
Reactor pressure: growth temperature relation for InN epilayers grown by high-pressure CVD

www.elsevier.com/locate/apcata
Applied Catalysis A: General 292 (2005) 229–243
Low temperature water-gas shift: Examining the efficiency
of Au as a promoter for ceria-based catalysts prepared
by CVD of a Au precursor
Gary Jacobs a, Sandrine Ricote a, Patricia M. Patterson a, Uschi M. Graham a,Alan Dozier b, Syed Khalid c, Elin Rhodus d, Burtron H. Davis a,*
a Center for Applied Energy Research, 2540 Research Park Drive, Lexington, KY 40511, USAb University of Kentucky Electron Microscopy Center, Chemical and Materials Engineering Department,
A004 ASTeCC Building 0286, Lexington, KY 40506, USAc National Synchrotron Light Source, Brookhaven National Laboratory, Upton, NY 11973, USA
d EBCE Program, Lafayette High School, 400 Reed Lane, Lexington, KY 40503, USA
Received 11 April 2005; received in revised form 3 June 2005; accepted 12 June 2005
Abstract
Increasing the Au loading had a significant positive impact on the catalytic activity. The partial reduction of ceria is necessary for
generating bridging OH groups on the surface of ceria, which serve as the active sites. The surface shell reduction process in H2 was monitored
by TPR, XANES, and in situ DRIFTS spectroscopy. Either the oxygen deficiencies are first formed, followed by dissociative adsorption of
H2O to generate the Type II bridging OH groups or, they may be formed directly by spillover of dissociated H2 from the metal to the ceria
surface. For each pair of bridging OH groups formed, two cerium atoms in the surface shell change from the Ce4+ to Ce3+ oxidation state.
Addition of Au facilitates the surface reduction process and thereby decreases the reduction temperature from 450 8C for the unpromoted
catalyst to 100 8C for the 5% Au/ceria catalyst. A systematic decrease in the required temperature for ceria surface shell reduction was
observed by increasing the Au promoter loading as follows: 0.1, 0.25, 0.5, 1, 2.5, 5%.
Gold and platinum promoted catalysts were compared in a suitable reaction temperature range after first ensuring that metal–oxide
interactions were overcome (ca. 5% metal loading). Approximately 20 times the amount of 5% Au/ceria catalyst was required to achieve a
lightoff curve similar to that of a 5% Pt/ceria in the temperature range 200–300 8C, and 5% Pt/ceria also exhibited higher steady-state activity
(about double that of Au) at 175 8C. This result suggests that, in addition to the role the metal plays in facilitating the formation of the active
site bridging OH groups, it also influences the intrinsic rate of the WGS reaction. Transient formate decomposition experiments carried out at
140 8C indicated that the rate of formate decomposition was approximately 20 times higher for 2.5% Pt/ceria than that of 2.5% Au/ceria,
suggesting that the metal (in addition to the promoting effect of H2O previously reported) plays a role in facilitating the decomposition of
surface formate intermediates, the proposed rate limiting step of the reaction mechanism.
# 2005 Published by Elsevier B.V.
Keywords: Au loading; Au/ceria catalyst; Pt/ceria catalyst
1. Introduction
The implementation of PEM fuel cells will require very
active and robust fuel processing catalysts for such reactions
as hydrocarbon steam reforming, low temperature water-gas
shift, and preferential oxidation [1–3]. The latter two reactions
* Corresponding author. Tel.: +1 606 257 0253; fax: +1 606 257 0302.
E-mail address: [email protected] (B.H. Davis).
0926-860X/$ – see front matter # 2005 Published by Elsevier B.V.
doi:10.1016/j.apcata.2005.06.003
are receiving widespread interest from catalyst developers for
the conversion of CO, which acts as a poison for fuel cell
electrode catalysts [4]. This study centers on comparing Au
relative to Pt, as a promoter of ceria-based low temperature
water-gas shift catalysts, as it has been claimed to be an
excellent water-gas shift catalyst [5–11].
There is much controversy in the literature regarding how
Au and Pt promoted ceria catalysts operate for water-gas
shift, and this probably stems in part from: (1) differences in

G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243230
Scheme 1. Surface formate mechanism.
Scheme 2. Ceria-mediated redox process.
feed conditions, as some researchers continue to utilize a
high temperature shift feed for low temperature shift studies;
(2) the absence of H2O, H2, or CO2 in many mechanistic
evaluations; and (3) lack of verification to determine
whether or not the surface of a particular catalyst is in an
active state.
Regarding the first point, the feed utilized in this study
is considered to approximate the exit stream of a high
temperature shift section of a fuel processor, with the caveat
that we do not include CO2 in this particular study. The reason
is that we are attempting to construct a mechanistic picture
for these catalysts on an incremental basis. The feed that we
have chosen includes: 3.75 cm3/min CO, 125 cm3/min H2O
(steam), 100 cm3/min H2, and a small amount of N2 for the
purpose of internal calibration. The feed matches that of our
previous study of the impact of Pt loading on low temperature
water-gas shift activity [12].
Considering the second point, H2O has been found in
many studies to promote the rate of the water-gas shift
reaction not only for metal promoted ceria systems [13–15],
but also for other common active materials, including ZnO
[16], MgO [17,18], ThO2 [19], and ZrO2 [18] containing
catalysts. Unfortunately, many mechanistic studies based on
IR spectroscopy have neglected the promoting effect of H2O
entirely. This is a critical point, especially considering that
the feed ratio for low temperature water-gas shift has a very
high H2O/CO ratio, and this is considered in Section 4. The
impact of CO2 to the rate also should be considered, since
excellent water-gas shift catalysts when excluding CO2 in
the feed can be inhibited when CO2 is present [20], leading
to potentially unfair comparisons among catalysts. Finally,
as low-temperature shift is essentially a hydrogen purifica-
tion step, adequate amounts of hydrogen were included in
the feed.
The final point also merits a brief discussion. It is
important for researchers comparing catalysts to ensure
that the catalyst is tested in a range of temperature that
ensures a suitably active surface is present. Most
researchers agree that defects (i.e., oxygen deficiencies)
in the surface shell of ceria play an important role in the
water-gas shift mechanism. Yet, it is necessary to use
techniques, like TPR or XANES, to make certain that the
defect-surface is generated. For example, in examining our
previous Pt loading study [12], 1% Pt/ceria is activated by
250 8C, while 5% Pt/ceria is activated at much lower
temperature (i.e., ca. 50 8C lower temperature). Therefore,
to benchmark Au, which typically is activated at a lower
temperature than Pt on an equivalent loading basis [21], to
Pt in a range where the Pt catalyst is not yet in an activated
state, should be avoided.
There are numerous inconsistencies in the literature
regarding the WGS mechanism over metal promoted ceria
catalysts, as well as the nature of the active sites involved.
Our results continue to support the initial surface formate
mechanism of Shido and Iwasawa [13] for Rh/ceria, which is
not unlike a mechanism considered by Tamaru [22] for ZnO-
based catalysts, and stems from the now classic studies of
formic acid decomposition [23]. In this mechanism [13],
depicted in Scheme 1, Type II bridging OH groups, defined
in [24,25], are the active sites and react with CO to generate
surface formate intermediates. The decomposition of
surface formates to unidentate carbonate is proposed to
be promoted by steam (leading to the inclusion of H2O in the
transition state complex by Shido and Iwasawa [13]) and is
considered to be the rate limiting step of the mechanism. On
the other hand, a support-mediated redox process has also
been proposed for precious metal loaded ceria catalysts (e.g.,
for Pd) [26,27], shown in Scheme 2, whereby adsorbed CO
on the metal promoter reacts with ceria to generate the
cerous oxide form and CO2, with H2O replenishing the
oxygen vacancy with liberation of H2. One basis for this
mechanism was the fact that the group [26] did not observe
surface formate species in their FTIR studies with Pd/ceria.
This mechanism, initially found support by another group to
describe the catalysis of Au/ceria [7], but their viewpoint has
recently changed to favor a mechanism relying on non-
metallic Au and Pt species in close association with ceria as
the active sites for the WGS reaction [5,6,8]. A basis for this
modification was that after a large fraction of metal promoter
was leached from the catalyst, the catalyst still retained its
activity. In this new viewpoint, one can infer that the metal is
proposed not to play an important role in the catalytic cycle.
Others support the view that the active sites are solely
associated with Au, in line with Haruta’s initial findings that
Au particles smaller than ca. 5 nm possessed intrinsic
activity for the CO oxidation reaction [28,29]. Recent work
by Haruta and co-workers, does, however, support a WGS
mechanism operating via formate intermediates [30].

G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243 231
In this contribution, the aim is to explore the efficiency of
Au as a promoter for low temperature water-gas shift relative
to its neighbor, Pt, and to make comparisons under
conditions where the defect concentrations in ceria are
similar, in order to shed light as to whether or not the metal is
involved in the actual catalytic mechanism. As such, we rely
on techniques such as TPR and XANES to make
determinations regarding the required temperature for,
and extent of, activation as well as reaction testing and in
situ DRIFTS spectroscopy to shed light onto the effective-
ness of each promoter. XANES is fast becoming a preferred
method for quantifying oxidation state changes for ceria
catalysts [31]. To ensure a small Au crystallite size, we
utilize a CVD technique previously employed for the
preparation of effective Pt dehydrocyclization catalysts [32].
Benefits of the technique are that small, uniform, well
dispersed, nano-crystallites are formed at typical low
promoter loadings, and that the precursor is virtually
completely loaded onto the substrate. It is very important to
note that this procedure is different from the deposition-
precipitation techniques used by other researchers [5–11].
