Molecular determinants of Akt-induced keratinocyte transformation
12
ORIGINAL ARTICLE Molecular determinants of Akt-induced keratinocyte transformation C Segrelles 1,5 , M Moral 1,5 , M Fernanda Lara 1 , S Ruiz 1,3 , M Santos 1 , H Leis 1,4 , R Garcı´a-Escudero 1 , AB Martı´nez-Cruz 1 , J Martı´nez-Palacio 1 , P Herna´ndez 1 , C Ballestı´n 2 and JM Paramio* ,1 1 Department of Cell and Molecular Biology, CIEMAT, Madrid, Spain and 2 Department of Pathology, Hospital ‘12 de Octubre’, Madrid, Spain The PI3K/PTEN/Akt signaling pathway has emerged in recent years as a main player in human cancers, increasing proliferation and decreasing apoptosis of transformed cells, and thus becoming a potential target for therapeutic intervention. Our previous data have demonstrated that Akt-mediated signaling is of a key relevance in the mouse skin carcinogenesis system, one of the best-known models of experimental carcinogenesis. Here, we investigated the involvement of several pathways as mediators of Akt- induced increased proliferation and tumorigenesis in keratinocytes. Tumors produced by subcutaneous injec- tion of Akt-transformed keratinocytes showed increased Foxo3a phosphorylation, but no major alterations in p21 Cip1/WAF1 , p27 Kip1 or mdm2 expression and/or localiza- tion. In contrast, we found increased expression and nuclear localization of DNp63, b-catenin and Lef1. Concomitantly, we also found increased expression of c- myc and CycD1, targets of the b-catenin/Tcf pathway. Such increase is associated with increased phosphoryla- tion and stabilization of c-myc protein as well as increased translation of c-myc and CycD1 due to mTOR activation. Using immunohistochemistry approaches in samples of oral dysplasias and human head and neck squamous cell carcinomas, we confirmed that increased Akt activation significantly correlates with increased DNp63 and CycD expression, c-myc phosphorylation and nuclear accumula- tion of b-catenin. Collectively, these results demonstrate that Akt is able to transform keratinocytes by specific mechanisms involving transcriptional and post-transcrip- tional processes. Oncogene (2006) 25, 1174–1185. doi:10.1038/sj.onc.1209155; published online 17 October 2005 Keywords: Akt; myc; CycD; keratinocyte; HNSCC; Wnt pathway Introduction Aberrant activation of the PI3K/PTEN/Akt pathway has been widely implicated in many human cancers. Multiple oncogenic insults can increase Akt activity, affecting a number of putative substrates, which includes a plethora of different signaling intermediates whose activity primarily modulates cell death and proliferation (reviewed in Lawlor and Alessi, 2001; Vivanco and Sawyers, 2002; Luo et al., 2003). As a consequence, possible therapies targeting the Akt path- way alone, or in conjunction with other chemother- apeutics, might be suitable cancer treatments (Luo et al., 2003). However, this possibility should be preceded by a complete characterization of Akt responses in model systems. Mouse cancer models have been invaluable in understanding the process of tumorigenesis: they allow defined tests of tumor genetics, close analysis of tumor pathology and validation of therapeutic targets. The mouse skin carcinogenesis system is one of the best- characterized models and has provided an important instrumental framework for understanding many of the concepts currently applied to human cancers. This system displays many parallelisms with certain human tumors, such as head and neck squamous cell carci- nomas (HNSCC) (reviewed in Slaga et al., 1996; Yuspa et al., 1996). Others and we have provided evidence on the particular relevance of the PI3K-Akt pathway in mouse skin carcinogenesis (Santos et al., 2002; Segrelles et al., 2002; Suzuki et al., 2003). However, it is important to consider that many genetic events and general changes in the expression profile occurred during the malignant progression of mouse skin tumors, thus making it difficult to ascertain what possible targets are fundamental and what are secondary outcome of the process. As a consequence, possible models that show increased tumorigenic properties upon a limited and controlled number of experimental alterations are also relevant. We have used the PB keratinocyte cell line for these purposes. These cells were obtained from a chemically induced mouse skin papilloma, but they do not show increased EGFR expression nor mutations in Ha-ras gene (Yuspa et al., 1986; Casanova et al., 2002). Moreover, when PB keratinocytes are used in xenograft experiments, they are poorly tumorigenic: few tumors are generated, and those obtained display very long latency, reduced growth rate and highly differentiated Received 20 January 2005; revised 5 July 2005; accepted 23 August 2005; published online 17 October 2005 *Correspondence: Dr JM Paramio, Department of Cell and Molecular Biology, CIEMAT (ed. 7), Ave. Complutense 22, Madrid E-28040, Spain. E-mail: [email protected] 5 These two authors contributed equally to this work. 3 Current address: Cancer Research Center and Institute of Cell and Molecular Biology of Cancer, CSIC-University of Salamanca, P Universidad de Coimbra s/n, Salamanca, Spain. 4 Current address: Genomics and Pharmacoproteomics Program, FVIB-CIPF, Camino de las Moreras, Valencia, Spain. Oncogene (2006) 25, 1174–1185 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc
Transcript of Molecular determinants of Akt-induced keratinocyte transformation

ORIGINAL ARTICLE
Molecular determinants of Akt-induced keratinocyte transformation
C Segrelles1,5, M Moral1,5, M Fernanda Lara1, S Ruiz1,3, M Santos1, H Leis1,4, R Garcıa-Escudero1,AB Martınez-Cruz1, J Martınez-Palacio1, P Hernandez1, C Ballestın2 and JM Paramio*,1
1Department of Cell and Molecular Biology, CIEMAT, Madrid, Spain and 2Department of Pathology, Hospital ‘12 de Octubre’,Madrid, Spain
The PI3K/PTEN/Akt signaling pathway has emerged inrecent years as a main player in human cancers, increasingproliferation and decreasing apoptosis of transformedcells, and thus becoming a potential target for therapeuticintervention. Our previous data have demonstrated thatAkt-mediated signaling is of a key relevance in the mouseskin carcinogenesis system, one of the best-known modelsof experimental carcinogenesis. Here, we investigated theinvolvement of several pathways as mediators of Akt-induced increased proliferation and tumorigenesis inkeratinocytes. Tumors produced by subcutaneous injec-tion of Akt-transformed keratinocytes showed increasedFoxo3a phosphorylation, but no major alterations inp21Cip1/WAF1, p27Kip1 or mdm2 expression and/or localiza-tion. In contrast, we found increased expression andnuclear localization of DNp63, b-catenin and Lef1.Concomitantly, we also found increased expression of c-myc and CycD1, targets of the b-catenin/Tcf pathway.Such increase is associated with increased phosphoryla-tion and stabilization of c-myc protein as well as increasedtranslation of c-myc and CycD1 due to mTOR activation.Using immunohistochemistry approaches in samples oforal dysplasias and human head and neck squamous cellcarcinomas, we confirmed that increased Akt activationsignificantly correlates with increased DNp63 and CycDexpression, c-myc phosphorylation and nuclear accumula-tion of b-catenin. Collectively, these results demonstratethat Akt is able to transform keratinocytes by specificmechanisms involving transcriptional and post-transcrip-tional processes.Oncogene (2006) 25, 1174–1185. doi:10.1038/sj.onc.1209155;published online 17 October 2005
Keywords: Akt; myc; CycD; keratinocyte; HNSCC;Wnt pathway
Introduction
Aberrant activation of the PI3K/PTEN/Akt pathwayhas been widely implicated in many human cancers.Multiple oncogenic insults can increase Akt activity,affecting a number of putative substrates, whichincludes a plethora of different signaling intermediateswhose activity primarily modulates cell death andproliferation (reviewed in Lawlor and Alessi, 2001;Vivanco and Sawyers, 2002; Luo et al., 2003). As aconsequence, possible therapies targeting the Akt path-way alone, or in conjunction with other chemother-apeutics, might be suitable cancer treatments (Luo et al.,2003). However, this possibility should be preceded by acomplete characterization of Akt responses in modelsystems. Mouse cancer models have been invaluable inunderstanding the process of tumorigenesis: they allowdefined tests of tumor genetics, close analysis of tumorpathology and validation of therapeutic targets. Themouse skin carcinogenesis system is one of the best-characterized models and has provided an importantinstrumental framework for understanding many of theconcepts currently applied to human cancers. Thissystem displays many parallelisms with certain humantumors, such as head and neck squamous cell carci-nomas (HNSCC) (reviewed in Slaga et al., 1996; Yuspaet al., 1996). Others and we have provided evidence onthe particular relevance of the PI3K-Akt pathway inmouse skin carcinogenesis (Santos et al., 2002; Segrelleset al., 2002; Suzuki et al., 2003). However, it isimportant to consider that many genetic events andgeneral changes in the expression profile occurredduring the malignant progression of mouse skin tumors,thus making it difficult to ascertain what possible targetsare fundamental and what are secondary outcome of theprocess. As a consequence, possible models that showincreased tumorigenic properties upon a limited andcontrolled number of experimental alterations are alsorelevant. We have used the PB keratinocyte cell line forthese purposes. These cells were obtained from achemically induced mouse skin papilloma, but they donot show increased EGFR expression nor mutations inHa-ras gene (Yuspa et al., 1986; Casanova et al., 2002).Moreover, when PB keratinocytes are used in xenograftexperiments, they are poorly tumorigenic: few tumorsare generated, and those obtained display very longlatency, reduced growth rate and highly differentiated
Received 20 January 2005; revised 5 July 2005; accepted 23 August 2005;published online 17 October 2005
*Correspondence: Dr JM Paramio, Department of Cell and MolecularBiology, CIEMAT (ed. 7), Ave. Complutense 22, Madrid E-28040,Spain.E-mail: [email protected] two authors contributed equally to this work.3Current address: Cancer Research Center and Institute of Cell andMolecular Biology of Cancer, CSIC-University of Salamanca, PUniversidad de Coimbra s/n, Salamanca, Spain.4Current address: Genomics and Pharmacoproteomics Program,FVIB-CIPF, Camino de las Moreras, Valencia, Spain.
