Microcrystalline chitosan
169
Transcript of Microcrystalline chitosan






Preparation of Microcrystalline chitosan(MCCh)/tricalcium phosphate complex withHydroxyapatite in sponge and fibre form for
hard tissue regeneration.
By
Luciano Pighinelli
Research Institute of Textile Chemistry/Physics, Dornbirn, Austria
Faculty of Chemistry and Pharmacy, Leopold-Franzens University of Innsbruck andInstitute of Biopolyners and Chemical Fibres, Lodz, Poland.
2012

ABSTRACT
Bone repair or regeneration is a common and complicated clinical problem in
orthopaedic surgery. The importance of natural polymers and calcium phosphates
composites has grown significantly over the last two decades due to their renewable and
biodegradable source, increasing the knowledge and functionality of composites in
technological and biomedical applications.
This work present chitosan and a new method to obtain derivatives such as
MCCh/ß-TCP complex with nano and micro size of calcium phosphates in different
forms (sponge, fibres), including chemical characterization, mechanical properties,
particle size, morphology, solution stability, biodegradation, bioactivity and also a new
method to obtain nanoceramic formation in chitosan salt solution.
All sponge preparations with MCCh/ß-TCP complex have a well-shaped 3-
dimensional interconnected and homogeneous pore structure with high porosity, to
ensure a biological environment conducive to cell attachment and proliferation
providing a passage of nutrient flow. In the fibre form, the presence of HAp/ -TCP
nanoparticles in the solution of chitosan has a beneficial effect on the production of
modified chitosan fibres with a lower cost of process, improving the mechanical
properties, such as tensile strenght, in wet conditions. The complex in sponge form is
susceptible to hydrolytic and enzymatic degradation with good mechanical properties
after 60 days of degradation, showing also a bacteriostatic activity and a bactericidal
activity against Escherichia coli and Staphylococcus aureus.
These materials can be used in the future for medical applications as a base for
scaffold production, as implants in regenerative medicine.

TABLE OF CONTENTS
Abstract……………………………………………………….……………...................2
Chapter 1 Introduction and literature review
1.1. Introduction……………………………………………………………08
1.2. Hard tissue regeneration………...……………..……………………..09
1.2.1. Bones histology and bone structure ………………….……………………...10
1.2.2. Tissue repair…………………………………………………………….…….13
1.2.3. Bone remodelling…………………………………..………………………....14
1.2.4. Regulations of bone cell function and bone turnover………………………15
1.2.5. Mechanical properties………………………………………………………..17
1.2.6. Biomaterials for hard tissue regeneration………………………………......18
1.3. Chitosan
1.3.1. Origin and general properties…………………………….….18
1.3.2. Microcrystalline chitosan…………………………………….24
1.3.3. Medical applications………………………………………….24
1.4. Calcium Phosphates
1.4.1. General properties of hydroxyapatite……………………….29
1.4.2. General properties of tricalcium phosphate………………...32
1.4.3. Medical applications………………………………………......36
1.4.4. References………………………………………………………37
1.5. Aim and scope of the thesis…………………………………………...48
Chapter 2 Properties of chitosan material
2.1. Introduction……………………………………………………….…..41
2.2. Materials……………………………………………………………....42
2.3. Methods

2.3.1 Preparation of chitosan hydrochloride salt…………………42
2.3.2. Analytical methods....................................................................43
2.3.3. Assessment of physical-mechanical properties of chitosan
hydrochloride salt in film form………………………………………………49
2.4. Results and discussions
2.4.1. Results of analytical methods of chitosan hydrochloride salt
solution…………………………………………………………………………49
2.4.2. Results for Mechanical properties from chitosan hydrochloride
salt in film form……………………………………………………………….50
2.5. Conclusions…………………………………………...……………….51
2.6. References…………………………………………………………......51
Chapter 3 Preparation of Microcrystalline chitosan (MCCh)/tricalcium phosphate
complex in sponge form.
3.1. Introduction………………………………………………………...….53
3.2. Materials…………………………………………………………...…..56
3.3. Methods
3.3.1 Schema of preparation MCCh/ tricalcium phosphate
complex………………………………………………………....56
3.3.2. Analytical methods…………………………………………….59
3.3.3. Preparation of MCCh/ ß-TCP composite in film form……...60
3.3.4. Preparation of MCCh/ ß-TCP complex in sponge form….....60
3.3.5. Infrared Spectroscopy of the complex………………………..60
3.3.6. Determination particles size, morphology of commercial ß-
TCP powder…………………………………………………………………………...61
3.4. Results and discussions
3.4.1. Elaboration of the quantitative and qualitative MCCh/ ß-TCP
complex………………………………………………………………………………...61
3.4.2. FTIR study..................................................................................61
3.4.3. Particles size, morphology of commercial ß-TCP powder….67
3.4.4. SEM of composite MCCh/ ß-TCP complex in film form…...69
3.4.5. SEM of MCCh/ ß-TCP complex in sponge form…………….72

3.5. Conclusions…………………………………………………………….73
3.6. References……………………………………………………………...74
Chapter 4 Preparation of Microcrystalline chitosan (MCCh)/tricalcium phosphate
complex with Hydroxyapatite in sponge form.
4.1. Introduction............................................................................................78
4.2. Materials.................................................................................................84
4.3. Methods
4.3.1 Preparation of composite in sponge form................................84
4.3.2. Powder complex preparation....................................................85
4.3.3. SEM study...................................................................................86
4.3.4. Infrared Spectroscopy................................................................86
4.3.5. Determination of Ca and P in commercial HAp and ß-TCP
powders and inthe composites………………………………………………………..86
4.3.6. Determination of particles size of commercial HAp poder…87
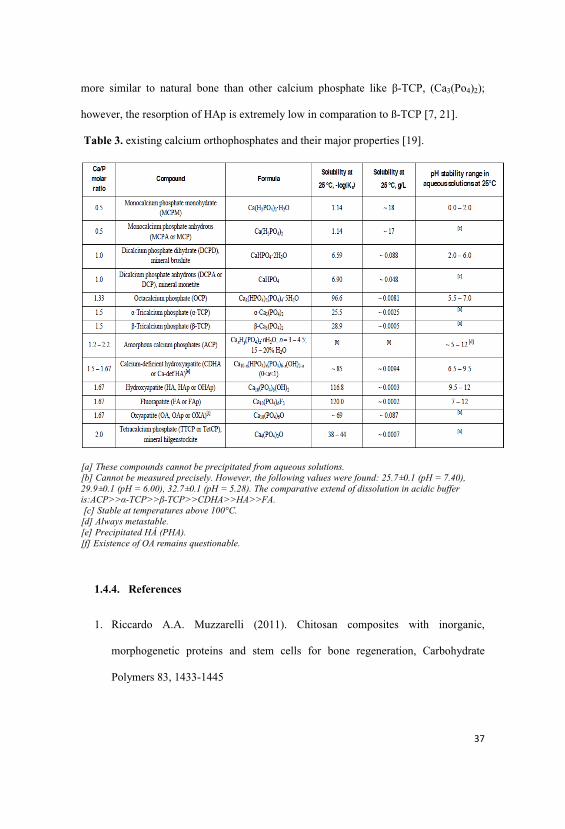
4.3.7. WAXS- Diffraction of ß-TCP and HAp poder………………87
4.3.8. Mechanical properties of composites........................................87
4.4. Results and discussions
4.4.1. Elaboration of the quantitative and qualitative of composites
in sponge form…………………………………………………………………………88
4.4.2. Composites with different ratios of ethanol to prepare sponge
form…………………………………………………………………………………….90
4.4.3. FTIR study of the commercial HAp………………………….91
4.4.4. FTIR study of the composites………………………………....93
4.4.5. WAXS investigation of ß-TCP and HAp powder…………... 95
4.4.6. Analysis of the particles size and morphology of the HAp
poder…………………………………………………………………………………...96
4.4.7. SEM study of the composites in sponge form……………..…98
4.4.8. Physical-mechanical tests of composite in sponge form…...105
4.4.9. Determination of Ca and P content in composites…………107
4.5. Conclusions...........................................................................................108
4.6. References.............................................................................................109

Chapter 5 Preparation of Microcrystalline chitosan (MCCh)/tricalcium phosphate
complex with Hydroxyapatite in fibre form.
5.1. Introduction..........................................................................................115
5.2. Materials...............................................................................................116
5.3. Methods
5.3.1 Methods of manufacture composite chitosan fibres………..116
5.3.2. Preparation of Chitosan Spinning Solution Containing HAp,
ß-TCP and HAp/ß-TCP Nanoparticles……………………………………………..117
5.3.3. Wet Spinning of Chitosan Fibres Containing HAp, ß-TCP
and HAp/ ß –TCP Nanoparticles…………………………………………………...118
5.3.4. Analytical Methods..................................................................118
5.4. Results and discussions
5.4.1. Preparation of Chitosan Solutions Containing -TCP, HAp
and HAp/ -TCP………………………………………………………………...……119
5.4.2. Rheology of Chitosan Solutions Modified with HAp/ -TCP
Nanoparticles………………………………………………………………………...124
5.4.3. Investigation into the Spinning of Chitosan Fibres Modified
with HAp, -TCP and HAp/ -TCP…………………………………………………125
5.4.4. Mechanical Properties of Chitosan Fibres Modified with
HAp, -TCP and HAp/ -TCP……………………………………………...……….126
5.4.5. FTIR Examination of Chitosan Fibres Modified with HAp, -
TCP and HAp/ -TCP Nanoparticles………………………………………...……..127
5.4.6. Morphology and Chemistry of Chitosan Fibres Modified with
HAp, -TCP and HAp/ -TCP Nanoparticles………………………………………131
5.5. Conclusions...........................................................................................134
5.6. References.............................................................................................136
Chapter 6 Degradation and bioactivity of the composite
6.1. Introduction..........................................................................................141
6.2. Experimental Section
6.2.1. Materials...................................................................................143
6.3. Methods

6.3.1 Preparation of the MCCh/ß-TCP complex…………………143
6.3.2. Preparation of the composites in sponge form……………..143
6.3.3. SEM study of the composites in sponge form………………144
6.3.4. Mechanical properties..............................................................144
6.3.5. Assessment of the degradability of the composites……..….144
6.3.6. Bioactivity.................................................................................145
6.4. Results and discussions........................................................................145
6.4.1. Biodegradation mass loss........................................................146
6.4.2. SEM study of the composites in sponge form………………147
6.4.3. Mechanical properties of the sponges………………………151
6.4.4. Bioactivity.................................................................................152
6.5. Conclusions...........................................................................................153
6.6. References.................................................................................154
Conclusions...................................................................................................................158

1.1. INTRODUCTION
Every year, millions of people are suffering from bone disease cause by trauma,
tumor, bone fractures or defects and unfortunately some of them are dying due to
insufficient optimal bone substitute and/or treatment.
Much attention has been given to the use of different materials that could be
used as a base material for scaffold devices and as modification tools for currently used
biomedical devices that improve hard and soft tissue regeneration and/or reinforcement
efficacy, also to expand the feasibility of combined controlled drug release and tissue
engineering, tissue formation in regenerative therapy in the field of periodontics,
orthopaedics, cancer and plastic surgery, and veterinary [1,2,3,4]. According to a new
market research report, ‘Global Biomaterial Market’ (2009-2014), published by Markets
and Markets (http://www.marketsandmarkets.com), the total global biomaterial market
is expected to be worth US$58.1 billion by 2014, growing by 15.0% from 2009 to 2014.
The U.S. market is the largest geographical segment for biomaterials and is expected to
be worth $22.8 billion by 2014, with 13.6% from 2009 to 2014. Europe is the second
largest segment and is expected to reach $17.7 billion by 2014 with 14.6%, while the
Asian market size is estimated to increase by 18.2% from 2009 to 2014. The biomaterial
market today has already exceeded $28 billion [7]. Biomaterials are defined as natural
or man-made materials that are used directly as a supplement and/or a replacement for
functions of the living tissues in human body. Two important criteria that a biomaterial
is required to have are biocompatibility and biofunctionality [5,6,8].
The properties of bone in health and disease attract much attention. With an ever
greater proportion of population needing medical devices for hard tissue regeneration
and/or replacement, health systems of all countries are pressured. Musculoskeletal
disorder affected by aging, diseases, micro and/or macrofractures and demineralization

contributes and improves the suitability and development of new materials and methods
for hard tissue engineering.
1.2. Hard Tissue Regeneration
The properties of bone in health and disease justifiably attract much attention.
With an ever greater proportion of the population surviving into the third and fourth age
groups, the pressure on the health and welfare systems of our countries is set to rise
even further. Aging is a musculoskeletal disorder, which is expected to happen as an
underlying trend, but when combined with other skeletal complications, it compounds
problems even further. [8]
The wide range of biomaterials available is a reflection of the current diversity
related to the use of several materials, whether naturals and/or artificial, and as for the
various synthesis techniques or processing methodologies. This condition allows the
obtaining of materials with various macro and microstructures, which can cause
different mechanical, physical and chemical properties for the same material. Despite
the huge scientific production involving bone repair in tissue engineering, there is no
consensus defining the best technique yet, or the best synthesis or processing material,
considering the difficulty in reproducing the bone tissue properties as a whole.
Therefore, the knowledge of how the bone tissue reacts before a new material
and how this material behaves during the preparation is extremely important in new
technologies assessment. This chapter emphasizes concepts that serves as support for
this study, as the detail of the issues concerning to bone issues; natural polymers such as
chitin and later the chitosan, calcium phosphates and other materials that can be
relevantly used to obtain biomaterials to human tissue repair.

1.2.1. Bones histology and bone structure
The bone is a connective tissue characterized by a mineralized extracellular matrix,
hematopoietic and adipose tissue, blood vessels and nerves. [1]
Bone formation, or osteogenesis, occurs through osteoprogenitor cells that are
derivated of mesenchymal stem cells. These ones can differentiate into several types of
cells involved in bone physiology:
Osteocytes: is the mature bone cell wrapped up by bone matrix and responsible by
keep it, synthesizing it or absorbing it when needed. Each osteocyte occupies its place
histologically known as gap and project cellular extensions by canals that connect to
gaps beside, so that it can produce a network of interconnected cells. The lifespan of an
osteocyte is estimated in 25 years. [1.2]
Osteoblast: is a bone-forming differentiated cells, which produces the bone matrix
organic part composed of type I collagen, glycoproteins and proteoglycan (phosphate
concentrated). It also acts some way in the mineralization of bone matrix [1,2]
Osteoclast: it participates in the bone resorption and remodeling process. It is huge
and multinucleated cells, widely branched, derived from monocytes that cross the blood
capillaries. The activity of this cell is controlled by calcitonin and parathormone and it
is not embryologically related to osteoblasts, but to the mononuclear hematopoietic
progenitor cells, which are monocytes same lineage. [1,2,5].
Bone matrix: bone matrix consists of a fundamental substance, highly mineralized,
in which numerous collagen fibers are embedded, usually arranged in parallel
arrangement. In mature bone, the matrix is moderately hydrated, being 10% of its mass
the water; in its dry weight, 60% is composed of inorganic materials, mineral salts
(hydroxyapatite and amorphous calcium phosphate), 30% of collagen and protein and
carbohydrates, especially in conjunction with glycoproteins. The proportions of several

components vary with the age, location and metabolic condition. The aspect that
differentiates bone from other tissues is the mineralization of its matrix, producing an
extremely rigid tissue capable to provide support and protection to the organism. The
mineral that is deposited in this matrix is calcium phosphate in the form of
hydroxyapatite crystals in addition to phosphorus, and other mineral elements, which
may undergo mobilization of the bloodstream by bone matrix [1,2,5,7]. This
mechanism serves both to maintain bone function and to regulate calcium and
phosphorus concentrations in the organism, thus maintaining the homeostatic regulation
[6].
In the early stages of bone formation, prior to mineralization, the matrix is named
osteoid. In adult bones, there is a small amount of osteoid, reflecting the bone local
remodeling in which the mineralization is performed and rapidly the deposition of the
organic matrix occurs. In some diseases, such as rickets, the mineralization is impaired,
and the amount of osteoids is much higher to healthy parameters [3.4].
Histologically, there are two variants of the bone tissue: compact bone tissue (dense
and cortical) and trabecular bone tissue (spongy and marrow) containing the same types
of cells and intercellular substance, being different from each other by the arrangement
of these elements and amount of no marrow space. Because they are widely
vascularized and innervated, the bones have great capacity for regeneration. The bone
tissue receives vascularization of about 20% of cardiac output [1,2,3,6].

Figure 1 - structure of spongy bone [1].
The compact bone has no marrow space, but it has canals which protect nerves andblood vessels known as Haversian canals which are perpendicularly interconnected bycanals known as Volkmann, thus forming what is known as Harvers or Harversiansystem. Around each Harvers canal it is possible to observe several concentriclamellae of intercellular substance and bone cells, as shown in Figure 2 [1].
Figure 2-Structure of compact bone.
Concentric lamellae which circulate a Harvers canal determine the basic unit of
bone named osteon. The spongy bone is composed of large marrow spaces, giving the
appearance of porous bone, formed by spicules and trabeculae [1,2].
Compact Bone spongybone

Anatomically there are two types of bones: the ones that are longs as tibia, femur
and humerus, or the ones that are short and flat: skull, scapula, mandible and
hipbones [1,2,3].
The maturity of mineralized bone tissue is reached around 30 years old, starting
after this period a progressive loss of mineralized tissue and developing the disease
known as osteoporosis [4.9].
The membrane covering the bone is called periosteum and contains
osteoprogenitor cells and may undergo transformation to osteoblasts and to assist bone
repair. The bone itself has a structural architecture to support compressive forces. The
intraosseous vessels are protected by the rigid structure of the bone, avoiding trauma
such as bone infarction by extrinsic compression [6.10].
1.2.2. Tissue repair
Bone tissue is among the most specialized tissues in the body due the presence
of unique characteristics, which combines structural resistance to the ability for
regeneration. The bone shows an ability to repair from fractures without the presence of
any scar. The mechanism of this repair pattern is considered a summary of osteogenesis
occurred in the embryo during the growth period [1,2,3,6].
The bone repair is a regenerative process highly complex and essentially a
repetition of developmental events. This process has a lot of similarities with the repair
of soft tissues. Both of them show similar phases in cell replacement process:
inflammation, proliferation and remodeling phases. The cells involved in this process
include polymorph nuclear leukocytes, macrophages, fibroblasts, endothelial cells and
bone matrix. There is also the participation of different types of proteins and an active
genetic expression restoring the natural integrity of the bones [4,8,9]. When the bone

suffers any injury, blood vessels produce a local hemorrhage which leads to clot
formation, promoting an intense local inflammatory reaction. During repairing, the cells
and the battered bone matrix and the clot are removed by phagocytosis. Concomitantly,
the periosteum is responsible for proliferation of connective tissue fibroblasts and
osteoprogenitor cells will form a new cell tissue. After one week of the trauma the
tissue formed around the lesion is transformed into immature bone tissue by the
connective tissue cells change by means of osteoblasts which start the production of
bone matrix [3,5,6,]. The action of osteoclasts in bone repair has several simultaneous
aspects and stages. One of them relates to the occurrence of variation in the pressure
zone, where the levels of oxygen and carbon dioxide are different than usual, due to
compression of blood vessels or tearing of them due the trauma. This makes the
osteoclasts come from other adjacent areas or are inactive stimulated to undergo
processing in monocytes, osteoclasts [1,3,5,6].
The cells found in the early stages of repair processes contain a range of
chemical mediators that are stored within cells and released during this process. The
release of these agents causes changes in cell membrane and in the covering in ions
transport into the cell. The release of chemical agents stimulates the presence of
fibroblasts and endothelial cells at the injured site. Endothelial cells are essential for the
formation of new capillaries, which transport nutrients and remove metabolites in the
repairing tissue [1,3,6].
1.2.3. Bone remodelling
Throughout adult life, the skeleton undergoes a continuous repair and renewal
process. Bone remodelling is a surface-dependent phenomenon: the turnover rate in
trabecular bone may be up to ten times greater than in cortical bone, reflecting the

largest surface area presented by the former tissue. Mineralised bone matrix is resorbed
by osteoclasts and replaced in plywood-like layers, or lamellae, by groups of
osteoblasts. This sequence of events is tightly coordinated both temporally and spatially.
Under normal circumstances in young adults, remodelling activity keeps overall
bone mass relatively constant. However Aging, the menopause and many other
pathophysiological states can alter the balance of the turnover process, such that
reabsorption begins to outstrip formation, leading to net bone loss and ultimately
osteoporosis. This could be due not only to enhance osteoclastic resorption but also to
reduce osteoblastic function. Trabecular bone sites, for example, in the vertebral bodies
or in the ends of the long bones, are particularly susceptible to remodelling imbalances,
due to the relatively high turnover rate [4,7,9].
Bone growth, turnover and repair involve high levels of cellular activity, and
require an effective blood supply. This is in contrast with adult cartilage, an essentially
primitive, avascular tissue with low cellularity and turnover rates. The importance of the
vascular supply of bone, with its attendant network of fine nerve fibres is perhaps
insufficiently recognised. Disruption of the blood supply to bone, for example because
of inflammation, infection, tumours or fractures will result in hypoxia and acidosis, and
may have profound negative consequences [4,6,8].
1.2.4. Regulations of bone cell function and bone turnover
Some of the classical systemic actions of hormones on bone may be mediated at
tissue level via local production of growth factors and cytokines, and these effects may
be mediated in turn by agents such as prostaglandins. The systemic and local actions on
bone cells of simple inorganic moieties such as hydrogen ions and molecular oxygen

(Arnett et al, 2003), phosphate (Yates et al, 1991) or nitric oxide (Ehrlich & Lanyon,
2001) also appear to be of considerable importance [1,2,4,6,9].
The strain resulting from mechanical loading is a key regulator of remodelling in
some parts of the skeleton. The long bones and the vertebral bodies appear to require
modest but regular loading cycles in order to maintain their mass. The mass and
strength of bones in normal individuals is ultimately determined by the need to resist the
loads and deformations resulting from the most extreme normal activities (for example,
jumping off a wall 1-2 metres high on to a hard surface) [9].
Growth factors are a group of biological mediators which regulate important
cellular events in tissue repair and cell proliferation, including differentiation,
chemotaxis and matrix formation. The TGF-pi and TGF-P2 proteins distinguish among
growth and transforming factors (TGF) involved in connective tissue repair, in general,
and bone regeneration, being its most important function the chemotaxis and
mitogenesis of osteoblasts precursors and its ability to stimulate collagen matrix
deposition in wound and bone repair [3,6.9].
The tension on the tissue is an important factor to proliferation and evolution of
the tissue to be repaired. Considering the bone tissue reaction to the movement, it can be
stated that the relative motion causes a tension, thus generating a deformation up to the
tolerance levels supported by the tissue repair [1,2,3,9].
When the motion increases and tension overcome the limits of the tissue, a
instability condition can be generated. If the tension is low, there is insufficient
mechanical induction tissue to tissue differentiation; this can often result in grafted
material resorption, for example. Several studies have shown decreased bone mass \

calcium concentration in the bone in gravityless environments, confirming these
discoveries [4.6].
In recent decades, experimental studies of cell culture have attempted to define
which molecule or system is responsible for translating the language of physical
stimulus into biological language. It is important to emphasize that although these
studies explain isolated phenomena, it cannot be extrapolated to what happens to the
tissue the organism as a whole [9].
1.2.5. Mechanical properties
Bone is a damageable, viscoelastic composite and most of all a living material
capable of self-repair and thus exhibits a complex repertoire of mechanical properties.
From a mechanical perspective, the rigidity and strength of a structure is determined not
only by the amount of material but even more importantly by the arrangement of the
material in its space. The cortical structure and microstructure contribute to the whole
bone mechanical competence and weakness or fragility and the cortical thickness and
area are strong predictors of bone strength and resistance to damages or fractures [3,6].
Collagen fibrils are responsible for tensile strength and toughness, while the
crystalline structure provides compressive strength, and the microdamage accumulated
during the years may weaken cortical bone tissue and contributes to increased
susceptibility to fracture [7]. Mechanical properties of cortical bone depend on the size
and distribution of the mineral crystals. Crystal size increases by the addition of ions
and by the circulating proteins, as well as bone diseases, drugs, diet and age. Generally
the crystals are smaller in young bones and larger in mature bones. As we find these

bones in the same organism, this represents the optimal situation for good resistance to
load [3,7].
Age affects the biomechanical properties of bone in animals and humans.
However, the so-called Aging effect is of primary importance to humans who suffer the
effects of senescence as they survive into old age [8,10].
1.2.6. Biomaterials for hard tissue regeneration
The success of a bone scaffold as measured in vivo is determined by its ability to
stimulate and aid in both the onset and completion of bone defect repair. Because the
only control parameters that can be affected prior to implantation are the incorporation
of growth factors, cell seeding and architecture modification, optimization of the
scaffold must be completed prior to use and must encompass at the very least
mechanical stability for its load bearing application. This optimization requires a
complete knowledge of the system the scaffold will be interacting with [8-10].
1.3. Chitosan
1.3.1. Origin and general properties
Chitin it is a natural (polysaccharide), abundant and renewable biomaterial,
being the second most abundant in nature after cellulose. It is the primary structural
component of the outer skeletons of crustaceans, and of many other species such as
molluscs, insects and fungi. The role played by chitin is similar to those played by
cellulose in plants and collagen in higher animals. It is a reinforcing material, which
occurs in three polymorphic forms, , and -chitin. Where hardness is needed, -chitin
is found; where flexibility is required, and -chitin occur. Chitin is inert in aqueous
environment [24].

The chitin derivative is chitosan, which appears to be a good candidate for
wound-dressing and for hard and soft tissue regeneration. Chitosan is prepared from
chitin to obtain a more reactive polymer. In preparing chitosan, ground shells are treated
with alkali and acid to remove proteins and minerals, respectively, after which the
extracted chitin is deacetylated to chitosan by alkaline hydrolysis at high temperature.
Preparation of chitosan from crustacean-shell waste, for example, from shrimp
(Pandalus borealis), is economically feasible and ecologically desirable because large
amounts of shell waste are available as a product and/or waste of the food industry.
Production of chitosan from these is inexpensive and easy [23, 24]. Structures of
cellulose, chitin and chitosan are showed in Figure 3.
Figure 3. Structures of cellulose, chitin and chitosan [23].
Like cellulose, it is a glucose-based unbranched polysaccharide. It differs from
cellulose at the C-2 carbon by having an acetamido group instead of a hydroxyl group.
Chitosan is a partially deacetylated polymer of acetyl glucosamine obtained after
alkaline deacetylation of chitin. Polyaminosaccharides, especially chitosan (poly(ß-

(1,4)-2-amino-2-deoxy-D-glucopyranose)) and its derivatives, are characterized by
excellent biostimulating properties that facilitate reconstruction and vascularisation of
damage tissues, also compensate the shortcomings of cells components, which are
conductive for small scar forming. This cationic property is the basis of many of the
potential applications of chitosan that can be considered as a linear polyelectrolyte with
a high charge density which can interact with negatively charged surfaces, like proteins
and anionic polysaccharides [23, 24, 25, 26, 27, 28, 29].
Currently, chitosan is been used also in water treatment, cosmetic, drug and
medicine manufacturing, food additives, semi-permeable membranes and the
development of biomaterials. One of the most important limits to determine chitosan is
the degree of acetylation and the molecular weight, which vary in molecular weight
(from about 10,000 to 2 million Dalton) this characteristic is directly related to the
hydrogen bonding existing in this biopolymer, affecting its structure, solubility,
reactivity and the viscosity [15, 23, 24, 30, 31].
Chitosan as well as its derivatives like MCCh are often used for the preparation
of biodregradeble biomaterials. Chitosan of acetylation degree over 80% and average
molecular weight around 350kDa demonstrated the highest level of activity. In the
Figure 4, a comparation between structures of the chisotan and cellulose monomers is
shown.