2. Experimental
2.1. Catalyst preparation
High surface area ceria was prepared via homogeneous
precipitation of the nitrate in urea with aqueous ammonia in
a procedure similar to Li et al. [33], whereby urea
decomposition is a slow process resulting in a more
homogeneous precipitation. Appropriate amounts of Ce(N-
O3)3�6H2O (Alfa Aesar, 99.5%) and urea (Alfa Aesar,
99.5%) were dissolved in 900 cm3 of deionized water, and to
this solution about 30 cm3 NH4OH (Alfa Aesar, 28–30%
NH3) was added drop wise (�1 cm3/min). The mixture was
then heated at 100 8C with constant stirring for 8 h. The
precipitate was filtered, washed with 600 cm3 of boiling
deionized water, and dried in an oven at 110 8C overnight.
The dried precipitate was then crushed and calcined in a
muffle furnace at 400 8C for 4 h.
Dimethyl(acetylacetonate gold III) was purchased from
Strem Chemicals (stock no. 79-1500). The precursor will
hereafter be referred to as AuAcAc, and was successfully
utilized previously in a vapor phase grafting procedure to
prepare nano-crystalline Au clusters [34]. The cerium oxide
(BET SA, 120 m2/g) was heated under high vacuum
(1 � 10�7 Torr) at 350 8C to drive water out of the pores
and subsequently cooled to room temperature. The sample
tube was backfilled with nitrogen and transferred to an inert
atmosphere, where the ceria was well mixed with the gold
precursor. A sample of the physical mixture was retrieved
for FTIR analysis. The physical mixture was loaded into a
sample tube and attached to the high vacuum line. The
sample was heated in stages (1 8C/min) to 80 8C, and finally,
100 8C with 15 min ramps between each temperature and
holding for 1 h at each temperature. Finally, the catalyst was
ramped to 130 8C, held for 15 min, and the catalyst was then
quenched to room temperature. The ceria is yellow in color,
but the sample retrieved after the sublimation procedure (for
the 1% Au catalyst) had a slightly orange, highly uniform
color. This sample was utilized for FTIR study of the
precursor decomposition, as well as the catalyst activation
step. Based on these experiments, a calcination temperature
of 250 8C in O2 was selected, while a temperature of 175 8Cin H2 was chosen as the initial temperature for catalyst
activation. The calcined catalysts ranged in color from a very
light, grayish-purple to dark black.
2.2. BET surface area
BET surface area measurements were carried out using a
Micromeritics Tristar 3000 gas adsorption analyzer. In each
trial, a weight of approximately 0.25 g of sample was used.
Nitrogen adsorption was carried out at its boiling temperature.
2.3. Temperature programmed reduction (TPR)
TPR was conducted on unpromoted and gold promoted
ceria catalysts in a Zeton-Altamira AMI-200 unit, which was
equipped with a thermal conductivity detector (TCD).
Argon was used as the reference gas, and 10% H2 (balance
Ar) was flowed at 30 cm3/min as the temperature was
increased from 50 to 1100 8C at a ramp rate of 10 8C/min.
2.4. X-ray absorption near edge spectroscopy (XANES)
XANES spectra at the Au LIII (11,919 eV) and Ce LIII
(5723 eV) edges were recorded at the National Synchrotron
Light Source (NSLS) at Brookhaven National Laboratory
(BNL) in Upton, New York at Beamline X-18b. Experi-
mental details for the beamline and the sample preparation
are provided in our previous investigation of Pt/ceria
catalysts [12]. After purging the cell for a long duration of
time with a high flow rate of helium to ensure removal of air,
the samples were treated in situ at the beamline with a
hydrogen/helium mixture (60 cm3/min H2 and 300 cm3/min
He) while heating at 10 8C/min in the temperature range 50–
300 8C. Scans were obtained in the transmission mode at
50 8C intervals to explore the partial reduction of ceria and
the reduction of the gold promoter. The UHP H2 and He
gases were mixed in a manifold and passed through an
oxygen/water trap prior to feeding to the cell. Raw data were
processed to give the normalized XANES spectra. Linear
combination XANES fits of treated catalysts with reference
standards were carried out using the WinXAS program [35].
2.5. High resolution transmission electron microscopy
(HRTEM)
High resolution transmission electron microscopy
(HRTEM) measurements were carried out using a JEOL
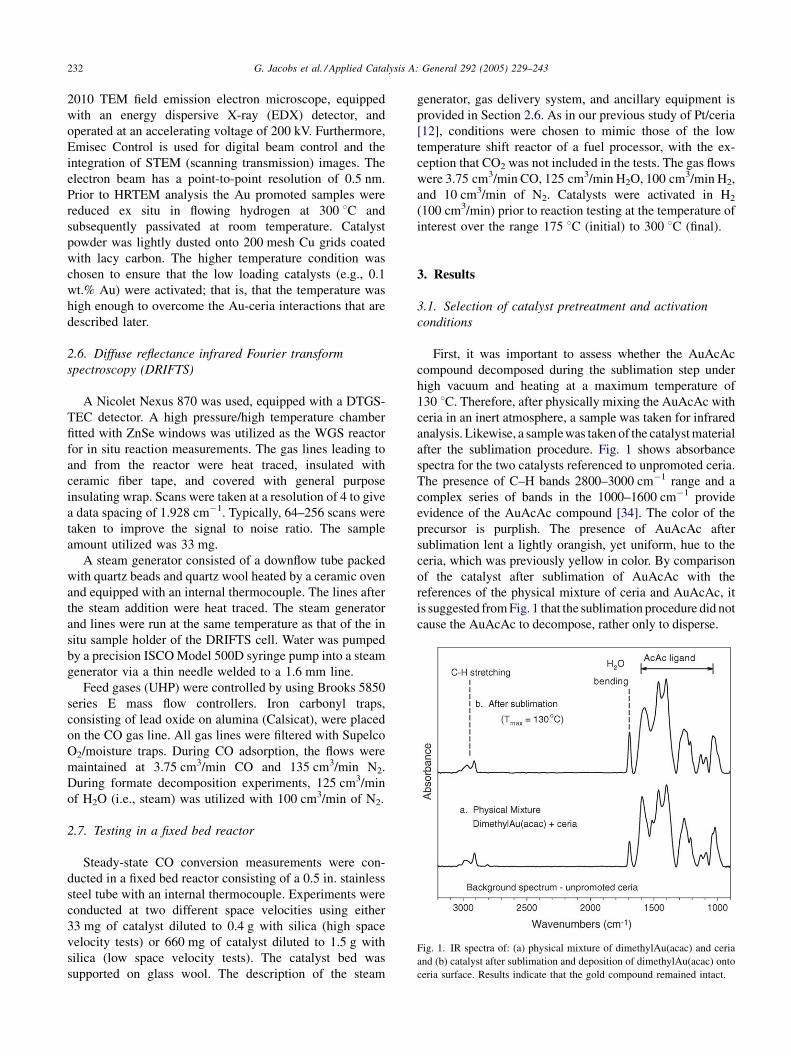
G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243232
Fig. 1. IR spectra of: (a) physical mixture of dimethylAu(acac) and ceria
and (b) catalyst after sublimation and deposition of dimethylAu(acac) onto
ceria surface. Results indicate that the gold compound remained intact.
2010 TEM field emission electron microscope, equipped
with an energy dispersive X-ray (EDX) detector, and
operated at an accelerating voltage of 200 kV. Furthermore,
Emisec Control is used for digital beam control and the
integration of STEM (scanning transmission) images. The
electron beam has a point-to-point resolution of 0.5 nm.
Prior to HRTEM analysis the Au promoted samples were
reduced ex situ in flowing hydrogen at 300 8C and
subsequently passivated at room temperature. Catalyst
powder was lightly dusted onto 200 mesh Cu grids coated
with lacy carbon. The higher temperature condition was
chosen to ensure that the low loading catalysts (e.g., 0.1
wt.% Au) were activated; that is, that the temperature was
high enough to overcome the Au-ceria interactions that are
described later.
2.6. Diffuse reflectance infrared Fourier transform
spectroscopy (DRIFTS)
A Nicolet Nexus 870 was used, equipped with a DTGS-
TEC detector. A high pressure/high temperature chamber
fitted with ZnSe windows was utilized as the WGS reactor
for in situ reaction measurements. The gas lines leading to
and from the reactor were heat traced, insulated with
ceramic fiber tape, and covered with general purpose
insulating wrap. Scans were taken at a resolution of 4 to give
a data spacing of 1.928 cm�1. Typically, 64–256 scans were
taken to improve the signal to noise ratio. The sample
amount utilized was 33 mg.
A steam generator consisted of a downflow tube packed
with quartz beads and quartz wool heated by a ceramic oven
and equipped with an internal thermocouple. The lines after
the steam addition were heat traced. The steam generator
and lines were run at the same temperature as that of the in
situ sample holder of the DRIFTS cell. Water was pumped
by a precision ISCO Model 500D syringe pump into a steam
generator via a thin needle welded to a 1.6 mm line.
Feed gases (UHP) were controlled by using Brooks 5850
series E mass flow controllers. Iron carbonyl traps,
consisting of lead oxide on alumina (Calsicat), were placed
on the CO gas line. All gas lines were filtered with Supelco
O2/moisture traps. During CO adsorption, the flows were
maintained at 3.75 cm3/min CO and 135 cm3/min N2.
During formate decomposition experiments, 125 cm3/min
of H2O (i.e., steam) was utilized with 100 cm3/min of N2.