Oncogene (2006) 25, 1174–1185& 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00
www.nature.com/onc

phenotypes (Yuspa et al., 1986; Casanova et al., 2002;Segrelles et al., 2002). On the other hand, increasedexpression of Akt, and thus increased activity, promoteda dramatic enhancement in tumorigenic behavior, withreduced latency, increased growth rate and less differ-entiated phenotypes (Supplementary Figure 1, see alsoSegrelles et al., 2002, 2004). These effects are due to cellautonomous effects, that is, augmented cell proliferationand decreased apoptosis, and to the interaction with thestroma–tumor environment, that is, increased angio-genic response (Segrelles et al., 2004).The aim of the present study is to investigate potential
targets of Akt that may mediate the cell autonomouseffects leading to increased tumorigenesis using the PBsystem. We report that Akt directly controls theexpression of specific targets both at the transcriptionaland post-transcriptional level. Interestingly, we alsofound that these alterations were also characteristic ofhuman HNSCC where Akt activity is deregulated.
Results
The specific features of the PB keratinocyte cell line inxenograft experiments (see Introduction) make thissystem a perfectly suited method to study molecularchanges due to Akt-induced transformation. Thesechanges can be further subdivided between thoseaffecting the tumoral injected cells, and those affectingthe crosstalk between these cells and the surroundingstroma. In this regard, the increased growth rate inxenografted tumors is associated with increased angio-genesis mediated by post-transcriptional regulation ofVEGF (Segrelles et al., 2004). In addition, others and wehave previously shown that Akt-mediated inhibition ofGSK3b modulates the levels and cellular distribution ofcyclin D1 (Diehl et al., 1998; Leis et al., 2002), whichmight explain increased proliferation. Here, we exploredother mechanisms that might increase the oncogenicpotential in a cell-autonomous manner.
Alterations in p21Cip1/WAF1, p27Kip1, mdm2 and Foxo3aAmong the many different Akt targets, the phosphory-lation of p21Cip1/WAF1 and p27Kip1, resulting in the alteredlocalization of these proteins, leads to cell cycleprogression (Zhou et al., 2001a; Blagosklonny, 2002;Liang et al., 2002; Mayo and Donner, 2002; Shin et al.,2002; Viglietto et al., 2002). In addition, Akt can alsophosphorylate mdm2, favoring mdm2–p53 associationand leading to subsequent p53 degradation (Mayo andDonner, 2001; Ogawara et al., 2002; Zhou and Hung,2002). Finally, Akt can also phosphorylate severalforkhead transcription factors, including Foxo3a, in-hibiting the expression of Foxo-regulated genes, whichcontrol the cell cycle and cell death (Burgering andKops, 2002). Consequently, one might expect that Aktthrough the direct modulation of the cyclin-dependentkinase inhibitors or indirectly through the downregula-tion of p53 or Foxo3a activity might increase the cellproliferation in keratinocytes.
We studied the expression and localization of theseproteins in tumors obtained with control PB cells orwith Akt-transfected cells upon subcutaneous injection.We found that in tumors generated by Akt-transfectedkeratinocytes, the localization and the expression ofp21Cip1/WAF1, p27Kip1 and mdm2 were similar to thatfound in control tumors (Figure 1a, and data notshown). On the contrary, we also found increasedphosphorylation of Foxo3a leading to cytoplasmiclocalization (Figure 1a, and data not shown). Asexpected, we found increased Akt expression(Figure 1a) and, concomitantly, increased Akt activityin those tumors generated by Akt-transfected keratino-cytes (Figure 1b). These results indicate that, in thissystem, the Akt-mediated modulation with p21Cip1/WAF1,p27Kip1 and/or mdm2 observed in other models is not amajor responsible factor for the observed increase intumorigenic behavior.
Increased activation of the Wnt signaling pathwaymediated by AktIt has been reported that Akt may interfere withelements of the Wnt pathway leading to increasedexpression and activity of Wnt effectors (Delcommenneet al., 1998; D’Amico et al., 2000; Desbois-Mouthonet al., 2001; Gotoh et al., 2003). Moreover, p63 a directtarget of PI3K pathway (Barbieri et al., 2003) can alsomodulate nuclear accumulation of b-catenin (Patturajanet al., 2002; Koga et al., 2003). To study the possiblecontribution of this pathway to Akt-induced transfor-mation, we monitored the expression and localization ofb-catenin, Lef1 and DNp63a. We found increasedexpression and increased nuclear accumulation of b-catenin (Figure 1c0 and f) and Lef1 (Figure 1d0 and f)compared with the correspondent controls (Figure 1c, dand f). With respect to DNp63a, we found a highnumber of the tumoral cells expressing this protein incontrol samples (Figure 1e). However, this is furtherincreased in Akt-derived samples, which also displayedincreased expression (Figure 1e0 and f).These data would suggest that Akt may induce the
transcriptional activation of b-catenin/Tcf-responsiveelements. To monitor this aspect, we performedluciferase reporter assays. We observed that Aktcotransfection induces activity from a reported plasmidcontaining Wnt-, but not mutant-, responsive elements(Figure 1g). These results confirmed that Akt is able toinduce the transcriptional activation of the Wnt path-way
Akt mediates increased expression and stability of c-mycinduced by AktAmong the multiple target genes induced by the b-catenin/Tcf pathway, c-myc and cyclin D1 (Roose andClevers, 1999; Barker and Clevers, 2000) might be ofparticular relevance. We monitored c-myc and cyclin D1gene expression in control and Akt-derived tumors. Asexpected, Northern blot analysis showed that bothgenes were expressed to a higher level in those tumorsgenerated by Akt-transfected keratinocytes (Figure 2a).