Figure 4. Structure of glucosamine (chitosan monomer) and glucose (cellulose
monomer) [20].
Chitosan is insoluble at neutral and alkaline pH, but forms water-soluble salts
with inorganic and organic acids including glutamic, hydrochloric, lactic and acetic
acids. Upon dissolution in acidic media, the amino groups of the polymer become
protonated rendering the molecule positively charged. Recently, a defined degree of
deacetylation and depolymerization has been attached to chitosan derivatives as
important, because of their significantly different physicochemical properties [23, 24,
27]. The degree of acetylation represents the proportion of N-acetyl-D-glucosamine
units with respect to the total number of reactive units. The properties of chitosan (pKa
and solubility) can be modified by changing the degree of deacetylation and formulation
properties such as the pH and ionic strength. At neutral pH, most chitosan molecules
will lose their charge and precipitate from solution. Chitosan exhibits a variety of
physicochemical and biological properties, therefore it has found numerous applications
in various fields such as waste and water treatment, agriculture, fabric and textiles,
cosmetics, nutritional enhancement, and food processing. In addition to its lack of
toxicity and allergenicity, its biocompatibility, biodegradability and bioactivity make it a
very attractive substance for diverse applications as a biomaterial in pharmaceutical and
medical fields [23, 31, 32, 43].
Chemical derivatization of chitosan provides good materials for promoting new
biological activities and for modifing its mechanical properties. The primary amino
groups on the molecule are reactive and provide a mechanism for side group attachment
using a variety of mild reaction conditions. The general effect of addition of a side chain
is to disrupt the crystal structure of the material and hence to increase the amorphous

fraction. This modification generates a material with lower stiffness and often altered
solubility, but the precise nature of changes in chemical and biological properties
depends on the nature of the side group. In addition, the characteristic features of
chitosan such as being cationic, hemostatic and insoluble at high pH, can be completely
reversed by a sulfation process, which can render the molecule anionic and water-
soluble, and also introduce anticoagulant properties [15, 23, 24,36].
The variety of groups which can be attached to chitosan is almost unlimited, and
side groups can be chosen to provide specific functionality, change biological properties
or modify physical properties. Due to its high molecular weight and a linear unbranched
structure, chitosan is an excellent viscosity-enhancing agent in acidic environments. It
behaves as a pseudoplastic material exhibiting a decrease in viscosity with increasing
rates of shear. The viscosity of chitosan solution increases with an increase in chitosan
concentration, decrease in temperature and with increasing degree of deacetylation,
which is a structural parameter also influencing physiochemical properties such as the
molecular weight, the elongation at break and the tensile strength. Viscosity also
influences biological properties such as wound-healing properties and osteogenesis
enhancement as well as biodegradation by lysozyme [23,24, 30, 31, 35].
Chitosan, which is polycationic in acidic environments, possesses an ability to
form gels because it is hydrophilic and can retain water in its structure. The acetylation
of chitosan in hydroalcoholic media allows the selective modification of the free amino
groups and is responsible for a process of gelation. It has been shown that the charge
density of the chain segments is an essential parameter for the formation of gels and all
factors that lower this parameter favor deswelling and reversibility. The high hydration,
the physicochemical and physical properties, as well as the polyelectrolyte behavior of

this kind of gel allow applications such as bioactive dressing for wound healing. Gels
can also be used as a slow release drug-delivery system [23, 24, 30, 33].
The solubility of chitosan can be sharply reduced by cross-linking the
macromolecules with covalent bonds using for example glutaraldehyde. Swelling of the
films, for exemple, decreases when increase the amount of cross-linking agent added.
When chitosan is intended for contact with serous liquids, sterility becomes
necessary. Heat is often employed to facilitate polymer processing and to sterilize the
pharmaceutical and medical products. However, exposure to high temperatures can
change the physical properties of chitosan, affecting its aqueous solubility, rheology,
and appearance. Chitosan films were found to be less hydrophilic when autoclaved at
121 0C for 1h 30min. This provoque a reduction in solubility that was related to the
formation of the anhydrous crystal polymorph observed in chitosan samples heated in
the presence of water. Unlike gamma irradiation, which caused main chain scissions and
a dose dependent decrease in viscosity [10, 19, 23, 24, 31, 34, 36].
While increasing the ionic strength, the counter-ions would screen the
protonated amine group and make the molecule contracted. Strong intramolecular
hydrogen bonding was formed in solution because of the large number of OH and acetyl
groups in the chitosan molecular chains. Additionally, the hydrophobic properties in
chitosan, acetyl groups and glucosidic rings can play a significant role on aggregation in
the formation of hydrophobic interaction
The conformation changes of chitosan in solution are attributed to the intra/inter
molecular forces interaction, and thus suggesting that the conformation change has a
relation with surface tension, charge surface distribution. This cationic property is the
basis of many of the potential applications of chitosan that can be considered as a linear

polyelectrolyte with a high charge density which can interact with negatively charged
surfaces, like proteins. The superior tissue compatibility, biofunctionality and non-
antigenic property make chitosan and derivatives an ideal material for medical
application in tissue regeneration [6, 18, 24, 36, 37].
Polysaccharides such as chitosan, in particular, have some excellent properties.
They have one of the largest and widest ranges of medical applications:
nontoxicity (monomer residues are not hazardous to health), water solubility or high
swelling ability by simple chemical modification, stability to pH variations, and so on.
There are some disadvantages, such as low mechanical properties, temperature
and chemical instability, which, in some cases, can appear as an advantage [35, 36].
1.3.2. Microcrystalline chitosan
Struszczyk H. M. 2003, showed “the preparation of microcrystalline chitosan
(MCCh), according to Polish Patent P-281975, 1989”. MCCh is a modified chitosan
form elaborated based on the aminoglucose macromolecule aggregation method; it’s
characterized by special properties of initial chitosan such as biocompatibility,
bioactivity, non-toxic, hydrophility with same extraordinary behavior like direct film-
forming and creation of molecular and super-molecular structure during its
manufacture. This form of chitosan is very suitable in medical application, especially
for wound dressings and drug delivery. However, application of microcrystalline
chitosan form shows resistance to dissolution at neutral pH, as well as prolongation of
the biodegradation due to the relatively high crystallinity of the formed biocomposites
[8, 16].
1.3.3. Medical applications
Medical Applications and bioactivity of chitosan (being the result of several
properties: biodegradability, biocompatibility, antibacterial, antifungal and antiviral

activity, high adhesivity, film-fibre forming, non-toxicity, high miscibility, high
chemical reactivity, high ability for creation of hydrogen and ionic bonds,
biostimulation of natural resistance, by controlling and improving bioactivity) make this
biomaterial an excellent substrate. Those properties make it susceptible for preparation
and modification of a modern generation of scaffolds for tissue regeneration. The
application of chitosan depends upon the useful form of the copolymer for different
places to use [24, 36, 38, 39, 40, 42, 43].
The major limitations in the use of the chitin and chitosan for designing medical
devices are [40].
· the collection of the raw material;
· difficulty to obtain reproducible products with different raw materials;
· constantly high cost of production;
· the absence of validated process and products of biopolymer manufacture;
· no standardization of product quality and product assessment methods for chitin
and chitosan.
In orthopaedic uses, the enzymatic degradability associated to its structural
similarity to extracellular matrix glycosaminoglycans makes chitosan an attractive
biopolymer for bone tissue repair. Numerous bone filling materials have been
developed in which chitosan is used in combination with calcium phosphates,
essentially as a binding agent, or associated to biological molecules. Additionally, its
versatility to be processed into injectable, porous and membrane forms without using
toxic solvents makes chitosan an interesting material to be used as a non-protein
temporary scaffold, for bone regeneration. Presently, an increasing number of
anchorage-dependent cells, including bone cells, are being cultured on 2-D and 3-D
chitosan-based matrices, for regenerative therapies.

Biocomposities as polymeric materials used in implants with the criteria of the
proper choice of polymers. These criteria cover the structure of a polymer and other
materials, porosity and surface properties and biodegradability process; they make the
chitosan and calcium phosphates a good choice to work as a scaffold [5, 6, 19, 38, 44].
Due to its high molecular weight and a linear unbranched structure, chitosan
shows to be an excellent viscosity-enhancing agent in acidic environments. It behaves
as a pseudoplastic material exhibiting a decrease in viscosity with increasing rates of
shear. The viscosity of chitosan solution increases with an increase in chitosan
concentration, decrease in temperature and with increasing degree of deacetylation,
which is a structural parameter also influencing physiochemical property such as the
molecular weight, the elongation at break and the tensile strength. Viscosity also
influences biological properties such as wound-healing properties and osteogenesis
enhancement as well as biodegradation by lysozyme [3]. Table 1 shows the biomedical
applications and bioactive properties of chitosan and derivatives.
Table 1. Biomedical applications and bioactive properties of chitosan [3].
ARTIFICIAL SKIN
Surgical sutures
Artificial blood vessels
Controlled drug release
Contact lens
Eye humour fluid
Bandages, sponges
Burn dressings
Blood cholesterol control

Anti-inflammatory
Tumor inhibition
Anti-viral
Dental plaque inhibition
Bone healing treatment
Wound healing accelerator
Hemostatic
Antibacterial
Antifungal
Weight loss effect
Tissue regeneration is related with cellular interactions of chitosan with
mammalian tissues, which have been positive from the tissue repair and regeneration
standpoint. One of chitosan’s most promising features is its excellent ability to be
processed into porous structures for use in cell transplantation and tissue regeneration.
Porous chitosan structures can be formed by freezing and lyophilizing chitosan
solutions in suitable molds. The mechanical properties of chitosan scaffolds depend
largely on the pore sizes and pore orientations [3]. Such scaffolds can enhance bone
repair by supporting the proliferation of osteoblastic cells as well as their differentiation.
Table 2, collects the main characteristic properties of the natural biopolymers of
interest in this area: it should be underlined that chitosan has the capacity to form
complexes with both inorganic and biochemical substances. In turn, the inorganic
complexes favor correct biomineralization, and chitosan-glycosaminoglycan complexes
concentrate and retain growth factors.

Table 2. Favorable and unfavorable properties of natural biopolymers prepared for
applications in regenerative medicine (pharmaceutical and medical grades) [1].
Chitosan Unique cationic behavior. Hydrophilic surface promoting cell adhesion,
proliferation and differentiation. High filmogenicity. Good biocompatibility and
good host response. High biochemical significance in hemostasis, angiogenesis,
macrophage activation, fibroblast proliferation control. Biodegradability by
lysozyme and other enzymes. Bactericidal/bacteriostatic activity. Mechanical
weakness. Capacity to maintain a predefined shape after cross-linking.
Silk fibroin Slow degradability, versatility in processing, remarkable mechanical strength.
Genetically tailorable composition and aminoacid sequence. Residual sericin may
cause biocompatibility problems.
Collagen Low antigenicity and good cell-binding properties. Collagen type I (the most
abundant extracellular matrix protein) supports cell adhesion and proliferation;
integrin-mediated adhesion to collagen type I enhances osteogenic differentiation
of human bone marrow mesenchymal stem cells. Low biomechanical stiffness
and rapid biodegradation. mesenchymal
Hyaluronan Absence of immunogenic properties. Easy chain size manipulation. Interactions
with cell-surface receptors. Production through large-scale microbial
fermentation. Its anionic surface does not promote cell attachment and tissue
formation. Very soluble in water. Quick degradation by lysozyme and other
enzymes
Alginate Cross-linking under very mild conditions. Suitable for gel injection. Mechanical
weakness. Difficulties in handling and sterilization. Variety of Structures
Starch Inexpensive. In vivo degradation has not been fully assessed yet
Bacterial cellulose High purity, nanofibrous structure, high tensile strength and good

biocompatibility. Small pore size. Unclear in vivo behavior
Dextran Susceptible to chemical modification, suitable for designing of scaffolds with
specific sites for cell recognition. Shortcomings typical of hydrogels. Needs
modification to enhance cell adhesion
Polymer-hydroxyapatite blends have been reported to be easily handled during surgery,
a moldable or injectable material being more easily applied than pure hydroxyapatite powder or
granules. Major disadvantages of those biodegradable systems are their considerably inferior
mechanical strength, when compared to natural bone. This limits application to high load
bearing parts of the human skeleton [7]. Several new forms of chitosan-based dressings are
elaborated in form of hydrogels, films, microspheres, sponges and so on. One of the
most promising chitosan derivatives is MCCh that is a modified chitosan form
elaborated and based on the aminoglucose macromolecule aggregation method. This
form of chitosan is very suitable for medical application, especially for wound dressings
and drug-delivery [32, 37, 41, 42, 44].
1.4. Calcium Phosphates
1.4.1. Hydroxyapatite general properties
Calcium phosphates, particularly hydroxyapatite and beta-tricalcium phosphate,
as biomaterials had a significant application in the past and, in those days, as materials
for bone replacement. Its applications include, for example, prostheses for replacement,
materials coating or support in parts of the "Scaffold" [1, 2, 10, 11].
The bone consists of 69 wt% calcium phosphate (mainly hydroxyapatite), 21%
collagen, 9% water and 1% other constituents. It has a composite nature which is built
up of mainly ceramic (hydroxyapatite) and polymer (collagen), with a complex

hierarchical microstructure very difficult to mimic, which gives most of the superior
mechanical properties to bone [3].
Recent developments in artificial bone field include ceramics, which are bioinert
(such as alumina and zirconia), resorbable (such as tricalciumphosphate), and bioactive
(hydroxyapatite). Different phases of calcium phosphate ceramics are used depending
upon whether a resorbable or bioactive material is desired. Many applications in hard
tissue have been repoted in the past and continue to be nowadays, such as, for example,
replacements for hips, knees, teeth, tendon and ligaments and repair for peridontal
disease, maxillofacial reconstruction, augmentation and stabilization of the jaw bone,
spinal fusion and bone repair after tumor surgery, the tissue bonding between ceramic
and soft tissues besides the hard tissue can be notice using bioactive ceramics [11, 12].
The most common biomaterial used in the past years in hard tissue regeneration
was Hydroxyapatite (HAp), because it is the major inorganic compound in mammalian
hard tissue and is highly recognized and used for its biocompatibility, not expensive and
abundant. It has been incorporated into a wide variety of biomedical devices including
dental implants, biodegradable scaffolds, and other types of orthopaedic implants in
different parts of the skeleton [1,13, 14].
Hydroxyapaptite (Ca10(Po4)3OH), has been widely used in the present due to its
chemical similarity to bone and good biocompatibility in the physiological
environmental, as well as compatibility with synthetic and natural polymers such as
polysaccharides and/or proteins like collagen, creating a functional biomaterial for
medical and veterinary application. HAp has been shown to stimulate osteoconduction
and is a material that can be integrated into bone without provoking an immune reaction
[15, 16, 17, 18, 19]. The biological response to HAp implants is influenced by factors
such as the properties of the HAp powder including the grain size or any decomposition

of the HAp powder [15, 16, 17, 18, 19]. The structural configuration of HAp including
the size and morphology of the particles within the fabricated scaffold has been shown
to be critical in allowing osteoconduction and bone growth into the scaffolds whilst also
allowing the transfer of nutrients through the scaffold [15, 16, 17, 18].
The challenge of hard tissue engineering is to develop a suitable bone scaffold
with sufficient porosity and mechanical strength to allow cell adhesion, migration,
growth and proliferation resulting in good integration with surrounding tissues. A
number of materials have been used for bone tissue engineering, including synthetic and
natural polymers, bioglass and a variety of calcium phosphate ceramics [15, 17, 18].
Surface with a positive charge promotes cell adhesion due to its negative charge,
it is able to chemically bond with positively charged polysaccharides and/or HAp with
negative charge like proteins and/or another calcium phosphate as a -TCP, forming a
stronger scaffold material [20, 21].
The biodegradation of calcium phosphates, including HAp, may represent a
combination of the following items [1, 11, 21, 22 ] :
1 - physical: abrasion, fracture, disintegration, shape, porosity, surface area,
crystallinity and grain size.
2 - chemical: dissolution, local increase of Ca and P on the surface, composition
of the material.
3 - method: reduction of pH caused by cellular activity, resulting in increased
rate of degradation due to dissolution.
4 - biological: includes pH involving cell involvement, infections or diseases,
degree of bone contact, bone type, specimens, sex, age, hormonal and genetic [1].
Studies suggest that the mechanism of degradation of dense ceramics of calcium

phosphate which exhibit high crystallinity is mainly dissolution by extracellular fluids
[1].
It has been claimed that the behavior of the apatite family upon immersion in a
simulated body fluid was structure and composition dependent. The powders dissolution
rate is dependent largely on the crystallinity level, phase composition, microstructure,
surface area and density [7, 10].
1.4.2. General properties of tricalcium phosphate
Calcium phosphates are chemical compounds of special interest in many
interdisciplinary fields of science, including geology, chemistry, biology and medicine.
According to the literature, the initial attempts to establish their chemical
composition were performed by J. Berzelius in the middle of the 19th century and
constitute a major family of inorganic materials currently in use in dental and
orthopaedic reconstructive medicine, specifically; hydroxyapatite (HAp) and -
tricalcium phosphate (ß-TCP) were developed as bioceramics in the early 1980s and,
nowadays, are the most common calcium phosphates used in medical applications.
Despite their relative importance, both ceramics show a number of drawbacks
that reduce their clinical performance. The biodegradation of HAp in physiological
environments is too low to achieve the optimal formation of bone tissue.
On the other hand, ß-TCP shows fast release of Ca2+ and PO43- ions when
exposed to physiological fluids and could be considered as bioactive [3, 19, 20].
The biological performance of biphasic mixtures is controlled by the dissolution
profile of the mixture. Selecting the appropriate blend of both calcium phosphates, the
mixture gradually dissolves in the physiological environment, releasing Ca2+ and PO43-
ions and inducing the bioactive behavior. The material that remains during dissolution

acts as a template for the newly formed bone. Tricalcium phosphate is called a
resorbable ceramic, and it is believed that it binds to bone and then is resorbed and
replaced by bone. It has been reported that the bioresorbability is due to dissolution and
phagocytosis. It has been considered that -TCP makes contact with bone directly,
suggesting mainly mechanical bonding. Synthetic -tricalcium phosphate (ß-TCP),
Ca3(PO4)2, is a material with a high potential for bioapplications. In particular,
composites made of ß-TCP and HA, the so-called biphasic calcium phosphates (BCPs),
which combine the excellent bioactivity of HA with the good bioresorbability of ß-TCP,
are interesting candidates for medical applications such as bone replacement [1,5] .
By definition, all calcium orthophosphates consist of three major chemical
elements: calcium (oxidation state +2), phosphorus (oxidation state +5) and oxygen
(reduction state. – 2), as a part of orthophosphate anions. These three chemical elements
are present in abundance on the surface of our planet: oxygen is the most widespread
chemical element of the Earth's surface (~ 47 mass %), calcium occupies the fifth place
(~ 3.3 – 3.4 mass %) and phosphorus (~ 0.08 .– 0.12 mass %) is among the first twenty
chemical elements most widespread on our planet [20]. In addition, the chemical
composition of many calcium orthophosphates includes hydrogen, either as part of an
acidic orthophosphate anion (for example, HPO42- or H2PO4-), hydroxide (for example,
Ca10(PO4)6(OH)2) and/or incorporated water (for example, CaHPO4·2H2O). Diverse
combinations of CaO and P2O5 (both in the presence of water and without it) provide a
large variety of calcium phosphates, which are distinguished by the type of the
phosphate anion: ortho- (PO43-), meta- (PO3
-), pyro- (P2O74-) and poly- ((PO3)n
n-). In
the case of multi-charged anions (orthophosphates and pyrophosphates), calcium
phosphates are also differentiated by the number of hydrogen ions attached to the anion.

Examples include mono- (Ca(H2PO4)2), di- (CaHPO4), tri- (Ca3(PO4)2) and tetra-
(Ca2P2O7) calcium phosphates [19, 20, 21].
-TCP ( -tricalcium phosphate, -Ca3(PO4)2; the chemically correct name is
calcium phosphate tribasic beta) cannot be precipitated from aqueous solutions. It is a
high temperature phase, which can only be prepared at temperatures above 800 ºC by
thermal decomposition of CDHA or by solid-state interaction of acidic calcium
orthophosphates, e.g., DCPA, with a base, e.g., CaO. Apart from the chemical
preparation routes, ion-substituted -TCP can be prepared by calcining of bones: such
type of -TCP is occasionally called. “bone ash”. In -TCP, there are three types of
crystallographically nonequivalent PO43- groups located at general points of the crystal,
each type with different intratetrahedral bond lengths and angles. At temperatures above
~ 1125 ºC, -TCP transforms into a high-temperature phase -TCP.
Being the stable phase at room temperature, -TCP is less soluble in water than
-TCP. Furthermore, the ideal structure contains calcium ion vacancies that are too
small to accommodate calcium ions, but allow for the inclusion of magnesium ions,
which thereby stabilize the structures. Pure -TCP never occurs in biological
calcifications. In biomedicine, -TCP is used in calcium orthophosphate bone cements.
In combination with HA, -TCP forms a biphasic calcium phosphate that are
widely used as a bone substitution bioceramics. Pure -TCP is added to some brands of
toothpaste as a gentle polishing agent. Multivitamin complexes with calcium
orthophosphate are widely available in the market and -TCP is used as the calcium
phosphate there. In addition, it serves as a texturizer, bakery improver and anti-
clumping agent for dry powdered food (flour, milk powder, dried cream, cocoa
powder). In addition, -TCP is added as a dietary or mineral supplement to food and

feed, where it is marked as E341 according to the European Classification of Food
Additives. Occasionally, it might be used as inert filler in pelleted drugs. Other
applications comprise porcelains, pottery, enamel, using as a component for mordants
and ackey, as well as a polymer stabilizer [19, 20, 21].
ACP (amorphous calcium phosphate, CaxHy(PO4)z·nH2O, n = 3 .– 4.5; 15 –
20% H2O) is often encountered as a transient phase during the formation of calcium
orthophosphates in aqueous systems. Usually, ACP is the first phase precipitated from a
supersaturated solution prepared by rapid mixing of solutions containing ions of
calcium and orthophosphate. ACP is thought to be formed at the beginning of the
precipitation due to a lower surface energy than that of OCP and apatites. The
amorphization level of ACP increases with the concentration increasing of Ca+2 and
PO4- containing solutions, as well as at a higher solution pH and a lower crystallization
temperature. A continuous gentle agitation of as precipitated ACP in the mother
solution, especially at elevated temperatures, results in a slow recrystallization and
formation of better crystalline compounds, such as CDHA. The lifetime of ACP in
aqueous solution was reported to be a function of the presence of additive molecules
and ions, pH, ionic strength and temperature.
In medicine, pure ACP is used in calcium orthophosphate cements and as a
filling material in dentistry. Bioactive composites of ACP with polymers have
properties suitable for use in dentistry and surgery. Due to a reasonable solubility and
physiological pH of aqueous solutions, ACP appeared to be consumable by some
microorganisms and, due to this reason; it might be added as a mineral supplement to
culture media. Non-biomedical applications of ACP comprise its using as a component
for mordants and ackey. In food industry, ACP is used for syrup clarification.

Occasionally, it is used as inert filler in pelleted drugs. In addition, ACP is used
in glass and pottery production and as a raw material for production of some organic
phosphates. For further details on ACP, interested readers are referred to specialized
reviews [19, 20, 21].
1.4.3. Medical applications
These substrates, when modified by the addition of cells or biomolecules, are
able to stimulate regeneration in a shorter time; they can function as hybrid materials,
further enhancing in vivo tissue formation femur, knee, teeth, tendons, ligaments,
materials for repairs due to problems of periodontics, neurosurgery and for filling bone
cavities after tumour surgery [2, 19, 20].
These substrates or supports can give two mechanisms to improve the
regeneration [1] :
1 - give support permissive for cell migration and adhesion and growth outside the
host;
2 - as a vehicle for controlled release of drugs that promote growth and survival
during regeneration.
Bone has a varied arrangement of material structures at many length scales
which work in concert to perform diverse mechanical, biological and chemical
functions; such as structural support, protection and storage of healing cells, and
mineral ion homeostasis. Scale is important to determine the ideal scaffold bone
architecture as the structure of the natural bone hierarchical and complex structure, in
Table 3, showed the the most used calcium phosphates for medical applications [33, 21,
23]. The calcium phosphates are fully supported by the physiological environment in
bone replacement, enabling rates of resorption and replacement very favorable. HAp is
more similar to natural bone than other calcium phosphate like -TCP, (Ca3(Po4)2);
however, the resorption of HAp is extremely low in comparation to ß-TCP [7, 21].
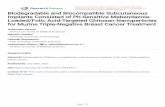
Table 3. existing calcium orthophosphates and their major properties [19].
[a] These compounds cannot be precipitated from aqueous solutions. [b] Cannot be measured precisely. However, the following values were found: 25.7±0.1 (pH = 7.40), 29.9±0.1 (pH = 6.00), 32.7±0.1 (pH = 5.28). The comparative extend of dissolution in acidic bufferis:ACP>> -TCP>> -TCP>>CDHA>>HA>>FA. [c] Stable at temperatures above 100°C.
[d] Always metastable. [e] Precipitated HÁ (PHA). [f] Existence of OA remains questionable.
1.4.4. References
1. Riccardo A.A. Muzzarelli (2011). Chitosan composites with inorganic,
morphogenetic proteins and stem cells for bone regeneration, Carbohydrate
Polymers 83, 1433-1445

2. Misiek D J, Kent JM, Carr RF. (1984). Soft tissue responses to hydroxyapatite
particles of different shapes. J Oral Maxillof Surg;42:150-60.
3. Oktay Yildirim. (2004). Preparation and Characterization of Chitosan /Calcium
Phosphate Based Composite Biomaterials, Master Of Science Dissertation, zmir
Institute of Technology zmir, Turkey.
4. Vert M, Li MS, Spenlehauer G, Guerin P. (1992). Bioresorbability and
biocompatibility of aliphatic polyesters. J Mater Sci;3:432-46.
5. Sevda Snel, Susan J. McClure. (2004). Potential applications of chitosan in
veterinary medicine, Advanced Drug Delivery Reviews 56, 1467- 1480.
6. Dorozhkin Sergey V. (2009). Review Nanodimensional and Nanocrystalline
Apatites and Other Calcium Orthophosphates in Biomedical Engineering,
Biology and Medicine, Materials, 2, 1975-2045.
7. Wilmington, Delaware, September 1 /PRNewswire/ - New market research
report, 'Global biomaterials Market (2009-2014)', published by Markets and
Markets(http://www.marketsandmarkets.com),(http://www.prnewswire.co.uk/cgi
/news/release?id=264557).
8. Muzzarelli C., Riccardo A.A. Muzzarelli. (2002). Natural and artificial chitosan-
inorganic composites, Journal of Inorganic Biochemistry 92 89-94.
9. Junqueira LC, Carneiro J, Long JA. (1986). Bone, Basic Histology. 5th Ed.
Norwalk, Conn: Appçeton-Cenury-Crofts;140-65.
10. Adler C. Bones and bone tissue: normal anatomy and histology. Bone Diseases.
2000. New York, NY: Springer-Verlag; 1-30.
11. McCarthy EF, Frassica FJ. Anatomy and physiology of the bone. Pathology of
Bone and Joint Disorders. Philadelphia, Pa: WB Saunders; 1998: 25-50

12. Mundy GR. (1987). Bone resorption and turnover in health disease. Bone;8
(suppl 1):S 9-16. [medline]
13. McHugh KP, Shen Z, Crotti TN. (2007). Role of cell-matrix interactions in
osteoclastc differentiation. Adv Exp Med Biol.;602:107-11. [medline]
14. Marks SC Jr, Propoff SN. (1988). Bone cell biology: the regulation of the
development, structure and function in the skeleton. Am J Anat.183(1):1-44.
[medline]
15. Leblond CP. (1989). Synthesis and secretion of collagen by cells of connective
tissue, bone and dentin. Anat Rec; 224(2): 123-28. [medline]
16. Bouxsein ML, Myburgh KH, van der Meulen MC, Lindenberger E, Marcus R.
(1994). Age-related differences in crosssectional geometry of the forearm bones
in healthy women. Calcif Tissue Int; 54: 113-8.
17. Burr DB, Martin RB, Schaffler MB, Radin EL. (1985). Bone remodelling in
response to in vivo fatigue microdamage. J Biomech; 18: 189-200.
18. Zioupus P. (2001). Aging Human Bone: actors Affecting Its Biomechanical
Properties and the Role of Collagen. JOURNAL OF BIOMATERIALS
APPLICATIONS Volume 15 - January; 187-229.
19. Dorozhkin Sergey V. (2011). Medical Application of Calcium Orthophosphate
Bioceramics, BIO, 1, 1-51.
20. Dorozhkin Sergey V. (2009). Review Calcium Orthophosphates in Nature,
Biology and Medicine, Materials, 2, 399-498.

