
(Masked)serinols: Molecules, Biomolecules, Building-Blocks, Supramolecules.Part (I): Syntheses Based...
33
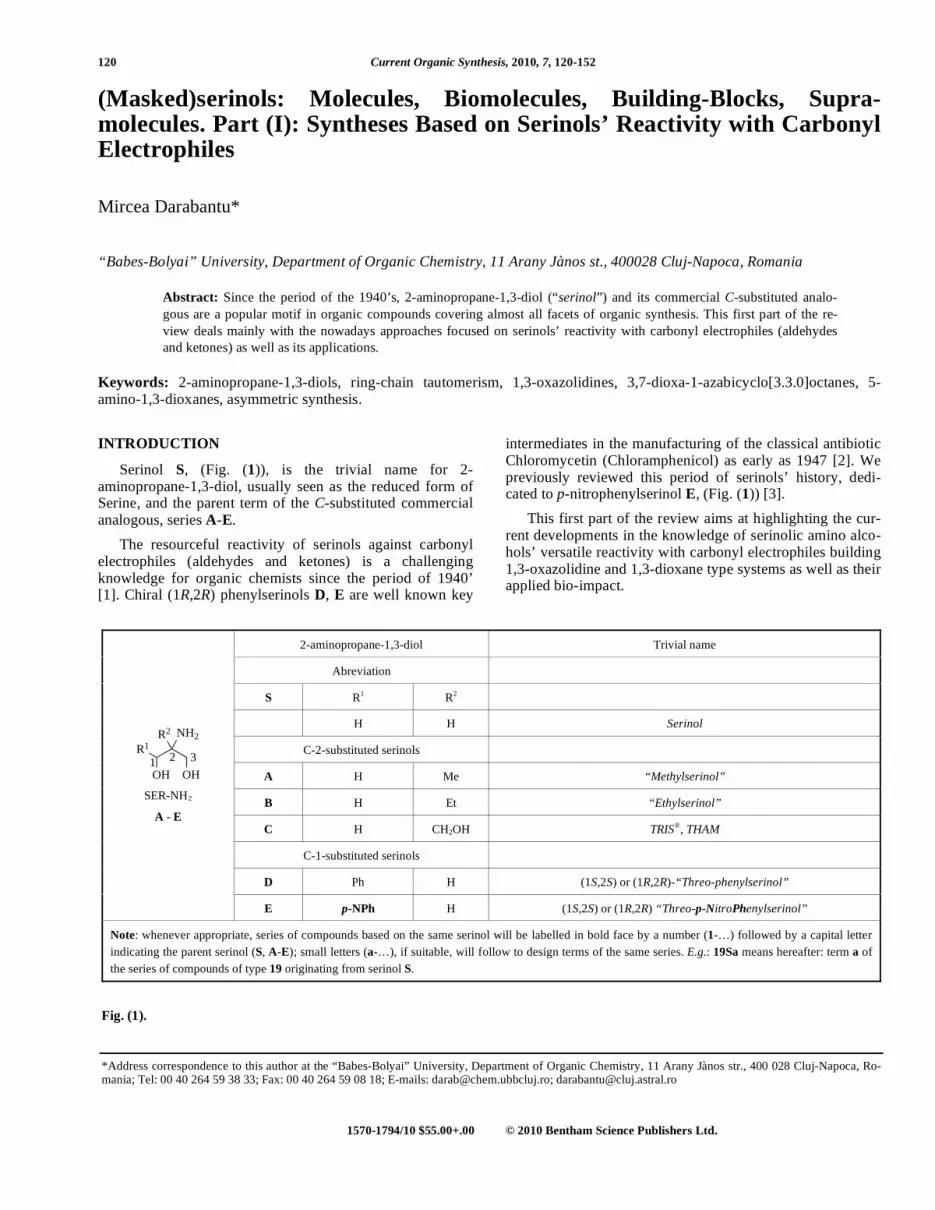
120 Current Organic Synthesis, 2010, 7, 120-152 1570-1794/10 $55.00+.00 © 2010 Bentham Science Publishers Ltd. (Masked)serinols: Molecules, Biomolecules, Building-Blocks, Supra- molecules. Part (I): Syntheses Based on Serinols’ Reactivity with Carbonyl Electrophiles Mircea Darabantu* “Babes-Bolyai” University, Department of Organic Chemistry, 11 Arany Jànos st., 400028 Cluj-Napoca, Romania Abstract: Since the period of the 1940’s, 2-aminopropane-1,3-diol (“serinol”) and its commercial C-substituted analo- gous are a popular motif in organic compounds covering almost all facets of organic synthesis. This first part of the re- view deals mainly with the nowadays approaches focused on serinols’ reactivity with carbonyl electrophiles (aldehydes and ketones) as well as its applications. Keywords: 2-aminopropane-1,3-diols, ring-chain tautomerism, 1,3-oxazolidines, 3,7-dioxa-1-azabicyclo[3.3.0]octanes, 5- amino-1,3-dioxanes, asymmetric synthesis. INTRODUCTION Serinol S, (Fig. (1)), is the trivial name for 2- aminopropane-1,3-diol, usually seen as the reduced form of Serine, and the parent term of the C-substituted commercial analogous, series A-E. The resourceful reactivity of serinols against carbonyl electrophiles (aldehydes and ketones) is a challenging knowledge for organic chemists since the period of 1940’ [1]. Chiral (1R,2R) phenylserinols D, E are well known key intermediates in the manufacturing of the classical antibiotic Chloromycetin (Chloramphenicol) as early as 1947 [2]. We previously reviewed this period of serinols’ history, dedi- cated to p-nitrophenylserinol E, (Fig. (1)) [3]. This first part of the review aims at highlighting the cur- rent developments in the knowledge of serinolic amino alco- hols’ versatile reactivity with carbonyl electrophiles building 1,3-oxazolidine and 1,3-dioxane type systems as well as their applied bio-impact. 2-aminopropane-1,3-diol Trivial name Abreviation S R 1 R 2 H H Serinol C-2-substituted serinols A H Me “Methylserinol” B H Et “Ethylserinol” C H CH2OH TRIS ® , THAM C-1-substituted serinols D Ph H (1S,2S) or (1R,2R)-“Threo-phenylserinol” OH OH R 1 NH 2 R 2 1 2 3 SER-NH2 A - E E p-NPh H (1S,2S) or (1R,2R) “Threo-p-NitroPhenylserinol” Note: whenever appropriate, series of compounds based on the same serinol will be labelled in bold face by a number (1-…) followed by a capital letter indicating the parent serinol (S, A-E); small letters (a-…), if suitable, will follow to design terms of the same series. E.g.: 19Sa means hereafter: term a of the series of compounds of type 19 originating from serinol S. Fig. (1). *Address correspondence to this author at the “Babes-Bolyai” University, Department of Organic Chemistry, 11 Arany Jànos str., 400 028 Cluj-Napoca, Ro- mania; Tel: 00 40 264 59 38 33; Fax: 00 40 264 59 08 18; E-mails: [email protected]; [email protected]
Transcript of (Masked)serinols: Molecules, Biomolecules, Building-Blocks, Supramolecules.Part (I): Syntheses Based...
120 Current Organic Synthesis, 2010, 7, 120-152
1570-1794/10 $55.00+.00 © 2010 Bentham Science Publishers Ltd.
(Masked)serinols: Molecules, Biomolecules, Building-Blocks, Supra-molecules. Part (I): Syntheses Based on Serinols’ Reactivity with Carbonyl Electrophiles
Mircea Darabantu*
“Babes-Bolyai” University, Department of Organic Chemistry, 11 Arany Jànos st., 400028 Cluj-Napoca, Romania
Abstract: Since the period of the 1940’s, 2-aminopropane-1,3-diol (“serinol”) and its commercial C-substituted analo-gous are a popular motif in organic compounds covering almost all facets of organic synthesis. This first part of the re-view deals mainly with the nowadays approaches focused on serinols’ reactivity with carbonyl electrophiles (aldehydes and ketones) as well as its applications.
Keywords: 2-aminopropane-1,3-diols, ring-chain tautomerism, 1,3-oxazolidines, 3,7-dioxa-1-azabicyclo[3.3.0]octanes, 5-amino-1,3-dioxanes, asymmetric synthesis.
INTRODUCTION
Serinol S, (Fig. (1)), is the trivial name for 2-aminopropane-1,3-diol, usually seen as the reduced form of Serine, and the parent term of the C-substituted commercial analogous, series A-E.
The resourceful reactivity of serinols against carbonyl electrophiles (aldehydes and ketones) is a challenging knowledge for organic chemists since the period of 1940’ [1]. Chiral (1R,2R) phenylserinols D, E are well known key
intermediates in the manufacturing of the classical antibiotic Chloromycetin (Chloramphenicol) as early as 1947 [2]. We previously reviewed this period of serinols’ history, dedi-cated to p-nitrophenylserinol E, (Fig. (1)) [3].
This first part of the review aims at highlighting the cur-rent developments in the knowledge of serinolic amino alco-hols’ versatile reactivity with carbonyl electrophiles building 1,3-oxazolidine and 1,3-dioxane type systems as well as their applied bio-impact.
2-aminopropane-1,3-diol Trivial name
Abreviation
S R1 R2
H H Serinol
C-2-substituted serinols
A H Me “Methylserinol”
B H Et “Ethylserinol”
C H CH2OH TRIS®, THAM
C-1-substituted serinols
D Ph H (1S,2S) or (1R,2R)-“Threo-phenylserinol”
OH OH
R1
NH2R2
1 2 3
SER-NH2
A - E
E p-NPh H (1S,2S) or (1R,2R) “Threo-p-NitroPhenylserinol”
Note: whenever appropriate, series of compounds based on the same serinol will be labelled in bold face by a number (1-…) followed by a capital letter
indicating the parent serinol (S, A-E); small letters (a-…), if suitable, will follow to design terms of the same series. E.g.: 19Sa means hereafter: term a of
the series of compounds of type 19 originating from serinol S.
Fig. (1).
*Address correspondence to this author at the “Babes-Bolyai” University, Department of Organic Chemistry, 11 Arany Jànos str., 400 028 Cluj-Napoca, Ro-mania; Tel: 00 40 264 59 38 33; Fax: 00 40 264 59 08 18; E-mails: [email protected]; [email protected]

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 121
1. SYNTHESES BASED ON REACTIVITY OF C-
SUBSTITUTED SERINOLS WITH CARBONYL
ELECTROPHILES IN RING-CHAIN OR RING-RING
TAUTOMERISM CONDITIONS
1.1. Overview
Seen as -aminoalcohols, when reactivity of C-substituted serinols with carbonyl electrophiles is examined, the current most complete approach appeared to us to take into account the ring-chain tautomerism of their imino-derivatives, namely the reversible intramolecular addition of a hydroxyl group to the >C=N- double bond, a 5-Endo-Trig cyclisation against the Baldwin’s rule, (Fig. (2)) [4-6].
NH2
OH
R
O
N
OH
R
..
..
..
NH
O
R..
..
..
1
23
4
5
Imino derivativeSchiff-Base (SB)
1,3-OXazolidine (OX)
5-Endo-Trig
C-substitutedserinol
Fig. (2).
If so, for a comprehensive hereafter discussion, a concise terminology should be adopted.
Thus, in serinols’ family, by simple ring-chain tautomer-ism conditions, we will refer henceforward to their reactivity against monocarbonyl electrophiles, (Figs. (2, 3)), whereas if dicarbonyl electrophiles are involved, our terminology will be double ring-chain-tautomerism.
One must note that, in the case of reaction with ketones, EQ-1, (Fig. (3)), cannot, so far, be detected but, in some situations, a regioisomeric and / or diastereomeric oxazolidi-nes spontaneous interconversion OX OX (EQ-II). We entitled this particular behaviour as simple ring-ring tautomerism [5], or, if appropriate diketones are concerned, double ring-ring tautomerism.
As summarised in Tables 1 and 2, the reactivity of C-2-substituted serinols S, A-C vs. (di)carbonyl electrophiles is very different if compared to that of C-1-substituted, D, E
although almost all equilibria in Fig. (3) can be conveniently shifted to right by, more or less, usual methods (e.g. in re-fluxing aromatic solvent with azeotropic removal of water in the presence of an acidic catalyst). Finally, in serinols’ series S, A-E, one must take into account not only the different number but also the different nature of hydroxyl groups im-plicated in tautomerism, homotopic in TRIS (C), enan-tiotopic in S, A and B but chemically non-equivalent in phenylserinols D, E [17b].
1.2. Simple Ring-Chain and Ring-Ring Tautomerism
1.2.2. C-2-substituted Serinols Based 1,3-oxazolidines
Systems
Only in the case of condensation between C-2-substituted serinols A-C and salicylaldehyde, (Fig. (4)), authentic chain-forms, stable indefinitely as Schiff-Bases SB, are evidenced by IR and NMR and isolated as such [e.g. the SB derivative of TRIS (C), 1Ca (SB)] [15b].
The actual explanation consists of the stabilisation of these Schiff-Bases by intramolecular hydrogen bonds as six membered chelates [15b], (Potapov et al., 1990 [18e]), (Star and Fuchs, 1999 [22a] and their later developments [22b, 22c]).
Completely different, monocondensates of TRIS (C) with other (het)arylaldehydes are isolable as OX species only, e.g. series 1Cb-i (OX) (Table 1, entry 1, Fig. (4)). They are men-tioned to be of interest as anti mycosis agents (Crozet and Nouguier [11]) or prodrugs as delivery drug systems, since, being much weaker bases (pKa = 5 – 7) than the initial -amino alcohol, were seen more lipophilic at physiological pH, for example oxazolidine 1Cb (OX) (Fig. (4)), (Buur and Bundgaard, 1987 [12a]).
OH OH
NH2R2
R1 R3 R4
O
OH OH
NR2
R1
O
R1CH2OH
R2
HN O
R1
HO R2
NOO
R2R1
123
45
6
8
R3R4
R5SER-NH2 R
1 R2
A H Me
B H Et
C H CH2OH
D* Ph H
E* p-NPh H
(p-NitroPhenyl)
Imino derivativesSchiff-Bases
(SB)
(Regioisomeric or diastereomeric)1,3-OXazolidines
(OX)
3,7-DiOxa-r-1-AzaBicyclo[3.3.0]-c-5-Octanes
(DOABO)
R5 R6
OR3 R4
R3 R4
EQ-I
EQ-IIIEQ-II
*valid for enantiomeric pure (e.g. 1S,2S, "threo", like)-phenylserinols D, E; for the D' (e.g. 1R,2S, "erythro", unlike) diastereomer of D, see discussion
R4
R3
R6
Fig. (3).

122 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
Table 1. Basic Reactivity of C-2-substituted Serinols A–C with Carbonyl Electrophiles in Simple Ring-Chain or Ring-Ring Tautomer-ism Conditionsa,b
Entry EQ-I EQ-II EQ-III Ref.
R3 = R5 = H, Alkyl, (Het)Aryl; R4 = R6 = H only
+ 1 eq. aldehyde + 1 eq. aldehyde
1
SB not detected: OX species only Not observed Dominant DOABO stereochemistry r-1-c-R2-c-R3-c-R5
[1, 7-15]
R3,R4 = -(CH2)n-CHR7-(CH2)m-; R7 = H, Me, t-Bu; n + m = 3 – 5 only
+ 1 eq. cyclanoneb + 1 eq. cyclanone
2
SB not detected: OX species only Diastereomeric ring-ring
tautomerism if R7 H No DOABO species observedc
[1c, 1d, 8,
16, 17a]
R3 = R4 or R3 R4 alkyl
+ 1 eq. ketone + 1 eq. ketone
3
SB not detected: (unstable) OX species
only
Not observed No DOABO species observedc
R3 = R4 or R3 R4 alkyl; R3,R4 = -(CH2)n-CHR7-(CH2)m-; R7 = H, Me, t-Bu, n + m
= 3 – 5 R5 = Aryl, H; R6 = H
[8]
+ 1 eq. (cyclan)one + 1 eq. aldehyde
4
SB not detected: OX species only Diastereomeric ring-ring
tautomerism if R7 H Dominant DOABO stereochemistry
r-1-c-R2-c-R5
[8, 15a, 16]
aAccording to literature, reactivity of the parent term, serinol S, is the less documented. bNo data with respect to arylalkylketones. cOur unpublished data.
Table 2. Basic Reactivity of C-1-substituted Serinols D, E with Carbonyl Electrophiles in Simple Ring-Chain or Ring-Ring Tautomer-ism Conditionsa
Entry EQ-I EQ-II EQ-III Ref.
R3 = R5 = H, Alkyl, (Het)Aryl; R4 = R6 = H
+ 1 eq. aldehyde + 1 eq. aldehyde
1
Ring–Chain tautomerism if R3 = (Het)Aryl, R4 = H
Both SB and OX species are detected
R3 = R4 = H; R3 = alkyl, R4 = H
Not observed
Not observed
Idem
Dominant DOABO stereochemistry r-1-c-R1-c-R3-c-R5
Idem
[18-20]
R3,R4 = -(CH2)n-CHR7-(CH2)m-; R7 = H, t-Bu; n + m = 3 – 5 only
+ 1 eq. cyclanone + 1 eq. cyclanone
2
SB not detected: OX species only Regioisomeric ring-ring
tautomerism
No DOABO species observed
[18a, 18d, 19a, 20d, 21b-e]
R3 = R4 or R3 R4 alkyl
+ 1 eq. ketone + 1 eq. ketone
3
SB not detected: (unstable) OX species only Regioisomeric ring-ring
tautomerism
No DOABO species observed
R3,R4 = -(CH2)n-CHR7-(CH2)m-; R7 = H, t-Bu R5 = Aryl, H; R6 = H
+ 1 eq. cyclanone + 1 eq. Arylaldehyde
4
SB not detected: OX species only Regioisomeric ring-ring
tautomerism
No DOABO species observed
aNo data with respect to arylalkylketones.

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 123
NOO
CH2OH
R R
NOO
CH2OH
R R
R SR* R*
s r
88
5
2
2
O
HN
C(CH2OH)3
O NH
OH
CH2OH
CH2OH
1Ca (SB)(in solid state as
well as in solution)
1Ca (OX)(unknown)
1
3C-"all cis" 3C-"trans"(meso form) (chiral form)
2Ca
3Ca
3Cb
3Cc
3Cd
3Cf
3Cg
3Ch
R
H
Ph
p-O2N-Ph
o-O2N-Ph
Ph-CO
Ph3-CH2
4-Py
5-nitrofurane-2-yl
Yield (%)
90
90
91
77
52
85
65
90
"all cis" : "trans"
-
87:13
0:100
78:22
95:5
95:5
27:73
0:100
O NH
(Het)Ar
CH2OH
CH2OH(Het)Ar (yield, %) in series 1Cb-i (OX):
1Cb (OX) (Ph, 41); 1Cc (OX) (p-O2N-Ph, 90)
1Cd (OX) (p-MeO-Ph, 80); 1Ce (OX) (p-Cl-Ph, 89)
1Cf (OX) (p-F-Ph, 65); 1Cg (OX) (m-F-Ph, 62);1Ch (OX) (m-O2N-Ph, 90); 1Ci (OX) (N-methyl-
5-nitroimidazol-2-yl, 92)1Cb-i (OX)
Fig. (4).
H2N-C(CH2OH)3
i) 3.4 eq. NaH / DME / reflux / 12 hrs.ii) 3.4 eq. Cl-CH2-O-CH2-CH2-OCH3 / r.t. / 12 hrs.
H2N
O
O
O OO
O
O
O
O86%
N
O
O
O OO
Lipophilic DOABO type derivative
2Cb
C
Fig. (5).
H2N
H2N N
N
OH
OH
O
O
H
H
NaIO4 / H2O pH = 3.5 / r.t.
H2N NH
CH(OH)2
O i) H2N-C(CH2OH)3 (C) / pH = 8.5
ii) CH3-(CH2)14-CO-O-Su* / t-BuOH / r.t.
N
O
O
CH2OH
H3C-(CH2)14-CO-HNHN
O
H3C-(CH2)14-CO-HN NH
O
N
O
O
CH(OH)2
H3C-(CH2)14-CO-HNHN
O
H3C-(CH2)14-CO-HN NH
ODess-Martin
periodinane
DCM / r.t. / 1h
58%
*CH3-(CH2)14-CO-O-Su: palmitic acid succinimidyl ester Amphiphilic aldehyde for the membrane immobilisation of unprotected peptides through hydrazone ligation
3Cj3Ci
81%
Fig. (6).
In reaction with excess of more electrophilic (het)aryl(alkyl)aldehydes, DOABO derivatives of type 2 and
3 are obtained, (Fig. (3, 4)), in a one-pot synthesis [1, 7-15] (Table 1, entry 1).
The dominant so called by us “all cis” diastereoselectiv-ity of this double cycloaminalisation (EQ-I, EQ-III, Fig. (3)) was supported by our DFT calculations [15a, 15f], (high-resolution) NMR experiments [8, 15a, 15f] and mo-lecular determined structures [13a, 13b, 15f-h]. They all re-vealed the cis-fused construction of the DOABO-skeleton, hence a heterofacial molecule [17b]. In contrast, the reason for the reversed diastereoselectivity observed in the synthesis of DOABO derivatives 3Cb, g, h is still not explained.
DOABO derivatives of C-2-substituted serinols A-C are also known as potent biocides and are used to prevent bacte-rial, fungal and microbial growth [(e.g. compound 2Ca, Fig. (4))] [12]. In this purpose, particular methodologies, focused on double cycloaminalisation of TRIS (C) with masked al-dehydes in basic conditions, were also reported, for example DOABO polyether 2Cb (Mattson and Norin, 1994 [12b]), (Fig. (5)), trans-alcohol 3Ci and trans-hydrated aldehyde 3Cj (Melnik et al., 2001 [14]), (Fig. (6)).
The spontaneous isomerisation implying the spiranic car-bon in spirooxazolidines OX, obtained from serinols A-C in reaction with some alicyclic ketones (Table 1, Entry 2, n + m = 4, Fig. (3)), EQ-II), we entitled, in 2000, ring-ring

