
The Influence of Aristide Cavaillé-Coll on French Romantic Organ Building and Organ Music
Upload
independentCategory
view
0download
0
plantation of fetal hematopoietic stem cells. Blood Cells 1991;17: 349.
30. Sanhadji K, Touraine JL, Aitouche A, Vicari A, Chargui J, Goil-lot E. Fetal liver cell transplantation in various murine mod-els. Bone Marrow Transplant 1992; 10: 77.
31. Hayes H, Petit E, Dutrillaux B. Comparison of RBG-bandedkaryotypes of cattle, sheep and goats. Cytogenet Cell Genet1991; 57: 51.
32. Zanjani ED, Silva MRG, Flake AW. Retention and multilineageexpression of human hematopoietic stem cells in human-sheepchimeras. Blood Cells 1994; 20: 331.
33. Pearce RD, Kiehm D, Armstrong DT, et al. Induction of hemo-poietic chimerism in the caprine fetus by intraperitoneal injec-tion of fetal liver cells. Experientia 1989; 45: 307.
34. Zanjani ED, Almeida-Porada G, Flake AW. Retention and mul-tilineage expression of human hematopoietic stem cells in hu-man-sheep chimeras. Stem Cells 1995; 13: 101.
35. Srour EF, Zanjani ED, Cornetta K, et al. Persistence of humanmultilineage, self-renewing lymphohematopoietic stem cells inchimeric sheep. Blood1993; 82: 3333.
36. Almeida-Porada GD, Hoffman R, Manalo P, Gianni AM, ZanjaniED. Detection of human cells in human/sheep chimeric lambswith in vitro human stroma-forming potential. Exp Hematol1996; 24: 482.
Received 11 February 1998.Accepted 27 October 1998.
0041-1337/99/6707-990/0TRANSPLANTATION Vol. 67, 990–998, No. 7, April 15, 1999Copyright © 1999 by Lippincott Williams & Wilkins, Inc. Printed in U.S.A.
LYMPHOPROLIFERATIVE DISORDERS AFTER ORGANTRANSPLANTATION IN CHILDREN
YIGAL DROR,1 MARK GREENBERG,1 GLENN TAYLOR,2 RICCARDO SUPERINA,3 DIANE HEBERT,4
LORI WEST,5 BAIRBRE CONNOLLY,6 LAURI SENA,6 UPTON ALLEN,7 AND SHEILA WEITZMAN1, 8
The Hospital for Sick Children, Toronto, Ontario M5G 1X8, Canada
Background. After organ transplant, patients are atrisk of posttransplant lymphoproliferative disorders(PTLD). The purpose of this study was to analyze 26pediatric cases of PTLD observed at our institutionbetween 1988 and 1996, and to evaluate the validity ofthe Society for Hematopathology Workshop (SHPW)1997 classification in our patient population.
Methods. Charts were reviewed for analysis of inci-dence, clinical course, and outcome. Tissue sampleswere classified by a pathologist according to SHPWrecommendations.
Results. By morphology, 20 were monomorphic, 5polymorphic, and 1 hyperplastic. Assessment of lin-eage by morphology, molecular studies, and immuno-phenotyping did not correlate in six cases. By immu-nophenotyping, 12 were B cell, 4 T cell, 8 mixed B/Tcells, and 2 undetermined. The 20 patients evaluablefor treatment efficacy were treated with various ther-apeutic combinations, including immunosuppressivedrug reduction, acyclovir/ganciclovir, interferon-a,immunoglobulins, surgery, and local irradiation. Nopatient received systemic chemotherapy. Thirteen pa-
tients achieved complete remission and 3, partial; 1died 5 days after starting therapy, and 3 of progressivedisease. Adverse prognostic factors included lowplatelet or neutrophil counts; stage III–IV and SHPWmorphology were marginally significant.
Conclusions. The majority of patients eligible fortreatment can be cured with immunosuppressive drugreduction and antiviral drugs, along with surgery andirradiation when indicated. Systemic chemotherapyor innovative approaches may have a role in unre-sponsive cases. Morphologic SHPW grouping is feasi-ble and seems to have clinical relevance. However,correlation with clonality and immunophenotyping isnot always possible, necessitating modifications in-cluding segregation of descriptive morphology fromclonality and cell origin.
After organ transplant, patients are at increased risk ofdeveloping malignant diseases, including posttransplantlymphoproliferative disorders (PTLD*) (1–3). PTLD is a het-erogeneous array of diseases ranging from reactive poly-clonal lymphoid hyperplasia to monoclonal malignant lym-phoma. As a consequence of prolonged administration ofimmunosuppressive drugs, a defect in T-cell regulation oc-
1 Department of Paediatrics, Division of Hematology/Oncology.2 Department of Pediatric Laboratory Medicine.3 Department of Surgery.4 Department of Paediatrics, Division of Nephrology.5 Department of Paediatrics, Division of Cardiology.6 Department of Diagnostic Imaging.7 Department of Paediatrics, Division of Infectious Disease.8 Address correspondence to: Dr. Sheila Weitzman, Division of
Hematology/Oncology, The Hospital for Sick Children, 555 Univer-sity Avenue, Toronto ON M5G 1X8, Canada.
* Abbreviations: CMVIG, cytomegalovirus hyperimmune globulin;CNS, central nervous system; CR, complete remission; CsA, cyclo-sporine; EBV, Epstein-Barr virus; H&E, hematoxylin and eosinstain; IFN, interferon-a; ISDR, immunosuppressive drug reduction;IVIG, intravenous immune globulin; PCR, polymerase chain reac-tion; PTLD, posttransplant lymphoproliferative disorder(s); Societyfor Hematopathology Workshop (SHPW) classification.