21. Dorozhkin Sergey V. (2009). Review Nanodimensional and Nanocrystalline
Apatites and Other Calcium Orthophosphates in Biomedical Engineering,
Biology and Medicine, Materials, 2, 1975-2045.
1.5. Aim and scope of research
The biomaterial development in hard tissue engineering is a multidisciplinary area,
which has been improving in the past years with many published literature and patents
related about new materials, methods of fabrications and applications. The aim of this
research is focusing in develop a new material and method to obtain an alternative
biomaterial containing micro and nano ceramic with natural polymer as a chitosan in
different shapes that can be used in regenerative medicine.

Chapter 2.
Properties of chitosan material
2.1. Introduction
Bone repair or regeneration is a common and complicated clinical problem in
orthopaedic surgery, and much attention has been given to the use of different materials
and methods that could be employed as a base material for scaffold devices and as
modification tools for currently used biomedical devices improving hard and soft tissue
regeneration and/or reinforcement efficacy, also to expand the feasibility of combined
controlled drug release and tissue engineering, tissue formation in regenerative therapy
in the field of periodontics, orthopaedics, cancer and plastic surgery, veterinary [1, 2, 3,
4].
Chitosan, presented in Chapter 1, has been used in biomedical applications and
exhibits a variety of physicochemical and biological properties, in addition to its lack of
toxicity and allergenicity, biocompatibility, biodegradability and bioactivity make it a
very attractive substance for diverse applications as a biomaterial in pharmaceutical and
medical fields, especially in tissue regenerative therapy. Chitosan is the N-deacetylated
derivative of chitin, the second most abundant polysaccharide in nature after cellulose.
Depending on the chitin source and the methods of hydrolysis, chitosan varies greatly in
its molecular weight (MW) and degree of deacetylation (DD). The typical DD of
chitosan is over 70%, making it soluble in some aqueous acidic solutions [5,6,7, 8].
While increasing the ionic strength, the counter-ions would screen the
protonated amine group and make the molecule contracted. Strong intra/intermolecular
hydrogen bonding was formed in solution because of the large number of OH- and
acetyl groups in the chitosan molecular chains. Additionally, the hydrophobic properties
in chitosan, acetyl groups and glucosidic rings can play a significant role on aggregation

in the formation of hydrophobic interaction. The conformation changes of chitosan in
solution are attributed to the intra/intermolecular force interaction, and thus suggested
that the conformation change has a relation with surface tension, charge surface
distribution [7, 8, 9, 10]. This cationic property is the basis of many of the potential
applications of chitosan that can be considered as a linear polyelectrolyte with a high
charge density which can interact with negative charged surfaces, like proteins. The
superior tissue compatibility, biofunctionality and non-antigenic property make chitosan
and its derivatives an ideal material for medical application in tissue regeneration [8, 9,
10]
The aim of this study was to prepare chitosan hydrochloride salt from two
different chitosan producers from Norway and Brazil, comparing results to continuous
studies using analytical methods for characterization and mechanical properties in the
film form.
2.2. Materials
- chitosan ChitoClear FG-90 (Primex, Norway (Pandalus borealis))
- chitosan Polymar (Brazil (Pandalus borealis))
- hydrochloric acid 37,8 %, pure p.a., manufactured by POCh
2.3. Methods
2.3.1 Preparation of chitosan hydrochloride salt
First, a suitable amount of inorganic acid (hydrochloric acid) was added to
chitosan in -form (2.4 g) suspended in distilled water (150 cm3) to give 0.2% final
concentration of chitosan hydrochloride salt. Next, 50 cm3 of distilled water were added
and stirred for 1 hour. To the chitosan solution, 1% aqueous NaOH to pH 5.5 volume of
240 cm3was added dropwise, slowly, under stirring.

Figure 1 shows that the batches of preliminary chitosan were transformed into
the salt form of chitosan according to method elaborated in the Institute of Biopolymers
and Chemical Fibres, Poland.
Figure 1. Scheme of chitosan salt preparation
2.3.2. Analytical methods
a) The average molecular weight of chitosan was determined according to the
viscometric method using 0.0365 ÷ 0.0400 g ± 0.0001 g on dry weight of chitosan;
sample prepared from microcrystalline chitosan or chitosan gel sample is dissolved
in 15 cm3 of the solvent in measuring flask with volume of 25 cm3 using a
laboratory shaker. The flask is filled up to 25 cm3 with the solvent and the obtained
solution is filtered using G-4 Schott filtering funnel. 10 cm3 of the chitosan solution
is transferred into dilution viscometer and conditioned for 15 min. at 25°C ± 0.1°C in
a thermostat. The flow time of the solution through the 1st capillary is measured.
Repeat the measurement 3 times. Further measurements are similarly done for four
consecutive dilutions of the chitosan solution. Each time, add 5 cm3 of the solvent to

the viscometer, mix thoroughly. Keep the vessel with the solvent in the thermostat to
avoid thermostatical conditioning of the diluted solutions. Elaborated on the base
of: R.A.A. Muzzarelli, “Chitin”, Pergamon Press, 1978.
For each concentration of the solution, calculate the reduced viscosity ηred from
0
0
tc
ttnred ×
−=η
where: tn - flow time of solution with the actual concentration, (sec)
t0 - flow time of pure solvent (sec)
c - amount of chitosan in 100 cm3 of diluted solution
The limiting viscosity number is expressed by the equation:
redc ηη 0lim][ →=
The limiting viscosity number is determined graphically from the dependence of
reduced viscosity and concentration of chitosan solution by extrapolating the curve up
to zero concentration. The viscometric average molecular weight of chitosan is
determined on the base of the Mark-Houwink equation:
[η] = K × ⎯Mva
where:
[η] - the limiting viscosity number
K, a - empirical constants, K = 8.93 × 10-4; a = 0,71
Measurement accuracy: ± 1.5%

b) The water retention value of chitosan (WRV) is determined by submerging 0.5g
± 0.0001g of chitosan in 50 cm3 of distilled water [6]. Next, it is centrifuged for
10 min at 4000 rpm. The weight of the sample is determined after centrifuging
(m1) and drying to constant weight after 20 hours at 105°C ± 1°C (m0). The
water retention value (WRV, %) is found from the equation: Measurement
accuracy: ± 1.0%. Elaborated on the base of: R. Ferrus, et al. Cell. Chem.
Technol. 11, 633, 1977.
WRV =0
01
m
mm − ×100
where:
m1- weight of samples after centrifuged [g],
m0 - weight of samples after drying [g]
c) Deacetylation degree of microcrystalline chitosan or chitosan gel is determined
by the potentiometric titration method using glass and calomel electrodes. 0.10-
0.15 g ± 0.0001 g (m) sample of chitosan, on a dry weight, prepared from
microcrystalline chitosan or chitosan gel is dissolved in 10 cm3 of 1 wt %
aqueous acetic acid in a measuring flask with volume of 100 cm3 (V). The flask
is filled up to 100 cm3 with anhydrous acetic acid. 20 cm3 of obtained solution
(v) is transferred into a measuring vessel; 20 cm3 of anhydrous acetic acid and
10 cm3 of 1,4-dioxane are added. The measuring vessel containing the
investigated solution is placed on the magnetic stirrer and the glass indicator
electrode is immersed. The modified calomel electrode is immersed in a second
measuring vessel with saturated solution of potassium chloride in anhydrous
acetic acid. Both measuring vessels are connected by a salt bridge.

Solution of perchloric acid in 1,4 dioxane with determined concentration of
approx. 0.1 mol/dm3 (n) is added from a burette to the investigated solution, and
the electromotive power (EMP) is determined. The value of perchloric acid
volume (V1) corresponding to the neutralization point of amine groups is found
from the dependence between EMP and the volume of perchloric acid solution
used. Content of amine groups in chitosan (-NH2) is calculated from the
following equation: Elaborated on the base of: K. H. Bodek; Acta Polonica
Pharmaceutica - Drug Research 52, 4, 337, 1995.
(-NH2) =vm
VVn
××× 1
where:
n - Determined normality of HClO4 (perchloric acid) solution (for example n =
0.1118 mol/dm3)
m - Chitosan sample weight (g) (± 0.0001 g)
V1 - Volume of perchloric acid solution at neutralization point (cm3)
V - Volume of dissolved chitosan solution (100 cm3)
v - Determined sample volume (20 cm3)
Deacetylation degree (DD) of chitosan as a percentage ratio between the actual
and theoretical content of amine group is calculated from the following
equation:
DD =( )
( )LTHEORETICANH
NH
2
2
−−
=( )
211.62NH−
x 100 [%]

Measurement accuracy: ± 2%
d) Chitosan ash content - The quartz crucible is heated in the furnace at 800 °C to
constant weight. After cooling to room temperature in the exsiccator, the
crucible is weighed on the analytical balance. Approx. 2 g ± 0.0001 g of dry
chitosan sample prepared from microcrystalline chitosan or chitosan gel is
placed in the crucible. The crucible with the sample is preheated on an electric
heater to about 200 °C. After preheating, the chitosan sample is burned in the
furnace at 800 °C for about 3 h, to the constant weight. After cooling to room
temperature, the burned sample is weighed. Chitosan ash content (X, %) is
calculated from the following equation: Elaborated on the base of: ISO 3451-1
(1997).
1002
1 ×=m
mX
where:
m1 - weight of ash (g), (± 0.0001 g)
m2 - weight of dry sample (g), (± 0.0001 g)
Measurement accuracy: ± 10%
e) The moisture content in chitosan and MCCh is determined by a weight method
drying samples to constant weight at temperature of 105 ± 1oC. The moisture
content (M) is calculated from the following equation:
M =1
01
m
mm −×100

where:
m1- weight of samples before drying [g],
m0 - weight of samples after drying [g]
f) Reaction medium (pH). Equipment, pH-meter CP-315 M (producer: Elmetron,
Poland), accuracy ± 0.01. The reaction medium is controlled using a suitable
pH-meter. Measurement accuracy: 0.01 pH ± 1 scale interval with
temperature control.
g) The determination of dynamic viscosity of polymer solutions by Brookfield was
performed by rotating the spindle immersed in a liquid measuring - Spindle
(cone). The spindle is coupled to a calibrated spindle. The strength of resistance,
resulting from the viscosity of the fluid stops the rotating spindle and causes
deformation of the spring, whose degree is measured electronically. For the
determination of dynamic viscosity of the initial chitosan solution to prepare 1%
solution of chitosan having moisture in 1% acetic acid or, in the case of
chitosan, after deproteinization 1% solution in dry weight in 1% acetic acid.
Before measuring viscosity, the viscometer has to be reset. After resetting the
viscometer spindle shall be secured by screwing it into the threaded ends of the
handle axis. Before measuring viscosity, adjust the distance between the cone
(stem) and the plate or the bottom of a measurement vessel. The test solution of
0.5 or 2 cm3 (depending on the stem) should be placed in the measurement
vessel for 15 min at 25 °C ± 1.0 °C. After 15 min, measure the viscosity of the
solution and adjust the speed so that the torque value was above 90% (the
smallest measurement error). The result marked the dynamic viscosity read from
the screen viscometer is given in [mPa • s] or [cp]. Tangential stresses are given

in [N/m2] or [dyna/cm2], shear rate is given in [1 / s]. Accuracy of the method:
± 2%.
2.3.3. Assessment of physical-mechanical properties of chitosan hydrochloride salt
in film form.
The mechanical properties of the film form was determined in the Accredited
Laboratory of Metrology at IBWCh (certificate No. AB 388) by using INSTRON (type
5544) in accordance with the following standards:
PN-ISO 2602:1994 (Statistical interpretation of test results)
PN-EN 527-3:1998 (tests conditions)
For product in film form:
ISO 4593:1999 (thickness)
PN-EN 527-1:1998 (breaking strength)
PN- EN ISO 1798:2009 (tensile, and elongation at break)
2.4. Results and discussions
2.4.1. Results of analytical methods of chitosan hydrochloride salt solution.
The analytical results comparing chitosan hydrochloride salt solution (2%) from
Primex and Polymar are showed in Table 1.
Table 1. Characterization of Chitosan: Primex FG-90 (Norway), and Polymar (Brazil).
Tests Norway, Primex,
Chito-Clear FG90
Brazil
Polymar
Viscosimetric average
molecular weight [kD]
342.1 113.3
Moisture content [%] 12.77 11.32
Water retention value [%] 43.45 49.76

Dynamic Viscosity [cP] 63.11 13.51
Deacetylation degree [%] 83.20 76.9
Ash content [%] 0.40 3.1
Salt solution concentration [%] 2.0 2.0
pH 0.95 0.93
Based on the results presented in Table 1, it has been found that ash content in
chitosan Polymar (Brazil) is too high, also the low molecular weight, which is directly
related to mechanical properties performance, the lower dynamic viscosity related also
with mechanical properties and processing and, finally, the lower deacetylation degree
related to bioactivity and degradability, giving the free amino groups available, these
results showed that the Primex FG-90 (Norway) was a good choice for further studies.
2.4.2. Results for Mechanical properties from chitosan hydrochloride salt in film
form.
The resuts in Table 2, present a higher mechanical properties of chitosan
hydrochloride salt from Primex, such as max load and tensile strength, are directly
related to the higher average molecular weight (Mv) showed in the literature. The
viscosity of chitosan solution increases with an increase in chitosan concentration;
decrease in temperature and with increasing degree of deacetylation, which is a
structural parameter (such as the molecular weight) and mechanical properties (such as
elongation at break and tensile strength) [1,7,9].
Table 2. Mechanical properties of films obtained from the chitosan salts
Producer Max Load
[N]
Elongation at
break [%]
Tensile strength
[MPa]
Film thickness
[mm]

Primex 33.8 13.0 49.03 0.042
Polymar 9.8 12.6 16.62 0.038
2.5. Conclusions
Based on the presented results, it has been found that the ash content in chitosan
Polymar (Brazil) is too high; therefore, the molecular weight and viscosity showed to
lower than the chitosan from Primex (Norway).
The mechanical properties of chitosan hydrochloride salt in film form depend on
the kind of initial chitosan used, molecular weight and kind of chemical degradability.
A better performance of Primex FG-90 (Norway) showed a good choice for future work
and continuous studies related to preparation of different chitosan forms (sponges and
fibres) as a base for scaffolds for hard tissue regeneration.
2.6. References
1) Oktay Yildirim (2004). Preparation and Characterization of Chitosan /Calcium
Phosphate Based Composite Biomaterials, Master Of Science Dissertation, zmir
Institute of Technology zmir, Turkey. August.
2) Misiek D J, Kent JM, Carr RF. (1984). Soft tissue responses to hydroxyapatite
particles of different shapes. J Oral Maxillof Surg;42:150-60.
3) Vert M, Li MS, Spenlehauer G, Guerin P. (1992). Bioresorbability and
biocompatibility of aliphatic polyesters. J Mater Sci;3:432-46.
4) Riccardo A.A. Muzzarelli. (2009). Chitins and chitosans for the repair of
wounded skin, nerve, cartilage and bone, Carbohydrate Polymers 76, 167-182.
5) Sevda Snel, Susan J. McClure. (2004). Potential applications of chitosan in
veterinary medicine, Advanced Drug Delivery Reviews 56, 1467- 1480.

6) Francis Suh J.-K., Howard W.T. Matthew. (2000). Application of chitosan-
based polysaccharide biomaterials in cartilage tissue engineering: a review,
Biomaterials 21, 2589-2598.
7) Marguerite Rinaudo. (2006). Chitin and chitosan: Properties and applications,
Prog. Polym. Sci. 31 603-632.
8) Alberto Di Martino, Michael Sittinger, Makarand V. Risbud. (2005). Review
Chitosan:A versatile biopolymer for orthopaedic tissue-engineering,
Biomaterials 26, 5983-5990.
9) Harish Prashanth K.V., R.N.Tharanathan. (2007). Chitin/chitosan: modifications
and their unlimited application potential-an overview, Trends in Food Science &
Technology 18, 117-131.
10)Struszczyk H. M. (2003). The effect of the preparation method on the
physicochemical properties of microcrystalline chitosan (MCCh). ed., Progress
on Chemistry and Application of Chitin and its Derivatives, PTChit., vol. IX,
p.179-186.

Chapter 3.
Preparation of Microcrystalline chitosan (MCCh)/tricalcium phosphate complex in
sponge form.
3.1. Introduction
Different types of materials have been developed from chitosan and calcium
phosphates that are biocompatible, osteoconductive, resorbable and osteoinductive. The
wide range of materials available for hard tissue regeneration reflects methods of
production and application to reconstruct part of the skeleton [1, 2, 3].
All methods may achieve restoration and/or replacement. They all possess
inherent limitations, such as use site, donor-site morbidity, an obligatory graft
resorption phase, contour irregularities, insufficient autogenous resources, disease
transmission, major histocompatibility, structural failure, stress shielding, infection and
many others problems [4,5].
Most of scaffolds can be classified based on their component materials: natural
scaffolds, synthetic scaffolds and mineral-based scaffolds [6, 7, 26, 27].
Natural scaffolds are a combination of naturally occurring materials such as
collagen, alginate, agarose, chitosan, fibrin glue, and so on. Natural scaffolds have
proven successful at stimulating new tissue growth, for example, bones. However, while
natural biodegradable materials integrate well with endogenous tissues, they lack the
structural integrity required for scaffolds used in load bearing locations. For example,
chitosan scaffolds are both biodegradable and biocompatible, but are mechanically
weak, limiting their use as scaffolds for bone regeneration.

Synthetic Scaffolds are most commonly used bioabsorbable polymers are alpha-
hydroxy acids such as polyglycolide (PGA), polylactides (PLA), polycaprolactone
(PCL), polydioxanone (PDS), and their copolymer variants. Alpha-hydroxy acid-based
materials are designed to hydrolytically degrade in situ and then be converted to CO2,
and water by tissue macrophages.
Mineral-Based Scaffolds, polycrystalline ceramic scaffolds are designed to
synthetically replicate the calcium phosphate found in the extracellular matrix of normal
bone, thus providing an osteoconductive substrate for bone formation. The chemical
anisotropic materials most often utilized to construct polycrystalline ceramics are
hydroxyapatite (HAp) and tricalcium phosphate (TCP). Bone formation invariably starts
on the surface of a ceramic and then proceeds through the interconnected pore system
toward the centre of the construct.
Tricalcium phosphate is called a resorbable ceramic, and it is believed that it
binds to bone and is then resorbed and replaced by bone. It has been reported that the
bioresorbability is due to dissolution and phagocytosis. It has been considered that ß-
TCP makes contact with bone directly, suggesting mainly mechanical bonding.
Synthetic ß-tricalcium phosphate (ß-TCP), Ca3(PO4)2 is a compound with a high
potential for bioapplications. In particular, composites made of ß-TCP and HAp, the so-
called biphasic calcium phosphates (BCPs), which combine the excellent bioactivity of
HAp with the good bioresorbability of ß-TCP, are interesting candidates for medical
applications such as, for example, materials for bone replacement [8, 9, 10, 11] .
Many factors can affect the performance of the scaffolds, such as mechanical
properties, including the chemical composition of the polymer, rate of organic/inorganic
part, molecular weight distribution, the crystalline/amorphous ratio, the size and shape

of the implant and the particles involved in the scaffold, polymer processing methods,
implantation site, and the age/metabolic condition of the patient. On the other hand,
mineral-based biomaterials, such as calcium phosphate ceramics, are attractive as bone
grafts substitutes due to their unlimited supply and ease of sterilization and storage [12,
13, 14]. The porosity of a scaffold influences fibrovascular infiltration and, in turn,
determines the extent of bone in growth, cells nutrient flow and attachment. Studies
showed that pore sizes smaller than 15 to 50 µm result in fibrous tissue invasion, pore
sizes from 50 to 150 µm encourage osteoid formation, and pore sizes greater than 150
µm encourage the formation of mineralized bone. The literature shows that the optimal
pore size is considered to be between 150 and 500 µm, that it’s also related with
mechanical properties, stability and inflammatory response of the scaffolds [1, 14, 15,
16].
It appeared that the resorption of calcium phosphate biomaterials is beneficial to
bone formation realising Calcium and phosphate ions that have been considered as the
origin of bioactivity working. The gradual degradation at the same rate in which the
new bone is formed, providing the appropriate initial mechanical properties, and thus
encouraging a new bone to form in the region, is the task of the scaffold studies in hard
tissue regeneration [17]. The release of ionically bound phosphate functionalities from
MCCh/complex, under alkaline conditions, possibly contributed to the formation of
calcium phosphate precursor sites, due to the chelation of calcium ions from the primer
solution. A multi-layered porous mineral structure of the composites with partially
carbonated and poorly crystalline apatite was formed on the surface and inner surface of
MCCh/ß-TCP complex [18, 19].

In orthopaedics, the enzymatic degradability associated to scaffold structure
similar to extracellular matrix with glucosamine groups makes MCCh an attractive
biopolymer for bone tissue repair. Numerous bone filling materials have been
developed in which chitosan and its derivatives are used in combination with calcium
phosphates, essentially as a binding agent, or associated to biological signalling
molecules. In addition, its versatility to be processed into different shapes makes MCCh
with calcium phosphates an interesting material to be used as a non-protein temporary
scaffold, for bone regeneration [18, 19, 20]. The aim of the research is to obtain a new
method and material combining a natural-/ mineral-based scaffold in sponge form that
could be a base for a scaffolds production in hard tissue regeneration. Characterized by
chemical analytical methods, structure of the raw material and the complex in sponge
form according with the Polish patent application P 393758, 2011 [21].
3.2. Materials
1) Chitosan ChitoClear FG-90 (Primex, Norway) characterized by deacetylation degree
(DD) = 83.2%, water retention value (WRV) = 43.45%, molecular weight =342.1 kDa,
Moisture content =12.77%.
2) Tricalcium phosphate (ß-TCP), (Ca3 (Po4)2) - Sigma Aldrich Lab., Germany.
3) Plasticizer -Glycerol (C3H8O3) 99%, pure p.a., Sigma-Aldrich, Germany.
4) Hydrochloridric acid, 37.8% p.a., manufactured by Fluka.
5) Composite with MCCh/ ß-TCP water retention value (WRV) = 480.77 %, Moisture
content = 12.06%.
3.3. Methods
3.3.1 Preparation of the MCCh/ tricalcium phosphate complex.

Tricalcium chitosan/orthophosphate microcrystalline complex and tricalcium
chitosan/orthophosphate microcrystalline complex production method according to the
invention and Polish patent application number P 393758, 2011 [21].
Tricalcium chitosan/orthophosphate microcrystalline complex is a gel-like
suspension of viscometrically determined average molecular weight of 100,000-
250,000, rate of secondary swelling of 500 - 600% and pH= 7.2-7.4, containing 0.5-4.5
wt% tricalcium chitosan/orthophosphate complex, including 0.05-1.93 wt% of
tricalcium orthophosphate together with the microcrystalline chitosan:tricalcium
orthophosphate ratio from 90:10 to 57:43 wt%. In the production, showed in the Figure
1, it is represented the scheme of preparation of the MCCh/ß-TCP complex according
to the invention; the following method has been used: the equivalent part quantity of
weight of tricalcium orthophosphate solution of 0.5-1.5 wt% concentration in
hydrochloric acid of 0.7-1.8 wt% concentration is entered into chitosan aqueous
suspension containing 2.0 wt% of polymer of viscometrically determined average
molecular weight of 200,000-400,000 and deacetylation degree of 70-90%. The mixture
is being homogenized for 10-15 minutes with rotary speed of 400-600 rev/min. The
obtained solution is filtrated, and the filtrate is entered into the mixer, to which then
(constantly mixing) 870-910 of weight of sodium hydroxide aqueous solution of 0.4
wt% concentration are added to obtain pH = 7.7-7.8. The tricalcium
chitosan/orthophosphate microcrystalline complex obtained in suspension form is left
for 24 hours at 5-7 °C temperature. Then, it is purified with demineralised water up to
pH = 7.2-7.4 showed in Figure 1.

Figure 1. Scheme of preparation MCCh/ ß-TCP complex [21].
Manufacture process of MCCh/ ß-TCP complex is based on several physical-
chemical and chemical phenomena, such as neutralization, coagulation, agglomeration
of glucosamine macromolecules with calcium phosphate affecting the properties of final
product. The choice of ß-TCP is due to its lower solubility and better overall
performance in the physiological environment in comparison to the -form [8, 3].

3.3.2. Analytical methods
a) The water retention value of microcrystalline chitosan (WRV) is determined by
submerging 0.5g ± 0.0001g of MCCh in 50 cm3 of distilled water. Next, it is
centrifuged for 10 min at 4000 rpm. The weight of the sample is determined
after centrifuging (m1) and after drying to constant weight after 20 hours at
105°C ± 1°C (m0). The water retention value (WRV, %) is found from the
equation: Measurement accuracy: ± 1.0%. Elaborated on the base of: R.
Ferrus, et al. Cell. Chem. Technol. 11, 633, 1977.
WRV =0
01
m
mm − ×100
where:
m1 - weight of samples after centrifugation [g],
m0 - weight of samples after drying [g]
b) The moisture content in MCCh is determined by a weight method drying
samples to constant weight at temperature of 105 ± 10C. The moisture content
(M) is calculated from following equation:
M =1
01
m
mm −×100
where:
m1- weight of samples before drying [g],
m0 - weight of samples after drying [g]

3.3.3. Preparation of MCCh/ ß-TCP composite in film form
The films before and after purification process were prepared from a mixture
composed of an aqueous suspension of MCCh/ß-TCP complex. The samples without
glycerol were poured on Teflon plates, and dried at room temperature to obtain samples
in film form. The film form was prepared because was an easy and inexpensive way to
study the dissolution of the ß-TCP particles before and after purification process and
also the possible high contraction of the volume and size in a dry complex film to study
the attraction forces between the characteristic charge surface of MCCh(+) and ß-TCP(-),
and to study the best concentration of glycerol content used as a plasticizer.
3.3.4. Preparation of MCCh/ ß-TCP complex in sponge form.
The sponges were prepared from a mixture composed of an aqueous suspension
of MCCh/ß-TCP, according with Table 1. The preparation was first carefully
homogenized and, next, freeze-dried in a lab freeze dryer ALFA 1-4 made by Christ Co
in the temperature range from (-25) to 10 0C and vacuum from 0.1 to 0.53 mbar during
20 to 24 hours depending on the charge size. Drying accomplished that way resulted in
the preparation of sponges with a smooth surface without defects.
3.3.5. Infrared Spectroscopy of the complex
Fourier transform infrared spectroscopy (FTIR) is a non-destructive technique
that was used to identify the functional groups through their chemical bonds, which
generate a spectrum of infrared bands characteristic of each connection type. The
infrared analysis was performed on a range from 500 to 4000 cm-1, resolution 4.0 cm-1
with a Spectrum Genesis Series. Samples were prepared for analysis with KBr, in the
form of tablets, to verify the presence of functional characteristic groups in the ß-TCP:

P-O, O-H, P-O-H, H-O-H and for MCCh material the functional groups: NH2, Amide I,
amide II, according with Table 1.
3.3.6. Determination of particles size and morphology of commercial ß-TCP
powder.
The particle size of ß-TCP powder was determined by a technique using laser
particle size analyser Sympatec Hellos H1330, type BF (sympatec GmbH, Clausthal,
Germany). This analysis was a cooperation made between Institute of Biopolymers and
Chemical Fibres (IBWCh) in Lodz, Poland, and Thuringian Institute of Textile and
Plastics Research (TITK), Rudolstadt, Germany. The morphology of the used powder
and distribution in the polymer matrix in sponge and film forms was observed using a
scanning electron microscope (SEM) - FEI Quanta 200, USA.
3.4. Results and discussions
3.4.1 Elaboration of the quantitative and qualitative MCCh/ ß-TCP complex
The composition of samples is showed in Table 1.
Table 1. The quantitative and qualitative composition of MCCh/ ß-TCP complex
Symbol of sample Weight proportion of components
MCCh/ ß-TCP(suspension)
Glycerol
Sample 1 1 0
Sample 2 1 0.5
3.4.2. FTIR study
The main peaks of energy vibration in transmission mode are showed in Figure
2, identified in the ß-TCP characteristic functional groups of orthophosphate, hydroxyl,
and phosphate, and the carbonate group characteristic of synthetic calcium phosphate

material showed in traces. The presence of carbonate suggests that, in some commercial
ß-TCP and other calcium phosphates such as a HAp, CaO and Ca(0H)2 are used to get
an ideal stoichiometric relation between Ca / P in the material. It is known that synthetic
ß-TCP is more stable and more soluble in human body than HAp, among the calcium
phosphates. These features can increase the regeneration of bone tissue in the presence
of an implant scaffold. Thus, it is desirable that the material replacements for bones
present some solubility to allow this regeneration, which can be achieved with
carbonated calcium phosphates [2, 6, 17].
In Table 2, it is showed the FTIR study in transmission mode of commercial ß-
TCP, the absence of bands at 460 and 740 (cm-1) and an isolated band, 600 cm-1;
characteristic of -TCP, indicating that the starting material is only composed of ß-TCP,
also shows a small amount of CO3-2 at 1454 cm-1. The ß-TCP is easily identified through
the presence of a broad band in 900-1200 cm-1, characteristic of the symmetric mode (P-
O-P) assigned to antisymmetric stretching of P-O. Since the peak at 1118 cm-1 is
characteristic of a non-degenerate deformation of hydrogen groups - OPO 3 - H, O-PO3,
of common ions HPO, the presence of this group may be a consequence of the
interaction of water molecules in the structure, changing some neighbors in the
crystalline part, showed in the spectrum of energy vibration [ 2, 6, 17].