124 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
tautomerism since, as in series 1Cb-i (OX), no tautomeric SB form can be observed on the 1H-NMR time scale (300–600 MHz) (Table 1, entry 2, Fig. (3)), EQ-I) [17a]. Depend-ing on the alicyclic ketone, the ring-ring tautomerism dis-closed a diastereomeric (cis trans) or homomeric OX
OX interconversion, spontaneous equilibria being solvent dependent (Fig. (7)).
For practical synthetic interest, shifting and blocking these equilibria was relevant when spirooxazolidines of type 4, (Fig. (7)), were treated with another equivalent of a car-bonyl electrophile such as an (aryl)aldehyde (Table 1, entry 4). Indeed, the excellent diastereoselectivities obtained, as deduced from NOE-Experiments [15a], we explained, again, by the cis fused shape of the resulting DOABO skeleton im-
posing a dominant “all cis” stereochemistry of the ligands at positions C-5, -8.
We note the order in which the carbonyl electrophiles should be used for the synthesis of this kind is successful [8, 15a, 17a]: i) the cyclanone, then ii) the (aryl)aldehyde. The reversed option would results in difficult to separate mix-tures of OX and DOABO species, based mainly on the (aryl)aldehyde (Fig. (3), EQ-III, Table 1, entries 3, 4).
We also note that, even with excess of a C5 – C7 cycla-none, cycloaminalisation of TRIS (C) is totally chemioselec-tive since just a single oxazolidine ring closure occurs. That is, double spiranic DOABO type structures are still unknown as well as trioxaazadispiranes (Table 1, entry 3), (Fig. (8)).
R1
NH
O
CH2OH
CH2OHN C(CH2OH)3
R1678
O
HN
R1
HOH2C CH2OH
Yield (%) Diastereomeric ring-ring tautomerism
20 4Ca-c (R1 = 6-Me-c) : 4Ca-t (R1 = 6-Me-t) = 100:0 ([D6]DMSO) 90:10 (CDCl3)
50 4Cb-t (R1 = 7-Me-t) : 4Cb-c (R1 = 7-Me-c) = 85:15 ([D6]DMSO) 82:18 (CDCl3)
80 4Cc-c (R1 = 8-Me-c) : 4Cc-t (R1 = 8-Me-t) = 60:40 ([D6]DMSO) 61:39 (CDCl3)
80 4Cd-c (R1 = 8-t-Bu-c) : 4Cd-t (R1 = 8-t-Bu-t) = 100:0 ([D6]DMSO) 75:25 (CDCl3)
4Ca-c; 4Cb-t; 4Cc-c; 4Cd-c; 4Ce 4Ca-t; 4Cb-c; 4c-t; 4Cd-t; 4Ce
1
1
678
+ 1 eq. R2-CH=O / H+ /
thermodynamic controll NOO
CH2OH
(m-, p-O2N)
NOO
CH2OH
R2
t-Bu
NOO
CH2OH
R2
t-Bu
13 73
17
5Ca-c* (R2 = H, 94%) 5Ca-t* (R2 = H, 6%) 5Cc (m-O2N, "all cis", 100%)
5Cb-c (R2 = n-C6H13, 100%) 5Cd (p-O2N, "all cis", 100%)
55-77%
*O-3 and t-Bu as references for the descriptors c(cis) and t(trans)
H2N-C(CH2OH)3 (C)Toluene / H+
Dean-Stark trap
(O-1 and R1 as references for the descriptors c(cis) and t (trans)
A value NH(eq.) vs. O-1(ax) = -2.72 kJ/mol [17a])
Homomeric ring-ring tautomerism (obscured by the conformational equilibrium)
86 4Ce (R1 = H) : rigid carbocycle in ([D6]DMSO) flipping carbocycle in (CDCl3).
5 5 75
8 8 84Cd or 4Ce
2 2 23
O
R1
Fig. (7).
O NH
CH2OH
CH2OH
( )n
n = 0 - 3
O ( )n
NOO
( )m( )n
CH2OH
n m or
n = m = 0 - 3
O
NH
O
O
( )n ( )m
n m or
n = m = 0 - 3
(MeO)3P
TolueneReflux / 6 hrs
O
NH
P-OCH3
O
O 90%
378 6
1
n = 1
5
14
6C
Fig. (8).

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 125
Solely in 1979, Sokolov et al. [16b] reported, besides spi-rooxazolidines of type 4C, (Fig. (7)), the synthesis of the trioxaaza-3-phosphapentadecane 6C whose identity was supported by elemental analysis, IR and MS spectra (Fig. (8)).
1.2.3. C-1-substituted Serinols Based 1,3-oxazolidines Systems
In C-1-substituted serinols series D, E, their mono-condensation products with aryl aldehydes had been known as authentic Schiff-Bases [SB, (Fig. (3)), (Table 2, entry 1)], both in solution and in solid state, since the period of the 1950’s [3, 18a-d] (UV and IR data) . Later investigations (1990) on these compounds went ahead from the same clas-sical assumption [18e, 18f].
Using high-resolution 1H-NMR analysis, we evidenced, in 1997 [19b], (Fig. (9)), the incidence of the ring-chain tautomerism SB OX [(Fig. (3)), EQ-I] of SB derivatives of serinol E and exploited it in synthesis [19c]. A related study focused on Schiff-Bases of phenylserinols D and D’
However, as early as 1956, Pedrazzoli and Tricerri expressed their doubts
[18d] on the real structure of these SB derivatives originating from of seri-nols D, E with respect to a possible occurrence of an EQ-I isomerisation (Fig. (3)); see also Bergmann’s 1953 review [1e] as well as Bergman and Resnick work [18a] for pioneering IR assignments.
was published by Fülöp et al. one year later [19d] which also exhaustively reviewed this phenomenon in 2003 [5].
Thus, at room temperature, the five terms slow equilibria involving 1D-E (SB) (100% E geometry) and two regioiso-meric 1D-E (OX-prim., -sec.) species, the latter as pairs of epimers at C-2, (Fig. (9)), could be 1H-NMR monitored. Hence, a linear correlation between log KX values of the equilibria SB OX (KX = [OX]/[SB]) and the Hammet-Brown parameters + of the substituents X on the C-2-aryl ring, log KX = + + KX = H, [19e-i], was found (see Table 3 for selected examples).
Resuming this versatile autoreactivity for diastereoselec-tive synthesis needs only, one can see that, when equilibria were established, (Fig. (3), EQ-I, Table 3), their composi-tions strongly depended on the configuration at positions C-1, -2 of phenylserinols E, D and D’ and aryl p-substitution X of their Schiff-Bases [19b, 19d]. Thus, no diastereoselectiv-ity regarding the configuration at the C-2 position in 1E (OX-sec.) followed the isomerisation of 1E (SB), but an almost complete regioselectivity as 1E (OX-sec.). In con-trast, although globally more shifted towards OX species, the equilibria showing the isomerisation of epimeric 1D, D’ (SB) derivatives was not regioselective (OX-sec. vs. OX-prim.) but somehow diastereoselective with respect to the configuration at OX C-2 position (italicised data in Table 3).
OH OH
N
Ar1 12 3
Ar2
S
D: Ar1 = Ph; (1S,2S) - "threo" series 1D
D': Ar1 = Ph; (1R,2S) - "erythro" series 1D'
E: Ar1 = p-NPh (1S,2S) - "threo" series 1E
O NH
CH2OH
Ar2
Ar1
45
2HN O
Ar2
Ar1
HO
2
4
Ar2 (X-C6H4, if suitable for Hammet-Brown monitoring) :
X = p-Me2N, p-MeO, p(m)-HO, p(m)-Me, H, p-F,
p-Cl, m-Br, p(m)-O2N
Ar2 if unsuitable for Hammet-Brown monitoring :
2(3)(4)-Pyridyl, o-Cl-Ph, o-O2N-Ph, -naphthyl
Ring-Chain tautomerism
8 eq. Ac2O
r.t.O N
CH2OAc
Ph
2
p-NPh
O
CH3O N
CH2OAc
Ph
2
p-NPh
CH3
O
1E (OX-sec.)-Acanti-2R,4S,5S syn-2R,4S,5S
59%
O N
CH2OH
Ph
p-NPh
Ph
2 eq. Ph-CHO
Benzene / H+ /
reflux / 8 hrs.
OH55%
O N
CH2OH
Ph
p-NPh
Ph
+N
OO
Ph Ph
Hp-NPh
123
4 5 6
78
3Ea
"all cis": 1R,2R,4S,5S,8S
45:55
p-NPh
NOO
R R
H
123
4 5 6
78
R (yield %): H (92, 2Ea); p-O2N-Ph (11, 3Eb); o-O2N-Ph (66, 3Ec); o-Cl-Ph (40, 3Ed); -naphthyl (22, 3Ee);
2-Py (61, 3Ef); 4-Py (62, 3Eg); 3-Py (67, 3Eh); Bn (50, 3Ei), i-Pr (63, 3Ej) [19b]; p-Me-Ph (89,
3Ek); p-MeO-Ph (49, 3El) [20f]
2Ea, 3Ea-l1E (OX-sec.)-Am 1E (OX-sec.)-Bcat
E
1D-E (OX-prim.) 1D-E (SB) 1D-E (OX-sec.)
1E (SB)
1E (OX-sec.)
1E (OX-prim.)
(Ar2 = Ph)
R R
R
4S 5S 4S 5S
Fig. (9).

126 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
A dominant cis relationship between ligands at C-2, -5 [in 1D, D’ (OX-sec.)] or C-2, -4 [in 1D, D’ (OX-prim.)] can be observed. As in the case of TRIS (C) and for the same rea-son, (Fig. (4)), the Schiff-Base of serinol E with salicyclic aldehyde exhibited no ring-chain tautomerism [18e, 19b].
Surprisingly, despite the lack of any OX-diastereoselectivity in 1E (SB) derivatives cyclisation (Table 3), when a second oxazolidine ring closure we attempted by employing the same (het)aryl aldehyde, C-1, -2, -8 trisubsti-tuted DOABO derivatives 3Ea-l were obtained with com-plete “all cis” (1R,2R,4S,5S,8S) stereochemistry (Fig. (9)) [19c]. The same was valid in reactions with formaldehyde (2Ea) and (aryl)alkyl aldehydes (3Ei, j) [19c, 20f, 20g]. A basic prerequisite for a successful synthesis of compounds 3E was, however, that, in the initial Schiff-Bases of E, sub-stituents X had positive + values and / or promoted KX = [OX]/[SB] values more than 0.200. Substituents X with negative + values caused the failure of the next ring-closure. Thus, starting from 1E (SB), no DOABO structure was ob-tainable if, in Ar2 moiety, X was p(o)-HO and p-Me2N but (unreacted) Schiff-Bases of E. Rationalisation of the above complete DOABO diastereoselective preparation we found, once more, in the cis-fused shape of the bicyclic skeleton, deduced from our NOE experiments [19c] in agreement with later AM-1 calculation (Zaitsev et al., 2006 [20f]) and mo-lecular determined structure (Zaitsev et al., 2006 [20g]. For an enlarged pharmaceutical interest in the synthesis of DO-ABO type compounds 3E of nitrophenylserinol E and re-lated structures, see Ref. [20].
On the other hand, in an earlier approach by us [19c], (Fig. (9)), we considered the second cycloaminalisation, for example that of 1E (OX-sec., Ar2 = Ph), as normally occur-ring via a hemi-aminal, 1E (OX-sec.)-Am, providing the stabilised benzyl carbocation 1E (OX-sec.)-Bcat. This in-termediate imposed the oxazolidine nitrogen to be involved in a partial double bond and, at this step of the reaction, (Am
Bcat), the adoption of a unique absolute configuration at
C-2 (S R) was required. Indeed, the parallel preparation of a ”model compound”, N-Acetyl-oxazolidine 1E (OX-sec.)-Ac, yielded, at room temperature, the almost equally populated mixture of its two blocked rotamers anti and syn, possessing the oxazolidine nitrogen involved in a partial double-bond but with the same R-configuration at C-2.
Finally, both us [19c] and Zaitsev et al. [20f] reached the same general conclusion, namely that the double cycloami-nalisation of p-nitrophenylserinol E with arylaldehydes should be performed under thermodynamic control, the “all cis” 3E (1R,2R,4S,5S,8S) diastereomer, (Fig. (9)), being the most stable from all four possible [not depicted in Fig. (9)]. Nevertheless, for the unexpected as versatile rearrangement of this preferred DOABO skeleton geometry, see later dis-cussion in Fig. (39) [27e].
Completely different from C-2-substituted serinols A-C series, (Fig. (7)), (Table 1, entry 4), DOABO derivatives of C-1-substituted serinols D and E bearing different substitu-ents at C-2, -8, (e.g. when operating with two different alde-hydes), are not known. That is, reaction of Schiff-Bases of D and E (Fig. (9)) with 1 eq. of a different aldehyde provided either the unreacted SB, if the subsequent aldehyde was less electrophilic than that already included in the starting mate-rial, or a double transaminalisation if the succeeding alde-hyde was more electrophilic than the previous one [21f]. In this last situation, rather complex DOABO reaction mixtures were formed. Consequently, no reaction occurred if ketones were used as electrophiles against Schiff-Bases of D and E.
As shown in Fig. (10), solely phosgene appeared to us a good subsequent ring-closure electrophile with respect to tautomeric equilibria involving Schiff-Bases of D and E, yielding optically pure 6,8-disubstituted-3,7-dioxa-1-azabicyclo[3.3.0]-octane-2-ones 7Da, 7Ea with complete regioselectivity but medium yields and diastereoselectivity concerning the “all cis” rule [21f].
Table 3. Tautomeric Ratios of Ring-Chain Tautomerism Five Component Equilibria of Some Schiff-Bases 1D-E (SB) Derived from
Serinols D, D’ and E (see Fig. (9))
Phenylserinol D (Ar1 = Ph) Phenylserinol D’ (Ar
1 = Ph) Phenylserinol E (Ar
1 = p-NPh)
X
(+) SB
OX-
prim.a
(S)c
OX-
prim.
(R)d
OX-
sec.b
(S)
OX-
sec.
(R)
SB
OX-
prim.
(S)
OX-
prim.
(R)
OX-
sec.
(S)
OX-
sec.
(R)
SB
OX-
prim.
(S)
OX-
prim.
(R)
OX-
sec.
(S)
OX-
sec.
(R)
p-O2N
(+0.79) 12.7 13.6 30.8 9.3 33.6 29.4 21.8 13.8 21.4 13.5 60.0 2.5 2.5 17.5 17.5
H
(0.00) 23.2 11.6 23.3 4.1 37.7 57.8 12.3 8.0 13.6 8.3 77.0 0 0 11.5 11.5
p-Me
(-0.31) 30.9 11.8 19.6 3.4 34.2 67.4 9.6 6.4 10.6 6.0 82.0 0 0 9.0 9.0
p-NMe2
(-1.70) 75.2 4.5 6.4 1.3 12.6 90.6 2.6 2.2 2.7 1.9 94.0 0 0 3 3
aOxazolidine ring closure with the primary hydroxyl group of SB. bOxazolidine ring closure with the secondary hydroxyl group of SB. cR configuration at C-2 oxazolidine carbon; dS configuration at C-2 oxazolidine carbon.

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 127
For related regioselective as 1,3-oxazolidinone ring clo-sure of p-nitrophenylserinol E, see Ref. [20e, 20h].
The regioisomeric ring-ring tautomerism exhibited by phenylserinols D and E based spirooxazolidines, (Table 2, entry 2, Fig. (3), EQ-II), (Fig. (11)), we observed and enti- tled as such in 1998 [21b], then re-examined in 2005 [21c] by starting from the pioneering structural assignments of Bergmann and Resnick on this class of compounds in 1956 (IR and UV data) [18a] . Shan et al. [21d, 21e] independ-
According to Bergmann and Resnick, ring closure largely dominant as OX-
sec. (IR spectra) was explained by an acidic autocatalysis promoted by the
ently reported similar results in the period of 2002-2003 as synthesis, stereochemistry and DFT calculation.
Thus, in cycloaminalisations of D and E, (Fig. (11)), car- ried out with cyclohexanone or 4-tert-butylcyclohexanone, 1-oxa-4-azaspiro[4.5]decanes 4D, E, a, b were isolated with medium to good yields as exclusive OX-sec. regioisomers,
most acid hydroxyl proton of E, the secondary one [18a]; we reached the same conclusion but in ring-chain tautomerism of SB derivatives of E (Fig. (9)) [19b]. Isomerisation also valid for derivatives of cyclopentanone, acetone and
butanone; they are less documented in Ref. [21d].
OH OH
N
Ph
Ar O NH
Ph
Ar CH2OH
NOO
Ar H
OPh
1 25 3
467
8 NOO
Ar H
OPh
1 25 3
46
7 8
cis (-c) trans (-t)
7Da-c 66 (45%*) 7Da-t 33 (22%)7Ea-c 85 (50%) 7Ea-t 15 (9%)
i
i: 1.2 eq. COCl2 / 2.5 eq. Et3N / THF / - 20 oC to r.t.
12 hrs; *isolated yields
Ar = Ph: 1D (SB)Ar = p-NPh: 1E (SB)
1D, E (OX-sec.)
Fig. (10).
OH OH
NH2
ArH+ / Aromaticsolvent / reflux18-21 hrs.
OR1 eq.
O NH
Ar CH2OH
R
HN O
Ar
HO
R
12 4
8
1
23
3
4
8
(R = H, t-Bu)
R = H: 4Da, Ea (OX-sec.) 4Da, Ea (OX-prim.)R= t-Bu: 4Db, Eb (OX-sec.)-c 4Db, Eb (OX-prim.)-c 4Db, Eb (OX-sec.)-t 4Db, Eb (OX-prim.)-t*O-1 (axial or equatorial) and R = t-Bu (equatorial) as references for the descriptors c(cis) and t(trans) respectively
78-88%
Ar = Ph (D)Ar = p-NPh (E)
Equilibrium Tautomeric Ratios According to the NMR solvent
[D6]
DMSO
[D6]
Acetone
[D8]
THF
[D3]
Chloroform
[D6]
Benzene
4Da (OX-sec.) (OX-prim.) 91:9 93:7 91:9 84:16 88:12
4Ea (OX-sec.) (OX-prim.) 90:10 93:7 91:9 83:17 84:16
4Db (OX-sec.)-c
4Db (OX-sec.)-t
4Db (OX-prim.)-c
4Db (OX-prim.)-t
100
0
0
0
94
0
6
0
100
0
0
0
68
13
14
5
90
7
3
0
4Eb (OX-sec.)-c
4Eb (OX-sec.)-t
4Eb (OX-prim.)-c
4Eb (OX-prim.)-t
50
50
0
0
46
46
4
4
45
45
5
5
41
41
11
7
37
37
13
13
Fig. (11).

