
Lipid-based nanoparticles with high binding affinity for amyloid-β 1–42 peptide
11
Lipid-based nanoparticles with high binding affinity for amyloid-b 1e42 peptide Marco Gobbi a, 1 , Francesca Re b, * ,1 , Mara Canovi a , Marten Beeg a , Maria Gregori b , Silvia Sesana b , Sandro Sonnino c , Doriano Brogioli b , Claudia Musicanti d , Paolo Gasco d , Mario Salmona a , Massimo E. Masserini b a Department of Biochemistry and Molecular Pharmacology, Mario Negri Institute for Pharmacological Research, Milano, Italy b Department of Experimental Medicine, University of Milano Bicocca, 20052 Monza, Italy c Center of Excellence on Neurodegenerative Diseases, University of Milan, 20090 Segrate, Italy d Nanovector S.r.l., 10144 Torino, Italy article info Article history: Received 3 March 2010 Accepted 21 April 2010 Available online 4 June 2010 Keywords: Nanoparticles Drug delivery Affinity Lipid Liposome Ab-peptide abstract The neurotoxic beta-amyloid peptide (Ab), formed in anomalous amounts in Alzheimer’s disease (AD), is released as monomer and then undergoes aggregation forming oligomers, fibrils and plaques in diseased brains. Ab aggregates are considered as possible targets for therapy and/or diagnosis of AD. Since nanoparticles (NPs) are promising vehicles for imaging probes and therapeutic agents, we realized and characterized two types of NPs (liposomes and solid lipid nanoparticles, 145 and 76 nm average size, respectively) functionalized to target Ab 1e42 with high affinity. Preliminary immunostaining studies identified anionic phospholipids [phosphatidic acid (PA) and cardiolipin (CL)] as suitable Ab 1e42 ligands. PA/CL-functionalized, but not plain, NPs interacted with Ab 1e42 aggregates as indicated by ultracentri- fugation experiments, in which binding reaction occurred in solution, and by Surface Plasmon Resonance (SPR) experiments, in which NPs flowed onto immobilized Ab 1e42 . All these experiments were carried out in buffered saline. SPR studies indicated that, when exposed on NPs surface, PA/CL display very high affinity for Ab 1e42 fibrils (22e60 nM), likely because of the occurrence of multivalent interactions which markedly decrease the dissociation of PA/CL NPs from Ab. Noteworthy, PA/CL NPs did not bind to bovine serum albumin. The PA/CL NPs described in this work are endowed with the highest affinity for Ab so far reported. These characteristics make our NPs a very promising vector for the targeted delivery of potential new diagnostic and therapeutic molecules to be tested in appropriate animal models. Ó 2010 Elsevier Ltd. All rights reserved. 1. Introduction Alzheimer’s disease (AD) is the most common neurodegenera- tive disorder in the elderly. AD causes continuous deterioration of higher nervous functions, due to progressive and irreversible degeneration of the limbic and other association cortices, leading to the total loss of autonomy and eventually to death. In the EU, about 5 million people have dementia, with AD accounting for over 3 million, and double of this figure is predicted by 2040 in Western Europe and treble in Eastern Europe. One in 20 people over 65, and one in five over 85, have AD: therefore, AD is considered as a major public health problem due to ageing of the population [1]. Although substantial progress has been made in the understanding of the molecular mechanisms underlying AD, there remains an urgent need to identify effective therapies and early detection strategies, in order to avert a financially overwhelming public health problem. One of the hallmarks of AD is the formation of anomalous amounts of b-amyloid (Ab) a 40e42 residues-long peptide produced by abnormal proteolytic cleavage of the Amyloid Precursor Protein (APP) [2,3].Ab peptide is released from cells in a soluble form, and progressively undergoes aggregation forming oligomers, multimers and fibrils, and ending with deposition of extracellular plaques, one of the histopathological hallmarks of AD, detectable in diseased brain [4,5]. The predominant and initial species deposited in the brain parenchyma is Ab 1e42 [6]. Oligomers have been indicated as the most toxic Ab species [7,8], likely appearing before plaque deposition in an early stage of AD pathology. However, proto- fibrillar and fibrillar aggregates of the peptide were also shown to be toxic [9e11]. Therefore, targeting of cerebral Ab 1e42 in all its aggregation forms has been suggested for therapeutic and/or diagnostic purposes of the disease [3,12e14]. Moreover, it has been * Corresponding author. Tel.: þ39 0264488228; fax: þ39 0264488251. E-mail address: [email protected] (F. Re). 1 These Authors contributed equally to the present investigation. Contents lists available at ScienceDirect Biomaterials journal homepage: www.elsevier.com/locate/biomaterials 0142-9612/$ e see front matter Ó 2010 Elsevier Ltd. All rights reserved. doi:10.1016/j.biomaterials.2010.04.044 Biomaterials 31 (2010) 6519e6529
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Lipid-based nanoparticles with high binding affinity for amyloid-β 1–42 peptide

lable at ScienceDirect
Biomaterials 31 (2010) 6519e6529
Contents lists avai
Biomaterials
journal homepage: www.elsevier .com/locate/biomater ia ls
Lipid-based nanoparticles with high binding affinity for amyloid-b1e42 peptide
Marco Gobbi a,1, Francesca Re b,*,1, Mara Canovi a, Marten Beeg a, Maria Gregori b, Silvia Sesana b,Sandro Sonnino c, Doriano Brogioli b, Claudia Musicanti d, Paolo Gasco d, Mario Salmona a,Massimo E. Masserini b
aDepartment of Biochemistry and Molecular Pharmacology, Mario Negri Institute for Pharmacological Research, Milano, ItalybDepartment of Experimental Medicine, University of Milano Bicocca, 20052 Monza, ItalycCenter of Excellence on Neurodegenerative Diseases, University of Milan, 20090 Segrate, ItalydNanovector S.r.l., 10144 Torino, Italy
a r t i c l e i n f o
Article history:Received 3 March 2010Accepted 21 April 2010Available online 4 June 2010
Keywords:NanoparticlesDrug deliveryAffinityLipidLiposomeAb-peptide
* Corresponding author. Tel.: þ39 0264488228; faxE-mail address: [email protected] (F. Re).
1 These Authors contributed equally to the present
0142-9612/$ e see front matter � 2010 Elsevier Ltd.doi:10.1016/j.biomaterials.2010.04.044
a b s t r a c t
The neurotoxic beta-amyloid peptide (Ab), formed in anomalous amounts in Alzheimer’s disease (AD), isreleased as monomer and then undergoes aggregation forming oligomers, fibrils and plaques in diseasedbrains. Ab aggregates are considered as possible targets for therapy and/or diagnosis of AD. Sincenanoparticles (NPs) are promising vehicles for imaging probes and therapeutic agents, we realized andcharacterized two types of NPs (liposomes and solid lipid nanoparticles, 145 and 76 nm average size,respectively) functionalized to target Ab1e42 with high affinity. Preliminary immunostaining studiesidentified anionic phospholipids [phosphatidic acid (PA) and cardiolipin (CL)] as suitable Ab1e42 ligands.PA/CL-functionalized, but not plain, NPs interacted with Ab1e42 aggregates as indicated by ultracentri-fugation experiments, in which binding reaction occurred in solution, and by Surface Plasmon Resonance(SPR) experiments, in which NPs flowed onto immobilized Ab1e42. All these experiments were carriedout in buffered saline. SPR studies indicated that, when exposed on NPs surface, PA/CL display very highaffinity for Ab1e42 fibrils (22e60 nM), likely because of the occurrence of multivalent interactions whichmarkedly decrease the dissociation of PA/CL NPs from Ab. Noteworthy, PA/CL NPs did not bind to bovineserum albumin. The PA/CL NPs described in this work are endowed with the highest affinity for Ab so farreported. These characteristics make our NPs a very promising vector for the targeted delivery ofpotential new diagnostic and therapeutic molecules to be tested in appropriate animal models.
� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
Alzheimer’s disease (AD) is the most common neurodegenera-tive disorder in the elderly. AD causes continuous deterioration ofhigher nervous functions, due to progressive and irreversibledegeneration of the limbic and other association cortices, leading tothe total loss of autonomy and eventually to death. In the EU, about5 million people have dementia, with AD accounting for over 3million, and double of this figure is predicted by 2040 in WesternEurope and treble in Eastern Europe. One in 20 people over 65, andone in five over 85, have AD: therefore, AD is considered as a majorpublic health problem due to ageing of the population [1]. Althoughsubstantial progress has been made in the understanding of the
: þ39 0264488251.
investigation.
All rights reserved.
molecular mechanisms underlying AD, there remains an urgentneed to identify effective therapies and early detection strategies, inorder to avert a financially overwhelming public health problem.One of the hallmarks of AD is the formation of anomalous amountsof b-amyloid (Ab) a 40e42 residues-long peptide produced byabnormal proteolytic cleavage of the Amyloid Precursor Protein(APP) [2,3]. Ab peptide is released from cells in a soluble form, andprogressively undergoes aggregation forming oligomers, multimersand fibrils, and ending with deposition of extracellular plaques, oneof the histopathological hallmarks of AD, detectable in diseasedbrain [4,5]. The predominant and initial species deposited in thebrain parenchyma is Ab1e42 [6]. Oligomers have been indicated asthe most toxic Ab species [7,8], likely appearing before plaquedeposition in an early stage of AD pathology. However, proto-fibrillar and fibrillar aggregates of the peptide were also shown tobe toxic [9e11]. Therefore, targeting of cerebral Ab1e42 in all itsaggregation forms has been suggested for therapeutic and/ordiagnostic purposes of the disease [3,12e14]. Moreover, it has been

M. Gobbi et al. / Biomaterials 31 (2010) 6519e65296520
reported that brain and blood Ab are in equilibrium, through thebloodebrainebarrier (BBB), and that peripheral sequestration of Abmay shift this equilibrium towards the blood, eventually drawingout the excess from the brain (“sink” effect) [15]. Therefore, for thetherapy of AD, also blood Ab could be worth targeting.