TRANSPLANTATION990 Vol. 67, No. 7

curs that allows for uncontrolled proliferation of B or other Tlymphocytes in response to a viral infection, particularlyEpstein-Barr virus (EBV). Compared to lymphoma arising inimmunocompetent patients, these disorders have a differenthistology, a more aggressive clinical course, less likelihood ofresponse to conventional chemotherapy, and a poorer out-come (4). PTLD is difficult to study, especially in children,because of the lack of consensus in the literature on thedefinitions of the clinical and pathological syndromes. Mostpublished series comprise adult patients with many differentprimary diseases and receiving a variety of transplant typeswith different immunosuppressive drugs. In an attempt tocategorize these disorders, the Society for HematopathologyWorkshop (SHPW) in 1997 developed and published a clas-sification based on morphology, immunophenotyping, andclonality (5).
We report herein 26 consecutive pediatric cases with PTLD,managed without systemic chemotherapy. Our purpose was toanalyze the clinical and histopathologic findings, treatment,and outcome, and to evaluate the validity of the SHPW 1997classification in our patient population. To our knowledge, thisis the largest pediatric series published to date.
PATIENTS AND METHODS
Between October 1988 and December 1996, 26 children developedPTLD at The Hospital for Sick Children, Toronto, Canada. Thesecases were reviewed retrospectively for the transplanted organ, sexof the patient, age at PTLD diagnosis, time interval between trans-plant and PTLD diagnosis, hemoglobin concentration and counts ofneutrophils, lymphocytes, and platelets, EBV status at transplantand at PTLD diagnosis, treatment, organ involvement, and outcome.Seven cases that had occurred after liver transplant (6) and threeafter renal transplant (7, 8) have been reported previously.
After transplantation, patients received cyclosporine (CsA), aza-thioprine, and prednisone for prevention of rejection. Some renal andcardiac recipients also received rabbit antithymocyte serum, anti-lymphocyte globulin, or OKT3 for the first 5–14 days after trans-plant. Liver recipients received high-dose methylprednisolone imme-diately after transplant. When CsA toxicity became unacceptable ortissue rejection occurred, patients after liver or heart transplantwere switched to tacrolimus. When rejection developed, patientswere treated with steroids, either by increasing the prednisone dos-age or by high-dose methylprednisolone administered intravenouslyfor 3 days—followed by OKT3, rabbit antithymocyte serum, or anti-lymphocyte globulin in failed cases.
Diagnosis of PTLD was based on examination of histologic mate-rial obtained by biopsy or at autopsy. All tissue samples were re-viewed for the current study and, whenever possible, reclassifiedaccording to the 1997 SHPW recommendations (5) and the Interna-tional Working Formulation schema (9). Formalin-fixed, paraffin-embedded tissue and snap-frozen tissue were immunostained forvarious combinations of T- and B-cell markers. Rearrangement stud-ies of the immunoglobulin heavy chain (JH) gene and the gene for
T-cell receptor b-chain were done by Southern blot analysis or bypolymerase chain reaction (PCR). Lesions were defined as monoclo-nal if either gene rearrangement or cytogenetic abnormalities intumor cells were demonstrated, or if one light chain predominated(.75%).
Tissue samples were examined with PCR for synthetic oligonucle-otides from the EBV genome, as previously described (10). As themajority of patients were multiply transfused, serologic studies werejudged positive if the results were positive for at least two out ofthree antibody titers: early antigen (positive$1:40), viral capsid an-tigen (positive$1:40), and EBV nuclear antigen (positive$1:10).
TABLE 1. Murphy staging system
Stage Criteria
Stage I A single tumor (extranodal or single anatomic area (nodal) with the exclusion of mediastinum or abdomenStage II A single tumor (extranodal) with regional node involvement; or two or more nodal areas on the same side of the diaphragm; or
a primary gastrointestinal tract tumor, usually in the ileocecal area, with or without involvement of associated mesentericnodes only
Stage III Two single tumors (extranodal) on opposite side of the diaphragm; or two or more nodal areas above and below thediaphragm; or all the primary intrathoracic tumors (mesenteric, pleural, thymic); or all extensive primary intra-abdominaldisease; or all paraspinal or epidural tumors, regardless of other tumor site(s)
Stage IV Any of the above with initial central nervous system and/or bone marrow involvement
FIGURE 1. Monomorphic posttransplant lymphoproliferative disor-der of small bowel. (A) Architectural destruction is evident, withsheets of transformed cells, extending to the submucosa and invadingthe lamina propria (hematoxylin and eosin stain [H&E]; original mag-nification, 310). (B) Most of the cells in the infiltrate are large, atypicaltransformed immunoblasts (H&E; original magnification, 3200).
DROR ET AL.April 15, 1999 991
Patients were staged whenever possible according to the Murphystaging system (Table 1) (11), by pathology review, and by the re-corded results of cerebrospinal fluid cytology and bone marrow bi-opsy or aspiration. All imaging studies were also reviewed. Standardresponse criteria were used to define complete remission (CR), con-tinuous complete remission, partial remission, no response, and pro-gressive disease (12).
A chi-square or Fisher’s exact test was used to test differences inproportions. Significance was defined as P,0.05.
RESULTS
Prevalence. Of 691 children who received solid-organtransplants at The Hospital for Sick Children (Toronto, Can-ada), 26 (3.8%) developed PTLD from October 1988 to Sep-tember 1996 inclusive: 14 of 135 liver recipients (10%), 6 of519 kidney recipients (1.2%), 5 of 34 heart recipients (15%),and 1 of 3 liver-kidney transplant recipients (33%). PTLDwas diagnosed in patients aged 1–17.5 years (median, 7.25),from 1.5 to 73 months (median, 5.25) after transplant, at amedian of 5.25 months after liver transplant, 4.25 monthsafter kidney transplant, and 9.5 months after heart trans-plant. The male to female ratio was 1.6:1.