128 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
clearly recognized as such if NMR spectra were recorded in [D6]DMSO [21b, 21c]. In other NMR solvents, quite similar to spirooxazolidines derived from C-2- substituted serinols 4C, (Fig. (7)), compounds 4D, E spontaneously isomerised on the NMR time scale as [OX-sec.] [OX-prim.], spi-rooxazolidines 4D, E (OX-sec.) remaining, however, major regioisomers. As shown by the 1H-NMR calculated composi-tions, (Fig. (11)), the ring-ring tautomerism was not only regioisomeric but, in the case of 4-tert-butylcyclohexanone derivatives 4Db, also cis trans diastereomeric.
On the other hand, one must distinguish the complete di-astereoselectivity in the synthesis of compound 4Db (OX-sec.), [100% 4D (OX-sec.)-c, in [D6]DMSO], meanwhile, if the process commenced from serinol E, the equimolar ratio between diastereomeric 4Eb (OX-sec.)-c and 4Eb (OX-sec.)-t, kept unaffected in all NMR solvents (Fig. (11)), was isolated. The single reason we found for this dissimilar re-sults consisted of the intrinsic nature of the benzyl S chiral centre in phenylserinols D and p-nitrophenylserinol E, epi-merisable in D but non epimerisable in E, (Fig. (12)) [21c].
Taking into account the thermodynamic conditions of the synthesis, (Fig. (11)), we postulated that there were, in fact, two different mechanisms of the above cycloaminalisation:
i) Via a Benzyl type carbocation 4Db-Bcat, originating from D, followed by a diastereoselective trans oxazolidine
ring closure, with respect to the ligands at C-2, -3, as 4Db (OX-sec.)-t.
ii) Through an Imonium cation 4Eb-Im originating from E, allowing the free rotation about the serinolic bond C-1-C-2, hence the nucleophilic attack of the secondary hydroxyl group of E, with equal probability, on both diastereotopic faces of the iminium double bond.
Upon treatment with a subsequent carbonyl electrophile [aldehyde or (alicyclic)ketone], (Table 2, entries 3, 4), in reaction with aldehydes, spirooxazolidines 4D and 4E un-derwent rather transaminalisation, yielding the previously discussed DOABO type 3 derivatives (Fig. (9)). No reaction occurred with (alicyclic) ketones (see, for comparison, Fig. (8)). As for the Schiff-Bases of D and E, (Fig. (10)), single phosgene was a successful electrophile able to convert com-pounds 4Da, b and 4Ea, b (OX-sec.) in cis-fused DOABO-2-one structures, the (1S,5S,6S)-spirooxazolidinones 7D, E, b, c, with satisfactory yields and complete regioselectivity (Fig. (13)) [21f].
In a more efficient approach, Shan et al. [21d] used spi-rooxazolidine 4Ea (OX-sec.), (Figs. (11, 14)) in a very sim-ple and efficient method for the kinetic resolution of racemic (1,1’-binaphthalene)-2,2’-diol, (rac-BINAPOL) with 100% e.e. As depicted in Fig. (14), the proposed chemistry consid-
t-Bu
O
HO
HO
NH2
Arequatorial
attack t-BuHN
OH
HOH2C
Ar OH
Ar = Ph (D)
t-BuHN
OH
HOH2C
Ph+
+ H+ / -H2O
Ar = p-NPh (E)
+ H+ / -H2O
t-BuHN
O
p-NPh
HOH2C 1
4
+ H+ / -H2O
t-BuHN
O
Ph
HOH2C 1
4
4Db (OX-sec.)-c - H+
- H+t-Bu
O
NHp-NPh
HOH2C
1
4
23
4Db-Bcat
t-BuHN+
HOH2C
p-NPh
OH
t-BuHN+
HOH2C
p-NPh
OH
4Eb-Im
4Eb-Im
1
2
4Eb (OX-sec.)-c4Eb (OX-sec.)-t
Fig. (12).
O NH
Ar CH2OH
R
R = H4Da (OX-sec.), 4Ea (OX-sec.)
R = t-Bu 4Db (OX-sec.) 4Eb (OX-sec.) (50:50 -c:-t)
1.2 eq. COCl2Et3N / THF /
- 20 oC r.t.
12 hrsN
OO
O
Ar H
R
1357
2
6
R = HAr = Ph, 7Db (60%*)Ar = p-NPh, 7Eb (63%)
R = t-BuAr = Ph, 7Dc-c** (63%)Ar = p-NPh, 7Ec-c : 7Ec-t (50:50) (48%)
*isolated yields; **R = t-Bu (equatorial) and O-7 (axial or equatorial) as references for descriptors c (cis) and t (trans) respectively
8
Fig. (13).

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 129
ered 4Ea (OX-sec.) as the single worthy mentioning regioi-somer.
1.3. Double Ring-Ring and Ring-Chain Tautomerism
1.3.1. 1,3-Oxazolidine Systems Based on Reactivity of C-2-
Substituted Serinols with 1,4-dicarbonyl Electrophiles in
Double Ring-Ring Tautomerism Conditions
Although well established and recently reviewed by Lázár and Fülöp [5], since only we were interested so far in the double ring-ring or double ring-chain tautomerism of the
condensates of serinols A-E with appropriate 1,4-dicarbonyl electrophiles and attempted to use it in synthesis [15a, 15b, 15f, 17a, 21b, 21c, 21f], this complex dynamic behaviour will be described only briefly.
Thus, dispirooxazolidines 8A-C, (Fig. (15)), were syn-thesised by double cycloaminalisation of 1,4-cyclo-hexandione with C-2-substituted serinols A-C with good yields [17a]. In [D6]DMSO, NMR detection of compounds 8A-C showed them to be unique and stable trans-diastereomers with respect to the carbocycle. In addition, if
OH
OH
rac-BINAPOL B(OH)3 4Ea (OX-sec.) 2 : 1 : 1
+ B(OH)3NHO
CH2OHp-NPh
+THF / reflux6 hrs.
O
O
O
O
BNH2
+O
CH2OHp-NPh
-
as adduct of (R)-BINAPOL : 4Ea (OX-sec.)
i) 1.2 N aq. HCl ii) crystallisation from toluene
OH
OH
i) evaporation of THF mother liquor ii) crystallisation from toluene
HO
HO
(S)-BINAPOL (R)-BINAPOL 61.5% yield 68.4% yield 100% e.e. 100% e.e.
precipitate
filtrate
Fig. (14).
H2N-CR(CH2OH)2
R = Me, A series AR = Et, B series BR = CH2OH, C series C
0.5 eq.
Toluene / reflux18-21 hrs.
75-81%
O
NH
O
HN
CH2OH
R
R
HOH2C
3
11
S
R
1
9
O
NH
O
HN
R
CH2OH
R
HOH2C
3
11R*
1
9
8A-, B-t*-u**; 8C-t 8A-, 8B-t-l
(meso form) (chiral form, racemate)(100% t-u in [D6]DMSO]) (0% t-l in [D6]DMSO)
(65% t-u in [D3]chloroform) (35% t-l in [D3]Chloroform)
2.1 eq. CH2O / H+
Aromatic solventReflux / 8 hrs.
O
N
O
NO
O
R
R
12
34
56
78
1'2'
3'4'
5' 6'
7'8'
O
N
O
NO
O
R
R
12
34
56
78
1'2'
3'4'
5' 6'
7'8'
9A-C-u (1S,5R,1'R,5'S) 9A-C-l (1S*,1'S*,5R*,5'R*)(meso form) 9A (31%*, 1:1 u:l) (chiral form, racemate) 9B (33%, 9:1 u:l) 9C (70%, 1:1 u:l)*isolated yields
YX
YX
CD
CDB
AA
B
XY
YX
DC
CDA
BA
B
R
R
R
R
unlike (u)(meso form)
1 (one) AA'XX' system (32 signals)*
2 (two) AX system (8 signals)*
like (l)(chiral form)
*nuclei labelled with the same letter are enantiotopic in unlike (u) (meso form and homotopic in like (l) (chiral form).
R*
R = Me, Et
NMR discrimination
between u vs. l forms
1,4-cyclohexandione
*t(trans) geometry with respect to oxazolidine oxygens O-1, -3.** for 8Aa and 8Ba: u(unlike), l(like) with respect to relative configurations at C-3, -11
Fig. (15).

130 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
the starting serinols had hydroxymethyl groups enantiotopic, as in A and B, the diastereomers were t-u (trans-unlike), meso forms, 8A-, 8B-t-u (see inset in Fig. (15)).
Dispirooxazolidines 8A-t and 8B-t revealed their double ring-ring tautomerism to be diastereomeric unlike like, spontaneous and solvent dependent, e.g. [D6]DMSO vs. [D3]Chloroform (Fig. (15)). Upon ensuing treatment with formaldehyde, tautomeric equilibria were shifted and blocked: equimolar mixtures of like-unlike dispiranic double DOABO systems 9A, C were obtained with moderate yields, unlike-diastereoselectivity being found only for serinol’ B
derivative, 9B.
1.3.2. 1,3-Oxazolidine Systems Based on Reactivity of C-1-
substituted Serinols with 1,4-dicarbonyl Electrophiles in Double Ring-Ring Tautomerism Conditions
On the contrary, in identical synthetic conditions, the double ring-ring tautomerism manifested by the dispirooxa-zolidines of phenylserinols D and E, 8D, E, (Fig. (16)), was both cis trans diastereomeric with respect to the cyclo-hexane ring and, depending on the hydroxyl group impli-cated in cyclisation, regioisomeric, OX-prim. OX-sec. [21b, 21c]. The six tautomers in series 8D, E could be de-duced successively, each of them been generated by a single isomerisation / equilibrium (regioisomeric or diastereo-meric). As already expected, equilibria were established spontaneously, solvent dependent and strongly shifted to-wards tautomers 8D-, E-t (OX-sec.-sec.). Tautomeric com-
positions could be calculated enough simply since 1H-NMR lines of certain protons, mainly benzyl (intra- vs. exocyclic) and hydroxyl (primary vs. secondary) were adequately sepa-rated. Moreover, 13C-NMR spectra, revealing the flipping or frozen nature of cyclohexane ring, allowed a rapid discrimi-nation between series of tautomers 8D, E-c against 8D, E-t respectively. Alike the monospirooxazolidines 4D, E, (Fig. (13)), only if phosgene was used as electrophile, shifting and blocking ring-ring equilibria of 8D, E were possible, with medium yields and total regio- and diastereoselectivity, (Fig. (16)), supported by NOE Experiments and the molecular determined structure of dispirooxazolidinone 10E [21f].
1.3.3. 1,3-Oxazolidine Systems Based on Reactivity of C-substituted Serinols with 1,4-dicarbonyl Electrophiles in
Double Ring-Chain Tautomerism Conditions
The incidence of the double ring-chain tautomerism of condensates formed in reaction between all the family of serinols A-E and terephthaldicarboxaldehyde we examined in 2002 comparatively [15b], (Fig. (17)), and attempted to utilise it in synthesis [15f, 21f].
In this purpose, combined techniques, IR (solid state) and NMR ([D6]DMSO) were exploited.
In C-2-substituted serinols series A-C, in solid state, only the double Schiff-Base of A, A (SB-SB) could be assigned unambiguously. Its evolution in solution (5 hrs.) indeed started from A (SB-SB) and reached slowly the equilibrium
OH OH
NH2
Ar
Ar = Ph, D series DAr = p-NPh, E series E
0.5 eq.
Toluene / H+
Reflux / 18-21 hrs.
79% (D), 85% (E)
O
NH
O
HN OH
Ar
HO
Ar
HN
O
O
HNHO
Ar
OHAr
O
NH
O
HN OH
Ar
Ar
HOH2C
HN
O
O
HN
OHAr
Ar
HOH2C
O
NH
O
HN
Ar
HOH2C
HN
O
O
HN
Ar
HOH2C
Ar
CH2OH
Ar
CH2OH
8D-, E-t* (OX-prim.-prim.**)
8D-, E-t (OX-sec.-prim.)
8D-, E-c (OX-prim.-prim.) 8D-, E-c (OX-sec.-prim.)
8D-, E-t (OX-sec.-sec.)8D-, E-c (OX-sec.-sec.)
*oxazolidine oxygens as references for the descriptors t(trans) or c(cis)** "prim." or "sec." with respect to the serinolic hydroxyl group (primary or secondary respectively) involved in cyclisation.
8D-, E-t (OX-sec.-sec.)
2.1 eq. COCl2 /
Et3N / THF /
- 20 oC r.t.
12 hrs.
O
N
O
N O
O
Ar
Ar
H
H O
O
(1S,1'S,5S,5'S,6S,6'S)10D (36%), 10E (45%)
16
5
1'
6'
5'
1,4-cyclohexandione
Fig. (16).

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 131
state, all expected type of species, A (SB-SB) A (SB-OX) A (OX-OX) being detectable by NMR. Very different, in
solid state, the condensate products of terephthaldicarboxal-dehyde with serinols B and C were the double oxazolidines B(C) (OX-OX) which, in solution, spontaneously provided the equilibrium states comprising B(C) (OX-OX) B(C) (SB-OX) B(C) (SB-SB) species. No change in composi-tion was observed.
Dynamic behaviour of the more conjugated D(E) (SB-SB) derivatives appeared accelerated with respect to that exhibited, in simple ring-chain tautomerism, by their mono Schiff-Bases with benzaldehyde (1D (SB), 1E (SB), Ar2 = Ph, Fig. (9)) since intermediates of type D(E) (SB-OX) were detected but in very small traces, unsuitable for pertinent assignments. The double ring-chain tautomerism of the dou-ble Schiff-Bases D(E) (SB-SB), identified as such in solid state, occurred almost spontaneously in the case of E (SB-SB) and about four times faster (24 vs. 105 hrs.) for D (SB-SB) vs. the isomerisation of its simple benzylideneamino analogue, 1D (SB) (Ar2 = Ph, Fig. (9)).
Attempts to shift and block the four terms equilibria in-volving species D(E) (SB-SB) and three epimeric D(E) (OX-OX), for example in reaction with phosgene, (Fig. (18)), provided, except complete regioselectivity, moderate stereoselective and quantitative results concerning oxazolidi-nones 11 and 12 [21f]
.
Similar to simple benzylideneamino derivatives’ behav-iour, 1D, E (SB), reactivity of double Schiff-Bases D(E) (SB-SB) with carbonyl compounds more electrophilic than terephthaldicarboxyaldehyde resulted in dominant double (trans)aminalisation, affording compounds of type 3 DO-ABO, (Fig. (9)). If less electrophilic, no reaction occurred [21f].
An essentially reversed situation we encountered when condensates of C-2
substituted serinols A-C with carbonyl electrophiles, series 4C (Fig. (7)), 8A-C (Fig. (15)) and (A-C) OX-OX (Fig. (17)), were treated with phos-gene. These attempts provided just complex reaction mixtures (our unpub-lished data).
OH OH
R1
NH2R2
R1 = H, R2 = Me, A series A
R1 = H, R2 = Et, B series B
R1 = H, R2 = CH2OH, C series C
R1 = Ph, R2 = H, D series D
R1 = p-NPh, R2 = H, E series E
2 eq. p-O=CH-C6H4-CH=Obenzene / (A, B), EtOH (C, D)i-PrOH (E) / reflux / 8-10 hrs.
92% (A), 59% (B), 87% (C)85% (D), 74% (E)
N
N
OH
OH
HO
HO
R1
R1
R2
R2
N
HO
HO R2
NH
OR2
CH2OH
R1 = H
R2 = Me (A)
Et (B)
CH2OH (C)
A-C (SB-OX)
[R2 = Me (A), Et (B): two epimers*)]
NH
O
O
HN
CH2OH
R2
HOH2C
R2
A-C (OX-OX)
[R2 = Me (A), Et (B): six epimers*
R2 = CH2OH (C): two epimers*]
R1 = Ph (D)
p-NPh (E)
R2 = H
O
HN
NH
O
R1
R1
CH2OH
HOH2C
D(E) (OX-OX) (three epimers*)
245 2' 5'
4'
A-E (SB-SB)
2'
4
24 4'
2
* ** **
*
*
Tautomeric Compositions (%)
[A-E (SB-SB) ] [A-E (SB-OX)] [A-E (OX-OX)]KI KII
Time (hrs.) KI KII Serinol
at “Time zero”a at equilibrium state
SB-SBb SB-OXc OX-OXc SB-SBb SB-OXc OX-OXc
A 82 18 0 36 47 17 5 1.31 0.36
B 13 30 57 13 30 57 - 2.31 1.90
C 0.25 14 85.75 0.25 14 85.75 - 56.00 6.13
D 100 0 0 70 0 30 24 - -
E 95 0 5 78 0 22 - - -
a"Time zero" defines the moment of the first display of the NMR spectrum. bIn some cases minor amounts of diastereomers E,Z and Z,Z (SB-SB) (5-10%) vs. E,E were detectable at “time zero” or at equilibrium state; the relevance of this isomerisation for the present discussion is still obscure. cAll type of mixture of epimers detectable on 300 – 600 MHz 1H NMR time scale.
Fig. (17).

132 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
Situation changed if the starting materials were the con-densates of terephthaldicarboxaldehyde with C-2-substituted serinols A-C (Fig. (19)) [15f, 15i].
Thus, in reaction with (aryl)aldehydes a-c, their initial complex epimeric mixtures, (Fig. (17)), gave separable “di-meric” DOABO of type 13 with satisfactory conversions together with transaminalisation “monomeric” DOABO side products (not shown in Fig. (19)).
One must differentiate the DOABO dimers 13 that they can exist both as meso (13-u, unlike series, Cs symmetric) or like forms (13-l, racemate series, centrosymmetric). In these situations, current NMR methods cannot distinguish between like vs. unlike global stereochemistry but the local stereo-chemistry of each DOABO unit, found, unsurprisingly, “all cis”. Diastereoselectivity results showed in Fig. (19), de-duced by 1H-NMR performed in the presence of a CSR (Chiral Shift Reagent), Eu(hfc)3 {europium tris[3-(hepta-fluoropropylhydroxymethylene)-(+)-camphorate), indicated an unlike long and total distance stereocontrol if a bulky sub-stituent (e.g. aryl) was present at the C-8, -8’-DOABO posi-tions (compounds 13Ab-u, 13Ac-u, 13Bb-u).
Nevertheless, in this analysis, the interaction between Eu(hfc)3 and the DOABO dimer of TRIS (C) 13Ca was ver-satile. Thus, in the presence of the CSR, only diastereomer 13Ca-l was assigned suggesting the existence of the above long distance stereocontrol even in the case of a small subse-quent electrophile, formaldehyde. This verdict was in obvi-ous contradiction with those taken for compounds 13Aa and 13Ba, identified as equimolar mixtures unlike-like. Con-versely, account been taken on the presence only in 13Ca-l of a typical chelatizing group, c-C-5-hydroxymethyl, we were suspicious that, actually, a rapid isomerisation of 13Ca-u into 13Ca-l in their initial equimolar mixture, (Fig. (19)), had occurred in the presence of Eu(hfc)3 (Fig. (20)) [15f].
As one can observe, the conversion of unlike into like global stereochemistry of 13Ca was dictated by the “all cis” local stereochemistry requirements in each DOABO unit. Thus, a stabilised iminium cation 13Ca-2, as a plausible open-chain intermediate, allowed the interaction with the rear nucleophile pro-R or front pro-S neither on its Re-face, nor on its Si-face respectively, in order to ensure the final DOABO-cis relationship between the phenylene ring and the pro-S ligand. Therefore, we believe that some free rotation about the bond N-Cbenzyl should occur, as in the benzyl car-
N
HO
HO
Ar
D(E) (SB-SB) 2
i: 2.6 eq. COCl2 / Py
0 oC / 4 hrs. / r.t. / 24 hrs.
NOO
Hp-NPh
O
O=HC
NOO
HPh
O
2
NOO
HPh
O
2
11E-t* (31%**)
12D-t (68%**) 12D-c (16%***)
*t(trans) or c(cis) with respect to the orientation of aromatic rings; **isolated yields; ***content inthe crude reaction mixture.
i
Fig. (18).
OH OH
NH2R1
R1 = Me, A
R1 = Et, B
R1 = CH2OH, C
i: 0.5 eq. p-O=CH-C6H4-CH=O
ii: 2.0 eq. R2-CH=O [R2 = H (a); Ph (b); p-NPh (c)]
Toluene / H+ / reflux / Dean-Stark trap
O N
O N
O
O
R1
R1
O N
N O
O
R1
O
R1
R2
R2
R2
R2
R*
S*
21 8
5'2'
1' 8'
1
1'
5 5
5'
2
2'
8
8'
R
SS
RR
S
R*
R*R*
S*
O NH
HN O
R1
CH2OH
CH2OH
R1
ii
A-C (OX-OX)
H
Ph
p-NPh
H
Ph
H
13Aa-u : 13Aa-l
13Ab-u : 13Ab-l
13Ac-u : 13Ac-l
13Ba-u : 13Ba-l
13Bb-u : 13Bb-l
13Ca-u : 13Ca-l
50:50
100:0
100:0
50:50
100:0
50:50
40
62
19
62
64
47
Productsmeso form : racemate Yield (%)R2R1
Me
Me
Me
Et
Et
CH2OH
i
13A-Cu (unlike) l (like)meso form racemate
Fig. 19
Fig. (19).