Akt-induced signaling during keratinocyte transformationC Segrelles et al
1175
Oncogene

At the protein level, others and we have previouslyshown that Akt mediates an increase in cyclin D1expression and nuclear localization through the inter-action with GSK3b (Diehl et al., 1998; Leis et al., 2002),concerning c-myc alterations, Western blot analyses andimmunostaining (Figure 2b, c and c0) showed that thelevel of expression of c-myc is high in those tumorsgenerated by Akt-transfected keratinocytes (Figure 2b,c0 and d). It is important to remark that c-mycexpression is not only regulated by transcriptionalmechanisms but also post-transcriptionally. These in-clude increased translation (Prats and Prats, 2002) andstability (Sears et al., 2000). Augmented translation of c-myc mRNA is mediated by p70S6K and mTOR. Asboth pathways are activated by Akt in this system(Segrelles et al., 2004), at least part of the increase in c-myc can be due to increased translation. Thus, wemonitored the possible increased c-myc stability due toAkt. In this regard, Ser62 phosphorylation of c-mycleads to augmented activity and stabilization, whereasphosphorylation in Thr58, mediated by GSK3b anddependent on the prior Ser62 phosphorylation, leads toc-myc degradation (Sears et al., 2000). Consequently, we
studied the possible alterations in c-myc phosphoryla-tion. Using an antibody reacting with both Ser62/Thr58-phosphorylated c-myc, we found increased c-mycphosphorylation in tumors obtained by subcutaneousinjection of Akt-transfected keratinocytes by Westernblot (Figure 2b) and immunostaining (Figure 2d0). Inparallel, we observed increased Ser9 phosphorylation ofGSK3b (Figure 2b). As this phosphorylation inactivatesGSK3b, the results might indicate that Akt mediates thisinactivation, thus suggesting that c-myc is stabilized. Toconfirm this, as it cannot be monitored in tumorsamples, we studied possible c-myc stabilization incultured control and Akt-transfected cells. Increasedlevels of c-myc and phosphorylated c-myc were observedin pooled clones of Akt-transfected keratinocytes whencompared with control cells (Figure 2e) similar to tumorsamples. In addition, the activity of Akt is also increasedin cultured cells (Figure 2e0). To investigate the stabilityof c-myc, protein synthesis was inhibited by treatmentwith cycloheximide (50 mM) and the levels of c-myc atdifferent times were determined by Western blotting. Inparental control cells, the levels of c-myc rapidlydecrease being o5% of the initial amounts after
Figure 1 Akt induces b-catenin, Lef1 and DNp63, and Foxo3a phosphorylation, but not p21Cip1/Waf, p27Kip1, mdm2 changes inkeratinocytes. (a) The expression of p21Cip1/Waf, p27Kip1, mdm2, Foxo3a and phosphorylated Foxo3a were analysed by Western blottingof whole extracts from tumors generated by subcutaneous injection of control pcDNA3- or Akt-transfected PB. The levels of Akt weresignificantly increased in the tumors generated by Akt-transfected keratinocytes (a) leading to increased activity of Akt kinase afterimmunoprecipitation (b). The expression and localization of b-catenin (c, c0), Lef1 (d, d0) and DNp63 (e, e0) were analysed byimmunohistochemical methods in tumors generated by subcutaneous injection of control-pcDNA3-transfected (c, d, e) or Akt-transfected (c0, d0, e0) PB keratinocytes. (f) Western blot analysis of whole tumor extracts confirming the increased expression observedby immunohistochemistry. (g) Transfection with Akt induces the b-catenin/Tcf transcriptional activation. Cultured cells werecotransfected with Akt or empty vector (control) and either TOPflash (closed bars) or mutant FOPflash (open bars). At 36 h aftertransfection, luciferase activity was monitored. Note the increase in b-catenin/Tcf activity promoted by Akt expression. Data comefrom triplicate experiments and are shown as mean7s.d. Actin was used in (a) and (f) to normalize the loading. Bars¼ 100mM.
Akt-induced signaling during keratinocyte transformationC Segrelles et al
1176
Oncogene

70min (Figure 2f and f0), thus giving a half-life of 37min(Figure 2f0). Conversely, in Akt-transfected PB cells,80min after protein synthesis inhibition, the levels of c-myc are 21% of those observed at time 0 (Figure 2f andf0), giving a half-life of 57min (Figure 2f0). These dataindicate that Akt mediates increased stabilization ofc-myc in keratinocytes.
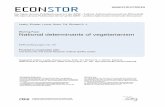
Increased activation of the Wnt signaling pathway inmouse skin carcinogenesisThe above-commented results provide information onthe changes downstream of Akt that occur in tumorsinduced by transfection of PB keratinocytes with an Aktexpression vector. To compare these data with thechanges in primary tumors generated in mouse skin bytwo-stage chemical carcinogenesis, we analysed theexpression of the above-commented targets (c-mycCycD and DNp63a) in parallel with Akt and phos-phorylated Akt in papillomas and squamous cellcarcinomas (SCCs). In addition, we included normalskin and 12-O-tetradecanoyl-phorbol-13-acetate (TPA)-treated skin samples. In agreement with our previousdata (Segrelles et al., 2002), we observed (Figure 3)increased activation of Akt, without increased expres-sion, in papillomas and SCC. Similarly, the expressionof c-myc, CycD and DNp63a was also increased intumors (Figure 3). Furthermore, the expression ofGSK3b was increased in papillomas and SCCs, and itsactivity was downregulated by Ser 9 phosphorylation(Figure 3), as we described previously (Leis et al., 2002).In agreement with such decreased GSK3b activity, wehave also found increased b-catenin and Lef1 expression(Figure 3). Collectively, the results show that thechanges observed in the PB keratinocyte system alsotake place in mouse skin upon two-stage chemicalcarcinogenesis protocol.
Increased expression and stability of targets is directlyinduced by AktThe multiple target proteins induced by increased Aktfound in tumors could be directly due to Akt or mightbe acquired during the extensive in vitro propagation ofthe cells, leading to increased malignancy. To discrimi-nate between these two possibilities, we carried out theanalysis of the above-mentioned elements using parentalcells 48 h after the expression of wild-type (wt) Akt.Western blot analyses (Figure 4) demonstrate thatincreased Akt expression leads to increased Akt activity,in parallel with increased cyclin D1, c-myc, b-catenin,Lef1 and DNp63. In addition, increased Akt activityalso leads to increased phosphorylation of c-myc andp70S6K. On the contrary, we did not observe alteredexpression or activity of Ha-ras, ERK1/2 or JNK1/2. Inaddition, we also monitored the effect of several specificinhibitors: PD98059 (inhibitor of ERK), SB203580(inhibitor of p38), Wortmanin (inhibitor of PI-3K) andrapamycin (inhibitor of mTOR). This experiment alsoallowed us to study how other pathways can influencethe observed molecular changes. As expected, the levelsof cyclin D1 and c-myc decrease in Akt-expressing cells
Figure 2 Akt induces c-myc expression and stabilization. (a) TotalmRNA from different tumor samples was probed for theexpression of c-myc and cyclin D1 by Northern blotting. Tocontrol loading, a 7S probe was used. (b) Western blot of totalprotein extracts from tumors generated by subcutaneous injectionof control or Akt-transfected PB keratinocytes probed against thequoted proteins. (c–d0) Immunohistochemical detection of c-myc(c, c0) and phosphorylated c-myc (d, d0) in control (c, d) and Akt-derived tumors (c0, d0). (e, e0) Protein extracts from pooled cellclones from control (pcDNA3) or Akt transfection were probedagainst the quoted antibodies (e). In parallel, the in vitro Akt kinaseactivity was monitored using histone 2b as a substrate (e0 lowerpanel). (f, f0) Akt induces higher half-life of c-myc protein. Culturesfrom Akt (f upper panel) or control (f lower panel) PBkeratinocytes were treated with cycloheximide (50mM) and proteinextracts were obtained at the quoted time points (in the presence ofcycloheximide) and probed for c-myc expression. (f0) Quantifica-tion of the blots shown in (f) demonstrating the increase in thec-myc half-life in Akt-transfected PB keratinocytes.
