRKs), leading to uncoupling of G proteins and the recruitment of β-arrestins (Figure 3) (65–67). The interaction of β-arrestins with GPCRs, which limits the ability of the GPCR to signal further through the G protein, is followed by the forma-tion of an endocytic complex that promotes the internalization of receptors via clathrin-coated pits (65, 67–69). However, the role of β-arrestins in GPCR regulation is not limited exclusively to desen-sitization. It has become apparent that apart from their canonical functionality through G proteins, GPCRs can also elicit cellular responses mediated by the formation of signaling protein com-plexes scaffolded by β−arrestins (70–72).
The D2-like dopamine receptors (including the D
2, D
3, and
D4 dopamine receptors) are a small family of GPCRs that couple
to the Gai/o
protein, whereby receptor activation leads to a reduc-tion in the production of the second messenger cyclic adenosine monophosphate (cAMP) (73–74). Prolonged stimulation of D
2
receptors in the mouse striatum causes the dephosphorylation and concomitant inhibition of Akt, which in turn results in the activa-tion of both GSK3 isoforms (Figure 4). Behavioral and biochemi-cal evidence has revealed that regulation of Akt/GSK3 signaling by the D
2 receptor is not affected by cAMP, but is instead mediated by
the formation of a protein complex that includes Akt, βArr2, and the heterotrimeric protein phosphatase 2A (PP2A), which facili-tates the dephosphorylation and concomitant deactivation of Akt by PP2A in response to dopamine (Figures 4 and 5) (30, 64, 77).
Lithium and the Akt·βArr2·PP2A Complex
In contrast to most previously described approaches to infer the molecular mechanism of lithium action, data supporting a role of the Akt·βArr2·PP2A complex were obtained primarily from in vivo models used to study the effect of mood-altering drugs. Acute lithium treatment in mice antagonizes the development of
Molecular Targets of Lithium
P
Akt
Akt
Akt
GSK3β
PP2A
β-arrestin 2
P
P
PDK1PI3K
D2RGPCR?
active
inactive
Akt
Akt
Akt
Akt
P
P
P
PP2A
Li+
Li+
Li+
Li+Li+
GSK3β
GSK3β
GSK3β
GSK3β
GSK3β
P
P
P
PDK1PI3K
active
inactive
Active
Inactive
β-arrestin 2
Figure 4. The effect of lithium on interactions among Akt, βArr2, and PP2A. Under basal conditions (upper panel) phosphorylated/activated Akt is efficiently dephosphorylated, leaving GSK3 mostly in an unphosphorylated/active state. During acute or chronic lithium treatment (lower panel), lithium destabilizes the Akt·βArr2·PP2A complex, augmenting the steady state of active Akt and promoting inactivation/phosphorylation of GSK3 in neurons.

236
dopamine-dependent locomotor behaviors by interfering with regulation of the Akt/GSK3 signaling through D
2 receptors (29, 30,
78). This interference can be replicated by preventing the forma-tion of the Akt·βArr2·PP2A protein complex. In βArr2-knockout mice, the absence of the Akt·βArr2·PP2A abolishes regulation of Akt/GSK3 signaling by D
2 receptors and an increases basal phos-
phorylation of Akt (Thr308) and GSK3β (Ser9), similar to obser-vations following lithium treatment (18, 30, 64). Furthermore, when administered acutely or chronically to βArr2-knockout mice, lithium fails to activate Akt and thus cannot inhibit GSK3 as it does in the striatum of wild-type animals (18). Accordingly, striatal β-catenin levels in the βArr2-knockout mice fail to rise in response to chronic lithium treatment (18). Significantly, all the βArr2-knockout characteristics described here correlate with the biochemical effects of lithium upon the formation of the Akt·βArr2·PP2A signaling complex. A series of co-immunopre-cipitation experiments have shown that therapeutically relevant lithium concentrations (0.5 to 1mM) are sufficient to destabilize the Akt·βArr2·PP2A complex in vitro as well as in the brains of living mice (18). Remarkably, lithium appears to be selective in destabilizing the Akt·βArr2·PP2A complex, as other protein com-plexes scaffolded by βArr2 and involved in GPCR desensitization or signaling are unaffected by lithium (18).
The exact mechanism through which lithium interferes with the formation of the Akt·βArr2·PP2A complex awaits further
investigation; however, preliminary in vitro experiments with recombinant purified Akt1 and βArr2 suggest that interactions of these two proteins required for complex formation is magnesium-dependent. Thus, competition between lithium and magnesium for binding to at least one of the components could underlie the instability of the complex in the presence of lithium (18, 47).
Lithium and the Akt·βArr2·PP2A Complex in Behavior
As discussed above, several behavioral studies implicate GSK3 inhibition in the effects of lithium (30, 51, 59–61). In addition, behavioral responses in βArr2-knockout mice indicate that regu-lation of GSK3 by the Akt·βArr2·PP2A protein complex is key. When evaluated for dopamine- or psychostimulant-induced loco-motor activity, βArr2-knockout mice are reminiscent of lithium-treated wild-type mice, displaying generally reduced responses (18, 30, 64). Furthermore, neither acute nor chronic lithium treat-ment has significant effects on the behaviors of βArr2-knockout mice (18). Intriguingly, despite their resistance to lithium, βArr2-knockout mice remain behaviorally responsive to a selective and direct inhibitor of GSK3 activity, thereby suggesting that the Akt·βArr2·PP2A protein complex is an upstream modulator of GSK3 in the signaling cascade (See Figures 3 and 4) that regulates lithium-sensitive behavioral responses (18).
Lithium and the Akt·βArr2·PP2A Protein Complex: Summary
The Akt·βArr2·PP2A protein complex appears to satisfy many of the prerequisites of a direct lithium target involved in the regula-tion of behavior. It is directly affected by lithium both in vitro and in the brain, and its elimination precludes both biochemical and behavioral effects of lithium. It is plausible that additional mechanisms contribute to the pharmacological effects of lithium on behavior. Furthermore, the relevance of lithium-mediated Akt·βArr2·PP2A complex disruption in the effects of lithium ther-apy remains to be established. A related question pertains to the selectivity of lithium for the Akt·βArr2·PP2A complex. Lithium does not affect general functions of βArr2 in GPCR desensitization and signaling; however, βArr2 has multiple interaction partners (79), and the possibility that lithium will undermine the interac-tion of other proteins with βArr2 must be considered. A second question relates to the regulation of the Akt·βArr2·PP2A complex by dopamine D
2 receptors; specifically, it is possible that other
GPCRs may also signal through the Akt·βArr2·PP2A protein complex. Indeed, depression-like behaviors that are affected by lithium have generally been linked to 5-HT- and norepinephrine-based neurotransmission (80). Finally, regulation of Akt activity by lithium raises the possibility that substrates other than GSK3 may also play a role in lithium action.
Review
GSK3 inhibitors
Akt Akt targets
Signal
Other GPCRs?
Dopamine Serotonin
D2R
GSK3targets
Behavioral responses
5-HT2R 5-HT1R
GSK3α/β
β-Arrestin 2PP2A
Activation
Inhibition
Activationor inhibition
Li+
Figure 5. Modulation of the Akt and GSK3 signaling pathway by vari-ous GPCRs. Signals from dopamine D2 receptors, serotonin receptors, and other GPCRs may modulate this pathway either through a β-arrestin-dependent or -independent mechanism. Lithium inhibits GSK3 either by enhancing Akt phosphorylation by interfering with the D2 receptor–dependent Akt·βArr2·PP2A complex or by acting as a direct inhibitor of GSK3.

237 October 2008
Volume 8, Issue 5
GSK3 substrates and behavioral regulation
The data discussed above establish that at least two potential mechanisms of lithium action converge on the modulation of the Akt/GSK3 signaling pathway. Although these enzymes are integra-tion hubs for many signaling modalities, the identity of the down-stream targets that may regulate behavior has remained elusive. Akt and GSK3 both have multiple substrates, including proteins involved in metabolism, cell survival and death, cytoskeletal orga-nization, and gene regulation (38, 43, 81, 82). Below we provide a brief overview of possible targets that have been investigated in the context of mood-stabilizing drug effects.
β-Catenin
β-catenin is a multifunctional protein that acts both as a transcrip-tion factor and a scaffolding molecule. In this latter function, β-catenin interacts with cadherins and a-catenin and thereby reg-ulates cytoskeletal organization of adherens junctions. Formation of such complexes may play a role in synaptic plasticity, because β-catenin is recruited to dendritic spines following depolarization (83). Moreover, a reduction in cytoplasmic levels of β-catenin and other members of the catenin·cadherin complex can reduce den-dritic arborization in cultured hippocampal neurons (84). In the Wnt signaling cascade, phosphorylation of monomeric β-catenin by GSK3 leads to its ubiquitination and proteosomal degradation (52). In this way, the modulation of GSK3 by monoamines and psychotropic drugs may lead to changes in β-catenin levels that affect synapse morphology and gene expression. Interestingly, chronic lithium treatment raises β-catenin expression in different regions of the mouse brain (18, 50, 51). Furthermore, overexpres-sion of β-catenin in transgenic mice recapitulates the effects of lithium hyperactivity and depression models in several relevant behavioral paradigms (50). However, the tissue-specific knockout of β-catenin in the adult mouse forebrain has little behavioral consequence (85) These studies collectively suggest that β-catenin may be important for the action of psychotropic drugs, whereas it may play a lesser role in regulation of normal behavior by mono-amine neurotransmitters.
Glutamate Receptors
Akt/GSK3 signaling appears to regulate synaptic plasticity and ion-otropic glutamate receptor functions. Importantly, these receptors, and glutamate neurotransmission in general, have been strongly implicated in the presentation of psychiatric disorders such as schizophrenia (86–89). Mood stabilizers have also been reported to affect glutamate neurotransmission (90). The expression and trafficking of the heteromultimeric ionotropic glutamate receptors
are closely regulated by complex networks of signaling molecules and scaffolding proteins (91). Dopamine and 5-HT may also affect synaptic plasticity by regulating the expression, phosphorylation, and trafficking of ionotropic glutamate receptors and associated proteins in neurons (92–94). In addition, to the extent that its activation level has been correlated to long-term potentiation and long-term depression, GSK3 has been linked to glutatmatergic activity (95–97) and affects the trafficking of NMDA receptor sub-units to the cell surface (95, 98).
Circadian Rhythm Proteins
The regulation of the fly GSK3 ortholog shaggy by 5-HT modu-lates circadian entrainment (99). Intriguingly, disruption of circa-dian rhythm has been proposed to play a role in diverse mental illnesses (100, 101). Exposure of cells to lithium appears to inter-fere with transcription of the clock gene Bmal1. Dopamine D
2
receptors, which regulate the activity of GSK3 (see above), also affect circadian rhythm regulated gene expression and behavior (102–104), and GSK3β has been shown to regulate mammalian circadian protein functions in cultured cells (105).
Arachidonic Acid
Chronic lithium treatment in rats has been shown to reduce brain arachidonic acid turnover by a mechanism involving downregula-tion of cycloxygenase 2 (COX-2) (106, 107), an effect also attrib-uted to chronic treatment with the mood stabilizers valproic acid and carmabazepine (108, 109). [Indeed, a recent clinical trial indi-cates that the COX-2 inhibitor clecoxib can potentiate the effects of antipsychotics and mood stabilizers in individuals with bipolar disorders (110).] Carmabazepine appears to affect arachidonic acid signaling by interfering with dopamine D
2 receptor functions
(111), and COX-2 expression is sensitive to GSK3 activity in some cell culture systems (112) and mouse peripheral tissues (113–114).
Conclusion
Since the realization, nearly sixty years ago, that lithium can exert mood-stabilizing effects, different molecular mechanisms have been proposed to explain its actions. Here, we have reviewed three of these putative mechanisms. Compelling lines of evidence suggest that lithium can reduce brain inositol, inhibit brain GSK3 activity, and disrupt the formation of the Akt·βArr2·PP2A in dop-aminoceptive brain neurons. For all three putative mechanisms, lithium may act by competing with magnesium binding. Lithium has also been suggested to affect the functions of G proteins in a magnesium-dependent way (115). However, mechanisms gov-erning the specificity of lithium/magnesium competition are still poorly understood (116), and the relevance of lithium/magnesium
Molecular Targets of Lithium

238
competition to the therapeutic effects of lithium awaits elucida-tion. Furthermore, the possible identification of other proteins or protein complexes affected by this mechanism in the regulation of behavior represents an interesting avenue for future research. doi:10.1124/mi.8.5.8
Acknowledgments This work was supported in part by a Canada Research Chair in Molecular Psychiatry and a Human Frontier Science Program Career Development Award (to J-MB) and National Institutes of Health Grants DA-13511, NS-19576, MH-73853 and MH-40159 (to MGC). Unrestricted gifts from the Lennon Family Foundation and Lundbeck Research USA to Duke University are greatly appreciated.
References 1. Cade, J.F. Lithium salts in the treatment of psychotic excitement. Med. J.
Aust. 2, 349–352 (1949).
2. Schou, M., Juel-Nielsen, N., Stromgren, E., and Voldby, H. The treat-ment of manic psychoses by the administration of lithium salts. J. Neurol. Neurosurg Psychiatry 17, 250–260 (1954).
3. De Montigny, C., Grunberg, F., Mayer, A., and Deschenes, J.P. Lithium induces rapid relief of depression in tricyclic antidepressant drug non-responders. Br. J. Psychiatry 138, 252–256 (1981).
4. Valenstein, M., McCarthy, J.F., Austin, K.L., Greden, J.F., Young, E.A., and Blow, F.C. What happened to lithium? Antidepressant augmentation in clinical settings. Am. J. Psychiatry 163, 1219–1225 (2006).
5. Bauer, M. and Dopfmer, S. Lithium augmentation in treatment-resistant depression: Meta-analysis of placebo-controlled studies. J. Clin. Psychopharmacol. 19, 427–434 (1999).
6. Cipriani, A., Pretty, H., Hawton, K., and Geddes, J.R. Lithium in the pre-vention of suicidal behavior and all-cause mortality in patients with mood disorders: A systematic review of randomized trials. Am. J. Psychiatry 162, 1805–1819 (2005).
7. Muller-Oerlinghausen, B., Felber, W., Berghofer, A., Lauterbach, E., and Ahrens, B. The impact of lithium long-term medication on suicidal behavior and mortality of bipolar patients. Arch. Suicide Res. 9, 307–319 (2005).
8. Carmichael, J., Sugars, K.L., Bao, Y.P., and Rubinsztein, D.C. Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxic-ity caused by the Huntington’s disease mutation. J. Biol. Chem. 277, 33791–33798 (2002).
9. Watase, K., Gatchel, J.R., Sun, Y. et al. Lithium therapy improves neu-rological function and hippocampal dendritic arborization in a spinocer-ebellar ataxia type 1 mouse model. PLoS Med. 4, e182 (2007).
10. Caccamo, A., Oddo, S., Tran, L.X., and LaFerla, F.M. Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am. J. Pathol. 170, 1669–1675 (2007).
11. Phiel, C. J., Wilson, C. A., Lee, V. M., and Klein, P. S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423, 435-439 (2003). This paper provides some of the initial evidence for an in vivo action of lithium on a neurodegenerative process. The article also demonstrates that lithium can inhibit brain GSK3a activity.
12. Feng, H.L., Leng, Y., Ma, C.H., Zhang, J., Ren, M., and Chuang, D.M. Combined lithium and valproate treatment delays disease onset, reduc-es neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 155, 567–572 (2008).
13. Fornai, F., Longone, P., Cafaro, L. et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 105, 2052–2057 (2008).
14. Shin, J.H., Cho, S.I., Lim, H.R., Lee, J.K., Lee, Y.A., Noh, J.S., Joo, I.S., Kim, K.W., and Gwag, B.J. Concurrent administration of Neu2000 and lithium produces marked improvement of motor neuron survival, motor function, and mortality in a mouse model of amyotrophic lateral sclero-sis. Mol. Pharmacol. 71, 965–975 (2007).
15. Berridge, M. J., Downes, C. P., and Hanley, M. R. Neural and develop-mental actions of lithium: a unifying hypothesis. Cell 59, 411-419 (1989). A review that elegantly summarizes the rationale and evidence for the inositol depletion hypothesis for the actions of lithium.
16. Stambolic, V., Ruel, L., and Woodgett, J.R. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 6, 1664–1668 (1996).
17. Klein, P. S., and Melton, D. A. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A 93, 8455-8459 (1996). References 16 and 17 provide some of the initial evidence for the direct inhibition of GSK3 by lithium.
18. Beaulieu, J. M., Marion, S., Rodriguiz, R. M., Medvedev, I. O., Sotnikova, T. D., Ghisi, V., Wetsel, W. C., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell 132, 125-136 (2008). This paper provides evidence for the ability of lithium to exert some of its actions by destabilizing a signaling protein complex.
19. Phiel, C.J. and Klein, P.S. Molecular targets of lithium action. Annu. Rev. Pharmacol. Toxicol. 41, 789–813 (2001).
20. Ohnishi, T., Ohba, H., Seo, K.C., Im, J., Sato, Y., Iwayama, Y., Furuichi, T., Chung, S.K., and Yoshikawa, T. Spatial expression patterns and bio-chemical properties distinguish a second myo-inositol monophosphatase IMPA2 from IMPA1. J. Biol. Chem. 282, 637–646 (2007).
21. Bone, R., Springer, J.P., and Atack, J.R. Structure of inositol monophos-phatase, the putative target of lithium therapy. Proc. Natl. Acad. Sci. U.S.A. 89, 10031–10035 (1992).
22. Pollack, S.J., Atack, J.R., Knowles, M.R., McAllister, G., Ragan, C.I., Baker, R., Fletcher, S.R., Iversen, L.L., and Broughton, H.B. Mechanism of inositol monophosphatase, the putative target of lithium therapy. Proc. Natl. Acad. Sci. U.S.A. 91, 5766–5770 (1994).
23. Brown, A.K., Meng, G., Ghadbane, H., Scott, D.J., Dover, L.G., Nigou, J., Besra, G.S., and Futterer, K. Dimerization of inositol monophosphatase Mycobacterium tuberculosis SuhB is not constitutive, but induced by binding of the activator Mg2+. BMC Struct. Biol. 7, 55 (2007).
24. Williams, R.S., Cheng, L., Mudge, A.W., and Harwood, A.J. A com-mon mechanism of action for three mood-stabilizing drugs. Nature 417, 292–295 (2002).
25. Shaldubina, A., Buccafusca, R., Johanson, R. A., Agam, G., Belmaker, R. H., Berry, G. T., and Bersudsky, Y. Behavioural phenotyping of sodi-um-myo-inositol cotransporter heterozygous knockout mice with reduced brain inositol. Genes Brain Behav 6, 253-259 (2007). The paper pro-vides evidence that genetic manipulations that reduce the levels of inositol do not reproduce the effect of lithium in vivo.
26. Allison, J.H., Boshans, R.L., Hallcher, L.M., Packman, P.M., and Sherman, W.R. The effects of lithium on myo-inositol levels in layers of frontal cerebral cortex, in cerebellum, and in corpus callosum of the rat. J. Neurochem. 34, 456–458 (1980).
27. Cryns, K., Shamir, A., Van Acker, N. et al. IMPA1 is essential for embryonic development and lithium-like pilocarpine sensitivity. Neuropsychopharmacology 33, 674–684 (2008).
28. Cryns, K., Shamir, A., Shapiro, J. et al. Lack of lithium-like behavioral and molecular effects in IMPA2 knockout mice. Neuropsychopharmacology 32, 881–891 (2007).
29. Cox, C., Harrison-Read, P.E., Steinberg, H., and Tomkiewicz, M. Lithium attenuates drug-induced hyperactivity in rats. Nature 232, 336–338 (1971).
30. Beaulieu, J. M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., and Caron, M. G. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A 101, 5099-5104 (2004). This paper established a potential molecular link for the ability of lithium to interfere with the dopamine system in vivo. It also pro-
Review