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 133
bocations 13Ca-1 and 13Ca-3. They also possess di-astereotopic faces. Only in this case, the two hydroxymethyl groups, having opposite prochiralities, could replace each other.
1.4. Revised Reactivity of Serinolic Oxazolidine Systems Against Brønsted and Lewis Acids – a Brief Survey in
Terms of Anomeric Effects on Ring-Chain and Ring-Ring Tautomerism
Actually, the above coordination ability of oxazolidine ring oxygens against metal cations was mentioned since 1945 by Senkus [1a]. He applied chemical methods to eluci-date the structure of some DOABO derivatives of serinols A-C by ring cleavage with Grignard reagents accessing more elaborated C-2-substituted serinols (Fig. (21)).
NOO
R1
R2 R2
R1 = Me, Et, CH2OH
R2 = H, n-Pr
i) n eq. R3MgX
(n = 2, 3) / Et2O / r.t.
ii) H2O
(50 - 82%)
NOHHO
R1
R3 R3
R2 R2
R3 = Et, n-Pr, n-Bu, Ph
Fig. (21).
In 1951, Pierce and Lunsford noticed an interesting ester-amide interchange when the ring-opening in alkaline and acidic two-step hydrolysis was performed with the O-protected DOABO compound 3Ca (Fig. (4)), 3Ca-OCOR in mild conditions (Fig. (22)) [1d].
Two not explained migrations of a variety of R-CO- groups, O N then N O were observed during ring cleavage 1Cb-O-COR (OX) O-protected TRIS (C).
If R = Me, (Fig. (22)), in 1987, Buur and Bundgaard, re-ported the same isomerisation [12a]. They also demonstrated the about 100 times faster hydrolysis of TRIS’ benzaldehyde based oxazolidine 1Cb (OX) (Fig. (4)), in comparison with TRIS’ cyclohexanone based spirooxazolidine 4Ce (Fig. (7)). An intramolecular catalysis, promoted by the two free gemi-nal hydroxymethyl groups, was suggested by the authors to support i) the N O migration accompanying hydrolysis 1Cb-N-COR (OX) O-protected TRIS (C) and ii) the gen-eral high instability towards hydrolysis of 2-aryl-1,3-oxazolidines based on TRIS (C).
It is of note that, in serinolic family, over the years, simi-lar isomerisations were also mentioned (see also Part II of this review) both as N O (Loncaric and Wulff, 2001 [23a]), (Smukste and Smithrud, 2001 [23b]) and O O’ acyl migrations (Ishii et al., 1999-2003 [23c, 23d]), (Neri and Williams, 2002 [23e]).
O N
O
HOH2C
R*
S*
R*
2
1/3 Eu3+
O-H2C
N
O
HOH2C
2
+H
(1/3Eu3+)
O-H2C
N
O
HOH2C
2
H
(1/3Eu3+)
+
pro-R
pro-S
x
Re faceH
pro-S
pro-R
N
O
CH2OH
2
H+
Si face
pro-R
pro-S
CH2O
(1/3Eu3+)
N
O
CH2OH
2
H
+
pro-R
pro-S
CH2O
(1/3Eu3+)
N O
O
H
2
CH2OH
S*S* R*
13Ca-unlike 13Ca-1 13Ca-2
13Ca-2 13Ca-3 13Ca-like
x
Fig. (20).
NOO
CH2-O-COR
Ph Ph
R = Me, n-C5H11, Ph
p-NPh, Ph-OCH2
H2O / H+
r.t. 100 oC
30 min.O NH2
+Cl-
Ph
CH2OH
CH2-O-COR
R = Me, n-C5H11, Ph
p-NPh, Ph-OCH2
1Cb-O-COR (OX) 1Cb-N-COR (OX)
NaOH or
Na2CO3
H2O / r.t.O N-COR
Ph
CH2OH
CH2-OH
R = Me, n-C5H11
p-NPh, Ph-OCH2
HCl / EtOH
r.t. 100 oC
30 - 90 min.
NH2
HOH2C O-CO-R
CH2OH
O-protected TRIS (C)3Ca-O-COR
17 - 49% 68 - 80% 69 - 95%
R = Ph-O-CH2, p-NPh
Fig. (22).

134 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
In a more advanced approach, in 2002, Pavia et al. [13a], then us, in 2004 [15b, 15f], rationalised in terms of a crossed endo-anomeric effect this type of serinolic oxazolidines’ reactivity.
So, both in solution and in solid state, in the aminalic DOABO sequence O-C-N-C-O, an increased basicity of the intracyclic oxygens was found and explained by two favour-able antiperiplanar orientations allowing delocalisations, lp(O) *(C-N) (weak hyperconjugation) and lp(N)
*(C-O) (strong hyperconjugation). They can be illustrated by using oxazanes’ bonding and non-bonding formulae ([17b], Fuchs et al. [22b]), (Fig. (23)).
For instance, in solid state, for almost all known DOABO molecular determined structures [13,15f], noticeable contrac-tions, (0.022 - 0.045 Å), of the appropriately oriented ami-nalic N-C bond, e.g. N-1-C-2, in comparison with the bridged N-1-C-5 one were observed.
Moreover, in solution, our DFT calculations and 1H-DNMR data [15f], confirmed the chiral flipping of the DO-ABO skeleton, (Fig. (23)). In this approach, the enantioforms (s-a) (a-s), allowing best the manifestation of the crossed endo-anomeric effect, were indeed 2.5-5.5 kJ/mol more sta-ble than the meso form (a-a) and 11-13 kJ/mol with respect to the alternative meso form (s-s).
If so, according to Pavia et al. [13c], hydrolysis of a C-2, -8 diphenyl DOABO compound, such as 3Ca, was achieved rapidly and quantitatively in smooth acidic conditions, (Fig. (24)), since the anomeric effect, increasing basicity of the heterocyclic oxygens, was existent both in the starting 3Ca and in the oxazolidine intermediate 1Cb (OX).
As Pavia et al. demonstrated, the coordination ability of serinolic oxazolidine oxygens with amplified basicity, could be selectively manipulated in synthesis by using a variety of Brønsted and Lewis acids, (Fig. (25)) [13c], e.g. starting from the DOABO derivative 14C.
NOO 12
37
8NO-O
+N-
O+O
non bonding bonding non bonding strong anomeric effect weak anomeric effect lpN-1 *(C-2-O-3) lpO-7 *(C-8-N-1)
NOO
NOO
NOO
NOO1 1 11
7 7 7 7
8 8 8 8
2 2 2 23 3 3 3
5
5 5 5 5
syn-syn* syn-anti* anti-anti anti-syn
(s-s) (s-a) (a-a) (a-s) (M,P)** (M,M) (P,M) (P,P)
*fiducial substituent [17b] at N-1 for the use of descriptors syn or anti
**descriptors M and P with respect to the torsion angles about the bonds N-1-C-2 and N-1-C-8 respectively
Fig. (23).
O
N
HOH2C
O
1
23
57
*
N
HOH2C
*HOH2C
H OH+ H+
CH2OH
NH2
HOH2C
HOH2CPh
Ph
Ph
O
N
HOH2C
HOH2C Ph
H
pyramidalinversion
C1Cb (OX)3Ca
Fig. (24).
NOO
OCO2Et
0.1 N HCl
NH3+Cl-
OHHO
OCO2Et
NBn2
OHHO
OCO2Et
PhPh
5 eq. of NaBH4-TFA (91%)*
or NaBH3CN-TFA (86%)
or NaBH4-AlCl3 (89%)
or NaBH3CN-AlCl3 (88%)
or 1.1 eq. BH3 (76%)
THF / 20 oC / 1hr.
NBn2
HO OH
OHO LiAlH4-AlCl3 .
THF / 20 oC / 1hr.
(80%)
NO O
O
Ph Ph
HOLiAlH4
THF / 20 oC / 1hr.
(95%)
14C
NaBH4 or NaBH3CN or NaBH4-AcOH
no reaction
Fig. (25).

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 135
In terms of anomeric effect governing oxazolidine ring-opening / ring-closure, a more subtle behaviour we observed. Thus, at least in the case of TRIS’ 2-aryl-1,3-oxazolidines, it could also explain the observed fast epimerisation at C-2, for example in the case of the two tautomers of double oxa-zolidines C (OX-OX) (unlike) C (OX-OX) (like), (see Fig. (17)), assisted by the intramolecular hydrogen bonds, easily developed by the two geminal hydroxymethyl groups, (Fig. (26)) [15b].
If so, similar rational cause should be the incidence of the ring-ring tautomerism, (Fig. (26)), encountered in the case of TRIS’ (di)spirooxazolidines, such as 4Ca-d-c, -t, (Fig. (7)),
On 300-600 MHz time scale, an important 3JH,H coupling pattern (H-2/NH),
9-12Hz, is found in almost all serinolic based 2-aryloxazolidines A-E.This fact should be consistent with a trans diaxial arrangement between these protons. It follows an equatorial orientation of the N-1 lone pair (donor) overlapping an excellent acceptor, *(C-2-O1) in an envelope conformation of the oxazolidine ring, see Ref. [13c, 15b, 15f, 19b].
disclosing a common origin of ring-chain and ring-ring tautomerism. Indeed, the latter was revealed each time when tautomers were first detected in a hydrogen bond acceptor solvent, ([D6]DMSO), then in a hydrogen bond donor one, (e.g. CDCl3), or viceversa, (Figs. (7, 11, 15, 16)).
1.5. Syntheses Based on Reactivity of Serinols with Other Dicarbonyl Electrophiles
1.5.1. 1,4-oxazin-2-ones and Benzo-1,4-oxazinones
In 1961, Hoffmann et al. [24a], (Fig. (27)), showed the against ring-chain tautomerism reactivity of TRIS (C) with respect to ethyl phenylglyoxalate, affording either the N-substituted amide 15C or the 5,6-dihydro-1,4-oxazin-2-one 17C via, presumably, the corresponding Schiff-Base, 16C.
In 1995, Fleury et al. [24b], (Fig (28)), which investi-gated the condensation between TRIS (C) and a hydroxy-ortho-quinone, noticed the pharmaceutical interest in ben-
H
N
CH2OH
CH2OH
HO
*
R*
H
ON
CH2OH
CH2OH
H
H
O
N CH2OH
H
HO
+
H
N
S*
H
O*
CH2OH
CH2OH
C (OX-OX) (unlike)(bonding) iminium cation
(non-bonding)
C (OX-OX) (like)(bonding)
N
CH2OH
CH2OH
HO
*
R
ON CH2OH
H
HO
+
iminium cation(non-bonding)
R
N
CH2OH
CH2OHO
*
R
H
4Ca-d-t(bonding)
ON
CH2OH
CH2OH
H
R4Ca-d-c(bonding)Ring-Chain tautomerism Ring-Ring tautomerism
-
-
Fig. (26).
H2NC(CH2OH)3 + Ph-CO-CO-OEt
110-115 oC
1 hr.Ph-CO-CO-NHC(CH2OH)3
NHOH2C
HOH2C
OH
O
Ph
OEt
i) AcOH (glac.) n-BuOH / reflux / 2 hrs.ii) pH 7-8
- EtOH
O
N
O
PhHOH2C
HOH2C
15C (90%)
16C 17C (60%)
12
3
4
5
6
C
Fig. (27).
OH
HO
HO
CO-Ph
10 eq. Et4N+ClO4-
MeOHor
N2 / Electrolysis / Pt electrode (400 mV)
OH
O
O
CO-Ph 1 eq. (C)
H2NC(CH2OH)3
MeOH
N
CO-PhO
HO
HO
HO
HO
N
O
O
HOPh
OH
CH2OHHOH2C
18C (70%)
Fig. (28).

136 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
zoxazinones of type 18C. The desired hydroxy-ortho-quinone was generated by electrochemical or chemical oxi-dation and reacted with TRIS to give the title benzoxazinone 18C following a prototropic tautomerism and a ring-chain one, which was not of type SB OX.
For related electrochemical behaviour of p-nitrophenyl-serinol E based saturated heterocycles, 1,3-oxazolidines and DOABO derivatives see Ref. [24c].
1.5.2. More Elaborated Heterocyclic Systems: 2,6-dioxa-10-azatricyclo[5.2.1.0.
4,10]decanes, 2,6-dioxa-11-azatricyclo
[5.3.1.0.4,11
]undecanes, 9,13-dioxa-14-azatetracyclo[6.5.1. 0
2,7.0
11,14]tetradeca-2,4,6-trienes, Chiral Pyrroles and
Isoindolines
Targeting new N-substituted pyrroles and dihydropyridi-nes with serinolic design, in 1976, Broadbent et al. [10a], explored the reaction between serinols S, A-E and some di-carbonyl electrophiles, 1,4(or 5)-dialkyldiketones and 1,2-phthaldicarboxaldehyde. As summarised in Fig. (29), the accessed products were very different than those initially planned, two new types of C-substituted dioxaazatricyclic derivatives being obtained instead, with moderate to satisfac-tory yield.
The isolated series of tricyclic compounds 19S, A-E, in-corporating a DOABO skeleton, resulted from two cones-
cutive totally diastereoselective “all cis” cycloaminalisations, (Fig. (3), Table 1, entry 1), with respect to rings’ condensa-tion (Oxazolidine route).
The classic envisaged Knorr-Pall route, implying exclu-sively the nucleofilicity of serinolic amino group, provided the expected pyrroles 20Sa, Sb, A, D, E, but mainly as side products in the synthesis of 19Sa, Sb, 19Aa, 19D and 19E. The incidence of pyrroles 20 was anyhow significant for serinol S (20Sb) and phenylserinols D, E (20D, 20E). In these last two cases, it was to presume that the secondary hydroxyl group of D (E) was a too bulky nucleophile to fol-low completely the Oxazolidine route. Dedicated attention was paid in exploiting the C-4-hydroxymethyl functionality of compound 19Ca whose etherification or transesterifica-tion gave series 21Ca-k, claimed for pharmaceutical apply.
In 2002, our similar study [15b], carried out with serinols A-E in reaction with 1,2-phthaldicarboxaldehyde, could en-tirely discriminate the nucleophilicity of hydroxyl groups in serinols
A-C vs. D, E, (Fig. (30)), though the non-isolable key in-termediate, of type 1,3-dihydroxyisoindoline, we postulated to be the same.
It also completed the above pioneering work of Broad-bent et al. [10a] then that of Schipchlander, in 1977 [10b], on the same topic.
OH OH
NH2R2
R1
R1 = R2 = H, series S
R1 = H, R2 = Me, series A
R1 = H, R2 = Et, series B
R1 = H, R2 = CH2OH, series C
R1 = p-NPh, R2 = H, series E
R3R3
O
Oi
NOO
R3 R3
R2
OH OH
N
R1
- 2 H2O
OH OH
N
R1
R3
HO
R3
OH
R3 R3
R2
Knorr - Paal
route
19Sa
19Sb
19Aa
19Ab
19Ba
19Bb
19Ca
19Cb
19D
19E
19S, A-E
R1 R2 R3 yield (%)
20Sa H H Me 13
20Sb H H Et 36
20A H Me Me 8
20D Ph H Me 18
20E p-NPh H Me 15R1
R2
i: toluene (cumene) or heptane cat. TsOH or AcOH / reflux /
1 eq.
( )n( )n
NOO
H
H H
NOO
H
H H
2 611
7
543
1
8
910
1
2
3 4 5
6
7
89
10R
S
rs
rsS
R
(1S,4r,7R,10s)-2,6-Dioxa-10-
azatricyclo[5.2.1.0.4,10]decane(1S,4r,7R,11s)-2,6-Dioxa-11-
azatricyclo[5.3.1.0.4,11]undecane
NOO
H3C CH3
CH2OH
NOO
H3C CH3
CH2O-R5 R5 (yield, %)
Et . HCl (22%) 21Ca
Ac (30%) 21Cb
CO-C(OH)Ph2 (60%) 21Cc
CO-C(OH)Ph(c-C6H11) . HCl (67%) 21Cd
CO-C(OH)Ph(c-C5H9) . HCl (42%) 21Ce
CO-CH(OEt)Et . HCl (33%) 21Cf
CO-t-Bu . HCl (52%) 21Cg
CO-NH-CH2Bn (43%) 21Ch
i
OOC
Me
(8%) 21Ci
N O . HCl (30%) 21CjOC
N O . HCl (30%) 21CkOC
Me
Me
i: 0.16 eq. Na / 1 eq. R4-CO-OMe or 1 eq. R4-CO-OEt
heptane / distillation, except 21Ca (Williamson)
19Ca 21Ca-k
Oxazolidine route
R1
H
H
H
H
H
H
H
H
Ph
p-NPh
R2
H
H
Me
Me
Et
Et
CH2OH
CH2OH
H
H
R3
Me
Et
Me
Et
Me
Et
Me
Me
Me
Me
n
1
1
1
1
1
1
1
2
1
1
20S, A, D, E
Fig. (29).

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 137
So, starting from serinols A-C, the 1,3-dihydroxyiso-indolines 22A-C, as sources of stable benzyl carbocations, could undergo two successive SN1 like mechanism etherifi-cation to produce the known 9,13-dioxa-14-azatetra-cyclo[6.5.1.02,7.011,14]tetradeca-2,4,6-trienes 23A-C [10a, 10b]. We elucidated the “all cis” fused shape of this tetra-cyclic system [15b]. In turn, 1,3-dihydroxyisoindolines 22D, E, originating from phenylserinols D, E, underwent a 1,4-dehydration yielding the isoindoline-1-ones 24D, E, in a variant of the traditional Knorr-Paal synthesis of pyrroles.
1.6. Applications of 3,7-dioxa-r-1-azabicyclo[3.3.0]-c-5-octane (DOABO) System in Coordinative Chemistry
1.6.1. -(3,7-Dioxa-r-1-azabicyclo[3.3.0]oct-c-5-ylmethoxy)
Diazines and s-triazines
Inspired from Broadbent’s earlier study [10a], (Fig. (29), series 21Ca-k), in 2004 [15f], we applied this strategy to alkaline alkoxides of 5-hydroxymethyl-DOABO derivatives 2Ca, 3Ca, (Fig. (4)), as efficient nucleophiles against alkyl and benzyl halides with promising results.
Nevertheless, the largest feasibility of this Williamson’s etherification we established in (poly)chlorodiazines and s-triazines series when they were reacted with 5-hydroxy-methyl-DOABO compounds as potassium or lithium alkox-ides respectively [25a-d]. Two of the most relevant series of derivatives, having 2Ca and 3Ca as starting materials, are depicted in Fig. (31).
We emphasize this very clean and simple “methoxide like chemistry”. Satisfactory to good yields and regioselec-tivities were obtainable, sometimes unexpected, e.g. com-pound 25Ch issued from the double replacement of chlorine at 2,4,6-trichloropyrimidine positions C-2, -4 and not C-4, -6. Potassium alkoxide of 3Ca was, overall, a less efficient nucleophile in comparison with the 2Ca analogue, presuma-bly because of a stronger solvation in THF. In reaction with cyanuric chloride, the successful selective nucleophilic sub-stitution of chlorine essentially depended on the type of
DOABO derivative and cation (K+ vs. Li+, compounds 26Ca-d). With this family of compounds in our hands and on extension of our previous know-how [25e] in halodiazi-nes’ reactivity in Stille’s cross-coupling conditions [25f], compounds 25Cb, g, h, j were reacted with 2,6-bis(tri-n-butylstannyl)pyrazine yielding the terdiazine series 27Ca-d, (Fig. (32)) [25g].
Their host-guest ability concerning two transition metals, Zn2+ and Eu3+, was examined comparatively by means of UV spectroscopy. Terpyrazine 27Ca was inert to both cations, most likely because of the lowest basicity of pyrazine nitro-gens in diazine series. UV spectra of the more basic terdiazi-nes 27Cb-d exhibited significant batochromic effects and two clear isosbestic points consistent with the s-cis-s-cis shifting of the two rotational equilibria about the central pyrazine C-2, -6 exocyclic bonds, 27Cb-d (s-trans-s-trans)
27Cb-d (s-cis-s-trans) 27Cb-d (s-cis-s-cis), required by the accommodation of the guest cation. Two selective complexations, Zn2+ vs. Eu3+, were observed i) due to the number of DOABO units (27Cb vs. 27Cc) and ii) due to the highest basicity of the pyridazine nitrogens (compound 27Cd) in the examined series.
1.6.2. The 3,7-dioxa-r-1-azabicyclo[3.3.0]oct-c-5-ylmethoxy
System Acting as Directed Ortho-Metallation Group in
Diazine Series
Next, selected DOABO alkoxylating diazines 25Ca-d, f, i-k, (Fig. (31)), we investigated in Directed ortho-Metallation (DoM) reaction [26a-c], with organolithiun rea-gents, (Fig. (33)). In this new approach [25g, 26d], the DO-ABO-CH2O motif was seen as a Directed ortho-Metallation Group (DoMG) of type “more sophisticated” methoxy [26e, 26f]. Indeed, by its c-C-5- CH2O sequence, it coordinated the lithium to the lone pair of its oxygen atom to bring the base into close vicinity of the ortho-diazine hydrogen and to pro-voke its removal (“Complex Induced Proximity Effect”, CIPE) [26a].
OH OH
NH2R2
R1
1 eq.
OO
R1 = H, R2 = Me, A
R1 = H, R2 = Et, B
R1 = H, R2 = CH2OH, C
R1 = Ph, R2 = H, D
R1 = p-NPh, R2 = H, E
i: A-C aromatic solvent / H+ / reflux
i: D, E MeOH / reflux / 4 hrs.
i
NHO OH
HO OH
R2
H+
- 2 H2OA-C
NHO
OH
D, E
NOO
R2
12
3
45
6
789
10 11 12
13
R SH H
rs
OH OH
R1
H - H2O
N
OH OH
R1
OHN
OH OH
R1
O
22A-C 23A (74%), 23B (37%), 23C (93%)
22D, E 24D (60%), 24E (33%)
H+
12
3
4
5 6
7
14
Fig. (30).