Nanoparticles (NPs) are an attractive mean for these tasks, beingsuitable vehicles for imaging probes and therapeutic agents and forthe possibility to functionalize their surface with target-specificligands. Moreover, multivalent interactions may strikingly increasethe affinity of functionalized NPs for their targets [16e18].
Within the present investigation we describe the preparationand characterization of NPs functionalized to target with highaffinity Ab1e42 peptide in all its aggregation forms. In particular, weused lipid based nanoparticles liposomes or solid lipid nano-particles (SLN), and focused our attention on amphipatic lipids ascandidate specific ligands for Ab1e42. In fact, the involvement ofmembrane lipids in AD has been extensively studied, and a numberof investigations reported their ability to interact with the peptide[19e23]. However, the majority of these studies was aimed toassess the influence of lipids upon Ab1e42 aggregation paradigm[24] whereas, to the best of our knowledge, the information aboutthe specificity of binding are circumscribed to a few molecules,among which gangliosides display the highest affinity [25e27].
2. Materials and methods
2.1. Materials
Monosialogangliosides GM1, GM2 and GM3; disialogangliosides GD1a, GD1band GD3; trisialoganglioside GT1b from bovine brain; sphingomyelin from bovinebrain (Sm); cholesterol (Chol); dimyristoylphosphatidic acid (PA); diphosphatidyl-glycerol (PG); cardiolipin (CL); 1-palmitoyl-oleoyl-phosphatidylcholine (PC); phos-phatidylethanolamine (PE)and Sephadex G75 were purchased from SigmaeAldrich(Milano, Italy). Phospholipon 90G (purified soy lecithin with a phosphatidylcholinecontent of at least 94% as by supplier) was supplied from Phospholipid (Cologne,Germany), distearoylphosphatidic acid was purchased from NOF (Tokyo, Japan).Stearic acid was purchased from Merck (Darmstad, Germany). Sodium taurocholatewas purchased from PCA (Basaluzzo, Italy). [3H]Dipalmitoyl-phosphatidylcholine,ECL reagents and Sepharose CL-4B were from Amersham (Castle Hill, NSW,Australia). PVDF membrane was from Millipore. Recombinant human Ab1e42, HFIPand DMSO were from SigmaeAldrich (Milano, Italy); in addition, also Ab1e42 syn-thetized in our labs as described below was utilized. Mouse monoclonal Ab1e42antibody mAb 6E10 was purchased from Signet (Dedham, MA). All other chemicalswere reagent grade. Ultrapure and deionized water were obtained from Direct-Q5system (Millipore, Italy).
2.2. Preparation of Ab1e42 in different aggregation forms
2.2.1. MonomersAb1e42 monomeric preparations were obtained from the commercial peptide or
from the in-house produced peptide as previously described [28,29].The commercial peptide was dissolved in HFIP at 1 mg/mL concentration to
monomerize pre-existing aggregates; HFIP was then allowed to evaporate and theresulting peptide filmwas stored at�20 �C. The peptidewas resuspended before usein DMSO at a concentration of 5 mM and bath sonicated for 10 min to obtain themonomeric preparation [28]. Alternatively, the monomeric preparation of in-houseproduced peptide was obtained from the depsi-peptide by a “switching” procedure,as previously described [29,30]. The depsi-peptide Ab1e42 is highly soluble and it hasa much lower propensity to aggregate. The presence of monomers in these prepa-rations has been previously confirmed and characterized and circular dicroismstudies did not show the presence of b-sheets structures [30,31]. The Ab1e42 peptideobtained with this procedure is here referred to as “monomers”.
2.2.2. OligomersTo obtain Ab1e42 oligomeric preparations, the monomeric peptide solutions
(both the commercial peptide and the in-house produced peptide) were diluted to100 mM in 50 mM phosphate buffer, 150 mM NaCl, pH 7.4, and incubated for 24 h at4 �C [32]. The presence of oligomers in these preparations has been previouslyconfirmed and characterized [30,31]. The oligomeric preparations of Ab1e42 peptideobtained with this procedure is here referred to as “oligomers”.
2.2.3. FibrilsTo obtain Ab1e42 fibrillar preparations, the monomeric peptide solutions (both
the commercial peptide and the in-house produced peptide) were diluted with
water to 100 mM, acidified to pH 2.0 with HCl and left for 24 h at 37 �C [27,28]. Kineticstudies with circular dicroism and thioflavin-T clearly indicated that these condi-tions enable to reach themaximal level of b-sheets structures, whereas AFM directlyconfirmed the presence of fibrillar species [30,31]. The fibrillar preparations ofAb1e42 peptide obtained with this procedure are therefore referred here to as“fibrils”.
2.3. Screening of Ab1e42/lipids interaction by TLC-immunostaining
Preliminary screening of Ab1e42/lipids interaction was carried out following theprocedure utilized for Cholera toxin B subunit and gangliosides [33] with smallmodifications. Briefly, equal amounts (1 nmole or 0.5 nmoles) of different lipids(gangliosides, Sm, PC, PE, Chol, PG, PA and CL) were applied on a TLC plate anddeveloped with a chloroform/methanol/CaCl2 0.2% (60:35:8 v/v) solvent. The TLCplate was incubated with polyisobutylmethacrylate in chloroform for 3 times. Thedried plate was incubated in the blocking solution [0.1 M Tris, 0.14 M NaCl, 1% bovineserum albimun (BSA)] at RT for 30 min. After washing with phosphate bufferedsaline (PBS), the platewas incubatedwithmonomeric Ab1e42 (2.5 mg/mL in 1% BSA inPBS) for 90 min at RT. After peptide incubation, the plate was incubated for 1 h at RTwith the mAb 6E10 diluted in 1% BSA in PBS 1� (1:1000) and then for 45 min at RTwith HRP-conjugated IgG anti-mouse in the same buffer (1:20000), followed by ECLdetection. Spots were digitally semi-quantitatively estimated by a Kodak Imagestation 2000R software. Parallel, a TLC plate was also prepared in which the lipids,after the chromatographic run, were visualized by iodine vapor. The purpose of thisplate was to compare the Rf of bands visualized by immunoblotting with thosevisualized by chemical staining of standard lipids.
2.4. Nanoparticles preparation
2.4.1. LiposomesLiposomes were composed of a matrix of Sm/Chol (1:1 molar ratio) mixed or not
with 5 or 20molar % of one of the following lipids: gangliosides, Sm, PC, PE, Chol, PG,PA or CL. Lipids were resuspended in chloroform/methanol (2:1, v:v) and driedunder gentle stream of nitrogen followed by a vacuum pump for 3 h to remove tracesof organic solvent. The resulting lipid film was rehydrated in 10 mM Tris, 150 mM
NaCl, 1 mM EDTA, pH 7.4, vortexed and then extruded 10 times at 40 �C througha stack of two polycarbonate filters (100-nm pore size diameter, Millipore Corp.,Bedford, MA) under 20 bar nitrogen pressure with an extruder (Lipex Bio-membranes, Vancouver, Canada). Liposomes were separated from possible unin-corporated material by passage through a Sepharose CL-4B [27]. Lipid recovery afterextrusion was assessed by assaying the individual components: phospholipidrecovery was determined by phosphorous assay using the method of Bartlett [34];glycolipid content was determined as lipid-bound sialic acid [35], Cholesterolcontent was determined as described in Ref. [36]. In the case of mixtures containingdifferent phospholipids, the recovery of the single components after extrusion wasassessed as follows: phospholipids were separated by HPTLC using the solventsystem chloroform/methanol/acetic acid/water, 60:45:4:2 (v/v/v/v). The bandscorresponding to each phospholipid were visualized with iodine, scraped off fromthe plate and submitted to phosphorous assay [34]. Identification of lipids afterseparation was assessed by co-migration with standard lipids loaded on the sameplate.
The presence of an aqueous core, indicative of the ability to trap hydrophilicmolecules, was established by using calcein using the procedure described inRef. [37], with small modifications. For this purpose, the rehydration of the lipid filmwas carried out with buffer containing 60mM calcein. To remove the unencapsulatedcalcein, the liposome suspension was passed through a Sephadex G75 column(25x1 cm) at 4 �C. Liposomes elution was assessed by DLS (Dynamic Light Scat-tering) and entrapment of calcein was assessed by dequenching of fluorescence(which is self-quenched at the high calcein concentrations inside liposomes) afterthe addition of Triton X-100.
In some instances, for studies of binding performed with ultracentrifugation,tritiated phosphatidylcholine was added as a tracer to follow lipid distribution byradioactivity counting.
2.4.2. Solid lipid nanoparticles (SLN)SLN were prepared by oil-in-water (O/W) warm microemulsion method [38]
using stearic acid (0.35 mM) as internal phase, phospholipon 90G as surfactant(0.070 mM), sodium taurocholate as cosurfactant (0.037 mM) and ultrapure water asexternal phase (55.55mM). Functionalized SLNwere prepared substituting 5%mol ofPhospholipon 90G with distearoylphosphatidic acid (PA). Stearic acid and phos-pholipon 90G were heated together at 70 �C then sodium taurocholate dissolved inultrapure water, and heated at the same temperature, was added to obtain anoptically clear system. The O/Wmicroemulsionwas dispersed 1:10 in cold ultrapurewatermaintained at 2e3 �C undermechanical stirring (700e900 rpm); the obtainedSLN aqueous dispersionwas washed three times by tangential filtration on Vivaflow50 module (Sartorius Stedim Biotech, Germany) with a 100,000 MWCO RCmembrane.