239 October 2008
Volume 8, Issue 5
vided direct evidence that genetic or pharmacological inhibition of GSK3 can mimic the behavioral effects of lithium.
31. Berry, G.T., Wu, S., Buccafusca, R., Ren, J., Gonzales, L.W., Ballard, P. L., Golden, J.A., Stevens, M.J., and Greer, J.J. Loss of murine Na+/myo-inositol cotransporter leads to brain myo-inositol depletion and central apnea. J. Biol. Chem. 278, 18297–18302 (2003).
32. Berry, G.T., Buccafusca, R., Greer, J.J., and Eccleston, E. Phosphoinositide deficiency due to inositol depletion is not a mechanism of lithium action in brain. Mol. Genet. Metab. 82, 87–92 (2004).
33. Shaldubina, A., Johanson, R.A., O’Brien, W.T., Buccafusca, R., Agam, G., Belmaker, R.H., Klein, P.S., Bersudsky, Y., and Berry, G.T. SMIT1 haploinsufficiency causes brain inositol deficiency without affecting lithium-sensitive behavior. Mol. Genet. Metab. 88, 384–388 (2006).
34. Bersudsky, Y., Shaldubina, A., Agam, G., Berry, G.T., and Belmaker, R.H. Homozygote inositol transporter knockout mice show a lithium-like phe-notype. Bipolar Disord. 10, 453–459 (2008).
35. Turski, W.A., Cavalheiro, E.A., Coimbra, C., da Penha Berzaghi, M., Ikonomidou-Turski, C., and Turski, L. Only certain antiepileptic drugs pre-vent seizures induced by pilocarpine. Brain Res. 434, 281–305 (1987).
36. Ryves, W.J. and Harwood, A.J. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem. Biophys. Res. Commun. 280, 720–725 (2001).
37. Embi, N., Rylatt, D.B., and Cohen, P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 107, 519–527 (1980).
38. Frame, S., and Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 359, 1–16 (2001).
39. Cross, D.A., Alessi, D.R., Cohen, P., Andjelkovich, M., and Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by pro-tein kinase B. Nature 378, 785–789 (1995).
40. Chen, M.J. and Russo-Neustadt, A.A. Exercise activates the phosphati-dylinositol 3-kinase pathway. Brain Res. Mol. Brain Res. 135, 181–193 (2005).
41. Andjelkovic, M., Alessi, D.R., Meier, R. et al. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 272, 31515–31524 (1997).
42. Alessi, D.R. and Cohen, P. Mechanism of activation and function of pro-tein kinase B. Curr. Opin. Genet. Dev. 8, 55–62 (1998).
43. Scheid, M.P. and Woodgett, J.R. PKB/AKT: Functional insights from genetic models. Nat. Rev. Mol. Cell Biol. 2, 760–768 (2001).
44. Jacinto, E., Facchinetti, V., Liu, D., Soto, N., Wei, S., Jung, S.Y., Huang, Q., Qin, J., and Su, B. SIN1/MIP1 maintains rictor-mTOR complex integ-rity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 (2006).
45. Li, X., Zhu, W., Roh, M.S., Friedman, A.B., Rosborough, K., and Jope, R.S. In vivo regulation of glycogen synthase kinase-3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacology 29, 1426–1431 (2004).
46. Beaulieu, J.M., Zhang, X., Rodriguiz, R.M., Sotnikova, T.D., Cools, M.J., Wetsel, W.C., Gainetdinov, R.R., and Caron, M.G. Role of GSK3beta in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. U.S.A. 105, 1333–1338 (2008).
47. Birch, N. J. Lithium and magnesium dependent enzymes. The Lancet (1974). Formulation of the idea that lithium might compete with magnesium for its action on various biological processes.
48. Gould, T.D. and Manji, H.K. Glycogen synthase kinase-3: A putative molecular target for lithium mimetic drugs. Neuropsychopharmacology 30, 1223–1237 (2005).
49. De Sarno, P., Axtell, R.C., Raman, C., Roth, K.A., Alessi, D.R., and Jope, R.S. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 181, 338–345 (2008).
50. Gould, T.D., Einat, H., O’Donnell K,C., Picchini, A.M., Schloesser, R.J., and Manji, H.K. beta-Catenin Overexpression in the Mouse Brain Phenocopies Lithium-Sensitive Behaviors. Neuropsychopharmacology (2007).
51. O’Brien, W. T., Harper, A. D., Jove, F., Woodgett, J. R., Maretto, S.,
Piccolo, S., and Klein, P. S. Glycogen synthase kinase-3beta haplo-insufficiency mimics the behavioral and molecular effects of lithium. J Neurosci 24, 6791-6798 (2004). In vivo evidence that a modest reduction in the activity of GSK3 by genetic means can recapitulate some of the effects of lithium.
52. Doble, B.W. and Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186 (2003).
53. De Sarno, P., Li, X., and Jope, R.S. Regulation of Akt and glycogen synthase kinase-3 beta phosphorylation by sodium valproate and lithium. Neuropharmacology 43, 1158–1164 (2002).
54. Chalecka-Franaszek, E., and Chuang, D. M. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci U S A 96, 8745-8750 (1999). Initial observation in neurons in culture that lithium can lead to an activation of Akt by promoting its phosporylation.
55. Zhang, F., Phiel, C.J., Spece, L., Gurvich, N., and Klein, P.S. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J. Biol. Chem. 278, 33067–33077 (2003).
56. Beaulieu, J.M. Not only lithium: Regulation of glycogen syn-thase kinase-3 by antipsychotics and serotonergic drugs. Int. J. Neuropsychopharmacol. 10, 3–6 (2007).
57. Beaulieu, J.M., Tirotta, E., Sotnikova, T.D., Masri, B., Salahpour, A., Gainetdinov, R.R., Borrelli, E., and Caron, M.G. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J. Neurosci. 27, 881–885 (2007).
58. Beaulieu, J.M., Gainetdinov, R.R., and Caron, M.G. The Akt-GSK-3 sig-naling cascade in the actions of dopamine. Trends Pharmacol. Sci. 28, 166–172 (2007).
59. Beaulieu, J.M., Zhang, X., Rodriguiz, R.M., Sotnikova, T.D., Cools, M.J., Wetsel, W.C., Gainetdinov, R.R., and Caron, M.G. Reply to Belmaker et al: GSK3β haploinsufficiency results in lithium-like effects in the forced swim test. Proc. Natl. Acad. Sci. U.S.A. 105, E24 (2008).
60. Gould, T.D., Einat, H., Bhat, R., and Manji, H.K. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int. J. Neuropsychopharmacol. 1–4 (2004).
61. Prickaerts, J., Moechars, D., Cryns, K., Lenaerts, I., van Craenendonck, H., Goris, I., Daneels, G., Bouwknecht, J.A., and Steckler, T. Transgenic mice overexpressing glycogen synthase kinase 3beta: A putative model of hyperactivity and mania. J. Neurosci. 26, 9022–9029 (2006).
62. McManus, E.J., Sakamoto, K., Armit, L.J., Ronaldson, L., Shpiro, N., Marquez, R., and Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 24, 1571–1583 (2005).
63. Hoeflich, K.P., Luo, J., Rubie, E.A., Tsao, M.S., Jin, O., and Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90 (2000).
64. Beaulieu, J. M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. An Akt/beta-arrestin 2/PP2A sig-naling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261-273 (2005). This article identifies the Akt/βArr-2/PP2A signaling complex as a regulator of Akt and GSK3 activity.
65. Shenoy, S.K. and Lefkowitz, R.J. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and sig-nalling. Biochem. J. 375, 503–515 (2003).
66. Gainetdinov, R.R., Premont, R.T., Bohn, L.M., Lefkowitz, R.J., and Caron, M.G. Desensitization of G Protein-Coupled Receptors and Neuronal Functions. Annu. Rev. Neurosci. 27, 107–144 (2004).
67. Lohse, M.J., Benovic, J.L., Codina, J., Caron, M.G., and Lefkowitz, R.J. beta-Arrestin: A protein that regulates beta-adrenergic receptor function. Science 248, 1547–1550 (1990).
68. Ferguson, S.S., Downey, W.E., 3rd, Colapietro, A.M., Barak, L.S., Menard, L., and Caron, M.G. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271, 363–366 (1996).
69. Laporte, S.A., Miller, W.E., Kim, K.M., and Caron, M.G. beta-Arrestin/AP-2 interaction in G protein-coupled receptor internalization:
Molecular Targets of Lithium

240
Identification of a beta-arrestin binging site in beta 2-adaptin. J. Biol. Chem. 277, 9247–9254 (2002).
70. Lefkowitz, R.J. and Shenoy, S.K. Transduction of receptor signals by beta-arrestins. Science 308, 512–517 (2005).
71. Luttrell, L.M., Ferguson, S.S., Daaka, Y. et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283, 655–661 (1999).
72. Luttrell, L.M., Roudabush, F.L., Choy, E.W., Miller, W.E., Field, M.E., Pierce, K.L., and Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc. Natl. Acad. Sci. U.S.A. 98, 2449–2454 (2001).
73. Kebabian, J.W. and Calne, D.B. Multiple receptors for dopamine. Nature 277, 93–96 (1979).
74. Kebabian, J.W. and Greengard, P. Dopamine-sensitive adenyl cyclase: Possible role in synaptic transmission. Science 174, 1346–1349 (1971).
75. Enjalbert, A. and Bockaert, J. Pharmacological characterization of the D2 dopamine receptor negatively coupled with adenylate cyclase in rat anterior pituitary. Mol. Pharmacol. 23, 576–584 (1983).
76. Missale, C., Nash, S.R., Robinson, S.W., Jaber, M., and Caron, M.G. Dopamine receptors: From structure to function. Physiol. Rev. 78, 189–225 (1998).
77. Andjelkovic, M., Jakubowicz, T., Cron, P., Ming, X.F., Han, J.W., and Hemmings, B.A. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 93, 5699–5704 (1996).
78. Ong, J.C., Brody, S.A., Large, C.H., and Geyer, M.A. An investigation of the efficacy of mood stabilizers in rodent models of prepulse inhibition. J. Pharmacol. Exp. Ther. 315, 1163–1171 (2005).
79. Xiao, K., McClatchy, D.B., Shukla, A.K., Zhao, Y., Chen, M., Shenoy, S.K., Yates, J.R., 3rd, and Lefkowitz, R.J. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. U.S.A. 104, 12011–12016 (2007).
80. O’Leary, O.F., Bechtholt, A.J., Crowley, J.J., Hill, T.E., Page, M.E., and Lucki, I. Depletion of serotonin and catecholamines block the acute behav-ioral response to different classes of antidepressant drugs in the mouse tail suspension test. Psychopharmacology (Berl.) 192, 357–371 (2007).
81. Cohen, P. and Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2, 769–776 (2001).
82. Woodgett, J.R. Judging a protein by more than its name: GSK-3. Sci STKE 2001, RE12 (2001).
83. Murase, S., Mosser, E., and Schuman, E.M. Depolarization drives beta-Catenin into neuronal spines promoting changes in synaptic structure and function. Neuron 35, 91–105 (2002).
84. Yu, X. and Malenka, R.C. Beta-catenin is critical for dendritic morpho-genesis. Nat. Neurosci. 6, 1169–1177 (2003).
85. Gould, T.D., O’Donnell, K.C., Picchini, A.M., Dow, E.R., Chen, G., and Manji, H.K. Generation and behavioral characterization of beta-catenin forebrain-specific conditional knock-out mice. Behav. Brain Res. 189, 117–125 (2008).
86. Sharp, F.R., Tomitaka, M., Bernaudin, M., and Tomitaka, S. Psychosis: Pathological activation of limbic thalamocortical circuits by psychomimet-ics and schizophrenia? Trends Neurosci. 24, 330–334 (2001).
87. Mohn, A.R., Gainetdinov, R.R., Caron, M.G., and Koller, B.H. Mice with reduced NMDA receptor expression display behaviors related to schizo-phrenia. Cell 98, 427–436 (1999).
88. Gainetdinov, R.R., Mohn, A.R., and Caron, M.G. Genetic animal models: Focus on schizophrenia. Trends Neurosci. 24, 527–533 (2001).
89. Carlsson, A., Waters, N., Holm-Waters, S., Tedroff, J., Nilsson, M., and Carlsson, M.L. Interactions between monoamines, glutamate, and GABA in schizophrenia: New evidence. Annu. Rev. Pharmacol. Toxicol. 41, 237–260 (2001).
90. Sanacora, G., Zarate, C.A., Krystal, J.H., and Manji, H.K. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat. Rev. Drug Discov. 7, 426–437 (2008).
91. Sheng, M. and Hoogenraad, C.C. The postsynaptic architecture of excitatory synapses: A more quantitative view. Annu. Rev. Biochem. 76, 823–847 (2007).
92. Yao, W.D., Gainetdinov, R.R., Arbuckle, M.I., Sotnikova, T.D., Cyr, M., Beaulieu, J. M., Torres, G.E., Grant, S.G., and Caron, M.G. Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 41, 625–638 (2004).
93. Svenningsson, P., Nishi, A., Fisone, G., Girault, J.A., Nairn, A.C., and Greengard, P. DARPP-32: An integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol. 44, 269–296 (2004).
94. Esteban, J.A., Shi, S.H., Wilson, C., Nuriya, M., Huganir, R.L., and Malinow, R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 6, 136–143 (2003).
95. Zhu, L.Q., Wang, S.H., Liu, D., Yin, Y.Y., Tian, Q., Wang, X.C., Wang, Q., Chen, J.G., and Wang, J.Z. Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J .Neurosci. 27, 12211–12220 (2007).
96. Peineau, S., Bradley, C., Taghibiglou, C., Doherty, A., Bortolotto, Z.A., Wang, Y.T., and Collingridge, G.L. The role of GSK-3 in synaptic plastic-ity. Br. J. Pharmacol. 153 Suppl 1, S428–437 (2008).
97. Peineau, S., Taghibiglou, C., Bradley, C. et al. LTP inhibits LTD in the hip-pocampus via regulation of GSK3beta. Neuron 53, 703–717 (2007).
98. Chen, P., Gu, Z., Liu, W., and Yan, Z. Glycogen synthase kinase 3 regu-lates N-methyl-D-aspartate receptor channel trafficking and function in cortical neurons. Mol. Pharmacol. 72, 40–51 (2007).
99. Yuan, Q., Lin, F., Zheng, X., and Sehgal, A. Serotonin modulates circa-dian entrainment in Drosophila. Neuron 47, 115–127 (2005).
100. Lewy, A.J., Lefler, B.J., Emens, J.S., and Bauer, V.K. The circadian basis of winter depression. Proc. Natl. Acad. Sci. U.S.A. 103, 7414–7419 (2006).
101. Mansour, H.A., Wood, J., Logue, T., Chowdari, K.V., Dayal, M., Kupfer, D.J., Monk, T.H., Devlin, B., and Nimgaonkar, V.L. Association study of eight circadian genes with bipolar I disorder, schizoaffective disorder and schizophrenia. Genes Brain Behav. 5, 150–157 (2006).
102. Yujnovsky, I., Hirayama, J., Doi, M., Borrelli, E., and Sassone-Corsi, P. Signaling mediated by the dopamine D2 receptor potentiates circadian regulation by CLOCK:BMAL1. Proc. Natl. Acad. Sci. U.S.A. 103, 6386–6391 (2006).
103. Yin, L., Wang, J., Klein, P.S., and Lazar, M.A. Nuclear receptor Rev-erbalpha is a critical lithium-sensitive component of the circadian clock. Science 311, 1002–1005 (2006).
104. Doi, M., Yujnovsky, I., Hirayama, J., Malerba, M., Tirotta, E., Sassone-Corsi, P., and Borrelli, E. Impaired light masking in dopamine D2 recep-tor-null mice. Nat. Neurosci. 9, 732–734 (2006).
105. Iitaka, C., Miyazaki, K., Akaike, T., and Ishida, N. A role for glycogen synthase kinase-3beta in the mammalian circadian clock. J. Biol. Chem. 280, 29397–29402 (2005).
106. Weerasinghe, G.R., Rapoport, S.I., and Bosetti, F. The effect of chronic lithium on arachidonic acid release and metabolism in rat brain does not involve secretory phospholipase A2 or lipoxygenase/cytochrome P450 pathways. Brain Res. Bull. 63, 485–489 (2004).
107. Bosetti, F., Rintala, J., Seemann, R., Rosenberger, T.A., Contreras, M.A., Rapoport, S.I., and Chang, M.C. Chronic lithium downregulates cycloox-ygenase-2 activity and prostaglandin E(2) concentration in rat brain. Mol. Psychiatry 7, 845–850 (2002).
108. Bosetti, F., Weerasinghe, G.R., Rosenberger, T.A., and Rapoport, S.I. Valproic acid down-regulates the conversion of arachidonic acid to eicosanoids via cyclooxygenase-1 and -2 in rat brain. J. Neurochem. 85, 690–696 (2003).
109. Ghelardoni, S., Tomita, Y.A., Bell, J.M., Rapoport, S.I., and Bosetti, F. Chronic carbamazepine selectively downregulates cytosolic phospho-lipase A2 expression and cyclooxygenase activity in rat brain. Biol. Psychiatry 56, 248–254 (2004).
110. Nery, F.G., Monkul, E.S., Hatch, J.P., Fonseca, M., Zunta-Soares, G.B., Frey, B.N., Bowden, C.L., and Soares, J.C. Celecoxib as an
Review

241 October 2008
Volume 8, Issue 5
Molecular Targets of Lithium
adjunct in the treatment of depressive or mixed episodes of bipolar disorder: A double-blind, randomized, placebo-controlled study. Hum. Psychopharmacol. 23, 87–94 (2008).
111. Basselin, M., Chang, L., Chen, M., Bell, J.M., and Rapoport, S.I. Chronic carbamazepine administration attenuates dopamine D2-like receptor-initiated signaling via arachidonic acid in rat brain. Neurochem. Res. 33, 1373–1383 (2008).
112. Takada, Y., Fang, X., Jamaluddin, M.S., Boyd, D.D., and Aggarwal, B.B. Genetic deletion of glycogen synthase kinase-3beta abrogates activation of IkappaBalpha kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by tumor necrosis factor. J. Biol. Chem. 279, 39541–39554 (2004).
113. Cuzzocrea, S., Crisafulli, C., Mazzon, E., Esposito, E., Muia, C., Abdelrahman, M., Di Paola, R., and Thiemermann, C. Inhibition of glyco-gen synthase kinase-3beta attenuates the development of carrageenan-induced lung injury in mice. Br. J. Pharmacol. 149, 687–702 (2006).
114. Cuzzocrea, S., Mazzon, E., Di Paola, R. et al. Glycogen synthase kinase-3beta inhibition attenuates the degree of arthritis caused by type II collagen in the mouse. Clin. Immunol. 120, 57–67 (2006).
115. Srinivasan, C., Toon, J., Amari, L., Abukhdeir, A.M., Hamm, H., Geraldes, C.F., Ho, Y.K., and Mota de Freitas, D. Competition between lithium and magnesium ions for the G-protein transducin in the guanos-ine 5′-diphosphate bound conformation. J. Inorg. Biochem. 98, 691–701 (2004).
116. Mota de Freitas, D., Castro, M.M., and Geraldes, C.F. Is competition between Li+ and Mg2+ the underlying theme in the proposed mechanisms for the pharmacological action of lithium salts in bipolar disorder? Acc. Chem. Res. 39, 283–291 (2006).
Jean-Martin Beaulieu, PhD, is Assistant Professor and Canada Research Chair in Molecular Psychiatry at Laval University/CRULRG in Québec City. His research on dopamine receptor sig-naling in vivo has led to the iden-tification of mechanisms of action for lithium and antipsychotics. He continues to research how changes
in signaling processes participate in the regulation of behavior by various psychiatric drugs.
Marc G. Caron, PhD, is James B. Duke Professor of Cell Biology and holds secondary appointments in Medicine and Neurobiology at Duke University Medical Center. His long-standing research interests have concerned G protein–coupled receptors and neurotransmitter transporters. His recent efforts have used genetic approaches to develop
animal models of neurobiological disorders.

Cellular Plasticity Cascades in the Pathophysiologyand Treatment of Bipolar Disorder
Robert J Schloesser1,3, Jian Huang2,3, Peter S Klein2 and Husseini K Manji*,1
1Laboratory of Molecular Pathophysiology, Mood and Anxiety Disorders Program, National Institute of Mental Health, NIH,
Bethesda, MD, USA; 2Department of Medicine (Hematology-Oncology), University of Pennsylvania School of Medicine,
Philadelphia, PA, USA
Bipolar disorder (BPD) is characterized by recurrent episodes of disturbed affect including mania and depression as well as
changes in psychovegetative function, cognitive performance, and general health. A growing body of data suggests that BPD
arises from abnormalities in synaptic and neuronal plasticity cascades, leading to aberrant information processing in critical
synapses and circuits. Thus, these illnesses can best be conceptualized as genetically influenced disorders of synapses and
circuits rather than simply as deficits or excesses in individual neurotransmitters. In addition, commonly used mood-
stabilizing drugs that are effective in treating BPD have been shown to target intracellular signaling pathways that control
synaptic plasticity and cellular resilience. In this article we draw on clinical, preclinical, neuroimaging, and post-mortem data
to discuss the neurobiology of BPD within a conceptual framework while highlighting the role of neuroplasticity in the
pathophysiology and treatment of this disorder.
Neuropsychopharmacology Reviews (2008) 33, 110–133; doi:10.1038/sj.npp.1301575; published online 3 October 2007
Keywords: bipolar disorder; neuroplasticity; intracellular signaling cascades; lithium; valproic acid; mood stabilizer
������������������������������������������
INTRODUCTION
Bipolar disorder (BPD) is a common, chronic, recurrentmental illness that affects the lives and functioning ofmillions of individuals worldwide. A growing number ofrecent studies indicate that for a majority of theseindividuals, outcome is quite poor. High rates of relapse,chronicity, lingering residual symptoms, subsyndromes,cognitive and functional impairment, psychosocial disabil-ity, and diminished well-being are unfortunately commonoccurrences in BPD (Belmaker, 2004). Furthermore, BPD isa systemic disease that is frequently associated with a widerange of physiological perturbations and medical problems,including cardiovascular disease, diabetes mellitus, obesity,and thyroid disease (Kupfer, 2005). Neurobiological studiesof mood disorders over the past 40 years have primarilyfocused on abnormalities of the monoaminergic neuro-transmitter systems, on characterizing alterations of in-dividual neurotransmitters in disease states, and on
assessing response to mood stabilizer and antidepressantmedications. The monoaminergic systems are extensivelydistributed throughout the network of limbic, striatal, andprefrontal cortical neuronal circuits thought to support thebehavioral and visceral manifestations of mood disorders(Drevets, 2000). Studies of cerebrospinal fluid chemistry,neuroendocrine responses to pharmacological challenge,and neuroreceptor and transporter binding have demon-strated a number of abnormalities in monoaminergicneurotransmitter and neuropeptide systems in mooddisorders (Goodwin and Jamison, 2007).
Unfortunately, these observations have not yet greatlyadvanced our understanding of the underlying biology ofrecurrent mood disorders, which must include an explana-tion for the predilection to episodic and often profoundmood disturbance that can become progressive over time.BPD likely arises from the complex interaction of multiplesusceptibility (and protective) genes and environmentalfactors, and the phenotypic expression of the diseaseincludes not only mood disturbance, but also a constellationof cognitive, motor, autonomic, endocrine, and sleep/wakeabnormalities. Furthermore, while most antidepressantsexert their initial effects by increasing intrasynaptic levelsof serotonin and/or norepinephrine, their clinical antide-pressant effects are observed only after chronic adminis-tration (over days to weeks), suggesting that a cascade ofdownstream events is ultimately responsible for theirtherapeutic effects. These observations have led to the idea
Received 16 July 2007; revised 1 August 2007; accepted 14 August2007
*Correspondence: Dr HK Manji, Laboratory of Molecular Pathophysio-logy, Mood and Anxiety Disorders Program, National Institute of MentalHealth, Porter Neuroscience Research Center, Building 35, Room 1C-917, 35 Convent Drive, Bethesda, MD 20892, USA, Tel: + 1 301 4969802, Fax: + 1 301 480 0123, E-mail: [email protected] Schloesser and Dr Huang share first authorship.
Neuropsychopharmacology REVIEWS (2008) 33, 110–133& 2008 Nature Publishing Group All rights reserved 0893-133X/08 $30.00
...............................................................................................................................................................
110 www.neuropsychopharmacology.org
REVIEW
..............................................................................................................................................
Neuropsychopharmacology REVIEWS