138 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
Our 1H-DNMR exploration, DFT calculations [15f] and molecular determined structures [15f, 25b, 25d], indicated the heterofacial [17b] DOABO part of the starting 25Ca-d, f, j, k (R1 = H) being flipping at room temperature in an over-all enantiomeric inversion, (see also Fig. (23)). It became frozen on a large domain of temperature (273 – 173 K) with its additional chelating sites, O-3, N-1 and O-7, oriented in a rigid non-chiral conformation (meso form, see also Fig. (23))). In contrast, in the ”all cis” analogue 25Ci (R1 = Ph), (Fig. (33)), the DOABO fragments were still flipping at 173 K.
Moreover, regardless the (non)substitution at C-2, -8 DOABO positions in 25Ca-d, f, i-k, by lowering the tem-perature, the orientation of the c-5-diazinyloximethyl moiety become near coplanar, s-trans-out, bisecting the DOABO
skeleton, hence an ideal stereochemistry to create the CIPE effect.
With these premises, metallation of pyrimidine deriva-tives 25Cf, i, (Fig. (31)), was accomplished comparatively with total ortho regioselectivity but in different conditions, (Fig. (34)). In the case of 25Cf ( 28Cf), no reaction oc-curred -78 oC. A slow progress was detected upon gently warming up to room temperature only. On the contrary, at -78 oC, the reaction starting from 25Ci reached completion at this temperature within two hours.
The general ability of the DOABO-CH2O moiety to act as an authentic “methoxy like” DoMG on pyrazine and pyri-dazine series is exemplified in Fig. (35).
NOO
CH2OH
(Ph)H H(Ph)
H: 2Ca (2H)DOABO-CH2OHPh: 3Ca (2Ph)DOABO-CH2OH
X
R1
R1
R1
n = 0.33, 0.66, 1.00
R1 = H, Cl, MeO
X = N: s-triazine
X = C: pyrazine
pyrimidine
pyridazine
n eq.
R2 - R4 = H, Cl, MeO,
(2H)DOABO-CH2O
(2Ph)DOABO-CH2ON,N,X
R4
R3
R2
Base* / THF / T (oC) / t (hrs.)
N,N
*equimolar KH for the replacement of chlorine in (poly)chlorodiazines series and disubstitution of chlorine in cyanuric chloride by 2Ca and trisubstitution by 3Ca; equimolar n-BuLi for the trisubstitution of chlorine in cyanuric chloride by 2Ca
2 85
Product Conditions Azine
R2 R3 R4 T (oC) t (hrs.)
No. Yield
(%)
N
N
R2 R3
(2H)DOABO-CH2O
(2H)DOABO-CH2O
(2H)DOABO-CH2O
(2H)DOABO-CH2O
(2Ph)DOABO-CH2O
H
Cl
MeO
(2H)DOABO-CH2O
Cl
-
-
-
-
-
40
r.t.
65
65, r.t.
65
16
6
24
3, 14
52
25Ca 85
25Cb 83
25Cc 33
25Cd 76
25Ce 34
N
N
R2 R3
R4
(2H)DOABO-CH2O
(2H)DOABO-CH2O
Cl
(2Ph)DOABO-CH2O
H
H
(2H)DOABO-CH2O
H
(2H)DOABO-CH2O
Cl
(2H)DOABO-CH2O
(2Ph)DOABO-CH2O
45
-78 r.t.
-78 r.t.
65
24
19
22
21
25Cf 81
25Cg 63
25Ch 76
25Ci 31
N NR2 R4
Cl
MeO
-
-
(2H)DOABO-CH2O
(2H)DOABO-CH2O
40
65
4
18
25Cj 86
25Ck 51
N
N
N
R2 R2
R3
(2H)DOABO-CH2O
(2H)DOABO-CH2O
(2Ph)DOABO-CH2O
(2Ph)DOABO-CH2O
Cl
(2H)DOABO-CH2O
Cl
(2Ph)DOABO-CH2O
-
-
-
-
65
-78 r.t.
0, 65
-78 r.t.
r.t., 65
36
20
1, 40
20
48, 4
26Ca 34
26Cb 82
26Cc 37
26Cd 29
Fig. (31).

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 139
N
Cl
N
N
N
(n-Bu)3Sn Sn(n-Bu)3
0.48 eq.
5% Pd(PPh3)4 / Toluene /reflux / 22 - 48 hrs. N
N
NNN N (R)n(R)n(R)n
25Cb, g, h, j 27Ca-d
R = (2H)DOABO-CH2O, n = 1, 2
Starting Material
Yield (%)
Main Product Salt and Bato-chromic Effect
( , nm)
Isosbestic Points
(nm)
Proposed Stoichiometry
L:M
N
N
Cl
R
25Cb
65
N
NN
N
NN
O ON
O
O
O
N
OZn2+, Eu3+
27Ca
Zn(BF4)2
0
EuCl3 6H2O
0
-
-
-
-
N N
R Cl
25Cg
60 N N
N
N
N N
OO
N OO
Zn2+, Eu3+
27Cb (s-cis-s-cis)
NOO
26
Zn(BF4)2
26
EuCl3 6H2O
16
226 305
230 311
1:1
1:1
25Ch
N N
R Cl
R
70 N N
N
N
N N
O
OO
NOO
NOO
N OO
NOO
O
Zn2+, Eu3+
27Cc (s-cis-s-cis)
6 2
Zn(BF4)2
49
EuCl3 6H2O
0
236 318
-
1:1
-
NNR
Cl
25Cj
22 NN
N
N
NNO O
NO
O
NO
O
Zn2+, Eu3+
27Cd (s-cis-s-cis)
6 2
Zn(BF4)2
50
EuCl3 6H2O
0
263 280
-
1:1.5
-
Fig. (32).
N
O
NOO
N
(Ph)H H(Ph)
O
N
O
N
O
N
N
O
N
N
OO
N
O
N
N
O O
R1
R1
R1
R1
R1
R1
N
O
N
N
O O
R1 = H
-78 oC Li
3
7
3
3
3
7
77
O-3-syn-O-7-anti
(M,M) chiral form
O-3-anti-O-7-syn
(P,P) chiral form
O-3-anti-O-7-anti
(P,M) meso form
R1 = H: G = 53.1 - 59.3 kJ / mol
R1 = Ph: still flipping at 173 K
R2 = H, Cl, MeO, (2H)DOABO-CH2O, (2Ph)DOABO-CH2O
? R2
R2
R2 R2
1
11
25Ca-d, f, i-k
Fig. (33).

140 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
N N
R R
In crude reaction mixture:
28Cf (64%) + 25Cf (36% )28Ci 100%
Ph OH
25Cf, i
Conditions
f or i
f: R = OCH2-DOABO(H) frozen (-78 oC) flipping (r.t.)
i: R = OCH2-DOABO(2Ph) flipping (-78 oC)
f: 4 eq. LTMP / THF / -78 oC / 2 hrs. / 4 eq. PhCHO / -78 oC r.t. / 12 hrs.
i: 4 eq. LTMP / THF / -78 oC / 2 hrs. / 4 eq. PhCHO / -78 oC / 2 hrs.
Fig. (34).
N
N
OCH2-DOABO(2H)
E1
R
25Ca-d
E2
i: 4 eq. LTMP / THF / -78 oC / 1 - 2 hrs. / 4 eq. E [Ph-CH=O, Me-CH=O,
Cl-Sn(n-Bu)3, Ph2S2] / - 78 oC or -78 oC r.t. / 2 - 12 hrs.
i
N
N
OCH2-DOABO(2H)
R
E2
E1
25Cj, ki
i: 4 eq. LTMP / THF / -78 oC / 1 - 1.5 hrs. / 4 eq. Ph-CH=O
-78 oC r.t. / 14 - 24 hrs.
O
O
N
O
N
N
HH
HO
H
P*,P*,R*
R*
29Ca
29Ca-k29Cl-n
*98% if the DoMG was O-CH2-DOABO(2Ph)
2.11 A
2.37 A
o
o
H
H
H
H
H
H
O-CH2-DOABO(2H)
Cl
Cl
MeO
MeO
29Ca
29Cb
29Cc
29Cd
29Ce
29Cf
29Cg
29Ch
29Ci
29Cj
29Ck
Ph-CHOH
MeCHOH
Et
I
Sn(n-Bu)3
SPh
Ph-CHOH
Ph-CHOH
H
Ph-CHOH
H
H
H
H
H
H
H
H
H
Ph-CHOH
H
Ph-CHOH
100
100
100
100
100
100
-
67
33
41
59
79*
41
80
68
73
73
61
52
26
29
41
R E1 E2 Regioselectivity Yield
(%) (%)
Cl
Cl
MeO
Ph-CHOH
H
H
H
Ph-CHOH
Ph-CHOH
63
37
100
53
31
78
R E1 E2 Regioselectivity Yield
(%) (%)
29Cl
29Cm
29Cn
Fig. (35).
In resulting pyrazine series 29Ca-f, a complete ortho re-gioselectivity was established with satisfactory to good yields, similar to those previously reported for methoxypyrazine [26e, 26f]. The higher chelating aptitude of DOABO-CH2O motif vs. methoxy was supported by the greater number of equivalents of LTMP (lithium-2,2,6,6-tetramethylpiperidide) required, 4 vs. 1.1 respectively, longer accumulation period of the lithiated species and reaction times. Competitions between chlorine and DOABO-CH2O as DoMGs provided parallel results as in the competition Cl against methoxy, both in pyrazine (regioisomeric 29Ch vs. 29Ci) and pyridazine (regioisomeric 29Cl vs. 29Cm) series: DOABO-CH2O was a better DoMG than chlorine. In con-trast, the “direct competition” between DOABO-CH2O and MeO as DoMGs was partially gained by the latter concern-ing regioisomeric pyrazines 29Cj vs. 29Ck but totally gained in the case of pyridazine 29Cn, found to be the unique re-gioisomer.
Finally, it is noteworthy that in the 1H-NMR spectra of this new family of chiral diarylmethanols, those having an ortho relationship between the DOABO heterofacial skeleton
and the stereogenic centre, 29Ca, g, h, j, l, exhibited a highly discriminating magnetic non-equivalence between the two condensed oxazolidine rings. The DFT calculations of a rep-resentative compound, 29Ca (see inset in Fig. (35)), revealed the most stable conformer being internally chelated by a tri-furcated hydrogen bond. That is, a certain relative configura-tion of the chiral centre required an univocally relative chiral conformation of the DOABO skeleton (e.g. R* P*,P*) and vice versa.
However, if in the above type of DoM reactions DOABO units having C-2 as stereogenic centre, (e.g. 5Ca, Fig. (7)), were used as DOMG in tandem with prochiral electrophiles, no diastereoselectivity was observed [25g, 26d].
2. SYNTHESES BASED ON REACTIVITY OF SERI-NOLS WITH CARBONYL ELECTROPHILES OUT-
SIDE OF RING-CHAIN TAUTOMERISM CONDI-TIONS
When serinolic primary amino functionality is blocked i) by N-protection or ii) temporarily, in strong acidic media, its nucleophilicity is suppressed, the reactivity being hereafter

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 141
due to the remainder nucleophiles, the hydroxyl groups, for example in acetalisation reactions. Both methodologies i), ii) are of interest, including, like it or not, noticeable as useful exceptions.
2.1. 5-Amino-4-aryl-1,3-dioxanes
2.1.1. Direct Acetalisation of Phenylserinols
Optically active N-formylated phenylserinol D can be acetalised with acetone, yielding the acetonide 30Da, (Fig. (36)), in a one pot three steps completely diastereospecific process [(1S,2S)-D (4S,5S)-30Da] (Nordin and Thomas, 1984 [27a]). This chemistry seemed to us inspired from the earlier data of Ohara et al. [3] with later improvements of Weinges et al. in 1980 [27b, 27c], which used 30Da as chiral auxiliary in the synthesis of optically active -amino acids since 1973 [27d]. We previously reviewed these contribu-tions connected to Chloromycetin synthesis [3]. Amino-1,3-dioxane (4S,5S)-30Da is nowadays commercially avail-able and a very attractive chiral tool (see dedicated Section 2.1.3.).
Nevertheless, a parallel strategy is not reproduced for (1S,2S)-p-nitrophenylserinol E acetalisation, but in the case of its racemate (rac)-E only, (Fig. (37)).
OH OH
NH2
p-NPhS*
S*
Me2CO / P2O5
HCl / 0 oCO O
NH2
p-NPh
Me Me(rac)-E(rac)-30Ea
(60-70%)
S*
S* 4 5
Fig. (37).
That is, in our hands [3, 28a], (4S,5S)-30Ea was too un-stable during work-up in identical conditions as those known for the isolation of its non-nitrated analogue (4S,5S)-30Da.
Fortunately, as early as 1994 [19a, 28a-c], we were de-lighted to see that (1S,2S)-p-nitrophenylserinol E readily underwent the so called by us “sulphuric acetalisation” with some non crotonisable (masked) (di)aldehydes, (Fig. (38)). We note the harsh conditions required, tenfold molar excess
For derivatisation of 30Da, a waste in Chloromycetin’ manufacturing,
including sequential epimerisations, other pathways to acces it, dedicated pharmaceutical interest, etc. see Ref. [3].
of concentrated sulphuric acid, to be the single suitable for complete cancellation of the nucleophilicity of amino group in E with respect to the depicted electrophiles. “Monomeric” 5-amino-1,3-dioxanes 30Eb, c could be obtained routinely in fairly to good yields in a total diastereospecific process. The N,N-dimethyl derivative 31Ea of E, accessible from E ac-cording to Sokolov et al. [28d], afforded 5-dimethylamino-1,3-dioxanes 30Ed, e, over again in “sulphuric acetalisation” only. That is, treatment of 31Ea with the same carbonyl partners in “conventional” protocols, e.g. in a Dean-Stark trap by refluxing in aromatic solvent and PTSA as catalyst, resulted in no reaction. Next, targeting a selective “cross sulphuric acetalisation” between E and 31Ea, “dimers” 30Ef-h were isolated, the disadvantage of the rather poor yields and selectivity obtained being offset by the structural profits gained. We mention the feasibility of “sulphuric acetalisation” to be restricted so far to p-nitropheylserinols E
and 31Ea. Phenylserinol D, as well as C-2-substituted seri-nols A-C, appears to be unstable in these conditions.
2.1.2. 1,3-Dioxanic Schiff-Bases
Surprisingly, even if we used excess of arylaldehydes as electrophiles in the “sulphuric acetalisation”, (Fig. (38)), just the corresponding Schiff-Bases 1E (SB), (Fig. (9)), we iso-lated with moderate yields [28a, 28e]. We were doubtful that, in fact, the desired 5-amino-2-aryl-1,3-dioxanes of E rapidly decomposed and partially isomerised to Schiff-Bases during the work-up. However, the reactivity of arylaldehydes could be oriented towards acetalisation if THF, seen in con-text as a base, was used as co-solvent. 1,3-Dioxanic Schiff-Bases (DX-SB) of E, (Fig. (39))
, could be prepared in poor
to moderate yields with complete (E,2R,4S,5S) diastereose-lectivity [28e].
Compounds 32Ea-e were stable in normal conditions and in refluxing anhydrous benzene in the presence of PTSA. Unexpectedly, if catalytic amounts of the same arylaldehyde as that already incorporated in the structure of 32Ea-e were added, a partial rearrangement of the (E,2R,4S,5S) (DX-SB) 32Ea-e skeleton into the corresponding (1R,2R,4S,5S,8S)-one of type DOABO 3E took place (see Fig. (9)). We note this isomerisation as being, besides diastereospecific, a much
Harsh conditions are also required for acetalisation of phenylserinol D with
benzaldehyde to give a 1,3-dioxanic Schiff-Base, according to Weinges et al. (1980) [27b] inspiring Rozwadowska in 1997 [28f].
OH OH
NH2
Ph
D
OH OH
NH-COH
Ph
1.5 eq. (MeO)2CMe2 / HBr
acetone / 0 oC / 2.5 hrs.
O O
NH-COH
Ph
Me Me
S
S 12
85% aq. H2N-NH2 H2O / reflux / 2 hrs.
O O
NH2
Ph
Me Me
30Da (78%)
S
S
4 5
1.2 eq. HCOOMeMeOH / r.t. / 3.5 hrs.
Thomas et al.
Weinges et al.
R-CO-R' / HCN
O O
HN
Ph
Me Me
R
CN
R'
H+
NH2
RHOOCR'
Fig. (36).

142 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
slow reaction in comparison with the direct double cy-cloaminalisation of E with the same arylaldehydes, (Fig. (9)), to infer us that the two processes were mechanistically different [28e].
Indeed, in some DOABO synthesis, involving 2 eq. of aryladehydes and 1 eq. of E, (Fig. (9)), some traces of prod-ucts of type (DX-SB) 32Ea-e we identified. In fact, since1956, Pedrazzoli and Tricerri [18d] mentioned them to be by-products as well in the preparation of several Schiff-Bases and DOABO derivatives of p-nitrophneylserinol E, 1E (SB), (Fig. (9)), The authors tentatively assigned these comppounds as (DX-SB) on the basis of UV and IR data corroborated to those of Edgerton et al. in 1954 [18b].
Additional types of (E,4S,5S)-1,3-dioxanic Schiff-Bases of p-nitrophenylserinol E could be conveniently prepared starting from amino-1,3-dioxanes 30Ea, b in reaction with arylaldehydes and even with -methyl-ketophenones (Fig. (40)) [28e]. No transaminalisation or rearrangement in syn-thesis of 32Ef-j series we observed.
One must note that, by applying the above classic type protocol, a much larger variety of useful (E,4S,5S)-1,3-dioxanic Schiff-Bases of phenylserinol D acetonide 30Da
are available (Section 2.1.3).
Standard condensation between amino-1,3-dioxanes of p-nitrophenylserinol E and 1,2-phthaldicarboxaldehyde
OH OH
N(R1)2
p-NPh
O O
N(R2)2
p-NPh
R3
O O
O O
N(R4)2
N(R5)2
p-NPh
p-NPh1 eq. CH2=O
(E 30Eb, 31Ea 30Ed)
or
1 eq. H2NCH2CH(OEt)2
(E 30Ec, 31Ea 30Ee)
30Eb: R2 = R3 = H (80%)
30Ec: R2 = H, R3 = CH2NH2 (65%)
30Ed: R2 = Me, R3 = H (70%)
30Ee: R2 = Me, R3 = CH2NH2 (84%)
30Ef: R4 = R5 = Me (35%)
30Eg: R4 = H, R5 = Me (11%)
30Eh: R4 = R5 = H (33%)
0.5 equiv. O=CH-CH(OH)2
(31Ea 30Ef, E 30Eh)
or
1.0 equiv. O=CH-CH(OH)2
(E + 31Ea 30Eg)
E: R1 = H
31Ea: R1 = Me
i) 10 eq. H2SO4 (c 96%)
24 h. (0 oC r.t.)
ii) NH3 / 0 oC / pH = 9
2 eq. H2COHCOOHreflux / 2 hrs.70%
12
S
SSS
SS
R
RR
i) 10 eq. H2SO4 (c 96%)
24 h. (0 oC r.t.)
ii) NH3 / 0 oC / pH = 9
Fig. (38).
OH OH
NH2
p-NPh
i) 2 eq. Ar-CH=O
10 eq. H2SO4 (c 96%)
2:1 v/v THF : H2SO4
0 oC r.t. / 24 hrs.
ii) NH3 / 0 oC / pH = 9O O
N
p-NPh
Ar
Ar
Ar (yield %): Ph (25, 32Ea); m-HO-Ph (34, 32Eb);o-O2N-Ph (7, 32Ec); o-Cl-Ph (22, 32Ed); -naphthyl (59, 32Ee)
traces Ar-CH=O / TsOHbenzene / reflux
NOO
p-NPh
Ar Ar
H
3Ea, 3Eb, 3Ed [Fig. (9)]3Em (Ar = m-HO-C6H4)3En (Ar = -naphthyl )
(DX-SB)*DOABO
K = = 1.5 - 1[DOABO]
[(DX-SB)]
*DioXanic Schiff-Base
32Ea-eE
S
R
S
S S
RSR
148
4
5
5
2
2
Fig. (39).
O O
NH2
p-NPh
R1 R1
4 5
R1 = Me, (rac)-30Ea (4S*,5S*)
R1 = H, 30Eb (4S,5S)
1 eq. R2-CO-C6H4-R3-p
EtOH / reflux (R2 = H)
or
Benzene / Ts-OH
reflux (R2 = Me)
O O
N
p-NPh
R1 R1
4 5
R2
R3
R1 R2 R3 yield (%)
32Ef H H H 85
32Eg Me H H 90
32Eh H Me H 15
32Ei H Me Br 53
32Ej H Me NO2 24
+ R2-CO-C6H4-R3-p
Ts-OH / benzene / refluxno rearrangement
Fig. (40).

