
Correlates of Perceived Pain-Related Restrictions among Women with Fibromyalgia
Upload
ua-birminghamCategory
view
1download
0

OSTEOARTHRITICJOINT PAIN
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

The Novartis Foundation is an international scienti¢c and educationalcharity (UK Registered Charity No. 313574). Known until September 1997as the Ciba Foundation, it was established in 1947 by the CIBA companyof Basle, which merged with Sandoz in 1996, to form Novartis. TheFoundation operates independently in London under English trustlaw. It was formally opened on 22 June 1949.
The Foundation promotes the study and general knowledge ofscience and in particular encourages international co-operation inscienti¢c research. To this end, it organizes internationallyacclaimed meetings (typically eight symposia and allied openmeetings and 15^20 discussion meetings each year) and publisheseight books per year featuring the presented papers and discussionsfrom the symposia. Although primarily an operational rather thana grant-making foundation, it awards bursaries to young scientiststo attend the symposia and afterwards work with one of the otherparticipants.
The Foundation’s headquarters at 41 Portland Place, London W1B 1BN,provide library facilities, open to graduates in science and allied disciplines.Media relations are fostered by regular press conferences and by articlesprepared by the Foundation’s Science Writer in Residence. The Foundationo¡ers accommodation and meeting facilities to visiting scientists and theirsocieties.
Information on all Foundation activities can be found athttp://www.novartisfound.org.uk

OSTEOARTHRITICJOINT PAIN
Novartis Foundation Symposium 260
2004

Copyright &Novartis Foundation 2004Published in 2004 byJohnWiley & Sons Ltd,
The Atrium, Southern Gate,Chichester PO19 8SQ, UK
National 01243 779777International (+44) 1243 779777e-mail (for orders and customer service enquiries): [email protected] our Home Page on http://www.wileyeurope.com
or http://www.wiley.com
All Rights Reserved. No part of this book may be reproduced, stored in a retrievalsystem or transmitted in any form or by any means, electronic, mechanical, photocopying,recording, scanning or otherwise, except under the terms of the Copyright, Designs andPatents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd,90 Tottenham Court Road, LondonW1T 4LP, UK, without the permission in writingof the Publisher. Requests to the Publisher should be addressed to the Permissions Department,JohnWiley & Sons Ltd,The Atrium, Southern Gate, Chichester,West Sussex PO19 8SQ,England, or emailed to [email protected], or faxed to (+44) 1243 770620.
This publication is designed to provide accurate and authoritative information in regard tothe subject matter covered. It is sold on the understanding that the Publisher is not engagedin rendering professional services. If professional advice or other expert assistance isrequired, the services of a competent professional should be sought.
OtherWileyEditorial O⁄ces
JohnWiley & Sons Inc., 111River Street, Hoboken, NJ 07030, USA
Jossey-Bass, 989 Market Street, San Francisco, CA 94103-1741, USA
Wiley-VCH Verlag GmbH, Boschstr. 12, D-69469 Weinheim, Germany
JohnWiley & Sons Australia Ltd, 33 Park Road, Milton, Queensland 4064, Australia
JohnWiley & Sons (Asia) Pte Ltd, 2 Clementi Loop #02-01, Jin Xing Distripark, Singapore129809
JohnWiley & Sons Canada Ltd, 22 Worcester Road, Etobicoke, Ontario, Canada M9W1L1
Wiley also publishes its books in a variety of electronic formats. Some content that appearsin print may not be available in electronic books.
Novartis Foundation Symposium 260ix+292 pages, 40 ¢gures, 13 tables
British Library Cataloguing in PublicationData
A catalogue record for this book is available from the British Library
ISBN 0 470 86761 2
Typeset in 101�2 on 121�2 pt Garamond by DobbieTypesetting Limited,Tavistock, Devon.
Printed and bound in Great Britain byT. J. International Ltd, Padstow, Cornwall.This book is printed on acid-free paper responsibly manufactured from sustainable forestry,in which at least two trees are planted for each one used for paper production.

Contents
Symposium onOsteoarthritic joint pain, held attheNovartis Foundation, London,1^3 July 2003
Editors: Derek J. Chadwick (Organizer) and Jamie Goode
This symposium is based on a proposal by Stuart Bevan and John Rediske
David Felson Chair’s introduction 1
Hans-Georg Schaible Spinal mechanisms contributing to joint pain 4Discussion 22
Blair D. Grubb Activation of sensory neurons in the arthritic joint 28Discussion 36
Kenneth D. Brandt Neuromuscular aspects of osteoarthritis: a perspective 49Discussion 58
Paul Creamer Current perspectives on the clinical presentation of joint pain inhuman OA 64Discussion 74
Walter Herzog, Andrea Clark andDavid Longino Joint mechanics inosteoarthritis 79Discussion 95
General discussion I Developing animal models of RA 100
Gunnar Ordeberg Characterization of joint pain in human OA 105Discussion 115
Bruce L. Kidd, Andrew Photiou and Julia J. Inglis The role of in£ammatorymediators on nociception and pain in arthritis 122Discussion 133
James L.Henry Molecular events of chronic pain: from neuron towhole animal inan animal model of osteoarthritis 139Discussion 145
v

C. S. McCabe, R. C. Haigh, N. G. Shenker, J. Lewis andD. R. BlakePhantoms in rheumatology 154Discussion 174
Peter A. Simkin Bone pain and pressure in osteoarthritic joints 179Discussion 186
Philip G. Conaghan andDavidT. Felson Structural associations of osteoarthritispain: lessons from magnetic resonance imaging 191Discussion 201
Martin Koltzenburg The role of TRP channels in sensory neurons 206Discussion 213
PatrickW. Mantyh and Stephen P. Hunt Mechanisms that generate and maintainbone cancer pain 221Discussion 238
N. G. Shenker, D. R. Blake, C. S. McCabe, R. Haigh and P. I. Mapp Symmetry,T cells and neurogenic arthritis 241Discussion 252
Laurence A. Bradley, Brian C. Kersh, Jennifer J. DeBerry, Georg Deutsch,
Graciela A. Alarco¤ n andDavid A. McLain Lessons from ¢bromyalgia:abnormal pain sensitivity in knee osteoarthritis 258Discussion 270
David Felson Chair’s summing up 277
Index of contributors 280
Subject index 282
vi CONTENTS

Participants
David R. Blake The Royal National Hospital for Rheumatic Diseases, UpperBoroughWalls, in conjunctionwithThe Department of Medical Sciences andThe Department of Pharmacy and Pharmacology, University of Bath, BathBA11RL, UK
Laurence A. Bradley Division of Clinical Immunology and Rheumatology,Universityof Alabama at Birmingham, 805 Faculty O⁄ceTower, 510 20th streetsouth, Birmingham, AL 35294, USA
KennethD. Brandt IndianaUniversity School ofMedicine, IndianaUniversityMultipurpose Arthritis and Musculoskeletal Diseases Center, 1110 WestMichigan Street, Room 545, Indianapolis, IN 46202-5100, USA
Philip G. Conaghan Academic Unit of Musculoskeletal Disease, University ofLeeds&Department ofRheumatology, LeedsGeneral In¢rmary,GreatGeorgeStreet, Leeds LS1 3EX, UK
Paul Creamer Southmead Hospital,Westbury onTrym, Bristol BS10 5NB, UK
Paul Dieppe Department of Social Medicine, University of Bristol, CanyngeHall,Whiteladies Road, Bristol BS8 2PR, UK
Christopher H. Evans Center for Molecular Orthopaedics, Harvard MedicalSchool, 221Longwood Avenue, BL-152, Boston, MA 02115, USA
DavidT. Felson (Chair) BostonUniversity School of Medicine, 715 AlbanyStreet, A203, Boston, MA 02118, USA
Janet K. Fernihough Novartis Institute for Medical Sciences, 5 Gower Place,LondonWC1E 6BN, UK
Alyson Fox Novartis Institute for Medical Sciences, 5 Gower Place, LondonWC1E 6BN, UK
vii

Blair D. Grubb Department of Cell Physiology and Pharmacology, Universityof Leicester, PO Box 138, Leicester LE19HN, UK
James L. Henry Department of Physiology and Pharmacology, University ofWestern Ontario, Medical Sciences Building, M221, London, Ontario,N6A 5C1, Canada
Walter Herzog Human Performance Laboratory, Faculty of Kinesiology,Department of Mechanical Engineering, Faculty of Engineering,The University of Calgary, 2500 University Drive NW B205, Calgary, AB,CanadaT2N1N4
StephenHunt Department of Anatomy and Developmental Biology, AnatomyBuilding, Gower Street, LondonWC1E 6BT, UK
David J. Hunter Clinical Epidemiology Research andTraining Unit, BostonMedical Center,715 Albany Street, RoomA-203, Boston, MA 02118-2526, USA
BruceL.Kidd Bone andJointUnit, Bart’s andTheLondonSchool ofMedicine,Charterhouse Square, London EC1M 6BQ, UK
MartinKoltzenburg Neural PlasticityUnit, Neural Plasticity, Institute of ChildHealth, 30 Guilford Street, LondonWC1N1EH, UK
Klaus E. Kuettner Department of Biochemistry, Rush Medical College, Rush-Presbyterian-St Luke’s Medical Center, 1653 W. Congress Parkway, Chicago,IL 60612, USA
Stefan Lohmander Department of Orthopaedics, University Hospital Lund,22185 Lund, Sweden
AndrewMackenzie Novartis Pharma AG,WSJ-386.10.35, CH-4002, Basel,Switzerland
Marzia Malcangio Chronic Pain Programme, Novartis Institute for MedicalScience, University College London, Gower Street, LondonWC1E 6BT, UK
AnthonyM.Manning Roche Bioscience, 3401HillviewAvenue, PaloAlto, CA94304-1397, USA
viii PARTICIPANTS

Gunnar Ordeberg Division of Orthopaedic Surgery, Karolinska Institutet,Danderyd Hospital, Stockholm, SE-18288, Sweden
David S. Pisetsky DukeUniversityMedical School,151GDurhamVAHospital,509 Fulton Street, Durham, North Carolina, 27705, USA
John Rediske Research, Arthritis and Bone MetabolismTherapeutic Area,Novartis Pharmaceuticals Corporation, 556Morris Avenue, Summit, NJ 07901,USA
Hans-Georg Schaible Institut fˇr Physiologie 1/Neurophysiologie,Teichgraben 8, 07740 Jena, Germany
Hua Shen (Novartis Foundation Bursar) Center of Experimental Rheumatology,andWHO Collaborating Center for Molecular Biology, and Novel TherapeuticStrategies for Rheumatic Diseases, UniversityHospital Zu« rich, Gloriastrasse 23,CH-8091Zu« rich, Switzerland
Peter A. Simkin Department of Medicine, University of Washington, Box356428, Seattle,WA 98195, USA
Wimvan den Berg Center of Rheumatology Research and AdvancedTherapeutics, Nijmegen Center of Molecular Life Sciences, Geert Grooteplein26-28, 6500 HBNijmegen,The Netherlands
Thasia G.Woodworth Arthritis/Bone, Novartis Pharma AG, CH-4002, Basel,Switzerland
KiranYashpal Department of Physiology and Pharmacology, University ofWestern Ontario, Medical Sciences Building, M221, London, Ontario,N6A 5C1, Canada
PARTICIPANTS ix

Chair’s introduction
David Felson
Clinical Epidemiology Research and Training Unit, Boston University School of Medicine,715 Albany Street, A 203, Boston, MA 02118-2526, USA
To begin with, I’d like to mention some results from Lois Verbrugge, who gave agroup of people in Detroit a diary to take home to record their daily healthsymptoms (Verbrugge 1979). In people aged 65 and over she tallied the mostcommon daily health symptom. In women over 65, it was knee trouble; in men itwas backache followed by knee trouble. The most common category of symptomsin older people was musculoskeletal problems. Is this just older people? No. Inwomen aged 45^64 the second most common daily symptom was knee trouble.Thus, pain in knees, often due to osteoarthritis (OA), is a remarkably prevalentproblem.Osteoarthritis is the most common form of arthritis. In elderly subjects the
prevalence of each of hand and hip OA is around 2^3% of the population aged 65and over. Prevalence in this case is de¢ned as symptoms onmost days and evidenceof structural OA as a cause of these symptoms. Knee OA (see below) is even morecommon. In the elderly, OA is a more common cause of lower extremity disabilitythan any other disease. It causes 1 in 8 days of restricted activity in US elders and isthe reason why most people have knee and hip replacements. The burden of thisdisease on society is truly remarkable.I’d also like to mention data on knee OA from a variety of recent studies in the
UK (Peat et al 2001). Of 10 000 people aged 55 and over, about 25% haveradiographic OA, but not necessarily symptoms. Irrespective of radiographs,about 25% of people in this over-55 age group have had an episode of knee painlasting at least 4 weeks. About half of these have radiographic OA. This 12.5% ofthe population would be the group that we would categorize as havingsymptomatic OA.The other thing to note is that arthritis is not going to decrease in prevalence in
western societies. Because of the demographics, it is only going to increase inprevalence. We are getting older, but we are also getting more overweight,which is a major risk factor for OA in weight-bearing joints. Add to this theincreasing number of sports injuries. The estimate is that the current level of 20million a¡ected in the USA will rise to 40 million by 2020.In the past the focus of OA research was on hyaline articular cartilage loss. I am
not going to suggest that cartilage loss is not pivotal; rather that the whole joint is
1
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

a¡ected in OA, with bony changes (sclerosis and osteophytes), cartilage loss, jointcapsule stretching and thickening, modest synovial in£ammation all often presentand weak periarticular muscles. If we were to discover this disorder today, weprobably would not call it OA, but in medical textbooks next to chronic hepaticfailure and chronic renal failure, there would be a chapter on ‘chronic joint failure’.What are the symptoms and signs of OA? In OA, pain is worse on use of the
joint,mild inmorning, severe after immobility. There is loss ofmovement, pain onmovement and restricted range of motion. The pain symptoms of OA will be afocus of this symposium.One of the central rationales for this symposium�and one of the reasons I am
excited about it� is that pain and disease structure do not necessarily go hand inhand. We have one group that has symptomatic knee pain, and only about half ofthem have radiographic OA. Then there is the population with radiographic OA,and a large percentage of them have no symptoms at all. Often people with severestructural disease have no symptoms. What accounts for this discordance betweensymptoms and structure?There is another problem: that of drug development in this challenging
situation. There is an interleukin (IL)1 inhibitor called diacerein that, in theory,would prevent cartilage loss. In a randomized trial of hip OA (Dougados et al2001), compared with placebo, the diacerein group experienced less joint spaceloss which signi¢es less cartilage loss. This suggests that diacerein did indeedprevent cartilage loss. But when the readout is pain improvement, the diacereingroup and placebo group had exactly the same scores. That is, IL1 inhibitionseemed to prevent cartilage loss but had no measureable e¡ect on pain. We have asituation where not only is there an observed discordance between pain andstructural change, but the results of this study suggest that drug developmentmay produce therapies which prevent cartilage loss but have no e¡ects on thesymptoms we are most concerned about.In OA treatments are limited and those that are widely used are dangerous,
expensive and suboptimal. Most treatments being developed focus onpreventing cartilage loss, and this may or may not have e¡ects on pain anddisability. What do we want to focus on at this meeting? Some of the largerperspective questions are as follows:
. What is the pathophysiology of OA joint pain?
. Why do some but not all people with OA get joint pain?
. Why do some people get severe pain and others mild, intermittent pain?
. Are there treatment opportunities that go along with a better understanding ofOA joint pain?
These are ambitious questions. Hopefully, wewill get some handle onwhere wemight go to answer those questions as we proceed.
2 FELSON

References
Dougados M, Nguyen M, Berdah L et al 2001 Evaluation of the structure-modifying e¡ects ofdiacerein in hip osteoarthritis: ECHODIAH, a three-year, placebo-controlled trial.Evaluation of the Chondromodulating E¡ect of Diacerein in OA of the Hip. ArthritisRheum 44:2539^2547
Peat G, McCarney R, Croft P 2001 Knee pain and osteoarthritis in older adults: a review ofcommunity burden and current use of primary health care. Ann Rheum Dis 60:91^97
Verbrugge LM 1979 Female illness rates and illness behavior: testing hypotheses about sexdi¡erences in health. Women Health 4:61^79
CHAIR’S INTRODUCTION 3

Spinal mechanisms contributing to
joint pain
Hans-Georg Schaible
Department of Physiology, Friedrich-Schiller-University of Jena, Teichgraben 8, D-07740Jena, Germany
Abstract.Nociceptive input from the joint is processed in di¡erent types of spinal cordneurons. A proportion of these neurons are only activated by mechanical stimulation ofthe joint and other deep tissue, e.g. adjacent muscles. Other neurons are activated bymechanical stimulation of joint, muscles and skin. The majority of the neurons are widedynamic-range neurons (small responses to innocuous pressure to deep tissue andstronger and graded responses to noxious mechanical stimulation). Importantly,neurons with joint input show pronounced hyperexcitability during development ofjoint in£ammation (enhanced responses to mechanical stimulation of the in£amed jointas well as to healthy adjacent deep structures, reduction of mechanical threshold in highthreshold neurons and expansion of the receptive ¢eld). Thus in£ammation inducesneuroplastic changes in the spinal cord which alter nociceptive processing. This state ofhyperexcitability is maintained during persistent in£ammation. The neurons are understrong control of descending inhibition which increases at least during the acute phaseof in£ammation. Several transmitters and mediators contribute to the generation andmaintenance of in£ammation-induced spinal hyperexcitability including glutamate,substance P, neurokinin A, CGRP, prostaglandins and probably others. The lattercompounds show enhanced release and an altered release pattern during in£ammationin the joint.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 4^27
Pain sensation in the joint
The major sensation in deep tissue such as joint and muscle is pain. Sensoryinformation from muscle and joint in£uences the motoric system and is involvedin the sense of movement and position but usually this does not reachconsciousness. In humans pain in the normal joint can be elicited particularlywhen noxious mechanical, thermal and chemical stimuli are applied to the ¢brousstructures such as ligaments and ¢brous cartilage (Lewis 1942, Kellgren& Samuel1950). In a normal joint pain is most commonly elicited by twisting or hitting the
4
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

joint. Stimulation of ¢brous structures with innocuousmechanical stimulation canevoke pressure sensations. No pain is elicited by stimulation of cartilage, andstimulation of normal synovial tissue rarely evokes pain (Kellgren & Samuel1950).Joint in£ammation is characterized by hyperalgesia and persistent pain at rest
which is usually dull and badly localized (Lewis 1938, 1942, Kellgren 1939,Kellgren & Samuel 1950). The application of noxious stimuli causes strongerpain than normal. Pain is even evoked by mechanical stimuli whose intensity isnormally not su⁄cient to elicit pain, i.e. movements in the working range andgentle pressure, e.g. during palpation. This heightened pain sensitivity resultsfrom peripheral sensitization (increase of sensitivity of nociceptive primarya¡erent neurons) and central sensitization (hyperexcitability of nociceptiveneurons in the central nervous system).Pain during degenerative osteoarthritis shows similarities and di¡erences to
arthritic pain. Similarly, as with arthritis, pain may increase when the joint is beingloaded. However, pain may also be reduced during walking, and pain may beparticularly severe during rest at night when the joint is immobile. It is likely thatmechanisms of pain during arthritis and osteoarthritis are di¡erent in some aspects.
Spinal cord neurons that respond to
mechanical stimulation of the joint
The articular nerves supplying the knee or elbow joint of rat, cat andmonkey enterthe spinal cord via several dorsal roots thus projecting to several spinal segments.Due to the widely distributed projection area joint a¡erents in£uence sensoryneurons and re£ex pathways in several spinal segments. Within the grey matterknee joint a¡erents project to the super¢cial lamina I and to the deep laminaeV^VII (c.f. Schaible & Grubb 1993). Figure 1A shows the spinal termination¢elds of horseradish peroxidase-labelled knee joint a¡erents in the segment L7 inthe cat spinal cord. Correspondingly, spinal cord neurons that are synapticallyactivated by joint a¡erents can be identi¢ed in the super¢cial and deep dorsalhorn and also in the ventral horn (Schaible et al 1986).
Receptive ¢elds and activation thresholds of neurons with joint input
In both cat and rat, mechanonociceptive inputs from the joint are processed indorsal horn neurons that respond solely to mechanical stimulation of deep tissue,or in neurons that respond to mechanical stimulation of both deep tissue and theskin. Receptive ¢elds of single sensory neurons (regions from which neurons canbe activated) are usually not restricted to the joint but more extended. Figure 1Cshows the receptive ¢eld of a spinal cord neuronwith convergent inputs from skin,
SPINAL MECHANISMS OF JOINT PAIN 5

6 SCHAIBLE
FIG
.1.
Spinalprojection
ofprim
arya¡erent¢
bresof
thekn
eejointand
respon
seprop
erties
ofspinalcord
neuron
swithinpu
tfrom
thekn
eejoint.(A
)Spinaltermination¢eldof
horseradishperoxidase-labelledprim
arya¡erent¢
bresof
thepo
steriorarticularnerveof
thekn
eejointinthe
grey
mattero
fthe
segm
entL
7inthecat.(B)R
espo
nsesof
awidedynamicrang
e(W
DR)n
euronwithinpu
tfromthekn
eejointtoinno
cuou
sand
noxiou
smov
ementsofthekn
eejoint.The
histog
ramssho
wthenu
mbero
factionpo
tentials/sthatwereelicited
bythemov
ements(binwidth1s).
Flex,£exion
ofthekn
eejoint;Ext,extension
ofthekn
eejoint;f.Ext,forcedextensionof
thekn
eejoint;OR,outwardrotation
ofthekn
eejoint
(sup
ination);n.O
R,noxious
outw
ardrotation
ofthekn
eejoint;IR
,inw
ardrotation
ofthekn
eejoint(pron
ation);n.IR,noxious
inwardrotation
ofthekn
eejoint.(C)R
eceptive
¢eldof
spinalcord
neuron
withinpu
tfromthekn
eejoint.Thisn
euronwasexcitedby
pressureappliedto
theskin
ofthepaw,the
deep
tissue
ofthigh(quadricepsmuscle)andlower
leg(gastrocnemius-soleus
muscle)andthestructures
ofthekn
eejoint.(D
)Receptive
¢eld
ofspinalcord
neuron
thatwas
only
excitedby
pressure
appliedto
deep
tissue
(muscles)andthekn
eejoint.(A
)reprintedwith
perm
ission
from
Craigetal1988;(B^D
)from
Schaibleetal1987a.

deep tissue and the knee joint. The neuron was activated by pressure applied to theknee joint (capsule, ligaments) and also by compression of the quadricepsmuscle inthe thigh and the gastrocnemius-soleus muscle in the lower leg, and in addition ithad a cutaneous receptive ¢eld at the paw. However, many neurons have receptive¢elds that are restricted to the deep tissue. Figure 1D shows the receptive ¢eld of aspinal cord neuronwith a receptive ¢eld in the deep tissue of the leg and in the kneejoint. Some neurons have bilateral receptive ¢elds (c.f. Schaible & Grubb 1993).Concerning mechanical thresholds, neurons are either nociceptive-speci¢c (NS)
or wide-dynamic-range (WDR) neurons. NS neurons respond only to intensepressure and/or to painful movements such as forceful supination and pronation.These stimuli elicit pain. WDR neurons respond to both innocuous pressure andnoxious pressure, encoding stimulus intensity by the frequency of actionpotentials. They may also be weakly activated by movements in the workingrange, and they show much stronger responses to painful movements. Figure 1Bdisplays the response pattern of a wide dynamic range neuronwith joint input. Theneuron exhibited small responses to £exion, extension, and outward rotation (OR)of the knee in its physiological range, but pronounced responses were elicited byforced extension (f. ext) and by noxious outward rotation (n. OR) exceeding theworking range of the joint. By and large, NS neurons have smaller receptive ¢eldsrestricted to deep tissue in joint andmuscle, and they donot have a receptive ¢eld inthe skin (c.f. Schaible & Grubb 1993).
Projections of spinal neurons with joint input
Neurons with joint input project to di¡erent supraspinal sites (cerebellum,spinocervical nucleus, thalamus, reticular formation) or to intraspinal(segmental) interneurons and motoneurons (c.f. Schaible & Grubb 1993).Ascending projections to the thalamus (in the spinothalamic tract) are importantto activate the thalamocortical systems that generate the conscious pain sensation.Segmental projections are important for the generation of motor and sympatheticre£exes. Furthermore, in the cat neurons have been identi¢ed that have cell bodiesin the ventral horn, belong to the spinoreticular tract and are predominantly orexclusively excited by noxious stimulation of deep tissue (Fields et al 1977,Meyers & Snow 1982).
Inhibition by heterotopic and descending inhibitory systems
Neurons with joint input are inhibited by heterotopic stimuli, in line with theconcept of di¡use noxious inhibitory controls (DNIC). The latter means thatpainful stimulation at one site of the body may reduce the pain at another site ofthe body (LeBars & Villanueva 1988). In addition, most spinal cord neurons with
SPINAL MECHANISMS OF JOINT PAIN 7

joint input are tonically inhibited by descending inhibitory systems that keep thespinal cord under continuous control (Cervero et al 1991, Schaible et al 1991). Theinterruption of descending inhibition can lower the excitation threshold of spinalcord neurons for mechanical input from the knee, substantially increase thereceptive ¢elds of neurons and cause (increased) ongoing discharges. Thus theresponse properties of neurons with joint input are controlled by the primarya¡erent input, by intrinsic properties of the spinal cord neurons, by local circuitsand by descending pathways.
Hyperexcitability in spinal cord neurons
during in£ammation in the joint
As described in the introduction, pain and hyperalgesia are usually elicited duringin£ammation of the joint. Hence experimental models have been used to studyneuronal mechanisms underlying these pain symptoms. In£ammation in the jointcan be induced by the intra-articular injections of crystals such as urate and kaolin,or by carrageenan. The injection of kaolin and carrageenan (K/C) into the jointproduces an oedema and granulocytic in¢ltration within 1^3 hours with a plateauafter 4^6 hours. Awake animals show limping of the injected leg and enhancedsensitivity to pressure onto the joint. By contrast, the injection of Freund’scomplete adjuvant (FCA) into one joint produces a monoarthritis that is presentfor 2 to 4 weeks. Usually the lesion is restricted to the injected joint, althoughbilateral e¡ects are observed sometimes. Hyperalgesia (limping or guarding ofthe leg, enhanced sensitivity to pressure onto the joint) develops within a day,reaches a peak within 3 days and is maintained to some degree up to severalweeks. When FCA is injected at a high dose into the tail base or lymph node, apolyarthritis develops (Schaible & Grubb 1993).
Generation of hyperexcitability
During the development of a K/C-induced in£ammation in the joint, both NSand WDR neurons with joint input show enhanced responses within 1^3 hoursto noxious stimuli applied to the in£amed joint (central sensitization). NSneurons exhibit a reduction in their mechanical threshold such that theapplication of innocuous stimuli to the in£amed joint is su⁄cient to excitethe neurons. Figure 2A shows the generation of hyperexcitability in a spinalcord neuron with joint input. Initially, while the joint was normal, theneuron responded only to noxious pressure applied to the knee (and adjacentmuscles in thigh and lower leg, Fig. 2B, left side). No responses were elicitedby pressure onto the ankle and the paw. After injection of kaolin and carrageenaninto the knee joint (K/C) the responses to noxious compression of the knee
8 SCHAIBLE

increased markedly, and at a latency of about half an hour the neuron startedalso to respond to pressure applied to the ankle and the paw. Thus the receptive¢eld expanded from the knee towards the paw (Fig. 2B, right side), and thepreviously high threshold neuron was then even activated by gentle innocuouspressure. The increased responses to stimuli applied to the in£amed joint resultmost likely from the enhanced synaptic input from a¡erent units which aresensitized during stimulation. However, the appearance of responses tostimulation of ankle and paw must result from a mechanism in the spinal cordbecause these regions were not in£amed. Thus nociceptive spinal cord neuronsobviously develop a state of hyperexcitability in which the responsiveness toboth inputs from in£amed and non-in£amed areas is increased (Dougherty et al1992, Neugebauer & Schaible 1990, Neugebauer et al 1993, Schaible et al 1987b).The increased responses to stimulation of the in£amed area are thought to be theneuronal mechanism of primary hyperalgesia (hyperalgesia at the site ofin£ammation) whereas the increased responses to stimuli applied to healthytissue are thought to be the neuronal mechanism underlying secondaryhyperalgesia (hyperalgesia in healthy tissue adjacent to and remote from in£amedtissue).Figure 2C and 2D show the working hypothesis how these changes are
produced. When the tissue is normal the neuron is only excited by stimuliapplied to the restricted receptive ¢eld (circle in Fig. 2C) but not by stimuliapplied to adjacent areas. When an in£ammation develops in the receptive ¢eld(shaded area, Fig. 2D), primary a¡erents in this region are sensitized and theyinduce a process of spinal sensitization. When the spinal neuron is hyperexcitableit shows stronger responses to stimuli applied to the original receptive ¢eld(stimulation sites 2 and 3), and in addition the neuron responds to inputs that arenormally tooweak to excite the neuron above threshold (stimulation sites 1 and 4).Hence the receptive ¢eld expands (Fig. 2D).This central sensitization can persist during chronic in£ammation. In rats with
unilateral arthritis (Grubb et al 1993) as well as in rats su¡ering from chronicpolyarthritis (Menetrey & Besson 1982) spinal cord neurons appear on averagemore sensitive and have expanded receptive ¢elds. Interestingly, the stimulationof primary a¡erents from deep tissue (muscle and joint) evokes more prolongedfacilitation of a nociceptive £exor re£ex than stimulation of cutaneous a¡erents(Woolf & Wall 1986), and capsaicin injection into deep tissue elicits moreprolonged hyperalgesia than injection of capsaicin into the skin (Sluka 2002)suggesting that deep input is particularly able to induce long term changes in thenociceptive system. However, spinal sensitization is counteracted to some extentby inhibitory in£uences. Descending inhibition (Schaible et al 1991) as well asheterotopic inhibitory in£uences (see above) are increased during in£ammation(Calvino et al 1987).
SPINAL MECHANISMS OF JOINT PAIN 9

10 SCHAIBLE

SPINAL MECHANISMS OF JOINT PAIN 11
FIG. 2. Development of in£ammation-evoked hyperexcitability in a spinal cord neuron withinput from the knee joint. (A) Histogram showing the responses (action potentials/response) ofthe neuron to noxious pressure applied to the knee joint, the ankle and the paw before and afterinjection of kaolin and carrageenan (K/C) into the ipsilateral knee joint. (B) Receptive ¢eld(shaded area) of the neuron before (control) and during knee joint in£ammation (3 h postK/C). C and D. Model showing the responses and the receptive ¢eld of a spinal cord neuronbefore in£ammation and (C) and after development of hyperexcitability (D). Beforein£ammation the neuron was only excited by pressure to the initial receptive ¢eld (stimulationsites 2 and 3). After in£ammation the neuron was activated from a larger area (stimulation sites1^4). A,B reprinted with permission from Neugebauer et al (1993).

Transmitters, mediators and receptors involved in
synaptic activation of spinal cord neurons with joint input
The generation and maintenance of central sensitization is produced by the actionof transmitter/receptor systems in the spinal cord. After sensitization primarya¡erent neurons release more transmitter from their spinal terminations uponperipheral stimulation (presynaptic component). Furthermore, spinal cordneurons are rendered more excitable by changes in receptor sensitivity(postsynaptic component).
Excitatory amino acids
Glutamate is themajor transmitter in the synaptic activation of spinal cord neuronswith joint input. On the postsynaptic site, glutamate activates N-methyl-D-aspartate (NMDA) receptors and non-NMDA receptors. The activation of non-NMDA receptors leads to basic excitation of neurons. By contrast, the activationof NMDA receptors leads to a calcium in£ux into neurons and causes processes ofneuronal plasticity in many neuronal circuits, such as long-term changes ofresponses. In our hands the ionophoretic application of antagonists at AMPA/kainate (non-NMDA) receptors close to neurons with joint input reduced theresponses to innocuous and noxious pressure whereas the application of NMDAreceptor antagonists reduced only the responses to noxious mechanicalstimulation. Thus, in our hands, NMDA receptors are only activated by noxiousstimulation (Neugebauer et al 1993).The ionophoretic application of NMDA antagonists at AMPA/kainate
and NMDA receptors to spinal cord neurons as well as systemic applicationof NMDA antagonists prevents the development of in£ammation-evokedspinal hyperexcitability (Neugebauer et al 1993). Figure 3 shows the e¡ect ofketamine, an antagonist at NMDA receptors. In six control neuronswithout ketamine the induction of in£ammation in the knee joint by injectionof K/C caused increases of the responses to noxious pressure appliedto the injected knee and the non-injected ankle. When ketamine wasadministered before and during induction of in£ammation, the induction ofin£ammation in the knee joint did not change responses in the fourneurons tested as long as the antagonist was applied (Fig. 3C,D). Importantly,antagonists at both receptor types can reduce responses of the neurons tomechanical stimulation of the joint also after in£ammation is established, andthis is even seen in a chronic model of in£ammation (Neugebauer et al 1993,1994a). Thus glutamate receptors play a key role in the generation andmaintenance of in£ammation-evoked spinal hyperexcitability even in the long-term range.
12 SCHAIBLE

Neuropeptides
Numerous joint a¡erents contain the neuropeptides substance P, neurokininA andCGRP that are coexpressed with glutamate. Noxious compression, but notinnocuous compression of the normal joint enhances the intraspinal release ofthese peptides above baseline. This pattern of release changes when the joint isin£amed. During acute in£ammation release occurs when the joint is stimulatedat innocuous intensity. Thus, under in£ammatory conditions a ‘cocktail’ oftransmitters and/or modulators is released in the spinal cord that changessynaptic processing under in£ammatory conditions (Hope et al 1990, Schaibleet al 1990, 1994).Excitatory neuropeptides facilitate the responses of spinal cord neurons. The
e¡ect of substance P is shown in Figs 4A,B. The WDR neuron in Fig. 4Ashowed graded responses to innocuous and noxious pressure applied to the kneejoint. A short ionophoretic application of substance P to the spinal cord neuroncaused reversible increases of ongoing discharges and responses to mechanicalstimulation. In the NS neuron in Fig. 4B substance P caused an increase ofresponses to noxious pressure and a small response to innocuous pressure and toankle stimulation.Ionophoretic application of antagonists at neurokinin 1, neurokinin 2 and
CGRP receptors attenuate the development of in£ammation-evokedhyperexcitability. Figure 4C shows the e¡ect of CP96,345, an antagonist at theneurokinin 1 receptor, on the development of in£ammation-evokedhyperexcitability. Compared to control neurons (top graph, induction ofin£ammation in the absence of the antagonist) the neurons treated with spinaladministration of the neurokinin 1 receptor antagonist showed a smaller increasein their responses after induction of in£ammation (middle graph). The inactiveenantiomer, CP96,344, did not attenuate the magnitude of in£ammation-evokedhyperexcitability (bottom graph). The antagonists also reduce hyperexcitabilitywhen it is established (Neugebauer et al 1995, 1996a,b). Probably, the activationof these peptide receptors enhances the sensitivity of glutamatergic synaptictransmission (Ebersberger et al 2000). However, it is important to point out thatthe antagonists at neuropeptide receptors are less antinociceptive than antagonistsat glutamate receptors.
Prostaglandins
Spinal prostaglandins (PGs) are synthesized inDRGneurons and in the spinal cordby both cyclooxygenases 1 and 2. PG receptors are located on primary a¡erentneurons and on spinal cord neurons indicating that PGs can act presynaptically
SPINAL MECHANISMS OF JOINT PAIN 13

14 SCHAIBLE

SPINAL MECHANISMS OF JOINT PAIN 15
FIG. 3. Blockade of the development of hyperexcitability in spinal cord neurons by i.v.administration of the NMDA receptor antagonist ketamine. (A, B) Changes of the responsesof spinal cord neurons to noxious pressure applied to the knee joint and the ankle duringdevelopment of in£ammation in the knee joint after the injection of K/C into the ipsilateralknee joint. (C, D) Same experimental approach as in A and B, but in these experimentsketamine was given i.v. during induction and in the initial period of in£ammation in the kneejoint. Reprinted with permission from Neugebauer et al (1993).

16 SCHAIBLE

SPINAL MECHANISMS OF JOINT PAIN 17
FIG. 4. E¡ect of substance P on the responses of spinal cord neurons tomechanical stimulationof the knee joint and e¡ect of a neurokinin 1 receptor antagonist on the development ofin£ammation-evoked hyperexcitability of spinal cord neurons. (A, B) The histograms (actionpotentials/s) shows that ionophoretic application of substance P at 70 nA or 100 nA enhancesresponses to mechanical stimulation. In the neuron in A substance P also caused enhancedongoing discharges. (C) Development of in£ammation-evoked hyperexcitability in spinal cordneurons (responses to innocuous pressure) in the absence of the antagonist (top), in the presenceof the neurokinin 1 receptor antagonist CP96,345 at the spinal cord neurons (middle) and in thepresence of the inactive enantiomer CP96,344 of the neurokinin 1 receptor antagonist (bottom).(A, B) reprinted with permission fromNeugebauer et al 1994b; (C) fromNeugebauer et al 1995.

18 SCHAIBLE

SPINAL MECHANISMS OF JOINT PAIN 19
FIG
.5.
Up-regu
lation
ofcycloo
xygenase
2(COX-2)in
thespinal
cord
during
in£ammationin
thekn
eejointandthee¡ectof
spinal
administration
ofindo
methacin
onthedevelopm
entof
in£ammation-evok
edhyperexcitability
ofspinal
cord
neuron
s.(A
,B)During
in£ammationin
thejointm
ainlyspinalCOX-2
show
san
increase.(C)T
hespinalapplicationof
indo
methacin,
ablockerof
COX-1
andCOX-2
attenu
ates
spinal
hyperexcitability.Opensquaresshow
thein£ammation-evok
edchangesof
respon
sesafterkaolin/carrageenan
injectionin
controlneuron
s,¢lledsquaresshow
thechangesof
therespon
sesdu
ring
developm
entof
in£ammationaftertopicaladministrationof
indo
methacinto
thespinal
cord.Top
graphs
show
respon
sesto
noxiou
spressure,bo
ttom
graphs
respon
sesto
inno
cuou
spressure.(A
,B)
reprintedwithperm
ission
from
Ebersbergeretal(1999);(C)from
Vasqu
ezetal(2001).

(in£uencing the release of synaptic mediators) and postsynaptically (in£uencingexcitability) (Vanegas & Schaible 2001). During in£ammation in the joint, thereis a tonic release of PGE2 within the dorsal and ventral horn (Ebersberger et al1999). This is likely to result from an upregulation of spinal COX-2 that isalready increased at 3 hours after induction of knee joint in£ammation (Figs5A,B). The application of PGE2 to the spinal cord surface facilitates theresponses of spinal cord neurons to mechanical stimulation of the joint, and thepattern of e¡ects is similar to that observed during peripheral in£ammation.When the cyclooxygenase inhibitor indomethacin is applied to the spinal cordbefore in£ammation, the development of hyperexcitability is signi¢cantlyattenuated compared to control rats in which only vehicle is applied to the spinalcord (Fig. 5C). Thus spinal PGs are involved in the generation of in£ammation-evoked spinal hyperexcitability (Vasquez et al 2001). The e¡ect of spinal PGE2 ismimicked by the application of agonists at EP1, EP2 and EP4 receptors to thespinal cord.
Concluding remarks
This review has its focus on the synaptic activation and development ofhyperexcitability of spinal cord neurons in the course of joint in£ammation. Thedata clearly show that the spinal cord undergoes neuroplastic changes duringin£ammation, and this will in£uence the expression of pain in patients. Thetransmission of spinal information to the thalamocortical cortex will generate thesubjective experience of pain with its di¡erent dimensions. However,hyperexcitability might also in£uence the in£ammatory process in the joint. Thegroup of Willis has recently reported that the spinal cord may in£uence theperipheral in£ammatory process through dorsal root re£exes (Willis 1999). Thiswill act in concert with other central nervous mechanisms that interact with thein£ammatory process (Straub & Cutolo 2001). Thus the nervous system does notonly act as a sensor for damaging stimuli, it also has an active part in the expressionof the in£ammatory lesion. It remains to be clari¢ed how important themechanisms described are in the generation and maintenance of pain duringdegenerative osteoarthritis.
References
Calvino B, Villanueva L, LeBars D 1987 Dorsal horn (convergent) neurons in the intactanaesthetized arthritic rat. II. Heterotopic inhibitory in£uences. Pain 31:359^379
Cervero F, Schaible H-G, Schmidt RF 1991 Tonic descending inhibition of spinal cord neuronsdriven by joint a¡erents in normal cats and in cats with an in£amed knee joint. Exp Brain Res83:675^678
20 SCHAIBLE

Craig AD, Heppelmann B, Schaible H-G 1988 The projection of the medial and posteriorarticular nerves of the cat’s knee to the spinal cord. J Comp Neurol 276:279^288
Dougherty PM, Sluka KA, Sorkin LS, Westlund KN,Willis WD 1992 Neural changes in acutearthritis in monkeys. I. Parallel enhancement of responses of spinothalamic tract neurons tomechanical stimulation and excitatory amino acids. Brain Res Brain Res Rev 17:1^13
Ebersberger A, Grubb BD, Willingale HL, Gardiner NJ, Nebe J, Schaible H-G 1999 Theintraspinal release of prostaglandin E2 in a model of acute arthritis is accompanied by anupregulation of cyclooxygenase-2 in the rat spinal cord. Neuroscience 93:775^781
Ebersberger A, Charbel Issa P, Vanegas H, Schaible H-G 2000 Di¡erential e¡ects of calcitoningene-related peptide and calcitonin gene-related peptide 8-37 upon responses to N-methyl-D-aspartate or (R, S)-alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate in spinalnociceptive neurons with knee input in the rat. Neuroscience 99:171^178
FieldsHL, Clanton CH,Anderson SD 1977 Somatosensory properties of spinoreticular neuronsin the cat. Brain Res 120:49^66
Grubb BD, Stiller RU, Schaible H-G 1993 Dynamic changes in the receptive ¢eld properties ofspinal cord neurons with ankle input in rats with unilateral adjuvant-induced in£ammation inthe ankle region. Exp Brain Res 92:441^452
Hope PJ, Jarrott B, Schaible H-G, Clarke RW, Duggan AW 1990 Release and spread ofimmunoreactive neurokinin A in the cat spinal cord in a model of acute arthritis. Brain Res533:292^299
Kellgren JH 1939 Some painful joint conditions and their relation to osteoarthritis. Clin Sci4:193^205
Kellgren JH, Samuel EP 1950 The sensitivity and innervation of the articular capsule. J BoneJoint Surg Am 32-B:84^91
LeBars D, Villanueva L 1988 Electrophysiological evidence for the activation of descendinginhibitory controls by nociceptive pathways. In: Fields HL, Besson J-M (eds) Prog BrainRes, Elsevier, Amsterdam, vol 77:275^299
Lewis T 1938 Suggestions relating to the study of somatic pain. Br Med J 1:321^325Lewis T 1942 Pain. Macmillan, LondonMenetrey D, Besson J-M 1982 Electrophysiological characteristics of dorsal horn cells in ratswith cutaneous in£ammation resulting from chronic arthritis. Pain 13:343^364
MeyersDER, SnowPJ 1982The responses to somatic stimuli of deep spinothalamic tract cells inthe lumbar spinal cord of the cat. J Physiol 329:355^371
Neugebauer V, Schaible H-G 1990 Evidence for a central component in the sensitization ofspinal neurons with joint input during development of acute arthritis in cat’s knee. JNeurophysiol 64:299^311
Neugebauer V, Lˇcke T, Schaible H-G 1993 N-methyl-D-aspartate (NMDA) and non-NMDAreceptor antagonists block the hyperexcitability of dorsal horn neurons during developmentof acute arthritis in rat’s knee joint. J Neurophysiol 70:1365^1377
Neugebauer V, Lˇcke T, Grubb BD, Schaible H-G 1994a The involvement of N-methyl-D-aspartate (NMDA) and non-NMDA receptors in the responsiveness of rat spinal neuronswith input from the chronically in£amed ankle. Neurosci Lett 170:237^240
Neugebauer V, Schaible H-G, Weiretter F, Freudenberger U 1994b The involvement ofsubstance P and neurokinin-1 receptors in the responses of rat dorsal horn neurons tonoxious but not to innocuous mechanical stimuli applied to the knee joint. Brain Res666:207^215
Neugebauer V, Weiretter F, Schaible H-G 1995 The involvement of substance P andneurokinin-1 receptors in the hyperexcitability of dorsal horn neurons during developmentof acute arthritis in rat’s knee joint. J Neurophysiol 73:1574^1583
Neugebauer V, Rˇmenapp P, Schaible H-G 1996a The role of spinal neurokinin-2 receptors inthe processing of nociceptive information from the joint and in the generation and
SPINAL MECHANISMS OF JOINT PAIN 21

maintenance of in£ammation-evoked hyperexcitability of dorsal horn neurons in the rat. EurJ Neurosci 8:249^260
Neugebauer V, Rˇmenapp P, SchaibleH-G1996b Calcitonin gene-related peptide is involved inspinal processing of mechanosensory input from the rat’s knee joint and in the generation andmaintenance of hyperexcitability of dorsal horn-neurons during development of acutein£ammation. Neuroscience 71:1095^1109
Schaible H-G, Grubb BD 1993 A¡erent and spinal mechanisms of joint pain. Pain 55:5^54Schaible H-G, Schmidt RF,Willis WD 1986 Responses of spinal cord neurons to stimulation ofarticular a¡erent ¢bres in the cat. J Physiol 372:575^593
Schaible H-G, Schmidt RF, Willis WD 1987a Convergent inputs from articular, cutaneous andmuscle receptors onto ascending tract cells in the cat spinal cord. Exp Brain Res 66:479^488
Schaible H-G, Schmidt RF, Willis WD 1987b Enhancement of the responses of ascending tractcells in the cat spinal cord by acute in£ammation of the knee joint. Exp Brain Res 66:489^499
Schaible H-G, Jarrott B, Hope PJ, Duggan AW1990 Release of immunoreactive substance P inthe spinal cord during development of acute arthritis in the knee joint of the cat: a study withantibody microprobes. Brain Res 529:214^223
Schaible H-G, Neugebauer V, Cervero F, Schmidt RF 1991 Changes in tonic descendinginhibition of spinal neurons with articular input during the development of acute arthritis inthe cat. J Neurophysiol 66:1021^1032
Schaible H-G, Freudenberger U, Neugebauer V, Stiller RU 1994 Intraspinal release ofimmunoreactive calcitonin gene-related peptide during development of in£ammation in thejoint in vivo�a study with antibodymicroprobes in cat and rat. Neuroscience 62:1293^1305
Sluka KA 2002 Stimulation of deep somatic tissue with capsaicin produces long-lastingmechanical allodynia and heat hypoalgesia that depends on early activation of the cAMPpathway. J Neurosci 22:5687^5693
Straub RH, Cutolo M 2001 Involvement of the hypothalamicpituitary adrenal/gonadal axis andthe peripheral nervous system in rheumatoid arthritis: viewpoint based on a systemicpathogenetic role. Arthritis Rheum 44:493^507
Vanegas H, Schaible H-G 2001 Prostaglandins and cyclooxygenases in the spinal cord. ProgNeurobiol 64:327-363 [Erratum in Prog Neurobiol 65:609]
Vasquez E, B�r K-J, Ebersberger A, Klein B, Vanegas H, Schaible H-G 2001 Spinalprostaglandins are involved in the development but not the maintenance of in£ammation-induced spinal hyperexcitability. J Neurosci 21:9001^9008
Willis WD Jr 1999 Dorsal root potentials and dorsal root re£exes: a double-edged sword. ExpBrain Res 124:395^421
Woolf CJ,Wall PD 1986 Relative e¡ectiveness of C primary a¡erent ¢bers of di¡erent origins inevoking a prolonged facilitation of the £exor re£ex in the rat. J Neurosci 6:1433^1442
DISCUSSION
Hunter:As someone who thinks a lot about the pathophysiology of OA, I aminterested in your thoughts as towhat e¡ect the sensitizationmay have, particularlyin the role of potential impairments to proprioception and trophic changes to themuscle.Youmentioned that the sensitizationmight have a role in changes inmotorre£exes. Could you expand on that?Schaible: There aren’t many studies that have addressed this. Usually, a noxious
stimulus would result in a nociceptive re£ex: you withdraw your limb from thenoxious stimulus. Some years ago we conducted a study in which we examined
22 DISCUSSION

these re£exes (He et al 1988). We induced joint in£ammation to see whether there£exes were altered. We found that the pattern is changing so that not all noxiousstimuli evoke the nociceptive re£ex. That is, there is some inhibition of the re£exes.This is important, because usually if you have an in£amed joint you don’t keep it inan extreme position, because this will activate the joint a¡erents the most. Whenthis re£ex is increasingly inhibited, you may be able to keep the joint in a positionwhere the joint a¡erents aren’t activated verymuch. This is an extremely important¢eld which has been neglected in recent years by neurophysiologists: we need toknow how this pathology in£uences the re£ex pattern.Hunter: Do the spinocortical tracts interact at all with the tracts which control
proprioception and any of the trophic response that it may have on the muscle?That is, does the sensitization of the upward ascension of pain ¢bres have anyinteraction with proprioceptive and trophic spinal ¢bres?Schaible: Intuitively, I would say yes. There are few data addressing this, though.
In some re£ex studies this has been seen, but it hasn’t been studied under theseconditions.Grubb: One of the things we use as a measure of central sensitization is the
expansion of the receptive ¢elds to nociceptive stimuli, and also to non-nociceptive stimuli, following stimulation of skin. This is most marked in the rat,but is also seen in larger animals such as the cat. When you examine patients withchronic OA, do you see changes in the responsivness well beyond the con¢nes ofthe joint itself, into the lower thigh or calf, for example, where we see theseexpanded receptive ¢elds? How well can the features we see be mapped intohuman?Creamer:We don’t know. I don’t think the equivalent studies have been done.
We know that pain often radiates from a¡ected joints, so that pain from the hip isoften felt in the thigh or pain in the knee is often felt lower down. But this is usuallythought to be because of radiation rather than increased sensitivity. We know thataround the knee people seem to be tender at points which don’t tend to correspondto particular pathological structures. There may be some sort of increased generalsensitivity to pain round the knee.Pisetsky:The data are clear-cut that, after a single stimulus, there are prolonged,
rapid changes in receptive ¢elds of neurons from in£ammation in the joint. Howmuch in£ammation in the joint do you need to get this kind of change? Thismodelinvolves very intense, acute in£ammationwhere there are potential systemic e¡ects(e.g. cytokine or glucocorticoid mediated). Would you see the same thing in achronic OA model, if you could do it in dogs? Or do you need a lot ofin£ammation over a short time scale to get remodelling?Schaible: It is di⁄cult to specify howmuch in£ammation is needed.What we see
is a robust phenomenon. The in£ammation does not need to be very thick toproduce these changes, and we see some variation. We also have the test
SPINAL MECHANISMS OF JOINT PAIN 23

stimulation (repeated mechanical stimulation of the joint), which could help topromote the sensitization processes. From what I know from all my experience, Iwould assume that the process of central sensitization is in principle the samewhenthere is a mild form of in£ammation. However, if we just use mechanicalstimulation and stimulate the knee in a noxious manner every ¢ve minutes forthree hours, we don’t get this central sensitization. The additional stimulus ofsome kind of sensitization by in£ammation is needed. The models ofin£ammatory pain that have been used in pain research are not very mild. We usein£ammatory processes which can be measured. But I would assume that even amild in£ammation would do the same job, with a slower time course and thephenomenon might be as large.Pisetsky: Could you block this with anti-tumour necrosis factor (TNF) or an
interleukin (IL)1 inhibitor? How much peripheral component is there to thecentral sensitization, which could be cytokine mediated?Schaible: We have some work on this, but the picture still isn’t clear. We can
partly reduce the sensitization by using COX inhibitors. We haven’t tried usinganti-TNF, but that is something we intend to do. We have tried to anaesthetizethe joint after in£ammation to see whether the central sensitization is stable. Inpain research we have two ways of thinking. One group says that there is a painmemory: you have one stimulus and the brain never forgets. The other groupthinks that there is sensitization as long as there is a process going on in theperiphery. From all of our recordings, I assume that the second is the case: if youreduce the peripheral input, you can get rid of the central sensitization. When weanaesthetized the joint wewere able to get rid of some of the activity. The problemis that the local anaesthetics don’t work very well in in£amed tissue, so we don’tknow the extent of the pain block.Dieppe: I want to come back to Blair Grubb’s point. One of the striking clinical
features of knee OA is the symmetry of the pain pattern. So to what extent do yousee bilateral spread across the spinal cord which might help explain this, as well asipsilateral spreading?Schaible: We have done spinal cord recordings. There are some spinal cord
neurons which show an altered response form. If they have a bilateral receptive¢eld, the responses of the neurons will change whether the ipsilateral orcontralateral side is stimulated. But there is also a bilateral e¡ect at the level of theprimary a¡erents.Wehave investigated receptor expression in dorsal root ganglion(DRG) primary a¡erents. If there is in£ammation on one side there is an up-regulation of receptors on both sides, but there is only pain on one side (Segondvon Banchet et al 2000). The reason why you don’t have pain on the other sidemight be because the ligands are not there if there is no in£ammatory process.Grubb: In reply to the earlier question about what type of stimuli the spinal cord
neurons are experiencing,we are possibly back to front in the order of papers in this
24 DISCUSSION

meeting. The primary a¡erents not only becomemore sensitive to noxious stimuli,but also many of them become spontaneously active. During this acutein£ammatory model which lasts hours, the spinal cord neurons experience aconstant a¡erent barrage around 1Hz from C ¢bres and the A-d pain a¡erents.The combination of this enhanced barrage from the increased mechanicalsensitivity and the increased spontaneous activity contributes to the centralsensitization that Professor Schaible describes.Henry:You described the distribution of projections in the spinal cord. It would
be interesting to knowwhat happens through the development of your RAmodel.We are seeing some contralateral e¡ects, which are a little surprising to us. Cananyone comment on the central distribution of primary a¡erent ¢bres in an RAmodel?Grubb: If you are talking about contralateral e¡ects, there was a panel at the
bottom of one of Professor Schaible’s slides showing that if you record theipsilateral side, in animals in which the in£ammation has been for 20 days, about20%have developed receptive ¢elds from the contralateral side. So it is a signi¢cantproportion of the WDR neurons in the deep dorsal horn that develop thesecontralateral receptive ¢elds in the rat. It may vary for other species and indi¡erent models.Kidd: I am very interested in the mirroring of in£ammationwithin the CNS.We
know of in£ammatory cells interacting with peripheral neurons. Might the sameprocesses be occurring at a central level which might then produce thesecontralateral changes? There is quite good evidence that the glial cells interactwith the neuron^neuron communication at a spinal level. What is the importanceof these glial^neuron communications at a spinal level?Schaible: I can’t comment directly on this because we haven’t done any work on
it. I know, however, that much of the COX is probably not located in the neuronsbut in the glial cells. Intuitively, I think that the glial cells are quite important forthe process of central sensitization.Simkin: I gather that all of these stimuli are starting in the synovium.Has anyone
done comparable work with stimuli in subchondral bone?Grubb: If you look at the peptidergic a¡erents that come from joint tissues, they
have been identi¢ed in almost every single joint tissue capsule, subchondral bone,cartilage, tendons and ligaments.Pisetsky:We are taught that cartilage has no nociceptive function.Grubb:There is one paper suggesting innervation on the edge of the cartilage. It
is a low density of innervation, but it has been reported (Schwab & Funk 1998).Pisetsky: Is any of this joint speci¢c? Or can you get this kind of sensitization
with in£ammation in some other structure?Schaible: It is possible. It has been demonstrated also for skin in£ammation.
However, the joint sensitization is very sensitive to mechanical stimulation. The
SPINAL MECHANISMS OF JOINT PAIN 25

skin in£ammation has mainly been studied with temperature stimulation. It hasalways been di⁄cult to do this with mechanical stimulation in skin.Lohmander: You pointed out that most of the relevant COX enzymes might be
located in the glial cells with receptors in the spinal cells. Would a systemicallydistributed COX inhibitor penetrate and be able to a¡ect the COX-relatedmechanisms in the spinal cord?Schaible: Yes, as long as the inhibitors can penetrate. This is the main problem:
most of the compounds we have are not useful for this.Lohmander:The point I am trying tomake is that the medications currently used
would not be very likely to a¡ect the central spinal mechanisms we are discussing.Schaible: In order to be sure we need to test each one. You cannot guarantee that
this is the case.Manning:There is certainly a good distribution of the COX inhibitors that are on
the market into the CNS. The challenge is to show that they are centrally acting.Rediske: In addition to playing a role in the activation of sensory neurons, a
number of in£ammatory mediators also have neurotrophic activities. Couldsome of the changes in the receptive ¢eld sensitivity be due to changes ininnervation patterns and increases in C ¢bre density?Schaible:There are changes like this in somemodels, although I must add that in
chronic in£ammation the number of C ¢bresmay be reduced. There are con£ictingdata in the periphery. In the spinal cord we don’t know whether there is anysprouting during in£ammation. This has not been tested to my knowledge. Thiswhole ¢eld of neurotrophic factors hasn’t been addressed that much. A peripheralapplication of nerve growth factor (NGF) will cause changes in primary a¡erentneurons. I am not sure how relevant this is for the central sensitization.Henry: I’d like to return to the mirror phenomenon. This bears on the question
of whether OA is an in£ammatory response or not. If it is, then mirror doesn’tnecessarily mean neural sprouting, but if it isn’t then it probably would be aneural-mediated e¡ect. We are seeing some evidence of bone sclerosis on thecontralateral side in our animal model of OA, which would suggest a systemicin£ammatory response.Schaible:How is that induced?Henry: Cutting the anterior cruciate ligament (ACL) and medial meniscus.Felson: There is evidence in human studies that limping and pain in one leg
changes the gait pattern so as to overload the contralateral side. The e¡ects youare seeing might be related to biomechanical changes in gait in the animal thathas been injured.Manning: I’d like to address some of your prostaglandin pharmacology data.
Cyclooxygenases produce a range of prostaglandins, not just PGE2. The use ofneutralizing antibodies to PGE2 has certainly substantiated that it is a key painmodulator, but PGI2 is also produced and that interacts with discrete inositol
26 DISCUSSION

phosphate (IP) receptors. A number of companies have developed IP receptorantagonists that look very good in pain models. Have you looked at PGI2 or IPreceptors in the spinal cord?Schaible:Not so far. We have measured prostaglandins in the spinal cord, and in
terms of quantity the most important are PGE2 and PGD2. I2 is not that high, butthe receptors are there. This needs to be measured.Grubb: In the periphery there is no doubt that both are extremely important for
the sensitization of the a¡erents in terms of primary hyperalgesia and allodynia.Mackenzie: You said that COX-2 inhibitors were more e¡ective than non-
selective COX inhibitors. Does this imply that at the spinal level COX-2 isconstitutively expressed?Schaible:This is de¢nitely known. Itmight not be true that indomethacin is non-
selective in the CNS. I recently read a report that considered indomethacin as morespeci¢c for COX-1 in the hippocampus. It really depends on the tissue as to howe¡ective these inhibitors are.Pisetsky:Does paracetamol work in this?Schaible:We haven’t studied this.Grubb:We did one set of experiments which we published in 1990 in which we
compared aspirin and paracetamol, and we got a modest reduction in primarya¡erent ¢ring, even though its IC50 for COX inhibition is very poor. There wasalso a story about COX-3, which may not exist in humans. There is a single basemissing in theDNAsequence and it appears that thewhole sequence is not in frame(Dinchuk et al 2003).Quitewhy paracetamolworks in humans I can’t tell you sincethe mechanism of action is unknown.Mackenzie: Experiments at our laboratories in Novartis have con¢rmed that
there is a single base missing in the human sequence.Grubb:One other possibility is that they got the sequence wrong in the original
paper in dogs, although this seems unlikely.
References
Dinchuk JE, Liu RQ, Trzaskos JM 2003 COX-3: In the wrong frame in mind. Immunol Lett86:121
HeX, Proske U, Schaible HG, Schmidt RF 1988 Acute in£ammation of the knee joint in the catalters responses of £exor motoneurons to leg movements. J Neurophysiol 59:326^340
SchwabW,FunkRHW1998 Innervation pattern of di¡erent cartilaginous tissues in the rat. ActaAnat (Basel) 163:184^190
Segond von Banchet GG, Petrow PK, Brauer R, Schaible HG 2000 Monoarticular antigen-induced arthritis leads to pronounced bilateral upregulation of the expression of neurokinin1 and bradykinin 2 receptors in dorsal root ganglion neurons of rats. Arthritis Res 2:424^427
SPINAL MECHANISMS OF JOINT PAIN 27

Activation of sensory neurons in the
arthritic joint
Blair D. Grubb
Department of Cell Physiology and Pharmacology, University of Leicester, PO Box 138,Leicester LE1 9HN
Abstract. Joints are richly innervated with a range of sensory nerve ¢bres that conveyinformation to the central nervous system about forces exerted on articular tissues byboth low and high threshold mechanical stimuli. High threshold nociceptive a¡erentsterminate primarily in the synovium and periosteum, and normally respond only tomovement of the joint beyond the working limits. Following joint damage, two factorscombine to alter the mechanical sensitivity of articular nociceptors. Firstly, physicalchanges (joint e¡usion and tissue oedema) alter the resting and movement-inducedforces exerted on the joint tissues and secondly, in£ammatory mediators released withinthe damaged tissue sensitize articular nociceptive a¡erents by binding to receptors on thenerve endings. These factors result in a reduction of the mechanical threshold foractivation of articular nociceptors such that manipulation of the joint within the normalrange is easily su⁄cient to activate them. Acute and chronic animal models of jointin£ammation have been used to study the mechanisms of articular nociceptorsensitization and a number of in£ammatory mediators and their receptors have beenimplicated. The focus of this paper will be to introduce some of the important issuesinvolved in the sensitization of nociceptive articular a¡erents.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 28^48
Joint innervation
Joints are innervated by distinct articular nerves although additional innvervationthrough accessory nerves is also known to be important. The network of primarya¡erent nerve ¢bres can detect both non-noxious and noxious mechanical stimuliapplied to the joint. In animal studies, most recordings from primary a¡erentarticular nociceptors have been made in either rat or cat where the anatomy of thearticular nerves is well understood and the physiological characteristics of theprimary a¡erent nerve ¢bres have been investigated. The precise location ofarticular nociceptors is an area of considerable interest and several studies haveidenti¢ed a¡erent nerve ¢bres in joint capsule, synovium, periosteum, proximal
28
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

ligament and tendon suggesting that damage to any part of the joint structure canexcite nociceptive a¡erents thus eliciting pain (Schaible & Grubb 1993).The two main systems of classi¢cation for primary a¡erent ¢bres based on
conduction velocity or ¢bre diameter, combined with the physiologicalproperties, have been long established. Articular a¡erents can easily be classi¢edusing the same methods and data from several studies have established the rangeof a¡erent nerve ¢bres innervating joint structures. Detailed morphologicalanalysis of articular nerves has shown that approximately 20% of articular a¡erent¢bres are myelinated with the majority of this group falling into the groupIII (Ad
«conduction velocity¼2.5^20m s�1) category of ¢nely myelinated
nociceptors. In addition, a relatively small number of large diameter myelinatedlow threshold group II (Ab
«conduction velocity¼20^65m s�1) ¢bres also
innervate each joint. The remaining 80% of articular ¢bres are unmyelinated andstudies suggest that approximately half of these are sensory group IV (C-¢bres,conduction velocity¼52.5m s�1) whilst the remainder are e¡erent sympatheticneurons innervating the joint. Low threshold a¡erents typically have largelycorpuscular endings with Ru⁄ni, Golgi and Paccinian endings all found in jointtissues (Johansson et al 1991). The high threshold mechanociceptive articulara¡erents show very little terminal specialization and can only be identi¢ed on thebasis of beaded varicosities containing accumulations of mitochondria. Thesenerve ¢bres are often found in association with blood or lymphatic vessels wherethey often form a dense network of nerve ¢bres running for considerable distancesalong each vessel (Heppelmann et al 1990).
Mechanical responses of articular a¡erents
Articular a¡erents have also been characterized according to their responseproperties with a range of mechanical sensitivities, from those activated byinnocuous manipulation or rotation of the joint, to those only activated bynoxious manipulation of the joint or rotation/£exion of the joint beyond thenormal working range. More precisely, the majority of articular a¡erentscharacterized as belonging to group II are low threshold mechanoreceptor andcan be either slowly or rapidly adapting (Dorn et al 1991). Interestingly, a few ofthese ¢bres are only activated by noxious stimuli applied to the joint indicating thattheymay have a role in nociception. Articular a¡erents belonging to groups III andIV typically have much higher threshold than group II ¢bres, and are most oftenactivated by noxious movements or manipulations of the joint (Schaible&Grubb1993). In addition, a further class of group III and IV nociceptors has beenreported, the ‘silent nociceptors’, which are mechano-insensitive in the normalanimal but which develop mechanical sensitivity following the development ofjoint in£ammation (Grigg et al 1986, Schaible & Schmidt 1988a).
JOINT SENSORY NEURONS 29

Phenotype classi¢cation of primary a¡erent nerve ¢bres
Primary a¡erent nerve ¢bres have also been classi¢ed according to theirneuropeptide phenotype which provides a guide to the subclasses of nociceptorsthat exist. In very general terms, primary a¡erent neurons have been classi¢ed intothree main groups:
. large diameter non-nociceptive neurons and nerve ¢bres that bind the RT97+veantibody (Bergman et al 1999) which recognizes phosphorylated epitopes onidenti¢ed neuro¢lament proteins (Johnstone et al 1997)
. isolectin B4 (IB4) positive neurons, which are non-peptidergic nociceptiveneurons (Silverman &Kruger 1990), and
. calcitonin gene-related peptide (CGRP)-expressing neurons (McCarthy &Lawson 1990), classed as peptidergic nociceptive neurons which can berecognized using selective CGRP antibodies. It should be noted that whilstCGRP is used to identify a distinct sub-population of primary a¡erentnociceptors, many other neuropeptides are also expressed in primary a¡erentneurons innervating joint structures. These include substance P, which ispresent in about half of the CGRP-positive neurons, and many othersincluding neurokinin A, galanin, opioid peptides and neuropeptide Y.
Approximately 25% of all dorsal root ganglion neurons contain CGRP in thenormal animal and neuroanatomical studies have clearly demonstrated CGRP-positive nerve terminals in a number of joint structures including synovium,intervertebral disks, ligaments, joint capsule and soft tissues. When Freund’sadjuvant monoarthritis is induced in the rat ankle joint, the proportion of CGRPpositive cell bodies in lumbar dorsal root ganglion (DRG) neurons increasesmarkedly presumably as a consequence of the increase in neuronal activation(Hanesch et al 1993). The precise functional consequence of this up-regulation ofCGRP expression in sensory nerves innervating the joint is unclear. It is known,however, that these neuropeptides are released fromboth the central andperipheralterminals of nociceptors where they are involved in central transmission andperipheral neurogenic in£ammation, respectively.
Models of in£ammation used to study
the properties of articular nociceptors
Osteoarthritis (OA) is a signi¢cant clinical problem for which suitable analgesictherapies are required. In recent years a number of animal models have beendeveloped which share similarities with the human disease process. These includenaturally occurring OA models in guinea pig and instability-induced OA in micewith a natural predisposition to spontaneous OA. In addition, there are also a
30 GRUBB

number of surgically induced models of OA in dog, rat and guinea pig (Bendele2002). Unfortunately, none of these models has been used to study the e¡ects ofin£ammation on the properties of articular a¡erents. The original studies, whichwere performed in the 1980s and early 1990s, were designed to look at the processof a¡erent sensitization without any speci¢c disease model in mind. Most studieshave used either:
. the carrageenan/kaolin model of acute arthritis which produces noticeablechanges in a¡erent activity in 1^2 hours and which is normally continued forup to 24 hours, or
. the complete Freund’s adjuvant (CFA)-induced mono- or poly-arthritis whereinjections of heat-killed Mycobacterium tuberculosis or Mycobacterium butyricuminduce an arthritic process lasting several days to months. The jointin£ammation normally peaks early at 2^5 days and then declines and eitherresolves (monoarthritic model) or enters a second phase that can last severalweeks (polyarthritic model).
Articular a¡erents and in£ammation
Following trauma to the joint we experience an increased sensitivity to loadbearing and to movement of the joint within the normal range (allodynia). Inaddition, there is also a markedly increased sensitivity to any further noxiousmechanical stimulation (hyperalgesia). These conscious manifestations of jointdamage are brought about by changes in the sensitivity of primary a¡erent nerve¢bres, by spinal processing of joint input and by processing within highercentres.Following the development of joint in£ammation, low threshold group II
articular a¡erents only show acute and relatively transient changes in theirresponses to joint manipulation which resolve within a few hours. By contrast,articular a¡erents belonging to groups III and IV often start to show ongoingspontaneous activity in the absence of joint movement, and show enhancedresponses to manipulation of the joint (rotation/extension/£exion of the joint orcapsular indentation using a blunt probe; Coggeshall et al 1983, Guilbaud et al1985, Schaible & Schmidt 1988a). Furthermore, many units which werepreviously mechanoinsensitive develop receptive ¢elds and may also showongoing spontaneous activity. These alterations in the ¢ring characteristics ofgroup III and IV a¡erents are the result of a marked reduction in the mechanicalthreshold for activation of articularmechanoreceptors and this contributes, in part,to the psychophysical measures of allodynia and hyperalgesia experienced inhumans.
JOINT SENSORY NEURONS 31

Mechanisms underlying articular mechanonociceptor sensitization
In£ammation is a complex process in which a number of in£ammatory mediatorsare released in response to tissue damage. In acute in£ammation, thesein£ammatory mediators are part of the normal process that is required for tissuerepair and regeneration. A number of these in£ammatory mediators areresponsible for the sensitization of primary a¡erent articular mechanonociceptorsand this serves to limit mobility and thus protect the joint so that it is not damagedfurther during the repair process.In chronic disease, the in£ammatory response may be protracted due to
abnormal pathology in the joint tissues. In the case of OA this is largely due tothe destruction of cartilage and bone remodelling (osteophytes or bony spurs)that is a response of bone to local damage. The loss of the normal articulatingsurfaces and the abnormal bone pathology results in chronic in£ammation thatcan last years. To understand the mechanisms underlying the sensitization ofarticular mechanonociceptors in chronic in£ammation it is vital to understandthe in£ammatory mediators that are present during each stage of the diseaseprocess. One problem is that the in£ammation is not constant, but ratherepisodic and dependent on a number of factors including activity. This can meanthat the in£ammatory mediators present in the joint will vary, thus making thetargeting of treatment potentially di⁄cult. Since speci¢c animal models of OAhave not been used to study mechanisms of sensitization of articularmechanonociceptors, the information available regarding their sensitization hasbeen generated using the animal models of joint in£ammation mentioned above.An important group of in£ammatory mediators that regulate the
mechanical sensitivity of articular mechanonociceptors are the prostaglandins.Prostaglandins are formed when membrane phospholipids are broken down bythe enzyme PLA2 to form arachidonic acid. This is then converted by the enzymecyclooxygenase (COX) to the biologically active prostaglandins, PGE2, PGI2,PGD2 and PGF2a. There is good evidence from the literature that non-selectiveCOX inhibitors (non-steroidal anti-in£ammatory drugs) such as indomethacinand the salicylic acid derivatives reverse the sensitization of articularmechanonociceptors seen in animal models of arthritis (Guilbaud & Iggo 1985,Heppelmann et al 1986). This evidence combined with ample evidence that COXinhibitors are clinically usefully treatments for arthritis in man shows that the jointtissues are a major target for this class of drugs. Further studies have shown thatPGE2 (and the related PGE1) and PGI2 seem to be particularly important for thesensitization of nerve ¢bres since these compounds are able to either directly excitearticular mechanonociceptors or sensitize them to mechanical stimuli(Heppelmann et al 1985, Schaible & Schmidt 1988b, Grubb et al 1991, Birrellet al 1991, Schepelmann et al 1992).
32 GRUBB

The COX enzyme exists in at least two forms in human (possibly three in dog,Chandrasekharan et al 2002). COX-1 (Funk et al 1991) is a constitutive enzyme thatis present in a large number of cell types and is seen as the housekeeping isoform.COX-2 (Hla&Neilson 1992) is only constitutively expressed in a small number oftissues, e.g. brain and spinal cord, but is readily up-regulated in many tissues inresponse to damage (Seibert et al 1994). In the periphery, an in£ammatory insultwill readily up-regulate COX-2 expression resulting in an increase in prostaglandinsynthesis. Whilst no a¡erent ¢bre studies have been published showing the rolesof COX-1 andCOX-2 in producing articularmechanonociceptor sensitization, theclinical success of coxibs, the new selective COX-2 inhibitors, suggests thatprostaglandins synthesized through this pathway are responsible in large part fornociceptor sensitization in chronic joint in£ammation.In addition to the prostaglandins, a number of other in£ammatory mediators
have been shown to excite and or sensitize articular mechanonociceptors.Bradykinin, for example, has been shown to directly excite the majority ofgroup III and IV articular a¡erents in cat knee and rat ankle joint (Kanaka et al1985, Birrell et al 1993) and to sensitize them to movements (Neugebauer et al1989). In addition, it is well established that prostaglandins (PGE2/PGI2)may augment the excitatory or sensitizing actions of bradykinin (Schaible &Schmidt 1988b, Birrell et al 1993, Schepelmann et al 1992). Likewise, anotherimportant in£ammatory mediator, serotonin, has been shown to exciteapproximately two-thirds of articular mechanonociceptors in the rat ankle andcat knee joint. This list is by no means extensive and there exists a large literatureon the e¡ects of leukotrienes, histamine, neuropeptides and other in£ammatorymediators on the sensitization of articular mechanonociceptors (Schaible &Grubb 1993).
In£uence of joint mechanics
An important aspect of joint damage is the e¡usion that accumulates in thejoint space. This is particularly important when considering the responses ofjoint mechanonociceptors to joint £exion or extension. Joint pressure isnormally subatmospheric and lowest when the joint is in resting or in the mid-range position. However, up to 70ml of e¡usate can be removed from thein£amed human knee joint (Jayson & Dixon 1970) and this markedly increasesthe baseline pressure within the joint. More importantly, however, upon £exionor extension, joint pressure increases dramatically, thus increasing stretching ofthe capsule and surrounding tissues. Since these are densely innervated byarticular mechanonociceptors, there can be little doubt that these physical changesin the condition of the joint will also contribute to the enhanced ¢ring of thesearticular a¡erents.
JOINT SENSORY NEURONS 33

Summary
A review of the recent literature shows that relatively little has been achieved inrecent years to push forward these early studies of articular mechanonociceptorsensitization in mammalian species. It is certainly true that the majority ofin£ammatory mediators have been tested in one of the animal models and there islittle doubt that they each have signi¢cant e¡ects on the baseline ¢ring and responseproperties of articular mechanonociceptors. From these we have a goodappreciation of the respective roles of each in£ammatory mediator and of someof the interactions that occur between them. Unfortunately, there are limitationsto the information that can be gleaned from these studies since direct recordingscannot be made from the very ¢ne nerve terminals in the joint.The way forward with this type of research is not entirely clear. It is obvious to
allwhowork on joints that articular a¡erents are distinct in that they are present in anumber of di¡erent locations, each designed to detect potential damage to the jointthrough di¡erent perturbations of the joint tissues. On the negative side, there isclearly a gap in the present knowledge regarding the location and responsivenessof joint mechanonociceptors in osteoarthritic joints. If the mechanisms ofjoint mechanonociceptor sensitization are also di¡erent in OA (or equivalentanimal models), i.e. di¡erent to the data obtained in more general models ofjoint in£ammation, then we will need to re-evaluate the role of these a¡erentsin OA.In addition, we still do not know which proteins/ion channels present in joint
a¡erent terminals are responsible for mechanosensation although a number havebeen suggested for di¡erent tissues. An important question, however, is whetherarticular a¡erents really are unique in terms of the basic mechanisms by which theydetect and respond to mechanical stimuli and to in£ammatory mediators. It isclear that there are relatively few data that would allow one to distinguish a jointa¡erent from a skin or muscle a¡erent in terms of phenotype.An interesting paradox is that many joint, skin and muscle a¡erents express the
heat-sensitive TRP channel (TRPV1) since they can respond to capsaicin. Thismeans that joint a¡erents have the capacity to respond to damaging or potentiallydamaging heat stimuli which they are unlikely to experience. A recent hypothesis(Liang et al 2001) suggests that the sensitization of skin a¡erents by bradykininproduces a reduction in the thermal threshold for opening of the vanilloidreceptor/channel complex that normally opens only in response to noxious heat.The threshold is reduced so far, indeed, that normal body temperature is su⁄cientto open these channels and excite the nociceptor. Suchmechanisms could also be anexplanation for the presence of these heat responsive channels in deep tissues andcould indicate that there is homogeneity in the mechanisms by which noxiousstimuli are detected in di¡erent tissues.
34 GRUBB