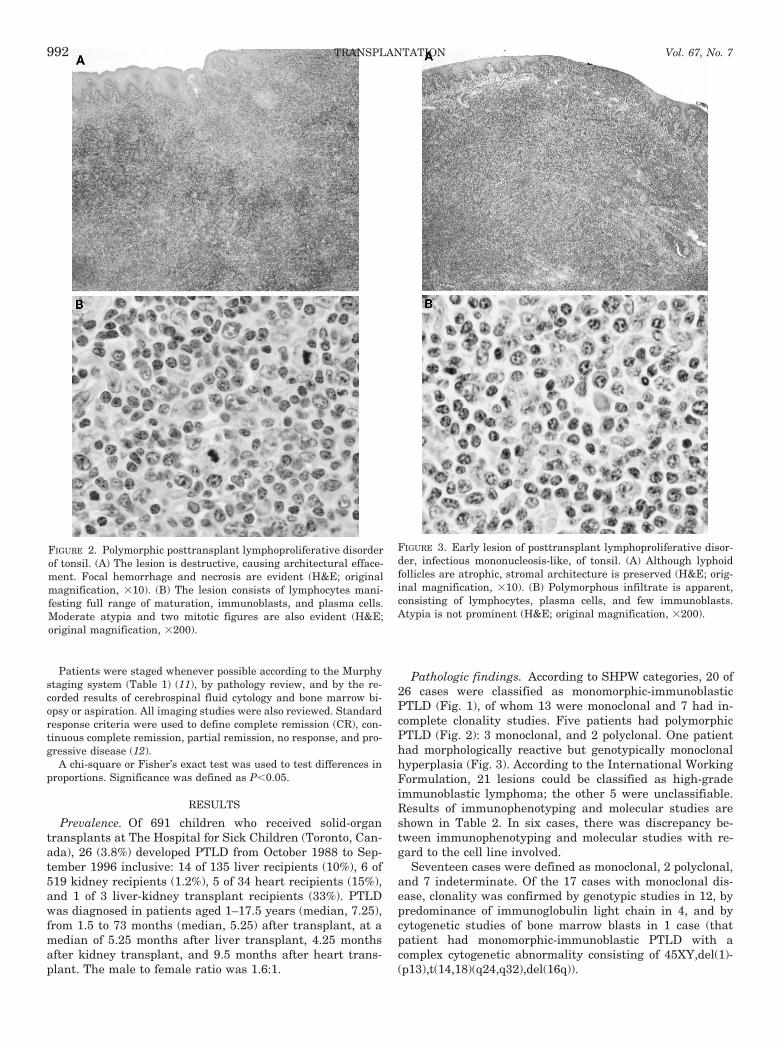
Pathologic findings. According to SHPW categories, 20 of26 cases were classified as monomorphic-immunoblasticPTLD (Fig. 1), of whom 13 were monoclonal and 7 had in-complete clonality studies. Five patients had polymorphicPTLD (Fig. 2): 3 monoclonal, and 2 polyclonal. One patienthad morphologically reactive but genotypically monoclonalhyperplasia (Fig. 3). According to the International WorkingFormulation, 21 lesions could be classified as high-gradeimmunoblastic lymphoma; the other 5 were unclassifiable.Results of immunophenotyping and molecular studies areshown in Table 2. In six cases, there was discrepancy be-tween immunophenotyping and molecular studies with re-gard to the cell line involved.
Seventeen cases were defined as monoclonal, 2 polyclonal,and 7 indeterminate. Of the 17 cases with monoclonal dis-ease, clonality was confirmed by genotypic studies in 12, bypredominance of immunoglobulin light chain in 4, and bycytogenetic studies of bone marrow blasts in 1 case (thatpatient had monomorphic-immunoblastic PTLD with acomplex cytogenetic abnormality consisting of 45XY,del(1)-(p13),t(14,18)(q24,q32),del(16q)).
FIGURE 2. Polymorphic posttransplant lymphoproliferative disorderof tonsil. (A) The lesion is destructive, causing architectural efface-ment. Focal hemorrhage and necrosis are evident (H&E; originalmagnification, 310). (B) The lesion consists of lymphocytes mani-festing full range of maturation, immunoblasts, and plasma cells.Moderate atypia and two mitotic figures are also evident (H&E;original magnification, 3200).
FIGURE 3. Early lesion of posttransplant lymphoproliferative disor-der, infectious mononucleosis-like, of tonsil. (A) Although lyphoidfollicles are atrophic, stromal architecture is preserved (H&E; orig-inal magnification, 310). (B) Polymorphous infiltrate is apparent,consisting of lymphocytes, plasma cells, and few immunoblasts.Atypia is not prominent (H&E; original magnification, 3200).
TRANSPLANTATION992 Vol. 67, No. 7

EBV studies. Pretransplant studies revealed seropositiv-ity in 5 (19%) of the 26 PTLD patients and seronegativity in21 (81%). EBV serology at the time of PTLD diagnosis waspositive in 16 cases (62%), negative in 9 (34%), and unavail-able in 1. The difference in EBV serology before transplantand at the time of PTLD diagnosis was statistically signifi-cant (P50.003). Eleven patients had seroconversion some-time between transplant and development of PTLD; theother 5 had been seropositive before transplant.
EBV sequences were detected in tumor tissue by PCR in 21(91%) of the 23 cases tested. Interestingly, EBV sequenceswere found in all four cases with immunophenotypically T-PTLD, and in one of two cases with pure T-cell rearrange-ment.
Murphy system staging. Pre- or postmortem examinationof cerebrospinal fluid cytology or bone marrow was not donefor seven patients. Of these, three had disseminated disease(at least stage III), which was classified as advanced (stageIII or IV) disease. Another three had localized disease, withno suggestion of bone marrow or CNS involvement by clini-cal, laboratory, and imaging studies. Because involvement of
these sites is very uncommon in childhood PTLD, particu-larly in the absence of positive clinical or laboratory findings,the latter trio of cases were classified as stage I or II, as ifbone marrow and lumbar puncture test results were nega-tive.
Considering the preceding, by Murphy staging there were8 patients with localized stage I–II disease and 17 withadvanced stage III–IV disease; 1 patient could not be stagedbecause of limited investigation.