Akt-induced signaling during keratinocyte transformationC Segrelles et al
1177
Oncogene
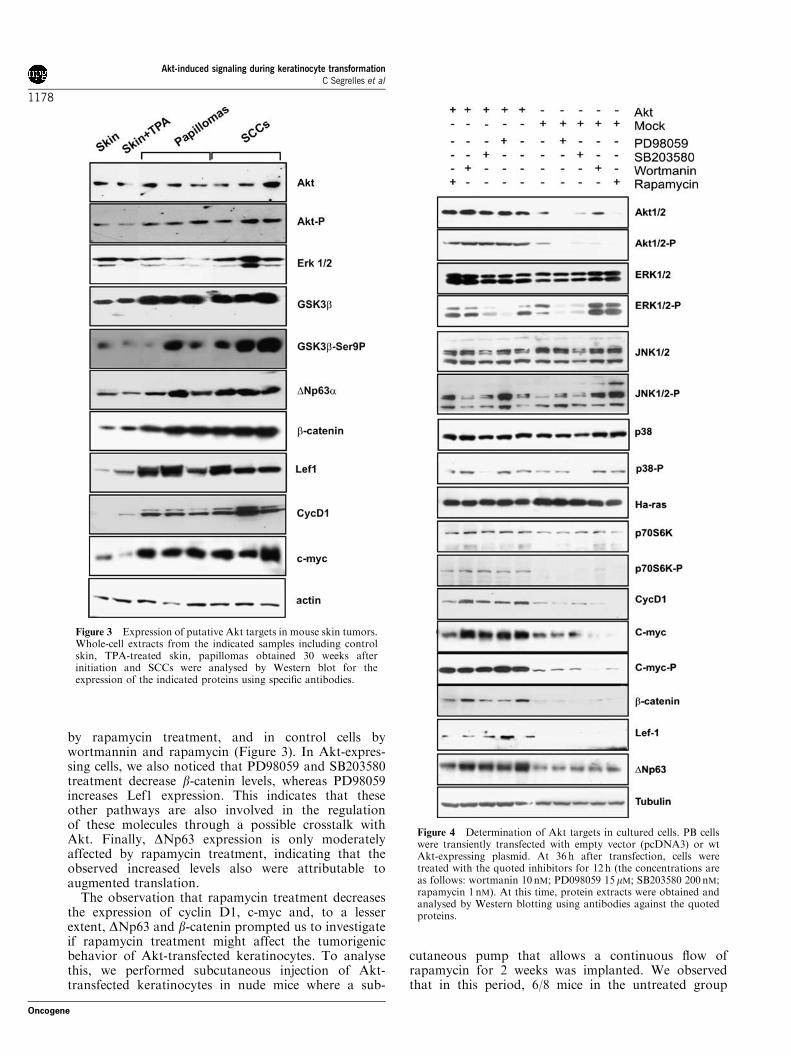
by rapamycin treatment, and in control cells bywortmannin and rapamycin (Figure 3). In Akt-expres-sing cells, we also noticed that PD98059 and SB203580treatment decrease b-catenin levels, whereas PD98059increases Lef1 expression. This indicates that theseother pathways are also involved in the regulationof these molecules through a possible crosstalk withAkt. Finally, DNp63 expression is only moderatelyaffected by rapamycin treatment, indicating that theobserved increased levels also were attributable toaugmented translation.The observation that rapamycin treatment decreases
the expression of cyclin D1, c-myc and, to a lesserextent, DNp63 and b-catenin prompted us to investigateif rapamycin treatment might affect the tumorigenicbehavior of Akt-transfected keratinocytes. To analysethis, we performed subcutaneous injection of Akt-transfected keratinocytes in nude mice where a sub-
cutaneous pump that allows a continuous flow ofrapamycin for 2 weeks was implanted. We observedthat in this period, 6/8 mice in the untreated group
Figure 3 Expression of putative Akt targets in mouse skin tumors.Whole-cell extracts from the indicated samples including controlskin, TPA-treated skin, papillomas obtained 30 weeks afterinitiation and SCCs were analysed by Western blot for theexpression of the indicated proteins using specific antibodies.
Figure 4 Determination of Akt targets in cultured cells. PB cellswere transiently transfected with empty vector (pcDNA3) or wtAkt-expressing plasmid. At 36 h after transfection, cells weretreated with the quoted inhibitors for 12 h (the concentrations areas follows: wortmanin 10 nM; PD098059 15mM; SB203580 200nM;rapamycin 1 nM). At this time, protein extracts were obtained andanalysed by Western blotting using antibodies against the quotedproteins.
Akt-induced signaling during keratinocyte transformationC Segrelles et al
1178
Oncogene
developed tumors with an average of 188773mm3
volume. In contrast, 0/8 mice developed tumors uponrapamycin treatment. These results indicate thatmTOR-mediated translation is essential for Akt-mediated keratinocyte transformation.
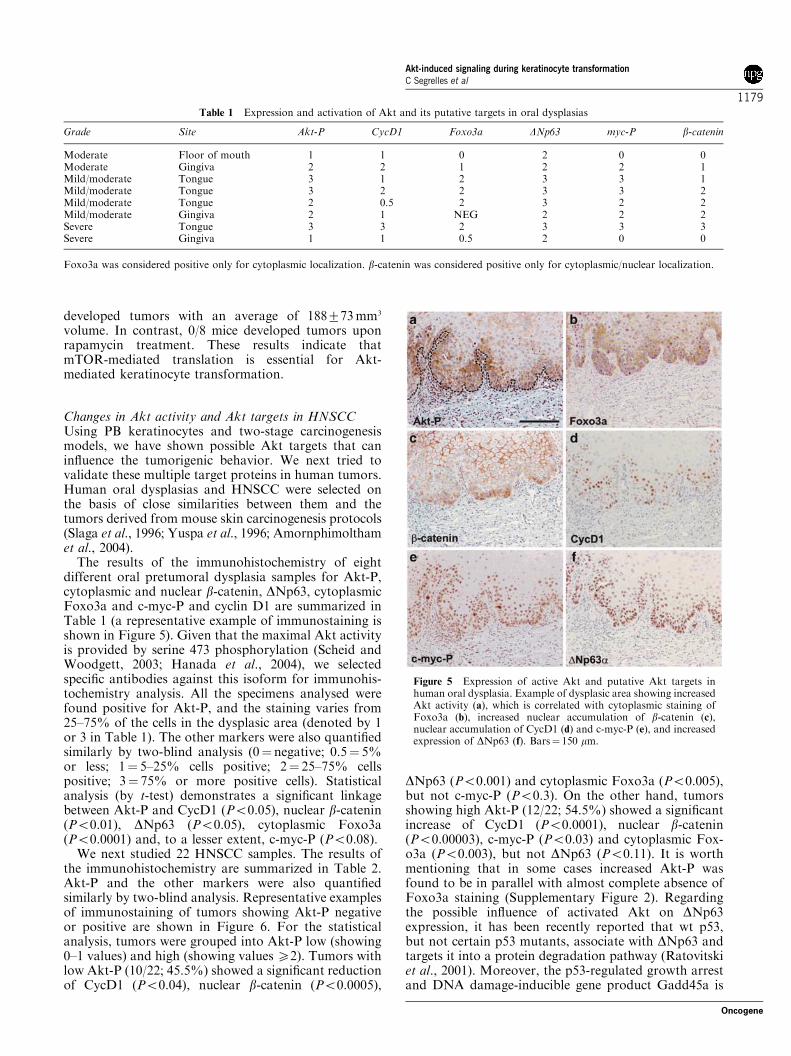
Changes in Akt activity and Akt targets in HNSCCUsing PB keratinocytes and two-stage carcinogenesismodels, we have shown possible Akt targets that caninfluence the tumorigenic behavior. We next tried tovalidate these multiple target proteins in human tumors.Human oral dysplasias and HNSCC were selected onthe basis of close similarities between them and thetumors derived from mouse skin carcinogenesis protocols(Slaga et al., 1996; Yuspa et al., 1996; Amornphimolthamet al., 2004).The results of the immunohistochemistry of eight
different oral pretumoral dysplasia samples for Akt-P,cytoplasmic and nuclear b-catenin, DNp63, cytoplasmicFoxo3a and c-myc-P and cyclin D1 are summarized inTable 1 (a representative example of immunostaining isshown in Figure 5). Given that the maximal Akt activityis provided by serine 473 phosphorylation (Scheid andWoodgett, 2003; Hanada et al., 2004), we selectedspecific antibodies against this isoform for immunohis-tochemistry analysis. All the specimens analysed werefound positive for Akt-P, and the staining varies from25–75% of the cells in the dysplasic area (denoted by 1or 3 in Table 1). The other markers were also quantifiedsimilarly by two-blind analysis (0¼ negative; 0.5¼ 5%or less; 1¼ 5–25% cells positive; 2¼ 25–75% cellspositive; 3¼ 75% or more positive cells). Statisticalanalysis (by t-test) demonstrates a significant linkagebetween Akt-P and CycD1 (Po0.05), nuclear b-catenin(Po0.01), DNp63 (Po0.05), cytoplasmic Foxo3a(Po0.0001) and, to a lesser extent, c-myc-P (Po0.08).We next studied 22 HNSCC samples. The results of
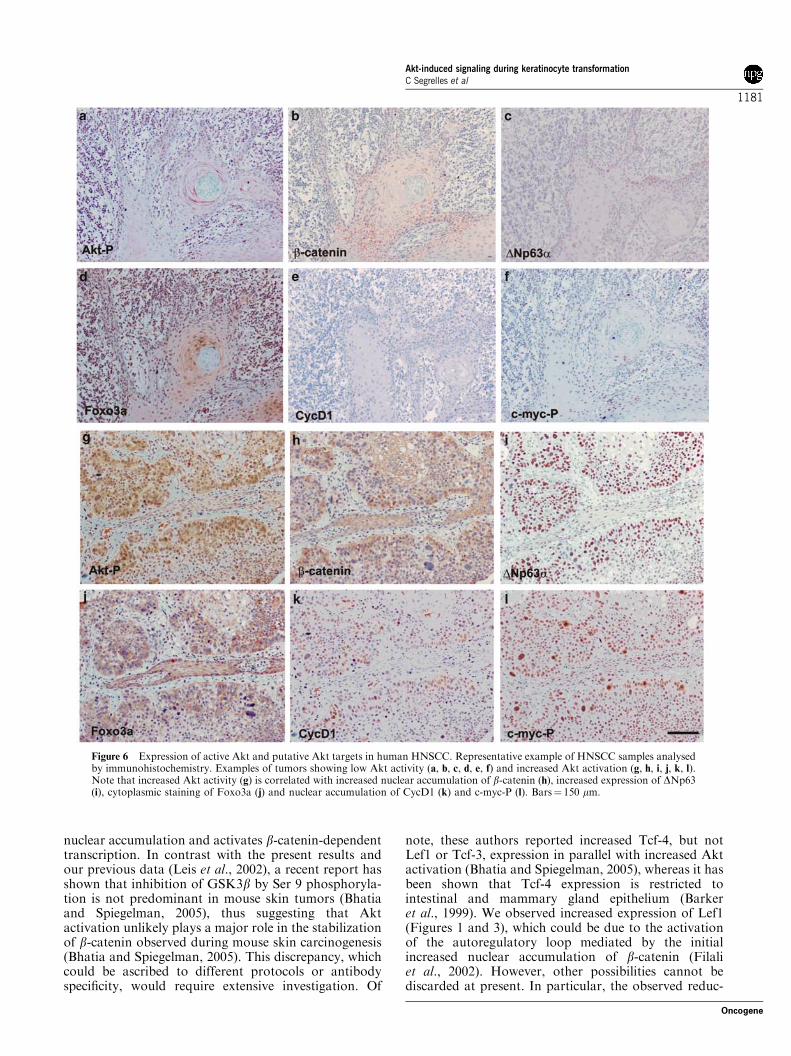
the immunohistochemistry are summarized in Table 2.Akt-P and the other markers were also quantifiedsimilarly by two-blind analysis. Representative examplesof immunostaining of tumors showing Akt-P negativeor positive are shown in Figure 6. For the statisticalanalysis, tumors were grouped into Akt-P low (showing0–1 values) and high (showing valuesX2). Tumors withlow Akt-P (10/22; 45.5%) showed a significant reductionof CycD1 (Po0.04), nuclear b-catenin (Po0.0005),
DNp63 (Po0.001) and cytoplasmic Foxo3a (Po0.005),but not c-myc-P (Po0.3). On the other hand, tumorsshowing high Akt-P (12/22; 54.5%) showed a significantincrease of CycD1 (Po0.0001), nuclear b-catenin(Po0.00003), c-myc-P (Po0.03) and cytoplasmic Fox-o3a (Po0.003), but not DNp63 (Po0.11). It is worthmentioning that in some cases increased Akt-P wasfound to be in parallel with almost complete absence ofFoxo3a staining (Supplementary Figure 2). Regardingthe possible influence of activated Akt on DNp63expression, it has been recently reported that wt p53,but not certain p53 mutants, associate with DNp63 andtargets it into a protein degradation pathway (Ratovitskiet al., 2001). Moreover, the p53-regulated growth arrestand DNA damage-inducible gene product Gadd45a is
Table 1 Expression and activation of Akt and its putative targets in oral dysplasias
Grade Site Akt-P CycD1 Foxo3a DNp63 myc-P b-catenin
