
Macrophage Stimulating Protein
29
Macrophage Stimulating Protein Edward 1. Leonard and Alla Danilkovitch Laboratory of Immunobiology NCl-Frederick Cancer Research and Development Center Frederick, Maryland 21 702 I. Introduction 11. Structural Aspects of Native and Recombinant Pro-MSP 111. Binding of MSP to its Receptor IV. A Model of MSP-Induced Receptor Dimerization V. Regulation of MSP Activity: Pathways for Pro-MSP Cleavage A. Pro-MSP Convertase Activity of Bovine or Human Serum B. Pro-MSP Cleavage Activity on the Surface of Murine Peritoneal Macrophages C. Pro-MSP Convertase Activity in Human Wound Exudates A. Activation of Ras-Dependent Pathways B. Activation of PUK Pathways C. Activation of Focal Adhesion Kinase D. Activation of Src Kinase E. Activation of JNK F. Possible Role of MSP-Ron Signaling Transduction Components in Tumor VI. MSP-Ron Signaling Progression VII. Modes of MSP Receptor Regulation or Activation A. Upregulation of Normal Receptor Expression as Part of a Host Defense Response B. Increased Ron Receptor Expression in Abnormal Cells C. Ligand-Independent Activation of Mutant Ron Receptors A. Macrophages B. Osteoclasts C. Cells of Ectodermal Origin D. Vascular Marrow Cells E. Bone Marrow Cells F, Insights from Message Expression and from Knockout Mice IX. Perspective References VIII. Target Cells for Macrophage Stimulating Protein I. INTRODUCTION MSP belongs to a family of proteins, typical members of which are serine proteases such as plasminogen or prothrombin. They are secreted as single- chain molecules that have no biological or enzymatic activity until they are proteolytically cleaved, generally at a single site to form a disulfide-linked ap Advances in CANCER RESEARCH 0065-23OWOO $30.00 Copyright 8 ZOO0 by Academic Press. All rights of reproduction in any form reserved.
Transcript of Macrophage Stimulating Protein

Macrophage Stimulating Protein
Edward 1. Leonard and Alla Danilkovitch Laboratory of Immunobiology
NCl-Frederick Cancer Research and Development Center Frederick, Maryland 21 702
I. Introduction 11. Structural Aspects of Native and Recombinant Pro-MSP
111. Binding of MSP to its Receptor IV. A Model of MSP-Induced Receptor Dimerization V. Regulation of MSP Activity: Pathways for Pro-MSP Cleavage
A. Pro-MSP Convertase Activity of Bovine or Human Serum B. Pro-MSP Cleavage Activity on the Surface of Murine Peritoneal Macrophages C. Pro-MSP Convertase Activity in Human Wound Exudates
A. Activation of Ras-Dependent Pathways B. Activation of PUK Pathways C. Activation of Focal Adhesion Kinase D. Activation of Src Kinase E. Activation of JNK F. Possible Role of MSP-Ron Signaling Transduction Components in Tumor
VI. MSP-Ron Signaling
Progression VII. Modes of MSP Receptor Regulation or Activation
A. Upregulation of Normal Receptor Expression as Part of a Host Defense Response B. Increased Ron Receptor Expression in Abnormal Cells C. Ligand-Independent Activation of Mutant Ron Receptors
A. Macrophages B. Osteoclasts C. Cells of Ectodermal Origin D. Vascular Marrow Cells E. Bone Marrow Cells F, Insights from Message Expression and from Knockout Mice
IX. Perspective References
VIII. Target Cells for Macrophage Stimulating Protein
I. INTRODUCTION
MSP belongs to a family of proteins, typical members of which are serine proteases such as plasminogen or prothrombin. They are secreted as single- chain molecules that have no biological or enzymatic activity until they are proteolytically cleaved, generally at a single site to form a disulfide-linked ap
Advances in CANCER RESEARCH 0065-23OWOO $30.00
Copyright 8 ZOO0 by Academic Press. All rights of reproduction in any form reserved.

140 Edward j . Leonard and Alla Danilkovitch
chain heterodimer (Fig. 1) (Francis and Marder, 1990). The family is char- acterized by a conserved triple disulfide loop structure (kringle) that occurs in multiple copies in the cc chain of the protein. MSP has a 45% sequence similarity to hepatocyte growth factor/scatter factor (HGF/SF) (Yoshimura et al., 1993). Hence, MSP is called hepatocyte growth factor-like protein by some investigators (Han et al., 1991). Whereas most of the proteins in this family are serine proteases, MSP and HGF have no enzymatic activity be- cause of amino acid substitutions in the catalytic triad. Instead they are thought to have evolved from an ancient coagulation protein (Donate et a/., 1994) to become growth and motility factors, while retaining the protease- dependent activation mechanism of the kringle protein family.
The receptor for MSP (called Ron in the human and Stk in the mouse) is
Hairpin
0 pro-MSP
I Q568 I c588 71 1 I I L - _ _ - _ - _ _ _ _ _ _ J
proteolysis by trypsin-like serine proteases
a chain (kringle p chain (serine containing subunit) proteinase-like subunit)
Fig. 1 Schematic representation of the structure and processing of pro-MSP to mature MSP. Pro-MSP is a single-chain, 71 1-residue protein, which is biologically inactive until it is cleaved at Arg483-Val484 (solid arrow) by trypsin-like serine proteases to form mature disulfide-linked a@-chain MSP. The interchain disulfide (dashed line) is formed by a-chain Cys468 and P-chain Cys.588. The a chain is a 53-kDa protein containing an N-terminal hairpin loop and four kringle domains (Kl-K4). The chain is a 25-kDa protein containing a serine protease-like domain, which is enzymatically inactive because the catalytic triad His, Asp, and Ser was replaced by Gln. Gln, and Tyr (open circles}. Five Arg residues (639, 641, 683, 687,689) forming positive- ly charged cluster and/or S1 substrate pocket Glu648 and Asn682 are hypothesized to play a role in binding of iMSP to its receptor. In some recombinant pro-MSP molecules, the interchain disulfide is absent, because Cys672 coopts Cys588 to form an intrachain disulfide.

Macrophage Stimulating Protein 141
one of a small family of receptor tyrosine kinases (RTKs) that comprises hu- man Met (Bottaro et al., 1991) and chicken Sea (Huff et al., 1993). Met and Sea can undergo neoplastic transformation. Met was first isolated from a hu- man osteosarcoma cell line as an oncogene fused with the tpr locus (Coop- er et al., 1984; Park et al., 1986), and oncogenic Sea causes avian erythro- blastosis (Huff et al., 1993). Hence, MSP/Ron is an appropriate subject for review in this series, because of the possibility of oncogenic receptor mutants.
11. STRUCTURAL ASPECTS OF NATIVE AND RECOMBINANT PRO-MSP
The MSP gene was cloned by screening a human genomic library with a probe coding for a kringle region of prothrombin (Han et al., 1991); the cDNA was cloned independently from a hepatocarcinoma cell line (Yoshimura et al., 1993). Development of a high production expression sys- tem in CHO cells provided recombinant pro-MSP for structural and biolog- ical studies (Yoshikawa et al., 1999). It was shown that MSP is a glycopro- tein, with N-linked sugars at the 3 Asn loci that add a total of about 5000 Da to the mass of the molecule. The oligosaccharides at these loci are het- erogeneous; four different sugars are bound to Am,,, six to and eight to Asn,,,.
Cleavage of pro-MSP to MSP was shown to occur at Arg,,,Val,,,. When recombinant pro-MSP was cleaved by kallikrein at the activation site to pro- duce the expected ap-chain disulfide-linked heterodimer, SDS-PAGE under nonreducing conditions showed not only the heterodimer, but also free a and p chains (Yoshikawa et al., 1999). Thus, the CHO cell recombinant pro-MSP was heterogeneous: Some molecules had the ap-chain disulfide linkage, oth- ers did not. In the aberrant molecules, p-chain Cys,,, formed an intrachain disulfide with Cys,,, instead of with a-chain Cys,,,. When Cys,,, was mu- tated to Ala, almost all recombinant pro-MSP had the interchain disulfide (Wahl et al., 1997). Interestingly, circular dichroism analysis of recombinant pro-MSP did not reveal detectable secondary structure (D. Xie and E. J. Leonard, unpublished data). This suggests that in the absence of a stable folded structure, formation in the CHO cell of either the interchain or in- trachain disulfide is a random event, which results in the secretion of both types of molecules.
The liver constitutively produces single-chain pro-MSP, which is released into the circulating blood. In contrast to the recombinant protein, isoelectric focusing studies show that the native material has secondary structure. The isoelectric point (PI) of the native protein in different samples of human serum was 5.5 to 6.2. In 6 M urea, the PI shifted to 7.6 (Leonard et al., 1982),

I42 Edward 1. Leonard and Alla Danilkovitch
a value close to the PI of 7.0 calculated from the amino acid composition. The PI shift suggests that the native protein is folded in a way that buries some of the alkaline amino acid residues. The difference between native and recombinant pro-MSP with respect to secondary structure injects a caution- ary note for studies that used the recombinant protein for pharmacokinetics (Leonard and Skeel, 1996) or for identifying specific pro-MSP convertases. It would be prudent to repeat these studies with natural pro-MSP.
111. BINDING OF MSP TO ITS RECEPTOR
The receptor for MSP is the human Ron gene product (Wang et al., 1994a; Gaudino et al., 1994), a transmembrane protein tyrosine kinase cloned from a keratinocyte cDNA library (Ronsin et al., 1993). The STK gene, cloned from hematopoietic stem cells of mouse bone marrow (Iwama et al., 1994), is the murine homolog of Ron (Wang et al., 1995). The Ron gene encodes a 190-kDa protein, the mature form of which is a disulfide-linked heterodimer comprising a 40-kDa extracellular a chain and a 150-kDa p chain (Ronsin et al., 1993). The p chain has an extracellular domain, a transmembrane seg- ment, and a large cytoplasmic tail with an intrinsic tyrosine kinase, the ac- tivity of which is increased by ligand-receptor binding.
In a study to identify the domains of MSP that interact with its receptor (Wang et al., 1997), five recombinant proteins were used, including pro-MSP, MSP, MSP a and f3 chains, and MSP-NK2 (an IgG Fc fusion protein com- prising the MSP N terminus including the first two kringles). The free a and p chains were purified from preparations of recombinant MSP molecules with the mismatched disulfide described earlier. Saturable binding of 1251- MSP was shown for various cells expressing the MSP receptor, including mouse and human epithelial cell lines as well as MDCK cells transfected with human Ron (MDCK-RE7 cells). The binding of radiolabeled MSP was com- pletely inhibited by a 10- to 15-fold molar excess of unlabeled MSP, partial- ly inhibited by a comparable excess of f3 chain, and not inhibited by a chain. Interaction of free p chain with the MSP receptor was confirmed by show- ing saturable binding of 12-51-p chain to MDCK-RE7 cells, in contrast to no specific binding of radiolabeled a chain or MSP-NK2. The estimated K , of 1.7 nM for f3 chain binding was higher than the K , of 0.6-0.8 nM for MSP, reflecting the differences in potency as competitive inhibitors noted earlier.
HGF and MSP are thought to have evolved from a common ancestor gene and, in addition to a 45% sequence similarity, they have comparable domain structures and mechanisms of activation. Furthermore, the receptors for HGF and MSP are in the same subfamily, and have many unique structural features in common (Ronsin et al., 1993; Huff etal., 1993; Park et al., 1987).