Therapeutic approaches. Of the 26 patients, 5 were iden-tified at postmortem examination only, and another diedbefore pathology results were available. The remaining 20were treated with various combinations of therapeutic mo-dalities at the discretion of the treating physician (Table 3)and were evaluable for treatment outcomes. All 20 had im-munosuppressive drug reduction (ISDR); 19 received eitheracyclovir (15 patients) or ganciclovir (4 patients). Fifteenpatients received either intravenous immune globulin (IVIG;8 patients) or cytomegalovirus hyperimmune globulin (CM-VIG, Cytogam, MedImmune, Inc. Gaithersburg, MD; 7 pa-tients). Eight patients with stage III–IV disease and two withstage II received interferon-a (IFN). A single intrathecalinjection of methotrexate was administered to one patientwith intracranial PTLD.
Eight patients underwent surgery for therapeutic pur-poses. Three patients had complete macroscopic resection oftheir tumor mass (kidney, bowel, spleen); all of them arealive and in CR. One patient with extrarenal disease hadnephrectomy of an uninfiltrated transplanted kidney be-cause of treatment-resistant systemic disease, achieved CR,and survived. Three patients with extrarenal disease hadresection of their transplant kidney that was also infiltratedby tumor; all three died. One patient with systemic diseasehad CR and survived after resection of intestinal diseasebecause of perforation.
Radiation therapy was given to two patients. One patienthad CNS disease that was treated with cranial irradiation(2400 cGy/24 fraction) after medical treatment failed. Thesecond patient had disease localized to the splenic area,which was macroscopically completely resected; he was sub-sequently treated with 1600 cGy/16 fractions to the whole
TABLE 2. Results of gene rearrangement studies according to thecell type of origina
Immunophenotype n Gene rearrangement
B cell (n512) 4 Ig1, TCR21 Ig1, TCR1 (discrepancy)7 Not done
T cell (n54) 2 Ig1, TCR2 (discrepancy)2 Not done
Mixed B/T (n58) 1 Ig1, TCR2 (discrepancy)1 Ig1, TCR12 Ig2, TCR1 (discrepancy)4 Not done
Indeterminate (n52) 1 Ig1, TCR21 Not done
Total 26 10 Ig1, 4 TCR1
a Ig, immunoglobulin; TCR, T-cell receptor; 1, positive; 2, nega-tive; discrepancy, case(s) of discrepancy of results between immuno-phenotyping and gene rearrangement studies: either B-PTLD lesionwith T-cell receptor gene rearrangement, or T-PTLD with Ig generearrangement, or mixed lesion with only one rearranged gene.
TABLE 3. Therapeutic modalities utilized for 26 patients with PTLD and outcomes
Therapy
TotalOutcome None ISDR,
CVISDR,CV,IVIG
ISDR,CV,IVIG,INF
ISDR,CV,Surgery
ISDR,CV,IVIG,Surgery
ISDR,IVIG, INF,Surgery,Radiotherapy
ISDR,CV,IVIG,INF,Surgery
ISDR, CV,IVIG,INF,Surgery,RadiotherapyIT-MTX
26
Early death, received no therapyb 6 6Early death, after starting therapy 1 1PD, died 2 1 3CR, diedc 1 2 1 4PR, alived 1 1 1 3CR, alive 1 3 1 1 1 1 1 9
a CV, acyclovir/ganciclovir; IVIG, regular intravenous immune globulin or cytomegalovirus hyperimmune globulin.b Diagnosed at postmortem examination or had early death without therapy.c Died of causes other than PTLD progression.d All three have slowly remitting disease.
DROR ET AL.April 15, 1999 993

abdomen, with an additional boost of 2000 cGy/20 fraction.Both patients have remained in CR at 34 and 60.5 months,respectively.
Remission duration and survival. Of the 20 treated pa-tients, 13 (65%) achieved CR (Table 3). Nine of the 13 arealive and in continuous CR at 0.7–6.8 years (median, 16.5months). The other four patients died in CR of unrelatedcauses (two liver rejections, one cerebral hemorrhage, andone seizures).
Three patients achieved partial remission; they are alivewith slowly remitting disease 9, 11, and 40 months, respec-tively, after presentation. One patient died 5 days after com-mencement of therapy. Three patients died with progressivedisease, 0.8–1 month after starting therapy. Overall, 60% (12of 20) of the treated patients are alive. Of the 12, 10 still havea functioning transplant (5 livers, 4 hearts, 1 kidney),whereas 2 required nephrectomy.
Patient characteristics and outcome. Sites of involvementwith disease are listed in Table 4. Two patients had bonemarrow involvement. One was diagnosed at autopsy. Theother was initially diagnosed with immune thrombocytopenicpurpura and was treated with IVIG and high-dose steroids.He died with progressive disease 24 days after diagnosis,despite treatment with ISDR, acyclovir, IVIG, and IFN.
Two patients had CNS involvement in addition to wide-spread systemic disease. One was diagnosed at autopsy. Theother had neurologic signs, positive cerebrospinal fluid testresults, and multiple nodular lesions revealed by computedtomography. Brain biopsy revealed monomorphic PTLD witha rearranged T-cell receptor gene. When initial treatmentwith nephrectomy, ISDR, acyclovir, and CMVIG failed, dexa-methasone and cranial radiation therapy were administered,followed by one intrathecal injection of methotrexate. Thispatient achieved CR and is a long-term survivor (.5 years).
Three patients had isolated gastrointestinal disease. Onepatient with gastric lymphoma was treated with ISDR, acy-clovir, and CMVIG; this individual achieved CR, but diedlater during a repeat transplantation attempt after trans-plant rejection. The other 2 patients presented with perfora-
tion of the bowel and were treated with ISDR, surgery, acy-clovir, and IVIG (one patient also received IFN); thesepatients survived. Another four patients had gastrointestinaltract disease as part of a generalized process; only one hashad CR and survived.
Four patients had parenchymal lung disease as part ofwidespread systemic disease, of whom only one is still alive.He achieved partial remission and has been treated for morethan 40 months.