In order to progress, we need to establish whether basic common principlesapply to all nociceptors irrespective of the tissue that they innervate. We need abetter understanding of the basic transduction mechanisms for thermal andmechanical stimuli, the mechanisms of sensitization including an appreciation ofthe G protein-coupled receptors, signalling pathways and the ion channels thatregulate nerve terminal excitability and ¢ring. Only by combiningelectrophysiological measurements from single cells, molecular biology, proteinbiochemistry, and modern neuroanatomical and imaging techniques can we hopeto start to understand the molecular mechanisms of articular mechanonociceptionin OA.
References
Bendele AM2002Animalmodels of osteoarthritis in an era ofmolecular biology. JMusculoskelNeur Interact 2:501^503
Bergman E, Carlsson K, Liljeborg A, Manders E, Hokfelt T, Ulfhake B 1999 Neuropeptides,nitric oxide synthase and GAP-43 in B4-binding and RT97 immunoreactive primary sensoryneurons: normal distribution pattern and changes after peripheral nerve transection andaging. Brain Res 832:63^83
Birrell GJ, McQueen DS, Iggo A, Coleman RA, Grubb BD 1991 PGI2-induced activation andsensitization of articular mechanoreceptors. Neurosci Lett 124:5^8
Birrell GJ, McQueen DS, Iggo A, Grubb BD 1993 Prostanoid-induced potentiation of theexcitatory and sensitizing e¡ects of bradykinin on articular mechanonociceptors in the ratankle joint. Neuroscience 54:537^544
Chandrasekharan NV, Dai H, Roos KLT et al 2002 COX-3, a cyclooxygenase-1 variantinhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, andexpression. Proc Natl Acad Sci USA 99:13926^13931
Coggeshall RE, Hong KAP, Langford LA, Schaible H-G, Schmidt RF 1983 Dischargecharacteristics of ¢ne medial articular a¡erents at rest and during passive movements ofin£amed knee joints. Brain Res 272:185^188
Dorn T, Schaible H-G, Schmidt RF 1991 Response properties of thick myelinated group IIa¡erents in the medial articular nerve of normal and in£amed knee joints of the cat.Somatosens Mot Res 8:127^136
Funk CD, Funk LB, Kennedy ME, Pong AS, Fitzgerald GA 1991 Human platelet/erythroleukemia cell prostaglandin G/H synthase: cDNA cloning, expression, and genechromosomal assignment. FASEB J 5:2304^2312
Grigg P, Schaible H-G, Schmidt RF 1986 Mechanical sensitivity of group III and IV a¡erentsfrom posterior articular nerve in normal and in£amed cat knee. J Neurophysiol 55:635^643
Grubb BD, Birrell GJ,McQueenDS, IggoA 1991 The role of PGE2 in the sensitization of jointmechanoreceptors in normal and in£amed ankle joints of the rat. Exp Brain Res 84:383^392
Guilbaud G, Iggo A 1985 The e¡ect of acetylsalicylate on joint mechanoreceptors in rats withpolyarthritis. Exp Brain Res 61:164^168
Guilbaud G, Iggo A, Tegner R 1985 Sensory receptors in ankle joint capsules of normal andarthritic rats. Exp Brain Res 58:29^40
Hanesch U, Pfrommer U, Grubb BD, Schaible H-G 1993 Acute and chronic phases of unilateralin£ammation in rat’s ankle are associatedwith an increase in the proportion of calcitonin gene-related peptide-immunoreactive dorsal root ganglion cells. Eur J Neurosci 5:154^161
Hla T,NeilsonK1992Human cyclooxygenase-2 cDNA. ProcNatl Acad SciUSA89:7384^7388
JOINT SENSORY NEURONS 35

Heppelmann B, Schaible H-G, Schmidt RF 1985 E¡ects of prostaglandins E1 and E2 on themechanosensitvity of group III a¡erents from normal and in£amed cat knee joints. AdvPain Res Ther 9:91^101
Heppelmann B, Pfe¡er A, Schaible H-G, Schmidt RF 1986 E¡ects of acetylsalicylic acid andindomethacin on single groups III and IV sensory units from acutely in£amed joints. Pain26:337^351
Heppelmann B, Messlinger K, Neiss WF, Schmidt RF 1990 Ultrastructural three-dimensionalreconstruction of group III and group IV sensory nerve endings (‘free nerve endings’) in theknee joint capsule of the cat: evidence for multiple receptive sites. J Comp Neurol 292:103^116
Jayson MIV, Dixon ASJ 1970 Intraarticular pressure in rheumatoid arthritis of the knee. I.Pressure changes during passive joint distension. Ann Rheum Dis 261^265
Johansson H, Sjolander P, Sojka P 1991 Receptors in the knee joint ligaments and their role inthe biomechanics of the joint. Crit Revs Biomed Eng 18:341^368
JohnstoneM,GooldRG, Fischer I, Gordon-Weeks PR 1997The neuro¢lament antibody RT97recognises a developmentally regulated phosphorylation epitope on microtubule-associatedprotein 1B. J Anat 191:229^244
Kanaka R, Schaible H-G, Schmidt RF 1985 Activation of ¢ne articular a¡erent units bybradykinin. Brain Res 327:81^90
Liang Y-F, Haake B, Reeh PW 2001 Sustained sensitization and recruitment of rat cutaneousnociceptors by bradykinin and a novel theory of its excitatory action. J Physiol 532:229^239
McCarthy PW, Lawson SN 1990 Cell type and conduction velocity of rat primary sensoryneurons with calcitonin gene-related peptide-like immunoreactivity. Neuroscience 34:623^632
Neugebauer V, SchaibleH-G, Schmidt RF 1989 Sensitization of articular a¡erents tomechanicalstimuli by bradykinin. P£ugers Arch 415:330^335
Schaible H-G, Schmidt RF 1988a Time course of mechanosensitivity changes in articulara¡erents during a developing experimental arthritis. J Neurophysiol 60:2180^2195
Schaible H-G, Schmidt RF 1988b Excitation and sensitization of ¢ne articular a¡erents fromcat’s knee joint by prostaglandin E2. J Physiol 403:91^104
Schaible H-G, Grubb BD 1993 A¡erent and spinal mechanisms of joint pain. Pain 55:5^54Schepelmann K, Messlinger K, Schaible H-G, Schmidt RF 1992 In£ammatory mediators andnociception in the joint: excitation and sensitization of slowly conducting a¡erent ¢bers ofcat’s knee by prostaglandin I2. Neuroscience 50:237^247
Seibert K, Zhang Y, Leahy K et al 1994 Pharmacological and biochemical demonstration of therole of cyclooxygenase 2 in in£ammation and pain. Proc Natl Acad Sci USA 91:12013^12017
Silverman JD, Kruger L 1990 Selective neuronal glycoconjugate expression in sensory andautonomic ganglia: relation of lectin reactivity to peptide and enzyme markers. JNeurocytol 19:789^801
DISCUSSION
Brandt: Earlier on, Peter Simkin asked a question about bone versus synovialspace. I was reminded of the elegant work by Arnoldi and Lemberg in the 1970s(Arnoldi et al 1972), who measured blood£ow through subchondral bone in OAjoints and showed that it is decreased. InOA, the CO2 concentration in blood fromthe medullary space is elevated, lactate concentration is increased andO2 tension isdecreased. Furthermore, Arnoldi and Lemberg demonstrated that the pain of
36 DISCUSSION

osteoarthritis could be relieved immediately and completely by an osteotomy, asurgical procedure that changes the pressure within the medullary space butwhich has nothing to do with treating the in£ammatory change directly. Thesynovium is easier to study than the bone, but I wouldn’t limit considerations ofOA pain to the articular space.Grubb: In the models that we have used to study nociceptors, there is little
damage to the bone. The studies of a¡erent ¢bre sensitization have been done inthe wrong sorts of models. They were done in animal models where the primaryaim was to look at a¡erent ¢bre sensitization, and they were not studies aimedparticularly at OA. The contribution of bone pain to OA and the contribution ofa¡erent ¢bres is simply a huge de¢cit in our knowledge.Kuettner: In clinical terms, we are studying OA in older individuals. In your
animal models, have you taken age as a component? Most animal work is doneon young animals.Grubb: It is almost all done on young animals.Kuettner: The question is, can we interpret data from young animals and apply
them to the adult population either in humans or animals? With the exception ofthe guinea pig, we have di⁄culties reproducing some of the animal models of OAin old animals.Grubb: That is a valid question and we don’t know the answer.Henry: This raises another issue: when does OA start? Does it start with the
clinical signs, or before?Dieppe:Goodquestion. I don’t have an answer, but it is probably long beforewe
see clinical signs.Moving to a completely di¡erent issue, one of the fascinating things about
musculoskeletal pain is that it is sensitive to barometric pressure: patients canpredict the weather. Is it possible from what you are saying that the increasedsensitization of mechanoreceptors is such that they can respond to theseminiscule changes in barometric pressure? The patients can certainly detect them.If it is not this, then what on earth is going on?Grubb: I don’t know the answer to that, but I can speculate. I understand that
people su¡ermore in damp, cold weather when the pressure is falling. It is true thata number of joint mechano-nociceptors are sensitive to gentle pressure on thecapsule in an in£amed joint. It may be that modest capsular stretching as a resultof a di¡erence between the articular and exterior pressure could be big enough toproduce some additional activation of the nociceptive nerve endings. It isinteresting that this only happens when pressure is falling, because this would bewhen any change in the capsule stretching would be occurring. I have no evidencefor this, though, and I stress that this is pure speculation.Koltzenburg: It may be that the source of pain in OA is muchmore the bone than
the joint itself. Reduction in bone pressure by bone marrow aspiration is also very
JOINT SENSORY NEURONS 37

painful. I don’t know how this relates to a fall in barometric pressure: this wouldbe interesting to discuss. I have a question about the TRPV1 (VR1) receptor. Howdo you think this is implicated in mechanosensation? I can see that this might beplaying an important role in heat sensitization, butwhen the vanilloid receptors areactivated they don’t change the mechanosensitivity in the same sensory neuronsand there was also no change in mechanically-evoked cutaneous pain in micelacking TRPV1.Grubb: All I can say is that many neurons respond to both mechanical and
thermal stimuli. All I am o¡ering in putting forward Peter Reid’s model is theidea that bradykinin present in the damaged tissue may be activating aproportion of the neurons through activation of the TRPV1 receptor, thusperhaps changing the background spontaneous activity of the a¡erent ¢bres.Whether this occurs in addition to the changes in mechanosensitivity is not clear.In our original experiments, we never thought to apply any thermal stimuli to deeptissues because youwould think that theywouldn’t normally experience this. But ifthey respond to capsaicin theymust contain the TRPV1 heat sensitive ion channel.If it is true that sensitization drops their heat threshold such that they are activatedby body temperature, then bradykinin could produce its sensitizing e¡ects in thisway. This is an interesting idea.Schaible: There is some work by Besson’s group on the polyarthritic rat.
They found that these animals are mechanically hypersensitive, but with respectto heat they are hypoalgesic (for reference see Schaible & Grubb 1993). I am notsure whether this Peter Reid model applies to the joint.Grubb: The problem with the CFA polyarthritis model is that it is a profound
arthritic model with arthritis in all joints, skin lesions and skin in£ammation. I amunsure what you can say about heat sensitivity of joint a¡erents in this model.Pisetsky:How are gravity, weight bearing and compression perceived?Grubb: There have been studies done on capsular a¡erents from way back,
looking at loading of the capsules in di¡erent planes. Loading the capsule axiallyproduces the most appropriate stimulus for activating these a¡erents.Pisetsky:What about just weight bearing?Grubb: I am not sure.Evans: I was interested in the suggested pathogenesis of OA that you showed.
You had one chain of arrows going through the in£ammatory route, and then youhad the particles o¡ to the side shunted directly into pain. Were you implying thatindependently of their in£ammatory properties they have the ability to inducepain? If so, how?Grubb: These articular nociceptors have become extremely mechanically
sensitive. If the subchondral bone has become damaged and the nociceptors arevery close to the surface, and you have particular matter in there, this could becontributing. Of course, the articular surfaces rubbing against each other could
38 DISCUSSION

be a more powerful stimulus. I just had them in there to suggest that theycontribute to the nociceptor activation.Henry:That would overlook the idea that there isn’t a 100% correlation between
the pathology and the pain.Brandt: Can you give us a quantitative sense of the duration of the latent period
that precedes ¢ring of a nociceptor in the joint versus that of a nerve ending thatdeals with proprioception?Grubb: The conduction velocities of C ¢bres are usually 2^2.5m/s, but large
¢bres will conduct at 30^100m/s. The nociceptors are slow conducting ¢bres andthe proprioceptors are much faster.Pisetsky: You de¢ned noxious movement in terms of the extent of movement.
What about the rate at which a joint is moved?Grubb: I presented a lot of work from Hans-Georg Schaible, and he has done
most of the work on joint movement, so I think he should answer that question.Schaible: Whenever we move our joints in the normal range there is not much
torque. But when we twist the joint against the resistance of the tissue this is whatwe call noxious movement. The rate of movement doesn’t matter. Moving thejoint quickly will only cause the low threshold mechanoreceptor ¢bres to ¢re.This will not activate a C ¢bre. A nociceptor is usually not even activated duringthe ramp of a stimulation, but when the stimulus is there, it is activated one or twoseconds after the joint has reached an extreme position. In the normal movementrange C ¢bres will not be activated.Pisetsky:Does duration of joint use matter?Schaible: It isn’t easy to answer this question. The tests were usually 15 seconds
long, and this short duration doesn’t change the response pattern.Hunter: In an abnormal joint which is mechanically unstable, would you expect
that this would produce a nociceptive stimulus in a lower range of movement thanis needed for a normal joint?Schaible: The question is whether mechanical changes alone would be su⁄cient
to change the response properties. Fromour experience so farwe believe that if youapply just mechanical stimuli, the response properties will not change a lot. Youneed something additional. We can’t really answer this sort of speci¢c questionbecause our studies have not addressed it.Felson: Can you give us a sense of the mechanical stimuli you have applied? The
local stresseswithin osteoarthritic joints are often very high. Is there someway thatyou can characterize the magnitude of the twisting beyond the normal range ofmotion that you apply?Schaible: It has been measured but I can’t recall the numbers.Fernihough:What is seen as a noxious twisting movement by ligaments may not
be seen at all by the synovium,which is a fully deformable tissue.Do you think thatsome of the areas that contribute to a pain response are also the response of the
JOINT SENSORY NEURONS 39

nerves within those tissues to di¡ering mechanical stresses? Following on fromthis, has anyone seen things like TRPV1 expression in di¡erent tissues within ajoint?Grubb: The responses are to the twisting of the joint and to £exion of the joint
beyond the normal range. As shown in work by Ferrell et al (1986), massivechanges in the ¢ring of the articular nerves are seen if there is an e¡usion. It reallymakes a huge di¡erence. This could contribute to the ¢ring of synovial a¡erents aswell. It depends on the volume of e¡usion, which varies in OA.Herzog:One of the interesting questions is raisedwhen you have unstable joints,
such as in some forms of OA where the anterior cruciate ligament (ACL) is absentand twistingmovements likemedial rotation becomemuch easier. TheACL is oneof the primary structures to prevent excessive anterior translation and medialrotation of the tibia relative to the femur. Therefore, if the ACL is absent (torn),anterior tibial translation and medial rotation encounter much less resistance thanin the intact knee (e.g. Maitland et al 1998). When you apply this noxious torque,and then take one of the structures out that primarily resist the torque (e.g. theACL) and do the same displacement but this time in the absence of torque, doyou still see the same nociceptive response? Does it come from a speci¢c tissue oris it a general joint response?Schaible: The anterior cruciate ligament seems to be quite important. I had a
collaboration with an orthopaedic surgeon, and they said if they treated the jointand theACLwasn’t properly repaired then therewere a lot of problems afterwards.We have studied the response properties of mechanoreceptors supplying the ACL(Krauspe et al 1992). On each movement they exactly indicate the torque and thestress which is applied to the joint. They ¢re all the time when you move the jointand increase the stress level. This proprioception is quite necessary. There is adisorder called Charcot’s joint, where proprioception is absent or reduced andthe joints are damaged. We need proprioceptive control all the time, and theACLwith its mechanoreceptors seems to be particularly important for this reason.Koltzenburg: Charcot joints are often a sequel of severe neuropathy. It is not
necessarily something speci¢c for a large ¢bre neuropathy and it seems to bemore frequent in patients that have demonstrable small ¢bre neuropathy.Brandt: Coming back to the issue of joint e¡usion, it is worth keeping in mind
that stresses and torques on joints are induced by muscle contraction. Aphenomenon called arthrogenous re£ex inhibition comes into play here. As wedevelop a knee e¡usion, contraction of our quadriceps muscle is inhibited. Thisprotects us from rupturing the joint.Herzog:That’s a goodpoint. In the presence of symptoms (joint pain, swelling of
the joint, loss of joint stability caused by ligament damage etc.), the muscles thatproduce joint movements have been shown to behave distinctly di¡erent (e.g. theforce they produce may be reduced, and the coordination among muscles of a
40 DISCUSSION

synergistic groupmay be disrupted) fromnormal. For example, in our catmodel ofOA (ACL transaction: Herzog et al 1993), we demonstrated that joint instability,caused by ligament transection, was associated with decreased knee extensorforces, and a completely di¡erent activation pattern of the knee £exors relative tosteps cycles during walking and running (Hasler et al 1998). Qualitatively, similarresults have been obtained for humans, where swelling was associated with adecrease in knee extensor activation compared to normal (e.g. de Andradne et al1965, Fahrer et al 1988, Stratford 1981), although the muscle forces in thesesituations were never measured, but were inferred through the electrical activitypatterns (measured using surface electromyograms).Regarding ‘re£ex inhibition’ of muscles in injured or painful joints, there is a
vast body of literature showing that OA, joint swelling, pain, joint instabilityand other joint pathologies are associated with a reduced ability for muscle forceproduction (e.g. Brandt 1997, Hurley et al 1994, Suter et al 1998, Suter & Herzog2000). Re£ex inhibition is measured by asking a subject to perform a maximalvoluntary contraction of the muscle group of interest, measuring the jointmoment exerted by the muscle contraction, and then superimposing an arti¢cialelectrical stimulation on the maximal e¡ort contraction (Merton 1954, Belanger& McComas 1981). The superimposed stimulation to the muscle or thecorresponding nerve will result in additional force production if the maximale¡ort did not recruit all motor units maximally. Such muscle inhibition is normal(i.e. in the knee extensors it varies from about 5^10%, depending on the knee angle,for normal subjects), but reaches high, pathological levels (50^80%) in somepatients with knee (joint) pathologies.Simkin: I am going to mention some results from Arnoldi’s work in my
paper which show that when a horse metacarpophalangeal joint £exes to 90degrees the intraosseus pressure soars dramatically (Arnoldi et al 1980). Anumber of studies in various species have been done, usually with a smalle¡usion, that show a very similar response of intraosseus pressure. This feedsinto Ken Brandt’s earlier point about intraosseus pressure and pain, and perhapsinto the sensitization.Grubb: One important issue that I haven’t covered is that if we really believe
that these a¡erents in the subchondral bone are particularly important for sensingpain in OA, then we need to develop a model for recording from them.What mayalso be interesting is that if the nociceptors are truly in the bone, do you getremodelling of the terminals within the bone as a result of the ongoing damagein OA? They may be sprouting or changing, constantly having to re-grow andsprout.Brandt: They may also be present in the walls of the blood vessels.Grubb: In the joint there is very good evidence that a lot of the a¡erents are
running along the blood vessel. This could be very true. As you get changes
JOINT SENSORY NEURONS 41

in the vascularization of the bone, you may get changes in the sensitivity of thenociceptors. Who knows? There may be a neuropathic component of OA.Brandt:With regard to the issue of innervation or not of articular cartilage, with
advanced disease when there is capillary invasion these capillaries do contain nerveendings.Fernihough: I think the innervation of cartilage is right at the edge where
osteophyte formation is seen (Schwab & Funk 1998).Creamer: I can see how local in£ammation causes peripheral sensitization, so that
mechanical stimuli are perceived as pain. If you take away the in£ammation, doesthis sensitization remain?Grubb: I have never studied a model of in£ammation where the in£ammation
has been allowed to resolve. But when we use enzyme inhibitors to take awaysome of the in£ammatory mediators, the sensitization goes away fairly rapidly,within the time period you would expect to see the breakdown of theprostaglandins. Those tend to be fairly quick e¡ects in terms of theprostaglandins. But I am not sure whether this is true for all cytokines andin£ammatory mediators.Creamer:Do you therefore think that a constant level of in£ammation is needed
in an OA joint for the maintenance of the sensitization?Grubb: That is an interesting question. I don’t know whether the damaged
articulating surfaces and the enhanced mechanical stimuli applied are su⁄cient toproduce activation of the nociceptors in the absence of in£ammation inOA.Aswehave heard, there is a poor correlation between in£ammation and pain. I wouldalways imagine that if you have that amount of damage, there must be somein£ammation in the background all the time.Pisetsky: Have people done these kinds of experiments in either transgenic or
knockout mice with relevant cytokines? For example, if an interleukin (IL)6transgenic mouse is exposed to in£ammation for a long time, is it more or lesslikely to be sensitized? There are enough mouse models around for this to beapproached.Lohmander: What is the degree of biological variability in the sensitization
response in these standardized animal models? Do you have any understanding atthis time of the biological basis for this variability, if it exists? And is there anythingin these models that could teach us about the basis of the variability that appears tobe present in the response of patients to what seems to be a standardized structuralchange in the joint?Grubb: I presented a model where there were two classes of pain a¡erents:
peptidergic and non-peptidergic. This is hugely simplistic. In skin, a¡erents havebeen characterized or broken down into a number of di¡erent types�nociceptorsin particular. There are many subgroups depending on their sensitivity to thermalor mechanical stimuli and so on. We know to some extent which ones exist within
42 DISCUSSION

the joint, but we don’t know how each of these di¡erent types of nociceptorrespond nor how each one is sensitized by in£ammatory mediators.Koltzenburg: At this point in time it is unclear whether, and if so how, the
phenotype of primary a¡erent neurons innervating the skin and joint di¡ers. Partof the di¡erence may simply arise because of the fact that the terminal endings areembedded in a di¡erent tissue so that for example a given mechanical stimulus actsdi¡erentially on the receptors.Lohmander: Is there a genotype for pain response? In other words, what
do we know about the in£uence of genetic variation on the variation in painresponse?Koltzenburg: The central termination patterns of cutaneous and deep somatic
a¡erents are clearly di¡erent. This would imply that the molecules that specifythis di¡erent speci¢city presumably are a result of di¡erential gene expressionpattern during development.Lohmander: Part of the reason I am asking is because of the current searches for
the ‘OAgene’, which are often linked toOApain as the phenotype. Arewe in thesestudies looking for the pain genotype instead of the OA genotype?Manning: There certainly are di¡erences between strains and within strains in
their response to various noxious stimuli. People such as Je¡ Mogul have beenphenotyping across strains and then looking for the genetic basis for thisphenotype.Koltzenburg:There is one caveat.We often assume that di¡erence in behaviour in
inbred mouse strains is attributed to the genetic di¡erences. However, the e¡ect ofupbringing in di¡erent inbred strains has not been evaluated for nociception. Theonus is on him to do cross-fostering experiments to show whether this change heattributes to genetics will hold.Manning: The observation is clear that there are variations within a population
and across strains. The basis for these is unclear at present.Noone has really lookedat this in the kinds of non-surgical OAmodels that we believe are themost relevantto human OA.Grubb: It is important to remember that the phenotypes of the di¡erent
nociceptors are not constant. This changes during in£ammation. Work doneyears ago shows that CGRP and substance P expression changes markedly inin£ammation. We see other changes in receptors and ion channels in thesemodels. Nociceptors are of many types and they are also plastic: they change theirphenotype in response to di¡erent insults, which makes studying themcomplicated.Koltzenburg:What do people think is the crucial tissue that generates the pain in
crystal-induced arthritis, rheumatoid arthritis and OA? Have people done localanaesthetic blocks or in¢ltration anaesthesia? The fact that joint replacement onOA eliminates pain suggests that the synovium isn’t so much involved.
JOINT SENSORY NEURONS 43

Felson: Most of us would be unsure which tissue is the source of pain ineveryone.Creamer:We looked at what happens when we inject local anaesthetic into OA
limbs. E¡ectively, pain is abolished in about 80% of people. On a visual analoguescore a lot of people will score zero, but for some people it appeared to have noe¡ect at all.We concluded thatmost people had a locally driven cause for their pain,but others had a very central cause. Identifying the tissue that is the cause of pain inOA is a bit of a holy grail. We interpreted those experiments as saying thatwhatever it is, it must be in contact with the intra-articular environment, orsomewhere that the anaesthetic could di¡use into.Koltzenburg: So not bone.Creamer: We weren’t really sure whether it would get through. In people with
established OA and lots of cartilage loss, the bone might be accessible to intra-articular £uid.Simkin: If I recall correctly your endpoint was 1 h.What was it like after 10min?
All intracapsular tissues should have been anaesthetized by then?Creamer:We didn’t measure this. Some e¡ects persisted for 24 h and even seven
days. The other thing that came out of this study is that we looked at whathappened on the uninjected knee. Interestingly, there was some symmetry ofresponse. These were all people who had bilateral knee pain, and their worseknee was injected with either saline or anaesthetic. In the group with anaestheticnot only did their index knee pain reduce, but it also decreased in the non-injectedknee.Felson: As someone who reads that paper often and cites it frequently, the n of
response is 6 out of 10 for me, not 8 out of 10. That is, 4 out of 10 did not have aresponse that was dramatic. I would interpret this as being related to bone originpain.Pisetsky: There is also an animal model side of this question. You create an
animal model of OA and look at cartilage. But, from what I am hearing, I shouldalso be looking at ligaments, tendons, capsule, innervation and so on. Everyonejust scores cartilage, but perhaps we should be expanding how we evaluate theanimal models.Hunter: The structural changes that occur in OA and the measurement of these
will probably go through a phase leap over the next two or three years, with the useand involvement of magnetic resonance imaging (MRI). This will give us a betterstructural characterization of which structures are important. Also, usingknowledge that we already have from MRI, we can show that bone is animportant source of pain in so far as bone marrow oedema is a signi¢cant sourceof pain in patients.Pisetsky: Even in human disease, much of the emphasis is on managing
cartilage.
44 DISCUSSION

Grubb: Joint replacement alleviates the pain. Immediately, this makes us thinkthat the pain must come from the articulating surfaces. If those grinding articularsurfaces generate an in£ammation which produces in£ammatory mediators, youmay have pain not only from the articulating surfaces but also from nociceptorsthat are close to the contents of the joint capsule itself. By getting rid of the sourceof the in£ammation, thismay eliminate the sensitization,which need not be limitedto the subchondral bone.Dieppe:As many people in this room know, I think OA is a bone disease. Carlo
Arnoldi and his group did critical experiments on bone, carrying outdecompression and showing that pain, particularly night pain, improved. I thinkwe are forgetting about the heterogeneity of the pain experience in OA. I don’tthink we have done enough work trying to dissect out the possibility thatdi¡erent experiences of pain might relate to di¡erent tissues generating it�night pain, for example, being related to intraosseous pressure. There is alsosome work suggesting that some types of pain might relate more to the presenceof e¡usion. To suggest that there is a single answer and tissue to explainOA pain isna|« ve, given the huge heterogeneity of the pain experience.Schaible: With regard to the reproducibility of these e¡ects, I think the
phenomenon of sensitization in the models that we use is very similar acrossmodels. However, this doesn’t mean that the molecular events that are leading tothis sensitization are always the same. We have experience from a chronicin£ammatory model lasting over 50 days, where we have a thick joint all the timebut we can see an ongoing disease process and an up- and down-regulation ofreceptors (Segond von Banchet et al 2000). It is possible that OA is di¡erent atdi¡erent time points in the same joint, and the pain process could also di¡er. Thisis another complication.Brandt: I agree with Paul Dieppe about the heterogeneity within tissues.
Heterogeneity between sites also exists. In the hip, a small e¡usion has a vastlygreater e¡ect on the intra-articular pressure than it does in a knee joint. No onehas yet mentioned periarticular pain, which is common in arthritis. In ourexperience, 1 of every 4 patients with knee OA has this, at least intermittently, asthe basis for their ‘knee pain’. It is associated with local sharp tenderness over theanserine bursa, which can be relieved by a local injection of anaesthetic or steroid.‘OA’ pain varies not only from patient to patient, but also within the same patientfrom visit to visit.Kidd: Coming back to age-related changes, there is evidence from the
psychophysical studies that as we get older, our response to capsaicin isdramatically reduced, and there is a nice linear decrease in capsaicinresponsiveness in humans over a period of time. Paradoxically, though, the painresponse is actually enhanced. So here you have a nice example of nociceptorresponse not being mirrored by the pain response. I have a question relating to
JOINT SENSORY NEURONS 45

the silent nociceptors which Dr Schaible described over a decade ago. This was apivotal observation that within the joint many of the a¡erents are silent. This fedback into observationsmade in the 1930s, where a group of heroic surgeons here inLondon went into joints unanaesthetized, poked and prodded them and got nopain response whatsoever. This raises the issues of whether we always need anin£ammatory mediator to get synovium-driven pain. Do you feel that there arean equal number of silent nociceptors supplying bone tissues? Pat Mantyhrecently showed a much denser innervation of bone than we had hithertosuspected. They seem to be unmyelinated a¡erents. Do you feel that we are likelyto have an equal proportion of silent nociceptors supplying the bone as are seen insynovium? This would imply that an in£ammatory response is needed to sensitizethese ¢bres and hence give pain?Schaible: It is always di⁄cult to say howmany silent nociceptors there are. They
need an additional stimulus such as sensitization to become responsive. They arefound in the skin at about 10%, which is not that much.My guess in the deep tissueof the joint is that about 30% are silent. It could well be that the density of silentnociceptors is important with regard to how strong pain is.Pisetsky: Is there any relatively simple operational way to ¢nd out whether a
patient’s joint has been sensitized?Schaible:We cannot di¡erentiate between a peripheral and a central sensitization.
You can just ¢nd out whether the patient is hypersensitive or not.Pisetsky: Are people with OA sensitized in that joint area?Schaible:We can’t tell.Pisetsky: People get better rapidly� sometimes the day after� following joint
replacement surgery.What does this imply about sensitization?Or is the pain froma diseased joint OA somuch greater than the pain from sensitization, so that whenthe joint is replaced, the pain essentially goes away even if there is somesensitization.Schaible: We can counteract sensitization by increasing inhibitory systems, for
example. This is another complication.Koltzenburg: It is much better understood for cutaneous pain and some forms
of neuropathic pain, where central sensitization in the absence of peripherala¡erent drive is very rare. In most experimental models you can show that alarge component of central sensitization is in fact a physiological re£ectionof the peripheral nociceptive a¡erent drive. There are some phenomena thatgo beyond this. Hyperalgesia to pinprick stimuli that is commonly found inthe capsaicin model is initiated by nociceptor a¡erent input but does notrequire primary a¡erent input for its maintenance. However, in most othercases of hyperalgesia, central sensitization will regress without primary a¡erentinput.Pisetsky:Over how long a period of time?
46 DISCUSSION

Koltzenburg: With capsaicin it is in the range of minutes. In some conditions itmay take much longer. As far as it has been tested we are talking hours at the most.Grubb: What proportion of OA patients respond to non-steroidal anti-
in£ammatory drugs (NSAIDs)? This is an indication of whether they aresensitized or not: if they respond to NSAIDs, there is at least prostaglandin-induced sensitization of the a¡erents.Brandt:About 30%havemarked improvement, a fewgetworse and themajority
exhibit modest improvement. If you add paracetamol to the NSAID someadditional analgesia is seen, but a very high proportion of OA patients treatedwith both agents still complain of residual pain.Grubb: Is the rest of the pain due to in£ammatory mediators other than
prostaglandin, or is it not due to sensitization? This is an interesting question.Hunter: If you block the sensitization with substance P or a NMDA inhibitor,
what impairment do you see on other neural re£exes and responses? In particular,what e¡ect do you think this may have on proprioception and motor control?Koltzenburg: First, substance P antagonists have not worked clinically. If you
give NMDA antagonists in an experimental setting, most subjects will complainabout psychotropic side-e¡ects. The majority of the e¡ects are not necessarily onproprioception but more on cognitive e¡ects. This has so far been the limitingfactor in the use of NMDA antagonists.
References
Arnoldi CC, Linderholm H, Mussbichler H 1972 Venous engorgement and intraosseoushypertension in osteoarthritis of the hip. J Bone Joint Surg Br 54:409^421
Arnoldi CC, Reimann I, Mortensen S et al 1980 The e¡ect of joint position on juxta-articularbone marrow pressure. Relation to intra-articular pressure and joint e¡usion�anexperimental study on horses. Acta Orthop Scand 51:893^897
Belanger AY, McComas AJ 1981 Extent of motor unit activation during e¡ort. J Appl Physiol51:1131^1135
Brandt KD 1997 Putting muscle into osteoarthritis. Ann Intern Med 127:154^155de Andradne JR, Grant C, Dixon ASJ 1965 Joint distension and re£ex muscle inhibition in theknee. J Bone Joint Surg Am 47:313^322
Fahrer H, Rentsch HU, Gerber NJ, Beyeler C, Hess CW, Gruenig B 1988 Knee e¡usion andre£ex inhibition of the quadriceps. J Bone Joint Surg Br 70:635^638
Ferrell WR, Nade S, Newbold PJ 1986 The interrelation of neural discharge, intra-articularpressure, and joint angle in the knee of the dog. J Physiol 373:353^365
Hasler EM, Herzog W, Leonard TR, Stano A, Nguyen H 1998 In-vivo knee joint loading andkinematics before and after ACL transection in an animal model. J Biomech 31:253^262
Herzog W, Adams ME, Matyas JR, Brooks JG 1993 A preliminary study of hindlimb loading,morphology and biochemistry of articular cartilage in the ACL-de¢cient cat knee.Osteoarthritis Cartilage 1:243^251
HurleyMV, Jones DV, NewhamDJ 1994 Arthrogenic quadriceps inhibition and rehabilitationof patients with extensive traumatic knee injuries. Clin Sci (Lond) 86:305^310
JOINT SENSORY NEURONS 47

KrauspeR, SchmidtM, SchaibleHG1992 Sensory innervation of the anterior cruciate ligament.An electrophysiological study of the response properties of single identi¢edmechanoreceptors in the cat. J Bone Joint Surg Am 74:390^397
Maitland ME, Leonard TR, Frank CB, Shrive NG, HerzogW 1998 Longitudinal measurementof tibial motion relative to the femur during passive displacements and femoral nervestimulation in the ACL-de¢cient cat model of osteoarthritis. J Orthop Res 16:448^454
Merton PA 1954 Voluntary strength and fatigue. J Physiol (Lond) 123:553^564Segond von Banchet GG, Petrow PK, Brauer R, Schaible HG 2000 Monoarticular antigen-induced arthritis leads to pronounced bilateral upregulation of the expression of neurokinin1 and bradykinin 2 receptors in dorsal root ganglion neurons of rats. Arthritis Res 2:424^427(available on http://arthritis-research.com/content/2/5/424)
Schaible HG, Grubb BD 1993 A¡erent and spinal mechanisms of joint pain. Pain 55:5^54Schwab W, Funk RH 1998 Innervation pattern of di¡erent cartilaginous tissues in the rat. ActaAnat (Basel) 163:184^190
Stratford P 1981 Electromyography of the quadriceps femoris muscles in subjects with normalknees and acutely e¡used knees. Phys Ther 62:279^283
Suter EW, Herzog W 2000 Does muscle inhibition after knee injury increase the risk ofosteoarthritis? Exerc Sport Sci Rev 28:15^18
Suter EW, Herzog W, DeSouza K, Bray RC 1998 Inhibition of the quadriceps muscles inpatients with anterior knee pain. J Appl Biomechan 14:360^373
48 DISCUSSION

Neuromuscular aspects of
osteoarthritis: a perspective
Kenneth D. Brandt
Multipurpose Arthritis and Musculoskeletal Diseases Center, and Performing Arts MedicineProgram, Indiana University School of Medicine, 1110 West Michigan Street, Indianapolis,IN 46202-5100, USA
Abstract.Osteoarthritis (OA) represents failure of the diarthrodial joint and may be dueto a primary abnormality in any of the tissues of the joint, e.g. articular cartilage,subchondral bone, synovium, periarticular muscle, or sensory nerves whose termini liewithin the joint. Neuropathic arthropathy, due to severe sensory neuropathy, causessevere and rapid breakdown of joints. We have shown that interruption of sensoryinput from the ipsilateral hind limb strikingly accelerates progression of OA afteranterior cruciate ligament transection in the dog; a clinical correlate exists in humanswith diabetic neuropathy who sustain even minor joint trauma. Knee OA in humans isaccompanied by defects in proprioception, although it is not clear whether theneurological abnormality is primary or a consequence of intra-articular pathology. Themagnitude of the load on a joint and, especially, the rate of impulsive loading, in£uencedevelopment of OA. It is relevant, therefore, that quadriceps weakness may precededevelopment of knee OA in some people, insofar as this may diminish the e¡ectivenessof protective muscular re£exes and thereby increase deleterious joint loading. Individualsvary with respect to how they load their joints, perhaps because of genetic di¡erences incentral program generators.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 49^63
Although the most striking pathologic changes in osteoarthritis (OA) are usuallyfound in the articular cartilage, OA is not a disease of any single tissue (e.g.cartilage) but a disease of an organ, the synovial joint. It is analogous, therefore,to heart disease, in which a primary problem in the endocardium, epicardium ormyocardiummayproduce a syndromeof congestive heart failure. In some cases thejoint may fail because of a primary problem in the articular cartilage; in other casesthe problemmay reside in the subchondral bone, synovium, periarticular muscles,supporting ligaments or the nerves whose sensory termini lie within the joint orsurrounding tissues.Although it is popular to categorize OA as being either primary (aetiopathic) or
secondary, in a general sense all OA is secondary. A genetic abnormality, such as a
49
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

point mutation in the cDNA coding for Type II collagen, may lead tospondyloepiphyseal dysplasia, resulting in wide-spread and early-onset OA andproviding an example of OA arising as the result of a primary cartilage disorder.Genetically based systemic metabolic abnormalities, such as haemochromotosis,Wilson’s disease and calcium pyrophosphate crystal deposition (CPPD) diseaseare also genetic conditions leading to chondropathies and to secondary OA.However, the OA phenotypes associated with these genetic disorders account foronly a very small proportion of the universe of clinical OA.Considering OA as a clinical entity, it is the major cause of chronic disability
among the elderly, in whom it is the most common cause of di⁄culty in climbingstairs and in walking. OA is chie£y a disorder of the elderly. In consideringdisability in OA, one must take into account not only the direct consequences ofthe structural changes in the joint due to the arthritis, but also the consequences ofageing and of inactivity (Stenstrom&Minor 2003; Fig. 1).
Adverse e¡ects of immobilization of a joint
Much of the discussion centring around the relationship between biomechanicsand OA relates to overloading of the joint, i.e. the concentration of peakdynamic loads. It is less widely recognized that a reduction in loading is alsodetrimental to joint tissues. For example, immobilization of the hind limb of anormal dog by application of an orthopaedic cast, restricting motion of the kneeand unloading of the joint during ambulation, rapidly results in striking changes inthe articular cartilage (Palmoski et al 1979). The cartilage thins, the net rate of
50 BRANDT
FIG. 1. Disability in patients withOA is due to the inactivity associated with the disease and tothe e¡ects of aging, as well as to the direct e¡ects of the intra-pathology. Reproduced withpermission from Stenstrom&Minor 2003.

proteoglycan synthesis is reduced by as much as 50%, and reductions of similarmagnitude occur in the proteogylcan concentration and proteogylcan content ofthe cartilage (Fig. 2). These changes are associated with striking disuseosteoporosis of the subchondral bone and atrophy of the periarticular muscles.If the period of immobilization is prolonged, ¢brofatty ankylosis of the joint
occurs. If, however, the period of immobilization is limited to a few months, theabove changes are completely reversible following removal of the cast andremobilization of the extremity. Immediately upon restoration of loading, thechondrocytes resume synthesis of matrix proteoglycans, which are produced at agreatly accelerated rate� comparable to, or greater than, that seen in OA. Theosteoporosis in the subchondral bone is also reversed. Within only a few weeks,biochemical, biomechanical, metabolic and ultrastructural analyses of the cartilageprovide no hint of the profound changes that had been present only a few weeksearlier� i.e. the changes are completely reversible (Fig. 2).
NEUROMUSCULAR ASPECTS 51
FIG. 2. Articular cartilage, stained with Safranin-O, from the femoral condyle of the left knee(left hand panel) of a dogwhose right hind limb had been immobilized in an orthopaedic cast for8 weeks (magni¢cation �35). Immobilization resulted in a marked decrease in the thickness ofthe articular cartilage of the immobilized knee, with depletion of proteoglycans and a decrease inthe net rate of proteoglycan synthesis. Note also the marked disuse osteoporosis that occurredduring the period of immobilization (right hand panel).

If, however, after articular cartilage atrophy has been induced byimmobilization, an attempt is made to accelerate recovery by loading of the limbduring remobilization, e.g. by a few weeks of low-intensity treadmill running,reversal of the atrophic changes in the cartilage is prevented (Palmoski & Brandt1981). Although proteoglycan synthesis resumes at a greatly augmented level, asdescribed above, the newly synthesized proteoglycans are not e¡ectively laid downin the cartilage matrix but are lost into the joint space, and the depletion of matrixproteoglycans appears to persist inde¢nitely. Notably, this ‘post-rehabilitationarthropathy’ occurs with levels of treadmill activity so low (e.g. 4 miles/h,30min/d; 5/d per week) that they produce no discernable changes in the normalcanine joint.Thus, after a period of disuse, such as may occur in some subjects with painful
hip or knee OA, atrophic changes develop in the articular cartilage, bone andperiarticular muscles. The chondrocytes, lying in their lacunae within thecartilage matrix, no longer have a proteoglycan cushion to protect them from thestresses of normal load bearing. We have shown that the cartilage atrophy thatfollows immobilization of the knee is due chie£y to the lack of normalcontraction of the periarticular muscles (hamstrings, quadriceps) in the stancephase of gait that results in loading of the knee joint, rather than to a lack of£exion/extension of the joint (Palmoski et al 1980). When knees of rabbits wereimmobilized in an orthopaedic cast, stimulation of the quadriceps by an electrodeinserted into the muscle through the cast prevented the atrophy of articularcartilage and subchondral bone that developed in the absence of musclestimulation (Burr et al 1984).
How do the experimental data presented
above relate to humans with OA?
Many patients with knee OA exhibit atrophy of the muscles surrounding theinvolved joint and, in particular, the quadriceps muscle. It has generally beenconsidered that this represents the consequences of disuse of the arthriticextremity as the patient avoids bearing load on the painful joint to minimizediscomfort. OA pain is classically worse with activity and relieved by rest.Several studies (see Slemenda et al 1997) have shown that the strength of amaximum voluntary contraction is some 30^40% lower in patients with kneeOA than that in age- and sex-matched controls. It is important torecognize, therefore, that the quadriceps weakness in patients with OA isreversible through strength-training�even among the elderly. The statement:‘Well, the 75 year old patient with OA isn’t going to be compliant with anexercise program, even if it should work,’ is nonsense. Adherence to therapeuticexercise programs by patients with OA is no poorer than their adherence to
52 BRANDT

prescribed dosing of non-steroidal anti-in£ammatory drugs (NSAIDs), and maybe better than the latter.More to the point are the questions: ‘Why should the patient with OA exercise?’
‘What good will it do?’ The answers are important: quadriceps strengtheningexercises may result in marked improvement in knee pain and function amongpatients with knee OA. In some cases, that improvement may be as great as orgreater than that achieved with NSAIDs.Furthermore, with respect to knee OA, in particular, quadriceps weakness may
not only be the result of joint pain, it plays an important role in pathogenesis ofstructural damage in the disease, i.e. it may be a risk factor for OA. Johansson et al(1991) and others have demonstrated the importance of the quadriceps muscle as astabilizer of the knee joint. Quadriceps contraction reduces anteroposteriordisplacement of the tibia on the femur. In further support of theaetiopathogenetic importance of quadriceps weakness in knee OA. In a three yearstudy of an elderly community-based cohort from central Indiana (Slemenda et al1997), found that incident radiographic knee OAwas associated with a decrease inthe baseline level of peak knee extensor torque. Notably, the quadriceps weaknessin these subjects was not the result of disuse atrophy. Indeed, because of obesity,lower extremity muscle mass in these subjects was increased, relative to thecontrols, and weakness was seen even among individuals who denied havingknee pain and considered themselves to be as active as, or more active than, theirpeers. In the absence of OA or knee pain, obesity is associated with an increase inlower extremity muscle mass. The obese subject needs more muscle to support theadiposity. The data suggest that, despite the increase in muscle bulk, for reasonsthat are unclear, the quadriceps muscle may be functionally impaired in subjectswith knee OA.Radin et al (1986) have shown that about 30%of individuals exhibit a gait pattern
that results in a generation of heel strike transient at the knee (Je¡erson et al 1990).Because thequadricepsmuscle is themainanti-gravitymuscle in the lowerextremityand serves as a brake to decelerate the leg during gait, thereby minimizing theimpact of heel strike, quadriceps weakness may lead to excessive impulsiveloading of the knee with gait and may predispose to joint pain and to OA (Radinet al 1991). The fact that a heel strike transient can be produced by the injection oflocal anaesthetic around the femoralnerveof subjectswhodonotnormallygeneratea heel strike transient (Radin et al 1986) provides biomechanical support for thatargument. Furthermore, subjects who generate a heel strike transient can betrained through biofeedback to alter their gait, so as to eliminate impulsiveloading of the knee (E. L. Radin, personal communication). Whether that willprotect against development of knee pain or OA remains to be shown.The neurophysiological mechanism underlying generation of a heel strike
transient during gait in some individuals may relate to di¡erences among us with
NEUROMUSCULAR ASPECTS 53

respect to central pattern generators (CPGs) in the spinal cord, as discussed byVilensky (2003). In any event, it is clear that periarticular muscle is a majorshock-absorber for joints. Radin et al (1984) showed that repetitive impulsiveloading of the hind limb of rabbits led to rapid breakdown of articular cartilageand bone in the ipsilateral knee when the load was delivered rapidly (50ms fromonset to peak) even if themagnitude of loadwas not excessive. In contrast, loads ofcomparable or even greater magnitude did not result in joint damage if the rate ofloading was ramped up more gradually (onset to peak¼500ms), permitting timefor the periarticular muscles to absorb the load by eccentric contraction.Notably, Mikesky et al (2000) found that the rate of loading of the knee was
much slower among strength-trained normal females (e.g. weight-lifters, rowers)than that among sedentary or aerobically trained women. Longitudinal follow-upof these subjects may ascertain whether the former group is protected fromdevelopment of knee OA.Figure 3 illustrates the relationships between immobilization, muscle weakness
and joint damage, as discussed above. In addition, it focuses on another aspect ofthe pathophysiology of knee OA�re£ex inhibition of the quadriceps muscle(arthogenous muscle inhibition)�which may provide an additional mechanismforweakness of themuscle surrounding the arthritic joint (Stokes&Young 1984).
E¡ects of therapeutic exercise in patients with knee OA
Several mechanisms may explain the salutory e¡ects of exercise on pain andfunction in patients with knee OA. Exercise may result in an increase in
54 BRANDT
FIG. 3. The ‘vicious circle of events’ that may lead to OA. Reproduced with permission fromStokes & Young (1984).

endurance, improvement in proprioceptive acuity, and a decrease in the severityof arthogenous muscle inhibition. In addition, it may result in improvementin comorbidity (e.g. obesity, cardiovascular disease) and psychological statusmay facilitate weight loss in the obese subject. It is of interest, therefore, thatSharma et al (2003) have suggested recently that quadriceps strength may bedetrimental to subjects with knee OA who have signi¢cant varus^valgusmalalignment or ligamentous laxity and that in the presence of these localabnormalities the exercise program for the patient with OA should beindividualized to reduce the risk of accelerating joint damage. More work isneeded in this area.The bene¢ts of exercise in patients with OA, furthermore, may extend beyond
the obvious improvements in joint pain and function.Analyses of the data from theFAST trial illustrate this point (Ettinger et al 1997). In this 18 month randomizedplacebo-controlled trial that compared the e¡ects of aerobic exercise andstrengthening exercise in subjects with knee OA, only modest improvements inknee strength and aerobic ¢tness were achieved. Only minimal di¡erences inoutcomes were seen upon comparison of the two exercise treatment groups,probably due to the relatively modest levels of exercise employed by theinvestigators in an attempt to minimize the drop-out rate. However, the resultsof the FAST trial were interesting with respect to the e¡ects of exercise ondepression (Penninx et al 2002) and to loss of activities of daily living (ADL)(Penninx et al 2001). Among subjects who were not depressed at the onset of thestudy and exhibited a high level of compliance with the exercise protocol, none ofthose in the aerobic exercise group but 26% of those in the strength-training groupand 31% of the controls developed evidence of depression during the trial.Furthermore, the risk of developing loss of ADL was only about 40% as greatamong subjects who were highly compliant with the exercise regimen as amongthose with poorer compliance.
Neuropathic arthropathy
It has been recognized for many years that impairment of sensory input for anextremity due to an underlying neurological disorder (e.g. diabetes mellitus,tabes dorsalis, syringomyelia) may result in rapid, severe breakdown of the jointsin that extremity. The mechanism most widely implicated in this phenomenon isthe accumulation of repeated microtrauma associated with usage of the joint, dueto impairment of proprioception or nociception, or both. However, a wide varietyof ablative neurosurgical procedures have failed to consistently produceneuropathic arthropathy experimentally.Some insight into this apparent inconsistency is provided by our studies in the
canine cruciate-de¢ciency model of OA. After transection of the anterior cruciate
NEUROMUSCULAR ASPECTS 55

ligament (ACL) in the neurologically intact dog, end-stage OA developsgradually, so that full-thickness ulceration of the articular cartilage is not seensooner than four years after the creation of joint instability (Brandt et al 1991).On the other hand, if sensory input from the ipsilateral extremity is markedlyreduced (e.g. by L4-S1 dorsal root ganglionectomy or transection of the articularnerves supplying the knee), advanced changes of OA, with full-thicknessulceration of the cartilage, may occur within only a few weeks (O’Connor et al1992). In the normal dog with a stable knee, the neurosurgical procedure alonedoes not lead to arthropathy even after follow-up periods as long as 2 years. Gaitstudies have shown that the combination of dorsal root ganglionectomy andACLT results in a marked alteration in knee kinematics, with hyperextension attouchdown, accounting for the rapid acceleration of joint destruction (Vilenskyet al 1997).A human correlate emphasizes the clinical relevance of the above ¢ndings:
among patients with severe diabetic neuropathy, neuropathic joint disease,usually in the foot, develops in about 10^12% of cases. Why not morefrequently? We have seen a series of patients with diabetic neuropathy who hadno evidence of joint disease until they sustained relatively minor trauma (e.g. asimple ankle sprain), following which neuropathic arthopathy developed acutelyand the involved joint broke down within a few weeks (Slowman et al 1990). Theanalogy to the results of the canine experiments described above is striking: in theexperimental model, dea¡erentation of the hind limb was achieved through anextensive neurosurgical procedure, whereas our patients presented with diabeticneuropathy. In the canine model, joint instability was established by ACLtransection, whereas the diabetic patients developed an unstable joint afterrelatively minor trauma. In both cases, however, the result was the same� in thepresence of an underlying neurosensory defect, joint traumawas followed by rapidbreakdown of the joint.Notably, in the dog, if ACL is performed prior to interruption of sensory input
from the limb, the development of knee OA is not accelerated. This suggests thatthe neurologically intact dog is able tomodify its CPGs tominimize the changes inloading of the knee induced by ACL transection. In contrast, when theneurosurgical procedure precedes ACL transection, the central nervous system(CNS) is unable to recognize the acute changes in loading and joint mechanicsresulting from ACL de¢ciency and is, therefore, unable to accommodate to thesealterations by modifying the pattern of gait (Vilensky 2003).
Proprioception
Awareness by the CNS of the position of the joint within space generates muscleactivity that results in the control of motion, stability of the joint and protection of
56 BRANDT

the joints from injury.Muscle is not only an e¡ector organ, essential formovementof a joint, it is an important sensory organ, containing specialized sensory nerveendings, muscle spindles (whose a¡erent ¢bres transmit proprioceptive impulsesthrough the dorsal root ganglion into the spinal cordwhere re£exes are establishedin which the e¡erent arm is a motor nerve to periarticular muscle.Several points may be made with respect to proprioception in OA: receptors in
muscle, tendons, joint capsule, ligaments, horns of the menisci and skin provideproprioceptive input that contributes to protective muscular re£exes and tovoluntary muscle activity. Proprioceptive activity diminishes with age.Furthermore, in subjects with unilateral knee OA, proprioception is lessaccurate� even in the apparently normal contralateral knee� than in non-arthritic control subjects, raising the possibility that a subclinical neurologicaldefect is of aetiological importance in some subjects with idiopathic knee OA�a view supported by the above discussion (Vilensky et al 1997).Proprioceptive impairment, furthermore, may be associated with impaired
physical function. It is notable, therefore, that proprioception may be improvedwith elastic bandages, knee sleeves, orthoses or exercise. However, the impact ofthe improvement in proprioception on joint pain, function, or progression ofstructural damage in OA is unclear.
References
Brandt KD, Braunstein EM, Visco DM, O’Connor B, Heck D, Albrecht M 1991 Anterior(cranial) cruciate ligament transection in the dog: a bona ¢de model of osteoarthritis, notmerely of cartilage injury and repair. J Rheumatol 18:436^446
Burr DB, Frederickson RG, Pavlinch C, Sickles M, Burkart S 1984 Intracast muscle stimulationprevents bone and cartilage deterioration in cast-immobilized rabbits. Clin Orthop Rel Res189:264^278
EttingerWH, Burns R,Messier SP et al 1997 A randomized trial comparing aerobic exercise andresistance exercise with a health education program in older adults with knee osteoarthritis.The Fitness Arthritis and Seniors Trial (FAST). J AmMed Assoc 277:25^31
Je¡erson RJ, Collins JJ, Whittle MW, Radin EL, O’Connor JJ 1990 The role of the quadricepsin controlling impulsive forces around heelstrike. Proc Inst Mech Eng 204:21^28
Johansson H, Sjolander P, Sojka P 1991 A sensory role for the cruciate ligaments. Clin Orthop268:161^178
Mikesky AE,Meyer A, Thompson KL 2000 Relationship between quadriceps strength and rateof loading during gait in women. J Orthop Res 18:171^175
O’Connor BL, ViscoDM, BrandtKD,Myers SL,Kalasinski L 1992Neurogenic acceleration ofosteoarthritis: the e¡ects of prior articular nerve neurectomy on the development ofosteoarthritis after anterior cruciate ligament transection in dogs. J Bone Joint Surg Am74:367^376
Palmoski MJ, Brandt KD 1981 Running inhibits the reversal of atrophic changes in canine kneecartilage after removal of a leg cast. Arthritis Rheum 24:1329^1337
Palmoski MJ, Perricone E, Brandt KD 1979 Development and reversal of a proteoglycanaggregation defect in normal canine knee cartilage after immobilization. Arthritis Rheum22:508^517
NEUROMUSCULAR ASPECTS 57

Palmoski MJ, Colyer RA, Brandt KD 1980 Joint motion in the absence of normal loading doesnot maintain normal articular cartilage. Arthritis Rheum 23:325^334
Penninx BW,Messier SP, RejeskiWJ et al 2001 Physical exercise and the prevention of disabilityin activities of daily living in older persons with osteoarthritis. Arch Intern Med 161:2309^2316
Penninx BW, Rejeski WJ, Pandya J et al 2002 Exercise and depressive symptoms: a comparisonof aerobic and resistance exercise e¡ects on emotional and physical function in older personswith high and low depressive symptomatology. J Gerontol B Psychol Sci Soc Sci 57:P124^P132
Radin EL, Martin RB, Burr DB, Caterson B, Boyd RD, Goodwin C 1984 E¡ects of mechanicalloading on the tissues of the rabbit knee. J Orthop Res 2:221^234
Radin EL, Whittle MW, Yang KH et al 1986 The heelstrike transient, its relationship with theangular velocity of the shank, and the e¡ects of quadriceps paralysis. In: Lantz SA, King AI(eds) Advances in bioengineering. American Society of Mechanical Engineering, New York,p 121^123
Sharma L, Dunlop DD, Cahue S, Song J, Hayes KW 2003 Quadriceps strength andosteoarthritis progression in malaligned and lax knees. Ann Intern Med 138:613^619
Slemenda C, Brandt KD, Heilman DK et al 1997 Quadriceps weakness and osteoarthritis of theknee. Ann Intern Med127:97^104
Slowman-Kovacs S, Braunstein EM, BrandtKD 1990 Rapidly progressive Charcot arthropathyfollowing minor joint trauma in patients with diabetic neuropathy. Arthritis Rheum 33:412^417
StenstromCH,MinorMA2003Evidence for the bene¢t of aerobic and strengthening exercise inrheumatoid arthritis. Arthritis Rheum 49:428^434
Stokes M, Young A 1984 The contribution of re£ex inhibition to arthrogenous muscleweakness. Clin Sci (Lond) 67:7^14
Vilensky J 2003Neuromuscular System. Innervation of the joint and its role in osteoarthritis. In:Brandt KD, Doherty M, Lohmander LS (eds) Textbook on osteoarthritis, 2nd edn. OxfordUniversity Press, p 161^167
Vilensky JA, O’Connor BL, Brandt KD, Dunn EA, Rogers PI 1997 Serial kinematic analysis ofthe canine hind limb joints after dea¡erentation and anterior cruciate ligament transection.Osteoarthritis Cartilage 5:173^182
DISCUSSION
Pisetsky: In your work onmuscle strength, there was a comparison of males andfemales. I think muscle strength in females showed a relationship withdevelopment of OA, but the males did not. Can you comment on that?Brandt: No, it was an observation. The numbers were too small to say more
about this.Fernihough: I believe Eric Radin was asked that question at a conference a long
time ago and he said it was because women wear silly shoes!Woodworth: Was body mass index (BMI) included, to determine whether this
recognized risk factor for OA progression might be an explanation for the e¡ectin women but not men?
58 DISCUSSION

Brandt:Yes, we adjusted for obesity and also for lean tissue mass (muscle) in thethigh, as determined by DXA.Herzog: I was interested in your two di¡erent categories of people according to
how they walk� the diggers and gliders. I noticed that even in the diggers wherethe impact occurred relatively fast, the total load was one times bodyweight. Iknow from people who run and contact the ground with their heel ¢rst that theinitial impact is much higher. Would this suggest that if you are a runner whostrikes the ground with your heel ¢rst, that you are more likely to developosteoarthritis?Brandt: The data would suggest that. It isn’t so much the magnitude of the load
as the rate of loading. The rabbit experiments support this.Herzog: We have just ¢nished a small study looking at loading of joints by
maximal muscular stimulation, and also by blunt impact. When you match theloading inside the joint to the pressure that goes across the joint, we ¢nd thatthe maximum muscular loading at the maximum rate will not produce celldeath whatsoever. However, if you reach the same load in 3ms by impactloading, there is an enormous amount of cell death. These preliminary datawould agree with your qualitative argument that the rate of joint loadinghas a greater in£uence on the adaptive/degenerative response of articularcartilage than the absolute load. However, we should keep in mind thatstudies in this area are still preliminary, and the precise link between in vivojoint loading, stress^strain ¢elds inside the joint, and the correspondingbiological response of joint structures (such as the articular cartilage) is notknown at present.Brandt: To come back to bone, rapid impulsive loading may also reactivate the
secondary centre of ossi¢cation, and initiate changes at the tide mark. It a¡ectsmore than only muscle and cartilage.Hunter: The heel strike is very interesting. Youmade a comment that you could
negate the e¡ect of the heel strike by anaesthetizing a femoral nerve. Was thatpurely by way of inactivating a quadriceps muscle? The quadriceps muscle ispresumably responsible for that heel strike impulse.Brandt: Yes, and it attenuates it.Hunter:Are there other ways that you think the heel strike could be attenuated?Brandt: Shoes may make a di¡erence. Radin has designed a study using
biofeedback to change diggers into gliders, by placing an accelerometer anchoredaround the knee that beeps if the knee is loaded too rapidly in gait.Hunter: I understood that their load throughmid-stance was greater, not just on
the heel strike.Brandt: Yes, but the most striking change is the heel strike transient. John
O’Connor estimated this to be some 15 times greater in the knees of diggers thanof gliders. This would ¢t the changes seen in the animal models.
NEUROMUSCULAR ASPECTS 59

Hunter: For a subject with symptomatic OA, do you think this has analogiesfrom that perspective also? Do these repeat stimuli coming from the foot withheel impact have any role to play in triggering symptoms?Brandt:The study I have justmentioned should answer that question.One thing
we can say is that exercise generally has a positive e¡ect on OA symptoms, withimprovement in pain and function.Rediske: Could that re£ect more the systemic response to exercise? For example,
could exercise trigger endorphin release?Brandt: It is not intensity related.Felson: I wanted to ask about the dark side of strength. You commented on
Sharma’s recent work which suggests that in malaligned joints, strong musclecontraction will cause loading in a very localized area. We published data prior tothis from the Framingham study showing that hand-grip strength was associatedwith amarked increase in the development of OA in proximal hand joints. Perhapsstrength isn’t so good. You have commented on exercise in general being salutary:I am wondering what the trade-o¡s are here. When is exercise good, and when isstrengthening and exercise bad?Brandt: Those are the questions that Leena’s data raise (Sharma et al 2003). We
need to segregate this out. Exercise in OA needs to pay attention to ¢tness(cardiovascular condition) as well as to strengthening and also range of motion.Pisetsky:Goingback to the diggers andgliders, towhat extent is thismale versus
female? Is it correlated with something else?Brandt: It is not correlated with gender. It is one-thirds diggers, two-thirds
gliders.Pisetsky: Is that common among populations?Brandt: As far as I know it hasn’t been studied in other populations.Dieppe: There has been a lot of epidemiological work on the issue of whether
doing a lot of exercise is a cause ofOA, but there has been precious little onwhetherexercise is a good or bad thing once you have OA. The trials have been largelyshort-term and have concentrated on pain relief. From what you are sayingtoday, Ken Brandt, one of the take home messages is that the evidence isbeginning to accumulate suggesting that exercise is a bad thing for damagedjoints. What is your perspective on this? All my patients ask me about it, and Ihave been telling them to keep active.Brandt: It is something we need to understand better. It is clear that exercise to
improve strength and cardiovascular ¢tness can be performed withoutexacerbating the symptoms. Whether we are doing something detrimentalstructurally, depending on the local mechanics and the speci¢cs of the exercise, isunclear.Felson: I am certain that it isn’t so simple. Ken Brandt brought up a nice way of
thinking about an una¡ected normal joint, and a vulnerable joint. Those two are
60 DISCUSSION

likely to be di¡erent entities with respect to the e¡ect of strengthening and exercise.I was hearing echoes of this in our earlier discussion about central sensitizationinduced by in£ammation which produces a very di¡erent joint in its response topain than one that would otherwise be normal. The same is probably true here ofthe e¡ects of strength of muscle contraction, and of exercise. The surprise to me inDr Sharma’s data was that the neutrally aligned joints weren’t also damaged. Inlong enough follow-up I think they will be. It is likely therefore that strengthprovokes damage, ultimately. It is in part because all of the joints she is studyingare more or less vulnerable joints.Dieppe: I would go along with that. Leena Sharma’s work is very nice, but it is a
very short-term study.Brandt: On the other hand, quadriceps weakness has some analogy to cruciate
ligament de¢ciency. The quadriceps is a stabilizer of the knee. It very much comesdown to the speci¢cs: what is being loaded and how?Felson: The non-vulnerable, normal joint is the one you study in those women
and men whom you follow longitudinally. Then, the e¡ect of strength may be inthe deceleration of the limb before heel strike and the distribution of a stabilizingforce in dynamic loading, as you said. These are the bene¢cial elements ofquadriceps contraction. Then when the joint becomes a vulnerable one, the darkside starts to overwhelm the favourable side of strength for a lot of biomechanicalreasons.Rediske: Could the kind of exercise that is contributing to increased quadriceps
strength be a contributing factor? Each form of exercise could have a di¡erentimpact on the a¡ected joint.Felson:They also have a di¡erent impact at di¡erent angulations of limbs, where
limbs have more or less joint space area to load across.Brandt: It also depends on the milieu of the joint. Studies looking at
marathon runners to see whether running leads to OA were looking at aselect population of people who had proved their ability to run 26 miles. Byde¢nition, the people able to do this have a favourable mechanical environmentin their joints. People who drop out with soft tissue injuries are protected bytheir aches and pains.Rediske: In these studies being discussed, what kinds of exercise contributed to
increased quad strength?Felson: One of the arguments against strength would be that aerobic exercise
makes people feel better and it doesn’t have any measureable e¡ect on strength.This would argue against strength playing a primary role in the salutary bene¢tsof exercise in knee OA. Resistance training also has a salutary e¡ect on symptoms,but the long-term e¡ects on structure aren’t clear.Pisetsky: In the dogmodels, have you looked at capsules, ligaments and tendons
around the joints to see what changes result from immobility?
NEUROMUSCULAR ASPECTS 61

Brandt: With the duration of immobility that we employed, few gross changeswere seen in these structures. If the immobilization is sustained for a considerablylonger period, ¢bro-fatty ankylosis of the joint will occur. It looks a bit like theaftermath of a badly treated Staphylococcus infection. In our experimentsconsiderable muscle atrophy occurs, but it is reversible. Tendons and ligamentswill tighten and shorten.Simkin: It would be worth mentioning occupational information. As I
understand it, the heavy lifters are at signi¢cantly increased risk.Felson:The data show that the stereotyped occupational activities which tend to
cause OA are those which require bending and lifting. Squatting increases loadingacross the tibiofemoral joint in particular, and lifting has a multiplying e¡ect onthis. If you do this for 8 h a day for many years, it more likely than notcauses OA. There are lots of occupational studies showing that people who usejoints in a stereotyped pattern for many years have a high risk of getting OA inthose joints.Herzog:What about people in some Asian countries who squat all the time?Felson:We have recently ¢nished a population prevalence study in Beijing. One
of the questions we asked subjects was the duration of squatting, which is franklyshocking there. Instead of standing and waiting for a bus they squat and wait, andthey often do their household chores in a squatting position. Especially amongwomen, this is associated with a higher than expected rate of tibiofemoraldisease. Interestingly, the Chinese women had more symptomatic knee OAthan women in the USA, even though they are substantially thinner than womenin the USA. One of the factors accounting for this is the high rate of squatting inChina.Fernihough: In the ACL model, do you see any behaviour in the animal that you
would report as pain? If there are any weight bearing changes, would you describethis as a pain behaviour?Brandt: Di¡erences may be seen by forceplate analysis, such as those in vertical
ground reaction force, a measure widely accepted as a surrogate for joint pain. I amnot sure that this is legitimate, because the animalmaybe reacting to a perception ofinstability, rather than the pain. The animal uses the limb with the unstable kneequite happily, however. We exercise them to promote the model, and they do thiswithout any obvious discomfort.Lohmander: In humans, pain is not a dominant symptom of the ACL de¢ciency.
Instability and insecurity are typically the dominant problems.Grubb: Is that true even in cases where there is profound damage to the knee?Lohmander: Then it is no longer an ACL-de¢cient knee, but an OA knee.Grubb: At what stage does the pain develop? Is there a relationship between the
pain and the damage in your model? There isn’t a good correlation in OA.Lohmander: I don’t think we can tell.
62 DISCUSSION

Brandt: In the dogmodel, we have shown a fairly good relationship between theseverity of chondropathy and the magnitude of vertical ground reaction forces.The more loading, the more cartilage damage.
Reference
Sharma L, Dunlop DD, Cahue S, Song J, Hayes KW 2003 Quadriceps strength andosteoarthritis progression in malaligned and lax knees. Ann Intern Med 138:613^619
NEUROMUSCULAR ASPECTS 63

Current perspectives on the clinical
presentation of joint pain in
humanOA
Paul Creamer
Southmead Hospital, Bristol, BS10 5NB, UK
Abstract. Pain is the commonest symptom of osteoarthritis (OA), the principal reasonwhy individuals seek medical care and a major determinant of other outcomes such asdisability and joint replacement. Most studies have examined knee OA: little is knownabout other sites. Community studies indicate only a modest relationship betweenstructural change on X-ray and reporting of pain. Many community subjects, forexample, fail to complain of pain despite extensive X-ray change, while others reportpain with normal X-rays. Pain severity of patients attending hospital is even less relatedtoX-ray change, beingmore dependent on bodymass index (BMI), coping strategies andpsychosocial variables. Many patients can identify more than one type of pain. It isincreasingly clear that OA pain is heterogeneous, being classi¢able on the basis oflocation, precipitating factors, response to anti-in£ammatory and steroid medicationand the e¡ects of local anaesthetic. This potential to classify OA pain represents a usefultool with which to test hypotheses regarding structural origin of pain.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 64^78
Osteoarthritis (OA) is a disorder of synovial joints characterized by destruction ofarticular cartilage and overgrowth of marginal and subchondral bone. Pain is theprincipal symptom of OA and the major reason why subjects seek medicalattention, which may include costly interventions such as joint replacement. Painis also the most signi¢cant determinant of disability. Given the current lack ofdisease modifying drugs in OA, the treatment of OA is essentially the treatmentof OA pain.Although OA may a¡ect many peripheral joints (knees, hands, hips, feet) most
of our knowledge of pain in OA derives from the knee. It is important to note thatmechanisms may vary from joint to joint and data from the knee may notnecessarily be transferable to other joints.
64
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

Knee pain in the community
Knee pain is usually assessed as a dichotomous variable (present or absent) using,for example, the NHANES-1 screening question: ‘Have you ever had pain in oraround the knee on most days for at least one month?’ Subtle changes in thephrasing of the question can result in large di¡erences in the apparent prevalenceof pain but in general about 24^28% of community dwellers aged 40^70 respondpositively to such a question (O’Reilly et al 1996). Prevalence of knee pain increaseswith radiographic severity of OA (Felson et al 1987, Hochberg et al 1989, Carman1989, Spector et al 1993, Lethbridge-Cejku et al 1995). In the NHANES-I study,for example, among subjects aged 65^74 knee pain was reported by 8.8% ofsubjects with normal X-rays, 20.4% with Kellgren and Lawrence (K+L) grade 1OA, 36.9% with grade 2 and 60.4% with grades 3^4 (Davis et al 1992). Similar¢ndings of a progressive increase in the risk of pain reporting with worseningradiographic change have been reported at other joint sites (Table 1).It is, however, clear that there are many subjects in whom X-ray changes and
reported pain are discordant. Pain may be reported in the absence of X-raychanges� the prevalence of self-reported knee pain with normal X-rays is about10.0%. There are several potential reasons for this. First, most studies utilize onlysupine or weight-bearing views of the tibio-femoral joint; failure to assess thepatellofemoral joint could result in a subject being classi¢ed as ‘X-ray negative’when in fact changes were present but not seen. Indeed, up to 24% of femalesreporting knee pain have isolated patellofemoral disease and if lateral views areincluded the predictive value of pain for radiographic change increases(McAlinden et al 1993). Second, a positive response to the NHANES-I kneequestion does not di¡erentiate between isolated knee pain and widespread pain ofwhich the knee is but a part. The prevalence of ‘widespread chronic pain’ is about
CLINICAL PRESENTATION OF JOINT PAIN IN HUMAN OA 65
TABLE 1 Prevalence (%) of reported pain by radiographic severity at 1st carpo-metacarpal (ICMC), distal (DIP) and proximal (PIP) interphalangeal, and hip joints
Radiographic severity
Joint site
(KL grade) ICMC a DIP/PIP a Hipb
0/1 10.6 15.2 8.0 (M) 12.0 (F)
2 34.2 48.7 10.0 (M) 14.0 (F)3�4 65.1 80.9 44.0 (M) 86.0 (F)
aHart et al 1994.bLawrence 1997.