Table 2. Localization of peaks of a commercial ß-TCP in transmission FTIR.
Figure 2. FTIR spectrum of the commercial ß-TCP
The commercial ß-TCP in powder form, showed in a symmetric stretching at
3338 cm-1 and stretching in 1650 cm-1, is presented by O-H, H-O-H group, referring a
water adsorbsion by the powder characteristic of calcium phosphate family. The band
observed at 968 cm-1 of low intensity, corresponding to non-degenerate symmetric
stretching of P-O bonds of phosphate groups. The bands 1041, 1081 and 1098 (cm-1)
represent the asymmetric stretch modes, respectively, the P-O bonds of phosphate
Mode of vibrations Wave number of peaks, cm-1 Mode of groups
Antisymmetric stretching
Symmetric stretching
1041, 1081, 1098
968, 1118
P-O of PO4
Symmetric stretching 3338 O-H
Antisymmetric stretching 1454, 1428 CO of C=O
Stretching 1650 H-O-H

groups. The peaks of MCCh/ ß-TCP complex, before purification process showed in
Table 3 and Figure 3, confirm the presence of functional groups NH2 (1539 cm-1),
Amide I (1648 cm-1), Amide II (1557 cm-1) in the material and the main peaks of energy
vibration (1026, 1086 cm-1) identified the C-O skeletal vibrations stretch of saccharide
structure showed, respectively, in Figure 3 before purification process and Figure 4 after
purification process.
Table 3. Localization of peaks of the MCCh/ ß-TCP complex before purification
process.
Mode of vibrations Wave number of peaks, cm-1 Mode of groups
Symmetric stretching 1648 Amide I C=O
bending 1539 -NH2
stretching 1506 -NH2
1557 Amide II
1318 Amide III
Anti-symmetric strechingbridge
Skeletal vibration stretchingof saccharide structure
1154
1026, 1086
C-O-C
C-O
stretching3298, 3500 N-H
stretching3434 O-H
stretching1000, 1100 P-O, PO

Figure 3. FTIR spectrum of the MCCh/ ß-TCP complex before purification process
In the spectrum of energy vibration in transmission mode showing the MCCh/ß-
TCP complex before purification process peaks that are observed in the Figure 3, the
peak with 1648 (cm-1) corresponds to a stretch modes of amide I, associated with energy
level and the kind of structure that are linked with the type of bound NH2 (1539, 1506
cm-1) and Amide II. In 3434 cm-1, it is also observed the stretch of OH- group peak
referring to the way they stretch and water absorption. The band observed at 1154 cm-1,
of high intensity, corresponding an anti-symmetric stretching of C-O-C bond groups.
Bands 1026 and 1086 (cm-1) represent the symmetric stretch modes,
respectively, with the C-O bonds being characteristic of the saccharide structure groups.
The Figure 4 shows the FTIR energy vibration in transmission mode, the spectra of
MCCh/ß-TCP complex after purification process and exhibits the same characteristic
bands, such as a strong and broad peak between 1154 cm-1 and 1026 cm-1 and 1086 cm-1
that refer to the skeletal vibrations stretch of saccharide structure and a peak at 1636 cm-
1 attributed to the free primary amino group (NH2) in MCCh. The main peaks of energy
vibration identified in the ß-TCP also was observed in the complex with characteristic
functional groups of orthophosphate at 1100 cm-1, hydroxyl, phosphate (HPO) at 1000

cm-1 , (OH)- at 3430 cm-1 , the last one referring a water absorption. A broad band
appears from 1665 cm-1 corresponding to the H-O-H stretching absorption band in the
samples, showing a fair agreement with the literature [23, 24, 25].
Table 4. Localization of peaks of the MCCh/ ß-TCP complex after purification process.
Mode of vibrations Wave number of peaks, cm-1 Mode of groups
stretching 1636 Amide I C=O
bending 1538 -NH2
stretching 1506 -NH2
1557 Amide II
1316 Amide III
antisymmetric stretching
Symmetric stretching
1000
1100
P-O
PO4-3
stretching 3430 O-H
Antisymmetric stretching 862 P-O-H
Stretching 1655 H-O-H

Figure 4. FTIR spectrum of the MCCh/ ß-TCP complex after purification process
The chemical interactions between the inorganic and organic constituents in the
composite probably take place via the ionic bonding between ion phosphate and the
amino group of chitosan. The FTIR study of complex before and after purification
process showed a very similar peak of the same product showed in Figure 5, suggesting
that the free amino groups from microcrystalline chitosan bonded with the phosphate
groups of tricalcium phosphate creating a complex and validating the method of
preparation of MCCh/ ß-TCP complex, also notice a little decrease in the initial calcium
phosphate amount, which comes from several washings (purification process) with
purified and demineralised water.
Figure 5. FTIR spectrum of the MCCh/ ß-TCP complex before and after purificationprocess
3.4.3. Particles size and morphology of commercial ß-TCP powder.
The aim of this study was to determine the particles size of commercial ß-TCP
in the form of powder. Before analysis, the material used was dried with vacuum at
105oC for 24h to remove moisture and was separated to measure the particles size

showed in Table 5 and to study the grain morphology of commercial ß-TCP, showed in
the figure 6.
Figure 6. Respective particles size and distribution of of the commercial ß-TCP and
SEM microphotography of grain morphology.
The measurements of particles size showed in Table 5, showing a specified
surface area covered by commercial ß-TCP at 2,53 e+04 cm2/g and average particle size
of 4.48 m. It was found less than 10% of the tricalcium phosphate particles in nano-
size, which also indicates a faster and easier release of calcium and phosphate ions in
physiological environmental following the literature [25].
Table 5. Particles size and distribution of commercial ß-TCP.
POWDERS ß-TCP
Particles size (90%), ( m) 12.78
Particles size (50%), ( m) 4.48
Particles size (10%), ( m) 1.35
Superficial area (cm2/g) 2.53 e+04
The analysis of particles size using laser particle size analyser Sympatec Hellos
H1330, type BF, showed that a standard deviation cannot be given so easy. The particle

size depends firstly on particle shape (aspect relation). Most calculation models refer
to ball-shaped bodies. The more the particles deviate from this model, the more the
deviation rises. Secondly, the broadness (width) of particle size distribution has an
influence on the standard deviation using this method. In a case where this distribution
covers only a narrow part, for instance 2 to 5 µm, then the deviation is lower than 10%.
However, the measurement itself is very precise. The measurement of the powders used
in this research had procedure of three-times for each sample and receives deviations of
results lower than 2 %, the samples of both powders had a range from 0.5 to 50 µm. The
analysis showed a ß-TCP grain size fits in the range 3.0 - 9.0 m with fair agreement
with the literature. Notice the morphology of the ß-TCP powder is more spherical
showing a great ability to agglomeration and cluster formation, which could be
explained by a non-homogenous charge distribution on the surfaces and high ability for
water adsorption characteristic of this material and showed in the FTIR study.
Typically, the very small particles tend to agglomerate due to the increased
intensity of the attractive forces over the repulsive forces, due to charge distribution on
the surfaces. There are many factors related with the performance of the biocomposite
for hard tissue regeneration that reflect in the preparation of suspension, mechanical
properties, calcium and phosphate ions released in the physiological environment,
particles size, shape, particles distribution and also ratio of inorganic part in the polymer
matrix [21, 22, 25].
3.4.4. SEM study of composite MCCh/ ß-TCP complex in film form.
The objective of the investigation was to study the dissolution of the ß-TCP
particles before and after purification process and also the possible high contraction of
the volume and size in a dry complex film, as well as to study the best concentration of

glycerol content used as a plasticizer and finally to estimate the suitability of the
MCCh/ß-TCP complex in film forms and the ß-TCP distribution in the polymer matrix
by SEM analysis. The Figure 7, 8, 9 and 10 showed presence of MCCh/ß-TCP before
and after purification.
x500 magnification x1000 magnification
Figure 7. SEM microphotographies, cross-section view of films, MCCh/ß-TCP complex
before purification process
x200 magnification x1000 magnification
Figure 8. SEM microphotographies, film surface, MCCh/ß-TCP complex before purification
process

x500 magnification x1000 magnification
Figure 9. SEM microphotographies, cross-section view of films, MCCh/ß-TCP complex after
purification process
Figure 10. SEM microphotographies, film surface, MCCh/ß-TCP complex after purification
process
The preparation of films showed in Figures 7, 8, 9, 10; there is presence of ß-TCP in the
composites before and after purification process with incomplete dissolution. The particles of
ß-TCP aggregates well in the polymer matrix with a homogeneous distribution and clusters
size reduction after purification process showed in Figure 9 and 10. In the suspension, no
precipitation was found, suggesting a good stability and an ionic bonding between the amino

groups and ion phosphate groups, also a good inter/intra hydrogen bonding in the complex.
Notice that films without glycerol used as a plasticizer reduces the size around 3X, suggesting
a very high electrostatic interaction between MCCh and ß-TCP in a complex formation. The
addition of glycerol (0.5 wt-%) in the complex suspension reduces the contraction, thus
improving the film elasticity; glycerol acts increasing the polymer chains mobility, thus
making the film less fragile with more elongation.
3.4.5. The SEM study of MCCh/ ß-TCP complex in sponge form.
The Figures 11 and 12 show the structure of MCCh/ ß-TCP complex after purification
process in sponge form according with Table 1 preparation.
x200 magnification x1000 magnification
Figure 11. SEM microphotographies, surface of MCCh/ß-TCP complex without glycerol.
x200 magnification x1000 magnification

Figure 12. SEM microphotographies, surface of MCCh/ß-TCP complex with glycerol.
The sponges showed the presence of ß-TCP particles in the polymer matrix after
purification process. The composite sponge without plasticizer did not show a smooth
surface with more agglomeration and bigger size of cluster formation in the entire area
of the polymer matrix. The sponges with glycerol as a plasticizer increase the
interconnected porosity with homogenous size and shape (round shape) of the pores
with good and homogenous distribution of the calcium phosphate in the polymer matrix.
The preparation of composites in sponge form with glycerol (0.5 wt-%) formed
smooth surface without defects and well-shaped 3-dimensional structure with highly
number of interconnected porous.
3.5. Conclusion
The FTIR study of the composites, according with Table 1, showed that both
components, MCCh and ß-TCP, existed in the composite before and after purification
process, which can be seen as well in the SEM study of the films, which suggests a
validation of the method to obtain MCCh/ ß-TCP complex.
The SEM analysis showed a ß-TCP more round shape and the grain size fits mainly
in the range 1.0 - 9.0 m, average particle size of 4.48 m ± 0.54 µm.
The SEM images showed that the composites have a homogeneous distribution of
the ß-TCP particles with reducing cluster size formation after purification process. The
calcium phosphate particles were uniformly dispersed in the amino-glucose
macromolecule aggregation (coagulation process) and no precipitation of the calcium
phosphate was noticed in the samples showing the good stability of the suspension. The
study showed the feasibility of freeze drying method in the preparation of the MCCh/ ß-

TCP complex sponges, resulting in a 3-dimensional porous network structure that can
be used in future as a base for scaffolds production.
3.6. References
1) Oktay Yildirim (2004). Preparation and Characterization of Chitosan /Calcium
Phosphate Based Composite Biomaterials, Master Of Science Dissertation, zmir
Institute of Technology zmir, Turkey.
2) Rumi Fujita, Atsuro Yokoyama, Yoshinobu Nodasaka, Takao Kohgo, Takao
Kawasaki. (2003). Ultrastructure of ceramic-bone interface using hydroxyapatite
and B-tricalcium phosphate ceramics and replacement mechanism of B-
tricalcium phosphate in bone, Tissue & Cell 35, 427-440.
3) Shinn-Jyh DING. (2006). Preparation and Properties of Chitosan/Calcium
Phosphate Composites for Bone Repair, Dental Materials Journal
25 4) 706 712.
4) Jue-Yeon Lee , Sung-Heon Nam , Su-Yeon Im , Yoon-Jeong Park ,Yong-Moo
Lee, Yang-Jo Seol, Chong-Pyoung Chung, Seung-Jin Lee. (2002). Enhanced
bone formation by controlled growth factor delivery from chitosan-based
biomaterials, Journal of Controlled Release 78, 187-197.
5) Barbosa M.A., P.L. Granja , C.C. Barrias, I.F. Amaral. (2005). Polysaccharides
as scaffolds for bone regeneration, ITBM-RBM 26, 212-217.
6) Huipin Yuan, J. D. de Bruijn, Yubao Li, Jianqing Feng, Zongjian Yang, K. de
Groot, Xingdong Zhang. (2001). Bone formation induced by calcium phosphate
ceramics in soft tissue of dogs: a comparative study between porous -TCP and
ß-TCP, Journal of Materials Science: Materials in medicine 12, 7-13.

7) Stephen M. Warren, MD, Kenton D. Fong, MD, Randall P. Nacamuli, MD,
HanJoon M. Song, MD, Tony D. Fang, MD, and Michael T. Longaker. (2003).
Materials for in one repair in Plastic Surgery, Operative Techniques in Plastic
and Reconstructive Surgery, 9, 1: pp 10-15.
8) Soon-Ho Kwon, Youn-Ki Jun, Seong-Hyeon Hong, Hyoun-Ee Kim. (2003).
Synthesis and dissolution behavior of -TCP and HA/ -TCP composite powder,
Journal of European ceramic society 23, 1039-1045.
9) Sanchez-Salcedo S., F. Balas, I. Izquierdo-Barba, M. Vallet-Regı. (2009). In
vitro structural changes in porous HA/ -TCP scaffolds in simulated body fluid,
Acta Biomaterialia 5, 2738-2751.
10)Chiau Liou SZ, San-Yuan Chen. (2002). Transformation mechanism of different
chemically precipitated apatitic precursor into B-tricalcium phosphate upon
calcinations, Biomaterials 23, 4541-4547.
11)Pena J., Vallet-Regi M. (2003). Hydroxyapatite, tricalcium phosphate and
biphasic materials prepared by a liquid mix technique, Journal of european
ceramic society 23, 1687-1696.
12)Homaeigohar S.Sh. (2006). The effect of reinforcement volume fraction and
particle size on the mechanical properties of B-tricalcium phosphate-high
density polyethylene composites, Journal of the European Ceramic Society 26,
273-278.
13)Alexandra E. Porter, Tom Buckland,Karin Hing,Serena M. Best,William
Bonfield. (2005). The Structure of the Bond Between Bone and Porous Silicon-
Substituted Hydroxyapatite Bioceramic Implants, Journal of Biomedical
Materials Research Part A pg. 25-33. DOI 10.1002/jbm.a.

14)Hutmacher D. W. (2000). Scaffolds in tissue engineering bone and cartilage,
Biomaterials 21, 2529-2543.
15)Clayton E. Wilson, Moyo C. Kruyt, Joost D. de Bruijn, Clemens A. van
Blitterswijk, F. Cumhur Oner, Abraham J. Verbout, Wouter J.A. Dhert. (2006).
A new in vivo screening model for posterior spinal bone formation: Comparison
of ten calcium phosphate ceramic material treatments, Biomaterials 27, 302-314.
16)Ranieri Cancedda, Beatrice Dozin, Paolo Giannoni, Rodolfo Quarto. (2003).
Tissue engineering and cell therapy of cartilage and bone, Matrix Biology 22,
81-91.
17)Dorozhkin Sergey V. (2011). Review, Biocomposites and hybrid biomaterials
based on calcium orthophosphates, Biomatter 1:1, 3-56.
18)Struszczyk H., A. Niekraszewicz, K.Wrze niewska-Tosik, S. Koch, O. Kivekas.
(1987). Process for preparing microcrystalline chitosan by the continuous
method, PL164247.
19)Niekraszewicz A., Kucharska M, Wisniewska-Wrona M., Wesolowska E.,
Struszckyk H. (2004). Chitosan in medical application, ed., Progress on
Chemistry and Application of Chitin and its Derivatives, PTChit, X, p.13-17.
20)Niekraszewicz A., Kucharska M, Wisniewska-Wrona M. (1998). Some Aspects
of Chitosan Degradation, in Struszczyk H. ed., Progress on Chemistry and
Application of Chitin and its Derivatives, PTChit, IV, p.55-63.
21)Pighinelli L, Kucharska M, Brzoza-Malczewska K, Gruchała B. (2011).
Complex microcrystalline chitosan/tricalcium orthophosphate and process for
preparing same. Poland patent application P 393758.

22)SZ-Chiau Liou, San-Yuan Chen. (2002). Transformation mechanism of different
chemically precipitated apatitic precursor into -tricalcium phosphate upon
calcinations, Biomaterials 23, 4541-4547.
23)Brugnerotto, J.; Lizardi, J.; Goycoolea, F.M.; ArguÈelles-Monal, W.;
DesbrieÁres, J.; Rinaudo, M. (2001). An infrared investigation in relation with
chitin and chitosan characterization. Polymer, 42, 3569-3580.
24)Skoog, D.A.; Holler, F.J.; Nieman, T.A. (2002). Principios de Analise
Instrumental, 5a ed.; Bookman: Porto Alegre, Brazil,; pp. 342-384.
25)Wawro, D.; Pighinelli, L. (2011). Chitosan Fibres Modified with HAp/ -TCP
Nanoparticles. Int. J. Mol. Sci., 12, 7286-7300
26) Council Directive 93/42/EEC of 14 June 1993 concerning medical devices (OJ
L 169, 12.7.1993, p. 1), with gives the directives for production, quality control
and commercialization.
27) http://www.meddev.info/, visited in 00/02/2011 at 11:35hs, Guidelines relating
to medical directives.

Chapter 4.
Preparation of Microcrystalline chitosan (MCCh)/tricalcium phosphate complex
with Hydroxyapatite in sponge form.
4.1. Introduction
In the last past years, much attention has been given to develop materials for
bone tissue regeneration such as polymer-based scaffolds containing bioactive
bioceramics, which have been under development in the last past years for hard tissue
regeneration and can be produced making bioceramics serving two purposes: (a)
making the scaffolds osteoconductive, and (b) reinforcing the scaffolds. With this
composite strategy, there are two approaches for making bioceramic-polymer composite
scaffolds [1, 2]:
(1) Incorporating bioceramic particles in the scaffold through a variety of
techniques;
(2) Coating a polymer scaffold with a thin layer of apatite through biomimetic
processes.
(3) To prepare a complex polymer/calcium phosphate
All strategies have been employed to develop usable scaffolds for bone tissue
engineering. One of the manufacturing process as a freeze-drying method was used at
the final stage to produce the porous structure. In production of polymer/apatite
scaffolds, same factors such as polymer solution concentration, porous type and size,
can be changed in the freeze-drying method. Same parameters, such as pressure,
temperature and time play a very important role in forming the scaffolds of desired

porous structures (pore geometry, pore size and size distribution, pore interconnectivity,
thickness of pore walls, etc.) and, hence, the mechanical performance [1,2,3].
The incidence of fractures in the natural bones increases rapidly with age. This is
partly due to extra osseous factors such as the impaired reflexes of the elderly, their
reduced proprioceptive efficiency, reduced cushioning by fat, and weakened
musculature, and by osseous factors such as the structural changes in the shape and size
of the bones like diseases and deterioration of the condition of the bone material itself.
The intrinsic bone changes can be subdivided into quantity and quality effects. Quantity
refers to a reduction in bone mass and an increased macroscopic porosity. Quality refers
to changes in the bone matrix material, which is related with chemical, compositional
and physical factors. In terms of mechanical effects, between 0 and 35 years of age,
human bone shows an increase in its mineral content, which benefits both the strength
and stiffness of the tissue. After 35, the elastic, ultimate and fracture properties of bone
tissue invariably deteriorate in men and women. The aging effects on cancellous and
trabeculae bone are nowadays primarily reduced into describing its architecture,
connectivity and level of porosity of the tissue [2, 3, 4,5].
The concepts of stiffness and strength are easy to comprehend. However,
toughness is less obvious and is here defined in terms of the amount of energy required
for fracture. This energy will be absorbed in three principal ways. Some energy is
absorbed prior to the generation of a major crack in the form of diffuse damage; this we
may call the “pre-fracture” toughness of the material, or its damage tolerance level [2,
7]. Some energy is required to start the final fracture crack, the rest is required to drive
the crack through the tissue in order to break the material in two (or more) pieces; this

being the conventionally defined “fracture” toughness of the material. Toughness is of
primary importance, especially in relation to fractures [2,3].
It has been shown that in older bone, the fatigue strength and two separate
toughness measurements (like the work of fracture and the energy absorbed in impact)
are considerably reduced. Age, as a separate variable, was able to explain well the trend
for work of fracture and impact. The bone behaves like a composite material. It is likely
that aging is a more general deteriorative process, whose increase affects reducing the
biochemical and biomechanical properties of bone in a number of ways [2].
Modern composite materials, on the other hand, have a positive relationship
between strength and toughness; the tougher materials are also stronger. Composites
benefit by having a heterogeneous structure and an ability to accept widespread and
regenerate microstructural damage without breaking. The intracortical porosity reflects
an imbalance between the rates of new bone deposition and bone resorption as well as
bone formation. Bulk density and mineral weight ratio values are the result of the
combined effects of maturation of individual elements and also their relative fractions.
In vivo fatigue microcracks give an appreciation of the loading history of the material;
these cracks accumulate in life as a result of everyday loading activities and may also
have a harmful effect on the residual properties of the bone material itself [2, 3, 5].
The size of individual cracks stayed well within limits, probably as a result of an
active remodelling process. In the worst of cases, the crack length simply doubled in
size between 35 and 92 years of age (note that the cracks at 35 are at most up to 500 m
long, while at 92, there are a few up to 1 mm long). However, their number dramatically
increases with age, especially for the cracks of small lengths truly related with the
mechanical properties [2, 3, 5]. There are basically two major options for biodegradable

and biocompatible scaffold: ceramic and polymer as defined by Vert. The ceramic,
usually made of calcium-phosphate, may be delicate to use in such demanding
mechanical environment as they present a brittle behavior. The polymers are ductile but
may not have enough mechanical properties to withstand the load. If the polymer
scaffolds have enhanced mechanical properties, they may be an ideal material for bone
substitute. Bone is a Nano-structure of HAp crystals and organic matrix. Because rigid
crystals such as HAp crystals cannot dissipate much energy, the organic matrix must be
involved in this process. Definitions given by Vert [4, 5]:
Biodegradable are solid polymeric materials and devices which break down due
to macromolecular degradation with dispersion in vivo but no proof for the elimination
from the body (this definition excludes environmental, fungi or bacterial degradation).
Biodegradable polymeric systems or devices can be attacked by biological elements so
that the integrity of the system and in some cases but not necessarily, of the
macromolecules themselves, gives fragments or other degradation by-products. Such
fragments can move away from their site of action but not necessarily from the body.
Bioresorbable are solid polymeric materials and devices which show bulk
degradation and further resorb in vivo; i.e. polymers which are eliminated through
natural pathways either because of simple filtration of degradation by-products or after
their metabolization. Bioresorption is thus a concept which reflects total elimination of
the initial foreign material and of bulk degradation by-products (low molecular weight
compounds) with no residual side elects. The use of the word ‘bioresorbable’ assumes
that elimination is shown conclusively. Bioerodible are solid polymeric materials or
devices, which show surface degradation and further, resorb in vivo.

Bioerosion is thus a concept, too, which reflects total elimination of the initial
foreign material and of surface degradation by-products (low molecular weight
compounds) with no residual side effects. Bioabsorbable are solid polymeric materials
or devices, which can dissolve in body fluids without any polymer chain cleavage or
molecular mass decrease. For example, it is the case of slow dissolution of water-
soluble implants in body fluids. A bioabsorbable polymer can be bioresorbable if the
dispersed macromolecules are excreted.
The incorporation of a tricalcium phosphate (ß-TCP), hydroxyapatite (HAp) and
basic salts into a polymer matrix produces a hybrid/composite material. These inorganic
fillers allow tailoring the desired degradation and resorption kinetics of the polymer
matrix. A composite material would also improve biocompatibility and hard tissue
integration in a way that ceramic particles, which are embedded into the polymer
matrix, allow for increased initial fast spread of serum proteins compared to the more
hydrophobic polymer surface [5, 6, 7].
The similarity of synthetic hydroxyapatite [Ca10(PO4)6(OH)2; HAp and ß-TCP
(Ca3(PO4)2) ] to bone mineral has led to the extensive use of HAp and/or ß-TCP as a
bone grafting material in hard tissue implants. Pores in apatite composite scaffolds has
been introduced clinically for applications such as spinal fusions, bone tumors,
fractures, and in the replacement of failed or loose joint prostheses. The development of
pores in apatite composite scaffolds stemmed from the results of studies highlighting
the role of porosity in acting as a promoter for mechanical fixation to the surrounding
tissue by providing a template for bone ingrowth. Furthermore, it has been suggested
that the quality of bone surrounding porous implants is superior to that around dense
implants. There is an increasing trend towards engineering bone grafts with

interconnected porosity between the macropores of the scaffolds. Pore interconnections
with size 30-100 m act as pathways between the macropores to favor cellular and
vascular penetration assuring bone ingrowth into the pores. Porosity is defined as
micrometer-sized porosity within the structure of the implant, and found that strut
porosity increases bone ingrowth, mineral apposition rates, and bone organization [3, 5,
10, 11, 12].
The scaffold or three-dimensional (3-D) construct provides the necessary
support for cells to proliferate and maintain their differentiated function, and its
architecture the ultimate shape of the new bone and cartilage. Skeletal tissue is usually
organized into 3-D structures in the body. For the repair and regeneration and/or
generation of new hard and ductile tissue, scaffolds need to have a high elastic modulus
in order to be retained in the space they were designated for; and also to provide the
tissue with adequate space and nutrient flow for growth. Therefore, one of the basic
problems from a scaffold design is that to achieve significant mechanical properties and
sufficiently high interatomic and intermolecular bonding and also allows for hydrolytic
attack and degradation [4, 5, 6].
Another point is that the porosity has to be also focused in the diffusion of
nutrients, gas exchange, and elimination of by-products into the 3-D scaffold. Although,
an interconnected macropore structure of 300-500 m enhances the diffusion rates to
and/from the center of a scaffold, transportation of the nutrients is not sufficient for
large scaffold volumes [5, 6]. The properties of bone in health and disease attract much
attention, with an ever greater proportion of the population in need for medical devices
for hard tissue regeneration and/or replacement. With this, pressure is put on health and
welfare systems of all countries. Aging, diseases, fractures, and so on, when combined

with other skeletal complications, as a demineralization, contribute and improve the
suitability and development in the hard tissue engineering. The aim of this study is to
prepare a quantitative and qualitative composite using the MCCh/ß-TCP complex with
another commercial calcium phosphate (HAp), studying chemical characterization and
mechanical properties performance in sponge form.
4.2. Materials
1) MCCh/ß-TCP complex characterized by: water retention value (WRV) = 560%,
polymer content = 3.25 %, Moisture content 12.06%, pH=7.40.
2) Tricalcium phosphate (ß-TCP), (Ca3(PO4)2) - Sigma Aldrich Lab., Germany.
3) Hydroxyapatite (Ca5(PO4)3OH) - Sigma Aldrich Lab., Germany.
4) Plasticizer -Glycerol (C3H8O3) 99%, pure p.a., Sigma-Aldrich, Germany.
5) Hydrochloridric acid, 37.8% p.a., manufactured by Fluka, Germany
6) Ethanol - C2H5OH, Fluka, Germany.
4.3. Methods
4.3.1. Preparation of composite in sponge form
The preparation of composites in sponge form using freeze-dried method was
made using ALFA 1-4 dryer made by Christ Co in the temperature range from (-25) to
10 0C and vacuum from 0.1 to 0.53 mbar during 20 to 24 hours depending on size of
the charge. Drying accomplished that way resulted in the preparation of sponges with a
smooth surface without defects. The freeze-dried method used for sponge preparation
is also showed in the scheme in Figure 1.