that while dysfunction within the monoaminergic neuro-transmitter systems is likely to play an important role inmediating some facets of the pathophysiology of BPD, itlikely represents the downstream effects of other, moreprimary abnormalities in signaling pathways (Table 1)(Goodwin and Jamison, 2007) (Figure 1a).
Why Consider Plasticity Cascades as Playinga Central Role in the Pathophysiology andTreatment of BPD?
Plasticity, the ability to undergo and sustain change, isessential for the proper functioning of our nervous system.This capacity for change allows organisms to adapt tocomplex alterations in both their internal and externalenvironments, a feature fundamentally important forsurvival and reproduction. Besides the evident need forsignificant adaptation mechanisms in learning and memoryas well as physiological homeostasis, all complex behavioralphenomenaFincluding mood and emotionFare dynamicprocesses that rely on plastic neural circuitry. The biologicalbasis of this capacity to adapt encompasses a diverse set ofcellular and molecular mechanisms that fall under the broadterm ‘neuroplasticity’. In this paper, we make the distinc-tion between synaptic plasticity and neuroplasticity.
Synaptic plasticity refers to the cellular process thatresults in lasting changes in the efficacy of neurotransmis-sion. More specifically, the term synaptic plasticity refers tothe variability of the strength of a signal transmittedthrough a synapse. The regulation of transmission at thesynapse may be mediated by changes in neurotransmitterlevels, receptor subunit phosphorylation, surface/cellularlevels or receptors, and conductance changes, amongothers.
Neuroplasticity is a broader term that encapsulateschanges in intracellular signaling cascades and generegulation (see McClung and Nestler, 2008 in this issue),modifications of synaptic number and strength, variationsin neurotransmitter release, modeling of axonal anddendritic architecture and, in some areas of the CNS, thegeneration of new neurons. Modifications arising fromneuroplastic mechanisms can be of short duration or longlasting, and this is determined by the qualitative, quanti-tative, and temporal characteristics of the precipitatingstimuli. For instance, compared to acute or single stimuli,chronic and repeated stimuli often lead to qualitativelydifferent, and often times long-lasting alterations (Hyman
and Nestler, 1996); furthermore, substantial life events thatoccur during development of the organism often have agreater impact than they would later in life.
In recent years, research has linked mood disorders withstructural and functional impairments related to neuroplas-ticity in various regions of the CNS. In addition, psycho-tropic drugs commonly used to treat these conditions targetmolecules and signaling cascades implicated in the controlof neuroplasticity. Research on the biological underpin-nings of mood disorders has therefore moved away fromfocusing on absolute changes in neurochemicals such asmonoamines and neuropeptides, and instead has begunhighlighting the role of neural circuits and synapses, and theplastic processes controlling their function. Thus, theseillnesses can best be conceptualized as genetically influ-enced disorders of synapses and circuits rather than simplyas deficits or excesses in individual neurotransmitters. Theintegration of knowledge derived from different physiolo-gical and phenomenological levels continues to help moveus toward a more conceptual understanding of the etiologyand pathophysiology of BPD. As we review in this paper, agrowing body of data supports the contention that BPDarises from abnormalities in cellular plasticity cascades,leading to aberrant information processing in synapses andcircuits mediating affective, cognitive, motoric, and neuro-vegetative functions (Catapano et al, in press; Post, 2007;Young, 2007). Indeed, in a recent whole-genome associationstudy of BPD, all of the highly significant associationsimplicated signaling cascades (Baum et al, 2007).
The role of cellular signaling cascades has the potential toexplain much of the complex neurobiology of BPD (Good-win and Jamison, 2007). Cellular signaling cascades regulatethe multiple neurotransmitter and neuropeptide systemsimplicated in the disorder, and are targets for the mosteffective treatments. Signaling pathways are also targets forhormones that have been implicated in the pathophysiologyof BPD. The highly integrated monoamine and prominentneuropeptide pathways are known to originate and projectheavily to limbic-related regions such as the hippocampus,hypothalamus, and brain stem, which are likely associatedwith neurovegetative symptoms. Abnormalities in cellularsignaling cascades that regulate diverse physiologic func-tions also likely explain the tremendous medical comorbid-ity associated with BPD. Furthermore, many of thesepathways play critical roles not only in synaptic (andtherefore behavioral) plasticity, but also in long-termatrophic processes (see below) (Figure 1b).
Targeting these cascades in the treatment of mooddisorders may stabilize the underlying disease process byreducing the frequency and severity of the profound moodcycling that contributes to morbidity and mortality.
In this paper, we focus upon the role of plasticity cascadesin the pathophysiology and treatment of BPD. The role ofneurotransmitter and neuropeptide systems has recentlybeen extensively covered elsewhere (Goodwin and Jamison,2007; Soares and Young, 2007), and is not addressedhere. There have also been tremendous advances in ourunderstanding of the fundamental processes underlyingsynaptic and neural plasticity; many of these advances arewell covered in accompanying papers (Bear et al, 2008;Martinowich and Lu, 2008; Citri and Malenka, 2008;McClung and Nestler, 2008), and are not discussed in detail
Table 1 Putative Roles for Signaling Pathways in Mood Disorders
Amplify, attenuate, and integrate multiple signals that form the basis forintracellular circuits and cellular modules
Regulate multiple neurotransmitter and peptide systems
Play critical role in cellular memory and long-term neuroplasticity
Regulate complex signaling networks that form the basis for higher order brainfunction, mood, and cognition
Act as major targets for many hormones implicated in mood disorders, includinggonadal steroids, thyroid hormones, and glucocorticoids
Act as targets for medications that are most effective in the treatment of mooddisorders
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
111
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

Neurotransmission:Neurotransmitters andNeuropeptidesSynaptic connectivity
MolecularSusceptibility genes
Protective genesTranscription factors
mRNA
CellularCell growth/survival/death
Cell morphology:dendritic remodeling
SystemsCritical neuronal circuitry
BehaviorCognitive/Affective/Sensorymotor
Environment
PKC & MARCKSGSK-3&substrates
CREB&BDNFERK MAP kinase
Bcl-2 family of proteins
Environmental factors(including external
environment: psychosocial stressors,
sleep deprivation,internal environment
gonadal/HPA steroids)
Dendritemorphology
Synapticstrength
Gene transcription
Intracellularsignallingcascades
Inactiveprotein kinase
Inactiveprotein kinase
Inactiveprotein kinase
Activeprotein kinase
Activeprotein kinase
Activeprotein kinase
GTPase
ATP ADP
P
ATP ADP
P
P
PATP ADPInactiveprotein
Activeprotein
Cellular response
Spinenumber
Neurotransmitterrelease
Figure 1 (a) A true understanding of the pathophysiology of BPD must encompass different systems on different physiological levels at which the diseasemanifests: molecular, cellular, and behavioral. (b) Biological mechanisms underlying neuroplasticity. The remarkable plasticity of neuronal circuits is achievedthrough different biological means including alterations in gene transcription and intracellular signaling cascades. These changes modify diverse neuronalproperties such as neurotransmitter release, synaptic function and even morphological characteristics of neurons.
Cellular plasticity cascades in BPDRJ Schloesser et al
...............................................................................................................................................................
112
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

here. Finally, the interested reader is referred to the chapterby Pittenger and Duman for an excellent, extensivediscussion of the role of plasticity cascades in depression,and as targets for antidepressants (Pittenger and Duman,2008).
CLINICAL EVIDENCE SUPPORTING THECONTENTION THAT ABNORMALITIES INSYNAPTIC AND CELLULAR PLASTICITYPLAY AN IMPORTANT ROLE IN BPD
Structural Neuroimaging Findings
As discussed in the introduction, considerable data nowshow that many forms of neural plasticity involve structuralbrain changes (Figure 2). Additionally, evidence hasaccumulated supporting the persistence of ‘structuralplasticity’ in the adult human brain. Thus, investigatorshave used neuroimaging and post-mortem human brainstudies to investigate potential alterations in structuralplasticity in mood disorders.
Computed tomography and magnetic resonance imaging(MRI) have revealed structural abnormalities in the brainsof patients with mood disorders. Overall, gray mattervolumes do not significantly differ in patients with BPD ascompared to healthy individuals (Brambilla et al, 2001;Schlaepfer et al, 1994; Zipursky et al, 1997); however,several studies have found region-specific reductions. Themajor region-specific structural imaging findings includeincreased ventricular size and decreased frontal corticalarea volumes. Several studies have reported that BPD
patients have enlarged ventricles (Kato et al, 1994; Nasrallahet al, 1982; Pearlson et al, 1984; Pearlson and Veroff, 1981;Strakowski et al, 2002), which suggests that decreases inbrain tissue volume may exist. Indeed, studies have revealeddecreases in specific cortical areas including the leftsubgenual region 24 (SG24), a structure in the anteriorgyrus ventral to the genu of the corpus callosum (Drevetset al, 1997). Other studies indicate reductions of gray mattervolume in the left dorsolateral prefrontal cortex (DLPC)(Brambilla et al, 2002), the ventral prefrontal cortex (PFC),and the orbital PFC (Frangou et al, 2002). Temporal lobestructures such as the hippocampus and the amygdala havenot been as thoroughly researched as the frontal lobes.
In contrast to findings in major depressive disorder(MDD), significant BPD-related volumetric differences havenot been consistently reported in the hippocampus; as wediscuss later, the possibility that mood stabilizers exert amitigating neurotrophic effect is an important potentialconfound. One study did, however, find that volumetricreductions were more prevalent in the right hippocampus ofthe affected individual in monozygotic twin sets discordantfor BPD (Noga et al, 2001).
Periventricular and deep hyperintensities in the subcor-tical white matterFwhite matter hyperintensities(WMH)Fhave consistently been identified with MRI inthe brains of elderly depressed patients and patients withBPD (Altshuler et al, 1995; Bearden et al, 2001; Lenox et al,2002; Stoll et al, 2000). Although the pathophysiological andfunctional meanings associated with WMH still need to beelucidated, they may have multiple causes includingcerebrovascular accidents, ischemia, demyelination, loss ofaxons, dilated perivascular space, minute brain cysts, andnecrosis. To assess the cellular pathophysiology of theselesions, a histopathological assessment was correlated withneuroimaging conducted in vitro using brain slices fromelderly depressed subjects and elderly controls. Deep WMHwere found to be ischemic in the depressed group whencompared against the control samples, and were localizedmainly in the DLPC, supporting the contention that thelesions were of vascular origin in these elderly depressedsubjects (Thomas et al, 2002).
Intriguingly, a growing body of data suggests that asignificant percentage of young bipolar patients (includingchildren) exhibit WMH (Lyoo et al, 2002; Pillai et al, 2002).In fact, these lesions have also been found to be increased inchildren with psychiatric disorders, though highest amongthose with BPD when compared to controls, particularly inthe frontal lobes (Lyoo et al, 2002) and also early in thecourse of BPD in adolescents (Pillai et al, 2002). Theyappear to be associated with poor treatment response inpatients with affective disorders (Lenox et al, 2002),particularly when they are located in subcortical ratherthan periventricular areas (Moore et al, 2001). Together,these results support the contention that WMH damage thestructure of brain tissue, and likely disrupt the neuronalconnectivity necessary for normal affective functioning.
Because these young BPD patients have no overtcerebrovascular risk factors, the findings are most consis-tent with the hypothesis that patients with BPD haveendogenous impairments of cellular resilience, leading tohypoxic-like changes even in the face of normal cerebro-vascular flow. Indeed, the relationship between reduced
Nucleusaccumbens
Prefrontalcortex
Cingulate Gyrus
Hippocampus
Thalamus
Amygdala
Figure 2 Neuroanatomical regions implicated in affective processes.Neuroimaging studies, observations on patients with selective CNS lesions,and data from animal behavioral experiments have elucidated several brainregions implicated in the perception and control of mood states andemotions. These include the PFC, amygdala, insula, hippocampus, anteriorcingulate cortex, and ventral striatum. Studies on BPD using neuroimagingand post-mortem pathological techniques have revealed several functionaland structural abnormalities in these regions.
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
113
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

intracellular pH and increased WMH in BPD supports thispossibility (Kato et al, 1998).
Magnetic Resonance Spectroscopy FindingsPertaining to Mitochondrial Function and CellEnergetics
It is clear that BPD is not a classical mitochondrial disorder.However, emerging data suggest that many upstreamabnormalities (likely nuclear genome coded) converge toregulate mitochondrial function, and that mitochondrialfunction is implicated in acute abnormalities of bothsynaptic and neural plasticity (Goodwin and Jamison,2007). Thus, in addition to the well-known function ofenergy production via oxidative phosphorylation, neuronalmitochondria also play an important role in apoptosis andin the regulation of intracellular calcium; increasingevidence suggests that the latter action may be criticallyimportant in regulating the release of, and response to,neurotransmitters. Furthermore, mounting evidence sug-gests that activation of mitochondrial-apoptotic cascadesmay lead to a process of ‘synaptic apoptosis’, in whichapoptotic processes are activated in a highly localizedmanner (Mattson, 2007).
A growing body of evidence suggests that mitochondriamay be integrally involved in the general processes ofsynaptic plasticity (Mattson, 2007; Yang et al, 2003). Inaddition, increased synaptic activity has been shown toinduce the expression of mitochondrial-encoded genes,suggesting that a long-lasting up-regulation of energyproduction may be triggered by synaptic activity, playinga role in the long-term regulation of synaptic strength(Williams et al, 1998). Thus, mitochondrial dysfunctionexerts major effects on ‘here and now’ neurotransmitterfunction, in addition to its better appreciated long-termcytoprotective effects.
Magnetic resonance spectroscopy (MRS) has increasinglybeen utilized in the study of neuropsychiatric disorders.N-acetyl-aspartate (NAA) is a predominant neurochemicalcompound that can be quantitatively assessed via MRS inthe normal adult human brain. Pertinent to the presentdiscussion is the fact that NAA is localized to matureneurons and synthesized within mitochondria.
Interestingly, inhibitors of the mitochondrial respiratorychain decrease NAA concentrations, effects that correlatewith reductions in adenosine triphosphate (ATP) andoxygen consumption. High-resolution 1H-MRS imagingstudies have found decreased levels of NAA in limbic andlimbic-related areas of the brain as well as decreased levelsin the hippocampus, independent of mood state (Bertolinoet al, 2003), in euthymic and medicated familial bipolar Ipatients (Diecken et al, 2003); in the DLPC in euthymic andunmedicated adult bipolar I and II patients (Winsberg et al,2000); in the orbitofrontal cortex in manic/mixed patients(Cecil et al, 2002); and in the DLPC in juvenile BPD patients(Chang et al, 2003). In addition, decreased levels of NAAwere also found in the basal ganglia in both the depressiveand euthymic states (Hamakawa et al, 1998). As discussedabove, these findings may be the expression of underlyingchanges in ATP spent and availability, in oxygen consump-tion, or in glutamatergic activity in BPD.
In addition to the NAA findings, studies that usedphosphorous-31 MRS (31P MRS), which permits thedetermination of abnormally high brain energy phosphatemetabolism, showed a decrease in phosphocreatine (PCr)and/or ATP levels in mood disorder patients (Diecken et al,1995; Kato et al, 1995; Volz et al, 1998). The most extensiveseries of studies investigating possible abnormalities inbrain energy regulation in mood disorders have beenconducted by Kato and colleagues. Consistent with thedecreased PCr and ATP levels discussed above, this researchgroup also found low pH levels (measured indirectly via31P MRS) in mood disorder patients compared to normalcontrols, in basal ganglia and the whole brain (Hamakawaet al, 2004); these observations originally led to the notionthat BPD may be associated with mitochondrial dysfunction(Hamakawa et al, 2004; Kato et al, 1998).
It is not currently known whether these neuroimagingresults are developmental abnormalities that confer vulner-ability to severe mood episodes, compensatory changes toother pathogenic processes, or the sequelae of recurrentaffective episodes (Carlson et al, 2006). Indeed, data suggestthat multiple factors may be operative. The reduced graymatter volumes, WMH, and reduced NAA levels describedabove often affect first-onset patients or children with BPD(Frazier et al, 2005; Sassi et al, 2005). While these studies donot demonstrate that the changes precede illness onset, theycertainly suggest that these changes do not represent thetoxic sequelae of decades of illness. Consistent with such acontention, a meta-analysis of imaging studies concludedthat volumetric abnormalities in the subgenual PFC,striatum, hippocampus, and amygdala are seen in first-episode bipolar subjects, children with BPD, and unaffectedsiblings, raising the possibility that this endophenotype mayconstitute a heritable vulnerability factor in these patients(Hajek et al, 2005).
However, some of the brain changes may be associatedwith duration of illness and the consequences of affectiveepisodes per se. Sheline et al (2003, 1996) measuredhippocampal volumes of subjects with a history of majordepressive episodes and found that the degree of hippo-campal volume reduction correlated with total duration ofMDD, and with duration of untreated depressive episodes.Another study found hippocampal volume reduction insubjects with multiple depressive episodes, but not in first-episode subjects (MacQueen et al, 2003). It is noteworthythat similar changes have not been reported in patients withBPDFthis difference may reflect distinct pathophysiolo-gies, or the neuroprotective effects of mood stabilizers (seebelow).
Post-Mortem Brain Findings
Neurons. In addition to accumulating neuroimaging find-ings, studies of post-mortem tissue from BPD patients haverevealed several abnormalities (Figure 3). Most studies havefocused on areas highlighted by structural and functionalimaging findings such as PFC, amygdala, hippocampus, andstriatum. In recent years, progress in techniques such asunbiased stereology, histopathology, and microscopy hashelped to advance efforts to provide a neuropathologicaldescription of BPD. Post-mortem observations have furthercorroborated the concept that impaired cellular resilience
Cellular plasticity cascades in BPDRJ Schloesser et al
...............................................................................................................................................................
114
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

and function play a role in the pathophysiology of BPD(Table 2).
Studies have found reduced subcortical nuclei volumes inpatients with recurrent mood disorders, with especiallystriking results in the BPD group (Baumann et al, 1999a, b;Bielau et al, 2005). Other studies have shown differentialneuron density and morphology, which appear to be layer-and cell-type specific. In a DLPC analysis using Nissl-stained sections from BPD and MDD brains, significantreductions in the density and soma size of some neurons inspecific layers were observed. Decreased soma size could becorrelated with less extensive or less active axodendritictrees, which could not be observed using standard Nisslstaining procedures. The density of neurons with largecell somas (most likely corresponding to glutamatergicpyramidal neurons) was significantly decreased in layers IIIand V (Rajkowska, 2000, 2002), and the size of neurons inlayer V and VI was found to be reduced (Cotter et al,2002b). In different anterior cingulate subregions of BPDindividuals, reduced cell densities were found in layers III–VI, nonpyramidal cell density was decreased in layer II, andneuron size was increased in layer V and in layer IInonpyramidal cells (discussed in Goodwin and Jamison,2007). In the hippocampus, decreased pyramidal cell somasize was found in the CA1 region in BPD (Liu et al, 2007).
GABAergic interneurons can be classified by theirimmunoreactivity for the calcium-binding proteins calbin-din, parvalbumin, and calretinin. Immunohistochemicalstudies in BPD patients using these markers founddecreased levels of calbindin- and parvalbumin-positivecells in the anterior cingulate cortex (Benes and Berretta,2001; Cotter et al, 2002a), the hippocampus (Benes et al,
2001), and reductions of parvalbumin-positive cells in theentorhinal cortex (Pantazopoulos et al, 2007). Clustering ofparvalbumin-positive neurons was increased in the anteriorcingulate cortex.
Most recently, Bezchlibnyk et al (2007) assessed the sizeand density of both neuronal and glial cells in discreteamygdalar nuclei in post-mortem sections from subjectswith MDD, BPD, schizophrenia, and from nonpsychiatriccontrol subjects. They found significantly decreased neuronsomal size in the lateral amygdalar nucleus (LAN) and theaccessory basal parvocellular nucleus in subjects with BPDrelative to control subjects. These changes in cellularmorphology were most prominent in the LAN in sectionsobtained from the left hemisphere (Bezchlibnyk et al, 2007).
Glial cells. In addition to the aforementioned abnormalitiesobserved in neuronal populations, prominent glial cellabnormalities have been identified in post-mortem BPDbrains. Glial cell density appears decreased in frontalcortical areas (Ongur et al, 1998; Rajkowska, 2000, 2002;Rajkowska et al, 2001), and is accompanied by increasednucleus size. Increased glial cell nuclei size has also beenobserved in reactive gliosis occurring over the course ofsome classic neurodegenerative diseases, but in these casesthe actual number of astrocytes markedly increases. Thisglial pathology might be more apparent in BPD individualswith a strong genetic component, because one study onlyfound striking reductions in the subgenual PFC (41%) inpatients with a clear family history of BPD (Ongur et al,1998). In the amygdala, glial cell numbers were decreased inMDD, but not in BPD. However, the only two BPD patientsin this study that had not been treated with mood stabilizers
Neurons
Pyramidal neuronF
indings on cell numbers, densities and size
more prevalent in deeper layers
Interneuron
Glia
Astrocyte Oligodendrocyte
Histopathological findings in BPD and MDD
Region specific Layer specific Cell type specific
I
II
IIa
IIb
IIc
IV
Va
Vb
VIa
VIbNucleus
accumbens
Prefrontalcortex
Cingulate Gyrus
Hippocampus
Thalamus
Amygdala
Figure 3 Histopathological findings in BPD research. Research on post-mortem brain tissue of BPD patients and cell biological findings in animal modelshave revealed several findings in areas implicated in emotion perception and control including hippocampus, amygdala, and prefrontal and cingulate cortices.
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
115
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