11% (Croft et al 1993). Such patients may answer a⁄rmatively about knee pain butthis would not necessarily imply local pathology. Third, X-rays are relativelyinsensitive: they may be normal when other diagnostic studies such asarthroscopy show clear evidence of OA (Fife et al 1991). X-rays do not allowvisualization of non-bony sources of pain, such as capsule, synovium orligaments. Finally, not all knee pain is due to OA: causes such as anserinebursitis, internal derangements and referred pain from hip or spine would not beidenti¢ed on X-rays of the knees.The second group (X-ray positive, pain negative) is larger. Pain reporting in
grade 3^4 OA ranges from 40^79%: thus, up to half the patients in thecommunity with, by any standard, established radiographic OA deny pain. Therelationship improves if osteophytes rather than global change are used (Spectoret al 1993, Lethbridge-Cejku et al 1995, Cicuttini et al 1996). The precise questionthat is asked may a¡ect the response in terms of pain reporting. The NHANES-Iquestion may underestimate prevalence: patients may have had pain but not on‘most days of a month’ or they may simply fail to recall previous episodes ofpain. Further, OA may be a phasic condition with episodes of pain separated byremissions: the question may fail to capture the painful episode.Another approach to examining the relationship between structural change and
pain is to consider the prevalence of X-ray change in those presenting with jointpain. A community survey of 4057 subjects aged 40^70 found a prevalence of kneepain of 28.3%. Of these, 74% had at least grade 1 osteophyte and 40.9% had at leastgrade 2 (O’Reilly et al 1996). In 195 subjects aged over 40 presenting to their GPwith a ¢rst episode of hip pain, Birrel et al (2000) found 44% had a KL grade52and 34%hadKL53. Aminimum joint space of42.5mmwas seen in 30%. By thetime subjects present to primary care with hip pain, therefore, a signi¢cant numberwill already have established OA change on X-ray.The risk factors for radiographic knee OA (age, sex, race, obesity) are di¡erent
from those for knee pain reporting in the community. In addition toX-ray change,psychological well-being and health status (Davis et al 1992), anxiety (in womenonly) (Creamer et al 1999a), feeling ‘low’ or ‘very low’ in spirits (Hochberg et al1989), hypochondriasis (Lichtenberg et al 1986) and ‘negative a¡ect’ (Dekker1993) have all been associated with higher levels of knee pain reporting. Lowereducational level is an independent risk factor for pain reporting (Hannan et al1992).
OA pain in the clinic
Some individualswith knee or hip pain elect to present tomedical care. The reasonsfor this choice are unclear but co-morbidity (especially psychosocial), copingbeliefs, social support, availability of services and degree of empowerment are all
66 CREAMER

likely to be more important than pain severity, radiographic change, age orfunctional limitation. A community study of subjects with hip or knee painfound that depression scores were signi¢cantly higher in those that had elected toseek medical care (Dexter & Brandt 1994). A similar role for psychological factorsin the promotion of healthcare seeking behaviour has been suggested in otherconditions such as ¢bromyalgia.It is safe to assume that almost all individuals with OA presenting to healthcare
will have pain. Although pain is clearly important to patients and is discussed at98% consultations, potential causes are discussed minimally or not at all in up to46% cases (Bellamy & Bradley 1996). Furthermore, physicians and patients maydisagree about the severity of their pain and e¡ect on life (Hogkins et al 1985).Suarez-Almazor et al (2001) in a study of 105 patients with musculoskeletaldisease found that intraclass correlation coe⁄cients (ICCs) were only 0.42 forpain. Physicians tended to rate their patients’ health status higher than thepatients themselves and were less willing to gamble on the risk of death versusperfect health. The importance of pain to patients with OA and the relationship
CLINICAL PRESENTATION OF JOINT PAIN IN HUMAN OA 67
FIG. 1. Circadian rhythm for pain in patients withOA of the hand. Self measurements/ratingswere made by 20 or 21 patients every 24 hours during waking for 10 days. Individual values hadtrends removed and were converted to a percentage of the mean before combining for groupanalysis by population mean cosinor. For rhythm characteristics P value is from the zeroamplitude test; amplitude¼half peak trough di¡erence of cosine; bathyphase¼ lowest point ofcosine (referenced from 0000). P50.001 for each variable from ANOVA for time e¡ect.Reproduced with permission from Bellamy et al (2002).

between pain severity and its importance has been little studied but clearly has greatrelevance.For patients presenting to healthcare, pain becomes a continuous variable�
pain severity. A major advance in OA pain research has been the adoption ofstandardized, validated questionnaires such as the WOMAC, Lequesne, McGillPain Questionnaire (MPQ) or a simple VAS. The WOMAC has recently beenshown to be more sensitive than the SF-36 (Davies et al 1999) and the LequesneIndex (Theiler et al 1999) and appears not to be in£uenced by anxiety anddepression as much as the MPQ (Creamer et al 1999b). It also allows painoccurring in di¡erent situations to be separately assessed. The risk factors forpain severity reporting are di¡erent from those for pain as a dichotomousvariable. In a group of hospital outpatients with knee OA (Creamer et al 1999b)risk factors for pain severity reporting di¡ered slightly according to the scale usedthough obesity, helplessness and education remained associated with pain severityafter adjustment for confounding variables. Age, disease duration and quality oflife were not related to severity of pain. Others have reported links between painseverity and psychological factors: Summers et al (1988) reporting on 65 patientswithOAof hip or knee found that depression (asmeasured by the BeckDepressionInventory) and anxiety correlated with somemeasures of theMPQ.Another studyof 61 patients with knee OA found signi¢cant correlations between MPQ andZung Anxiety and Depression Inventory scores (Sala⁄ et al 1991). In thecommunity chronicity and severity of knee pain were associated with higherpsychosocial disability (as measured by subscales of the Sickness Impact Pro¢le)compared to age- and sex-matched controls from the same community(Hopman-Rock et al 1996).A number of studies have shown that, in hospital patients, radiographic change
is not related to pain severity (Creamer et al 1999b, Bruyere et al 2002). It may bethat a threshold needs to be reached for joints to become painful but beyond that,other factors (coping strategies, depression, co-morbidity, BMI) determine theperceived severity for an individual.
The nature of OA pain
Generally quoted descriptions of OA pain are largely anecdotal, supported bysurprisingly little patient-based evidence. ‘Typical’ OA pain is said to beinsidious, variable and intermittent (‘good days and bad days’); mainly occurringon use, movement or weight bearing and later in the day. Nearly all symptomaticpatients have use-related pain but many also have rest or night pain. Knee pain isgenerally anterior or medial; hip pain classically is felt in the groin but may radiateto the knee. Thumb base OA is more likely to cause pain than interphalangeal OAand may be felt di¡usely ‘around the wrist’. A diurnal variation has been described
68 CREAMER

at both the knee (Bellamy et al 1990; see Fig. 1) and the hand (Bellamy et al 2002)with pain worse in the evenings and easier in mornings. The reason for good andbad days is unclear: in£uences of weather or barometric pressure are often cited bypatients and may have some validity. Strusberg et al (2002) found that in OA, paincorrelated with low temperature (r¼�0.23, P50.001) and high humidity(r¼0.24, P50.001). Seasonal variation (worse in winter) is often reported butthis may be more due to perception than reality since reported symptoms do notnecessarily agree with measured clinical scores (Hawley et al 2001). Pain may alsobe reported more strongly at weekends (Bellamy et al 1990).Although the cause remains uncertain it is increasingly clear that pain in OA is
heterogeneous, varying between individuals and with di¡erent phases of thedisease. Recently e¡orts have been made to identify di¡erent patterns of pain, inthe hope that theymay indicate di¡erent pathological or anatomical processes. Thelocation of pain at the knee, for example, is not random, but falls into two wellde¢ned groups: generalized anterior pain and localized inferomedial pain. Thesedi¡erences are not explicable by radiographic change and may represent localbony or soft tissue sources (Creamer et al 1998a). Another example is theresponse to local anaesthetic (Creamer et al 1996, Hassan et al 2002): in manypatients this will abolish pain temporarily whilst in others no e¡ect is seen. Insimple terms, some patients may have local sources of pain whilst in others thepain is centrally driven. The complexity of pain mechanisms is furtheremphasised by the fact that intra-articular anaesthetic can also abolish pain incontralateral, untreated joints, implying central or spinal mechanisms (Creameret al 1996).Finally, the e¡ect of intra-articular steroids overall is short lived, but individual
patients derive sustained bene¢t�do they have a more in£ammatory cause fortheir pain?Night pain (often used by orthopaedic surgeons as an indicator of the need for
joint surgery) is said to be an unusual feature, limited to advanced disease. Wefound (Creamer et al 1998b) that 43% subjects with knee OA reported pain of530mm on a VAS for night pain and 14.7% actually felt the night to be themost painful time. 27.9% felt that resting in bed made their pain worse. Using thelatter de¢nition, we were unable to con¢rm a relationship between night pain anddisease severity as assessed by pain severity, disability, examination ¢ndings orradiographic change. A modest relationship with disease duration was seen, butmost signi¢cantly, night pain was associated with high levels of helplessness andworse perceived quality of life, perhaps due to underlying fatigue.Such clinical observations allow testable hypotheses to be generated. Night
pain, for example is often thought to be due to raised intraosseous pressure: theability of MRI to detect focal changes in subchondral bone linked with pain(Felson et al 2001) allows this to be investigated further. If inferomedial knee
CLINICAL PRESENTATION OF JOINT PAIN IN HUMAN OA 69

pain is due to collateral ligament pathology, again thismay be detected bymagneticresonance imaging (MRI). If failure of intra-articular anaesthetic to abolish painindicates a central source this may be associated with higher depression orhelplessness. Such studies have the potential to allow a more tailored, individualapproach to pain treatment.Longitudinal studies show thatmost patients feel that their pain gets worse with
time though there is considerable variability. In the Bristol OA 500 study (Dieppeet al 2000), for example, the proportion of subjects with knee OA reporting theirpain to be ‘severe’ was 25% at baseline, 17% at 3 years and 27% at 8 years.However,80% of patients felt they had worsened overall.
E¡ect of pain in disease
We have considered the risk factors for pain reporting but what about the e¡ectpain may have on the underlying disease? Reduction in pain, for example by intra-articular local anaesthetic, results in increased maximum voluntary contraction(MVC) of quadriceps (Hassan et al 2002). The in£uence of pain on otherpotential risk factors such as proprioception and balance is unclear: Hassan et al(2002) reported that pain reduction did not result in improvements inproprioception or static postural stability. Jadelis et al (2001), examined dynamicbalance in a cross sectional study of older patients with kneeOA. Balancewasmoststrongly related to quadriceps strength, but in those subjects with weak quadricepspain severity became an independent predictor of poor balance.We do not know if long term pain reduction can reduce progression of disease
but there is some evidence that pain predicts incident knee OA and that subjectswith pain progress faster than those with similar radiographic change withoutpain. Hart (Hart et al 1999) found odds ratios of 1.91 (95% con¢dence interval1.18^3.09) for knee pain predicting development of osteophyte at follow up.Cooper et al (2000) in a follow up study of 354 community subjects found thatbaseline knee pain predicted incident knee OA at 5 years (odds ratio 2.9 [1.2^6.7]for KL 51; odds ratio 1.3 [0.6^2.7] for KL 52). Knee pain also predictedprogression over 5 years.
Causes of pain in OA
The anatomic cause of pain in OA remains unknown. Any theory has to considerthat the principal structure involved (cartilage) possesses few pain-sensitive ¢bres.Bone pain may be a factor in many subjects: perhaps via osteophyte growth withstretching of periosteum, raised intraosseous pressure or microfractures. Felsonet al (2001) examined the relationship between ‘bone marrow lesions’ (thought torepresent oedema) on MRI and knee pain. Lesions were found in 77.5% persons
70 CREAMER

with painful knees compared with 30% with no knee pain (P50.001). ‘Large’lesions were present almost exclusively in persons with knee pain (35.9% vs. 2%;P50.001). Although lesions were associated with more severe radiographicchange in general, the relation with pain persisted even after adjustment forseverity of radiographic disease, e¡usion, age and sex. No relation was seen withpain severity.Other sources of pain include ligament damage, capsular tension, meniscal
injury and synovitis. In£ammation may be present in OA and may cause paineither by direct stimulation of primary a¡erent peripheral a¡erent nociceptive¢bres (PANs) or by sensitizing PANs to mechanical or other stimuli. Systemicmarkers of in£ammation such as C reactive protein (CRP) are raised in manypatients with OA and may predict future progression of disease (Spector et al1997). In addition there is a central component to pain and in£uences such asanxiety, depression and comorbidity are likely to operate in some patients asdescribed above.
Conclusions
Many questions remain about OA pain. What makes a person with OA pain seekmedical attention? Is it worsening of the disease (little evidence for this)? Loss ofcoping skills? Socioeconomic or ¢nancial factors?Whatwould be the e¡ect of earlyaggressive pain control in reducing intensity or duration of chronic pain? In otherwords, does control of pain a¡ect the natural history of the disease? Towhat extentis pain protective and to what extent does it reduce function and result in physicaldeconditioning? How can we improve our understanding of our patients’ healthperceptions and risk-bene¢t preferences so that we may suggest more appropriateinterventions?Many individuals with radiographic OA do not report pain and perhaps we
should ask not ‘why is OA painful?’ but ‘why is it so often pain free?’Much e¡ort is being expended on ¢nding drugs capable ofmodifying the disease
process, notably on cartilage loss. We would expect an e¡ective disease-modifyingdrug to also have an e¡ect on pain but, given the poor correlation currently seenbetween structural change (at least on X-ray) and symptoms, a word of cautionmight reasonably be sounded. There are grounds to at least consider the wisdomof investing large resources in expensive technologies designed to reducestructural change when this may not, in fact, a¡ect the problems that areimportant for the patient.
References
Bellamy N, Bradley L 1996 Workshop on chronic pain, pain control and patient outcomes inrheumatoid arthritis and osteoarthritis. Arthritis Rheum 39:357^362
CLINICAL PRESENTATION OF JOINT PAIN IN HUMAN OA 71

BellamyN, Sothern R, Campbell J 1990 Rhythmic variations in pain perception in osteoarthritisof the knee. J Rheumatol 17:364^372
Bellamy N, Sothern R, Campbell J, Buchanan WW 2002 Rhythmic variations in pain, sti¡nessand manual dexterity in hand osteoarthritis. Ann Rheum Dis 61:1075^1080
Birrell F, Croft P, Cooper C, Hosie G, Macfarlane GJ, Silman A 2000 Radiographic change iscommon in new presenters in primary care with hip pain. PCR Hip Study Group.Rheumatology (Oxford) 39:772^775
BruyereO,Honore A, Rovati LC et al 2002 Radiologic features poorly predict clinical outcomesin knee osteoarthritis. Scand J Rheumatol 31:13^16
Carman WJ 1989 Factors associated with pain and osteoarthritis in the Tecumseh CommunityHealth Study. Semin Arthritis Rheum 18:S10^S13
Cicuttini FM, Baker J, Hart DJ, Spector TD 1996 Association of pain with radiological changesin di¡erent compartments and views of the knee joint. Osteoarthritis Cartilage 4:143^147
Cooper C, Snow S, McAlindon TE et al 2000 Risk factors for the incidence and progression ofradiographic knee osteoarthritis. Arthritis Rheum 43:995^1000
Creamer P, Hunt M, Dieppe P 1996 Pain mechanisms in osteoarthritis of the knee: e¡ect ofintraarticular anaesthetic. J Rheumatol 23:1031^1036
Creamer P, Lethbridge-Cejku M, Hochberg MC 1998a Where does it hurt? Pain localization inosteoarthritis of the knee. Osteoarthritis Cartilage 6:318^323
Creamer P, Lethbridge-Cejku M, Hochberg M 1998b What is the signi¢cance of night pain inknee osteoarthritis? Br J Rheumatol 37:S153
Creamer P, Lethbridge-Cejku M, Costa P, Tobin J, Herbst JH, Hochberg MC 1999a Therelationship of anxiety and depression with self reported knee pain in the community: datafrom the Baltimore Longitudinal Study of Aging. Arthritis Care Res 12:3^7
Creamer P, Lethbridge-Cejku M, Hochberg MC 1999b Determinants of pain severity in kneeosteoarthritis: e¡ect of demographic and psychosocial variables using 3 pain measures.J Rheumatol 26:1785^1792
Croft P, RigbyAS, Boswell R, Schollum J, SilmanA1993The prevalence of chronicwidespreadpain in the general population. J Rheumatol 20:710^173
Davies GM, Watson DJ, Bellamy N 1999 Comparison of the responsiveness and relative e¡ectsize of the Western Ontario and McMaster Universities Osteoarthritis Index and the short-formMedical Outcomes Study Survey in a randomised, clinical trial of osteoarthritis patients.Arthritis Care Res 12:172^179
Davis M, EttingerW, Neuhaus JM, Barclay JD, Degal MR 1992 Correlates of knee pain amongUS adults with and without radiographic knee osteoarthritis. J Rheumatol 19:1943^1949
Dekker J, Tola P, Aufdemkampe G, Winckers M 1993 Negative a¡ect, pain and disability inosteoarthritis patients: the mediating role of muscle weakness. Behav Res Ther 31:203^206
Dexter P, BrandtK 1994Distribution and predictors of depressive symptoms in osteoarthritis. JRheumatol 21:279^286
Dieppe P, Cushnaghan J, TuckerM, Browning S, Shepstone L 2000The Bristol ‘OA500’ study:progression and impact of the disease after 8 years. Osteoarthritis Cartilage 8:63^68
Felson DT, Naimark A, Anderson J, Kazis L, Castelli W, Meenan RF 1987 The prevalence ofknee osteoarthritis in the elderly: the Framingham Osteoarthritis Study. Arthritis Rheum30:914^918
FelsonDT, Chaisson CE,Hill CL et al 2001 The association of bonemarrow lesions with pain inknee osteoarthritis. Ann Internal Med 134:541^549
Fife RS, Brandt KD, Braunstein EM et al 1991 Relationship between arthroscopic evidence ofcartilage damage and radiographic evidence of joint space narrowing in early osteoarthritis ofthe knee. Arthritis Rheum 34:377^382
72 CREAMER

Hannon MT, Anderson JJ, Pincus T, Felson DT 1992 Educational attainment andosteoarthritis: di¡erential associations with radiographic changes and symptom reporting. JClin Epidemiol 45:139^147
Hassan BS, Doherty SA,Mockett S, DohertyM 2002 E¡ect of pain reduction on postural sway,proprioception, and quadriceps strength in subjects with knee osteoarthritis. AnnRheumDis61:422^428
Hart DJ, Spector TD, Egger P, Coggon D, Cooper C 1994 De¢ning osteoarthritis of the handfor epidemiological studies: the Chingford Study. Ann Rheum Dis 53: 220^223
Hart DJ, Doyle DV, Spector TD 1999 Incidence and risk factors for radiographic kneeosteoarthritis in middle-aged women: the Chingford Study. Arthritis Rheum 42:17^24
Hawley DJ, Wolfe F, Lue FA, Moldofsky H 2001 Seasonal symptom severity in patients withrheumatic diseases: a study of 1,424 patients. J Rheumatol 28:1900^1909
Hochberg MC 1996 Prognosis of osteoarthritis. Ann Rheum Dis 55:685^688Hochberg MC, Lawrence RC, Everett DF, Cornoni-Huntley J 1989 Epidemiologicalassociations of pain in osteoarthritis of the knee: data from the National Health andNutrition Examination Survey and the National Health and Nutrition Examination-IEpidemiologic Follow-up Survey. Semin Arthritis Rheum 18:S4^S9
HodgkinsM,AlbertD,DaltroyL 1985Comparing patients’ and their physicians’ assessments ofpain. Pain 23:273^277
Hopman-Rock M, Odding E, Hofman A, Kraaimaat FW, Bijlsma JW 1996 Physical andpsychosocial disability in elderly subjects in relation to pain in the hip and/or knee. JRheumatol 23:1037^1044
Jadelis K, Miller ME, Ettinger WH Jr, Messier SP 2001 Strength, balance, and the modifyinge¡ects of obesity and knee pain: results from the Observational Arthritis Study in Seniors(OASIS). J Am Geriatr Soc 49:884^891
Lawrence JS 1977 Rheumatism in populations. William Heinemann, LondonLethbridge-CejkuM, Scott WW Jr, Reichle R et al 1995 Association of radiographic features ofosteoarthritis of the knee with knee pain: data from the Baltimore Longitudinal Study ofAging. Arthritis Care Res 8:182^188
Lichtenberg PA, Swensen CH, Skehan MW 1986 Further investigation of the role ofpersonality, lifestyle and arthritic severity in predicting pain. J Psychosom Res 30:327^337
McAlindon TE, Cooper C, Kirwan JR, Dieppe PA 1993 Determinants of disability inosteoarthritis of the knee. Ann Rheum Dis 52:258^262
O’Reilly SC, Muir KR, Doherty M 1996 Screening for pain in knee osteoarthritis: whichquestion? Ann Rheum Dis 55:931^933
Sala⁄ F, Cavalieri F, Nolli M, Ferraccioli G 1991 Analysis of disability in knee osteoarthritis.Relationship with age and psychological variables but not with radiographic score. JRheumatol 18:1581^1586
SpectorTD,HartDJ, Byrne J,Harris PA,Dacre JE,DoyleDV1993De¢nition of osteoarthritisfor epidemiological studies. Ann Rheum Dis 52:790^794
Spector TD, Hart DJ, Nandra D et al 1997 Low-level increases in serum C-reactive protein arepresent in early osteoarthritis of the knee and predict progressive disease. Arthritis Rheum40:723^727
Strusberg I, Mendelberg RC, Serra HA, Strusberg AM 2002 In£uence of weather conditions onrheumatic pain. J Rheumatol 29:335^338
Suarez-Almazor ME, Conner-Spady B, Kendall CJ, Russell AS, Skeith K 2001 Lack ofcongruence in the ratings of patients’ health status by patients and their physicians. MedDecis Making 21:113^121
Summers MN, Haley WE, Reveille JD, Alarcon GS 1988 Radiographic assessment andpsychological variables as predictors of pain and functional impairment in osteoarthritis ofthe knee or hip. Arthritis Rheum 31:204^209
CLINICAL PRESENTATION OF JOINT PAIN IN HUMAN OA 73

Theiler R, Sangha O, Schaeren S et al 1999 Superior responsiveness of the pain and functionsections of the Western Ontario and McMaster Universities Osteoarthritis Index(WOMAC) as compared to the Lequesne-Algofunctional Index in patients withosteoarthritis of the lower extremities. Osteoarthritis Cartilage 7:515^519
DISCUSSION
Bradley: I want to comment on your data on anxiety. The role of anxiety in painreporting is greatly underestimated. With regard to the McGill PainQuestionnaire, one reason why you ¢nd low correlations between the WOMACand the visual analogue scale from the McGill is that, unlike the WOMAC painscale, the McGill is multidimensional and contains a large subgroup of wordsdealing with a¡ect and emotion. When you look at ethnic group di¡erences onthe McGill, do you ¢nd variation as a function of the types of words chosen todescribe pain? My reason for asking this is that when we apply quanti¢ed stimuliin the laboratory to patients, we ¢nd that African^Americans tend to use higherintensity a¡ective words compared with Caucasians, even if their sensory intensityresponses are the same. Do you ¢nd similar phenomena in your larger populationstudies?Creamer: There are di¡erences between English English and American English.
In the more detailed study we didn’t have enough African Americans to reallyaddress this. They did report higher McGill pain scores, but most of this e¡ectdisappeared when we adjusted for BMI. There was a sense that the words chosenmight have been di¡erent. And with the McGill we have a sense that it isn’t reallymeasuring what I want it to measure. On theMcGill I also looked at whether therewere words people would choose given the opportunity that aren’t on theMcGill.There aren’t many, because the McGill has 76 words, but there were a few. Thereare somewords that are never chosen by people.McGill was developed for all sortsof pain, including cancer and dental pain. Itmay not be the best tool to look at someof these issues in OA.Schaible:What is the minimum set of symptoms that you need to diagnose OA?
In e¡ect, one could ask if someone reports pain but you don’t ¢nd anything in thejoint, why dowe call itOApain?And if someone has joint changes visible byX-raybut the pain doesn’t correlate, is this OA?Creamer: It depends on how you de¢ne OA. OA can be de¢ned pathologically,
radiographically or clinically. There is some overlap, but there are also somedi¡erences. In the community, pain is de¢ned as ‘yes’ or ‘no’. Patients are asked aquestion about whether they have pain, and we try to de¢ne this on the basis of itbeing experienced on most days for at least a month. When we are looking at painseverity then it is much more arbitrary, but sometimes people try to develop
74 DISCUSSION

cut-o¡s for studies to ensure that they have people in the study with adequate pain,so a bene¢t can be shown for whatever intervention is used.Schaible: For a clinician, if someone reports joint pain, is this su⁄cient to make
the diagnosis of OA in the absence of any visible in£ammation?Creamer: Not all knee pain is due to OA. One of the explanations for the knee-
pain-positive people who areX-ray negative is that they have another pathology inthe knee joint. The other thing is thatX-rays are relatively insensitive.Arthroscopymay well reveal cartilage changes long before the X-ray has changed. Some peoplepropose that we should talk less about knee OA and more about knee pain.Dieppe: I think the sub-setting of pain is a very important concept. I thinkwe are
not getting this sorted because we don’t know what hypotheses to test and wedon’t know what questions to ask. We have got into this habit of thinking aboutnight pain, rest pain and walking pain as being entities. I think this is based onnothing. Similarly, the other methods of subtyping that are being attempted arepotentially unhelpful. I have been rather impressed bywhat the social scientists cano¡er ¢elds like this. We should be doing qualitative research before we doquantitative research. We should be doing in-depth, unstructured interviewswith people with OA, however de¢ned, trying to take out themes from this sortof qualitative research as to what the issues are, and then derive the hypotheses anddo the sorts of studies you have done having ¢rst got some hypotheses about painsubgroups.Creamer: I agree with you. Qualitative research is ¢endishly di⁄cult, so it is
much easier to go for the tools that have already been developed. But at least thissort of work shows that potentially there are di¡erences.Dieppe: Yes, your sort of work stimulates me to think we really should go for
this.Kuettner: We are mixing the di¡erent forms of OA. Aren’t knee and hip OA
totally di¡erent in their aetiology, and don’t they require di¡erent clinicalapproaches?Felson: There is no clear cut answer to this.Pisetsky: There is another way you could subset: those patients who have
surgery and those who don’t. If you look at the people who have operations,how are they describing their pain as opposed to those who don’t? Do you getany insight by dividing the patients up in this way?Dieppe: We have been looking at those issues. We have been studying the
barriers and facilitators to people seeking medical help in the ¢rst place, and wehave been trying to take this through to referrals and surgery. A lot of this isbeing done with qualitative research, so the numbers of people we haveinformation on is small. Where we are so far suggests to me that healthcareutilization for OA has little to do with pain or disease severity, but that othersociocultural factors are determining it. Some of my social scientist colleagues
CLINICAL PRESENTATION OF JOINT PAIN IN HUMAN OA 75

go as far as saying that there is no disease here, and that it is all purely a socioculturalphenomenon!Brandt: With respect to the issue of OA pain and progression, there was some
nice work from Hurwitz et al (2000) measuring gait and pain in patients witharthritic medial compartment knee OA. They showed that when the patientswere taking pain medication they increased the loading of the medialcompartment. When the pain medication was washed out and joint pain becamemore severe, the subjects changed their gait so as to protect the damaged cartilage.However, long-termdata are not available to showwhether this results in analgesicarthropathy. But this also relates to Leena’s study (Sharma et al 2003), because shedidn’t measure joint pain. One of the possibilities that needs to be considered iswhether those people who were stronger had less pain and therefore loaded theirknee more than others.Felson: Pain is a protective mechanism. I’d like to ask an almost rhetorical
question. In RA, it is my understanding that anxiety and depressive symptomscontribute to pain severity also. Yet therapies for RA seem to have terri¢c e¡ectson pain. Does this mean that we can address pain anyway without grappling withthis concern? In RA, a third of the patients don’t have morning sti¡ness, forexample, so there is the same variability in pain description and reporting thatyou have described in OA. Yet we don’t seem to have too much trouble indeveloping therapies for RA while we ignore the qualitative aspects of pain. Willthis be true for OA also?Creamer:Do you think that in RAwe have a more de¢ned pathology and site of
origin of pain? There is synovitis and in£ammation.Felson: Is that where the pain comes from in RA?Creamer: Treatments such as a steroid injection into an in£amed knee are highly
e¡ective ways of reducing pain in RA. I have a better feel for the pathology of RAthanOA, and in£ammation seems to bewhat is drivingmost of the pain inRA.Butyour point is well made.Grubb: A number of us are interested in the development of animal models for
the study of OA, and what worries me is that we have this clear lack of correlationbetween the radiological scores of the disease and pain. How can we develop amodel if we don’t have a clear idea of what typical OA is? What features shouldwe be looking for in an animal model that would well represent human OA? Wecan’t develop a good animal model of human OA without that correlation.Brandt: It also depends onwhat youwant to use themodel for. If youwant to use
it to study a drug that might inhibit cartilage loss, then you want a model thatdemonstrates a certain rate of cartilage loss. If you want a model to evaluate pain,this imposes an entirely di¡erent set of requirements. This is challenging.Grubb: That is what many of us here are interested in� the pain aspect. It is not
clear to me what we should be doing here.
76 DISCUSSION

Brandt: There is an obvious di⁄culty in evaluating OA pain. It is, however, noeasier to assess structural damage. While there are similarities in pathology, noanimal models have been clearly shown to predict the e¡ects of‘chondroprotective’ drugs in humans.Schaible: This comes down to the question of nociception and pain. A model is
urgently required to ¢nd out whether there is any change in nociception, orwhether there is nociception at all in degenerative processes in a joint. Then thereis a discussion about what this means for pain. I wouldn’t be too negative aboutthis.Grubb: I am not being negative. Complete Freund’s adjuvant (CFA)
polyarthritis is a very good animal model with a lot of joint pathology in whichchanges in nociception are seen. It is not, however, a model of OA.Schaible: I am biased. We should say that there is a de¢ned process in the joint,
and we should answer the question about whether this evokes nociception. This issomething we could answer and should answer.Henry: The idea of subgroupings raises a lot of issues. The people looking for
biomarkers must feel lost as well. The real answer lies in making a stab atdeveloping animal models. When we develop an animal model, what can welearn about the process? From this we might stumble across one model that willbe particularly useful in terms of understanding nociception. Even humans don’thave a good model of OA. Some have pain without clinical signs, and some haveclinical signswithout pain.What are the basic scientists trying tomodel? It is not assimple as it was a few years ago when we had OA and models.Pisetsky: I have a question about the value of pathology. There are many
operative specimens in OA. Are we getting the most information out of them?Given the heterogeneity of the disease, should we be doing more pathology? Inthe RA world where there is not much surgery any more, when people didpathological studies, di¡erent forms of RA were histologically distinguishable.Not all people are alike, and subsets that were informative could be identi¢ed.Kuettner: If you explant the cartilage from di¡erent animals, you get distinctive
responses to di¡erent mediators. It becomes very di⁄cult to say, for example, thatthe rabbit is a good model for the human disease. Even within the human, thedi¡erent cartilages from di¡erent joints respond quite di¡erently.Pisetsky: I am asking, for example, should I be looking at nerve ¢bres in
capsules?Lohmander: The problem with surgical specimens in OA is that they represent
end-stage disease in most cases, and also that the patients receiving surgery havebeen ¢ltered through the ¢lters we have heard about here, so they might not berepresentative.Pisetsky: But, even bearing these limitations in mind, we have the opportunity
to get pathological tissue.
CLINICAL PRESENTATION OF JOINT PAIN IN HUMAN OA 77

Kuettner: You can also get normal tissue from tissue donor banks.Hunter: I’d like to address the qualitative research issues. You presented some
nice data fromNick Bellamy showing huge diurnal changes within subjects. Thereis also that huge dichotomy between people who have structural OA and thosepeople who are symptomatic. Surely there is room for research into thoseparticular subjects who are asymptomatic with structural changes and thosepeople who have big diurnal changes, to try to explore what is going on there.Do you have any ideas what this may be?Dieppe: I agree with you. Unfortunately qualitative research is di⁄cult,
expensive and time-consuming. The only data we have are more to do withaccessing healthcare utilization, so I don’t have any useful qualitative data on pain.Mackenzie: You tried to control for pain threshold di¡erences in a structured
way. How clear are you that this really re£ects the ability of di¡erent patients totolerate pain in the real world in very di¡erent ways?Creamer: It can only ever be a surrogate. It is coming from the ¢bromyalgia
literature where there has been some work on the pain threshold in general. Thiswas just an attempt to get a bit of a handle on this.Mackenzie: You can perhaps get a handle on this by trying to understand the
di¡erences in the way in which people deal with pain.Creamer: Some of the brain imaging studies might be relevant here.Bradley:One thing that comes out from the imaging literature is that we haven’t
paid as much attention as we should to the emotional/a¡ective dimension of pain.This a¡ective dimension is very important in the way patients present in the clinic.However, even in the laboratory, psychological factors have a much greaterassociation with pain tolerance tasks as compared to pain threshold tasks. In theimaging world, where people are only just beginning to study pain throughneuroimaging, most of the e¡ort is focused on mapping the neural correlates ofintensity, and much less attention is paid to the neural correlates of a¡ect. So,neuroimaging of pain a¡ect responses is a very important question.
References
Hurwitz DE, Ryals AR, Block JA, Sharma L, Schnitzer TJ, Andriacchi TP 2000 Knee pain andjoint loading in subjects with osteoarthritis of the knee. J Orthop Res 18:572^579
Sharma L, Dunlop DD, Cahue S, Song J, Hayes KW 2003 Quadriceps strength andosteoarthritis progression in malaligned and lax knees. Ann Intern Med 138:613^619
78 DISCUSSION

Joint mechanics in osteoarthritis
Walter Herzog, Andrea Clark and David Longino
Faculties of Kinesiology, Engineering, and Medicine, University of Calgary, 2500 UniversityDrive NW, Calgary, AB T2N 1N4, Canada
Abstract. The primary goal of our research has been to quantify the in vivo loading ofnormal and osteoarthritic (OA) joints, and to determine the corresponding biologicalresponses. Much of the research in this area has been performed using articular cartilageexplants. We feel that, although critically important to our understanding of cartilagemechanics and biology, these experiments may not be directly transferable tointerpreting the in vivo joint mechanics and elucidating the detailed mechanisms of onsetand progression of OA. Therefore, we have attempted to measure the loading of the kneein freely moving feline and lapine models of OA. We have found that, upon anteriorcruciate ligament transection in the cat, knee joints are more £exed, muscle forces aredecreased and muscle control patterns are destroyed. Articular cartilage initiallybecomes thicker, softer and more permeable, resulting in generally increased jointcontact areas and decreased peak pressures in the initial stages of joint degenerationcompared to control values. Based on our results, we speculate that unloading of thejoint (rather than overloading), combined with poor muscular control and weakness,might constitute risks for the onset of joint degeneration.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 79^99
Biomechanics is the science that deals with the external and internal forces actingon a de¢ned biological system, and the e¡ects that are produced by these forces.When we consider joint mechanics in osteoarthritis (OA), it is a biomechanicalproblem. The biological system is the joint. The external forces may berepresented by gravitational, inertial and contact forces; and the internal forcesare the forces acting on and within joint structures, such as the articular cartilage,ligaments, muscles, tendons, menisci, etc. The speci¢c e¡ect we are interested in, atleast in the context of this chapter, is OA.OA is a joint disease of primarily unknown origin. Its end-stage is de¢ned by
complete cartilage erosion from all, or parts, of the articular surfaces. As such, it isoften considered a disease of the articular cartilage. However, we would like tode¢ne it in much broader terms. OA is a joint disease associated with damage tothe articular surfaces, accompanied by osteophyte formation and changes in thesubchondral bone structure, the ligaments, menisci, the synovial £uid, etc.
79
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

It has typically (one might even say always) been assumed that OA is associated,in oneway or another, with joint loading. However, to our knowledge this has notbeen proven with absolute certainty. Nevertheless, we will adopt the notion thatjoint loading, which can be quanti¢ed through the in vivo joint mechanics, triggersthe onset of OA, and once triggered, the disease continues and, at present, cannotbe stopped or reversed. This progression of the disease, typically over years, mayalso be associated with joint loading. However, this notion is muchmore tentativethan the one assuming that it merely triggers the onset of OA.If we adopt the premise that a certain type of joint loading produces
joint degeneration leading to OA, the following basic scienti¢c questionsemerge:
. What is the type of loading that is harmful to the joint in terms ofmagnitude, rateof application, frequency, and other mechanical descriptors?
. What is the biological response to this load that produces the OA response?
. Andprobablymost importantly, what are the pathways (ormechanisms) linkingthe load to the degenerative response?
In order to answer the ¢rst two questions, it seems imperative that the normal invivo joint loading is quanti¢ed and compared to situations that are known toproduce end-stage OA, and that the biological responses to such in vivo loadingare measured. Theoretically, such experiments should be straightforward, butvery little progress has been made towards these goals, probably because thework involved is extremely time consuming and therefore expensive. The thirdquestion is the one that really needs answering. However, with little systematicknowledge concerning the ¢rst two questions, solutions to the third question arelikely far away. Impressive research on the third question has been made in studiesusing articular cartilage explants that are exposed to well-de¢ned loadingconditions in a laboratory setting (e.g. Burton-Wurster et al 1993, Quinn et al1998, Sah et al 1989, Torzilli et al 1997). However, the boundary conditions ofexplants under these arti¢cial loading conditions are far removed from real in vivojoint loading, therefore, the results from these studies, although interesting from amaterials property point of view, may have little relevance for the scenariooccurring in human or animal OA. Of course, there is the possibility thatarticular cartilage cylinders with perfectly £at-shaved surfaces subjected tocon¢ned, or uncon¢ned, in vitro loading conditions between two metal platesmay behave similarly to the naturally rounded cartilage surfaces attached to theirnative bone that are exposed to the gliding and compression loading from theiradjacent cartilage surfaces. However, if so, this must be demonstrated. Here, wewill not discuss the vast number of excellent studies on articular cartilage explants,partly because of a lack of relevance in the current context, and partly because
80 HERZOG ET AL

those studies have received much attention and are summarized in a series ofexcellent reviews (e.g. Guilak et al 1997, Hasler et al 1999, Mow et al 1994, Sahet al 1992).Instead, we will focus our attention on studies aimed at quantifying the internal
and external forces occurring in diarthrodial joints of an experimental model ofbona ¢de end-stage OA. This will be done by presenting research on the groundreaction forces and kinematic patterns of the anterior cruciate ligament (ACL)transected cat, by discussing work on the corresponding muscle forces andactivation patterns, and by showing results on the associated degenerativeresponses. Following this discussion, we would like to present preliminary workon the biological response of articular cartilage to controlled in vivo loading ofjoints, and on an experimental model of muscle weakness. The ¢rst of thesestudies is a natural extension of the ¢rst part of our work, aimed at approaching asolution to the second question above. The second of these studies is motivated bythe idea that muscle forces provide the dominant loads on joints (e.g.Crowninshield & Brand 1981), and that muscle weakness and the associatedadaptations in neuromuscular control of movement, might constitute powerfulrisk factors for joint degeneration and OA.
Methods, techniques and approaches
The cat model of OA
There are only a few experimental animal models of OA that have been followedlongitudinally over years. Two such models are the ACL transected dog (Brandtet al 1991) and cat (Herzog et al 2003, Suter et al 1998). Both thesemodels have beenfound to present a history of onset and progression of OA similar to the humandisease, and have led to complete erosion of articular cartilage from speci¢c areas ofthe articular surfaces. We chose ACL transection of the cat as our model of choicefor in vivo investigation for two primary reasons:
. the cat hind limb is the best known mammalian system in terms of muscularanatomy and in vivo force production, and neurophysiological movementcontrol and
. the cat knee does not su¡er fromnaturally occurringOA, therefore degenerativechanges in joint structure, control andmovement can be associated directly withtargeted intervention, even when animals are followed over years where age-e¡ects might play a role in other experimental models, such as the dog. Thedetailed approach to arthroscopic and open joint ACL transection surgery hasbeen described previously (Herzog et al 1993, 1998).
JOINT MECHANICS 81

External forces and kinematics
The external ground reaction forces for walking animals, before and afterexperimental intervention, were measured using a pair of animal-sized(10�15 cm) force-platforms (AMTI, Amherst, USA) embedded in a speci¢callydesigned walkway (Suter et al 1998). These platforms measure the three-dimensional forces and moments acting from the ground on the animal’s paws,and simultaneously provide the location of the centre of pressure of the resultantground reaction force. Hind limb kinematics were measured using a high speedvideo system (Motion analysis, Sta. Rosa, CA, 200Hz), with a correctionalgorithm to account for skin marker movement (Goslow et al 1973).
Muscle forces and activation patterns
Forces in selected ankle extensor muscles were measured using E-shaped, externaltendon force transducers based on a strain-gauged design (Walmsley et al 1978).Forces in the knee extensors (patellar tendon) were quanti¢ed using an omega-shaped, implantable force transducer based on the design of Xu et al (1992) andtested and adapted by Herzog et al (1996) and Hasler et al (1998). Activationpatterns of selected knee and ankle extensor and £exor muscles were measuredusing indwelling, Te£on coated, bipolar, ¢ne wire electrodes embedded into themid-belly of the target muscles with an approximate inter-electrode distance of5mm, and aligned in the approximate direction of the muscle ¢bres (Guimaraeset al 1995).
Joint pressure distribution
In situ joint pressure distributions in normal and ACL transected knees weremeasured using Fuji low and medium grade, pressure sensitive ¢lms (Herzog et al1998). Films were packaged in 12�120mm strips for multiple measurements, andwere sealed in polyethylene adhesive layers for moisture proo¢ng (Liggins et al1995). Film strips were inserted into the joint space through a lateral opening(approximately 20mm). Once inserted, controlled knee extensor activation wasproduced by stimulating the femoral nerve via a bipolar cu¡ electrode and aGrass (S88) stimulator. Knee extensor force was measured using a speci¢callydesigned tibial restraining bar, and the force was adjusted by changing thevoltage and frequency of stimulation. Fuji ¢lm calibration and analysis wereperformed as outlined in detail by Clark et al (2002).
Results and discussion
Osteoarthritis
Transection of the catACL causes increases in thickness, softness, and permeabilityof the articular cartilage; and produces osteophyte formation, increases in thickness
82 HERZOG ET AL

andmass of the medial collateral ligament andmedial joint capsule, and atrophy ofthe hind limbmusculature within weeks (Herzog et al 1993, 1998). Over time, site-speci¢c articular surfaces are eroded away, cartilage becomes soft and loses internalstructure, joint space becomes narrower, and osteophytes increase in number andsize (Fig. 1).
Mechanics
Mechanically, ACL transection is associated with an immediate instability of theknee that results in an excessive anterior translation and internal rotation of thetibia relative to the femur for a given amount of force or moment, respectively.Muscle forces, external ground reaction forces and knee angle decreaseimmediately following ACL transection, and activation patterns of the musclesshow two distinct features:
. a burst-like (rather than the normal, continuous) ¢ring pattern of the kneeextensors and
. an additional phase of activation of the knee £exors (semitendinosus) to, as itappears, compensate for the loss of mechanical function associated with ACLtransection (Fig. 2).
Within approximately four months of ACL transection, joint stability in theanterior^posterior and internal rotation direction are fully re-established(Maitland et al 1998). This result is in contrast to the notion that continuing jointinstability, particularly the pronounced anterior shift of the tibia relative to thefemur in many animal species (Tashman et al 1995) and humans, is responsible, atleast in part, for the progression of joint degeneration. In fact, despite accuratemeasurements of relative movements between tibia and femur using asonomicrometry (Sonometrics Corporation, London, ON, Canada) system witha 16 mm spatial resolution, we have been unable to detect the anterior tibial shifttypically observed in ACL de¢cient humans, dogs and sheep at the instant of footcontact during cat locomotion (e.g., Korvick et al 1994, Tashman et al 1995). Wespeculate that the activation of the knee £exors just prior to paw contact in the cat(Fig. 2) might prevent the anterior shift of the tibia relative to the femur. If so, onecould argue that proper neuromuscular adaptation o¡sets the mechanical loss offunction associated with ACL de¢ciency. However, the crucial clinical questionof whether such an adaptation is meaningful in terms of preventing or slowingdown joint degeneration has never been addressed to our knowledge (although itis an intuitively appealing idea).Similarly, the reason for the quick re-establishment of knee stability following
ACL transection in the cat is not known. We speculate that the initial increase in
JOINT MECHANICS 83

84 HERZOG ET AL
FIG
.1.
Tibialp
lateau
andretrop
atellararticularcartilage
samples
from
ananterior
cruciatelig
amentde¢cient
cat,¢v
eyearspo
stintervention
.Leftp
anel,experim
entalside;righ
tpanel,con
tralateralside.N
ote,thecompleteerosionof
articularcartilage
from
themedialtibialp
lateau
ofthe
experimentalhind
limb(arrow
),andtheloss
ofstructureand¢ssuring
inthesurfacezone
(arrow
)of
theexperimentalarticularcartilage.The
contralateraltibialp
lateau
andarticularcartilage
(right
panel)look
relativelyno
rmal.

articular cartilage thickness, the strengthening of themedial capsule, the increase inmass of the medial collateral ligament, and the appearance of osteophytes at thejoint margins might contribute to the observed increase in joint stability(Maitland 1996). However, despite the apparent lack of anterior translation ofthe tibia during locomotion, and the reduction of anterior translation and medialrotation to normal values within four months of ACL transection, the cat kneecontinues to degenerate. Therefore, joint instability might be a factorcontributing to the onset and progression of osteoarthritis. However, the resultsof our studies would indicate that joint instability is not a necessary, but possibly asu⁄cient factor for joint degeneration and OA.We hinted above at the idea that ground reaction forces decrease immediately
following ACL transection. However, as with the stability of the knee discussed
JOINT MECHANICS 85
FIG. 2. Gastrocnemius (Gastroc) and knee extensor (Quad) forces, as well as semitendinosus(ST) and vastus lateralis (VL) EMG before (Intact knee) and ten days after anterior cruciateligament transection (ACLT knee). The values shown are for slow walking (0.4m/s) on amotor driven treadmill. Observe the decrease in muscle force, and the change in EMGpatterns and continuity from before to after ACL transection. Vertical line TD shows thetouch down of the hindlimb during the step cycle; the corresponding vertical line PO indicatesthe instant of paw o¡.

above, the ground reaction-force patterns recover to normal values within about16 weeks for static (quiet standing) tasks, and within about 6 months for dynamic(straight walking and running) tasks (Fig. 3). These results are of interest insofaras in the ACL transected dog, neither the ground reaction forces nor knee stabilityappear to recover following intervention, not even after 54months. Therefore, wehave twomodels of experimentally inducedOAwhose pathogenesis is very similar(e.g. Adams 1989, Herzog et al 1993) but the long-term external mechanics arecompletely di¡erent. In the dog, the ACL transected joint remains unstable overyears, while in the cat, such instability is never observed during walking, and inanterior ormedial rotation displacement tests, the instability (of the passive joint) isabolished within four months of ACL transection. Similarly, the ACL transecteddog shows reduced vertical ground reaction forces over years, while such forcereductions are all but gone in the cat after 4^6 months, depending on the task.Although the interpretation of the pathogenesis of these two animal models of
OA, in conjunction with the directly measured mechanics, is wide open, thefollowing one appears the most obvious, and although by no means proven,might stimulate new thinking about the idea of how altered joint mechanics mayproduce OA. First, one has to acknowledge that the mechanical changes to thejoint are similar in the cat and dog in the early stages following intervention, and
86 HERZOG ET AL
FIG. 3. Peak vertical ground reaction forces (means �1 SD, n¼7) as a percentage of bodyweight for the ACL transected (open symbols) and the corresponding contralateral, intacthindlimb (¢lled symbols) during cat walking and running before (pre) and at various timeperiods after unilateral ACL transection (1 week^12 months). Note that the forces di¡erstatistically at 1 week, 3 weeks and 3 months post-intervention from those observed pre-intervention, but not for the other time points. A minimum of 10 step cycles for bothhindlimbs was used at each time point and each animal to calculate the mean values shown.

the pathogenesis is similar too. Then, one has to consider that, after approximately4^6 months, the mechanics start to di¡er substantially, but the progression of thedisease remains similar. This scenario suggests that ACL transection in the cat anddog disrupts the mechanics of the knee, and that this event is responsible fortriggering events that lead to degeneration and OA of the joint. Obviously, weare not sure what these events are, but in general, we see less muscular forces, lesscontact pressures, and less external ground reaction forces, thus leading to thehypothesis that there is an initial ‘unloading’ of the joint. Therefore, unloadingthe joint, rather than the more commonly accepted ‘overloading’ might actuallybe the key to initiating the degenerative events.Furthermore, once these degenerative processes have been initiated, the
stabilization of the joint, and the normalization of its mechanics, do not appear toin£uence the progress of the disease, suggesting that once degenerative processesare initiated, the progression of the disease is not very sensitive to changes in jointloading. If so, this result would imply that rehabilitation strategies aimed atestablishing normal movement patterns and joint loading would be mostsuccessful early in the rehabilitation process. Early, here, refers to the idea of asearly as possible following a traumatic event that might pose the joint at risk forOA. Unfortunately, this result, if correct, would also imply that rehabilitationstrategies based on physiotherapy principles would likely not work late in thedegenerative process. This is tragic insofar as most degenerative joint diseases aretypically diagnosed at an advanced state only, when, according to ourinterpretation of the results, any mechanical or neuromuscular intervention is toolate to be very e¡ective. Or, in other words, diagnosing the possibility for jointdegeneration needs to be done immediately upon an insult or trauma. Therefore,the early detection of joint degeneration and OA is likely of prime importance forthe successful ¢ght against this disease.
Muscle forces and EMG
As indicated above (Fig. 2), ankle and knee extensor forces are dramaticallyreduced in the ¢rst few weeks following ACL transection in the cat. No muscleforce measurements in any experimental model of OA have ever been mademonths or years after intervention. However, it is trivial to show that a givendirection and magnitude of the ground reaction force is associated with givenmuscle force patterns, assuming that there is no substantial amount of co-contraction. Therefore, based on the fact that external ground reaction forces andmoments are, on average, the same at about six months following intervention inthe cat, we may assume that the individual muscle forces are also similar prior toand about six months following ACL transection. Electromyographical patternsof eight muscles in the cat hind limb are indistinguishable in terms of timing
JOINT MECHANICS 87

relative to the step cycle, the magnitude and the frequency content, six monthsfollowing ACL transection compared to normal.We feel that one result concerning muscle forces and electromyograms (EMGs)
deserves attention. Figure 2 shows that gastrocnemius and quadriceps forces arereduced following ACL transection. But not only are the muscle forces reduced,they are also ‘jerky’ rather than ‘smooth’. When comparing the EMG patterns inthe vastus lateralis (VL, one of the quadriceps muscles) before and afterintervention, it is apparent that the continuous EMG during the stance phaseprior to intervention changes to a 3^5 burst pattern during stance followingintervention. This result suggests that the ‘jerkiness’ of the knee extensor forcepatterns following intervention is caused by this burst-like (rather than thecontinuous) activation pattern. We assume that these individual bursts duringstance represent a ¢ght between an excitatory extensor mechanism that sends thecommands to extend the knee so that the animal does not collapse, and aninhibitory extensor mechanism of unknown origin, possibly associated with theinstability of the knee in the early phase following intervention. We furtherspeculate that this perturbed activation pattern, which results in poorlycoordinated muscle activation patterns, and thus presumably poorly controlled¢ne mechanics of joint movement, might be the mechanism for triggering someof the degenerative responses that are seen quickly following joint perturbation.
Pressure patterns
In the most perfect world, joint loading could be accurately described by theinstantaneous and time-evolving stress and strain states of all the individualtissues that make up a joint. However, that is not possible. An acceptablecompromise to this perfect scenario would be the instantaneous and timeevolving pressure distribution patterns in diarthrodial contact surfaces duringnormal movement. However, even such measurements could not be made todate, although pressure-time histories in arti¢cial joints have been made onisolated patients with instrumented joint prostheses (e.g. Bergmann et al 1993,Krebs et al 1991). We have measured the joint surface pressure distributions inthe in situ cat knee using Fuji pressure sensitive ¢lm for a variety of knee anglesand knee extensor forces representing those observed during normal locomotion(Herzog et al 1998, 2000), in normal and ACL-de¢cient knees.Among the many results, arguably the most important was that pressure
distribution in a given knee (for a given knee extensor force and knee angle)changed dramatically from pre- to post-intervention. In 38 measurements ofjoint contact patterns in ¢ve cats, we found that the contact area in the patello-femoral joint increased by 22% (�37%), and the corresponding peak pressure(n¼34) decreased by 55% (�21%) at 16 weeks post ACL transection compared
88 HERZOG ET AL

to the normal, contralateral joint (Fig. 4). This result demonstrated for the ¢rsttime that joint contact loading changes very quickly following ACL transection,and that a given joint loading (produced bymuscular forces via nerve stimulation)for a given joint kinematics (knee angle and angular velocity) might producecompletely di¡erent local loading conditions on the articular surfaces, dependingon the progression of joint disease.The increase in contact area and the decrease in peak pressure observed in this
study with the progression of disease could readily be explained by the alteredmechanical properties of the articular cartilage associated with joint degeneration(Herzog et al 1998). This result should illustrate at least two points: ¢rst,measurement of ground reaction forces, and even the muscular forces, says littleabout the local loading (stress strain states) of the joint. However, it is likely thatthe detailed local loading is responsible for the di¡erentiated local degeneration ofthe joint. Therefore, understanding the local, in vivo joint loading might be anessential piece of information that scientists should try to obtain in the near
JOINT MECHANICS 89
FIG. 4. Pressure distribution in the patellofemoral joint of a cat 16 weeks post ACLtransection. The contact area in the intact joint (Intact, right) is smaller, and the peak pressure(grey scale) is greater compared to the ACL transected joint (ACLT, left) for the same forceacross the joint. This result was statistically signi¢cant across measurements in ¢ve di¡erentcats (38 measurements total).

future, and although we, like many others, have attempted to do this usingtheoretical approaches, it is our ¢rm belief that meaningful results must comefrom experimental measurement. Second, the local loading changes continuouslyin a degenerating joint, thus the biological responses for a given load are likely tochange in accordance.Of course, we have not even considered the possibility that aprecisely identical load given to a normal and a diseased articular cartilage mightproduce completely di¡erent biological responses, because of adaptations in themechanisms that relate loading to the biological response as the jointdegenerates. If this should happen (which seems reasonable), the relationshipbetween how, and how fast, a joint will adapt to mechanical stimuli becomeseven more complex than for the situation in which the relationship betweenmechanical loading and biological response remains constant during an adaptive/degenerative process.
Final comments
Physiological joint loading and biological response
In order to elucidate the detailed mechanisms underlying joint degenerationleading to OA, it seems essential that the in vivo local loading of the target joint isknown. Aside from the standard measurements in humans and animal models ofOA involving gait analysis, measurement of the external ground reaction forces,and possibly EMG, quanti¢cation of the muscle forces and pressure distributionpatterns are possible and provide much needed insight into the joint mechanics.For lack of space, we restricted our focus primarily on this aspect, and we usedthe cat ACL transection as the model for illustration for the simple reasonbecause, as far as we know, it is the only bona ¢de model of end-stage OA forwhich systematic measurements of individual muscle forces, EMGs and jointpressure distributions have been made in combination with all the other standardmeasurements of joint kinematics and external ground reaction forces.Because of this focus, we have neglected two issues that we intended to discuss
provided that space was available. The ¢rst of these is related to the idea ofobtaining the biological response to controlled in vivo joint loading. Usingarti¢cial electrical nerve or direct muscle stimulation, a muscle, or group ofmuscles, can be activated to produce a precisely de¢ned force, through varyingthe frequency and current of stimulation, for a precisely de¢ned joint kinematics(joint angle and angular displacement). We have started such work in the rabbitknee. An example of a one hour loading protocol (2 s of knee extensorstimulation every 30 s at about 50% of the total maximal isometric force) wasproduced and, following the loading protocol, the mRNA expression of somekey proteins and proteinases was measured (Fig. 5). In principle, this approach
90 HERZOG ET AL

JOINT MECHANICS 91
FIG. 5. Normalized mRNA levels of metalloproteinase 3 (MMP3) from articular cartilage ofcentral and peripheral regions of patella and femoral groove. The experimental samples wereloaded in vivo for one hour as shown in the top panel. The contralateral knee, and control kneesfrom normal animals, were used as unloaded controls.

92 HERZOG ET AL
FIG. 6. Normal articular cartilage from the retropatellar surface of a control animal (A), andarticular cartilage from the retropatellar surface of an experimental animal with quadricepsweakness (about 70% loss of force) four weeks past intervention (B). The articular cartilagefrom the experimental animal has surface ¢ssures, and a great loss of structure. It received aMankin score of 11 (out of 14) from a blinded observer, while the cartilage from the controlanimal was scored perfectly normal.

can be re¢ned to mimic precise movement conditions and muscle forces (and thusjoint loading) that occur during unrestrainedmovements, such as locomotion.Wefeel that this presents a powerful approach, as physiologically relevant jointloading conditions can be simulated experimentally, the corresponding articularsurface pressure distributions can be quanti¢ed, and the expected biologicalresponses can be measured. It seems imperative that these physiologicallyrelevant loading conditions and biological responses are compared toexperiments performed on isolated cartilage explants that are subjected toarti¢cial in vitro loading conditions, and to test whether or not the in vitro resultsare meaningful for the physiological system.The second issue is related to muscle force and weakness. We have described
here the important role that muscles play in joint loading, and have hinted thatneuromuscular malfunctioning might produce fertile conditions for the onset ofOA. However, there is another aspect to the story of muscles: the one of muscleweakness. Muscle weakness has been implicated as a risk factor for jointdegeneration (Slemenda et al 1997, 1998). However, there is no direct evidencelinking muscle weakness to joint degeneration. We have developed a rabbitmodel of knee extensor weakness, and have found that within four weeks ofmuscle weakness, there are gross morphological and histological signs (Fig. 6) ofknee and articular cartilage changes that are consistent with a degenerativeresponse (Longino 2003). Realizing that muscle weakness was associated withchanges in loading patterns similar to those in the ACL de¢cient cat, the idea that‘unloading’ or ‘under loading’ may predispose a joint to degeneration, becomes arecurrent theme.Here, we could only summarize the salient features of selectedwork on the in vivo
loading of joints in experimental animal models of joint degeneration and OA.Nevertheless, we hope that we might inspire additional work in this exciting andwide-open ¢eld of scienti¢c investigation. It is our belief that ultimateunderstanding of human OA will not come from theoretical models of jointcontact mechanics and loading, nor from the experimental analysis of articularcartilage explants subjected to loading and boundary conditions far removedfrom those of the native joint, but must come from the precise understanding ofthe in vivo loads in diarthrodial joints and the corresponding biological response.
References
Adams ME 1989 Cartilage hypertrophy following canine anterior cruciate ligament transectiondi¡ers among di¡erent areas of the joint. J Rheumatol 16:818^824
Bergmann G, Graichen F, Rohlmann A 1993 Hip joint loading during walking and running,measured in two patients. J Biomech 26:969^990
JOINT MECHANICS 93

Brandt KD, Braunstein EM, Visco DM, O’Connor B, Heck D, Albrecht M 1991 Anterior(cranial) cruciate ligament transection in the dog: a bona ¢de model of osteoarthritis, notmerely of cartilage injury and repair. J Rheumatol 18:436^446
Burton-Wurster N, Vernier-Singer M, Farquhar T, Lust G 1993 E¡ect of compressive loadingand unloading on the synthesis of total protein, proteoglycan, and ¢bronectin by caninecartilage explants. J Orthop Res 11:717^729
Clark AL, Herzog W, Leonard TR 2002 Contact area distribution in the feline patellofemoraljoint under physiologically meaningful loading conditions. J Biomech 35:53^60
Crowninshield RD, Brand RA 1981 The prediction of forces in joint structures: distribution ofintersegmental resultants. Exercise and Sport Sciences Reviews. The Franklin Institute Press,Philadelphia, p 159^181
GoslowGE,ReinkingRM,StuartDG1973The cat step cycle: hind limb joint angles andmusclelengths during unrestrained locomotion. J Morphol 141:1^41
Guilak F, Sah RL, Setton LA 1997 Physical regulation of cartilage metabolism. In: Mow VC,Hayes WC (eds) Basic orthopaedics biomechanics. Lippincott-Raven Publishers,Philadelphia, p 179^207
Guimaraes ACS, Herzog W, Allinger TL, Zhang YT 1995 The EMG-force relationship of thecat soleus muscle and its association with contractile conditions during locomotion. J ExpBiol 198:975^987
Hasler EM, Herzog W, Leonard TR, Stano A, Nguyen H 1998 In-vivo knee joint loading andkinematics before and after ACL transection in an animal model. J Biomech 31:253^262
Hasler EM, Herzog W, Wu JZ, Muller W, Wyss U 1999 Articular cartilage biomechanics:theoretical models, material properties, and biosynthetic response. Crit Rev Biomed Eng27:415^488
Herzog W, Adams ME, Matyas JR, Brooks JG 1993 A preliminary study of hindlimb loading,morphology and biochemistry of articular cartilage in the ACL-de¢cient cat knee.Osteoarthritis Cartilage 1:243^251
HerzogW,Hasler EM, Leonard TR 1996 In-situ calibration of the implantable force transducer.J Biomech 29:1649^1652
HerzogW,Diet S, Suter E et al 1998Material and functional properties of articular cartilage andpatellofemoral contact mechanics in an experimental model of osteoarthritis. J Biomech31:1137^1145
Herzog W, Hasler EM, Leonard TR 2000 Experimental determination of in vivo pressuredistribution in biologic joints. J Musculoskel Res 4:1^7
Herzog W, Longino C, Clark A 2003 The role of muscles in joint adaptation and degeneration.Langenbecks Arch Surg 388:305^315
Korvick DL, Pijanowski GJ, Schae¡er DJ 1994 Three-dimensional kinematics of the intact andcranial cruciate ligament-de¢cient sti£e of dogs. J Biomech 27:77^87 [Erratum in: J Biomech27:1295]
KrebsDE,ElbaumL,Riley PO,HodgeWA,MannRW1991Exercise and gait e¡ects on in vivohip contact pressures. Phys Ther 71:301^309
Liggins AB, Hardie WR, Finlay JB 1995 Spatial and pressure resolution of Fuji pressure-sensitive ¢lm. Exp Mech 35:166^173
Longino D 2003 Botulinum toxin and a new animal model of muscle weakness. MSc thesis,University of Calgary, Calgary, AB, Canada
Maitland ME 1996 Longitudinal measurement of tibial motion relative to the femur duringpassive displacements and femoral nerve stimulation in the ACL-de¢cient cat model ofosteoarthritis. PhD thesis, University of Calgary, Calgary, AB, Canada
Maitland ME, Leonard TR, Frank CB, Shrive NG, HerzogW 1998 Longitudinal measurementof tibial motion relative to the femur during passive displacements in the cat before and afteranterior cruciate ligament transection. J Orthop Res 16:448^454
94 HERZOG ET AL

Mow VC, Bachrach NM, Setton LA, Guilak F 1994 Stress, strain, pressure and £ow ¢elds inarticular cartilage and chondrocytes. In: Mow VC, Guilak F, Tran-Son-Tray R, HochmuthRM (eds) Cell mechanics and cellular engineering. Springer Verlag, New York, p 345^379
Quinn TM, Grodzinsky AJ, Buschmann MD, Kim YJ, Hunziker EB 1998 Mechanicalcompression alters proteoglycan deposition and matrix deformation around individual cellsin cartilage explants. J Cell Sci 111:573^583
Sah RL, Kim YL, Doong J-YH, Grodzinsky AJ, Plaas AHK, Sandy JD 1989 Biosyntheticresponse of cartilage explants to dynamic compression. J Orthop Res 7:619^636
Sah RL, Grodzinsky AJ, Plaas AHK, Sandy JD 1992 E¡ects of static and dynamic compressionon matrix metabolism in cartilage explants. In: Kuettner K, Peyron JG, Schleyerbach R,Hascall VC (eds) Articular cartilage and osteoarthritis. Raven Press, New York, p 373^392
Slemenda C, Brandt KD, Heilman DK et al 1997 Quadriceps weakness and osteoarthritis of theknee. Ann Intern Med 127:97^104
Slemenda C, Heilman DK, Brandt KD et al 1998 Reduced quadriceps strength relative to bodyweight. A risk factor for knee osteoarthritis in women? Arthritis Rheum 41:1951^1959
Suter E, Herzog W, Leonard TR, Nguyen H 1998 One-year changes in hindlimb kinematics,ground reaction forces and knee stability in an experimental model of osteoarthritis. JBiomech 31:511^517
Tashman S, DuPre¤ K, Goitz H, Lock T, Kolowich P, Flynn M 1995 A digital radiographicsystem for determining 3D joint kinematics during movement. American Society ofBiomechanics, p 249^250
Torzilli PA, Grigiene R, Huang C et al 1997 Characterization of cartilage metabolic response tostatic and dynamic stress using a mechanical explant test system. J Biomech 30:1^9
Walmsley B,Hodgson JA, Burke RE 1978 Forces produced bymedial gastrocnemius and soleusmuscles during locomotion in freely moving cats. J Neurophysiol 41:1203^1216
Xu WS, Butler DL, Stou¡er DC, Grood ES, Glos DL 1992 Theoretical analysis of animplantable force transducer for tendon and ligament structures. J BiomechEng 114:170^177
DISCUSSION
Brandt:With your botox model, where you reduce muscle strength around thejoint, do you see osteophytes? And what does the subchondral bone look like?Herzog: We haven’t looked at that yet. By gross morphology, the osteophytes
don’t appear to be there after four weeks. It is really an adaptation model at themoment, and not anything that I would like to say much more about until wehave followed it longer-term. What we do see after four weeks of inducing severemuscle weakness through botox is a reddening of parts of the joint surfaces (medialtibial plateau), and degenerative signs (based on theMankin score) on the articularcartilage, assessed in histological sections.Felson:Ken Brandt, do youwant to say something about the Bole animal model
for OA?Brandt: Giles Bole, who was the Head of Rheumatology at the University of
Michigan, tried, some 20 years ago, to establish OA in the guinea pig with aprocedure that did not invade the joint. He performed gluteal tenotomy andmyotomy, producing OA in both hips and both knees. This resulted in gaitabnormalities that he was able to demonstrate elegantly by dipping the paws of
JOINT MECHANICS 95

the animal into India ink and having it then walk across a piece of paper. It issomewhat similar to what Walter Herzog is doing with botox�destablizingmuscle forces.Herzog:This is a very interesting animalmodel ofOA that I was not aware of. In
the past two years, we looked at many di¡erent possibilities for inducing muscleweakness, including denervation of the muscle, immobilizing the joint, unloadingthe hindlimb (for example as done in rats through hindlimb suspension where ratsare hung up by their tails, and the hindlimbs are thus lifted o¡ the ground and noloading through ground contact can occur). Although, it is possible in thehindlimb suspension model that muscle loading occurs through the activation ofmuscles.We decided on the botoxmodel becausewewanted to control the amountof muscle weakness, and in this ¢rst study, wanted enough muscle strength left sothat the animal could function normally if it wanted to.We played aroundwith thenumber of botox units. At the end, we injected 3.5 units per kilogram and thatseemed to be the magic ¢gure: it reduced muscle force by about 70^80%, andthus left about 20^30% of muscle force, enough to walk and run around, if theanimals wanted to.Brandt:Were there any changes in the contralateral joint after four weeks?Herzog:No.Henry: There are some of us in the roomwho are curious about how long it will
take for your cats to develop pain.Herzog: That’s a good question. After ¢ve years the joints look terrible. But
having worked for 18 years with cats, I am 99% sure that even after ¢ve yearsthese joints are causing no pain, because the cats walk normally and climbaround. When cats have pain, they let you know. I think I have a fantastic modelof very bad joint disease with apparently no associated pain. I would be verycurious to hear from pain experts about this.Felson: In the break we were talking about structural equivalents of pain. This
raises a question about the cat model at 5 years. When you say there are all thechanges of OA, would you include synovitis and bone marrow oedema? Did yousee this in the cat model after 5 years?Herzog:No, we have not looked at that.Conaghan:Have you scored the synovium for severity?Herzog:No.Pisetsky: If I understood correctly, you are saying that the muscle is doing
something in the ACL model to compensate.Herzog:Yes, we think the knee £exormuscles are doing something. In the intact
cat knee (as in human knees), the ACL prevents anterior translation of the tibiarelative to the femur. In animal models (except the cat), and in humans, it hasbeen reported that after the loss of the ACL, the tibia slips forward relative to thefemur at ¢rst foot contact in the stance phase of the step cycle. In the cat, this has
96 DISCUSSION

never been observed, neither by us nor other people. However, what we haveobserved is that following ACL transection in the cat, the knee £exor muscles areactivated at the instant of ¢rst foot contact (they are not activated at that time in theintact hindlimb).Knee £exor activity and forcemechanically prevent the tibia fromslipping forward on the femur, because the knee £exors are pulling the tibiabackward. Therefore, it appears that following the loss of the ACL, the knee£exors take over the mechanical stabilization of the knee that is normallyprovided by the ACL. The interesting question here is: why does this happen inthe cat and not in other animals, and not in humans?Pisetsky: If it is not pain, what is the driving force for themuscle to change? And
is this compensation somehow a¡ecting the time until the onset of the arthritis? It isa very long model.Herzog: It is a long model. Except for Ken Brandt, I don’t think I know of
anyone here who has run an animal model for several years. I don’t know whatthe muscle senses to make it contract. My hunch would be that the initialinstability would cause some stretch of the muscles that was not there in theintact knee, and that this stretch might activate the muscles. If this is the case, itwould all happen very quickly, as wemeasure this adapted activation pattern of theknee £exors within three days of intervention.Pisetsky: Is that pain?Herzog: In the ¢rst few days the animals have pain. It could be pain, although it is
more likely to be a spinal re£ex mediated response. We must also consider thatthese animals are on pain killers for the ¢rst few days following intervention,thus pain as the source for these adaptations seems rather unlikely.Pisetsky: Can you add a drug to slow or speed up this model?Herzog:We haven’t tried this.Grubb: In your ACL transection model, where you saw the decreased loading of
the quadriceps muscle, was there any evidence of guarding?Was the loading largeron the contralateral side?Herzog: Yes. The loading on the contralateral side is initially greater than
normal, and greater than on the experimental hindlimb. For static loading(standing still), these di¡erences disappear after approximately 3^4 months(Herzog et al 1993). For dynamic loading (walking), di¡erences in loadingbetween the experimental and the contralateral hindlimbs seem to disappear afterabout 6 months (Suter et al 1998).Grubb: So were there also gait abnormalities?Herzog: Yes. The results I showed were initial results from 2^4 weeks after
intervention. There was a distinct limp and an overloading of the muscles andthe ground reaction forces on the contralateral limb. This seemed to recover afterabout six months. Incidentally, the anterior drawer (i.e. the anterior displacementof the tibia relative to the femur for a precisely controlled anterior force), even in
JOINT MECHANICS 97

the passive sense, goes away in the cat. It seems that the passive structures in thatknee take up the slack within about 4 months (Maitland et al 1998).Evans:Why did the cartilage in the femoral groove remain intact?Herzog:There are a couple of possible answers from amechanical point of view.
At any given instant in time, the pressure exerted from one cartilage on the otherhas to be equal and opposite. If youmeasure the pressure distribution in the patellaat di¡erent knee angles, you realise that themid-part of the patella is always loaded,whereas there is no area of the femoral condyle that is always loaded. In the patellathere is one area that is always loaded, but there is not in the femur. A secondexplanation could be that the articular cartilage properties di¡er from one side ofthe joint to the other. For example, we know that the retropatellar surface articularcartilage is always about twice as thick as the one on the femoral groove side. Athird explanation might be that it is advantageous having a convex versus aconcave surface.Dieppe:Coming back to your painless cats, I would like to suggest that you have
a very good model of human elbow OA. There is evidence from pathologicalstudies that elbow OA is extremely common, but it is hardly ever symptomatic.Here we have one human joint which seems to get the disease but which doesn’tget pain, which is what you seem to bemodelling in the cat. This also reinforces theearlier point about the heterogeneity between joints, which we are lumpingtogether under the single banner of OA.Herzog: I didn’t know that. I should add that the reason I am holding on to these
long-term animals is that I would love to see them develop clinical signs (i.e. pain),to see why after such a long time, when these joints ¢nally might become painful,these joints might be di¡erent from the joints that look terrible at ¢ve years, andwhat might have been responsible for the sudden appearance of clinical signs.Kuettner: We have been looking at the ankle and knee joints, and we recently
extended this study to the elbow and the shoulder. We ¢nd that the elbowbehaves similarly to the knee joint, whereas the shoulder is more like the anklejoint, with regard to the capacity ex vivo to synthesize matrix components.Brandt:Did all of your animals acquire a compensatory hamstring gait?Herzog: Yes.Brandt: In humans who have an ACL rupture, physical therapists try to teach
that, but some people simply can’t learn it.Herzog: I would suggest that in humans it is not quite as e¡ective because we
(humans) essentially walk with a straight knee. In contrast, the cat knee anglegoes from about 80^120 degrees during walking, and the semitendinosus (a knee£exorwhose activity patternswemeasured in our animalmodel ofOA) is attachingabout a third down on the tibia. In mid-stance it will perfectly pull the tibia backrelative to the femur, whereas in human gait this is never really the case because wewalk with an extended joint.
98 DISCUSSION

Hunter:You showed quite a large di¡erence in the muscle size and strength afterthe botox treatment. Does botox have any e¡ect on muscle quality in addition tomuscle size, and if so does this have any relationship to the joint changes you seesubsequently?Herzog: I can’t answer that. But we do know that there is a disparity between the
musclemass and the loss of force. The initial loss of force is about twice as big as theloss of mass. This presumably has to do with the fact that botox reacts at the neuraljunction to the muscle, inhibiting acetylcholine release. Therefore what wepresumably see is that we have an initial very high inhibition of acetylcholinerelease. The muscle then adapts by atrophying and losing mass. Over the nextfew weeks it will probably lose even more muscle mass before it recovers again.Then of course the e¡ect of botox is reversible, as the beauty industry knows:people who use it to reduce wrinkles have to inject it every three or four months,because after this time there is enough sprouting of new terminal axons that themuscle will be virtually fully re-innervated. Similarly, in the animals, we know thatthe e¡ect of botox wears o¡ and themuscles recover virtually their full strength. Infact, one of the beautiful aspects of the botox model is that you can control quiteprecisely the time frame ofmuscle weakness. Repeat injections can keep the muscleforce low for inde¢nite periods, whereas a single injection loses most of its e¡ectswithin 3^4 months.Grubb: Botulinum toxin injected into muscle classically causes neuromuscular
preterminal sprouting but loss of junctional contact. It has always been assumedthat the muscle simply atrophies for the period where it is inactive, and then whenthe botox disappears the new neuromuscular junctions form very well fromresprouting of the e¡erents into the neuromuscular or junctional ¢eld. Then youget recovery. I don’t think there is any muscular involvement.
References
Herzog W, Adams ME, Matyas JR, Brooks JG 1993 A preliminary study of hindlimb loading,morphology and biochemistry of articular cartilage in the ACL-de¢cient cat knee. OsteoarthCartilage 1:243^251
Maitland ME, Leonard TR, Frank CB, Shrive NG, HerzogW 1998 Longitudinal measurementof tibial motion relative to the femur during passive displacements in the cat before and afteranterior cruciate ligament transection. J Orthop Res 16:448^454
Suter E,HerzogW, Leonard TR,NguyenH 1998One-year changes in the hindlimb kinematics,ground reaction forces and knee stability in an experimental model of osteoarthritis. JBiomech 31:511^517
JOINT MECHANICS 99

General discussion I
Developing animal models of RA
Felson: I wanted to reintroduce a question that was posed earlier by BlairGrubb.What is osteoarthritis (OA) and how are we ever going to design an animal modelto deal with this particular set of problems?Howwould it be reasonable to developor study animal models for drug development or other therapeutic purposes thatmight have painful OA, and not just OA?Grubb:Oneof the things thatmademe ask the question is that there seems to be a
very poor correlation between pain and the radiological evidence. In the discussionafter my paper, Paul Dieppe made a strong point that the synovium probablywasn’t involved in the pain of OA; that it was probably subchondral bone. Whatis the evidence concerning the source of pain in OA, and why do clinicians think itis possibly from subchondral bone? And why don’t you think that it is from otherstructures?Felson: Let me defer you for a minute. There will be lots of discussion later on
about bone as a potential source of pain in OA.Grubb: From the point of view of an animal model of OA, if you want one that
produces the same pain as OA, you want to know where the pain is coming from.Kuettner: Should we call it an animal model of OA, or should we say it is an
animal model of degenerative joint disease? Calling it OA already has clinicalimplications. If we call it an animal model for degenerative joint disease then weaddress certain questions.Dieppe: I agree. Also, given the facts that we have alluded to: that hip OA is
di¡erent from knee OA or elbow OA, to have a single model doesn’t seem to bethe right way to go. I think we are muddling up three things and calling them allOA. There is a group of people, relatively young, whose joints fall to pieces andwho need joint replacement. This is a terrible disease that is relatively uncommon.Then there is something that happens to joints that involves adaptation tomechanical change. Then there is a huge burden of pain in the community. Ithink they are potentially quite di¡erent things, yet we are muddling them upand calling them all OA. Unless we start to unpack this muddle, I don’t see howwe can go forward. To say we will have a model of OA seems to be going in thewrong direction, because it is confounding that muddle rather than unpacking it.
100
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