Moderate Floor of mouth 1 1 0 2 0 0Moderate Gingiva 2 2 1 2 2 1Mild/moderate Tongue 3 1 2 3 3 1Mild/moderate Tongue 3 2 2 3 3 2Mild/moderate Tongue 2 0.5 2 3 2 2Mild/moderate Gingiva 2 1 NEG 2 2 2Severe Tongue 3 3 2 3 3 3Severe Gingiva 1 1 0.5 2 0 0
Foxo3a was considered positive only for cytoplasmic localization. b-catenin was considered positive only for cytoplasmic/nuclear localization.
Figure 5 Expression of active Akt and putative Akt targets inhuman oral dysplasia. Example of dysplasic area showing increasedAkt activity (a), which is correlated with cytoplasmic staining ofFoxo3a (b), increased nuclear accumulation of b-catenin (c),nuclear accumulation of CycD1 (d) and c-myc-P (e), and increasedexpression of DNp63 (f). Bars¼ 150 mm.
Akt-induced signaling during keratinocyte transformationC Segrelles et al
1179
Oncogene

an important negative regulator of DNp63 andb-catenin (Hildesheim et al., 2004). Thus, we investi-gated whether tumors showing augmented DNp63expression display positive staining for p53, suggestiveof mutated form. We observed increased DNp63 (Figure6a and b) in tumors showing negative (Figure 6a0) ornuclear staining for p53 (Figure 6b0), regardless Aktactivation status. This result might suggest that multiplefactors regulate the DNp63 increased expression inHNSCC.Overall, the results indicate that Akt activation is a
frequent event in HNSCC. In addition, we showed thatmany of the targets downstream of Akt activationobserved in the PB and mouse skin tumorigenesismodels were also activated early in these tumors, whichreinforces that Akt and some of these targets can beconsidered possible objectives in future preclinicalstudies.
Discussion
The activation of the PI3K/PTEN/Akt pathway is acommon feature in many human cancers, making thisan attractive target for molecular therapies. However, acomplete characterization to validate these therapeutictargets is a necessary prerequisite. Here, we presentevidence for specific mechanisms, due to augmented Aktactivity that might mediate increased tumorigenesis inkeratinocytes. These mechanisms would be of particularrelevance in similar types of tumors in human popula-tions, such as HNSCC. In this regard, previous datahave reported activation of Akt in these tumors inhumans (Leethanakul et al., 2000; Wang et al., 2001;
Brown et al., 2003; Amornphimoltham et al., 2004). Inagreement, we have found increased Akt activation byimmunohistochemistry in oral dysplasias and HNSCC(Figures 5 and 6 and Tables 1 and 2).Our present data support the existence of cell type-
specific mechanisms. We failed to detect alterations inp21Cip1/WAF1, p27Kip1 and/or mdm2 (Figure 1), proteinsthat have been described as putative Akt targets inbreast cancers (Zhou et al., 2001a, b; Liang et al., 2002;Shin et al., 2002; Viglietto et al., 2002). However, themajority of HNSCC also display increased p21Cip1/WAF1
and p27Kip1 expression, indicating the existence ofmechanisms by which some growing tumor cells maytolerate these cell cycle inhibitors (Erber et al., 1997;Warnakulasuriya et al., 1998). In addition, mdm2protein is expressed in HNSCC mostly at advancedstages (Ambrosch and Ruschenburg, 2000). Therefore, itis conceivable that these are not relevant targets inHNSCC.Wnt pathway is highly relevant in human tumorigen-
esis (reviewed in Novak and Dedhar, 1999; Roose andClevers, 1999; Giles et al., 2003) including HNSCC(Leethanakul et al., 2000; Rhee et al., 2002). Our dataalso support such involvement downstream of Aktactivation in the PB model system (Figure 1) and inmouse skin tumorigenesis (Figure 3). However, themolecular mechanism responsible is not known. Onemight speculate that the increased DNp63 levels,attributable to increased Akt activity (Barbieri et al.,2003), may induce inhibition of GSK3b (Patturajanet al., 2002). This effect, together with the direct abilityof Akt to phosphorylate and inhibit GSK3b (Crosset al., 1995; Diehl et al., 1998; Desbois-Mouthon et al.,2002; Leis et al., 2002) can mediate a decrease inphosphorylation levels of b-catenin, which induces
Table 2 Expression and activation of Akt and its putative targets in HNSCC
Site Grade Akt-P Cyc-D1 Foxo3a DNp63 myc-P bcatenin
Gingiva Well diff SCC 1.0 1.0 0.5 2.0 0.5 1.0Floor of mouth Well diff SCC 0.5 1.0 0.5 1.0 0.0 0.5Floor of mouth Well diff SCC 1.0 1.0 0.5 2.0 0.5 0.5Retromolar Well diff SCC 3.0 2.0 3.0 2.0 3.0 2.0Tongue Well diff SCC 0.0 1.0 0.0 1.0 0.0 0.0Tongue Well diff SCC 0.0 1.0 0.0 1.0 0.0 0.0Tongue Well diff SCC 2.0 2.0 2.0 2.0 2.0 2.0Tongue Well diff SCC 2.0 1.0 NEG 1.0 1.0 0.0Tongue Well diff SCC 3.0 2.0 NEG 1.0 2.0 0.0Tongue Well diff SCC 1.0 1.0 0.0 1.0 1.0 0.0Tongue Well diff SCC 2.0 1.0 0.5 3.0 3.0 0.5Tongue Well diff SCC 3.0 3.0 2.0 3.0 3.0 1.0Tongue Well diff SCC 3.0 2.0 NEG 3.0 3.0 2.0Tongue Well diff SCC 1.0 0.0 0.0 2.0 1.0 0.0Tongue Well diff SCC 3.0 1.0 0.0 1.0 2.0 1.0Tongue Well diff SCC 3.0 2.0 2.0 2.0 3.0 0.5Tongue Well diff SCC 1.0 2.0 0.5 1.0 1.0 0.5Tongue Well diff SCC 1.0 1.0 0.0 2.0 2.0 0.0Tongue Well diff SCC 1.0 3.0 0.0 1.0 2.0 0.0Floor of mouth Mod Diff SCC 0.5 1.0 0.5 1.0 0.0 0.0Retromolar Mod Diff SCC 2.0 2.0 1.0 2.0 1.0 2.0Tongue Poor Diff SCC 3.0 3.0 3.0 3.0 3.0 1.0
Foxo3a was considered positive only for cytoplasmic localization. b-catenin was considered positive only for cytoplasmic/nuclear localization.