Macrophage Stimulating Protein 143
The finding that MSP p chain, not a chain, bound to Ron was therefore com- pletely unanticipated, because the high-affinity binding site of HGF for its receptor is in the a chain. A critical region for HGF binding is a hairpin loop in the N-terminal domain, deletion of which eliminates binding (Matsumo- to et al., 1991; Okigaki et al., 1992). An energy minimization model of the hairpin loops of HGF and MSP revealed a three-arginine positively charged face of the HGF loop, in contrast to only one arginine for the corresponding region of MSP (Donate et al., 1994). A role for the arginines in HGF recep- tor binding was suggested by diminished activity of an HGF mutant in which alanines were substituted for the 3 arginines (though the authors did not rule out the possibility of agonist instability to account for their results) (Sakata et al., 1997). The absence of a highly charged MSP hairpin loop region and undetectable MSP a-chain binding to Ron are consistent with this suggest- ed role.
We then considered the possibility that binding of the MSP p chain to Ron might also be via a positively charged region. Accordingly, an energy-mini- mized model of the MSP p chain was constructed (M. Miller, A. Danil- kovitch, and E. J. Leonard, unpublished data) using coordinates from HGF modeling (Donate et al., 1994) based on the structures of three mammalian serine proteases. This revealed a cluster of five arginines on surface loops of the MSP p chain. A critical role of this region for binding was established when substitution of one of the arginine residues for alanine caused com- plete loss of p-chain binding to Ron.
N. A MODEL OF MSP-INDUCED RECEPTOR DIMERIZATION
It is generally accepted that ligand binding to transmembrane receptor pro- tein tyrosine kinases causes receptor dimerization and autophosphorylation (Ullrich and Schlessinger, 1990). Two different examples of receptor dimer- ization by growth factors may be relevant to activation of Ron by MSP. One is the binding of stem cell factor (SCF) to its receptor, kit: SCF dimers bind to pairs of Kit receptors, the stoichiometry being 2:2 (Philo et al., 1996; Lem- mon et al., 1997). In the case of human growth hormone (HGH), the stoi- chiometry is 1:2: One region of an HGH monomer binds with high affinity to its receptor (Rl), after which another region of HGH binds to a second receptor (R2). Thus, the HGH monomer is bivalent with respect to receptor binding. Although the second site binding affinity is lower, the ligand-re- ceptor complex is stabilized by an RUR2 interaction as the receptors are brought into proximity by HGH (Cunningham et al., 1991; de Vos et al., 1992).

I44 Edward 1. Leonard and Alla Danilkovitch
The following considerations lead us to favor the HGH model for MSP- induced dimerization of Ron (Miller and Leonard, 1998). Although the MSP p chain binds to cells expressing Ron, it does not induce a cellular response. Only the MSP a@-chain disulfide-linked heterodimer induces biological ac- tivity and presumably receptor dimerization. Additional evidence for the im- portance of both CL and p chains for biological activity of MSP has recently been published (Bezerra et af., 1998). Although the requirement for intact MSP might reflect the formation of ligand dimers, there is no evidence by gel filtration of MSP dimers at physiologic concentrations. An alternative is that MSP, like HGH, is bivalent, with a high-affinity @-chain binding site and a second low-affinity binding site for Ron on the 01 chain. The converse might apply to HGF, for which a high-affinity binding site has been demonstrated on the 01 chain, and for which a low-affinity site on the @ chain can be pos- tulated, since critical amino acid substitutions in the @ chain cause decreas- es in biological activity to 2% of wild-type HGF.
Detection of MSP a-chain binding to Ron would provide support for the HGH model. We did not observe any specific binding of 1251-01 chain to Ron- expressing cells (Wang et a/., 1997). However, after equilibration of unla- beled a chain with MDCK-RE7 cells at 4"C, followed by washing of fluid phase a chain, we detected by Western blot bound (Y chain in immunopre- cipitated cell lysates. No 01 chain was adsorbed to parental MDCK cells that did not express Ron. This finding supports the idea that MSP may be bi- valent with respect to receptor binding, so that one ligand molecule is capa- ble of inducing receptor dimerization. When soluble receptor becomes avail- able, it should be possible to determine if 1igand:receptor stoichiometry is indeed 1:2.
V. REGULATION OF MSP ACTIVITY: PATHWAYS FOR PRO-MSP CLEAVAGE
Like kringle proteins of the coagulation system, MSP is constitutively syn- thesized by hepatic parenchymal cells. In a study of the biochemical basis for liver-specific transcription of the MSP gene, both positive and negative reg- ulatory elements have been found. The latter are thought to account for in- hibition of transcription in nonhepatic cells (Ueda et al., 1998). The gene product is secreted by the liver into the circulating blood as biologically in- active pro-MSP. The mean concentration of pro-MSP in the plasma of a se- ries of normal human subjects is 4 nM, which is in the range for optimal bi- ological activity (Nanney et d., 1998). The level is not changed in the course of an acute phase reaction (Wang et al., 1993). To act on target cells in ex- travascular sites, this protein of hepatic origin must diffuse into tissues and

Macrophage Stimulating Protein I45
be cleaved by a pro-MSP convertase to biologically active MSP. Although R,,,-V,,, is a cleavage site, a pro-MSP mutant with that site blocked (R483E) has biological activity, which indicates that cleavage at an alterna- tive locus can yield a functional protein (Waltz et al., 1997).
A. Pro-MSP Convertase Activity of Bovine or Human Serum
The R,,,-V,,, scissile bond is a typical cleavage site for trypsin-like ser- ine proteases. Indeed, several such purified proteases of the coagulation sys- tem, including factors XIa and XIIa and serum kallikrein cleave pro-MSP to active MSP (Wang et al., 199413). Cleavage does not occur when blood clots, indicating that pro-MSP is not a preferred substrate for these enzymes (Wang et al., 1996~) . However, as noted later, serum has pro-MSP convertase ac- tivity.
CHO cells transfected with the pro-MSP cDNA have been used to express recombinant protein. When the cells were grown in serum-free medium, no cleavage of secreted pro-MSP was detected in culture fluid sampled over a 36-hr period. In contrast, in cultures containing 10% heat-activated fetal bovine serum, partial cleavage of pro-MSP to MSP was detected after 16 hr in culture, which was almost complete at 36 hr. Cleavage was prevented by inhibitors of trypsin-like serine proteases (aprotinin, leupeptin, and soybean trypsin inhibitor) (Wang et al., 1994a). The cleavage product was biologi- cally active. In a cell-free system, fetal bovine serum, normal calf serum, and normal human serum partially cleaved 1251-pro-MSP to MSP within 3 hr at 37°C (A. Skeel and E. J. Leonard, unpublished data). These results indicate that fetal bovine serum accounts for pro-MSP cleavage in transfected CHO cell cultures.
Although cleavage of pro-MSP in v i m by serum is important in the labo- ratory, it probably does not relate to the requirement for convertase activity in local tissue sites. Two pro-MSP convertase activities of potential in vivo relevance have been described.
B. Pro-MSP Cleavage Activity on the Surface of Murine Peritoneal Macrophages
When labeled pro-MSP was incubated with murine peritoneal macro- phages and the culture fluids sampled within 3 hr and analyzed by im- munoprecipitation and radioautography, several cleavage products were de- tected (Wang et al., 1996~) . The a and p chains of mature MSP (about 53 and 30 kDa) comprised only a small fraction of the total; most of the cleav-