Felson: I disagree with you. I’m nervous about doing this because you aresomeone I look to for ideas that I then slavishly follow! I think OA is a spectrumof a single end-stage disorder. I would take rheumatoid arthritis (RA) and otherforms of rheumatic disease such as ankylosing spondylitis as examples where somepeople have mild structural disease and no reason for severe pain but who areplagued by pain, and other people who have much more structural disease butlittle pain, and we seem to be able to treat all these people with new agents. Thisis what OA is going to be to. I would argue that the elbow that isn’t symptomaticwould be if we walked on our hands. The cat may not be symptomatic because it issomehow adapted in ways we don’t understand andwalks on the other three paws.So much of the symptom relation is mechanical: the incipient symptoms arereproducibly mechanical. I am trying to be a lumper more than a splitter. I’minclined to say let’s make it easier for people trying to develop therapies and notput impossible barriers in front of them by splitting OA into 20 di¡erent diseases,each of which needs to be treated di¡erently. I think it is ultimately onemechanically driven disease for which we need to understand the aetiology ofsymptoms better.Pisetsky: In the animal models of RA, I don’t think pain has ever been a
requirement for validation. Nor can I remember anyone saying that this shouldbe part of an assessment. Investigators have looked for pathology such asin£ammation. The other thing investigators have studied is whether drugs thatwork in human disease work in animal disease. If you said that non-steroidalanti-in£ammatory drugs (NSAIDs), for example, had an e¡ect in human OA, itwould be of interest to test it in your projected animal models to see whetherthere is a bene¢t. In lupus, another question has been whether there is a di¡erenceinmales versus females in disease. One of the attractions of theNZB/Wmice is thatdisease is much worse in females. Are there any animal models of OA where thedisorder is worse in female animals?Brandt: We are looking at somewhat di¡erent things. The drugs developed
recently for RA, that are proving to be so e¡ective in this disease, were developedwith some understanding of the pathogenesis of the disease. They are not generalanti-in£ammatories. The drugs we use today for symptomatic therapy of OA aregeneral analgesics.Hunter: That is an important point. You may help structure but you may not
necessarily help symptoms. This is a lesson that has been learned in humans. Anyanimal model development that occurs needs to marry any structural abnormalitywith any symptomatic consequence that may be revealed. It is important to try toclarify the disease as opposed to trying to divide the disease as much as possible.Animal models are a means of trying to eliminate the pyschosocial elements andjust concentrate on the biology. I believe that in the next two to three years a lot ofanswers will come through from some big longitudinal cohort studies that are
GENERAL DISCUSSION I 101

about to begin, investigating various abnormalities with structurally moresensitive tools than we currently have available to us. It would be reasonable tohave a prioritization list of structures within the knee that have a¡erentabnormalities, which people can then walk away with and then develop ananimal model to help them explore each structural abnormality further.Rediske: There were a couple of abstracts at the recent national In£ammation
Research Association meeting (October 2002) by groups at P¢zer that describedthe joint pain component of the mouse iodoacetate model of OA. They used apressure plate device, and showed a di¡erence between the OA knee versus theuna¡ected knee in terms of weight-bearing sensitivity. It was this sensitivity thatcould be reduced byNSAIDswhich eliminates any neuropathic component. Thereis some potential in thismodel. AtRoche, TonyManning has also started to look atthis question of joint pain in OA animal models. Could he comment on hisongoing studies?Manning: Two points. Are there pain measurements that can be made in animal
models? Yes there are, and groups are beginning to take these and apply them tocaustic and surgical models. Some of the most interesting work has been done intheHarltey guinea pig, a spontaneousmodel. The quandary is that we are in a catch22 situation. If the clinicians don’t know how to diagnose the clinical endpointsneeded for the registration of new disease-modifying products, how can theresearchers then re¢ne their models to ask speci¢c questions about therapies?Any advances in standardizing magnetic resonance imaging (MRI) or biomarkersor classi¢cation of human disease would be helpful. At this point it is unrealistic toask which is a better animal model of disease that we haven’t yet de¢ned.Felson: Paul Dieppe was pessimistic about OA as a monolithic disease. What if I
were to tell you that ¢ve years from now we won’t have any MRI or biochemicalmarkers that help us understand human painful OA?Manning: Until then, you cannot build a better animal model and you will not
have better therapeutic opportunities. It will be a long time until we havetreatments that protect the structure and the symptoms.Felson:Why is that?You saidwe can evaluate painful paws and look atOA.Why
can’t you test therapies?Manning: The challenge is as you just said: which patients do you do this in? If
you pick patients with radiological OA, howmany of these truly have pain? If partof the registration requirements are to impact symptoms as well as joint structure,this places an additional burden.Fernihough: What we have with the animal models is a very complicated test-
tube, so we can only look at the mechanistic side of things. Since the animalmodel is induced, and is controlled to be consistent, it cannot re£ect the fullrange of features that characterize painful human OA. Therefore, we can onlyanswer questions regarding the biochemistry and physiology of nociception in a
102 GENERAL DISCUSSION I

whole animal setting.We can look at nerves at the level of the dorsal root ganglion(DRG) and there are no huge changes between normal andOA rats.We can look inthe knee, but you see nerves everywhere in a rat knee joint. We are required todemonstrate that we are looking at OA from a histological point of view, so wewant amodel of OA. Butwhere it falls down is that clinicians can talk to patients ina psychosocial environment and ask them questions about what pain is importantto them, but this doesn’t give the animal model people a handle on themechanisticnociception. There may be subsets such as activity related pain that we can mirror,but the communication is not good at the moment as to what the clinicians wouldde¢ne as more important from a mechanistic point of view, and separating thisfrom the social environment.Conaghan: I liked David Felson’s use of the phrase ‘joint failure’. What if we said
that an individual animal model is just a model of joint failure with predominantsubchondral bone damage, or synovitis, or cartilage failure? This is what we havethe models of at present. I would agree with Paul Dieppe that there is an importantdi¡erence between the people who seek help and the people who don’t, but assomeone who has run a very busy knee service for a long time, I don’t see thisdiscrepancy between community OA, the young person with premature failure,and a 60 year old with age-related joint failure. I agree with David Felson thatthis is a common end-stage process. I think the animal models are still valid forparticular parts of the joint failure model.Pisetsky: It would be very hard to develop a drug for OA without an animal
model. Registration organizations would not be happy, and nor would peopletesting the drugs. There are so many mouse knockouts available that we shouldbe able test di¡erent combinations of strains and knockouts to produce a suitableanimal model.Kuettner:But shouldwe concentrate on stopping the degeneration, or try to ¢nd
a drug that will restore and repair the tissue? The surgeon is going inwhen say 60%of the cartilage is gone. If we want to use a drug, we need to use it way before thisand stop the degeneration earlier, possibly in tandem with a repair mechanism.Pisetsky: There are recent studies in RA suggesting that early anti-tumour
necrosis factor (TNF) can actually lead to repair. One of the speculations is that,at early stages of disease, there is a repair capability such that, if the in£ammation isblocked, repair ensues. If you go too long before treatment starts, then you lose therepair process.Manning: The challenge is to identify those patients very early so there is an
opportunity to intervene. In RA we are in a slightly better situation of being ableto detect those patients early. We need better diagnostics in OA.Creamer: Klaus Kuettner said that by the time people get to surgery they have
lost 60% of their cartilage and it is then too late. But there are plenty of people whohave lost this much cartilage who don’t seem to have any problem.
GENERAL DISCUSSION I 103

Fernihough: From the animal model perspective we are only worried about thehistology to demonstrate that it is an OA or degenerative joint process. Workingon an animal pain model of OA I don’t care whether the histology gets better orworse�we are focused on the pain. But we don’t have good readouts for that in aclinical environment to keep our model relevant.Felson: I’m confused as to why you are struggling with the clinical readout
issues. There are well-validated scales and all kinds of criteria for approval oftherapies that relate to signs and symptoms.Fernihough: One of the comments early on was that the psychosocial
environment and qualitative analysis of patients is much more important. But Ican’t have an in-depth unstructured interview with a rat!Manning:There are good, well-validated readouts. I don’t think there is somuch
of a problem here. My comments were more concerned with the pursuit of thehypothesis that protecting joint structure will impact signs and symptoms. Thisis as yet unproven, even in the animal models.Felson: There are drugs that improve symptoms and signs of OA that have been
well validated. WOMAC and Lequesne Index have been used and they work OK.Brandt: The problem is that you can say that an animal model mirrors the
chondroprotective e¡ects of a new drug for OA only after you demonstrate thee⁄cacy of that drug in humans. Only then can you say that the model is apredictor of the e¡ect of the drug in humans. No animal model of OA has yetbeen shown to have predictive value.Pisetsky: Is there anything di¡erent about the biomechanics of the animals that
has been a hindrance here? I am impressed that the cats go a long time withoutdeveloping problems, but the dogs get problems in a month or two. Should webe thinking of a particular animal that is more likely to have some correspondenceto humans?Brandt:The dogs do not develop joint pain, as far aswe know.They change their
gait, as do the cats, but they don’t look as though they are in discomfort.
104 GENERAL DISCUSSION I

Characterization of joint pain
in humanOA
Gunnar Ordeberg
Division of Orthopaedic Surgery, Karolinska Institutet, Danderyd Hospital, Stockholm,SE-18288, Sweden
Abstract. Characterization and di¡erentiation of joint pain is di⁄cult. Thoughdegenerative changes in joints are frequent causes of pain in hip and knee, these changesare not always painful, and other possible causes of pain must also be considered. Indegenerative changes in the spine, the problem is even more complex, as peripheralneuropathic pain, caused by mechanical compression and/or leakage of cytokinesirritating nerve roots may be di⁄cult to di¡erentiate from nociceptive pain fromintervertebral joints, discs or muscles. We know now that nociceptive pain has oftenreferred to areas of pain with numbness and parestesthethic sensations, previouslyregarded as characteristic for neurogenic pain. Furthermore, in patients with painfulcoxarthrosis quantitative sensory testing (QST) has shown disturbed sensory thresholdsnot only in regions adjacent to the a¡ected hip but also contralaterally. These sensorydisturbances, previously noted in neuropathic pain, normalized after successful surgerywith relief of pain, thus con¢rming the relation to the hip joint. Patients with painfulcoxarthrosis also have moderately increased substance P activity in cerebrospinal £uid.Thus the ¢ndings show some similarities with ¢bromyalgic patients with highlyincreased substance P in cerebrospinal £uid and sensory disturbances. In conclusion,joint pain has a profound impact on the sensory system and need a multimodal approach.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 105^121
Pain is usually characterized as nociceptive, neuropathic, idiopathic orpsychogenic. Di¡erent receptors and pain transmitters are involved, andresponses to analgesic agents di¡er in these categories as does the pattern of paindistribution. Pain is also characterized regarding its quality (stabbing, aching,shooting or paresthetic), whether it is permanent or occasional, or whether it isrelated to the time of the day, exercise, strain and physical or mental stress.Pain in osteoarthritis (OA) is most frequent in hip and knee, i.e. the big joints
under mechanical load. Degenerative changes concomitant with pain are alsoextremely common in the spine; however there is often controversy as towhether the pain is produced from OA in the intervertebral joints, degenerationof discs or in other structures such as muscles and ligaments (Schwarzer et al 1994).
105
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

Furthermore osteophytes, synovitis and capsular thickening in OA of theintervertebral joints� as well as herniation from the degenerated disc�withmechanical and chemical irritation of the nerve structures may cause pain ofperipheral neurogenic origin that is sometimes hard to di¡er from degenerativenociceptive pain (Brisby et al 2002).Pain in OA may start either from subchondral bone, as when OA develops as a
cause of an avascular necrosis in the femoral head (Arlet et al 1978), from primarylesions in the cartilage (Broom et al 2001) or from joint swelling and in£ammatoryreactions with distension of the capsule.As the hip is a ball and socket joint, hip OA will in most cases follow a fairly
similar course, as there is only one joint compartment. Though the localizationof pain in hip OA may vary, as I will come to later, the surgical treatment in hipOA is also fairly uniform; total hip replacement with endoprosthesis is by far themost common surgical treatment. The questions aremainly of design of prosthesisand whether bone cement or other ¢xation is to be preferred. Osteotomy may stillbe used in a few young patients with focal degeneration, but it is becomingincreasingly rare (Millis & Kim 2002).In the knee there are three compartments froma functional view (McAlinder et al
1992): themedial and the lateral femurotibial joints and the femuropatellar joint. Inpatients with advanced knee OA usually all three compartments are engaged.However, in patients with moderate knee OA symptoms di¡er according towhich compartment is primarily engaged. Femuropatellar joint ¢brillation ordegeneration of patellar cartilage is common even in young individuals,especially in athletes, and pain is provoked when the knee is under load in£exion, as in climbing stairs, squatting or in sports. In most cases pain from thisjoint is moderate; in patients with very severe pain and with malalignment in thefemuropatellar joint this can be addressed surgically either by a lateral release of thecapsule or transfer of the tibial tubercle (Aderinto & Cobb 2002, Wang 2001).Arthroscopic lavage or smoothening of the cartilage has been used widely inthese cases as in cases with slight or moderate OA in other compartments.However, in a recent double-blind investigation (Bradley et al 2002) thistreatment turned out to be no better than a sham operation with only skinincision. Results from surgery with patellar prosthesis in patients with isolatedOA in the femuropatellar compartment have also been questioned, and have notgained widespread use. In contrast a patellar prosthesis is often used in total kneereplacement with replacement of all three compartments (Waters&Bentley 2003).In most cases clinical symptoms in knee OA start in the medial femurotibial
compartment (McAlinder et al 1992). Lateral femurotibial onset is seen mainly inpatients with deformities or previous fractures, especially compression fractures ofthe lateral tibial condyle, which is a common fracture in elderly women. Painusually starts in the part of the knee ¢rst a¡ected; there is also localized tenderness
106 ORDEBERG

of the a¡ected joint space with provocation of pain in passive extension androtation of the knee. In isolated medial femurotibial OA in younger cases,proximal tibial osteotomy with shift of the load axis to the lateral compartment issometimes performed. This operation used to be more common before theevolution of knee endoprosthesis. Interestingly this operation had the reputationof relieving pain at rest. It may be speculated that part of the e¡ect was caused byrelief of increased intramedullary pressure in the tibia by the fenestrationperformed with the surgery (Arnoldi et al 1980).Today surgical implantation of arti¢cial joint components is by far the most
common surgery in knee OA. The most common procedure is a total kneereplacement with prosthetic replacement of both medial and lateral femurotibialcompartments, with or without replacement of the patellofemoral joint (Waters& Bentley 2003), even though isolated replacement of the medial or lateralfemurotibial compartment is sometimes performed.Symptoms of hip OA usually start with localized pain either in the groin or
trochanteric region. Pain in the ventral part of the thigh and knee is alsocommon and occasionally a patient with hip OA may present with painpredominantly in the knee. The true source of pain is revealed by the clinicalexamination with restriction and pain with hip rotation and normal physicalexamination of the knee. In the beginning the patient usually has pain only whenthe joint is under load. Later, pain at rest may follow. Furthermore, in thebeginning the pain can be eliminated or at least signi¢cantly reduced by di¡erentkinds of analgesics but later the response may become insu⁄cient or absent. Thearea with pain will often spread to the thigh and knee, but pain localization in theleg or even widespread pain all over the body is not uncommon. In fact, havingstarted to use these pain drawings in patients with clinical signs of hip OA, I havebeen surprised howmanypatients havewide pain distribution, and also despite thismost patients report almost total pain relief following surgery with total hipreplacement. Of course, enlargement of the area of pain could be due toconcomitant OA in other joints or in the spine. However, we think thatsensitization is the most common cause, as this mechanism has been reported inanimal experiments, and increased pain area has also been reported in patientswith pain from burns (Pedersen 2000).The wide variation of pain intensity in hip OA, with poor correlation with the
degree of radiological severity as well as of the histological evidence of synovitis,turned our interest to the plasticity of the nervous system and especially to factorssuch as central sensitization and activation of endogenous pain inhibitorymechanisms for the modulation of pain sensation. We studied an endogenouspain inhibitory mechanism, namely di¡use noxious inhibitory control (DNIC).In normal individuals, DNIC-like heterotopic noxious condition stimulation caninduce reduction of pain sensitivity in other parts of the body. However, in a
MECHANISMS IN JOINT PAIN 107

previous study, Eva Kosek showed that ¢bromyalgia patients exhibiteddysfunction of this mechanism (Kosek & Hansson 1997) as well as hyperalgesia/allodynia not restricted to painful areas (Kosek et al 1996).In the study of DNIC in OA, 15 patients with painful hip OA underwent
examination and, as 13 of them were later operated on, these were also examined¢ve months after surgery, when they were almost pain-free. Comparison was alsomadewith sex- and age-matched controls.We used a submaximal e¡ort tourniquettest to induce ischaemic pain. A blood pressure cu¡ was applied on the upper armipsilateral to the painful hip and in£ated to 170mmHg. The subjects wereinstructed to perform a standardized exercise which gave rise to ischaemic pain inthe arm. The pain remained as long as the cu¡ was in£ated. Quantitative sensorytesting (QST) was performed contralaterally to the painful area in the leg in thepatients and pressure pain thresholds were determined with pressure algometry(Jensen et al 1986), while perception thresholds to light touch were assessed by
108 ORDEBERG
FIG. 1. Mean pressure pain thresholds (PPTs) (�SEM) in OA patients and healthy controls.Before surgery no statistically signi¢cant change in PPTwas seen during tourniquets in patients.In controls, PPT increased during tourniquet (P50.002), decreased following tourniquet(P50.001), and returned to baseline values. Following surgery PPT increased duringtourniquet in patients (P50.02), decreased following tourniquet in patients (P50.03) andreturned to baseline values. In controls PPT increased during tourniquet (P50.001) anddecreased following tourniquet (P50.002), but remained slightly elevated compared tobaseline values (P50.01). With permission from Lindh et al (1997).

von Frey ¢laments (Aestesiometer; Weinstein 1962). The thermal sensitivity wasanalysed by a Thermotest with cold and warm stimuli (Hansson et al 1988).Registrations were performed before, during and 45 minutes after the tourniquettest.As shown in Fig. 1, patients had a tendency to lower pressure pain thresholds
(PPTs) compared to controls before tourniquet (P¼0.05), while with appliedtourniquet the PPTs were signi¢cantly lower (P50.001) in patients than incontrols, and after tourniquet the PPTs were also signi¢cantly lower in patients(P50.002). However, at the second registration, when the patients had beenoperated on, PPTs increased during tourniquet in patients and controls alike,and there was no statistically signi¢cant di¡erence in PPTs between patients andcontrols either before, during or after tourniquet.Thus the main ¢nding was that no modulation of pressure pain sensitivity was
found in patients before surgery, in contrast to controls, thus suggesting adysfunction of DNIC mechanisms. However following surgery this dysfunction
MECHANISMS IN JOINT PAIN 109
FIG. 2. Mean light-touch perception thresholds (LTTs) (�SEM) in OA patients and healthycontrols before and following surgery. Before surgery, LTTs increased during tourniquet inboth groups (P50.001) and decreased following tourniquet in controls (P50.001), but not inpatients. After tourniquet LTTs returned to baseline in controls, but therewas a strong tendencyto elevated LTTs in patients (P50.05). Following surgery, LTTs increased signi¢cantly duringtourniquet in both groups (P50.001), decreased following tourniquet in both groups(P50.001) and returned to baseline values. With permission from Lindh et al (1997).

had subsided. As shown in Fig. 2 light-touch perception thresholds (LTTs) weremore similar in patients and controls; before surgery the thresholds increased inboth groups and the only di¡erence was a tendency towards elevated LTTs inpatients after tourniquet. Following surgery no di¡erences in LTTs were seenbetween patients and controls. In the registration of temperature thresholds therewas no e¡ect of tourniquet on thresholds to either innocuous warmth or heat pain,before and after surgery.Perception thresholds to innocuous cold increased during tourniquet in patients
as well as in controls, decreased in both groups after tourniquet before and aftersurgery (Fig. 3), and no signi¢cant di¡erences were found between the groups.In the study of hyperalgesia in 14 of the patients with painful hip OA,
quantitative sensory testing was performed in the most painful area, in mostpatients in the trochanteric region, and in their respective controls (see Table 1)before and (in the 12 patients who had subsequent operation) after surgery.Pressure pain sensitivity was assessed with a pressure algometer and thermalsensitivity with a Thermotest. Compared to controls, patients had increasedsensitivity to pressure pain (P50.002), innocuous warmth (P50.03), cold pain
110 ORDEBERG
FIG. 3. Mean thresholds to innocuous cold (CTs) (�SEM) in OA patients and healthycontrols before and following surgery. Before, as well as following surgery, CTs increasedduring tourniquet in both groups (P50.001), decreased following surgery in both groups(P50.001) and returned to baseline. With permission from Kosek & Ordeberg (2000b).

(P50.05) and a tendency to increased sensitivity to heat pain (P¼0.054) beforesurgery. However, at the second examination after surgery, no signi¢cantdi¡erences in sensitivity of any kind were seen between patients and controls.Furthermore patients also had reduced PPTs on the contralateral side beforesurgery. This was also normalized following operation. Interestingly, the sametype of sensory aberrations seen before surgery in these patients with hip OAhave previously also been registered by my collaborator Eva Kosek in patientswith ¢bromyalgia (Kosek et al 1996). However, these abnormalities have notbeen found in similar studies of patients with rheumatoid arthritis and withtrapezius myalgia (Le¥er et al 2002a,b).
MECHANISMS IN JOINT PAIN 111
TABLE 1 Quantitative sensory testing in patients with OA of the hip and healthycontrols (mean �SEM)
Before surgery (n¼14) Following surgery (n¼12)
Pain side Contralateral side Pain side Contralateral side
PPT (kPa)
Patients 198.8�22.7**{ 252.8�30.1 290.8�44.7{ 298.7�52.7
Controls 338.7�26.3 333.2�22.0 333.8�32.3 315.4�29.8
LTT log 10 (mg)
Patients 3.25�0.4 3.11�0.2 3.01�0.3{{ 3.03�0.2
Controls 3.06�0.2 3.00�0.2 3.03�0.3 2.97�0.2
CT (D8C)
Patients 1.4�0.2 1.6�0.2 1.6�0.3 1.8�0.4
Controls 1.6�0.3 1.6�0.2 1.5�0.2 1.6�0.1
WT (D 8C)
Patients 2.5�0.4* 3.0�0.5* 3.8�0.5{ 3.8�0.8{
Controls 4.2�0.7 3.8�0.5 3.2�0.3 3.2�0.3
CT+WT (D 8C)
Patients 3.8�0.5 4.6�0.6 5.5�0.7 5.6�1.1
Controls 5.8�0.9 5.4�0.6 4.6�0.4 4.8�0.3
HPT (8C)
Patients 41.2�0.8 41.2�0.7 43.1�1.0 43.3�0.9
Controls 42.9�0.6 42.8�0.8 42.8�0.7 43.0�0.6
CT, perception threshold to innocuous cold; HPT, heat pain threshold; LTT, light-touch perceptionthreshold; PPT, pressure pain threshold; WT, perception threshold to innocuous warmth; (CT+WT),sum of innocuous thermal thresholds. Statistically signi¢cant di¡erences are indicated (a) between groups*P50.05; **P50.01; (b) between treatments {P50.05; {{P50.01; (c) between sides {P50.05. Withpermission from Kosek &Ordeberg (2000a).

It is known from several studies that patients with ¢bromyalgia have increasedsubstance P-like activity (SPLI) in cerebrospinal £uid (CSF) (Vaeroy et al 1988 ). Inearlier investigations we have also analysed CSF samples from 11 patients withpainful hip or knee OA for SPLI and compared them to a group of nine pain-freecontrols and nine patients with rhizopathic pain from a herniated lumbar disc(Lindh et al 1997). The SPLI in CSF from the OA patients was increased incomparison to the controls (Fig. 4) but also in comparison to the patients withrhizopathic pain. As shown in Fig. 5 there was a correlation between SPLI andpain score as recorded pre-operatively, but the increased SPLI was less than thatseen in patientswith ¢bromyalgia (Russell et al 1994). TheOApatients in the studywere all candidates for surgical joint replacement and had another CSF sample 5months after the operation. SPLI had decreased, but was still higher than in thecontrols.Thus these ¢ndings suggest a gradual transition in OA from uncomplicated
nociceptive pain to secondary sensory disturbances having similarities with¢ndings in patients with ¢bromyalgia. An interesting question is of course whenthese disturbances are reversible; it is obvious that they were reversible in thestudies above; however we all know that widespread pain for example in patients
112 ORDEBERG
FIG. 4. sP levels in cerebrospinal £uid from patients with hip OA (coxarthrosis) and ischialgia(radiating pain from herniated lumbar disc) (mean �SEM). ***Patient groups compared tocontrol group (P50.001). Postoperative activity compared to preoperative values (P50.01).With permission from Kosek &Ordeberg (2000b).

with diagnosis of ¢bromyalgia is very di⁄cult to counteract. In the orthopaedicliterature, especially regarding patients with back pain, wide pain distribution isoften regarded as a sign of behavioural symptoms (Waddell & Richardson 1992).Accordingly there is a reluctance to treat patients with widespread pain surgically,even in the presence of a morphological, potentially pain-inducing lesion.However, we have recently taken part in a study of fusion surgery in lower backpain of segmental origin, where preoperative pain distribution was evaluated frompain drawings. In this study the clinical result two years postoperatively was thesame in the patients with wide pain distribution on their pain drawings as in thosewith more localized pain (H�gg et al 2003).In conclusion, though our knowledge of painmechanisms inOA and other pain
conditions is still very fragmentary, improved classi¢cation and understanding ofunderlying mechanisms have already increased treatment options in recent years.We believe that further research will give additional improvements in thetreatment of pain.
References
Aderinto J, Cobb AG 2002 Lateral release for patellofemoral arthritis. Arthroscopy 18:399^403Arlet J, Ficat P, Mazieres B 1978 Coxarthroses due to ischemia. Ischemic coxarthropathy.(French) Rev RhumMal Osteoartic 45:549^560
MECHANISMS IN JOINT PAIN 113
FIG. 5. Correlation between CSF levels of SPLI and pain scores as recorded preoperatively.y¼1.78 + 13.9 r 2¼ 0.51 (P50.01). With permission from Kosek &Ordeberg (2000b).

Arnoldi CC, Djurhuus JC, Heerfordt J, Karle A 1980 Intraosseous phlebography, intraosseouspressure measurements and 99mTC-polyphosphate scintigraphy in patients with variouspainful conditions in the hip and knee. Acta Orthop Scand 51:19^28
Bradley JD, Heilman DK, Katz BP, Gsell P, Wallick JE, Brandt KD 2002 Tidal irrigation astreatment for knee osteoarthritis: a sham-controlled, randomized, double-blinded evaluation.Arthritis Rheum 46:100^108
Brisby H, Olmarker K, Larsson K, Nutu M, Rydevik B 2002 Proin£ammatory cytokines incerebrospinal £uid and serum in patients with disc herniation and sciatica. Eur Spine J11:62^66
Broom N, Chen MH, Hardy A 2001 A degeneration-based hypothesis for interpreting ¢brillarchanges in the osteoarthritic cartilage matrix. J Anat 199:683^698
H�ggO, Fritzell P, Hedlund R,Moller H, Ekselius L, Nordwall A 2003 Swedish Lumbar SpineStudy. Pain-drawing does not predict the outcome of fusion surgery for chronic low-backpain: a report from the Swedish Lumbar Spine Study. Eur Spine J 12:2^11
Hansson P, Ekblom A, Lindblom U, Marchettini P 1988 Does acute intraoral pain altercutaneous sensibility? J Neurol Neurosurg Psychiatry 51:1032^1036
Jensen K, Andersen HO, Olesen J, Lindblom U 1986 Pressure-pain threshold in humantemporal region. Evaluation of a new pressure algometer. Pain 25:313^323
Kosek E, Ordeberg G 2000a Abnormalities of somatosensory perception in patients withpainful osteoarthritis normalizes following successful treatment. Eur J Pain 4:229^238
Kosek E, Ordeberg G 2000b Lack of pressure pain modulation by heterotopic noxiousconditioning stimulation in patients with painful osteoarthritis before, but not following,surgical pain relief. Pain 88:69^78
Le¥er AS, Kosek E, Lerndal T, Nordmark B, Hansson P 2002a Somatosensory perception andfunction of di¡use noxious inhibitory controls (DNIC) in patients su¡ering from rheumatoidarthritis. Eur J Pain 6:161^176
Le¥er AS, Hansson P, Kosek E 2002b Somatosensory perception in a remote pain-free area andfunction of di¡use noxious inhibitory controls (DNIC) in patients su¡ering from long-termtrapezius myalgia. Eur J Pain 6:149^159
Lindh C, Liu Z, Lyren�s S, OrdebergG,Nyberg F 1997 Elevated cerebrospinal substance P-likeimmunoreactivity in patients with painful osteoarthritis, but not in patients with rhizopaticpain from a herniated lumbar disc. Scand J Rheumatol 26:468^472
LindhC, LiuZ,WelinM,OrdebergG,Nyberg F 1999Lowcalcitonin gene-related, peptide-likeimmunoreactivity in cerebrospinal £uid from chronic pain patients.Neuropeptides 6:517^521
McAlindon TE, Snow S, Cooper C, Dieppe PA 1992 Radiographic patterns of osteoarthritis ofthe knee joint in the community: the importance of the patellofemoral joint. Ann RheumDis51:844^849
Millis MB, Kim YJ 2002 Rationale of osteotomy and related procedures for hip preservation: areview. Clin Orthop 405:108^121
Pedersen JL 2000 In£ammatory pain in experimental burns in man. Dan Med Bull 47:168^195Russell IJ, Orr MD, Littman B et al 1994 Elevated cerebrospinal £uid levels of substance P inpatients with the ¢bromyalgia syndrome. Arthritis Rheum 37:1593^1601
Schwarzer AC, Aprill CN, Derby R, Fortin J, Kine G, Bogduk N 1994 The relativecontributions of the disc and zygapophyseal joint in chronic low back pain. Spine 19:801^806
Vaeroy H, Helle R, Forre O, Kass E, Terenius L 1988 Elevated CSF levels of substance P andhigh incidence of Raynaud phenomenon in patients with ¢bromyalgia: new features fordiagnosis. Pain 32:21^26
Waddell G, Richardson J 1992 Observation of overt pain behaviour by physicians duringroutine clinical examination of patients with low back pain. J Psychosom Res 36:77^87
Wang CJ 2001 Management of patellofemoral arthrosis in middle-aged patients. Chang GungMed J 24:672^680
114 ORDEBERG

Waters TS, Bentley G 2003 Patellar resurfacing in total knee arthroplasty. A prospective,randomized study. J Bone Joint Surg Am 85A:212^217
Weinstein S 1962 Tactile sensitivity of the phalanges. Percept Mot Skills 14:351^154
DISCUSSION
Lohmander: You have some very impressive data. I have a question about thenormalization of the pain threshold after OA surgery. The data you showed weregroup averages, yet we know that not all patients respond equally well to what wethink is standardized and perfect surgery. Have you looked behind these groupaverage data and tried to determine whether those patients who do not respondas well to surgery as you might expect also do not normalize with regards to theirpain thresholds? Is there any sign of a relationship between the di¡erent ways ofmonitoring the outcome?Ordeberg:We tried to do this. In this group there were 15 patients, two of whom
were excluded from operation because of medical reasons. So the post-operative¢gures are from the 13 patients who were operated on. Fortunately (orunfortunately) they were all well. It would be interesting to address the point youraised.Kidd: You showed that the DNICs were reduced in the painful OA pre-
operatively. Interestingly, this was reversed following joint replacement surgery.What clinical relevance does this reduction in DNIC have for the patient?Ordeberg: I can only speculate. It may be that theDNIC is one of the reasons why
we feel less pain when we are highly occupied with something else. It may be thatpatients with sensory disturbances don’t have that relief from other activities.Grubb: Yesterday we discussed whether the sensitization in OA is central or
peripheral. The data you presented on the changes in skin thresholds are veryinteresting. Are the skin threshold measures simple and easy to apply?Ordeberg: The instruments are described in our papers and are commercially
available. However calibration and instructions to patients are crucial. We thinkall registrations in a study should be done by the same investigator, as was done inour study.Fox: Are these sensory measurements relieved by analgesics? Is the reduced
threshold altered?Ordeberg: I can’t answer that. From what I remember there have been a few
reports with con£icting results. This may be di¡erent in controls and patientswith sensory disturbances.Bradley:We did a small placebo-controlled trial using sertraline (ZoloftTM) and
we found that this did produce a signi¢cant increase in pain thresholds at the tenderpoints in our ¢bromyalgia patients compared to placebo (Alberts et al 1998). This isan unusual ¢nding. When you look at most of the studies of serotonin inhibitors
MECHANISMS IN JOINT PAIN 115

you don’t see much e¡ect on pain sensitivity. One reason for this may be that inmost of the previous studies, the investigators were simply content to look atchanges in the number of tender points. By virtue of the fact that one of theclassi¢cation criteria for ¢bromyalgia is extreme tenderness at 11 or more of 18anatomic sites, it is very unlikely that any pharmacological treatment willsubstantially reduce the number of sites that are found to be extremely tender. Itprobably is more likely that an e¡ective treatment will increase pain thresholdlevels at multiple sites, even if most of those sites are still tender. At least our datasuggest that it is really worthwhile to examine the value of using quanti¢ablemeasurements of pain sensitivity, such as pain threshold or tolerance levels,rather than simply counting the number of tender points, to assess changeproduced by a pharmacological intervention. Since patients with ¢bromyalgiaare sensitive to a wide array of stimuli at multiple body sites, several othermeasures of pain sensitivity may also be used to assess the e¡ects of treatmentinterventions, such as magnitude estimates of the intensity or unpleasantness ofpressure, thermal, or ischaemic stimuli or temporal summation tasks involvingpressure or thermal stimuli (e.g. Edwards & Fillingim 1999, Staud et al 2003a).Felson: You commented on your study and contrasted it with others. Would it
be fair to say that in ¢bromyalgia trials, by and large, the trials that show e⁄cacioustherapy with respect to self-reported pain don’t show any e¡ect on tender points?Bradley: That is absolutely right. It is the number of tender points that doesn’t
really change. It would be very unusual for a single pharmacological interventionto reduce signi¢cantly the number of tender points. But certainly it couldpotentially reduce sensitivity at those points.Fox: It is potentially important from an industry perspective. We are trying to
design more sensitive clinical trials which will pick up e⁄cacy. We are relying onthe VAS score all the time. It may be that if these sorts of changes are consistent,and they are quanti¢able in the way that VAS is, then they may be more sensitive.This could encourage companies to do another trial with their compound ratherthan discard it.Hunter: Do they have solid standardization sessions where they actually go in
and anatomically localize where they are going to take these measurements from?Bradley: We do that by training our research assistants to reliably identify the
tender points and to show a high level of consistency with one another in theirpain threshold measurements on the same individuals (Aaron et al 1996). I don’tknow to what extent other investigators do that. One other variable to consider inthese trials is that they will involve multiple study sites. How can we ensurereliability of our measurements across these sites? In addition, even within asingle site, the reliability of pressure stimuli delivered manually by one researchassistant may vary across multiple trials. There are at least two investigators Iknow of who have tried to solve these problems by building automated
116 DISCUSSION

piston-driven devices that apply consistent levels of pressure stimulation acrosstrials. One group is at the University of Florida with Don Price and RolandStaud (Staud et al 2003b) and the other is at the University of Michigan(Gracely et al 2002). These devices are used to apply pressure to ¢ngertips.There also are devices that deliver thermal stimuli in a reliable manner thatare commercially available. However, even with these devices, you get intothe question of the extent to which they will be used consistently across studycentres. So, I think there is still a need to focus on the inter-site and inter-judgereliability in planning these treatment trials.Creamer:Yesterday I mentioned that we have looked at pain threshold in OA of
the knee, and it didn’t correlate with reported pain severity.Pisetsky: In your studies, were patients allowed to be on medication in the
preoperative state?Ordeberg: From what I remember, not in the days immediately preceding the
operation.Pisetsky: Generally, is it necessary to wash people o¡ their medications before
this kind of assessment?Ordeberg: We had no speci¢c washout. Most patients did not use analgesics as
they reported that the e¡ect had subsided. Those who did use analgesics wereinstructed not to do so on the day of investigation.Pisetsky: Would existing medicines, such as a non-steroidal anti-in£ammatory
drug (NSAID) or a tricyclic, a¡ect these measurements?Ordeberg: It is likely that they would. On the other hand at the follow-up, if they
were onmedication and had some concentration left at the time of the operation, itwould a¡ect the results in the opposite way. After surgery they were not on drugs.Dieppe: I was intrigued by your emphasis on avascular necrosis. This raises an
issue that we didn’t discuss yesterday when we were talking about pain features ofOA, and this is that inmany patients it is episodic.Many people talk about £ares ofOApain. The tendency in the literature is to assume that £ares and episodes of painare related to synovitis, although the evidence for this is almost non-existent. Thesort of thing you have shown raises the possibility that £ares and episodes of painare actually vascular in origin, andminor events of avascular necrosis. The vascularhypothesis of OA and OA pain is something we have only talked about withrespect to raised intraosseus pressure. In the 1950s, when they were developingsome of these ideas, Trueta and others talked about the possibility that minorepisodes of tiny events of avascular necrosis could be very important inpathogenesis and pain. This vascular hypothesis is worthy of further investigation.Ordeberg: The problem is how we should register these small ¢ndings. Can they
be registered by MRI?Dieppe: I don’t know. There is a bit of pathological work in the literature which
is supportive of this idea, but I have no idea how you could do this in vivo.
MECHANISMS IN JOINT PAIN 117

Felson:There is avascular necrosis noted in most pathological specimens in OA,and there is work in the spine by LeenaKauppila and others that shows associationof vascular insu⁄ciency with back pain in spinal disease.Hunter: There have been some interesting pathological studies done on joint
replacements, suggesting that the pathological changes that occur in the bonemarrow of people with end-stage OA are not dissimilar to what you wouldexpect with a person who has ischaemic necrosis otherwise. Potentially there aresome reported vascular changes which you can measure reasonably well usingstandard MRI.Ordeberg: I would like to add that when hip prostheses are ¢xed with cement,
they show immediate pain relief, whereas with many of the constructs with other¢xation to the bone marrow it takes some time for pain relief after surgery.Implants without bone cement may have some initial motion in the interfacebetween prosthesis and bone marrow, which may cause pain from receptors inthe bone.Hunter:Are you suggesting with your substance P ¢ndings within the CSF that
this is a process similar to what Professor Schaible was talking about yesterday interms of a central sensitization? With the blood pressure cu¡, is this really just adistraction that allows it to increase the sensitivity?Ordeberg: I think it is very much a question of sensitization.Fox: It seems that your attention is going towards the spinal cord. Do your
studies imply that substance P would be a good target?Ordeberg: It is a marker. To call it a target would be taking a step further.Fox: There has been at least one published trial with NK1 antagonists.Grubb:With regard to the issue of central or peripheral sensitization, I like your
mapping diagrams. Earlier, we were discussing whether this was truly nociceptivepain or a sensitization process going on in the joint. Can you say from your datawhether all patients showed what looked like extended receptive ¢elds? Do allpatients show such marked changes, or is it often quite restricted to the jointregion itself?Ordeberg: I think it varies quite widely. Most patients have some degree of pain
distribution, but on the other hand those patients selected for these studies all hadpain at rest.Schaible: I would be very pleased to see whether substance P is in fact a marker.
This would be some way of getting at the issue of whether there is a centralsensitization in pain states, even if substance P and substance P receptors are nota very good target for therapy.Ordeberg:On the other hand, we don’t know whether substance P has an action
inCSForwhether its presence represents an out£ow from the dorsal root ganglion.Kidd: It would seem logical that substance P would be a good marker. When
released in the periphery, it is quickly degraded by degradative enzymes so it
118 DISCUSSION

can’t di¡use. But from your work and the other work that Blair Grubb has done itwould seem that the purpose of substance P in the spinal cord is to di¡use. It islogical that it is going to spill over into the CSF. I would think that it is quite agood surrogate marker. It is as good as anything else.Fox: If it is a marker, isn’t it a surprise that the apparent central sensitization,
which is perhaps leading to this referred pain and expanded receptive ¢elds, isrelieved so quickly by surgery?Ordeberg: It was six months afterwards. There was still some elevation of
substance P compared with controls.Fox: But the pain goes very quickly.Grubb: Hans-Georg Schaible, in your carrageenan/kaolin model of OA where
you see expansion of receptive ¢elds, did the receptive ¢elds recede at the same timeas you saw the changes in neuronal ¢ring when you applied the NK1 antagonist inthe spinal cord?Schaible: Yes, they became a bit smaller. If we then take away the compound
there is again an expansion. Gunnar Ordeberg, how stable is the increase of thesubstance P concentration that you see? Is it a big increase comparedwith normals?Ordeberg: It is not as big as has been previously reported in ¢bromyalgia patients,
for example. In these studies I was limited in that I had only 11 patients.Hunter: It looked very much like the cold sensitivity was being a¡ected in the
sameway as the pain thresholds were. Is there an analogous way ofmeasuring this?Ordeberg: These systems are quite di¡erent. The e¡ects on the di¡erent
thresholds might be varying with the di¡erent pain source.Hunter: I understand that pain ¢bres may also act as the temperature
¢bres.Grubb: There are C cold ¢bres and there are di¡erent ¢bres which respond to
warmer temperatures (432 8C). There are also C ¢bres that are mechano^heatsensitive, showing that there is considerable variety in the responsiveness ofa¡erent nerve ¢bres. There have been many attempts to try to divide these upinto subgroups in skin. I was in a lab recently where this has been done and theyhave a list of 14 di¡erent types of skin C ¢bres, grouped according to their di¡erentmechanical and heat thresholds.When considering human receptive ¢elds for deeptissues such as the joint, we are also thinking about convergent inputs onto spinalcord neurons. These neurons are receiving inputs from joint mechanoreceptorsand from skin mechanoreceptors which may also be thermally sensitive. Thismeans that the situation is complex.Bradley: Most investigators who look at pain sensitivity in patients with OA
have primarily focused on mechanical pressure. Our group has looked atdi¡erences in heat thresholds and have not found a substantial di¡erence betweenpatients and controls. It would be interesting to perform a temporal summationparadigm using both mechanical pressure as well as thermal stimulation.
MECHANISMS IN JOINT PAIN 119

Looking for changes or di¡erences in wind-upwould allow us to take a better lookat the question of identifying sensitivity markers for central sensitization.Regardless of what we ¢nd, it will be interesting. Will patients respond to bothpressure and heat/cold, or will they respond primarily to pressure?Kidd: There is an Australian study looking at pain threshold for mechanical
pressure over OA joints and reference sites over the forearms, showing changesin the threshold (Farrell et al 2000). The correlation with pain came from thereference sites. This argues in favour of a central component. Your temporalsummation idea would be another way of testing this.Felson: I’d like to ask about capsaicin. Its e⁄cacy in OAmight relate to some of
what you commented on. Presumably capsaicin has as its mechanism of action thisdistraction of substance P, similar maybe to the tourniquet that you wereadministering. Is this a reasonable parallel?Henry: I don’t think so. What we saw earlier was a heterosegmental inhibitory
mechanism, whether it loops through the brainstem or lies within the spinal cord.But capsaicin activates the vanilloid receptors,which after awhile desensitize. Thusafter a while you don’t get activation of the C ¢bres that are releasing substance Pand CGRP. This is a totally di¡erent mechanism. It is a desensitization, because atthe beginning the e¡ects of capsaicin are not anti-nociceptive but pro-nociceptive.If you eat ameal with a lot of chilli pepper in it, there is a fairly rapid desensitizationso 10 minutes into the meal it won’t have the same burning sensation as thatexperienced initially.Malcangio:You said that capsaicin disrupts C ¢bres. It doesn’t do this unless it is
given to neonates.Fox: It is very well established that capsaicin produces a functional
desensitization of C ¢bres, which underlies its analgesic activity.Rediske: Along this theme of central versus peripheral sensitization, is there
much known about the e⁄cacy of centrally acting analgesics in OA? Does thisgive us some insights into what is happening mechanistically?Schaible:Has anyone tried dipyrone in OA? This is a compound that hasn’t been
used much because it causes some problems.When we tested it for other reasons itreduced spinal activity a lot, but it doesn’t have much e¡ect on a¡erent ¢bres. Hasanyone used this? It might be worth trying.Bradley: I have a comment about the paper you publishedwithDrKosek. This is
one of the papers that drove my interest in pain in people with OA.When we lookat DNIC, one factor we need to take into account is whether there are sexdi¡erences in the response. The reason I mention this is that in the ¢bromyalgialiterature there was a recent paper by Roland Staud and Don Price where theyfound, just as you did, that there was an abnormal di¡use noxious inhibitorycontrol (DNIC) response in ¢bromyalgia patients (Staud et al 2003b). Theirsample was primarily composed of women. When they performed the same
120 DISCUSSION

experimentwith healthy female controls, there was no di¡erence inDNIC betweenthe patients and controls. In contrast, healthy men showed a normal DNICresponse. So the question that is now raised is to what extent is abnormal DNICresponse related to female sex. I don’t remember the ratio ofmen towomen in yoursample.Ordeberg: There were 9 men and 6 women.Bradley: If you looked at themen andwomen separately youmight ¢nd a greater
abnormalDNIC response in themen, and perhaps a greater response to the surgicalintervention.
References
Aaron LA, Bradley LA, Alarco¤ n GS et al 1996 Psychiatric diagnoses are related to health care-seeking behavior rather than to illness. Arthritis Rheum 39:436^445
Alberts KR, Bradley LA, Alarco¤ n GS et al 1998 Sertraline hydrochloride (Zoloft) alters painthreshold, sensory discrimination ability, and functional brain activity in patients with¢bromyalgia (FM): a randomized, controlled trial (RCT). Arthritis Rheum 41(Suppl):S259
Edwards RR, Fillingim RB 1999 Ethnic di¡erences in thermal pain responses. PsychosomMed61:346^354
Farrell M, Gibson S, McMeeken J, Helme R 2000 Pain and hyperalgesia in osteoarthritis of thehands. J Rheumatol 27:441^447
Gracely RH, Petzke F, Wolf JM, Clauw DJ 2002 Functional magnetic resonance imagingevidence of augmented pain processing in ¢bromyalgia. Arthritis Rheum 46:1333^1343
Staud R, Cannon RC, Mauderli AP, Robinson ME, Price DD, Vierck CJ Jr 2003a Temporalsummation of pain from mechanical stimulation of muscle tissue in normal controls andsubjects with ¢bromyalgia syndrome. Pain 102:87^95
Staud R, Robinson ME, Vierck CJ Jr, Price DD 2003b Di¡use noxious inhibitory controls(DNIC) attenuate temporal summation of second pain in normal males but not in normalfemales or ¢bromyalgia patients. Pain 101:167^174
MECHANISMS IN JOINT PAIN 121

The role of in£ammatorymediators on
nociception and pain in arthritis
Bruce L. Kidd, Andrew Photiou and Julia J. Inglis
Bone & Joint Unit, Bart’s & The London, Queen Mary School of Medicine & Dentistry,Charterhouse Square, London EC1M 6BQ, UK
Abstract. Pain is the most common complaint of individuals with osteoarthritis but thecause of symptoms in this disorder remains unclear. Quantitative sensory testing revealsthat in patients with chronic joint disease there is di¡use and persistent alteration ofnociceptive (pain) pathways, irrespective of the level of activity of the underlyingdisease. In£ammatory mediators contribute to this plasticity either by directly activatinghigh threshold receptors or more commonly by sensitizing nociceptive neurons tosubsequent everyday stimuli. This involves early post-translational modi¢cation ofreceptors/ion channels and later, longer-lasting transcription-dependent mechanismsinvolving changes to the chemical phenotype of the neuron. Included amongst thesechanges are the increased production and release of various pro- and anti-in£ammatoryneuropeptides which have diverse actions on both circulating and resident cellpopulations. These neurally derived mediators act synergistically with cytokines andgrowth factors to contribute to ongoing tissue injury. It is becoming apparent that theinteraction between a damaged joint and the sensory nervous system is far fromstraightforward and that activity arising from such interactions may produce not onlypain but may also in£uence the subsequent course of the underlying disease.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 122^138
Pain is themost common complaint of individuals with osteoarthritis (OA) but thecause of symptoms in this disorder remains unclear. The broad aim of this review isto examine the complex relationship between tissue injury and the sensory nervoussystem.More speci¢cally, the emerging concept of neural plasticity, its importanceto symptoms and the in£uence of in£ammatorymediatorswill be described. This isfollowed by a discussion concerning the likely contribution of sensory nerves tothe outcome of chronic joint disease.
Neural plasticity and pain
At the outset, it is useful to identify several processes as being associated with painincluding nociception, pain perception (interoception) and a number of secondaryevents including communication of distress and disability. For the purposes of thisreview, nociception is de¢ned as the detection of noxious stimuli by nerves and the
122
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

subsequent transmission of encoded information to the brain. Interoception, onthe other hand, is a perceptual and ultimately private experience that arises inresponse to nociceptive activity but is separate from it.In contrast to interoception, verbal and non-verbal communications can be
observed and therefore measured, as can disability, although these all appear tobe as strongly in£uenced by a range of cultural, social, demographic andenvironmental factors as by internal factors. It follows that both the context inwhich symptoms occur as well as the underlying nociceptive process need to betaken into account when assessing an individual’s report of pain.A characteristic feature of all disorders involving persistent or chronic pain is
that there is an exaggerated response to everyday stimuli. In the most obviousexample, simple mechanical stimuli such as walking produce varying levels ofdiscomfort in patients with any persistent arthropathy. Whereas in past centuriesit was believed that an immutable nervous system delivered a faithfulrepresentation of the damaged tissue to a single ‘pain’ centre in the brain, it is nowappreciated that the entire nociceptive system is capable of considerable functionalchange, or plasticity. The extent of this change varies according to di¡erentconditions and appears to be a key determinant of symptoms arising from tissuedisease.Minor injuries produce short-lived excitation of specialized high threshold
nociceptors with brief, spatially localized pain. More severe tissue damage resultsin excitationof relevant nociceptors aswell as longer lasting changes in the responseto subsequent stimuli (peripheral sensitization) (Raja et al 1999). Heightened skinsensitivity following sunburn provides a convenient example. In turn, sustained orrepetitive activity within peripheral ¢bres leads to substantial changes to thefunction and activity of central nociceptive pathways. At a spinal level, thisinvolves exaggerated responses to normal stimuli together with expansion ofreceptive ¢eld size producing tenderness and referred pain in areas away from thesite of injury (spinal sensitization) (Coderre et al 1993). Functional imaging studiesof the brain following noxious stimuli using functional magnetic resonanceimaging (fMRI) and positron emission tomography (PET) show a complexpattern with discrete areas of activity throughout both cerebral hemispheres(Gracely et al 2002) and it is likely that sensitization also occurs at a supraspinallevel (cortical senstization). The consequences of cortical sensitization remainunclear but may produce states of hypervigilance and other more generalphenomena observed in patients with chronic pain.
Assessing nociception
Given the importance of plasticity in the genesis of persistent pain it is perhapssurprising that relatively few studies have directly assessed nociceptive
PAIN AND INFLAMMATION 123

mechanisms in chronic human diseases. It is well recognized that studies ofreported pain may not necessarily be reliable guides of underlying nociceptivemechanisms. Quantitative sensory testing (QST) and related procedures attemptto overcome these shortcomings by using psychophysical methods and multiplestimulus modalities to provide a systematic analysis of nociceptive function. Thetechniques have proved useful for making individual clinical decisions as well asfor basic investigation of underlying mechanisms (Gracely et al 2003).The clinical observation that pain may be referred away from OA joints and
reports of increased tenderness over apparently normal tissues have led tospeculation that changes in central modulation of nociceptive input mightcontribute to symptoms in OA. Recent quantitative psychophysical studiesevaluating pain mechanisms in OA lend support to this idea. Cutaneous and deephyperalgesia to thermal and mechanical stimuli in subjects with OA have beentested over symptomatic carpometacarpal joints and control sites in the forearm(Farrell et al 2000). Signi¢cantly, variance in movement-related pain ratings waspredicted by forearm pain thresholds. Similar results were obtained in a study inwhich muscle hyperalgesia was assessed by intramuscular infusion of 6%hypertonic saline (Bajaj et al 2001). OA subjects had increased pain intensity withsigni¢cantly larger referred and radiating pain areas than matched controls. It ishighly unlikely that local changes to nociceptive activity account for either set ofresults and points to the presence of enhanced central mechanisms.In patients with other musculoskeletal disorders, capsaicin-based techniques
have been used to quantify cutaneous axon-re£ex skin £ares as an indirect markerof peripheral sensory activity. Capsaicin is the active ingredient in hot chilli peppersand has proved a useful tool for nociceptive research as it selectively excitesunmyelinated sensory ¢bres concerned with nociceptive transmission. Indegenerative arthritis of the spine, capsaicin-induced skin £ares were found to bereduced (LeVasseur et al 1990), whereas selective increases over in£amed jointshave been reported in rheumatoid arthritis (RA) (Jolli¡e et al 1995). At otherreference sites, away from in£amed joints, skin £ares were similar to thoseobserved in normal controls, arguing against a generalized up-regulation ofperipheral sensory ¢bre activity in this condition.Capsaicin-based assessments have also been used to explore abnormal central
nociceptive activity in patients with chronic joint disease. In addition toproducing skin £ares, intradermal injections of capsaicin also producemeasurable areas of hyperalgesia and allodynia around the injection site as a resultof enhanced sensitization of spinal neurons (Torebjork et al 1992). Pinprickhyperalgesia induced by capsaicin has been shown to be substantially greater overthe forearms ofRApatients compared to normal controls (Morris et al 1997; Fig. 1)Peripheral sensory activity asmeasured by von Frey threshold over the forearms ofrheumatoid patients was normal. In the rheumatoid patients the maximal area of
124 KIDD ET AL

PAIN AND INFLAMMATION 125
A
B
FIG. 1. (A) Comparison of the area of capsaicin-induced pinprick hyperalgesia in normal andrheumatoid subjects (solid line indicates mean value) demonstrating enhanced centralnociceptive (pain) processing in arthritic disease. (B) Comparison of sensory thresholds forpinprick stimuli using von Frey hairs of increasing force in normal (circles) and rheumatoid(squares) subjects (n¼35 each group) illustrating similar peripheral nociceptive processing atcontrol sites over the forearm. (FromMorris et al 1997 with permission.)

hyperalgesia was found to correlate with a composite score of upper limbtenderness, but not with overall pain score or a systemic marker of in£ammation.Taken together, these studies show that in patients with chronic joint disease
there is a di¡use and persistent alteration of central nociceptive processing,irrespective of the level of activity of the underlying in£ammatory disease.Paradoxically, the studies also reveal a selective increase of peripheral activityover diseased joints but not at control sites elsewhere. These results raise theintriguing possibility that prior stimuli can produce long term and di¡usechanges within central nociceptive pathways. They also lead to the question as towhat the consequences of peripheral neurogenic (axon re£ex) activity on the long-term outcome of joint disease are likely to be.
E¡ects of in£ammatory mediators on nociception
During in£ammation a range of pro- and anti-in£ammatory mediators are releasedwith the balance between these mediators playing a critical role in the subsequentactivity of nociceptive ¢bres. Characteristically, the terminals of nociceptive ¢bresexpress multiple receptors for in£ammatory mediators that become active acrossrelatively narrow ranges of stimulus intensity. The cellular mechanisms by whichthese changes occur involve early post-translational changes to receptors/ionchannels and later, longer-lasting transcription-dependent mechanisms involvingchanges to the chemical phenotype of the cell (Kidd & Urban 2001; Fig. 2).
Acute in£ammation
Whilst somemediators such as bradykinin contribute to pain by directly activatingnociceptors others are generally considered to be sensitizing agents. This may ariseas a result of changes to the sensitivity of receptor molecules or via modulation ofvoltage-gated ion channels. Prostaglandins, for example, have been shown to up-regulate receptors on primary nociceptive ¢bres using an adenylyl cyclase^proteinkinase A pathway (Segond von Banchet et al 2003). A second example is providedin the vallinoid VR1 (TRPV1) receptor which is involved in the transduction ofnoxious heat stimuli and is also the receptor for capsaicin, discussed earlier.Mutation analysis of the TRPV1 receptor has revealed an inhibitory binding sitefor phosphatidylinositol 4,5-bisphosphate (PIP2) which can be hydrolyzedfollowing activation of phospholipase C (PLC) by mediators includingprostaglandins, bradykinin and NGF. This releases TRPV1 from basal inhibitionand lowers the threshold for activation (Prescott & Julius 2003).
126 KIDD ET AL

Nerve growth factor
Neurotrophin growth factors, including NGF, make signi¢cant and long lastingcontributions to the changes of neuron sensitivity observed during in£ammation.NGF mRNA and/or protein has been identi¢ed in various cell types and a largenumber of in£ammatory mediators act to increase production, particularlyinterleukin (IL)1b and tumour necrosis factor (TNF)a (Woolf et al 1997). Duringthe acute stages of an in£ammatory response, nerve growth factor (NGF), actingvia neuronal TrkA receptors, produces tyrosine phosphorylation of intracellulartargets including ion channels. Over the longer term, NGF exerts a more globalin£uence by regulating gene expression leading to prolonged changes in neuronfunction (Levine & Reichling 1999).In studies using primary cultures of adult dorsal root ganglia neurons, treatment
with NGF has been shown to increase both TRPV1 mRNA expression andcapsaicin-invoked release of CGRP in a dose-dependent fashion (Winston et al2001). Other studies have shown NGF regulation of mRNA for the
PAIN AND INFLAMMATION 127
FIG. 2. In£uences on primary nociceptive neurons leading to ‘peripheral sensitization’. Undernormal circumstances, high intensity stimuli are encoded by specialized membrane-boundreceptors. Conduction of message to central terminals is mediated by ion channels andexcitatory amino acids respectively. During acute in£ammation (A) prostaglandins (PG) andother mediators change the sensitivity of receptors and reduce activation threshold for ion-channels. Longer-term changes include transcriptional events (B) mediated by cytokines andgrowth factors resulting in enhanced production of receptors, ion channels and transmitters/modulators. (From Kidd & Urban 2001.)

neuropeptides substance P and CGRP as well as the tetrodotoxin resistant sodiumchannel SNS/PN3. More recently, NGF has also been shown to exert long termin£uences on nociceptor activity in a transcription-independent fashion via the p38mitogen-activated protein kinase (MAPK) (Ji et al 2002).
Cytokines
TNFa, IL1b and IL6 have been shown to induce both heat and mechanicalhypersensitivity when injected peripherally (Opree & Kress 2000, Sa¢eh-Garabedian et al 1995). Antisera to these cytokines reduce hyperalgesia inin£ammatory models (Woolf et al 1997) and the use of novel anti-TNF therapiesin rheumatoid arthritis is accompanied by a signi¢cant reduction in pain scores(Andreakos et al 2002).Exact mechanisms of underlying cytokine-induced hyperalgesia remain
controversial. As cytokines exert e¡ects on numerous cell types, theirhyperalgesic actions have largely been attributed to indirect mechanismsinvolving production of prostaglandins and other mediators. Consistent withthis, IL1b-, TNFa- and IL6-induced hyperalgesia has been shown to besigni¢cantly attenuated by pre-treatment with indomethacin (Cunha et al 1992,Samad et al 2001). Furthermore, in£ammation of peripheral tissues inducesCOX-2 expression in the CNS and endothelial cells of the blood^brain barrierthrough an IL1-dependent pathway (Samad et al 2001, Ek et al 2001).Evidence is also accumulating for more direct cytokine e¡ects on nociceptive
neurons. Electrophysiological studies of the hyperalgesic actions ofsubcutaneous TNFa have shown ectopic activity in nociceptive neurons 2minafter injection, whilst signi¢cant mechanical hyperalgesia is present within 15min (Junger & Sorkin 2000). Likewise, heat-evoked CGRP release fromnociceptive neurons was increased 15min following incubation with IL1b,TNFa and IL6 incubation (Opree & Kress 2000). TNFa increases the sensitivityof cultured sensory neurons to capsaicin, whilst IL1b increases substance Psynthesis in cultured dorsal root ganglion (DRG) (Nicol et al 1997). In our own(unpublished) studies we have shown low-level expression of TNF receptors onprimary nociceptive neurons and a subsequent bilateral increased expressionfollowing tissue in£ammation. This increased expression of receptors for TNFduring in£ammation may result in hypersensitivity to the cytokine and mayexplain the rapid analgesic properties of anti-TNF therapy.
Neurogenic in£ammation
The nervous system in£uences the immune responses systemically via humoralsubstances such as cortisol following activation of the hypothalamic^pituitary^
128 KIDD ET AL

adrenal (HPA) axis. A second and potentially more selective mechanism by whichneuro^immune interactions may occur is via local sensory and autonomic nerves.Such interactions, which arewell documented in disorders a¡ecting the respiratoryand gastrointestinal systems (Joos&Pauwels 2000, Chavolla-Calderon et al 2003),may also in£uence the clinical features ofmany rheumatic disorders. For example, aneurogenic role in the production of symmetrical joint disease has been suggestedfollowing the observation that in£ammation in one joint can result in aneurogenically mediated response in the contralateral joint (Kidd et al 1995).Neurogenic in£ammation is mediated, at least in part, by the neuropeptides,
substance P (SP) and neurokinin A (NKA) (Maggi 1997). These are members ofthe tachykinin family of peptides which are encoded by the pre-pro-tachykinin A(PPT-A) gene and are produced by small diameter sensory nerves as well as manytypes of immune cell (Ho et al 1997, Joos & Pauwels 2000). They act via theneurokinin family of receptors that includes neurokinin-1 (NK-1) andneurokinin-2 (NK-2) with SP having the greatest a⁄nity for the NK-1 receptorwhereas NKA acts primarily via the NK-2 receptor. Both receptors are presenton leukocytes and many other cell populations (Maggi 1997, Joos & Pauwels2000). Given the presence of PPT mRNA and SP-like immunoreactivity in manyimmune cells which also express neurokinin receptors the possibility exists thatin£ammatory cells use tachykinins as a paracrine or autocrine signallingmechanism to propagate in£ammation (Chavolla-Calderon et al 2003).In vitro studies indicate that SP stimulates the release of IL1, IL6 and TNF from
human monocytes (Lotz et al 1988). SP also mediates mast cell degranulation andproduction of PGE2 and collagenase from synoviocytes (Maggi 1997). It has alsobeen shown to be chemotactic for a number of cell types including neutrophils,eosinophils, monocytes and more recently, lymphocytes (Hood et al 2000). Theexpression of vascular endothelial cell adhesion molecules is also up-regulated(Quinlan et al 1999). Importantly, disruption of the NK-1 receptor protectsagainst the injury induced by antigen-antibody complex formation in theairways, revealing that neurokinins serve an important role in this type of injury(Chavolla-Calderon et al 2003).The true arthrogenic potential of substance P and other neuropeptides in
naturally occurring human arthropathy is unknown, but we and others haveassessed their contribution to the pathogenesis of experimental arthritis in anumber of models (Cruwys et al 1995, Kidd et al 2003). For example, mice with adisruption of the NK-1 receptor had signi¢cantly less footpad swelling andmechanical hyperalgesia than wild-type animals in a complete Freund’s adjuvant(CFA) arthritismodel (Kidd et al 2003; Fig. 3).Histological and radiological scoreswere markedly reduced in the knock-out group. The di¡erential e¡ect wasparticularly marked with respect to scores for synovial hyperplasia andin£ammatory cell in¢ltrate. We are currently exploring the in£uence of SP on
PAIN AND INFLAMMATION 129

130 KIDD ET AL
FIG. 3. Footpad diameter (a) and mechanical hyperalgesia (b) in wild-type (circles) and NK-1knockout (squares) mice following adjuvant-induced in£ammation (10mg/ml) demonstratingpro-in£ammatory e¡ects of substance P during persistent disease. *Indicates signi¢cantdi¡erences (P50.05) (from Kidd et al 2003 with permission).

leukocyte recruitment by endothelium using intra-vital microscopy. In ongoing(unpublished) studies we have shown that in NK-1 knockout mice, neutrophilmigration across the endothelial cell barrier is substantially reduced. Furtherstudies have suggested that SP acts to promote leukocyte migration by amechanism involving the chemokine, MCP-1.
Summary
A characteristic feature of symptomatic OA is an exaggerated response to everydaystimuli occurring as a result of functional changes within the nociceptive system.Quantitative sensory testing reveals that in patients with chronic joint diseasesthere is a di¡use and persistent alteration of central nociceptive processing,irrespective of the level of activity of the underlying disease. In£ammatorymediators contribute to pain either by directly activating high thresholdreceptors or more commonly by sensitizing these receptors to mechanical andother stimuli. Sensory sensitization involves early post-translational changes toreceptors/ion channels and later, longer-lasting transcription-dependentmechanisms involving changes to the chemical phenotype of the sensory neuron.This includes increased production and release of various pro- and anti-in£ammatory neuropeptides which have diverse actions on immune and othercells in the periphery. It is therefore apparent that the interaction between adamaged joint and the sensory nervous system is far from straightforward andthat activity arising from such interactions may produce not only pain but mayalso in£uence the subsequent course of the underlying disease.
References
Andreakos ET, Foxwell BM, Brennan FM, Maini RN, Feldmann M 2002 Cytokines and anti-cytokine biologicals in autoimmunity: present and future. Cytokine Growth Factor Rev45:299^313
Bajaj P, Bajaj P, Graven-Nielsen T, Arendt-Nielsen L 2001 Osteoarthritis and its associationwith muscle hyperalgesia: an experimental controlled study. Pain 93:107^114
Chavolla-Calderon M, Bayer MK, Fontan JJP 2003 Bone marrow transplantation reveals anessential synergy between neuronal and hemopoietic neurokinin production in pulmonaryin£ammation. J Clin Invest 111:973^980
Coderre T, Katz J, Vaccarino A, Melzack R 1993 Contribution of central neuroplasticity topathological pain: review of clinical and experimental evidence. Pain 52:259^285
Cruwys SC, Garrett NE, Kidd BL 1995 Sensory denervation with capsaicin attenuatesin£ammation and nociception in arthritic rats. Neurosci Lett 193:205^207
Cunha FQ, Poole S, Lorenzetti BB, Ferreira SH 1992 The pivotal role of tumour necrosis factoralpha in the development of in£ammatory hyperalgesia. Br J Pharmacol 107:660^664
Ek M, Engblom D, Saha S, Blomqvist A, Jakobsson PJ, Ericsson-Dahlstrand A 2001In£ammatory response: pathway across the blood-brain barrier. Nature 410:430^431
Farrell M, Gibson S, McMeeken J, Helme R 2000 Pain and hyperalgesia in osteoarthritis of thehands. J Rheumatol 27:441^447
PAIN AND INFLAMMATION 131

Gracely RH, Petzke F, Wolf JM, Clauw DJ 2002 Functional magnetic imaging evidence ofaugmented pain processing in ¢bromyalgia. Arthritis Rheum 46:1333^1343
Gracely RH,GrantMA,Giesecke T 2003 Evoked pain measures in ¢bromyalgia. Best Pract ResClin Rheumatol 17:593^609
Hood VC, Cruwys SC, Urban L, Kidd BL 2000 Di¡erential role of neurokinin receptors inhuman lymphocyte and monocyte chemotaxis. Regul Pept 96:17^21
Ji R-R, Samad TA, Jin S-X, Schmoll R, Woolf CJ 2002 p38 MAPK activation by NGF inprimary sensory neurons after in£ammation increases TRPV1 levels and maintains heathyperalgesia. Neuron 36:57^68
Jolli¡e VA, Anand P, Kidd BL 1995 Assessment of cutaneous sensory and autonomic axonre£exes in rheumatoid arthritis. Ann Rheum Dis 54:251^255
Joos GF, Pauwels RA 2000 Pro-in£ammatory e¡ects of substance P: new perspectives for thetreatment of airways disease? Trends Pharmacol Sci 21:131^133
Junger H, Sorkin LS 2000 Nociceptive and in£ammatory e¡ects of subcutaneous TNFalpha.Pain 85:145^151
Kidd BL, Urban LA 2001 Mechanisms of in£ammatory pain. Br J Anaesth 87:3^11Kidd BL, Cruwys SC, Garrett NE et al 1995 Neurogenic in£uences on contralateral responsesduring rat monoarthritis. Brain Res 688:72^76
Kidd BL, Inglis JJ, Vetsika E et al 2003 Inhibition of in£ammation and hyperalgesia in NK-1receptor knock-out mice. Neuroreport 14:2189^2192
LeVasseur SA, Gibson SJ, Helme RD 1990 The measurement of capsaicin-sensitive sensorynerve ¢bre function in elderly patients with pain. Pain 41:19^25
Levine JD, Reichling DB 1999 Peripheral mechanisms of in£ammatory pain. In: Wall PD,Melzac R (eds) Textbook of pain, 4th edn. Churchill Livingstone, Edinburgh, p 59^84
Lotz M, Vaughan JH, Crason DA 1988 E¡ect of neuropeptides on production of in£ammatorycytokines by human monocytes. Science 241:1218^1221
Maggi CA 1997 The e¡ects of tachykinins on in£ammatory and immune cells. Regul Pept 70:75^90
HoWZ, Lai JP, ZhuXH,UvaydovaM,Douglas SD 1997Humanmonocytes andmacrophagesexpress substance P and neurokinin^1 receptor. J Immunol 159:5654^5660
Morris VH, Cruwys SC, Kidd BL 1997 Characterisation of capsaicin-induced mechanicalhyperalgesia as a marker for altered nociceptive processing in patients with rheumatoidarthritis. Pain 71:179^186
Nicol GD, Lopshire JC, Pa¡ord CM 1997 Tumor necrosis factor enhances the capsaicinsensitivity of rat sensory neurons. J Neurosci 17:975^982
Opree A, Kress M 2000 Involvement of the proin£ammatory cytokines tumor necrosis factor-alpha, IL-1b and IL-6 but not IL-8 in the development of heat hyperalgesia: e¡ects on heat-evoked calcitonin gene-related peptide release from rat skin. J Neurosci 20:6289^6293
Prescott ED, Julius DA 2003 Modular PIP2 binding site as a determinant of capsaicin receptorsensitivity. Science 300:1284^1288
Quinlan KL, Song IS, Naik SM et al 1999 VCAM-1 expression on human dermal microvascularendothelial cells is directly and speci¢cally up-regulated by substance P. Am J Immunol162:1656^1661
Raja S,Meyer R, KingkampMJNC 1999 Peripheral neural mechanisms of nociception. In:WallPD, Melzac R (eds) Textbook of pain, 4th edn. Churchill Livingstone, Edinburgh, p 11^58
Sa¢eh-Garabedian B, Poole S, Allchorne A, Winter J, Woolf CJ 1995 Contribution ofinterleukin-1 beta to the in£ammation-induced increase in nerve growth factor levels andin£ammatory hyperalgesia. Br J Pharmacol 115:1265^1275
Samad TA,Moore KA, Sapirstein A et al 2001 Interleukin-1beta-mediated induction of COX-2in the CNS contributes to in£ammatory pain hypersensitivity. Nature 410:471^475
132 KIDD ET AL

Segond vonBanchetG, ScholzeA, SchaibleH-G2003 ProstaglandinE2 increases the expressionof the neurokinin1 receptor in adult sensory neurones in culture: a novel role ofprostaglandins. Br J Pharmacol 139:672^680
Torebjork H, Lundberg LER, LaMotte RH 1992 Central changes in processing ofmechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. J Physiol448:765^780
Winston J, Toma H, Shenoy M, Pasricha P 2001 Nerve growth factor regulates VR-1 mRNAlevels in cultures of adult dorsal root ganglia neurons. Pain 89:181^186
Woolf CJ, Allchorne A, Sa¢eh-Garabedian B, Poole S 1997 Cytokines, nerve growth factor andin£ammatory hyperalgesia: the contribution of tumor necrosis factor alpha. Br J Pharmacol121:417^424
DISCUSSION
Blake:What is the de¢nition of interoception?Kidd:What I have gone to some lengths to di¡erentiate is the internal unpleasant
experience that we all feel, and the external expression of that distress.Interoception is a term used to de¢ne the internal and essentially privateexperience we have. It is a useful term to di¡erentiate what we experience as aresult of nociceptive activity from what we can measure. The word ‘pain’, like‘in£ammation’, covers a multitude of sins. I think we need to be more speci¢cwith our terminology if we are ever going to progress.Fernihough: TNF expression at day 7 after CFA treatment is quite striking. Do
you think that this kind of cytokine up-regulation in this phenotypic switch innerves is then leading to an up-regulation of all the usual candidates, whichmechanistically makes in£ammatory pain no di¡erent from neuropathic pain orany other pain? Or do you think that the phenotypic switch is directly linked toion channel changes?Kidd: When we get down to a molecular level the de¢nitions we use for
neuropathic pain begin to break down. Nerve injury is followed by the ectopicexpression of receptors. I have shown you that in£ammation is also followed bythe ectopic expression of receptors. So you could perhaps argue that one is the sameas the other. How you resolve that dilemma is that in neuropathic pain you getectopic expression of di¡erent sorts of receptors. Maybe it is the cluster ofreceptors that are expressed that matters, and we now need to de¢ne what theseclusters are.Pisetsky: Your results on TNF explain a lot of the clinical bene¢t of anti-TNF.
These agents result in an extraordinarily rapid improvement in symptoms, and canwork far faster than you would expect clinically. Do you have any information onother cytokines, such as IL1?Kidd: We haven’t looked at IL1 receptor expression speci¢cally, but we would
presume that it would increase. It seemed to me that there was a big problem withTNF. There just wasn’t any TNF receptor expression on sensory nerves in normal
PAIN AND INFLAMMATION 133

situations. There is some IL1 receptor expression, and it too may increase within£ammation.Pisetsky: Just as there are pro-in£ammatory cytokines, there are anti-
in£ammatory ones. Do you think cytokines like IL10 will have another e¡ect onneurons, just as they do on other cells such as lymphocytes?Kidd: As a rheumatologist, the more I look at the nervous system the more it
seems to resemble the immune system. It is completely under the control of bothpositive and negative in£uences. There may well be receptors for anti-in£ammatory cytokines. Nancy Watkins has shown IL10 receptor expression andsome impressive analgesic e¡ects using IL10.Mackenzie: By using anti-TNF antibodies have you been able to show an anti-
nociceptive e¡ect in your CFA model?Kidd: This is work we are doing at the moment. We have shown that anti-TNF
can knock this e¡ect down. We are in the process of quantifying this.Schaible:Wealso have some data showingTNF receptors on root ganglia. I have
a question about the TRPV1 receptor up-regulation: is this the reason why you seean increased capsaicin response in the skin?Kidd: The argument goes that intradermal capsaicin acting through TRPV1
gives an intense a¡erent barrage into the nervous system which then inducessecondary sensitization and hence the areas that we have shown. If there is pre-existing up-regulation of TRPV1 then those areas will be increased, and so thealternative explanation of these data is that they are not showing centralsensitization at all, but are just re£ecting some change in the periphery. Theanswer to your question is my original observation that i.v. capsaicin did notincrease £are areas over the control sites. I can say with con¢dence therefore thatTRPV1 was not up-regulated at the control site. This is pretty strong evidence tosupport a central e¡ect rather than a peripheral one.Henry: You would need a change in TRPV1 receptors to see this. The TRPV1
receptor is simply a Ca2+ channel, and an increase in intracellular Ca2+will cause therelease of whatever is releasable from the terminals, both central and peripheral. Ifthere is an up-regulation of substance P or CGRP that can produce those changes,then simply activating the TRPV1 receptor is going to cause the release of moresubstance P or CGRP both centrally and peripherally.Fox: I think I’mmissing something. The capsaicin response is greater, implying
that you think this seems to be an ongoing sensory process. If this is the case, whyweren’t they hyperalgesic to start with?Kidd: The £are area was greater in the in£amed joints. We didn’t test over the
in£amed joints because we reasoned that the data would not give us meaningfulresults, because the interpretation would be di⁄cult for the reasons we have justbeen discussing. We had to go to an area where we believed that peripheralnociceptive function was not modi¢ed. Our hypothesis was that there was
134 DISCUSSION

pre-existing central sensitiztion in RA. To test this, we had to have an unmodi¢edsite in the periphery. This is why we looked at forearms, rather than over the jointsthemselves.Fox:TRPV1 expression has certainly been shown to be increased in human skin.Shen: I am interested in substance P. Where does the substance P come from at
the local level?Kidd: I was slightly disingenuous to the extent that I just talked about substance
P being produced within DRG cells. It is quite clear that substance P is alsoproduced both in resident macrophages and circulating leukocytes. There is abody of evidence now coming out suggesting that this peripheral release ofsubstance P is important. The peripheral cells that have substance P, or have beenshown to have the precursor to substance P, also have NK-1 receptors. There isnow evidence that these peripheral cells act in an autocrine or paracrine fashion toamplify the in£ammatory cascade. Most of this work has been done in the lung.The nerves release substance P, and acting through an NK-1 mechanism, furtherenhance substance P release from resident macrophages, which then ampli¢es theresponse. This is further evidence to indicate that the nervous system is animportant player in persistence of an in£ammatory response.Grubb:I’mvery interested in the central sensitization argument and the use of the
capsaicin model either in a na|« ve ¢eld, away from the RA joint, or over the joint.Let us think about the model that Hans-Georg Schaible proposed yesterday, withthese skin inputs which are normally very small and insu⁄cient to excite theneuron. When the neuron becomes sensitized by a constant input, minor inputsfrom the skin may be su⁄cient to depolarize and activate these neurons. If youbelieve that this is required and if you choose a site well away from the RA joint,is this really central sensitization? Why didn’t you do it over the joint?Kidd: We had already shown that there was up-regulation of peripheral
nociceptive neurons over diseased joints. We know that the capsaicin responsesare critically dependent on the state of the peripheral activity. If we were workingin an area where there was already pre-existing activity and then showed enhancedhyperalgesia we couldn’t make a claim that this was telling us about centralsensitization.Grubb: But if these neurons in the na|« ve areas don’t have inputs to the same
neurons that receive inputs in the RA areas, there must be something very di¡use.Kidd: Yes, we feel that there is likely to be a di¡use e¡ect. We come back to
substance P and these other neuromodulators in the CNS which di¡use widely.One has to think that there is a di¡use up-regulation of spinal activity: this is theonly way to explain the data.Dieppe: As I see it there are some people with OA where the peripheral or
noxious stimulus and the peripheral sensitization are probably critical, and thereare others where the interoception and the context are critical. Myworry is that we
PAIN AND INFLAMMATION 135

don’t know how to di¡erentiate them. Can you give us some clues: if thishypothesis is in any way correct, how can we di¡erentiate what are potentiallytwo quite di¡erent pain scenarios?Kidd: Disease in a joint does result in nociceptive activity. I’ve argued that we
can nowmeasure that using quantitative sensory testing. We now need to go backto a group of people who we have de¢ned as having OA pain and assess thenociceptive mechanisms. I would argue that we would identify a group who hadnociceptive activity and symptoms, and a group in whom there is less nociceptiveactivity. We would speculate that this group would have more of a psychosocialcontextual input. This would be the way to sort it out. We have to look atnociception and we have to look at context. Trying to do one on its own won’tgive the picture. The answer is to go back and do more quantitative sensory tests.Blake: I think Paul Dieppe is fundamentally wrong. This comes back to the
comment made before that the visual analogue scale is an unreliable measure ofpain. Intrinsically, it has to be because we are using a linear system to measure anon-linear event. A study that was recently sponsored byGlaxo involved someonewith a burn injury. Using a MRI machine they showed a correlation between thevisual analogue score and accelerated uptake around the limbic system, which was0.9. The only thing that lights up is the hypothalamus, cingulate gyrus and limbicsystem. The sensory cortex doesn’t come into play. The fact that the correlation is0.9 tells us that the visual analogue scale (VAS) is your emotional response towhatever, and the lighting up of the cingulate system is an index of youremotional response. This indicates that the distinction between these things iszilch.Kidd: I’d say that you are wrong! The fMRI studies are showing nociception not
pain. You are failing to address the di¡erence between what the patient is telling usand what the patient is experiencing. The fMRI studies are no more valid than acapsaicin skin test in telling us about nociceptive activity. How the patient thenchooses to communicate the resulting distress is an entirely di¡erent matter and iscritically dependent on context. The VAS score does not just assess the emotionalresponse; it tells us about the sensory discriminative aspect also.Bradley: I’d like to a⁄rmwhat Bruce Kidd has just said.What you get in a fMRI
study looking at a correlation between brain activity and VAS depends in part onhow you instruct the subject to use the VAS. It is possible with about an hour oftraining to teach people to di¡erentiate between the intensity of whatever it is theyare experiencing versus the unpleasantness of what they are experiencing. I willshow some data in my paper on ethnic group di¡erences in emotional painresponses, and how this correlates with brain imaging. Another simple way oflooking at this is as follows. We are working on a study now in which we areexposing patients with ¢bromyalgia and controls to stressors in the laboratory.Beforehand, we train people to use the VAS in one way to measure the intensity
136 DISCUSSION

of the sensation produced by a thermal stimulus and in another way tomeasure theunpleasantness. What we are ¢nding is that stressors in the laboratory don’tdi¡erentially a¡ect perceptions of pain intensity between patients and controls,but patients with ¢bromyalgia show much greater increases in perceptions ofpain unpleasantness after exposure to the stressors in the laboratory. We also ¢ndsome reductions in blood levels of cortisol as a function of the stressors. Withquanti¢ed sensory testing we can train people to di¡erentiate between di¡erentdimensions of the pain experience, and you see di¡erential e¡ects when you haveproperly trained subjects.Malcangio:Do antidepressant drugs relieveOApain? Fromwhat you say there is
some emotional component.Yashpal: When you look at intracellular mechanisms of action, it is becoming
clearer that there are some proteins thatmight be common to pain and epilepsy. Ananti-epileptic such as gabapentin is very e¡ective for some chronic pain patients.On the other hand, we have published a paper showing that NK-1 antagonists arevery e¡ective antidepressants. The emerging theory is that some of the intracellularmechanisms might be common.Fox:What you say is correct but I think it only applies to certain pain states.We
are used to recognizing that anticonvulsants and the newer anti-epileptic drugswork in neuropathic pain, as do SSRIs and tricyclics. It would be new to us if weknew that these agents were working in nociceptive or in£ammatory painmechanisms. This would be a real surprise. The other thing about the NK-1story is that one of the studies showed NK-1 antagonists are e¡ective indepression, but the same drug was ine¡ective in chronic pain.Pisetsky: Clinically, antidepressants are commonly used in OA.Felson: Although frankly there is no evidence that they work.Dieppe: The clinical answer is that there isn’t evidence, but occasional patients
respond extremely well. The problem is that we don’t know how to pick who isgoing to respond to an anxiolytic or antidepressant and a non-steroidal anti-in£ammatory drug (NSAID). This is because we don’t know how to sort out theheterogeneity of the pain in OA.Yashpal: We don’t know the intracellular mechanism by which gabapentin
works, but it is commonly prescribed. It works pretty well in quite a few chronicpain patients.Creamer: You said that one of the ¢rst things we should do is to look at
quantifying pain responses in OA. Could you expand on how we might begin todo this?Kidd: The algometer applies mechanical pressure and measures thresholds for
mechanical stimuli. You can also measure thermal thresholds. There is a bigliterature on this. These studies need to be done carefully so they can’t be used ina big population. The tools are there.
PAIN AND INFLAMMATION 137