For the preparation of functionalized SLN, 20% PA was introduced in previousformulation by substituting the same percentage of Phospholipon 90G. As

M. Gobbi et al. / Biomaterials 31 (2010) 6519e6529 6521
amphiphilic molecule, PA, is going to locate at interphase of microemulsionwith thehydrophilic moiety (phosphate group) towards the aqueous phase and the lipophilicmoiety (acyl chains) towards the oil phase, allowing the hydrophilic portion nega-tively charged to remains exposed on the surface of SLN after quenching of themicroemulsion in cold water.
2.5. Characterization of nanoparticles
The size, polydispersity and z-potential of the liposome and SLN were deter-mined using a ZetaPlus particle sizer and z-potential analyzer (Brookhaven Instru-ments Corporation, Holtsville, NY, U.S.A.). The size and polydispersity measurementswere performed at 25 �C. Liposomes, prepared in 10 mM Tris, 150 mM NaCl, 1 mM
EDTA, pH 7.4, were diluted at 0.25 mM total lipid concentration. SLN samples werediluted 1:100 with deionized water. The particle size was assessed by DLS witha 652 nm laser beam. Particle size and polydispersity index were obtained from theintensity autocorrelation function of the light scattered at a fixed angle of 90� . Thecorrelation function was analyzed by means of a two cumulant expansion. The z-potential was measured at 25 �C. Each measurement was performed on freshlyprepared liposomes samples diluted 1:25 with deionized water. The liposomemeasurements have been performed with ZetaPALS device, which is basically aninterferometic doppler velocimetry. The reported data are the mean of at least fivedifferent measurements. The SLN z-potential was measured by laser doppler elec-trophoresis using a Zetasizer 3000HSA (Malvern Instruments, UK) with a capillarycell. Each measurement was performed under an electrical field of 29.7 V/cm.Standard deviations were calculated from at least threemeasurements. Stability wasmeasured in buffer by following size and polydispersity index by DLS for 48 h.
2.6. Binding of liposomes to Ab1e42, investigated by Surface Plasmon Resonance(SPR)
For these studies we used the Proteon XPR36 (Biorad) apparatus, which has sixparallel flow channels that can be used to uniformly immobilize strips of six“ligands” on the sensor surface. Ab1e42 monomers, oligomers or fibrils wereimmobilized in parallel-flow channels of a GLC sensor chip (Biorad) using amine-coupling chemistry. Briefly, after surface activation the peptide solutions (10 mM inacetate buffer pH 4.0) were injected for 5 min at a flow rate of 30 mL/min, and theremaining activated groups were blocked with ethanolamine, pH 8.0. The finalimmobilization levels were similar, about 2500 Resonance Units (1 RU ¼ 1 pgprotein/mm2). Before performing experiments with liposomes, we previouslychecked that all the Ab species immobilizedwere bindingwith high affinity the anti-Ab antibody 6E10, and that only fibrils were binding the b-sheet specific ligandCongo-Red. In some cases, bovine serum albuminwas immobilized in a parallel flowchannel, as a reference protein. Another “reference” surface was always prepared inparallel using the same immobilization procedure but without addition of thepeptide (empty surface). The fluidic system of Proteon XPR36 can automaticallyrotate 90� so that up to six different analytes (e.g. different nanoparticles prepations,or different concentrations of the same nanoparticle) could be injected simulta-neously over all the different immobilized molecules [39].
2.7. Binding of liposomes to Ab1e42, investigated by ultracentrifugation ona discontinuous sucrose density gradient
0.5 mM of Ab1e42 monomers, oligomers or fibrils were incubated for differenttimes with liposomes (incorporating or not PA, CL or GM1) at different concentra-tions (2.5 mM total lipids for monomers and oligomers; 2.5 mM and 0.5 mM total lipidsfor fibrils), containing a tritiated lipid tracer (less than 0.0001% of total lipids), ina final volume of 450 mL of 10mM Tris,150mMNaCl,1 mM EDTA pH 7.4 at 37 �C, undercontinuous agitation. After incubation the peptide bound to liposomes was sepa-rated from free peptide by flotation in a discontinuous sucrose density gradientperformed as follows. Incubation mixtures of Ab1e42 with liposomes, were mixedwith 1350 mL 80% sucrose in 10 mM Tris, 150 mM NaCl, 1 mM EDTA, pH 7.4, to obtaina final 60% sucrose concentration. On top of this suspension, 1350 mL of 50% sucrosein the same buffer and 1350 mL of sucrose-free buffer were layered in the order. Thesamples were centrifuged in a Beckman MLS 50 rotor at 140,000 � g for 2 h inpolycarbonate tubes; 10 fractions of 450 mL each were collected from the top of thegradient and assayed for lipid and peptide content [40,41]. The distribution of lipidsalong the gradient was followed by phosphorous assay orby counting the liposome-associated radioactivity by liquid scintillation. The distribution of the peptide wasfollowed by dot-blot procedure: briefly, 5 ml of each gradient fraction were spottedon a PVDFmembrane (pre-wet in methanol), washed for 5 min inwater, followed byTBS and left to air dry. Membranewas incubated for 90min at RT with themAb 6E10diluted in non-fat milk in TBS 1� (1:1000) and then for 2 h at RT with HRP-conju-gated IgG anti-mouse in the same buffer (1:20000), followed by ECL detection.Chemiluminescent spots were digitally semi-quantitatively estimated by a KodakImage station 2000R using Kodak Molecular Imaging Software Version 4.0.
Fractions of the gradient carrying both lipids and peptide (usually fractions 1e5)were considered as “liposome-bound peptide”, and fractions containing onlypeptide (usually fractions 6e10) were considered as “unbound peptide”. The
proportion of peptide bound was expressed as the % ratio between the amount ofpeptide in the fractions 1e5 over the total peptide amount.
3. Results
3.1. Screening of Ab1e42/lipids interaction by TLC-immunostaining
To collect some preliminary information on the interaction ofAb1e42 with lipids, we utilized a TLC-immunostaining technique.Lipids under testing were separated by TLC and the plate wasincubated with monomeric Ab1e42, followed by anti-Ab antibodiesand ECL detection. When 0.5 nmol of each lipid were spotted on theplate, chemoluminescence was detected in correspondence of allthe gangliosides, PA and CL, and in correspondence with ganglio-sides and PA using 0.1 nmol of each lipid (Fig. 1). No apparentassociation of peptide was detectable with PC, Sm, PE, Chol and PG,under these experimental conditions.
3.2. Characterization of NPs
3.2.1. LiposomesLiposomes were prepared by extrusion procedure. After extru-
sion, either the total recovery of lipids or the recovery of each lipidcomponent of the mixture were assessed. The total lipid recoverywas about 90% for all the samples. The different lipid componentsof the mixtures were recovered with equal efficiency after theextrusion procedure and always reflected the proportion in thestarting mixture. The samples used for binding experiments wereassayed for lipid content before the experiments. The final prepa-rations of liposomes were monodispersed (Table 1). z-Potentialdistributions of liposomal preparations were also measured and arereported in Table 1. Phospholipid composition predominantlyinfluenced surface charge of liposomes, and, as expected,z-potential decreased with increasing concentrations of anionicphospholipids.
As a further characterization of themost relevant liposomes (seefollowing results), we investigated the stability of liposomes con-taining PA or CL, following their physical features by DLS uponincubation in phosphate buffered saline for up to 48 h. Sizeremained constant, within the experimental error (Table 2), Inadditional experiments, the liposomes were prepared in the pres-ence of calcein and submitted to column chromatography. Calceinfluorescence was detected also in the fractions coinciding withliposomes and increased after Triton treatment (data not shown),indicating its entrapment.
3.2.2. SLNz-potential of functionalized SLN (Table 1) were more negative
after substitution with PA of surfactant Phospholipon 90G, sug-gesting increasing percentage of incorporation of the anionicmoiety of PA on the surface of SLN. Formulations with PA showedhigher average diameter of SLN dispersion.
3.3. Binding of liposomes to Ab1e42, investigated by SurfacePlasmon Resonance (SPR)
In the first SPR experimental session, we flowed liposomes ofdifferent compositions over an empty sensor surface, used asreference (Fig. 2A) and, in parallel over a sensor surface coated withAb1e42 fibrils (Fig. 2B). No or negligible binding was detected whenflowing liposomes composed of Sm/Chol alone or embedding 20%PE, 20% PC or 5% PG, even at the highest concentration tested (1 mM
total lipids), whereas some binding was observed with liposomescontaining 5% GM1, confirming previous results [27].