did show decreases in glial number, indicating thattreatment with mood stabilizers had a possible protectiveeffect (Bowley et al, 2002).
Glial cells, including astrocytes, oligodendrocytes, andmicroglia, constitute a diverse group of nonneuronal cellseach with different functions. Astrocytes play importantroles in neurotransmitter catabolism, but have also beenimplicated in synapse formation and as providers of trophicsupport to neurons. The main function of oligodendrocytesis axon myelination in the CNS. Microglia are highly mobilecells constituting the major immune cells of the brain. Thetype of glial cells most affected in BPD remains unclear.Findings in MDD have suggested that astrocytes may beuniquely affected; however, a different morphological typeof glial cell may be targeted. A proteomic study on MDDand BPD brains found disease-specific alterations in levelsof glial-fibrillary acidic protein, a protein that is abundantlyexpressed in astrocytes (Johnston-Wilson et al, 2000). Otherstudies have found reductions in oligodendrocyte number(Uranova et al, 2004), as well as in the expression of keyoligodendrocyte and myelin-related genes in the DLPC inindividuals with BPD (Tkachev et al, 2003).
Overall, cell loss and atrophy (both neurons and glia)likely represent etiologic factors as well as the consequenceof disease progression in BPD. There is almost no doubtthat these atrophic brain changes contribute to illness
pathophysiology by disrupting the circuits that mediatenormal affective, cognitive, motoric, and neurovegetativefunctioning. Furthermore, these findings suggest that theneurotrophic effects of mood stabilizers (see below) may bevery relevant to the treatment of BPD.
Post-mortem gene expression of key mitochondrialproteins in BPD. Interestingly, additional evidence ofdysregulated mitochondrial processes in BPD comes froman elegant series of recent post-mortem brain microarraystudies. Konradi et al (2004), using gene arrays thatanalyzed mRNA expression in hippocampus, studied12 558 nuclear genes analyzed in three different groups(healthy controls, BPD, and schizophrenia patients). Usingstringent statistical analysis, they found that the expressionof only 43 genesF42% of which coded for mitochondrialproteinsFdecreased in BPD compared with schizophrenia.Notably, this gene expression was involved in regulatingoxidative phosphorylation in the mitochondrial innermembrane, which included subunits of complexes I andthe ATP-dependent process of proteasome degradation((NADH dehydrogenase in one gene), IV (cytochrome coxidase in one gene), and V (ATP synthase in five genes)).Based on these findings, and additionally on the decreasedexpression of the enzymes glutamic acid decarboxylase 67and somatostatin, the authors suggested that a subset ofhippocampal interneurons is abnormal in BPD, particularlythose that affect mitochondrial energy metabolism (Heckerset al, 2002; Konradi et al, 2004). Most recently, Buttner et al(2007) investigated whether or not apoptosis is associatedwith GABAergic interneurons in the anterior cingulatecortex in schizophrenia and BPD. A double-labelingtechnique using the Klenow method of in situ end-labelingof single-stranded DNA breaks was combined with an insitu hybridization localization of mRNA for the 67 kDaisoform of glutamate decarboxylase (GAD67); an increase inKlenow-positive, GAD67-negative nuclei was observed inlayer V/VI of patients with BPD, but not schizophrenics(Buttner et al, 2007). These findings suggest that there ismore DNA fragmentation in cells showing no detectableGAD67 mRNA in patients with BPD than in patients withschizophrenia or controls, and further, that non-GABAergiccells may be selectively vulnerable to oxidative stress inpatients with BPD.
Nevertheless, interpreting the results of post-mortembrain studies requires caution due to the numerouspotentially confounding factors (including ante-mortemmedication history and/or substance abuse, post-morteminterval, and cause of death). Notably, a study usinglymphoblastoid cell lines from BPD patients and healthycontrols reported a decreased expression level of NDUFV2gene (a nuclear-encoded mitochondrial complex I subunitgene) in patients with bipolar I disorder (Washizuka et al,2003). Finally, a whole-genome association study from theWellcome Trust in the United Kingdom (Wellcome TrustCase Control Consortium, 2007) investigated 2000 bipolarsubjects and 3000 controls. This study found a highlysignificant association between complex I and BPD;together with the data demonstrating that mood stabilizersenhance mitochondrial function (see below), these datasuggest that mitochondrially mediated plasticity may be
Table 2 Post-mortem Morphometric Brain Studies in MoodDisorders Demonstrating Cellular Atrophy and/or Loss
Reduced volume
Cortical thickness of rostral orbitofrontal cortex in MDD
Laminar cortical thickness in layers III, V, and VI in subgenual anterior cingulatecortex in BPD
Volume of subgenual prefrontal cortex in MDD and BPD
Volumes of nucleus accumbens and basal ganglia in MDD and BPD
Parahippocampal cortex size in suicide
Reduced neuronal size and/or density
Neuronal size in layer V and VI in prefrontal cortex in MDD and BPD
Pyramidal neuronal density, layers III and V in dorsolateral prefrontal cortex inBPD and MDD
Neuronal density and size in layers II–VI in orbitofrontal cortex in MDD
Neuronal density in layers III, V, and VI in subgenual anterior cingulate cortexin BPD
Neuronal size in layer VI in anterior cingulate cortex in MDD
Layer-specific interneurons in anterior cingulate cortex in BPD and MDD
Nonpyramidal neuronal density in layer II in anterior cingulate cortex in BPD
Nonpyramidal neuronal density in the CA2 region in BPD
Reduced glia
Density/size of glia in dorsolateral prefrontal cortex and caudal orbitofrontalcortex in MDD and BPD
Glial cell density in layer V in prefrontal cortex in MDD
Glial number in subgenual prefrontal cortex in familial MDD and BPD
Glial cell density in layer VI in anterior cingulate cortex in MDD
Glial cell counts, glial density, and glia : neuron ratio in amygdala in MDD
Abbreviations: MDD, major depressive disorder; BPD, bipolar disorder.
Cellular plasticity cascades in BPDRJ Schloesser et al
...............................................................................................................................................................
116
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

integral to BPD. Indeed, Stork and Renshaw (2005) havealso posited that the many facets of the complex neurobio-logy of BPD can be fit into a more cohesive bioenergetic andneurochemical model. Specifically, they propose that theexistence of mitochondrial dysfunction in BPD involvesimpaired oxidative phosphorylation, a resultant shifttoward glycolytic energy production, a decrease in totalenergy production and/or substrate availability, and alteredphospholipid metabolism (Stork and Renshaw, 2005).
Subcellular markers for neuroplasticity. Further studieshave examined protein and mRNA levels of moleculesimplicated in neuronal and glial cell function. Severalindicator proteins for synaptic number and function, suchas GAP-43 and the synapsins, have been studied in the post-mortem brains of BPD patients. The neuronal plasticitymarker GAP-43 is highly expressed in axonal growth conesduring development (Strittmatter, 1992) and is implicatedin the regulation of axonal morphology and synapticplasticity in the mature brain (Benowitz et al, 1990).Expression of GAP-43 increases after in vitro antidepressanttreatment (Chen et al, 2003) and decreased levels of thisprotein have been associated with depression and suicide(Hrdina et al, 1998). In BPD, GAP-43 protein levels arereduced in cingulate cortex (Eastwood and Harrison, 2001)and hippocampus (Tian et al, 2007).
Several proteins implicated in the machinery regulatingsynaptic vesicle and neurotransmitter release have beenstudied. Synapsins are a family of proteins that bindsynaptic vesicles to the cytoskeleton, thereby preventingtheir transport to the presynaptic membrane and subse-quent neurotransmitter release (Hilfiker et al, 1999).Synaptic vesicle docking and actual neurotransmitterrelease are regulated by a complex of proteins that includesSNAP-25, syntaxin, and synaptobrevin. Synaptophysin isanother protein of unknown function that might interactwith synaptobrevin during neurotransmitter release. In BPDpost-mortem brain samples, reductions of synapsins(Vawter et al, 2002) in the hippocampus, and increases in
SNARE complex proteins in the DLPC have been found(Scarr et al, 2006). Another study showed decreases ofsynaptobrevin and synaptophysin in visual associationcortex (Beasley et al, 2005). Finally, mRNA levels ofnetrinsFa class of proteins implicated in axon guidan-ceFwere found to be reduced in the CA3 region of thehippocampus and the entorhinal cortex of individuals withBPD (Eastwood and Harrison, 2007).
EVIDENCE THAT CELLULAR PLASTICITYCASCADES ARE THE TARGETS OFMOOD-STABILIZING AGENTS
Despite substantial advances in mood disorder pharma-cotherapeutics in the past 20 years, lithium remains themost effective therapy for BPD. However, the direct targetsand mechanisms of its action remain elusive. Several directtargets of lithium have been identified and extensivelystudied (reviewed in Gould and Manji, 2005; Gurvich andKlein, 2002; Li et al, 2002). These include inositolmonophosphatase (IMPase) and structurally related phos-phomonoesterases, phosphoglucomutase, and glycogensynthase kinase-3 (GSK-3). Given the broad range ofbiological functions for pathways regulated by phospho-monoesterases and GSK-3, even this limited set of directlithium targets is likely to regulate numerous and diversedownstream effectors. Here, we discuss these two best-characterized primary direct targets, as well as thosedownstream targets (ie those consistent with a clinicaltemporal profile) that have been demonstrated to exertmajor effects on neural plasticity.
Inositol Monophosphatase
Inositol monophosphatase dephosphorylates inositolmonophosphate, an intermediate in the turnover of thesecond messenger inositol 1,4,5-tris-phosphate (IP3) toinositol, and is inhibited by lithium at concentrations
Cell membraneCell membrane
neuronal growth cone spreading, hippocampal LTP,stress induced cognitive impairments
IP3 and DAG mediated signaling
DAG
GPCR
IP3
PA
PKCIMP PIP2
MARCKS
Ras PIP4 ...
DGK
IMPase
Lithium
PLCMyoinositol
IPPase
PKC Inhibitors:Anti-manic
Figure 4 Phosphatidylinositol signaling pathway. Therapeutic levels of lithium directly inhibit several key enzymes that regulate recycling of inositol-l,4,5-trisphosphate (IP3). Inositol-monophosphatase (IMPase) is the final, common step for conversion of monophosphorylated inositols into myo-inositol, andtherefore inhibition of IMPase can reduce the level of myo-inositol. The inositol depletion hypothesis proposes that inhibition of this step may interfere withthe synthesis of phosphatidylinositol (PI), although this has not been demonstrated in vivo. The PI signaling cascade starts with surface receptor-mediatedactivation of phospholipase C (PLC). Activated PLC catalyzes the hydrolysis of PIP2 to diacylglycerol (DAG) and IP3. DAG activates protein kinase C (PKC)that, among many other functions, activates myristoylated aknine-rich C kinase substrate (MARCKS). Lithium and valproate both decrease levels ofphosphorylated and total MARCKS.
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
117
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

similar to those achieved clinically in the treatment ofBPD (Berridge et al, 1989; Hallcher and Sherman, 1980).The inositol depletion hypothesis (Figure 4) proposes thatlithium interferes with the regeneration of inositol and,under conditions where inositol limits phosphatidylinositol(PI) synthesis, depletes the cell of PI (Berridge et al, 1989).Because PI is an obligate precursor for phosphatidylinositolbisphosphate (PIP2), the hypothesis posits that inhibition ofIMPase could disrupt PIP2/IP3-mediated signaling.
Several other enzymes involved in inositol turnover,including the inositol polyphosphate 1-phosphatase (IP-Pase) and 30 (20) phosphoadenosine-50-phosphate (PAP)phosphatase (which also hydrolyzes inositol-l,4-bispho-sphate (Lopez-Coronado et al, 1999; Spiegelberg et al,1999)), have been postulated as targets for the therapeuticaction of lithium in BPD. IMPase (encoded by IMPA1 andIMPA2), IPPase (encoded by INPP1), and PAP phosphataseare all inhibited by lithium at or below therapeuticallyrelevant concentrations. Loss of function mutations in thegenes encoding IPPase (ipp in Drosophila) and IMPase (ttxin C. elegans) demonstrate synaptic phenotypes that arephenocopied by lithium treatment, strongly supportingthese inositol phosphatases as targets of lithium in thesesettings (Acharya et al, 1998; Tanizawa et al, 2006).
Lithium has been shown to reduce inositol levels(Maslanski et al, 1992; O’Donnell et al, 2000; Shaldubinaet al, 2006) and to inhibit IMPase in vivo (Hedgepeth et al,1997), although the magnitude of inhibition is generallymodest. Furthermore, the effects of lithium in severalexperimental systems can be reversed by adding millimolarconcentrations of exogenous myoinositol, which has beeninterpreted to indicate reversal of inositol depletion. Forexample, lithium and other drugs used to treat BPD,including valproate (VPA) and carbamazepine, can stabilizegrowth cones, thereby increasing growth cone spreading insensory neurons in culture (Williams et al, 2002). Theseeffects were reversed by the addition of 1 mM inositol,suggesting that these drugs may act through inositoldepletion (although inositol levels were not actuallymeasured). Exogenous inositol also corrects the thermotaxisdefect observed in ttx/IMPase mutants in C. elegans(Tanizawa et al, 2006).
Although the inositol depletion hypothesis provides anelegant potential mechanism to explain lithium action, a fewimportant issues remain unresolved. First, therapeuticlithium does not deplete PIP2 in vivo, an essentialcomponent of the hypothesis, nor does it reduce IP3 invivo (Dixon et al, 1994). Second, although lithium inhibitsIMPase in vivo (Hedgepeth et al, 1997; Maslanski et al, 1992;O’Donnell et al, 2000; Shaldubina et al, 2006), the inhibitionis incomplete, and it is not clear that the modest reductionin inositol is sufficient to impair PI or PIP2 synthesis;indeed, marked reduction in inositol through other meansdoes not reduce PI levels in vivo in rodents (Berry et al,2004), and potent alternative IMPase inhibitors do notmimic lithium action (Klein and Melton, 1996). Thus, inmice lacking the sodium myoinositol cotransporter 1 gene(SMIT1), a 92% reduction of intracellular inositol in fetalbrain had no effect on PI levels (Berry et al, 2004). This islikely because basal intracellular inositol concentrations inthe brain are in the 4–8 mM range, which is orders ofmagnitude above the limiting concentrations that may be
found in cultured cells and neuronal slices. The 25%reduction in inositol caused by lithium in vivo therefore isunlikely to affect global PIP2 or IP3 levels.
An interesting hypothesis that has not been extensivelyexplored posits that lithium may affect functions of inositolor inositol phosphates that are independent of PI and PIP2
(Lee et al, 2007; Odom et al, 2000; Seeds et al, 2005;Shaldubina et al, 2002). In addition, specific regions of thebrain or compartments within the cell could have uniquelylow basal inositol concentrations in vivo, rendering theseregions exquisitely sensitive to small reductions in inositol.Alternatively, inhibition of other lithium-sensitive phos-phomonoesterases may be responsible for the therapeuticresponse in BPD, although these potential targets have notyet been as thoroughly explored as IMPase and IPPase. Inspite of these controversies, IMPase and structurally relatedmolecules remain important potential targets for lithiumaction in BPD.
Notably, a recent whole-genome association study of BPDhas further implicated the overall phosphoinositide/proteinkinase C (PKC) pathway. This study identified a molecule inthe diacylglycerol (DAG) limb of the phosphoinositidecascade as a putative BPD susceptibility gene. This study,utilizing North American bipolar pedigrees as a test sample,and German bipolars as the replication sample found astatistically highly significant association (pB10�8) ofdiacylglycerol kinase Z (DGKH) with BPD (Baum et al, inpress). DAG is the major activator of PKC, and these geneticfindings receive indirect support from the preclinical datasuggesting that PKC is a target for antimanic agents(reviewed in Goodwin and Jamison, 2007), and thepreliminary clinical efficacy of nonslective PKC inhibitorsin the treatment of mania (Bebchuk et al, 2000; Zarate et al,2007).
Glycogen Synthase Kinase-3
Glycogen synthase kinase-3 is also inhibited by clinicallyrelevant concentrations of lithium, unlike most otherprotein kinases (Klein and Melton, 1996; Davies et al,2000) (Figure 5). First identified in mammals as an inhibitorof glycogen synthase, GSK-3 is a ubiquitous, constitutivelyactive, multisubstrate serine/threonine kinase encoded bytwo closely related genes, Gsk-3a (51 kDa) and Gsk-3b(46 kDa) (Woodgett, 1990). As a key component of manysignaling pathways including insulin, neurotrophin, andWnt pathways, GSK-3 plays a critical role in multiplecellular processes, including metabolism, proliferation,differentiation, axonogenesis and synaptogenesis, develop-ment, and apoptosis (Cohen and Frame, 2001; Doble andWoodgett, 2003; Gould et al, 2006; Gurvich and Klein, 2002;Hall et al, 2000; Huang and Klein, 2006; Jope and Johnson,2004; Kim and Kimmel, 2000; Kim et al, 2006; Salinas, 1999;Shaltiel et al, 2007).
As a general rule, GSK-3 antagonizes canonical signalingpathways, including the insulin and Wnt pathways. Forexample, GSK-3 phosphorylates and inhibits glycogensynthase, a downstream effector of insulin action; further-more, GSK-3-mediated phosphorylation of b-catenin,a downstream component of the canonical Wnt pathway,causes rapid degradation of b-catenin. Thus, to activatethese downstream components, the pathways must inhibit
Cellular plasticity cascades in BPDRJ Schloesser et al
...............................................................................................................................................................
118
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

GSK-3. This explains why lithium can activate glycogensynthesis, Wnt/b-catenin-dependent transcription, andother pathways normally inhibited by GSK-3.
The best-characterized mode of GSK-3 inhibition ismediated by N-terminal phosphorylation of GSK-3. Insulinactivates the protein kinase Akt/PKB, which directlyphosphorylates GSK-3 (at serine-21 in GSK-3a and serine-9in GSK-3b) converting the N-terminal sequence of GSK-3into a pseudosubstrate that autoinhibits the enzyme (Frameet al, 2001; Doble and Woodgett, 2003). Akt activation viaother receptor tyrosine kinases (such as neurotrophinreceptors) or through G-protein-coupled receptors (suchas serotonin receptors) also causes phosphorylation andinhibition of GSK-3. Several other protein kinases (includ-ing PKA, PKC, p70S6kinase, and p90rsk) can also phosphor-ylate GSK-3 in vitro and may inhibit GSK-3 in vivo (Dobleand Woodgett, 2003).
Wnt signaling also inhibits GSK-3 enzymatic activity(Cook et al, 1996; Ruel et al, 1999), preventing b-catenindegradation, which in turn activates transcription of Wnt-dependent target genes. Wnt-mediated inhibition of GSK-3is independent of serine-21/9 phosphorylation, as micelacking serine-21 and -9 have no defects in Wnt signaling(McManus et al, 2005). Instead, the pool of GSK-3 involvedin Wnt signaling is associated with the Axin/APC complexand is inaccessible to N-terminal protein kinases (Dinget al, 2000). This has been proposed as a mechanism toinsulate Wnt signaling from crosstalk with other GSK-3-regulated signaling pathways.
Lithium directly inhibits GSK-3 at therapeutically relevantconcentrations in vitro (Klein and Melton, 1996) and in vivoin diverse cell types, including cultured neurons and rodentbrain (Gould et al, 2004a; Hedgepeth et al, 1997; Hong et al,1997; Lovestone et al, 1999; Munoz-Montano et al, 1997;Noble et al, 2005; O’Brien et al, 2004; Stambolic et al, 1996).Many of the known effects of lithium can be explained interms of GSK-3 inhibition based on parallels to otherphysiological (ie insulin- or Wnt-induced effects), genetic(Gsk-3 loss of function), or pharmacological (ie smallmolecule inhibitors of GSK-3) modes of GSK-3 inhibition,including the effects on glycogen synthesis, early develop-ment (Klein and Melton, 1996), neurogenesis, neuronal
survival (Chalecka-Franaszek and Chuang, 1999; Chuang,2004; Gould et al, 2006; Li et al, 2002), and behavior(Beaulieu et al, 2004; Gould et al, 2004b; Kaidanovich-Beilinet al, 2004; O’Brien et al, 2004).
For example, lithium increases neuronal growth cone area(Goold et al, 1999; Hall et al, 2000; Lucas et al, 1998;Williams et al, 2002), alters synaptogenesis (Hall et al, 2000;Salinas, 1999), and stimulates hippocampal neurogenesis(Chen et al, 2000); all of these effects mimic Wnt signaling(Burden, 2000; Lie et al, 2005; Salinas, 1999). Furthermore,N-terminal-phosphorylated GSK-3b localizes to growthcones, and agents that induce growth cone collapse, suchas sema-3A and lysophosphatidic acid, also induce depho-sphorylation of GSK-3 (Eickholt et al, 2002; Sayas et al,1999).
GSK-3 also regulates neuronal polarity in developinghippocampal neurons. Local GSK-3 inactivation specifiesaxon site formation, and global GSK-3 inhibition inducesmultiple axons (Jiang et al, 2005; Kim et al, 2006;Yoshimura et al, 2005). The effects of lithium on growthcone stability and on neuronal polarity may be mediated viachanges in phosphorylation of microtubule-associatedproteins that are known targets of GSK-3, including MAP-1B, APC, and CRMP-1 (Cole et al, 2004; Goold et al, 1999;Lucas et al, 1998; Yoshimura et al, 2005; Zhou et al, 2004,2005). Taken together, these observations indicate thatinhibition of GSK-3 by lithium may regulate multipleaspects of axonal morphogenesis.
However, inhibition of GSK-3 by lithium (IC50 B1–2 mMin vitro) is achieved at the higher end of its therapeuticrange (0.5–1.5 mEq/l), raising some questions aboutwhether this level of inhibition is sufficient to havesignificant biological effects. On the other hand, IC50 isstrongly affected by in vitro assay conditions; lithiuminhibits GSK-3 competitively with respect to magnesiumand therefore inhibition is reversed by raising the [Mg2 + ] tosuperphysiological levels (Gurvich and Klein, 2002; Ryvesand Harwood, 2001). Intracellular [Mg2 + ] is significantlylower than that generally used for in vitro kinase assays, andwhen GSK-3 is assayed at physiological [Mg2 + ], theobserved IC50 is 0.8–1.0 mM. Nevertheless, this degreeof inhibition might still be insufficient to explain the
Frizzled Receptor TrkB ReceptorWnt
?
Cell membrane
Nuclear membrane
DNA
Transcription
Proliferation, DifferentiationAxonogenesis, Synaptogenesis,
Development, Apoptosis
BDNF
AktOther targets of GSK3:• Micotubule associated proteins• Tau• Glycogen Synthase • Rev-erb alpha
GSK-3
β-catenin
GSK-3
Lithium
Figure 5 Wnt pathway and GSK-3. MSDs inhibit glycogen synthase kinase 3 (GSK-3). In the Wnt signaling pathway, Wnt glycoproteins interact with thefrizzled family of receptors to stimulate the disheveled-mediated inactivation of GSK-3. Inhibition of GSK-3 prevents b-catenin phosphorylation and therebyinhibits degradation of this molecule that can act as a transcription factor binding to lef/tcf. Wnt proteins are implicated in the regulation of neuronmorphology, neurotransmission, and synaptogenesis.
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
119
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