Rediske: I thought your last point was provocative, that mediators of theneurogenic in£ammatory processes could be playing a role in OA. Are you awareof studies looking for the potential disease modifying activity of analgesics thattarget some of the potential neurotransmitters?Kidd: I think it is the other way round. The data are that NSAIDs are
catastrophic for the joints. There have been no long-term studies. How wouldyou do it? How would you reduce peripheral e¡erent activity? It would be verydi⁄cult.Rediske: With substance P antagonists, which have been tried in a number of
clinical situations.Kidd: My understanding of those studies is that they have just looked at pain
responses.
138 DISCUSSION

Molecular events of chronic pain:
from neuron to whole animal in an
animal model of osteoarthritis
James L. Henry
Department of Physiology and Pharmacology, Medical Sciences Building, University of WesternOntario, London, Ontario, N6A 5C1, Canada
Abstract. A novel animal model of osteoarthritis has been established for studies on thedisease process as well as on mechanisms underlying the symptoms of osteoarthritis,principally pain and fatigue. The model is established by cutting the anterior cruciateligament in the rat to introduce instability of the joint, removing the medial meniscus toinduce further derangement of the joint, and exercising the rat on a modi¢ed rota-rod togenerate weight-bearing mobility of the joint; this exercise can be regulated in terms ofduration to govern severity of the model. The model exhibits many of the characteristicsof osteoarthritis in humans, including bone and cartilage remodelling, in¢ltration of thejoint tissues by immune cells and alterations in sensory mechanisms. This model will ¢ndapplication in development of novel interventions for the treatment of osteoarthritis andits symptoms as well as development of diagnostic tools for early detection.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 139^153
An astounding 18% of the population in the western world is expected to su¡erfrom some type of arthritis by the year 2020 (Lawrence et al 1998). Osteoarthritis(OA) remains the most common form of arthritis, a¡ecting 12% of US adults. OAis the secondmost common diagnosis, after chronic heart disease, leading to SocialSecurity disability payments due to long-term absence from work. Although it ismore prevalent in older patients, it has been shown to be di¡erent than the ageingprocess (Aurich et al 2002). The features of OA constitute a group of conditionsthat are diagnosed upon commonpathological and radiological characteristics. It isprevalent in the knees, hands, hips and apophyseal joints of the spine. The cause ofOA is likely multifactorial, although there are two general concepts. One theory isthat there is material failure of the cartilage network leading to tissue breakdown(Poole 1999). The second theory is directed toward injury of chondrocytes withincreased degradative responses (Aigner & Kim 2002). Underlying factorscontributing to OA include obesity, developmental and anatomic abnormalities,
139
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

aging and immune responses. Recent studies have suggested genetic contributionsto OA (Loughlin 2001). Despite a myriad of causative factors, clinical andradiological features of the disease are similar.Clinical signs, such as radiological features, are useful for the diagnosis and
prognosis of OA. However, patients complain of pain and fatigue.Pain that is a medical problem is actually a family of disorders that can be broken
down based on origin: nociceptive pain, due to activation of pain sensoryreceptors; neuropathic pain, due to activation of the axons of pain ¢bres; andidiopathic pain, for which there is no discernable aetiology. Recent advanceshave led to our understanding of pain as a process rather than a phenomenon(Stucky et al 2001). The earliest pains are brought about by a set of processes thattrigger additional mechanisms, including plastic changes in the nervous system.Thus, those processes that underlie acute pain can eventually trigger mechanismsthat lead to chronic pain, where pain becomes unnecessary and debilitating (Woolf& Salter 2000). In the past, it was not understood that to treat pain we need to treatthe mechanisms that produce the pain, rather than just the disease itself. Previousthought was that if the disease were treated successfully, the pain would disappear.This is not consistent with current thought among pain specialists, whowould citephantom limb pain as a striking example; the pain persists even after amputation ofthe a¡ected site (Flor 2003, Haigh et al 2003). Chronic pain is seen as a disorder ofthe nervous system. There is an obvious relationship between OA and thesymptoms of OA, but we need to identify the mechanisms triggering thesesymptoms, in particular, pain and fatigue. These concepts underlie thehypotheses that follow.To recap, diseases can trigger changes in the nervous system that then generate
ongoing persistent pain. Because of the nature of signal transduction mechanisms,untreated or even undertreated acute pain, if allowed to persist, can develop intochronic pain that arises by di¡erentmechanisms (Woolf& Salter 2000, Ji&Woolf2001, Stucky et al 2001). These longer-term mechanisms are usually moreinsurmountable, and thus present greater medical challenges.Early intervention is thus important. Irrespective of what triggers the original
pain, a virtual soup of chemicals participates in the activation of pain neurons inperipheral tissues. There is a similar soup of chemicals participating in conductionof the pain signal at the ¢rst sensory synapse, where nerve cells projecting from theperiphery pass information to the next neuron in the pathway. At both peripheraland central nerve terminals, certain chemicals not only act over the short term tocreate and transfer the pain signal but also alter the properties of the pain neuronsthemselves (Herbert&Schmidt 2001, Zimmerman 2001). Thus, these neurons canbe seen as being plastic or malleable. It is important to block the pain signal beforeit can act long enough to induce these plastic changes. Once established, thechronic pain that results is di⁄cult to overcome (Goucke 2003, Pohl et al 2003).
140 HENRY

Early intervention requires early detection. In fact, detection of the diseaseprocess before it can initiate pain is important in preventing plastic changes. Tothis end, there is a search for early markers of pain. There are typically threecomponents to a comprehensive pain assessment: pain diagnosis based on inferredpathophysiology, identi¢cation of contributing factors and identi¢cation ofbarriers. We always try to identify ¢rst the primary cause of pain. However, byapplying a single-dimensional pain assessment to a case of chronic pain, one risksslipping into a quagmire of failed therapies, increasing symptoms, angry patientsand frustrated physicians. Reliable surrogate markers of pain may ¢nd inestimableusefulness in objectifying and quantifying assessment measurements. Surrogatescan be used in a comparative way to characterize the disease (diagnostic value) andto measure disease progression (prognostic value) (Wang et al 2002). Surrogatemarkers also o¡er ameans to determine e⁄cacy of painmanagement interventions.The contribution of the nervous system to OA has been the subject of only
limited qualitative assessment, surprising in view of the obvious involvement ofthe nervous system in OA pain.One more concept that needs to be introduced is how sensory nerves in£uence
the health of peripheral tissues. Sensory nerves can initiate and maintainin£ammation by the release of mediators from a¡erent terminals. Simpleexamples are the £are and wheal of the triple response that is seen after a scratchto the skin. The £are and the wheal are eliminated by prior anaesthesia of thesensory nerves, and are absent in animal experiments in which sensory nerveshave been cut and allowed to degenerate (Herbert & Schmidt 2001). Neurogenicin£ammation has been implicated in a number of conditions, includingin£ammatory arthritis (Sharif et al 2004). Sharif et al (2003) provide evidence thatdi¡erent nerve ¢bre types regulate joint physiology in di¡erent directions, withsome acting in an anti-in£ammatory capacity, while others are pro-in£ammatory.
Hypotheses
Primary sensory neurons pick up the sensory signal in peripheral tissues througheither specialized or undi¡erentiated peripheral nerve terminals, called sensoryreceptors. These receptors can transduce only three types of adequatestimulus�mechanical, thermal and chemical. Our primary hypothesis is that thepain of OA arises as a result of chemical activation of sensory receptors thatspeci¢cally carry the pain signal. This hypothesis demands identifying thechemicals that are altered in and around the joint at di¡erent stages of OA, as oneor more of these may generate the pain. A second hypothesis is that joint chemicalsin OA enter the circulation and lead to fatigue. A third hypothesis is thatneurotransmitters released from the peripheral terminals of sensory ¢bresregulate disease progression and may also participate in the initiation of OA.
ANIMAL MODEL OF OSTEOARTHRITIS 141

Objectives
Our objectives are to (i) understand aetiology of OA and its symptoms of pain andfatigue, (ii) identify biomarkers and other tools for early diagnosis before onsetof clinical signs, (iii) discover targets for development of innovativeintervention strategies, and (iv) provide reliable markers of prognosis inlongitudinal studies. The proposed programme will provide a richtransdisciplinary environment for the training of basic and clinical scientists. Aswell, this programme will provide novel research models, reagents and techniquesfor academia and industry.We have established an animal model of OA to enable longitudinal studies that
can focus on the earliest stages ofOA, including a period before the onset of clinicalsigns. The long-term objective using this model is to investigate the earliest signsof OA, and to identify biomarkers and gene markers of the disease and itssymptoms�pain and fatigue�to develop methods of early detection; to gatherfundamental knowledge regarding factors that will modify disease progression;to identify mechanisms underlying the pain of OA; to develop novelinterventions based on mechanisms, and to measure outcomes of these and otherinterventions under development.AnimalmodelsofOAallowlongitudinal studies tobeginbeforemanifestationof
any clinical signs. Early detection and intervention are of critical importance tocircumvent the debilitation of OA, if we assume that clinical signs becomedetectable only when the condition is already at an advanced stage. There areseveral animal models of OA available, including horse, rabbit, dog, guinea pigand rat (Smith & Ghosh 2001, Brandt 2002). In selecting the model, we wereaware that any animal model has its shortcomings when compared to the humancondition. These allowances aside, the rat model was chosen for several reasons:
. High-content A¡ymetrix microarray analyses can be done only in rat or mouse.Electrophysiological studies of the type required cannot be done in mouse.Therefore, the rat is the only species where function can be tied directly withgene chip analysis.
. Physiological data from the rat for pain and fatigue are abundantly available inthe published literature.
. Electrophysiological data on rat spinal and peripheral sensory mechanisms arealso extensive.
. The rat is a common species for studies on transcription factors,neurotransmitters, cytokines, etc.
. In our model, the severity of the disease can be controlled (see below).
. Small animals such as rats are preferred by industry asmodels for preclinical drugdevelopment.
142 HENRY

The model
We have developed a novel OA model in the rat through modi¢cation of apreviously published procedure (Williams et al 1982, Stoop et al 2001). The kneejoint is destabilized by cutting the anterior cruciate ligament (ACL) and removingthe medial meniscus (Karahan et al 2001). Next, we developed three methods toregulate severity. One is through forced weight-bearing articulation of the OAjoint; to achieve this, a rota-rod device was modi¢ed in-house. The rotating rodis elevated to one metre as a disincentive for the rat to fall. The rotating rod is alsocovered with carpeting to increase grip by the rat. A DC motor was installed toslow the turning rate to a level that the rat can remain comfortably on the rod,but is required to walk continuously. The diameter of the walking surface is6.7 cm so that the hind legs are continuously walking uphill. Each rat is exposedto forced weight-bearing articulation for one hour. Pilot studies indicate thatforced movement exacerbates cartilage degradation in the ACL/medial meniscusmodel. A second method is forced articulation without loading, achieved by daily5min periods of swimming in a circular tank ¢lled with water to a depth of 27 cm.The third method is voluntary weight-bearing articulation in an open ¢eld. In thiscase, rats are exposed for daily 10min periods in an open ¢eld with rat toys. In allcases, severity can be controlled by changing the duration of the activity.Themodel we have developed exhibits a number of signs that allowquantitative
measurement of disease progression and its associated symptoms of pain andfatigue. The OA joint exhibits sclerosis of bone, osteophytes, altered 3Dconformation and decreased gap. Other data indicate decreased cartilagethickness in the OA joint, cartilage lesions, increased sensitivity of nociceptiveneurons and decreased threshold for activation of nociceptive neurons arisingfrom the joint.The overarching strategy for developing a new model in this way is that the
severity can be regulated by the duration of the activity paradigm. A commoncomplaint about preclinical animal models is that they are so severe that it isdi⁄cult to assess responses to interventions. On the other hand, it is important tobe able to quantify physiological parameters of the model (particularly pain andfatigue) and to identify biomarkers. The ability to regulate severity wasconsidered very important for this study, and thus this model stands distinctfrom other animal models of OA.
Results
Signs of osteoarthritis
Radiological examination using micro-computed tomography reveals joint spacenarrowing, osteoarthritis and osteophyte formation in the arthritic joint.
ANIMAL MODEL OF OSTEOARTHRITIS 143

Histological examination indicates in¢ltration of immune cells. Injection of EvansBlue dye intravenously and analysis of joint tissue demonstrated extravasation ofplasma proteins, indicating loss of integrity of the endothelial barrier. Behaviouraltests indicate a withdrawal response to von Frey hair stimulation at intensitiesbelow those that provoke withdrawal in normal animals, indicating thedevelopment of an animal equivalent to allodynia. Exploration in open ¢eldtesting was also reduced.
Electrophysiological experiments
Pilot electrophysiological experiments have been run on this model. Recordingsfrom single dorsal horn neurons have demonstrated increased receptive ¢eld size,reorganization of receptive ¢elds to include both deep and cutaneous territories,spontaneous discharge developing in silent neurons following passive articulationof the arthritic joint, and reversal of the e¡ects of g-amino butyric acid (GABA)applied by iontophoresis from inhibition to excitation.
Summary and conclusions
This model therefore demonstrates several signi¢cant features, generallyremodelling of bone and cartilage, in¢ltration of immune cells, disruption ofhomeostatic mechanisms maintaining the integrity of soft tissues in the joint,alterations in sensory withdrawal thresholds and remodelling of spinal painmechanisms. Our results thus indicate that this novel animal model of OA mayprove useful as one step in understanding mechanisms underlying initiation anddevelopment of OA, and has potential applicability to identifying biomarkers andgene markers of disease onset and disease progression. This mechanistic approachnotwithstanding, it is important to appreciate that this is only one approach togetting control of OA and its symptoms, and many important issues remain to beanswered.Among these are the development of innovative biosocial approaches tomanagement of the disease, understanding comorbidity of OA and the causalrelationships with and impact of other signs, the basis for gender di¡erences inthe propensity to develop OA, optimization of social support, and factorsin£uencing access to care.
References
Aigner T, Kim HA 2002 Apoptosis and cellular vitality: issues in osteoarthritic cartilagedegeneration. Arthritis Rheum 46:1986^1996
Aurich M, Poole AR, Reiner A et al 2002 Matrix homeostasis in aging normal human anklecartilage. Arthritis Rheum 46:2903^2910
Brandt KD 2002 Animal models of osteoarthritis. Biorheology 39:221^235Flor HJ 2003 Cortical reorganisation and chronic pain: implications for rehabilitation. J RehabilMed (41 suppl):S66^S72
144 HENRY

Goucke CR 2003 The management of persistent pain. Med J Aust 178:444^447HaighRC,McCabeCS,Halligan PW,BlakeDR2003 Joint sti¡ness in a phantom limb: evidenceof central nervous system involvement in rheumatoid arthritis. Rheumatology (Oxford)42:888^892
Herbert MK, Schmidt RF 2001 Sensitization of group III articular a¡erents to mechanicalstimuli by substance P. In£amm Res 50:275^282
Ji RR, Woolf CJ 2001 Neuronal plasticity and signal transduction in nociceptive neurons:implications for the initiation and maintenance of pathological pain. Neurobiol Dis 8:1^10
Karahan S,Kincaid SA,Kammermann JR,Wright JC 2001Evaluation of the rat sti£e joint aftertransection of the cranial cruciate ligament and partial medial meniscectomy. Comp Med51:504^512
Lawrence RC, Helmick CG, Arnett FC et al 1998 Estimates of the prevalence of arthritis andselected musculoskeletal disorders in the United States. Arthritis Rheum 41:778^799
Loughlin J 2001Genetic epidemiology of primary osteoarthritis. CurrOpin Rheumatol 13:111^116
Pohl M, Meunier A, Hamon M, Braz J 2003 Gene therapy of chronic pain. Curr Gene Ther3:223^238
Poole AR 1999 An introduction to the pathophysiology of osteoarthritis. Front Biosci 4:D662^D670
Sharif RN, Cahill CM, Ribeiro da Silva A, Menard HA, Henry JL 2004 Remodelling ofsomatosensory synapses. A mechanism for allodynia in arthritis. Submitted
Smith MM, Ghosh P 2001 Experimental models of osteoarthritis. In: Moskowitz RW,Buckwalter JA, Altman RD, Howell DS, Goldberg VM (eds) Osteoarthritis: diagnosis andmedical/surgical management, 3rd edn. Saunders, Philadelphia
StoopR, BumaP, van derKraan PMet al 2001Type II collagen degradation in articular cartilage¢brillation after anterior cruciate ligament transection in rats. Osteoarthritis Cartilage 9:308^315
Stucky CL, Gold MS, Zhang X 2001 Mechanisms of pain. Proc Natl Acad Sci USA 98:11845^11846
Wang H, Sun H, Della Penna K et al 2002 Chronic neuropathic pain is accompanied by globalchanges in gene expression and shares pathobiology with neurodegenerative diseases.Neuroscience 114:529^546
Williams JM, Felten DL, Peterson RG, O’Connor BL 1982 E¡ects of surgically inducedinstability on rat knee articular cartilage. J Anat 134:103^139
Woolf CJ, Salter MW 2000 Neuronal plasticity: increasing the gain in pain. Science 288:1765^1769
Zimmermann M 2001 Pathobiology of neuropathic pain. Eur J Pharmacol 429:23^37
DISCUSSION
Schaible:Your results with the nerve cuts seem to be at variance with the earlierresults of Levine. Is that true? He claimed that cutting of nerves will make arthritisbetter.Henry: Yes. I can’t account for his results.Blake: You don’t have to. In a clinical setting, all of us who have tried to
reproduce this have got it to swing in both directions. The most commonobservation is that it reduces it. Likewise, in rheumatoid arthritis (RA) in aclinical setting, if someone has an ulnar nerve lesion they do not get the RA in
ANIMAL MODEL OF OSTEOARTHRITIS 145

that area, by and large. Everything depends on the time of the injury. Lesions cansuppress it prior, aggravate it after, and mild switches which we don’t understandcan £ick it one way or the other.Henry: I agree: the timing is critically important. Patients who have stroke with
RA are a good example. We have to make sure that we don’t see the peripheralchanges as being due to sensory neurons only. In stroke the sensory neuronsaren’t altered, it is the motor neurons and autonomic neurons. We should avoidtaking a simplistic approach.Kuettner: I was surprised to see your pictures of the receptor for substance P on
chondrocytes, calci¢ed cartilage and newly formed bone. Do you have any idea ofthe function of substance P speci¢cally during cartilage remodelling? Do you seethat the collagen breakdown products are derived from type 2 collagen or are theygelatin?Henry:They are type 2. Those slides were fromRA animals, so this suggests that
there is probably an up-regulation of the receptors in those tissues. Keep in mindalso that despite the fact that there is an abundance of degrading enzymes £oatingaround, in this area there are fewer enzymes to break down substance P, whichmeans that it can then di¡use to other places. The issue of sprouting of neuronswas raised yesterday, and whether this could account for the pain of OA. I don’tknow, but if substance P can di¡use from one site to another, then you don’t haveto have the hard wiring because of the di¡usion. I’ll be able to tell you more nextyear.Herzog: Coming back to cutting the nerve, you were talking primarily about
trophic e¡ects and the release of chemicals from the nerve. How do youdistinguish this from all the events that happen mechanically when the nerve iscut, such as muscle function loss?Henry: We haven’t yet. But by giving the antagonists we can eliminate the
sensory neurons without altering gait.Grubb: This experiment depends on when and how you cut the nerve. The
temporal aspects are important. There are two interpretations. If you cut thenerve you could say that you are getting rid of the sensory nerves and any thingsthey might be protecting. But also if you cut the nerve there will be a massiverelease of substance P for a short period from that dying nerve terminal. Insteadof getting rid of the nerve this may actually exacerbate nerve activity transiently.Henry:Wemade our measurements well after the cutting of the nerve.Mackenzie:With regard to the discrepancy between the Levine experiments and
yours, I think the models are actually di¡erent. If I remember correctly, the Levineexperiments were in adjuvant polyarthritis where they were looking at thedevelopment of the secondary lesions which ¢rst occur about 10 or 12 days afterinjection of adjuvant. Your experiments involve the injection of adjuvant directlyinto one joint.
146 DISCUSSION

Henry:Ours is a model of mono-arthritis a¡ecting one joint.Rediske: Have you looked to see whether chondrocytes are also a source of
substance P?Henry:No.Evans: A recent paper showed that they are (Millward-Sadler et al 2003). They
have the capacity to make it under the right conditions.Pisetsky: I have a question about this model, and trying to distinguish e¡ects of
local versus systemic in£ammation. Even if you are putting complete Freund’sadjuvant (CFA) in a joint, it is likely to be doing a lot of things peripherally,given what it contains. How do you distinguish e¡ects that are local from thoseon general in£ammation?Henry: We have looked contralaterally and we don’t see any evidence of joint
destruction.Pisetsky: There are local problems plus systemic problems. You wouldn’t see
contralateral joint damage, but these animals probably have high levels of TNFand other cytokines. How do you distinguish the systemic e¡ect which may beacting on the brain from the local e¡ects?Henry:We don’t. Until we have these data I am assuming that there are going to
be systemic e¡ects beyond the local joints. The way I see the biological system, thishas to be occurring. But this is also why I was alluding earlier to commonaetiology. I would expect there to be other e¡ects.Pisetsky: I am just trying to ¢gure out how you extrapolate this to anOAmodel,
where the amount of in£ammation is much less.Felson: Can you comment on the OA model? Do you have some preliminary
data from that?Henry: Yesterday I also raised the question of whether OA is a systemic disease.
It is very early for us to comment on our results, but we have seen an osteophytecontralaterally in our OA model. This stunned us. I can’t explain this.Fernihough: The model has comparisons with Geza Pap’s overuse model (Pap
et al 1998). This is an established model of OA and has histological features ofthe disease. In conjunction with pain, the importance of time has been discussed.How does the pain sensation change with time? Do all of your animals share acommon range of pain sensation?Henry: I can’t comment on theOAmodel. In the RAmodel, however, there was
a very consistent reduction in pain that started immediately. By day 2 and 3 it wasdown to baseline and was stable after that. So there is a very rapid change insensitivity of the nociceptive mechanisms. There wasn’t a lot of variability fromone animal to another.Fox: There will be some di⁄culty in interpreting the behavioural data
from animals. This is a perennial problem. We have been doing similar models ofRA and Janet Fernihough has produced some beautiful histology showing
ANIMAL MODEL OF OSTEOARTHRITIS 147

changes in the knee. There are changes in nociceptive markers in DRGs and suchlike. This is ongoing. But behaviourally it is very di⁄cult to distinguishnociception changes compared with changes in movement, just because the jointis now unstable. This will be a real challenge. I will be interested to see how youintend measuring this.Henry:We don’t intend to do it in isolation, for these very reasons.Van den Berg: I have some di⁄culty with the discussion, linking your ¢ndings
to either an RA or OAmodel. What you have done in these acute models is simplymaking in£ammation. Whatever stimulus you use, you will probably get the samekind of data out of it.Whether you use Freund’s or something else, it doesn’t reallymatter: you are making an in£amed site somewhere in the body. The only thingyou can learn from this is what in£ammation is doing in terms of feedback on yourpain system. In the discussion about cutting the ligament, it might be criticalwhether you cut the ligament before or after the nerve. Do you recall what youdid in this situation?Dieppe: I wanted to pick up on your throwaway remark about the bilateral
osteophyte. It is quite a common experience in animal models of arthritis to seesomething on the contralateral side if you look for it. This relates a bit to ourearlier discussion about contralateral pain representation and spreading. I’d liketo hear from the neurophysiologists and pain people what they think is going onwith these bilateral e¡ects on animal models of arthritis.Henry: There are two elements. One is mirror pain, which is not uncommon.
The osteophyte on the other side could simply be due to changed gait. It is notnecessarily a systemic disease, but we can’t eliminate the possibility of it being asystemic disease.Dieppe: It probably is some sort of systemic e¡ect. If you do an in£ammation
model you will see synovitis the other side.Henry: We haven’t seen any evidence of synovitis on the other side in our RA
model.Blake: You will if you look carefully enough and in the right genetic
strain.Henry:We have been using Sprague-Dawley.Blake: If you use Wistars you will see it. Contralaterality in these systems is
almost invariable providing that you have the right tool and you know what youare looking for. It goes back, as Paul is alluding to, to a lot of the basic clinicalobservations made on selective upper motor neuron lesions. The people whoreally understand this are the ophthalmic surgeons who deal with sympatheticuveitis. This is where someone injures one eye and in the worst cases they canend up going blind in both. They have techniques where they can show that thismirroring occurs in almost every instance. Blindness is not 100%, butelectrophysiological disturbance in the other side is.
148 DISCUSSION

Lohmander: I wanted to follow up on Paul Dieppe’s comment and point out thatwe see similar ¢ndings in our corresponding human post-injury models of OA. Inthese patients we have a higher than expected rate of OA in the other knee,although patients claim that they haven’t injured it. Of course, there are issuesabout overloading of the other, un-injured, side, but my gut feeling is that thereis more to it than that.Felson:TedPincu has tried to improve the e⁄ciency ofOA clinical trial outcome
measurement by asking about pain in the signal knee, and avoiding having to askabout the other joints. He found that it was more e⁄cient to ask about both kneesbecause patients entering the trials often switched side where the knee was mostpainful. Having completed a longitudinal study of knees, we had to keep track ofwhich sidewe got the initialMRI on. Subjects would come back for repeatedMRIsand they wouldn’t remember which side they had had the initial MRI on.Grubb: I have a more direct answer to Paul Dieppe’s question about the
contralateral issue and the hard wiring of the neurons. There is no doubt that ifyou record from wide dynamic range neurons in rat spinal cord (these are likelypresent in the deep dorsal horn) that they can have quite large receptive ¢elds onthe ipsilateral limb. But even in some normals, occasionally you can ¢nd a hardwired input from the other side, the contralateral limb. We don’t see many ofthese far away from the region where we are trying to identify the receptive ¢eld.Mostof theneuronswehave looked for have the centre of the receptive¢eld aroundthe ankle joint, because we were interested in the rat ankle joint. But occasionallythey had receptive ¢elds running right up the limb, and just occasionally we got aneuron that responded in a normal animal to squeezing the paw on the other side.But when we induced the in£ammation peripherally on this ipsilateral side, thenumber of neurons responding to contralateral pressure increases to about 18^20%. There are ¢bres that go across, into the deep dorsal horn on the other side,and have an in£uence on those deep dorsal horn neurons. The fact that the centralneurons become a bit more sensitive and a bit depolarized by the constant a¡erentinput from the in£amed region means that they then respond more to thesecontralateral inputs because they are more sensitive. Central sensitization hasoccurred, but because the contralateral inputs are there, their in£uence becomesmore obvious and you see more cells responding to that stimulus.Henry: However, some but not all of the contralateral e¡ects can be due to
crossed projections and sprouting of primary a¡erent or even second-orderneurons. It is interesting that in some models there are even changes inexpression in DRG cells contralaterally. There are changes in mRNA levels, forexample, on the contralateral DRG.Schaible: We have done a similar study. One has to make a distinction between
the segmental bilateral increase and the systemic increase. A good control might beto study the ganglion at di¡erent levels.
ANIMAL MODEL OF OSTEOARTHRITIS 149

Pisetsky: I have a question about the e¡ects of cytokines on nociception in thesemodels. Clinically, if we give people cytokines, they feel sick. Howmuch of this iscentral? Should we consider this in these models?Schaible: I mentioned earlier Linda Watkins who has talked a lot about sickness
response. This implies that interleukins will act in the CNS to organize acoordinated response to a noxious stimulus. So there is some good evidencefrom her studies that these cytokines can induce a general sickness response. Wedon’t know whether that is happening in these models. She can evoke such asickness response with LPS injection and so on, and she can demonstratepathways involved.Kidd:Oneof the problemswith the cytokine story is that the blood^brain barrier
is impermeable to many cytokines. Linda Watkins was hypothesizing the role ofthe vagus nerve for a long time, but it is now becoming apparent that in situationsof in£ammation the blood^brain barrier can break down and it becomes muchmore permeable to cytokines. It might be that in an in£ammatory situation thereis an entirely di¡erent physiology, and under these circumstances IL6, IL1 andTNF may gain access to the brain receptors.Henry: Yes, the blood^brain barrier should not be seen as a ¢xed barrier. It is
under functional control.Malcangio: In any case, sensory neurons can produce IL and TNF themselves, so
the cytokines don’t need to cross the blood^brain barrier. And in some othermodels microglia produce cytokines within the spinal cord. Jim Henry, you talkabout the ‘good guy’ that might be released by sensory neurons. Do you think thatsomatostatin could be protective?Henry: We haven’t looked at somatostatin, but it was a surprise to us that
neurokinin A (NKA) seems to have opposite e¡ects to substance P.Malcangio: By which receptor?Henry:Wehaven’t looked at that yet, but it could be the NK-2 receptor which is
in peripheral tissues. The reason why this is surprising is because substance P andneurokinin A are derived from the same precursor. So you have one neuronreleasing both a good guy and a bad guy from the terminal, which suggests thatthe regulatory controls�whether the release is having a bene¢cial e¡ect or adamaging e¡ect to peripheral tissues�depend on the enzymes that are presentbreaking them down.Malcangio: This is in the periphery, not centrally.Henry: That is correct.Mackenzie: There was an intriguing series of experiments published some years
ago from Sergio Ferreira’s laboratory (Ferreira et al 1988, Cunha et al 1992) inwhich minute quantities of TNF and IL1 were injected into rat footpads. Hethen looked at pressure hyperalgesia. He demonstrated that not only was therehyperalgesia in the injected paw, but also in the contralateral paw. The quantities
150 DISCUSSION

used were so tiny that it is very unlikely that this is a systemic e¡ect. The onlyconclusion that can be drawn is that this was a neuronal e¡ect. The ¢ndings havebeen reproduced in a number of laboratories, so I think it is a genuine phenomenonthat has never been fully explained mechanistically. It might be important in termsof sensitization through the release of cytokines.Henry: Cytokines are obviously one of the areas on which we will be focusing. I
can add that we have an animal model of neuropathic pain where the animalsbecome extremely sensitive. We get a contralateral e¡ect there also.Rediske: Your OA model has a lot of promise. It underscores some of the
designer nature of OA models, because you build in two instabilities plus chronicloading. Instead of building in those kinds of stimuli, have you ever thought ofbuilding in a chronic neurogenic or neuropathic component into a destabilizationmodel?Henry: It is interesting that you ask about a neuropathic element. Part of me as a
neurophysiologist wonders how much of the pain of OA is neuropathic pain. Idon’t know. As we develop this model we are going to be adding other things.We can address indirectly the issue of neurogenic in£ammation by givingantagonists long-term to some of these chemicals that we believe areparticipating in neurogenic in£ammation. Substance P is the ¢rst one we will dobecause it seems to be the most obvious.Fox: Thinking about extrapolating frommodels to humans, preclinical models
regard chronic pain as lasting a day whereas clinicians are looking at months andyears.What happens to thewide dynamic range neurons in the spinal cord after thein£ammation has resolved in one of these models? Presumably inOA there is not apersistent in£ammation that might be constantly driving the input to the spinalcord, yet we are talking in terms of a prolonged central sensitization.Henry: As far as we know, our RA model goes on forever. Our model of
neuropathic pain also goes on for ever: we have measured that up to 150 days.We haven’t got to this stage with the OA model yet.Evans:Wewere talking earlier about the contralateral e¡ect and communication
through the nervous system. Iwant to point out that there are otherways that thesejoints can communicate aside from general systemic means. We have done thereverse contralateral experiment using bilaterally arthritic animals. When weinhibited the arthritis in one joint through gene transfer, the other joint gotbetter. This seems to be connected with the tra⁄cking of cells that we are stilltrying to identify, but we think they are DCs that migrated from the joint weinjected to the joint we didn’t inject. They may be moving under the in£uence ofchemokines released from the nervous system. I am not saying that the nervoussystem is not involved, but it doesn’t always have to be hard wiring.Grubb:Hard wiring does exist, though.Blake: Could you block this by sciatic nerve section?
ANIMAL MODEL OF OSTEOARTHRITIS 151

Evans: That is on the list of things to do.Felson: Peter Simkin, I want to ask you about the bilaterality of gout. How
related is this very in£ammatory but episodic disorder?Simkin: The localization of gout, to me, is much more associated with the
presence of OA. The big toe is hit because this is the bunion joint, for instance(Simkin 1977, Simkin et al 1983). OA tends to be bilateral, and I think this iswhat causes bilaterality when it is seen in gouty arthritis.Hunter: Iwanted to get you to comment on structure rather than pain.With your
OA model, where you have forced articulation, can you comment on thedi¡erences that you saw between the rats that did the weight bearing versus thenon-weight bearing exercise?Henry: If we go at a certain distance and time, we seemore osteophytes andmore
bone degradation sclerosis in the articulated animals than in the surgically treatednon-articulated animals. This work is so new that I can’t answer directly whetherthere is more at 2 months in the articulated animal than we ever see in the non-articulated model.Pisetsky: In RAmodels in rodents, there is a big impact of genetics. Is there any
genetics in OA models so far, in terms of the likelihood that one or another strainwill develop these?Henry:My feeling is that we are sadly lacking in good OAmodels.Pisetsky: Are the rat strains that get adjuvant arthritis more likely to get OA in
these models?Henry: There is such a lack of activity in OAmodels that we haven’t got to that
stage yet.Van denBerg: There is a lot of genetics in spontaneous OA. However, when you
use an instability model you can cause OA in any animal, and the geneticbackground is not that relevant.Pisetsky: Unless the in£ammatory component has more of a role in the disease.Lohmander: We have some recent data which are from a repeat of a study that
Paul Dieppe was involved in about 20 years ago. This was the ¢rst published studyin the area and when we publish ours, it will be the second! We showed that if youhave hand OA as a marker of a genetic trait of OA, your post injury OA will beworse andmore frequent than if you don’t have handOA.This shows that geneticsdoes feed into human post-injury OA.Dieppe: It is nice to hear that this study has been repeated. I was worried about it
because our early report contained statistical £aws!Jim Henry, you said you worry about how much OA pain might be
neuropathic. Perhaps there is another clinical model we should be thinking aboutwhichmight get us someway to that.We have been doing quite a lot of work withsurgical colleagues on joint replacement. For most people, pain disappears afterjoint replacement, but there are about 10% of people where it doesn’t. This is
152 DISCUSSION

fairly consistent across all the studies that the pain stays in this 10% even though thejoint has been replaced. Could those be the people who have the neuropathic pain?Henry: That is one of the two possibilities that come to mind. The other is
phantom limb pain.Dieppe: They might be quite an interesting group to study in terms of our
understanding of pain in OA.Grubb:Have treatments for neuropathic pain been tried in OA?Dieppe:Not that I know of.Blake:There is a third explanation given tome by a knee surgeon, that there was
a form of CRPS (complex regional pain syndrome) developing in those knees.Dieppe: I have never seen re£ex sympathetic dystrophy in that situation.Blake: They are much more aware of it than we are, and they are not using the
same clinical criteria that we are using. One thing that is for certain is that the painisn’t necessarily the same as it was before the operation.Fox:Do you know that there hasn’t been some sort of neuronal activation just
because of the operation?Blake: That is very rare. You can usually dissect these from a decent history and
examination.Ordeberg: It is a multifactorial problem. We see change in pain even in those
patients who bene¢t from the operation. Going back to my presentation, therewere patients who had sensory aberrations. These are patients where theanalgesia is no longer su⁄cient in the treatment. The pain is not purelynociceptive: it may be neuropathic or neurogenic. I am mainly a spine surgeon.Among those patients with failed hip arthroplasties there are quite a few whohave L3 or L4 root pain. This seems fairly natural as the distribution of L3 andL4 root pain is very much the same as in pain from hip osteoarthritis.
References
Cunha FQ, Poole S, Lorenzetti BB, Ferreira SH 1992 The pivotal role of tumour necrosis factoralpha in the development of in£ammatory hyperalgesia. Br J Pharmacol 107:660^664
Ferreira SH,Lorenzetti BB,BristowAF, Poole S 1988 Interleukin-1 beta as a potent hyperalgesicagent antagonized by a tripeptide analogue. Nature 334:698^700
Millward-Sadler SJ,Mackenzie A,WrightMO et al 2003 Tachykinin expression in cartilage andfunction in human articular chondrocyte mechanotransduction. Arthritis Rheum 48:146^156
Pap G, Eberhardt R, Sturmer I et al 1998 Development of osteoarthritis in the knee joints ofWistar rats after strenuous running exercise in a running wheel by intracranial self-stimulation. Pathol Res Pract 194:41^47
Simkin PA 1977 The pathogenesis of podagra. Ann Intern Med 86:230^233Simkin PA, Campbell PM, Larson EB 1983 Gout in Heberden’s nodes. Arthritis Rheum 26:94^97
ANIMAL MODEL OF OSTEOARTHRITIS 153

Phantoms in rheumatology
C. S. McCabe, R. C. Haigh*, N. G. Shenker, J. Lewis and D. R. Blake1
The RoyalNational Hospital for Rheumatic Diseases, Upper BoroughWalls in conjunction withThe Department of Medical Sciences and The Department of Pharmacy and Pharmacology,University of Bath, Bath BA1 1RL, and *Royal Devon & Exeter Hospital (Wonford),Exeter EX2 5DW, UK
Abstract. This paper examines rheumatology pain and how it may relate to amputeephantom limb pain (PLP), speci¢cally as experienced in rheumatoid arthritis,¢bromyalgia and complex regional pain syndrome (CRPS). Clinical ¢ndings, whichsuggest cortical sensory reorganization, are discussed and illustrated for each condition.It is proposed that this sensory reorganization generates pain and altered body image inrheumatology patients in the same manner as has previously been hypothesized foramputees with PLP; that is via a motor/sensory con£ict. The correction of this con£ictthrough the provision of appropriate visual sensory input, using a mirror, is tested in apopulation of patients with CRPS. Its analgesic e⁄cacy is assessed in those with acute,intermediate and chronic disease. Finally, the hypothesis is taken to its naturalconclusion whereby motor/sensory con£ict is arti¢cially generated in healthy volunteersand chronic pain patients to establish whether sensory disturbances can be created whereno pain symptoms exists and exacerbated when it is already present. The ¢ndings of ourstudies support the hypothesis that a mismatch between motor output and sensory inputcreates sensory disturbances, including pain, in rheumatology patients and healthyvolunteers. We propose the term ‘ominory’ to describe the central monitoringmechanism and the resultant sensory disturbances as a dissensory state.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 154^178
Pain is the predominant complaint of patients with a rheumatological condition. Itmay be intermittent or continuous and vary in nature depending on the cause andcourse of the disease. In the majority of cases clinical ¢ndings provide supportingevidence for the source of this pain such as swollen joints in rheumatoid arthritis(RA) or bony overgrowth in osteoarthritis (OA). However, there are someconditions in rheumatology where a patient’s pain cannot be matched to physical¢ndings or relieved by traditional therapeuticmeasures. It is pain of this nature thatthis paper addresses, speci¢cally the types of pain experienced in rheumatoid
154
1This paper was presented at the symposium byD. R. Blake to whom correspondence should beaddressed.
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

arthritis (RA), ¢bromyalgia and complex regional pain syndrome (CRPS), andhow these may relate to amputee phantom limb pain.
Amputee phantom limb pain
Phantom limb pain (PLP) is a phenomenon that occurs in approximately 70% ofpatients after amputation (Jensen et al 1985). For the amputee ‘these painmemoriesare vivid, perceptually integrated experiences which incorporate both emotionaland sensory aspects of the pre-amputation pain’ (Hill et al 1996). Tingling is themost common complaint but pins and needles, shooting, burning or crushing painhave all been reported (Melzack 1971). Phantom sensations are also described by90^100% of amputees (Melzack 1990). Post-operatively an amputee will perceive aphantom limb that has all the same sensations andmobility of the real limb prior toamputation and is so strikingly real to the individual that it feels an integral part ofthem. The phantom appears to ‘inhabit the body’ (Melzack 1990) when the eyes areopen andmoves appropriatelywith other limbs. It initially feels perfectly normal insize and shape but may alter over time so that the phantom gradually becomes lessapparent and may eventually fade away (Katz &Melzack 1990). For those peoplewho wear a prosthesis the phantom limb can appear to ¢ll it or telescope upinto the remaining stump (Melzack 1990). It has been proposed that it is acombination of the duration and intensity of such pre-operative pain thatdetermines whether long-term central nervous system processes are altered withresulting persistent phantom sensations (Katz & Melzack 1990). A long-lastingmild sensation such as a watch on a wrist or a sock on a foot may be just ase¡ective at developing somatosensory memories as the intense short-term pain ofgangrene.Referred sensations (RS) have also been described in amputees. These are
somatosensory feelings that are perceived to emanate from a body part other thanbut in association with the body part being stimulated. They have not only beenreported following limb amputation (Ramachandran et al 1992), but alsosomatosensory dea¡erentation (Clarke et al 1996), local anaesthesia (Gandevia &Phegan 1999), stroke (Turton & Butler 2001) and spinal cord injury (Moore et al2000). Collectively these studies have shown that the referred sites (the body partnot physically touched) are non-random and often closely correspond to thecortical topographical map representing the body structure ¢rst described byPen¢eld and Rasmussen (1950). In the case of an amputated upper limb, patientsreport sensation in their phantom when parts of the face are lightly stroked(Ramachandran et al 1992). This is thought to be because the hand is positionedadjacent to the face on Pen¢eld’s map. These aberrant somatosensory, but reliablesensations were interpreted as resulting from central sensory reorganization
PHANTOMS 155

following disconnection or dysfunction of sensory pathways (Ramachandran et al1992).In conclusion, amputees report a variety of sensations that are not supported by
conventional notions of clinical pathology. The nature of the sensations described,provide the ¢rst link to the rheumatology patient with unexplained pain.
Pain in rheumatology
Rheumatoid arthritis
Rheumatoid Arthritis (RA) a¡ects one per cent of the population and is a chronicdisabling disease which occurs two thirds more frequently in women than men(Walker 1995). The peak age of onset is between 40 and 50 years, its aetiology isuncertain and there is, as yet no cure. The main symptoms of this disease arepain, sti¡ness, fatigue and joint swelling but other organs in the body mayalso be involved (Gordon & Hastings 1995). The pain that these patientsexperience is ‘chronic, unpredictable and frequently severe’ (Parker et al1989) and combined with joint destruction results in progressive disability overtime.A key feature of RA is a pattern of remissions and £ares that are a result of the
£uctuations in disease activity. During these £ares the joints, particularly the smalljoints of the hands and feet, become swollen and tender. This swelling is due toincreased activity in the joint caused by an inappropriate in£ammatory response.As a result of prolonged or frequent episodes of this in£ammation the synoviumlining the joint, becomes permanently thickened and bony erosions may occur(Gordon &Hastings 1995).Pain is synonymouswith the disease of rheumatoid arthritis and the types of pain
that su¡erers of this disease experience are complex and varied. The descriptionsthat they use may alter depending on the time of day, the duration of their disease,the joints that are involved and whether those joints are moving or at rest(Papageorgiou & Badley 1989).A less well-reported quality of pain that someRApatients describe is where they
feel their joints to be excessivelymore swollen than they look. They describe all thesensations associated with swollen joints but they are clinically not swollen andindeed when the subject looks at the a¡ected joints they too are aware that theyare not swollen (Blake et al 2000). Interestingly this perception of swelling isnot isolated to the joints, the patient will report that they feel their whole digitto be a¡ected (Fig. 1). These sensations are similar to the e¡ects you may haveafter an injection in your mouth at the dentist. The anaesthetic leaves you feelingthat your lip is huge and yet you look in the mirror and ¢nd that it is actually itsnormal size.
156 McCABE ET AL

The characteristics of this ‘phantom swelling’ and how it di¡ers from routinereports of RA joint swelling, were identi¢ed in a cross-sectional study involving10 patients with RA (McCabe 1999). Five of the subjects reported ‘phantomswelling’ and ¢ve did not. The two groups did not di¡er signi¢cantly in age,disease duration or disease activity, as measured by in£ammatory markers andjoint activity. Using a modi¢ed McGill Pain Questionnaire (MPQ), each subjectwas asked to describe the sensations they currently experienced in all their joints atrest and on movement. A semi-structured interview was used to collect additionalinformation on duration and severity of disease in each joint and the impact ofvision on the sensations that they reported.The subjects with ‘phantom swelling’ reported that their a¡ected joints felt
excessively hot (‘burning’, ‘scalding’) and hugely swollen (‘massive’). Theirremaining RA-a¡ected joints were described in exactly the same manner as thecontrol group described theirs, ‘warm’ and ‘slightly pu¡y’. When the phantomswollen joints were viewed by the subjects the perception of swellingdisappeared but the lesser sensation of ‘slight pu⁄ness’ in their other jointsremained on visualization. Phantom swelling was only present in those jointsthat had been most severely a¡ected by RA and for the longest duration which
PHANTOMS 157
FIG. 1. Rheumatoid arthritis patient’s drawing of ‘phantom swelling’ a¡ecting their hands.The shaded areas depict perceived swelling over the joints and the outer lines, perceivedswelling of the digits.

is very reminiscent of Katz and Melzacks’ theory that it takes a certain durationand intensity of pain to alter central processing resulting in persistentsensations.Interestingly the nature and cause of sti¡ness in RA, another pain related
symptom, is not well explained, even though it is a well established and de¢ningsymptom of the disease (Arnett 1988). Objective measures of sti¡ness do not relateto the subjective experience and indeed, compared with non-arthritic controls,objective sti¡ness can be reduced in RA joints (Helliwell et al 1988). We thereforehypothesised that the central nervous system is capable of generating a feedback-dependent state which can result in pathological sensations such as pain andsti¡ness in RA, that are to some extent independent of the initial peripheralpathology. We sought clinical evidence to support this proposal by investigatingthe clinical presentation of perceived sti¡ness in RA patients who had undergonelimb amputation but nevertheless retained an experience of a phantom limb (Haighet al 2003).Three patients with a current diagnosis of RA and lower limb amputation were
identi¢ed from the local Arti¢cial Limb Centre database and investigated todetermine the nature and pattern of pain and sti¡ness in their phantom and intactlimb. In addition to standard physical examination, pain and sti¡ness severity weremeasured using visual analogue scales (VAS) for both limbs. The duration andtiming of sti¡ness was also recorded for each limb. In all three cases, the patternof perceived RA sti¡ness was similar for the intact and phantom limb. All threepatients described sti¡ness in their phantom limb which mirrored that of physicalRA joint symptoms in terms of quality, frequency, diurnal variation, location,distribution and response to medication (non-steroidal anti-in£ammatory drugs,corticosteroid, opiate and disease-modifying drugs). Unilateral exercise (orattempted exercise) relieved sti¡ness only in the limb being exercised. Thus, theextent to which the subjective experience of perceived sti¡ness could bedissociated from the assumed original peripheral source was strikingly illustratedin RA patients with phantom limbs.Accordingly, we proposed that the experience of peripherally located sti¡ness
results from impairment to central brain processes. Conditions are present in RAto produce inaccurate sensory informationwhichmay lead to con£ict with plannedoutput from motor systems. These include peripheral and central proprioceptiveabnormality, cortical reorganization, neurogenic in£ammation and circulatingcytokines with central e¡ects. Such con£ict of information is ultimately perceivedas ‘sti¡ness’ by the patient with RA.RA is not the only rheumatological condition where phantom swelling and
sti¡ness are described. Clinical experience has long shown that patients with¢bromylagia also report sti¡ness and perceive body areas to be subjectivelyswollen when objectively they are not.
158 McCABE ET AL

Fibromylagia
Fibromyalgia (FMS) is a chronic pain condition where su¡erers report widespreadpain, fatigue and psychological distress all ofwhich have amajor impact upon theirdaily lives (Wolfe et al 1990). Although hyperalgesia and allodynia are commonlyreported at speci¢c trigger points these sensations often spread far beyond theseareas with su¡erers describing generalized sensitivity (Staud et al 2001). For themajority of patients there is no known initiating event or observable physicalpathology and symptoms are frequently resistant to therapeutic initiatives.In addition to the symptoms described above it has long been observed, but only
recently systematically recorded, that these patients also experience phantomswelling sensations in the same manner as those with RA (C. McCabe, D. Blake,unpublished work). The sensation most commonly a¡ects the hands, bilaterallyfrom the wrist to the ulna styloid, or the feet, bilaterally from the toes to theankle joints. The subject is most aware of the perceived swelling when they havetheir eyes closed and it decreases or disappears completely when they view thea¡ected area. With regular viewing on a daily basis the sensation can bediminished permanently. When phantom swelling is reported it is commonlyassociated with the patient feeling that they are clumsy or less aware of wheretheir limbs are in space. This reduction in limb position sense will be discussedfurther towards the end of this paper. Phantom swelling in FMS is a clearexample of a sensation being reported without supporting underlying clinicalpathology and CRPS is another such condition where the cause of thecharacteristic symptomology is ambiguous.
Complex regional pain syndrome
Complex regional pain syndrome (CRPS) is a painful, debilitating condition. Thisdiagnostic term embraces several syndromes including re£ex sympatheticdystrophy, causalgia, and algodystrophy. The pain that a patient with CRPS willreport shares many similar characteristics to amputee phantom limb pain:mislocalized, intense and burning. Clinical features include sensory disturbancessuch as burning pain with allodynia and hyperanalgesia, motor disturbances suchas weakness, tremor andmuscle spasms, and changes in vascular tone, temperatureand oedema (Scadding 1999). Over time functional loss and trophic changes mayoccur. The syndrome can occur spontaneously or following trauma (CRPSType 1)or in association with peripheral nerve damage (CRPS Type 2).A characteristic feature of CRPS is that signs and symptoms spread beyond the
site of initial insult. Severe pain may occur seemingly out of proportion to theoriginal pathology. It may persist over long periods and is frequently resistant toa wide range of treatments. Theories abound on the cause of this pain and its
PHANTOMS 159

underlying pathology. Traditionally, interrupting the sympathetic supply to thepainful area was thought to treat such pain. However, the e¡ectiveness of thisapproach is not supported by randomised controlled trials (Jadad et al 1995).Neural plasticity occurs in a variety of pain syndromes (Harris 1999, Lenz&Byl
1999). We predicted that referred sensations would be present in patients withCRPS type 1 as evidence of sensory cortical reorganization. The resultant sensorymislocalizations could then provide the inappropriate sensory feedback required tocreate painful sensations (McCabe et al 2003a). Furthermore, we hypothesized thatthese referred sensations would be perceived to emanate from the body structuresimmediately adjacent to the stimulated site and in keepingwith their topographicallocation on the Pen¢eld homunculus as in phantom and allied pain states. Wespeci¢cally selected those patients with CRPS Type 1 as we wished to discoverwhether central reorganization occurs even where there is no evidence of localperipheral nerve damage.Over two years, 16 subjects (13 female, 3 male) who met the entry criteria were
recruited. Five showed evidence of referred sensations (Table 1). There was nodi¡erence in age, disease duration, levels of pain, or severity of disease (Table 2)between those who presented with RS and those who did not. All ¢ve patientsreported referred sensations during examination with their eyes closed (Fig. 2).They were experienced in real time and disappeared when stimulation ceased orvision was permitted. When the subjects viewed the area being touched thesensations were either diminished (Case 5) or not present and when thesymptoms of CRPS resolved (Cases 1, 2 and 4), referred sensations were lost.Sensations were referred in a modality-speci¢c manner with touch referred in allcases and pinprick also referred in two (Cases 1 and 2). Vibration was neverreferred. All referred sites were located on body parts immediately adjacent, onPen¢eld’s homunculus, to the stimulated site.The location of the referred sites, in our study population, was consistent with
previous reports in other pain conditions (Ramachandran et al 1992, Flor et al1997) and ¢t particularly well with predicted cortical changes that have beenshown to occur within the somatosensory body map in amputees (Halligan et al1993). Ramachandran (Ramachandran & Hirstein 1998) proposed that due to thelocation and speedwith which referred sensations occur in amputees, such ‘ectopicrepresentations’ following functional remapping were probably due to theunmasking of latent synapses within the cortex, as previously described inprimates (DeFelipe et al 1986, Jones 1990). These synapses are suppressed whenthere is simultaneous input from two connected receptors but with reduced orimpaired sensory activation in one area, the connection becomes disinhibited.Recent imaging studies, using magnetoencephalography, in six patients withupper limb CRPS Type 1 have also shown changes in the cortical somatosensorymap though it was not reported whether these were associated with referred
160 McCABE ET AL

sensations (Juottonen et al 2002). There was a signi¢cantly shorter distancebetween the areas representing the thumb and little ¢nger on the somatosensorycortex contralateral to the a¡ected limb than the ipsilateral side. Interestingly, therewas no signi¢cant correlation between the distance of thumb and ¢nger and thelevel or duration of pain. Hand dominance was also not an in£uencing factor.Alternatively, referral of sensationsmay occur at the spinal level. A large body of
evidence shows that sensitization of wide dynamic range neurons at level V of thedorsal horn results in ipsilateral and contralateral enlarged receptive ¢eldswhich donot rely on a cortical homunculus (Ji & Woolf 2001). In addition, experimentalmodels of peripheral neuropathic pain demonstrate bilateral spinal cord changesafter unilateral nerve damage (Koltzenburg et al 1999). However, all of ourpatients had CRPS 1, so therefore had no precipitating neural trauma. Theirsensations were not referred bilaterally, either from the stimulated site to its
PHANTOMS 161
TABLE 1 Location, direction and type of sensations referred in subjects with adiagnosis of CRPS Type 1 (Cases 1^5). Loss of detection of referred sensations isshown in relation to current disease duration and future status (resolved or chronic)
PatientPainsite
Diseaseduration
Areatouched (1)Referralsite (2,3)
Directionofreferral
Typeofsensation
Loss ofreferredsensation
Resolutionof CRPS(wks)
Case 128 years F(Fig. 1)
Lefthand
3 weeks L 3rd¢ngertip (1)L lowerjaw (2)
1^2 Light touchand pinprick
3 weeks 6
Case 234 years F(Fig. 2)
Leftankle
8 weeks L forefoot(1)L patella (2)
1^2 and2^1
Light touchand pinprick
3 weeks 4
Case 324 years M(Fig. 3)
Leftknee
3 years L patella (1)L forefoot(2)
1^2 and2^1
Light touch No Chronic
Case 441 years F(Fig. 4)
Rightfoot
6 years R forefoot(1)R patella (2)
2^1 Light touch 4 weeks Chronic
Case 557 years F(Fig. 5)
(Fig. 6)
Lefthand
4 years L shoulder(1)L ear (2)L hand (3)L cheek (1)L hand (2)
1^21^3
1^2
Pulling,light touchand handmovement
Light touch
Nochange
Chronic

contralateral partner (i.e. left hand to right hand) or mirrored on the contralateralside (i.e. from stimulated site to referral site on the una¡ected limb). In addition, thespeed of referral (both in terms of disease duration, response time on stimulationand resolution as the condition improved), combined with the magnitude of thesensations all detract from a purely spinal route. Contemporary theories suggestthat CRPS is a disorder involving both CNS and peripheral nervous systemcomponents (Baron et al 2002, Janig & Baron 2002). This is based on the
162 McCABE ET AL
TABLE 2 Demographics of total study population to compare di¡erences in age,disease duration and levels of pain between subjects who experienced referredsensations and those who did not
Case Age GenderDiseaseduration
A¡ectedlimb
Pain level onmovement atpresentation
1** 28 years F 3 weeks Left hand 8
2** 34 years F 8 weeks Left ankle 8
3** 24 years M 3 years Left knee 8
4** 41 years F 6 years Right foot 9
5** 57 years F 4 years Left hand 5
Mean** 36.8 years 4F:1M 2.6 years 7.6
6 38 years F 6 weeks Left ankle 9
7 35 years F 5 months Right arm 5
8 40 years F 1 year Right arm 6
9 38 years F 3 years Left leg 5
10 27 years M 2 years Left leg 8
11 51 years F 2 years Right arm 7.5
12 68 years F 1 year Left arm 5
13 54 years M 4 years Left foot 9
14 38 years F 7 years Left foot 10
15 22 years F 4 years Left foot 9
16 59 years F 10 years Left foot 9.5
Mean 42.7 years 9F:2M 3.1 years 8
FIG. 2. Artist’s impression of Cases 1^5 illustrating location of stimulus and direction ofreferred sensations (area touched¼1, referred site/s¼2, 3). (a) to (d) correspond to Cases 1 to4, (e) & (f) correspond to Case 5. Shaded area (1) depicts area stimulated by examiner, shadedareas (2) & (3) depict where referred sensations were felt. The arrows illustrate direction ofreferral. Reprinted with permission fromMcCabe et al (2003b).

PHANTOMS 163

evidence that some patients respond positively to sympathetic blockade, therebyimplicating involvement of the sympathetic nervous system but conversely,sympathetically maintained pain involves the deep somatic tissue (asdemonstrated by our patients’ report of increased pain on movement) which isthe domain of the autonomic system. Therefore isolating one clear route forreferred sensations is at present problematic.
The power of vision and mirror visual feedback
Visual feedback strongly in£uences the experience of referred sensations in patientswith CRPS. Recent studies have shown this also to be the case in amputees wherestimulation of the intact limb evoked sensory changes in the phantom only whenthe subjects’ eyeswere closed (Hunter et al 2003). Touch and vision are inextricablylinked. Touch is known to in£uence vision such as dispelling the visual illusion of athree-dimensional object when it is drawn on a £at surface. Equally, in someclinical conditions such as somatosensory loss after stroke, visual feedback of thea¡ected limb during testing can signi¢cantly improve reported perception(Halligan et al 1997). In addition, recent ¢ndings by Taylor-Clarke et al (2002)showed that the enhancing e¡ect of vision modulated somatosensory corticalprocessing. Gregory (1998) points out that vision evolved from the simplerprocesses for touch and that it is possible that the somatosensory map is inverted(the feet above the hand) in order to correspond with the inverted visual image onthe retina. This ensures that the link between vision and touch is as short aspossible. Consequently, when our subjects viewed their limbs being stimulated itwould appear that the more powerful sense of vision overruled the referredsensations.It has already been stated that vision is able to dismiss the sensation of phantom
swelling inRAandFMSbut in recent studies onPLPvision has been shown to alsoprovide an analgesic bene¢t. Ramachandran & Rogers-Ramachandran (1996)superimposed the image of amputees’ normal limbs, by means of a mirror, on thespace that their phantom limbs occupied. Viewing the mirror image of theirresidual limb, the amputees moved their normal limbs and attempted to movetheir abnormal side. Subjects reported that sensation in their abnormal limbreturned towards normal during the exercises and their pain diminished. Harris(1999) subsequently hypothesized that the reason for this analgesic e¡ect was thatPLP is generated by a discordance inmotor intention and predicted proprioceptivefeedback and that when this mismatch is corrected, through appropriate visualfeedback via the mirror, pain is relieved.Objective evidence of the cortical e¡ects of this mismatch was provided by Fink
and colleagues (Fink et al 1999) using PET imaging and healthy volunteers. Theydemonstrated that when congruent and incongruent movements were performed,
164 McCABE ET AL

whilst viewing only one limb in a mirror, cortical activity varied depending on themovement. When the limbs moved incongruently and yet were seen, by means ofmirror imaging, to move congruently, cortical activity was unilateral, unlikevisually observed congruent and actual congruent movement, where bilateralcortical activity was produced. When unilateral cortical activity occurred it wasin the right dorsolateral pre-frontal cortex and it was this area that Fink andcolleagues concluded was speci¢cally involved in the monitoring of con£ictbetween motor intention and its sensory/perceptual consequences.The existence of referred sensations in CRPS and evidence of changes in cortical
representation (Juottonen et al 2002) suggest that pain in CRPSmay also be drivenby a mismatch between motor output and sensory input as Harris proposed forPLP. We hypothesized that if this were the case then the provision of appropriatesensory input should correct the mismatch and reduce pain. ModifyingRamachandran’s methodology for the relief of PLP, we too used a mirror toprovide congruent visual feedback, from the moving una¡ected limb, to restorethe integrity of cortical processing aiming to relieve pain and restore function inthe a¡ected limb (McCabe et al 2003a).Eight subjects were recruited aged 24^40 years (mean 33 years) with disease
duration of 3 weeks to 3 years (three subjects early disease 48 weeks, twointermediate, 5 months and 1 year and the remaining three long standing diseaseof52 years). All presented with a single limb a¡ected by allodynia, hyperalgesia,reduced movement with related pain and sti¡ness, and vasomotor disturbances(Table 3).All subjects reported no relief of pain on movement when both limbs were
visualized without a device or when a non-re£ective surface was viewed (Fig. 3).Indeed, movement exacerbated pain. All three subjects with early CRPS(48 weeks) reported a striking reduction in their VAS for pain, during and aftervisual feedback of their re£ected moving, una¡ected limb as provided by themirror (Fig. 4). A marked analgesic e¡ect was observed within a few minutes ofmirror usage, followed by an abrupt return of pain when the mirror was removedinitially. With repeated usage (4^9�daily, week 1), the period of analgesiaprogressively extended from a few minutes to hours, requiring less mirror useover the six-week study period. At six weeks there was a reversal of vasomotorchanges as measured by infrared thermal imaging, a return to normal functionand no pain at rest or on movement. All three subjects felt they no longerrequired analgesic relief from the mirror and had stopped prior to assessment atsix weeks (Case 3, week 4, Cases 1 and 2, week 6).The two subjects with intermediate disease duration, 5months and 1 year (Cases
4& 5), reported that themirror immediately eased theirmovement related sti¡nessbut therewas no analgesic e¡ect in Case 5. They both reported that this reduction insti¡ness facilitated movement and the e¡ect lasted for increasing periods after
PHANTOMS 165

166 McCABE ET ALTABLE3
Patientcharacteristicsandthee¡ectofthecontrolandinterventionphasesontheirpain
atpresentation;thefrequencyof
mirroruse
onfollow-upand¢nalpain
scoresat6weekswithinfra-redtherm
aldi¡erencesanduna¡ectedlimbs
Atpresentation
Followup
*Mean
temperature
Control
Phase1
Lookingat
bothlim
bs(N
odevice)
Control
Phase2
Painful
limb
hidden
Intervention
Mirror
visual
feedback
Frequentm
irrorusage�perday
(Durationofeachtreatment10min)
At6
weeks
Subject
(painful
limb)
Symptom
duration
di¡erence(8C
)[non-painful�
painfullimb]
Pain
VASat
rest
Pain
VASon
movement
Pain
VASon
movement
Pain
VASon
movement
Week
12
34
56
Pain
VAS
Mean
temperature
di¡erence(8C
)
Treatment
duration
(weeks)
Case1
(leftleg)
38yearsF
6weeks
1.1
99
92
83
33
20
00.2
6
Case2
(leftarm
)28
yearsF
3weeks
2.0
78
83
44
33
20
00.4
6
Case3
(leftleg)
34yearsF
8weeks
2.7
68
82
94
30
00
00.8
4
Case4
(right
arm
35yearsF
5mon
ths
1.9
05**
5**
3**
54
54
54
2**
0.3
6
Case5
(right
arm)
40yearsF
1year
0.5
46
66
54
54
43
10.4
6
Case6
(leftleg)
24yearsM
2years
1.4
78
88
55
50
00
81.3
Unresolved
Case7
(leftleg)
38yearsF
3years
Not
performed***
45
55
44
00
00
5Not
performed
Unresolved
Case8
(leftleg)
27yearsM
2years
2.1
78
88
44
00
00
82.6
Unresolved
LA,leftarm
;LL,leftleg;R
A,right
arm;F
,fem
ale;M,m
ale;n.d.,n
otdo
ne.*Regionof
interestconstant
Sign
i¢cant
di¡erenceif40.48C
13.**Sti¡ness.***Case7had
widespreadulceration
onherleftlegwhich
madethermalim
ageinterpretation
impo
ssible.
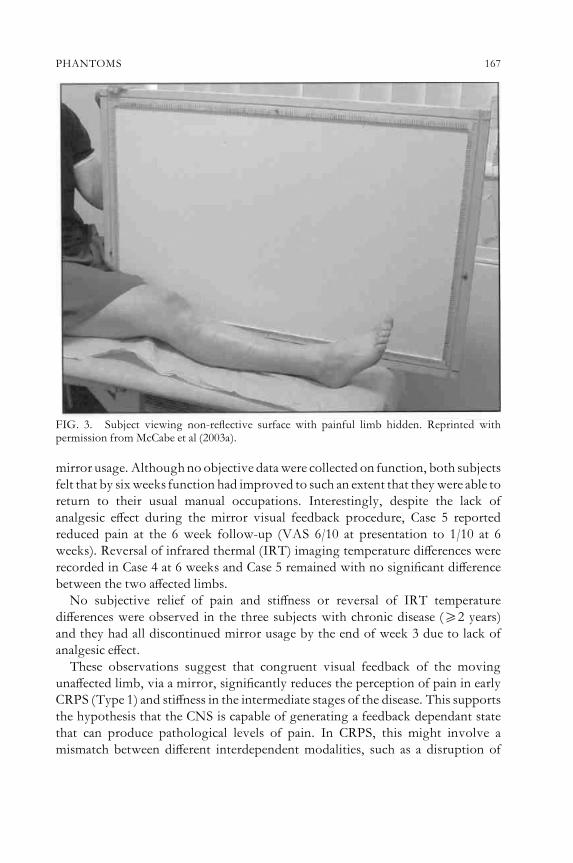
mirror usage.Althoughnoobjective datawere collected on function, both subjectsfelt that by sixweeks function had improved to such an extent that theywere able toreturn to their usual manual occupations. Interestingly, despite the lack ofanalgesic e¡ect during the mirror visual feedback procedure, Case 5 reportedreduced pain at the 6 week follow-up (VAS 6/10 at presentation to 1/10 at 6weeks). Reversal of infrared thermal (IRT) imaging temperature di¡erences wererecorded in Case 4 at 6 weeks and Case 5 remained with no signi¢cant di¡erencebetween the two a¡ected limbs.No subjective relief of pain and sti¡ness or reversal of IRT temperature
di¡erences were observed in the three subjects with chronic disease (52 years)and they had all discontinued mirror usage by the end of week 3 due to lack ofanalgesic e¡ect.These observations suggest that congruent visual feedback of the moving
una¡ected limb, via a mirror, signi¢cantly reduces the perception of pain in earlyCRPS (Type 1) and sti¡ness in the intermediate stages of the disease. This supportsthe hypothesis that the CNS is capable of generating a feedback dependant statethat can produce pathological levels of pain. In CRPS, this might involve amismatch between di¡erent interdependent modalities, such as a disruption of
PHANTOMS 167
FIG. 3. Subject viewing non-re£ective surface with painful limb hidden. Reprinted withpermission fromMcCabe et al (2003a).

normal interaction between motor intention and sensory feedback. In those withan inherent vulnerability to this incongruence it can lead, in some, to referred,intractable pain following trauma or, in others, promote CRPS with a centralnervous system origin. This might explain why some types of CRPS occurwithout discrete peripheral injury.If the correction of a sensory/motor mismatch produces an analgesic response
then the reverse should also be true. That is to say when expected sensory input isdeliberately falsi¢ed, sensory abnormalities should be generated in healthyvolunteers and exacerbated in patients with chronic pain of unknown aetiology.
Generating pain
In a recent studywe invited healthy volunteers and patientswith FMS andCRPS tomove their upper and lower limbs whilst undergoing normal and altered visualsensory feedback as provided via a mirror (McCabe et al 2003c,d). Motor/sensorycon£ict was at its optimum when the subjects moved their limbs in opposingdirections whilst viewing, via the mirror, their limbs apparently movingtogether. The primary aim of this study was to comprehensively capture, using a
168 McCABE ET AL
FIG. 4. Subject viewing non-painful limb in mirror with painful limb hidden. Reprinted withpermission fromMcCabe et al (2003a).

qualitative methodology, the range of sensory experiences that subjects describedas they underwent thesemanoeuvres. Each assessmentwas conducted ¢rst with thesubjects viewing the control side (a whiteboard) and moving their limbscongruently and incongruently and then repeating the movements whilstviewing the intervention side (a mirror).41 healthy volunteers were recruited (9 males, 32 females) aged 23^65 years
(mean 40.4 years). They reported sensory changes at all stages of the protocol,control (congruent movement n¼6 [15%], incongruent movement n¼4 [10%])and intervention. However, the maximum number of reports occurred when thesubjects moved their limbs incongruently but perceived, via mirror imaging, thatthey were moving them congruently (congruent movement n¼10 [25%],incongruent movement n¼23 [56%]). The healthy volunteers reporteddiscomfort (‘pins and needles’, ‘shooting pain’), changes in temperature and/orweight (‘£oaty sensation’ or ‘my arm was so heavy I was unable to lift it’),perceived loss of or additional limbs and disorientation (‘dizzy’, ‘strange’)(Table 4). Altered sensations were described predominantly in the hidden limbthough this sometimes automatically conferred sensations on to the visualizedlimb, such as a hidden limb felt heavier and therefore the visualised limb wasperceived as lighter. All altered sensations faded rapidly after limb movementhad ceased and the hidden limb was visualized by the subject.Data collection in the patient population is still ongoing with 24 patients (7
CRPS Type 1, 17 FMS) recruited to date (3 males, 21 females) aged 23^73 (mean
PHANTOMS 169
TABLE 4 Type and incidence of sensory changes reported by healthy volunteers inhidden limb during congruent and incongruent movement whilst viewing awhiteboard (control) and mirror (intervention)
Type of sensation
Whiteboardcongruentmovement
Whiteboardincongruentmovement
Mirrorcongruentmovement
Mirrorincongruentmovement
Discomfort/pain 1 (2%) 1 (2%) 4 (10%) 7 (17%)
Temperature change 0 0 0 2 (5%)
Weight change 2 (5%) 0 3 (7%) 6 (15%)
Perceived ‘‘loss’’ of limb 4 (10%) 2 (4.9%) 8 (20%) 11 (27%)
Perceived ‘‘extra’’ limb 0 0 1 (2%) 9 (22%)
Disorientation 3 (7%) 4 (10%) 15 (37%) 13 (32%)
Total number of subjectsexperiencing anysensory disturbances
6 (15%) 4 (10%) 10 (24%) 23 (56%)
n¼41 (male¼8, female¼33).