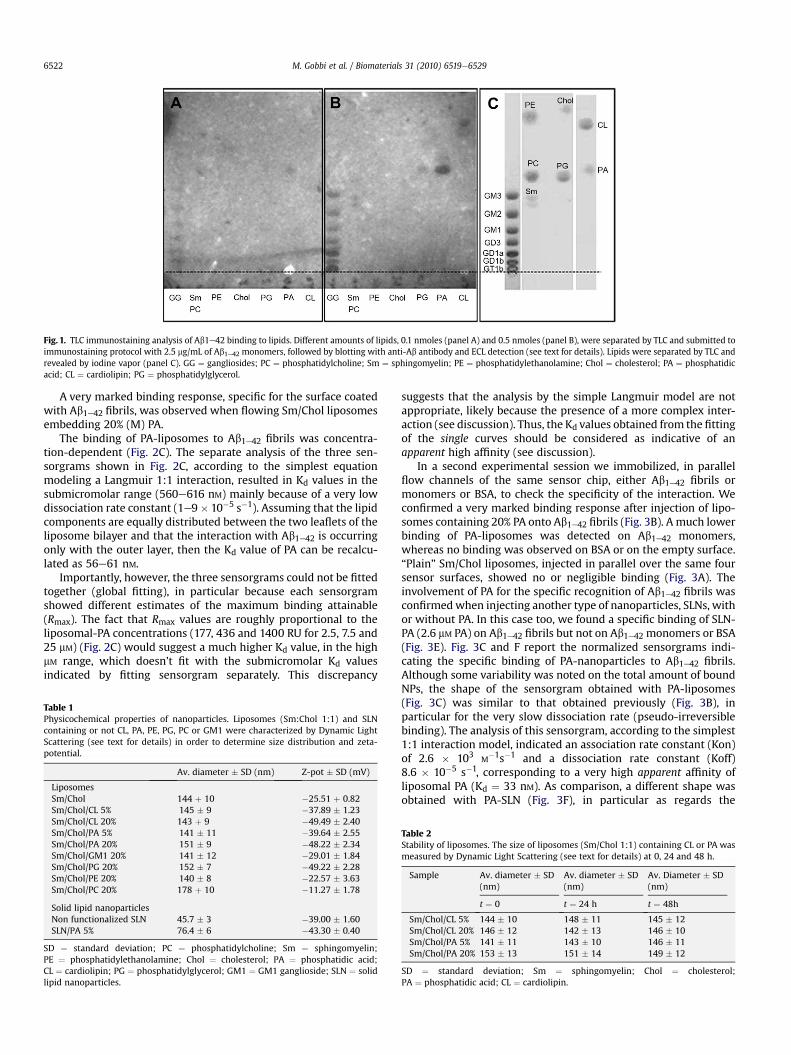
Fig. 1. TLC immunostaining analysis of Ab1e42 binding to lipids. Different amounts of lipids, 0.1 nmoles (panel A) and 0.5 nmoles (panel B), were separated by TLC and submitted toimmunostaining protocol with 2.5 mg/mL of Ab1e42 monomers, followed by blotting with anti-Ab antibody and ECL detection (see text for details). Lipids were separated by TLC andrevealed by iodine vapor (panel C). GG ¼ gangliosides; PC ¼ phosphatidylcholine; Sm ¼ sphingomyelin; PE ¼ phosphatidylethanolamine; Chol ¼ cholesterol; PA ¼ phosphatidicacid; CL ¼ cardiolipin; PG ¼ phosphatidylglycerol.
M. Gobbi et al. / Biomaterials 31 (2010) 6519e65296522
A very marked binding response, specific for the surface coatedwith Ab1e42 fibrils, was observed when flowing Sm/Chol liposomesembedding 20% (M) PA.
The binding of PA-liposomes to Ab1e42 fibrils was concentra-tion-dependent (Fig. 2C). The separate analysis of the three sen-sorgrams shown in Fig. 2C, according to the simplest equationmodeling a Langmuir 1:1 interaction, resulted in Kd values in thesubmicromolar range (560e616 nM) mainly because of a very lowdissociation rate constant (1e9� 10�5 s�1). Assuming that the lipidcomponents are equally distributed between the two leaflets of theliposome bilayer and that the interaction with Ab1e42 is occurringonly with the outer layer, then the Kd value of PA can be recalcu-lated as 56e61 nM.
Importantly, however, the three sensorgrams could not be fittedtogether (global fitting), in particular because each sensorgramshowed different estimates of the maximum binding attainable(Rmax). The fact that Rmax values are roughly proportional to theliposomal-PA concentrations (177, 436 and 1400 RU for 2.5, 7.5 and25 mM) (Fig. 2C) would suggest a much higher Kd value, in the highmM range, which doesn’t fit with the submicromolar Kd valuesindicated by fitting sensorgram separately. This discrepancy
Table 1Physicochemical properties of nanoparticles. Liposomes (Sm:Chol 1:1) and SLNcontaining or not CL, PA, PE, PG, PC or GM1 were characterized by Dynamic LightScattering (see text for details) in order to determine size distribution and zeta-potential.
Av. diameter � SD (nm) Z-pot � SD (mV)
LiposomesSm/Chol 144 þ 10 �25.51 þ 0.82Sm/Chol/CL 5% 145 � 9 �37.89 � 1.23Sm/Chol/CL 20% 143 þ 9 �49.49 � 2.40Sm/Chol/PA 5% 141 � 11 �39.64 � 2.55Sm/Chol/PA 20% 151 � 9 �48.22 � 2.34Sm/Chol/GM1 20% 141 � 12 �29.01 � 1.84Sm/Chol/PG 20% 152 � 7 �49.22 � 2.28Sm/Chol/PE 20% 140 � 8 �22.57 � 3.63Sm/Chol/PC 20% 178 þ 10 �11.27 � 1.78
Solid lipid nanoparticlesNon functionalized SLN 45.7 � 3 �39.00 � 1.60SLN/PA 5% 76.4 � 6 �43.30 � 0.40
SD ¼ standard deviation; PC ¼ phosphatidylcholine; Sm ¼ sphingomyelin;PE ¼ phosphatidylethanolamine; Chol ¼ cholesterol; PA ¼ phosphatidic acid;CL ¼ cardiolipin; PG ¼ phosphatidylglycerol; GM1 ¼ GM1 ganglioside; SLN ¼ solidlipid nanoparticles.
suggests that the analysis by the simple Langmuir model are notappropriate, likely because the presence of a more complex inter-action (see discussion). Thus, the Kd values obtained from the fittingof the single curves should be considered as indicative of anapparent high affinity (see discussion).
In a second experimental session we immobilized, in parallelflow channels of the same sensor chip, either Ab1e42 fibrils ormonomers or BSA, to check the specificity of the interaction. Weconfirmed a very marked binding response after injection of lipo-somes containing 20% PA onto Ab1e42 fibrils (Fig. 3B). A much lowerbinding of PA-liposomes was detected on Ab1e42 monomers,whereas no binding was observed on BSA or on the empty surface.“Plain” Sm/Chol liposomes, injected in parallel over the same foursensor surfaces, showed no or negligible binding (Fig. 3A). Theinvolvement of PA for the specific recognition of Ab1e42 fibrils wasconfirmedwhen injecting another type of nanoparticles, SLNs, withor without PA. In this case too, we found a specific binding of SLN-PA (2.6 mM PA) on Ab1e42 fibrils but not on Ab1e42 monomers or BSA(Fig. 3E). Fig. 3C and F report the normalized sensorgrams indi-cating the specific binding of PA-nanoparticles to Ab1e42 fibrils.Although some variability was noted on the total amount of boundNPs, the shape of the sensorgram obtained with PA-liposomes(Fig. 3C) was similar to that obtained previously (Fig. 3B), inparticular for the very slow dissociation rate (pseudo-irreversiblebinding). The analysis of this sensorgram, according to the simplest1:1 interaction model, indicated an association rate constant (Kon)of 2.6 � 103 M
�1s�1 and a dissociation rate constant (Koff)8.6 � 10�5 s�1, corresponding to a very high apparent affinity ofliposomal PA (Kd ¼ 33 nM). As comparison, a different shape wasobtained with PA-SLN (Fig. 3F), in particular as regards the
Table 2Stability of liposomes. The size of liposomes (Sm/Chol 1:1) containing CL or PA wasmeasured by Dynamic Light Scattering (see text for details) at 0, 24 and 48 h.
Sample Av. diameter � SD(nm)
Av. diameter � SD(nm)
Av. Diameter � SD(nm)
t ¼ 0 t ¼ 24 h t ¼ 48h
Sm/Chol/CL 5% 144 � 10 148 � 11 145 � 12Sm/Chol/CL 20% 146 � 12 142 � 13 146 � 10Sm/Chol/PA 5% 141 � 11 143 � 10 146 � 11Sm/Chol/PA 20% 153 � 13 151 � 14 149 � 12
SD ¼ standard deviation; Sm ¼ sphingomyelin; Chol ¼ cholesterol;PA ¼ phosphatidic acid; CL ¼ cardiolipin.