Figure 1. Scheme of freeze-dried method used to prepare composites in sponge form.
4.3.2. Powder complex preparation
The composition for powder preparation was MCCh/ß-TCP complex
suspension (1 wt-%) /HAp (1 wt-%) /glycerol (1 wt-%) composite content 4% was with
freeze-dried method in lab, ALFA 1-4 dryer made by Christ Co in the temperature range
from (-25) to 10 0C and vacuum from 0.1 to 0.53 mbar during 20 to 24 hours depending
upon size of the charge. In the Figure 2, there is a schematic illustration of the method
to obtain a composite powder.
Figure 2. Scheme of preparation of composite powder used to prepare composites insponge form.
Composite
MCCh/ß-TCP complex (1 p.w)
HAp (1 p.w)
Glycerol (1 p.w)
Composite Powder
MCCh - 26.7 %
ß-TCP - 6.7 %
HAp - 33.3 %
Glycerol - 33.3 %
Freeze drying method
sponge
Teflon pin
Freeze-dried plate
Water and ethanol
Water and ethanol
Lyophilizing
Chamber
Surface A
Surface B

4.3.3. SEM study
The HAp particle morphology and distribution of the calcium phosphates in the
polymer matrix in a sponge and film forms were observed using a scanning electron
microscopy (SEM) - FEI Quanta 200, USA.
4.3.4. Infrared Spectroscopy
Fourier transform infrared spectroscopy (FTIR) is a non-destructive technique
that was used to identify the functional groups through their chemical bonds, which
generate a spectrum of infrared bands characteristic of each connection type. The
infrared analysis was performed on a range from 500 to 4000 cm-1, resolution 4.0 cm-1
with a Spectrum Genesis Series. Samples were prepared for analysis with KBr, in the
form of tablets, to verify the presence of functional characteristic groups in the ß-TCP:
P-O, O-H, P-O-H, H-O-H and for MCCh material the functional groups: NH2, Amide I,
amide II. The FTIR study of the ß-TCP and the MCCh/ ß-TCP complex are showed in
chapter 3.
4.3.5. Determination of Ca and P in commercial HAp and ß-TCP powders and in
the composites.
The aim of this study is to determine and compare the quantity of Ca and P
remaining after film and sponge preparation of the composites, by ICP-OES, Germany,
through a method of microwave digestion according to SOP2.5L126. Quantification of
Calcium and Phosphorus was made by (DIN EN ISO 11885). This analysis was a
cooperation made between Institute of Biopolymers and Chemical Fibres (IBWCh) in
Lodz, Poland, and Thuringian Institute of Textile and Plastics Research (TITK),
Rudolstadt, Germany).

4.3.6. Determination of particles size of commercial HAp powder
The technique used was by laser particle size analyser by Sympatec Hellos
H1330, type BF (sympatec GmbH, Clausthal, Germany), to determine HAp range size
of particles and distribution. This analysis was a cooperation made between Institute of
Biopolymers and Chemical Fibres (IBWCh) in Lodz, Poland, and Thuringian Institute
of Textile and Plastics Research (TITK), Rudolstadt, Germany).
4.3.7. WAXS- Diffraction of ß-TCP and HAp powder
Two calcium phosphates in form of powder have been investigated by means of
WAXS-diffraction in transmission mode. Both powders, HAp and ß-TCP, were simply
placed between two polypropylene films for running the WAXS-scan in transmission
mode. The following parameters were adjusted: X-ray radiation - CuK -doublet, non-
monochromatic -portion suppressed by Ni-filter, mode of operation - Transmission,
anode Voltage - 40kV, tube current - 40mA, Steo width - 0,0250, scan rate - 5 sec/step,
aperture slit - 1mm, anti-diffusion slit - 1mm, detector slit - 1mm. This analyse was a
cooperation between Institute of Biopolymers and Chemical Fibres (IBWCh) in Lodz,
Poland, and Thuringian Institute of Textile and Plastics Research (TITK), Rudolstadt,
Germany).
4.3.8. Mechanical properties of composites
The mechanical properties of the composites in sponge and film forms were
determined in the Accredited Laboratory of Metrology at IBWCh (certificate No. AB
388) by using dynamometer Instron (type 5544) in accordance with the following
standards (Polish Standard PN-EN-ISO 527-1., Polish Standard PN-EN-ISO 527-3,
Polish Standard PN-ISO 4593).

4.4. Results and discussions
4.4.1. Elaboration of the quantitative and qualitative of composites in sponge form
The main objective of this study was the elaboration of qualitative and
quantitative composition of MCCh/ß-TCP complex with HAp showed in Table 1, to
find the best concentration of complex resulting in the preparation of sponges with a
smooth surface without defects, the samples A, B, C the glycerol content is related
with wt-% complex content (MCCh/ß-TCP complex) and wt-% PC content.
Table 1. Quantitative and qualitative composite preparation in sponge form
Symbol ofsamples Concentration Composition wt-% %
A 5.0%Complex
PC Glyc.
(1.0)(4.0)(2.5)
MCCh - 32.00% ß-TCP - 8.00% HAp - 26.66%
Glycerol - 33.33%
B 8.0%Complex
PC Glyc.
(1.0)(4.0)(2.5)
MCCh - 32.00% ß-TCP - 8.00% HAp - 26.66%
Glycerol - 33.33%
C 10.0%Complex
PC Glyc.
(1.0)(4.0)(2.5)
MCCh - 32.00% ß-TCP - 8.00% HAp - 26.66%
Glycerol - 33.33%
* (MCCh/ß-TCP) complex,
** PC-Powder composite- (MCCh/ß-TCP complex (1.0 wt-%) / HAp (1.0 wt-%) / Glycerol (1.0 wt-%).
The decision to prepare the powder composite with HAp and mix with
the complex suspension to finally obtain sponges was made to minimize the
agglomeration and size of clusters formation. It was notice that preparation of the
complex suspension by mechanical mixing process using HAp powder was a challenge,
considering that the small particle size in a liquid suspension tends to move rapidly and

randomly, which is explained by micro Brownian movement, due to the impact between
the particles themselves, or with molecules of the suspension, or also against the surface
of the container, facilitating agglomeration, making the mixture more difficult and, at
the same time, a challenge. As a consequence, there is increase of the mechanical
degradation of the polymer. The small particles of the powder absorb more water and
reduce their free energy and charge distribution on surface, and typically the very small
particles tend to agglomerate due to the increased intensity of the attractive forces over
the repulsive forces, due to charge distribution on the surfaces, considering the cationic
nature of the HAp and microcrystalline chitosan and anionic nature of the ß-TCP also
responsible for high electrostatically bonding that reflects in the structure and suggests a
ceramic composite formation by the two calcium phosphates involved. This
phenomenon presents a remarkable influence on the rheological behaviour of
suspensions, as specific surface areas are highly conducive to cluster formation.
When the HAp powder is directly mixed in the complex suspension, it is noticed
that the smoothest surface of composites began to be disturbed with incorporation and
increasing amount of HAp powder, gradually resulting in a rough surface, which was
not smooth anymore. This phenomenon did not happen while the following steps
weren’t completed:
A) Preparation of the composite suspension by mechanical mixing ((MCCh/ß-TCP
complex (1.0 wt-%) / HAp (1.0 wt-%) / Glycerol (1.0 wt-%)).
B) Use the suspension to prepare sponges by freeze-dried method.
C) Crushing the sponge composite to a powder.

D) Mix the powder composite into complex (MCCh/ß-TCP), suspension by
mechanical mixing.
E) The final result of this mixture was a suspension used to obtain the final
composite sponges by freeze-dried method.
Another problem was to find the best concentration for the samples to obtain the
smoothest surface without defects, considering the composite samples in Table 1. It was
noticed that the concentration at 5 and 8% was difficult to prepare sponges. The way to
solve this problem was to increase the composite concentration by 10%, keeping the
same preparation method. In this 10% concentration, the composite showed a smooth
surface without defects, thus making continuous studies possible.
4.4.2. Composites with different ratios of ethanol to prepare sponge form
The set of samples in Table 2 shows the qualitative and quantitative analyses of
sample preparation, resulting in the sponges with a smooth surface without defects.
Table 2. Quantitative analysis of composite preparation with ethanol in sponge form
Symbolof
samplesContent of
compositionEthanolcontent
Composition wt-% Materials (%)
A1 10.0% 0%Complex
PC Glyc.
(1.0)(4.0)(2.5)
MCCh - 32.00% ß-TCP - 8.00% HAp - 26,.66%
Glycerol - 33.33%
B1 10.0% 1%Complex
PC Glyc.
(1.0)(4.0)(2.5)
MCCh - 32.00% ß-TCP - 8.00% HAp - 26.66%
Glycerol - 33.33%
C1 10.0% 2%Complex
PC Glyc.
(1.0)(4.0)(2.5)
MCCh - 32.00% ß-TCP - 8.00% HAp - 26.66%
Glycerol - 33.33%

D1 10.0% 5%Complex
PC Glyc.
(1.0)(4.0)(2.5)
MCCh - 32.00% ß-TCP - 8.00% HAp - 26.66%
Glycerol - 33.33%
* (MCCh/ß-TCP) complex,
** PC-Powder complex - (MCCh/ß-TCP complex (1.0 wt-%) / HAp (1.0 wt-%) / Glycerol (1.0 wt-%)
The glycerol used in samples A1, B1, C1 and D1 is related with wt-% complex
content (MCCh/ß-TCP complex) and wt-% of PC content. The different ratios of
ethanol are related only with the content of composition (10%) to study the viability to
increase the number and shape of pores by freeze-dried method.
4.4.3. FTIR study of the commercial HAp
The main peaks of energy vibration mode identified in the ß-TCP powder and
MCCh/ ß-TCP complex are showed in chapter 3.
Table 3 shows the main peaks of energy vibration mode identified in the
commercial HAp (Figure 3), the characteristic functional groups of orthophosphate,
hydroxyl, phosphate and probably pyrophosphate, the latter one in trace amount,
characteristic of HAp material. The presence of carbonate was also observed in the HAp
material as well as in ß-TCP (Chapter 3), which suggests that in some commercial HAp
the CaO and Ca (0H)2 also are used to get an ideal stoichiometric relation between Ca /
P in the material [25,26, 27,33].
Table 3. Localization of peaks of energy vibration of commercial HAp in transmission
FTIR.

Figure 3. Transmission FTIR spectrum of energy vibration of commercial HAp
In the transmission FTIR, spectra of the commercial HAp powder are observed
in Figure 3 at 839 (cm1), corresponding to deformation modes of phosphate groups (O-
P-H) bonds. The antisymmetric and symmetric stretching of OH- group are observed
3570 cm-1 and 3464 cm-1, respectively, referring the way they stretch and also
characteristic of the material in water adsorption. The band observed at 962 cm-1 of low
intensity corresponds to non-degenerate symmetric stretching of P-O bonds of
Mode of vibrations Wave number of peaks, cm-1 Mode of groups
antisymmetric stretching
symmetric stretching
1040, 1093, 962 P-O of PO4
antisymmetric, symmetricstretching
3570, 3464 O-H
Antisymmetric stretching 876, 1456 CO of C-O
Stretching 1635 H-O-H
Antisymmetric stretching 839 P-O-H
Tra
nsm
itta
nce
%

phosphate groups. The bands 1040 and 1093 (cm-1) represent the asymmetric stretch
modes, respectively, the P-O bonds of phosphate groups. There is the presence of water
in the starting material for the existence of peaks associated in the regions of 1590, 1635
(cm-1) and the peaks around 3400 cm-1, in fair agreement with the literature [13, 25, 26].
4.4.4. FTIR study of the composites
The Figure 4 and 5 show the peaks of energy vibration of the selected
composites with MCCh/ß-TCP complex with HAp. The peaks of composite preparation
according with Table 2 with different ratios of ethanol confirm the presence of the
functional groups of amino groups of microcrystalline chitosan (NH2) at 1539 cm-1,
Amide I at 1648 cm-1, amide II at 1557 cm-1, the OH- group peak 3434 cm-1 and then N-
H peak 3298 and 3500 cm-1, referring to the way they stretch. The main peaks of energy
vibration at 1026 and 1086 cm-1 identified the C-O skeletal vibration stretching of
saccharide structure, also in fair agreement of FTIR study of MCCh/ß-TCP complex
showed in chapter 3 and literature [14, 15, 36]. The characteristic functional groups of
HAp, between 840 to 1100 cm-1, also are showed in Figure 4 and 5. The presence of
ethanol in the final sponge didn’t show a significant variation in the FTIR spectrum of
composite, suggesting no interaction between ethanol, polymer and calcium phosphates
involved.

Figure 4. Absorbance FTIR spectrum of energy vibration of the composite sample A1
Figure 5. Absorbance FTIR spectrum of energy vibration of the composite sample B1
The composites with HAp are characterized by the presence of mainly
coordinate bonds between the phosphate and calcium ions and the amide and hydroxide
groups of the microcrystalline chitosan and by the forming intra and intermolecular
hydrogen bonds between amino and hydroxide groups of the chitosan chains, with a
very high energy making the structure of the composite durable resulting in an excellent
stability of temperature, pH compared to known forms of chitosan and derivatives.
Sample A1
Sample B1

Another advantage of the method to obtain the composite with HAp is the
simple procedure providing unique properties like high content bound of Ca and P ions,
high water retention value, and two different calcium phosphates with high bioactivity.
4.4.5. WAXS-Diffraction investigation of ß-TCP and HAp powder
The aim of this study is to confirm the crystalline and amorphous phases of the
commercial HAp and ß-TCP powders. The crystalline phase of powders is directed
related with calcium phosphate dissolution and less time for new tissue stimulation and
formation. Crystalline HAp is considered to be the final, stable product in the
precipitation of calcium and phosphate ions from neutral or basic solutions like in the
physiological environmental. The possible role that amorphous calcium phosphate may
play as a precursor to HAp in biological calcification places it in the mainstream of
calcium phosphate chemistry [23, 26]. However, the WAXS-diffraction of the calcium
phosphate powders showed the intensity of peak at 32,5o and the intensities of peaks at
26o and 40o showing a characteristic peaks of HAp, Figure 6, in red color. Regarding
the peak values observed, the structure is hydroxyapatite, and no different new crystal
phase was observed. The increased sharpness and intensities and decreased broadness of
mentioned peaks can be explained by the increase in crystal sizes. The ß-TCP was
found to be completely amorphous.
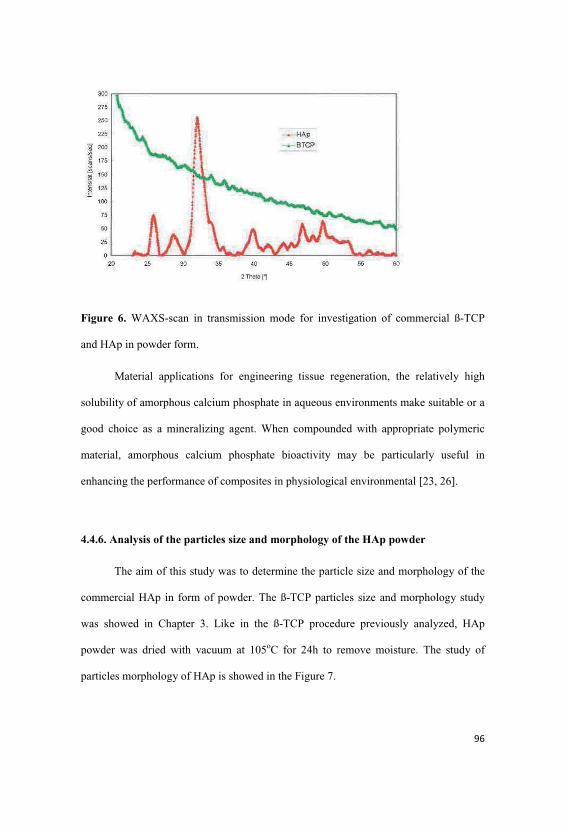
Figure 6. WAXS-scan in transmission mode for investigation of commercial ß-TCP
and HAp in powder form.
Material applications for engineering tissue regeneration, the relatively high
solubility of amorphous calcium phosphate in aqueous environments make suitable or a
good choice as a mineralizing agent. When compounded with appropriate polymeric
material, amorphous calcium phosphate bioactivity may be particularly useful in
enhancing the performance of composites in physiological environmental [23, 26].
4.4.6. Analysis of the particles size and morphology of the HAp powder
The aim of this study was to determine the particle size and morphology of the
commercial HAp in form of powder. The ß-TCP particles size and morphology study
was showed in Chapter 3. Like in the ß-TCP procedure previously analyzed, HAp
powder was dried with vacuum at 105oC for 24h to remove moisture. The study of
particles morphology of HAp is showed in the Figure 7.
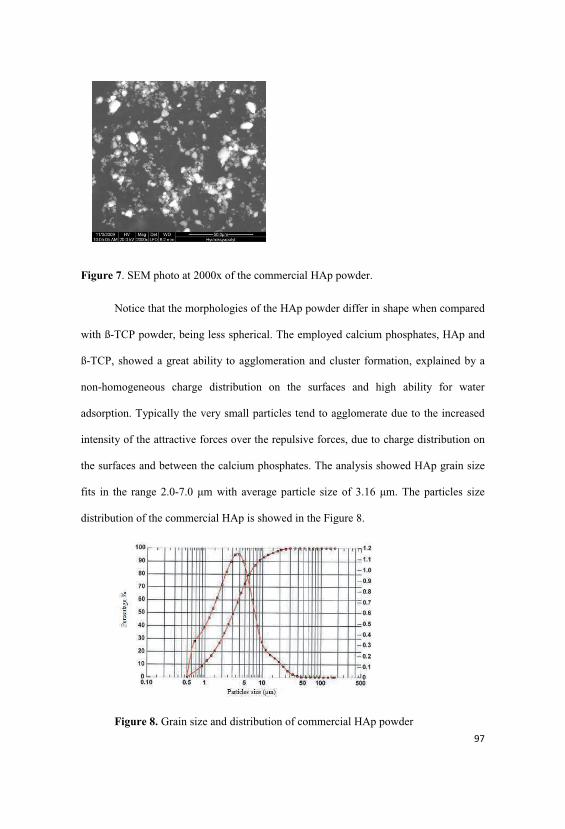
Figure 7. SEM photo at 2000x of the commercial HAp powder.
Notice that the morphologies of the HAp powder differ in shape when compared
with ß-TCP powder, being less spherical. The employed calcium phosphates, HAp and
ß-TCP, showed a great ability to agglomeration and cluster formation, explained by a
non-homogeneous charge distribution on the surfaces and high ability for water
adsorption. Typically the very small particles tend to agglomerate due to the increased
intensity of the attractive forces over the repulsive forces, due to charge distribution on
the surfaces and between the calcium phosphates. The analysis showed HAp grain size
fits in the range 2.0-7.0 m with average particle size of 3.16 m. The particles size
distribution of the commercial HAp is showed in the Figure 8.
Figure 8. Grain size and distribution of commercial HAp powder

In Table 4, it is the particles size of commercial HAp; the specified surface area
covered by HAp is 3.41 +04 cm2/g. Also it was found around 10% of the HAp particles
used in nano size, which also indicates a faster and easier release of calcium and
phosphate ions in physiological environmental. In Table 5, it is showed the particles
size of commercial HAp and ß-TCP following the literature.
Table 4. Particles size of commercial HAp.
POWDERS HAp
Particles size (90%), 8.81 m
Particles size (50%), 3.16 m
Particles size (10%), 0.97 m
Superficial area (cm2/g) 3.41 e+04
Table 5. Particles size of commercial HAp and ß-TCP [6, 7, 8, 12, 21].
POWDERS HAp ß-TCP
Particles size (90%), ( m) 7.37 9.77
Particles size (50%), ( m) 2.83 2.67
Particles size (10%), ( m) 0.80 0.60
Superficial area (cm2/g) 45.7 + 0.2 45.7 + 0.1
A great number of materials have been used for bone tissue engineering
including polymers, bioglass and a variety of calcium phosphate ceramics, and the
performance of the biocomposite also reflects in the method of preparation, particles
size, shape and ratio of inorganic part in the polymer matrix [6,7,8, 12, 21].
4.4.7. SEM study of the composites in sponge form.

The objective of the investigation was to study the HAp, suitability and
distribution in the MCCh/ß-TCP complex in the sponge form preparations.
The literature shows that scaffolds should possess an interconnected and diffuse
porosity (usually over 90%), if cell adhesion, ingrown and reorganization are to be
sustained in vitro, and room for neovascularisation has to be provided in vivo; pore
interconnections influence the diffusion of nutrients to the cells. Because excessive pore
size means decreased internal surface area, a compromise is necessary: for instance, for
regenerating bone tissue in vitro, some authors preferred pore size 200-400 m, while
others used scaffolds with 500 m nominal pore size. When pore diameter is too small,
pore occlusion prevents cellular penetration into the scaffold: as a consequence, pore
size 75-100 m resulted in ingrown non-mineralized osteoid tissue, while even smaller
pores were penetrated only by fibrous tissue. Because highly porous materials have
limited mechanical strength, the void volume must be tuned to allow for the
accommodation of a large number of cells and the preservation of the structural strength
particularly in load-bearing tissues. A proper porosity may improve mechanical
interlocking between the scaffold and the surrounding host tissue, providing necessary
mechanical stability at this critical interface. Scaffold surface properties such as
morphology, hydrophilicity, zeta-potential and surface energy influence cell adhesion,
migration, phenotype maintenance and intracellular signaling in vitro, as well as cell
recruitment at the tissue / scaffold interface in vivo [6, 7, 19, 37].
All composites of MCCh/ß-TCP complex with HAp in sponge form showed a
well-shaped 3-dimensional structure with high interconnected porous, suggesting a
good biological environment to cell attachment and proliferation, providing the passage
of nutrient flow through micro and macro interconnected porous. Those samples can be

used in future as a base for scaffolds production. The HAp powder aggregates well and
showed a homogenous distribution in all MCCh/ ß-TCP complex matrix that could be
explained also by the lyophilization process reducing the time for agglomeration and
cluster formation to obtain sponges and the mixing process to obtain the suspension. All
samples were obtained with smooth surface without defects, the pore morphology
structure showed more round shape, also affected by the glycerol used as a plasticizer
increasing the mixing process in the preparation of the suspension and directed related
with the homogenous interconnected pores formation.
The literature shows that more round shape porous, great number of porous and
smaller size decrease the chance for inflammatory reactions [9, 10, 14, 38].
There is a possibility to improve the number and shape of pores in the structure
of sponges, changing parameters in the freeze-dried method such as temperature,
pressure, time and adding ethanol for a faster removal of water in the samples and
method of liophylization showed in Figure 1. Figures 9-20 show the structure on surface
and cross-section of sponges according with Table 2.
x90 magnification x200 magnification x1000 magnification
Figure 9. SEM photographies cross-section of sample A 1, sponge conc. 10%.

x200 magnification x1000 magnification
Figure 10. SEM photographies surface A, of sample A 1, sponge conc. 10%.
x200 magnification x1000 magnification
Figure 11. SEM photographies surface B, of sample A 1, sponge conc.10%.
x90 magnification x200 magnification x1000 magnification
Figure 12. SEM photographies cross-section of sample B 1, sponge conc. 10% and 1%

of ethanol.
x200 magnification x1000 magnification
Figure 13. SEM photographies surface A, of sample B 1, sponge conc. 10% and 1% of
ethanol.
x200 magnification x1000 magnification
Figure 14. SEM photographies surface B, of sample B 1, sponge conc. 10% and 1% of
ethanol.

x90 magnification x200 magnification
x1000 magnification
Figure 15. SEM photographies cross-section of sample C 1, sponge conc. 10% and 2%
of ethanol.
x200 magnification
x1000 magnification
Figure 16. SEM photographies surface A, of sample C 1, sponge conc. 10% and 2% of
ethanol.