47.5). Preliminary ¢ndings suggest patients perceive the same sensations as thehealthy controls but the intensity and frequency of these sensations is greater.For example discomfort is reported as ‘crampy’, ‘sharp’ and ‘extremely painful’,temperature changes as ‘very hot’, ‘burning’. Importantly these sensory changesare described in addition to the subjects’ current symptoms at all stages of theprotocol and for those with CRPS in their a¡ected and una¡ected limbs. Theother striking di¡erence between the two study populations is that the patientsreport far more sensory disturbances than the healthy volunteers during thecontrol stages (Table 5). It would appear that when sensory disturbances arealready present simply hiding a limb from view is su⁄cient to exacerbate existingsymptoms and generate new ones.
Summary
Our clinical observations and research studies support the conjecture put forwardby Harris (1999) that when motor intentions to move a limb or series of joints nolonger matches the corresponding sensory feedback then the subsequent‘misrouting of information’ activates a central monitoring mechanism that £agsup such incongruity as pain. However, we would now like to extend Harris’theory and propose that this monitoring mechanism is one of many monitoringmechanisms that act as alerts to warn the body that there is a problem withinformation processing and that pain may be only one of a broad range ofsensory disturbances that subsequently occur. These central mechanisms we have
170 McCABE ET AL
TABLE 5 Type and incidence of sensory changes reported by patients with CRPStype 1 and ¢bromyalgia in hidden limb during congruent and incongruentmovement whilst viewing a whiteboard (control) and mirror (intervention)
Type of sensation
Whiteboardcongruentmovement
Whiteboardincongruentmovement
Mirrorcongruentmovement
Mirrorincongruentmovement
Discomfort/pain 11 (45.8%) 13 (54.1%) 10 (41.6%) 11 (45.8%)
Temperature change 2 (8.3%) 3 (12.5%) 4 (16.6%) 5 (20.8%)
Weight change 7 (29.2%) 8 (33.3%) 5 (20.8%) 10 (41.7%)
Perceived ‘‘loss’’ of limb 5 (20.8%) 8 (33.3%) 13 (54.2%) 14 (58.3%)
Perceived ‘‘extra’’ limb 0 0 0 0
Disorientation 0 6 (25%) 7 (29.2%) 7 (29.2%)
Total number of subjectsexperiencing any sensorydisturbances
20 (83.3%) 15 (62.5%) 15 (62.5%) 16 (66.7%)
n¼24 (male¼3, female¼21, ¢bromyalgia¼17, CRPS¼7).

termed ‘ominory’ from the Latin word ominor meaning to prophesy, predict,foreboding. Our studies have focused on the mechanism that monitors motor/sensory con£ict but a separate ominory mechanism could generate motionsickness when there is discordance between body position, balance andequilibrium. These mechanisms may be triggered by externally induced con£ict(e.g. incongruent movement whilst viewing the mirror) or internally (e.g. diseasedamage in RA leading to inaccurate execution of movement and/or alteredproprioception). The key feature of these mechanisms is that when they aretriggered they generate sensory disturbances such as nausea with motionsickness, pain in a phantom limb, phantom swelling and sti¡ness in RA andFMS. These resultant states we have termed dissensory from the Latin worddissensio meaning con£ict, disagreement. These are feedback dependent states inthat the sensory/motor con£ict will continue to trigger the ominory mechanismand ultimately either via duration or intensity of this state the subject will su¡erpain. If however, an intervention is targeted to correct the initial source ofcon£ict, the ominory mechanism is suppressed and ideally pain is prevented oralleviated as with mirror visual feedback in early CRPS or the individualvisualizing their phantom swollen joints in RA and FMS.We propose that the threshold at which a person either triggers the ominory
mechanism or becomes aware of the subsequent sensory disturbances isindividually determined but there will be some who are more sensitive thanothers. This we assume will relate to the standard variables of genetic factors,age, gender and sex hormone state. This was demonstrated by our healthyvolunteer study; not all subjects experienced sensory disturbances. In addition,the preliminary patient data show that where sensory disturbances are alreadypresent a far lower stimulus is required to intensify the problem. Simply hiding alimb from view was su⁄cient to exacerbate sensory disturbances.This paper has only addressed three rheumatological conditions, RA, FMS and
CRPS, but the same ominory mechanism may apply to the pain of osteoarthritis(OA) and indeed the development of pathological changes in the joint. Sharma et al(1997) and Pai et al (1997) have both shown that patients with unilateral knee OAhave worse proprioception in their a¡ected and una¡ected joints than elderlycontrols without OA. The fact that both knees have reduced proprioceptioneven when only one is diseased supports the theory of ‘mirror imaging’ acrossthe body (Shenker et al 2004, this volume). The abnormal proprioception in thecontralateral knee will be su⁄cient to continually trigger the ominory mechanismand perpetuate the problem. The subsequent dissensory state may explain theclinical observation that some individuals report high levels of pain when onlyminimal changes suggestive of OA are seen on X-ray imaging. This continuoussensory imbalance in the contralateral knee may increase the risk of injury andultimately of generating OA (Hurley 1997). If targeted exercise is used to
PHANTOMS 171

improve proprioception the initial trigger is removed and the ominorymechanismsuppressed thereby perhaps preventing the onset of pain. Interestingly, patientswith OA often report in clinic that their pain is worse at night and this may be adirect result of reduced corrective sensory input exacerbating the dissensory state.A darkened room diminishes visual feedback and immobilised limbs reduceproprioceptive input.In conclusion, a mismatch between motor output and sensory input triggers a
warning, ominory mechanism in rheumatology patients and healthy volunteers.This generates the dissensory state and the individual will experience sensorydisturbances that may include pain.
Acknowledgements
C. S. McCabe is supported as an Arthritis Research Campaign Lecturer in RheumatologicalNursing. N. G. Shenker is supported as an Arthritis Research Campaign Clinical ResearchFellow. J. M. Lewis is supported on an Arthritis Research Campaign ICAC award. D. R. Blakeholds an endowed Chair� ‘The Glaxo Wellcome Chair in Locomotor Sciences’. An ArthritisResearch Campaign ICAC award supports the Royal National Hospital for Rheumatic Diseases,Bath.
References
Arnett FC, Edworthy SM, Bloch DA et al 1988 The American Rheumatism Association 1987revised criteria for the classi¢cation of rheumatoid arthritis. Arthritis Rheum 31:315^324
Baron R, Fields HL, JanigW, Kitt C, Levine JD 2002 National Institutes of HealthWorkshop:re£ex sympathetic dystrophy/complex regional pain syndromes� state of the science.Anaesth Analg 95:1812^1816
Blake DR, McCabe CS, Skevington SM, Haigh R 2000 Cortical origins of pathological pain.Lancet 355:318^319
Clarke S, Regli L, Janzer RC, Assal G, de Tribolet N 1996 Phantom face: conscious correlate ofneural reorganization after removal of primary sensory neurones. Neuroreport 7:2853^2857
DeFelipe J, Conley M, Jones EG 1986 Long-range focal collateralisation of axons arising fromcorticocortical cells in monkey sensory-motor cortex. J Neurosci 6:3749^3766
Flor H, Braun C, Elbert T, Birbaumer N 1997 Extensive reorganization of primarysomatosensory cortex in chronic back pain patients. Neurosci Lett 224:5^8
Fink GR, Marshall JC, Halligan PW et al 1999 The neural consequences of con£ict betweenintention and the senses. Brain 122:497^512
Gandevia SC, Phegan CML 1999 Perceptual distortions of the human body image produced bylocal anaesthesia, pain and cutaneous stimulation. J Physiol 514:609^616
Gordon DA, Hastings DE 1995 Clinical features of rheumatoid arthritis: early, progressive andlate disease. In: Klippel JH, Dieppe, PA (eds) Practical rheumatology. Times MirrorInternational Publishers, London, p 169^182
Gregory RL 1998 Eye and brain. The psychology of seeing. Oxford University Press, p 53Haigh RC, McCabe CS, Halligan P, Blake DR 2003 Joint sti¡ness in a phantom limb: evidenceof central nervous system involvement in rheumatoid arthritis. Rheumatology (Oxford)42:888^892
172 McCABE ET AL

Halligan PW, Marshall JC, Wade DT, Davey J, Morrison D 1993 Thumb in cheek? Sensoryreorganisation and perceptual plasticity after limb amputation. Neuroreport 4:233^236
Halligan PW, Marshall JC, Hunt M, Wade DT 1997 Somatosensory assessment: can seeingproduce feeling? J Neurol 244:199^203
Harris AJ 1999 Cortical origins of pathological pain. Lancet 354:1464^1466Helliwell PS, Howe A, Wright V 1988 Lack of objective sti¡ness in rheumatoid arthritis. AnnRheum Dis 47:754^758
Hill A, Niven CA, Knussen C 1996 Pain memories in phantom limbs: a case study. Pain 66:381^384
Hunter JP, Katz J, Davis KD 2003 The e¡ect of tactile and visual sensory inputs on phantomlimb awareness. Brain 126:579^589
Hurley MV 1997 The e¡ects of joint damage on muscle function, proprioception andrehabilitation. Man Ther 2:11^17
Jadad AR, Carroll D, Glynn CJ, McQuay HJ 1995 Intravenous regional sympathetic blockadefor pain relief in re£ex sympathetic dystrophy: a systematic review and a randomized, double-blind crossover study. J Pain SymptomManage 10:13^20
Janig W, Baron R 2002 Complex regional pain syndrome is a disease of the central nervoussystem. Clin Auton Res 12:150^164
Jensen TS, Krebs B, Nielsen J Rasmussen P 1985 Immediate and long-term phantom limb painin amputees: incidence, clinical characteristics and relationships to pre-amputation limb pain.Pain 21:267^278
Ji RR, Woolf CJ 2001 Neuronal plasticity and signal transduction in nociceptiveneurons: implications for the initiation and maintenance of pathological pain. NeurobiolDis 8:1^10
Jones EG 1990The role of a¡erent activity in themaintenance of primate neocortical function. JExp Biol 153:155^176
Juottonen K, Gockel M, Silen T, Hurrir H, Hari R, Forss N 2002 Altered central sensorimotorprocessing in patients with complex regional pain syndrome. Pain 98:315^323
Katz J,Melzack R 1990 Painmemories in phantom limbs: review and clinical observations. Pain43:319^336
Koltzenburg M, Wall PD, McMahon SB 1999 Does the right side know what the left is doing?Trends Neurosci 22:122^127
Lenz F, Byl NN 1999 Reorganisation in the cutaneous core of the human thalamic principalsomatic sensory nucleus (Ventral caudal) in patients with dystonia. J Neurophysiol82:3204^3212
McCabe CS 1999 An exploratory study into the experience of pain in rheumatoid arthritis. MScthesis, University of Bath, Bath, UK
McCabe CS, Haigh RC, Ring EFR, Halligan PW, Wall PD, Blake DR 2003a A controlled pilotstudy of the utility of mirror visual feedback in the treatment of Complex Regional PainSyndrome (Type 1). Rheumatology (Oxford) 42:97^101
McCabe CS, Haigh RC, Halligan PW, Blake DR 2003b Referred sensations in patients withcomplex regional pain syndrome type 1. Rheumatology (Oxford) 42:1067^1073
McCabe CS, Haigh RC, Halligan PD, Blake DR 2003c Generating sensory disturbance inhealthy controls. Rheumatology (Oxford) 42:63
McCabe CS, Haigh RC, Halligan PD, Blake DR 2003d Distorting proprioception in chronicpain patients exacerbates sensory disturbances� implications for pathology. Rheumatology(Oxford) 242:145
Melzack R 1971 Phantom limb pain. Anesthesiology 35:409^419Melzack R 1990 Phantom limbs and the concept of a neuromatrix. Trends Neurosci 13:88^92Merskey H, Bogduk N 1994 Classi¢cation of chronic pain. IASP Press, Seattle
PHANTOMS 173

Moore CI, Stern CE, Dunbar C, Kostyk SK, Gehi A, Corkin S 2000 Referred phantomsensations and cortical reorganisation after spinal cord injury in humans. Proc Natl Acad SciUSA 97:14703^14708
Pai YC, Rymer WZ, Chang RW, Sharma L 1997 E¡ect of age and osteoarthritis on kneeproprioception. Arthritis Rheum 40:2260^2265
Papageorgiou AC, Badley EM 1989 The quality of pain in arthritis: the words patients use todescribe overall pain and pain in individual joints at rest and on movement. J Rheumatol16:106^112
Parker JC, Smarr KL, Buescher KL et al 1989 Pain control and rational thinking. Implicationsfor rheumatoid arthritis. Arthritis Rheum 32:984^990
Pen¢eld W, Rasmussen TL 1950 The cerebral cortex of man: a clinical study of localization offunction. Macmillan, New York
Ramachandran VS, Rogers-RamachandranD 1996 Synaesthesia in phantom limbs induced withmirrors. Proc R Soc Lond B Biol Sci 263:377^386
Ramachandran VS, HirsteinW 1998 The perception of phantom limbs. TheD.O.Hebb lecture.Brain 121:1603^1630
Ramachandran VS, Stewart M, Rogers-Ramachandran D 1992 Perceptual correlates of massivecortical reorganisation. Neuroreport 3:583^586
Scadding JW1999 Complex regional pain syndrome. In:Wall PD,Melzack R (eds) Textbook ofpain. 4th edn. Churchill Livingston, Edinburgh, p 835^850
Sharma L, Pai YC, Holkamp K, Rymer WZ 1997 Is knee joint proprioception worse in thearthritic knee versus the una¡ected knee in unilateral knee osteoarthritis? Arthritis Rheum40:1518^1525
Shenker NG, Blake DR, McCabe C, Haigh R, Mapp P 2003 Symmetry, T cells and neurogenicarthritis. In: Osteoarthritic joint pain. Wiley, Chichester (Novartis Found Symp 260)p 241^257
Staud R, Vierck CJ, Cannon RL, Mauderli AP, Price DD 2001 Abnormal sensitization andtemporal summation of second pain (wind-up) in patients with ¢bromyalgia syndrome.Pain 91:165^175
Taylor-Clarke M, Kennett S, Haggard P 2002 Vision modulates somatosensory corticalprocessing. Curr Biol 12:233^236
Turton AJ, Butler SR 2001 Referred sensations following stroke. Neurocase 7:397^405Walker DJ 1995Rheumatoid arthritis. In: Butler RC, JaysonMIV (eds) Collected reports on therheumatic diseases. Arthritis and Rheumatism Council, p 39^44
Wolfe F, Smythe HA, Yunus MB et al 1990 The American College of Rheumatology 1990criteria for the classi¢cation of ¢bromyalgia. Report of the Multicenter Criteria Committee.Arthritis Rheum 33:160^172
DISCUSSION
Pisetsky:What is the currentworking knowledge of referred pain in general? Is itcommon in OA?Blake: I’m not sure I’m the best person to answer that question. Clearly you can
get referral at various levels: spinal, midbrain, brainstem or cortical. It is not just asensation.Pisetsky:Does it re£ect sensitization, or is it neural spread?Why do some people
have it?
174 DISCUSSION

Blake: Over and above some of the explanations that have been given whereyou can have two peripheral events that look like referred sensation, like backpain and hip pain, but which aren’t, I would imagine that practically all of itre£ects some degree of central sensitization. But since I am saying that everysingle pain centrally sensitizes if you know how to look for it, the distinction isnot so important to me.Felson: The common clinical explanation is that a given joint is innervated by
several di¡erent routes, and referred pain can go in that distribution.Blake: It has to be more complex than that. This is the simplest pathway, but
there are clearly explanations in Halligan’s work that are over and above thatsystem.Felson: There is clashing nomenclature here, and I wondered if you would help
us a bit. The word ‘proprioception’ is the problem. Yesterday there was discussionof proprioceptive inaccuracy inOA.The last thing you talked aboutwith ‘arms notconjugate if you try to make them conjugate in ¢bromyalgia’ is similar to theproprioceptive inaccuracy discussed yesterday. But you seemed to be using theterm proprioceptive in several di¡erent ways. Could you de¢ne it for us?Blake: I’m embracing the entire concept of your appreciation of yourself in
space. This breaks down into lots of di¡erent contributory factors which varywith time of day and so on. This would involve ocular and vestibular inputs andwould relate much more to so-called joint position sense, which is what we havetraditionally de¢ned as proprioception. Quite clearly, proprioception is vastlymore than knowing whether your toe is pointing up or down in anextraordinarily crude examination by a clinician who doesn’t understand whatproprioception is. My de¢nition at the moment encompasses all those factorsthat relate to localization of the body in space. What is good about the trick wehave learned directly from Ramachandran’s work is that by throwing out onethat we don’t tend to use very much�a visual one�we can magnify inherentdefects or what we presume were inherent defects.Koltzenburg:What if you just ask people to close their eyes and then tell them to
mimic onemovement that you show themhow to doonone side, on the other side?The question is, if you do this for 10 minutes, will they have weakness or pain thatwill interfere with that? You could be causing that relationship.Blake:Possibly, except that closing your eyes and looking at themirrorwhen the
movement is coming in backward at you is not the same thing. We have doneclosed eye experiments to show this. Likewise, the di¡erent referred sensationsthat we have reported vary between eyes open and eyes closed. The test that weare doing is not mimicked by just closing your eyes.Koltzenburg: I am not talking about the defect, but rather the inference that these
are proprioceptive de¢cits, and if there is a structural reason for such aproprioceptive de¢cit this should be evidenced in the ¢rst test that you do. The
PHANTOMS 175

fact that you have to do it several times suggests to me there may be other factorsthat need to be activated.Blake: I have no doubt that this is the case.Hunter: For those people who have chronic pain states, is there any biological
rationale in terms of their cortical distribution for pain sensation, in terms of whatpsychological disturbances they may subsequently develop?Blake: I can’t answer that. A major cognitive disturbance can create an
environment where someone is sensitized at a point in time. We are right at theedges of our knowledge of this. It appears to me that since we repeatedly hearthat a major cognitive in£uence appears to precipitate something that it is morelikely that they are telling the truth than they are fabricating it. And all of thoseof us who specialize in RA are aware that the patients can develop bizarre £ares intheir arthritis which are described di¡erently from more classical in£ammatory£ares, particularly following grief reactions. The delay seems to be extremelyvariable.Koltzenburg: Is it possible that this is something congruent with a patient’s desire
to anchor an event? It is correlative. If you ask most patients what you thoughtbrought their £are on, they will come up with an explanation.Blake:That is the traditional approach thatmedicinehas taken to theseproblems.
I am suggesting that we need to be more tolerant than that and embrace thesecognitive events as likely inciting factors. For any individual, you could easilystatistically dismiss the association by denying it in many other people. This ishow medicolegal practice is conducted in this country. But it doesn’t make theindividual observation incorrect, particularly when you have an explanation for it.Felson: There were a couple of large-scale longitudinal studies in RA. One was
by Alex Zautra, a psychologist at the University of Connecticut, and another wasby John Mason in our group. These studies looked at stressful life activities and£ares of RA, and both were well done and well powered but they showed nothing.Why is that?Blake: I think it is because we are not adopting the right models. It is similar to
theway that doctors dismissed the ability of patients to relate £ares in their arthritisto changes in theweather. Exactly the same kind of cross-sectional studywas done,and found negative results.Felson: These were longitudinal diary studies.Blake: This was also done in the study I’m referring to. You can use this to say
that this event doesn’t occur. But I am not so sure that the fault isn’t with ourmodelling system. As anyone with OA of the knee knows, you can detectweather change because the joint becomes barometric. The problem is that thetools we are using to study it are poor. It is a very hard position to argue from. Itcertainly allows the traditionalist view that we have just had, which then gainsfurther supportwhen it becomes amedical legal issue, but I amnot sure it is correct.
176 DISCUSSION

Dieppe: You said that some people undergoing these clever incongruityexperiments will experience sti¡ness. Do you regard the sensation of sti¡ness aspart of the pain experience?Blake:Yes. It is clearly not a peripheral event. There is a paper coming out bymy
colleague RichardHague on this.Whenwe had our ¢rst phantom pain patient, thething that distressed her most was her phantom sti¡ness. Eachmorning she wouldwake up and mentally engage her phantom limb. Then she would exercise herphantom limb in parallel with her normal limb. What we were interested in wasexactly the question you are asking, which is totally pivotal to what people thinksti¡ness is. The general feeling is that it is a peripheral event. This is rubbish. Itmaystart that way but it doesn’t end up that way. We did a whole series of similarpatients and they all said exactly the same thing. Then we started taking historiesof sti¡ness very di¡erently. We ask people when they get up in the night to use thetoilet whether they have a full quantum of sti¡ness or do they have to wait for acertain amount of rest ¢rst. They get a full quantum ever so quickly once they havehit a certain stage of sleep. They can get a full quantum of sti¡ness within an hour.This leads us on to the issues of night pain and rest pain. Is it rest, or is it changes insensory perception which are altered when people down-regulate their brains? Isthatwhat changes the quality ofOApain at night? It is not there all night but comesat speci¢c times and is extraordinarily distressing.Herzog: I was very intrigued by the pain you induced in normal people with
incongruent movements. I was wondering if you had ever tried to measure inpeople with incongruent movements without the mirror and with the mirrorwhether the muscular control of the movement is the same. My hunch would bethat since you get thewrong feedback, itmight be quite an exercise for these peopleto do what you are asking them and there could be a lot of co-contraction. Thiscould be measured by electromyogram. Some of the pain might be due to this.Blake: It is not too much exercise. We have done it on ourselves thousands of
times. I used 10 min as a simple system. You can get this in some people in one ortwo minutes. The ones that are going to go, go quite quickly.Herzog:Have you ever measured electromyographic activity?Blake: No. There are a lot of experiments we need to do, linking these
observations back to monitors of the incongruence centre. Are the ones that areexperiencing pain the ones that are ¢ring o¡ in the right hemisphere? We presumethey are, butwe don’t know. In some peoplewe can do this to them in seconds. It isnot a fatigue event.Herzog: I just assume that the wholemotor control may get the wrong feedback.
I am not sure what this would do to the mechanics of the system.Blake: You are alluding now to phenomena such as repetitive strain injury. We
have many more subtle tests where we are making people do false movements thatare essentially seen as incongruent when viewed on a computer screen. These are
PHANTOMS 177

verymuchmore tiny, ¢nemovements. This allows us to look at the position of theaxial skeleton, which seems to impinge on this.Herzog: If you just left the muscle on because of wrong control at quite a high
level, whichwould prevent it having an adequate blood supply, within 20^30 s youwould feel a certain tingling sensation and pain.Blake: In terms of where they are feeling the pain, it seems to me to be much
wider than this. It becomes a very di¡use type of pain, very much akin to¢bromyalgia.Herzog: If I understood correctly, you said that in normal people youmade your
observations very systematically, that is, all normal people undergoing yourexercise regime would end up with pain.Blake: There certainly was a self-similarity in those that reported it with what
people experience in repetitive strain injury. It seemed to cross multiple musclegroups. We have lots of work to do with this system. This is a very crude way ofcreating this incongruent input and I am sure there will be many better ways. Weare working on this.Conaghan:How did you come to the four month CRPS rule?Blake: In our series of 15, it is roughly about four months. SPET scanning of
CRPS localizes where uptake is maximal. There are uptake changes in themidbrainand they change at about 4^6months. There is a threshold change in the populationat around that time. It is not thatwe don’tmake the chronic ones better: we actuallymake them worse, because they are incapable of doing congruent movement.Felson:Another issue you raisedwas the denervated synoviumofRA.Thiswas a
surprise to me. Why, then, is synovectomy such an e¡ective surgery for peoplewith RA?Blake: I am talking microns, whereas surgeons are talking feet and inches! It is a
very di¡erent process. I am referring to the very super¢cial synovium. In patientswith palindromic RA, the joints are fantastically painful. This is when thesynovium is innervated, and this goes.Grubb: What is the evidence that it is truly denervated and you haven’t simply
lost the peptide content? There are non-peptidergic a¡erents that would be a lotharder to see.Blake: We did this in Julia Pollock’s lab, who is an expert on this. As far as we
could see, in terms of what we have marked for, the nerves have gone.
178 DISCUSSION

Bone pain and pressure in
osteoarthritic joints
Peter A. Simkin
Professor of Medicine, Division of Rheumatology, University of Washington, Box 356428,Seattle WA 98195, USA
Abstract. Intraosseous hypertension has been associated with a deep aching bone pain,particularly at rest, in subsets of patients with osteoarthritis of the hip and knee. Thepathophysiology of this problem remains uncertain, but intraosseous phlebographyimplicates out£ow impairment at relatively distal venous sites. Although the issue hasbeen controversial, intraosseous pressures rise normally, and painlessly, whenepiphyseal bone is loaded and these pulses may be mechanically meaningful in thedistribution and transmission of impact energy. Increased out£ow resistance mayamplify the episodic pressure response with subsequent intravasation of epiphyseal fatleading to ‘marrow oedema’ and altered mechanics. The relationship between persistingpain and pressure is an old but convincing association. Its precise mechanism inosteoarthritis remains in need of an adequate explanation.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 179^190
This symposium was convened to consider the problem of pain in patients withosteoarthritis (OA). Where are the relevant receptors, and what triggers them, dothe patterns di¡er in a meaningful (and therefore interpretable) way, and why isthere such variance in the pain perceived by individuals who appear to havecomparable degrees of degradation in articular cartilage and in the hard and softtissues of the underlying subchondrium? For themost part, these questions remainunanswered and our deliberations focus properly more on opportunities forinvestigation than on meaningful current data. A conspicuous exception,however, lies in the extensive, older studies of hypertension in bones adjacent tolarge, painful OA human joints. This paper will review some of those ¢ndings andwill consider further the possible role of intraosseous pressure in articularphysiology and pathophysiology.
Intraosseous hypertension in osteoarthritis
The foremost champion of the pressure/pain relationship in OA has been CarlArnoldi of Copenhagen. In a series of papers on the subject, Arnoldi and his
179
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

colleagues focused on rest pain, especially that occurring at night (Arnoldi 1991,Lemperg & Arnoldi 1978, Arnoldi et al 1980). They reported that thissymptom was best described as a deep, throbbing ache; that it was usuallyaccompanied by an increase in intraosseous pressure; and that decompressivesurgical procedures such as simple fenestration or a tibial osteotomy led toe¡ective pain relief along with abolition of the intraosseous hypertension (Deyet al 1989). When intraosseous phlebography was done, there was impairedclearance of the radiopaque dye as well as impressive dilation of sinusoids andcollecting veins. Further evidence of obstructed out£ow was provided byimpaired clearance of injected radio-iodide (Hernborg 1969). Most of these basic¢ndings have been shown both in hips and in knees and were perhapsdemonstrated most convincingly in the patellae of patients with anterior kneepain (Bj˛rkstr˛m et al 1980, Schneider et al 2000, Waisbrod & Treiman 1980,Ficat & Hungerford 1977).Osteoarthritis is not unique in its disposition toward intraosseous hypertension.
Avascular necrosis of bone is perhaps the best studied entity with persuasiveevidence of obstructed venous out£ow in each of the disparate entities associatedwith this lesion. Thus steroid therapy, alcoholism and Gaucher’s disease lead tohyperplasia and or hypertrophy of intraosseous cells with resulting compressionof out£ow vessels while sickle cell disease and disbaric decompression obstructthe same vessels through intravascular processes. All of these patients are thensubject to the same ischaemic catastrophes in the £exible convex members oflarge, human joints (Ficat & Arlet 1980).Like OA, the remaining entities with bony hypertension (re£ex sympathetic
dystrophy, fatigue fracture, regional osteoporosis, etc.) have less logical vascularpathophysiology. It is intriguing to note, however, that each of them is associatedwith ‘marrowoedema’ bymagnetic resonance imaging (MRI). Since the volume ofeach bone is ¢xed by its mineralized shell, it seems axiomatic that its £uid contentsmust obey zero sum principles. This means that more water (bony oedema) meansless fat. It seems reasonable to hypothesize that increased local pressure in somewaypromotes intravasation of bone fat cells, with resultant vascular clearance thatparallels the increase in intraosseous water. Parenthetically, it is also reasonable toexamine the extent to which intraosseous hypertension is also present, and acontributing cause of pain, in ankylosing spondylitis, rheumatoid arthritis, andother in£ammatory and traumatic conditions associated with marrow oedema(Arndt et al 1996, Visuri 1997, Yamamoto & Bullough 1999, McGonagle et al1999, 2003).Thus, intraosseous hypertension may be a more pervasive mechanism in human
disease than rheumatologists now recognize, and it is appropriate to consider someof the outstanding questions that must be addressed before we can understand andinterpret this ¢nding.
180 SIMKIN

What is the normal intraosseous pressure?
This question has been addressed in a number of studies but there is considerablevariation in the ¢ndings both in humans and in experimental animals (Stein et al1957). Virtually all data have been obtained under anaesthesia and this practice aswell as the speci¢c agent used, could be important factors in the observed variation.One study in vertebral bodies found the intraosseous pressure to be comparable tothat in adjacent veins, and similar equilibrium pertains in the sternum, skull andother ‘£at bones’. Almost all studies in appendicular bones, however, have foundhigher pressures, usually in the 15^20mmHg range that prevails in the normalhuman eye�an unquestionably turgid tissue. Ficat and Arlet, in their bookIschemia and Necrosis of Bone (Ficat & Arlet 1980) cite an extensive study of youngAfrican subjects (Table 1) (Kabakele 1972, cited by Ficat & Arlet). In accord withtheir own data from the proximal femur, these ¢ndings indicate that intraosseouspressure rises as the points of observation move away from the shaft and into thesubchondral regions of the epiphyses. They then point out that ‘notwithstandingthe direction of this gradient, its existence presupposes the presence of adaptablebarriers and permanent di¡erences in neurovascular control.’
What regulates intraosseous pressure?
Overall, the resting pressure in each bony compartment must be determined byvascular factors a¡ecting the balance between arterial in£ow and venous out£ow.Speci¢cally, osseous pressures greater than these in adjacent veins means thatintraosseous vascular resistances must be in action (Wilkes & Visscher 1975). Toinvestigate possible controlling factors, infusions of epinephrine andnorepinephrine have been studied by many investigators (Stein et al 1958,Schneider et al 1998, Shim 1968). The consensus ¢nding of such work is anincrease in vascular resistance with a concomitant fall in intraosseous pressure.Thus, in this case, arteriolar, a¡erent constriction explains the results well.
PRESSURE AND BONE PAIN 181
TABLE 1 Mean tibial intramedullary pressures
Child Adult
Epiphysis 26.1 (24)* �
Metaphysis 22.3 (69)* 19.9 (14)
Diaphysis 13.6 (124)* 11.8 (26)
Pressures are in mmHg with the number of observations in parenthesis.Children were 3^17 years old.*Single, high outlying values were deleted from each of the three bonyregions in children.

In OA however, the compartmental pressure increases with decreased £ow andconcomitant dilatation of sinusoids. This combination ¢ts only with an e¡erentconstrictor analogous to those present in erectile tissues. At present, no suchconstrictor has been identi¢ed clearly, although at least two electron microscopicstudies have identi¢ed apparent sphincters at the out£ow site where sinusoidsempty into collecting venules (Ohtani et al 1982, Kessel & Kardon 1979). Weknow almost nothing about the controls of these putative sphincters but onestudy has found an increase in pressure with a decrease in £ow with ephedrineinfusion, and re£ex neurological and/or metabolic controls deserve consideration(Bˇnger et al 1982, Holm et al 1990). Somewhat surprisingly, exercise leads toincreased resistance with decreased £ow while muscle contraction (in di¡erentstudies) leads to a modest increase in intraosseous pressure (Gross et al 1979).These ¢ndings are consistent with an epiphyseal pressurization during activeexercise, but more experimental con¢rmation is clearly needed.Finally, an intriguing ¢nding from the clinic may be relevant. Transplant
patients taking cyclosporine, a known vasoconstrictor, are subject to severe,episodic knee pain in the absence of any apparent articular pathology. Thisphenomenon is readily controlled, however, by administration of a vasodilator:nifedipine (Barbosa et al 1995, Kart-Koseoglu et al 2002). To date, there is nodirect con¢rmation of increased intraosseous pressure in this syndrome, but the¢ndings are considered most consistent with the possibility of drug-inducedvasoconstriction in venous, e¡erent vessels. It may be relevant to note that thispain is perceived in the distal femoral epiphysis, rather than the shaft of thefemur, and the putative vasoconstriction would presumably be localized there.
What happens with joint use?
Within narrow limits, the volume of each normal bone remains constant at a valuedetermined by its enveloping walls of semi-rigid, calci¢ed tissue. Under load,however, those walls, and the trabecular framework within them, will £ex. In sodoing, the bone serves as a complex series of springs which are capable of storingand distributing loading energy which then can be recovered when eachcompressed spring re-expands to resume its resting position. Most human jointsare comprised of a sti¡, in£exible, concave component such as the glenoid fossaopposed by a £exible convex member such as the humeral head (Simkin et al1980). In vitro, and presumably in vivo, this £exion causes a signi¢cant boost in thehumeral intraosseous pressure (Downey et al 1988). This process of compressionunder load with subsequent re-expansion must necessarily impact on the cells,interstitial £uid, and other soft tissue elements contained within theintertrabecular space, and the hydrostatic pressure must rise accordingly. Thisrise will be greatest in the bony compartments that are immediately subchondral,
182 SIMKIN

but as their walls and £oors then £ex as well, a descending pressure gradientdevelops in which each trabeculum is burdened primarily by the pressuredi¡erence between adjoining soft tissue spaces.There has been long-standing controversy regarding the possibility that
articular loading might induce su⁄cient pressure to provide a hydrauliccontribution to the mechanics of trabecular bone, but it is necessary toacknowledge that most bioengineers currently feel this possibility has beenexcluded. Most of the available, mechanical data are derived, however, fromtesting, in vitro, of small pieces of excised bone, often obtained from elderlysubjects and/or from bones (such as vertebrae) or animals (such as cows) wherehydraulic support is less likely. The results may di¡er with more appropriate (butmore di⁄cult) testing in vivo.
Does trabecular tension cause pain?
It is not controversial to say that impact events may raise subchondral intraosseouspressure by amounts well in excess of 100mmHg (Downey et al 1988). Injection of5 ml of saline into human femoral necks is reported to be highly painful when thepressure rises just as pressure causes pain in an obstructed bowel, a blocked ureter, agouty great toe, or a simple old fashioned boil. Such a distension-induced pain cannot be excluded in OA, particularly in the use-pain that many patients report. Thisexplanation would not seem to ¢t, however, with the nocturnal rest-pain that hasbeen most closely related to the ¢nding of intraosseous hypertension. It also seemsunlikely in the epiphyses where I believe that intermittent, load-induced pressurepulses are a painless part of normal articular physiology.
Does ischaemia cause intraosseous pain?
This important question must be asked although there has been little work thataddresses the possibility directly. Metabolic evidence of ischaemia (decreased pO2
and increased venous lactate) was found to accompany increased pressure in onestudy of OA femoral heads (Kiaer et al 1988). Further intraoperative studies couldprovide important support for or against the presence of this mechanism. Indirectevidence may be found, however, in studies of post-traumatic compartmentsyndromes in human extremities (Mars & Hadley 1998). There, a persistingpressure greater than 35 mmHg will regularly induce pain and poses a signi¢cantrisk of ischemic infarction for motor nerves traversing such a space. Intraosseouspressures in OA regularly exceed this level, and it seems logical to attribute theattendant pain to a bony compartment syndrome (Fricker et al 1995). Theprompt pain relief provided by decompressive fenestration of bone would be
PRESSURE AND BONE PAIN 183

entirely consistent with this interpretation just as decompressive fasciotomyrelieves the pain of a soft tissue compartment syndrome.
Is intraosseous in£ammation important?
Although in£ammation has become increasingly important in studies ofOA,mostof this work has focused on the synovium and the chonodrocyte. In£ammatorycells are present in the subchondral bone of OA patients, however, and it isplausible (in the absence of evidence to the contrary) to think thatproin£ammatory products of these cells could play a role in bone pain. Theunusual human syndrome of pancreatitic arthritis provides an example of aspeci¢c mechanism that could be relevant (Simkin et al 1983). There, thepresence of circulating pancreatic enzymes appears to activate and to amplify thee¡ects of local tissue lipases with a resultant £ood of fatty acids into the involvedtissue. When the fatty acid release exceeds the binding capacity of availablealbumin, the excess of free fatty acids causes intense local in£ammation. Thisphenomenon is known best, and recognized most readily when it occurs focallyin subcutaneous fat. The same events may occur, however, in the fattysubchondrium with devastating pain and rapid osteolysis. Were a similaractivation of tissue lipase to be triggered by ischaemic infarction in hypertensivebone, the resultant fatty acid-induced in£ammation could result in substantiallocal pain and marrow oedema.
Conclusion
The organizers of this symposium advised each contributor that ‘your talk shouldform a provocative basis for the discussion’ and that it was appropriate ‘to presentnew or preliminary results and to speculate on their signi¢cance.’ In fact, most ofthe information I’ve reviewed here is old, but I’d like to exercise the licence I’vebeen given to speculate anyway and to present the following working hypotheses:
(1) Appendicular bones are normally pressurized through control systems thatremain unknown. That pressure is greatest in the subchondral regionswhere pressurization enhances trabecular elasticity just as in£ation enhancesthe bounce of a basketball or a rubber tyre.
(2) The baseline pressure in intermittently ampli¢ed during loading, and thisprocess serves to distribute impact energy down a steep pressure gradientthroughout the extensive system of subchondral trabeculae. In so doing, itenlists the entire trabecular framework to serve as one, extensive spring.
184 SIMKIN

(3) This process serves to protect both the overlying cartilage and the supportingcortical bone against excessive energy burdenswhile also permitting recoveryof the stored energy to perform useful work.
(4) Under a variety of clinical circumstances, including OA, the neurovascularcontrol of this system breaks down with a resultant rise in resting pressurethat causes pain, most probably on an ischaemic basis.
(5) A more e¡ective understanding of the normal system may help us tounderstand how and why it fails and may lead ultimately to newer and moree¡ective strategies for controlling this part of the arthritis pain spectrum.
References
Arndt WF 3rd, Truax AL, Barnett FM, Simmons GE, Brown DC 1996 MR diagnosis of bonecontusions of the knee: comparison of coronal T2-weighted fast spin-echo with fat saturationand fast spin-echo STIR images with conventional STIR images. Am J Roentgenol 166:119^124
Arnoldi CC 1991 Patellar pain. Acta Orthopaedica Scandinavica 62(suppl 244):1^29Arnoldi CC, Djurhuus JC, Heerfordt J, Karle A 1980 Intraosseous phlebography, intraosseouspressure measurements and 99mTC-polyphosphate scintigraphy in patients with variouspainful conditions in the hip and knee. Acta Orthop Scand 51:19^28
Barbosa LM,Gauthier VJ, Davis CL 1995 Bone pain that responds to calcium channel blockers.A retrospective and prospective study of transplant recipients. Transplantation 59:541^524
Bj˛rkstr˛m S, Goldie IF, Wetterqvist H 1980 Intramedullary pressure of the patella inChondromalacia. Arch Orthop Traumat Surg 97:81^85
Bˇnger C, Harving S, Bˇnger EH 1982 Intraosseous pressure in the patella in relation tosimulated joint e¡usion and knee position: an experimental study in puppies. Acta OrthopScand 53:745^751
Dey A, Sarma UC, Dave PK 1989 E¡ect of high tibial osteotomy on upper tibial venousdrainage: study by intraosseous phlebography in primary osteoarthritis of knee joint. AnnRheum Dis 48:188^193
Downey DJ, Simkin PA, Taggart R 1988 The e¡ect of compressive loading on intraosseouspressure in the femoral head in vitro. J Bone Joint Surg Am 70:871^877
Ficat RP, Hungerford DS 1977 Disorders of the patello-femoral joint. 1st edn. Williams &Wilkins, Baltimore
Ficat RP, Arlet J 1980 Ischemia and necroses of bone. Williams &Wilkins, BaltimoreFricker C, Bucher K, Stuker G 1995 Are degenerative joint diseases chronical compartmentsyndromes? (German) Schweiz Arch Tierheilkd 137:137^140
Gross PM,Heistad DD,MarcusML 1979Neurohumoral regulation of blood £ow to bones andmarrow. Am J Physiol 237:H440^H448
Hernborg J 1969 Elimination of Na-131-I from the head and the neck of the femur in una¡ectedand osteoarthritic hip joints. Arthritis Rheum 12:30^33
Holm IE, Ewald H, Bulow J, Bunger C 1990 Vasoactive substances in subchondral bone of thedog knee. J Orthop Res 8:205^212
Kabakele M 1972 Contribution au diagnostic pre¤ coce de l’oste¤ one¤ crose dre¤ panocytaire. The' sed’agre¤ gation, Kinshasa, Zaire
Kart-Koseoglu H, Yucel AE, Isyklar I, Turker I, Akcaly Z, Haberal M 2003 Joint pain andarthritis in renal transplant recipients, and correlation with cyclosporine therapy. RheumatolInt 23:159^162
PRESSURE AND BONE PAIN 185

Kessel RG, Kardon RH 1979 Tissues and organs: a text-atlas of scanning electron microscopy.W.H. Freeman, San Francisco
Kiaer T, Gronlund J, Sorensen KH 1988 Subchondral pO2, pCO2, pressure, pH, and lactate inhuman osteoarthritis of the hip. Clin Orthop 149^155
Lemperg RK,Arnoldi CC 1978 The signi¢cance of intraosseous pressure in normal and diseasedstates with special reference to the intraosseous engorgement-pain syndrome. ClinOrthopRelRes 136:143^156
Mars M, Hadley GP 1998 Raised intracompartmental pressure and compartment syndromes.Injury 29:403^411
McGonagle D, Conaghan PG, O’Connor P et al 1999 The relationship between synovitis andbone changes in early untreated rheumatoid arthritis: a controlled magnetic resonanceimaging study. Arthritis Rheum 42:1706^1711
McGonagle D, Marzo-Ortega H, Benjamin M, Emery P 2003 Report on the Secondinternational Enthesitis Workshop. Arthritis Rheum 48:896^905
Ohtani O, Gannon B, Ohtsuka A, Murakami T 1982 The microvasculature of bone andespecially of bone marrow as studied by scanning electron microscopy of vascular casts� areview. Scan Electron Microsc (Pt 1):427^434
Schneider T, Drescher W, Becker C et al 1998 The impact of vasoactive substances onintraosseous pressure and blood £ow alterations in the femoral head: a study based onmagnetic resonance imaging. Arch Orthop Trauma Surg 118:45^49
Schneider U, Breusch SJ, ThomsenM,WenzW,Graf J, Neithard FU 2000Anew concept in thetreatment of anterior knee pain: patellar hypertension syndrome. Orthopedics 23:581^586
Shim SS 1968 Physiology of blood circulation of bone. J Bone Joint Surg Am 50:812^824Simkin PA,GraneyDO, Fiechtner JJ 1980 Roman arches, human joints, and disease: di¡erencesbetween convex and concave sides of joints. Arthritis Rheum 23:1308^1311
Simkin PA, Brunzell JD,WisnerD, Fiechtner JJ, Carlin JS,Willkens RF 1983 Free fatty acids inthe pancreatitic arthritis syndrome. Arthritis Rheum 26:127^132
Stein AH Jr, Morgan HC, Reynolds FC 1957 Variations in normal bone-marrow pressures. JBone Joint Surg Am 39:1129^1134
Stein AH Jr, Morgan HC, Porras RF 1958 The e¡ect of pressor and depressor drugs onintramedullary bone-marrow pressure. J Bone Joint Surg Am 40:1103^1110
Visuri T 1997 Stress osteopathy of the femoral head. 10military recruits followed for 5^11 years.Acta Orthop Scand 68:138^141
Waisbrod H, Treiman N 1980 Intra-osseous venography in patellofemoral disorders: apreliminary report. J Bone Joint Surg Br 62:454^456
Wilkes CH, Visscher MB 1975 Some physiological aspects of bone marrow pressure. J BoneJoint Surg Am 57:49^57
Yamamoto T, Bullough PG 1999 Subchondral insu⁄ciency fracture of the femoral head: adi¡erential diagnosis in acute onset of coxarthrosis in the elderly. Arthritis Rheum 42:2719^2723
DISCUSSION
Pisetsky: I’d like to ask about the relationship between OA and osteoporosis.Where does bone loss ¢t into yourmodel in terms of the ability to handle pressure?Simkin:The inverse relationship that you are speaking of is intriguing. It would
support my position if osteoporosis does not lead to perforations of the trabecularwalls in the convex areas that I think are hydraulically supported. The factthat osteoporotic bone is more compliant under load could actually facilitate
186 DISCUSSION

load-bearing by the marrow content. In our studies of femoral head loadingin vitro the highest intraosseous pressures were found in two very osteoporoticspecimens (Downey et al 1988).Pisetsky: What about women and their post-menopausal bone loss problems?
How does this ¢t in?Simkin: It is the same issue: if someone has lost bone, I would think it would be
lost in a way that preserves the ba¥e system. Those women are at risk of fracturingtheir femoral necks but they don’t crush their femoral heads when they fall (Toddet al 1972, Wicks et al 1982).Fernihough: Earlier we discussed the di⁄culty of getting a good pain model of
OA, particularly in rat. Although the rat walks with its knee at 90 degrees of£exion, which would suggest a high intraosseous pressure, it is quite strikingthat their growth plate is not fused and remains cartilagenous. How do you thinkthiswould a¡ect intraosseous pressure, and how is this related to the pain response?Simkin: I don’t have any data on intraosseous pressure in animal models of OA.
In humans, we don’t expect to see changes of OA in children with open growthplates. Perhaps their intraosseous springs are more e¡ective. As I’ve listened to thepapers over the last two days I have wondered whether there is intraosseoushypertension in the cat and also the human elbow. It may be very useful to knowthis in sites where there are degenerative changes without accompanying pain.Lohmander:You have done some seminal work in joint £uid physiology, and on
the turnover of joint £uid. Listening to your talk on the hydraulics of bone makesme want to ask you about your opinion on the communication between thesubchondral hydraulic space and the joint space. What are the possibilities forcommunication between subchondral cells and cartilage cells?Simkin: There is no disagreement that in immature individuals that pathway is
open. We have done some work on this in mature sheep, and when we use highpressures analogous to those that occur within normal joints we can drive £uidacross (Simkin & Peterson 2001). The evidence against osteochondralcommunication is largely based on di¡usion experiments rather than convectionexperiments.Felson:What Stefan Lohmander is implying is that your view is unconventional:
it is generally felt that there is no communication between bone marrow and thecartilagenous and articular surface, except in pathology where there might be cyststhat communicate. Would this be a fair statement?Simkin: Yes, it’s fair. In the child though, there is clearly di¡usion across the
cartilage/bone interface. In the adult, it is thought there is no such di¡usion. Theexperiments that I am alluding to indicate that in fact there is convection whenthe pressure is high.Kuettner: Cartilage is as hydraulic as your bone. There are two hydraulic systems
sitting on top of each other under normal conditions. What you presented was the
PRESSURE AND BONE PAIN 187

bone excluding the cartilage, but your lines could have continued into the cartilage.There is a connection. It may not be a liquid £ow, but a biomechanicalsupplementation of the two tissues. Would you agree?Simkin: Yes. The other thing I would add is that I suspect the permeability we
see with convective £ow, is in fact, ‘semipermeability’. Water will cross; the largermolecules will not. I think the feature that we all know as the tidemark is the debrisof apoptotic chondrocytes. These macromolecules may be driven downward bythrough-£owing water and be sieved out at the border of calci¢ed cartilage.Herzog: I can accept one or the other of your stories, but not both. The dilemma I
have is that on the one hand, to have good load distribution you need a viscoelasticsystem.On the other hand, you are saying that this is very good for the recovery ofenergy, but for the recovery of energy it would be much better if you had a purelyelastic system. If you do the indentation test on an entire joint, youwill see that it isviscoelastic, with a tremendous loss of energy. I can believe in your viscoelasticargument, but not so much in your bouncy element.Simkin: The key thing is the time factor. Someone who hops twice a second is
operating at the elastic end of the viscoelastic curve. This is very fast and permitsrecovery of most of the energy. If you hop once every ¢ve seconds you getsigni¢cant £ow within the bone, this disspates energy and it can’t be recovered.Herzog: This is correct if we think about one hop or one step, but if we think
about someone doing a 2 hour run then the viscoelastic e¡ect will over timedissipate all the energy that is put in.Simkin: You keep recovering with each step.Herzog:We know that you don’t. If we do stress^relaxation testing where we go
to the same displacement each time, we lose pressure. If we go to the same pressurewe lose displacement.Simkin: Are the experiments you are talking about done in living vascularized
bone?Herzog: These are in situ joints from freshly harvested animals.Simkin: So there is no blood £ow: I think thismakes a big di¡erence. In our own
studies of femoral head loading in vitro, we maintained a slow, constant infusion ofsaline and observed no decay in the viscoelastic behaviour (Downey et al 1988).Herzog: You started by talking about bone pressure. You talked about arterial
pressures, but then towards the end of the talk I had the feeling you were talkingabout pressure within the £uid of the bone. It wasn’t clear tomewhether they weremixed from the beginning to the end.Simkin: The original data I showed were from living bone, but some of the
mechanical data were from intact bones studied in vitro. I didn’t really talk aboutarterial pressures.Herzog: So you are saying that if we did indentation testing in an anaesthetized
but fully vascularized animal, that we would not see much loss of energy. In fact,
188 DISCUSSION

you would argue that the system would behave virtually elastically and allenergy put into the joint would be fully recovered. If that was really the case, thatwould challenge much of the current thinking on joint mechanics. I think I willtry this experiment, as we are perfectly set up to do precisely these types ofexperiments.Simkin: Yes, but it wouldn’t be perfect. As with a bouncing rubber ball some
energy would be lost with each impact.Herzog: That would be a tremendous change in mechanical thinking if that was
correct, and it is easily testable.Felson:WalterHerzog, does bone deformduringweight bearing?Thiswould be
part of this hypothesis.Herzog: From extreme studies, on humans doing the triple jump, I know that
bone can deform to quite an extent. I guess the issue that you are bringing up is thatthe elastic modulus of articular cartilage is about 1000 times smaller than that ofbone. So, articular cartilage would deform under a given amount of compressionmuch more than bone.Simkin: But, as Eric Radin pointed out, the cartilage is very thin compared to the
subchondrium (Radin et al 1970). When we measured compliance in canineshoulders in vitro, the overall de£ection was much greater in bone than incartilage (Simkin et al 1985).Lohmander:Accepting your suggestion of convective £owbetween cartilage and
subchondral space, could that be related to what the magnetic resonance imaging(MRI) people observe as signal changes in bone marrow oedema, which in turn isconnected back to pain in OA?Simkin: I wouldn’t think so. The amount of £uid we are talking about is
relatively small. The subchondrium is surprisingly vascular and I suspect that itsinterstitial water turns over quite rapidly.Pisetsky: What happens in core decompression? What would happen to the
cartilage and bone when pressure goes?Simkin: I would think that the normal spring would be compromised.
But it is not a normal spring that you are decompressing: it is one that isalready sti¡ened by disease. This is certainly a procedure that we believe to beuseful.Pisetsky: Is there any sense that this makes cartilage worse?Simkin: I can’t say.Dieppe: I wanted to come back to the homage we have been paying to Carlo
Arnoldi throughout this meeting concerning decompression experiments andthere importance in pain. There is another phenomenon one sees with so-calledatrophic OA, when you get massive destruction of bone. You can wipe out mostof the femoral head of the hip. These people don’t have a little pain, they have ahuge amount of pain. It doesn’t make any sense that this could have anything to do
PRESSURE AND BONE PAIN 189

with intraosseous pressure because they have lost thewhole subchondrium and lotsmore bone.Simkin: I agree. Clearly, the study that I showed initially in which they selected
50 patellae out of 136 painful knees is re£ective of the idea that I am speaking ofwhich concerns a subset of OA patients. This is a heterogeneous lesion, however,and I believe it makes sense to actively seek out subsets in the hope, and theexpectation, that di¡erences in pathophysiology may lead to useful di¡erences inmanagement.Hunter:With those atrophic people there hasn’t been a lot of documentation of
their bone density. I would suspect that they may be having a lot of microfracturesand underlying remodelling as a result of this. As an aside, I don’t think oedema is agood term to describe these lesions, because there isn’t a great deal of oedema onpathological specimens. There isn’t a great deal of longitudinal improvement inbone marrow lesions, to suggest that this is going to account for variability inpain on its own. Intraosseus blood pressure or pressure changes in addition tothose bone marrow lesions may potentially account for some of that variability. Iwould be interested in your thoughts about the aetiology of some of the structuraland remodelling changes, and the relationship that this intraosseous blood£owmay have with those remodelling changes that are occurring in the subchondralbone.Simkin: The key issue is time. It may be that the ‘oedema’ that we recognize
happened within a single event and then it persists. Disordered mechanics thenpersist because of the loss of that substance. We shouldn’t think of these as thingsthat must be concurrent.
References
Downey DJ, Simkin PA, Taggart R 1988 The e¡ect of compressive loading on intraosseouspressure in the femoral head in vitro. J Bone Joint Surg Am 70:871^877
Radin EL, Paul IL, LowyM 1970A comparison of the dynamic force-transmitting properties ofsubchondral bone and articular cartilage. J Bone Joint Surg Am 52:444^456
Simkin PA, Peterson JR 2001 The cartilage/bone interface is permeable to saline underphysiologic pressures. Arthritis Rheum 44:548(abstr)
Simkin PA, Houglum SJ, Pickerell CC 1985 Compliance and viscoelasticity of canine shouldersloaded in vitro. J Biomech 18:735^743
ToddRC, FreemanMAR, Pirie CJ 1972 Isolated trabecular fatigue fractures in the femoral head.J Bone Joint Surg Br 54:723^728
WicksM,Garrett R,Vernon-Roberts B, FazzalariNL1982Absence ofmetabolic bone disease inthe proximal femur in patients with fracture of the femoral neck. J Bone Joint Surg Br 64:319^322
190 DISCUSSION

Structural associations of osteoarthritis
pain: lessons frommagnetic resonance
imaging
Philip G Conaghan and David T. Felson*
Academic Unit of Musculoskeletal Disease, University of Leeds & Department ofRheumatology, Leeds General In¢rmary, Great George Street, Leeds LS1 3EX, UK and*Multipurpose Arthritis and Muscoskeletal Diseases Center, Boston University School ofMedicine, 715 Albany Street, A 203, Boston, MA 02118-2526, USA
Abstract. Formany years the search for structural associations of osteoarthritis (OA) painwere based on conventional radiographic imaging that predominantly visualizes bone.Aswell as being tomographic, magnetic resonance imaging (MRI) has the ability to directlyvisualize all the structures of a joint, including soft tissue and cartilage. InitialMRI studiesfocused on cartilage assessment, but recently there has been a growing body of workexamining the correlation of structural ¢ndings with pain in OA and their relation tostructural progression. Painful OA knees have more MRI-detected abnormalities andthese pathologies are often correlated making individual contributions di⁄cult toassess. However, in large cohort studies, both synovial hypertrophy and large synoviale¡usionswere demonstrated to bemore frequent in patients withOAknee pain. SimilarlyMRI-determined subchondral bone marrow oedema lesions (BME), particularly largeones, are associated with OA knee pain. Meniscal tears in OA knees, although common,have not been linked with pain. Improved, reliable quanti¢cation of the structuralfeatures and the rapid advances in MRI technology can only improve structure^painunderstanding.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 191^205
It is currently accepted that the osteoarthritis (OA) process involves the wholejoint organ including the cartilage, synovium, subchondral bone, menisci andligaments (Felson et al 2000). However traditional research into structure^painassociations relied on the conventional radiograph which predominantly imagesonly bone and a surrogate measure of cartilage thickness, the joint space.Although the odds for knee pain generally increase with radiographic severity ofOA (Felson et al 1987, Lethbridge-Cejku et al 1995), signi¢cant discordancebetween clinical and radiographic changes has been described in communitybased cohorts (Dieppe et al 1997, Creamer &Hochberg 1997, Hannan et al 2000).
191
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

Magnetic resonance imaging (MRI) provides the ability to visualize not only allthe relevant structures within a joint, but its tomographic nature allows forimaging in three dimensions. Hence whole organ evaluation of this complexdisease process is possible (Peterfy 2002). Until recently, much of the OA MRIliterature focused on cartilage with attention to scoring cartilage defects andanalysing cartilage volume. Since the cartilage tissue is not supplied with nerves,it seems an unlikely primary source of joint pain (even though it can produceproin£ammatory molecules); consequently this review will focus on therelatively few, recent OA MRI studies pertaining to non-cartilage structures andpain. Preliminary data from whole OA joint evaluation will be presented beforereviewing information on individual structures. Of course, structure^painstudies cannot fully explain the complex and personal pain process, and thispresentation should be viewed in the context of the entire Symposiumproceedings.
Structure^pain study limitations
Before proceeding it is worthwhile considering the limitations of all studies onstructure^pain associations (and the majority to date concern the knee).
Howwas subject pain identi¢ed and quanti¢ed?
The phasic or episodic nature of pain may interfere in detecting associations, soinclusion criteria specifying duration and frequency of pain may in£uence studyresults. Which pain measure is chosen (examples include global, night or weight-bearing pain) may also in£uence the outcome.
Howwas the structure visualized?
Even using the conventional radiograph of the knee, many studies have imagedonly the tibiofemoral joint and not the patellofemoral joint. Arthroscopy maynot fully visualize the posterior portion of a joint. Studies employing MRI bringa host of novel variables to the analysis, including di¡erent magnet strengths,di¡erent sequences, lack of standardized de¢nitions of structures and, for non-cartilage structures, no consensus on quanti¢cation of structural abnormalities.This area of MRI is rapidly expanding and improving but care should be taken ininterpreting and comparing studies.
Whole organ evaluation
Recently preliminary data have been reported from large studies employing MRIto assess the whole joint. Although there is no consensus yet on scoring methods
192 CONAGHAN & FELSON

for assessing the multiple structures within a joint, these studies have employed awhole-organ semi-quantitative OA knee scoring system (the WORMS score)(Peterfy et al 2004). This complex scoring system assesses 14 anatomical featuresat multiple intra-articular sites. The features are scored with semi-quantitativescales varying from 0^1 to 0^7.Some important messages are emerging from these novel cohorts. It is clear that
the radiographically ‘normal’ knee may be far from normal in structure. A recentreport evaluated subjects from the Health ABC study (a biracial community basedcohort aged 70^79), many of whom had normal knee radiographs (Taouli et al2002). This study demonstrated that over 75% had some cartilage abnormalitieswhile 30^60% had meniscal tears, bone marrow oedema (BME), bone cysts andsynovitis. There was at least a moderate correlation between the presence ofcartilage defects and the other structural abnormalities. Importantly, whenpainful and contralateral painless knees in the cohort were comparted, highertotal WORMS scores were found in painful knees (Wildy et al 2002), especiallyin the tibiofemoral compartments. Surprisingly, the MRI abnormalities wereequally present in both painful and non-painful patellofemoral compartments,suggesting that patellofemoral compartments may not be the source of pain inmany persons.
Synovitis, e¡usions and synovial cysts
Synovitis in OA, although secondary, is common; synovial abnormalities arepresent from the earliest stages of OA and the severity of synovitis is generallyrelated to the severity of chondropathy in the a¡ected joint (see Fig. 1; Myers et al1990, Smith et al 1997, Loeuille et al 2002). This synovitis is the source of manypro-in£ammatory cytokines and pain mediators (Pelletier et al 2001). Synovitis iscommonly assessed with MRI using the intravenous, paramagnetic-enhancingagent gadolinium (�stergaard et al 1997), although non-gadolinium sequencescan be optimized for this purpose (Peterfy et al 1994). Studies have correlated theMRI synovial hypertrophy seen in OA with microscopic synovial in£ammation(Fernandez-Madrid et al 1995, �stergaard et al 1997).One recent cross-sectional MRI studied synovitis, e¡usions and popliteal cysts
in a large cohort of OA knee subjects comparing persons with symptomatic kneeOA with persons with radiographic OA but without symptoms recruitedpredominantly from Veterans A¡airs medical centres in Boston, USA (Hill et al2001). The study employed semi-quantitative scoring systems for assessinge¡usions and cysts which were graded as absent, small, moderate or large andcorrespondingly scored 0^3. Synovial hypertrophy was assessed as present orabsent (scored 0 or 1) at three sites: the infrapatellar fat pad, intercondylar spaceand anterior horn of the lateral meniscus. There was a signi¢cant increase in the
OA PAIN AND MRI 193

frequency of both e¡usions (moderate or large) and synovial hypertrophy in thepainful knees compared to those without pain, after adjustment for theradiographic OA severity. In subjects with knee pain and radiographic OA, therewas an association between synovitis and pain severity. Popliteal cysts werecommon (420%) amongst those people without knee pain and not surprisinglythey were associated with e¡usions; they were not associated with pain.Another clue to the importance of e¡usions in OA pain derives from a large,
cross-sectional study of painful OA knees studied with ultrasonography(D’Agostino et al 2003). This study demonstrated that the presence of e¡usion
194 CONAGHAN & FELSON
FIG. 1. Sagittal MRI image of an osteoarthritic knee demonstrating a large amount ofsynovitis in the supra-patella pouch and a large anterior tibial osteophyte (courtesy of DrAndrew Grainger, Leeds, UK).

correlated with sudden aggravation of pain in the week prior to ultrasonography.However not all imaging studies have demonstrated synovial associations withpain. Another large OA knee study employed arthroscopy of the medialcompartment and graded the synovium according to macroscopic appearance asnormal, reactive or in£ammatory (Ayral et al 2002). There was no associationbetween this synovial score and subject pain. The di¡erent methods inclassi¢cation of synovitis and the di¡erent sites evaluated may account for thesediscrepant ¢ndings.
Bone marrow oedema
MRI allows the evaluation of the subchondral bone, for many years consideredimportant in the pain and structural progression of OA (Dieppe 1999, Bollet2001). The commonest MRI subchondral abnormality is BME, ill-de¢ned highsignal areas seen on fat-suppressed T2-weighted or STIR sequences (see Fig 2;Peterfy 2002). It is not known exactly what these lesions represent, and this MRIfeature is not speci¢c for OA, with similar appearances seen in trauma,osteomyelitis and rheumatoid arthritis (Adalberth et al 1997, Bollet 2001,Conaghan et al 2003). Two small studies have matched the histological ¢ndingsfrom tibial plateau bone with the site of MRI BME in OA patients (Bergman et al1994, Zanetti et al 2000). In the larger of these studies (16 patients) abnormal tissuewas only seen in half the sites corresponding to BME, with marrow necrosis,¢brosis and abnormal, remodelled bony trabeculae being the commonestabnormalities and actual oedema being a very uncommon ¢nding (Zanetti et al2000). A link with structural deterioration has been demonstrated for BMElesions: in 256 OA knees followed for up to 30 months, there was a strongassociation between progressive radiographic joint space loss and the presence ofBME in the same (medial or lateral) compartment of the knee, even when adjustedfor a known risk factor, knee alignment (Felson et al 2003).Just as importantly, these BME lesions have been linked to OA pain in recent
studies. One interesting study described a range of subchondral tibialabnormalities (using T1-weighted images and therefore not directly comparableto other BME studies) in OA patients selected for recent onset of symptoms(mean duration 6 months) and followed for up to 4.5 years (Lotke et al 2000).The lesions described by widespread MRI subchondral changes extending intothe metaphysis and with MRI appearance similar to that of osteonecrosis (whichtypically demonstrates BME) were predominantly associated with persistence ofpain over the follow-up period.The most convincing study on the importance of BME and pain involved 401
radiographic OA knee participants, 50 of whom had no knee pain (Felson et al2001). Subjects had coronal T2-weighted fat-suppressed MRI scans and BME
OA PAIN AND MRI 195

lesions were graded 0^3 depending on size. Frequency of BME lesions increasedwith radiographic grade (Kellgren Lawrence, KL, graded on posteroanteriorradiographs only) of OA: 48% of KL grade 0 had BME compared with 100% ofthose with KL grade 4. The BME lesions were present in 78% of the painful kneescompared with 30% of the non-painful group (P50.001), but remarkably largelesions (graded 2 or 3) were present in 36% versus 2% respectively (P50.001).However, this study did not demonstrate an association of BME with painseverity. Another recent study looked at 120 women divided into 4 groups onthe basis of presence or absence of knee pain and presence or absence ofradiographic changes (Sowers et al 2003). This study used appropriate proton
196 CONAGHAN & FELSON
FIG. 2. Axial fat-suppressedMRI image of an osteoarthritic knee demonstratingmassive bonemarrow oedema in a femoral condyle and to a lesser extent in the patella (courtesy of DrDamienLoeuille, Nancy, France).

density, fat-suppressed sequences to identify BME, which was graded by size on a0^2 (grade 0 absent, grade 151 cm2, grade 241 cm2) scale. Although BME wascommon and its frequency similar in painful and non-painful OA groups, largerlesions (41 cm2) were more frequent in the painful OA knee group (36 versus14% in the painless OA group, P50.05). In line with the radiographic ¢ndingsin the previous study (Felson et al 2001), women with these large BME lesionswere more likely to have full thickness cartilage defects; the painful radiographicOA group with full thickness cartilage defects, adjacent subchondral cortical boneabnormalities and BME had signi¢cantly greater likelihood of painful OA(OR 3.2).
Periarticular lesions
It is certainly possible that knee pain in OA subjects may arise from extra-articularstructures, such as the bursae located around the joint. This concept may besupported by a unique study using local anaesthetic to examine knee pain(Creamer et al 1996). In this study, 6 out of 10 subjects receiving intra-articularlocal anaesthetic had complete relief of pain at 1 hour, suggesting that not allstructures giving rise to joint pain were in contact with the joint cavity. Hill andcolleagues looked at the prevalence and pain relationship of MRI-detectedperiarticular lesions in their Boston cohort (Hill et al 2003). They categorizedabnormalities as being peripatellar (prepatellar, super¢cial or deep infrapatellabursitis) or periarticular (including semimembranosus-tibial collateral ligamentbursitis, anserine bursitis, iliotibial band syndrome or tibio¢bular cyst). Thefrequency of peripatellar lesions was not signi¢cantly di¡erent betweenparticipants with radiographic OA with and without knee symptoms (12%versus 21% respectively). However periarticular pathology was seen morefrequently in the radiographic OA knee pain group than in the pain-free group(15% versus 4% respectively, P¼0.004). Neither peripatellar nor periarticularlesions were seen in subjects without pain or radiographic OA.
Menisci
MRI has long been seen as the best non-invasive test for evaluating meniscalpathology (Cheung et al 1997); meniscal damage, and in particularmeniscectomy, has also been associated with subsequent increased risk ofsymptomatic and radiographic OA (Roos et al 2001, Englund et al 2003).Abnormalities of the menisci are very common in OA: the prevalence of meniscaltears in a large elderly community-based cohort (the Health ABC study referred toabove) with radiographic knee OA subjects was 83% for men and 73% for women(Guermazi et al 2002). Another study also demonstrated a high prevalence of
OA PAIN AND MRI 197

meniscal tears even in elderly patients without symptoms, and that tears weremorecommon in symptomatic OA knees compared with asymptomatic controls (91%versus 76% respectively, P50.005) (Bhattacharyya et al 2003). This latter studyalso showed that the frequency of meniscal tears increased with higher KLradiographic grade, but that both pain and function were no di¡erent betweenOA patients with and without meniscal tears. This suggests that meniscal tearsare not contributing to pain in OA knees, although they may occasionallycontribute to mechanical (locking) symptoms.
Ligaments
The association of anterior cruciate ligament (ACL) tears with subsequentdevelopment and progression of radiographic OA is well described, andcombined injuries involving collateral ligaments result in higher incidence of OA(Lundberg & Messner 1997, Gillquist & Messner 1999). Modern MRI cohortshave again suggested a surprisingly high frequency of ligamentous abnormalitiesin OA patients, with the Health ABC study demonstrating partial or completeligament tear (cruciate or collateral ligaments) frequencies of 27% in men and30% in women (Guermazi et al 2002). Preliminary data from a Boston cohortdemonstrated a complete ACL tear in 26% of 234 subjects with painful,radiographic OA knee (Amin et al 2003). Analysis of this cohort demonstratedthat the ACL tears were not associated with worse pain or disability whencompared to those subjects without ACL tears, nor was it associated with greaterprogression of pain over a 30 month follow-up.
Osteophytes
Osteophytes are integral to radiographic de¢nitions of OA, andMRI studies havedemonstrated correlations between radiographic osteophytes and cartilage defectsin all compartments of the knee (Boeg�rd et al 1998a,b). Not surprisingly, themulti-planar nature of MRI has resulted in a marked sensitivity to osteophytedetection (see Fig. 1) and the Health ABC cohort reported osteophytes in 72% ofmen and 67%of women (Taouli et al 2002). As yetMRI studies have not suggestedan association of osteophytes with joint pain, although radiographic studies havepreviously demonstrated this association (Cicuttini et al 1996).
Summary
This presentation could not attempt to address the rapid improvements in MRItechnology that will improve joint evaluation. The remarkable ability of MRI tovisualize all the structures in the OA joint has already dramatically and rapidly
198 CONAGHAN & FELSON

increased our understanding of potential sources of OA pain, although suchstudies have been limited largely to the knee. Important candidate featuresinclude synovitis and e¡usions, BME and periarticular lesions such as anserinebursitis. The knowledge emerging from MRI cohorts will help us betterunderstand pain and structural progression and their complex inter-relationship,ultimately allowing for a rational basis for therapeutic strategies.
References
Adalberth T, Roos H, Lauren M et al 1997 Magnetic resonance imaging, scintigraphy, andarthroscopic evaluation of traumatic hemarthrosis of the knee. Am J Sports Med 25:231^237
Amin S, LaValley MP, Niu J et al 2003 Complete anterior cruciate ligament (ACL) tear and riskfor radiographic and symptom progression in subjects with knee osteoarthritis. ArthritisRheum 48:S70
Ayral X, Pickering EH, Woodworth TG, Loose LD, MacKillop N, Dougados M 2002Synovitis is not correlated with the level of symptomatic severity in painful kneeosteoarthritis patients. Ann Rheum Dis 61:S37
Bergman AG, Willen HK, Lindstrand AL, Pettersson HT 1994 Osteoarthritis of the knee:correlation of subchondral MR signal abnormalities with histopathologic and radiographicfeatures. Skeletal Radiol 23:445^448
Bhattacharyya T, Gale D, Dewire P et al 2003 The clinical importance of meniscal tearsdemonstrated by magnetic resonance imaging in osteoarthritis of the knee. J Bone JointSurg Am 85:4^9
Boeg�rd T, Rudling O, Petersson IF, Jonsson K 1998a Correlation between radiographicallydiagnosed osteophytes and magnetic resonance detected cartilage defects in thepatellofemoral joint. Ann Rheum Dis 57:395^400
Boeg�rd T, Rudling O, Petersson IF, Jonsson K 1998b Correlation between radiographicallydiagnosed osteophytes and magnetic resonance detected cartilage defects in the tibiofemoraljoint. Ann Rheum Dis 57:401^407
Bollet AJ 2001 Edema of the bone marrow can cause pain in osteoarthritis and other diseases ofbone and joints. Ann Intern Med 134:591^593
Cheung LP, Li KCP, Hollett MD, Bergman AG, Herfkens RJ 1997 Meniscal tears of the knee:accuracy of detection with fast spin-echo MR imaging and arthroscopic correlation in 293patients. Radiology 203:508^512
Cicuttini FM, Baker J, Hart DJ, Spector TD 1996 Association of pain with radiological changesin di¡erent compartments and views of the knee joint. Osteoarthritis Cartilage 4:143^147
Conaghan PG, O’Connor P, McGonagle D et al 2003 Elucidation of the relationship betweensynovitis and bone damage: a randomisedMRI study of individual joints in patientswith earlyrheumatoid arthritis. Arthritis Rheum 48:64^71
Creamer P, Hochberg MC 1997 Why does osteoarthritis of the knee hurt� sometimes? Br JRheumatol 36:726^728
Creamer P, Hunt M, Dieppe P 1996 Pain mechanisms in osteoarthritis of the knee: e¡ect ofintraarticular anesthetic. J Rheumatol 23:1031^1036
D’Agostino MA, Le Bars M, Schmidely N et al 2003 Interest of ultrasonography to detectsynovitis in painful knee osteoarthritis in daily practice. Arthritis Rheum 48:S80
Dieppe PA, Cushnaghan J, Shepstone L 1997 The Bristol ‘OA500’ Study: progression ofosteoarthritis (OA) over 3 years and the relationship between clinical and radiographicchanges at the knee joint. Osteoarthritis Cartilage 5:87^97
OA PAIN AND MRI 199

Dieppe P 1999 Subchondral bone should be the main target for the treatment of pain and diseaseprogression in osteoarthritis. Osteoarthritis Cartilage 7:325^326
Englund M, Roos E, Lohmander LS 2003 Impact of meniscal tear on radiographic andsymptomatic knee osteoarthritis: a sixteen year followup of meniscectomy with matchedcontrols. Arthritis Rheum 48:2178^2187
Felson DT, Naimark A, Anderson J, Kazis L, Castelli W, Meenan RF 1987 The prevalence ofknee osteoarthritis in the elderly: the Framingham Osteoarthritis Study. Arthritis Rheum30:914^918
Felson DT, Lawrence RC, Dieppe PA et al 2000 Osteoarthritis: new insights. Part 1: the diseaseand its risk factors. Ann Intern Med 133:635^646
FelsonDT, Chaisson CE,Hill CL et al 2001 The association of bonemarrow lesions with pain inknee osteoarthritis. Ann Intern Med 134:541^549
Felson DT, McLaughlin S, Goggins J et al 2003 Bone marrow edema and its relation toprogression of knee osteoarthritis. Ann Intern Med 139:330^336
Fernandez-Madrid F, Karvonen RL, Teitge RA et al 1995 Synovial thickening detected by MRimaging in osteoarthritis of the knee con¢rmed by biopsy as synovitis. Magn Reson Imaging13:177^183
Gillquist J, Messner K 1999 Anterior cruciate ligament reconstruction and the long-termincidence of gonarthrosis. Sports Med 27:143^156
Guermazi A, Taouli B, Lynch JA et al 2002 Prevalence of meniscus and ligament tears and theircorrelation with cartilage morphology and other MRI features in knee osteoarthritis (OA) inthe elderly. The Health ABC Study. Arthritis Rheum 46:S567
Hannan MT, Felson DT, Pincus T 2000 Analysis of the discordance between radiographicchanges and knee pain in osteoarthritis of the knee. J Rheumatol 27:1513^1517
Hill CL, Gale DG, Chaisson CE et al 2001 Knee e¡usions, popliteal cysts, and synovialthickening: association with knee pain in osteoarthritis. J Rheumatol 28:1330^1337
Hill CL, Gale DR, Chaisson CE et al 2003 Periarticular lesions detected on magnetic resonanceimaging: prevalence in knees with and without symptoms. Arthritis Rheum 48:2836^2844
Lethbridge-CejkuM, Scott WW Jr, Reichle R et al 1995 Association of radiographic features ofosteoarthritis of the knee with knee pain: data from the Baltimore Longitudinal Study ofAging. Arthritis Care Res 8:182^188
LoeuilleD, Toussaint F, Champigneulles J et al 2002MR evaluation of synovial in£ammation inknee OA: histological correlation. Arthritis Rheum 46:S566
Lotke PA, Ecker ML, Barth P, Lonner JH 2000 Subchondral magnetic resonance imagingchanges in early osteoarthrosis associated with tibial osteonecrosis. Arthroscopy 16:76^81
Lundberg M, Messner K 1997 Ten-year prognosis of isolated and combined medial collateralligament ruptures. A matched comparison in 40 patients using clinical and radiographicevaluations. Am J Sports Med 25:2^6
Myers SL, Brandt KD, Ehlich JW et al 1990 Synovial in£ammation in patients with earlyosteoarthritis of the knee. J Rheumatol 17:1662^1669
�stergaard M, Stoltenberg M, Lovgreen-Nielsen P, Volck B, Jensen CH, Lorenzen I 1997Magnetic resonance imaging-determined synovial membrane and joint e¡usion volumes inrheumatoid arthritis and osteoarthritis. Comparison with the macroscopic and microscopicappearance of the synovium. Arthritis Rheum 40:1856^1867
Pelletier JP, Martel-Pelletier J, Abramson SB 2001 Osteoarthritis, an in£ammatory disease.Potential implication for the selection of new therapeutic targets. Arthritis Rheum 44:1237^1247
Peterfy CG 2002 Imaging of the disease process. Curr Opin Rheumatol 14:590^596Peterfy CG, Majumdar S, Lang P, van Dijke CF, Sack K, Genant HK 1994 MR imaging of thearthritic knee: improved discrimination of cartilage, synovium, and e¡usion with pulsedsaturation transfer and fat-suppressed T1-weighted sequences. Radiology 191:413^419
200 CONAGHAN & FELSON

Peterfy CG, Guermazi A, Zaim S et al 2004 Whole-Organ Magnetic Resonance Imaging Score(WORMS) of the knee in osteoarthritis. Osteoarthritis Cartilage 12:177^190
Roos EM, Ostenberg A, Roos H, Ekdahl C, Lohmander LS 2001 Long-term outcome ofmeniscectomy: symptoms, function, and performance tests in patients with or withoutradiographic osteoarthritis compared tomatched controls. Osteoarthritis Cartilage 9:316^324
Smith MD, Trianta¢llou S, Parker A, Youssef PP, Coleman M 1997 Synovial membranein£ammation and cytokine production in patients with early osteoarthritis. J Rheumatol24:365^371
Sowers MF, Hayes C, Jamadar D et al 2003 Magnetic resonance-detected subchondral bonemarrow and cartilage defect characteristics associated with pain and X-ray-de¢ned kneeosteoarthritis. Osteoarthritis Cartilage 11:387^393
Taouli B, Guermazi A, Zaim S et al 2002 Prevalence and correlates of knee cartilage defects,meniscal lesions and other abnormalities evaluated by MRI in a population sample of kneeswith normal x-rays. The Health ABC Study. Arthritis Rheum 46:S148
Wildy KS, Nevitt MC, Kwoh CK et al 2002MRI ¢ndings associated with knee pain: analysis ofdiscordant knee pairs in Health ABC. Arthritis Rheum 46:S148
Zanetti M, Bruder E, Romero J, Hodler J 2000 Bone marrow edema pattern in osteoarthriticknees: correlation between MR imaging and histologic ¢ndings. Radiology 215:835^840
DISCUSSION
Brandt: Are there any studies looking for the presence of e¡usion by bothultrasound and MRI in the same patient?Conaghan: Yes. Back in 1995 Mikkel �stergaard looked at OA or rheumatoid
arthritis (RA) knees. He did correlations and e¡usion was the feature thatcorrelated best (�stergaard et al 1995). Ultrasound detected e¡usions 30% moreoften than clinical examination.Brandt: What about the presence and size of e¡usions, as judged by ultrasound
versus MRI of the same knee?Conaghan: When e¡usions are seen by both modalities they correlate very well,
but MRI is more sensitive.Creamer: I have a question about osteophytes on MRI: are they all the same?
There was a thought that perhaps growing osteophytes might be painful as theyirritate the periosteum, whereas more established osteophytes may not be. WhenMRI ¢rst came out we noticed some osteophytes that were brighter.Conaghan: It would be nice to go back and look at some of the cohorts according
to bone oedema in the osteophytes alone. This hasn’t really been looked at. Thereare a lot of data to be gained out of the cohorts we have. I suspect that while theosteophytes are remodelling we might have some activity. This is why I say thatBME isn’t BME, because the bone oedema in a remodelling osteophyte is probablygoing to be quite di¡erent to the bone oedema of microfractured trabeculae.Felson: I think the radiographic studies are pretty consistent in suggesting a
correlation between the presence of osteophytes or their size and pain.Creamer: Is that correlation with pain with the presence or absence of pain?
OA PAIN AND MRI 201