Akt-induced signaling during keratinocyte transformationC Segrelles et al
1180
Oncogene
nuclear accumulation and activates b-catenin-dependenttranscription. In contrast with the present results andour previous data (Leis et al., 2002), a recent report hasshown that inhibition of GSK3b by Ser 9 phosphoryla-tion is not predominant in mouse skin tumors (Bhatiaand Spiegelman, 2005), thus suggesting that Aktactivation unlikely plays a major role in the stabilizationof b-catenin observed during mouse skin carcinogenesis(Bhatia and Spiegelman, 2005). This discrepancy, whichcould be ascribed to different protocols or antibodyspecificity, would require extensive investigation. Of
note, these authors reported increased Tcf-4, but notLef1 or Tcf-3, expression in parallel with increased Aktactivation (Bhatia and Spiegelman, 2005), whereas it hasbeen shown that Tcf-4 expression is restricted tointestinal and mammary gland epithelium (Barkeret al., 1999). We observed increased expression of Lef1(Figures 1 and 3), which could be due to the activationof the autoregulatory loop mediated by the initialincreased nuclear accumulation of b-catenin (Filaliet al., 2002). However, other possibilities cannot bediscarded at present. In particular, the observed reduc-
Figure 6 Expression of active Akt and putative Akt targets in human HNSCC. Representative example of HNSCC samples analysedby immunohistochemistry. Examples of tumors showing low Akt activity (a, b, c, d, e, f) and increased Akt activation (g, h, i, j, k, l).Note that increased Akt activity (g) is correlated with increased nuclear accumulation of b-catenin (h), increased expression of DNp63(i), cytoplasmic staining of Foxo3a (j) and nuclear accumulation of CycD1 (k) and c-myc-P (l). Bars¼ 150 mm.
Akt-induced signaling during keratinocyte transformationC Segrelles et al
1181
Oncogene

tion in b-catenin and increased Lef1 as a consequence ofPD098059 and SB203580 treatment of Akt-transfectedkeratinocytes would require further analysis. Impor-tantly, p38 has been recently involved as a negativemodulator of DNp63 and, subsequently, b-catenin(Hildesheim et al., 2004).Among the possible Wnt response genes, cyclin D1
and c-myc are significant in human tumors. Increasedcyclin D1 has been associated with mouse skintransformation (Robles and Conti, 1995; Zhang et al.,1997; Rodriguez-Puebla et al., 1998, 1999), whereas c-myc is able to induce skin tumors and also modulatesepidermal stem cell homeostasis (Pelengaris et al., 1999;Arnold and Watt, 2001; Waikel et al., 2001), the primarytarget in mouse skin cancer (DiGiovanni, 1992; Slagaet al., 1996). Together with our previous data (Leis et al.,2002), the results (Figures 2 and 3) indicate that Akt-mediated transformation of keratinocytes proceedsthrough the increased expression of c-myc and CycD1.Interestingly, such increased expression is not only dueto transcriptional processes but also post-transcriptionalevents leading to increased translation and stabilizationof c-myc and increased translation and nuclear localiza-tion of CycD1 (Leis et al., 2002). The suggestion that theincreased levels of c-myc and CycD1 are also due toincreased translation is based on experiments in Akt-expressing keratinocytes. We observed that rapamycindecreases c-myc and CycD1 levels (Figure 4), inagreement with previous reports showing that, in bothcases, translation is dependent on mTOR/eIF4E path-way (Rosenwald et al., 1995; De Benedetti and Harris,1999; Bjornsti and Houghton, 2004; Gera et al., 2004).We also observed that rapamycin treatment leads to apartial decrease in DNp63. As mTOR pathway is also atarget of Akt (Nave et al., 1999; Aoki et al., 2001; Geraet al., 2004), and in this system it can mediate theincreased translation of VEGF responsible for theincreased angiogenic response (Segrelles et al., 2004),possible drugs affecting this pathway can be consideredas attractive targets in cancer therapies. In addition, thereduced tumorigenic potential upon rapamycin treatmenthighlights the dependence on translational processesin Akt-induced transformation.HNSCC represent the sixth most common cancer and
a major cause of morbidity and mortality due to itsrelatively poor prognosis. Molecular studies havedemonstrated activation of various oncogenes (Ras,Myc, EGFR and cyclin D1) and tumor suppressor geneinactivation (TP53 and p16) in HNSCC. In agreementwith previous results (Amornphimoltham et al., 2004;Pedrero et al., 2005), we provide evidence of theinvolvement of Akt activation in the development andprogression of HNSCC. We also demonstrate that thePB model system recapitulates many of the alterationsfound in these tumors. In human dysplasias, weobserved a predominant activation of Akt, which iscorrelated with nuclear b-catenin, cytoplasmic Foxo3a,CycD1 and DNp63, whereas minor correlation wasfound with phosphorylated c-myc. Interestingly, inpapillomas produced during two-stage skin carcinogen-esis protocols, activation of Akt, increased b-catenin,
CycD1 and c-myc is also a common event (Figure 3). Inhuman HNSCC, we found similar correlation, particu-larly in those tumors showing Akt activation with asignificant increase in nuclear b-catenin, cytoplasmicFoxo3a, CycD1 and phosphorylated c-myc. We alsofound positive staining for DNp63 in some samplesshowing reduced Akt activation, whereas low Aktactivation is well correlated with low DNp63 levels.These results are in agreement with previous reportsshowing that DNp63 is overexpressed in the vastmajority of HNSCC (Sniezek et al., 2004; Gwosdzet al., 2005; Thurfjell et al., 2005), and might indicatethat, besides Akt, alternative pathways influence suchoverexpression. Although p53 may be actively involvedin this process (Hildesheim et al., 2004), our dataindicated that p53 is not relevant in the modulation ofDNP63 expression (Figure 7). In this regard, amplifica-tion of chromosome 3q21–29, where the p63 gene islocated, has also been reported in HNSCC (Thurfjellet al., 2005). In summary, we confirm that Aktactivation is a frequent event in HNSCC and, interest-ingly, in pretumoral dysplasias. Additional studies,including large sample collections and high-throughputtechniques, would be required to evaluate whether activeAkt represents a marker of diagnostic or prognosticvalue in HNSCC patients. This would allow one toassess the relevance of possible antitumoral treatmentsusing Akt or its downstream elements as putative targetseither at early or late stages of tumor development.
Materials and methods
Tumor induction protocolsTo generate tumors in nude mice, upon transfection withcontrol (pcDNA3) or Akt (pcDNA3 Akt) plasmids, PB cellswere grown (see below). Upon trypsinization, cells frompooled clones (20–60 different clones) were washed twice andresuspended in PBS (1� 106 cells/0.1ml). Afterwards, cell
Figure 7 Coexpression of DNp63 and p53 in human HNSCC.Representative examples of tumors showing high expression ofDNp63 (a, b) and the corresponding fields showing completeabsence (a0) or strong positive staining (b0) of p53. Bar¼ 100 mm.