x200 magnification
x1000 magnification
Figure 17. SEM photographies surface B, of sample C 1, sponge conc. 10% and 2% of
ethanol.
Figure 18. SEM photographies cross-section of sample D 1, sponge conc. 10% and 5% o

x200 magnification x1000 magnification
Figure 19. SEM photographies surface A, of sample D 1, sponge conc. 10% and 5% of
ethanol.
x200 magnification
x1000 magnification
Figure 20. SEM photographies surface B, of sample D 1, sponge conc. 10% and 5% of
ethanol.
The SEM observation of the composite sponge showed that the sample B 1 with
1 % of ethanol, showed in Figure 12, presents great network porosity with more
homogenous shape and size of pores and clusters. The estimate pore size ranges from 20
to 150 m were fabricated, suggesting that ethanol increases the evaporation of water
speed and directed related with the porosity. In general, the micro and macrostructure is
controlled by varying the polymer material, polymer concentration, solvents, processing
parameters (temperature, pressure and time), it is user and technique sensitive and the
processing parameters have to be well controlled.
4.4.8. The physical-mechanical tests of composite in sponge form.
From a biomechanical and clinical point of view, the hard tissue-
engineered implant should allow also for a mechanically stable fixation as well as
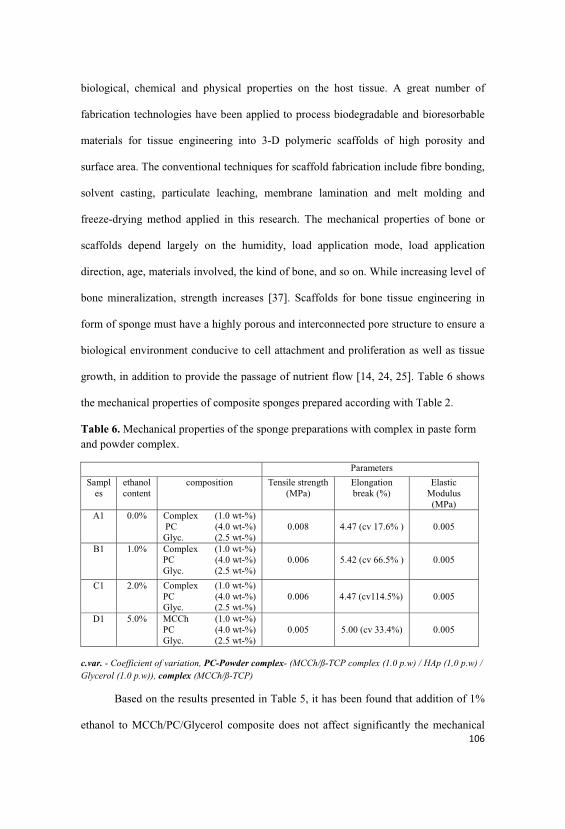
biological, chemical and physical properties on the host tissue. A great number of
fabrication technologies have been applied to process biodegradable and bioresorbable
materials for tissue engineering into 3-D polymeric scaffolds of high porosity and
surface area. The conventional techniques for scaffold fabrication include fibre bonding,
solvent casting, particulate leaching, membrane lamination and melt molding and
freeze-drying method applied in this research. The mechanical properties of bone or
scaffolds depend largely on the humidity, load application mode, load application
direction, age, materials involved, the kind of bone, and so on. While increasing level of
bone mineralization, strength increases [37]. Scaffolds for bone tissue engineering in
form of sponge must have a highly porous and interconnected pore structure to ensure a
biological environment conducive to cell attachment and proliferation as well as tissue
growth, in addition to provide the passage of nutrient flow [14, 24, 25]. Table 6 shows
the mechanical properties of composite sponges prepared according with Table 2.
Table 6. Mechanical properties of the sponge preparations with complex in paste formand powder complex.
Parameters
Samples
ethanolcontent
composition Tensile strength(MPa)
Elongationbreak (%)
ElasticModulus(MPa)
A1 0.0% Complex (1.0 wt-%)PC (4.0 wt-%)Glyc. (2.5 wt-%)
0.008 4.47 (cv 17.6% ) 0.005
B1 1.0% Complex (1.0 wt-%)PC (4.0 wt-%)Glyc. (2.5 wt-%)
0.006 5.42 (cv 66.5% ) 0.005
C1 2.0% Complex (1.0 wt-%)PC (4.0 wt-%)Glyc. (2.5 wt-%)
0.006 4.47 (cv114.5%) 0.005
D1 5.0% MCCh (1.0 wt-%)PC (4.0 wt-%)Glyc. (2.5 wt-%)
0.005 5.00 (cv 33.4%) 0.005
c.var. - Coefficient of variation, PC-Powder complex- (MCCh/ß-TCP complex (1.0 p.w) / HAp (1,0 p.w) /Glycerol (1.0 p.w)), complex (MCCh/ß-TCP)
Based on the results presented in Table 5, it has been found that addition of 1%
ethanol to MCCh/PC/Glycerol composite does not affect significantly the mechanical

properties of sponge, on the other hand, it showed a increase in the homogenous
structure with higher porosity directed related with no variation of tensile strength and
higher elongation at break. The MCCh/ß-TCP complex with HAp samples can be
summarized: the addition of PC powder to complex causes decrease of tensile strength
by the addition of calcium phosphate, but, on other hand, it could improve the
elongation at break by addition of glycerol, which could certainly alter the physical and
mechanical properties by enhancing the mobility of the polymer chains. According with
literature, the mechanical property tests have a glycerol and HAp influence in the
preparations. The presence of glycerol in causing significant differences in tensile
strength and elongation at break, without glycerol, presented greater tensile strength and
lower elongation. Glycerol interfered with MCCh chains, decreasing intermolecular
attraction and increasing polymer mobility which facilitate elongation [6, 19, 21, 38].
The literature also shows that the deacetylation degree was also a significant
factor on the mechanical properties. In the absence of additives, chitosan with lower
deacetylation (60.9% DD) presented higher tensile strength and higher elongation than
citosan of 96% DD. Significant interaction was observed only between glycerol and
deacetylation degree. Thus, glycerol effect was more intense on chitosan with lower DD
than on those with high DD [6].
4.4.9. Determination of Ca and P content in composites
The aim of this analysis is to determine the content of Ca and P that remains
after sponges preparation. This relation is also mentioned in the literature that minerals
in study are the most important elements for hard tissue regeneration activity and are
directly related with calcium phosphate dissolution and less time for new tissue

stimulation [7, 8, 9, 10, 12]. Table 7 shows the proportion of the Ca and P in
composites.
Table 7. Weight proportion of Ca and P in the raw material and composites.
The results showed a relation between Ca and P of complex and composite
approximately 2/1 wt-%, respectively, which shows a high content of Ca and P in all
samples.
4.5. Conclusions
The FTIR analysis of the composites showed that, in both cases, the components
(MCCh, ß-TCP and HAP) existed in the samples. The presence of ethanol in the final
sponge composite didn’t show a significant variation in the FTIR spectrum of
composite, suggesting a no interaction between ethanol, polymer and calcium
phosphates involved.
The WAXS-diffraction of the calcium phosphate powders showed the
characteristic crystalline peaks of commercial HAp, the ß-TCP was found to be
completely amorphous. The particles analysis showed HAp grain size fits in the range
2.0 - 7.0 m with average particles size 3.16 m with a highly tendency to agglomerate
and water absorption.
Symbol ofsample
Proportion of Ca and P
composition Ca(ppm)
P(ppm)
HAp powder 447200 186500ß-TCP powder 425400 199000MCCh 108 186MCCh/ß-TCPcomplex
53600 28200
(Sample A1) Complex (1.0 wt-%)PC (4.0 wt-%)Glyc. (2.5 wt-%)
183000 80800

All composites of MCCh/ß-TCP complex with HAp in sponge form showed a
well-shaped 3-dimensional structure with high interconnected porous, suggesting a
good biological environment to cells attachment and proliferation, providing the
passage of nutrient flow thought the micro and macro interconnected porous. Those
samples can be used in future as a base for scaffolds production.
Addition of 1% ethanol to MCCh/PC/Glycerol composite does not affect
significantly the mechanical properties of sponge, in another hand showed a increase in
the homogenous structure with higher porosity related with no variation of tensile
strength and higher elongation at break. The MCCh/ß-TCP complex with HAp samples
can be summarized: the addition of PC powder in to complex causes decrease of tensile
strength by the addition of calcium phosphate but in another hand could enhancing the
elongation at break by addition of glycerol could certain alter physical and mechanical
properties by enhancing the mobility of the polymer chains. The relation between Ca
and P approximately 2/1 wt-% respectively, that shows a high content of Ca and P in all
samples.
ACKNOWLEDGEMENTS
The research and work presented in this study were conducted with financial
support from the European Community's Seventh Framework Programme (“Marie Curie
Initial Training Network), FP7/2007-2013.
The particle size measurements, X-ray diffraction and quantification of calcium
and phosphate content were conducted using equipment provided by the TITK-
Thuringian Institute of Textile and Plastics Research in Rudolstadt, Germany.
4.6. References

1. Min Wang. (2006). Composite Scaffolds for Bone Tissue Engineering, American
Journal of Biochemistry and Biotechnology 2 (2): 80-84, ISSN 1553-3468.
2. Zioupus P. (2001). Aging Human Bone: actors affecting Its Biomechanical
Properties and the Role of Collagen. journal of biomaterials applications, 15; 187-229.
3. Porter Alexandra E.,Tom Buckland,Karin Hing,Serena M. Best,William Bonfield.
(2003). The Structure of the Bond Between Bone and Porous Silicon-Substituted
Hydroxyapatite Bioceramic Implants, Journal of Biomedical Materials Research Part A
pg. 25-33. DOI 10.1002/jbm.a.
4. Vert M, Li MS, Spenlehauer G, Guerin P. (1992). Bioresorbability and
biocompatibility of aliphatic polyesters. J Mater Sci;3:432-46.
5. Hutmacher D. W. (2000). Scaffolds in tissue engineering bone and cartilage,
Biomaterials 21, 2529-2543.
6. Cyster L.A., D.M. Grant, S.M. Howdle, F.R.A.J. Rose, D.J. Irvine, D. Freeman, C.A.
Scotchford, K.M. Shakesheff. (2005). The influence of dispersant concentration on the
pore morphology of hydroxyapatite ceramics for bone tissue engineering, Biomaterials
26, 697-702.
7. Oktay Yildirim. (2004). Preparation and Characterization of Chitosan /Calcium
Phosphate Based Composite Biomaterials, Master Of Science Dissertation, zmir
Institute of Technology zmir, Turkey.
8. Rumi Fujita, Atsuro Yokoyama, Yoshinobu Nodasaka, Takao Kohgo, Takao
Kawasaki. (2003). Ultrastructure of ceramic-bone interface using hydroxyapatite and ß-
tricalcium phosphate ceramics and replacement mechanism of B-tricalcium phosphate
in bone, Tissue & Cell 35, 427-440.
9. Shinn-Jyh DING. (2006). Preparation and Properties of Chitosan/Calcium Phosphate
Composites for Bone Repair, Dental Materials Journal 25 4):706 712.

10. Sanchez-Salcedo S., F. Balas, I. Izquierdo-Barba, M. Vallet-Regı. (2009). In vitro
structural changes in porous HA/ß-TCP scaffolds in simulated body fluid, Acta
Biomaterialia 5, 2738-2751.
11. SZ-Chiau Liou, San-Yuan Chen. (2002). Transformation mechanism of different
chemically precipitated apatitic precursor into ß-tricalcium phosphate upon calcinations,
Biomaterials 23, 4541-4547.
12. Soon-Ho Kwon, Youn-Ki Jun, Seong-Hyeon Hong, Hyoun-Ee Kim. (2003).
Synthesis and dissolution behavior of ß-TCP and HA/ß-TCP composite powder, Journal
of European ceramic society 23, 1039-1045.
13. Pena J., M. Vallet-Regi. (2003). Hydroxyapatite, tricalcium phosphate and biphasic
materials prepared by a liquid mix technique, Journal of european ceramic society 23,
1687-1696.
14. Jue-Yeon Lee , Sung-Heon Nam , Su-Yeon Im , Yoon-Jeong Park ,Yong-Moo Lee,
Yang-Jo Seol, Chong-Pyoung Chung, Seung-Jin Lee. (2002). Enhanced bone formation
by controlled growth factor delivery from chitosan-based biomaterials, Journal of
Controlled Release 78, 187-197.
15. Barbosa M.A., P.L. Granja , C.C. Barrias, I.F. Amaral. (2005). Polysaccharides as
scaffolds for bone regeneration, ITBM-RBM 26, 212-217.
16. Niekraszewicz A., Kucharska M, Wisniewska-Wrona M. (1998). Some Aspects of
Chitosan Degradation, in Struszczyk H. ed., Progress on Chemistry and Application of
Chitin and its Derivatives, PTChit, IV, p.55-63.
17. Niekraszewicz A., Kucharska M, Wisniewska-Wrona M., Wesolowska E.,
Struszckyk H. (2004). Chitosan in medical application, ed., Progress on Chemistry and
Application of Chitin and its Derivatives, PTChit, X, p.13-17.

18. Struszczyk H. M. (2006). Global Requirements for Medical Applications of Chitin
and its derivatives. ed., Progress on Chemistry and Application of Chitin and its
Derivatives, PTChit, XI, p.95-102.
19. Kucharska M., A. Niekraszewicz, M. Wisniewska-Wrona, E. Wesolowska, H.
Struszczyk. (2003). Dressing Sponges Made of Chitosan and Chitosan-Alginate Fibrids,
ed., Progress on Chemistry and Application of Chitin and its Derivatives, PTChit, IX,
p.69-72.
20. Klinkaewnarong J. (2009). Nanocrystalline hydroxyapatite powders by a chitosan-
polymer complex solution route: Synthesis and characterization, Solid State Sciences
11, 1023-1027.
21. Homaeigohar S.Sh. (2006). The effect of reinforcement volume fraction and particle
size on the mechanical properties of ß-tricalcium phosphate-high density polyethylene
composites, Journal of the European Ceramic Society 26, 273-278.
22. Wisniewska-Wrona M., Niekraszewicz A., Ciechanska D., Pospieszny H.,
Orlikowski B.L. (2007). Biological Properties of Chitosan Degradation Products, ed.,
Progress on Chemistry and Application of Chitin and its Derivatives, PTChit, XII, 149-
156.
23. Skrtic D., J. M. Antonucci and E. D. Eanes. (2003). Amorphous Calcium
Phosphate-Based Bioactive Polymeric Composites for Mineralized Tissue
Regeneration, Journal of Research of the National Institute of Standards and
Technology,108, 3.
24. Boriniec S., Strobin G., Struszczyk H., Niekraszewicz A., Kucharska M. (1984).
GPC Studies of Chitosan Degradation, Int. J. polym. Anal. & Charact. 3, 359-368.
25. Misiek D J, Kent JM, Carr RF. (2007). Soft tissue responses to hydroxyapatite
particles of different shapes. J. Oral Maxillof Surg;42:150-60.

26. Higashi T., Okamoto H. (1996). Influence of particles size of hydroxyapatite as a
capping agent on cell proliferation of culture fibroblasts, Journal of Endodontics
(U.S.A.), 22,5.
27. Dorozhkin Sergey V. (2011). Review, Biocomposites and hybrid biomaterials based
on calcium orthophosphates, Biomatter 1:1, 3-56.
28. Dorozhkin Sergey V. (2011). Medical Application of Calcium Orthophosphate
Bioceramics, BIO, 1, 1-51.
29. Dorozhkin Sergey V. (2009). Review Calcium Orthophosphates in Nature, Biology
and Medicine, Materials, 2, 399-498.
30. Dorozhkin Sergey V. (2009). Review Nanodimensional and Nanocrystalline
Apatites and Other Calcium Orthophosphates in Biomedical Engineering, Biology and
Medicine, Materials, 2, 1975-2045.
31. De Paulo B.O ET AL. Compatibilidade de Compósitos Biologicamente Ativos em
Implantes de Tecido Osteotraumatico, Coletanea, Belo Horizonte, v.2-2, 102-111, 2008.
32. Trommer R. M., L. A. dos Santos, C. P. Bergmann. (2007). Técnica Alternativa
Para Obter Recobrimentos De Hidroxiapatita, Cerâmica 53, 153-158.
33. Skoog, D.A.; Holler, F.J.; Nieman, T.A. (2004). Principios de Analise Instrumental,
5a ed.; Bookman:Porto Alegre, Brazil, 2002; pp. 342–384.34. Ribeiro C.C., Barrias
C.C., Barbosa M.A. Calcium Phosphate-alginate microspheres as enzyme delivery
matrices, biomaterials 25, 4363-4373.
35. Maachou H., K.E. Bal, Y. Bal, A. Chagnes, G. Cote, D. Alliouche. (2008).
Characterization and In Vitro Bioactivity of Chitosan/Hydroxyapatite Composite
Membrane Prepared by Freeze-Gelation Method, Trends Biomater. Artif. Organs, 22
(1).

36. Henryk Pospieszny, Wojciech Folkman. (2004). Factors modyfying a biological
activity of chitin derivatives. ed. Progress on Chemistry and Application of Chitin and
its Derivatives, PTChit. X, p.07-12.
37. Qiaoling Hu*, Baoqiang Li, Mang Wang, Jiacong Shen. (2004). Preparation and
characterization of biodegradable chitosan/hydroxyapatite nanocomposite rods via in
situ hybridization: a potential material as internal fixation of bone fracture, Biomaterials
25, 779-785.
38. Young Rho-Jae, Liisa Kuhn-Spearing, Peter Zioupos. (1998). Mechanical properties
and the hierarchical structure of bone, Medical Engineering & Physics 20, 92-102.

Chapter 5
Preparation of chitosan with nano calcium phosphates composite in fibre form.
5.1. Introduction
Fibres made of chitosan are widely used in new generation biomaterials. The
formation of chitosan fibres is a rather simple process usually accomplished by wet
spinning from a polymer solution. A variety of fibrous chitosan materials are available,
ranging from fine microfibrids [1] to very strong fibres and pseudo-dry-spun fibres [2]
and multifilament chitosan yarns [3,4]. Wet spinning from a solution of the polymer
enables modifications by adding various functional substances to the solution, notably
nanoparticles, carbon nanotubes (CNT) [5-7], nanosilver [8], calcium phosphates [9],
calcium sulphite [10] and a number of polymers such as fibroin and keratin proteins
[11,12]. Chitosan lends itself to use in biomaterials thanks to properties such as its
biodegradability, biosorption and ability to accelerate wound healing.
To confer functionality upon chitosan fibres, they are frequently coated with
other biomaterials such as collagen or calcium phosphates [13-17]. Following coating,
they are used to reinforce composite implants containing hydroxyapatites (HAp) [18].
Calcium phosphates are known for their compatibility, osteoconductivity and
easy absorption by the human body, which makes them suitable for the preparation of
composite dental implants [19] and, in combination with chitosan, artificial bones
[20,21]. Chitosan multifilament yarn, which is used in the construction of textile
scaffolds or as culture medium for cell growth, requires the addition of calcium
phosphates like HAp or ß-TCP and has been successfully used in medical applications
such as artificial bones, cartilage and glues for hard tissue engineering [3,13-15,20-23].
Calcium phosphates are not easy to dissolve in chitosan solutions; dissolution
depends on their chemical composition, crystallinity and dissolving conditions. For this

reason HAp nanoparticles can be used in the preparation of chitosan gels [24] or can be
introduced into the fibre matrix. Calcium phosphates HAp and ß-TCP have different
dissolving properties, and hence their release from composites or chitosan fibres may
differ. For this reason they are usually employed in a mixture to maintain a favorable
calcium-to-phosphorus ratio of 1.67 [25].
The aim of this study was to prepare novel chitosan fibres containing
nanoparticles of calcium phosphates for medical use, particularly for textile scaffolds
and cell growth. We also investigated how nanoparticles of ß-TCP, HAp and of the
complex HAp/ ß-TCP affect the rheology of the chitosan spinning solution, spinning
conditions and mechanical properties of the resulting HAp/ ß-TCP-modified fibres.
5.2. Materials
Tricalcium phosphate (ß-TCP) (Ca3(PO4)2), Hydroxyapatite (Ca10(PO4)6OH)2, supplied
by Sigma Aldrich, Germany.
Glycerol as a plasticizer (C3H8O3), 99% pure, Sigma Aldrich, Germany.
Hydrochloric acid 37.8% pure, manufactured by Fluka, Germany.
Acetic acid 80% and NaOH made by POCh S.A., Gliwice, Poland.
Chitosan derived from the northern shrimp (Pandalus borealis), supplied by Primex Co.,
Norway. Characteristics properties of chitosan such as Viscometric average molecular
mass [kD] 342; Moisture content [%] 5.58; Water retention value [%] 43.5; Dynamic
viscosity, 1 % chitosan in 1% acetic acid at 20 °C[CP] 63.1; Degree of deacetylation
[%] 83.2; Ash content [%] 0,40; Nitrogen content [%] 6.84.
5.3. Methods
5.3.1. Methods of manufacture composite chitosan fibres
The composite chitosan fibres production method is according to the invention,
polish patent application number. P. 393022, 2010 (Wawro, D.; Pighinelli, L.;

St plewski, W.) presents as follows: a homogeneous aqueous acid solution of 40 - 70
°C temperature, containing 0,1 - 10 wt% of hydroxyapatite and/or tricalcium phosphate
nano-particles, and 0,1 - 3 wt% of chitosan is entered, during mixing, into the spinning
solution of 20 - 55 °C temperature, containing chitosan of average molecular weight of
150 000 - 700 000, deacetylation degree of 80 - 95 %, poly-dispersion degree between 2
- 5, and concentration of 2 - 7 wt% in acetic acid aqueous solution of 1 - 3 wt%
concentration, containing 0,1 - 15 wt% of glycerine in relation to chitosan. Then, after
venting, the obtained spinning solution is being embossed through multi-hole spinning
nozzle into the coagulation bath of 25 - 45 °C temperature, which is a sodium hydroxide
aqueous solution of 1 - 5 wt% concentration. Obtained fibres are being stretched from
15 to 67 % in sodium hydroxide aqueous solution of 0,1 - 0,5 % concentration in
temperature of 80 - 95 °C. Then they are rinsed in water, finished in bath of 30 - 75 °C
temperature containing hydro-alcoholic emulsion with surfactants, and they are being
dried in temperature of 40 °C.
5.3.2. Preparation of Chitosan Spinning Solution Containing HAp, ß-TCP and
HAp/ß-TCP Nanoparticles
A 5.0 wt% weight chitosan solution (A) was prepared in aqueous 3.0 wt%
weight acetic acid. The dissolution of chitosan took 60 minutes, during which time
glycerol was introduced to the solution to a volume of 10%. After filtration and
deaeration at ambient temperature, the chitosan solution was blended with 2.0 wt%
chitosan solution containing ß-TCP particles (solution B), a chitosan solution containing
HAp (solution C) or a mixture of chitosan solutions containing HAp/ß-TCP
nanoparticles (solution B/C). Solution B was an aqueous 2.0 wt% solution of chitosan in
0.9 wt% solution of hydrochloric acid with a 2.0 wt% weight content of ß-TCP

nanoparticles. Solution C was a diluted aqueous 2.5 wt% chitosan solution in 1.5 wt%
acetic acid containing HAp particles at a concentration of 2.0 wt%.
5.3.3. Wet Spinning of Chitosan Fibres Containing HAp, ß-TCP and HAp/ ß -TCP
Nanoparticles
Chitosan fibres modified with HAp, ß-TCP and HAp/ß-TCP nanoparticles were
prepared by wet spinning on a pilot line equipped with a spinning head holding a
rhodium/platinum spinneret with 150 holes, each having a diameter of 0.08 mm. The
spinning solution is 35 °C, the same as a coagulation bath (aqueous 3.0 wt% sodium
hydroxide at 35 °C). The spun fibres were washed first in a water bath at 40 °C then in a
water-ethanol (60% v/v) bath and were then dried without tension in a loose bundle.
The spinning speed for the chitosan fibres modified with HAp/ß-TCP nanoparticles was
31 m/min.
5.3.4. Analytical Methods
The rheological properties of the chitosan solutions were measured using a
digital viscometer Brookfield model RV DV-II+, with the Rheocalc V3.1-1 programat
20, 25, 30, 35 and 40 °C. A Biolar ZPO polarizing microscope equipped with an
advanced image analysis system (MultiScan V. 14.02) was used to prepare images of
the aqueous solution of chitosan containing HAp and ß-TCP particles.
Images of the cross-section and surface of the fibres were obtained using a
scanning electron microscope SEM/ESEM, Quanta 200 (W), FEI Co., USA.
The size of the nanoparticles of HAp, ß-TCP and HAp/ß-TCP in chitosan solution was
measured using a ZETASIZER 2000 (Malvern Instruments) apparatus.
The amount of chitosan in the spinning solutions was determined using the
gravimetric method. A film was formed from a weighed amount of the solution by

evaporation of water and drying at 60 °C. This was then coagulated in an aqueous
solution of 3.0 wt% sodium hydroxide, rinsed and dried to constant weight at 105 °C.
The water retention value (WRV) was determined according to Standard
ISO/FDIS 23714. The mechanical properties of the chitosan fibres were tested
according to Standards PN-ISO-1973:1997 and PN-EN ISO 5079:1999 in an air-
conditioned room at 65 ± 4% relative humidity and 20 ± 2 °C.
Spectrophotometric spectra in the infrared range were prepared using FTIR
apparatus produced by Unicam Co. equipped with the control program Winfirst ATI
Mattson. Samples were prepared in the form of pressed cubes in potassium bromide
(KBr Aldrich Co.).
Samples of the fibres were mineralized at 575 °C in 6 M HCl to determine the
calcium content, which was measured in the mineralized residue by flame atomic
absorption spectrometry (FAAS) at a wavelength of 422.7 nm. A SCAN-1 f-my Thermo
Jarrell Ash atomic absorption spectrometer was used for the analysis.
The ash content in the chitosan fibres was estimated according to standard EN
ISO3451-1.
5.4. Results and discussions
5.4.1. Preparation of Chitosan Solutions Containing -TCP, HAp and HAp/ -TCP
A 5.16 wt% aqueous chitosan solution (A) was prepared in 3.0 wt% acetic acid. The
solution was blended with a chitosan solution containing -TCP, HAp, or a mixture of
solutions containing HAp/ -TCP nanoparticles in the appropriate proportions. Table 1
presents the properties and the composition and some properties of these solutions. The
5.16 wt% chitosan solution (A) had a temperature of 52 °C and a high dynamic
viscosity of 19000 Pa. The dissolution of chitosan and the blending of the particular

solutions caused no problems. Admixing of the solution with calcium phosphate
resulted in a decrease in the concentration of the acid and chitosan and in the dynamic
viscosity. Small particles of calcium phosphate ( -TCP) were found in the chitosan
solutions upon microscopic observation. A clear, stabile chitosan solution suitable for
spinning was obtained by blending solutions A and B (code MCT 6).
Table 1. Some properties of chitosan solutions containing hydroxyapatite (HAp) and
tricalcium phosphate ( -TCP) particles.
Solutioncode
Percentage ofsolution used,
%
Concentration ofDynamic
viscosity/temp.-TCP HAp Acetic acid Chitosan
A B C wt% wt% wt% wt% Pa/°CChit 58 100 - - - - 3.00 5.16 19000/52MCT 6 83.3 16.7 - 0.333 - 2.75 4.50 7500/49MCT 7 83.3 - 16.7 - 0.330 2.75 4.63 9250/49MCT 8 71.4 14.3 14.3 0.286 0.283 2.36 4.46 4500/52MCT 11 62.5 25.0 12.5 0.707 0.252 2.10 4.01 1750/51
A chitosan solution (A) is 5.0 wt% chitosan was prepared in aqueous 3.0 wt%weight acetic acid; Solution B was an aqueous 2.0 wt% solution of chitosan in 0.9wt% solution of hydrochloric acid with a 2.0 wt% weight content of -TCPnanoparticles; Solution C was a diluted aqueous 2.5 wt% chitosan solution in 1.5wt% acetic acid containing HAp particles at a concentration of 2.0 wt%.
Comparison of microscopic images of the chitosan solutions denoted Chit 58 (A) and
MCT 6 indicated that the introduction of the chitosan solution containing -TCP
nanoparticles reduced the number of undissolved particles in the MCT 6 solution.
Hydroxyapatite is less soluble than -TCP; this was apparent in the course of
preparing solution C, when only a small amount of HAp dissolved while the majority
appeared as an opalescent suspension. Blending of the HAp-containing chitosan
solution with the chitosan spinning solution (A) only slightly improved the solubility of
HAp in the chitosan acetate solution. Hence, the chitosan acetate solution denoted MCT
7 contained HAp particles with a diameter of up to a few micrometres. Though not
negating the solution’s spinnability, this feature may negatively influence the quality of

the prepared calcium-containing fibre. The reverse was the case when chitosan acetate
solution was mixed with chitosan B and C solutions containing HAp and -TCP
(solution B contained 0.9 wt% hydrochloric acid). The result was the complete
dissolution of the hydroxyapatite and a clear solution denoted MCT 11.
It is likely that mixing of the chitosan solution containing HAp with the chitosan
solution containing -TCP in hydrochloric acid at a ratio of 2:1 provided adequate
conditions for the dissolution of HAp. The dissolution of HAp in chitosan solution is the
subject of further investigations and of a patent application [26]. The mechanism of
salts and amino acids upon the dissolution of HAp has been reported previously [27].
Mixing of solutions B and C in the ratio 1:1 did not cause HAp to dissolve (MCT 8).
Microscopic analysis of the MCT 7 and MCT 11 chitosan acetate solutions
confirmed the presence of HAp particles in the MCT 7 solution, while the B/C mixture
(MCT 11) clearly contained HAp/ -TCP nanoparticles. The microscopic image of the
MCT 11 solution was optically pure. The chitosan solution (B) containing -TCP
nanoparticles was also clear and optically pure. Hydroxyapatite added to chitosan acetate
solution (C) produced a suspension with only some of it going into solution.
Agglomerates with a size of 1500 nm that are present in the spinning solution may
negatively influence the mechanical properties of the spun fibres. The situation was
slightly improved when mixing the chitosan B/C solution with the chitosan A solution
in a 1:1 ratio, which resulted in a chitosan solution (MCT 8) with fewer HAp particles.
Another solution of the B/C mixture was prepared with more of the -TCP particle-
containing chitosan solution (2:1). During the blending of the HAp and -TCP particle-
containing chitosan solutions, the HAp particles dissolved immediately, resulting in a
clear solution (MCT 11). Comparison of the images of the chitosan solutions MCT 7
and MCT 11 revealed that mixing the HAp- and -TCP-containing chitosan solutions

had a beneficial effect by limiting the size of the nanoparticles within the range of 12.8-
58.0 nm in MCT 11. Figure 1(a-c), presents the particle size distribution in the chitosan
solutions containing HAp or -TCP and HAp and -TCP.
Figure 1. (a) Particle size distribution of -TCP in chitosan solution in hydrochloric
acid (solution B); (b) HAp particles in chitosan acetate solution (solution C); (c) The
HAp/ -TCP blend in chitosan (solution B/C).
(a)
(b)

(c)
Table 2 shows selected parameters of the solutions analyzed by means of the
ZetaSizer analyzer. The results presented in Table 2 and Figure 1 confirm. The smallest
nanoparticles found in the B/C mixture were much smaller than those in chitosan
solutions B and C before blending. As can be seen in the Figure 2 the pictures of the
solutions B, C and the blend solution B/C.
Figure 2. The cup number 1 shows the blend solution B/C (2:1), the cup number
2 shows the solution B and cup number 3 shows solution C.