mood-stabilizing effects of lithium, as clinical efficacy canbe observed at the low end of the response window.
To address this issue, Jope and colleagues have proposedthat a secondary mode of inhibition enhances the directinhibition by GSK-3 (De Sarno et al, 2002). Thus, inaddition to directly inhibiting GSK-3, lithium may alsoinduce N-terminal phosphorylation, enhancing its moredirect inhibitory effects (as discussed above) (Bhat et al,2000; Chalecka-Franaszek and Chuang, 1999; De Sarno et al,2002; Hall et al, 2002; Lochhead et al, 2006; Noble et al,2005; Roh et al, 2005; Song et al, 2002; Zhang et al, 2003).Proposed mechanisms of secondary inhibition (Figure 6)include inhibition of the phosphatase that dephosphorylatesGSK-3 (which is itself regulated by GSK-3) (Zhang et al,2003), and activation of Akt through an unknown mechan-ism (Chalecka-Franaszek and Chuang, 1999). In either casethis indirect inhibition would arise through direct inhibi-tion of GSK-3 itself, shown with alternative GSK-3inhibitors and genetic disruption of Gsk-3b, revealing apositive autoregulatory loop for GSK-3 (Zhang et al, 2003)(Figure 6). In support of this indirect mode of inhibition,evidence for increased N-terminal phosphorylation of GSK-3in peripheral blood mononuclear cells has been reported inpatients treated with lithium (Li et al, 2007). In addition,lithium prevents an activating autophosphorylation ontyrosine of newly translated GSK-3, suggesting an additionalmode of inhibition that might show a delayed effect, as itwould only affect protein translated in the presence oflithium and therefore would not be manifested until existingGSK-3 had turned over sufficiently (Lochhead et al, 2006).
Behavioral Effects of VPA and Lithium that maybe Mediated by the GSK-3 and/or IMPaseCascades
Preclinical work has found that chronic lithium treatmentin mice reduces immobility time in the forced swim test(FST) and reduces exploratory behavior in the hole-boardapparatus without affecting overall activity in the open fieldtest (OFT) or other general measures of the state of theanimal (O’Brien et al, 2004). Similarly, mice treated withVPA for 5–10 days display reduced immobility in the FST(Semba et al, 1989) and reduced exploratory behaviorwithout an accompanying reduction in locomotor activityor rearing (File and Aranko, 1988; Rao et al, 1991). Lithiumand VPA also attenuate amphetamine- and chlordiazep-oxide-induced hyperlocomotion (Cao and Peng, 1993;Murphy, 1977). The hypothesis that these behavioral effectscould be mediated by direct or indirect inhibition of GSK-3is supported by pharmacological and genetic evidence. Forinstance, two structurally distinct GSK-3 inhibitorsFAR-A014418 (Gould et al, 2004b) and the peptide L803-mts(Kaidanovich-Beilin et al, 2004)Freduce immobility in theFST and attenuate amphetamine-induced hyperactivity. Inaddition, GSK-3 overexpression reportedly increases activ-ity (although this transgenic line also displayed compensa-tory changes in Akt signaling that may complicate analysisof their behavior (Prickaerts et al, 2006)). The specificbehavioral responses to lithium and other GSK-3 inhibitorsare observed in animals lacking one copy of the Gsk-3b gene(Beaulieu et al, 2004; O’Brien et al, 2004), establishingstrong genetic and pharmacological support that thesebehavioral effects of lithium are mediated by inhibition ofGSK-3.
In contrast, genetic perturbations that reduce inositol inrodent brain do not affect lithium-sensitive behaviors. Asdiscussed above, lithium and VPA partially reduce in vivobrain inositol levels, and in invertebrate model systems,mutations that interfere with inositol turnover cause cleardefects in synaptic function, including at the neuromuscularjunction in Drosophila and in synaptic function regulatingthermotaxis behavior in C. elegans. However, in adult miceheterozygous for the inositol transporter SMIT1, inositollevels in the brain are reduced to an even greater extent (33–37% reduction) than in lithium-treated wild-type siblings(22–25%); nevertheless, this reduction in inositol has noeffect on the FST (Shaldubina et al, 2006) or other lithium-sensitive behaviors (Shaldubina et al, 2007), indicating thatinositol reduction is not responsible for the behavioraleffects of lithium in mice. Knockout of the IMPA2 gene inmice reportedly also did not yield lithium-like behaviors,but apparently these mice also did not show a reduction inbrain inositol, perhaps because of redundancy with IMPA1(Cryns et al, 2007).
Reduction of GSK-3 activity by lithium is predicted toincrease b-catenin signaling and activation of Wnt-depen-dent gene expression. This was confirmed by examining atransgenic mouse line expressing a Wnt reporter (BAT-Gal);when these reporter mice were treated with lithium (1 mEq/l)or VPA, Wnt/b-catenin reporter activity enhanced specifi-cally in the dentate gyrus (DG), medial amygdala, and,weakly, in the hypothalamus (O’Brien et al, 2004; Freidman,O’Brien, and Klein, unpublished data). Furthermore,
GSK-3
Inhibition of protein kinase:GSK-3 phosphorylation ofIRS-1 may reduce Aktactivation. Lithium would“activate” Akt under theseconditions.
Activation of protein phosphatase:GSK-3 phosphorylation of I-2 activatesPP1. Lithium prevents activation of PP1.
AKT P
P
P
P
GSK-3
I–2
IRS–1 PI3K
PP1
Figure 6 Glycogen synthase kinase 3 (GSK-3) autoregulatory loop. Inaddition to directly inhibiting GSK-3, lithium (as well as other inhibitors)induces N-terminal phosphorylation of GSK-3. This inhibitory phosphoryla-tion may enhance the effect of direct inhibitors and appears to be achievedboth through activation of Akt and through inhibition of proteinphosphatase 1 (PP1). These observations reveal an autoregulatory loopin which GSK-3 maintains itself in an active state, which is then interruptedby direct inhibition of GSK-3 (‘P’ indicates phosphorylation; IRS-1, insulinreceptor substrate-1; PI3K, phosphatidylinositol-3 kinase; PP1, proteinphosphatase 1; I-2, PP1-specific inhibitor-2; - -| indicates inhibitory step; and- -4 indicates activating step).
Cellular plasticity cascades in BPDRJ Schloesser et al
...............................................................................................................................................................
120
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

overexpression of a stabilized form of b-catenin leads toreduced immobility in the FST (Gould et al, 2007),paralleling the effects of lithium, other GSK-3 inhibitors,and Gsk-3b+ /� mice.
Another interesting observation concerns circadian per-iod lengths, which are shortened in patients with BPD andlengthened with lithium treatment. This behavioral effect isparalleled by changes in GSK-3 phosphorylation in bothmice and cultured cells. In Drosophila, both lithium andVPA can prolong the circadian period (Dokucu et al, 2005;Kaladchibachi et al, 2007; Padiath et al, 2004) (reviewed inGould and Manji, 2005; Martinek et al, 2001; McClung,2007). Moreover, a dominant-negative mutation in themouse CLOCK gene results in hyperactivity, decreasedsleep, increased mobility in the FST, and heightened rewardvalue for cocaine or sucrose. These effects have beeninterpreted to indicate a manic state, and interestingly,these alterations can be reversed by chronic lithiumtreatment (Roybal et al, 2007). The Bmal1/CLOCK complexregulates expression of the nuclear hormone receptor rev-erba; the rev-erba protein is in turn stabilized by GSK-3phosphorylation and represses Bmal1 expression. Lithiumtherefore destabilizes rev-erba and results in activation ofBmal1 (Yin et al, 2006), which could play a central role inthe circadian response to lithium.
Because both inositol phosphates and GSK-3 affectmultiple downstream pathways, inhibition of IMPase andGSK-3 could affect overlapping targets, thereby yieldingsimilar cellular or morphological readouts in specificsettings, such as growth cone spreading/collapse orneuronal survival. In support of this idea, interesting recentwork from Greenberg and colleagues showed that GSK-3 isrequired for optimal myoinositol-3 phosphate synthaseactivity and de novo inositol biosynthesis (Azab et al, 2007)in budding yeast. If similar regulation is present inmammals, this could provide an additional mechanism forinositol reduction caused by lithium, and, more generally,suggest interplay between GSK-3 and inositol pathways.
Cellular Signaling Cascades Converge toRegulate Synaptic Plasticity and BehavioralPlasticity
As discussed previously, it is now clear that certainintracellular signaling cascades play important roles in thepathophysiology and treatment of severe mood disorders.An important consideration to address at this point is howcan changes in intracellular molecules bring about complexbehavioral changes? These signaling cascades undoubtedlyconverge to regulate synaptic plasticity, and therebyinformation processing in critical circuits mediating theaffective, cognitive, motoric, and somatic manifestations ofmood disorders.
In this context, it is now clear that modification of thelevels of synaptic AMPA receptors, in particular by receptorsubunit trafficking, insertion, and internalization, is acritically important mechanism for regulating various formsof synaptic plasticity (Malinow and Malenka, 2002). Thus,through phosphorylation of specific sites on AMPA receptorsubunit GluR1, GluR1 trafficking is regulated by proteinkinase A (PKA), Ca2 + /calmodulin-dependent protein ki-nase II, and PKC (Du et al, 2004b, 2006). Phosphorylation/
dephosphorylation of the receptor subunits regulates boththe intrinsic channel properties of the receptor and theinteraction of the receptor with associated proteins thatmodulate the membrane trafficking and synaptic targetingof the receptor. N-methyl-D-aspartate (NMDA) receptorsubunits are regulated by similar mechanisms. It wastherefore postulated that the effects of mood stabilizers/antidepressants ultimately converge to regulate AMPA andNMDA synaptic transmission. The regulation of transmis-sion at the synapse may be mediated by changes inneurotransmitter levels, receptor subunit phosphorylation,surface/cellular levels or receptors, and conductancechanges, among others. For the purposes of generaldiscussion, we use the term ‘throughput,’ as we discussthe studies that identified AMPA and NMDA receptorsas targets for the actions of mood stabilizers andantidepressants.
Chronic lithium and VPA regulate surface/synaptic GluR1and GluR2 levels. A series of studies was undertaken to testthe hypothesis that lithium and VPA’s effects on intracel-lular signaling cascades converge to regulate surface and/orsynaptic AMPA receptors (Du et al, 2003, 2004a, b). Usingthree independent assays, it was demonstrated that chronictreatment of rats with therapeutically relevant concentra-tions of lithium or VPA reduced hippocampal synaptosomalGluR1 and GluR2 levels via a reduction of surface GluR1and GluR2 distribution onto the neuronal membrane (Duet al, 2003, 2004a, b). In addition, both agents induced adecrease in GluR1 phosphorylation at a specific PKA site(GluR1p845) known to facilitate AMPA receptor insertionand opening of the sodium channel. Notably, in strikingcontrast to the effects observed with lithium and VPA,antidepressants, psychostimulants, dopamine agonists, andsleep deprivation have all been shown to increase phos-phorylation and/or synaptic levels of GluR1 receptors (Duet al, 2004a, 2007). Complementary behavioral studiessuggest that attenuating hippocampal AMPA throughputattenuates manic-like behaviors (Du et al, submitted).
Anticonvulsants with a predominantly antidepressantprofile enhance surface/synaptic GluR1 and GluR2receptors. Anticonvulsants are being increasingly used inthe treatment of BPD. They were initially thought to exertpredominantly antimanic effects by suppressing neuronalexcitability; however, recent data suggest that some antic-onvulsantsFnotably lamotrigine and riluzoleFmay havean antidepressant profile (Zarate et al, 2005; Zarate et al,2006b). It was found that lamotrigine and riluzolesignificantly enhanced the surface expression of GluR1and GluR2 in a time and dose-dependent manner incultured hippocampal neurons; by contrast, the antimanicanticonvulsant VPA significantly reduced surface expres-sion of GluR1 and GluR2 (Du et al, 2007). Concomitant withthe GluR1 and GluR2 changes, the peak value of depolarizedmembrane potential evoked by AMPA was significantlyhigher in lamotrigine- and riluzole-treated neurons, sup-porting the surface receptor changes. Phosphorylation ofGluR1 at the PKA site (S845) was enhanced in bothlamotrigine- and riluzole-treated hippocampal neurons,but reduced in VPA-treated neurons. In addition, lamo-trigine and riluzole, as well as the traditional antidepressant
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
121
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

imipramine, increased GluR1 phosphorylation at GluR1(S845) in the hippocampus after chronic in vivo treatment(Du et al, 2007).
Ketamine’s antidepressant effects involve NMDA/AMPAreceptor interactions. Building upon these preclinical data,recent clinical trials have investigated the clinical effects ofglutamatergic agents in subjects with mood disorders. Thesestudies have demonstrated effective and rapid antidepres-sant action of glutamatergic agents, including ketamine, anNMDA receptor antagonist (Zarate et al, 2006a, b). It wasfound that a single intravenous dose of the noncompetitiveNMDA antagonist ketamine produced a rapid (within 2 h),robust (70% response on day 1) and relatively sustained(approximately 1 week) antidepressant effect in patientswith treatment-resistant major depression (Zarate et al,2006a). Preclinical studies have revealed that in addition toits antidepressant effects in humans ketamine producesantidepressant-like behavioral effects in rodent models. Asingle administration of ketamine facilitated animal recov-ery from behavioral despair within 24 h in the learnedhelplessness paradigm. A single administration of ketaminealso reduced animal immobility in the FST, with effectslasting for at least 2 weeks. A selective NR2B antagonist alsoexerted antidepressant-like effects (Maeng et al, 2007).Pretreatment with NBQX, an AMPA receptor antagonist,attenuated both ketamine-induced antidepressant-like be-havioral alterations and regulation of hippocampal phos-phorylations of GluR1 AMPA receptors at serine 845(Maeng et al, 2007). These and other data have led to thehypothesis that alterations in neural plasticity in criticallimbic and reward circuits, mediated by increasing thepostsynaptic AMPA to NMDA throughput, may represent aconvergent mechanism for antidepressant action (Zarateet al, 2006b). This line of research holds considerablepromise for developing new treatments for both MDDand BPD.
Indirect, Long-Term Targets of Mood-StabilizingAgents Involved in Neuroplastic Processes
Several clinical observations of patients undergoing treat-ment for BPD are noteworthy when considering alterationsin neuroplastic processes as an underlying mechanism ofmood stabilizer action. These include the time course ofonset for antimanic and antidepressant effects of moodstabilizers, the efficiency of mood stabilizer maintenancetreatment in episode prevention, and observations on thetime to recurrence after treatment withdrawal (Goodwinand Jamison, 2007). The mood stabilizers discussed in thisarticle, namely lithium and the anticonvulsants, do notappear to have rapid effects (within minutes or hours), butinstead require days to weeks to achieve their maximalresponse (Goodwin and Jamison, 2007). Further support forlong-term neuroplastic adaptations comes from observa-tions regarding episode relapse after lithium cessation.Particularly when done rapidly, discontinuation of lithiummaintenance is clearly associated with increased rates ofrelapse (Davis et al, 1999; Suppes et al, 1991). Interestingly,and again consistent with longer-term neuroplastic changes,new episodes are not experienced immediatelyFas wouldbe expected for an acute withdrawal mechanismFbut,
rather, are seen approximately 3–9 months after medicationwithdrawal. The recurrence of episodes during the course ofBPD is a defining feature and occurs in a nonrandomfashion. Therefore, treatments for BPD must not onlyaddress symptoms of mania and depression during acuteepisodes, but also the underlying vulnerability predisposingto their recurrence. Indeed, maintenance treatment withmood stabilizers seems capable of preventing the onset ofnew episodes and attenuating the symptoms experiencedupon eventual relapse (Baastrup and Schou, 1967; Gyulaiet al, 2003).
REGULATION OF CELL SURVIVAL ANDRESILIENCE PATHWAYS BY MOODSTABILIZERS
Lithium and VPA Activate the ERK SignalingCascade
In view of the important role that the extracellular receptor-coupled kinase (ERK) signaling cascade plays in mediatinglong-term neuroplastic events (Figure 7), a series of studieswere undertaken to investigate the effects of lithium andVPA on this signaling cascade (Chen and Manji, 2006; Yuanet al, 2001). These studies showed that lithium and VPA, attherapeutically relevant concentrations, robustly activatethe ERK MAPK cascade in human neuroblastoma SH-SY5Ycells (Chen and Manji, 2006; Yuan et al, 2001). Recentfollow-up studies showed that similar to the effectsobserved in neuroblastoma cells in vitro chronic lithiumand VPA also robustly increase the levels of activated ERKin areas of the brain that have been implicated in thepathophysiology and treatment of BPDFthe anteriorcingulate cortex and hippocampus (Chen and Manji, 2006).
Animal behavioral studies have shown that chemicalinhibition of brain ERK pathway in rats reduces immobilityin the FST and increased locomotive/explorative activity inthe large OFT. These studies also show that ERK1 (one oftwo ERK subtypes) knockout mice have a brain region-specific functional deficit of the ERK pathway and exhibitreduced immobility in the FST, increased activity in theOFT, persistently increased home-cage wheel runningactivity for at least 30 days, and enhanced response topsychostimulants (reviewed in Chen and Manji, 2006). Veryrecent studies have therefore examined the role of the ERKpathway as a behavioral modulator in the left anteriorcingulate cortex, one of the brain regions being implicatedin the pathophysiology of mood disorders by human brainimaging and post-mortem studies; these pharmacologicaland genetic studies have shown that attenuating ERKactivity in the ACC may be associated with manic-likebehaviors (Chen and Manji, 2006).
However, it would undoubtedly be an oversimplication tosuggest that ERK inhibition in the brain always results inmanic-like behavior. Recent data from several laboratoriesclearly show that certain molecules with functions related toneuronal plasticity (eg BDNF, CREB, and ERK) can havedivergent effects on rodent behavior, depending on thebrain region manipulated (Berton et al, 2006; Duman et al,2007; Eisch et al, 2003). Notably, recent studies from theDuman (Duman et al, 2007) and Manji laboratories (Shaltiel
Cellular plasticity cascades in BPDRJ Schloesser et al
...............................................................................................................................................................
122
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

et al, 2007) raise the possibility that ERK may exertregionally selective effects on affective-like behaviors.Moreover, emerging evidence suggests that BDNF playsdifferent and perhaps opposing roles in the brain stresssystem including the hippocampus and hypothalamus–pituitary–adrenocortical axis, and the brain reward systemincluding the nucleus accumbens and the ventral tegmentalarea (Berton et al, 2006; Eisch et al, 2003). It is clear thatspecific circuits and brain regions are undoubtedly involvedin the pathophysiology of mood disorders; defining theeffects of altering-specific signaling pathways, in specificbrain regions of rodents, will allow for translational studiesof similar brain regions/functions in humans (ie studies ofhuman endophenotypes).
Bcl-2 is a Therapeutic Target for the Actions ofLithium and VPA
One of the major downstream targets of the ERK MAPKcascades is arguably one of the most important neuropro-tective proteins, bcl-2. Bcl-2 is expressed in the rodent andmammalian nervous system and is localized to the outermitochondrial membrane, endoplasmic reticulum, andnuclear membrane. It is now clear that bcl-2 is a proteinthat inhibits both apoptotic and necrotic cell death inducedby diverse stimuli (Adams and Cory, 1998; Bruckheimeret al, 1998; Merry and Korsmeyer, 1997, and referencestherein). Several cellular mechanisms are likely involved inmediating bcl-2’s protective effects, including sequesteringthe proforms of caspases, inhibiting the effects of caspaseactivation, antioxidant effects, enhancing mitochondrialcalcium uptake, and attenuating the release of calciumand cytochrome c from mitochondria (reviewed in Adamsand Cory, 1998; Bruckheimer et al, 1998; Li and Yuan, 1999;Sadoul, 1998). The role for bcl-2 in protecting neurons fromcell death is now supported by abundant evidence; bcl-2 has
been shown to protect neurons from a variety of insults invitro including growth factor deprivation, glucocorticoids,ionizing radiation, and oxidant stressors such as hydrogenperoxide, tert-butylhydroperoxide, reactive oxygen species,and buthionine sulfoxamine (Adams and Cory, 1998;Bruckheimer et al, 1998). In addition to these potent invitro effects, bcl-2 also prevents cell death in numerousstudies in vivo.
In the absence of pharmacological means of increasingCNS bcl-2 expression (until recently), all the studies havehitherto utilized transgenic mouse models or viral vector-mediated delivery of the bcl-2 gene into the CNS. In thesemodels, bcl-2 overexpression has been shown to preventmotor neuron death induced by facial nerve axotomy andsciatic nerve axotomy, to save retinal ganglion cells fromaxotomy-induced death, to protect cells from the deleter-ious effects of MPTP or focal ischemia, and to protectphotoreceptor cells from two forms of inherited retinaldegeneration; interestingly, neurons that survive ischemiclesions or traumatic brain injury in vivo show upregulationof bcl-2 (Bonfanti et al, 1996; Chen et al, 1997; Lawrenceet al, 1996; Merry and Korsmeyer, 1997; Raghupathi et al,1998; Sadoul, 1998; Yang et al, 1998, and references therein).
Overexpression of bcl-2 has also recently been shown toprolong survival and attenuate motor neuron degenerationin a transgenic animal model of amyotrophic lateralsclerosis (Kostic et al, 1997). Not only does bcl-2 over-expression protect against apoptotic and necrotic cell death,it can also promote regeneration of axons in the mamma-lian CNS, leading to the intriguing postulate that bcl-2 actsas a major regulatory switch for a genetic program thatcontrols the growth of CNS axons (Chen et al, 1997).Because bcl-2 has also recently been shown to promoteneurite sprouting, increasing CNS bcl-2 levels may repre-sent a very effective therapeutic strategy for the treatment ofmany neurodegenerative diseases (Chen et al, 1997).
Lithium
VPA
Lithium
VPA
BDNFCell membrane
Nuclear membrane
DNA
Transcription
Ras
Rsk
Raf
MEK
Erk
Mitochondrion
bcl-2
bcl-2CREB
Figure 7 Erk pathway and bcl-2. Lithium and valproate (VPA), at therapeutically relevant concentrations, robustly activate the extracellular receptor-coupled kinase (ERK) MAPK cascade. One of the major downstream targets of the ERK MAPK cascades is arguably one of the most importantneuroprotective proteins, bcl-2. Bcl-2 is expressed in the rodent and mammalian nervous system and is localized to the outer mitochondrial membrane,endoplasmic reticulum, and nuclear membrane. It is now clear that bcl-2 is a protein that inhibits both apoptotic and necrotic cell death induced by diversestimuli. Bcl-2 attenuates apoptosis by sequestering proforms of caspases, preventing the release of mitochondrial apoptogenic factors such as calcium andcytochrome c, and by enhancing mitochondrial calcium uptake.
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
123
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