146 Edward 1. Leonard and Alla Danilkovitch
age product comprised 47- and 3.5-kDa fragments. When the experiment was repeated with a panel of serine protease inhibitors, AEBSF and soybean trypsin inhibitor prevented the degradative cleavage, so that the predomi- nant product was mature MSP. It thus appeared that this macrophage pop- ulation had two pro-MSP cleavage activities that could be differentially in- hibited by selected protease inhibitors. Low concentrations of murine or human serum had effects similar to that of STI. The protein in human serum that inhibited the degradative cleavage of pro-MSP by macrophages is al- antichymotrypsin (A. Skeel and E. J. Leonard, unpublished data). These re- sults suggest an interesting mode of localizing the site of MSP action, in which the peritoneal macrophage, one of the target cells for MSP, also has the capacity to cleave pro-MSP to active MSP. An additional levei of control is apparently provided by a pro-MSP degrading enzyme and its inhibitor, a, -antichymotrypsin. Blood monocytes have neither the MSP receptor nor pro-MSP cleavage activities, indicating that both properties are late events in mononuclear phagocyte maturation. We recently showed that all macro- phages in normal human dermis express the MSP receptor (Nanney et al., 1998). It would be important to know if these cells, and tissue macrophages in general, have pro-MSP convertase activity, providing a mechanism for physiologic regulation of this growth and motility factor.
C. Pro-MSP Convertase Activity in Human Wound Exudates
In contrast to the undetectable or minimal amount of mature MSP in nor- mal human blood plasma, MSP comprises about half the total pro-MSP + MSP found in surgical drainage fluid or human burn wound fluid (Nanney et al., 1998). We conclude that the circulating pro-MSP that diffuses into the wound site is cleaved by cell-associated or fluid phase enzymes or both. In a large series of tested wound exudates, all cleaved labeled pro-MSP to MSP. The enzymatic activity was inhibited by leupeptin and aprotinin, but not by STI, a,-macroglobulin or C1-inhibitor. This inhibition profile is the same as for human tissue kallikrein, which brought to mind the fact that in a series of serine proteases tested for pro-MSP convertase activity, two murine tissue kallikreins were the most potent (Wang et al., 1 9 9 4 ~ ) . How- ever, in studies of four wound fluids, Chao showed that the kallikrein in- hibitor kallistatin was present in great molar excess over tissue kallikrein; and highly purified tissue kallikrein (KLK1) did not cleave pro-MSP (J. Chao, unpublished data). If we succeed in our current efforts to purify and characterize the wound fluid pro-MSP convertase activity, it may be possi- ble to determine if this is a product of one of the cell types at the site of tis- sue injury.
Macrophage Stimulating Protein
- - z z
. 2 = 2
I47
- z z 42 2
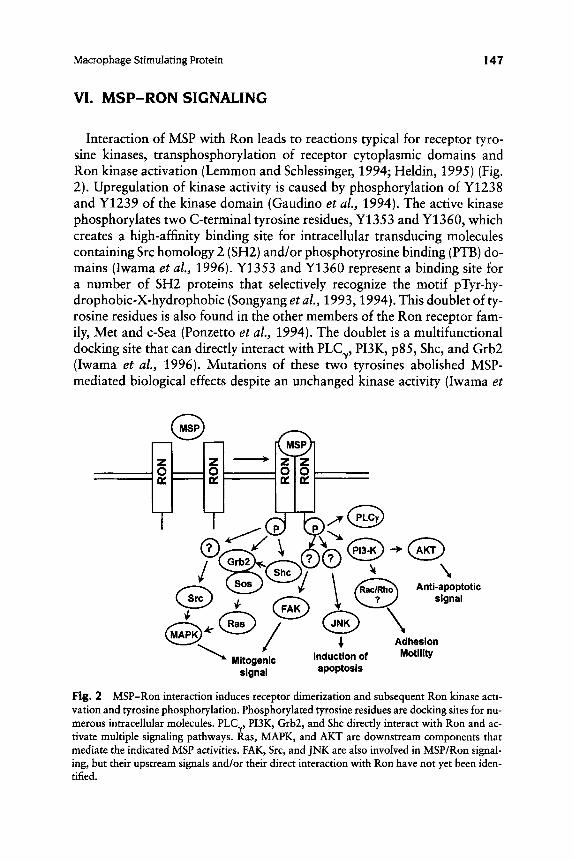
VI. MSP-RON SIGNALING
Interaction of MSP with Ron leads to reactions typical for receptor tyro- sine kinases, transphosphorylation of receptor cytoplasmic domains and Ron kinase activation (Lemmon and Schlessinger, 1994; Heldin, 1995) (Fig. 2). Upregulation of kinase activity is caused by phosphorylation of Y1238 and Y1239 of the kinase domain (Gaudino et al., 1994). The active kinase phosphorylates two C-terminal tyrosine residues, Y 1353 and Y1360, which creates a high-affinity binding site for intracellular transducing molecules containing Src homology 2 (SH2) and/or phosphotyrosine binding (PTB) do- mains (Iwama et al., 1996). Y1353 and Y1360 represent a binding site for a number of SH2 proteins that selectively recognize the motif pTyr-hy- drophobic-X-hydrophobic (Songyang et al., 1993,1994). This doublet of ty- rosine residues is also found in the other members of the Ron receptor fam- ily, Met and c-Sea (Ponzetto et al., 1994). The doublet is a multifunctional docking site that can directly interact with PLCy, PUK, p85, Shc, and Grb2 (Iwama et al., 1996). Mutations of these two tyrosines abolished MSP- mediated biological effects despite an unchanged kinase activity (Iwama et

I48 Edward I . Leonard a n d AIla Danilkovitch
al., 1996). Although the cytoplasmic part of Ron contains 14 tyrosine residues that might serve as phosphorylation sites, there are no data about the functional role of most of them, and it is proposed that the pleotropic re- sponse induced by MSP results from the activation of multiple signaling pathways by recruitment of intracellular components to the Y 1353/Y 1360 multifunctional docking site.
A. Activation of Ras-Dependent Pathways
Ras is a 21-kDa protein belonging to the small molecular weight GTP- binding protein family. Ras proteins regulate cell proliferation and differen- tiation (Heimbrook et al., 1997). Ras is active in GTP-bound form and acts through protein-protein interactions involving the Ras effector loop (amino acid residues 32-40) (Marshall, 1995; Hwang et al., 1996). The Ras-de- pendent pathway in cells involves activation of the MAPK cascade by li- gands of protein tyrosine kinases. GTP-bound RAS binds cytoplasmic Raf- 1, the first member of the MAPK cascade that terminates with activation of transcriptional factors required for the S phase of the cell cycle (Marshall, 1995). Ras can also regulate Raf-independent signals via Rac/Rho and PUK (Rodriguez-Viciana et al., 1994; Qiu et al., 1995a,b).
In MDCK cells with overexpressed Ron receptor, the SH2-domain con- taining adaptor protein Grb2 bound to activated Ron @ chain (Li et al., 1995). Grb2, which recognizes phosphotyrosine-X-N sequences (Songyang et al., 1994), may therefore bind to Ron at its C-terminal Y1360/N1362 motif (Iwama et d., 1996). Binding of Grb2 to Ron was associated with increased GTP-Ras due to activation of SOS (Li et al., 1995), a protein that catalyzes the exchange of GDP for GTP on Ras and activates Ras-depen- dent signaling pathways (Aronheim et af., 1994). MSP caused transloca- tion of Grb2-SOS complexes from cytosol to plasma membrane, where the target Ras is located. In contrast to the absence of binding of the Shc adap- tor protein to Ron (Li et al., 1995), association of Shc with activated Stk (the rnurine MSP receptor) was detected in murine hematopoietic cell lines stably transfected with Stk (Iwama et al., 1996). Thus, it is possible for Sch to recruit Grb2 to Stk, and it has been shown that Grb2 recognizes phos- phorylated Shc (Clark et al., 1992). Experiments with overexpression of dominant negative N17Ras demonstrated that Ras does not mediate MSP- induced cell motility (Wang et al., 1996). Activated Ras is implicated in MSP mitogenic signaling (A. Danilkovitch, unpublished observations), al- though it is possible that in cells where activation of PI3K is Ras depen- dent, Ras might also mediate MSP-induced motility. The role of PI3K in MSP signaling is discussed next.

Macrophage Stimulating Protein 149
B. Activation of PI3K Pathways
Phosphatidylinositol-3-kinase (PI3K) is a heterodimeric protein consisting of an 85-kDa regulatory subunit with two SH2 domains and one SH3 domain, and a 110-kDa catalytic subunit. PI3K phosphorylates the D-3 po- sition of the inositol ring of phosphatidylinositol, phosphatidylinositol-4- phosphate, and phosphatidylinositol-4,5-bisphosphate (Cantley et al., 1991). PI3K is associated with numerous activated growth factor receptors via SH2 domains and is involved in growth factor-mediated signaling (Kapeller and Cantley, 1994; Hu et al., 1992).
MSP induces PI3K tyrosine phosphorylation, and PI3K catalytic activity is necessary to transduce MSP signaling: Wortmannin, a specific inhibitor of PUK activity, inhibits MSP-dependent adhesion, motility, and cell shape change. Addition of MSP to epithelial cells expressing Ron causes PI3K to associate directly with Ron (Wang etal., 199610). Studies of the binding speci- ficity of the p85 SH2 domains reveal a core phosphotyrosine containing mo- tif p Tyr-X-X-Met on target proteins (Varticovski et d., 1994), and Ron has such a motif around Y1317 (Ronsin et al., 1993). On the other hand, it has been shown that PI3K may interact with Y1353 and Y1360 of the multi- functional docking site (Iwama et al., 1996). In contrast to signaling by many other growth factors, where PI3K activation is Ras dependent (Rodriguez- Viciana et al., 1994), it appears that activated Ron binds PI3K directly and stimulates catalytic activity. The lack of inhibition of MSP-mediated cell motility by dominant negative N17Ras supports this idea (Wang et al., 1996b).
Downstream components of PI3K signaling include ribosomal protein ki- nase ~ 7 0 ~ ~ ~ (Chung et d., 1994), Rho family GTP-binding protein Rac (Parker, 1995), PKC (Akimoto et al., 1996; Moriya et al., 1996), and AKT (Franke et al., 1997). Rac is important for PUK-dependent actin rearrange- ment (Reif et al., 1996), and thus it may mediate MSP-induced cell shape change, adhesion, and motility. AKT is a serinehhreonine-specific kinase that is important for protection of cells from apoptosis (Kauffmann-Zeh et al., 1997; Songyang et al., 1997; Dudek et al., 1997). We found that MSP may protect cells from apoptosis via PI3K and AKT. MSP induces PI3K-depen- dent AKT activation, and dominant-negative AKT abolishes the MSP anti- apoptotic effect (A. Danilkovitch and E. J. Leonard, unpublished data). Thus, PI3K is important for MSP-induced cell survival as well as adhesion and motility. PUK is required for HGF-induced mitogenic signals in epithe- lial cells (Rahimi et al., 1996). We found that PUK is also involved in MSP mitogenic signaling, but this effect is related to the ability of PI3K to protect cells from apoptosis via its downstream component AKT kinase (A. Danil- kovitch and E. J. Leonard, unpublished data).

150 Edward 1. Leonard a n d Alla Danilkovitch
C. Activation of Focal Adhesion Kinase
Focal adhesion kinase (FAK) is a nonreceptor cytoplasmic tyrosine kinase implicated in cell growth, survival, adhesion, and motility (Hanks and Polte, 1997). FAK is spatially and functionally associated with the p chain of inte- grins, cell surface receptors that are responsible for the interaction of cells with extracellular matrix (ECM) (Schaller et al., 1992; Hanks et al., 1992). ECM-induced integrin receptor aggregation is accompanied by FAK tyrosine phosphorylation and kinase activation, followed by activation of down- stream signals (Hanks and Polte, 1997). FAK tyrosine phosphorylation and activation may also be regulated by growth factors (Abedi and Zachary, 1997; Rankin and Rozengun, 1994; Baron et al., 1998; Zhu et al., 1998). MSP, like HGF (Matsumoto et al., 1994), induces FAK tyrosine phosphory- lation and activation (Danilkovitch and Leonard, 1997). FAK is a compo- nent of the signaling path that mediates MSP-induced cell proliferation: In- hibition of FAK activity via overexpression of dominant-negative FAK leads to significant reduction of MSP-induced mitogenic signaling (Danilkovitch and Leonard, 1998, also unpublished data). These data demonstrate that there is crosstalk between signal transduction pathways coming from two different receptors, Ron and integrins.
D. Activation of Src Kinase
Src and Src related kinases are included in the large family of cytoplas- mic tyrosine kinases on the basis of their common domain structure: acety- lated amino terminus, SH2 and SH3 domains, and highly conserved cat- alytic domain (Williams et al., 1998). Src kinase has little activity in cells in the absence of an activating signal, but many stimuli increase Src enzy- matic activity via phosphorylation mechanisms (Cooper and Howell, 1993). Src is involved in growth factor receptor signaling: Src interacts with PDGF (Kypta et al., 1990), EGF (Maa et al., 1995), bFGF (Zhan et al., 1994), and CSF-1 (Courtneidge et a/. , 1993) receptors. Ligand-dependent association of Src with growth factor receptors is mediated through the Src SH2 domain (Williams et al., 1998; Erpel and Courtneidge, 1995). Src is involved in HGF/Met signaling; it directly interacts with tyrosine residues of the multifunctional docking site (Ponzetto et al., 1994). Although asso- ciation of Src with Ron has not yet been shown, Src is a component of MSP-Ron signaling. In bone marrow-derived osteoclast-like cells, MSP caused ruffled border formation, which was associated with a rapid redis- tribution of Src from a diffuse cytoplasmic distribution to a peripheral lo- cation near the plasma membrane (Kurihara et al., 1996). Src also medi- ates MSP-induced cell proliferation, and MAPK activity is regulated by Src