Felson:No, it was correlated with severity.Schaible: I think this is an important study. Presumably you had a good reason
for doing MRI scans of the patients. Did all of them come because they had somepain or knee problems?Conaghan:There are a number of di¡erent cohorts, but of the two largest I talked
about, one of them is a community based cohort, so these were not justsymptomatic cohorts. The problems with the smaller cohorts is that they are justsymptomatic presentations. I think we learn more from the community-basedcohorts.Schaible: In the community cohorts are there people with no pain in which you
check whether they have any knee abnormalities?Felson: I was involved in both cohorts that Philip described. The Health ABC
cohort is a population-based cohort of people in theUSAaged 70^79.Roughly halfof theMRIswere frompeople in that large population-based cohortwhohappenedto have knee pain, and the other half were in people without any knee pain. Wedesigned the pain MRI study so we could control for confounders of pain such asactivity andmedication.We compared a unilateral painful kneewith the other kneethat wasn’t painful to see whether the MRI features were di¡erent. In the BOKSstudy, the comparison was between a person who has symptomatic OA andother people who have radiographic OA without symptoms. The comparisonsin the BOKS study were between a large number of people with knee pain and amuch smaller number that don’t have any symptoms, but have radiographicdisease.Pisetsky: I have a comment about synovitis. We have drugs from RA that work
on synovitis. When you see this amount of synovitis in OA, do you do anythingtherapeutically di¡erent?Conaghan: That is exactly why we are looking at methods of determining this.
What clinicians want out of this is an algorithm that enables us to pick thesubgroups that we can target appropriate therapies to. Perhaps even hyaluronanscould work if we can target them to the right group.Hunter:Another aspect that needs to be taken into account is the way that pain is
measured. In the Health ABC study they are looking at WOMAC global pain, notspeci¢cally activity-related pain.Koltzenburg: Are there techniques such as voxel-based morphometry that you
can apply in longitudinal studies? Can you use gadolinium to gauge the intensityof the synovitis, or other contrast media?Conaghan:There is a lot of work on the technology that I didn’t present. There is
also a lot of work on cartilage volume. Cartilage reliability and precision is muchworse in areas where there are curved surfaces, so the knee is better than the hip towork on. In terms of quantitating, we have done work on synovial volume. Thereis quite a lot of published work on this.
202 DISCUSSION

Koltzenburg: Have people looked at the e¡ects of non-steroidal anti-in£ammatory drugs (NSAIDs) on synovial thickness?Conaghan:Not at that sort of level in OA. In RA we have looked at this, but we
haven’t published this yet. We are able to see di¡erences in synovial volume.Hunter: There are a number of studies including a recent one from Canada
looking at the use of intra-articular steroids (Raynauld et al 2003). This suggestedthat there was little if any measurable change on synovitis scored semi-quantitatively.Conaghan: Yes, there are six or seven studies on intra-articular steroid studies
using MRI as an outcome. But there are no published studies for NSAIDs.Brandt:We have performed a pilot study in patients with knee OAwho were on
NSAIDs, whom we studied with a battery of imaging procedures. The thingsthat were most closely related to the increase in joint pain after they were washedout of their NSAIDs were changes in synovial e¡usion volume and in synovialvolume.Van den Berg: I was intrigued with what you said about the patellar^femoral
lesions not relating to pain. Let me try to make a parallel with animal modelstudies. If you make an OA model through direct enzymatic injury, for example,you see damage at multiple sites in the joint. If you make a model withbiomechanical insults, inducing joint instability, you will normally only seelesions in the tibial^femoral area. Your selective pain story might suggest thatonly such biomechanically induced lesions will give an e¡usion at load bearingareas that will relate to pain, which you don’t have if you make OA in a non-biomechanical way. There might be a relationship between biomechanicalinstability and joint pain, and having lesions of the tibial^femoral area.Conaghan: I think that is right: position is important and the location of bone
oedema is giving us a clue to what is important. But I think if you have patellar^femoral chondropathy, you don’t get synovitis there but somewhere else.We haveto look at the whole joint. I ¢nd these data really interesting. Theworst group for aclinician to deal with in the knee is patellar^femoral arthritis. This is the areawe cando the least for.Herzog: If you do make instabilities in the tibial^femoral joint in some animal
models you can clearly see an e¡ect on the patella^femoral joint.Van den Berg: The patella^femoral area is not showing the dominant lesions,
though. I am not saying that it is not there. However, if you make OA lesionswithout an instability, then normally the lesions in the tibial^femoral joint areless dominant compared with the patella^femoral area. This is what we see inmice, which might of course be di¡erent in other species.I have a comment about synovitis. It is interesting to look at this in people and
inducedmodels ofOA, but to be honest, if you look in spontaneousOA in animals,there is hardly any synovitis. Why does it have such prominence in human disease,
OA PAIN AND MRI 203

while it is not necessarily involved in the animal lesions which go to fulldestruction?Conaghan: I have done some work with animal models and I used to think this
was because of weight bearing. But even the big animal models don’t seem to getthe degree of synovitis that humans get later on. I can’t explain this.Creamer: With regard to the common appearance of synovitis in human OA
whereas it is rarely seen in animal OA, this may be a presentation bias. If it iscausing symptoms, it may be that these are the people that we are seeing inhuman OA, whereas there are lots of people out there with OA who we neverget to see.Pisetsky:Have you correlated thosewith synovitis with any biomarker such as C
reactive protein (CRP)?Conaghan:No.Felson: There were two very nice large-scale papers on CRP and its association
with synovitis and OA at the 2002 American College of Rheumatology (ACR)meeting. One was from the Health ABC study (Nevitt et al 2002) with a verylarge n and lots of synovitis on MRI. There was a univariate correlation withCRP, but CRP is very strongly correlated with weight, as is OA. Once weightwas adjusted for there was nothing left. This was also seen in a radiographicstudy from North Carolina (Jordan et al 2002). The best current data thereforesuggest that CRP and OA are not on their own correlated with one another, butthat this is mediated through a relationship with obesity.Pisetsky: In that study, were those who had synovitis any di¡erent to those who
did not?Felson: The CRP study from Health ABC was a synovitis�CRP correlative
study.Van den Berg: If you want to push synovitis as being something related to pain,
then why don’t painkillers work in OA? There is a tendency now to believe thatsynovitis is not only important in RA but also OA. Perhaps we should even treatOAwith anti-tumour necrosis factor (TNF) and anti-interleukin (IL)1 if this is thecase. But then if you think the synovitis is really contributing, you would expectthat the pain would be rather similar in RA and OA, but it is not. The OA pain isprobably more related to oedema in the bone as compared with things taking placein the synovial tissue.Conaghan: I think they are both important. I didn’t present the data on synovitis
and progression of chondropathy: this is now pretty clear cut. Two large studieshave shown that synovitis is related to progression of chondropathy both at thepatellar femoral joint and in the medial tibiofemoral joint. For progression alone,synovitis is worth treating. For pain, I think there are some parameters we need totweak. One of them is how we measure pain, and secondly our measures ofsynovitis so far have not been very thorough. They have been global measures of
204 DISCUSSION

synovitis rather than compartment speci¢c measures. I think this literature willcome out in the next year.Dieppe: I sense there are dangerous concepts of causality creeping into this
conversation. You could interpret all these data as saying that bad OA doesbadly: everything is worse in the worse people. Let’s beware of linking thesecausally in casual conversation.
References
Jordan JM, Luta G, Stabler T 2002 Serum C-reactive protein (CRP) and osteoarthritis (OA).Arthritis Rheum 46(suppl):S148
Nevitt M, Felson D, Peterfy C et al 2002 In£ammation markers (CRP, TNF-a, IL-6) are notassociated with radiographic or MRI ¢ndings of knee OA in the elderly: the Health ABCStudy. Arthritis Rheum 46(suppl):S148
�stergaard M, Court-Payen M, Gideon P et al 1995 Ultrasonography in arthritis of the knee. Acomparison with MR imaging. Acta Radiologica 36:19^26
Raynauld JP, Buckland-Wright C, Ward R et al 2003 Safety and e⁄cacy of long-termintraarticular steroid injections in osteoarthritis of the knee: a randomized, double-blind,placebo-controlled trial. Arthritis Rheum 48:370^377 [Erratum in Arthritis Rheum 48:3300]
OA PAIN AND MRI 205

The role of TRP channels in sensory
neurons
Martin Koltzenburg
Neural Plasticity Unit, Institute of Child Health, 30 Guildford Street, London WC1N 1EH,UK
Abstract. Two parallel processes characterize the contemporary pain ¢eld. Firstly,enormous progress is being made in the discovery of the cellular and molecularmechanisms responsible for the pathogenesis of pain and secondly, there is a growingappreciation that multiple mechanisms contribute to common clinical pain syndromes.The aim of this chapter is to provide a short overview how transient receptor potential(TRP) channels could contribute to acute and chronic pain states. TRP channels of thevanilloid family (TRPV1, TRPV2, TRPV3, TRPV4) are excited by heat stimuli whereasTRPM8 and ANKTM1 are cold responsive. TRPV1 and ANKTM1 are mediating thepungency of nociceptor-speci¢c chemicals such as capsaicin or mustard oil. Sensitizationof TRPV1 is an important mechanisms for heat hyperalgesia and thus the generation ofchronic pain symptoms.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 206^220
Several TRP channels are expressed in primary sensory neurons
Transient receptor potential (TRP) channels were ¢rst described in thephotoreceptors of the fruit £y Drosophila. In mammals TRP channels ful¢ldiverse functions and to date six subfamilies have been recognized on the basis oftheir structural homology (Minke & Cook 2002, Clapham 2003). Following theseminal discovery of the capsaicin receptor TRPV1 (Caterina et al 1997) it has beenrecognized that several other TRP channels are expressed in dorsal root ganglia orperipheral tissues innervated by sensory neurons, including TRPV2 (Caterina et al1999), TRPV3 (Peier et al 2002a, Smith et al 2002,Xu et al 2002 ), TRPV4 (Trost etal 2001, Guler et al 2002, Nilius et al 2003, Vriens et al 2004), TRPM8 (McKemyet al 2002, Peier et al 2002b) and ANKTM1 (Story et al 2003, Jordt et al 2004).
The sensitivity of nociceptors in normal tissues
There is broad consensus that acute pains as studied under laboratory conditionsare evoked by excitation of nociceptors with thin myelinated or unmyelinated
206
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

axons (Meyer et al 1994, Handwerker 1996, Raja et al 1999). Usingmicroneurographic recordings of primary a¡erents and psychophysicalmagnitude estimation techniques simultaneously in conscious humans,investigators have shown that cutaneous nociceptors can encode the intensity ofpainful heat (Gybels et al 1979), mechanical (Koltzenburg & Handwerker 1994,Schmidt et al 2000, Schmelz et al 2003) or chemical stimuli (Schmelz et al 2003).However, it is also agreed that ¢ring of a single nociceptor cannot necessarily beequated with the perception of pain. In human microneurographic experiments, itis not uncommon to activate individual nociceptors with painless stimuli.Moreover, mechano-heat sensitive C-¢bres (also known as polymodalnociceptors) innervating the hairy skin have thresholds around 41^43 8C, but thepsychophysical heat pain threshold of individuals is often considerably higher(LaMotte et al 1984). Correspondingly, increases of skin temperature that evokea low discharge rate of less than 0.3 impulses per second over short periods areusually painless (Van Hees & Gybels 1981). Furthermore, brief mechanicalimpact stimuli can elicit bursts of activity with instantaneous frequenciesexceeding 10Hz without being called painful (Koltzenburg & Handwerker1994). Moreover, there is also often a considerable time lag between the ¢ring ofnociceptors and the appearance of pain following application of algesic chemicals(Adriaensen et al 1980). In aggregate, these results led to the conclusion that bothtemporal and spatial summation in a population of nociceptors is important forencoding the magnitude of pain (Raja et al 1999).Apart from anatomical studies that have localized many TRP channels to
sensory neurons there is now a large body of evidence that TRP channels are themolecular correlate for many of the receptive properties of nociceptors.Heterologous expression studies have shown that TRPV1 (Caterina et al 1997),TRPV2 (Caterina et al 1999), TRPV3 (Peier et al 2002a, Smith et al 2002, Xu et al2002) and TRPV4 (Guler et al 2002) respond to heat. Mutant mice lacking TRPchannels have been very informative in revealing the contribution ofTRP channelsfor the functional properties of sensory neurons. Capsaicin has long been known tospeci¢cally excite nociceptors and mice lacking TRPV1 are completely insensitiveto this and related irritants and display a strongly reduced response to protonsindicating that TRPV1 is a main transducer in the peripheral pain pathway(Caterina et al 2000, Davis et al 2000). TRPV1 knockout mice also show areduced sensitivity to strong noxious heat stimuli. However, while the heat-induced currents of dorsal root ganglia in culture are completely abolished thereremains a signi¢cant heat sensitivity in sensory neurons recorded in situ (Caterinaet al 2000). The discrepancy for this observation is not fully understood. However,one possible explanation that would be compatible with these ¢nding is that thereare other heat transducing receptors in the target tissue that could releasemediatorsand thereby indirectly excite heat-sensitive nociceptors by a TRPV1-independent
TRP CHANNELS IN SENSORY NEURONS 207

mechanism. Indeed, other heat-sensitive TRP channels such as TRPV3 andTRPV4 are found in keratinocytes or other epithelial cells, but although wellpositioned for such a task their relatively low heat thresholds are not compatiblewith a role in nociception (Caterina 2003, Chung et al 2003).TRPV2 is a very strong candidate to mediate the heat sensitivity of thin
myelinated nociceptors. TRPV2 is found in neurons that do not express TRPV1with larger cell sizes than those of TRPV1-containing neurons, and it is co-expressed with markers for myelinated ¢bres (Caterina et al 1999). Importantly,the properties in the heterologous system resemble the heat currents in somecapsaicin-insensitive sensory neurons recorded in culture (Nagy & Rang 1999).This has led to the suggestion that TRPV2 is the key heat transducing moleculeon myelinated capsaicin-insensitive nociceptors whereas TRPV1 is the importantheat sensor on thin myelinated and unmyelinated capsaicin-sensitive nociceptors(Caterina & Julius 2001).Because both TRPV3 and TRPV4 are readily activated by innocuous
temperatures they are unlikely to play a signi¢cant role in nociception inisolation. However, because TRPV3 is found in many capsaicin-sensitiveneurons it is possible that it modulates the sensitivity of TRPV1, possibly byforming heteromultimers (Smith et al 2002). TRPV4 shows a wide spectrumof sensitivity including heat, arachidonic acid derivates, endocannabinoidsand hypo-osmolar stimuli (Guler et al 2002, Nilius et al 2003, Suzuki et al2003, Watanabe et al 2003) and animals lacking TRPV4 have reportedly amild impairment in their mechanical nociception (Suzuki et al 2003).Interestingly, hypo-osmolar stimuli, leading to swelling of neurons, havebeen used in culture to simulate mechanical stimuli (Viana et al 2001) andcells responding to this stimulus express TRPV4 (Alessandri-Haber et al2003).TRP channels have also been identi¢ed as the crucial molecules in cold sensation
(Jordt et al 2003). TRPM8 is found in a small percentage of sensory neurons in thetrigeminal or dorsal root ganglia. In heterologous expression studies TRPM8 isactivated by small reduction of temperatures and is sensitized by the coolingcompound menthol (McKemy et al 2002, Peier et al 2002b). The properties ofthis channel suggest that it is mediating the excitation of non-nociceptive coldsensitive neurons that signal innocuous cold (Konietzny 1984). However, manysensory neurons in the skin are also excited by noxious cold stimuli and it isgenerally thought that these neurons do not respond to menthol. Another cold-sensitive TRP channel, ANKTM1, has many properties that make it a strongcandidate for mediating the excitation of cold sensitive nociceptors. Thisincludes the restricted distribution amongst dorsal root ganglion neurons thatalso express markers for nociceptors (Story et al 2003), a cold activationthreshold in the noxious temperature range (Story et al 2003) and the response to
208 KOLTZENBURG

the irritant mustard oil (Jordt et al 2004) which is known to speci¢cally excitenociceptors (Reeh et al 1986).
The sensitivity of nociceptor changes during tissue in£ammation
The properties of nociceptors change profoundly following tissue injury andin£ammation. The release of in£ammatory mediators usually activatesnociceptors and protons, bradykinin, serotonin and prostaglandins (Reeh &Kress 1995) are amongst the most potent substances that excite nociceptiveterminals whereas non-nociceptive a¡erents are typically not a¡ected (Reeh &Kress 1995, Handwerker 1996, Raja et al 1999).While part of a persistent change in excitability is undoubtedly the consequence
of the maintained availability of mediators, there is also evidence that chronicin£ammation leads to long-lasting changes in the receptive properties ofnociceptors. Nerve growth factor (NGF) appears to play a prominent role in thisprocess of acute and long-term sensitization. NGF tissue concentration increasesrapidly in in£ammatory lesions (Donnerer et al 1993, McMahon & Bennett 1997)or after application of proin£ammatory cytokines notably interleukin 1b ortumour necrosis factor a (Sa¢eh-Garabedian et al 1995, Woolf et al 1997).Moreover, application of NGF to rodents results in profound hyperalgesia(Lewin & Mendell 1993). In the adult nervous system many nociceptors expressreceptors for NGF.While all small-diameter sensory neurons require NGF duringearly development (Crowley et al 1994), it is only the subpopulation of peptidergicneuroneswhich continues to express theNGF receptorsTrkA andp75 throughoutadult life. Non-peptidergic neurones express the receptor elements for thetransforming growth factor (TGF)b-related neurotrophic factor glial cell line-derived neurotrophic factor (GDNF), ret and GFRa (Snider & McMahon 1998,Airaksinen et al 1999). This suggests that NGF could sensitize the subpopulationof peptidergic nociceptors.During tissue in£ammation the hallmark of an altered excitability of polymodal
nociceptors is the ongoing activity and the strong sensitization to thermal, butusually not to mechanical stimuli. At the onset of an in£ammation there is alsothe excitation of mechanically insensitive nociceptors that appear to beparticularly important to signal some aspects of mechanical hyperalgesia(Schmidt et al 1994, Schmelz et al 2000, Koltzenburg et al 2002). The intensity ofthe ongoing discharge of these nociceptors correlates with the magnitude ofpersistent pain and hyperalgesia to heat in humans (Treede et al 1992,Koltzenburg 1995). Electrophysiological recordings of the receptive propertiesof thin myelinated and unmyelinated nociceptors innervating normal hairy skinor carrageenan-in£amed skin have shown a close correlation between nociceptorexcitability and NGF (Koltzenburg et al 1999). Following carrageenan
TRP CHANNELS IN SENSORY NEURONS 209

in£ammation, about half of the nociceptors displayed ongoing activity that wasonly rarely observed in nociceptors innervating non-in£amed skin.Spontaneously active ¢bres were sensitized to heat and displayed a more thantwofold increase in their discharge to a standard noxious heat stimulus, but therewere no changes in the same ¢bres tomechanical stimuli. Furthermore, the numberof nociceptors responding to the algesic mediator bradykinin increasedsigni¢cantly. When the NGF-neutralizing molecule TrkA^IgG was co-administered with carrageenan at the onset of the in£ammation, primary a¡erentnociceptors did not sensitize and displayed essentially normal response propertiesalthough the in£ammation as evidenced by tissue oedema developed normally.Thus, endogenous NGF is an important factor in the sensitization of nociceptorsand it is tempting to suggest that the nociceptors that developed NGF-mediatedongoing activity are peptidergic neurons that express theTrkA receptor. Studies ofcultured dorsal root ganglion cells have extended these ¢ndings and have shownthat NGF regulates the capsaicin and bradykinin sensitivity (Bevan & Winter1995, Petersen et al 1998, Nicholas et al 1999).Several studies also investigated the relative contribution of both NGF
receptors and have shown that the relative importance of TrkA or p75 maydepend on the functional context. Whereas p75 knockout mice develop a normalacute heat hyperalgesia to systemic injection of recombinant human NGF(Bergmann et al 1998), dorsal root ganglion cells from these animals do not showthe up-regulation of bradykinin binding sites that can be normally induced byNGF (Petersen et al 1998). In agreement, several studies suggest that the acutee¡ects of NGF resulting in heat hyperalgesia are mediated by TrkA (Lewin &Barde 1996). TRPV1 is an essential component for the behavioural manifestationof heat hyperalgesia, because mice lacking TRPV1 show a complete abolition ofthe behavioural correlates of hyperalgesia to heat, but not to mechanical stimuli(Caterina et al 2000, Davis et al 2000). The strong link between TRPV1 andin£ammatory mediators for the generation of heat hyperalgesia is also apparenton a cellular level and there are at least two possible explanations for thisinteraction that are not mutually exclusive. In the model of bradykinin-inducedhyperalgesia it appears that the activation of bradykinin type 2 receptors leads tothe membrane translocation of protein kinase C epsilon (PKCe) (Cesare et al 1999)and subsequent phosporylation of TRPV1. Another possibility for TRPV1sensitization is the diminution of phosphatidylinositol-bisphosphate throughphospholipase C (PLC)-mediated hydrolysis. Thus, the excitatory PKCe e¡ectsand the disinhibition mediated by PLC would in aggregate result in thesensitization of TRPV1 and hence of nociceptors.In conclusion there is an increasing body of evidence implicating TRP channels
as a key foundation for the di¡erent functional properties seen in subpopulations ofsensory neurons. While several TRP channels of the vanilloid family appear to be
210 KOLTZENBURG

important for the signalling of normal heat, TRPM8 and ANKTM1 are coldsensors. TRPV1 and ANKTM1 mediate the pungency of many irritant chemicalsincluding capsaicin and mustard oil. The role of TRP channels formechanotransduction is currently less clear although some studies haveimplicated TRPV4 for this important function. The important role of TRPsunder pathological conditions leading to chronic pain channels is demonstratedby the crucial speci¢c involvement of TRPV1 in the generation of heathyperalgesia.
References
Adriaensen H, Gybels J, Handwerker HO, Van Hees J 1980 Latencies of chemically evokeddischarges in human cutaneous nociceptors and of the concurrent subjective sensations.Neurosci Lett 20:55^59
Airaksinen MS, Titievsky A, SaarmaM 1999 GDNF family neurotrophic factor signaling: fourmasters, one servant? Mol Cell Neurosci 13:313^325
Alessandri-Haber N, Yeh JJ, Boyd AE et al 2003 Hypotonicity induces TRPV4-mediatednociception in rat. Neuron 39:497^511
Bergmann I, Reiter R, Toyka KV, Koltzenburg M 1998 Nerve growth factor evokes hyper-algesia in mice lacking the low-a⁄nity neurotrophin receptor p75. Neurosci Lett 255:87^90
Bevan S,Winter J 1995Nerve growth factor (NGF) di¡erentially regulates the chemosensitivityof adult rat cultured sensory neurons. J Neurosci 15:4918^4926
Caterina MJ 2003 Vanilloid receptors take a TRP beyond the sensory a¡erent. Pain 105:5^9Caterina MJ, Julius D 2001 The vanilloid receptor: a molecular gateway to the pain pathway.Annu Rev Neurosci 24:487^517
CaterinaMJ, Le¥erA,MalmbergAB et al 2000 Impaired nociception and pain sensation inmicelacking the capsaicin receptor. Science 288:306^313
Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D 1999 A capsaicin-receptorhomologue with a high threshold for noxious heat. Nature 398:436^441
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D 1997 Thecapsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389:816^824
Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA 1999 Speci¢c involvement ofPKC-epsilon in sensitization of the neuronal response to painful heat. Neuron 23:617^624
Chung MK, Lee H, Caterina MJ 2003 Warm temperatures activate TRPV4 in mouse 308keratinocytes. J Biol Chem 278:32037^32046
Clapham DE 2003 TRP channels as cellular sensors. Nature 426:517^524Crowley C, Spencer SD, Nishimura MC et al 1994 Mice lacking nerve growth factor displayperinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergicneurons. Cell 76:1001^1011
Davis JB, Gray J, Gunthorpe MJ et al 2000 Vanilloid receptor-1 is essential for in£ammatorythermal hyperalgesia. Nature 405:183^187
Donnerer J, Schuligoi R, Stein C, Amann R 1993 Upregulation, release and axonal transport ofsubstance P and calcitonin gene-related peptide in adjuvant in£ammation and regulatoryfunction of nerve growth factor. Regul Pept 46:150^154
Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M 2002 Heat-evoked activation ofthe ion channel, TRPV4. J Neurosci 22:6408^6414
Gybels J, Handwerker HO, Van Hees J 1979 A comparison between the discharges of humannociceptive nerve ¢bres and the subject’s ratings of his sensations. J Physiol (Lond) 292:193^206
TRP CHANNELS IN SENSORY NEURONS 211

Handwerker HO 1996 Sixty years of C-¢ber recordings from animal and human skin nerves:historical notes. Prog Brain Res 113:39^51
Jordt SE, McKemy DD, Julius D 2003 Lessons from peppers and peppermint: the molecularlogic of thermosensation. Curr Opin Neurobiol 13:487^492
Jordt SE, Bautista DM, Chuang HH et al 2004 Mustard oils and cannabinoids excite sensorynerve ¢bres through the TRP channel ANKTM1. Nature 427:260^265
Koltzenburg M 1995 The stability and plasticity of the encoding properties of peripheral nerve¢bres and their relationship to provoked and ongoing pain. Semin Neurosci 7:199^210
Koltzenburg M, Handwerker HO 1994 Di¡erential ability of human cutaneous nociceptors tosignal mechanical pain and to produce vasodilatation. J Neurosci 14:1756^1765
Koltzenburg M, Bennett DL, Shelton DL, McMahon SB 1999 Neutralization of endogenousNGF prevents the sensitization of nociceptors supplying in£amed skin. Eur J Neurosci11:1698^1704
Koltzenburg M, Handwerker HO, Koerber HR 2002 Diversity of nociceptors�which areimportant for the generation of clinically relevant persistent pains? 10th World Congress ofPain (abstr 710)
Konietzny F 1984 Peripheral neural correlates of temperature sensation inman. HumNeurobiol3:21^32
LaMotte RH, Torebj˛rk HE, Robinson CJ, Thalhammer JG 1984 Time-intensity pro¢les ofcutaneous pain in normal and hyperalgesic skin: a comparison with C-¢ber nociceptoractivities in monkey and human. J Neurophysiol 51:1434^1450
Lewin GR, Mendell LM 1993 Nerve growth factor and nociception. Trends Neurosci 16:353^359
Lewin GR, Barde Y-A 1996 Physiology of the neurotrophins. Ann Rev Neurosci 19:289^317McKemyDD,NeuhausserWM, Julius D 2002 Identi¢cation of a cold receptor reveals a generalrole for TRP channels in thermosensation. Nature 416:52^58
McMahon SB, Bennett DLH 1997Growth factors and pain. In: Dickenson A, Besson J-M (eds)The pharmacology of pain. Springer Verlag, Berlin, p 135^165
Meyer RA, Campbell JN, Raja SN 1994 Peripheral neural mechanisms of nociception. In: WallPD, Melzack R (eds) Textbook of pain. Churchill Livingstone, Edinburgh, p 13^44
Minke B, Cook B 2002 TRP channel proteins and signal transduction. Physiol Rev 82:429^472Nagy I, Rang H 1999 Noxious heat activates all capsaicin-sensitive and also a sub-population ofcapsaicin-insensitive dorsal root ganglion neurons. Neuroscience 88:995^997
Nicholas RS,Winter J,Wren P, Bergmann R,Woolf CJ 1999 Peripheral in£ammation increasesthe capsaicin sensitivity of dorsal root ganglion neurons in a nerve growth factor-dependentmanner. Neuroscience 91:1425^1433
Nilius B, Watanabe H, Vriens J 2003 The TRPV4 channel: structure-function relationship andpromiscuous gating behaviour. P£ˇgers Arch 446:298^303
Peier AM, Reeve AJ, Andersson DA 2002a A heat-sensitive TRP channel expressed inkeratinocytes. Science 296:2046^2049
Peier AM, Moqrich A, Hergarden AC et al 2002b A TRP channel that senses cold stimuli andmenthol. Cell 108:705^715
PetersenM, Segond vB,HeppelmannB,KoltzenburgM1998Nerve growth factor regulates theexpression of bradykinin binding sites on adult sensory neurons via the neurotrophin receptorp75. Neuroscience 83:161^168
Raja SN, Meyer RA, Ringkamp M, Campbell JN 1999 Peripheral neural mechanisms ofnociception. In: Wall PD, Melzack R (eds) Textbook of pain. Churchill Livingstone,Edinburgh, p 11^57
Reeh PW, Kress M 1995 E¡ect of classic algogens. Semin Neurosci 7:221^226Reeh PW, Kocher L, Jung S 1986 Does neurogenic in£ammation alter the sensitivity ofunmyelinated nociceptors in the rat? Brain Res 384:42^50
212 KOLTZENBURG

Sa¢eh-Garabedian B, Poole S, Allchorne A, Winter J, Woolf CJ 1995 Contribution ofinterleukin-1 beta to the in£ammation-induced increase in nerve growth factor levels andin£ammatory hyperalgesia. Br J Pharmacol 115:1265^1275
Schmelz M, Schmid R, Handwerker HO, Torebj˛rk HE 2000 Encoding of burning painfrom capsaicin-treated human skin in two categories of unmyelinated nerve ¢bres. Brain123:560^571
SchmelzM, Schmidt R,Weidner C,HilligesM, TorebjorkHE,HandwerkerHO2003 Chemicalresponse pattern of di¡erent classes of C-nociceptors to pruritogens and algogens. JNeurophysiol 89:2441^2448
Schmidt R, SchmelzM,TorebjorkHE,HandwerkerHO2000Mechano-insensitive nociceptorsencode pain evoked by tonic pressure to human skin. Neuroscience 98:793^800
Schmidt RF, Schaible H-G, Messlinger K, Heppelmann B, Hanesch U, Pawlak M 1994 Silentand active nociceptors: structure, functions and clinical implications. In: Gebhart GF,Hammond DL, Jensen TL (eds) Proceedings of the 7th World Congress on Pain. IASPPress, Seattle, p 213^250
Smith GD, Gunthorpe MJ, Kelsell RE et al 2002 TRPV3 is a temperature-sensitive vanilloidreceptor-like protein. Nature 418:186^190
Snider WD, McMahon SB 1998 Tackling pain at source: new ideas about nociceptors. Neuron20:629^632
Story GM, Peier AM, Reeve AJ et al 2003 ANKTM1, a TRP-like channel expressed innociceptive neurons, is activated by cold temperatures. Cell 112:819^829
Suzuki M, Mizuno A, Kodaira K, Imai M 2003 Impaired pressure sensation in mice lackingTRPV4. J Biol Chem 278:22664^22668
Treede R-D, Meyer RA, Raja SN, Campbell JN 1992 Peripheral and central mechanisms ofcutaneous hyperalgesia. Prog Neurobiol 38:397^421
Trost C, Bergs C, Himmerkus N, Flockerzi V 2001 The transient receptor potential, TRP4,cation channel is a novel member of the family of calmodulin binding proteins. Biochem J355:663^670
VanHees J, Gybels J 1981 C nociceptor activity in human nerve during painful and non painfulskin stimulation. J Neurol Neurosurg Psychiat 44:600^607
Viana F, de la PE, Pecson B, Schmidt RF, Belmonte C 2001 Swelling-activated calciumsignalling in cultured mouse primary sensory neurons. Eur J Neurosci 13:722^734
Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B 2004 Cell swelling, heat,and chemical agonists use distinct pathways for the activation of the cation channel TRPV4.Proc Natl Acad Sci USA 101:396^401
Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B 2003 Anandamide andarachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 424:434^438
Woolf CJ, Allchorne A, Sa¢eh-Garabedian B, Poole S 1997 Cytokines, nerve growth factor andin£ammatory hyperalgesia: the contribution of tumour necrosis factor alpha. Br J Pharmacol121:417^424
Xu H, Ramsey IS, Kotecha SA et al 2002 TRPV3 is a calcium-permeable temperature-sensitivecation channel. Nature 418:181^186
DISCUSSION
Schaible: I have looked at the literature on the TTX-resistant Na+ channels.Many people say they are only expressed in primary a¡erent neurons that are
TRP CHANNELS IN SENSORY NEURONS 213

nociceptive, but there is also a big literature on TTX-resistant Na+ channels in theCNS. What is your view?Koltzenburg: Some of these ion channels, in particular the SNS channels, were
initially cloned only from dorsal root ganglion (DRG) neurons. Of course, thereare TTX-resistant Na+ channels outside the ganglion neurons, but they are notSNS. There are studies that show that some ion channels are up-regulated inpathological conditions in the CNS, and there are certainly also TTX-resistantNa+ channels in the heart but these are not identical to those neuronal ion channels.Grubb: I have been through the arguments before about the acid sensitivity of
ion channels. Howmuch acid is required to activate many of these ion channels? Isit within the physiological range?Koltzenburg:There is quite an important species di¡erence. The pH sensitivity in
the rat is much stronger than in the mouse. This could suggest that there may bedi¡erent stoichiometries of ion channels expressed in sensory neurons in the rat andmouse. There is consensus that in themouse the sustained proton current is mainlymediated by TRPV1.Grubb: You showed very elegantly the B2 receptor modulation of the vanilloid
receptor being mediated by PKC. For one of the inward rectifying K+ channelsthat we are interested in, people have shown in heterologous expression systemsthat this can be modulated not just by PKC, but also by bg subunits of the Gprotein-coupled receptor and phosphatidylinositol-4,5-bisphosphate (PIP2). Thischannel regulates membrane potential and we think it is present in nociceptors. Sothere are actually three ways of modulating this ion channel. Has this been shownfor the TRPV1 receptor?Koltzenburg: Peter McNaughton and David Julius would probably argue that
the two hypotheses that I put forward are exclusive. I don’t see them necessarilybeing exclusive, but I ¢nd it di⁄cult to see why they come to such di¡erentconclusions. David Julius works primarily on heterologous expression systemsand this may be one reason for discrepancies. The mechanisms he described areclearly possible for cellular activation, and phosphorylation and sensitization ofthe TRPV1 channel. I would be surprised if there were not more than onemechanism capable of modulating the function of this channel.Grubb: If you inhibit PKCe, does this completely abolish the B2 sensitization of
the TRPV1 receptor?Koltzenburg: That is an interesting question. To my knowledge this has never
been systematically investigated. One of the problems is that some of the phorbolesters that are used for PKC activation actually appear to bind and displace RTX,suggesting that they can competewith the vanilloid binding site. This is, of course,a confounding problem.Pisetsky: Can anyone clarify the di¡erence between acute and chronic
in£ammation in terms of this pattern? On the one hand, we hear that acute
214 DISCUSSION

in£ammation sensitizes; on the other hand, the histology shows that it causesdenervation, at least in the joint or synovium. Is the synovium special, or shouldwe look in another tissue?Grubb: I ¢nd this idea that the tissue becomes denervated in in£ammation very
unusual. All the evidence that Hans-Georg Schaible has provided in the joint andother people have provided in the skin shows that there is an increased sensitivityand there is no loss.Pisetsky:Do you see new nerves come into those sites?Koltzenburg:We looked in skin biopsies in patientswith acute contact dermatitis.
There we see a mild increase of GAP43, which is a protein associated with axonalsprouting.Schaible: Both phenomena have been reported, which makes it di⁄cult to sort
out.Kidd: I would not use theword denervation. I thinkwhat happens is that there is
minor retraction for a very small area. It could be a volume e¡ect related to oedema.I don’t believe that there is denervation in synovium. I think there is retraction oroedema to reduce the density.We do not see a neuropathic picture. It is very easy tolook for damaged nerves by looking in the DRG. If you have denervation youwould expect to see a neuropathic picture in the DRG, which we don’t see.Schaible: The only receptor which is missing now is the warmth receptor, or
would you say that there is already a candidate?Koltzenburg: I think there are two receptors, TRPV3 and TRPV4. The Novartis
group says that TRPV3 is not expressed on DRG neurons, but there are two otherpublications that suggest this is the case. If I understand correctly, the Novartisgroup originally cloned it from DRG libraries, so it is probably in DRG, butpossibly in low amounts. The interesting question is why this channel hasn’tbeen seen in animals lacking TRPV1. In the heterologous expression system forTRPV3, repetition of the stimulus often sensitized the current and it also hasvery strong outward rectifying properties. When we retrospectively looked atsome of our patch clamp recordings in the TRPV1 knockout animal, wesometimes see that there are small inward currents, but they are so small, inboth capsaicin-sensitive and -insensitive neurons that we think they are caused bynon-speci¢c e¡ects of the heating rather than re£ecting activation of TRPV3.Henry:Your HCN seems to be correlated with IB4, which is the presumed non-
nociceptive C ¢bre. All three seem to be more highly correlated with IB4 in thecapsaicin. This would suggest that if you are able to nullify the activation of thatchannel, you might modify alterations in non-nociceptive mechanisms.Koltzenburg:Recent reports have shown that there is an increase of the Ih current
in myelinated ¢bres after peripheral nerve damage (Yao et al 2003, Chaplan et al2003). To my knowledge this hasn’t been investigated in in£ammation and thesetwo studies focused on non-nociceptive a¡erents. Chaplan et al (2003) proposed
TRP CHANNELS IN SENSORY NEURONS 215

that the increased Ih current contributed to the ectopic ¢ring of large myelinated¢bres after nerve injury. Speci¢c blockers of Ih had some antihyperalgesicproperties in the nerve-injury model.Henry:Do those channels shift in animal models or human tissues?Koltzenburg: I think they will shift. There is only one publication out that has
looked speci¢cally in the rat at the expression of HCN isoforms after nerve injuryusing quantitative PCR (Chaplan et al 2003). They actually found a reduction inmRNA levels, but they showed an increase in functional properties of the channelsas well as an increase of the protein, implying signi¢cant post-transcriptionalregulation.Henry: In relation to types of pain, acute pain is not a big hurdle for medical
treatment� the real problem is chronic, non-cancer pain. It would be interestingto see over time how many of these ion channels start to correlate with di¡erenttypes of pain.Koltzenburg: If one looks at the TTX-resistant Na+ channel Nav1.8, there
is some indication that animals lacking this channel exhibit only a mildphenotype for acute cutaneous nociceptive pain. In models of visceral pain,the contribution of Nav1.8 may be stronger. There is a recent publicationindicating that spontaneous activity developing in axotomized neurons isactually much lower in animals lacking this TTX-resistant Na+ channel (Rozaet al 2003).Fox:Wehave also found that TRPV1blockers are e¡ective in all sorts ofmodels
of pain.What is interesting is that this includes in£ammatory pain and neuropathicpain: this was a bit of a surprise to us. The end point we used was mechanicalhyperalgesia as well as thermal hyperalgesia. It is not just the neurons that arepolymodal; these ion channels are responding to all sorts of things, perhaps inways that we don’t yet understand.Koltzenburg: I’d have thought that TRPV1 was not directly involved in
mechanotransduction.Fox: Somehow the ligand it is responding to endogenously must be, in some
way, doing something to the neuron that is responsive to it.Henry: We have also found that TRPV1 receptor inhibitor or antagonist is
e¡ective in our rheumatoid arthritis models.Schaible: A potentially important question is whether we should block ion
channels or mediators and mediator receptors.Koltzenburg: Blockade of conduction in a nociceptive neuron would prevent
signalling, regardless of which receptors it expresses at the receptive terminal.Thus, this would provide analgesia for a large number of stimuli or algesicmediators. On the other hand, if you have an in£ammatory cascade involvingmolecules such as TNFa, blockade of a key component upstream of the synthesisof other in£ammatorymediatorsmay also be very e¡ective. In rheumatoid arthritis
216 DISCUSSION

it appears that if you block the crucialmolecule, you also inhibit all the downstreamsignalling and the in£ammation. It probably depends on the pathophysiology. Itwould be ideal if we had a mechanism that blocked exclusively the action potentialpropagation in nociceptors. We all know that local anaesthetics are the bestanalgesics. One of the reasons we don’t give them more often is because ofmotor side e¡ects and complete sensory loss. But if you have something thatwould exclusively block nociceptors you would have an ideal analgesic,especially if you could modify it so that it just reduces the frequency of ¢ring. Infact ideally you want to have a reduction in ¢ring that results in anti-hyperalgesia,but not in anaesthesia.Grubb: Regulating the frequency is key. In a C ¢bre, when we elicit an action
potential, the cell after-hyperpolarizes and then it takes ages for its restingmembrane potential to return to normal� longer than a second. We areinterested in which ion channels regulate that repolarization. In repetitive ¢ringthis after-hyperpolarization gets deeper and longer. This is regulated byprostaglandins, histamine and bradykinin. One of the candidate channels is thesmall-conductance Ca2+-activated K+ channel.Rediske: Do you think this TRPV family could play a role as noxious stimuli
receptors in non-neuronal cells?Koltzenburg: They certainly produce an inward current in keratinocytes. It is
possible. The reason why I am not too keen on that particular hypothesis isbecause heat sensitivity is restricted to those neurons that express the capsaicinreceptor. If there were a generalized secondary e¡ect of TRPV channel activationby heat-sensitive keratinocytes and a release of a mediator, I would assume a morewidespread heat activation of nociceptors unless one assumes that TRPV1 and areceptor for the elusive keratinocyte mediator were expressed on the sameneuron.Pisetsky: Many of the agents that work in the animal models of rheumatoid
arthritis are anti-in£ammatory and analgesic. Are there any data on the e¡ects ofjust analgesics in thesemodels, in terms of the in£ammation and structural damage?Is there a way that this may make things worse?Koltzenburg: In some conditions there is a discrepancy between oedema
and nociceptor in£ammation. This can be seen in models of carrageenan wherethere is frequently profound oedema, but sometimes no sensitization. It mayalso depend on which tissue you study. Many studies have focused on hairy skinand there is room for the tissues to expand, whereas in more con¢ned spaces itwould be di⁄cult and the resulting pressure would contribute to nociceptoractivation. Conversely, if NGF is sequestered in the carrageenan model there isreduction of in£ammation, but the increased excitability of the nociceptors isstrongly reduced. Thus both phenomena of nociceptor sensitization and oedemacan be dissociated.
TRP CHANNELS IN SENSORY NEURONS 217

Schaible:Most of these mediators act on nerve cells and other cells. What is youropinion of the importance of the opioid receptors?Koltzenburg:The opioid receptors and other potentially analgesicmolecules such
as cannabinoid receptors are expressed there. It seems that the opioid receptorsbecome primarily functional under in£ammatory conditions.Pisetsky:Do opioids make arthritis worse?Grubb: There are studies where opiates have been injected intra-articularly. In
humans they produce a degree of analgesia, but I have no idea whether thisa¡ects disease progression.Mackintosh: There is evidence that they reduce cellular in£ux. If you
count that as a component of in£ammation then they are anti-in£ammatory.Schaible: Theoretically they would be ideal for treating pain with because it
doesn’t matter how the pain is produced.Felson: It is probably time to bring in an entity called the analgesic hip. There
was a study looking at people on the waiting list for total hip replacement,comparing people treated with potent anti-in£ammatories with those whoweren’t. The people treated with anti-in£ammatories had more degradationand degeneration of their hip and worse pathology at surgery. Thissuggested that pain is protective and functions to help us know when toprotect our joints. Too much suppression of pain may not be healthy. Iguess this calls into question this whole symposium! One wonders whether,by messing around too much with nature, we might not be helpingpeople.Koltzenburg: It is very clear that we don’t want to have an anaesthetic condition.
This is probably best illustrated in people with congenital analgesia. The organsthat are most e¡ectively traumatized tend to be joints.Felson: We talked about Charcot arthropathy yesterday. This is probably more
than just pain and temperature loss. It is almost certainly vibration and positionsense loss. This would be an example of pharmacological-induced Charcotarthropathy.Kidd: I think we might be unnecessarily damning analgesia here. I think you are
confusing analgesia with anti-in£ammatory. Non-steroidal anti-in£ammatorydrugs (NSAIDs) do more than just act as antihyperalgesics. What we need is acomparable study with comparable algesia to see whether the same e¡ect isachieved. We must also remember that the NSAIDs are not giving you ananaesthetic joint, they are just reducing the sensitization. They areantihyperalgesic as opposed to analgesic in the true sense of the word. If we aregoing to incriminate NSAIDs let’s look at alternative mechanisms for e¡ects onblood £ow and so on, not focusing exclusively on the analgesic e¡ect. There ismore than one explanation of that study.
218 DISCUSSION

Dieppe: I agree with that. The NSAIDs also have a signi¢cant e¡ect on bone.There is some other evidence that this so-called indomethicin or analgesic hip isactually a bone problem.Grubb: In a way, the holy grail is to leave the in£ammation alone and let it try to
do the repair process that it is trying to do, and block one of these important ionchannels that Martin Koltzenburg has been talking about. We don’t know whatthe mechanical transducer is. If we can ¢nd this, and it turns out to be selectivelyexpressed in small C ¢bres, then we are on to a winner.Fernihough: Do we know the expression pattern of TRPV1 in structures within
the joint?Koltzenburg: There is one anatomical study that found TRPV1 in a¡erents
innervating the facet joint of the rat. The percentage of joint a¡erents expressingTRPV1 was reported to be much lower than for the DRG population at large(Ohtori et al 2001). TRPV1 has also been found in non-neuronal cells such asurothelium. However, if one studies the expression level withimmunohistochemistry it is not very high (Birder et al 2002).Schaible: In our previous recordings in the cat we could block a lot of these joint
a¡erents with a higher dose of capsaicin (He et al 1990). At that time it was morefashionable to study desensitization. I would assume that many of them arecapsaicin sensitive, but this has not been shown histologically.Kidd: From my rheumatological rather than pain perspective, listening to this
debate on TRPV1 in which you have told us that it is a promiscuous receptor inthat it responds to heat but also protons and other things, in the joint heat is notsuch an important thing so could it be that protons are the most importantactivator? When I get a sore back I get a burning sort of pain, so do you thinkTRPV1 could explain a lot of the symptoms that people get? Acidicenvironments might be triggering this.Koltzenburg: If you induce spontaneous activity in capsaicin-sensitive
nociceptors, this results in pain and also central sensitization. Depending on howstrong this particular component is in deep somatic tissue, this would then bean important aspect. If one looks in skeletal muscle, it appears that theresponses to capsaicin and the proton responses via TRPV1 are rather small,compared with some of the acid-sensing ion channels (Benson et al 1999). Atthis moment in time we have little information about the relative importanceof TRPV1 and ASICs in skeletal muscle and joint. Alyson Fox referred tosome of the work done at Novartis where they used antagonists. Theyfound a much wider e⁄cacy than you would expect from the studies onmice lacking TRPV1. The knockout studies are quite clear that you wouldnot predict an involvement in cutaneous mechanical hyperalgesia andneuropathic pain. However, from what Alison has said it appears that if youuse TRPV1 antagonists you observe a wider-spread e¡ectiveness. This can be
TRP CHANNELS IN SENSORY NEURONS 219

explained in a couple of ways. It could be that compensatory changes occur inthe transgenic model. It is also possible that there are other non-TRPV1e¡ects of these drugs.
References
Benson CJ, Eckert SP, McCleskey EW 1999 Acid-evoked currents in cardiac sensory neurons: apossible mediator of myocardial ischemic sensation. Circ Res 84:921^928
Birder LA, Nakamura Y, Kiss S et al 2002 Altered urinary bladder function in mice lacking thevanilloid receptor TRPV1. Nat Neurosci 5:856^860
Chaplan SR, Guo HQ, Lee DH et al 2003 Neuronal hyperpolarization-activated pacemakerchannels drive neuropathic pain. J Neurosci 23:1169^1178
He X, Schepelmann K, Schaible HG, Schmidt RF 1990 Capsaicin inhibits responses of ¢nea¡erents from the knee joint of the cat to mechanical and chemical stimuli. Brain Res530:147^150
Ohtori S, Takahashi K, Chiba T, Yamagata M, Sameda H, Moriya H 2001 Brain-derivedneurotrophic factor and vanilloid receptor subtype 1 immunoreactive sensory DRGneurons innervating the lumbar facet joints in rats. Auton Neurosci 94:132^135
Roza C, Laird JM, Souslova V, Wood JN, Cervero F 2003 The tetrodotoxin-resistant Na+
channel Nav1.8 is essential for the expression of spontaneous activity in damaged sensoryaxons of mice. J Physiol 550:921^926
YaoH,DonnellyDF,MaC, LaMotte RH 2003Upregulation of the hyperpolarization-activatedcation current after chronic compression of the dorsal root ganglion. J Neurosci 23:2069^2074
220 DISCUSSION

Mechanisms that generate and
maintain bone cancer pain
Patrick W. Mantyh and Stephen P. Hunt*1
Departments of Preventive Sciences, Neuroscience, Psychiatry, and the Cancer Center,University of Minnesota, 18-208 Moos Tower, 515 Delaware Street SE, Minneapolis, MN55455, USA and *Department of Anatomy and Developmental Biology, Medawar Building,University College London, Gower Street, LondonWC1E 6BT, UK
Abstract. Although bone cancer pain can be severe and is relatively common, as itfrequently arises from metastases from breast, prostate, and lung tumours, very little isknown about the basicmechanisms that generate andmaintain this chronic pain. Tobeginto de¢ne themechanisms that give rise to bone cancer pain, we have developedmouse andrat models using the intramedullary injection and containment of tumour cells into thefemur. These tumour cells induced bone remodelling as well as ongoing and movementevoked pain behaviours similar to that found in patients with bone cancer pain. Inaddition there is a signi¢cant reorganization of the spinal cord that received sensoryinput from the cancerous bone and this reorganization generated a neurochemicalsignature of bone cancer pain that is both dramatic and signi¢cantly di¡erent from thatobserved in mouse and rat models of chronic neuropathic or in£ammatory pain. Thesemodels have provided insight into the mechanisms that drive cancer pain and have begunto allow the development ofmechanism-based therapies. Together these advances shouldreduce tumour-induced pain and su¡ering and signi¢cantly improve the quality of life ofcancer patients.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 221^240
The negative impact that cancer pain has on quality of life cannot be overestimated.As advances in cancer detection and therapy are extending the life expectancy ofcancer patients, there is increasing focus on improving the patients’ quality of life.Many patients present with pain as the ¢rst sign of cancer and 30^50% of all cancerpatients will experience moderate to severe pain (Mercadante 1997, Mercadante &Arcuri 1998, Portenoy & Lesage 1999, Portenoy et al 1999). Cancer-associatedpain can be present at any time during the evolution of the disease, but thefrequency and intensity of cancer pain tends to increase with advancing stages of
221
1This paper was presented at the symposium by Stephen P. Hunt.
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

cancer. 75^95% of patients with metastatic or advanced cancer will experiencesigni¢cant, life-altering cancer-induced pain (Mercadante 1997, Mercadante &Arcuri 1998, Portenoy & Lesage 1999, Portenoy et al 1999).The treatment of cancer pain can involve a variety of modalities. Therapies
targeted at decreasing tumour size are often e¡ective and include radiation,chemotherapy, and/or surgery, but these can be burdensome to administer andare accompanied by signi¢cant unwanted side e¡ects. Medications targeted atdecreasing in£ammation and pain such as non-steroidal anti-in£ammatory drugs(NSAIDs) or opiates can also be very useful, but these too are accompanied byunwanted side e¡ects.The relative ine¡ectiveness of current treatments re£ects the fact that therapies
have not changed for decades (Coyle et al 1990, Payne 1997, Payne et al 1998,Hoskin 2000). Largely because of treatment-associated side e¡ects, it has beenreported that 45% of cancer patients have inadequate and under-managed paincontrol (de Wit et al 2001, Meuser et al 2001). A formidable obstacle to thedevelopment of new therapies is the fact that the current neurobiological basisfor pharmacological treatments is largely empirical, and is based on scienti¢cadvances in painful conditions other than those induced by cancer.Recently, the ¢rst animal models of cancer pain have been developed. In the
mouse femur model, bone cancer pain is induced by injecting murine osteolyticsarcoma cells into the intramedullary space of the murine femur (Fig. 1) (Schweiet al 1999). Critical to this model is that the tumour cells are con¢ned within themarrow space of the injected femur and the tumour cells donot invade adjacent softtissues which would directly impact the joints of the muscle making behaviouralanalysis problematic (Schwei et al 1999, Honore et al 2000, Luger et al 2001).Following injection the tumour cells proliferate, and ongoing, movement, andtouch-evoked pain related behaviours develop that increase in severity with time.These pain behaviours correlate with the progressive tumour-induced bonedestruction that ensues and that appears to mimic the condition in patients withprimary or metastatic bone cancer (Fig. 1). These models have allowed us to gainmechanistic insights into how cancer pain is generated and how the sensoryinformation it initiates is processed as it moves from sense organ to the cerebralcortex under a constantly changing molecular architecture. As detailed below,these insights promise to fundamentally change the way cancer pain is controlled.
Primary a¡erent sensory neurons
Primary a¡erent sensory neurons are the gateway by which sensory informationfrom peripheral tissues is transmitted to the spinal cord and brain (Fig. 2), andthese neurons innervate the skin and every internal organ of the body includingmineralized bone, marrow, and periosteum. The cell bodies of sensory ¢bres that
222 MANTYH & HUNT

BONE CANCER PAIN 223
FIG. 1. Progressive destruction ofmineralized bone inmice with bone cancer. (A) Low poweranterior^posterior radiograph of mouse pelvis and hindlimbs after a unilateral injection ofsarcoma cells into the distal part of the femur and closure of the injection site with an amalgamplug (arrow) which prevents the tumour cells from growing outside the bone (Honore et al2000). Radiographs of murine femora (B) show the progressive loss of mineralized bonecaused by tumour growth. These images are representative of the stages of bone destruction inthe murine femur. At week 1 there is a minor loss of bone near the distal head (arrow); at week 2substantial loss of mineralized bone at both the proximal and distal (arrow) head; and at week 3loss of mineralized bone throughout the entire bone and fracture of distal head of the femur(arrow). Scale bar¼2mm. (Modi¢ed from Schwei et al 1999.)

innervate the head and body are housed in the trigeminal and dorsal root gangliarespectively, and can be divided into to two major categories: myelinated A ¢bresand smaller diameter unmyelinated C ¢bres. Nearly all large diameter myelinatedAb ¢bres normally conduct non-noxious stimuli applied to the skin, joints, andmuscles and thus these large sensory neurons usually do not conduct noxiousstimuli (Djouhri et al 1998). In contrast, most small-diameter sensory ¢bres�unmyelinated C ¢bres and ¢nely myelinated myelinated A ¢bres� are specializedsensory neurons known as nociceptors whose major function is to detect andconvert environmental stimuli that are perceived as harmful into electrochemicalsignals that are then transmitted to the central nervous system. Unlike primarysensory neurons involved in vision or olfaction which are required to detect onlyone type of sensory stimulus (light or chemical odorants, respectively), individualprimary sensory neurons of the pain pathway have the remarkable ability to detect a
224 MANTYH & HUNT
FIG. 2. Sensory neurons and detection of noxious stimuli due to tumour cells.Nociceptors usea diversity of signal transduction mechanisms to detect noxious physiological stimuli and manyof these signal transduction mechanisms may be involved in driving cancer pain. Thus, whennociceptors are exposed to products of tumour cells, tissue injury, or in£ammation theexcitability is altered and this nociceptive information is relayed to the spinal cord and thenonto higher centres of the brain. Some of the mechanisms that appear to be involved ingenerating and maintaining cancer pain include: activation of nociceptors by factors such asextracellular protons (+), endothelin 1 (ET-1), interleukins (ILs), prostaglandins (PG) andtumour necrosis factor (TNF).

wide range of stimulus modalities including those of physical and chemical nature(Basbaum & Jessel 2000, Julius & Basbaum 2001). To accomplish this,nociceptors express an extremely diverse repertoire of transduction moleculesthat can sense forms of noxious stimulation (thermal, mechanical, and chemical),albeit with varying degrees of sensitivity.In the past few years remarkable progress has been made toward understanding
the signalling mechanisms and speci¢c molecules that nociceptors use to detectnoxious stimuli. For example, the vanilloid receptor (VR-1, or TRPV1) that isexpressed by most nociceptors detects heat (Kirschstein et al 1999) and alsoappears to detect acid (Welch et al 2000), extracellular protons (Bevan andGeppetti 1994, Caterina et al 2000) and lipid metabolites (Tominaga et al 1998,Nagy & Rang 1999). In order to detect noxious mechanical stimuli, nociceptorsexpress mechanically gated channels which initiate a signalling cascade uponexcessive stretch (Price et al 2001). The cells also express several purinergicreceptors capable of sensing ATP, which is released from cells upon excessivemechanical stimulation (Krishtal et al 1988, Xu &Huang 2002).To sense noxious chemical stimuli, nociceptors express a complex array of
receptors capable of detecting in£ammation-associated factors released fromdamaged tissue including protons (Bevan & Geppetti 1994, Caterina et al 2000),endothelins (Nelson & Carducci 2000), prostaglandins (Alvarez & Fy¡e 2000),bradykinin (Alvarez & Fy¡e 2000) and nerve growth factor (McMahon 1996).Aside from providing promising targets for the development of more selectiveanalgesics, identi¢cation of receptors expressed on the nociceptor surface hasincreased our understanding how di¡erent tumours generate cancer pain in theperipheral tissues they invade and destroy.In addition to expressing channels and receptors that detect tissue injury,
sensory neurons are highly ‘plastic’, in that they can change their phenotype inthe face of a sustained peripheral injury. Following tissue injury, sensory neuronsub-populations alter patterns of signalling peptide and growth factor expression(Woolf & Salter 2000). This change in phenotype of the sensory neuron in partunderlies peripheral sensitization, whereby the activation threshold ofnociceptors is lowered so that what would normally be perceived as a mildnoxious stimulus is perceived as highly noxious (hyperalgesia). Damage to aperipheral tissue has also been shown to activate previously ‘silent’ or ‘sleeping’nociceptors which then become highly responsive to both normally non-noxious(allodynia) and noxious (hyperalgesia) stimuli.There are several examples of nociceptors that undergo peripheral sensitization
in experimental cancer models (Schwei et al 1999, Honore et al 2000a,b,c, Lugeret al 2001). In normal mice, the neurotransmitter substance P is synthesized bynociceptors and released in the spinal cord in response to a noxious but not anon-noxious palpation of the femur. In mice with bone cancer, normally non-
BONE CANCER PAIN 225

painful palpation of the a¡ected femur now induces the release of substance P fromprimary a¡erent ¢bres that terminate in the spinal cord. Substance P in turn bindsto and activates the neurokinin 1 receptor that is expressed by a subset of spinalcord neurons (Mantyh et al 1995a,b, Hunt & Mantyh 2001). Similarly, normallynon-noxious palpation of tumour-bearing limbs of mice with bone cancer alsoinduces the expression of c-Fos protein in spinal cord neurons. In normal animalsthat do not have cancer, only noxious stimuli will induce the expression of c-Fos inthe spinal cord (Hunt et al 1987). Peripheral sensitization of nociceptors may beinvolved in the generation and maintenance of bone cancer pain.
Properties of tumours that excite nociceptors
Tumour cells and tumour-associated cells that include macrophages, neutrophils,and T lymphocytes secrete a wide variety of factors that have been shown to
226 MANTYH & HUNT
FIG. 3. Development of chronic pain in mice with bone cancer. Time course of thedevelopment of ongoing, ambulatory, and touch-evoked pain behaviours in na|« ve, sham andsarcoma-injected animals. Note that the development of pain behaviours closely follows thetime course of tumour growth and tumour-induced bone remodelling seen in Fig. 1. Greyshading indicates a signi¢cance of P50.05 vs. sham.

sensitize or directly excite primary a¡erent neurons (Fig. 3). These includeprostaglandins (Nielsen et al 1991, Galasko 1995), endothelins (Nelson &Carducci 2000, Davar 2001), interleukins 1 and 6 (Watkins et al 1995, Opree& Kress 2000, DeLeo & Yezierski 2001), epidermal growth factor (Stoscheck &King 1986), transforming growth factor (Poon et al 2001, Roman et al 2001), andplatelet-derived growth factor (Daughaday & Deuel 1991, Radinsky 1991, Silver1992) and receptors for many of these factors have been shown to be expressed byprimary a¡erent neurons.While each of these factors may play an important role inthe generation of pain in particular forms of cancer, two that are currentlyapproved for use in humans with other non-cancer indications are theprostaglandins and the endothelins.Prostaglandins are pro-in£ammatory lipids that are formed from arachidonic
acid by the action of cyclooxygenase (COX) and other downstream synthetases.There are two distinct forms of the COX enzyme, COX-1 and COX-2.Prostaglandins have been shown to be involved in the sensitization and/or directexcitation of nociceptors by binding to several prostanoid receptors expressed bynociceptors which sensitize or directly excite nociceptors (Vasko 1995). Severaltumour cells and tumour-associated macrophages have been shown to expresshigh levels of COX-2 and produce high levels of prostaglandins (Dubois et al1996, Molina et al 1999, Kundu et al 2001, Ohno et al 2001, Shappell et al 2001).The COX enzymes are a major target of current medications, and COX
inhibitors such as aspirin or ibuprofen are commonly administered for reducingboth in£ammation and pain. A major problem with mixed COX inhibitors suchas aspirin or ibuprofen to block cancer pain is that these compounds inhibit bothCOX-1 and COX-2, and inhibition of the constitutively expressed COX-1 cancause bleeding and ulcers. In contrast, the new COX-2 inhibitors preferentiallyinhibit COX-2 and avoid the side-e¡ects of COX-1 inhibition, which may allowtheir use in treating cancer pain. Other experiments have suggested that COX-2 isinvolved in angiogenesis and tumour growth (Masferrer et al 2000, Moore &Simmons 2000, Sabino et al 2002), so in cancer patients COX-2 inhibitors mayhave the added advantage that in addition to blocking cancer pain, a COX-2inhibitor may also reduce the growth and metastasis of the tumour (Sabino et al2002). COX-2 antagonists show signi¢cant promise for alleviating at least someaspects of cancer pain, although clearly more research is required to fully de¢nethe actions of COX-2 in di¡erent types of cancer.A second pharmacological target for treating cancer pain, this one a peptide
possibly responsible for sensitizing and/or activating primary a¡erent neurons, isendothelin 1. Several tumours including prostate cancer express very high levels ofendothelins (Shankar et al 1998, Kurbel et al 1999, Nelson & Carducci 2000) andclinical studies have reported a correlation between the severity of the pain inpatients with prostate cancer and plasma levels of endothelins (Nelson et al 1995).
BONE CANCER PAIN 227

Endothelins could contribute to cancer pain by directly sensitizing or excitingnociceptors, as a subset of small unmyelinated primary a¡erent neurons expressreceptors for endothelin (Pomonis et al 2001) and direct application ofendothelin to peripheral nerves induces activation of primary a¡erent ¢bres andan induction of pain behaviours (Davar et al 1998). Like prostaglandins,endothelins that are released from tumour cells are also thought be involved inregulating angiogenesis (Dawas et al 1999) and tumour growth (Asham et al1998), suggesting again that use of endothelin antagonists may be useful not onlyin inhibiting cancer pain but in reducing the growth and metastasis of the tumour.
Tumour-induced release of protons and acidosis
Tumour cells become ischaemic and undergo apoptosis as the tumour burdenexceeds its vascular supply (Helmlinger et al 2002). Local acidosis, a state wherean accumulation of acid metabolites is present, is a hallmark of tissue injury(Reeh & Steen 1996, Julius & Basbaum 2001). In the past few years, the conceptthat sensory neurons can be directly excited by protons or acidosis has generatedintense research and clinical interest. Studies have shown that subsets of sensoryneurons express di¡erent acid-sensing ion channels (Olson et al 1998, Julius &Basbaum 2001). The two major classes of acid-sensing ion channels expressed bynociceptors are VR-1 (TRPV1)(Caterina et al 1997, Tominaga et al 1998) and theacid-sensing ion channel 3 (ASIC-3) (Bassilana et al 1997, Olson et al 1998,Sutherland et al 2000). Both of these channels are sensitized and excited by adecrease in pH. More speci¢cally, the VR-1 is activated when the pH falls below6.0, while the pH that activates ASIC-3 appears to be highly dependent on the co-expression of other ASIC channels in the same nociceptor (Lingueglia et al 1997).There are several mechanisms by which a decrease in pH could be involved in
generating and maintaining cancer pain. As tumours grow, tumour-associatedin£ammatory cells invade the neoplastic tissue and release of protons thatgenerate local acidosis (Helmlinger et al 2002). A second mechanism by whichacidosis may occur is apoptosis of the tumour cells. Release of intracellular ionsmay generate an acidic environment that activates signalling by acid-sensingchannels expressed by nociceptors.Tumour-induced release of protons and acidosis may be particularly important
in the generation of bone cancer pain. In both osteolytic (bone destroying) andosteoblastic (bone forming) cancers there is a signi¢cant proliferation andhypertrophy of osteoclasts (Clohisy et al 2000a,b, 2001). Osteoclasts areterminally di¡erentiated, multinucleated, monocyte lineage cells that are uniquelydesigned to resorb bone by maintaining an extracellular microenvironment ofacidic pH (4.0^5.0) at the osteoclast-mineralized bone interface (Delaisse & Vaes1992).Most sensory neurons that innervate bone express theVR-1 (Guo et al 1999)
228 MANTYH & HUNT

and/orASICs (Olson et al 1998), and these sensory neuronswill be depolarized andtransmit pain signals to the spinal cord if exposed to the osteoclast’s acidicextracellular microenvironment. Recent experiments in a murine model of bonecancer pain reported that osteoclasts play an essential role in cancer-induced boneloss, and that osteoclasts contribute to the aetiology of bone cancer pain (Honoreet al 2000, Luger et al 2001). Based on these reports it has recently been shown thatosteoprotegerin (Honore et al 2000) or a bisphosphonate (Fulfaro et al 1998,Mannix et al 2000), both of which are known to induce osteoclast apoptosis, aree¡ective at decreasing osteoclast-induced bone cancer pain. Similarly, VR-1 orASIC antagonists may be used to reduce pain in patients with soft tumours orbone cancer by blocking excitation of the acid sensitive channels on sensoryneurons.
Release of growth factors by tumour cells
One of the most important discoveries in the past decade has been thedemonstration that the biochemical and physiological status of sensory neuronsis maintained and modi¢ed by factors derived from the innervated tissue.Changes in the periphery associated with in£ammation, nerve injury, or tissueinjury are mirrored by changes in the phenotype of sensory neurons (Honore et al2000). After peripheral nerve injury, expression of a subset of neurotransmittersand receptors by damaged sensory neurons is altered in a highly predictablefashion. These changes are caused, in part, by a change in the tissue level ofseveral growth factors, including nerve growth factor, bradykinin and tumournecrosis factor (Hunt &Mantyh 2002).While tumour invasion alters the invaded tissue, it is also clear that the tissue
also in£uences the phenotype of the invading tumour cell (Mundy 2002).Because the local environment can in£uence what tumour cells express andrelease, it follows that the same tumour in the same individual may be painful atone site of metastasis but not at another. Based on clinical observations, pain fromcancer can be quite perplexing, as the size, location, or type of cancer tumour doesnot necessarily predict symptoms.Di¡erent patientswith the same cancermay havevastly di¡erent symptoms. Kidney cancer may be painful in one person andasymptomatic in another. Metastases to bone in the same individual may causepain at the site of a rib lesion, but not at the site of a humeral lesion. Small cancerdeposits in bone may be very painful, while very large soft tissue cancers may bepainless (Mantyh et al 2002). Important areas for future research includeidenti¢cation of tissue-speci¢c mechanisms of cancer pain, i.e. soft tissue vs.bone; site-speci¢c mechanisms of cancer pain, i.e. £at bones (rib) vs. tubularbones (femur); and patient-speci¢c factors that in£uence disease progression andits relationship to pain perception.
BONE CANCER PAIN 229

Tumour-induced distention and destruction of sensory ¢bres
In reports, data have suggested that tumours are not highly innervated by sensoryor sympathetic neurons (O’Connell et al 1998, Seifert & Spitznas 2001, Terada &Matsunaga 2001). However, in a large number of cancers rapid tumour growthfrequently entraps and injures nerves, causing mechanical injury, compression,ischaemia or direct proteolysis (Mercadante 1997). Proteolytic enzymes producedby the tumour can also cause injury to sensory and sympathetic ¢bres, causingneuropathic pain.The capacity of tumour to injure and destroy peripheral nerve ¢bres has been
directly observed in an experimental model of bone cancer. Following injectionand containment of lytic murine sarcoma cells in the intramedullary of the mousefemur, tumour cells grow in the marrow space and disrupt innervating sensory¢bres. As the tumour cells grow they ¢rst compress and then destroy both thehaematopoietic cells that normally comprise the marrow as well as the sensory¢bres that normally innervate the marrow, mineralized bone and periosteum(Schwei et al 1999).While the mechanisms by which any neuropathic pain is generated and
maintained are still not well understood, several therapies that have proveduseful in the control of other types of neuropathic pain may also be useful intreating tumour-induced neuropathic pain. For example gabapentin, originallydeveloped as an anticonvulsant but whose mechanism of action still remainsunknown, has been shown to be e¡ective in treating several forms of neuropathicpain and may also be useful in treating cancer pain of neuropathic origin(Ripamonti & Dickerson 2001).
Central sensitization in cancer pain
Acritical question iswhether the spinal cord and forebrain also undergo signi¢cantneurochemical changes as a chronic cancer pain state develops. In the murinecancer pain model it was observed that there was extensive neurochemicalreorganization within spinal cord segments that receive input from primarya¡erent neurons innervating the cancerous bone (Honore et al 2000, Luger et al2001). These changes included astrocyte hypertrophy (Fig. 4) and up-regulationof the pro-hyperalgesic peptide dynorphin. Spinal cord neurons that normallywould only be activated by noxious stimuli were activated by normally non-noxious stimuli. These spinal cord changes were attenuated by blocking thetumour-induced tissue destruction and pain (Honore et al 2000, Luger et al2001). Together these neurochemical changes suggest that cancer pain inducesand is at least partially maintained by a state of central sensitization in which anincreased transmission of nociceptive information allows normally non-noxiousinput to be ampli¢ed and perceived as noxious stimuli.
230 MANTYH & HUNT

BONE CANCER PAIN 231
FIG. 4. Cancer-induced reorganization of the central nervous system. Chronic cancer pain notonly sensitizes peripheral nociceptors, but also can induce signi¢cant neurochemicalreorganization of the spinal cord which may participate in the phenomenon of centralsensitization i.e. an increased responsiveness of spinal cord neurons involved in transmissionof pain. Confocal image of a coronal section of the mouse L4 spinal cord showing glial¢brillary acidic protein (GFAP)-positive astrocytes (white) which have undergonehypertrophy on the side ipsilateral to the tumour-bearing bone. (B, C) Higher magni¢cationphotos ipsilateral (B) and contralateral (C) to the dorsal horn seen in (A) where the GFAPstaining is present with staining for the neuron speci¢c antibody NeuN. Note that while theastrocytes (spindle-shaped cells) have undergone a massive hypertrophy, there does not appearto be any signi¢cant loss of NeuN-positive neurons (modi¢ed from Schwei et al 1999). Scalebars¼A, 200 mm; B, C, 30 mm.