2.7. Testing in a fixed bed reactor
Steady-state CO conversion measurements were con-
ducted in a fixed bed reactor consisting of a 0.5 in. stainless
steel tube with an internal thermocouple. Experiments were
conducted at two different space velocities using either
33 mg of catalyst diluted to 0.4 g with silica (high space
velocity tests) or 660 mg of catalyst diluted to 1.5 g with
silica (low space velocity tests). The catalyst bed was
supported on glass wool. The description of the steam
generator, gas delivery system, and ancillary equipment is
provided in Section 2.6. As in our previous study of Pt/ceria
[12], conditions were chosen to mimic those of the low
temperature shift reactor of a fuel processor, with the ex-
ception that CO2 was not included in the tests. The gas flows
were 3.75 cm3/min CO, 125 cm3/min H2O, 100 cm3/min H2,
and 10 cm3/min of N2. Catalysts were activated in H2
(100 cm3/min) prior to reaction testing at the temperature of
interest over the range 175 8C (initial) to 300 8C (final).
3. Results
3.1. Selection of catalyst pretreatment and activation
conditions
First, it was important to assess whether the AuAcAc
compound decomposed during the sublimation step under
high vacuum and heating at a maximum temperature of
130 8C. Therefore, after physically mixing the AuAcAc with
ceria in an inert atmosphere, a sample was taken for infrared
analysis. Likewise, a sample was taken of the catalyst material
after the sublimation procedure. Fig. 1 shows absorbance
spectra for the two catalysts referenced to unpromoted ceria.
The presence of C–H bands 2800–3000 cm�1 range and a
complex series of bands in the 1000–1600 cm�1 provide
evidence of the AuAcAc compound [34]. The color of the
precursor is purplish. The presence of AuAcAc after
sublimation lent a lightly orangish, yet uniform, hue to the
ceria, which was previously yellow in color. By comparison
of the catalyst after sublimation of AuAcAc with the
references of the physical mixture of ceria and AuAcAc, it
is suggested from Fig. 1 that the sublimation procedure did not
cause the AuAcAc to decompose, rather only to disperse.
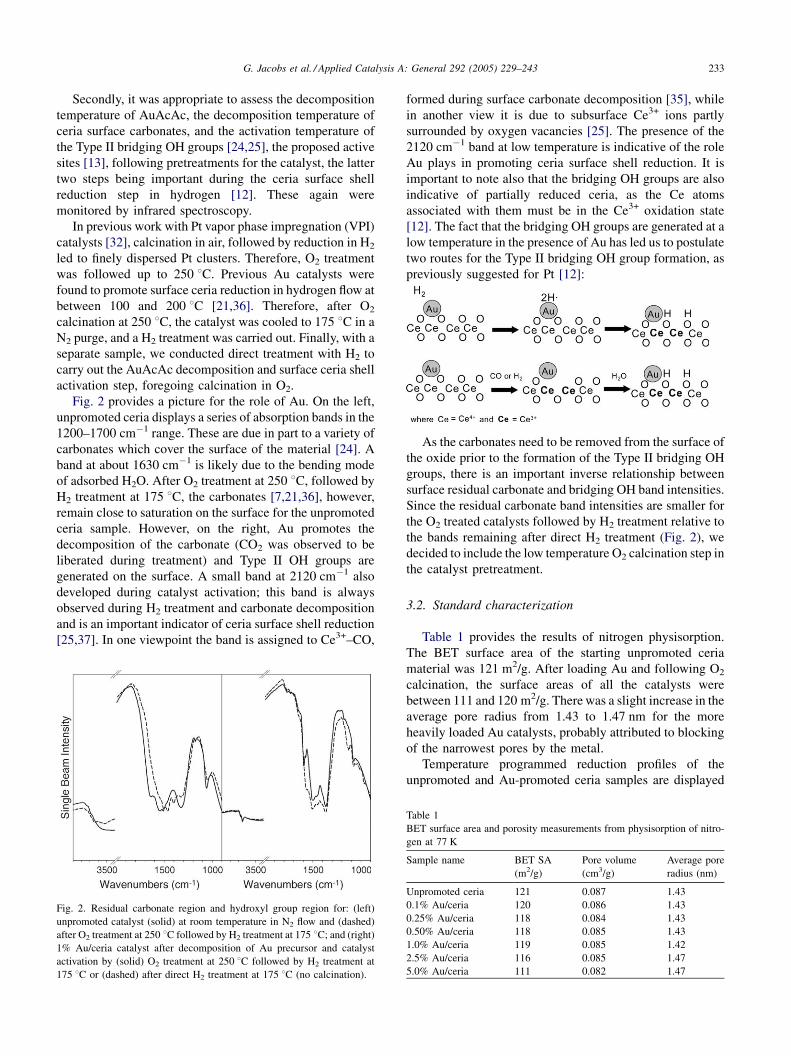
G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243 233
Secondly, it was appropriate to assess the decomposition
temperature of AuAcAc, the decomposition temperature of
ceria surface carbonates, and the activation temperature of
the Type II bridging OH groups [24,25], the proposed active
sites [13], following pretreatments for the catalyst, the latter
two steps being important during the ceria surface shell
reduction step in hydrogen [12]. These again were
monitored by infrared spectroscopy.
In previous work with Pt vapor phase impregnation (VPI)
catalysts [32], calcination in air, followed by reduction in H2
led to finely dispersed Pt clusters. Therefore, O2 treatment
was followed up to 250 8C. Previous Au catalysts were
found to promote surface ceria reduction in hydrogen flow at
between 100 and 200 8C [21,36]. Therefore, after O2
calcination at 250 8C, the catalyst was cooled to 175 8C in a
N2 purge, and a H2 treatment was carried out. Finally, with a
separate sample, we conducted direct treatment with H2 to
carry out the AuAcAc decomposition and surface ceria shell
activation step, foregoing calcination in O2.
Fig. 2 provides a picture for the role of Au. On the left,
unpromoted ceria displays a series of absorption bands in the
1200–1700 cm�1 range. These are due in part to a variety of
carbonates which cover the surface of the material [24]. A
band at about 1630 cm�1 is likely due to the bending mode
of adsorbed H2O. After O2 treatment at 250 8C, followed by
H2 treatment at 175 8C, the carbonates [7,21,36], however,
remain close to saturation on the surface for the unpromoted
ceria sample. However, on the right, Au promotes the
decomposition of the carbonate (CO2 was observed to be
liberated during treatment) and Type II OH groups are
generated on the surface. A small band at 2120 cm�1 also
developed during catalyst activation; this band is always
observed during H2 treatment and carbonate decomposition
and is an important indicator of ceria surface shell reduction
[25,37]. In one viewpoint the band is assigned to Ce3+–CO,
Fig. 2. Residual carbonate region and hydroxyl group region for: (left)
unpromoted catalyst (solid) at room temperature in N2 flow and (dashed)
after O2 treatment at 250 8C followed by H2 treatment at 175 8C; and (right)
1% Au/ceria catalyst after decomposition of Au precursor and catalyst
activation by (solid) O2 treatment at 250 8C followed by H2 treatment at
175 8C or (dashed) after direct H2 treatment at 175 8C (no calcination).
formed during surface carbonate decomposition [35], while
in another view it is due to subsurface Ce3+ ions partly
surrounded by oxygen vacancies [25]. The presence of the
2120 cm�1 band at low temperature is indicative of the role
Au plays in promoting ceria surface shell reduction. It is
important to note also that the bridging OH groups are also
indicative of partially reduced ceria, as the Ce atoms
associated with them must be in the Ce3+ oxidation state
[12]. The fact that the bridging OH groups are generated at a
low temperature in the presence of Au has led us to postulate
two routes for the Type II bridging OH group formation, as
previously suggested for Pt [12]:
As the carbonates need to be removed from the surface of
the oxide prior to the formation of the Type II bridging OH
groups, there is an important inverse relationship between
surface residual carbonate and bridging OH band intensities.
Since the residual carbonate band intensities are smaller for
the O2 treated catalysts followed by H2 treatment relative to
the bands remaining after direct H2 treatment (Fig. 2), we
decided to include the low temperature O2 calcination step in
the catalyst pretreatment.
3.2. Standard characterization
Table 1 provides the results of nitrogen physisorption.
The BET surface area of the starting unpromoted ceria
material was 121 m2/g. After loading Au and following O2
calcination, the surface areas of all the catalysts were
between 111 and 120 m2/g. There was a slight increase in the
average pore radius from 1.43 to 1.47 nm for the more
heavily loaded Au catalysts, probably attributed to blocking
of the narrowest pores by the metal.