Macrophage Stimulating Protein 151
in MSP stimulated cells: Dominant negative Src significantly reduced MSP- dependent proliferation and MAPK activation (Danilkovitch and Leonard, 1998).
E. Activation of JNK
The c-Jun amino-terminal kinases (JNKs) are members of the MAPK fam- ily, which are activated by cytokines and/or environmental stress (Whit- marsh and Davis, 1996). The JNK pathway regulates cell proliferation, apoptosis, and tissue morphogenesis (Ip and Davis, 1998). JNK activation is mediated by dual phosphorylation on Thr and Tyr by two MAP kinases, MKK4 and MKK7 (Whitmarsh and Davis, 1996). Rho family GTPases and PI3K also may serve as intermediates in pathways leading to JNK activation (Joneson et al., 1996; Lamarche et al., 1996; Logan et al., 1997; Lopez- Ilasaca et al., 1997). MSP and HGF both activate the JNK pathway (Ro- drigues et d., 1997). MSP-induced JNK activation in a murine erythro- leukemia cell line transfected with Stk was associated with induction of apoptosis (Iwama et al., 1996). MSP may simultaneously activate two dif- ferent pathways leading to either cell growth or apoptosis; cells undergo apoptosis if the apoptotic pathway overcomes the mitogenic signals. It has been proposed that MSP-induced JNK activation is related to expression of two unidentified proteins, p61 and p65. These proteins interact directly with the multifunctional docking site at the C terminus of Ron in a ligand-de- pendent manner (Iwama et al., 1996). In contrast to the above hematopoi- etic cell line, MSP-induced JNK activation in epithelial cells does not induce apoptosis (A. Danilkovitch, unpublished data). However MSP has been re- ported to induce apoptosis in selected human pulmonary carcinoma cell lines (Willett et al., 1997).
F. Possible Role of MSP-Ron Signal Transduction Components in Tumor Progression
Growth factor receptor tyrosine kinases can activate multiple pathways that lead to mitogenesis. There are many examples of growth factor recep- tor overexpression in tumor cells, and their constitutive activation by vari- ous mechanisms is related to tumor formation or progression (Karunagaran et al., 1996; Tsujimura, 1996; Empereur et al., 1997; Borset et al., 1996; Moscatello et al., 1996; Schmidt et al., 1997; Santoro et al., 1995). Many of the above-described MSP-Ron signaling molecules are known to cause cell transformation. For example, Ras genes are mutated in 20-30% of human cancers and the encoded proteins are believed to contribute to the patho-

152 Edward 1. Leonard and Alla Danilkovitch
genesis of these malignancies (Lowy and Willumsen, 1993; Bos, 1989). Con- stitutively active Ras leads to cell transformation, which is related to activa- tion of the Ras downstream component MAPK (Oldham et al., 1998). Ac- tive PI3K is also an essential component for efficient Ras transformation (Rodriguez-Viciana et al., 1997). Activation of c-Src kinase occurs with high frequency in cells with malignant potential (Cartwright et al., 1990), in pri- mary tumors (Cartwright et al., 1994), and in metastatic tumors (Talamon- ti et al., 1993). Analysis of human tumors with an invasive phenotype re- vealed FAK overexpression (Owens et al., 1995; Weiner et al., 1993), and this is highly correlated with invasive potential (Partin et al., 1989). Thus, FAK may be involved in tumor invasion and metastasis, and it may also be useful as a marker of tumor cell invasion. Activation of the JNK pathway is essential for cell transformation by the tpr-Met oncoprotein and, thus, JNK may also be implicated in tumor development and progression (Rodrigues et al., 1997). The relationship of Ron gene mutations to tumor formation or invasiveness is reviewed in Section VII.
VII. MODES OF MSP RECEPTOR REGULATION OR ACTIVATION
Control of MSP actions on cells can be at the level of ligand or receptor (Table I ) . Generation of active MSP from pro-MSP has been reviewed in Sec- tion V. Receptor-based controls or activation are summarized in this sec- tion.
Table I Modes of Ron Receptor Activation
1. Activation of pro-MSP (Section V) Cleavage of plasma pro-MSP in extravascular sites
Cell-bound pro-MSP convertase Fluid phase pro-MSP convertase
Cleavage of locally produced pro-MSP: testis; embryonic development?
Human wounds (Section VII,A) Response to cytokines or growth factors (Sections VII,A and VII,B)
Primary cancers Cancer cell lines
Tpr-Ron A-Ron Ron point mutations akin to those in Kit and Ret
11. Upregulation of receptor expression as part of host defense response
111. Increased receptor expression in abnormal cells (Section VII,B)
IV. Ligand-independent activation of mutant receptors (Section VI1,C)

Macrophage Stimulating Protein I53
A. Upregulation of Normal Receptor Expression as Part of a Host Defense Response
An example is from a recent study that evaluated Ron expression in his- tologic sections of normal skin and of human burn wounds (Nanney et al., 1998). Ron was detected immunohistochemically with a monoclonal anti- body against the extracellular domain of the receptor. Ron expression by keratinocytes was undetectable in four of six specimens of normal skin. In contrast, Ron was strongly expressed in epithelial cells of human burn wounds. The cellular distribution of increased Ron expression was variable in different wound specimens, occurring in proliferative and/or differentiat- ed populations, the latter including cells in accessory structures such as sweat ducts and hair follicles. Frequency of Ron detection on endothelial cells of dermal capillaries was also higher in burn wounds than in normal skin, sug- gesting the possibility of an angiogenic effect of MSP. All dermal macrophages in both normal and burn wound skin expressed Ron. Inasmuch as biologically active MSP was found in wound exudates, the findings are consistent with a host defense response in which MSP interacts with the Ron receptor on macrophages, keratinocytes and vascular endothelium-all par- ticipants in wound healing.
It is reasonable to assume that Ron upregulation observed in the preced- ing study is caused by one or more of the many cytokines that are generat- ed in response to tissue injury (Martin, 1997). Chen et al. (1997) tested the effects of a series of cytokines and growth factors on MSP receptor protein expression in murine Hep 1-6 cells (a cell line derived from a murine hepa- tocellular carcinoma). The receptor was upregulated by IL-1, IL-6, TNFa, and HGF, but not by TGF-P or EGF (Chen et al., 1997).
B. Increased Ron Receptor Expression in Abnormal Cells
Maggiora et al. (1998) recently studied Ron expression in 74 human pri- mary breast carcinomas, 12 benign tumors, and 8 normal mammary glands. Solubilized protein from surgical specimens was immunoprecipitated with an anti-Ron antibody and analyzed by Western blot after SDS-PAGE. Whereas Ron protein was barely detectable in blots from normal or benign tumor tissue, Ron was overexpressed in 35 of the 74 carcinomas; in 12 of the 35, the increase was more than 20-fold. In a number of cases, Ron was phosphorylated, indicating that the receptor was activated. High expression was confirmed by Ron immunoreactivity in paraffin sections of selected pa- tients. Because Southern blots showed no evidence of gene amplification, it was presumed that overexpression was mediated at the transcriptional lev-

I54 Edward 1. Leonard and AIla Danilkovitch
el. In a similar evaluation of three normal liver samples and seven primary hepatocellular carcinomas, Ron was overexpressed in two of the carcinomas (Chen et al., 1997).
The cause of Ron overexpression in these tumors could be paracrine or autocrine. The paracrine possibility would be another example of a host re- sponse to tissue pathology in which cytokine release by normal cells that par- ticipate in the host response causes upregulation of the receptor on the tu- mor cells (item I1 in Table I). On the other hand, because Ron has been readily detected in a number of cancer cell lines that are removed from the host tissue environment (Wang et af., 1996b; Gaudino et af., 1994; Maggio- ra et al., 1998), Ron expression may be enhanced in autocrine fashion by factors secreted by the tumor cells.
What are the functional effects of Ron overexpression in these cancer cells? In the case of Met, cells of the murine C127 cell line that are engineered to overexpress normal Met and HGF/SF become morphologically transformed and tumorigenic in vivo (for review, see Jeffers et al., 1996). In contrast, there are no examples to date of tumorigenicity for cells that overexpress normal Ron (see later section). The in vitro effects of MSP on tumor cell lines in- clude proliferation, stimulation of motility, and capacity to migrate through an artificial ECM (Wang et al., 1996a,b; Maggiora et al., 1998), properties that might contribute to a metastatic potential in vivo provided that active MSP is present at the site.
C. Ligand-Independent Activation of Mutant Ron Receptors
Three mutant Ron receptors have been described with constitutively acti- vated tyrosine kinase. Of these three mutants, only A-Ron occurs naturally; the other two mutants were engineered.
1 . A-RON
In a screening of tumor cell lines for Ron, KATO-111, a gastric carcinoma cell line, was unique in that the predominant Ron band under reducing con- ditions was 165-kDa uncleaved, truncated Ron. This was in contrast to the products of other cells, which showed 170-kDa uncleaved Ron and the 150- kDa p chain of the normal disulfide-linked heterodimer. The unique A-Ron band was the product of an alternatively spliced mRNA that resulted in dele- tion of 147 bp of message coding for a segment of Ron (Collesi et al., 1996). When the cDNA of A-Ron was transfected into COS-1 cells, the product was uncleaved A-Ron, which was not expressed on the cell surface and which