Six patients had isolated allograft disease. The two pa-tients with isolated transplanted-kidney disease achieved CRand survived, whereas all four patients with isolated trans-planted-liver PTLD died. Nephrectomy in children with kid-ney transplant who developed PTLD was performed in fivecases, mainly to enable withdrawal of immunosuppressivetherapy.
Prognostic factors (Table 5). Platelet counts less than803109/L and neutrophil counts under 1.03109/L correlatedsignificantly with lack of response. Age, sex, type of trans-planted organ, time to PTLD, hemoglobin level, lymphocytecount (data not shown), clonality, frequency of mitoses, de-gree of necrosis, and degree of atypia were not prognosticallysignificant. Children with Murphy stage III or IV diseaseshowed a worse prognosis (P50.05). All patients with poly-morphic disease or hyperplasia by SHPW criteria achievedCR, compared with 53% of the patients with monomorphicdisease; although the difference did not reach statistical sig-nificance (P50.114).
DISCUSSION
Prevalence. The incidence of PTLD in adults is 1.4–2.5%after renal transplant (3, 13, 16), 2.1–2.8% after liver trans-plant (14, 16), 1.8–6.3% after heart transplant (3, 13, 14, 16),4.5–10% after lung transplant (13, 14), and 33% after heartand lung transplantation (16). This incidence is graduallydecreasing (15). The incidence of PTLD in children is con-stant and higher than in adults (15); however, the exactincidence among the various transplant types in children hasnot been well defined. Cox and associates (17) reported a2.9% prevalence of PTLD after liver transplant in childrenunder 5 years of age who received CsA therapy and 19% inthose who received tacrolimus. In our series, the prevalenceof PTLD was 1.2% among kidney recipients, 10% among liverrecipients, and 15% among heart recipients.
The reason for higher incidence of PTLD in children isprobably related to the higher incidence of primary EBVinfection observed in children after transplant (13, 15, 18),likely because of their lower incidence of pretransplant EBVseropositivity as compared with adults (15). In our study,EBV seropositivity at PTLD diagnosis was significantly morecommon than before transplant; the incidence of seroconver-sion among PTLD patients was also high. However, determi-nation of EBV status by serology alone is not reliable, be-cause of secondary immunodeficiency and transfusions ofblood products. Therefore, in our study, EBV sequences weresought in the tumor, and were detected by PCR in over 90%of the lesions tested. As shown in our series, the associationwith EBV is not confined to B-PTLD, and can occur also inT-PTLD (13).
Pathology. A variety of histopathologic classificationshave been used to describe PTLD lesions (19). Because theInternational Working Formulation refers to malignant lym-
TABLE 4. Sites of disease involvement
Organ na
Bone 1Bone marrow 2Bowel, large 3Bowel, small 6Central nervous system 2Diaphragm 3Kidney 8Liver 10Lung 4Lymph node 19Ovary 1Paraspinal area 1Salivary gland 1Skin 3Spleen 4Stomach 1Testes 1Tonsil 8Uterus 1
a Most patients had involvement of more than one site.
TRANSPLANTATION994 Vol. 67, No. 7

phomas and does not include categories for hyperplastic andpolymorphic lesions, a substantial number of cases withPTLD cannot be classified according to this system. At therecent SHPW, several distinctive categories of PTLD wereagreed upon (Table 6), on the bases of 20 submitted cases andthe participants’ combined experience (5).
In our series, tissue samples from all cases were reviewedand reclassified according to SHPW categories. Reclassifica-tion of the cases, however, posed some difficulties. First,because of discrepancy between immunophenotyping andmolecular studies, division into B- and T-PTLD was some-times difficult. Second, tumor cells of one of the five polymor-phic cases were stained exclusively for B-cell markers, andhad immunoglobulin but not T-cell receptor gene rearrange-ment, thus calling into question the assumption that poly-morphic lesions cannot be subclassified as B or T. Third, twocases with immunoblastic PTLD were exclusively of T-cellimmunophenotype. This is in contrast to the workshop clas-sification, which groups only B-cell lesions under the immu-noblastic category, but is in keeping with other reports (18),20). Fourth, cases of polyclonal lymphoma have been re-ported previously (18), 21). In the present study, two patientswith monomorphic-immunoblastic PTLD had neither immu-noglobulin nor T-cell receptor gene rearrangement. Thus, theSHPW assumption that all monomorphic lesions are mono-clonal may be inaccurate. Fifth, as has already been de-scribed (22), one patient in the current study was diagnosed
with reactive, yet monoclonal, hyperplasia. These reserva-tions have caused us to suggest modifications to the SHPWclassification (Table 6), including segregation of descriptivemorphology from clonality and cell of origin.
In some studies, specific histopathology findings have cor-related with prognosis (13 14, 23, 24) but not in others (18).Although not statistically significant, there was a trend to-ward better survival for patients with polymorphic PTLD/hyperplasia (of whom all survived) than for patients withmonomorphic-immunoblastic type (of whom just over halfsurvived). Specific morphological characteristics such as de-gree of atypia and necrosis and mitosis rate did not correlatewith outcome.
Clonality has been found to correlate with poor prognosisin some studies (18 25, 26) but not in others (16, 27). Con-temporary exclusion of monoclonality is complex, necessitat-ing the evaluation of both g and k chains, immunoglobulinand T-cell receptor genes, karyotype, and EBV sequencing—without all of which monoclonality can not be entirely ex-cluded. In the present study, monoclonality based upon ge-notypic and light-chain predominance did not imply poorprognosis.