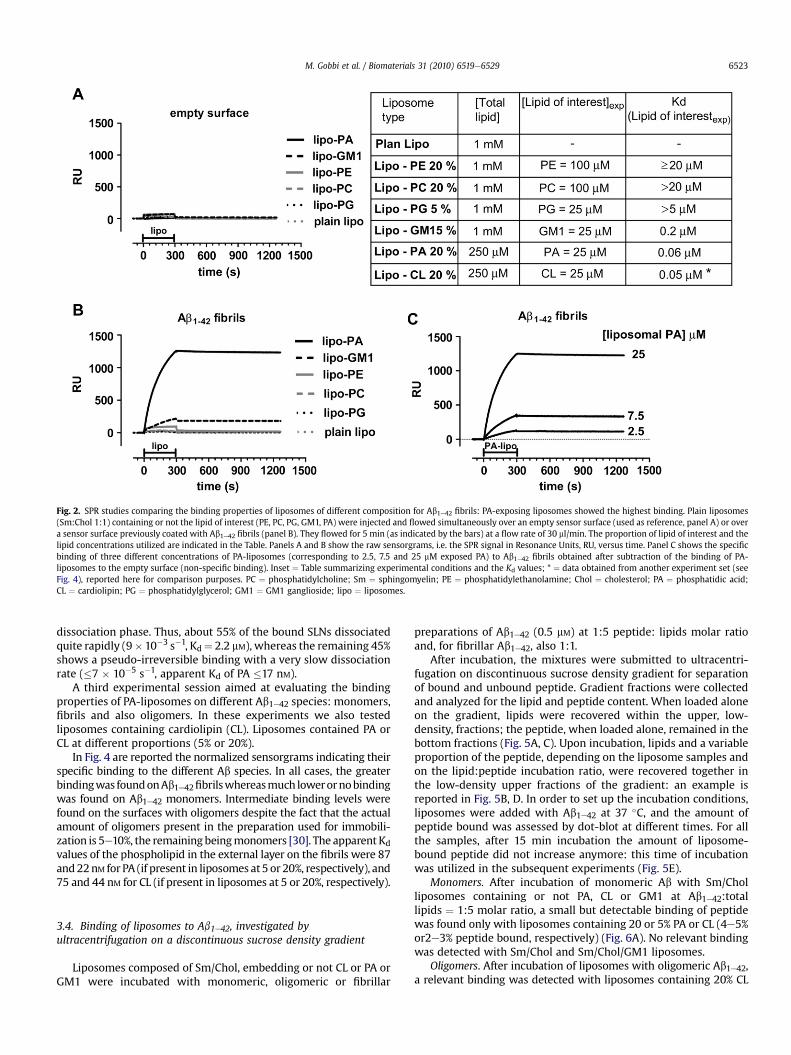
Fig. 2. SPR studies comparing the binding properties of liposomes of different composition for Ab1e42 fibrils: PA-exposing liposomes showed the highest binding. Plain liposomes(Sm:Chol 1:1) containing or not the lipid of interest (PE, PC, PG, GM1, PA) were injected and flowed simultaneously over an empty sensor surface (used as reference, panel A) or overa sensor surface previously coated with Ab1e42 fibrils (panel B). They flowed for 5 min (as indicated by the bars) at a flow rate of 30 ml/min. The proportion of lipid of interest and thelipid concentrations utilized are indicated in the Table. Panels A and B show the raw sensorgrams, i.e. the SPR signal in Resonance Units, RU, versus time. Panel C shows the specificbinding of three different concentrations of PA-liposomes (corresponding to 2.5, 7.5 and 25 mM exposed PA) to Ab1e42 fibrils obtained after subtraction of the binding of PA-liposomes to the empty surface (non-specific binding). Inset ¼ Table summarizing experimental conditions and the Kd values; * ¼ data obtained from another experiment set (seeFig. 4), reported here for comparison purposes. PC ¼ phosphatidylcholine; Sm ¼ sphingomyelin; PE ¼ phosphatidylethanolamine; Chol ¼ cholesterol; PA ¼ phosphatidic acid;CL ¼ cardiolipin; PG ¼ phosphatidylglycerol; GM1 ¼ GM1 ganglioside; lipo ¼ liposomes.
M. Gobbi et al. / Biomaterials 31 (2010) 6519e6529 6523
dissociation phase. Thus, about 55% of the bound SLNs dissociatedquite rapidly (9�10�3 s�1, Kd¼ 2.2 mM), whereas the remaining 45%shows a pseudo-irreversible binding with a very slow dissociationrate (�7 � 10�5 s�1, apparent Kd of PA �17 nM).
A third experimental session aimed at evaluating the bindingproperties of PA-liposomes on different Ab1e42 species: monomers,fibrils and also oligomers. In these experiments we also testedliposomes containing cardiolipin (CL). Liposomes contained PA orCL at different proportions (5% or 20%).
In Fig. 4 are reported the normalized sensorgrams indicating theirspecific binding to the different Ab species. In all cases, the greaterbindingwas foundonAb1e42fibrilswhereasmuch lowerornobindingwas found on Ab1e42 monomers. Intermediate binding levels werefound on the surfaces with oligomers despite the fact that the actualamount of oligomers present in the preparation used for immobili-zation is 5e10%, the remaining beingmonomers [30]. The apparent Kdvalues of the phospholipid in the external layer on the fibrils were 87and22nM for PA (if present in liposomes at5 or 20%, respectively), and75 and 44 nM for CL (if present in liposomes at 5 or 20%, respectively).
3.4. Binding of liposomes to Ab1e42, investigated byultracentrifugation on a discontinuous sucrose density gradient
Liposomes composed of Sm/Chol, embedding or not CL or PA orGM1 were incubated with monomeric, oligomeric or fibrillar
preparations of Ab1e42 (0.5 mM) at 1:5 peptide: lipids molar ratioand, for fibrillar Ab1e42, also 1:1.
After incubation, the mixtures were submitted to ultracentri-fugation on discontinuous sucrose density gradient for separationof bound and unbound peptide. Gradient fractions were collectedand analyzed for the lipid and peptide content. When loaded aloneon the gradient, lipids were recovered within the upper, low-density, fractions; the peptide, when loaded alone, remained in thebottom fractions (Fig. 5A, C). Upon incubation, lipids and a variableproportion of the peptide, depending on the liposome samples andon the lipid:peptide incubation ratio, were recovered together inthe low-density upper fractions of the gradient: an example isreported in Fig. 5B, D. In order to set up the incubation conditions,liposomes were added with Ab1e42 at 37 �C, and the amount ofpeptide bound was assessed by dot-blot at different times. For allthe samples, after 15 min incubation the amount of liposome-bound peptide did not increase anymore: this time of incubationwas utilized in the subsequent experiments (Fig. 5E).
Monomers. After incubation of monomeric Ab with Sm/Cholliposomes containing or not PA, CL or GM1 at Ab1e42:totallipids ¼ 1:5 molar ratio, a small but detectable binding of peptidewas found only with liposomes containing 20 or 5% PA or CL (4e5%or2e3% peptide bound, respectively) (Fig. 6A). No relevant bindingwas detected with Sm/Chol and Sm/Chol/GM1 liposomes.
Oligomers. After incubation of liposomes with oligomeric Ab1e42,a relevant binding was detected with liposomes containing 20% CL
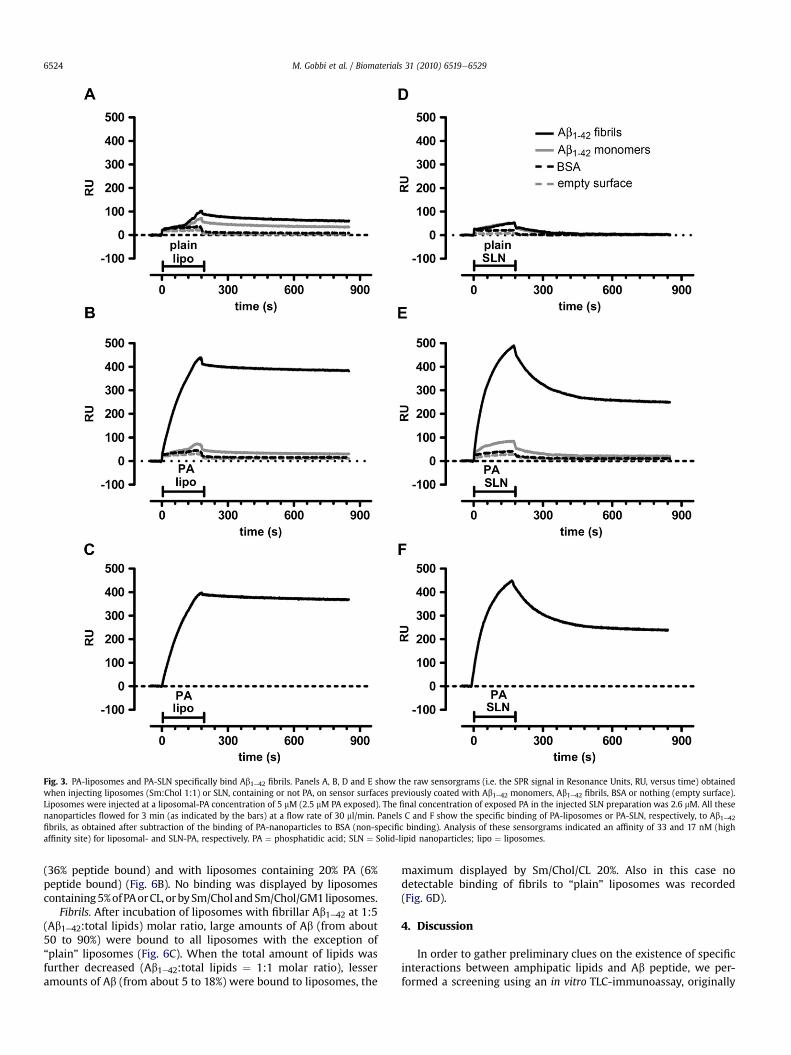
Fig. 3. PA-liposomes and PA-SLN specifically bind Ab1e42 fibrils. Panels A, B, D and E show the raw sensorgrams (i.e. the SPR signal in Resonance Units, RU, versus time) obtainedwhen injecting liposomes (Sm:Chol 1:1) or SLN, containing or not PA, on sensor surfaces previously coated with Ab1e42 monomers, Ab1e42 fibrils, BSA or nothing (empty surface).Liposomes were injected at a liposomal-PA concentration of 5 mM (2.5 mM PA exposed). The final concentration of exposed PA in the injected SLN preparation was 2.6 mM. All thesenanoparticles flowed for 3 min (as indicated by the bars) at a flow rate of 30 ml/min. Panels C and F show the specific binding of PA-liposomes or PA-SLN, respectively, to Ab1e42fibrils, as obtained after subtraction of the binding of PA-nanoparticles to BSA (non-specific binding). Analysis of these sensorgrams indicated an affinity of 33 and 17 nM (highaffinity site) for liposomal- and SLN-PA, respectively. PA ¼ phosphatidic acid; SLN ¼ Solid-lipid nanoparticles; lipo ¼ liposomes.
M. Gobbi et al. / Biomaterials 31 (2010) 6519e65296524
(36% peptide bound) and with liposomes containing 20% PA (6%peptide bound) (Fig. 6B). No binding was displayed by liposomescontaining5%ofPAorCL, orbySm/CholandSm/Chol/GM1liposomes.