Macrophage Stimulating Protein 155
was tyrosine phosphorylated. In both transfected cells and KATO cells, Ron oligomers were found by SDS-PAGE under nonreducing conditions, which became monomers under reducing conditions. The authors suggest that an uneven number of cysteines in A-Ron allows a free cysteine to make a disul- fide linkage with another receptor molecule, since oligomerization was pre- vented by treating cells with mercaptoethanol. Furthermore, there are three highly conserved cysteines in the 49-residue deletion fragment that normal- ly contribute to intramolecular disulfide bond formation. When one of these cysteines in normal Ron was mutated to alanine, the receptor was retained in the intracellular compartment and was tyrosine phosphorylated, presum- ably due to oligomerization.
Several standard assays have been used to test for transformation of cells transfected with mutated receptors: (1) proliferation rates in low-serum medium, with estimation of a rate decrease at confluence; (2) focus forma- tion; (3) anchorage-independent growth in soft agar; and (4) formation of tumors in nude mice. In addition, the Met/Ron/Sea receptor family can in- duce complex patterns of motility and morphogenesis: ( 5 ) increased motili- ty or scatter of MDCK epithelial cells, one of the original assays for the Met ligand, HGF/SF; (6) chemotaxis to ligand or increased motility of cells with constitutively activated receptor, measured as migration through pores of polycarbonate membranes; (7) “invasive” behavior, measured as migration through matrigel, an extracellular matrix mixture of collagen IV, laminin, and glycosaminoglycans; (8) in vivo invasive behavior, measured by the ca- pacity of intravenously injected cells to colonize lungs of nude mice; (9) mor- phogenetic responses in three-dimensional collagen gels, typically the for- mation of multicellular branched tubular structures. By criteria 1 and 2, cells transfected with activated A-Ron were not transformed. However, they did have an invasive phenotype by tests 6 and 7 .
2. TPR-RON
The oncogenic potential of Met was revealed by the discovery of Tpr-Met, in which the extracellular, transmembrane, and intracellular juxtamembrane portions of Met are replaced by the N-terminal sequence of the Tpr gene product (Cooper et al., 1984; Park et al., 1986). Tpr-Met is constitutively dimerized (Rodrigues and Park, 1993), which causes ligand-independent transphosphorylation and activation of the Met kinase. Stable transfectants of Tpr-Met in murine NIH 3T3 cells are transformed (focus formation, growth in soft agar). To test for the oncogenic potential of the other mem- bers of the Met family, transfectants of Tpr-Ron and Tpr-Sea were generat- ed. Whereas NIH 3T3 cells transfected with Tpr-Sea were transformed, the Tpr-Ron transfectants were not. Cells with all three chimeras had higher pro-

I56 Edward 1. Leonard and AIla Danilkovitch
liferation rates than the medium-2% FCS control, but the rate for Tpr-Ron cells reached a plateau between 5 and 7 days, indicative of contact-inhibit- ed growth (Santoro et al., 1996).
The absence of Tpr-Ron transforming ability was possibly related to a five- fold lower catalytic efficiency (Vmax/Km) of the Ron kinase relative to the Met kinase. From focus forming and anchorage-independent growth exper- iments on cells with domain swapping, it was apparently not related to the C-terminal multifunctional docking site, but to the kinase. For example, a Tpr-Ron with a Met kinase domain became transforming, and a Tpr-Met with a Ron C terminus retained its transforming ability.
Although Tpr-Ron did not induce transformation, transfected 3T3 cells migrated spontaneously through 8-pm pores in a transwell assay as well as through matrigel. And expression in MDCK cells induced scattering, as well as tube formation, in three-dimensional collagen gels. Thus, Tpr-Ron is sim- ilar to A-Ron in its capacity to induce cellular motility and morphogenesis.
3 . RON POINT MUTATIONS
Santoro et al. (1998) showed that Ron does indeed have oncogenic po- tential, by engineering specific point mutations in the tyrosine kinase do- main. The approach was based on the fact that a number of human neo- plastic syndromes are associated with activating point mutations in highly conserved loci in the TK domains of Kit and Ret receptors (Santoro et al., 1995). Mutations in these receptors are found in mast cell leukemia and multiple endocrine neoplasia respectively. Likewise, similar point mutations in Met have been found in hereditary and sporadic human renal papillary carcinomas (Schmidt et al., 1997). Two different point mutations were con- structed in the TK domains of both Ron and Tpr-Ron, a Kit-type mutant (D1232V) and a Ret-type mutant (M1254T). Both mutations converted Ron and Tpr-Ron to tramforming genes, as shown by NIH 3T3 transfec- tants that caused focus formation and tumors when injected into nude mice. All transfectants also colonized murine lungs after intravenous injection. Comparison of the TK catalytic efficiency of nontransforming Tpr-Ron with the Tpr-Ron mutants showed that the latter were three to four times higher than Tpr-Ron. This observation lends support to the suggested im- portance of TK activity noted in the comparison of nontransforming Tpr- Ron and oncogenic Tpr-Met. Also of interest was the detection of two phos- phorylated proteins in lysates from cells transfected with mutant, but not wild-type, receptors. This is the first demonstration that the Ron gene shares with other members of the Met family the potential to become an oncogene. It should provide impetus to use an efficient screening program to determine if there are any human cancers caused by mutations in this receptor tyro- sine kinase.

Macrophage Stimulating Protein 157
VIII. TARGET CELLS FOR MACROPHAGE STIMULATING PROTEIN
A. Macrophages
Expression of Ron or Stk is restricted to specific subpopulations of the mononuclear phagocyte lineage and is a late maturational event. Whereas murine resident peritoneal macrophages responded to MSP, acute exudate macrophages, which are recent arrivals from the circulation, did not (Leo- nard and Skeel, 1980; Iwama et al., 1995). Functional responses correlated with expression of Stk, as well as with expression of F4/80, a marker of macrophage maturation. By FACS analysis, resident peritoneal macrophages were Stkhigh-F4/80high; acute exudate macrophages were Stknegative-F4/ 80'"". Within 3 days after induction, the exudate macrophages acquired the mature expression pattern of resident macrophages. Other Stk-negative murine mononuclear phagocyte subpopulations by FACS analysis included gated cells from bone marrow, blood, spleen, and bronchoalveolar lavage (Iwama et al., 1995). In humans, peripheral blood monocytes do not express Ron and do not respond to MSP. In contrast, Ron is detectable by immuno- staining in all resident macrophages of normal human dermis (Nanney et al., 1998). It will be of interest to test for Ron expression by macrophages at oth- er interfaces between host and external environment, notably lung and in- testinal tract.
The cellular responses to MSP are remarkably diverse. In the case of murine resident peritoneal macrophages they can be grouped into two broad categories: motility and mediator production. Effects on motility have been documented in various assays. Before MSP was purified, activity in serum was detected as induction of macrophage responsiveness to the complement- derived chemoattractant, C5a (Leonard and Skeel, 1976). When added to the bottom wells of a multiwell chemotaxis chamber, purified MSP is an attractant for murine macrophages (Skeel and Leonard, 1994). Unlike chemoattractants that can recruit circulating leukocytes to extravascular sites, MSP is incapable of this action because of the absence of Ron or Stk receptors on circulating monocytes. Addition of MSP to macrophages in tis- sue culture wells induces a shape change characterized by cytoplasmic pro- jections that is fully developed within an hour (Leonard and Skeel, 1976). This reflects induction of a complex motility pattern that can be observed by time-lapse photography (K. Fuller and E. J. Leonard, unpublished data). Signs of membrane stimulation are evident within 15 min, as spiky projec- tions (filipodia) extend and withdraw from the cell surface. This is followed shortly by broader projections (lamellipodia), which appear to envelop flu- id droplets (macropinocytosis) that enter the cell and migrate to the interior.

158 Edward 1. Leonard and Alla Danilkovitch
Membrane activity is accompanied by cellular motility, characterized by ex- tension and withdrawal of cell projections, unaccompanied by significant translational movement. Although this cellular motility program is initiated by MSP, it persists for at least 2 hr without a requirement for continuing MSP stimulation. This conclusion is based on the fact that MSP action is depen- dent on PI3K, and can be prevented by prior administration of the PUK in- hibitor wortmannin. However, once MSP induces the cellular motility pat- tern, addition of wortmannin has no effect.
The functional importance of this MSP-induced macrophage membrane motility is unclear. Even unstimulated resident macrophages have a remark- ably high plasma membrane turnover; they internalize the equivalent of all of their cell surface every 33 min (Steinman et al., 1976). This is associated with rnicropinocytosis, which can be quantified by uptake of a nonbinding solute such as Lucifer Yellow (Knight et al., 1992). Our preliminary data show that Lucifer Yellow uptake by macrophages is not increased by MSP (Casas-Finet and E. J. Leonard, unpublished data). Is it possible that MSP enhances phagocytosis of certain types of nonopsonized particles, which might enhance wound debridement? In the case of opsonized particles, the effect of MSP on uptake via the CR3 receptor of erythrocytes coated with C3bi is unequivocal: In the absence of MSP, these cells adhere to the macrophages, but are not ingested. Addition of MSP causes ingestion with- in minutes (Skeel et al., 1991). The receptor specificity of these phagocytic events is striking. In contrast to the C3bi opsonin, erythrocytes coated with IgC are ingested via the macrophage Fc receptor without a requirement for MSP.
The other broad category of MSP actions on macrophages relates to me- diator production. Endotoxin, or combinations of proinflammatory cy- tokines, causes expression of murine macrophage-inducible nitric oxide syn- thase, an effect that can be detected by Northern blots for the mRNA or by measurement of nitrate in the culture fluid. MSP prevents induction of NO- synthase by any of the above stimuli (Wang et al., 1994d). The inhibitory action of MSP is confined to this specific mediator. MSP did not inhibit en- dotoxin-induced expression of mRNA for monocyte chemoattractant pro- tein-1. Furthermore, IVSP caused secretion of IL-6 (but not IL-1 or TNFa) within 6 hr, and did not inhibit endotoxin-induced secretion of IL-1, IL-6, or TNFa (A. Skeel and E. J. Leonard, unpublished data). The in vitro mod- ulation by MSP of endotoxin-induced N O production now has an in vivo counterpart. Concentrations of nitrate in serum of Stk-’- mice that received endotoxin intravenously were higher than in serum of comparably treated normal mice; and at a critical endotoxin dose, only 20% of the Stk-’- mice survived, compared to 80% survival for normal mice (Correll et al., 1997). If MSP plays a role in the host response to endotoxemia, pro-MSP must be cleaved to biologically active MSP. Within 4 hr after i.v. administration of