Most cases of PTLD have a B-cell origin, with only 11.7–12.5% reported to originate from T cells (13, 18). Our resultsshow that distinguishing B- from T-PTLD can be difficult andmay not be prognostically important, as both of our patientswith T-cell receptor gene rearrangement are in continuous
TABLE 5. Prognostic significance (among 20 patients treated) of various factors present at diagnosis of PTLDa
Factor Division CR % No CR % P-value
Age ,9 years 7 54 6 46 0.329$9 years 6 86 1 14
Sex Males 8 67 4 33 1.00Females 5 62 3 38
Transplant type Liver 5 56 4 44 0.615Renal 5 71 2 29Heart 4 80 1 20
Time to diagnosis ,6 months 6 67 3 33 1.00$6 months 7 64 4 36,1 year 9 69 4 31 0.651$1 year 4 57 3 43
Hemoglobin ,90 g/L 6 67 3 33 1.00$90 g/L 7 64 4 36
Neutrophil count ,109/L 13 93 1 7 ,0.001$109/L 0 6 100
Platelet count ,803109/L 0 3 100 0.03$803109/L 13 76 4 24
Murphy system Stage I–II 6 100 0 0.051Stage III–IV 7 50 7 50
IWF category Immunoblastic 9 56 7 44 0.249Unclassifiable 4 100 0
SHPW category Monomorphic 8 53 7 47 0.114Poly/hyper 5 100 0 0
Clonality Monoclonal 8 67 4 33 0.740PC or UD 4 50 4 50
Mitosis 0–2/HPF 9 75 3 25 0.356.2/HPF 4 50 4 50
Necrosis Focal 6 60 4 40 1.000Diffuse 7 70 3 30
Atypia 0 or 1 6 75 2 25 0.64211 or 111 7 58 5 42
a IWF, International Working Formulation; Poly/hyper, polymorphic or hyperplastic; PC, polyclonal; UD, undetermined; HPF, high-powerfield (3400).
DROR ET AL.April 15, 1999 995

CR, 14 and 60.5 months after PTLD diagnosis, and one of ourfour patients with T-cell immunophenotyping is in continu-ous CR 26 months after diagnosis. This is in contrast tostudies in which all patients with T-cell PTLD died (13, 18).
Clinical characteristics. Site involvement by PTLD maybe clinically important. Involvement of the CNS has beenreported (13, 18) to be an adverse prognostic factor. CNSinvolvement in children, which usually presents as a solitaryintracranial lesion, is estimated (18) to be very low (1%)compared to adults (26.7%); however, it implies a very graveprognosis. Two patients in the current series had CNS in-volvement in addition to widespread systemic disease; one ofthem currently survives disease-free at .5 years. Bone mar-row involvement has also been reported (13, 18) to be anadverse prognostic factor. This was confirmed in our study, inwhich both patients with marrow involvement died. In thecurrent study low platelet and neutrophil counts correlatedwith poor outcome. This might in part be related to bonemarrow involvement by PTLD cells. Alternatively, it mightbe related to higher viral load and suppression of hematopoi-esis.
Small intestine involvement has been reported to occur in47% of adult PTLD patients, and large bowel and stomach,each in 6% (16). Thirty-six percent of children have beendescribed as having gastrointestinal involvement (25). Pa-tients with gastrointestinal disease that is either localizedand/or resectable seem to have a better prognosis, with long-term survival in 80% (16). All three patients in our studywith isolated gastrointestinal disease achieved CR, in con-trast to only one continuous CR among four other patients
with gastrointestinal tract disease as part of a generalizedprocess.
Pulmonary disease, characterized by multiple nodular le-sions or localized infiltrative process, is described in 15–38%of adult PTLD patients, with the highest incidences amongpatients who had heart, heart-lung, or bone marrow trans-plantation (13, 16, 18, 27). These patients seem to havedisseminated disease with poor outcome. Of four patients inour study who had parenchymal lung disease as part ofwidespread systemic disease, only one is still alive, withslowly remitting disease 40 months after diagnosis.
In patients with isolated allograft involvement, outcomemay depend upon the specific type of graft. In the presentstudy, both patients with isolated disease in the transplantedkidney achieved CR and survived with medical treatment;one of them had nephrectomy as well. In contrast, none of thefour patients with isolated liver disease achieved CR, andthree of them died.
Treatment. PTLD represents the outcome of functionalover-immunosuppression, which makes ISDR necessary (16,27, 28). If ISDR reverses the lymphomatous proliferation, anincreased risk of rejection would be acceptable. In our study,all 20 of the treated patients had their immunosuppressivedrugs reduced or discontinued. Ten patients experienced 21episodes of graft rejection that required treatment, but only 3required a second transplant.
All 20 treated patients in our series received either acyclo-vir or ganciclovir. As studies reporting the use of acyclovir organciclovir in PTLD are mostly case reports involving smallnumbers of patients (29–32), combined with other therapies
TABLE 6. Categories of PTLD recognized by the 1997 SHPW, and suggested modifications
SHPW categories Recommended modificationsa
Early lesions HyperplasiaReactive plasmacytic hyperplasia Reactive plasmacytic hyperplasiaInfectious mononucleosis-like Infectious mononucleosis-like
Polymorphic PTLD Polymorphic PTLDPolyclonalMonoclonal
Monomorphic PTLD Monomorphic PTLDB-cell lymphoma Diffuse, large-cell immunoblastic
1. Diffuse large-cell lymphoma (immunoblastic, centroblastic, anaplastic) Diffuse, large-cell anaplastic2. Burkitt’s/Burkitt’s-like lymphoma Diffuse, large-cell centroblastic
Small non-cleaved cell lymphomaPeripheral T-cell lymphoma
T-cell lymphoma1. Peripheral T-cell lymphoma, unspecified type (usually large-cell)2. Anaplastic large-cell lymphoma (T or null)3. Other types (e.g., T-NK)b
Other OtherT-cell-rich/Hodgkin’s disease-like large B-cell lymphoma Hodgkin’s-like lesion
Plasmacytoma-likeMyeloma
Plasmacytoma-likeMyeloma
Lesions can be further subclassified as:P, polyclonalM, monoclonalB, B-cell typeT, T-cell typeN, nullOther cell origins (e.g., NK) can be specified
a Lesions can be classified, for example, as diffuse large-cell immunoblastic PTLD-B,M for specification of B-cell type and monoclonality.b NK, natural killer cells.