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 143
yielded N-substituted amidines of the isoindoline-1-ones 32Ek, l, (Figs. (41, 30)) [28e].
One can observe the unpredictedable diastereoselectivity if the reaction implicated (rac)-30Ea since construction of (rac)-32Ek skeleton was made only from 5-amino-1,3-dioxane 30Ea building-blocks having the same relative con-figuration (“matched pair” [28g]), e.g. 4S*,5S*,4’S*,5’S*. The unfavourable steric course of this reaction towards the alternative two possible diastereomers of 32Ek, 4S*,5S*,4’R*,5’R* or 4R*,5R*,4’S*5’S* (“mismatched pairs”) we discussed in detail elsewhere [28e].
Finally, it is noteworthy that, besides phenylserinols D, E, solely Gabler and Hauptmann, in 1968 [28h], (Fig. (42)), claimed the syntheses of some (E)-1,3-dioxanic Schiff-Bases of TRIS (C), in enzymatic conditions (see also Fig. (4)). Un-fortunately, little structural evidence supported the identity of the prepared compounds, mostly in the imine series 32Ca-d.
2.1.3. Use of (4S,5S)-5-amino-4-phenyl-1,3-dioxane and its Schiff-Bases as Chiral Tools
In 1986, Bertz et al. [29], investigated 49 optically active amines as chiral auxiliaries in the enantioselective 1,4-addition of phenylamidocuprates to 2-cyclohexenone. In a “serinolic approach”, (Fig. (43)), the phenylamidocuprate of aminodioxane 30Da was prepared and the R vs. S enantiose-lectivity in the synthesis of 3-phenylcyclohexanone de-pended on the solvent and the base used in the phenylami-docuprate generation.
In 1980, Weinges and Blackholm [27c] described the asymmetric nucleophilic addition of cyanide anion to several E-iminoderivatives, generated in situ by condensation of phenylserinol D acetonide 30Da, (Fig. (36)), with a diversity of methylketones. The chiral aminonitrile series 33Da-o, (Fig. (44)), was obtained with good yields and incipient enantiopurity.
O O
NH2
p-NPh
R1 R1
4 5
R1 = Me, (rac)-30Ea (4S*,5S*)
R1 = H, (4S,5S)-30Eb
0.5 eq. o-O=CH-C6H4-CH=OEtOH / reflux
NN
OO
O
O
R1
R1R1
R1
NO2
O2N
45
5'4'
(rac)-32Ek: R1 = Me (4S*,5S*,4'S*,5'S*) (50%)
32El: R1 = H (4S,5S,4'S,5'S) (55%)
Fig. (41).
H2NC(CH2OH)3
R-CH=OAldehyde-dehydrogenase
r.t.O
O
R
HOH2C
N
R
R-CH=N-C(CH2OH)3
R (yield, %) = Me, 32Ca (35); Et, 32Cb (45) i-Pr, 32Cc (66); Ph, 32Cd (21)
R (yield, %) = o-HO-Ph 1Ca (SB) (89) o-hydroxynaphthyl, 1Cj (SB) (84)
C
Fig. (42).
O O
NH2
Ph
Me Me
1 eq. MeLi or n-BuLi
O O
NHLi
Ph
Me Me
(PhLi + CuI)
PhCu + LiI
- 50 0 oC
O O
NCuPh
Ph
Me Me
O i) 1 eq.
- 78 oC / 1 hr.
ii) aq. NH4Cl
Ph O
30Da
Solvent R/S Yield
ratio (%)
Et2O 75:25 62
Et2O 75:25 29*
THF 50:50 26Hexane 35:65 51
Me2S 55:45 53
Et2S 50:50 56
*PhMgBr in lieu of PhLi1
2
3
3-Phenylcyclohexanone
Fig. (43).

144 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
Some basic conformational concepts apply in this type of syntheses. Thus, 30Da was an anancomeric structure be-cause of its overwhelmingly one-sided conformational equi-librium [17b], due to the adoption of an equatorial position by the phenyl ring (bisectional rotamer [28e]) located at C-4 (A value 11.93 kJ/mol) [17b, 30]. This A value was greater by far than the A value of the C-5 amino group (5.15–7.10 kJ/mol) [17b, 30]. Hence, the diastereotopic faces of the ax-ial E-imine double bond in Weinges’ Schiff-Bases were strongly heterotopic as demonstatred by the initial excellent diastereoselectivities, (Fig. (44)). They were the result of the cyanide anion nucleophilic attack occurring largely on the >C=N- Re face. The absolute configuration of the newly created chiral centre was established by 1H-NMR only, on the 90 MHz time scale. In CDCl3, its partial epimerisation was detected within 24 hrs, except aminonitriles 33Dl and 33Do.
In 1995, Enders et al. used 5-amino-4-phenyl-1,3-dioxane 30Da as chiral auxiliary in order to convert , -unsaturated aldehydes into optically active primary amines, (Fig. (45)) [31a, 31b].
First, upon treatment of 30Da with the title aldehydes, se-ries 32Da-d of 1,3-dioxanic Schiff-Bases of 30Da were pre-pared in high yield with variable E/Z diastereoselectivity. Regardless the E- or Z-configuration of the imine double bond, the 1,2-nucleophilic addition of organocerium reagents on the Si face in 32Da-d occurred with 100% regioselectiv-ity and good to excellent d.e. values, yielding the highly elaborated (S,4S,5S)-5-amino-1,3-dioxanes 30Db-i. Chelatiz-ing aptitude of cerium against dioxanic O-atoms explained the resulted diastereoselectivities. Next, the cycloacetalic skeleton of 30Db-i was removed in four steps, not shown in Fig. (45), to provide the optically active primary allyl-a, b or propargylamines c-h. Of a special interest was the silylated 1,3-dioxanic Schiff-Base 32Dd. Its manipulation via 1,2- diastereoselective nucleophilic methylcerium addition (
30Dj) followed by desilylation ( 30Dk) and Sonogashira cross-coupling with haloderivatives ( 30Dl, m) afforded, after acetalic part removal, the propargyl- or (E)-4-ene-2-inyl amines i, j with high e.e. values. The absolute configuration and the enantiomeric excess of the primary amines a-j was established by 1H-NMR carried on their amides obtained with the use of Mosher Acid [(R)-MTPA] chloride [(R)-MTPA-Cl], (R)-3,3,3-Trifluoro-2-methoxy-2-phenylpropio-nic acid chloride [31c, 31d].
In 1997, Yamashita et al., (Fig. (46)) [32a], reported the enantioselective synthesis of some chiral 2,2-dialkylbutenals via the E,E-imino derivatives 32De, f of 30Da as chiral auxi-liary. Compounds 32De, f were issued from the high yield condensation between 30Da and two -substituted- , -unsaturated aldehydes, (E)-2-methyl- and (E)-2-ethylbutenal. Next, 1,3-dioxanic Schiff-Bases 32De, f were deprotonated with LDA then treated with bromo- or iododerivatives as electrophiles to give, simultaneously with the double bond migration, the corresponding chiral (E)- , -disubstituted-
, -unsaturated imines. They were hydrolysed in situ yield-ing the target aldehydes a-e in acceptable to good yields and enantiomeric excess. Particularly, starting from imine 32De
and geranyl chloride as electrophile, the above strategy al-lowed the synthesis and the absolute configuration assign-ment of the natural sesquiterpene of a beefsteak plant f.
Some (het)arylideneamino derivatives of 30Da were ef-fective chiral catalyst in the enantioselective synthesis of 1-aryl-1-propanols by addition of diethyl zinc to aryl alde-hydes, as communicated by the same Yamashita et al., in 1998, (Fig. (47)) [32b]. From the four tested E-imines 32Dg-j, 32Dj, having not only an easily deprotonable hydroxyl group ortho to the imine double bond but also the bulky t-Bu substituent in meta position of the benzylidene fragment, provided the best results as yields and enantioselectivity (see inset in Fig. (47)).
OO
NH2
CH3
CH3
S
S4
5
OO
N
CH3
CH3
R
Me
PhSi face(33Do)
Re face(33Da-n)
OO
NH
CH3
CH3
R
N C
Ph
H3C
OO
NH
CH3
CH3
R
H3C
Ph
N C
Compound Topicity* Epimerisation Yield
(%)
n = 0
2
3
R = o-(p-)R'-C6H4-CH2
R' = p-O2N
o-O2N
p-Cl
o-Cl
p-Me
o-Me
R = C(Me)2OH
C(Me)2OMe
C(Me)2OPh
CH2OH
CH2OMe
CH2OPh
S R 61:39
S R 82:18
S R 80:20
S R 84:16
S R 74:26
S R 73:27
S R 76:24
S R 66:34
S R 66:34
R S 63:37
R S 84:16
R S 100:0
R S 63:37
R S 85:15
S R 100:0
S
S
S
S
S
S
S
S
S
R
R
R
R
R
S
33Da-n 33Do
Re
Re
Re
Re
Re
Re
Re
Re
Re
Re
Re
Re
Re
Re
Si
53
72
70
61
63
68
62
82
83
64
60
69
66
50
38
30Da
*Attacked face Resulted absolute configuration: ul (unlike) 33Da-h but lk (like) 33Dj-o see Ref. [28g]
(H2C)n
33Da
33Db
33Dc
33Dd
33De
33Df
33Dg
33Dh
33Di
33Dj
33Dk
33Dl
33Dm
33Dn
33Do
R =
i: 1 eq. R-CO-Me / 1.1 eq. NaCN / AcOH / MeOH / 60 oC
i
Fig. (44).

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 145
Among other (chiral) 1,2-diamines, in 2000, Strotmann and Butenschön [33] also made use of aminodioxane 30Da
in the chemistry depicted in Fig. (48), designed to prepare chiral amine carbonyl chromium (0) complexes with [(nor-bornadiene)Cr(CO)4] as the complexation reagent.
30Da was routinely converted into the bis-1,3-dioxane diamine 30Dn, by means of the double E,E-Schiff-Base 32Dk. Although 30Dn could be easily complexed with tetra-carbonyl-(norbornadiene)chromium (0), there are no data in the cited paper with respect to the ability of complex 30Do
to insert diastereoselectively the Cr(CO)3 fragment on the diastereotopic faces of some selected chiral methoxylated arenes.
In the end of this Section we note that none of the above strategies applied, so far, to (E)-1,3-dioxanic Schiff-Bases 32Ea-l, derived from p-nitrophenylserinol E.
2.2. 5-Acylamino-1,3-dioxanes
Since the period of 1950’, (a)cetalisation of Chloromice-tyn was mentioned to be a difficult synthetic task, requiring harsh conditions as strong dehydrating reagents (P2O5, CaH2, etc.), and / or high temperature. We previously reviewed this facet of p-nitrophenylserinol E chemistry [3]. Highlighted by the excellent review of Meeskens (1981) [34a], focused on
Our unpublished data and, to our knowledge, there is no report in the litera-
ture on this topic.
R1-CH=O
O O
NH2
Ph
Me Me
30Da
1 eq. / cyclohexane
O
O
N
Me
Me
Ph
R1
32Da-d
R1 (yield, %; E:Z diastereoselectivity)
32Da (E)-Ph-CH=CH (98, 100:0)
32Db Ph-C C (98; 60:40)
32Dc t-Bu-C C (90; 70:30)
32Dd Me3Si-C C (92; 60:40)
R2Li / CeCl3
THF
- 100 oC r.t.
O
O
HN
Me
Me
Ph
30Db-i
R1
R2
SS
R1, R2 (yield, %; d.e. as 4S,5S,S %)
30Db (E)-Ph-CH=CH, Me (81; 86)
30Dc (E)-Ph-CH=CH, n-Bu (82; 97)
30Dd Ph-C C, Me (84; 98)
30De Ph-C C, Et (91; 98)
30Df Ph-C C, n-Bu (88; 92-98)
30Dg Ph-C C, (t-Bu)Me2SiO(CH2)3 (65; 98)
30Dh Ph-C C, H2C=CH-CH2 (91; 95)
30Di t-Bu-C C, Me (79; 98)
4
5
R1 R2
NH2 R1, R2 (global yield from 30Da %;
e.e. as S %)
a (E)-Ph-CH=CH, Me (40; 98)
b (E)-Ph-CH=CH, n-Bu (26; 98)
c Ph-C C, Me (37; 98)
d Ph-C C, Et (41; 98)
e Ph-C C, n-Bu (36; 97)
f Ph-C C, (t-Bu)Me2SiO(CH2)3 (22; 98)*
g Ph-C C, H2C=CH-CH2 (32; 98)
h t-Bu-C C, Me (44; 98)
S
S
i) HCl / Et2O
ii) NaOH / Et2O
iii) NaIO4 / SiO2
H2O / Et2O
iv) KOH / EtOH
*isolated as R2 : HO(CH2)3
32Da-c
MeLi / CeCl3 (3.5 eq.)
THF / - 100 oC r.t.32Dd O
O
HN
Me
Me
PhR3
Me
30Dj, R3 = Me3Si (78%, d.e. as 4S,5S,S, 93%)Bu4NF / THF
30Dk, R3 = H (95%)
Cl-CH=CH-Cl or 2-iodothiophene (2.2-5 eq.)
Pd[(PPh3)]4 (7.5%), CuI (0.2 eq) / Piperidine
or n-butylamine
O
O
HN
Me
Me
PhR4
Me
R4, (yield, %; d.e. as 4S,5S,S %)
30Dl (E)-Cl-CH=CH (79; 98)
30Dm thien-2-yl (83; 98)
i) HCl / Et2O; ii) NaOH / Et2Oiii) NaIO4 / SiO2; H2O / Et2O iv) KOH / EtOH Me
NH2
R4
S
R4 (yield, %; e.e. as S)
i (E)-Cl-CH=CH (79; 98)
j thien-2-yl (71; 98)
a-e
i, j
Fig. (45).

146 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
updated acetalisation methods, in 1984, Meslard et al. [34b] found mild conditions in order to obtain (1R,2R)-Chloromicetyn’s (4R,5R)-1,3-dioxanes 34Ea-i, (Fig. (49)).
In spite of its obvious availability, the efficiency of Mes-lard’s methodology was restricted to acetone ( 34Ea), cyclanones ( 34Ed, e) and arylaldehydes ( 34Eg-i). If the carbonyl electrophile was located in a bulky ketone envi-
R1 = Me, Et
O O
NH2
Ph
Me Me
1 eq.
F3C-COOH / benzenereflux / 5 hrs.
O O
N
Ph
Me Me
CH3
R1
R1 = Me, 32De (85%)
R1 = Et, 32Df (86%)
i) 1 eq. LDA / THF
-78 oC / 1 hr.
ii) 1 eq. R2X / -78 oC
r.t. / 20 hrs.
iii) H2O / r.t.
O O
N
Ph
Me Me
2N HClr.t. / 5 hrs.
H3C O
R1
R1
R2
O
R1 R2 R1 R2 e.e. (%) Yield (%)
a Me Bn
b Me Me2C=CH-CH2
c Me n-C5H11
d Me n-C9H17
e Et n-C5H11
Sa-e
32De
Cl
O
f
(73% yield, 53% e.e.)
30Da
40
61
81
76
63
69
42
78
75
56
Fig. 46.
O O
N
Ph
Me Me
(Het)Ar
(Het)Ar = (2-Py), 32Dg [(6-Me)-2-Py], 32Dh
(2-OH-Ph), 32Di
(2-OH-3-t-Bu-Ph), 32Dj32Dg-j
Ph-CH=O
i) 0.05 eq. 32Dj
1 eq. n-BuLi / Toluene / -78 oC / 20 min.
ii) 1 eq. Ti(O-i-Pr)4 / -78 oC / 20 min.
iii) 2 eq. Et2Zn / Hexane / -35 oC / 1 hr.
iv) -35 oC / 24 hrs.
v) 2N HCl
Ph Et
OH
S
94% yield82% e.e.
EtHO
82% yield78% e.e.
EtHO
R
R yield (%) e.e. (%)3-Me 88 854-Me 92 813-MeO 91 824-MeO 93 80
S
S
Fig. (47).
O O
NH2
Ph 0.5 eq. 30% aq. glyoxal
MeOH / 0 oC r.t. / 2 hrs.N N
O
OO
OMe Me
Ph
Me
MeMe
Me 2.4 eq. NaBH4
MeOH / 50 oC / 3 hrs.
Ph
NH HN
O
OO
O
Ph
Me
MeMe
Me
Ph
Cr(CO)4
1 eq.
THF / reflux / 45 min.
NH HN
O
OO
O
Ph
Me
MeMe
Me
Ph
Cr(CO)4
99% 99%
76%
30Da 32Dk
30Dn 30Do
Fig. (48).

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 147
ronment, a dramatic decreasing of the yields was noticed (compounds 34Eb, c and f).
In more “classic” acetalisation conditions, the N-dichloroacetamido analogue of the less crowded methylseri-nol A was investigated by Pihlaja et al. in 1988 [34c], (Fig. (50)).
The series of 5-acylamino-1,3-dioxanes 34Aa-m were obtained simply with moderate to excellent yields as a single diastereomer. Although the diastereoselectivity was clearly evidenced by 2D-1H,1H-NOESY and 2D-1H,13C-COSY Ex-periments and found in agreement with known A conforma-tional energies data [30], compounds 32Aa-m rapidly epi-merised in solution, e.g. in the slightly acidic CDCl3 (no data in the cited paper). For the synthesis and conformational analysis of 5-substituted 1,3-dioxanes based on serinol S, see Juaristi et al. (1997, [34d]) and Part II of this review.
Targeting new building-blocks for the protein kinase C, in 2000, Peng et al. [34e] described a stereoselective transacetalisation of some N-benzoylated aminodiols ob-tained from L-amino acids, including L-serine, with 1,1,3,3-tetramethoxypropane (Fig. (51)).
Transacetalisation of N-benzoylserinol S with an equimo-lar amount of 1,1,3,3-tetramethoxypropane in various condi-tions gave, throughout, a separable by column chromatogra-phy mixture of four diastereomeric 2,5-disubstituted-1,3-dioxanes, “monomeric” (34Sa-t, -c) and “dimeric” (34Sb-c-c, -t-c). According to the authors, the high stereoselective
character of the depicted syntheses consisted of the dominant incidence of compounds 34Sa-c and 34Sb-t-c (italicised data in Fig. (51)).
Under kinetic control, the selective transacetalisation with ethanol or glycol of 34Sa-c was individually explored providing useful results since all formed diastereomers were isolable materials by column chromatography (Fig. (52)).
For the synthesis of TRIS N-Boc acetonide and its use in the synthesis of other C-substituted 2-aminopropane-1,3-diols, see Ref. [35a, 35b] and Part II of this review. For the synthesis of serinol S N-Cbz derivative preparation and acetalisation see Ref. [34d, 35c] and Part II of this review. For N-acylation of phenylserinolic 5-amino-1,3-dioxanes 30Da, (Fig. (36)), (rac)-30Ea, 30Eb, (Fig. (38)) see Ref. [21a, 35d] and Part II of this review.
2.3. Six Membered Cyclisation of Phenylserinols with Other C-, S- and P-electrophiles
The very few data about the six membered ring closures of phenylserinols D, E with other electrophiles than typical carbonyles (aldehydes and ketones) refer to their “tradi-tional” N-dichloroacetylated derivatives, in Chloromicetyn’s chemistry context.
For example, in an old and peculiar strategy of Chloro-micetyn manufacturing, (Fig. (53)) [36a], cyclisations of the N-dichloroacetylated (rac)-phenylserinol D with phosgene or thionyl chloride were carried out, yielding the carbonate
OH OH
NH-CO-CHCl2
p-NPh
Chloromicetyn
(Chloramphenicol)
2 M R1-CO-R2 / THF / r.t. / 24 hrs.
0.32 g / mL molecular sieves 4 A
+ 0.09 g / mL Amberlyst A 15
O O
R1 R2
p-NPh
NH-CO-CHCl2
R1 R2 (yield %)
Me
Et
i-Bu
-(CH2)5-
-(CH2)4-
Et
Ph
p-MeO-Ph
m-HO-Ph
89
22*
traces
64
80
4
59
80
82
Me
Me
Me
Ph
H
H
H
34Ea
34Eb
34Ec
34Ed
34Ee
34Ef
34Eg
34Eh
34Ei
*isolated as a non-separable diastereomeric mixture 70 (2S,4R,5R) : 30 (2R,4R,5R).
34Ea-i
12
45
Fig. (49).
OHOH
NH2
Me
1.5 eq. Cl2CHCO2Et
reflux / 5 hrs.
82% OHOH
NH-CO-CHCl2
Me
OO
R2
R1Me
Cl2CH-CO-NH1.1 eq. HC(OEt)3
1.1 eq. R1-CO-R2
Ts-OH / reflux / 2 - 8 hrs.
34Aa-m
34Aa 34Ab 34Ac 34Ad 34Ae 34Af
34Ag 34Ah
34Ai 34Aj
34Ak
34Al
34Am
MeEtEtc-C5H9
c-C6H11
c-C7H13
Ph-CH=CHPh
o-EtO-Phm-Cl-Ph
p-MeO-PhMeMe
Me
EtMe
c-C5H9
c-C6H11
c-C7H13
HHHHH
Php-Br-Ph
707250446528
4697
8789
856877
R1 R2 Yield (%)
A
Fig. (50).

148 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
35Da or the sulphite 35Db respectively with good yields. Both 35Da and 35Db were seen as simple O,O,N-protected forms of (rac)-D before nitration. However, as shown in Fig. (53), their subsequent manipulation provided unpromising quantitative results with respect to nitrocarbonate 35Ea or nitrosulphite 35Eb of Chloromicetyn.
Much later on, in 1993, Neidlein et al. [36b] intended to valorise the antibiotic as a chiral auxiliary in the preparation of optically active 1-hydroxyaminophosphonates by a pre-liminary cyclocondensation between optically active
Chloromicetyn and 1-benzyloxyiminophosphonyl dichlo-rides in mild conditions, (Fig. (54)).
It was shown that, though the selected -alkylated (E)-1- benzyloxyiminophosphonyl dichlorides could be conven iently prepared, in three steps, from the corresponding E- oxime-diethylesters via bis-TMS intermediates as unique diastereomers, cyclisations, giving epimeric 1,3,2- dioxaphosphorinanes 35Ec-e, were promising neither from diastereoselectivity nor from global conversion point of view.
CH2OH
H2N H
COOH
L-serine
SOCl2 / MeOH
98%CH2OH
H2N H
COOMePhCOCl
81%CH2OH
Ph-CO-HN H
COOMe
1.25 eq. NaBH4THF / r.t. / 24 hrs. OH
OH
NH
97%
Ph-CO-(MeO)2C C(OMe)21 eq.
cat. conc. HCl / CHCl3 / 40 oC / 12 hrs.
OO
CH2CH(OMe)2HNPh-CO-
OO
NHPh-CO-
CH2CH(OMe)2
OO
OO
HN NH-CO-Ph
34Sa-t* 34Sa-c
Ph-CO-
OO
OO
NH-CO-Ph
HNPh-CO-
34Sb-c-c 34Sb-t-c
1 2 3
2
5
25
55
2 2' 2 2'5'5'
*descriptors -c(cis) and -t(trans) with respect to the orientation of the ligands at positions 2(') and 5(').
T (oC) Time (hrs.) Cat. 34Sa-t 34Sa-c 34Sb-c-c 34Sb-t-c
6040
2040
4040
40
1.0*
1.95.2
2.51.4
1.02.9
HCl
HClHCl
TsOHH3PO4
TFAH2SO4
8
1224
1224
368
2.6
6.3
8.6
9.7
4.4
2.44.2
2.21.0
1.01.0
1.02.6
1.0
6.6
3.32.3
2.52.6
8.3
1.8
N-benzoylserinol S
*contents as molar ratios
Fig. (51).
O O
NH-CO-Ph
CH2CH(OMe)2
34Sa-c
8.5 eq. EtOH / cat. HCl
CHCl3 / 20 oC / 10 hrs.O O
NH-CO-Ph
CH2CH(OMe)(OEt)
34Sd-c, -t (cis:trans 82 : 18, 82%)
1 eq. glycol / cat. HCl
CHCl3 / 20 oC / 10 hrs.O O
NH-CO-Ph
O
O
34Sc-c, -t(cis:trans 67 : 33, 90%*) *global conversion
Fig. (52).