Macrophage Stimulating Protein I59
endotoxin to normal human volunteers, mature MSP was detected in EDTA plasma (E. J. Leonard and A. F. Suffredini, unpublished data). In view of a proposed role of NO in inflammation (Grigoriadis et al., 1994), it was also of interest that DTH responses to oxazalone, measured as ear swelling, were more intense in Stk-’- mice than in normal mice (Correll et al., 1997).
B. Osteoclasts
It has been suggested that macrophages and osteoclasts are derived from a common hematopoietic precursor (Grigoriadis et al., 1994). Of great interest, therefore, is the transient expression of Stk mRNA in foci of bone formation in mouse embryos (Gaudino et al., 1995; Quantin et al., 1996). Localization of Stk in cells that also contained tartrate-resistant acid phos- phatase (TRAP) suggested that expression was in differentiated osteoclasts (Quantin et al., 1996). Stk protein was detected by immunohistochemical staining in multinucleated osteoclasts isolated from femurs of 14-day-old mice (Kurihara et al., 1996). Together, these findings suggest that MSP could play a role in remodeling of developing bone. However reported functional effects of MSP on osteoclasts-stimulation of membrane ruffling and pit for- mation-have been limited to osteoclast-like cells derived from cultured bone marrow (Kurihara et al., 1996). In contrast to effects of HGF on osteoclasts isolated from rat (Fuller et al., 1995) or mouse long bones, MSP did not stim- ulate formation of pits in bone slices, and there was no effect on motility as determined by time-lapse video (K. Fuller and E. J. Leonard, unpublished data). Likewise, MSP did not increase release of 45Ca from fetal rat long bones in tissue culture (L. Raisz and E. J. Leonard, unpublished data).
C. Cells of Ectodermal Origin
Ron, the MSP receptor, was cloned from the cDNA of a human ker- atinocyte library (Ronsin etal., 1993). This led to studies showing that MSP binds with high affinity to human (Wang et al., 1996b) and murine epithe- lial cell lines (Wang et af., 1996a). MSP stimulated thymidine incorporation into A549 lung carcinoma cells, T47D mammary carcinoma cells, PC12 pheochromocytoma cells (Gaudino et al., 1994, 1995), and CMT-93 rectal carcinoma cells (Waltz et al., 1997). When tested on BK-1, a murine normal keratinocyte cell line, MSP caused an increase in cell number over a 10-day culture period, with an efficacy comparable to that of EGF (Wang et al., 1996a). Induction by MSP of migration in chemotaxis chambers was ob- served for A549 cells (Gaudino et al., 1994), and for human and murine neo- plastic cell lines as well as HK-NOC, a cell line established from normal hu-

160 Edward J . Leonard and Alla Danilkovitch
man foreskin (Wang et al., 1996a,b). Stimulation of motility is dependent on PI3K (Wang et al., 1996b), as elaborated in Section VI. In response to hu- man wounds, Ron is upregulated in keratinocytes and in accessory epithe- lial structures including sweat ducts and hair follicles. This finding, along with the presence of active MSP in wound fluid, suggests a possible role for MSP in wound healing (Nanney et al., 1998).
In immunolocalization studies of ciliated epithelium, Ron was detected in nasal and bronchial epithelium and in normal oviduct (Sakamoto et al., 7 997). Primary cultures of normal human bronchial epithelium expressed Ron as assessed by flow cytometry and bound MSP with a K , of 0.5 nM. MSP caused tyrosine phosphorylation of the receptor. Ron and Met were both expressed in ciliated bronchial epithelium, the latter in a basolateral lo- cation, the former at the apical surface just below the base of the cilia. This led to the finding that MSP caused a transient increase of about 20% in the ciliary beat frequency of nasal mucosal cells. In view of Ron mRNA expres- sion in sperm and MSP mRNA in epithelium of epididymis (Ohshiro et al., 1996), the authors suggest a possible role for MSP in sperm motility as well as in mucociliary transport.
D. Vascular Endothelial Cells
Ron protein was detected by immunolocalization on vascular endothelial cells in human dermis. The frequency of detection was higher in burn wounds than in normal skin (Nanney et al., 1998). HGF has been reported to be angiogenic. MSP is also angiogenic by the mouse cornea assay (Y. Cao and E. J. Leonard, unpublished data).
E. Bone Marrow Cells
MSP was evaluated for effects on erythroid and myeloid progenitor cells of human bone marrow, and no colony stimulating activity was detected. However when marrow progenitors were maximally stimulated by colony stimulating factor plus either steel factor or Flt3 ligand, MSP inhibited by about 50% the formation of granulocyte-macrophage colonies (Broxmeyer et al., 1996).
MSP stimulated maturation of human megakaryocyte cell lines as well as primary bone marrow megakaryocytes. The criterion for maturation was an increase in ploidy, which was quantified as DNA content per cell, determined by flow cytometry. MSP stimulated increased secretion of IL-6 by these cells. Since the effect on ploidy was abolished by antibodies to IL-6, it appears that MSP acts via IL-6 in this system (Banu et al., 1996). MSP also stimulates se-

Macrophage Stimulating Protein 161
cretion of IL-6 by murine resident peritoneal macrophages, in amounts com- parable to that induced by endotoxin (A. Skeel and E. J. Leonard, unpub- lished data).
E Insights from Message Expression and from Knockout Mice
Two papers on Stk mRNA in mouse embryos show that it is expressed transiently in specific locations in the developing central nervous system (Quantin et af., 1996; Gaudino et af., 1995). Receptor message is expressed in mucosal cells of stomach, small intestine, and colon in both embryonic and adult mice (Quantin et af., 1996). The possibility of paracrine effects of MSP should be considered in light of low MSP message expression in organs other than liver, the locus of constitutive secretion of pro-MSP into the cir- culation. This includes kidney and pancreas (Yoshimura et af., 1993), ep- ithelium of rat epididymis (Ohshiro et af., 19961, rat lung, adrenal and pla- centa (Degen et al., 1991) and central nervous system of the chick embryo (Thery et af., 1995). MSP mRNA was also detected by RT-PCR in some, but not all, samples of nonneoplastic human lung tissue adjacent to surgi- cally removed lung tumor as well as selected lung carcinoma cell lines (Wil- lett et al., 1998).
In contrast to developmental defects in mice with targeted mutations in Met (Bladt et af., 1995) or its ligand (Uehara et al., 1995; Schmidt et af., 1995), Stk-l- mice developed normally (Correll et af., 1997). Abnormali- ties reported to date relate to endotoxin challenge and DTH reactions as not- ed in section VIIIA. MSP-l- mice also grow to adulthood without obvious abnormalities except for lipid-containing vacuoles in hepatocytes. Notable is the fact that these animals healed an incisional wound as rapidly as nor- mal mice (Bezerra et af., 1998). This should be considered in relation to the generation of active MSP and upregulation of Ron in human burn wounds (Nanney et af., 1998). The possibilities are that the human data are epiphe- nomena unrelated to wound healing, that there is redundancy in the systems, that an unidentified additional ligand can activate Ron, or that a more chal- lenging wound model is required to reveal a defect in the MSP-l- mice.
IX. PERSPECTIVE
From the structural and in vitro biological data summarized in this review, we can conclude that MSP is a growth and motility factor that activates a typical cell membrane protein tyrosine kinase receptor. However, despite an

162 Edward 1. Leonard and Alla Danilkovitch
increasing body of knowledge about MSP and Ron in vitro, we do not know the role of MSP/Ron in vivo. “In vivo veritas” was posted on the office door of the first chief of our laboratory, an adaptation for medical research of the ancient insight (“in vino veritas”) into the effects of the Roman equivalent of the two-martini lunch.
We hope to elicit this in vivo truth incrementally, by using the in vitro data to design animal experiments and to make clinical correlations in patholog- ical conditions. For example, the inhibition by MSP of endotoxin-induced macrophage synthesis of NO synthase (Wang et al., 1994d) provided a ra- tional basis for studying the response of S t k k - mice to endotoxin challenge (Correll et al., 1997). And the study of MSP and Ron in human wounds (Nanney et al., 1998) derived from a postulated role in tissue injury, which was based on the evolution of MSP from coagulation proteins (Donate et al., 1994) and on the presence of Ron on keratinocyte cell lines (Wang et al., 1996a,b). We hope eventually to know if MSP has a physiologic role in nor- mal turnover of cells in skin and intestinal mucosa, and whether any of the actions described in Section VIII on target cells have in vivo relevance. Fi- nally, the recent demonstration that cells transfected with an appropriately mutated Ron are tumorigenic (Santoro et al., 1998) will doubtless stimulate a search for similar mutations in human cancer.
Note added in proof: A vital role for the RodStk receptor in development was recently established by Muraoka et al. (1999) who reported that Stk-’- murine embryos remained viable through the blastocyst stage, but failed to survive thereafter. Hemizygous mice had abnormally high nitric oxide pro- duction in response to endotoxin, confirming the results of Correll et d., (1 997).
REFERENCES
Abedi, H., and Zachary, I. (1997). J. Biol. Chem. 272,15442-15451. Akimoto, K., Takahashi, R., Moriya, S., Nishioka, N., Takayanagi, J., Kimura, K., Fukui, Y.,
Osada, S., Mizuno, K., Hirai, S . , Kazlauskas, A., and Ohno, S. (1996). EMBOJ. 15, 788- 798.
Aronheim, A., Engelberg, D., Li, N., al-Alawi, N., Schlessinger, j., and Karin, M. (1994). Cell (Cambridge, Mass.) 78, 949-961.
Banu, N., Price, D. J., London, R., Deng, B., Mark, M., Godowski, P. J., and Avraham, H. (1996). J. Immunol. 156,2933-2940.
Baron, V., Calleja, V., Ferrari, P., Alengrin, F., and Van Obberghen, E. (1998). J. Biol. Chem.
Bezerra, J. A., Carrick, T. L., Degen, J. L., Witre, D., and Degen, S. F. (1998). 1. Clin. Invest. 273,7162-7168.
101,1175-1183.