Fibrils. After incubation of liposomes with fibrillar Ab1e42 at 1:5(Ab1e42:total lipids) molar ratio, large amounts of Ab (from about50 to 90%) were bound to all liposomes with the exception of“plain” liposomes (Fig. 6C). When the total amount of lipids wasfurther decreased (Ab1e42:total lipids ¼ 1:1 molar ratio), lesseramounts of Ab (from about 5 to 18%) were bound to liposomes, the
maximum displayed by Sm/Chol/CL 20%. Also in this case nodetectable binding of fibrils to “plain” liposomes was recorded(Fig. 6D).
4. Discussion
In order to gather preliminary clues on the existence of specificinteractions between amphipatic lipids and Ab peptide, we per-formed a screening using an in vitro TLC-immunoassay, originally

Fig. 4. CL- and PA-liposomes preferentially bind to Ab1e42 fibrils and oligomers. Liposomes (Sm:Chol 1:1) containing 5% or 20% PA or CL (panel A and B ¼ 5%; C and D ¼ 20%)corresponding to a concentration of exposed PA or CL of 12.5 and 50 mM were flowed for 3 min (as indicated by the bars) simultaneously over a sensor surface previously coatedwith Ab1e42 monomers, oligomers or fibrils. All the panels show the specific binding of CL- and PA-liposomes to Ab1e42 species, obtained after subtraction of the binding of the samenanoparticles to the empty surface (non-specific binding). PA ¼ phosphatidic acid; CL ¼ cardiolipin; lipo ¼ liposomes.
M. Gobbi et al. / Biomaterials 31 (2010) 6519e6529 6525
described for gangliosides and Cholera toxin [33]. The results sug-gested that, among the different lipids tested, PA, CL and ganglio-sides could be the best candidate Ab1e42 ligands. Then, we utilizedamphipatic lipids for the preparation of lipid-based nanoparticles,namely liposomes and SLN, able to target with high affinityAb1e42peptide in all its aggregation forms.
Liposomes and SLN are biocompatible nanoscale materials thathave been extensively studied in the recent years, showing inter-esting features, such as stealth characteristics (ability to avoid thereticulo-endothelial system), biocompatibility and high physicalstability [42], supporting their use for therapy and diagnosis ofhuman diseases [43,44]. Moreover, and particularly useful in ourcase, the insertion of amphipatic lipids, beside those alreadypresent in their formulations, is easily carried out during thepreparation of these NPs.
We utilized liposomes composed by a matrix of Sm and Chol, inequimolar ratio, for the following reasons: i) such liposomesembedding gangliosides have been utilized for assessing theaffinity of these glycolipids for Ab1e42: the Kd in this system provedto be in the micromolar range [25]; ii) Sm/Chol liposomes havebeen repeatedly utilized in vivo for therapeutic purposes, display-ing good circulation times in blood, biocompatibility, resistance tohydrolysis, low ion permeability [45e48]; iii) Sm/Chol bilayers areknown to form raft-like or liquid-ordered domains that are repre-sentative of a native cellular membrane where Ab1e42 accumulates[49]; iv) the presence of cholesterol was shown to strengthen theAb-membrane interaction [50]. It should be pointed out that alsoother lipid matrices have been utilized for pharmacologic purposesand it will be interesting to test them in future experiments [43].
Within this investigation we performed some experiment alsowith SLN. Stearic acid has been chosen as the lipid matrix becauseof SLN of small size are obtained, and because of the few compo-nents required. This kind of formulation, with same excipients butloaded with drugs, showed improvement of drug intracellularaccumulation in in vitro models [51], and showed to improvepharmacokinetic parameters in in vivo models. Same formulationas well has been previously tested for PEG surface functionalisation,obtaining good circulation time and BBB overcoming in animalmodel [52,53].
For determination of the affinity constants of binding betweenNPs and Ab1e42 we utilized the SPR technique, flowing NPs overimmobilized peptide. This is a very suitable approach to charac-terize the binding properties of functionalized nanodevices,providing very useful information on the binding constants and theavidity effects (see below) [17,18]. All experiments were performedin a buffer containing a physiological concentration of salt (150 mM
NaCl) and with a pH of 7.4 in order to mimic the biological fluids.Our SPR data indicated that “plain” Sm/Chol liposomes do not
bind to immobilized Ab fibrils. The highest binding was found withliposomes containing the anionic phospholipids PA or CL, sug-gesting the involvement of electrostatic interactions. However, thevery low binding displayed by liposomes containing PE, PC or PG, inspite of the presence of localized anionic charges on these lipids,calls also for the involvement of hydrophobic interactions. Anintermediate binding was found with liposomes containing GM1ganglioside, with estimated affinities in the micromolar range. Thisfinding confirms previous data reported in literature [25,27], sug-gesting that non-electrostatic interactions are involved also in the
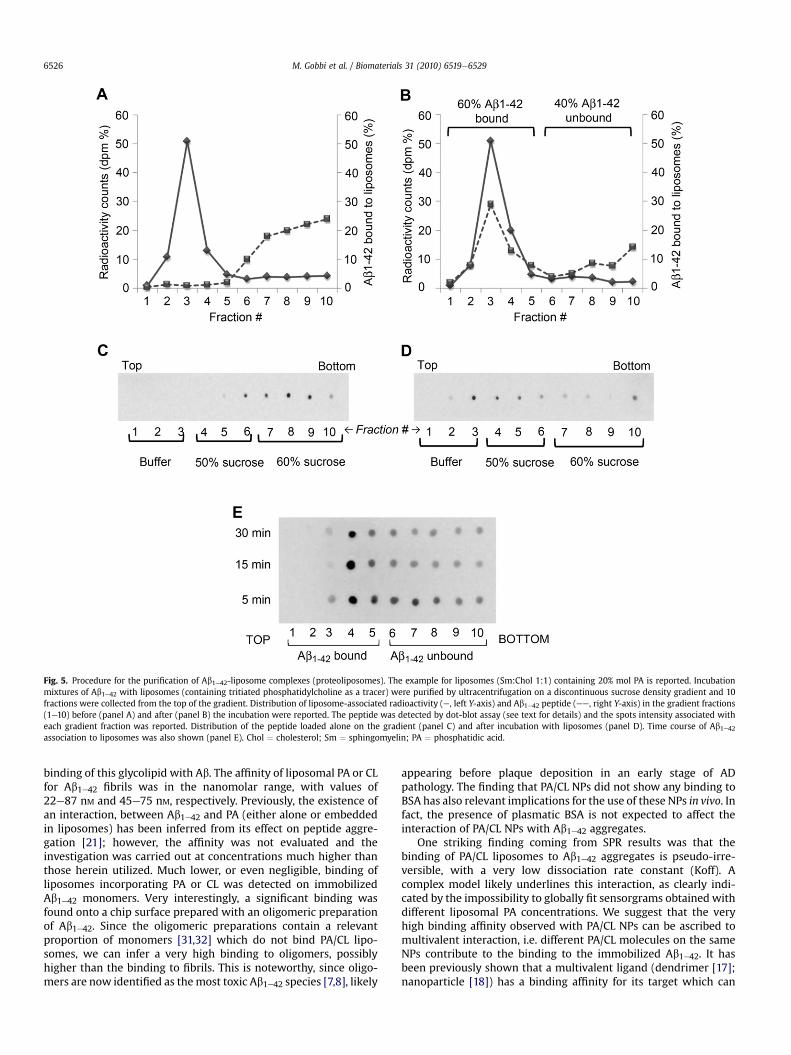
Fig. 5. Procedure for the purification of Ab1e42-liposome complexes (proteoliposomes). The example for liposomes (Sm:Chol 1:1) containing 20% mol PA is reported. Incubationmixtures of Ab1e42 with liposomes (containing tritiated phosphatidylcholine as a tracer) were purified by ultracentrifugation on a discontinuous sucrose density gradient and 10fractions were collected from the top of the gradient. Distribution of liposome-associated radioactivity (e, left Y-axis) and Ab1e42 peptide (ee, right Y-axis) in the gradient fractions(1e10) before (panel A) and after (panel B) the incubation were reported. The peptide was detected by dot-blot assay (see text for details) and the spots intensity associated witheach gradient fraction was reported. Distribution of the peptide loaded alone on the gradient (panel C) and after incubation with liposomes (panel D). Time course of Ab1e42association to liposomes was also shown (panel E). Chol ¼ cholesterol; Sm ¼ sphingomyelin; PA ¼ phosphatidic acid.
M. Gobbi et al. / Biomaterials 31 (2010) 6519e65296526
binding of this glycolipid with Ab. The affinity of liposomal PA or CLfor Ab1e42 fibrils was in the nanomolar range, with values of22e87 nM and 45e75 nM, respectively. Previously, the existence ofan interaction, between Ab1e42 and PA (either alone or embeddedin liposomes) has been inferred from its effect on peptide aggre-gation [21]; however, the affinity was not evaluated and theinvestigation was carried out at concentrations much higher thanthose herein utilized. Much lower, or even negligible, binding ofliposomes incorporating PA or CL was detected on immobilizedAb1e42 monomers. Very interestingly, a significant binding wasfound onto a chip surface prepared with an oligomeric preparationof Ab1e42. Since the oligomeric preparations contain a relevantproportion of monomers [31,32] which do not bind PA/CL lipo-somes, we can infer a very high binding to oligomers, possiblyhigher than the binding to fibrils. This is noteworthy, since oligo-mers are now identified as themost toxic Ab1e42 species [7,8], likely
appearing before plaque deposition in an early stage of ADpathology. The finding that PA/CL NPs did not show any binding toBSA has also relevant implications for the use of these NPs in vivo. Infact, the presence of plasmatic BSA is not expected to affect theinteraction of PA/CL NPs with Ab1e42 aggregates.