Temperature programmed reduction profiles of the
unpromoted and Au-promoted ceria samples are displayed
Table 1
BET surface area and porosity measurements from physisorption of nitro-
gen at 77 K
Sample name BET SA
(m2/g)
Pore volume
(cm3/g)
Average pore
radius (nm)
Unpromoted ceria 121 0.087 1.43
0.1% Au/ceria 120 0.086 1.43
0.25% Au/ceria 118 0.084 1.43
0.50% Au/ceria 118 0.085 1.43
1.0% Au/ceria 119 0.085 1.42
2.5% Au/ceria 116 0.085 1.47
5.0% Au/ceria 111 0.082 1.47
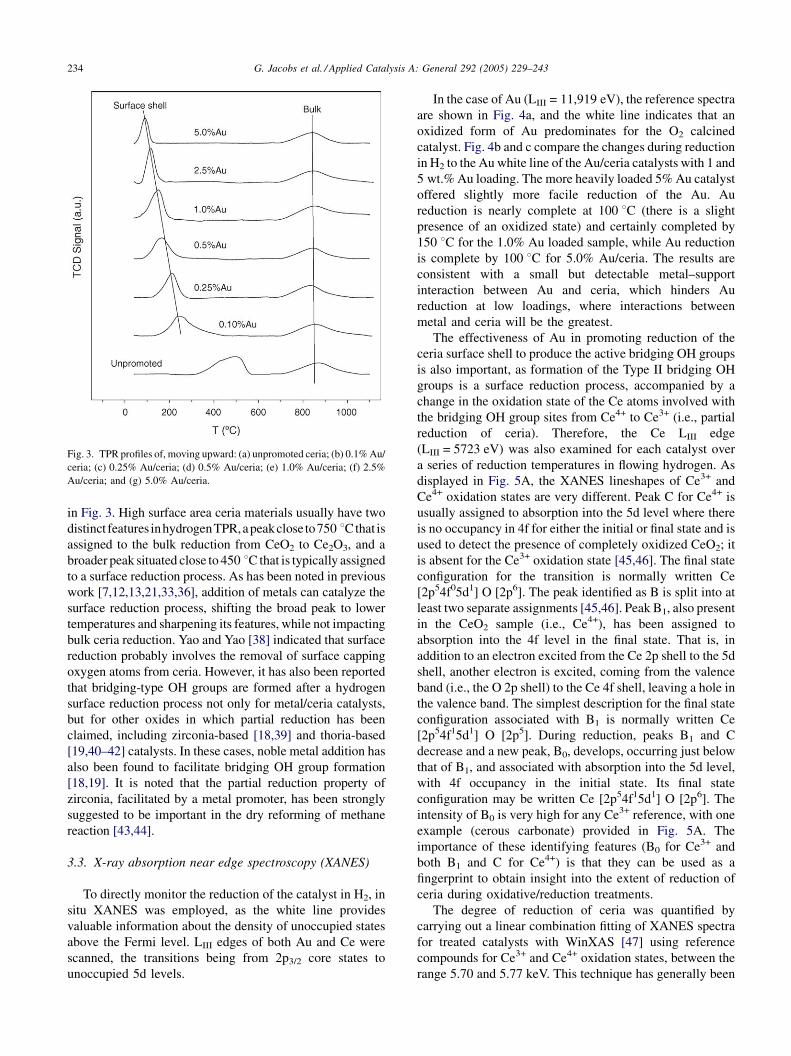
G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243234
Fig. 3. TPR profiles of, moving upward: (a) unpromoted ceria; (b) 0.1% Au/
ceria; (c) 0.25% Au/ceria; (d) 0.5% Au/ceria; (e) 1.0% Au/ceria; (f) 2.5%
Au/ceria; and (g) 5.0% Au/ceria.
in Fig. 3. High surface area ceria materials usually have two
distinct features inhydrogenTPR,apeakclose to750 8Cthat is
assigned to the bulk reduction from CeO2 to Ce2O3, and a
broader peak situated close to 450 8C that is typically assigned
to a surface reduction process. As has been noted in previous
work [7,12,13,21,33,36], addition of metals can catalyze the
surface reduction process, shifting the broad peak to lower
temperatures and sharpening its features, while not impacting
bulk ceria reduction. Yao and Yao [38] indicated that surface
reduction probably involves the removal of surface capping
oxygen atoms from ceria. However, it has also been reported
that bridging-type OH groups are formed after a hydrogen
surface reduction process not only for metal/ceria catalysts,
but for other oxides in which partial reduction has been
claimed, including zirconia-based [18,39] and thoria-based
[19,40–42] catalysts. In these cases, noble metal addition has
also been found to facilitate bridging OH group formation
[18,19]. It is noted that the partial reduction property of
zirconia, facilitated by a metal promoter, has been strongly
suggested to be important in the dry reforming of methane
reaction [43,44].
3.3. X-ray absorption near edge spectroscopy (XANES)
To directly monitor the reduction of the catalyst in H2, in
situ XANES was employed, as the white line provides
valuable information about the density of unoccupied states
above the Fermi level. LIII edges of both Au and Ce were
scanned, the transitions being from 2p3/2 core states to
unoccupied 5d levels.
In the case of Au (LIII = 11,919 eV), the reference spectra
are shown in Fig. 4a, and the white line indicates that an
oxidized form of Au predominates for the O2 calcined
catalyst. Fig. 4b and c compare the changes during reduction
in H2 to the Au white line of the Au/ceria catalysts with 1 and
5 wt.% Au loading. The more heavily loaded 5% Au catalyst
offered slightly more facile reduction of the Au. Au
reduction is nearly complete at 100 8C (there is a slight
presence of an oxidized state) and certainly completed by
150 8C for the 1.0% Au loaded sample, while Au reduction
is complete by 100 8C for 5.0% Au/ceria. The results are
consistent with a small but detectable metal–support
interaction between Au and ceria, which hinders Au
reduction at low loadings, where interactions between
metal and ceria will be the greatest.
The effectiveness of Au in promoting reduction of the
ceria surface shell to produce the active bridging OH groups
is also important, as formation of the Type II bridging OH
groups is a surface reduction process, accompanied by a
change in the oxidation state of the Ce atoms involved with
the bridging OH group sites from Ce4+ to Ce3+ (i.e., partial
reduction of ceria). Therefore, the Ce LIII edge
(LIII = 5723 eV) was also examined for each catalyst over
a series of reduction temperatures in flowing hydrogen. As
displayed in Fig. 5A, the XANES lineshapes of Ce3+ and
Ce4+ oxidation states are very different. Peak C for Ce4+ is
usually assigned to absorption into the 5d level where there
is no occupancy in 4f for either the initial or final state and is
used to detect the presence of completely oxidized CeO2; it
is absent for the Ce3+ oxidation state [45,46]. The final state
configuration for the transition is normally written Ce
[2p54f05d1] O [2p6]. The peak identified as B is split into at
least two separate assignments [45,46]. Peak B1, also present
in the CeO2 sample (i.e., Ce4+), has been assigned to
absorption into the 4f level in the final state. That is, in
addition to an electron excited from the Ce 2p shell to the 5d
shell, another electron is excited, coming from the valence
band (i.e., the O 2p shell) to the Ce 4f shell, leaving a hole in
the valence band. The simplest description for the final state
configuration associated with B1 is normally written Ce
[2p54f15d1] O [2p5]. During reduction, peaks B1 and C
decrease and a new peak, B0, develops, occurring just below
that of B1, and associated with absorption into the 5d level,
with 4f occupancy in the initial state. Its final state
configuration may be written Ce [2p54f15d1] O [2p6]. The
intensity of B0 is very high for any Ce3+ reference, with one
example (cerous carbonate) provided in Fig. 5A. The
importance of these identifying features (B0 for Ce3+ and
both B1 and C for Ce4+) is that they can be used as a
fingerprint to obtain insight into the extent of reduction of
ceria during oxidative/reduction treatments.
The degree of reduction of ceria was quantified by
carrying out a linear combination fitting of XANES spectra
for treated catalysts with WinXAS [47] using reference
compounds for Ce3+ and Ce4+ oxidation states, between the
range 5.70 and 5.77 keV. This technique has generally been

G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243 235
Fig. 4. XANES spectra at Au LIII edge for: (a) Au reference compounds; (b) 1% Au/ceria; and (c) 5.0% Au/ceria ranging from 50 to 150 8C in hydrogen.
found to be more consistent than alternate methods, like
XPS [31]. Results of the fitting procedure are reported in
Table 2. As displayed in Fig. 5B, for unpromoted ceria, the
ceria remains relatively unreduced with H2 treatment at
300 8C, and only about 3% of cerium atoms are in the Ce3+
oxidation state. However, with just 0.1% Au, changes are
observed consistent with enhanced partial reduction of ceria,
with 10.8% Ce3+ detected by 300 8C (Fig. 5C). With
increasing Au promoter loading, the reduction of the ceria
surface shell is further facilitated (i.e., shifts to lower
temperature). For 0.25% Au, 21.1% Ce3+ is present at
300 8C (Fig. 5D). An upper limit of about 1/4 of Ce atoms in
the 3+ oxidation state is obtained with 0.5% Au at 300 8C
(Fig. 5E). Above that loading, this limit is obtained at lower
and lower temperatures, with approximately 1/4 of the atoms
in the Ce3+ oxidation state at 250 8C for 1.0% Au, 200 8C for
2.5% Au, and 150 8C for the 5.0% Au loading (Fig. 5F–H,
respectively).
The results are consistent with the hypothesis that
enhanced partial reduction of ceria is attributed to the
promotion by Au. If one examines the XANES spectra of the
Au and Ce edges closely for each of the catalysts, partial
reduction of ceria is not facilitated until Au0 is present, and
we therefore suggest that the partial reduction of ceria may
be due to the result of the spillover of dissociated H2 from
Au0 to the surface of ceria, although one cannot rule out a

G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243236
Fig. 5. XANES spectra at Ce LIII edge for: (A) references and, the reduction of Au/ceria catalysts, including (B) unpromoted, (C) 0.1% Au/ceria, and (D) 0.25%
Au/ceria. Note that the shifts in energy are not real. The spectra are staggered for ease of viewing the lineshape differences. XANES spectra at Ce LIII edge for
the reduction of Au/ceria catalysts continued, including (E) 0.5% Au/ceria, (F) 1.0% Au/ceria, (G) 2.5% Au/ceria and (H) 5.0% Au/ceria.
chemical effect, whereby O removal is facilitated by the
presence of the metal, allowing for H2O to dissociate at the
vacancy. The former would allow the bridging OH groups to
be formed directly, although both processes must be
accompanied by reduction of the cerium ions in the surface
shell from Ce4+ to Ce3+, as discussed previously, and similar
to what we proposed for Pt/ceria [12].