Macrophage Stimulating Protein I63
Bladt, F., Riethmacher, D., Isenmann, S., Aguzzi, A., and Birchmeier, C. (1995). Nature (Lon-
Borset, M., Lien, E., Espevik, T., Helseth, E., Waage, A., and Sundan, A. (1996). J. Biol. Chem.
Bos, J. L. (1989). Cancer Res. 49,4682-4689. Bottaro, D. P., Rubin, J. S., Faletto, D. L., Chan, A. M., Kmiecik, T. E., Vande Woude, G. F.,
Broxmeyer, H. E., Cooper, S., Li, Z.-H., Lu, L., Sarris, A., Wang, M.-H., Chang, M.-S., Don-
Cantley, L. C., Auger, K. R., Carpenter, C., Duckworth, B., Graziani, A., Kapeller, R., and
Cartwright, C. A., Meisler, A. I., andEckhart, W. (1990). Proc. Natl. Acad. Sci. U.S.A. 87,558-
Cartwright, C. A., Coad, C. A., and Egbert, B. M. (1994). J. Clin. Invest. 93,509-515. Chen, Q., Seol, D. W., Carr, B., and Zarnegar, R. (1997). Hepatology 26,59-66. Chung, J., Grammer, T. C., Lemon, K. P., Kazlauskas, A., and Blenis, J. (1994). Nature (Lon-
Clark, S . G., Stern, M. J., and Horvitz, H. R. (1992). Nature (London) 356,340-344. Collesi, C., Santoro, M. M., Gaudino, G., and Comoglio, P. M. (1996). Mol. Cell.. Biol. 16,
Cooper, C. S., Park, M., Blair, D. G., Tainsky, M. A., Huebner, K., Croce, C. M., and Vande,
Cooper, J. A., and Howell, B. (1993). Cell (Cambridge, mass.) 73,1051-1054. Correll, P. H., Iwama, A., Tondat, S., Mayrhofer, G., Suda, T., and Bernstein, A. (1997). Genes
Courtneidge, S. A., Dhand, R., Pilat, D., Twamley, G. M., Waterfield, M. D., and Roussel,
Cunningham, B. C., Ultsch, M., de Vos, A. M., Mulkerrin, M. G., Clauser, K. R., and Wells,
Danilkovitch, A., and Leonard, E. J. (1997). J. Leukocyte Biol., Suppl., p. 19 (abstr.). Danilkovitch, A., and Leonard, E. J. (1998). J. Leukocyte Biol., Suppl., p. 32 (abstr.). Degen, S. J., Stuart, L. A., Han, S., and Jamison, C. S. (1991). Biochemistry 30, 9781-
de Vos, A. M., Ultsch, M., and Kossiakoff, A. A. (1992). Science 255,306-312. Donate, L. E., Gherardi, E., Srinivasan, N., Sowdhamini, R., Aparicio, S . , and Blundell, T. L.
(1994). Protein Sci. 3,2378-2394. Dudek, H., Datta, S. R., Franke, T. F., Birnbaum, M. J., Yao, R., Cooper, G. M., Segal, R. A.,
Kaplan, D. R., and Greenberg, M. E. (1997). Science 275,661-665. Empereur, S., Djelloul, S., Di Gioia, Y., Bruyned, E., Mareel, M., Van Hengel, J., Van Roy, F.,
Comoglio, P., Courtneidge, S., Paraskeva, C., Chastre, E., and Gespach, C . (1997). BY. J. Can- cer 75,241-250.
don) 376,768-771.
271,24655-24661.
and Aaronson, S. A. (1991). Science 251, 802-804.
ner, D. B., and Leonard, E. J. (1996). Ann. Hematol. 73,l-9.
Soltoff, S . (1991). Cell (Cambridge, Mass,) 64,281-302.
562.
don) 370,71-75.
551 8 -5526.
W. G. (1984). Nature (London) 311,29-33.
Funct. 1, 1-15.
M. F. (1993). EMBO J. 12,943-950.
J. A. (1991). Science 254, 821-825.
9791.
Erpel, T., and Courtneidge, S. A. (1995). Cum Opin. Cell biol. 7, 176-182. Francis, C. W., and Marder, V. J. (1990). In "Hematology" (W. J. Williams, E. Beutler, A. J. Er-
Franke, T. F., Kaplan, D. R., and Cantley, L. C. (1997). Cell (Cambridge, Muss.] 88,435-437. Fuller, K., Owens, J., and Chambers, T. J. (1995). Biochem. Biophys. Res. Commun. 212,334-
Gaudino, G., Follenzi, A., Naldini, L., Collesi, C., Santoro, M., Gallo, K. A., Godowski, P. J.,
Gaudino, G., Avantaggiato, V., Follenzi, A., Acampora, D., Simeone, A., and Comoglio, P. M.
slev, and M. A. Lichtman, eds.), p. 1313. McGraw-Hill, New York.
340.
and Comoglio, P. M. (1994). EMBO J. 13,3524-3532.
(1995). Oncogene 11,2627-2637.

164 Edward J . Leonard and Alla Danilkovitch
Grigoriadis, A. E., Wang, Z. Q., Cecchini, M. G., Hofstetter, W., Felix, R., Fleisch, H. A., and
Han, S., Stuart, L. A,, and Degen, S. J. F. (1991). Biochemistry 30, 9768-9780. Hanks, S. K., and Polte, T. R. (1997). BioEssays 19, 137. Hanks, S. K., Calalb, M. B., Harper, M. C., and Patel, S. K. (1992). Proc. Natl. Acad. Sci. U.S.A.
Heimbrook, D. C., Oliff, A., and Gibbs, J. B. (1997). In “Cancer Principles and Practice of On- cology” (V. DeVita, S. Hellman, and S. Rosenberg, eds.), pp. 35-43. Lippincott-Raven press, Philadelphia.
Wagner, E. F. (1994). Science 266,443-448.
89,8487-8491.
Heldin, C. H. ( 1995). Cell (Cambridge, Mass.) 80,213-223. Hu, P., Margolis, B., Skolnik, E. Y., Lammers, R., Ullrich, A., and Schlessinger, J. (1992). Mol.
Huff,J. L., Jelinek, M. A., Borgman, C. A., Lansing, T. J., and Parsons, J. T. (1993). Proc. Nutl.
Hwang, ,M. C., Sung, Y. J., and Hwang, Y. W. (1996). J . Biol. Chem. 271, 8196-8202. Ip, Y. T., and Davis, R. J. (1998). Curr. Opin. Cell Biol. 10, 205-219. Iwarna, A., Okano, K., Sudo, T., Matsuda, Y., and Suda, T. (1994). Blood 83,3160-3169. Iwama, A.. Wang, M.-H., Yamaguchi, N., Okano, K., Sudo, T., Gervais, F., Morissette, C.,
Iwama, A., Yamaguchi, N., and Suda, T. (1996). EMBO J . 15,5866-5875. Jeffers, M., Rong, S., and Woude, G. F. (1996). J . Mol. Med. 74, 505-513. Joneson, T., McDonough, iM., Bar-Sagi, D., and Van Aelst, L. (1996). Science 274, 1374-1376. Kapeller, R., and Cantley, L. C. (1994). BioEssays 16, 565. Karunagaran, D., Tzahar, E., Beerli, R. R., Chen, X., Graus-Porta, D., Ratzkin, B. J., Seger, R.,
Kauffmann-Zeh, A., Rodriguez-Viciana, P., Ulrich, E., Gilbert, C., Coffer, P., Downward, J., and
Knight, K. R., Vairo, G., and Hamilton, J. A. (1992). J . Leukocyte Biol. 51, 350-359. Kurihara, N., Iwama, A., Tatsumi, J., Ikeda, K., and Suda, T. (1996). Blood 87, 3704-3710. Kypta, R. M., Goldberg, Y., Ulug, E. T., and Courtneidge, S. A. (1990). Cell 62,481-492. Lamarche, N., Tapon, N., Stowers, L., Burbelo, P. D., Aspenstrom, P., Bridges, T., Chant, J.,
Lemmon, M. A.. and Schlessinger, J. (1994). Trends Biochem. Sci. 19,459-463. Lemmon, M. A,, Pinchasi, D., Zhou, M., Lax, I., and Schlessinger, J. (1997).]. Biol. Chem. 272,
Leonard, E. J., and Skeel, A. (1976). Exp. Cell Res. 102,434-438. Leonard, E. J., and Skeel, A. (1980). J . Retrculoendothel. Soc. 28,437-447. Leonard, E. J., and Skeel, A. (1996). 1. Leukocyte Biol. 60,453-458. Leonard, E. J., Skeel, A., and Allenmark, S. (1982). Arch. Biochem. Biophys. 214, 12-16. Li, B. Q., Wang, M. H., Kung, H. F., Ronsin, C., Breathnach, R., Leonard, E. J., and Kamata,
Logan, S. K., Falasca, hi., Hu, P., and Schlessinger, J. (1997). Mol. Cell. Biol. 17,5784-5790. Lopez-Ilasaca, M., Li, W., Uren, A., Yu, J. C., Kazlauskas, A., Gutkind, J. S., and Heidaran,
Lowy, D. R., and Willurnsen, B. M. (1993). Annu. Rev. Biochem. 62,851-891. Maa, M. C., Leu, T. H., McCarley, D. J., Schatzman, R. C., and Parsons, S. J. (1995). Proc.
Maggiora, P., Marchio, S., Stella, M., Giai, M., Belfiore, A., De Bortoli, M., Di Renzo, M.,
Marshall, M. S. (1995). FASEB]. 9, 1311-1318. Martin, P. (1997). Science 276, 75-81.
Cell. Biol. 12, 981-990.
Acad. Sci. U.S.A. 90, 6140-6144.
Leonard, E. J., and Suda, T. (1995). Blood 86, 3394-3403.
Hynes, N. E., and Yarden, Y. (1996). EMBO 1. IS, 254-264.
Evan, G. (1997). Nature (London) 385,544-548.
and Hall, A. (1996). Cell (Cambridge, Mass.) 87,519-529.
631 1-6317.
T. (1995). Biochem. Biophys. Res. Commun. 216, 110-118.
Sf. A. (1997). Biochem. Biophys. Res. Commun. 232,273-277.
Nutl. Acad. Sct. U.S.A. 92, 6981-6985.
Costantino, A., Sismond, P., and Comoglio, P. M. (1998). Oncogene 16,2927-2933.