One striking finding coming from SPR results was that thebinding of PA/CL liposomes to Ab1e42 aggregates is pseudo-irre-versible, with a very low dissociation rate constant (Koff). Acomplex model likely underlines this interaction, as clearly indi-cated by the impossibility to globally fit sensorgrams obtained withdifferent liposomal PA concentrations. We suggest that the veryhigh binding affinity observed with PA/CL NPs can be ascribed tomultivalent interaction, i.e. different PA/CL molecules on the sameNPs contribute to the binding to the immobilized Ab1e42. It hasbeen previously shown that a multivalent ligand (dendrimer [17];nanoparticle [18]) has a binding affinity for its target which can
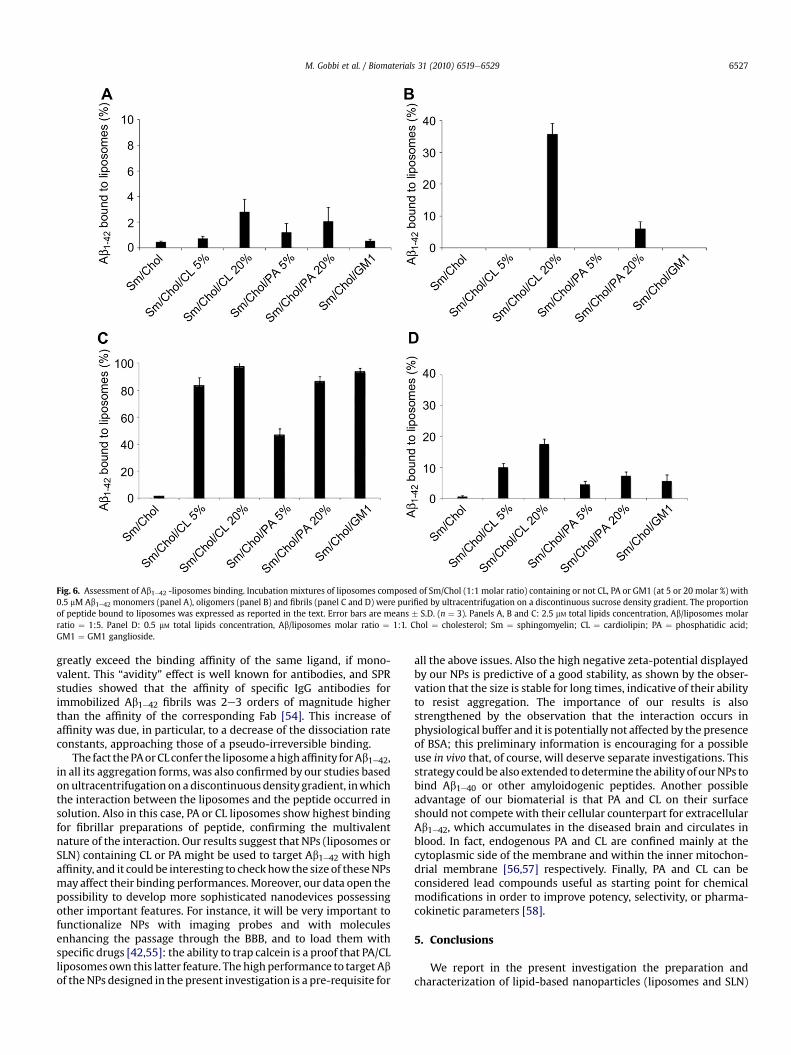
Fig. 6. Assessment of Ab1e42 -liposomes binding. Incubation mixtures of liposomes composed of Sm/Chol (1:1 molar ratio) containing or not CL, PA or GM1 (at 5 or 20 molar %) with0.5 mM Ab1e42 monomers (panel A), oligomers (panel B) and fibrils (panel C and D) were purified by ultracentrifugation on a discontinuous sucrose density gradient. The proportionof peptide bound to liposomes was expressed as reported in the text. Error bars are means � S.D. (n ¼ 3). Panels A, B and C: 2.5 mM total lipids concentration, Ab/liposomes molarratio ¼ 1:5. Panel D: 0.5 mM total lipids concentration, Ab/liposomes molar ratio ¼ 1:1. Chol ¼ cholesterol; Sm ¼ sphingomyelin; CL ¼ cardiolipin; PA ¼ phosphatidic acid;GM1 ¼ GM1 ganglioside.
M. Gobbi et al. / Biomaterials 31 (2010) 6519e6529 6527
greatly exceed the binding affinity of the same ligand, if mono-valent. This “avidity” effect is well known for antibodies, and SPRstudies showed that the affinity of specific IgG antibodies forimmobilized Ab1e42 fibrils was 2e3 orders of magnitude higherthan the affinity of the corresponding Fab [54]. This increase ofaffinity was due, in particular, to a decrease of the dissociation rateconstants, approaching those of a pseudo-irreversible binding.
The fact thePAorCL confer the liposomeahighaffinity forAb1e42,in all its aggregation forms, was also confirmed by our studies basedonultracentrifugation on a discontinuous density gradient, inwhichthe interaction between the liposomes and the peptide occurred insolution. Also in this case, PA or CL liposomes show highest bindingfor fibrillar preparations of peptide, confirming the multivalentnature of the interaction. Our results suggest that NPs (liposomes orSLN) containing CL or PA might be used to target Ab1e42 with highaffinity, and it could be interesting to checkhowthe size of theseNPsmay affect their binding performances. Moreover, our data open thepossibility to develop more sophisticated nanodevices possessingother important features. For instance, it will be very important tofunctionalize NPs with imaging probes and with moleculesenhancing the passage through the BBB, and to load them withspecific drugs [42,55]: the ability to trap calcein is a proof that PA/CLliposomes own this latter feature. The highperformance to target Abof theNPs designed in the present investigation is a pre-requisite for
all the above issues. Also the high negative zeta-potential displayedby our NPs is predictive of a good stability, as shown by the obser-vation that the size is stable for long times, indicative of their abilityto resist aggregation. The importance of our results is alsostrengthened by the observation that the interaction occurs inphysiological buffer and it is potentially not affected by the presenceof BSA; this preliminary information is encouraging for a possibleuse in vivo that, of course, will deserve separate investigations. Thisstrategycouldbe also extended todetermine the ability of ourNPs tobind Ab1e40 or other amyloidogenic peptides. Another possibleadvantage of our biomaterial is that PA and CL on their surfaceshould not compete with their cellular counterpart for extracellularAb1e42, which accumulates in the diseased brain and circulates inblood. In fact, endogenous PA and CL are confined mainly at thecytoplasmic side of the membrane and within the inner mitochon-drial membrane [56,57] respectively. Finally, PA and CL can beconsidered lead compounds useful as starting point for chemicalmodifications in order to improve potency, selectivity, or pharma-cokinetic parameters [58].
5. Conclusions
We report in the present investigation the preparation andcharacterization of lipid-based nanoparticles (liposomes and SLN)

M. Gobbi et al. / Biomaterials 31 (2010) 6519e65296528
targeting with very high affinity aggregated forms of Ab1e42 (fibrilsand oligomers), opening the possibility to develop more sophisti-cated nanodevices to be exploited for diagnostic or therapeuticpurposes. Currently, there are no efficient diagnostic and thera-peutic tools for AD and our results could be important to set upa further experimental investigation in order to functionalizeliposomes with imaging probes and with ligands able to allow theBBB passage.
Acknowledgements
The research leading to these results has received funding fromthe European Community’s Seventh Framework Programme (FP7/2007e2013) under grant agreement no 212043.
References
[1] Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, et al. Globalprevalence of dementia: a Delphi consensus study. Lancet 2005;366(9503):2112e7.
[2] Selkoe DJ. Alzheimer’s disease results from the cerebral accumulation andcytotoxicity of amyloid beta-protein. J Alzheimers Dis 2001;3(1):75e80.
[3] Findeis MA. The role of amyloid beta peptide 42 in Alzheimer’s disease.Pharmacol Ther 2007;116(2):266e86.
[4] Nussbaum RL, Ellis CE. Alzheimer’s disease and Parkinson’s disease. N Engl JMed 2003;348(14):1356e64.
[5] Soto C. Unfolding the role of protein misfolding in neurodegenerativediseases. Nat Rev Neurosci 2003;4(1):49e60.
[6] Mann DM, Iwatsubo T, Ihara Y, Cairns NJ, Lantos PL, Bogdanovic N, et al.Predominant deposition of amyloid-beta 42(43) in plaques in cases of Alz-heimer’s disease and hereditary cerebral hemorrhage associated with muta-tions in the amyloid precursor protein gene. Am J Pathol 1996;148(4):1257e66.
[7] Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessonsfrom the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 2007;8(2):101e12.
[8] Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloidbeta peptide concentration as a predictor of synaptic change in Alzheimer’sdisease. Am J Pathol 1999;155(3):853e62.
[9] Crouch PJ, Harding SM, White AR, Camakaris J, Bush AI, Masters CL. Mecha-nisms of A beta mediated neurodegeneration in Alzheimer’s disease. Int JBiochem Cell Biol 2008;40(2):181e98.
[10] Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem2008;283(44):29639e43.
[11] Cappai R, Barnham KJ. Delineating the mechanism of Alzheimer’s disease Abeta peptide neurotoxicity. Neurochem Res 2008;33(3):526e32.
[12] DeFelice FG, Ferreira ST. Beta-amyloid production, aggregation, clearance astargets for therapy in Alzheimer’s disease. Cell Mol Neurobiol 2002;22(5e6):545e63.
[13] Greenberg SM, Grabowski T, Gurol ME, Skehan ME, Nandigam RN, Becker JA,et al. Detection of isolated cerebrovascular beta-amyloid with Pittsburghcompound B. Ann Neurol 2008;64(5):587e91.
[14] Look GC, Jerecic J, Cherbavaz DB, Pray TR, Breach JC, Crosier WJ, et al.Discovery of ADDLetargeting small molecule drugs for Alzheimer’s disease.Curr Alzheimer Res 2007;4(5):562e7.