3.4. High resolution transmission electron microscopy
(HRTEM)
Fig. 6 presents STEM images with a 5 nm scale for most
of the Au/ceria samples after reduction in H2 at 300 8C,
including 0.25% Au, 0.5% Au, 1% Au, 2.5% Au, and 5.0%
Au. The ceria agglomerates are composed of 5–8 nm
diameter ceria particles which exhibit a strong tendency to
align and form elongated ridges and channels, resulting in
what appears to be an almost corrugated surface. The Au
nanoparticles are clearly visible in Fig. 6 (their presence was
confirmed by an EDX analysis). For the 0.25% Au and 0.5%
Au samples, very small, well-dispersed Au crystallites are
observed, mainly in the 1–2 nm diameter and 1–3 nm
diameter range, respectively. For the 1% Au/ceria catalyst,
some larger clusters in the 5–8 nm range were also found (in
addition to the smaller crystallites). For the 2.5% Au/ceria
and 5.0% Au/ceria catalysts, a heterogeneous distribution

G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243 237
Fig. 5. (Continued ).
Table 2
Linear combination fits of XANES spectra with the Ce3+ and Ce4+ reference compounds
T (o.c.) No Au 0.1% Au 0.25% Au 0.5% Au 1.0% Au 2.5% Au 5.0% Au
Ce4+ Ce3+ Ce4+ Ce3+ Ce4+ Ce3+ Ce4+ Ce3+ Ce4+ Ce3+ Ce4+ Ce3+ Ce4+ Ce3+
50 100 0 99.3 0.7 99.5 0.5 99.2 0.8 99.6 0.4 99.5 0.5 99.3 0.7
100 – – 98.4 1.6 99.3 0.7 97.9 2.1 99.2 0.8 98.7 1.3 81.2 18.8
150 – – 97.9 2.1 93.6 6.4 86.8 13.2 87.9 12.1 79.1 20.9 76.1 23.9
200 – – 94.7 5.3 86.9 13.1 78.7 21.3 78.2 21.8 76.5 23.5 74.4 25.6
250 – – 92.1 7.9 81.8 18.2 77.0 23.0 76.5 23.5 75.1 24.9 73.7 26.3
300 97.0 3.0 89.2 10.8 78.9 21.1 74.7 25.3 75.8 24.2 75.2 24.8 73.3 26.6
Relative percentages as a function of reduction temperature. Reduction carried out in hydrogen.
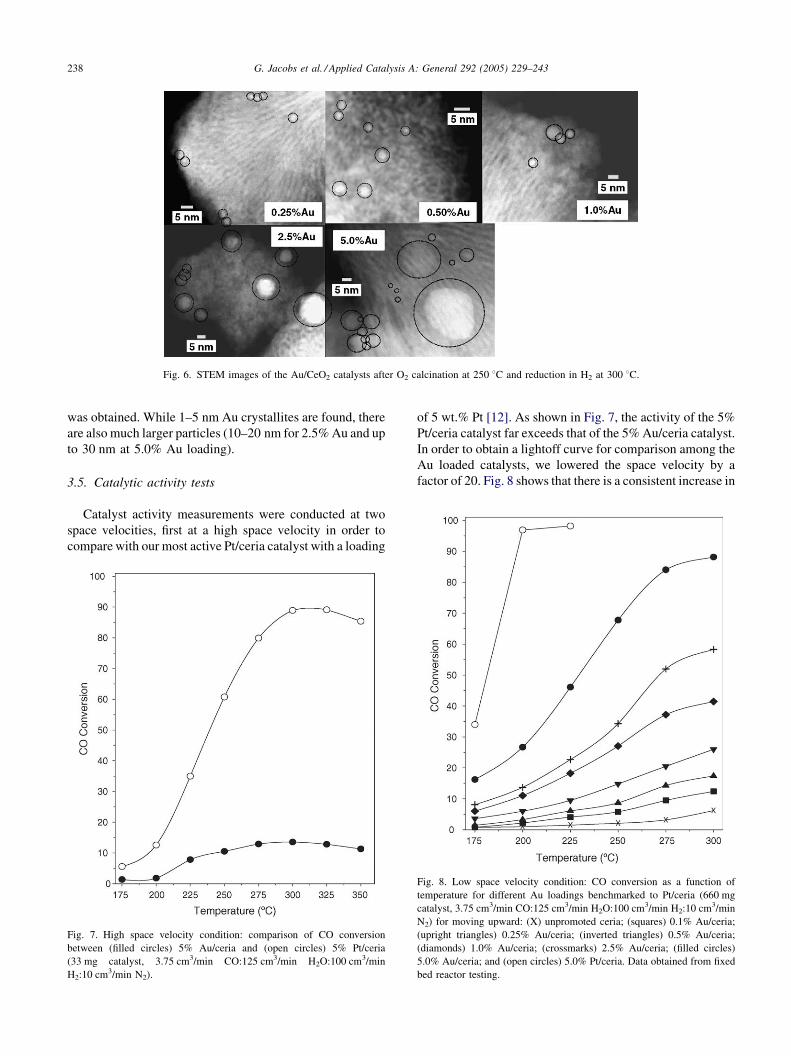
G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243238
Fig. 6. STEM images of the Au/CeO2 catalysts after O2 calcination at 250 8C and reduction in H2 at 300 8C.
was obtained. While 1–5 nm Au crystallites are found, there
are also much larger particles (10–20 nm for 2.5% Au and up
to 30 nm at 5.0% Au loading).
3.5. Catalytic activity tests
Catalyst activity measurements were conducted at two
space velocities, first at a high space velocity in order to
compare with our most active Pt/ceria catalyst with a loading
Fig. 7. High space velocity condition: comparison of CO conversion
between (filled circles) 5% Au/ceria and (open circles) 5% Pt/ceria
(33 mg catalyst, 3.75 cm3/min CO:125 cm3/min H2O:100 cm3/min
H2:10 cm3/min N2).
of 5 wt.% Pt [12]. As shown in Fig. 7, the activity of the 5%
Pt/ceria catalyst far exceeds that of the 5% Au/ceria catalyst.
In order to obtain a lightoff curve for comparison among the
Au loaded catalysts, we lowered the space velocity by a
factor of 20. Fig. 8 shows that there is a consistent increase in
Fig. 8. Low space velocity condition: CO conversion as a function of
temperature for different Au loadings benchmarked to Pt/ceria (660 mg
catalyst, 3.75 cm3/min CO:125 cm3/min H2O:100 cm3/min H2:10 cm3/min
N2) for moving upward: (X) unpromoted ceria; (squares) 0.1% Au/ceria;
(upright triangles) 0.25% Au/ceria; (inverted triangles) 0.5% Au/ceria;
(diamonds) 1.0% Au/ceria; (crossmarks) 2.5% Au/ceria; (filled circles)
5.0% Au/ceria; and (open circles) 5.0% Pt/ceria. Data obtained from fixed
bed reactor testing.
G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243 239
the catalyst activity as a function of the Au loading. At
175 8C, the 5% Pt catalyst exhibited CO conversion
at slightly more than double that of the 5% Au loaded
catalyst.
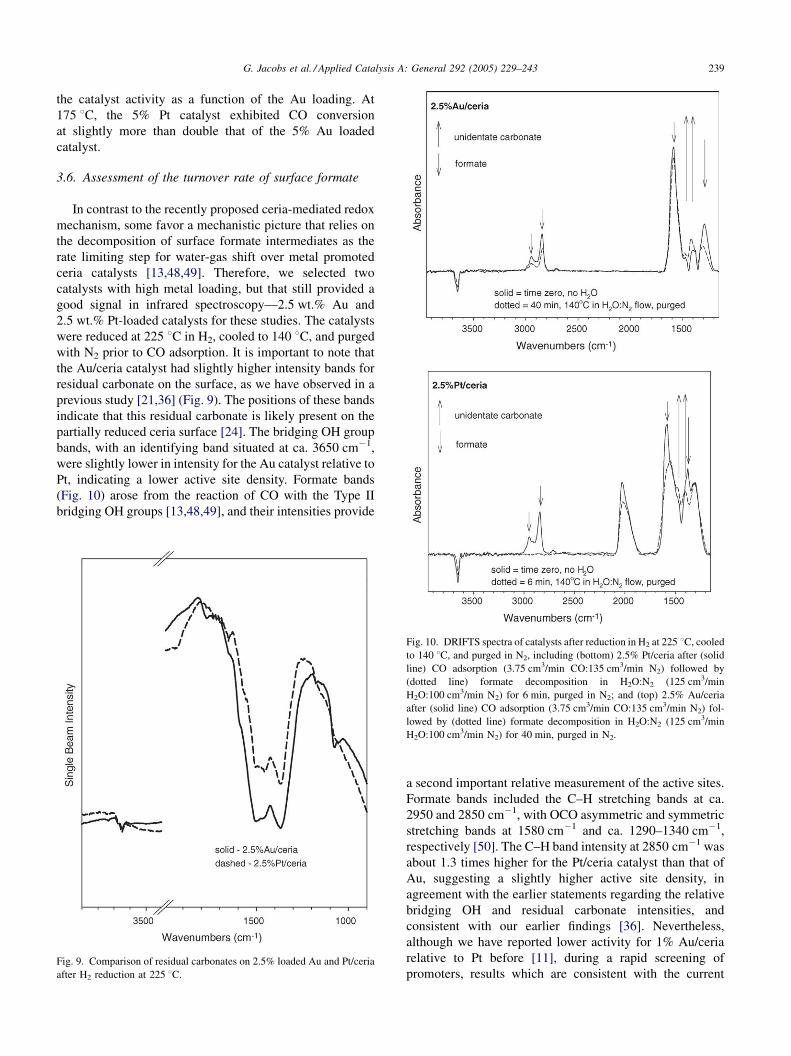
3.6. Assessment of the turnover rate of surface formate
In contrast to the recently proposed ceria-mediated redox
mechanism, some favor a mechanistic picture that relies on
the decomposition of surface formate intermediates as the
rate limiting step for water-gas shift over metal promoted
ceria catalysts [13,48,49]. Therefore, we selected two
catalysts with high metal loading, but that still provided a
good signal in infrared spectroscopy—2.5 wt.% Au and
2.5 wt.% Pt-loaded catalysts for these studies. The catalysts
were reduced at 225 8C in H2, cooled to 140 8C, and purged
with N2 prior to CO adsorption. It is important to note that
the Au/ceria catalyst had slightly higher intensity bands for
residual carbonate on the surface, as we have observed in a
previous study [21,36] (Fig. 9). The positions of these bands
indicate that this residual carbonate is likely present on the
partially reduced ceria surface [24]. The bridging OH group
bands, with an identifying band situated at ca. 3650 cm�1,
were slightly lower in intensity for the Au catalyst relative to
Pt, indicating a lower active site density. Formate bands
(Fig. 10) arose from the reaction of CO with the Type II
bridging OH groups [13,48,49], and their intensities provide
Fig. 9. Comparison of residual carbonates on 2.5% loaded Au and Pt/ceria
after H2 reduction at 225 8C.