Table 2. Selected parameters of chitosan solutions containing HAp, -TCP and HAp/ -
TCP.
Chitosan solutionRange of particle
sizeSize of fraction with
highest volume contentPercentage of
volumePotential Zeta
nm nm % mVSolution B 28.9-164.9 65.2 11.5 43.0 ± 2.3Solution C 417.3-1495 745.4 34.2 45.3 ± 2.3Blend of chitosansolutions B/Cin 2:1 ratio
12.8-58 22.9 19 52.9 ± 4.0
The chitosan solutions characterized in Table 2 were prepared for the spinning of
chitosan fibres. Addition of the calcium phosphate nanoparticles to the chitosan
solutions resulted in a slight drop in the chitosan content and concentration of acetic
acid, an insignificant increase in the hydrochloric acid content in the blended mixture,
and a radical decrease in the apparent dynamic viscosity. The Zeta potential showed
greater stability for the solution MCT-11 (solution A + B/C(2:1)) in this ratio than for
other solutions.
5.4.2. Rheology of Chitosan Solutions Modified with HAp/ -TCP Nanoparticles
The rheology of chitosan solution MCT 11 modified with HAp/ -TCP nanoparticles
was examined at 20, 25, 30, 35 and 40 °C. The impact of shearing speed and
temperature upon the dynamic viscosity of the chitosan solution modified with calcium
phosphate is presented in Figure 3. The flow curves were drawn for the rise and fall in
shearing speed.
Figure 3. Dependence of the apparent dynamic viscosity on the shearing rate and
temperature of the acetate chitosan solution modified with HAp/ -TCP (MCT 11).

It can be inferred from the curves that chitosan modified with HAp/ -TCP
nanoparticles has the attributes of a non-newtonian fluid. In the examined temperature
range, the viscosity of the solution decreased with increasing shearing speed. The flow
curves overlapped with each other both at increasing and decreasing shearing speed.
Chitosan solutions thus showed the features of a pseudo-plastic fluid (they are diluted by
shearing) typical of polymer solutions. An increase in temperature from 20 to 25 °C
distinctly reduced the dynamic viscosity. Further increases in temperature and shearing
speed caused much greater decreases in viscosity. Given that fibres are spun from
solutions of chitosan acetate and that the spinning process takes a relatively long time,
the stability of solutions as confirmed by the rheological examinations is of particular
importance.
5.4.3. Investigation into the Spinning of Chitosan Fibres Modified with HAp, -TCP and HAp/ -TCP
To allow comparison, regular chitosan fibres and those modified with calcium
phosphate were spun under the same conditions at a spinning speed of 31.0 m/min and a

draw ratio of 34%. The fibres were spun into a coagulation bath containing aqueous
3.0% NaOH without ethanol. The spinning process of both regular chitosan fibres and
those modified with -TCP, HAp and HAp/ -TCP was stable in the adopted conditions
with no disturbance in the spinneret zone. The addition of calcium phosphates to the
spinning solution had a beneficial effect on spinning stability, with fewer breaks in the
elementary filaments.
5.4.4. Mechanical Properties of Chitosan Fibres Modified with HAp, -TCP and
HAp/ -TCP
The mechanical properties of chitosan fibres modified with HAp/ -TCP are
presented in Table 3. The impact of the calcium phosphate on the mechanical properties
of the fibres varied depending on the type and amount of additive used. The addition of
-TCP had little effect on tenacity (MCT 6) while the same amount of HAp reduced
both the tenacity and elongation (MCT 7). The large amount of HAp agglomerates in
the fibres may be the underlying cause. Blending of the chitosan solutions with HAp
and -TCP led to an improvement in the solution with higher amounts of calcium
phosphates (some of the HAp particles dissolved), with a slight increase in fibre
tenacity (MCT 8). Doubling of the amount of chitosan solution B (containing -TCP) in
the mixture led to a homogenous solution of HAp/ -TCP (MCT 11) nanoparticles along
with the highest level of calcium phosphates, and resulted in fibres with the same
tenacity as that of regular chitosan fibres.
Table 3. Impact of HAp, -TCP and HAp/ -TCP concentration in the chitosan
solution on the mechanical properties of fibres.
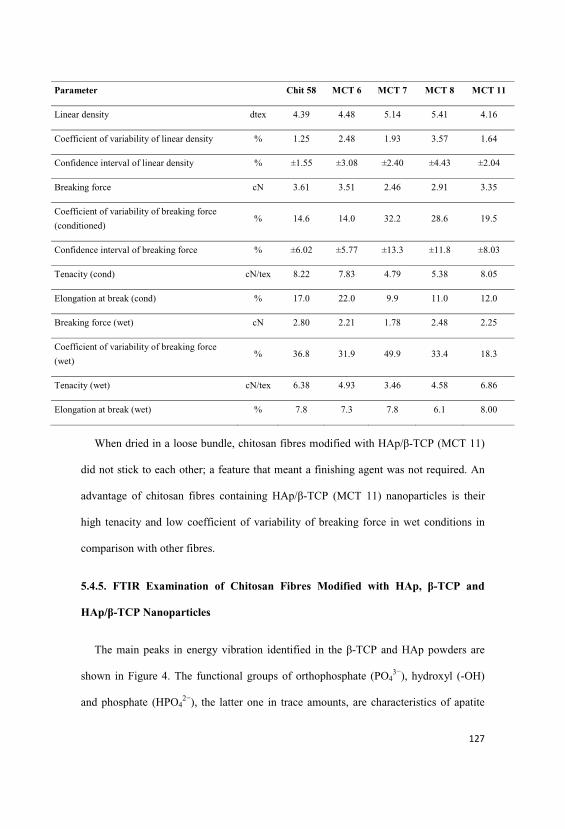
Parameter Chit 58 MCT 6 MCT 7 MCT 8 MCT 11
Linear density dtex 4.39 4.48 5.14 5.41 4.16
Coefficient of variability of linear density % 1.25 2.48 1.93 3.57 1.64
Confidence interval of linear density % ±1.55 ±3.08 ±2.40 ±4.43 ±2.04
Breaking force cN 3.61 3.51 2.46 2.91 3.35
Coefficient of variability of breaking force
(conditioned)% 14.6 14.0 32.2 28.6 19.5
Confidence interval of breaking force % ±6.02 ±5.77 ±13.3 ±11.8 ±8.03
Tenacity (cond) cN/tex 8.22 7.83 4.79 5.38 8.05
Elongation at break (cond) % 17.0 22.0 9.9 11.0 12.0
Breaking force (wet) cN 2.80 2.21 1.78 2.48 2.25
Coefficient of variability of breaking force
(wet)% 36.8 31.9 49.9 33.4 18.3
Tenacity (wet) cN/tex 6.38 4.93 3.46 4.58 6.86
Elongation at break (wet) % 7.8 7.3 7.8 6.1 8.00
When dried in a loose bundle, chitosan fibres modified with HAp/ -TCP (MCT 11)
did not stick to each other; a feature that meant a finishing agent was not required. An
advantage of chitosan fibres containing HAp/ -TCP (MCT 11) nanoparticles is their
high tenacity and low coefficient of variability of breaking force in wet conditions in
comparison with other fibres.
5.4.5. FTIR Examination of Chitosan Fibres Modified with HAp, -TCP and
HAp/ -TCP Nanoparticles
The main peaks in energy vibration identified in the -TCP and HAp powders are
shown in Figure 4. The functional groups of orthophosphate (PO43−), hydroxyl (-OH)
and phosphate (HPO42−), the latter one in trace amounts, are characteristics of apatite

materials. Trace amounts of (CO32−) groups were observed at 1428 cm−1, for both -
TCP and HAp, indicating that in some commercial calcium phosphate powders, CaO
and Ca(OH)2 are added to achieve the ideal stoichiometric proportion between Ca/P
[23,25,28-30]. In the Figure 4 show the FTIR spectrum of the commercial calcium
phosphate.
Figure 4. FTIR spectrum of the commercial HAp and -TCP.
0
0,5
1
1,5
2
2,5
3
5001000150020002500300035004000
Absorbance
Wavenumber cm-1
-TCP
HAp
The absence of bands at 460 and 740 cm−1 and an isolated band at 600 cm−1,
characteristic of -TCP, indicate that the starting material was composed of -TCP only.
This calcium phosphate is easily identified by a broad band at 900-1200 cm−1, and the
presence of a peak at 724 cm−1, characteristic of the symmetric mode of (P-O-P), the
distortion of P-O. The peak at 1211 cm−1 is characteristic of the non-degenerate
deformation of hydrogen groups (H-OPO3, O-PO3, HPO42−), which may reflect the
interaction with water molecules in the structure [23,29,31,32-34]. Figure 3 shows the
spectrum of HAp with a peak at 839 cm−1 that corresponds to the deformation modes of
the phosphate group (O-P-H) bonds, which are associated with the energy levels and the

rotational type of (O-H) bond. The (-OH) group peaks are also apparent at 3570 cm−1
and 3464 cm−1, reflecting the way they stretch.
The low intensity peak visible at 962 cm−1 corresponds to the non-degenerate
symmetric stretching of P-O bonds in the phosphate groups. The peak at 1040 and 1093
cm−1 represent the asymmetric stretch modes of the P-O bonds in the phosphate groups
[23,31-34]. Figure 5 shows the FTIR spectra of the MCT 7 HAp composite. spectrum
exhibits characteristic bands such as: a strong and broad peak between 1032 cm−1 and
1086 cm−1 and 1160 cm−1 that reflects the skeletal vibrations stretch of the saccharide
structure and a peak at 1559, 1543 cm−1 attributed to the free primary amino group
(NH2), amide I (1637 cm−1) indicating that chitosan used in this research is partially
deacetylated (83.2%), amide II (1559 cm−1) and amide III (1319 cm−1). The anti-
symmetric stretch bridge C-O-C (1160 cm−1), stretch N-H (3298, 3500 cm−1), stretch O-
H (3445 cm−1) and the main peaks of energy vibration (1032, 1086 cm−1) identified the
chitosan. The peak at 1243 cm−1 represents the free amino groups and the C2 position of
glycosamine [23,25,27,30-32,35]. The main peaks of energy vibration identified in HAp
are characteristic of the functional groups of orthophosphate (PO43−) at 1100 cm−1,
hydroxyl (-OH) at 3700 and 2600 cm−1 and phosphate (HPO42−) at 1000 cm−1.
Figure 5. FTIR spectra of chitosan fibres (Chit 58) and modified -TCP (MCT 6),
and those modified with HAp (MCT 7), HAp/ -TCP (1:1) (MCT 8) and HAp/ -
TCP (1:2) (MCT 11).
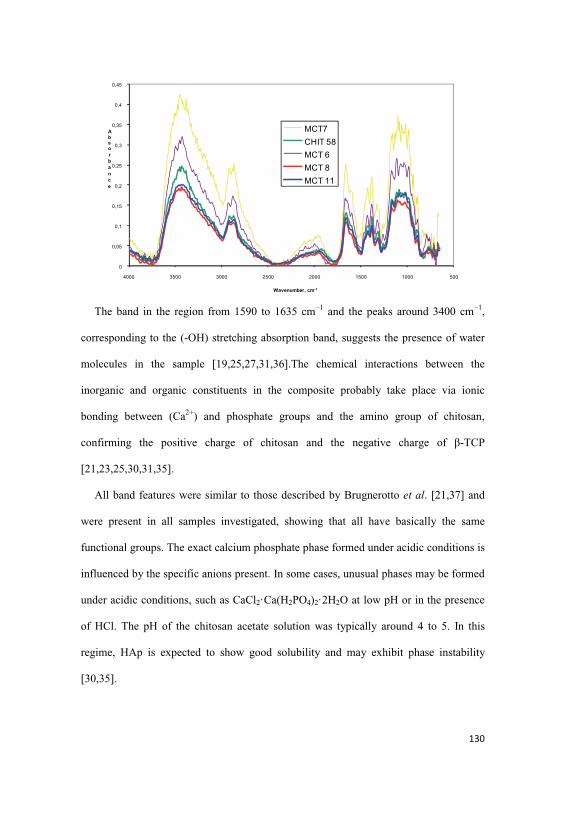
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
5001000150020002500300035004000
Absorbance
Wavenumber, cm-1
MCT7CHIT 58MCT 6MCT 8MCT 11
The band in the region from 1590 to 1635 cm−1 and the peaks around 3400 cm−1,
corresponding to the (-OH) stretching absorption band, suggests the presence of water
molecules in the sample [19,25,27,31,36].The chemical interactions between the
inorganic and organic constituents in the composite probably take place via ionic
bonding between (Ca2+) and phosphate groups and the amino group of chitosan,
confirming the positive charge of chitosan and the negative charge of -TCP
[21,23,25,30,31,35].
All band features were similar to those described by Brugnerotto et al. [21,37] and
were present in all samples investigated, showing that all have basically the same
functional groups. The exact calcium phosphate phase formed under acidic conditions is
influenced by the specific anions present. In some cases, unusual phases may be formed
under acidic conditions, such as CaCl2·Ca(H2PO4)2·2H2O at low pH or in the presence
of HCl. The pH of the chitosan acetate solution was typically around 4 to 5. In this
regime, HAp is expected to show good solubility and may exhibit phase instability
[30,35].

High turbidity was observed in solutions containing HAp, which indicates a strong
adsorption interaction between chitosan and the HAp surface. Chitosan has a relatively
high zeta potential and can enhance the stability of HAp [30,35].
5.4.6. Morphology and Chemistry of Chitosan Fibres Modified with HAp, -TCP
and HAp/ -TCP Nanoparticles
The WRV of wet-spun regular- and HAp- and -TCP modified chitosan fibres
ranged from 150% to 331% depending on the calcium phosphate content.
MCT 8 had the highest WRV of 331%, which may be attributed to the high content
of apatite agglomerates capable of forming water-retaining pores. The amount of
calcium phosphate in the chitosan spinning solutions was estimated according to the
calcium and ash content (Table 4).
Table 4. Water retention value (WRV) content of calcium and ash in HAp/ -TCP
modified chitosan fibres.
Fibre codeWRV Ash Calcium content
% % g/kgChit 58 158 0.1 0.14MCT 6 163 0.4 0.35MCT 7 154 3.2 8.45MCT 8 331 4.8 9.95MCT 11 210 4.8 14.35
Cross-sections of selected HAp/ -TCP-modified chitosan fibres are illustrated in
SEM images in Figure 6.
Figure 6. (a,b) SEM images of the surface and cross-section of chitosan fibres
modified with HAp/ -TCP (MCT 8); (c,d) chitosan fibres modified with HAp/ -
TCP nanoparticles (MCT 11) and (e) picture of the final fibres.

(a) (b)
(c) (d)
(e)
The amount and size of HAp/ -TCP particles has a significant influence on the shape
of the fibre cross-section. In MCT 11 it is oval and uniform with an undeveloped brim.
The surface is characteristic of chitosan fibres with distinct recesses and grooves. The

white spots that can be seen in the fibre cross-sections in Figure 6 (a,b) represent
HAp/ -TCP agglomerates. In contrast, chitosan fibres spun from solutions containing
HAp/ -TCP nanoparticles have a pure cross-section with no traces of anything other
than the fibre-forming polymer substances (Figures 6 (c,d)).
As can be seen in the pictures represented below the clear chitosan/ -TCP/HAp
solution with nanoparticles of the respective calcium phosphates
Figure 7. Solution with chitosan and nano calcium phosphates composite used to
prepare fibre (solution MCT-11).

Figure 8. Wet-Spinning process using the solution with chitosan and nano calcium
phosphates composite used to prepare fibre (solution MCT-11).
5.5. Conclusions
The presence of HAp/ß-TCP nanoparticles in the solution of chitosan had a
beneficial effect on the production of the modified chitosan fibres. It allowed a smooth
run in the spinning process at a speed of 31 m/min. The addition of the HAp or ß-TCP
particles resulted in a decrease in the tenacity of the modified chitosan fibres. Only the
complex of chitosan with HAp/ ß-TCP nanoparticles had the same tenacity as regular
chitosan fibres. Chitosan fibres were prepared with a calcium content of 14.35 g/kg and
an ash content of 4.8%. The HAp/ ß-TCP-modified chitosan fibres had higher WRV
values than the regular ones. Their more hydrophilic character may contribute to the
higher susceptibility of fibres to enzymatic degradation.
When we compare the HAp/ ß-TCP-modified chitosan fibres with the regular
chitosan fibres (Chit 58) we found similar mechanical properties with higher content of
calcium, which is the main mineral responsible for hard tissue regeneration. The

nanoparticles of ceramic calcium phosphates in the modified chitosan fibres could
probably improve the speed of the release of minerals.
This resea rch also shows a method to obtain ceramic nanoparticles in chitosan
solution from commercial calcium phosphates.
The resulted Composite chitosan fibres have a Polish Patent Application No
393022 related with the composite chitosan fibres production method, according to the
invention, presents as follows: a homogeneous aqueous acid solution of 40 - 70 °C
temperature, containing 0,1 - 10 wt% of hydroxyapatite and/or tricalcium phosphate
nano-particles, and 0,1 - 3 wt% of chitosan is entered, during mixing, into the spinning
solution of 20 - 55 °C temperature, containing chitosan of average molecular weight of
150 000 - 700 000 Da, deacetylation degree of 80 - 95 %, poly-dispersion degree
between 2 - 5, and concentration of 2 - 7 wt% in acetic acid aqueous solution of 1 - 3
wt% concentration, containing 0,1 - 15 wt% of glycerine in relation to chitosan. Then,
after venting, the obtained spinning solution is being embossed through multi-hole
spinning nozzle into the coagulation bath of 25 - 45 °C temperature, which is a sodium
hydroxide aqueous solution of 1 - 5 wt% concentration. Obtained fibres are being
stretched from 15 to 67 % in sodium hydroxide aqueous solution of 0,1 - 0,5 %
concentration in temperature of 80 - 95 °C. Then they are rinsed in water, finished in
bath of 30 - 75 °C temperature containing hydro-alcoholic emulsion with surfactants,
and they are being dried in temperature of 40 °C.
Acknowledgments
This research was funded by the Ministry of Science and Higher Education in 2009-
2012 as research project No. N N508 445636.

The authors received funding from the European Community’s Seventh Framework
Program [FP7/2007-2013] under grant agreement no. PITN-GA-2008-214015.
Analysis of the HAp and -TCP particle size in the chitosan solutions was carried out
using equipment provided by the Institute of Polymer and Dye Technology of the
Technical University, Lodz, Poland.
5.6. References
1. Wawro, D.; Ciecha ska, D.; St plewski, W.; Bodek, A. Chitosan microfibrids:
preparation, selected properties and application. Fibres Text. East. Eur. 2006, 14,
97-101.
2. Notin, L.; Viton, C.; Lucas, J.-M.; Domard, A. Pseudo-dry-spinning of chitosan.
Acta Biomater. 2006, 2, 297-311.
3. Niekraszewicz, A.; Kucharska, M.; Wawro, D.; Struszczyk, M.H.; Kopias, K.;
Rogaczewska, A. Development of a manufacturing method for surgical meshes
modified by chitosan. Fibres Text. East. Eur. 2007, 15, 105-107.
4. Wawro, D. Multifilament chitosan yarn. Fibres Text. East. Eur. 2011, 19, 101.
5. Spinks, G.M.; Shin, S.R.; Whitten, P.G.; Kim, S.I.; Kim, S.J.; Wallace, G.G.
Mechanical properties of chitosan/CNT microfibres obtained with improved
dispersion. Sens. Actuators 2006, 115, 678-684.
6. Wawro, D.; St plewski, W.; Ciecha ska, D.; Kruci ska, I.; Surma, B.; Lipp-
Symonowicz, B. Methods of production high tenacity chitosan fibres. Pol. Pat.
Appl. P. 387234, 2009.
7. Wawro, D.; Kruci ska, I.; Ciecha ska, D.; Niekraszewicz, A.; St plewski, W. Some
Functional Properties of Chitosan Fibres Modified with Nanoparticles. In

Proceedings of the 10th International Conference of the European Chitin Society,
EUCHIS’11, St. Petersburg, Russia, 20-24 May 2011.
8. Sarkar, S.; Jana, A.D.; Samanta, S.K.; Mostafa, G. Facile synthesis of silver
nanoparticles with highly efficient antimicrobial property, 2007, 26, 4419-4426.
9. Zargarian, S.S.; Haddadi-Asl V. A Nanofibrous composite scaffold of PCL/
hydroxyapatite-chitosan/PVA prepared by electrospinning. Iran. Polym. J. 2010,
19, 457-468.
10. Lee, C.-Y.; Lee, J.-S.; Chen, S.-C. Dechlorinating Chitosan Fibres and Method for
Manufacturing the same. U.S. 2010/0084336 A1, 8 April 2010.
11. Strobin, G.; Ciecha ska, D.; Wawro, D.; St plewski, W.; Jó wicka, J.; Sobczak, S.;
Haga, A. Chitosan fibres modified by fibroin. Fibres Text. East. Eur. 2007, 15, 64-
65.
12. Wawro, D.; St plewski, W.; Wrze niewska-Tosik, K. Preparation of keratin-
modified chitosan fibres. Fibres Text. East. Eur. 2009, 17, 37-42.
13. Heineman, C.; Heineman, S.; Bernhard, A.; Worch, H.; Hanke, T. Novel textile
chitosan scaffolds promote spreading, proliferation, and differentiation of
osteoblasts. Biomacromolecules 2008, 9, 2913-2920.
14. Heineman, C.; Heineman, S.; Bernhard, A.; Lode, A.; Worch, H.; Hanke, T. In
vitro evaluation of textile chitosan scaffolds for tissue engineering using human
bone marrow stromal cells. Biomacromolecules 2009, 10, 1305-1310.
15. Heineman, C.; Heineman, S.; Bernhard, A.; Lode, A.; Worch, H.; Hanke, T. In
vitro osteoclastogenesis on textile chitosan scaffolds. Eur. Cells Mater. 2010, 19,
96-106.

16. Tuzlakoglu, K.; Alves, C.M.; Mano, J.F.; Reis, R.L. Production and
characterization of chitosan fibres and 3-D fibre mesh scaffolds for tissue
engineering applications. Macromol. Biosci. 2004, 4, 811-819.
17. Tuzlakoglu, K.; Reis, R.L. Formation of bone-like apatite layer on chitosan fibre
mesh scaffolds by a biomimetic spraying process J. Mater. Sci. Mater. Med. 2007,
18, 1279-1286.
18. Lian, Q.; Li, D.C.; He, J.K.; Wang, Z. Mechanical properties and in-vivo
performance of calcium phosphate cement-chitosan fibre composite. Proc. Inst.
Mech. Eng. H 2007, 222, 347-353.
19. Kent, J.N.; Block, M.S.; Finger, I.M.; Guerra, L.; Larsen, H.; Misiek, D.J.
Biointegrated hydroxyapatite coated dental implants: 5 Year clinical observation. J.
Am. Dent. Assoc. 1990, 121, 138-144.
20. Yokoyama, A.; Yamamoto, S.; Kawasaki, T.; Kogho, T.; Nakasu, M. Development
of calcium phosphate cement using chitosan and citric acid for bone substitute
materials. Biomaterials 2002, 23, 1091-1101.
21. Pighinelli, L. Properties of Microcrystalline Chitosan/Calcium Phosphate Complex,
Potential Use of Chitosan-Based Materials in Medicine. In Proceedings of
Conference 7th ISNaPol and XII IMC, Gramado, Brazil, 7-10 September 2010.
22. Ratajska, M.; Haberko, K.; Ciecha ska, D.; Niekraszewicz, A.; Kucharska, M.
Hydroxyapatite-chitosan biocomposites. Prensent at the VII International Polymer
Seminar, Gliwice, Poland, 26 June 2008.
23. Murugan, R.; Ramakrishna, S. Bioresorbable composite bone paste using
polysaccharide based nano hydroxyapatite. Biomaterials 2004, 25, 3829-3835.
24. Wilsonjr, O.; Hull, J.R. Surface modification of nanophased hydroxyapatite with
chitosan Materials. Sci. Eng. C 2008, 28, 434-437.

25. Kwon, S.-H.; Jun, Y.-K.; Hong, S.-H.; Kim, H-E. Synthesis and dissolution
behavior of -TCP and HA/ -TCP composite powder. J. Eur. Ceram. Soc. 2003,
23, 1039-1045.
26. Wawro, D.; Pighinelli, L.; St plewski, W. Methods of manufacture composite
chitosan fibres. Pol. Pat. Appl. P. 393022, 2010.
27. Matsumoto, T.; Okazaki, M.; Inoune, M.; Hamada, Y.; Taira, M.; Takahashi, J.
Crystallinity
and solubility characteristics of hydroxyapatite adsorbed amino acid. Biomaterials
2002, 23, 2241-2247.
28. Shinn-Jyh, D. Preparation and properties of chitosan/calcium phosphate composites
for bone repair. Dent. Mater. 2006, 25, 706-712.
29. Liou, S.C.; Chen, S.Y. Transformation mechanism of different chemically
precipitated apatitic precursor into -tricalcium phosphate upon calcinations.
Biomaterials 2002, 23, 4541-4547.
30. Pena, J.; Vallet-Regi, M. Hydroxyapatite, tricalcium phosphate and biphasic
materials prepared by a liquid mix technique. J. Eur. Ceram. Soc. 2003, 23, 1687-
1696.
31. Fujita, R.; Yokoyama, A.; Nodasaka, Y.; Kohgo, T.; Kawasaki, T. Ultrastructure of
ceramic-bone interface using hydroxyapatite and B-tricalcium phosphate ceramics
and replacement mechanism of -tricalcium phosphate in bone. Tissue Cell 2003,
35, 427-440.
32. Skrtic, D.; Antonucci, J.M.; Eanes, E.D. Amorphous calcium phosphate-based
bioactive polymeric composites for mineralized tissue regeneration. J. Res. Natl.
Inst. Stand. Technol. 2003, 108, 167-182.

33. Fathi, M.H.; Hanifi, A.; Mortazavi, V. Preparation and bioactivity evaluation of
bone-like hydroxyapatite nanopowder. J. Mater. Process. Technol. 2008, 202, 536-
542.
34. Skoog, D.A.; Holler, F.J.; Nieman, T.A. Principios de Analise Instrumental, 5a ed.;
Bookman: Porto Alegre, Brazil, 2002; pp. 342-384.
35. Wilson, O.C., Jr.; Hull, J.R. Surface modification of nanophase hydroxyapatite
with chitosan. Mater. Sci. Eng. C 2008, 28, 434-437.
36. Maachou, H.; Bal, K.E.; Bal, Y.; Chagnes, A.; Cote, G.; Alliouche, D.
Characterization and
in vitro bioactivity of chitosan/hydroxyapatite composite membrane prepared by
freeze-gelation method. Trends Biomater. Artif. Organs 2008, 22, 16-27.
37. Brugnerotto, J.; Lizardi, J.; Goycoolea, F.M.; ArguÈelles-Monal, W.; DesbrieÁres,
J.; Rinaudo, M. An infrared investigation in relation with chitin and chitosan
characterization. Polymer 2001, 42, 3569-3580.