TRANSPLANTATION996 Vol. 67, No. 7

as well, effectiveness is hard to evaluate. The same difficul-ties apply to IVIG and CMVIG (4, 31). In the current series,most patients who received IVIG or CMVIG achieved CR.Because the CMVIG product, Cytogam, contains higher lev-els of antibodies against EBV (30), we recommend its use inpreference to the standard IVIG products. Of the 20 treatedpatients in our series, 10 were treated with IFN; 6 of themachieved CR. This response rate is equivalent to others re-ported in the literature (4, 18, 32, 33).
When possible, surgical resection of the tumor appeared tobe important (18). The tumors of seven patients in our studywere resected. Patients were cured when complete resectionwas achieved. When only debulking of tumor mass was done,no patients achieved CR, which emphasizes the importanceof complete resection or resectability of the tumor. Debulkingsurgery for airway obstruction by large tonsils may be re-quired, as documented in one patient in the present study.
Involved field radiation therapy has been tried in patientswith intestinal or CNS PTLD with limited success (13, 18)).However, because controlled studies are lacking, its role inunresponsive localized disease and CNS disease is still un-clear. In the present series, two patients received radiationtherapy, one after complete gross resection; both went intoCR and survived.
Several studies (13, 26, 37, 38) have indicated that anti-Bcell monoclonal antibodies directed against the CD21 andCD24 antigens might be effective in controlling B-cell PTLD.Monoclonal antibodies were not used in our series, withoutcompromising outcome; Benkerrou and associates (26) re-ported continuous CR in 65% of their subjects utilizing thismodality, compared to 60% in our series. The role of mono-clonal antibodies for progressive disease, unresponsive toother therapies, remains to be determined.
Aggressive chemotherapy has been used in the past for themanagement of PTLD (13, 27, 34) with conflicting results.Swinnen and colleagues (27) treated 11 adult patients (in-cluding 2 with monoclonal PTLD) with aggressive chemo-therapy; 4 achieved CR and survived. Leblond et al. (13)treated eight adults with PTLD with various chemotherapyregimens; only two achieved CR and survived at 4 and 12months, respectively. Gallego-Melcon and co-workers (34)treated three children with Burkitt’s-like lymphoma (twomonoclonal and one polyclonal) with the LMB89 protocol; allachieved CR and survived at 22, 35, and 36 months. Becauseof generally poor survival reported with systemic chemother-apy, as well as the high incidence of death secondary to sepsis(35, 36), no patients in the current series received systemicchemotherapy without apparent adverse effect on outcome. Itis possible, however, that the use of chemotherapy either forrejection occurring in response to ISDR or for progressivedisease may prevent the loss of the allograft and improvesurvival over the 60% seen in this series.
CONCLUSIONS
From this, the largest pediatric series published, we con-clude that the majority of patients, including those withmonomorphic or monoclonal lesions, who are diagnosed earlyenough to be eligible for treatment can be cured with ISDR,acyclovir/ganciclovir, CMVIG, and IFN, with the addition ofsurgery when total resection is possible and irradiation whenindicated. Systemic chemotherapy or anti-B-cell monoclonalantibodies should be reserved for unresponsive cases. Low
platelet count and low neutrophil count have been identifiedas adverse prognostic factors. A trend toward better outcomeof patients with hyperplastic or polymorphic lesions bySHPW and of patients with Murphy stage I–II was noted. Asthe first published series independent of SHPW to be classi-fied by the new SHPW categories, we found that groupingaccording to morphology was feasible in all cases and seemsto have clinical relevance. However, correlation with clonal-ity and cell origin did not fit all cases, necessitating modifi-cations including segregation of descriptive morphology fromclonality and cell origin.
Acknowledgments. The authors thank Lori Acal, Enza DeLuca,and Camilla Cook, liver transplantation coordinator nurses; JaneGildner, heart transplant coordinator nurse; and Rita Pool andMoira Korus, kidney transplantation coordinator nurses, for theirhelp in gathering the clinical data for this study. This paper wasprepared with the assistance of Editorial Services, The Hospital forSick Children, Toronto, Ontario, Canada.
REFERENCES
1. Penn I. The price of immunotherapy. Curr Probl Surg 1981; 18:681.
2. Hoover R, Fraumeni JF. Risk of cancer in renal transplantrecipients. Lancet 1973; 2: 55.
3. Opelz G, Henderson R, for the Collaborative Transplant Study:incidence of non-Hodgkin lymphoma in kidney and hearttransplant recipients. Lancet 1993; 342: 1514.
4. Magrath IT, Rowe M, Filipovich AH, et al. Advances in under-standing of EBV associated lymphoproliferative disorders. InAblashi DV, ed. EBV and human disease. Clifton, NJ: HumanPress, 1991; 243.
5. Harris NL, Ferry JA, Swerdlow SH. Post transplant lymphopro-liferative disorders: summary of Society for HematopathologyWorkshop. Semin Diagn Pathol 1997; 14(1): 8.
6. Morgan G, Superina R. Lymphoproliferative disorders post livertransplant. J Pediatr Surg 1994; 29 (9): 1192.
7. Cockfield SM, Preiksaitis J, Harvey E, et al. Is sequential use ofALG and OKT3 in renal transplants associated with an in-creased incidence of fulminant posttransplant lymphoprolif-erative disorder? Transplant Proc 1991; 23: 1106.
8. Houle AM, McLorie GA, Churchill BM, Khoury AE, Harvey E,Hebert D. Rapid development of an immunoblastic lymphomaand death in children following cadaver renal transplantation.J Pediatr Surg 1992; 27: 626.
9. Anonymous. National Cancer Institute sponsored study of clas-sification of non-Hodgkin lymphoma: summary and descrip-tion of a working formulation for clinical usage; The Non-Hodgkin Lymphoma Pathologic Classification Project. Cancer1982; 49: 2112.
10. Sixby JW, Shirly PS. Viral diagnosis using DNA-based probes:enzymatic amplification of target DNA. NY Acad Sci 1988; 158.
11. Murphy SB. Classification, staging and end results of treatmentof childhood non-Hodgkin lymphoma: dissimilarities with lym-phomas in adults. Semin Oncol 1980; 7: 332.