Macrophage Stimulating Protein 165
Matsumoto, K., Takehara, T., Inoue, H., Hagiya, M., Shimizu, S . , and Nakamura, T. (1991).
Matsumoto, K., Nakamura, T., and Kramer, R. H. (1994). J. Biol. Chem. 269,31807-31813. Miller, M., and Leonard, E. J. (1998). FEBS Lett. 429, 1-3. Moriya, S., Kazlauskas, A., Akimoto, K., Hirai, S., Mizuno, K., Takenawa, T., Fukui, Y., Watan-
Moscatello, D. K., Montgomery, R. B., Sundareshan, P., McDanel, H., Wong, M. Y., and Wong,
Muraoka, R. S., Sun, W. Y., Colbert, M. C., Waltz, S . E., Witte, D. P., Degen, J. L., and Degen,
Nanney, L. B., Skeel, A., Luan, J., Polis, S., Richmond, A., Wang, M.-H., and Leonard, E. J.
Ohshiro, K., Iwama, A., Matsuno, K., Ezaki, T., Sakamoto, O., Hamaguchi, I., Takasu, N., and
Okigaki, M., Komada, M., Uehara, Y., Miyazawa, K., and Kitamura, N. (1992). Biochemistry
Oldham, S. M., Cox, A. D., Reynolds, E. R., Sizemore, N. S., Coffey, R. J. J., and Der, C. J. (1998). Oncogene 16,2565-2573.
Owens, L. V., Xu, L., Craven, R. J., Dent, G. A., Weiner, T. M., Kornberg, L., Liu, E. T., and Cance, W. G. (1995). Cancer Res. 55,2752-2755.
Park, M., Dean, M., Cooper, C. S., Schmidt, M., O’Brien, S. J., Blair, D. G., and Vande, W. G. (1986). Cell (Cambridge, Mass.) 45,895-904.
Park, M., Dean, M., Kaul, K., Braun, M. J., Gonda, M. A., and Vande Woude, G. (1987). Proc. Natl. Acad. Sci. U.S.A. 84, 6379-6383.
Parker, P. J. (1995). Curr. Biol. 5,577-579. Partin, A. W., Schoeniger, J. S., Mohler, J. L., and Coffey, D. S. (1989). Proc. Natl. Acad. Sci.
Philo, J. S., Wen, J., Wypych, J., Schwartz, M. G., Mendiaz, E. A., and Langley, K. E. (1996).
Ponzetto, C., Bardelli, A., Zhen, Z., Maina, F., dalla Zonca, P., Giordano, S., Graziani, A.,
Qiu, R. G., Chen, J., Kirn, D., McCormick, F., and Symons, M. (1995a). Nature (London) 374,
Qiu, R. G., Chen, J., McCorrnick, F., and Symons, M. (1995b). Proc. Natl. Acud. Sci. U.S.A.
Quantin, B., Schuhbaur, B., Gesnel, M.-C., DollC, P., and Breathnach, R. (1996). Dev. Dyn. 204,
Rahimi, N., Tremblay, E., and Elliott, B. (1996). J. Biol. Chem. 271,24850-24855. Rankin, S., and Rosengurt, E. (1994). J. Biol. Chem. 269, 704-710. Reif, K., Nobes, C. D., Thomas, G., Hall, A., and Cantrell, D. A. (1996). Curr. Biol. 6, 1445-
Rodrigues, G. A., and Park, M. (1993). Mol. Cell. Biol. 13,6711-6722. Rodrigues, G. A., Park, M., and Schlessinger, J. (1997). EMBO J. 16, 2634-2645. Rodriguez-Viciana, P., Warne, P. H., Dhand, R., Vanhaesebroeck, B., Gout, I., Fry, M. J., Wa-
terfield, M. D., and Downward, J. (1994). Nature (London) 370,527-532. Rodriguez-Viciana, P., Warne, P. H., Khwaja, A., Marte, B. M., Pappin, D., Das, P., Waterfield,
M. D., Ridley, A. and Downward, J. (1997). Cell (Cambridge, Mass.) 89,457-467. Ronsin, C., Muscatelli, F., Mattei, M. G., and Breathnach, R. (1993). Oncogene 8, 1195-
1202. Sakamoto, O., Iwama, A., Amitani, R., Takehara, T., Yamaguchi, N., Yamamoto, T., Masuya-
ma, K., Yamanaka, T., Ando, M., and Suda, T. (1997). J. Clin. Invest. 99, 701-709.
Biochem. Biophys. Res. Commun. 181,691-699.
abe, Y., Ozaki, S., and Ohno, S. (1996). Proc. Natl. Acad. Sci. U.S.A. 93, 151-155.
A. J. (1996). Oncogene 13,85-96.
S. J. (1999). J. Clin. Invest. 103,1277-1285.
(1998). J. Invest. Dermatol. 111,573-581.
Suda, T. (1996). Biochem. Biophys. Res. Commun. 227,273-280.
31,9555-9561.
U.S.A. 86, 1254-1258.
1. Biol. Chem. 271,6895-6902.
Panayotou, G., and Comoglio, P. M. (1994). Cell (Cambridge, Mass.) 77,261-271.
457-459.
92,11781-11785.
383-390.
1455.

I66 Edward 1. Leonard and Alla Danilkovitch
Sakata, H., Stahl, S. J., Taylor, W. G., Rosenberg, J. M., Sakaguchi, K., Wingfield, P. T., and Ru- bin, J. S. (1997). J . Biol. Chem. 272, 9457-9463.
Santoro, M., Carlomagno, F., Romano, A., Bottaro, D. P., Dathan, N. A., Grieco, M., Fusco, A., Vecchio, G., Matoskova, B., and Kraus, M. H. (1995). Science 267,381-383.
Santoro, M., Penengo, L., Minetto, M., Orecchia, S., Cilli, M., and Gaudino, G. (1998). Onco- gene 17, 741-749.
Santoro, M. M., Collesi, C., Grisendi, S., Gaudino, G., and Comoglio, P. M. (1996). Mol. Cell. Biol. 16,7072-7083.
Schaller, M. D., Borgman, C. A., Cobb, B. S., Vines, R. R., Reynolds, A. B., and Parsons, J. T. (1992). Proc. Natl. Acad. Sci. U.S.A. 89, 5192-5196.
Schmidt, C., Bladt, F., Goedecke, S., Brinkmann, V., Zschiesche, W., Sharpe, M., Gherardi, E., and Birchmeier, C. (1995). Nature (London) 373, 699-702.
Schmidt, L., Duh, F. M., Chen, F., Kishida, T., Glenn, G., Choyke, P., Scherer, S. W., Zhuang, Z., Lubensky, I., Dean, M., Allikmets, R., Chidambaram, A., Bergerheim, U. R., Feltis, J. T., Casadevall, C., Zamarron, A., Bernues, M., Richard, S., Lips, C. J., Walther, M. M., Tsui, L. C., Geil, L., Orcutt, M. L., Stackhouse, T., and Zbar, B. (1997). Nut. Genet. 16, 68-73.
Skeel, A., and Leonard, E. J. (1994). J . lmmunol. 152,4618-4623. Skeel, A., Yoshimura, T., Showalter, S . , Tanaka, S., Appella, E., and Leonard, E. (1991). J. Exp.
Med. 173,1227-1234. Songyang, Z., Shoelson, S. E., Chaudhuri, M., Gish, G., Pawson, T., Haser, W. G., King, F.,
Roberts, T., Ratnofsky, S., and Lechleider, R. J. (1993). Cell (Cambridge, Mass.) 72, 767- 778.
Songyang, Z., Shoelson, S . E., McGlade, J., Olivier, P., Pawson, T., Bustelo, X. R., Barbacid, M., Sabe, H., Hanafusa, H., and Yi, T. (1994). Mol. Cell. Biol. 14, 2777-2785.
Songyang, Z., Baltimore, D., Cantley, L. C., Kaplan, D. R., and Franke, T. F. (1997). Proc. Natl. Acad. Sci. U.S.A. 94, 11345-11350.
Steinman, R. M., Brodie, S . E., and Cohn, Z. A. (1976). J. Cell. Biol. 68, 665-687. Talamonti, M. S., Roh, M. S., Curley, S. A,, and Gallick, G. E. (1993). J . Clin. Invest. 91, 5 3 -
Thery, C., Sharpe, M. J., Batley, S. J., Stem, C. D., and Gherardi, E. (1995). Dev. Genet. 17,
Tsujimura, T. (1996). Pnthol. lnt. 46, 933-938. Ueda, A., Takeshita, F., Yamashiro, S., and Yoshimura, T. (1998). J Biol. Chem. 273,
Uehara, Y., Minowa, O., Mori, C., Shiota, K., Kuno, J., Noda, T., and Kitamura, N. (1995).
Ulirich, A., and Schlessinger, J. (1990). Cell (Cambridge, Mass.) 61,203-212. Varticovski, L., Harrison-Findik, D., Keeler, M. L., and Susa, M. (1994). Biochim. Biophys.
Wahl, R. C., Costigan, V. J., Batac, J. P., Chen, K., Cam, L., Courchesne, P. L., Patterson, S . D.,
Waltz, S. E., McDowell, S. A., Muraoka, R. S., Air, E. L., Flick, L. M., Chen, Y. Q., Wang,
Wang, M.-H., Skeel, A., Yoshimura, T., Copeland, T. D., Sakaguchi, K., and Leonard, E. J.
Wang, M.-H., Ronsin, C., Gesnel, M.-C., Coupey, L., Skeel, A., Leonard, E. J., and Breathnach,
Wang, M.-H., Yoshimura, T., Skeel, A., and Leonard, E. J. (1994b). J . Biol. Chem. 269,3436-
Wang, M.-H., Gonias, S. L., Skeel, A., Wolf, B. B., Yoshimura, T., and Leonard, E. J. (1994~).
60.
90-101.
19339-1 9347.
Nature (London) 373,702-705.
Acta 1226, 1-11.
Zhang, K., and Pacifici, R. E. (1997).J. Biol. Chem. 272,15053-15056.
M. H., and Degen, S. J. (1997). J . Biol. Chem. 272,30526-30537.
(1993). J . Leukocyte B i d . 54,289-295.
R. (1994a). Science 266, 117-119.
3440.
J . Biol. Chem. 269,13806-13810.

Macrophage Stimulating Protein 167
Wang, M.-H., Cox, G. W., Yoshimura, T., Sheffler, L. A., Skeel, A., and Leonard, E. J. (1994).
Wang, M.-H., Iwama, A., Skeel, A., Suda, T., and Leonard, E. J. (1995). Proc. Natl. Acad. Sci.
Wang, M.-H., Dlugosz, A. A., Sun, Y., Skeel, A., Yuspa, S. H., Suda, T., and Leonard, E. J.
Wang, M.-H., Montero-Julian, F. A., Dauny, I., and Leonard, E. J. (1996b). Oncogene 13,
Wang, M.-H., Skeel, A., and Leonard, E. J. (1996~). J. Clin. Invest. 97, 720-727. Wang, M.-H., Julian, F. M., Breathnach, R., Godowski, P. J., Takehara, T., Yoshikawa, W.,
Hagiya, M., and Leonard, E. J. (1997). J. Biol. Chem. 272,16999-17004. Weiner, T. M., Liu, E. T., Craven, R. J., and Cance, W. G. (1993). Lancet 342, 1024-1025. Whitmarsh, A. J., and Davis, R. J. (1996). J. Mol. Med. 74,589-607. Willett, C. G., Smith, D. I., Shridhar, V., Wang, M. H., Emanuel, R. L., Patidar, K., Graham,
S. A., Zhang, F., Hatch, V., Sugarbaker, D. J., and Sunday, M. E. (1997). J. Clin. Invest. 99,
Willett, C. G., Wang, M. H., Emanuel, R. L., Graham, S. A., Smith, D. I., Shridhar, V., Sugar-
Williams, J. C., Wierenga, R. K., and Saraste, M. (1998). Trends Biochem. Sci. 23, 179-184. Yoshikawa, W., Hara, H., Takehara, T., Shimonishi, M., Sakai, H., Shimizu, N., Shimizu, S.,
Wang, M.-H., Hagiya, M., Skeel, A., and Leonard, E. J. (1999). Arch. Biochem. Biophys.
Yoshimura, T., Yuhki, N., Wang, M.-H., Skeel, A., and Leonard, E. J. (1993). J. Biol. Chem.
Zhan, X., Plourde, C., Hu, X., Fried, R., and Maciag, T. (1994). J. Biol. Chem. 269,20221-
Zhu, T., Goh, E. L., and Lobie, P. E. (1998). J. Biol. Chem. 273, 10682-10689.
J. Biol. Chem. 269,14027-14031.
U.S.A. 92,3933-3937.
(1996a). Exp. Cell Res. 226,39-46.
21 67-21 75.
2979-2991.
baker, D. J., and Sunday, M. E. (1998). Am. 1. Respir. Cell Mol. Biol. 18,489-496.
363,356 - 360.
268,15461-15468.
20224.




