Chapter 6
Degradation and bioactivity of the MCCh/ -TCP complex
6.1. Introduction
The recent developments in the area of artificial bone materials involve
ceramics, which are bio-inert like alumina and zirconia, resorbable like a tri-calcium
phosphate, and bioactive like a hydroxyapatite. Ceramic applications in hard tissue
regeneration and replacement are well documented. The main fields of their applications
are the replacements of hips, knees, teeth, tendons, and ligaments and repair for
periodontal disease, maxillofacial reconstruction, augmentation and stabilisation of the
jaw bone, spinal fusion, and bone repair after tumour surgery [1-3].
Polysaccharides, such as chitosan and its derivatives like microcrystalline
chitosan (MCCh), have some excellent properties for medical applications: non-toxicity
(monomer residues are not harmful to health); water solubility or high swelling ability
after simple chemical modification; stability to pH variations; biocompatibility;
antibacterial, antifungal and antiviral activity; high adhesiveness; non-toxicity; and
extensive chemical reactivity. Moreover, these materials evidence a strong ability to
create hydrogen and ionic bonds and bio-stimulation of natural resistance by controlling
and improving bioactivity. These properties make chitosan a good candidate for the
preparation and modification of modern generation of scaffolds for tissue regeneration
[2,4-8,17-20]. Much attention has been given to different materials or composites
including organic and inorganic materials that can be used as a base material for
scaffold devices, as modification tools for currently used biomedical devices that
improve hard and soft tissue regeneration and/or reinforcement efficacy. Additionally,

they are applicable as a tissue formation precursor in regenerative therapy in the field of
periodontics, orthopaedics, cancer, plastic surgery, and veterinary applications [9-13].
The degradation behaviour of chitosan and dissolution of calcium phosphates in
physiological environmental, plays a crucial role in the long-term performance of a
tissue engineered cell/material construction. The degradation kinetics may affect many
cellular processes, including cell growth, tissue regeneration, and host response, the
polymer in case of microcrystalline chitosan also stimulates the forming of monocytes
and inhibits the growth of bacteria and fungi thus lowering the risk of wound
inflammation. Some characteristics of the scaffold material such as porosity, particle
size and particle shape were reported to have a significant influence on the
inflammatory response and reparative bone formation. Irregularly shaped, sharp-edged
particles prompted higher inflammatory response than round particles of the same size
[8,9,10,15-17]. The solution parameters of initial pH, ionic concentration and
temperature have a large effect on the rate of scaffold dissolution and even type of
calcium phosphate precipitated. Ionic concentration, and therefore pH, will obviously
change with time as dissolution progresses and this will in turn affect the dissolution
rate. If pH rises above a critical value, cytoxicity will occur [22]. This study compares
the hydrolytic and enzymatic biodegradation, bioactivity, structure and mechanical
properties in sponge form of microcrystalline chitosan and microcrystalline chitosan/ß-
TCP complex composites, the last one according with earlier investigation in the
formulation and method to prepare a new complex which is described in Polish patent
application P 393758, 2011, that can be potentially useful in the field of bone tissue
engineering.

6.2. Experimental Section
6.2.1. Materials
The following materials were used:
Microcrystalline chitosan (MCCh paste) - average molecular weight (Mw) = 330
kD, deacetylation degree (DD) = 82%, ash content = 0.7%, water retention value
(WRV) = 598%, polymer content = 2.79%, pH = 7.38.
MCCh/ß-TCP complex paste - water retention value (WRV) = 560%, complex
content = 3.76%, content of MCCh: 3.008% and ß -TCP: 0.752%, pH = 7.40.
Tri-calcium orthophosphate (ß-TCP) powder, (Ca3(PO4)2) – Sigma Aldrich Lab.,
Germany, with 425400 ppm Ca and 199000 ppm P.
Hydroxyapatite powder (Ca10(PO4)6(OH)2 – Sigma Aldrich Lab., Germany, with
447200 ppm Ca and 186500 ppm P.
Plasticiser - Glycerol (C3H8O3), 99%, pure p.a., Sigma-Aldrich, Germany.
Phosphate buffer – pH = 7.40
Lysozyme – muramidase from chicken protein, EC 3.2.1.17, by Merck Co. with
an activity of 50000 U/mg.
6.3. Methods
6.3.1. Preparation of the MCCh/ß-TCP complex
The MCCh/ß-TCP complex was prepared according to Poland Patent Application P
393758 [21].
6.3.2. Preparation of the composites in sponge form
Sponges were prepared via a freeze-drying method using an ALFA 1-4 dryer
made by Christ Co. in the temperature range from -25 to 10°C with a vacuum ranging
from 0.1 to 0.53 mbar for 20 to 24 hours, depending upon the size of the charge. Freeze-
drying resulted in the preparation of sponges with a smooth surface that was free of

defects. The compositions of the samples (MCCh and the MCCh/ß-TCP complex
sponges) are shown in Table 1.
6.3.3. SEM study of the composites in sponge form
The pores morphology, their size and distribution in the polymer sponge were
observed using a scanning electron microscope (SEM, FEI Quanta 200, USA).
6.3.4. Mechanical properties
The mechanical properties of the composites in sponge form were determined in
the Accredited Laboratory of Metrology at Institute of Biopolymers and Chemical
Fibers (IBWCh) (certificate No. AB 388) using a dynamometer Instron (type 5544) in
accordance with the following standards: Polish Standard PN-EN-ISO 527-1, Polish
Standard PN-EN-ISO 527-3, and Polish Standard PN-ISO 4593.
6.3.5. Assessment of the degradability of the composites
Tested composites were put into a phosphate buffer at pH = 7.41 and bath
module 1:300 w/w and sterilised in an autoclave at 121°C for 25 minutes. Then,
samples were dried under vacuum for 15 minutes. The susceptibility of the bio-
composites to biodegradation was assessed in the buffer solution with the addition of
lysozyme in amount of 200 µg/cm3. The test was conducted in an incubator at 37°C
under static conditions according to standard PN-81C-06504 ”preparation of buffer
solutions”. Bio-composites in the form of sponges were removed from bath after 20, 40,
and 60 days. Next, the samples were filtered in a Buchner funnel, washed with distilled
water at 50°C, poured over 70% ethanol, filtered after 5 minutes, and dried under
vacuum at 70°C until a constant mass. The estimation of the hydrolytic and enzymatic
degradation progress of the tested preparations was based on the change of the pH,

measurements of the concentration of aminosaccharides (products of degradation) in the
bath, and the mass loss of the samples.
6.3.6. Bioactivity
A quantity of 0.4 g of tested samples and 0.4 g of reference samples (cotton)
were put into separate vessels and sterilised in an autoclave at 121°C for 25 minutes.
After sterilisation and drying, all samples were inoculated with a suspension of bacteria
for the determination of antibacterial activity using a quantitative test according to
standard JIS L 1902:2002. Escherichia coli (ATCC 11229) and Staphylococcus aureus
(ATCC 6538) were used as testing microorganisms.
6.4. Results and Discussion
The Table 1 showed the composition of samples prepared, named SMC for
microcrystalline chitosan and SMC-TCP for (microcrystalline chitosan/ß-TCP
complex), to compare the biodegradation, bioactivity, structure and mechanical
properties in form of sponges.
Table 1. Samples composition.
Sample symbol Dry sample composition *[wt%]
SMC MCCh:66.7 Glycerol: 33.3
SMC-TCP MCCh: 53.4ß-TCP: 13.3
Glycerol: 33.3
* moisture not taken into account
The glycerol was used as a plastisizer for a better manufacture of the samples
increasing the mixing process in the preparation of the suspension and directed related
with the homogenous interconnected pore morphology structure showing, smooth

surface without defects. Notice the addition of glycerol increase the number and round
shape of pores in freeze-dried method in all samples.
6.4.1. Biodegradation mass loss
The assessment of hydrolytic and enzymatic degradation process of the SMC
and SMC-TCP in sponge form, was estimated in the course of the degradation: pH,
percentage of mass loss of the samples (calculated on the microcrystalline chitosan
contained in the biocomposite) and concentration of aminosugars (products of
degradation contained in the citric-phosphate buffer). Considering it that the
preparations only assist in the joining of bones and their role in the organism is short
lasting, the testing time was limited to 60 days. The results are showed in Table 2 a,b.
Table 2a, showed the hydrolytic degradation process by immersion the sample
into the buffer solution of pH 7.4 at 37°C. Notice that mass loss of SMC-TCP complex
after 60 days, gives evidence that samples are very susceptible for degradation. The
mass loss of SMC-TCP complex is slightly above 16 % and SMC is around 15 % with
forming of reductive aminosugars around 1.3 % and 2.0% respectively.
Table 2a. Percentage mass loss and saccharification before and after hydrolyticdegradation
Time in days /Sample
Polymer contentin sample before
degradation[g]
Polymer content insample afterdegradation
[g]
Mass loss[%]
Saccharification[%]
pH
0 / SMC 0.2273 0.1937 0 0 7.500 / SMC-TCP 0.3132 0.2842 0 0 7.39
60 / SMC 0.2237 0.1885 15.70 1.99 7.4760 / SMC-TCP 0.3373 0.2828 16.15 1.31 7.44
The results of enzymatic degradation of biocomposites are estimated by the
same parameters and compiled in Table 2 b. As can be seen the biocomposites are also

susceptible to enzymatic degradation after 60 days whose intensity depends on the
applied lysozyme concentration, at 200 µg/cm3, the mass loss of the SMC-TCP around
18 % and SMC biocomposite is around 20 % with higher concentration of aminosugars
as a result of degradation amounted 7.09 % and 10.68 % respectively. This result
suggest that in SMC-TCP complex sample, the calcium phosphate (ß-TCP) shields a
little the polymer against the lysozyme by crosslinking and probably ionic bonds
between ion phosphate and the amino group of polymer affecting the time and amount
of degradation in the microcrystalline chitosan.
Table 2b. Percentage mass loss and saccharification before and after enzymaticdegradation
Time in days /Sample
Polymer contentin sample before
degradation[g]
Polymer content insample afterdegradation
[g]
Mass loss[%]
Saccharification[%]
pH
0 / SMC 0.2273 0.1937 0 0 7.500 / SMC-TCP 0.3132 0.2842 0 0 7.39
60 / SMC 0.1878 0.1507 19.75 10.68 7.4760 / SMC-TCP 0.3159 0.2594 17.90 7.09 7.47
6.4.2. SEM study of the composites in sponge form
In this section, the structure and morphology of the samples before and after the
biodegradation process are presented according to the preparation conditions listed in
Table 1. Figures 1 a and b are SEM pictures of SMC before the degradation process,
and Figure 2 c is the digital picture of sample SMC.
Figure 1. SEM pictures of SMC before degradation process, a-200X, b-1000X, c-
digital picture.

The Figures 2 a and b are SEM pictures of the SMC-TCP before the degradation
process, and Figure 2 c is the digital picture of sample SMC-TCP.
Figure 2. SEM pictures of SMC-TCP complex before degradation process, a-200X, b-
1000X, c- digital picture.
The Figures 3 a and b are SEM pictures of SMC after 60 days of the hydrolytic
degradation process, and Figure 3 c is a digital picture of sample 60 / SMC.
Figure 3. SEM pictures of SMC after 60 days of hydrolytic degradation process, a-
200X, b-1000X, c- digital picture.

The Figures 4 a and b are SEM pictures of the of SMC-TCP complex after 60
days of the hydrolytic degradation process, and Figure 4 c is a digital picture of sample
60 / SMC-TCP.
Figure 4. SEM pictures of SMC-TCP complex after 60 days of hydrolytic degradation
process, a-200X, b-1000X, c- digital picture.
The Figures 5 a and b are SEM pictures of the SMC after 60 days of the
enzymatic degradation (concentration of lysozyme - 200 µg/cm3) and Figure 5 c is a
digital picture of sample 60 / SMC.
Figure 5. SEM pictures of the SMC after 60 days of enzymatic degradation process, a-
200X, b-1000X, c- digital picture.

The Figures 6 a and b are SEM pictures of SMC-TCP complex after 60 days of
the enzymatic degradation (concentration of lysozyme - 200 µg/cm3) and Figure 6 c is
the digital picture of sample 60 / SMC-TCP complex.
Figure 6. SEM pictures of SMC-TCP complex after 60 days of enzymatic degradation
process, a-200X, b-1000X, c- digital picture.
The morphology of the microcrystalline chitosan and complex sponges changed
progressively with time. In the case of the complex, the sample contained pores that
became more compact over time.
Comparing the samples before and after degradation process indicates that under
the action of the enzyme at 200 µg/cm3 concentration the surface structure of the sponge
undergoes changes with the forming of new and interconnected wider pores. The

complex structure reveals a homogenous distribution of micro and nanosized particles
and clusters formation of ß-TCP in the polymer matrix. The white colour of the sample
in the digital picture results from the calcium phosphate particles in the complex.
6.4.3. Mechanical properties of the sponges
The assessment of mechanical properties of the SMC and the SMC-TCP
complex in sponge form are presented in (Tables 3 a, b) in respect of strength,
elongation and elastic modulus, qualities that are crucial for implants in orthopedical
surgery. The addition of glycerol interfered with MCCh chains, decreasing
intermolecular attraction and increasing polymer chains mobility maintaining the
homogenous porous network structure which facilitates elongation, in case of complex
with -TCP had little effect on improving the elongation at break in another hand a little
decrease in tensile strength, but showing the good interaction (ionic and covalent
bonding) between the calcium phosphate and the MCCh according with the literature
[21]. After biocomposite materials were affected by 60 days of enzymatic and
hydrolytic degradation, similar behaviour of mechanical properties was observed, with
lower values of tensile strength, elastic modulus and with higher elongation at break due
the process. The SMC-TCP preparation feature by best mechanical parameters qualifies
for further investigations. In the case of enzymatic and hydrolytic degradation, similar
behaviour was observed, with lower values of tensile strength, elastic modulus with
higher elongation at break due the process
Table 3a. Mechanical properties of sponges before and after hydrolytic degradation
Parameters
Samples Degradationtime
[days]
Composition Tensilestrength[MPa]
Elongation atbreak [%]
Elastic modulus[MPa]

SMC 0 MCCh: 66.7 Glycerol: 33.3
0.1720 1.53 1.400
SMC-TCP 0 MCCh: 53.4ß-TCP: 13.3
Glycerol: 33.3
0.0150 2.41 1.000
SMC 60 MCCh: 66.7 Glycerol: 33.3
0.0091 3.19 0.010
SMC-TCP 60 MCCh: 53.4ß-TCP: 13.3
Glycerol: 33.3
0.0148 5.65 0.005
Table 3b. Mechanical properties of sponges before and after enzymaticdegradation
Parameters
Samples Degradationtime
[days]
Composition Tensilestrength[MPa]
Elongation atbreak [%]
Elastic modulus[MPa]
SMC 0 MCCh: 66.7 Glycerol: 33.3
0.172 1.53 1.40
SMC-TCP 0 MCCh: 53.4ß-TCP: 13.3
Glycerol: 33.3
0.015 2.41 1.00
SMC 60 MCCh: 66.7 Glycerol: 33.3
0.0073 1.88 0.005
SMC-TCP 60 MCCh: 53.4ß-TCP: 13.3
Glycerol: 33.3
0.0055 3.14 0.015
6.4.4. Bioactivity
The results of the bioactivity investigations are presented in (Table 4a,b), the
following test was estimated in the course of antibacterial activity (bacteriostatic and
bactericidal activity) using a quantitative test according to standard JIS L 1902:2002
with Escherichia coli and Staphylococcus aureus. Both samples SMC and SMC-TCP
complex showed a bacteriostatic activity and a bactericidal activity against Escherichia
coli, showing the SMC higher activity than SMC-TCP complex, suggesting more free
amino groups from SMC and lower free amino groups from complex that probably are
bonded with the phosphate groups of the calcium phosphate reducing the activity.
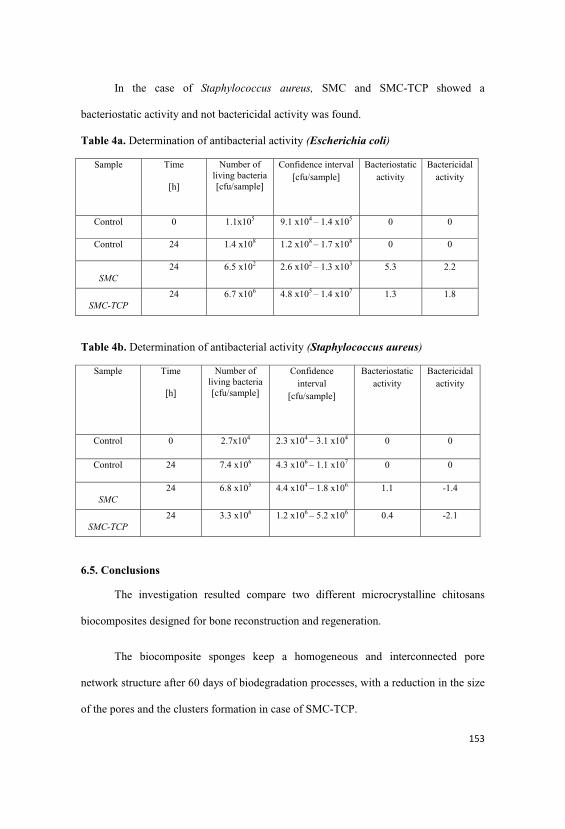
In the case of Staphylococcus aureus, SMC and SMC-TCP showed a
bacteriostatic activity and not bactericidal activity was found.
Table 4a. Determination of antibacterial activity (Escherichia coli)
Sample Time
[h]
Number ofliving bacteria[cfu/sample]
Confidence interval[cfu/sample]
Bacteriostaticactivity
Bactericidalactivity
Control 0 1.1x105 9.1 x104 – 1.4 x105 0 0
Control 24 1.4 x108 1.2 x108 – 1.7 x108 0 0
SMC24 6.5 x102 2.6 x102 – 1.3 x103 5.3 2.2
SMC-TCP24 6.7 x106 4.8 x105 – 1.4 x107 1.3 1.8
Table 4b. Determination of antibacterial activity (Staphylococcus aureus)
Sample Time
[h]
Number ofliving bacteria[cfu/sample]
Confidenceinterval
[cfu/sample]
Bacteriostaticactivity
Bactericidalactivity
Control 0 2.7x104 2.3 x104 – 3.1 x104 0 0
Control 24 7.4 x106 4.3 x106 – 1.1 x107 0 0
SMC24 6.8 x105 4.4 x104 – 1.8 x106 1.1 -1.4
SMC-TCP24 3.3 x106 1.2 x106 – 5.2 x106 0.4 -2.1
6.5. Conclusions
The investigation resulted compare two different microcrystalline chitosans
biocomposites designed for bone reconstruction and regeneration.
The biocomposite sponges keep a homogeneous and interconnected pore
network structure after 60 days of biodegradation processes, with a reduction in the size
of the pores and the clusters formation in case of SMC-TCP.

The SMC and SMC-TCP in sponges form are susceptible to hydrolytic and
enzymatic degradation process.
The hydrolytic degradation by immersion the sample into the buffer solution of
pH 7.4 at 37°C showed the mass loss of SMC-TCP complex is slightly above 16 % and
SMC is around 15 % with forming of reductive aminosugars around 1.3 % and 2.0%
respectively.
The enzymatic degradation in the presence of lysozyme, under the action of the
enzyme at a 200 µg/cm3 concentration, the mass loss of the SMC-TCP around 18 % and
SMC biocomposite is around 20 % with higher concentration of aminosugars as a result
of degradation amounted 7.09 % and 10.68 % respectively.
The mechanical properties after 60 days of enzymatic and hydrolytic
degradation, similar behaviour was observed, with lower values of tensile strength,
elastic modulus and with higher elongation at break showing that can be processed in
elastic forms which have the potential to be used as fillers in hard tissue regeneration.
Both samples SMC and SMC-TCP complex showed a bacteriostatic activity and
a bactericidal activity against Escherichia coli. In the case of Staphylococcus aureus,
SMC-TCP complex and SMC showed only a bacteriostatic activity.
Acknowledgements
The research and work presented in this study were conducted with financial
support from the European Community's Seventh Framework Programme (“Marie Curie
Initial Training Network), FP7/2007-2013.
6.6. References

1. Liou SC, Chen SY. (2002). Transformation mechanism of different chemically
precipitated apatitic precursor into B-tricalcium phosphate upon calcinations.
Biomaterials;23:4541–7.
2. Zhang Y, Zhang M. (2001). Microstructural and mechanical characterization of
chitosan scaffolds reinforced by calcium phosphates. J Non Cryst Solids;282:159–64.
3. Dorozhkin SV. (2009). Nanodimensional and nanocrystalline apatites and other
calcium orthophosphates in biomedical engineering, biology and medicine.
Materials;2:1975–2045.
4. Pillai CKS, Paul W, Sharma CP. (2009). Chitin and chitosan polymers: chemistry,
solubility and fiber formation. Prog Polym Sci 2009;34:641–78.
5. Suh JK, Matthew HW. (2000). Application of chitosan-based polysaccharide
biomaterials in cartilage tissue engineering: a review. Biomaterials;21:2589–98.
6. Kim IY, Seo SJ, Moon HS, Yoo MK, Park IY, Kim BC. (2008). Chitosan and its
derivatives for tissue engineering applications. Biotechnol Adv;26:1–21.
7. Muzzarelli RA. (2011). Chitosan composites with inorganics, morphogenetic proteins
and stem cells, for bone regeneration. Carbohydr Polym;83:1433–45.
8. Niekraszewicz A, Kucharska M, Wisniewska-Wrona M, Cienchanska D, Ratajska M,
Haberko K. (2009). Surgical biocomposites with chitosan. Progress on Chemistry and
Application of Chitin and Its Derivatives;14:167–78.
9. Misiek DJ, Kent JM, Carr RF. (1984). Soft tissue responses to hydroxyapatite
particles of different shapes. J Oral Maxillofac Surg;42:150–60.
10. Yildirim O. (2004). Preparation and characterization of chitosan/calcium phosphate
based composite biomaterials [dissertation]. Izmir, Turkey: Izmir Institute of
Technology.

11. Vert M, Li MS, Spenlehauer G, Guerin P. (1992). Bioresorbability and
biocompatibility of aliphatic polyesters. J Mater Sci;3:432–46.
12. Senel S, McClure SJ. (2004). Potential applications of chitosan in veterinary
medicine. Adv Drug Deliv Rev;56:1467–80.
13. Dorozhkin SV. (2009). Calcium orthophosphates in nature, biology and medicine.
Materials;2:399–498.
14. Ren D, Yi H, Wang W, Ma X. (2005). The enzymatic degradation and swelling
properties of chitosan matrices with different degrees of N-acetylation. Carbohydr
Res;340:2403–10.
15. Muzzarelli C, Muzzarelli RA. (2002). Natural and artificial chitosan–inorganic
composites. J Inorg Biochem;92:89–94.
16. Fathi MH, Hanifi A, Mortazavi V. (2008). Preparation and bioactivity evaluation of
bone-like hydroxyapatite nanopowder. J Mater Process Technol;202:536–42.
17. Wang M. (2006). Composite scaffolds for bone tissue engineering. Am J Biochem
Biotechnol;2:80–4.
18. Kong L, Gao Y, Lu G, Gong Y, Zhao N, Zhang X. (2006). A study on the bioactivity
of chitosan/nano-hydroxyapatite composite scaffolds for bone tissue engineering. Eur.
Polym. J;42:3171–9.
19. Struszczyk HM. (2006). Global requirements for medical applications of chitin and
its derivatives. Progress on Chemistry and Application of Chitin and Its
Derivatives;11:95–102.
20. Viala S, Freche M, Lacout JL. (1998). Preparation of a new organic-mineral
composite-chitosan and hydroxyapatite. Ann Chim Sci Mat;23:69–72.

21. Pighinelli L, Kucharska M, Brzoza-Malczewska K, Gruchała B. (2011). Complex
microcrystalline chitosan/tri calcium orthophosphate and process for preparing same.
Poland patent application P 393758.
22. Hench L. L, Jones J. R, Sepulveda P. (2002) Bioactive Materials for Tissue
Engineering Scaffolds, Future Strategies for Tissue and Organ Replacement, Imperial
College Press, London-UK, chapter 1, 3-24.
23. Wawro, D., & Pighinelli, L. (2011). Chitosan fibers modified with HAp/ß–TCP
nanoparticles. International Journal of Molecular Sciences, 12, 7286–7300.
24. Wawro, D.; Pighinelli, L.; St plewski, W. (2010). Methods of manufacture
composite chitosan fibers. Pol. Pat. Appl. P. 393022.

Conclusions
Bone in healthy and diseased tissue attract much attention in the last decades, a
revolution in orthopaedics occurred which has led to a remarkable improvement of
quality of life for millions patients who needs those medical devices for hard tissue
regeneration and/or replacement. Significant results have been obtained and published
in the last past years in form of articles and patents showing the efforts are being made
to develop methods and materials in artificial tissues, including bones, cartilage, nerve,
blood vessels, and skin to restore the functions of damaged tissues in musculoskeletal
disorder. The material chemistry and the biochemical technology have progressed from
use of biomaterials like natural polymers (chitosan and its derivatives) and calcium
phosphates like HAp and -TCP to repair and/or replace wounded tissues to the
implantable scaffolds with excellent results.
One significant result of the research was reported in article published in 2011
and in Patent Application P. 393758 in Poland, about new method to obtain a
microcrystalline chitosan /tri-calcium orthophosphate complex showing the calcium
phosphate particles were uniformly dispersed in the amino-glucose macromolecule
aggregation (coagulation process) and no precipitation of the calcium phosphate was
noticed in the samples showing the good stability of the suspension. The study showed
the feasibility of freeze drying method in the preparation of the MCCh/ ß-TCP complex
in sponge form, resulting in a 3-dimensional interconected porous network structure that
can be used in future as a base for scaffolds production. The complex in sponge form is
susceptible to hydrolytic and enzymatic degradation with good mechanical properties
after 60 days of degradation, showing also a bacteriostatic activity and a bactericidal
activity against Escherichia coli and Staphylococcus aureus.

Another important result was publishing in 2010 in another Patent Application P.
393022 in Poland, resulting a article published in 2011 about new method of
manufacture nanocomposite chitosan fibres, showing a better mechanical properties in
wet conditions than regular chitosan fibres with a lower cost process, because no
finishing agent in the process was required. Their more hydrophilic character may
contribute to the higher susceptibility of fibers to enzymatic degradation. This research
also shows a method to obtain ceramic nanoparticles in chitosan solution from
commercial calcium phosphates.
The challenge of hard tissue engineering continue the development of suitable
material bone scaffold with sufficient interconnected porosity and mechanical strength
to allow cell adhesion, migration, growth and proliferation resulting in good integration
with surrounding tissues. These concepts will combine the understanding about
developments of materials, manufacturing technologies and surgical techniques creating
a new generation of scaffolds for skeletal reconstruction used in the regenerative
medicine.


Buy your books fast and straightforward online - at one of world’s
fastest growing online book stores! Environmentally sound due to
Print-on-Demand technologies.
Buy your books online at
www.get-morebooks.com
Kaufen Sie Ihre Bücher schnell und unkompliziert online – auf einer
der am schnellsten wachsenden Buchhandelsplattformen weltweit!
Dank Print-On-Demand umwelt- und ressourcenschonend produzi-
ert.
Bücher schneller online kaufen
www.morebooks.deVDM Verlagsservicegesellschaft mbH
Heinrich-Böcking-Str. 6-8 Telefon: +49 681 3720 174 [email protected] - 66121 Saarbrücken Telefax: +49 681 3720 1749 www.vdm-vsg.de