[15] Matsuoka Y, Saito M, LaFrancois J, Gaynor K, Olm V, Wang L, et al. Noveltherapeutic approach for the treatment of Alzheimer’s disease by peripheraladministration of agents with an affinity to beta-amyloid. J Neurosci 2003;23(1):29e33.
[16] Montet X, Funovics M, Montet-Abou K, Weissleder R, Josephson L. Multivalenteffects of RGD peptides obtained by nanoparticle display. J Med Chem2006;49(20):6087e93.
[17] Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker Jr JR, Banaszak Holl MM. Thebinding avidity of a nanoparticle-based multivalent targeted drug deliveryplatform. Chem Biol 2007;14(1):107e15.
[18] Tassa C, Duffner JL, Lewis TA, Weissleder R, Schreiber SL, Koehler AN, et al.Binding affinity and kinetic analysis of targeted small molecule-modifiednanoparticles. Bioconjugate Chem 2010;21(1):14e9.
[19] Matsuzaki K. Physicochemical interactions of amyloid beta-peptide with lipidbilayers. Biochim Biophys Acta 2007;1768(8):1935e42.
[20] Verdier Y, Penke B. Binding sites of amyloid beta-peptide in cell plasmamembrane and implications for Alzheimer’s disease. Curr Protein Pept Sci2004;5(1):19e31.
[21] Chauhan A, Ray I, Chauhan VP. Interaction of amyloid beta-protein withanionic phospholipids: possible involvement of Lys28 and C-terminusaliphatic amino acids. Neurochem Res 2000;25(3):423e9.
[22] Cordy JM, Hooper NM, Turner AJ. The involvement of lipid rafts in Alzheimer’sdisease. Mol Membr Biol 2006;23(1):111e22.
[23] Taylor DR, Hooper NM. Role of lipid rafts in the processing of the pathogenicprion and Alzheimer’s amyloid-beta proteins. Semin Cell Dev Biol 2007;18(5):638e48.
[24] Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progressand problems on the road to therapeutics. Science 2002;297(5580):353e6.
[25] Kakio A, Nishimoto S, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Interactions ofamyloid beta-protein with various gangliosides in raft-like membranes:importance of GM1 ganglioside-bound form as an endogenous seed for Alz-heimer amyloid. Biochemistry 2002;41(23):7385e90.
[26] Lin MS, Chiu HM, Fan FJ, Tsai HT, Wang SS, Chang Y, et al. Kinetics andenthalpy measurements of interaction between beta- amyloid and liposomesby surface plasmon resonance and isothermal titration microcalorimetry.Colloids Surf B Biointerfaces 2007;58(2):231e6.
[27] Ariga T, Kobayashi K, Hasegawa A, Kiso M, Ishida H, Miyatake T. Character-ization of high-affinity binding between gangliosides and amyloid beta-protein. Arch Biochem Biophys 2001;388(2):225e30.
[28] Dahlgren KN, Manelli AM, Stine Jr WB, Baker LK, Krafft GA, LaDu MJ. Oligo-meric and fibrillar species of amyloid-beta peptides differentially affectneuronal viability. J Biol Chem 2002;277(35):32046e53.
[29] Taniguchi A, Sohma Y, Hirayama Y, Mukai H, Kimura T, Hayashi Y, et al. “Clickpeptide”: pH-triggered in situ production and aggregation of monomerAbeta1-42. Chembiochem 2009;10(4):710e5.
[30] Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, et al. Syntheticamyloid-b oligomers impair long-term memory independently of cellularprion protein. Proc Natl Acad Sci USA 2010;107(5):2295e300.
[31] Bulbarelli A, Lonati E, Cazzaniga E, Re F, Sesana S, Barisani D, et al. TrkApathway activation induced by amyloid-beta (Abeta). Mol Cell Neurosci2009;40(3):365e73.
[32] Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al.Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent centralnervous system neurotoxins. Proc Natl Acad Sci USA 1998;95(11):6448e53.
[33] Wu GS, Ledeen R. Quantification of gangliotetraose gangliosides with choleratoxin. Anal Biochem 1988;173(2):368e75.
[34] Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem 1959Mar;234(3):466e8.
[35] Svennerholm L. Quantitative estimation of sialic acids. II. A colorimetric resor-cinol-hydrochloric acid method. Biochim Biophys Acta 1957;24(3):604e11.
[36] Palestini P, Botto L, Guzzi F, Calvi C, Ravasi D, Masserini M, et al. Develop-mental changes in the protein composition of sphingolipid- and cholesterol-enriched membrane domains of rat cerebellar granule cells. J Neurosci Res2002;67(6):729e38.
[37] Zhang L, Rozek A, Hancock RE. Interaction of cationic antimicrobial peptideswith model membranes. J Biol Chem 2001;276(38):35714e22.
[38] Gasco MR. Solid lipid microspheres having a narrow size distribution andmethod for producing them. Eur Patent No EP052666; 1993.
[39] Bravman T, Bronner V, Lavie K, Notcovich A, Papalia GA, Myszka DG. Exploring“one-shot” kinetics and small molecule analysis using the ProteOn XPR36array biosensor. Anal Biochem 2006;358(2):281e8.
[40] Re F, Sesana S, Barbiroli A, Bonomi F, Cazzaniga E, Lonati E, et al. Prion proteinstructure is affected by pH-dependent interaction with membranes: a study ina model system. FEBS Lett 2008;582(2):215e20.
[41] Sesana S, Re F, Bulbarelli A, Salerno D, Cazzaniga E, Masserini M. Membranefeatures and activity of GPI-anchored enzymes: alkaline phosphatase recon-stituted in model membranes. Biochemistry 2008;47(19):5433e40.
[42] Roney C, Kulkarni P, Arora V, Antich P, Bonte F, Wu A, et al. Targeted nano-particles for drug delivery through the blood-brain barrier for Alzheimer’sdisease. J Controlled Release 2005;108(2e3):193e214.
[43] Schwendener RA. Liposomes in biology and medicine. Adv Exp Med Biol2007;620:117e28.
[44] Uner M, Yener G. Importance of solid lipid nanoparticles (SLN) in variousadministration routes and future perspectives. Int J Nanomedicine 2007;2(3):289e300.
[45] Webb MS, Harasym TO, Masin D, Bally MB, Mayer LD. Sphingomyelin-cholesterol liposomes significantly enhance the pharmacokinetic and thera-peutic properties of vincristine in murine and human tumour models. Br JCancer 1995;72(4):896e904.
[46] Boehlke L, Winter JN. Sphingomyelin/cholesterol liposomal vincristine: a newformulation for an old drug. Expert Opin Biol Ther 2006;6(4):409e15.
[47] Gelmon KA, Tolcher A, Diab AR, Bally MB, Embree L, Hudon N, et al. Phase Istudy of liposomal vincristine. J Clin Oncol 1999;17(2):697e705.
[48] Thomas DA, Sarris AH, Cortes J, Faderl S, O’Brien S, Giles FJ, et al. Phase II studyof sphingosomal vincristine in patients with recurrent or refractory adultacute lymphocytic leukemia. Cancer 2006;106(1):120e7.
[49] Choucair A, Chakrapani M, Chakravarthy B, Katsaras J, Johnston LJ. Preferentialaccumulation of Abeta(1-42) on gel phase domains of lipid bilayers: an AFMand fluorescence study. Biochim Biophys Acta 2007;1768(1):146e54.
[50] Qiu L, Lewis A, Como J, Vaughn MW, Huang J, Somerharju P, et al. Cholesterolmodulates the interaction of beta-amyloid peptide with lipid bilayers. BiophysJ 2009;96(10):4299e307.
[51] Serpe L, Guido M, Canaparo R, Muntoni E, Cavalli R, Panzanelli P, et al.Intracellular accumulation and cytotoxicity of doxorubicin with differentpharmaceutical formulations in human cancer cell lines. J Nanosci Nano-technol 2006;6(9e10):3062e9.
[52] Podio V, Zara GP, Carazzonet M, Cavalli R, Gasco MR. Biodistribution of stealthand non-stealth solid lipid nanospheres after intravenous administration torats. J Pharm Pharmacol 2000;52(9):1057e63.
[53] Zara GP, Cavalli R, Bargoni A, Fundaro A, Vighetto D, Gasco MR. Intravenousadministration to rabbits of non-stealth and stealth doxorubicin-loaded solid

M. Gobbi et al. / Biomaterials 31 (2010) 6519e6529 6529
lipid nanoparticles at increasing concentrations of stealth agent: pharmaco-kinetics and distribution of doxorubicin in brain and other tissues. J DrugTarget 2002;10(4):327e35.
[54] Gardberg AS, Dice LT, Ou S, Rich RL, Helmbrecht E, Ko J, et al. Molecular basisfor passive immunotherapy of Alzheimer’s disease. Proc Natl Acad Sci USA2007;104(40):15659e64.
[55] Kingsley JD, Dou H, Morehead J, Rabinow B, Gendelman HE, Destache CJ.Nanotechnology: a focus on nanoparticles as a drug delivery system. J Neu-roimmune Pharmacol 2006;1(3):340e50.
[56] Houtkooper RH, Vaz FM. Cardiolipin, the heart of mitochondrial metabolism.Cell Mol Life Sci 2008;65(16):2493e506.
[57] Gascard P, Tran D, Sauvage M, Sulpice JC, Fukami K, Takenawa T, et al.Asymmetric distribution of phosphoinositides and phosphatidic acid in thehuman erythrocyte membrane. Biochim Biophys Acta 1991;1069(1):27e36.
[58] Zhang Z, Zhu M, Tang W. Metabolite identification and profiling in drugdesign: current practice and future directions. Curr Pharm Des 2009;15(19):2220e35.




