Akt-induced signaling during keratinocyte transformationC Segrelles et al
1182
Oncogene

suspensions were subcutaneously injected into nu/numice (five-to six-week-old females; C57Bl/6 strain background) asdescribed (Segrelles et al., 2002, 2004). In a set of experiments2 days after inoculation, a continuous flow pump (Alzet 2002;Alza Corporation, Palo Alto, CA, USA) was surgicallyimplanted on the flank of every mouse near the site of tumorinoculation. The pump was filled with either vehicle or 2mg ofrapamycin (Sigma) in 250 ml PBS supplemented with 5mg/mlBSA and operated at a flux of 0.5 ml/h for 15 days. Tumorsthat formed were measured, excised and processed forhistopathology analysis, RNA and protein extraction. Simi-larly sized pieces from different tumors, with similar stroma–tumor cell content as determined by histopathology, andobtained at the same time points upon subcutaneous injection,were used in biochemical analyses. For two-stage carcinogen-esis, dorsal skin of FVB females was shaved in the resting stageof the hair cycle (8–10 weeks of age) and initiated 3 days laterwith a single dose of 7, 12-dimethyl-benz(a)anthracene(DMBA, Sigma) (100mg/200 ml in acetone). At 10 days afterinitiation, mice were subsequently treated twice a week withTPA (Sigma) (5 mg/200ml in acetone) during 10 weeks. Tumorsamples were harvested 30 weeks after initiation and frozen inliquid nitrogen for protein analysis or fixed for histopatho-logical procedures.
Protein extracts and Western blottingTumors were ground with a mortar on liquid nitrogen andhomogenized in buffer P (Tris, pH 7.5, NaCl 150mM, EDTA1mM, EGTA 1mM, b-glycerophosphate 40mM, sodiumorthovanadate 1mM, PMSF 0.1mM, aprotinin 2mg/ml,leupeptin 2mg/ml, NP-40 1%). Cells were pelleted andlysed in the above-mentioned buffer by freeze-thawing cycles.The supernatants after centrifugation at 10 000 r.p.m. for10min at 41C were stored at �701C or used directly. Proteinconcentration in each sample was determined with theBio-Rad protein assay system (Bio-Rad, Richmond, CA,USA). Cell and tumor proteins were resolved in SDS–PAGEgels and transferred to nitrocellulose (Amersham, IL, USA).Membranes were blocked with 5% nonfat milk in TBS,incubated with appropriate antibodies diluted in TBSTB(0.5% BSA, 0.1% Tween 20 in TBS). Antibodies used wereas follows: Akt, mdm2, p21Cip1/WAF1, p27Kip1, cyclin D1, Erk1/2phospho-Erk 1/2, p38, phospho-p38, DNp63 and c-myc (SantaCruz Biotechnology, diluted 1/500); b-catenin, JNK, GSK3b-Ser9-P and GSK3b (Transduction Labs, diluted 1/250); c-myc-Ser58-P-Thr62-P, p70S6K, phospho-JNK, p70S6K-P andphospho Foxo3a (Cell Signaling Technology, diluted 1/500);and Lef1 and Foxo3a (Upstate, diluted 1/300). Actin ortubulin (Santa Cruz Biotechnology, diluted 1/1000) was usedthroughout the experiments to confirm equal loading. Second-ary antibodies anti-rabbit, anti-mouse or anti-goat IgG werepurchased from Jackson Immunoresearch and used 1/2000 inTBSTB. WestPicoSignal (Pierce, Rockford, IL, USA) wasused to detect the bands according to the manufacturer’srecommendations.
Northern blottingTotal RNA from frozen tumors was isolated by Trizolextraction. Northern blots containing total RNA (20 mg/lane)were probed for the expression of CycD1 and c-myc. DNAprobes were prepared by random primed reactions using thecomplete sequences as described previously (Paramio et al.,1999). The membranes were also hybridized with a 7S RNAprobe to verify that equal amounts of mRNA were loaded andtransferred.
Akt kinase assayAkt activity in tumors extracts was determined after immu-noprecipitation with anti-Akt (Santa Cruz, C-20 antibody 1ml/25 mg protein) essentially as described (Paramio et al., 2001;Santos et al., 2002; Segrelles et al., 2002) using histone 2B(Roche Molecular Biochemicals) as substrate. Autoradiogramswere subsequently quantified using a Phosphorimager (Bio-Rad).
Cell culture and transfectionPooled clones (>40) of PB keratinocytes transfected withplasmid coding for wt Akt (pcDNA3Akt) or control(pcDNA3) (Segrelles et al., 2002, 2004), selected by growingin the presence of G418 (0.5mg/ml) for 3–4 weeks, werecultured in Dulbecco’s modified Eagle’s medium (DMEM)(Life technologies) supplemented with 10% of fetal calfserum (BioWhitakker, Walkersville, MD, USA). Cyclohex-imide was purchased from Sigma and used at 50mM (finalconcentration).Luciferase assays were performed upon transfection essen-
tially as described (Ruiz et al., 2004). Briefly, cells werecultured in DMEM (GibcoBRL) supplemented with 10% fetalbovine serum and antibiotics. Transient transfections wereperformed with the Lipofectamine reagent (GibcoBRL)according to the manufacturers’ protocol. At 36 h aftertransfection, cells were harvested for luciferase assays (Prome-ga Dual-Luciferase Kit). Firefly luciferase values werestandardized to Renilla luciferase values (pRL-SV40; Prome-ga) to account for differences in transfection efficiency betweensamples. As a control, the a-catenin/Lef1 reporter (TOPflash)was replaced by a mutant (FOPflash) reporter to ensure thatvalues were dependent on the Lef1/Tcf-binding sites in thepromoter. Plasmids coding for wt Akt, TOPflash andFOPflash were provided by Dr JS Gutkind (NIDCR/NIH).
ImmunohistochemistryFormalin-fixed, paraffin-embedded tissue specimens from headand neck cancer patients were retrieved from the archives ofDepartment of Pathology, ‘12 de Octubre’ University Hospital.Formalin- or ethanol- (70%) fixed, paraffin-embedded tumorsamples upon subcutaneous injection, or formalin-fixed hu-man HNSCC samples were sectioned (6 mm thick). All tissueswere stained H&E for pathological diagnosis. Sections weredewaxed and treated by microwave boiling in citrate buffer,blocked by incubation in 10% nonimmune horse serum andincubated with the primary antibodies overnight at 41C (seeWestern blotting section). After exhaustive washing in PBST,sections were incubated with appropriate biotin-coupledsecondary antibodies (all 1/1000 in PBST) followed byavidin–peroxidase (ABC elite kit Vector, Burlingame, CA,USA). Positive staining was determined using diaminobenzi-dine as a substrate (DAB kit Vector, Burlingame, CA, USA)following the manufacturer’s recommendations. Sections werethen counterstained with hematoxylin and mounted. At least10 different tumors for xenograft experiments of each typewere analysed. Controls omitting primary antibodies or afterthe preincubation of the antibodies with the immunizingpeptide were routinely performed (not shown). Statisticalanalyses were performed using SPSS software.
Abbreviations
SCC, squamous cell carcinoma; TPA, 12-O-tetradecanoyl-phorbol- 13-acetate; DMBA, 7, 12-dimethyl-benz(a)anthracene.
Akt-induced signaling during keratinocyte transformationC Segrelles et al
1183
Oncogene

Acknowledgements
We thank S Gutkind for providing materials, the animalfacility personnel of the CIEMAT for the excellent care of the
animals and the Histology facility for their work involving thehistological preparations. This work was partially supportedby Grant SAF2002-01037 from the CICYT, La CaixaFoundation and GR/SAL/0103/2004 from CAM.
References
Ambrosch P, Ruschenburg I. (2000). Anticancer Res 20: 3151–3155.
Amornphimoltham P, Sriuranpong V, Patel V, Benavides F,Conti CJ, Sauk J et al. (2004). Clin Cancer Res 10:4029–4037.
Aoki M, Blazek E, Vogt PK. (2001). Proc Natl Acad Sci USA98: 136–141.
Arnold I, Watt FM. (2001). Curr Biol 11: 558–568.Barbieri CE, Barton CE, Pietenpol JA. (2003). J Biol Chem278: 51408–51414.