Fig. 10. DRIFTS spectra of catalysts after reduction in H2 at 225 8C, cooled
to 140 8C, and purged in N2, including (bottom) 2.5% Pt/ceria after (solid
line) CO adsorption (3.75 cm3/min CO:135 cm3/min N2) followed by
(dotted line) formate decomposition in H2O:N2 (125 cm3/min
H2O:100 cm3/min N2) for 6 min, purged in N2; and (top) 2.5% Au/ceria
after (solid line) CO adsorption (3.75 cm3/min CO:135 cm3/min N2) fol-
lowed by (dotted line) formate decomposition in H2O:N2 (125 cm3/min
H2O:100 cm3/min N2) for 40 min, purged in N2.
a second important relative measurement of the active sites.
Formate bands included the C–H stretching bands at ca.
2950 and 2850 cm�1, with OCO asymmetric and symmetric
stretching bands at 1580 cm�1 and ca. 1290–1340 cm�1,
respectively [50]. The C–H band intensity at 2850 cm�1 was
about 1.3 times higher for the Pt/ceria catalyst than that of
Au, suggesting a slightly higher active site density, in
agreement with the earlier statements regarding the relative
bridging OH and residual carbonate intensities, and
consistent with our earlier findings [36]. Nevertheless,
although we have reported lower activity for 1% Au/ceria
relative to Pt before [11], during a rapid screening of
promoters, results which are consistent with the current

G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243240
investigation, it is apparent that the difference in relative
active site density cannot adequately account for the
significant difference in activity between the two promoters.
Therefore, in addition to the role that the metal promoter
plays in activating the catalyst at lower temperature, it
appears that the metal promoter may also be directly
involved in the mechanism [11,12,36].
Since the forward decomposition of formates rapidly
occurs in the presence of H2O at shift temperatures, the very
low temperature of 140 8C was selected so that the formate
decomposition could be monitored at a measurable rate. The
data in Fig. 10 are consistent with our earlier transient
formate decomposition study [15]. Over 2.5% Pt/ceria, the
formate decomposed to unidentate carbonate (OCO vibra-
tions at ca. 1460 and 1390 cm�1, respectively [50]) rapidly
(in approximately 6 min), and a band for gas phase CO2 was
observed, indicating that some WGS product was liberated
from the catalyst. The intense C–H stretching band of
formate at 2850 cm�1 was used to assess the changes. In
contrast, for the 2.5% Au/ceria catalyst, the formate
decomposition proceeded at a much slower rate, and even
after 40 min, after purging in N2 and using the C–H
stretching band at 2850 cm�1 as a reference, about 63% of
the original formate intensity was still observed.
4. Discussion
In this work, the goal was to fairly compare Au relative to
Pt as a promoter for the low temperature water-gas shift
reaction. As such, conditions are chosen (with the exception
of added CO2) to mimic the feed entering the low
temperature water-gas shift section of a fuel processor for
PEM fuel cell applications. That is, the feed is assumed to be
close to that of the tail gas exiting the high temperature shift
reactor, which is typically operated at a temperature
resulting in close to equilibrium conversion at that higher
reaction temperature [4]. In this case, the feed conditions
contain a very high H2O/CO ratio. An appropriate amount of
hydrogen was also included in the feed. In order to make a
fair comparison of Au with Pt, we therefore include the
5 wt.% Pt-loaded catalyst in the study, because at 5 wt.%
loading, the metal–oxide interactions are overcome by
200 8C. This is supported by a comparison of the TPR and
XANES results with those of our earlier loading study of Pt/
ceria catalysts [12]. That is to say, one should not select a Pt
catalyst with a low Pt loading, and test it in a low
temperature shift range where the catalyst is not in an active
state. For example, a 1 wt.% Pt catalyst is fully activated at
about 250 8C, whereas the 1% Au catalyst is fully activated
at a temperature that is about 50–100 8C lower.
Therefore, on the basis of the 5% metal loading, the first
important finding in this work is that, for the Au/ceria
catalysts prepared by CVD in this work Au was found to be
far less effective than Pt as a promoter for the low
temperature water-gas shift reaction, as only 1/20th of the
amount of Pt/ceria catalyst (33 mg) was needed to achieve a
lightoff curve comparable to that of Au (using 660 mg). This
is in contrast to recent reports that maintain that Au/ceria is
an excellent low temperature water-gas shift catalyst relative
to Pt [5,6,8]. In fairness, however, differences in preparation
procedure may play an important role in determining the
cluster size and/or the nature of the active site, which could
conceivably alter the mechanistic picture (e.g., Au cationic
site [6–8]). Regarding cluster size, for example, Tabakova
et al. reported highly active Au/ceria catalysts with clusters
<1 nm were more active than larger clusters [9]. Rather, we
found that in the range 200–300 8C, Au/ceria is only
marginally active relative to Pt/ceria, in agreement with our
earlier findings [21]. It is only at the 175 8C condition that
Au performs relatively better, yet still maintaining only a
little less than half of the activity of the 5% Pt/ceria catalyst.
It is important to note that low temperature shift catalysts
considered for commercial applications are typically
screened at conditions close to those exhibited by our high
space velocity condition.
A first glance at the TPR and XANES results shows that
Au appears to be somewhat of an anomaly. Au is certainly an
excellent promoter for the reduction of the surface shell of
ceria, as previously observed [7,11,36]. Yet if Au promotes
reduction of ceria, then why are the catalysts so much less
active than Pt/ceria? To obtain answers to these questions, a
characterization of these materials using TPR, in situ
XANES, and in situ FTIR spectroscopy was conducted.
Also, we have good evidence that a role of the metal
promoter is to assist in generating the active sites for WGS at
low temperature. However, a fundamental question that has
remained elusive and requires a reasonable explanation is
whether the metal promoter participates in the WGS
reaction. And, if this is indeed the case, then at which
step in the mechanism does the metal play a role, and exactly
how does it function?
In addressing these questions, however, it is helpful to
cast the answers in terms of a particular mechanism, the two
most often cited being the recently claimed ceria-mediated
redox process [7,26,27] and the earlier, surface formate
mechanism [13,14,48,49]. We select the latter mechanism to
describe our results, as all of our previous findings point to a
mechanism involving Type II bridging OH groups as active
sites [12] and surface formate intermediate decomposition
as the rate limiting step, in agreement with a mechanism like
that proposed by Shido and Iwasawa for Rh/ceria [13].
Arguments to support this mechanism are provided in our
earlier work and those of others, and are therefore not
detailed here. We refer the reader to the TPD and kinetic
studies of Shido and Iwasawa [13,14]; as well as our steady-
state dynamic studies of the formate coverage with respect to
the WGS reaction [48,49]; our steady-state [51] and
transient [15] kinetic isotope effect studies; and most
recently, our steady-state isotopic transient kinetic analyses
utilizing 12C and 13C isotopes in conjunction with in situ
DRIFTS spectroscopy for both the forward shift [52] and

G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243 241
reverse water-gas shift [53] reactions. One interesting note
from the work at hand, however, is the fact that Au/ceria
possesses water-gas shift activity [4–11], and yet there is no
evidence for a Au–CO band by either this contribution or
those of others (e.g., see Fig. 11 in [54]). Recall that it is the
reaction of Au–CO with CeO2 in the Ce4+ oxidation state
that was proposed by proponents of the ceria-mediated
redox process [7] to generate CO2. This observation suggests
that one feature of a redox mechanism is not present.
Returning to the formate perspective, we consider first the
activation of the catalysts in hydrogen. Reduction of the
catalyst does not lead to free oxygen vacancies when H2O or
H2 is present, but rather to the formation of Type II bridging
OH groups, which are strongly related to vacancies, and we
have noted that the Ce atoms involved with either vacancies
or the Type II bridging OH groups must be in the Ce3+
oxidation state (i.e., in the ceria surface shell). They are
produced either via oxygen vacancy formation followed by
dissociation of H2O or via hydrogen dissociation on a metal
promoter and spillover to the oxide surface, as described
previously. Both the TPR profiles and the XANES spectra
provide information on the temperature at which activation
of the surface occurs, as well as the extent of the reduction as
a function of the temperature.
As mentioned earlier, low loadings of Au activate the
reduction process at a temperature that is lower than that of
equivalent Pt-loaded catalysts (e.g., compare with [12]).
Examining results of XANES for 0.5 wt.% metal loadings,
for example [12], the 1/4 ceria reduction threshold is reached
by 300 8C for 0.5% Pt/ceria, while the same extent of
reduction occurs at roughly 200 8C for 0.5% Au/ceria. This
is why a fair comparison of Au and Pt/ceria catalysts should
only be made in a temperature range where the two catalysts
exhibit comparable extents of ceria reduction.