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 149
ABBREVIATIONS
A = 2-amino-2-methylpropane-1,3-diol (Methylserinol) and its series of de-rivatives
A (value) = conformational energy
AM-1 = Austin Model 1
B = 2-amino-2-hydroxymethylbutan-1-ol (Ethylserinol) and its series of de-rivatives
C = 2-amino-2-hydroxymethylpropane-1,3-diol (TRIS
®, THAM) and its se-
ries of derivatives
CIPE = Complex Induced Proximity Effect
D = (1S,2S)- or (1R,2R)-2-amino-1-phenylpropane-1,3-diol (Threo-phenylserinol) and its series of derivatives
D’ = (1R,2S)-2-amino-1-phenylpropane-1,3-diol (Erythro-phenylserinol) and its series of derivatives
DFT = Density Functional Theory
DOABO = 3,7-dioxa-r-1-azabicyclo[3.3.0]-c-5-octane type derivatives
(2H)DOABO-CH2OH = 5-hydroxymethyl-3,7-dioxa-r-1-azabicyclo[3.3.0]-c-5-octane
(2Ph)DOABO-CH2OH = (1r,2R,5s,8S)-5-hydroxymethyl-2,8-diphenyl-3,7-dioxa-1-azabicyclo[3.3.0] octane
DoM = Directed ortho-Metallation
DoMG = Directed ortho-Metallation Group
DX-SB = 1,3-Dioxanic Schiff-Base type de-rivatives
E = (1S,2S)- or (1R,2R)-2-amino-1-(4-nitrophenyl)-propane-1,3-diol (Threo-nitrophenylserinol) and its series of derivatives
EQ = Tautomeric equilibrium
(R)-MTPA-Cl = (R)-3,3,3-Trifluoro-2-methoxy-2-phenylpropionic acid chloride
p-NPh = para-nitrophenyl
OX = 1,3-oxazolidine type derivatives
OX-OX = double 1,3-oxazolidine type deriva-tives
S = 2-aminopropane-1,3-diol (Serinol) and its type of derivatives
SB = Schiff-Base type derivatives
SB-OX = Schiff-Base - 1,3-oxazolidine type derivatives
SB-SB = double Schiff-Base type derivatives
REFERENCES
[1] a) Senkus, M. Some new derivatives of amino hydroxy compounds. J. Am. Chem. Soc., 1945, 67, 1515-1519; b) Tilford, H.C.; Van Campen, Jr., M.G.; Shelton, R.S. Aminoesters of substituted ali-cyclic carboxylic acids. J. Am. Chem. Soc., 1947, 69, 2902-2906; c) Pierce, J.S.; Wotiz, J. Tris-(hydroxymethyl)-aminomethane deriva-tives. II. Alkylation products. J. Am. Chem. Soc., 1951, 73, 2594;
OH OH
NH-CO-CHCl2
Ph
OX
O
NH-CO-CHCl2
Ph
OX
O
NH-CO-CHCl2
p-NPh
X = C, 35Da (87%) X = S, 35Db (85%)
X = C, 35Ea (19%)X = S, 35Eb (18%)
OH OH
NH-CO-CHCl2
p-NPh
(rac)-N-dichloroacetylatedlike (l) ("threo")-D
(rac)-Chloromicetyn
like (l) ("threo")
R*
R* R*R*
O O
i: (X = C) 2 eq. COCl2 / Py / 0 - 5 oC
(X = S) 9 eq. SOCl2 / r.t.
i
Cl-X(=O)-Cl
ii: (X = C) 27 eq. HNO3 / -20 oC
(X = S) 43 eq. HNO3 / -30 oC
ii
HNO3 100%iii
H+
iii: HCl / EtOH / r.t.
Fig. (53).
R P(O)Cl2
NBnO
O
P O
NH-CO-CHCl2
p-NPh
O
R
NBnO
O
P O
p-NPh
NH-CO-CHCl2
O
R
NBnO
24
52 4
5
1 eq.
3 eq. Et3N / THF
r.t. / 3 hrs.
OH OH
p-NPh
Cl2CH-CO-NH
Chloromicetyn
c R = Me 30.2%* 12.0% d R = Et 35.4% 14.7%e R = i-Pr 25.3% 9.0%
35Ec-e E-2R,4R,5R E-2S,4R,5R
*partial conversions of Chloromicetyn
Fig. (54).

150 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
d) Pierce, J.S.; Lunsford, C.D.; Raiford, Jr., R.W.; Rush, J.L.; Riley, D.W. Tris-(hydroxymethyl)-aminomethane derivatives. III. Oxamides, ureas, oxazolidines and 1-aza-3,7-dioxabicyclo(3.3.0)octanes. J. Am. Chem. Soc., 1951, 73, 2595-2596; e) Pierce, J.S.; Lunsford, C.D. Tris-(hydroxymethyl)-aminomethane derivatives. IV. Substituted 4-hydroxymethyloxazolidines; ester and amide interchange. J. Am.
Chem. Soc., 1951, 73, 2596-2598; f) Bergmann, E.D. The oxa-zolidines. Chem. Rev., 1953, 53, 309-353.
[2] a) Ehrlich, J.; Bartz, Q.R.; Smith, R.M.; Josylyn, D.A.; Burkholder, P.R. Chloromycetin, a new antibiotic from a soil Actinomycete. Science, 1947, 106, 417; b) Smadel, J.E.; Jackson, E.B. Chloromy-cetin, an antibiotic with chemotherapeutic activity in experimental rickettsial and viral infections. Science, 1947, 106, 418-419; c) Rebstock, M.C.; Harry, M.; Crooks, M.H.; Controulis, J.; Bartz, R.Q. Chloramphenicol (Chloromycetin).1.IV.1a Chemical studies. J. Am. Chem. Soc., 1949, 71, 2458-2462; d) Controulis, J.; Rebstock, M.C.; Crooks, H.M. Chloramphenicol (Chloromycetin).1.V. Syn-thesis. J. Am. Chem. Soc., 1949, 71, 2463-2468; e) Long, M.L.; Troutman, H.D. Chloramphenicol1 (Chloromycetin). VI. A syn-thetic approach. J. Am. Chem. Soc., 1949, 71, 2469-2472; f) Long, M.L.; Troutman, H.D. Chloramphenicol1 (Chloromycetin). VII. Synthesis through p-nitroacetophenone. J. Am. Chem. Soc., 1949, 71, 2473-2475.
[3] Darabantu, M.; Mager, S.; Plé, G.; Puscas, C. Heterocyclic satu-rated compounds as derivatives or precursors of Chloromycetine and of some related structures. Heterocycles, 1995, 41, 2327-2356 and the literature cited therein.
[4] Valters, R.; Flitsch, W. Ring-Chain Tautomerism, Plenum Press: New-York, 1985.
[5] Lázár, L.; Fülöp, F. Recent developments in the ring-chain tautomerism of 1,3-heterocycles. Eur. J. Org. Chem., 2003, 16, 3025-3042.
[6] a) Baldwin, J.E. Rules for ring closure. J. Chem. Soc. Chem. Comun., 1976, 18, 734-736; b) Baldwin, J.E.; Cutting, J.; Dupont, W.; Kruse, L.; Silberman, L.; Thomas, R.C. 5-Endo-trigonal reac-tions: a disfavoured ring closure. J. Chem. Soc. Chem. Commun., 1976, 18, 736-738.
[7] Laurent, P.A.; Riehl, M.; Frazão, C.S. Recherches sur les dérivés d’ammonium quaternaries des méthyl-5, éthyl-5 et hydroxyméthyl-5 (3,7)-dioxa-1-aza [3.3.0]octanes. Bull. Soc. Chim. Fr., 1967, 10, 3868-3872.
[8] Crabb, T.A.; Hall, M.J.; Williams, R.O. Proton magnetic resonance studies of compounds with bridgehead nitrogen atoms-XXVI: con-figurational studies with derivatives of perhydro-oxazolo[3,4-c]oxazole and the conformational analysis of perhydro-imidazo[1,5-c]thiazole. Tetrahedron, 1973, 29, 3389-3398.
[9] Howe, R.A.; Crowther, A.F.; Stephenson, J.S.; Rao, B.S.; Smith, L.H. Beta.-Adrenergic blocking agents. I. Pronethalol and related N-alkyl and N-aralkyl derivatives of 2-amino-1-(2-naphthyl)ethanol. J. Med. Chem., 1968, 11, 1000-1008.
[10] a) Broadbent, H.S.; Burnham, W.S.; Sheely, R.M.; Olsen, R.K. Novel heterotricyclic systems: 2,6-Dioxa- and 2-oxa-6-thia-10-azatricyclo-[5.2.1.04,10]decanes; 2,6-dioxa-11-azatricyclo[5.3.1.0. 4,11]undecane; and 9,13-dioxa-14-azatetracyclo[6.5.1.02,7.011,14]tetra-deca-2,4,6-triene (1a, b). J. Heterocycl. Chem., 1976, 13, 337-348; b) Schipchlander, M.T. The reaction of -aminoalcohols with o-phthalaldehyde. J. Heterocycl. Chem., 1977, 14, 305-306.
[11] a) Crozet, M.; Nouguier, R. Réactivité du tris(hydroxyméthyl) aminométhane avec des nitro-aldéhydes aromatiques. Synthèse de nouveaux composés antimicrobiens. Tetrahedron Lett., 1985, 26, 5523-5524; b) Vanelle, P.; De Meo, M.P.; Maldonado, J.; Nouguier, R.; Crozet, M.P.; Laget, M.; Dumenil, G. Genotoxicity in oxazolidine derivatives: influence of the nitro group. Eur. J. Med. Chem., 1990, 25, 241-250.
[12] a) Buur, A.; Bundgaard, H. Prodrugs as drug delivery systems 66. Hydrolysis of various oxazolidines and N-acylated oxazolidines–potential prodrug types for -aminoalcohols or carbonyl-containig drugs. Arch. Pharm., Sci. Ed., 1987, 15, 76-86; b) Mattson, A.; Norin, T. A convenient synthesis of 5-hydroxymethyl-1-aza-3,7-dioxabicyclo[3.3.0]octane and its -methoxyethoxy-methyl deriva-tive. Synth. Commun., 1994, 24, 1489-1491.
[13] a) Monge, S.; Sélambaron, J.; Carré, F.; Verducci, J.; Roque, J-P.; Pavia, A.A. Anomeric effects in non-carbohydrate compounds: conformational differences between the oxazolidine rings of a cis-fused bicyclic system. Carbohydr. Res., 2000, 328, 127-133; b) Sé-
lambaron, J.; Carré, F.; Fruchier, A.; Roque, J-P.; Pavia, A.A. Stereochemical study of 1,3-N,X-heterocycles derived from -aminoacids and formaldehyde. Structural evidence for the existence of the anomeric effect. Tetrahedron, 2002, 58, 4439-4444; c) Sé-lambaron, J.; Monge, S.; Carré, F.; Roque, J-P.; Pavia, A.A. Stereoelectronic control of oxazolidine ring-opening: structural and chemical evidences. Tetrahedron, 2002, 58, 9559-9566.
[14] Bonnet, D.; Pascal, J.; Gras-Masse, H.; Melnyk, O. Synthesis of an amphiphilic aldehyde using as a key step the condensation of a lipophilic glyoxylic acid amide derivative with tris(hydroxymethyl)aminomethane. Tetrahedron Lett., 2001, 42, 1875-1877.
[15] a) Darabantu, M.; Plé, G.; Maiereanu, C.; Silaghi-Dumitrescu, I.; Ramondenc, Y.; Mager, S. Synthesis and stereochemistry of some 1,3-oxazolidine systems based on TRIS ( , , -trimethylolamino-methane) and related aminopolyol-skeletons (II). 1-Aza-3,7-dioxabicyclo[3.3.0]octanes. Tetrahedron, 2000, 56, 3799-3816; b) Maiereanu, C.; Darabantu, M.; Plé, G.; Berghian, C.; Condamine, E.; Ramondenc, Y.; Silaghi-Dumitrescu, I.; Mager, S. Ring-chain tautomerism and other versatile behaviour of 1,4-diimino- and 1,2-phenylene derivatives of some C-substituted serinols. Tetrahedron, 2002, 58, 2681-2693; c) Maiereanu, C.; Silaghi-Dumitrescu, I.; Berghian, C.; Pintea, M.; Fazekas, M.; Darabantu, M. Stereocon-trolled synthesis by anomeric effects of substituted 1-aza-3,7-dioxabicyclo [3.3.0]octanes. Stud. Univ. “Babes-Bolyai”, Serie
Chem. XLVIII, 2003, 48, 103-111; d) Maiereanu, C.; Condamine, E.; Silaghi-Dumitrescu, I.; Darabantu, M. 1-Aza-5-hydroxymethyl-3,7-dioxabicyclo[3.3.0]octanes: chelating properties related to their conformational chirality. Stud. Univ. “Babes-Bolyai”, Serie
Chem. XLVIII, 2003, 48, 91-101; e) Sarbu, C.; Casoni, D.; Dara-bantu, M.; Maiereanu, C. Quantitative structure-retention activity relationship of some 1,3-oxazolidine systems by RP-HPTLC and PCA. J. Pharm. Biomed. Anal., 2004, 35, 213-219; f) Darabantu, M.; Maiereanu, C.; Silaghi-Dumitrescu, I.; Toupet, L.; Condamine, E.; Ramondenc, Y.; Berghian, C.; Plé, G.; Plé, N. 3,7-Dioxa-1-azabicyclo[3.3.0]octanes substituted at C-5 position. From local to global stereochemistry. Eur. J. Org. Chem., 2004, 12, 2644-2661; g) Brush, J.R.; Magee, R.J.; O’Connor, M.J.; Teo, S.B.; Geue, R.J.; Snow, M.R. Nature of the copper(II) complex formed in the reaction of formaldehyde with bis((S)-serinato)copper(II). J. Am. Chem. Soc., 1973, 95, 2034-2035; h) Geue, R.J.; Snow, M.R.
Crystal studies of the reaction products of chelate complexes. I. The structure of bis(dihydro-1H,3H,5H-oxazolo[3,4-c]oxazole-7a-carboxylato)copper(II). Acta Cryst., 1977, B33, 510-516; i) Maiereanu, C.; Darabantu, M.; Condamine, E.; Plé, G.; Ramon-denc, Y.; Fazekas, M.; Pintea, M.; Berghian, C. First example of long distance stereocontrolled synthesis in 1-aza-3,7-dioxabicyclo[3.3.0.]octane series. Heterocyclic Commun., 2005, 11, 305-310.
[16] a) Levkovskaya, G.G.; Guseva, S.A.; Kazimirovskaya, V.B.; Kholdeeva, L.N.; Voronkov, M.G.; Mirskova, A.N. 2-Hydroxyalkylammonium arylihtoacetates and their effects on the functional activity of thrombocytes. Pharm. Chem. J., 1986, 20, 180-184 (Translated from Khim. Farm. Zh., 1986, 20, 295-300); b) Sokolov, V.B.; Krykov, L.N.; Koloniets, A.A.; Sololsky, G.A. In-teraction of trimethylolaminomethane (TMAM) with cyclanones. Zh. Org. Khim., 1979, 15, 1777-1779.
[17] a) Darabantu, M.; Plé, G.; Silaghi-Dumitrescu, I.; Maiereanu, C.; Turos, I.; Silberg, I.A.; Mager, S. Synthesis and stereochemistry of some 1,3-oxazolidine systems based on TRIS ( , , -trimethylolaminomethane) and related aminopolyol-skeletons (I). (Di)spirooxazolidines. Tetrahedron, 2000, 56, 3785-3798; b) Eliel, L.E.; Wilen, H.S. Stereochemistry of the Organic Compounds, John Wiley & Sons, Inc.: New-York, 1994, pp. 21, 466, 696, 699, 700, 704-706, 769-771, 1199, 1191.
[18] a) Bergman, E.D.; Resnick, H. The -phenylserine series. Part IV. J. Chem. Soc., 1956, 1662-1665; b) Edgerton, W.H.; Fisher, J.R. The preparation of several substituted 1-phenyl-1,3-propanediol amines. J. Org. Chem., 1954, 19, 593-598; c) Edgerton, W.H.; Fisher, J.R.; Moersch, G.W. Chloramphenicol. 2,4,8-substituted-1-aza-3,7-dioxabicyclo[3.3.0]octanes. J. Am. Chem. Soc., 1957, 79, 6487-6490; d) Pedrazzoli, A.; Tricerri, S. Derivate des l-(p-nitrophenyl)-2-amino-1,3-propandiols: Schiff'sche basen; l-aza-2,8-diaryl-6-(p-nitrophenyl)-3,7-dioxabicyclo-[3,3,0]-octane; oxa-zolidine. Helv. Chim. Acta., 1956, 39, 965-976; e) Potapov, V.M.; Demyanovich, V.M.; Zaitsev, V.P.; Sharipova, S.Kh. Synthesis and