Lithium and VPA Robustly Increase theExpression of bcl-2
Chronic treatment of rats with therapeutic doses of lithiumand VPA doubled bcl-2 levels in the frontal cortex, an effectprimarily due to a marked increase in the number of bcl-2immunoreactive cells in layers II and III of the anteriorcingulated cortex (Chen et al, 1999; Manji et al, 1999, 2000).Interestingly, the importance of neurons in the anteriorcingulate has recently been emphasized in neuroimagingstudies of BPD, particularly because these areas provideconnections with other cortical regions and are targets forsubcortical input (Rajkowska, 2000). Chronic lithium wasalso found to markedly increase the number of bcl-2immunoreactive cells in the DG and striatum (Manji et al,1999). Subsequent to these findings, lithium was shown toincrease bcl-2 levels in C57BL/6 mice (Chen et al, 1999), inhuman neuroblastoma SH-SY5Y cells in vitro (Manji et al,2000), and in rat cerebellar granule cells in vitro (Chen et al,1999).
Overall, the data clearly show that chronic lithiumrobustly increases levels of the neuroprotective proteinbcl-2 in areas of rodent frontal cortex, hippocampus, andstriatum in vivo, and in cultured cells of both rodent andhuman neuronal origin in vitro. Furthermore, at least incultured cell systems, lithium reduces levels of theproapoptotic protein p53. As demonstrated recently,repeated electroconvulsive shocks also significantly increaseprecursor cell proliferation in the DG of the adult monkey,an effect that appears to be due to increased expression ofbcl-2 (Perera et al, 2007). These results suggest thatstimulation of neurogenesis and enhanced expression ofbcl-2 may contribute to the therapeutic actions of ECT.
Behavioral studies have also been undertaken to deter-mine if bcl-2 plays a role in the pathogenesis and treatmentof depression (Yuan et al, unpublished data). Bcl-2 + /� miceand wild-type littermates were also treated with theantidepressant citalopram (10 mg/kg/day) acutely andchronically, and responses in the tail suspension andlearned helplessness tests were examined. In the learnedhelplessness test, bcl-2 + /� mice demonstrated a signifi-cantly higher rate of escape failures in the bcl-2 + /� mice.Furthermore, while chronic citalopram decreased escapefailures in the wild-type mice, it did not affect the bcl-2 + /�
mice. Similarly, citalopram was effective in wild-type micein the tail suspension test, but did not affect the bcl-2 + /�
mice. These data demonstrate that Bcl-2 + /� mice areinsensitive to the SSRI antidepressant citalopram in twoanimal models of depression, suggesting that some of thetherapeutic effects of antidepressants may be mediatedthrough the actions of bcl-2. In total, these observationssuggest that regulation of bcl-2-mediated plasticity is likelyto play an important role in regulating synaptic throughputin the circuitry-mediating complex behaviors (Yuan et al,unpublished data).
BAG1 (bcl-2-Associated Athanogene) is alsoa Long-Term Target for Mood Stabilizers
Recent microarray studies with stringent validating criteriaidentified bcl-2-associated athanogene (BAG1) as a targetfor the actions of mood stabilizers (Zhou et al, 2005). BAG1
interacts with glucocorticoid receptors, Bcl-2, Hsp70, andRaf, thereby regulating cell survival pathways and gluco-corticoid function. The potential role of BAG1 in regulatingaffective-like behavior was investigated in mice selectivelyoverexpressing BAG1 in the brain. BAG1-overexpressingmice displayed less anxiety on the anxiety-related tests.The mice did not differ from controls on measures ofimmobility in the FST or helplessness in the learnedhelplessness paradigm; however, the BAG1 mice showedhigher spontaneous recovery rates from the helplessnessbehavior. On mania-related tests, BAG1-overexpressingmice recovered much faster in the amphetamine-inducedhyperlocomotion test, and displayed a clear resistance tococaine-induced behavioral sensitization. BAG1-overex-pressing mice had specific hippocampal neurochemicalalterations including increased Hsp70 and decreasedFKBP51 levels. These data suggest that BAG1, a novel targetfor the actions of mood stabilizers, plays an important rolein affective resilience (Maeng et al, 2007).
Lithium Exerts Robust Neuroprotective Effectsin Preclinical Paradigms
In view of its major effects on GSK-3, bcl-2, and BAG1, it isnot surprising that recent studies have investigatedlithium’s potential neuroprotective effects in a variety ofpreclinical paradigms. Lithium demonstrated robust neuro-protective properties against a variety of insults (reviewedin Bachmann et al, 2005; Chuang and Priller, 2006; Manjiet al, 2000) (Table 3). Notably, lithium pretreatmentprotected cerebral and cerebellar neurons in primaryculture from glutamate-induced, NMDA receptor-mediatedapoptosis (reviewed in Chuang and Priller, 2006). ExcessiveNMDA throughput is likely involved in stress-inducedhippocampal atrophy, and has been implicated in thepathogenesis of a variety of neurodegenerative diseasessuch as stroke, Huntington’s disease, ALS, spinal cordinjury, brain trauma, and cerebellar degeneration. Incultured neurons, lithium-induced neuroprotection againstglutamate excitotoxicity occurred within the therapeuticconcentration range of this drug and required 5–6 dayspretreatment for maximal effects. Lithium neuroprotectioninvolved BDNF induction and activation of its receptor,TrkB, and was associated with upregulation of theantiapoptotic protein bcl-2, downregulation of the proa-poptotic proteins p53 and Bax, and inhibition of caspase-3.Treatment of cultured neurons with other GSK-3 inhibitorsor transfection with GSK-3 siRNA mimicked the neuropro-tective effects of lithium (Liang and Chuang, 2007), againsuggesting a critical role for GSK-3 in mediating neuro-protection.
Lithium has also showed beneficial effects in a number ofanimal models of neurodegenerative diseases. For example,pre- or post-insult treatment with lithium suppressedcerebral ischemia-induced brain infarction, caspase-3 acti-vation, and neurological deficits in rats, and theseneuroprotective effects were associated with induction ofheat shock protein 70 and decreased expression of Bax (Renet al, 2003; Xu et al, 2003). Several independent studiesdemonstrated that lithium has neuroprotective effects inanimal and cellular models of Alzheimer’s, Huntington’s,and Parkinson’s diseases, retinal degeneration, spinal cord
Cellular plasticity cascades in BPDRJ Schloesser et al
...............................................................................................................................................................
124
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

injury, and HIV infection (reviewed in Chuang and Priller,2006). Notably, therapeutic concentrations of lithium, byacting on GSK-3, block the production of Ab peptides byinterfering with amyloid precursor protein (APP) cleavageat the g-secretase step (Phiel et al, 2003; Rockenstein et al,2007; Ryder et al, 2003; Su et al, 2004). Importantly, lithiumalso blocks the accumulation of Ab peptides in the brains ofmice that overproduce APP (Phiel et al, 2003; Rockensteinet al, 2007; Ryder et al, 2003; Su et al, 2004). Similarly,lithium administration has been shown to significantlylower levels of phosphorylation at several epitopes oft known to be hyperphosphorylated in Alzheimer’s diseaseand to significantly reduce levels of aggregated, insolublet (Munoz-Montano et al, 1997; Noble et al, 2005).Furthermore, levels of aggregated t correlate strongly withthe degree of axonal degeneration, and lithium-treated miceshowed less degeneration if administration was startedduring early stages of tangle development.
Lithium is also neuroprotective in APP transgenic mice(Rockenstein et al, 2007). Thus, mice treated with lithiumdisplayed improved performance in the water maze,preservation of the dendritic structure in the frontal cortexand hippocampus, and decreased t phosphorylation (Rock-enstein et al, 2007). Chronic lithium treatment alsoprotected against neurodegeneration and improved spatiallearning deficits in rats perfused with Ab fibrils (De Ferrari
et al, 2003). Interestingly, some preliminary retrospectivehuman studies show that long-term lithium treatment mayhave protective effects against Alzheimer’s disease and/orcognitive deficits (Angst et al, 2007; Nunes et al, 2007; Teraoet al, 2006).
Mood Stabilizers Increase Neurogenesis
Neurogenesis persists in the mammalian brain into adult-hood and is stimulated by several mood stabilizers. Seminalwork by the Duman laboratory demonstrated that variousclasses of antidepressants enhance adult hippocampalneurogenesis (Pittenger and Duman, 2008). Lithium stimu-lates progenitor cell proliferation in cultured neurons(Hashimoto et al, 2003) and increases BrdU-positive cellsin the adult rat hippocampus by approximately 25% (Chenet al, 2000; Son et al, 2003). VPA has similar effects onneurogenesis, specifically increasing the proliferation of ratcerebral cortical cells in culture and the number of BrdU-positive neurons in the mouse DG (Hao et al, 2004).Interestingly, the Wnt ligands Wnt3a, Wnt7a, and Wnt7bare expressed in the subgranular zone of the DG, andcanonical Wnt signaling appears to be critical for postnatalhippocampal neurogenesis (Lie et al, 2005; Maekawa et al,2005). In addition, a Wnt-dependent transcription reporteris active in the DG (Lie et al, 2005; Maekawa et al, 2005) andis further enhanced in vivo by lithium treatment (O’Brienet al, 2004). Moreover, recent findings suggest canonicalWnt signaling in general, and GSK-3 specifically, are majorregulators of stem cell self-renewal and proliferation (Reyaand Clevers, 2005; Trowbridge et al, 2006).
HUMAN EVIDENCE FOR THENEUROTROPHIC EFFECTS OF LITHIUM
While the body of preclinical data demonstrating neuro-trophic and neuroprotective effects of lithium is striking,considerable caution must clearly be exercised in extra-polating to the clinical situation with humans. In view oflithium’s robust effects on the levels of the cytoprotectiveprotein bcl-2 in the anterior cingulate, Drevets andcolleagues re-analyzed older data demonstrating B40%reductions in subgenual PFCPFC volumes in familial mooddisorder subjects (Drevets, 2001). Consistent with neuro-trophic/neuroprotective effects of lithium, they found thatpatients treated with chronic lithium or VPA exhibitedsubgenual PFC volumes that were significantly higher thanthose in non-lithium- or non-VPA-treated patients, and notsignificantly different from controls. To investigate thepotential neurotrophic effects of lithium in humans moredefinitively, Moore et al (2000a) used proton MRS anddemonstrated that treatment of bipolar patients withlithium for 4 weeks increased the level of NAA, a markerof neuronal viability, in the cerebral cortex. A follow-upvolumetric MRI study demonstrated that 4 weeks of lithiumtreatment also significantly increased total gray mattercontent in the human brain (Moore et al, 2000b), suggestingthe possibility of an increase in the volume of the neuropil(the moss-like layer comprised of axonal and dendriticfibers that occupies much of the cortex gray mattervolume). A subsequent study by Sassi et al (2002)
Table 3 Neurotrophic and Neuroprotective Effects of Lithium
Protects (human and rodent) brain cells in vitro from
Glutamate and NMDA toxicity
Calcium toxicity
Thapsigargin (which mobilizes MPP+ and Ca2+) toxicity
b-Amyloid toxicity
Aging-induced cell death
Growth factor and serum deprivation
Glucose deprivation
Low K+
C2-ceramide
Ouabain
Aluminum toxicity
HIV regulatory protein, Tat
Demonstrates following effects in rodent brain (in vivo)
Enhanced hippocampal neurogenesis
Protection against cholinergic lesions
Protection against radiation injury
Protection against medial cerebral artery occlusion (stroke model)
Protection against quinolinic acid (Huntington’s model)
Demonstrates following effects in human brain
Increased gray matter volumes in lithium-treated bipolar patients
Increased N-acetylaspartate (NAA) levels in lithium-treated bipolar patients
Protection against reduced subgenual prefrontal cortex volumes
Larger anterior cingulate volumes in lithium-treated bipolar patients
Protection against reduced glial numbers or glia : neuron ratio in the amygdala
Abbreviations: NMDA, N-methyl-D-aspartate; HIV, human immunodeficiencyvirus.
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
125
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

confirmed a similar increase by lithium in gray mattervolume, compared to the brains of untreated patients andhealthy subjects.
Another study of familial pediatric BPD revealed thatsubjects with BPD with past exposure to lithium or VPAtended to have greater amygdala gray matter volume thansubjects with BPD without such exposure (Chang et al, 2005).Yucel et al (2007) compared the volume of the hippocampus,hippocampal head (Hh), and body/tail in three groups withno history of medication use before entry into the study: (1) agroup of patients treated with lithium for 1–8 weeks and thenscanned, (2) a group comprising patients who wereunmedicated at the time of scan, and (3) a group of patientstreated with either VPA or lamotrigine for 1–8 weeks. Theyobserved a bilateral increase in hippocampal and Hh volumesin the lithium-treated group compared to the unmedicatedgroup, an effect that was apparent even over a brief treatmentperiod (Yucel et al, 2007).
Another study used high-resolution MRI and corticalpattern matching methods to map gray matter differencesin 28 BPD patients, 20 of whom were lithium treated, and 28healthy were controls (Bearden et al, 2007). Their resultsshowed that gray matter density was significantly greater indiffused cortical regions in bipolar patients than in healthysubjects; the differences were most pronounced in thebilateral cingulated and paralimbic cortices, which are areasused in attention, motivation, and emotion. In addition,their data revealed greater gray matter density in the rightanterior cingulate in lithium-treated patients relative to thebipolar subjects not taking lithium. Their lithium-treatedsample included subjects who were on the drug for varyingtime durations and their dosages were not uniform. Thelack of difference in the gray mater density between theuntreated patients and healthy controls, as well as thegrowing evidence that lithium exerts major effects on anumber of cellular proteins and pathways (see above)known to regulate cell atrophy/death lend support to theview that the gray matter enlargement is mediated throughthe trophic actions of lithium in the brain (Chuang andManji, 2007).
FUTURE RESEARCH DIRECTIONS
As we have reviewed here, a considerable body of evidencesupports abnormalities in the regulation of cellularplasticity cascades as integral to the underlying neurobio-logy of BPD. Many of these pathways play critical roles notonly in ‘here and now’ synaptic plasticity (and thereforeprofound changes in mood), but also in long-term cellgrowth/atrophy and cell survival/cell death. Indeed, theatrophic changes observed in multiple cell types (neuronsand glia), as well as the reversibility of the changes withtreatment support a role for intracellular plasticity cascades.It is likely that the major defect is in the ability to regulateneuroplastic adaptations to perturbations (both physiolo-gical and pathophysiological)Fan inability to handle‘normal loads’ (neurochemical, hormonal, stress-induced,pharmacologically induced, etc) without failing or invokingcompensatory adaptations that overshoot and predispose tooscillations. Indeed, the allostatic load would thus con-tribute to long-term disease progression (and potentially to
cycle acceleration). Many of the very same cascadesinvolved in regulating synaptic plasticity also play a criticalrole in cell atrophy and cellular resilience. These observa-tions serve to explain the atrophic aspect of the illness insome patients, as well as the presence of stigmata normallyassociated with ischemic/hypoxic insults, such as WMH.
In the past decade several studies have questioned thecommon belief that BPD is associated with a favorableprognosis and good long-term outcome; this has led bothclinicians and researchers to focus on the early stages ofthe illness to study psychopathological prodromes and bio-logical developmental abnormalities that could guide newtreatment algorithms in high-risk populations. Neuroima-ging studies have shown widespread cortical and subcorticalinvolvement in BPD even in patients experiencing their firstepisode; prospective studies have also suggested that afteronset, new brain pathological remodeling takes place inareas involved in BPD pathophysiology, with gray matterloss, white matter abnormalities, and new functional andcognitive deficits. Evidence also suggests that somewhatakin to the treatment of conditions such as hypertensionand diabetes, early and sustained treatment may benecessary to adequately prevent the deleterious long-termsequelae associated with mood disorders (Leverich et al,2007; Post, 2007). However, careful selection of the subjectswho might benefit from early intervention and appropriatestudy designs for a correct evaluation of the outcome areneeded; prospective large-scale studies from high-riskpopulations with appropriate biological markers could helpidentify ‘real’ high-risk subjects and develop new treatmentalgorithms.
Unfortunately, there has been little progress in developingtruly novel medications specifically for the treatment ofBPD, and most recent additions to the pharmacopeia arebrain penetrant drugs developed for the treatment ofepilepsy or schizophrenia. This era may now be over asthere are a number of pharmacological ‘plasticity-enhan-cing’ strategies that may be of considerable utility in thetreatment of BPD. Indeed, these next-generation drugs, inaddition to treating the core symptoms of BPD, might beable to target other important aspects of the illness. Theseinclude enhancing cognition independent of any improve-ment in mood symptoms, and preventing or reversingepigenetic factors that may have long-term negative impactson the course of the illness.
?? Responsiblefor “comorbidities”
Cell loss andatrophy
Impairments ofcellular resilience
Targets for themost effective
treatments
Synaptic and cellularplasticity cascades
SerotoninNorepinephrine
DopamineNeuropeptides
GABA
We are optimistic that the advances outlined here willultimately lead to the discovery of new approaches for the
Cellular plasticity cascades in BPDRJ Schloesser et al
...............................................................................................................................................................
126
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

prevention and treatment of some of mankind’s mostdevastating and least understood illnesses. The develop-ment of novel therapeutics holds much promise for thelong-term treatment of severe mood disorders and forimproving the lives of the many who suffer from them.
ACKNOWLEDGEMENTS
We thank Ioline Henter for outstanding editorial assistance.RJS and HKM are supported by the Intramural Program ofthe NIMH. PSK is supported by grants from the NationalInstitute of Mental Health and the American Federation forAging Research. JH is supported by a fellowship from theLeukemia and Lymphoma Society.
DISCLOSURE/CONFLICT OF INTEREST
The authors report no conflicts of interest.
REFERENCES
Acharya JK, Labarca P, Delgado R, Jalink K, Zuker CS (1998).Synaptic defects and compensatory regulation of inositolmetabolism in inositol polyphosphate 1-phosphatase mutants.Neuron 20: 1219–1229.
Adams JM, Cory S (1998). The Bcl-2 protein family: arbiters of cellsurvival. Science 281: 1322–1326.
Altshuler LL, Curran JG, Hauser P, Mintz J, Denicoff K, Post RM(1995). T2 hyperintensities in bipolar disorder: magneticresonance imaging comparison and literature meta-analysis.Am J Psychiatry 152: 1139–1144.
Angst J, Gamma A, Gerber-Werder R, Zarate CA, Manji HK (2007).Does long-term medication with lithium prevent or attenuatedementia in bipolar and depressed patients? Int J Psychiatry ClinPract 11: 2–8.
Azab AN, He Q, Ju S, Li G, Greenberg ML (2007). Glycogensynthase kinase-3 is required for optimal de novo synthesis ofinositol. Mol Microbiol 63: 1248–1258.
Baastrup PC, Schou M (1967). Lithium as a prophylactic agents. Itseffect against recurrent depressions and manic-depressivepsychosis. Arch Gen Psychiatry 16: 162–172.
Bachmann RF, Schloesser RJ, Gould TD, Manji HK (2005). Moodstabilizers target cellular plasticity and resilience cascades:implications for the development of novel therapeutics. MolNeurobiol 32: 173–202.
Baum A, Akula N, Cabenero M, Cardona I, Corona W, Klemens Bet al (2007). A genome-wide association study implicatesdiacylglycerol kinase eta (DGKH) and several other genes inthe etiology of bipolar disorder. Mol Psychiatry; advance onlinepublication 8 May 2007; doi:101038/sjmp4002012.
Baumann B, Danos P, Krell D, Diekmann S, Leschinger A, Stauch Ret al (1999a). Reduced volume of limbic system-affiliated basalganglia in mood disorders: preliminary data from a postmortemstudy. J Neuropsychiatry Clin Neurosci 11: 71–78.
Baumann B, Danos P, Krell D, Diekmann S, Wurthmann C, BielauH et al (1999b). Unipolar-bipolar dichotomy of mood disordersis supported by noradrenergic brainstem system morphology.J Affect Disord 54: 217–224.
Bear M, Dolen G, Osterweil E, Nagarajan N (2008). Fragile X:translation in action. Neuropsychopharmacology Rev 33: 84–87.
Bearden CE, Hoffman KM, Cannon TD (2001). The neuropsycho-logy and neuroanatomy of bipolar affective disorder: a criticalreview. Bipolar Disord 3: 106–150.
Bearden CE, Thompson PM, Dalwani M, Hayashi KM, Lee AD,Nicoletti M et al (2007). Greater cortical gray matter density in
lithium-treated patients with bipolar disorder. Biol Psychiatry62: 7–16.
Beasley CL, Honer WG, Bergmann K, Falkai P, Lutjohann D, BayerTA (2005). Reductions in cholesterol and synaptic markers inassociation cortex in mood disorders. Bipolar Disord 7: 449–455.
Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR,Gainetdinov RR et al (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthasekinase 3 signaling cascade. Proc Natl Acad Sci USA 101: 5099–5104.
Bebchuk JM, Arfken CL, Dolan-Manji S, Murphy J, Hasanat K,Manji HK (2000). A preliminary investigation of a protein kinaseC inhibitor in the treatment of acute mania. Arch Gen Psychiatry57: 95–97.
Belmaker RH (2004). Bipolar disorder. N Engl J Med 351: 476–486.Benes FM, Berretta S (2001). GABAergic interneurons: implica-
tions for understanding schizophrenia and bipolar disorder.Neuropsychopharmacology 25: 1–27.
Benes FM, Vincent SL, Todtenkopf M (2001). The density ofpyramidal and nonpyramidal neurons in anterior cingulatecortex of schizophrenic and bipolar subjects. Biol Psychiatry 50:395–406.
Benowitz LI, Perrone-Bizzozero NI, Neve RL, Rodriguez W (1990).GAP-43 as a marker for structural plasticity in the mature CNS.Prog Brain Res 86: 309–320.
Berridge MJ, Downes CP, Hanley MR (1989). Neural and develop-mental actions of lithium: a unifying hypothesis. Cell 59: 411–419.
Berry GT, Buccafusca R, Greer JJ, Eccleston E (2004). Phospho-inositide deficiency due to inositol depletion is not a mechanismof lithium action in brain. Mol Genet Metabolism 82: 87–92.
Bertolino A, Frye MA, Callicott JH, Mattay VS, Rakow R, Shelton-Repella J et al (2003). Neuronal pathology in the hippocampalarea of patients with bipolar disorder: a study with protonmagnetic resonance spectroscopic imaging. Biol Psychiatry 53:906–913.
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, RussoSJ et al (2006). Essential role of BDNF in the mesolimbicdopamine pathway in social defeat stress. Science 311: 864–888.
Bezchlibnyk YB, Sun X, Wang J, MacQueen GM, McEwen BS,Young LT (2007). Neuron somal size is decreased in the lateralamygdalar nucleus of subjects with bipolar disorder. J PsychiatryNeurosci 32: 203–210.
Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CWet al (2000). Regulation and localization of tyrosine216phosphorylation of glycogen synthase kinase-3beta in cellularand animal models of neuronal degeneration. Proc Natl Acad SciUSA 97: 11074–11079.
Bielau H, Trubner K, Krell D, Agelink MW, Bernstein HG, Stauch Ret al (2005). Volume deficits of subcortical nuclei in mooddisorders: a postmortem study. Eur Arch Psychiatry ClinNeurosci 255: 401–412.
Bonfanti L, Strettoi E, Chierzi S, Cenni MC, Liu XH, Martinou J-Cet al (1996). Protection of retinal ganglion cells from natural andaxotomy-induced cell death in neonatal transgenic mice over-expressing bcl-2. J Neurosci 16: 4186–4194.
Bowley MP, Drevets WC, Ongur D, Price JL (2002). Low glialnumbers in the amygdala in major depressive disorder. BiolPsychiatry 52: 404–412.
Brambilla P, Harenski K, Nicoletti MA, Mallinger AG, Frank E,Kupfer DJ et al (2001). Differential effects of age on brain graymatter in bipolar patients and healthy individuals. Neuropsycho-pharmacology 43: 242–247.
Brambilla P, Nicoletti MA, Harenski K, Sassi RB, Mallinger AG,Frank E et al (2002). Anatomical MRI study of subgenualprefrontal cortex in bipolar and unipolar subjects. Neuropsycho-pharmacology 27: 792–799.
Bruckheimer EM, Cho SH, Sarkiss M, Hermann J, McDonnell TJ(1998). The Bcl-2 gene family and apoptosis. Adv Biochem EngBiotechnol 62: 75–105.
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
127
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