Next consider the metal–oxide interaction. As shown in
both the TPR and XANES studies of Pt/ceria [12] and those
in this investigation of Au/ceria, ceria interactions with the
promoter are important and increasing the loading assists in
decreasing this interaction. This suggests that moving to a
larger cluster size assists in overcoming the interaction. That
is, the larger metal clusters for the more heavily loaded
catalysts are more easily reduced, and once Pt or Au is
reduced, hydrogen can dissociate and spillover to the surface
of ceria, to facilitate reduction from Ce4+ to Ce3+ in the
surface shell by bridging OH group formation. This
explanation is supported by the XANES results at the Au
LIII edge, which show that the gold, appearing close to 1+
oxidation state (by comparison with reference spectra) when
initially loaded into the cell, is virtually completely reduced
to Au0 by 100 8C for the 1 and 5 wt.% Au loaded catalysts,
and this reduction occurs below the temperature at which
major changes in the cerium surface shell reduction occurs
(e.g., in the range of 150–200 8C for 1 wt.% Au/ceria). It is
often noted that Au/ceria catalysts require no activation, but
it is probably more appropriate to say that the WGS feed,
through surface reduction, activates the catalyst, since the
reduction process occurs at such a low temperature. The
results do not support a recent proposal [5,6,8] that active
non-metallic species are responsible for WGS activity,
although differences regarding preparation methods used
among researchers may change the nature of the active site.
For example, in certain cases, Au in a cationic state, strongly
interacting with ceria, has been proposed to be an active
center for WGS. Rather, for our catalysts from the XANES
results, though cationic after calcination, Au and Pt [12] are
in a reduced form when they promote the surface shell
reduction of ceria. We do not rule out the possibility that a
fraction of Au, not detectable by XANES, might be in
cationic form, as suggested by others [5,6,8]. Au species
with greater than zero oxidation state have been reported for
catalysts for the selective oxidation of CO by Calla and
Davis [55], although the oxide support in that case was
Al2O3. However, using XANES, the authors observed only
metallic Au after activation.
The comparison of the 5% loaded Pt and Au catalysts,
with equivalent extents of ceria surface shell reduction,
raises questions. Why is the 5% Pt/ceria catalyst 20 times
more active in the temperature range between 200 and
300 8C than gold? The result suggests that the metal plays a
role not only in activating the Type II bridging OH groups,
but also in turning over the intermediates involved in water-
gas shift. This conclusion is in contrast to recent reports that
suggest metallic species play no role in the catalysis [5,6,8].
For example, after cyanide leaching of metal (i.e., Au and Pt)
from the catalyst, one group [5,6,8] found that the water-gas
shift activity is not changed, leading them to the conclusion
that active non-metallic species in close association with
ceria are responsible for catalytic activity. As discussed
earlier, this is not supported by our XANES results, either for
Au in this work or Pt from our earlier study [12]. Therefore,
to test the possibility of a participation of the metal in the
reaction mechanism, we examined the turnover rate of
formate by transient decomposition with H2O.
To preface the decision to use formate decomposition, we
are aware that recently, two different groups have suggested
that formates are not intermediates in the water-gas shift
reaction. For example, Tibiletti et al. [56] have used steady-
state isotopic transient kinetic analysis of the RWGS
reaction with 12C and 13C isotopes for labeling CO2. They
have observed that Pt carbonyl exchanges faster than the
formate (which exchanged very slowly) in RWGS. They
have claimed that, therefore, formates are merely spectators
on the surface, and that a redox mechanism is operating. By
the principle of microscopic reversibility, they further claim
that formates are not intermediates to the forward shift,
either. We have recently repeated their study of RWGS, and
obtained the same data. However, the principle of
microscopic reversibility must be invoked with caution,
as reactants or products can promote the rate of the reaction.
Shido and Iwasawa [13] have shown, and we have confirmed
[15], that while formate is quite stable in the absence (or
under low concentrations) of H2O, when H2O is present, the

G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243242
forward decomposition of formate to unidentate carbonate
and H2 occurs rapidly, even at a low temperature. In fact, we
recently performed the RWGS experiments in the presence
of co-adsorbed H2O, and the formate exchanges very
rapidly, as it does for the forward shift reaction as well
[52,53]. Moreover, the Pt carbonyl exchanges rapidly for
both 12CO2 to 13CO2 switching and 12CO to 13CO switching
even when the second reactant (i.e., H2 for RWGS and H2O
for forward shift) is absent, being replaced by inert gas,
suggesting that the Pt-carbonyl exchange occurs rapidly
even in the absence of the RWGS or forward WGS reactions
[52,53]. There has been, more recently [54], a second claim
that formates are responsible for deactivation, as CO + H2
led to their appearance in in situ spectroscopy, and, more-
over, switching from WGS, to a CO + H2 feed, and back to
WGS led to decreased activity. Again, however, when H2O is
present [13,15], formates decompose very rapidly at shift
temperatures to CO2 and H2. We have found (unpublished
results) that the Pt/ceria catalysts have some activity for
Fischer–Tropsch synthesis, so it is more likely that the latter
authors were generating hydrogenation products (and there-
fore, carbonaceous residue), by running a mixture of only
CO + H2 that likely led to the decreased activity.
Returning to the formate decomposition experiments,
therefore, we are in agreement with Shido and Iwasawa that,
while formates are quite stable when CO is reacted with the
Type II bridging OH groups, they are likely intermediates to
the WGS reaction when H2O is present. In our previous
switching experiments, one can observe the rapid formation
of CO2 when the stabilized formates are decomposed with
H2O by FTIR spectroscopy [15,57] at shift temperatures.
And, in fact, for Pt/ceria, we had to lower the temperature to
140 8C in order to slow the formate forward decomposition
rate enough so that we could make measurements by FTIR to
monitor their decomposition rate [15]. In those studies, the
intensity of the Pt-carbonyl band remained little affected,
and it was suggested that intermediate carbonate species
(i.e., precursor to CO2) were produced from the formate
decomposition in steam.
In good agreement with our previous results for Pt/ceria,
the formate decomposition by H2O was complete in about
6 min. Here the result is slightly faster than previously
observed, since we are using a 2.5% Pt/ceria catalyst versus
a 1% Pt/ceria [15], which took about 8 min to complete the
decomposition. However, even after 40 min, a considerable
amount of formate (about 63%) remained on the surface of
the 2.5% Au/ceria catalyst after the H2O was purged from
the reactor with N2. This is the first direct evidence that
suggests that the metal does play a role in the mechanism,
and apparently during the proposed rate limiting step [13],
the forward decomposition of formate, which involves C–H
bond breaking. A relative rate comparison of the two
catalysts, based on these measurements, can be estimated, as
follows:
Xð1�0:63Þ=40 ¼ ð1�0Þ=6; Pt : Au ¼ X ¼ 18
The results suggest that on a relative basis, the formate
decomposes at approximately 18 times faster for the 5 wt.%
Pt/ceria catalyst, than it does for the 5 wt.% Au/ceria
catalyst.
It is still not clear how Pt accelerates the rate of the
formate decomposition in the presence of H2O. We consider
at least three possibilities. (1) Pt may assist in dehydro-
genating the formate intermediate. Certainly, Pt has been
found to exhibit far greater hydrogenation/dehydrogenation
ability than Au, leading to the extensive study of Au for
partial hydrogenation reactions, including acetylene [58]
and butadiene [59,60]. If the formate intermediate is a
mobile species and the metal assists in formate C–H bond
breaking, the hypothesis suggests that the promoter type and
loading should impact the WGS rate. (2) Another possibility
is an electronic effect, which may impact the tilting of the
formate, proposed to be associated with the transition state
of formate decomposition [13]. (3) In re-considering the
work of Tamaru, H+ may promote the decomposition rate of
formate. It is not clear at this time whether or not Pt may be
able to promote the heterolytic dissociation of H2O, but
dissociation of a fraction of adsorbed H2O on Pt (1 1 1) has
been suggested recently [61].
5. Conclusions
Increasing the Au loading had a significant positive
impact on the catalytic activity for WGS. The partial
reduction of ceria is necessary for generating bridging OH
groups on the surface of ceria, which serve as the active sites.
During this surface shell reduction process in hydrogen,
bridging OH groups are formed, and two cerium atoms for
each pair of Type II OH groups change from the Ce4+ to Ce3+
oxidation state. Addition of Au to ceria catalyzes the surface
reduction process and reduces the peak reduction tempera-
ture from 450 8C for the unpromoted catalyst to 100 8C for
the 5 wt.% Au/ceria catalyst. A systematic decrease in the
temperature required for ceria surface shell reduction was
observed by increasing the Au promoter loading as follows:
0.1, 0.25, 0.5, 1.0, 2.5, and 5.0 wt.%.
After ensuring that we accounted for metal–oxide
interactions and obtained similar extents of ceria surface
shell reduction to provide a valid comparison, 5 wt.% Au/
ceria was found to have about 1/20th the activity of 5 wt.%
Pt/ceria in the range 200–300 8C, and 5 wt.% Pt/ceria
exhibited a higher steady-state activity (about double that of
Au) at 175 8C. Transient formate decomposition experi-
ments carried out showed that the rate of formate
decomposition was approximately 20 times faster for Pt/
ceria than that observed with Au/ceria, suggesting that the
metal (in addition to the rate promotion of H2O) is also
involved in promoting the formate decomposition, which is
the proposed rate limiting step of the formate reaction
mechanism. It is suggested that Pt accomplishes this higher
turnover rate by nature of its greater dehydrogenating ability

G. Jacobs et al. / Applied Catalysis A: General 292 (2005) 229–243 243
over Au, or via an electronic influence, or that it may
possibly be involved in dissociating H2O to promote the
decomposition of formate intermediate.