(Masked)serinols: Molecules, Biomolecules, Building-Blocks Current Organic Synthesis, 2010, Vol. 7, No. 2 151
chiral-optical characteristics of Schiff bases from (+)-threo-1-(4-nitrophenyl)-2-amino-1,3-propandiol. Zh. Org. Khim., 1990, 26, 2574-2579; f) Zaitsev, V.P.; Sharipova, S.K.; Zhuravleva, I.I. Levomycetin and threo-2amino-1-(4-nitrophenyl)-1,3-propandiol. Pharm. Chem. J., 1998, 32, 157-160.
[19] a) Darabantu, M.; Mager, S.; Puscas, C.; Bogdan, M.; Cotora, E.; Plé, G.; Bratu, I. Heterocyclic saturated compounds (1,3-dioxanes and 1,3-oxazolidines) by diastereospecific ring closure of threo-2-amino-1-(p-nitrophenyl)-propane-1,3-diol (I). Rev. Rom. Chim., 1994, 39, 955-965; b) Darabantu, M.; Plé, G.; Mager, S.; Cotora, E.; Gaina, L.; Costas, L.; Mates, A. Synthesis and stereochemistry of some heterocyclic saturated compounds based on l-p-nitrophenylserinol skeleton (I). Ring-chain tautomerism of some Schiff-bases of l-p-nitrophenylserinol. Tetrahedron, 1997, 53, 1873-1890; c) Darabantu, M.; Plé, G.; Mager, S.; Gaina, L; Cotora, E.; Mates, A.; Costas, L. Synthesis and stereochemistry of some heterocyclic saturated compounds based on l-p-nitrophenylserinol skeleton (II). 1-Aza-3,7-dioxabicyclo[3.3.0]octanes. Tetrahedron, 1997, 53, 1891-1908; d) Martinek, T.; Làzàr, L.; Fülöp, F.; Riddell, F.G. Five-component equilibria of ring-chain tautomeric mixtures derived from 2-amino-1-phenyl-1,3-propanediol diastereomers. Tetrahedron, 1998, 54, 12887-12896; e) Brown, H.C.; Okamoto, Y.; Inukai, T. Rates of solvolysis of the m- and p-phenyl-, m- and p-methylthio-, and m- and p-trimethylsilylphenyldimethylcarbinyl chlorides. Steric inhibition of resonance as a factor in electrophilic substituent constants. J. Am. Chem. Soc., 1958, 80, 4964-4968; f) Okamoto, Y.; Inukai, T.; Brown, H.C. Rates of solvolysis of phen-yldimethylcarbinyl chlorides containing meta directing substitu-ents. J. Am. Chem. Soc., 1958, 80, 4969-4972; g) Okamoto, Y.; Ynukai, T.; Brown, H.C. Rates of solvolysis of substituted phen-yldimethylcarbinyl chlorides in methyl, ethyl and isopropyl alco-hols. Influence of the solvent on the value of the electrophilic sub-stituent constant. J. Am. Chem. Soc., 1958, 80, 4972-4976; h) Brown, H.C.; Okamoto, Y. Rates of solvolysis of phenyldimethyl-carbinyl chlorides containing substituents (-NMe3
+, -CO2-) bearing
a charge. J. Am. Chem. Soc., 1958, 80, 4976-4979; i) Brown, H.C.; Okamoto, Y. Electrophilic substituent constants. J. Am. Chem.
Soc., 1958, 80, 4979-4987. [20] a) Couquelet, J.; Madesclaire, M.; Leal, F.; Zaitsev, V.P.; Shari-
pova, S. Kh. Regioselectivity in the reaction of paraformaldehyde with (1S,2S)-2-aryl(hetaryl)methylamino-1-(4-nitrophenyl)-1,3-propanediols. Chem. Heterocycl. Compd., 1999, 35, 474-478 (Translated from Khim. Geterotsikl. Soedin., 1999, 4, 536-541); b) Couquelet, J.; Madesclaire, M.; Leal, F.; Zaitsev, V.P.; Sharipova, S.Kh. Synthesis and stereochemistry of (1S,3S,4S,7R,11R)-3-(4-nitrophenyl)-11-aza-2,6-dioxatricyclo[5.3.1.04,11]undecane. Chem. Heterocyclic Compds., 2001, 37, 898-902; c) Madesclaire, M.; Couquelet, J.; Leal, F.; Zaitsev, V.P.; Sharipova, S. Kh. Hydrazi-nolysis of compounds containing oxazolidine ring. Chem. Hetero-
cycl. Compd., 2002, 38, 71-73; d) Madesclaire, M.; Coudert, P.; Zaitsev, V.P.; Zaitseva, V.Yu. Regioselectivity of the interaction of (1S,2S)-2-amino-1-(4-nitrophenyl)-1,3-propanediol with some symmetrical ketones. Chem. Heterocycl. Compd., 2004, 40, 1310-1314 (Translated from Khim. Geterotsikl. Soedin., 2004, 10, 1518-1523); e) Madesclaire, M.; Coudert, P.; Zaitsev, V.P.; Zaitseva, J.V. Synthesis of 2-oxazolidinones from (1S,2S)-2-amino-1-(4-nitrophenyl)-1,3-propanediol. Chem. Heterocycl. Compd., 2006, 42, 506-511 (Translated from Khim. Geterotsikl. Soedin., 2006, 4, 579-584); f) Madesclaire, M.; Coudert, P.; Gaumet, V.; Zaitsev, P.V.; Zaitseva, V.Yu. Diastereoselective reaction of (1S,2S)-2-amino-1-(4-nitrophenyl)-1,3-propanediol with aromatic aldehydes. Synthesis of (1R,2R,4S,5S,8S)-2,8-diaryl-4-(4-nitrophenyl)-1-aza-3,7-dioxabicyclo[3.3.0]octanes. Chem. Heterocycl. Compd., 2006,
42, 665-670 (Translated from Khim. Geterotsikl. Soedin., 2006, 5, 757-763); g) Madesclaire, M.; Coudert, P.; Goumet, V.; Zaitsev, V.P.; Zaitseva, J.V. X-ray Structural analysis of (1R,2R,4S,5S,8S)-2,8-bis(4-chlorophenyl)-4-(4-nitrophenyl)-1-aza-3,7-dioxabicyclo [3,3,0]octane. Chem. Heterocycl. Compd., 2006, 42, 813-817
(Translated from Khim. Geterotsikl. Soedin., 2006, 6, 930-934. An erratum to this article is available at http://dx.doi.org/10.1007/ s10593-009-0240-8); h) Madesclaire, M.; Zaitsev, V.P.; Zaitseva, J.V.; Sharipova, S.Kh. Synthesis of isomeric 2-oxazolidinones from (1R,2R)- and (1S,2S)-2-amino-1-(4-nitrophenyl)-1,3-propanediols. Chem. Heterocycl. Compd., 2007, 43, 1325-1332 (Translated from Khim. Geterotsikl. Soedin., 2007, 10, 1562-1570).
[21] a) Darabantu, M.; Mager, S.; Puscas, C.; Bogdan, M.; Plé, G.; Cotora, E.; Kovacs, D. Heterocyclic saturated compounds (1,3-dioxanes and 1,3-oxazolidines) by diastereospecific ring closure of threo-2-amino-1-(p-nitrophenyl)-propane-1,3-diol (II). Amino- and 5-acylamino derivatives. Rev. Rom. Chim., 1995, 40, 453-461; b) Darabantu, M.; Plé, G.; Gaina, L.; Maiereanu, C.; Silaghi-Dumitrescu, I. Synthesis and ring-ring tautomerism of some spi-rooxazolidines based on l-p-nitrophenylserinol skeleton. Stud. Univ.“ Babes-Bolyai”, Serie Chem., 1998, 43, 179-192; c) Maiere-anu, C.; Darabantu, M.; Plé, G.; Ramondenc, Y.; Berghian, C. Ring-ring tautomerism of some spirooxazolidines derived from l-phenylserinols. Rev. Rom. Chim., 2005, 50, 29-40; d) Shan, Z.; Wan, B.; Wang, G. A Highly efficient chemoselective cyclocon-densation of threo-(1S,2S)-2-amino-1-(4-nitrophenyl)-1,3-propanediol with ketones and isomerization of the condensates. Helv. Chim. Acta, 2002, 85, 1062-1068; e) Liu, D.; Shan, Z.; Liu, F.; Xiao, C.; Lu, G.; Qin, J. A new and practical method for prepar-ing enantiomerically pure [1,1 -Binaphthalene]-2,2 -diol: resolu-tion of racemic [1,1 -binaphthalene]-2,2 -diol with threo-(1S,2S)-2-amino-1-(4-nitrophenyl)propane-1,3-diol cyclohexanone conden-sate. Helv. Chim. Acta, 2003, 86, 157-163; f) Maiereanu, C.; Dara-bantu, M.; Plé, G.; Ramondenc, Y.; Toupet, L. 3,7-Dioxa-1-azabicyclo[3.3.0]octan-2-ones based on l-phenylserinols via the ring-ring or ring-chain tautomerism of their imino-derivatives. Rev. Rom. Chim., 2005, 50, 103-112.
[22] a) Star, A.; Fuchs, B. Mechanism of formation and stabilities of the new dioxadiazadecalin systems. Ring-chain tautomerism. J. Org.
Chem, 1999, 64, 1166-1172; b) Star, A.; Goldberg, I.; Lemcoff, N.G.; Fuchs, B. New supramolecular host systems. The stereoiso-meric diaminobutanediol and dioxadiazadecalin systems: synthesis, structure, stereoelectronics, and conformation-theory vs. experi-ment. Eur. J. Org. Chem., 1999, 9, 2033-2043; c) Star, A.; Gold-berg, I.; Fuchs, B. Dioxadiazadecalin / salen tautomeric macrocy-cles and complexes: prototypal dynamic combinatorial virtual li-braries. Angew. Chem., Int. Ed. Engl., 2000, 39, 2685-2689.
[23] a) Loncaric, C.; Wulff, D.W. An efficient synthesis of ( )-Chloramphenicol via asymmetric catalytic aziridination: a com-parison of catalysts prepared from triphenylborate and various lin-ear and vaulted biaryls. Org. Lett., 2001, 3, 3675-3678; b) Smukste, I.; Smithrud, D.B. Condensation reactions of calix[4]arenes with unprotected hydroxyamines, and their resulting water solubilities. Tetrahedron, 2001, 57, 9555-9561; c) Sugiyama, S.; Watanabe, S.; Ishii, K. Asymmetric synthesis of the 4-hydroxymethyl-2-oxazolidinone from the serinol derivative and chloroformates. Tet-rahedron Lett., 1999, 40, 7489-7492; d) Sugyama, S.; Inoue, T.; Ishii, K. One-pot acylation and fractional crystallization: prepara-tion of optically active serinol monobenzoates. Tetrahedron Asym-
metry, 2003, 14, 2153-2160; e) Neri, C.; Williams, J.M.J. Prepara-tion of 2-oxazolidinones by enzymatic desymmetrisation. Tetrahe-
dron Asymmetry, 2002, 13, 2197-2199. [24] a) Biekert, E.; Hoffmann, D.; Enslein, L. Über 1.4-oxazine, IV.
Kondensation von 1,2-amino-alkoholen mit -ketocarbonsäure-estern zu 5,6-dihydro-1.4-oxazinonen-(2). Chem. Ber., 1961, 94, 2778-2785; b) Biekert, E.; Sonnenbichler, J. Über 1.4-oxazine, V. 3-Phenyl-5,6-dihydro-1.4-oxazinone-(2) und phenylglyoxylsäure-[2-hydroxy-alkylamide]. Chem. Ber., 1961, 94, 2785-2791; c) Largeron, M.; Dupuy, H.; Fleury, M.B. Novel 1,4-Benzoxazine de-rivatives of pharmacological interest. Electrochemical and chemi-cal syntheses. Tetrahedron, 1995, 51, 4953-4968; d) Cristea, C.V.; Moinet, C.; Jitaru, M.; Darabantu, M. Electroreduction of (1S,2S)-2-amino-1-(4-nitrophenyl)-propane-1,3-diol derivatives. Behaviour of electrogenerated species and application to organic synthesis. J. Appl. Electrochem., 2005, 35, 845-849.
[25] a) Berghian, C.; Maiereanu, C.; Plé, N.; Plé, G.; Darabantu, M. First example of selective nucleophilicity of 1-aza-5-hydroxymethyl-3,7-dioxabicyclo[3.3.0]octanes in alkoxide form. Stud. Univ. “Babes-Bolyai”, Serie Chem., 2003, 48, 113-125; b) Berghian, C.; Darabantu, M.; Lameiras, P.; Plé, N.; Turck, A. First examples of the conformation chirality of heterobicy-clo[3.3.0]octanes: 3,7-dioxa-r-1-azabicyclo[3.3.0]oct-c-5-yl-methoxypyrazines.Heterocycl. Commun., 2005, 11, 517-522; c) Berghian, C.; Plé, N.; Turck, A.; Darabantu, M. First report on 3,7-dioxa-r-1-azabicyclo[3.3.0]oct-c-5-ylmethoxy system substituting s-triazine. Stud. Univ.“Babes-Bolyai” Serie Chem., 2005, 1, 201-207; d) Berghian, C.; Lameiras, P.; Toupet, L.; Condamine, E.; Plé, N.; Turck, A.; Maiereanu, C.; Darabantu, M. -(3,7-Dioxa-r-1-

152 Current Organic Synthesis, 2010, Vol. 7, No. 2 Mircea Darabantu
azabicyclo[3.3.0]oct-c-5-ylmethoxy)-diazines (I): Synthesis and stereochemistry. Extension to s-triazine series. Tetrahedron, 2006, 62, 7319-7338; e) Darabantu, M.; Boully, L.; Turck, A.; Plé, N. Synthesis of new polyaza heterocycles. Diazines. Part 42. Tetrahe-dron, 2005, 61, 2897-2905; f) Stille, J.K. The Palladium-catalyzed cross-coupling reactions of organotin reagents with organic elec-trophiles [New synthetic methods (58)] Angew. Chem., Int. Ed.
Engl., 1986, 25, 508-524; g) Berghian, C.; Condamine, E.; Plé, N.; Turck, A.; Silaghi-Dumitrescu, I.; Maiereanu, C.; Darabantu, M. -(3,7-Dioxa-r-1-azabicyclo[3.3.0]oct-c-5-ylmethoxy)-diazines (II): Functionalisation via directed ortho-metallation and cross-coupling reactions. Tetrahedron, 2006, 62, 7339-7354.
[26] a) Snieckus, V. Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aro-matics. Chem. Rev., 1990, 90, 879-933; b) Turck, A.; Plé, N.; Mongin, F.; Quéguiner, G. Advances in the directed metallation of azines and diazines (pyridines, pyrimidines, pyrazines, pyridazines, quinolines, benzodiazines and carbolines). Part 2: Metallation of pyrimidines, pyrazines, pyridazines and benzodiazines. Tetrahe-
dron, 2001, 57, 4489-4505; c) Quéguiner, G.; Marsais, F.; Sniekus, V.; Epsztajn, J. Directed metalation of pi-deficient azaaromatics: strategies of functionalization of pyridines, quinolines and diazines. Adv. Heterocycl. Chem., 1991, 52, 187-304; d) Berghian, C.; Maiereanu, C.; Plé, N.; Turck, A.; Condamine, E.; Darabantu, M. First example of 1-aza-3,7-dioxabicyclo[3.3.0]octane-5-yl-methoxy system as directed ortho-metallation group. Stud. Univ. “Babes-Bolyai”, Serie Chem., 2003, 48, 127-138; e) Ward, J.S.; Merritt, L. The convergent syntheses of pyrazinyl and quinoxalinyl phen-ylmethanones from 2-lithio pyrazines and quinoxalines. J. Hetero-
cycl. Chem., 1991, 28, 765-768; f) Turck, A.; Trohay, D.; Mojovic, L.; Plé, N.; Quéquiner, G. Metalation of diazines: IV. Lithiation of sym-disubstituted pyrazines. J. Organomet. Chem., 1991, 412, 301-310.
[27] a) Nordin, I.C.; Thomas, J.A. An improved synthesis of (4S,5S)-2,2-dimethyl-4-phenyl-1,3-dioxan-5-amine. Tetrahedron Lett., 1984, 25, 5723-5724; b) Weinges, K.; Klotz, K.-P.; Droste, H. Asymmetrische synthesen, V. Optisch aktive 5-amino-4-phenyl-1,3-dioxane und deren einfluß auf die stereoselektivität der asym-metrischen Strecker-synthese. Chem. Ber., 1980, 113, 710-721; c) Weinges, K.; Blackholm, H. Asymmetrische synthesen, VII. Her-stellung optisch aktiver aminonitrile. Chem. Ber., 1980, 113, 3098-3201; d) Weinges, K.; Stemmle, B. Asymmetrische synthesen, III. Die asymmetrische Strecker-synthese von aliphatischen -methyl-
-aminosäuren. Chem. Ber., 1973, 106, 2291-2297. [28] a) Darabantu, M.; Mager, S.; Puscas, C.; Plé, G.; Bogdan, M.;
Cotora, E. Heterocyclic saturated compounds (1,3-dioxanes and 1,3-oxazolidines) by diastereospecific ring closure of threo-2-amino-1-(p-nitrophenyl)-propane-1,3-diol (III). Some N, N’, N”-bis(tris)amides and Schiff-bases. Rev. Rom. Chim., 1995, 40, 907-915; b) Darabantu, M.; Maiereanu, C.; Plé, G.; Berghian, C.; Con-damine, E.; Ramondenc, Y. Convenient synthesis of chiral 5-amino-1,3-dioxanes built on some l-p-nitrophenylserinols skele-tons. Heterocycl. Commun., 2001, 7, 593-598; c) Fazekas, M.; Pin-tea, M.; Lameiras, P.; Lesur, A.; Berghian, C.; Silaghi-Dumitrescu, I.; Plé, N.; Darabantu, M. Serinolic amino-s-triazines: iterative syn-thesis of N-substituted amino-1,3-dioxane derivatives from l-(p-nitrophenyl)serinols and rotational stereochemistry phenomena. Eur. J. Org. Chem., 2008, 14, 2473-2494; d) Sokolov, L.B.; Nichu-govskaya, K.M.; Karpenko, M.P. Quaternary salts of D-( )-1-(p-nitrophenyl)-2-dimethylamino-propandiol-1,3. Pharm. Chem. J., 1978, 12, 593-594 (Translated from Khim.-Farm. Zh., 1978, 12, 45-47) ; e) Darabantu, M.; Plé, G.; Mager, S.; Puscas, C.; Cotora, E. Synthesis and stereochemistry of some heterocyclic saturated compounds based on l-p-nitrophenylserinol skeleton (III). 1,3-Dioxanic Schiff-bases. Tetrahedron, 1997, 53, 1909-1922; f) Roz-wadowska, M.D. Reaction of (1S,2S)-2-amino-1-phenyl-1,3-
propanediol with thioacids under the Mitsunobu reaction condition. Tetrahedron, 1997, 53, 10615-10622; g) Gawley, R.E.; Aubé, J. Principles of Asymmetric Synthesis, Pergamon, Tetrahedron Or-
ganic Chemistry Series, Elsevier Science Ltd: Oxford, 1996, vol. 14, p. 11; h) Gabler, W.; Hauptmann, S. Zur reaktion von tris-(hydroxymethyl)-aminomethan (Tris) mit einigen aldehyden. Z. Naturforsch, 1968, 23b, 111-112.
[29] Bertz, S.H.; Dabbagh, G.; Sundararajan, G. Asymmetric induction with amidocuprates. J. Org. Chem., 1986, 51, 4953-4959.
[30] Anteunis, M.J.O.; Tavernier, D.; Borremans, F. Conformational analysis of 1,3-dioxanes. Heterocycles, 1976, 4, 293-371.
[31] a) Enders, D.; Schankat, J. 79. Enantioselektive synthese von allyl-, propargyl- und 4-en-2-inyl-aminen durch 1,2-addition von or-ganocer-reagenzien an chorale aldimine. Helv. Chim. Acta, 1995, 78, 970-992; b) Enders, D.; Breuer, K.; Runsink, J.; Teles, H. The first asymmetric intramolecular Stetter reaction. Preliminary com-munication. Helv. Chim. Acta, 1996, 79, 1899-1902; c) Dale, J.A.; Dull, D.L.; Mosher, H.S. -Methoxy- -trifluoromethylphenylacetic acid, a versatile reagent for the determination of enantiomeric com-position of alcohols and amines. J. Org. Chem., 1969, 34, 2543-2549; d) Dale, J.A.; Mosher, H.S. Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear mag-netic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and -methoxy- -trifluoromethylphenylacetate (MTPA) esters J. Am. Chem. Soc., 1973, 95, 512-519.
[32] a) Mino, T.; Saitoh, M.; Yamashita, M. Enantioselective synthesis of 2,2-dialkyl-3-butenals by alkylation of (4S,5S)-ADPD-imines. J.
Org. Chem., 1997, 62, 3981-3983; b) Mino, T.; Oishi, K.; Yama-shita, M. Enantioselective addition of diethylzinc to aryl aldehydes catalized by ADPD imine catalysts. Synlett, 1998, 956-966.
[33] Strotmann, M.; Butenschön, H. Preparation of chiral amine car-bonyl Chromium(0) complexes with [(Norbornadiene)Cr(CO)4] as the complexation reagent. Eur. J. Org. Chem., 2000, 12, 2273-2284.
[34] a) Meeskens, F.A.J. Methods for the preparation of acetals from alcohols or oxiranes and carbonyl compounds. Synthesis, 1981, 1, 501-522; b) Meslard, J.C.; Subira, F.; Vairon, J.P.; Guy, A.; Gar-reau, R. Synthèse d’acétals cycliques dans des conditions douces. Application à l’acétalisation du chloramphénicol. Bull. Soc. Chim.
Franc. 1985, 1, 84-89; c) Pihlaja, K.; Hellman, J.; Mattinen, J.; Göndös, G.; Wittman, G.; Gera, L.; Bartók, M.; Pelczer, I.; Dombi, G. NMR structural study and stereoselective synthesis of 2-substituted and 2,2-disubstituted 5-dichloroacetamido-5-methyl-1,3-dioxanes. Acta. Chem. Scandinavica, 1988, B42, 601-604; d) Juaristi, E.; Diaz, F.; Cuéllar, G.; Jiménez-Vázquez, H.A. Confor-mational analysis of 5-substituted 1,3-dioxanes. 7. Effect of lithium bromide addition. J. Org. Chem., 1997, 62, 4029-4035; e) Bi, L.; Zhao, M.; Wang, C.; Peng, S. Stereoselective transacetalization of 1,1,3,3,-tetramethoxypropane and N-benzoylaminodiols. Eur. J.
Org. Chem., 2000, 14, 2669-2676. [35] a) Matsumura, Y.; Maki, T.; Murakami, O.; Onomura, O. Copper
ion-induced activation and asymmetric benzoylation of 1,2-diols: kinetic chiral molecular recognition. J. Am. Chem. Soc., 2003, 125, 2052-2053; b) Ooi, H.; Ishibashi, N.; Iwabuchi, Y.; Ishihara, J.; Ha-takeyama, S. A concise route to (+)-Lactacystin. J. Org. Chem., 2004, 69, 7765-7768; c) Sørbye, K.; Carlssen, P.H.J. Preparation of protected Serinol. Synth. Commun., 1997, 27, 2813-2816; d) Chênevert, R.; Voyer, N. Synthesis of chiral crown ethers from (1S,2S)-(+)-2-amino-1-phenyl-1,3-propanediol. Synthesis, 1985, 981-982.
[36] a) Nitrophenyl amino diol derivatives. Parke, Davis and Co. Brit.
Pat., 700710 (Dec. 9, 1953); conf. Chem. Abstr. 1955, 49, P15963h; b) Neidlein, R.; Keller, H.; Boese, R. Mild preparation of 1-benzyloxyiminoalkylphosphonic dichlorides: application to the synthesis of cyclic phosphonic diesters and cyclic monoester am-ides. Heterocycles, 1993, 35, 1185-1203.
Received: 07 October, 2009 Revised: 05 December, 2009 Accepted: 11 January, 2010




