Burden SJ (2000). Wnts as retrograde signals for axon and growthcone differentiation. Cell 100: 495–497.
Buttner N, Bhattacharyya S, Walsh J, Benes FM (2007).DNA fragmentation is increased in non-GABAergic neuronsin bipolar disorder but not in schizophrenia. Schizophr Res 93:33–41.
Cao BJ, Peng NA (1993). Magnesium valproate attenuateshyperactivity induced by dexamphetamine-chlordiazepoxidemixture in rodents. Eur J Pharmacol 237: 177–181.
Carlson PJ, Singh JB, Zarate CA, Drevets WC, Manji HK (2006).Neural circuitry and neuroplasticity in mood disorders: insightsfor novel therapeutic targets. NeuroRx 3: 22–41.
Catapano L, Chen G, Du J, Zarate CA, Manji HK (in press). Cellularplasticity cascades: genes to behavior pathways in the patho-physiology and treatment of bipolar disorder. In: Charney DS,Nestler EJ (eds). Neurobiology of Mental Illness. OxfordUniversity Press: New York.
Cecil KM, DelBello MP, Morey R, Strakowski SM (2002). Frontallobe differences in bipolar disorder as determined by proton MRspectroscopy. Bipolar Disord 4: 357–365.
Chalecka-Franaszek E, Chuang DM (1999). Lithium activates theserine/threonine kinase Akt-1 and suppresses glutamate-inducedinhibition of Akt-1 activity in neurons. Proc Natl Acad Sci USA96: 8745–8750.
Chang K, Adleman N, Dienes KA, Barnea-Goraly N, Reiss A, KetterTA (2003). Decreased N-acetylaspartate in children with familialbipolar disorder. Biol Psychiatry 53: 1059–1065.
Chang K, Barnea-Goraly N, Karchemskiy A, Simeonova DI, BarnesP, Ketter TA et al (2005). Cortical magnetic resonance imagingfindings in familial pediatric bipolar disorder. Biol Psychiatry 58:197–203.
Chen B, Wang JF, Sun X, Young LT (2003). Regulation of GAP-43expression by chronic desipramine treatment in rat culturedhippocampal cells. Biol Psychiatry 53: 530–537.
Chen DF, Schneider GE, Martinou JC, Tonewaga S (1997). Bcl-2promotes regeneration of severed axons in mammalian CNS.Nature 385: 434–439.
Chen G, Manji HK (2006). The extracellular signal-regulated kinasepathway: an emerging promising target for mood stabilizers.Curr Opin Psychiatry 19: 313–323.
Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK (2000).Enhancement of hippocampal neurogenesis by lithium.J Neurochem 75: 1729–1734.
Chen G, Zeng WZ, Yuan PX, Huang LD, Jiang YM, Zhao ZH et al(1999). The mood-stabilizing agents lithium and valproaterobustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J Neurochem 72: 879–882.
Chuang DM (2004). Neuroprotective and neurotrophic actions ofthe mood stabilizer lithium: can it be used to treat neurodegen-erative diseases? Crit Rev Neurobiol 16: 83–90.
Chuang DM, Manji HK (2007). In search of the holy grail for thetreatment of neurodegenerative disorders: has a simple cationbeen overlooked? Biol Psychiatry 62: 4–6.
Chuang DM, Priller J (2006). Potential use of lithium inneurodegenerative disorders. In: Bauer M, Grof P, Muler-Oerlingausen B (eds). Lithium in Neuropsychiatry: The Compre-hensive Guide. Taylor & Francis: London. pp 381–397.
Citri A, Malenka RC (2008). Mechanisms of plasticity in excitatorysynapses. Neuropsychopharmacology Rev 1 (in press).
Cohen P, Frame S (2001). The renaissance of GSK3. Nat Rev MolCell Biol 2: 769–776.
Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ,Connolly CN et al (2004). GSK-3 phosphorylation of theAlzheimer epitope within collapsin response mediator proteinsregulates axon elongation in primary neurons. J Biol Chem 279:50176–50180.
Cook D, Fry MJ, Hughes K, Sumathipala R, Woodgett JR, Dale TC(1996). Wingless inactivates glycogen synthase kinase-3 via an
intracellular signalling pathway which involves a protein kinaseC. EMBO J 15: 4526–4536.
Cotter D, Landau S, Beasley C, Stevenson R, Chana G, MacMillan Let al (2002a). The density and spatial distribution of GABAergicneurons, labelled using calcium binding proteins, in the anteriorcingulate cortex in major depressive disorder, bipolar disorder,and schizophrenia. Biol Psychiatry 51: 377–386.
Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP(2002b). Reduced neuronal size and glial cell density in area 9 ofthe dorsolateral prefrontal cortex in subjects with majordepressive disorder. Cereb Cortex 12: 386–394.
Cryns K, Shamir A, Shapiro J, Daneels G, Goris I, VanCraenendonck H et al (2007). Lack of lithium-like behavioraland molecular effects in IMPA2 knockout mice. Neuropsycho-pharmacology 32: 881–891.
Davies SP, Reddy H, Caivano M, Cohen P (2000). Specificity andmechanism of action of some commonly used protein kinaseinhibitors. Biochem J 351: 95–105.
Davis JM, Janicak PG, Hogan DM (1999). Mood stabilizers in theprevention of recurrent affective disorders: a meta-analysis. ActaPsychiatr Scand 100: 406–417.
De Ferrari GV, Chacon MA, Barria MI, Garrido JL, Godoy JA,Olivares G et al (2003). Activation of Wnt signaling rescuesneurodegeneration and behavioral impairments induced bybeta-amyloid fibrils. Mol Psychiatry 8: 195–208.
De Sarno P, Li X, Jope RS (2002). Regulation of Akt and glycogensynthase kinase-3beta phosphorylation by sodium valproate andlithium. Neuropharmacology 43: 1158–1164.
Diecken RF, Fein G, Weiner MW (1995). Abnormal frontal lobephosphorous metabolism in bipolar disorder. Am J Psychiatry152: 915–918.
Diecken RF, Pegues MP, Anzalone S, Feiwell R, Soher B (2003).Lower concentration of hippocampal N-acetylaspartate infamilial bipolar I disorder. Am J Psychiatry 160: 873–882.
Ding VW, Chen RH, McCormick F (2000). Differential regulationof glycogen synthase kinase 3beta by insulin and Wnt signaling.J Biol Chem 275: 32475–32481.
Dixon JF, Los GV, Hokin LE (1994). Lithium stimulates glutamate‘release’ and inositol 1,4,5-trisphosphate accumulation viaactivation of the N-methyl-D-aspartate receptor in monkeyand mouse cerebral cortex slices. Proc Natl Acad Sci USA 91:8358–8362.
Doble BW, Woodgett JR (2003). GSK-3: tricks of the trade for amulti-tasking kinase. J Cell Sci 116: 1175–1186.
Dokucu ME, Yu L, Taghert PH (2005). Lithium- and valproate-induced alterations in circadian locomotor behavior in Droso-phila. Neuropsychopharmacology 30: 2216–2224.
Drevets WC (2000). Neuroimaging studies of mood disorders. BiolPsychiatry 48: 813–829.
Drevets WC (2001). Neuroimaging and neuropathological studiesof depression: implications for the cognitive–emotional featuresof mood disorders. Curr Opin Neurobiol 11: 240–249.
Drevets WC, Price JL, Simpson Jr JR, Todd RD, Reich T, VannierM et al (1997). Subgenual prefrontal cortex abnormalities inmood disorders. Nature 386: 824–827.
Du J, Creson T, Gray NA, Falke CA, Wei Y, Wang YM et al(submitted). The role of hippocampal GluR1 and GluR2receptors in manic-like behaviors.
Du J, Gray NA, Falke CA, Chen W, Yuan P, Szabo ST et al (2004a).Modulation of synaptic plasticity by antimanic agents: the role ofAMPA glutamate receptor subunit 1 synaptic expression.J Neurosci 24: 6578–6589.
Du J, Gray NA, Falke CA, Yuan PX, Szabo ST, Manji HK (2003).Structurally dissimilar antimanic agents modulate synapticplasticity by regulating AMPA glutamate receptor subunit GluR1synaptic expression. Ann NY Acad Sci 1003: 378–380.
Du J, Machado-Vieira R, Maeng S, Martinowich K, Manji HK,Zarate CA (2006). Enhancing AMPA to NMDA throughput as a
Cellular plasticity cascades in BPDRJ Schloesser et al
...............................................................................................................................................................
128
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

convergent mechanism for antidepressant action. Drug DiscovToday: Ther Strat 3: 519–526.
Du J, Quiroz J, Gray NA, Szabo ST, Zarate Jr CA, Manji HK(2004b). Regulation of cellular plasticity and resilience by moodstabilizers: the role of AMPA receptor trafficking. Dialog ClinNeurosci 6: 143–157.
Du J, Wei Y, Chen Z, Falke CA, Zarate Jr CA, Manji HK (2007). Theanticonvulsants lamotrigine, riluzole and valproate differentiallyregulate AMPA receptor trafficking: relationship to clinicaleffects in mood disorders. Neuropsychopharmacology 32:793–802.
Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS(2007). A role for MAP kinase signaling in behavioral models ofdepression and antidepressant treatment. Biol Psychiatry 61:661–670.
Eastwood SL, Harrison PJ (2001). Synaptic pathology in theanterior cingulate cortex in schizophrenia and mood disorders.A review and a western blot study of synaptophysin, GAP-43 andthe complexins. Brain Res Bull 55: 569–578.
Eastwood SL, Harrison PJ (2007). Decreased mRNA expression ofnetrin-G1 and netrin-G2 in the temporal lobe in schizophreniaand bipolar disorder. Neuropsychopharmacology; advance onlinepublication, 16 May 2007; doi:101038/sjnpp1301457.
Eickholt BJ, Walsh FS, Doherty P (2002). An inactive pool of GSK-3at the leading edge of growth cones is implicated in Semaphorin3A signaling. J Cell Biol 157: 211–217.
Eisch AJ, Bolanos CA, de Wit J, Simonak RD, Pudiak CM, Barrot Met al (2003). Brain-derived neurotrophic factor in the ventralmidbrain–nucleus accumbens pathway: a role in depression. BiolPsychiatry 54: 994–1005.
File SE, Aranko K (1988). Sodium valproate decreases exploratorybehaviour in mice: development of tolerance and cross-tolerancewith chlordiazepoxide. Eur J Pharmacol 151: 293–299.
Frame S, Cohen P, Biondi RM (2001). A common phosphatebinding site explains the unique substrate specificity of GSK3and its inactivation by phosphorylation. Moi Cell 7: 1321–1327.
Frangou S, Raymont V, Bettany D (2002). The Maudsley bipolardisorder project. A survey of psychotropic prescribing patternsin bipolar I disorder. Bipolar Disord 4: 378–385.
Frazier JA, Ahn HS, DeJong S, Bent EK, Breeze JL, Giuliano AJ(2005). Magnetic resonance imaging studies in early-onsetbipolar disorder: a critical review. Harv Rev Psychiatry 13:125–140.
Goodwin FK, Jamison KR (2007). Manic-Depressive Illness: BipolarDisorders and Recurrent Depression. Oxford University Press:New York.
Goold RG, Owen R, Gordon-Weeks PR (1999). Glycogen synthasekinase 3beta phosphorylation of microtubule-associated protein1B regulates the stability of microtubules in growth cones. J CellSci 112: 3373–3384.
Gould TD, Chen G, Manji HK (2004a). In vivo evidence in the brainfor lithium inhibition of glycogen synthase kinase-3. Neuro-psychopharmacology 29: 32–38.
Gould TD, Einat H, Bhat R, Manji HK (2004b). AR-A014418, aselective GSK-3 inhibitor, produces antidepressant-like effects inthe forced swim test. Int J Neuropsychopharmacol 7: 387–390.
Gould TD, Einat H, O’Donnell KC, Picchini AM, Schloesser RJ,Manji HK (2007). Beta-catenin overexpression in the mousebrain phenocopies lithium-sensitive behaviors. Neuropsycho-pharmacology; advance online publication, 14 February 2007;doi:101038/sjnpp1301338.
Gould TD, Manji HK (2005). Glycogen synthase kinase-3: aputative molecular target for lithium mimetic drugs. Neuro-psychopharmacology 30: 1223–1237.
Gould TD, Picchini AM, Einat H, Manji HK (2006). Targetingglycogen synthase kinase-3 in the CNS: implications for thedevelopment of new treatments for mood disorders. Curr DrugTargets 7: 1399–1409.
Gurvich N, Klein PS (2002). Lithium and valproic acid: parallelsand contrasts in diverse signaling contexts. Pharmacol Ther 96:45–66.
Gyulai L, Bowden CL, McElroy SL, Calabrese JR, Petty F, Swann ACet al (2003). Maintenance efficacy of divalproex in theprevention of bipolar depression. Neuropsychopharmacology28: 1374–1382.
Hajek T, Carrey N, Alda M (2005). Neuroanatomical abnormalitiesas risk factors for bipolar disorder. Bipolar Disord 7: 393–403.
Hall AC, Brennan A, Goold RG, Cleverley K, Lucas FR, Gordon-Weeks PR et al (2002). Valproate regulates GSK-3-mediatedaxonal remodeling and synapsin I clustering in developingneurons. Mol Cell Neurosci 20: 257–270.
Hall AC, Lucas FR, Salinas PC (2000). Axonal remodeling andsynaptic differentiation in the cerebellum is regulated byWNT-7a signaling. Cell 100: 525–535.
Hallcher LM, Sherman WR (1980). The effects of lithium ion andother agents on the activity of myo-inositol-1-phosphatase frombovine brain. J Biol Chem 255: 10896–10901.
Hamakawa H, Kato T, Murashita J, Kato N (1998). Quantitativeproton magnetic resonance spectroscopy of the basal ganglia inpatients with affective disorders. Eur Arch Psychiatry ClinNeurosci 248: 53–58.
Hamakawa H, Murashita J, Yamada N, Inubushi T, Kato N, Kato T(2004). Reduced intracellular pH in the basal ganglia and wholebrain measured by 31P-MRS in bipolar disorder. Psychiatry ClinNeurosci 58: 82–88.
Hao Y, Creson T, Zhang L, Li P, Du F, Yuan P et al (2004).Mood stabilizer valproate promotes ERK pathway-dependentcortical neuronal growth and neurogenesis. J Neurosci 24:6590–6599.
Hashimoto R, Senatorov V, Kanai H, Leeds P, Chuang DM (2003).Lithium stimulates progenitor proliferation in cultured brainneurons. Neurosci 117: 55–61.
Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM (2002).Differential hippocampal expression of glutamic acid decarbox-ylase 65 and 67 messenger RNA in bipolar disorder andschizophrenia. Arch Gen Psychiatry 59: 521–529.
Hedgepeth C, Conrad L, Zhang Z, Huang H, Lee V, Klein P (1997).Activation of the Wnt signaling pathway: a molecular mechan-ism for lithium action. Dev Biol 185: 82–91.
Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ,Greengard P (1999). Synapsins as regulators of neurotransmitterrelease. Philos Trans R Soc Lond B Biol Sci 354: 269–279.
Hong M, Chen DC, Klein PS, Lee VM-Y (1997). Lithium reducestau phosphorylation by inhibition of glycogen synthase kinase-3.J Biol Chem 272: 25326–25332.
Hrdina P, Faludi G, Li Q, Bendotti C, Tekes K, Sotonyi P et al(1998). Growth-associated protein (GAP-43), its mRNA, andprotein kinase C (PKC) isoenzymes in brain regions of depressedsuicides. Mol Psychiatry 3: 411–418.
Huang HC, Klein PS (2006). Multiple roles for glycogen synthasekinase-3 as a drug target in Alzheimer’s disease. Curr DrugTargets 7: 1389–1397.
Hyman SE, Nestler EJ (1996). Initiation and adaptation: aparadigm for understanding psychotropic drug action. Am JPsychiatry 153: 151–162.
Jiang H, Guo W, Liang X, Rao Y (2005). Both the establishmentand the maintenance of neuronal polarity require activemechanisms: critical roles of GSK-3beta and its upstreamregulators. Cell 120: 123–135.
Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, ShoreAD, Torrey EF et al (2000). Disease-specific alterations in frontalcortex brain proteins in schizophrenia, bipolar disorder, andmajor depressive disorder. The Stanley Neuropathology Con-sortium. Mol Psychiatry 5: 142–149.
Jope RS, Johnson SL (2004). The glamour and gloom of glycogensynthase kinase-3. Trends Biochem Science 29: 95–102.
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
129
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H (2004). Rapid antidepressive-like activity ofspecific glycogen synthase kinase-3 inhibitor and its effect onbeta-catenin in mouse hippocampus. Biol Psychiatry 55:781–784.
Kaladchibachi SA, Doble B, Anthopoulos N, Woodgett JR,Manoukian AS (2007). Glycogen synthase kinase 3, circadianrhythms, and bipolar disorder: a molecular link in thetherapeutic action of lithium. J Circadian Rhythms 5: 3.
Kato T, Murashita J, Kamiya A, Shiori T, Kato N, Inubushi T(1998). Decreased brain intracellular pH measured by 31P-MRSin bipolar disorder: a confirmation in drug-free patients andcorrelation with white matter hyperintensity. Eur Arch Psychia-try Clin Neurosci 248: 301–306.
Kato T, Shiori T, Murashita J, Hamakawa H, Inubushi T,Takahashi JS (1994). Phosphorus-31 magnetic resonance spec-troscopy and ventricular enlargement in bipolar disorder.Psychiatry Res 55: 41–50.
Kato T, Shiori T, Murashita J, Hamakawa H, Takahashi Y,Inubushi T et al (1995). Lateralized abnormality of high energyphosphate metabolism in the frontal lobes of patients withbipolar disorder detected by phase-encoded 31P-MRS. PsycholMed 25: 557–566.
Kim L, Kimmel AR (2000). Regulation of a cyclin-CDK-CDKinhibitor complex by inositol pyrophosphates. Curr Opin GenetDev 10: 508–514.
Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura Tet al (2006). Essential roles for GSK-3s and GSK-3-primedsubstrates in neurotrophin-induced and hippocampal axongrowth. Neuron 52: 981–996.
Klein PS, Melton DA (1996). A molecular mechanism for theeffect of lithium on development. Proc Natl Acad Sci USA 93:8455–8459.
Konradi C, Eaton M, MacDonald ML, Walsh J, Benes FM, HeckersS (2004). Molecular evidence for mitochondrial dysfunction inbipolar disorder. Arch Gen Psychiatry 61: 300–308.
Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M,Przedborski S (1997). Bcl-2: prolonging life in a transgenicmouse model of familial amyotrophic lateral sclerosis. Science277: 559–562.
Kupfer DJ (2005). The increasing medical burden in bipolardisorder. JAMA 293: 2528–2530.
Lawrence MS, Ho DY, Sun GH, Steinberg GK, Sapolsky RM (1996).Overexpression of Bcl-2 with herpes simplex virus vectorsprotects CNS neurons against neurological insults in vitro and invivo. J Neurosci 16: 486–496.
Lee YS, Mulugu S, York JD, O’Shea EK (2007). Regulation of acyclin-CDK-CDK inhibitor complex by inositol pyrophosphates.Science 316: 845–846.
Lenox RH, Gould TD, Manji HK (2002). Endophenotypes inbipolar disorder. Am J Med Genet 114: 391–406.
Leverich GS, Post RM, Keck PE, Altshuler LL, Frye MA, Kupka RWet al (2007). The poor prognosis of childhood-onset bipolardisorder. J Pediatrics 150: 485–490.
Li H, Yuan J (1999). Deciphering the pathways of life and death.Curr Opin Cell Biol 11: 261–266.
Li X, Bijur GN, Jope RS (2002). Glycogen synthase kinase-3beta,mood stabilizers, and neuroprotection. Bipolar Disord 4: 137–144.
Li X, Friedman AB, Zhu W, Wang L, Boswell S, May RS et al(2007). Lithium regulates glycogen synthase kinase-3beta inhuman peripheral blood mononuclear cells: implication in thetreatment of bipolar disorder. Biol Psychiatry 61: 216–222.
Liang MH, Chuang DM (2007). Regulation and function ofglycogen synthase kinase-3 isoforms in neuronal survival. J BiolChem 282: 3904–3917.
Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio Aet al (2005). Wnt signalling regulates adult hippocampalneurogenesis. Nature 437: 1370–1375.
Liu L, Schulz SC, Lee S, Reutiman TJ, Fatemi SH (2007).Hippocampal CA1 pyramidal cell size is reduced in bipolardisorder. Cell Mol Neurobiol 27: 351–358.
Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, CleghonV (2006). A chaperone-dependent GSK3beta transitional inter-mediate mediates activation-loop autophosphorylation. Mol Cell24: 627–633.
Lopez-Coronado JM, Belles JM, Lesage F, Serrano R, Rodriguez PL(1999). A novel mammalian lithium-sensitive enzyme with adual enzymatic activity, 30-phosphoadenosine 50-phosphatephosphatase and inositol-polyphosphate 1-phosphatase. J BiolChem 274: 16034–16039.
Lovestone S, Davis DR, Webster MT, Kaech S, Brion JP, Matus Aet al (1999). Lithium reduces tau phosphorylation: effects inliving cells and in neurons at therapeutic concentrations. BiolPsychiatry 45: 995–1003.
Lucas FR, Goold RG, Gordon-Weeks PR, Salinas PC (1998).Inhibition of GSK-3beta leading to the loss of phosphorylatedMAP-1B is an early event in axonal remodelling induced byWNT-7a or lithium. J Cell Sci 111: 1351–1361.
Lyoo IK, Lee HK, Jung JH, Noam GG, Renshaw PF (2002). Whitematter hyperintensities on magnetic resonance imaging of thebrain in children with psychiatric disorders. Comp Psychiatry 43:361–368.
MacQueen GM, Campbell S, McEwen BS, Macdonald K, Amano S,Joffe R et al (2003). Course of illness, hippocampal function, andhippocampal volume in major depression. Proc Natl Acad SciUSA 100: 1387–1392.
Maekawa M, Takashima N, Arai Y, Nomura T, Inokuchi K, Yuasa Set al (2005). Pax6 is required for production and maintenance ofprogenitor cells in postnatal hippocampal neurogenesis. GenesCells 10: 1001–1114.
Maeng S, Zarate CA, Du J, Schloesser RJ, McCammon J, Chen Get al (2007). Cellular mechanisms underlying the antidepressanteffects of ketamine: role of AMPA receptors. Biol Psychiatry [20July 2007; E-pub ahead of print].
Malinow R, Malenka RC (2002). AMPA receptor trafficking andsynaptic plasticity. Annu Rev Neurosci 25: 103–126.
Manji HK, Moore GJ, Chen G (1999). Lithium at 50: have theneuroprotective effects of this unique cation been overlooked?Biol Psychiatry 46: 929–940.
Manji HK, Moore GJ, Chen G (2000). Lithium up-regulates thecytoprotective protein Bcl-2 in the CNS in vivo: a role forneurotrophic and neuroprotective effects in manic depressiveillness. J Clin Psychiatry 61(Suppl 9): 82–96.
Martinek S, Inonog S, Manoukian AS, Young MW (2001). A rolefor the segment polarity gene shaggy/GSK-3 in the Drosophilacircadian clock. Cell 105: 769–779.
Martinowich K, Lu B (2008). Interaction between BDNF andserotonin: role in mood disorders. NeuropsychopharmacologyRev 33: 73–83.
Maslanski JA, Leshko L, Busa WB (1992). Lithium-sensitiveproduction of inositol phosphates during amphibian embryonicmesoderm induction. Science 256: 243–245.
Mattson MP (2007). Mitochondrial regulation of neuronalplasticity. Neurochem Res 32: 707–715.
McClung CA (2007). Circadian genes, rhythms and the biology ofmood disorders. Pharmacol Ther 114: 222–232.
McClung CA, Nestler EJ (2008). Neuroplasticity mediated by alteredgene expression. Neuropsychopharmacology Rev 33: 3–17.
McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N,Marquez R et al (2005). Role that phosphorylation of GSK3 playsin insulin and Wnt signalling defined by knockin analysis.EMBO J 24: 1571–1583.
Merry DE, Korsmeyer SJ (1997). Bcl-2 gene family in the nervoussystem. Annu Rev Neurosci 20: 245–267.
Moore GJ, Bebchuk JM, Hasanat K, Chen G, Seraji-Bozorgzad N,Wilds IB et al (2000a). Lithium increases N-acetyl-aspartate in
Cellular plasticity cascades in BPDRJ Schloesser et al
...............................................................................................................................................................
130
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