Acknowledgments
The work was supported by the Commonwealth of
Kentucky. We would like to thank Joel Young at the
University of Oklahoma’s Department of Physics for
fabrication of the in situ X-ray spectroscopy cell. Research
carried out (in part) at the NSLS, at BNL, is supported by the
U.S. Department of Energy, Division of Materials Science
and Division of Chemical Sciences, under Contract No. DE-
AC02-98CH10886.
References
[1] National Hydrogen Energy Roadmap, US DOE, 2002.
[2] Fuel Cell Report to Congress, US DOE, 2003.
[3] D.C. Dayton, M. Ratcliff, R. Bain, Fuel Cell Integration—A Study of
the Impact of Gas Quality and Impurities, NREL/MP-510-30298,
2001.
[4] A.F. Ghenciu, Curr. Opin. Solid State Mater. Sci. 6 (2002) 389.
[5] Q. Fu, W. Deng, H. Saltsburg, M. Flytzani-Stephanopoulos, Appl.
Catal. B: Environ. 56 (2005) 57.
[6] Q. Fu, H. Saltsburg, M. Flytzani-Stephanopoulos, Science 301 (2003)
935.
[7] Q. Fu, A. Weber, M. Flytzani-Stephanopoulos, Catal. Lett. 77 (2001) 87.
[8] Q. Fu, S. Kudriavtseva, H. Saltsburg, M. Flytzani-Stephanopoulos,
Chem. Eng. J. 93 (2003) 41.
[9] T. Tabakova, F. Boccuzzi, M. Manzoli, J.W. Sobczak, V. Idakiev, D.
Andreeva, Appl. Catal. B: Environ. 49 (2004) 73.
[10] T. Tabakova, F. Boccuzzi, M. Manzoli, V. Idakiev, D. Andreeva, Appl.
Catal. A: Gen. 252 (2003) 385.
[11] D. Andreeva, V. Idakiev, T. Tabakova, L. Ilieva, P. Falaras, A.
Bourlinos, A. Travlos, Catal. Today 72 (2002) 51.
[12] G. Jacobs, U.M. Graham, E. Chenu, P.M. Patterson, B.H. Davis, J.
Catal. 229 (2005) 499.
[13] T. Shido, Y. Iwasawa, J. Catal. 141 (1993) 71.
[14] T. Shido, Y. Iwasawa, J. Catal. 136 (1992) 493.
[15] G. Jacobs, P.M. Patterson, U.M. Graham, D.E. Sparks, B.H. Davis,
Appl. Catal. A: Gen. 269 (2004) 63.
[16] T. Shido, Y. Iwasawa, J. Catal. 129 (1991) 343.
[17] T. Shido, K. Asakura, Y. Iwasawa, J. Catal. 122 (1990) 55.
[18] E. Chenu, G. Jacobs, A.C. Crawford, R.A. Keogh, P.M. Patterson, D.E.
Sparks, B.H. Davis, Appl. Catal. B: Environ. 59 (2004) 45.
[19] G. Jacobs, A.C. Crawford, L. Williams, P.M. Patterson, B.H. Davis,
Appl. Catal. A: Gen. 267 (2004) 27.
[20] M.W. Balakos, M.R. Madden, T.L. Walsh, J.P. Wagner, Water gas
shift: alternatives for fuel processing, in: Proceedings of the AICHE
Spring National Meeting, New Orleans, LA, 2004.
[21] G. Jacobs, E. Chenu, P.M. Patterson, L. Williams, D.E. Sparks, G.
Thomas, B.H. Davis, Appl. Catal. A: Gen. 258 (2004) 203.
[22] K. Tamaru, Dynamic Heterogeneous Catalysis, Academic Press, 1977.
[23] P. Mars, J.J.F. Scholten, P. Zwietering, Adv. Catal. 14 (1963) 35.
[24] A. Laachir, V. Perrichon, A. Badri, J. Lamotte, E. Catherine, J.C.
Lavalley, J. El Fallah, L. Hilaire, F. Le Normand, E. Quemere, G.N.
Sauvion, O. Touret, J. Chem. Soc., Faraday Trans. 87 (1991)
1601.
[25] C. Binet, M. Daturi, J.C. Lavalley, Catal. Today 50 (1999) 207.
[26] S. Hilaire, X. Wang, T. Luo, R.J. Gorte, J. Wagner, Appl. Catal. 215
(2001) 271.
[27] T. Bunluesin, R. Gorte, G. Graham, Appl. Catal. B 15 (1998) 107.
[28] M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M.J. Genet, B.
Delmon, J. Catal. 144 (1993) 175.
[29] M. Haruta, S. Tsubota, T. Kobayashi, S. Iijima, J. Catal. 115 (1989)
301.
[30] H. Sakurai, T. Akita, S. Tsubota, M. Kiuchi, M. Haruta, Appl. Catal. A:
Gen. 291 (2005) 179.
[31] F. Zhang, P. Wang, J. Koberstein, S. Khalid, Siu-Wai Chan, Surf. Sci.
563 (2004) 74.
[32] S. Jongpatiwut, P. Sackamduang, T. Rirksomboon, S. Osuwan, D.E.
Resasco, J. Catal. 218 (2003) 1.
[33] Y. Li, Q. Fu, M. Flytzani-Stephanopoulos, Appl. Catal. B 27 (2000)
179.
[34] M. Okumura, S. Tsubota, M. Haruta, J. Mol. Catal. A: Chem. 199
(2003) 73.
[35] T. Ressler, WinXAS 97 Version 1.0, 1997.
[36] G. Jacobs, P.M. Patterson, L. Williams, E. Chenu, D.E. Sparks, G.
Thomas, B.H. Davis, Appl. Catal. A: Gen. 262 (2004) 177.
[37] F. Bozon-Verduraz, A. Bensalem, J. Chem. Soc., Faraday Trans. 90
(1994) 653.
[38] H.C. Yao, Y.F.Y. Yao, J. Catal. 86 (1984) 254.
[39] K. Jung, A.T. Bell, J. Catal. 193 (2000) 207.
[40] J. Lamotte, J.C. Lavalley, E. Druet, E. Freund, J. Chem. Soc., Faraday
Trans. 1 79 (1983) 2219.
[41] J. Lamotte, J.C. Lavalley, V. Lorenzelli, E. Freund, J. Chem. Soc.,
Faraday Trans. 1 81 (1985) 215.
[42] J. Lamotte, V. Moravek, M. Bensitel, J.C. Lavalley, React. Kinet.
Catal. Lett. 36 (1988) 113.
[43] S.M. Stagg-Williams, D.E. Resasco, Stud. Surf. Sci. Catal. 130 (2000)
3663.
[44] F.B. Noronha, C. Taylor, E.C. Fendley, S.M. Stagg-Williams, D.E.
Resasco, Catal. Lett. 90 (2003) 13.
[45] S. Overbury, D. Huntley, D. Mullins, G. Glavee, Catal. Lett. 51 (1998)
133.
[46] J. El Fallah, S. Boujani, H. Dexpert, A. Kiennemann, J. Majerns, O.
Touret, F. Villain, F. Le Normand, J. Phys. Chem. 98 (1994)
5522.
[47] T. Ressler, WinXAS 97 Version 1.0, 1997.
[48] G. Jacobs, L. Williams, U. Graham, D.E. Sparks, B.H. Davis, J. Phys.
Chem. B 107 (2003) 10398.
[49] G. Jacobs, L. Williams, U. Graham, D.E. Sparks, G. Thomas, B.H.
Davis, Appl. Catal. A 252 (2003) 107.
[50] A. Holmgren, B. Andersson, D. Duprez, Appl. Catal. B: Environ. 22
(1999) 215.
[51] G. Jacobs, S. Khalid, P.M. Patterson, L. Williams, D.E. Sparks, B.H.
Davis, Appl. Catal. 268 (2004) 255.
[52] G. Jacobs, A.C. Crawford, B.H. Davis, Catal. Lett. 100 (2005)
147.
[53] G. Jacobs, B.H. Davis, Appl. Catal. A: Gen. 284 (2005) 31.
[54] C.H. Kim, L.T. Thompson, J. Catal. 230 (2005) 66.
[55] J.T. Calla, R.J. Davis, Catal. Lett. 99 (2005) 21.
[56] D. Tibiletti, A. Goguet, F.C. Meunier, J.P. Breen, R. Burch, Chem.
Commun. 14 (2004) 1636.
[57] G. Jacobs, P.M. Patterson, L. Williams, D.E. Sparks, B.H. Davis,
Catal. Lett. 96 (2004) 97.
[58] T.V. Choudhary, C. Sivadinarayana, A.K. Datye, D. Kumar, D.W.
Goodman, Catal. Lett. 86 (2003) 1.
[59] P.A. Sermon, G.C. Bond, J. Chem. Soc., Faraday Trans. I 74 (1978)
385.
[60] M. Okumura, T. Akita, M. Haruta, Catal. Today 74 (2002) 265.
[61] T. Jacob, W.A. Goddard III, J. Am. Chem. Soc. 126 (2004)
9360.
Copyright © 2022 FDOKUMEN






![CVd`]feZ`_ V]fUVd :_UZR 4YZ_R eR]\d - Daily Pioneer](https://static.fdokumen.com/doc/165x107/633c1d2ea028126067032bb8/cvdfez-vfuvd-uzr-4yzr-erd-daily-pioneer.jpg)






![HR_X¶d gZdZe cRZdVd Y`aVd W`c V]fdZgV =24 cVd`]feZ`](https://static.fdokumen.com/doc/165x107/63243881be5419ea700ef43e/hrxd-gzdze-crzdvd-yavd-wc-vfdzgv-24-cvdfez.jpg)