12. Miller AB, Hoogstraten B, Staquer M, et al. Reporting results ofcancer treatment. Cancer 1981; 47: 207.
13. Leblond V, Sutton L, Dorent R, et al. Lymphoproliferative dis-orders after organ transplantation: a report of 24 cases ob-served in a single institution. J Clin Oncol 1995; 13 (4): 961.
14. Walker RC, Marshall WF, Stickler JG, et al. Pretransplant as-sessment of the risk of lymphoproliferative disorders. ClinInfect Dis 1995; 20: 1346.
15. Ho M, Jaffe R, Miller G, et al. The frequency of Epstein-Barrvirus infection and associated lymphoproliferative syndromeafter transplantations in children. Transplantation 1988; 45
DROR ET AL.April 15, 1999 997

(4): 719.16. Starzl TE, Nalesnik MA, Porter KA, et al. Reversibility of lym-
phomas and lymphoproliferative lesions developed after cy-closporine-steroid therapy. Lancet 1984; 1: 583.
17. Cox KL, Lawrence-Miyasaki LS, Garcia-Kennedy R, et al. Anincrease incidence of Epstein-Barr virus infection and lympho-proliferative disorder in young children on FK506 after livertransplantation. Transplantation 1995; 59 (4): 524.
18. Morrison VA, Dunn DL, Manivel JC, et al. Clinical characteristicof post transplant lymphoproliferative disorders. Am J Med1994; 97: 14.
19. Swerdlow SH. Classification of the posttransplant lymphoprolif-erative disorders: from the past to the present. Semm DiagnPathol 1997; 14 (1): 2.
20. Wu TT, Swerdlow SH, Locker J, et al. Recurrent Epstein-Barrvirus–associated lesions in organ transplant recipients. HumPathol 1996; 27 (2): 157.
21. Shapiro RS, McClain K, Frizzera G, et al. Epstein-Barr virusassociated B cell lymphoproliferative disorders following bonemarrow transplantation. Blood 1988; 72 (5): 1234.
22. Nelson BN, Naleznik MA, Locker JD, Swerdlow SH. Posttrans-plant lymphoproliferative disorders in the adult: the Pitts-burgh experience [Abstract]. Mod Pathol 1997; 10 (1): 131A.
23. Chadburn A, Cesarman E, Knowles DM. Classification of post-transplantation lymphoproliferative disorders based on mor-phologic and molecular genetic features has clinical relevance[Abstract]. Mod Pathol 1997; 10 (1): 121A.
24. Collins MH, Mortone KT, Hodinka RL, et al. Posttransplantlymphoproliferative disorder in children: clinicopathologic cor-relation of fourteen cases [Abstract]. Mod Pathol 1997; 10 (1):156A.
25. Fisher A, Blanche S, Le Bidois J, et al. Anti-B cell monoclonalantibodies in the treatment of severe B-cell lymphoprolifera-tive syndrome following bone marrow and organ transplanta-tion. N Engl J Med 1991; 324: 1451.
26. Benkerrou M, Durrandy A, Fischer A. Therapy for transplant-related lymphoproliferative disease. Hematol Oncol Clin NorthAm 1993; 7 (2): 467.
27. Swinnen LJ, Mullen GM, Carr TJ, et al. Aggressive treatmentfor postcardiac transplant lymphoproliferation. Blood 1995; 86(9): 3333.
28. Hanto DW, Fizzera KJ, Gajl-Peczalska, et al. Acyclovir therapyof Epstein-Barr virus–induced posttransplant lymphoprolif-erative diseases. Transplant Proc 1985; 17 (1): 89.
29. Pirsch JD, Stratta RJ, Sollinger HW, et al. Treatment of Ep-stein-Barr virus–induced lymphoproliferative syndrome withganciclovir: two cases after solid organ transplantation. Am JMed 1989; 86: 241.
30. Delone P, Corkill J, Jordan M, et al. Successful treatment ofEpstein-Barr virus infection with ganciclovir and cytomegalo-virus hyperimmune globulin following kidney transplantation.Transplant Proc 1995; 27 (5) (Suppl 1): 58.
31. Oettle H, Wilborn F, Schmidt CA, et al. Treatment with ganci-clovir and Ig for acute Epstein-Barr virus infection after allo-geneic bone marrow transplantation. Blood 1993; 82 (7): 2257.
32. Dellemijn PLI, Brandenburg A, Niesters HGM, et al. Successfultreatment with ganciclovir of presumed Epstein-Barr menin-goencephalitis following bone marrow transplant. Bone Mar-row Transplant 1995; 16: 311.
33. Shapiro RS, Chauvenet A, McGuire W, et al. Treatment of B-celllymphoproliferative disorders with interferon alpha and intra-venous gamma globulin. N Engl J Med 1988; 318: 1334.
34. Gallen-Melcon S, De Toledo JS, Martinez V, et al. Non-Hodgkinlymphoma after liver transplantation: response to chemother-apy. Med Pediatr Oncol 1996; 27: 156.
35. Nalesnick MA, Makowka L, Starzl TE. The diagnosis and treat-ment of post-transplant lymphoproliferative disorders. CurrProbl Surg 1988; 25: 365.
36. Cohen J Epstein-Barr virus lymphoproliferative disease. Medi-cine 1991; 70: 137.
37. Blanche S, Le Deist F, Veber F, et al. Treatment of severeEpstein-Barr virus–induced polyclonal B-lymphocyte prolifer-ation by anti-B-cell monoclonal antibodies. Ann Intern Med1988; 108: 199.
38. Lazarovits AI, Tibbles LA, Grant DR, et al. Anti-B cell antibodiesfor the treatment of monoclonal Epstein-Barr virus–inducedlymphoproliferative syndrome after multivisceral transplanta-tion. Clin Invest Med 1994; 17 (6): 621.
Received 14 April 1998.Accepted 10 September 1998.
TRANSPLANTATION998 Vol. 67, No. 7
Copyright © 2022 FDOKUMEN




