the human brain: in vivo evidence in support of bcl-2’sneurotrophic effects? Biol Psychiatry 48: 1–8.
Moore GJ, Bebchuk JM, Wilds IB, Chen G, Manji HK (2000b).Lithium-induced increase in human brain grey matter. Lancet356: 1241–1242.
Moore PB, Schepherd DJ, Eccleston D, Macmillan IC, Goswami U,McAllister VL et al (2001). Cerebral white matter lesions inbipolar affective disorder: relationship to outcome. Br JPsychiatry 178: 172–176.
Munoz-Montano JR, Moreno FJ, Avila J, Diaz-Nido J (1997).Lithium inhibits Alzheimer’s disease-like tau protein phospho-rylation in neurons. FEBS Lett 411: 183–188.
Murphy DL (1977). Animal models for mania. In: Hanin I, Usdin E(eds). Animal Models in Psychiatry and Neurology. PergamonPress: Oxford. pp 211–223.
Nasrallah HA, McCalley-Whitters M, Jacoby CG (1982). Corticalatrophy in schizophrenia and mania: a comparative CT study.J Clin Psychiatry 43: 439–441.
Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F et al(2005). Inhibition of glycogen synthase kinase-3 by lithiumcorrelates with reduced tauopathy and degeneration in vivo. ProcNatl Acad Sci USA 102: 6990–6995.
Noga JT, Vladar K, Torrey EF (2001). A volumetric magneticresonance imaging study of monozygotic twins discordant forbipolar disorder. Psychiatry Res 106: 25–34.
Nunes PV, Forlenza OV, Gattaz WF (2007). Lithium and the riskfor Alzheimer’s disease in elderly patients with bipolar disorder(abstract). Br J Psychiatry 190: 359–360.
O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo Set al (2004). Glycogen synthase kinase-3beta haploinsufficiencymimics the behavioral and molecular effects of lithium.J Neurosci 24: 6791–6798.
O’Donnell T, Rotzinger S, Nakashima TT, Hanstock CC, Ulrich M,Silverstone PH (2000). Chronic lithium and sodium valproateboth decrease the concentration of myo-inositol and increase theconcentration of inositol monophosphates in rat brain. BrainRes 880: 84–91.
Odom AR, Stahlberg A, Wente SR, York JD (2000). A role fornuclear inositol 1,4,5-trisphosphate kinase in transcriptionalcontrol. Science 287: 2026–2029.
Ongur D, Drevets WC, Price JL (1998). Glial reduction in thesubgenual prefrontal cortex in mood disorders. Proc Natl AcadSci USA 95: 13290–13295.
Padiath QS, Paranjpe D, Jain S, Sharma VK (2004). Glycogensynthase kinase 3beta as a likely target for the action of lithiumon circadian clocks. Chronobiol Int 21: 43–55.
Pantazopoulos H, Lange N, Baldessarini RJ, Berretta S (2007).Parvalbumin neurons in the entorhinal cortex of subjectsdiagnosed with bipolar disorder or schizophrenia. Biol Psychia-try 61: 640–652.
Pearlson GD, Garbacz DJ, Tompkins RH, Ahn HS, Gutterman DF,Veroff AE et al (1984). Clinical correlates of lateral ventricularenlargement in bipolar affective disorder. Am J Psychiatry 141:253–256.
Pearlson GD, Veroff AE (1981). Computerised tomographic scanchanges in manic-depressive illness. Lancet 2: 470.
Perera TD, Coplan JD, Lisanby SH, Lipira CM, Arif M, Carpio C et al(2007). Antidepressant-induced neurogenesis in the hippocampusof adult nonhuman primates. J Neurosci 27: 4894–4901.
Phiel CJ, Wilson CA, Lee VM, Klein PS (2003). GSK-3alpharegulates production of Alzheimer’s disease amyloid-betapeptides. Nature 423: 435–439.
Pillai JJ, Friedman L, Stuve TA, Trinidad S, Jesberger JA, Lewin JSet al (2002). Increased presence of white matter hyperintensities inadolescent patients with bipolar disorder. Psychiatry Res 114: 51–56.
Pittenger C, Duman R (2008). Stress, depression, and neuroplas-ticity: a convergence of mechanisms. NeuropsychopharmacologyRev 33: 88–109.
Post RM (2007). Kindling and sensitization as models for affectiveepisode recurrence, cyclicity, and tolerance phenomena. Neu-rosci Biobehav Rev 31: 858–873.
Prickaerts J, Moechars D, Cryns K, Lenaerts I, van CraenendonckH, Goris I et al (2006). Transgenic mice overexpressing glycogensynthase kinase 3beta: a putative model of hyperactivity andmania. J Neurosci 26: 9022–9029.
Raghupathi R, Fernandez SC, Murai H, Trusko SP, Scott RW,Nishioka WK et al (1998). BCL-2 overexpression attenuatescortical cell loss after traumatic brain injury in transgenic mice.J Cereb Blood Flow Metab 18: 1259–1269.
Rajkowska G (2000). Postmortem studies in mood disordersindicate altered numbers of neurons and glial cells. BiolPsychiatry 48: 766–777.
Rajkowska G (2002). Cell pathology in bipolar disorder. BipolarDisord 4: 105–116.
Rajkowska G, Halaris A, Selemon LD (2001). Reductions inneuronal and glial density characterize the dorsolateral pre-frontal cortex in bipolar disorder. Biol Psychiatry 49: 741–752.
Rao S, Rajesh KR, Joseph T (1991). Effect of antiepileptic drugsvalproic acid, carbamazepine and ethosuccimide on exploratorybehaviour in mice. Ind J Exper Biol 29: 127–130.
Ren M, Senatorov VV, Chen RW, Chuang DM (2003). Postinsulttreatment with lithium reduces brain damage and facilitatesneurological recovery in a rat ischemia/reperfusion model. ProcNatl Acad Sci USA 100: 6210–6215.
Reya T, Clevers H (2005). Wnt signalling in stem cells and cancer.Nature 434: 843–850.
Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JBet al (2007). Neuroprotective effects of regulators of the glycogensynthase kinase-3beta signaling pathway in a transgenic modelof Alzheimer’s disease are associated with reduced amyloidprecursor protein phosphorylation. J Neurosci 27: 1981–1991.
Roh MS, Eom TY, Zmijewska AA, De Sarno P, Roth KA, Jope RS(2005). Hypoxia activates glycogen synthase kinase-3 in mousebrain in vivo: protection by mood stabilizers and imipramine.Biol Psychiatry 57: 278–286.
Roybal K, Theobold D, Graham A, Dinieri JA, Russo SJ, KrishnanV et al (2007). Mania-like behavior induced by disruption ofCLOCK. Proc Natl Acad Sci USA 104: 6406–6411.
Ruel L, Stambolic V, Ali A, Manoukian AS, Woodgett JR (1999).Regulation of the protein kinase activity of Shaggy(Zeste-white3)by components of the wingless pathway in Drosophila cells andembryos. J Biol Chem 274: 21790–21796.
Ryder J, Su Y, Liu F, Li B, Zhou Y, Ni B (2003). Divergent roles ofGSK3 and CDK5 in APP processing. Biochem Biophys ResCommun 312: 922–929.
Ryves WJ, Harwood AJ (2001). Lithium inhibits glycogen synthasekinase-3 by competition for magnesium. Biochem Biophys ResCommun 280: 720–725.
Sadoul R (1998). Bcl-2 family members in the development anddegenerative pathologies of the nervous system. Cell Death Differ5: 805–815.
Salinas PC (1999). Wnt factors in axonal remodelling andsynaptogenesis. Biochem Soc Symp 65: 101–109.
Sassi RB, Nicoletti M, Brambilla P, Mallinger AG, Frank E, KupferDJ et al (2002). Increased gray matter volume in lithium-treatedbipolar disorder patients. Neurosci Lett 329: 243–245.
Sassi RB, Stanley JA, Axelson D, Brambilla P, Nicoletti MA,Keshavan MS et al (2005). Reduced NAA levels in thedorsolateral prefrontal cortex of young bipolar patients. Am JPsychiatry 162: 2109–2115.
Sayas CL, Moreno-Flores MT, Avila J, Wandosell F (1999). Theneurite retraction induced by lysophosphatidic acid increasesAlzheimer’s disease-like tau phosphorylation. J Biol Chem 274:37046–37052.
Scarr E, Gray L, Keriakous D, Robinson PJ, Dean B (2006).Increased levels of SNAP-25 and synaptophysin in the dorso-
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
131
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

lateral prefrontal cortex in bipolar I disorder. Bipolar Disord 8:133–143.
Schlaepfer TE, Harris GJ, Tien AY, Peng LW, Lee S, Federman EBet al (1994). Decreased regional cortical gray matter volume inschizophrenia. Am J Psychiatry 151: 842–848.
Seeds AM, Bastidas RJ, York JD (2005). Molecular definition of anovel inositol polyphosphate metabolic pathway initiated byinositol 1,4,5-trisphosphate 3-kinase activity in Saccharomycescerevisiae. J Biol Chem 280: 27654–27661.
Semba J, Kuroda Y, Takahashi R (1989). Potential antidepressantproperties of subchronic GABA transaminase inhibitors in theforced swimming test in mice. Neuropsychobiology 21: 152–156.
Shaldubina A, Buccafusca R, Johanson RA, Agam G, Belmaker RH,Berry GT et al (2007). Behavioural phenotyping of sodium-myo-inositol cotransporter heterozygous knockout mice with reducedbrain inositol. Genes Brain Behav 6: 253–259.
Shaldubina A, Johanson RA, O’Brien WT, Buccafusca R, Agam G,Belmaker RH et al (2006). SMIT1 haploinsufficiency causesbrain inositol deficiency without affecting lithium-sensitivebehavior. Mol Genet Metab 88: 384–388.
Shaldubina A, Ju S, Vaden DL, Ding D, Belmaker RH, GreenbergML (2002). Epi-inositol regulates expression of the yeast INO1gene encoding inositol-1-P synthase. Mol Psychiatry 7: 174–180.
Shaltiel G, Chen G, Manji HK (2007). Neurotrophic signalingcascades in the pathophysiology and treatment of bipolardisorder. Curr Opin Pharmacol 7: 22–26.
Sheline YI, Gado MH, Kraemer HC (2003). Untreated depressionand hippocampal volume loss. Am J Psychiatry 160: 1516–1518.
Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier M(1996). Hippocampal atrophy in recurrent major depression.Proc Natl Acad Sci USA 93: 3908–3913.
Soares JC, Young A (2007). Bipolar Disorder: Basic Mechanismsand Therapeutic Implications. Marcel Dekker, Inc.: New York.
Son H, Yu IT, Hwang SJ, Kim JS, Lee SH, Lee YS et al (2003).Lithium enhances long-term potentiation independently ofhippocampal neurogenesis in the rat dentate gyrus. J Neurochem85: 872–881.
Song L, De Sarno P, Jope RS (2002). Central role of glycogensynthase kinase-3beta in endoplasmic reticulum stress-inducedcaspase-3 activation. J Biol Chem 277: 44701–44708.
Spiegelberg BD, Xiong JP, Smith JJ, Gu RF, York JD (1999).Cloning and characterization of a mammalian lithium-sensitivebisphosphate 30-nucleotidase inhibited by inositol 1,4-bispho-sphate. J Biol Chem 274: 13619–13628.
Stambolic V, Ruel L, Woodgett J (1996). Lithium inhibits glycogensynthase kinase-3 activity and mimics wingless signalling inintact cells. Curr Biol 6: 1664–1668.
Stoll AL, Renshaw PF, Yurgelen-Todd DA, Cohen BM (2000).Neuroimaging in bipolar disorder: what have we learned? BiolPsychiatry 48: 505–517.
Stork C, Renshaw PF (2005). Mitochondrial dysfunction in bipolardisorder: evidence from magnetic resonance spectroscopyresearch. Mol Psychiatry 10: 900–919.
Strakowski SM, DelBello MP, Zimmerman ME, Getz GE, Mills NP,Ret J et al (2002). Ventricular and periventricular structuralvolumes in first- versus multiple-episode bipolar disorder. Am JPsychiatry 159: 1841–1847.
Strittmatter SM (1992). GAP-43 as a modulator of G proteintransduction in the growth cone. Perspect Dev Neurobiol 1: 13–19.
Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P et al (2004). Lithium,a common drug for bipolar disorder treatment, regulatesamyloid-beta precursor protein processing. Biochem 43:6899–6908.
Suppes T, Baldessarini RJ, Faedda GL, Tohen M (1991). Risk ofrecurrence following discontinuation of lithium treatment inbipolar disorder. Arch Gen Psychiatry 48: 1082–1088.
Tanizawa Y, Kuhara A, Inada H, Kodama E, Mizuno T, Mori I(2006). Inositol monophosphatase regulates localization of
synaptic components and behavior in the mature nervoussystem of C. elegans. Genes Dev 20: 3296–3310.
Terao T, Nakano H, Inoue Y, Okamoto T, Nakamura J, Iwata N(2006). Lithium and dementia: a preliminary study. ProgNeuropsychopharmacol Biol Psychiatry 30: 1125–1128.
Thomas AJ, O’Brien JT, Davis S, Ballard C, Barber R, Kalaria RNet al (2002). Ischemic basis for deep white matter hyperinten-sities in major depression: a neuropathological study. Arch GenPsychiatry 59: 785–792.
Tian SY, Wang JF, Bezchlibnyk YB, Young LT (2007). Immuno-reactivity of 43 kDa growth-associated protein is decreased inpost mortem hippocampus of bipolar disorder and schizophre-nia. Neurosci Lett 411: 123–127.
Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T,Jones PB et al (2003). Oligodendrocyte dysfunction in schizo-phrenia and bipolar disorder. Lancet 362: 798–805.
Trowbridge JJ, Xenocostas A, Moon RT, Bhatia M (2006). Glycogensynthase kinase-3 is an in vivo regulator of hematopoietic stemcell repopulation. Nat Med 12: 89–98.
Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI(2004). Oligodendroglial density in the prefrontal cortex inschizophrenia and mood disorders: a study from the StanleyNeuropathology Consortium. Schizophr Res 67: 269–275.
Vawter MP, Thatcher L, Usen N, Hyde TM, Kleinman JE, Freed WJ(2002). Reduction of synapsin in the hippocampus ofpatients with bipolar disorder and schizophrenia. Mol Psychiatry7: 571–578.
Volz HP, Rzanny R, Riehemann S, May S, Hegewald H, Preussler Bet al (1998). 31P magnetic resonance spectroscopy in the frontallobe of major depressed patients. Eur Arch Psychiatry ClinNeurosci 248: 289–295.
Washizuka S, Kakiuchi C, Mori K, Kunugi H, Tajima O, Akiyama Tet al (2003). Association of mitochondrial complex I subunitgene NDUFV2 at 18p11 with bipolar disorder. Am J Med Genet120B: 72–78.
Wellcome Trust Case Control Consortium (2007). Genome-wideassociation study of 14 000 cases of seven common diseases and3,000 shared controls. Nature 447: 661–678.
Williams JM, Thompson VL, Mason-Parker SE, Abraham WC,Tate WP (1998). Synaptic activity-dependent modulation ofmitochondrial gene expression in the rat hippocampus. BrainRes Mol Brain Res 60: 50–56.
Williams RS, Cheng L, Mudge AW, Harwood AJ (2002). A commonmechanism of action for three mood-stabilizing drugs. Nature417: 292–295.
Winsberg ME, Sachs N, Tate DL, Adalsteinsson E, Spielman D,Ketter TA (2000). Decreased dorsolateral prefrontal N-acetylaspartate in bipolar disorder. Biol Psychiatry 47: 475–481.
Woodgett JR (1990). Molecular cloning and expression of glycogensynthase kinase-3/factor A. EMBO J 9: 2431–2438.
Xu J, Culman J, Blume A, Brecht S, Gohlke P (2003). Chronictreatment with a low dose of lithium protects the brain againstischemic injury by reducing apoptotic death. Stroke 34: 1287–1292.
Yang F, He XP, Russell J, Lu B (2003). Ca2+ influx-independentsynaptic potentiation mediated by mitochondrial Na(+)-Ca2+exchanger and protein kinase C. J Cell Biol 163: 511–523.
Yang L, Matthews RT, Schulz JB, Klockgether T, Liao AW,Martinou J-C et al (1998). 1-Methyl-4-phenyl-1,2,3,6-tetrahy-dropyride neurotoxicity is attenuated in mice overexpressingBcl-2. J Neurosci 18: 8145–8152.
Yin L, Wang J, Klein PS, Lazar MA (2006). Nuclear receptor Rev-erbalpha is a critical lithium-sensitive component of thecircadian clock. Science 311: 1002–1005.
Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A,Kaibuchi K (2005). GSK-3beta regulates phosphorylation ofCRMP-2 and neuronal polarity. Cell 120: 137–149.
Young LT (2007). Is bipolar disorder a mitochondrial disease?J Psychiatry Neurosci 32: 160–161.
Cellular plasticity cascades in BPDRJ Schloesser et al
...............................................................................................................................................................
132
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW

Yuan PX, Huang LD, Jiang YM, Gutkind JS, Manji HK, Chen G(2001). The mood stabilizer valproic acid activates mitogen-activated protein kinases and promotes neurite growth. J BiolChem 276: 31674–31683.
Yucel K, Taylor VH, McKinnon MC, Macdonald K, Alda M, YoungLT et al (2007). Bilateral hippocampal volume increase inpatients with bipolar disorder and short-term lithium treatment.Neuropsychopharmacology [E-pub ahead of print; doi:10.1038/sj.npp.1301405].
Zarate Jr CA, Quiroz JA, Singh JB, Denicoff KD, De Jesus G,Luckenbaugh DA et al (2005). An open-label trial of theglutamate-modulating agent riluzole in combination withlithium for the treatment of bipolar depression. Biol Psychiatry57: 430–432.
Zarate Jr CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R,Luckenbaugh DA et al (2006a). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant majordepression. Arch Gen Psychiatry 63: 856–864.
Zarate CA, Singh JB, Carlson PJ, Quiroz J, Jolkovsky L,Luckenbaugh D et al (2007). Efficacy of a protein kinase C
Inhibitor (Tamoxifen) in the treatment of acute mania: a pilotstudy. Bip Disord 9: 561–570.
Zarate Jr CA, Singh JB, Manji HK (2006b). Cellular plasticitycascades: targets for the development of novel therapeutics forbipolar disorder. Biol Psychiatry 59: 1006–1020.
Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS (2003). Inhibitoryphosphorylation of glycogen synthase kinase-3 (GSK-3) inresponse to lithium. Evidence for autoregulation of GSK-3.J Biol Chem 278: 33067–33077.
Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD (2004). NGF-induced axon growth is mediated by localized inactivation ofGSK-3beta and functions of the microtubule plus end bindingprotein APC. Neuron 42: 897–912.
Zhou R, Gray NA, Yuan P, Li X, Chen J, Chen G et al (2005). Theanti-apoptotic, glucocorticoid receptor cochaperone proteinBAG-1 is a long-term target for the actions of mood stabilizers.J Neurosci 25: 4493–4502.
Zipursky RB, Seeman MV, Bury A, Langevin R, Wortzman G,Katz R (1997). Deficits in gray matter volume are present inschizophrenia but not bipolar disorder. Schizophr Res 26: 85–92.
Cellular plasticity cascades in BPDRJ Schloesser et al...............................................................................................................................................................
133
..............................................................................................................................................
Neuropsychopharmacology REVIEWS
REVIEW




