Barker N, Clevers H. (2000). Bioessays 22: 961–965.Barker N, Huls G, Korinek V, Clevers H. (1999). Am J Pathol154: 29–35.
Bhatia N, Spiegelman VS. (2005). Mol Carcinogen 42:213–221.
Bjornsti MA, Houghton PJ. (2004). Nat Rev Cancer 4:335–348.
Blagosklonny MV. (2002). Cell Cycle 1: 391–393.Brown J, O’Prey J, Harrison PR. (2003). Carcinogenesis 24:171–177.
Burgering BM, Kops GJ. (2002). Trends Biochem Sci 27:352–360.
Casanova ML, Larcher F, Casanova B, Murillas R, Fernan-dez-Acenero MJ, Villanueva C et al. (2002). Cancer Res 62:3402–3407.
Cross DA, Alessi DR, Cohen P, Andjelkovich M, HemmingsBA. (1995). Nature 378: 785–789.
D’Amico M, Hulit J, Amanatullah DF, Zafonte BT,Albanese C, Bouzahzah B et al. (2000). J Biol Chem 275:32649–32657.
De Benedetti A, Harris AL. (1999). Int J Biochem Cell Biol 31:59–72.
Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, DedharS. (1998). Proc Natl Acad Sci USA 95: 11211–11216.
Desbois-Mouthon C, Blivet-Van Eggelpoel MJ, Beurel E,Boissan M, Delelo R, Cadoret A et al. (2002). Hepatology36: 1528–1536.
Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ,Bertrand F, Cherqui G, Perret C et al. (2001). Oncogene 20:252–259.
Diehl JA, Cheng M, Roussel MF, Sherr CJ. (1998). Genes Dev12: 3499–3511.
DiGiovanni J. (1992). Pharmacol Ther 54: 63–128.Erber R, Klein W, Andl T, Enders C, Born AI, Conradt C
et al. (1997). Int J Cancer 74: 383–389.Filali M, Cheng N, Abbott D, Leontiev V, Engelhardt JF.(2002). J Biol Chem 277: 33398–33410.
Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JHet al. (2004). J Biol Chem 279: 2737–2746.
Giles RH, van Es JH, Clevers H. (2003). Biochim Biophys Acta1653: 1–24.
Gotoh J, Obata M, Yoshie M, Kasai S, Ogawa K. (2003).Carcinogenesis 24: 435–442.
Gwosdz C, Balz V, Scheckenbach K, Bier H. (2005). AdvOtorhinolaryngol 62: 58–71.
Hanada M, Feng J, Hemmings BA. (2004). Biochim BiophysActa 1697: 3–16.
Hildesheim J, Belova GI, Tyner SD, Zhou X, Vardanian L,Fornace AJ. (2004). Oncogene 23: 1829–1837.
Koga F, Kawakami S, Kumagai J, Takizawa T, Ando N, AraiG et al. (2003). Br J Cancer 88: 740–747.
Lawlor MA, Alessi DR. (2001). J Cell Sci 114: 2903–2910.Leethanakul C, Patel V, Gillespie J, Pallente M, Ensley JF,Koontongkaew S et al. (2000). Oncogene 19: 3220–3224.
Leis H, Segrelles C, Ruiz S, Santos M, Paramio JM. (2002).Mol Carcinogen 35: 180–185.
Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK,Han K et al. (2002). Nat Med 8: 1153–1160.
Luo J, Manning BD, Cantley LC. (2003). Cancer Cell 4:257–262.
Mayo LD, Donner DB. (2001). Proc Natl Acad Sci USA 98:11598–11603.
Mayo LD, Donner DB. (2002). Trends Biochem Sci 27:462–467.
Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR.(1999). Biochem J 344(Part 2): 427–431.
Novak A, Dedhar S. (1999). Cell Mol Life Sci 56:523–537.
Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T,Tanaka K et al. (2002). J Biol Chem 277: 21843–21850.
Paramio JM, Navarro M, Segrelles C, Gomez-Casero E,Jorcano JL. (1999). Oncogene 18: 7462–7468.
Paramio JM, Segrelles C, Ruiz S, Jorcano JL. (2001). Mol CellBiol 21: 7449–7459.
Patturajan M, Nomoto S, Sommer M, Fomenkov A, Hibi K,Zangen R et al. (2002). Cancer Cell 1: 369–379.
Pedrero JM, Carracedo DG, Pinto CM, Zapatero AH,Rodrigo JP, Nieto CS et al. (2005). Int J Cancer 114:242–248.
Pelengaris S, Littlewood T, Khan M, Elia G, Evan G. (1999).Mol Cell 3: 565–577.
Prats AC, Prats H. (2002). Prog Nucleic Acid Res Mol Biol 72:367–413.
Ratovitski EA, Patturajan M, Hibi K, Trink B, Yamaguchi K,Sidransky D. (2001). Proc Natl Acad Sci USA 98:1817–1822.
Rhee CS, Sen M, Lu D, Wu C, Leoni L, Rubin J et al. (2002).Oncogene 21: 6598–6605.
Robles AI, Conti CJ. (1995). Carcinogenesis 16: 781–786.Rodriguez-Puebla ML, LaCava M, Gimenez-Conti IB,Johnson DG, Conti CJ. (1998). Oncogene 17: 2251–2258.
Rodriguez-Puebla ML, Robles AI, Conti CJ. (1999). MolCarcinogen 24: 1–6.
Roose J, Clevers H. (1999). Biochim Biophys Acta 1424:M23–37.
Rosenwald IB, Kaspar R, Rousseau D, Gehrke L, Leboulch P,Chen JJ et al. (1995). J Biol Chem 270: 21176–21180.
Ruiz S, Segrelles C, Santos M, Lara MF, Paramio JM. (2004).Dev Dyn 230: 410–418.
Santos M, Paramio JM, Bravo A, Ramirez A, Jorcano JL.(2002). J Biol Chem 277: 19122–19130.
Scheid MP, Woodgett JR. (2003). FEBS Lett 546: 108–112.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR.(2000). Genes Dev 14: 2501–2514.
Segrelles C, Ruiz S, Perez P, Murga C, Santos M, BudunovaIV et al. (2002). Oncogene 21: 53–64.
Segrelles C, Ruiz S, Santos M, Martinez-Palacio J, Lara MF,Paramio JM. (2004). Carcinogenesis 25: 1137–1147.
Akt-induced signaling during keratinocyte transformationC Segrelles et al
1184
Oncogene

Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga Jet al. (2002). Nat Med 8: 1145–1152.
Slaga TJ, Budunova IV, Gimenez-Conti IB, Aldaz CM.(1996). J Invest Dermatol Symp Proc 1: 151–156.
Sniezek JC, Matheny KE, Westfall MD, Pietenpol JA. (2004).Laryngoscope 114: 2063–2072.
Suzuki A, Itami S, Ohishi M, Hamada K, Inoue T, KomazawaN et al. (2003). Cancer Res 63: 674–681.
Thurfjell N, Coates PJ, Boldrup L, Lindgren B, Backlund B,Uusitalo T et al. (2005). Adv Otorhinolaryngol 62: 49–57.
Viglietto G, Motti ML, Bruni P, Melillo RM, D’Alessio A,Califano D et al. (2002). Nat Med 8: 1136–1144.
Vivanco I, Sawyers CL. (2002). Nat Rev Cancer 2: 489–501.Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR.(2001). Nat Genet 28: 165–168.
Wang XQ, Sun P, Paller AS. (2001). J Biol Chem 276:44504–44511.
Warnakulasuriya KA, Tavassoli M, Johnson NW. (1998).J Oral Pathol Med 27: 376–381.
Yuspa SH, Dlugosz AA, Denning MF, Glick AB. (1996).J Invest Dermatol Symp Proc 1: 147–150.
Yuspa SH, Morgan D, Lichti U, Spangler EF, Michael D,Kilkenny A et al. (1986). Carcinogenesis 7: 949–958.
Zhang SY, Liu SC, Goodrow T, Morris R, Klein-Szanto AJ.(1997). Mol Carcinogen 18: 142–152.
Zhou BP, Hung MC. (2002). Semin Oncol 29: 62–70.Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC.(2001a). Nat Cell Biol 3: 245–252.
Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC.(2001b). Nat Cell Biol 3: 973–982.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
Akt-induced signaling during keratinocyte transformationC Segrelles et al
1185
Oncogene




