Once nociceptive information has been transmitted to the spinal cord byprimary a¡erent neurons there are multiple ascending ‘pain’ pathways thatproject from the spinal cord to higher centres of the brain. Classically, mainemphasis in examining the ascending conduction of pain has been placed onspinothalamic tract neurons. However, recent data have necessitated areassessment of this position, for recent clinical studies have shown thatsigni¢cant attenuation of some forms of di⁄cult to control visceral cancer paincan be achieved following lesion of the axons non-spinothalamic tract neurons(Willis et al 1999, Nauta et al 2000). Together these data suggest that one reasonthat cancer pain is frequently perceived as such an intense and disturbing pain isthat it ascends to higher centres of the brain viamultiple parallel neuronal pathways(Hunt & Mantyh 2001). Importantly for cancer patients, it is clear that highercentres of the brain can modulate the ascending conduction of pain so that thegeneral mood and attention of the patient may in itself be a signi¢cant factor indetermining the intensity and degree of unpleasantness of the pain. Since manycancer patients are frequently anxious and/or depressed, these descendingpathways, which modulate the ascending conduction of cancer pain, maythemselves play an important role either enhancing or inhibiting the perceptionof cancer pain by the patient.
A changing set of factors may drive
cancer pain as the disease progresses
Cancer pain frequently becomes more severe as the disease progresses and controlof cancer pain is more di⁄cult to fully achieve without encountering signi¢cantunwanted side e¡ects (Payne 1998, Foley 1999, Portenoy & Lesage 1999). Whiletolerance may contribute to the escalation of the dose of analgesics required tocontrol cancer pain, a compatible possibility is that with the progression of thedisease di¡erent factors assume a greater importance in driving cancer pain. Forexample, in the mouse model of bone cancer, tumour cells ¢rst begin toproliferate pain-related behaviours that are present long before any signi¢cantbone destruction is evident and this pain may be due to pro-hyperalgesic factorssuch as prostaglandins and endothelin that are released by the growing tumourcells that activate nociceptors in the marrow and this pain could be attenuated byCOX-2 inhibitors and endothelin antagonists. As the tumour continues to grow,sensory neurons innervating the marrow are compressed and destroyed causing aneuropathic pain to develop which may best respond to treatment with drugswhich attenuate neuropathic pain such as gabapentin. When the tumour beginsto induce proliferation and hypertrophy of osteoclasts, the pain due to excessiveosteoclast activity could be largely blocked by anti-osteoclastogenic drugs such asbisphosphonates or osteoprotegerin (Fig. 5). As the tumour cells completely ¢ll
232 MANTYH & HUNT

the intramedullary space they begin to die, generating an acidic environment,antagonists to VR-1 or ASICs should attenuate the pain induced by this acidosis.Finally, as bone destruction ensues and the mechanical strength of the bone iscompromised, antagonists that block the mechanically gated channels and/orATP receptors in the richly innervated periosteum may attenuate the movementevoked pain.While the above pattern of tumour-induced tissue destruction and nociceptor
activation may be unique to bone cancer, an evolving set of nociceptive eventsprobably occurs in other cancers. This may, in part, explain why cancer pain isfrequently di⁄cult to treat and why it is so heterogeneous in nature and severity.That the tumour-induced tissue injury, nociceptor activation, and the CNS brainareas involved in transmitting these nociceptive signals are changing as the diseaseprogresses suggests that di¡erent therapies will be e⁄cacious at particular stages ofthe disease. Understanding how tumour cells di¡erentially excite nociceptors atdi¡erent stages of the disease, and how the phenotype of nociceptors and CNSneurons involved in nociceptive transmission change as the disease progressesshould allow a mechanistic approach to designing more e¡ective therapies totreat cancer pain.
Progress and future directions
For the ¢rst time, animal models of cancer pain are now available that mirror theclinical picture of human patients with cancer pain. Information generated from
BONE CANCER PAIN 233
FIG. 5. Attenuation of bone cancer pain by osteoprotegerin (OPG). Histograms showing thatadministration of OPG which was started 6 days following tumour implantation attenuatedboth spontaneous (A) and palpation-evoked (B) pain in mice at day 17 following tumourimplantation (modi¢ed from Honore et al 2000). OPG is a naturally occurring protein that is asecreted decoy receptor that inhibits osteoclast di¡erentiation, proliferation, and hypertrophyresulting in reduced osteoclast activity and bone resorption.

these models should provide insight into the mechanisms that generate andmaintain the di¡erent types of cancer pain. Since many of the cancer modelshave been developed in mice, human tumours can be implanted inimmunocompromised mouse strains, which should allow examination of thepain that di¡erent human tumours generate. These animal models may also o¡erinsight into one of themajor conundrums of cancer pain: that the severity of cancerpain is so variable from patient to patient, tumour to tumour, and even site to site.Newer molecular techniques using microarrays and proteomics should providedata about what speci¢c features of di¡erent tumours are important in inducingcancer pain. Once the mechanisms by which the di¡erent types of cancer inducepain have been elucidated, molecular targets can be identi¢ed and mechanismbased therapies developed. Ultimately, the key will be to integrate tumourbiology and the host’s response to neoplasia with our understanding of howchronic pain is generated and maintained. These studies should lead toimprovement in the quality of life for all those who su¡er from cancer pain.
References
Alvarez FJ, Fy¡e RE 2000 Nociceptors for the 21st century. Curr Rev Pain 4:451^458Asham EH, Loizidou M, Taylor I 1998 Endothelin-1 and tumour development. Eur J SurgOncol 24:57^60
Basbaum AI, Jessel TM 2000 The perception of pain. In: Kandel ER, Schwartz JH, Jessell TM(eds) Principles of neural science. McGraw-Hill, New York, p 472^490
Bassilana F, Champigny G, Waldmann R et al 1997 The acid-sensitive ionic channel subunitASIC and the mammalian degenerin MDEG form a heteromultimeric H+-gated Na+
channel with novel properties. J Biol Chem 272:28819^28822Bevan S,Geppetti P 1994 Protons: small stimulants of capsaicin-sensitive sensory nerves. TrendsNeurosci 17:509^512
Caterina MJ, Schumacher MA, Tominaga M et al 1997 The capsaicin receptor: a heat-activatedion channel in the pain pathway. Nature 389:816^824
CaterinaMJ, Le¥erA,MalmbergAB et al 2000 Impaired nociception and pain sensation inmicelacking the capsaicin receptor. Science 288:306^313
Clohisy DR, Perkins SL, Ramnaraine ML 2000a Review of cellular mechanisms of tumorosteolysis. Clin Orthop Rel Res 373:104^114
Clohisy DR, Ramnaraine ML, Scully S et al 2000b Osteoprotegerin inhibits tumor-inducedosteoclastogenesis and bone growth in osteopetrotic mice. J Orthop Res 18:967^976
Clohisy DR, O’Keefe PF, Ramnaraine ML 2001 Pamidronate decreases tumor-inducedosteoclastogenesis in mice. J Orthop Res 19:554^558
Coyle N, Adelhardt J, Foley KM, Portenoy RK 1990 Character of terminal illness in theadvanced cancer patient: pain and other symptoms during the last four weeks of life [seecomments]. J Pain SymptomManage 5:83^93
DaughadayWH, Deuel DF 1991 Tumor secretion of growth factors. Endocrinol Metab Clin NAm 20:539^563
Davar G 2001 Endothelin-1 and metastatic cancer pain. Pain Med 2:24^27Davar G, Hans G, FareedMU et al 1998 Behavioral signs of acute pain produced by applicationof endothelin-1 to rat sciatic nerve. Neuroreport 9:2279^2283
234 MANTYH & HUNT

Dawas K, Laizidou M, Shankar A et al 1999 Angiogenesis in cancer: the role of endothelin-1.Ann R Coll Surg Engl 81:306^310
deWit R, vanDam F, Loonstra S et al 2001 The Amsterdam PainManagement Index comparedto eight frequently used outcome measures to evaluate the adequacy of pain treatment incancer patients with chronic pain. Pain 91:339^349
Delaisse J-M, Vaes G 1992 Mechanism of mineral solubilization and matrix degradation inosteoclastic bone resorption. In: Rifkin BR, Gay CV (eds) Biology and physiology of theosteoclast. CRC, Ann Arbor, p 289^314
DeLeo JA, Yezierski RP The role of neuroin£ammation and neuroimmune activation inpersistent pain. Pain 90:1^6
Djouhri L, Bleazard L, Lawson SN 1998 Association of somatic action potential shape withsensory receptive properties in guinea-pig dorsal root ganglion neurones. J Physiol513:857^872
Dubois RN, Radhika A, Reddy BS, Entingh AJ 1996 Increased cyclooxygenase-2 levels incarcinogen-induced rat colonic tumors. Gastroenterology 110:1259^1262
Foley KM 1999 Advances in cancer pain. Arch Neurol 56:413^417Fulfaro F, Casuccio A, Ticozzi C, Ripamonti C 1998 The role of bisphosphonates in thetreatment of painful metastatic bone disease: a review of phase III trials. Pain 78:157^169
Galasko CS 1995 Diagnosis of skeletal metastases and assessment of response to treatment. ClinOrthop Rel Res 312:64^75
Guo A, Vulchanova L, Wang J et al 1999 Immunocytochemical localization of the vanilloidreceptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 bindingsites. Eur J Neurosci 11:946^958
Helmlinger G, Sckell A, Dellian M et al 2002 Acid production in glycolysis-impaired tumorsprovides new insights into tumor metabolism. Clin Cancer Res 8:1284^1291
Honore P, Luger NM, Sabino MA et al 2000a Osteoprotegerin blocks bone cancer-inducedskeletal destruction, skeletal pain and pain-related neurochemical reorganization of thespinal cord. Nat Med 6:521^528
Honore P, Menning PM, Rogers SD et al 2000b Neurochemical plasticity in persistentin£ammatory pain. Prog Brain Res 129:357^363
Honore P, Rogers SD, Schwei MJ et al 2000cMurine models of in£ammatory, neuropathic andcancer pain each generates a unique set of neurochemical changes in the spinal cord andsensory neurons. Neuroscience 98:585^598
Honore P, Schwei J, Rogers SD et al 2000 Cellular and neurochemical remodeling of the spinalcord in bone cancer pain. Prog Brain Res 129:389^397
Hoskin P 2000 Radiotherapy. In: Body J-J (ed) Tumor bone diseases and osteoporosis in cancerpatients. Marcel Dekker, New York, p 263^286
Hunt SP,Mantyh PW 2001 Themolecular dynamics of pain control. Nat RevNeurosci 2:83^91Hunt SP, Pini A, Evan G 1987 Induction of c-fos-like protein in spinal cord neurons followingsensory stimulation. Nature 328:632^634
Julius D, Basbaum AI 2001 Molecular mechanisms of nociception. Nature 413:203^210Kirschstein T,Gre¡rathW, BusselbergD, Treede RD 1999 Inhibition of rapid heat responses innociceptive primary sensory of rats by vanilloid receptor antagonists. J Neurophysiol82:2853^2860
Krishtal OA, Marchenko SM, Obukhov AG 1988 Cationic channels activated by extracellularATP in rat sensory neurons. Neuroscience 27:995^1000
KunduN,YangQY,DorseyR, FultonAM2001 Increased cyclooxygenase-2 (cox-2) expressionand activity in a murine model of metastatic breast cancer. Int J Cancer 93:681^686
Kurbel S, Kurbel B, Kovacic D et al 1999 Endothelin-secreting tumors and the idea of thepseudoectopic hormone secretion in tumors. Med Hypotheses 52:329^333
BONE CANCER PAIN 235

Lingueglia E, Weille JR, Bassilana F et al 1997 A modulatory subunit of acid sensing ionchannels in brain and dorsal root ganglion cells. J Biol Chem 272:29778^29783
Luger NM, Honore P, Sabino MAC et al 2001 Osteoprotegerin diminishes advanced bonecancer pain. Cancer Res 61:4038^4047
Luger NM, Sabino MAC, Schwei MJ et al 2002 E⁄cacy of systemic morphine suggests afundamental di¡erence in the mechanisms that generate bone cancer vs. in£ammatory pain.Pain 99:397^406
Mannix K, Ahmedzai SH, Anderson H, Bennet M, Lloyd-Williams M, Wilcock A 2000 Usingbisphosphonates to control the pain of bone metastases: evidence based guidelines forpalliative care. Palliat Med 14:455^461
Mantyh PW, Allen CJ, Ghilardi JR et al 1995a Rapid endocytosis of a G protein-coupledreceptor: substance P evoked internalization of its receptor in the rat striatum in vivo. ProcNatl Acad Sci USA 92:2622^2626
Mantyh PW, DeMaster E, Malhotra A et al 1995b Receptor endocytosis and dendrite reshapingin spinal neurons after somatosensory stimulation. Science 268:1629^1632
Mantyh PW, ClohisyDR,KoltzenburgM,Hunt SP 2002Molecular mechanisms of cancer pain.Nat Rev Cancer 2:201^209
Masferrer JL, Leahy KM, Koki AT et al 2000 Antiangiogenic and antitumor activities ofcyclooxygenase-2 inhibitors. Cancer Res 60:1306^1311
McMahon SB 1996 NGF as a mediator of in£ammatory pain. Phil Trans R Soc Lond B Biol Sci351:431^440
Mercadante S 1997 Malignant bone pain: pathophysiology and treatment. Pain 69:1^18Mercadante S, Arcuri E 1998 Breakthrough pain in cancer patients: pathophysiology andtreatment. Cancer Treat Rev 24:425^432
Meuser T, Pietruck C, Radbruch L et al 2001 Symptoms during cancer pain treatment followingWHO-guidelines: a longitudinal follow-up study of symptom prevalence, severity andetiology. Pain 93:247^257
Molina MA, Sitja-Arnau M, Lemoine MG et al 1999 Increased cyclooxygenase-2 expression inhuman pancreatic carcinomas and cell lines: growth inhibition by nonsteroidal anti-in£ammatory drugs. Cancer Res 59:4356^4362
Moore BC, Simmons DL 2000 COX-2 inhibition, apoptosis, and chemoprevention bynonsteroidal anti-in£ammatory drugs. Curr Med Chem 7:1131^1344
Mundy GR 2002 Metastases to bone: causes, consequences, and therapeutic opportunities. NatRev Cancer 2:584^593
Nagy I, Rang H 1999 Noxious heat activates all capsaicin-sensitive and also a sub-population ofcapsaicin-insensitive dorsal root ganglion neurons. Neuroscience 88:995^997
Nauta HJW, Soukup VM, Fabian RH et al 2000 Punctate midline myelotomy for the relief ofvisceral cancer pain. J Neurosurg 92:S125^S130
Nelson JB, Carducci MA 2000 The role of endothelin-1 and endothelin receptor antagonists inprostate cancer. BJU Int 85:S45^S48
Nelson JB, Hedican SP, George DJ et al 1995 Identi¢cation of endothelin-1 in thepathophysiology of metastatic adenocarcinoma of the prostate. Nat Med 1:944^949
Nielsen OS, Munro AJ, Tannock IF 1991 Bone metastases: pathophysiology and managementpolicy. J Clin Oncol 9:509^524
O’Connell JX, Nanthakumar SS, Nielsen GP, Rosenberg AE 1998 Osteoid osteoma: theuniquely innervated bone tumor. Mod Pathol 11:175^180
Ohno R, Yoshinaga K, Fujita T et al 2001Depth of invasion parallels increased cyclooxygenase-2 levels in patients with gastric carcinoma. Cancer 91:1876^1881
Olson TH, Riedl MS, Vulchanova L et al 1998 An acid sensing ion channel (ASIC) localizes tosmall primary a¡erent neurons in rats. Neuroreport 9:1109^1113
236 MANTYH & HUNT

Opree A, Kress M 2000 Involvement of the proin£ammatory cytokines tumor necrosis factor-alpha, IL-1 beta, and IL-6 but not IL-8 in the development of heat hyperalgesia: e¡ects onheat-evoked calcitonin gene-related peptide release from rat skin. J Neurosci 20:6289^6293
Payne R 1997 Mechanisms and management of bone pain. Cancer 80:S1608^S1613Payne R 1998 Practice guidelines for cancer pain therapy. Issues pertinent to the revision ofnational guidelines. Oncology 12:169^175
Payne R, Mathias SD, Pasta DJ et al 1998 Quality of life and cancer pain: satisfaction and sidee¡ects with transdermal fentanyl versus oral morphine. J Clin Oncol 16:1588^1593
Pomonis JD, Rogers SD, Peters CM et al 2001 Expression and localization of endothelinreceptors: implication for the involvement of peripheral glia in nociception. J Neurosci21:999^1006
Poon RT, Fan ST,Wong J 2001 Clinical implications of circulating angiogenic factors in cancerpatients. J Clin Oncol 19:1207^1225
Portenoy RK, Lesage P 1999 Management of cancer pain. Lancet 353:1695^1700Portenoy RK, Payne D, Jacobsen P 1999 Breakthrough pain: characteristics and impact inpatients with cancer pain. Pain 81:129^134
Price MP, McIlwrath SL, Xie JH et al 2001 The DRASIC cation channel contributes to thedetection of cutaneous touch and acid stimuli in mice. Neuron 32:1071^1083
Radinsky R 1991 Growth factors and their receptors in metastasis. Semin Cancer Biol 2:169^177
Reeh PW, Steen KH 1996 Tissue acidosis in nociception and pain. Prog Brain Res 113:143^151Ripamonti C, Dickerson ED 2001 Strategies for the treatment of cancer pain in the newmillennium. Drugs 61:955^977
Roman C, Saha D, Beauchamp R 2001 TGF-beta and colorectal carcinogenesis. Microsc ResTech 52:450^457
SabinoMAC, Ghilardi JR, Jongen JLM et al 2002 Simultaneous reduction of cancer pain, bonedestruction, and tumor growth by selective inhibition of cyclooxygenase 2. Cancer Res62:7343^7349
Schwei MJ, Honore P, Rogers SD et al 1999 Neurochemical and cellular reorganizationof the spinal cord in a murine model of bone cancer pain. J Neurosci 19:10886^10897
Seifert P, Spitznas M 1998 Tumours may be innervated. Virchows Archiv 438:228^231Shankar A, LoizidouM, Aliev G et al 1998 Raised endothelin 1 levels in patients with colorectalliver metastases. Br J Surg 85:502^506
Shappell SB, Manning S, Boeglin WE et al 2001 Alterations in lipoxygenase andcyclooxygenase-2 catalytic activity and mRNA expression in prostate carcinoma. Neoplasia3:287^303
Silver BJ 1992 Platelet-derived growth factor in human malignancy. Biofactors 3:217^227Stoscheck CM, King Jr LE 1986 Role of epidermal growth factor in carcinogenesis. Cancer Res46:1030^1037
Sutherland SP, Cook SP, and McCleskey EW 2000 Chemical mediators of pain due to tissuedamage and ischemia. Prog Brain Res 129:21^38
Terada T, Matsunaga Y 2001 S-100-positive nerve ¢bers in hepatocellular carcinoma andintrahepatic cholangiocarcinoma: an immunohistochemical study. Pathol Int 51:89^93
Tominaga M, Caterina MJ, Malmberg AB et al 1998 The cloned capsaicin receptor integratesmultiple pain-producing stimuli. Neuron 21:531^543
Vasko MR 1995 Prostaglandin-induced neuropeptide release from spinal cord. Prog Brain Res104:367^380
Watkins LR,Goehler LE, Relton J et al 1995Mechanisms of tumor necrosis factor-alpha (TNF-a) hyperalgesia. Brain Res 692:244^250
BONE CANCER PAIN 237

Welch JM, SimonSA,Reinhart PH2000The activationmechanismof rat vanilloid receptor 1 bycapsaicin involves the pore domain and di¡ers from the activation by either acid or heat. ProcNatl Acad Sci USA 97:13889^13894
Willis WD, Al-Chaer ED, Quast MJ, Westlund KN 1999 A visceral pain pathway in the dorsalcolumn of the spinal cord. Proc Natl Acad Sci USA 96:7675^7679
Woolf CJ, Salter MW 2000 Neuronal plasticity: increasing the gain in pain. Science 288:1765^1769
XuGY,HuangLYM2002 Peripheral in£ammation sensitizes P2X receptor-mediated responsesin dorsal root ganglion neurons. J Neurosci 22:93^102
DISCUSSION
Blake: The COX-2 inhibition is quite surprising in terms of its extent. Couldsome of that e¡ect which probably was generated at decent drug levels beattributed to promoting thrombosis in a hypoxic tumour?Hunt: My understanding is that there is some literature on reduced tumour
growth anyway from COX-2 inhibitors. We don’t know the mechanism andyour suggestion is one possibility. Would prostaglandins drive tumour growth?There is some evidence that this is the case.Blake:This seems themore plausible option. It is just that the extent of the e¡ect
you got seemed remarkable.Fox:We see the same e¡ect with COX-2 inhibitors.Blake:Do you see increased thrombosis?Fox:No, we see reduction in tumour size.Felson: How tumour speci¢c are all the mechanisms you talked about? Colon
cancer is the one that is supposed to be COX-2 sensitive. Some tumours causeblastic lesions in bone, not lytic lesions, and are associated with pain.Hunt: I can’t answer that. The only experience I have is with bone cancer.Kuettner: How e¡ective is the bisphosphonate treatment? You can theoretically
make the bone so hard that it cannot be resorbed by either tumour cells orosteoclasts. Is it an e¡ective treatment?Hunt: I think it is. The advantage of OPG is that it is a drug that has been
developed already. It is one of those drugs that could be used if they could bebothered to do the trials. Companies are more interested in the osteoporosismarket than they are in the bone cancer market. Unless it is speci¢cally tested onthis group of patients it may not be widely used and they’ll carry on usingbisphosphonates. OPG is much simpler.Schaible: You mentioned changes in gene expression. Which gene groups are
changing?Hunt: I don’t know if you are familiar with the bioinformatics programs. They
are elaborate programswith a large do-it-yourself component. If you are interestedin a particular pathway, such as the MAP kinase pathway, you have to ¢nd it,
238 DISCUSSION

annotate it and put it into your databank. Then you can ask which of thesepathways is activated. We are in the process of doing this. It produces abewildering array of data.Kidd: What are the implications for normal bone of the osteoclast^nerve cell
interaction you described? What is to prevent an osteoclast just blundering acrossa nerve cell in normal bone?Hunt:My understanding is that there are very few osteoclasts in normal bone. I
would imagine that any slight activation of osteoclasts has quite severeconsequences for the nerve supply.Grubb: You raised the issue of markers of neuronal damage being up-regulated
in these models of sarcoma. Does this mean that this pain is neuropathic in nature?Are these animals responsive to gabapentin, a drug frequently used to treatneuropathic pain?Hunt: They are responsive to gabapentin. If you make even a fairly severe
in£ammation you never see ATF3 in the sensory neurons. It has to be beyondthat. I don’t know what the line you have to cross is, whether it is qualitative orquantitative, but you can make a massive in£ammation and not get ATF3.Blake: When you set up rodent models of arthritis you can more or less
completely block the so-called T cell-mediated complete Freund’s adjuvant(CFA) model with non-selective or selective COX-2 inhibitors. But when this istranslated to rheumatoid arthritis in humans, the e¡ect is very small, apart from abit of a suppression of in£ammation and a bit of analgesia. Then you look at awhole host of very di¡erent acting drugs in the animal model of arthritis, andthey all seem to have quantitatively a very similar e¡ect on the model. Yet whenyou translate this to human they don’t. So my question, which is probablyunanswerable, is what is di¡erent about a rodent who is turning up 131 genes andturning down 73 that allows it to respond to single and very disparate treatmentoptions in a very similar fashion? When we translate this into human we don’t seethe same e¡ect.Hunt: I was talking to Bruce Kidd earlier about this. There seems to be a serious
problem in that a lot of the experimental work doesn’t seem to translate to human.Think of the time course of these events. In humans it takes years to get a bad knee.In the animals we are doing this acutely. Perhaps it is the rapidity that changes themechanism.Fox: I think the time course is key. It is unrealistic that these rapidmodels can be
extrapolated exactly into human. The outcome measures we have in animals arealso imperfect. All drug e¡ects are being squashed into this window.Mackenzie:One of the big di¡erences is between the di¡erentmodels of arthritis.
In adjuvant arthritis COX-2 inhibitors and non-selective COX inhibitors areextremely e¡ective, but they are much less e¡ective in monoarticular antigen-induced arthritis. One of the di¡erences is that in adjuvant arthritis there is
BONE CANCER PAIN 239

massive bone remodelling. It is verymuch like the picture in the sarcomamodel. Itmay be that the primary thing the COX inhibitors are doing is to inhibit the boneremodelling.Felson: You commented on preventing osteoclast activation, and showed that
this prevented pain. Therewas a decrement of pain but it wasn’t all that impressive.What does this mean?Hunt: I would say that we are talking about components. I think what this work
shows is that you can break up pain into di¡erent components, but there is nosingle pathology that generates the signal. Palpation is a particularly severestimulus for these animals. If you can get 40% decrease in pain that is prettyacceptable.Felson:Does anyone want to comment on osteoclast activation in osteoarthritis
(OA)? There is subchondral bone remodelling in OA, which would involveosteoclasts. One of the most rapid osteoclast activation syndromes is womengoing through the menopause and there is no obvious bone pain there. Theother one which might be relevant is transient regional osteoporosis, which is adisorder in which bone is resorbed quite rapidly associated with pain. This painmay be because of microcracks or microfractures.Pisetsky:One other condition to consider is acute vertebral collapses, which are
very painful. A lot of that pain is attributed to the fracture, but you could also saythat this is osteoclast activation. I have heard that people use calcitonin for this kindof fracture.Felson: Clinically, the observation is that calcitonin isn’t a very e¡ective
osteoporosis medication, but it is an e¡ective pain medication.Blake: Another relevant disease state here is hypertrophic pulmonary
osteoarthropathy. This presents clinically in people with lung cancer as aneurogenically mediated osteoclastic resorption at the wrist where there is clearlyno tumour at the event. This provides yet another way of looking at some of thesephenomena that are less tumour speci¢c in terms of destruction and much moresecondary in terms of e¡ects that we don’t understand. This is a fantastic humanclinical model.Pisetsky: Is there clinical experience with gabapentin in people with cancer pain
that would give credence to this idea that it is neuropathic depending on howwellgabapentin works? Are people using it?Hunt: I think so.Ordeberg: We have used it, but our patients with cancer pain (with skeletal
metastases) also have bone destruction and spinal metastasis with encroachmenton neural structures with a mixture of pain of di¡erent mechanisms.
240 DISCUSSION

Symmetry, T cells and neurogenic
arthritis
N. G. Shenker*{, D. R. Blake*{1, C. S. McCabe*, R. Haigh{ and P. I. Mapp{
*Department of Rheumatology, Royal National Hospital for Rheumatic Diseases, Bath,BA1 1RL, {Department of Medical Sciences, University of Bath, Claverton Down,BA2 7AY and {Department of Rheumatology, Royal Devon&Exeter Hospital, Exeter, UK
Abstract. Symmetry in clinical disease occurs more commonly than expected by chanceand is unexplained. In this paper we focus on symmetry in arthritis and describe theneurogenic hypothesis. Neuropeptides are anatomically relevant to systemic arthritisand have been shown to have modulating e¡ects on both the immune and circulatorysystems. Neural networks project bilaterally and are involved in the development andpropagation of in£ammatory disease. These putative pathological neuro-feedback loopsmay derive from the existence of biologically protective symmetrical mechanisms.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 241^257
Symmetry in clinical disease
Symmetry in clinical disease is common and can be seen in a wide spectrum ofconditions. Symmetrical diseases are found within the realms of rheumatology,dermatology, ophthalmology and neurology, as might be expected given theanatomical duplicity of the systems with which these specialities deal. Theconsistency of the presence of this clinical ¢nding points to an underlyingmechanism.Bilateral expression of a unilateral stimulus has best been studied in the
neurosciences and Koltzenburg et al (1999) have reviewed this phenomenon.They summarised the work of several teams who had independently describedsymmetrical, topographically precise, time-dependent changes in response tospeci¢c local neurological insults. Neural connections between left and rightsides of the spinal cord are hypothesized to account for these changes (Fig. 1).
241
1This paper was presented at the symposium byD. R. Blake to whom correspondence should beaddressed.
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

Detailed histological examination in the spinal cords of primates and othervertebrates (Light & Perl 1979, Culberson et al 1979) demonstrate posteriordecussation of signi¢cant numbers of neurons at all levels of the spinal cord.These ¢bres terminate in the contralateral substantia gelatinosa (Rexed’s laminaeI^II) as well as in the deeper layers (laminae III^IV) and their function has yet to bedescribed.The implication that bilateral neurally mediated pathways exist and determine
symmetrical disease patterns is important because conditions that exhibitsymmetry are common and their aetiology largely unknown. Skin psoriasis, forexample, is much more symmetrical than expected by chance (Farber et al 1986).Other dermatological symmetrical conditions include vitiligo and pityriasisrosacea. The classic in£ammatory example seen in ophthalmology is sympatheticophthalmia. This is a bilateral uveitis complicating a unilateral perforatingwound.Interestingly, acute demyelinating plaques seen in the brains of patients withmultiple sclerosis are associated with subtle contralateral changes seen on
242 SHENKER ET AL
FIG. 1. Scheme demonstrating routes identi¢ed from histological studies of sensory ¢bresdecussating via the dorsal commissure.

di¡usion MRI (Werring et al 2000). Glomerulonephritis and pulmonary ¢brosisnever occur unilaterally and this is noteworthy in itself.
Symmetry in degenerative arthritis
With respect to the musculoskeletal system, both degenerative and in£ammatoryarthritides are symmetrical. The Heberden’s nodes of osteoarthritis (OA), thecommonest arthritis of them all, are symmetrical. This is emphasised by a recentepidemiological survey from the Framingham population (Niu et al 2003) thatdemonstrated that symptomatic OA is remarkably symmetrical, especially inwomen. Proprioceptive feedback is abnormal in a radiologically normal kneethat is contralateral to an osteoarthritic knee. This contralateral sensory de¢cit ismore abnormal than expected when compared to age-matched patients who haveno OA (Sharma et al 1997, Garsden & Bullock-Saxton 1999). These observationssuggest that neural de¢cits exist before the symmetrical expression of OA occurs.Further evidence that the neural supply to the joint is fundamental to theexpression of this disease is that neural disruption prevents the development ofHeberden’s nodes (Table 1).
Symmetry in in£ammatory arthritis
A symmetrical pattern of involvement is clinically so important in rheumatoidarthritis that it is a feature of its diagnostic criteria (Arnett et al 1988). There have
NEUROGENIC ARTHRITIS 243
TABLE1 Reports in the literature of the development of arthritic diseases in partiallyparalysed patients
Disease Neural injury Outcome suppressed
OA Hemiplegia Heberden’s nodes
OA Poliomyelitis OA knee and hip
OA Nerve injury OA and Heberden’s nodes
RA Hemiplegia Digital vasculitis, RA and RA nodules
RA Hemiplegia Arthritis
RA Hand dominance Radiological changes of arthritis
RA Poliomyelitis Arthritis
Gout Hemiplegia Arthritis and tophi deposits
Modi¢ed from Dixon (1989).

been recent publications noting that in£ammatory arthritis is symmetricalirrespective of rheumatoid factor (Bukhari et al 2002) and that some of theseronegative arthritides, such as psoriatic (Helliwell et al 2000), are moresymmetrical than would be expected by chance.The pathophysiology of in£ammatory arthritis currently centres on T
lymphocyte-dominated autoimmune processes. Yet, how could the immunesystem, e¡ected by blood-borne agents, manifest clinically symmetrical disease?This paper supports a neurogenic hypothesis to explain disease symmetry, andwith reference to T cell-mediated disease, presents possible mechanisms for theinterface between some neuropeptides and the immune system.
Role of the neuropeptides substance P and CGRP in arthritis
Hench’s classical report of symmetrical palindromic rheumatism dates back morethan 50 years and in it he describes wheals and £ares occurring over a¡ected joints.The similarity between this observation and Lewis’ triple £are-wheal response wasquickly made. The £are-wheal response is dependent upon an intact nerve supplyand,more speci¢cally, upon the release of the co-localized neuropeptides substanceP (SP) and calcitonin gene-related peptide (CGRP) contained within populationsof unmyelinated neurons. These are the commonest neuropeptides found in smalldiameter nerve ¢bres. It is now known that some of the unmyelinated ¢bres thatinnervate joint structures also have sprouting peripheral terminals that project intothe surrounding skin. The skin changes that Hench described could well beattributed to the vasodilatory e¡ects, known to be caused by the release of SP andCGRP, via an antidromic route. The role of these neuropeptides in thein£ammatory processes of arthritis is therefore of interest.Experimental work on a variety of animal species utilizing a number of insults to
generatemono- and systemic arthritides have demonstrated that there is a release ofSP in response to an arthritic insult (Garrett et al 1992, Holzer 1988). There is acorresponding increase in the production of SP and CGRP in the nerve cellbodies located in the rat dorsal root ganglion (Mapp et al 1993, Hanesch et al1995). The ¢nding that joints ‘primed’ with substance P demonstrate a moresevere subsequent arthritis (Levine et al 1984) supports the importance of therole of substance P in in£ammatory arthritis. Furthermore, it can be shown thatthe depletion of these neuropeptides through denervation (Levine et al 1985) orexposure to capsaicin (Donaldson et al 1995) attenuates the progression andseverity of an acute polyarthritis. Thus the presence of SP worsens, and itsabsence improves, experimental arthritis.There is a persistent increase in SP in the synovial £uid of patients with RA
compared with normal controls (Westermark et al 2001). This is a consistent¢nding reported by several groups independently and SP levels are raised in the
244 SHENKER ET AL

synovial £uid from patients with in£ammatory arthritis when compared to bothplasma concentrations and patients with non-in£ammatory arthritis (Garrett et al1992). Histochemical studies support the release of neuropeptides from arthriticsynovium by ¢nding a reduction in the presence of neural SP (Garrett et al 1992).Interestingly, bothCGRP and SP are produced by peripheral lymphocytes and thisroute may also contribute to the presence of neuropeptide release in the in£amedsynovial joint (Qian et al 2001, Wang et al 2002).Although SP and CGRP are the most prevalent neuropeptides and have been
best studied, other neuropeptides such as vasoactive intestinal peptide (VIP) havee¡ects on the course of experimental arthritis (Delgado et al 2001).
In£uence of neuropeptides on T cells
Neuropeptide-containing nerves are in contact with immune cells throughouttheir lifespan, from the thymus to the peripheral circulation and also the lymphnode. There is a rich neuropeptide innervation to the blood vessels within lymphnodes and there appears to be an especially strong anatomical link to the T cellsrather than the B cells (Weihe & Krekel 1991).SP and CGRP are located perivascularly in the synovium (Garrett et al 1992).
CGRP has powerful vasodilatory e¡ects (Brain et al 1985) and will thereforeincrease synovial circulation signi¢cantly. These neuropeptides are alsoimportant in the migration of lymphocytes from the circulation to the joint.Capsaicin, a SP- and CGRP-depleting agent, reduces the in£ux of T cells into theacutely arthritic joint of guinea pigs (Hood et al 2001). This is a speci¢c e¡ect onthese T cells because macrophage in£ux was una¡ected in the same model.Substance P binds to a particular subset of T lymphocytes via speci¢c receptors
and causes proliferation of these lymphocytes (McGillis et al 1990). Furthermore,Levite’s work suggests that neuropeptides can cause T lymphocytes to produce arather di¡erent and distinctive pattern that is described as ‘forbidden’. Th0 cells aredriven by antigenic stimulation to induce rather ¢xed patterns of cytokinesecretion characterized by the ‘pro-in£ammatory’ Th1 (e.g. interleukin [IL]2,interferon [IFN]g) and the ‘pro-antibody’ Th2 (e.g. IL4, IL10) cytokine pro¢les.The ‘forbidden’ pattern that is induced by some neuropeptides combines both thecharacteristic Th1 responses with the additional secretion of IL4 and IL10, and thecharacteristic Th2 responses with the additional secretion of IL2 and IFNg (Levite2001). These in vitro studies need con¢rmation in vivo. Neuropeptide-mediated Thelper responses however obviate the need for antigen and may well contributeto the immune dysregulation seen in rheumatoid arthritis.The exact role of neuropeptides in in£ammatory arthritis is not known.
Substance P and other neuropeptides are anatomically, physiologically andpharmacologically relevant to the mechanisms seen in the pathological processes
NEUROGENIC ARTHRITIS 245

of in£ammatory arthritis. Neuropeptides have an important role in both thetra⁄cking and the subsequent immunomodulation of T cells in in£ammatoryarthritis.
The nervous system in arthritis
The role that the nervous system has in rheumatoid arthritis in humans is welldocumented. There are numerous examples of both lower motor and uppermotor neuron lesions ameliorating the progression of this deforming jointdisease. Denervation also prevents deformities caused by osteoarthritis, gout andseronegative arthritides. Table 1 summarizes some of the identi¢ed case reports.The presence of an intact nervous system is important to allow the full expressionof these arthritides.A putativemechanism can be proposedwhereby an in£ammatory insult, such as
a monoarthritis, triggers changes in the ipsilateral neural network which are thenmirrored to the contralateral side to predispose this side to a similar in£ammatorystate. This is an example of an autoregulatory loop and this hypothesis is testable.
Contralateral in£uences of neuro-in£ammatory insults
Rotshenker & Tal (1985) reported in both frogs and mice that sprouting in thecontralateral neuromuscular junction followed nerve section and degeneration ofthe ipsilateral neuromuscular junctions. These contralateral ¢ne extra sprouts were¢rst seen 5 days after the sciatic nerve was proximally sectioned and this sproutingoccurred much more frequently than in control rats. Associated contralateralincreases in GAP43, neuropeptide mRNA and expression, and proliferation ofmicroglia were also seen (Koltzenburg et al 1999).An in£ammatory lesion induced in a rat knee using latex spheres resulted in a
thickening bilaterally of the synovial intimal layer and this was accompanied by abilateral cellular in¢ltrate (Kidd et al 1995). No changes were detectable in any ofthe other joints. There were also contralateral changes in bradykinin-inducedextravasation from the knee joint, as measured using Evans’ blue, whichfollowed the course of this latex monoarthritis. This somatotopic distantresponse to a local in£ammatory insult can only be mediated through the neuralnetwork. Systemic circulatory factors cannot display such topographical precisionand biomechanical in£uences fail to explain the lack of response in other load-bearing joints. In further support of a neurogenic mechanism, the expression ofsubstance P in the dorsal root ganglion was increased within 3 days bilaterally inthis monoarthritis model.There exists a body of published evidence to corroborate the ¢ndings that a
unilateral insult induces contralateral topographically-precise changes associated
246 SHENKER ET AL

with appropriately located changes at the spinal level. We have recently reviewedthis literature and this is summarized in Table 2.It is important to note that although contralateral changes in the periphery can
be seen from these models, the earliest changes on the contralateral side occurwithin the spinal cord. These are seen at the cellular level with changes inneuropeptide expression in the contralateral nerve cell bodies (Kidd et al 1995).It has been documented that unilateral capsaicin injections, the pungent extract
of chilli that induces the release of SP andCGRP, alters contralateral depolarizationthresholds and re£exes (Gjerstad et al 2000). We were able to demonstratecontralateral central pain responses in human by detecting contralateral allodyniaand hyperalgesia within 30 minutes following a unilateral intradermal injection ofcapsaicin (Fig. 2). In a group of 20 normal control subjects, 9 (45%) demonstratedcontralateral sensory changes that were between 5^50% of the hypersensitive areainduced by the capsaicin injection. In a group of 21 subjects with rheumatoidarthritis, 11/21 (52%) demonstrated contralateral sensory changes (R. Haigh,unpublished data).The prior development of changes within the central nervous system implies
that this is driving any contralateral peripheral change. This physiological loopmay well be important in conferring survival bene¢t, as will be discussed later,but ‘physiology begets pathology’. It is therefore possible that the nervoussystem not only underlies the symmetry of some diseases, but may be principle intheir aetiopathogenesis as well.
Charcot’s joints
Charcot was the ¢rst to note the possible role of the nervous system in arthritis andhis name has become synonymous with the neuroarthropathic joint. It is thereforeno coincidence that the clinical history of these joints re£ects the surmised role thatneuropeptides have in their aetiology.Sella & Barrette (1999) were able to identify ¢ve stages of neuroarthropathy in
the feet of diabetics from the careful evaluation of clinical histories and radiologicaltests. In stage 0 the patient presents with a locally swollen, warm, and often painfulfoot. It is this stage that is often missed clinically as patients tend to present later.Radiographs are negative whereas a technetium 99 bone scan is markedly positive.In addition to these clinical ¢ndings there are also periarticular cysts, erosions andlocalized osteopenia radiologically in stage 1. Stages 2^4 demonstrate theprogressive joint subluxations, dislocations and destruction of the joints in thediabetic foot. Clinically in these stages, there is no temperature gradient betweenthe two feet. Radiologically, there is bony trabeculation across joint spacesindicative of mature fusion.
NEUROGENIC ARTHRITIS 247

248 SHENKER ET AL
TABLE 2 The contralateral e¡ects of localized unilateral in£ammation
Lesions inducing arthritis Contralateral e¡ect
CFA (1 mg) in knee Decrease in anabolism of cartilage for 6^72 h
Freund’s adjuvant in knee(0.5ml of 1mg/ml)
Increase in SP, CGRP and NPY in knee for 2^24 hIncrease in NK-A in knee at 2 h and at 24 h
Carrageenan 2% (0.05ml) in knee Increase in CGRP and NPY for 2^24 hIncrease in SP at 2 h and at 24 h; and NK-A at 24 h
Human recombinant IL1 (0.05mlof 1mg/ml) in knee
Increase in CGRP and NPY for 2^24 hIncrease in NK-A at 2^6 h and SP at 2 h
SP (0.05ml of 10�5M) in knee Increase in SP for 2^24 h, CGRP for 6^24 h andNPY for 2^6 hNo increase in NK-A
SP in knee (0.2, 1, 2, 10, 20 mg in 50 ml) Decrease in anabolism of cartilage for 6^72 h
500 ml of mBSA in knee of rats pre-immunized with 1ml CFA/2mgMTb
Histopathological and biochemical evidence of jointdestruction up to 80 d
500 mg (50 ml) mBSA in knee of pre-sensitized rats
Increased proteoglycan loss and mechanical hyperalgesia upto 9 days
100 ml of 1% latex spheres in knee Bradykinin-induced plasma extravasation enhanced between10^21 days. Macrophage in¢ltration noted between 3^10days
Lesions inducing hindpaw oedema Contralateral e¡ect
CFA (100 ml) TNFa levels elevated up to 120 h
Carrageenan (0.1ml of 2%) CGRP levels increase in hindpaw perfusate at 3^4 hThermal andmechanicalwithdrawal latencies reduced at 3^4 h
Carrageenan 100 ml of 2% Mechanical hyperalgesia between 3^5 days
Formaldehyde 50 ml (0.1%, 5%, 10%) Licking responses occur at 10^60 minutes
Bee venom 100 ml (0.2mg) Heat (not mechanical) withdrawal latency reduced for 2^48 h
Repeated saline injection150 ml on 3 consecutive days
Reduction in mechanical withdrawal latencies for 3^5 hIncrease in paw thickness for 3^5 h
Urate, pyrophosphate and oxalatecrystals (3mg in 150 ml)
Swelling observed, maximal at 5 h
CGRP 100 ml (300 pmol) Oedema induced at 5^24 h
NGF injections (3 days of 4 mg/day)in hindpaw or ear or forepaw
Increased expression of preprotachykinin andpreproCGRP mRNA in nerve (sciatic; trigeminal;or brachial plexus)
IL1b (10 ng) B1 receptor-mediated mechanical hyperalgesia for1^6 h
Thermal stimuli (55 8C for 15 s) Thermal foot withdrawal latencies reduced at 24 h
Thermal stimuli (55 8C for 15^20 s) Thermal foot withdrawal latencies reduced at 24 h
Thermal stimuli to decerebrate rat(75 8C for 60 s)
Reduction in the £exor re£ex threshold to mechanical andthermal stimuli at 1 h
All of these studies were performed in rats, unless otherwise indicated.CFA, complete Freund’s adjuvant; SP, substance P; NPY, neuropeptide Y; CGRP, calcitonin gene relatedpeptide; NK-A, neurokinin A; IL1, interleukin 1; MBSA, methylated bovine serum albumin; MTb,Mycobacterium tuberculosis; TNF, tumour necrosis factor; NGF, nerve growth factor; RNA, ribonucleic acid.

Histological examination of Charcot joints has been di⁄cult because tissue fromearly disease is rare andmost samples have been removed fromdestroyed joints thatare late in the disease process. Most of these specimens show non-speci¢c changesconsistent with an in£ammatory process but there are isolated reports that showscattered lymphocytes (Clement et al 1984).As can be seen from the experimental evidence presented above, neuropeptides
from damaged nerve terminals increase both the synovial blood supply and thein£ux of activated macrophages and lymphocytes into the joint. These cells alterthe complex neurochemical intra-articular matrix substantially leading to massiverapid destruction of the joint. Later in the disease, however, neural damage hassu⁄ciently depleted neuropeptides so that their vasodilatory e¡ects arenegligible. Interestingly Charcot joints are not uncommonly bilateral(Armstrong et al 1997).
Survival hypothesis of an auto-neuroin£ammatory loop
There is evolutionary pressure for rapid appropriate responses to be available aspart of an organism’s defence against noxious environs (Kidd et al 1989).
NEUROGENIC ARTHRITIS 249
FIG. 2. Rheumatoid subject demonstrating mirroring of painful stimulus. IpsilateralCapsaicin injection in the left forearm. Using Semmes Weinstein ¢laments, the sensorychanges are then mapped using di¡erent colours to represent 10 minute intervals between 10^60minutes. Changes in allodynia are markedwith an ‘x’. Changes tomechanical hyperalgesia aremarked with ‘^’. Note the development of both allodynia and hyperalgesia in a topographicallyprecise contralateral region.

Bilaterality has evolved so that there is functional capacity should one part beirreparably damaged. It is therefore vitally important that the contralaterallyhomonymous area should be protected following focal damage. A neuro-in£ammatory loop whereby protective responses are contralaterally rapidly up-regulated would therefore be a useful addition to any symmetrical organism’sprotection and long-term survival. The advantage that this precise response hasover any systemic mechanism is that it is economic with the energy expended andlimits widespread self-damage caused by inappropriate in£ammation. Similarly,pain withdrawal re£exes are primed through previously documentedelectrophysiological changes in the spinal cord so that the contralateral limb canbe withdrawn more quickly. There is more chance of damage limitation from adangerous environment if such responses existed and it is argued that survival isenhanced through use of such pathways.As our control over the environment has reduced the risk of noxious exposure in
modern society, these pathways may now unfortunately be more associated withdisease states. There is insu⁄cient evolutionary pressure to down-regulate thesepathways because these diseases have a limited e¡ect on reproductive capacity astheir peak onset is after childbearing years.
Conclusion
Chronic in£ammatory disease is symmetrical where possible. This symmetry ismediated neurogenically through neuropeptide mechanisms that de¢ne a neuro-in£ammatory loop that is insult speci¢c and topographically precise. Theimmune system can fundamentally be a¡ected by the release of suchneuropeptides through their e¡ects on T cells. This interaction may be importantnot only in the expression of symmetry in diseases but also in theiraetiopathogenesis.
References
Armstrong DG, ToddWF, Lavery LA, Harkless LB, Bushman TR 1997 The natural history ofacute Charcot’s arthropathy in a diabetic foot speciality clinic. Diabet Med 14:357^363
Arnett FC, Edworthy SM, Bloch DA et al 1988 The American Rheumatism Association 1987revised criteria for the classi¢cation of rheumatoid arthritis. Arthritis Rheum 31:315^324
Brain SD, Williams TJ, Tippins JR, Morris HR, MacIntyre I 1985 Calcitonin gene-relatedpeptide is a potent vasodilator. Nature 313:54^56
Bukhari M, Lunt M, Harrison BJ, Scott DG, Symmons DP, Silman AJ 2002 Erosions inin£ammatory polyarthritis are symmetrical regardless of rheumatoid factor status: resultsfrom a primary care-based inception cohort of patients. Rheumatology (Oxford) 41:246^252
Clement GB, Grizzard K, Vasey FB, Germain BF, Espinoza LR 1984 Neuropathic arthropathy(Charcot joints) due to cervical osteolysis: a complication of progressive systemic sclerosis. JRheumatol 11:545^548
250 SHENKER ET AL

Culberson JL, Haines DE, Kimmel DL, Brown PB 1979 Contralateral projection of primarya¡erent ¢bers to mammalian spinal cord. Exp Neurol 64:83^97
Delgado M, Abad C, Martinez C, Leceta J, Gomariz RP 2001 Vasoactive intestinal peptideprevents experimental arthritis by downregulating both autoimmune and in£ammatorycomponents of the disease. Nat Med 7:563^568
Dixon A 1989 Hemiplegia, hand dominance and asymmetry of expression of arthritis in relationto intra-articular pressure. In: Dixon A, Hawkins C (eds) Raised intra-articular pressure�clinical consequences. Bath Institute for Rheumatic Diseases, Bath, p 65
Donaldson LF, McQueen DS, Seckl JR 1995 Neuropeptide gene expression and capsaicin-sensitive primary a¡erents: maintenance and spread of adjuvant arthritis in the rat. J Physiol486:473^482
FarberEM,Nickolo¡BJ,Recht B, Fraki JE1986 Stress, symmetry and psoriasis: possible role ofneuropeptides. J Am Acad Dermatol 14:305^311
Garsden LR, Bullock-Saxton JE 1999 Joint reposition sense in subjects with unilateralosteoarthritis of the knee. Clin Rehabil 13:148^155
GarrettNE,MappPI, Cruwys SC,KiddBL,BlakeDR1992Role of substance P in in£ammatoryarthritis. Ann Rheum Dis 51:1014^1018
Gjerstad J, Tjolsen A, Svendsen F, Hole K 2000 Inhibition of spinal nociceptive responses afterintramuscular injection of capsaicin involves activation of noradrenergic and opioid systems.Brain Res 859:132^136
Hanesch U, Blecher F, Stiller RU, Emson PC, Schaible HG, Heppelmann B 1995 The e¡ect of aunilateral in£ammation at the rat’s ankle joint on the expression of preprotachykinin-AmRNA and preprosomatostatin mRNA in dorsal root ganglion cells� a study using non-radioactive in situ hybridization. Brain Res 700:279^284
Helliwell PS, Hetthen J, Sokoll K et al 2000 Joint symmetry in early and late rheumatoid andpsoriatic arthritis: comparison with a mathematical model. Arthritis Rheum 43:865^871
Herzburg U, Murtaugh MP, Mullet MA, Beitz AJ 1995 Electrical stimulation of sciatic nervealters neuropeptide content and lymphocyte migration in the subcutaneous tissues of the rathind paw. Neuroreport 6:1773^1777
Holzer P 1988 Local e¡ector functions of capsaicin-sensitive sensory nerve endings:involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides.Neuroscience 24:739^768
Hood VC, Cruwys SC, Urban L, Kidd BL 2001 The neurogenic contribution to synovialleucocyte in¢ltration and other outcome measures in a guinea pig model of arthritis.Neurosci Lett 299:201^204
Kidd BL, Mapp PI, Gibson SJ et al 1989 A neurogenic mechanism for symmetrical arthritis.Lancet 2:1128^1130
Kidd BL, Cruwys SC, Garrett NE,Mapp PI, Jolli¡e VA, Blake DR 1995 Neurogenic in£uenceson contralateral responses during experimental rat monoarthritis. Brain Res 688:72^76
Koltzenburg M, Wall PD, McMahon SB 1999 Does the right side know what the left side isdoing? Trends Neurosci 22:122^127
Levine JD, Clark R, Devor M, Helms C, Moskowitz MA, Basbaum AI 1984 Intraneuronalsubstance P contributes to the severity of experimental arthritis. Science 226:547^549
Levine JD, Dardick SJ, Basbaum AI, Scipio E 1985 Re£ex neurogenic in£ammation. I.Contribution of the peripheral nervous system to spatially remote in£ammatory responsesthat follow injury. J Neurosci 5:1380^1386
Levite M 2001 Nervous immunity: neurotransmitters, extracellular potassium and T-cellfunction. Trends Immunol 22:2^5
Light AR, Perl ER 1979 Reexamination of the dorsal root projection to the spinal dorsal hornincluding observation of the di¡erential termination of coarse and ¢ne ¢bres. J CompNeurol186:117^131
NEUROGENIC ARTHRITIS 251

Mapp PI, Terenghi G, Walsh DA et al 1993 Monoarthritis in the rat knee induces bilateral andtime-dependent changes in substance P and calcitonin gene-related peptide immunoreactivityin the spinal cord. Neuroscience 57:1091^1096
McGillis JP, Mitsuhashi M, Payan DG 1990 Immunomodulation by tachykinin neuropeptides.Ann NY Acad Sci 594:85^94
Niu J, Zhang Y, LaValley M, Chaisson CE, Aliabadi P, Felson DT 2003 Symmetry andclustering of symptomatic hand osteoarthritis in elderly men and women: the Framinghamstudy. Rheumatology (Oxford) 42:343^348
Qian BF, Zhou GQ, Hammarstrom ML, Danielsson A 2001 Both substance P and its receptorare expressed in mouse intestinal T lymphocytes. Neuroendocrinology 73:358^368
Rotshenker S, Tal M 1985 The transneuronal induction of sprouting and synapse formation inintact mouse muscles. J Physiol 360:387^396
Sella EJ, Barrette C 1999 Staging of Charcot neuroarthropathy along the medial column of thefoot in the diabetic patient. J Foot Ankle Surg 38:34^40
Sharma L, Pai YC, Holtkamp K, Rymer WZ 1997 Is knee joint proprioception worse in thearthritic knee versus the una¡ected knee in unilateral knee osteoarthritis? Arthritis Rheum40:1518^1525
Wang H, Xing L, Li W, Hou L, Guo J, Wang X 2002 Production and secretion of calcitoningene-related peptide from human lymphocytes. J Neuroimmunol 130:155^162
Weihe E, Krekel J 1991 The neuroimmune connection in human tonsils. Brain Behav Immun5:41^54
Werring DJ, Brassat D, Droogan AG et al 2000 The pathogenesis of lesions and normal-appearing white matter changes in multiple sclerosis: a serial di¡usion MRI study. Brain123:1667^1676
Westermark T, Rantapaa-Dahlqvist S, Wallberg-Jonsson S, Kjorell U, Forsgren S 2001Increased content of bombesin/GRP in human synovial £uid in early arthritis: di¡erentpattern compared with substance P. Clin Exp Rheumatol 19:715^720
DISCUSSION
Hunt:What does bilaterality tell you about the ipsilateral in the original cause ofthe disease?Blake: The whole thing is basically a neural discharge event. I’m talking here
about rheumatoid arthritis (RA). If someone had taught me immunology sayingthat the primitive system was neuropeptidergic, the secondary evolution of thesystem is antigen-driven, and then I had heard Bruce Kidd’s lecture on the natureof the pain response and the axon re£ex in palindromic RA, I would by preferencechoose to adopt the idea that it is a neuropeptide discharge state that creates thisunusual degree of symmetry. Then the subsequent disruption creates a secondaryautoimmune reaction sequence that is very diverse. This is whatwe see.My guess isthat arthritis might be better dissected by neurologists.Hunt:We are talking about very precise topography here: one joint on one side
and one on the other. Yet the work on placebo by Benedetti involving capsaicininjections, placebo response is very precise (Benedetti et al 1999). So what ishappening peripherally can be adjusted precisely. It is not clear how, because the
252 DISCUSSION

descending systems themselves from the brain to the spinal cord don’t look verytopographically organized. They appearmore di¡use. I don’t know how it is done.Blake: Nevertheless, you have a deep suspicion it is done. That is a powerful
argument in terms of the upper control of the whole system.Rediske: Earlier in the meeting there was some discussion about how patients
with OA in one knee develop OA in the second knee with time. The argumentgiven was that this was more of a compensatory gait phenomenon. How muchcould this bilateral phenomenon play a role in evolution of secondary OA? Whenyou compare the location of the cartilage injuries in the two knees, how similar arethey?Felson: David Blake was on target when he showed the Herberden’s nodes
bilaterally. This is nicely described by Cyrus Cooper, and then we published apaper last year in Rheumatology which corroborated it in Framingham (Niu et al2003). There is a remarkable symmetry in the occurrence of OA in hands inpeople. If you have a ¢fth DIP on one side the odds ratio for getting a ¢fth DIPin the exact joint on the other side is 15. There is remarkable symmetry in OA also.The question is the one you raised, John: are there other explanations in OA otherthan some kind of tra⁄cking or neuropeptide release that crosses sides? We havethought about anatomic similarities between sides. David Hunter might want tocomment on the genetics of hand OA and how it might relate to anatomy.Hunter: We have recently done a factor analysis on the symmetry using the
Framingham family data. This demonstrates that the predisposition is purelysymmetrical. The predisposition to disease is largely heritable. We don’t knowwhether that symmetry is being predisposed to a heritable element in the brain oran anatomical element at the joint. This has some impressive LOD scores in termsof linkage.Pisetsky: Is the heritability joint speci¢c? It is symmetrical but is it joint speci¢c?
A person could inherit some factor that predisposes to base of thumb arthritis butwhich doesn’t increase the risk of DIP or PIP arthritis. This suggests that this isanatomical.Blake: One does not exclude the other options. We can equally have lots of
pathways that create this, and it would be foolish to say this explains everything.Clearly, walking unevenly can create symmetrical events for a completelydi¡erent reason. This is a partial explanation of some kinds of pathologies. Thefact is that the joint system as I would describe it is built up by serial divisionsusing fractal-based mathematics. What you have just described is a fractalpattern, and what you are saying is that this is a di¡erent genetic pattern fromthat. That’s ¢ne: this then impacts upon that. Then you have to say to yourselfthat nodal OA of the ¢ngers relates to OA of the hip, which it seems to do.Could it be that the DIP joints are actually the same fractal zone that embracesthe hips? Then if you start going back in the evolutionary sense, and look at
NEUROGENIC ARTHRITIS 253

amphibians, then what lies behind what now becomes a hand system is the hip. Sowhat you are describing are genetically determined fractal zones. Then it all ¢tstogether.Pisetsky:There are some forms of arthritis that are classically asymmetrical.How
do you interpret them? The other thing is that we have a variety of e¡ectiveinterventions that are asymmetric, such as joint injections and joint replacement.Blake:You are playing intomy hands here. The whole purpose of my paper was
to describe asymmetry. For example, look at the ¢sh model where there is overtin£ammation on one gill. But destroy the organism with a gaseous system on theother side and clinically you see asymmetry. The processes are occurring in bothgills but you just see it in one because it isworking.Asymmetry should be the normif this system is working. It is pretty pointless us having two kidneys, two eyes andtwo testes if every time a disease gets one it gets the other. The fact that they arebilaterally located in mirror image spots across the spinal cord seems more than alittle coincident. But the whole process from the evolutionary point of view is tosay that ‘I’m under attack, don’t go down that route’. Then one presumes likeeverything else in biology that there is a genetic in£uence that promotes theextent of bilaterality. It will be a small subpopulation that have a very dominantmirror-imaging system that then undergo the process. Exactly the same is true insympathetic uveitis, where most people get away with a metal object in their eyeand 5% go blind. But there are all points in between. So asymmetry is thephysiological state.Lohmander: Iwould argue thatwe need to accept that all of thementioned factors
would feed into the systemwith regards to symmetry or asymmetry ofOA.Clearly,unilateral OAwill change gait and loading on the other side. There are neurogenicmechanisms and there are likely systemic mechanisms by which arthritis could betransferred. Finally, we have the genetic aspects and anatomical aspects. Anexample which would ¢t into David’s argument is that of the fairly few speci¢cmutations we know about in connective tissue-related genes that are associatedwith bona ¢de OA. These di¡erent mutations do not generate the same forms ofOA. Each mutation is fairly speci¢c in the form of OA that it brings about.Matrilin 3 mutations are di¡erent from collagen 2 mutations and so forth. I thinkthis speaks to the developmental/fractal theory that David is proposing.Dieppe:There is some oldwork from the Leeds biomechanical group (Swann&
Seedhom 1993) showing that there were very symmetrical patterns ofdeformability between joints. If you have one type of mechanical feature of oneknee, you had exactly the same on the other side. This suggests that there is a lotof symmetry of theway that joints are built and theway they respondmechanically.I have a question about CRPS,which you have been looking at.My experience is
that this is asymmetrical, but I would have thought from your thesis that itshouldn’t be.
254 DISCUSSION

Blake: As it happens it is not asymmetrical. It is surprisingly symmetrical whenyou have the right tools to look at it.Simkin: It is relevant tomention that the vascular physiology of the synovium is
strikingly symmetrical and di¡ersmarkedly fromone pair of joints to the next. Theshoulders are not like the knees which are not like the wrists (Simkin & Benedict1990a,b). I am not sure what this has to do with the pain of OA, but it may havequite a bit to do with the patterns that we see in symmetrical joint disease.Hunter: Perhaps I am misinterpreting what you are saying. It seems you are
suggesting that asymmetry is the rule and only 10^20% of people will ¢t thissymmetrical-type pattern. Irrespective of whatever tool I have tried to use toimage OA, the symmetry is still striking.Blake: I am just making these ¢gures up, but I am talking about 10% as a total
physiological event for all processes in human society. But one can understand thatthere might be a disease or process like OA where there is an underlying zonalfractal system that determines pattern, and which somehow is also in£uenced bya secondary genetic system that evokes symmetry but which can be disrupted in astroke model. The onset of this in normality could be a fractal genetic aberration,whereas the mirror-imaging process that is meta and paraceptively modi¢ed isanother process that comes into the game. We often end up in these symposia byfeeling that one explanation provides all explanations.We only have to realise howmany genes are going up and down in that cancer model to realise this is not true.These things have to work together.Hunter: If you want to put a ballpark ¢gure on it, it sounds like your belief for
RA is that this whole process accounts for the majority of the disease even on theipsilateral side. If you want to put a ballpark ¢gure on the in£uence of yourmetaceptive system for OA, for the majority of the community, what would it be?Blake:When people crack the genes for this fractal patterning then you have the
answer�basically, it’s what’s left.Evans: Coming back to the T cell and antigen-driven events, and how the
nervous system can in£uence them, we have an interesting observation relatingto what I talked about yesterday with this contralateral suppression of disease.We take rabbits and sensitize them to two di¡erent antigens, ovalbumin andBSA. Then we have the choice of giving the rabbits bilateral knee disease withthe same antigen or di¡erent antigens. We only see the contralateral suppressionof disease if the contralateral knee is su¡ering from the same antigen-inducedarthritis. We don’t get contralateral suppression of arthritis when a di¡erentantigen is driving it. I don’t know how to explain this, but it suggests that thiswhole relationship between the immune system and the nervous system is goingto be fascinating to tease apart.Grubb: In neuropathic and in£ammatory pain we see sets of neuropeptides
and cell markers going up and down, but we see modest contralateral e¡ects
NEUROGENIC ARTHRITIS 255

quite often. Do you have a feeling for what the mechanism is in the nervoussystem?Hunt: I’ve always ignored contralateral events as being artefacts. If you cut the
sciatic nerve on one side the animal is obviously moving in a di¡erent way. Butthere is a lot of literature on this, and some people have done some veryinteresting experiments. But it always comes out that the ¢nal reason for this is soobscure.Grubb: The fact that the capsaicin treatment seems to prevent some of these
changes is interesting. We know there is a certain amount of hard wiring betweenleft and right. We discussed the development of contralateral inputs. Neurons thatshow central sensitization develop receptive ¢elds from the contralateral side. Wecan see cross spinal connections, but we can’t imagine how that gets back to theDRG on the other side.Fox: How frequent are these changes? The contralateral e¡ects aren’t terribly
robust, are they?Blake:They are very robust providing that you look at them in the rightmodels.
It depends what you are looking at. For the example I gave on the eye, if your onlyinvestigative tool was blindness, this is a very infrequent event. But if you use anERG it is an incredibly frequent event.Hunt: In general, for chip studies I would never use the other side. I think it does
change.Blake: That is an extraordinarily sensitive technique for looking at the
phenomenon.Fox: We have done this. I can’t quote the numbers, but the changes were not
signi¢cant.Blake:My whole point is that the changes were small. This is a down-regulated
system. We are looking for a small subtle change.Felson: I want to bring up another potential use for this contralateral e¡ect. We
talked earlier about looking for models of early disease that might correspond toearly ways of identifying some of this going-to-develop disease. The contralateralphenomenon is so common in clinical OA that it might not be a bad example, sortof like taking someone from a family with a mutation who hasn’t yet developeddisease but who is likely to.
References
Benedetti F, Arduino C, Amanzio M 1999 Somatotopic activation of opioid systems by target-directed expectations of analgesia. J Neurosci 19:3639^3648
Niu J, Zhang Y, LaValley M, Chaisson CE, Aliabadi P, Felson DT 2003 Symmetry andclustering of symptomatic hand osteoarthritis in elderly men and women: the FraminghamStudy. Rheumatology 42:343^348
256 DISCUSSION

Simkin PA, Benedict RS 1990a Iodide and albumin kinetics in normal canine wrists and knees.Arthritis Rheum 33:73^79
Simkin PA, Benedict RS 1990b Hydrostatic and oncotic determinants of microvascular £uidbalance in normal canine joints. Arthritis Rheum 33:80^86
Swann AC, Seedhom BB 1993 The sti¡ness of normal articular cartilage and the predominantacting stress levels: implications for the aetiology of osteoarthrosis. Br J Rheumatol 32:16^25
NEUROGENIC ARTHRITIS 257

Lessons from ¢bromyalgia: abnormal
pain sensitivity in knee osteoarthritis
Laurence A. Bradley, Brian C. Kersh*, Jennifer J. DeBerry, Georg Deutsch,Graciela A. Alarco¤ n and David A. McLain{
Division of Clinical Immunology andRheumatology, University ofAlabama at Birmingham, 805Faculty O⁄ceTower, 510 20th Street South, Birmingham,AL35294, *NewMexicoVeteransAdministration Health Care System, 1501 San Pedro Drive, SE Building 41, Albuquerque,NM 87108 and {Brookwood Medical Center, Homewood, AL 35209, USA
Abstract. Fibromyalgia (FM) is a disorder that is characterized by widespread,musculoskeletal pain and abnormal pain sensitivity at multiple anatomic sites.Laboratory studies involving psychophysical and neuroimaging methods suggest thatcentral augmentation of low intensity stimulation may contribute to abnormal painsensitivity in FM. Recently, several investigators, using similar laboratory methods,have shown that patients with knee or hip osteoarthritis (OA) exhibit abnormal painsensitivity or abnormal pain inhibition at anatomic sites distal to a¡ected joints.Consistent with animal models of central sensitization, di¡erences between patients andhealthy controls in pain processing and pain inhibition at these distal sites are eliminatedafter nociceptive input is eliminated following total joint replacement surgery. This paperreviews these ¢ndings from our laboratory and those of independent investigators. It alsopresents verbal, psychophysical and neuroimaging data concerning ethnic groupdi¡erences in a¡ective and cognitive pain responses among patients with knee OA. Wesuggest that central sensitization as well as centrally-mediated cognitive and a¡ectivefactors in£uence the pain responses of patients with knee OA. In addition, ethnic groupdi¡erences in pain cognition and a¡ect may contribute to di¡erences among these groupsin preferences for healthcare interventions such as total joint replacement.
2004 Osteoarthritic joint pain. Wiley, Chichester (Novartis Foundation Symposium 260)p 258^276
Fibromyalgia (FM) is a disorder that is characterized by (a) persistent, widespread,musculoskeletal pain, and (b) abnormal pain sensitivity (i.e. allodynia) evoked bylow intensity stimuli (Table 1). Neither psychiatric morbidity nor healthcareseeking behaviour accounts for abnormal pain sensitivity in FM. For example,pain-free healthy controls exhibit signi¢cantly higher pain thresholds in responseto pressure stimulation than both rheumatology clinic patients with FM andcommunity residents who meet criteria for FM but do not seek medical care fortheir pain (i.e. non-patients) (Aaron et al 1996, Bradley et al 1999). However, the
258
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

FM patients are characterized by signi¢cantly greater lifetime psychiatricmorbidity than both the non-patients with FM and the controls (Aaron et al1996, Bradley et al 1999). Moreover, the FM non-patients do not di¡er from thecontrols in psychiatric morbidity.Patients with FM, compared to controls, also show abnormal pain thresholds in
response to thermal stimuli (e.g. Gibson et al 1994) as well as enhanced wind-upresponses to phasic mechanical and thermal stimulation (Staud et al 2001, 2003a).However, several centrally mediated pain inhibition mechanisms in patients withFM are intact. It has been found that the abnormal pain responses described aboveare inhibited by the NMDA antagonist ketamine (Graven-Nielsen et al 2000) andfentanyl (Price et al 2002). In addition, combining counter-irritation procedures,such as hand immersion in a hot water bath, with distraction attenuates thermalwind-up responses in women with FM (Staud et al 2003b).
Central correlates of abnormal pain sensitivity in ¢bromyalgia
Neuroimaging studies indicate that abnormal pain sensitivity in patients with FMis associated with altered patterns of regional cerebral blood £ow (rCBF) duringrest and in response to phasic pressure stimulation. At rest, patients with FM,compared to controls, show signi¢cantly lower levels of rCBF in the thalamus orcaudate nucleus (Mountz et al 1995, Bradley et al 1999, Kwiatek et al 2000). Similar
ABNORMAL PAIN SENSITIVITY 259
TABLE 1 The American College of Rheumatology classi¢cation criteria for¢bromyalgia
I. History of Widespread Pain (53 months)
. Left and right sides of body
. Above and below the waist
. Axial skeletal pain
. cervical spine, or
. anterior chest, or
. thoracic spine, or
. low back
II.Tender point pain sensitivity
. Pain, on digital palpation (44 kg), must be present in at least 11 of 18 speci¢c tender pointsites
Occiput Lateral epicondyle
Lower cervical Gluteal
Trapezius Greater trochanter
Supraspinatus Knees

resting state abnormalities in brain rCBF are displayed by patients with persistentneuropathic pain and patients with metastatic cancer pain (Iadarola et al 1995,Di Piero et al 1991).In addition, recent investigations indicate that low intensity stimuli evoke
abnormal pain perceptions and augmented neural input among patients with FMand those with neuropathic pain syndromes (e.g. Gracely et al 2002, Petrovic et al1999). For example, Gracely and colleagues (2002) found that it was necessary toapply di¡erent levels of phasic pressure stimulation to the left thumbnail of patientswith FM (2.4 kg) and healthy controls (4.2 kg) for both groups to produce a meanrating of 11 on a 20 point scale of pain intensity. Consistent with their verbal painresponses, both patients and controls exhibited signi¢cant increases in functionalmagnetic resonance imaging (fMRI) signal in the same seven brain regions (e.g.somatosensory cortices, insula, putamen, cerebellum) in response to thestimulation. However, when the controls received pressure stimulation at thesame intensity level delivered to patients, they showed signi¢cant fMRI signalincreases in only two brain regions; neither of these regions overlapped withthose activated within the patient group. These ¢ndings suggest that abnormalpain sensitivity in persons with FM is associated with dysregulation of centralprocessing of sensory input.
Mechanisms underlying abnormal pain sensitivity
Several investigators have suggested that central sensitization may contribute tothe abnormal pain sensitivity associated with FM. The mechanisms underlyingcentral sensitization involve hyper-excitability of spinal dorsal horn neurons thattransmit nociceptive input to the brain. Speci¢cally, intense or prolongednociceptive input from Ad and C a¡erents su⁄ciently depolarizes the dorsal hornneurons thatMg2+ exits NMDA-linked ion channels. This is followed by an in£uxof extracellular Ca2+ and production of nitric oxide which di¡uses out of the dorsalhorn neurons. Nitric oxide, in turn, promotes the exaggerated release of excitatoryamino acids and substance P from presynaptic a¡erent terminals and causes thedorsal horn neurons to become hyperexcitable (Schaible et al 2002). As aconsequence, low intensity stimuli delivered to the skin or deep muscle tissuegenerate high levels of nociceptive input to the brain and the perception of pain.However, central sensitization generally is diminished following cessation ofnociceptive input from Ad and C a¡erents.Recently, Watkins and colleagues (2001) have provided evidence that dorsal
horn glia cells also play a role in producing and maintaining abnormal painsensitivity. Synapses within the central nervous system are encapsulated by gliathat normally do not respond to nociceptive input from local sites. However,following the initiation of central sensitization, spinal glial cells are activated by a
260 BRADLEY ET AL

wide array of factors that contribute to hyperalgesia such as immune activationwithin the spinal cord, substance P, excitatory amino acids, nitric oxide, andprostaglandins. Once activated, glial cells release several pro-in£ammatorycytokines (e.g. tumour necrosis factor [TNF], interleukin [IL]6, IL1), substanceP, nitric oxide, prostaglandins, excitatory amino acids, ATP and fractalkine that,in turn, (a) further increase the release of excitatory amino acids and substance Pfrom the Ad and C a¡erents that synapse in the dorsal horn and (b) enhance thehyperexcitability of the dorsal horn neurons (Milligan et al 2003). It must be
emphasized, however, that investigators have not been able to reliably
identify abnormalities in deep muscle tissue or skin among persons with
FM that might produce the prolonged nociceptive input that is necessary
to initiate the events underlying the development of central sensitization
or spinal glial cell activation (Sprott et al 1998).
Abnormal pain sensitivity in knee osteoarthritis
Patients with knee osteoarthritis (OA) generally identify pain as the primarysymptom of knee or hip OA. Although knee OA is associated with abnormalitiessuch as pressure on exposed subchondral bone ormicrofractures that might inducenociception and pain, patients with knee OA report pain that often is poorlylocalized and is not highly correlated with abnormalities in joint structureidenti¢ed by radiographs or MR. This intriguing similarity in pain reportsbetween patients with knee OA and those with FM have led several investigatorsto assess whether abnormalities in central processing of sensory input may also beassociated with knee OA.Indeed, recent studies have provided evidence that pain associated with knee or
hip OA is associated with abnormal central processing of sensory input. Forexample, patients with OA of the lower extremities, compared to healthycontrols, exhibit longer periods of hyperalgesia and larger areas of the leg withreferred pain following infusion of hypertonic saline into the tibialis anteriormuscle (Bajaj et al 2001). Similarly, Kosek & Orderberg (2000) have shown thatpatients with unilateral hip OA, compared to healthy controls, display lower painthresholds for pressure stimulation both at the a¡ected hip and at the una¡ected,contralateral hip. Unlike the controls, the patients fail to show an increase in painthreshold at the una¡ected, contralateral hip in response to ischaemic counter-stimulation. However, 5 months after the patients undergo hip replacementsurgery, there is no di¡erence between patients and controls in pain thresholdlevels at the contralateral hip. Moreover, the abnormality in patients’ painmodulation is reversed. That is, the patients now display a signi¢cant increase inpain threshold at the contralateral hip in response to ischaemic counter-stimulation. This suggests that cessation of nociceptive input from the a¡ected
ABNORMAL PAIN SENSITIVITY 261

hip allowed the patients’ pain thresholds and central pain inhibitory functions toreturn to normal.Our laboratory is now completing a series of studies of pain sensitivity in 11
patients with knee OA and 11 age-matched healthy controls, all of whom wereright-handed (DeBerry et al 2002, Kersh et al 2001). Given that most of ourpatients were characterized by bilateral knee OA, we sought to determinewhether they would display abnormal pain sensitivity at sites quite distant fromthe knees, such as the arms and shoulders. Figure 1 shows that the patients,compared to the controls, reported signi¢cantly higher levels of pain intensity inresponse to a series of phasic pressure stimuli applied to four sites on the left andright shoulders and arms (P¼0.045). This e¡ect was due primarily to the patients’responses to the higher stimulus intensities (i.e. 5.5 and 7.0 kg/cm2). However,there was no di¡erence in the responses of patients and controls to thermal heatstimulation at the same sites. This suggests that patients with knee OA mayexhibit abnormal, generalized pain sensitivity only in response to stimulation ofdeep muscle tissue. Moreover, the consistent ¢ndings across laboratories of (a)abnormal, generalized pain sensitivity and (b) normalization of pain sensitivityand pain modulation after termination of nociceptive input strongly suggeststhat central sensitization and perhaps glial cell activation may contribute to thepain experiences of persons with knee OA.
262 BRADLEY ET AL
FIG. 1. Mean pain intensity ratings of phasic pressure stimulation delivered to 4 sites on theright and left shoulders and arms. Patients with knee osteoarthritis (OA) produced signi¢cantlyhigher ratings than age-matched healthy controls (P¼ 0.045).
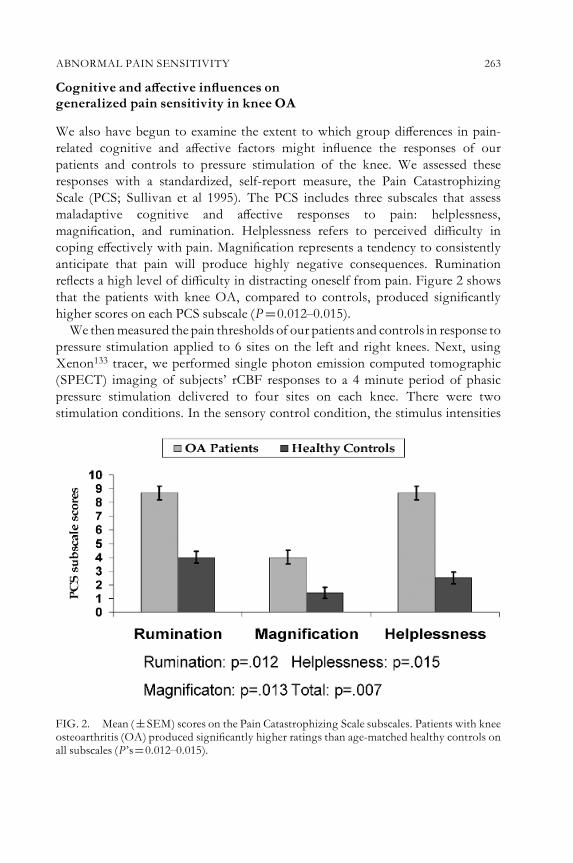
Cognitive and a¡ective in£uences on
generalized pain sensitivity in knee OA
We also have begun to examine the extent to which group di¡erences in pain-related cognitive and a¡ective factors might in£uence the responses of ourpatients and controls to pressure stimulation of the knee. We assessed theseresponses with a standardized, self-report measure, the Pain CatastrophizingScale (PCS; Sullivan et al 1995). The PCS includes three subscales that assessmaladaptive cognitive and a¡ective responses to pain: helplessness,magni¢cation, and rumination. Helplessness refers to perceived di⁄culty incoping e¡ectively with pain. Magni¢cation represents a tendency to consistentlyanticipate that pain will produce highly negative consequences. Ruminationre£ects a high level of di⁄culty in distracting oneself from pain. Figure 2 showsthat the patients with knee OA, compared to controls, produced signi¢cantlyhigher scores on each PCS subscale (P¼0.012^0.015).We thenmeasured the pain thresholds of our patients and controls in response to
pressure stimulation applied to 6 sites on the left and right knees. Next, usingXenon133 tracer, we performed single photon emission computed tomographic(SPECT) imaging of subjects’ rCBF responses to a 4 minute period of phasicpressure stimulation delivered to four sites on each knee. There were twostimulation conditions. In the sensory control condition, the stimulus intensities
ABNORMAL PAIN SENSITIVITY 263
FIG. 2. Mean (�SEM) scores on the Pain Catastrophizing Scale subscales. Patients with kneeosteoarthritis (OA) produced signi¢cantly higher ratings than age-matched healthy controls onall subscales (P’s¼0.012^0.015).

were kept constant at 1 kg. However, in the experimental condition, the intensitiesof the pressure stimuli were calibrated to each subject’s respective pain thresholdlevel (3 kg/cm24pain threshold). Immediately following stimulation, each subjectrated the intensity of the sensory and a¡ective (i.e. unpleasantness) dimensions ofhis or her pain using the McGill Pain Questionnaire (MPQ; Melzack 1975).Figure 3 shows that, consistent with our expectations, the patients displayed
signi¢cantly lower pain threshold levels than the controls (P¼0.025). Inaddition, Table 2 reveals that calibrating the intensity of the pressure stimulationto subjects’ pain threshold levels evoked similar MPQ ratings of sensory intensityat the right and left knee among the patients and controls. Nevertheless, compared
264 BRADLEY ET AL
TABLE 2 Mean (�SEM)MPQ sensory and a¡ective subscale scores of patients withknee osteoarthritis (OA) and healthy controls
MPQ subscaleOApatients(n¼11)
Healthy controls(n¼11) P value
Right kneeSensory 17.6�2.0 15.17�2.4 0.45
A¡ective 6.95�1.9 1.76�0.6 0.02
Left kneeSensory 18.3�2.0 17.3�2.5 0.76
A¡ective 7.0�2.5 1.46�0.7 0.003
MPQ¼McGill Pain Questionnaire.
FIG. 3. Mean (�SEM) pain thresholds for pressure stimulation delivered to 6 sites on theright and left knees. Patients with knee osteoarthritis (OA) displayed signi¢cantly lower painthreshold levels than age-matched healthy controls (P¼0.025).

to the controls, the patients produced signi¢cantly higherMPQ ratings of a¡ectiveintensity (i.e. unpleasantness) at both the right (P¼0.02) and left (P¼0.003) knee(Table 2). Moreover, statistically controlling for the in£uence of subjects’ PCShelplessness scale scores on their MPQ responses eliminated the signi¢cant groupdi¡erences in a¡ective intensity evoked by stimulation of each knee. This suggeststhat the di¡erence between patients and controls on this measure of pain-relatedcognition and a¡ect mediated the di¡erence in their MPQ pain-a¡ect scores.Using statistical parametric mapping, we compared the SPECT images of brain
rCBF in the sensory control and experimental conditions within the patient andcontrol groups. We found that, during the experimental condition, the patientsand controls generally exhibited reliable increases in rCBF in similar brainregions (e.g. somatosensory cortex) in response to stimulation of both the rightand left knee. However, only the patients with knee OA exhibited reliableincreases in brain rCBF in the left and right anterior cingulate cortex (ACC) inresponse to stimulation of each knee (P¼0.05). Activation of the ACC issigni¢cantly associated with individuals’ reports of pain unpleasantness and othermeasures of the a¡ective component of pain (e.g. Rainville et al 1997). In addition,among healthy volunteers, more robust activation of the ACC is associated withhigher levels of thermal pain sensitivity (Coghill et al 2003).Finally, we performed an exploratory analysis of possible ethnic group
di¡erences among our patients on their PCS, MPQ and brain rCBF responses.Figure 4 shows that our ¢ve African-American patients tended to produce higher
ABNORMAL PAIN SENSITIVITY 265
FIG. 4. Mean (�SEM) scores on the Pain Catastrophizing Scale subscales. African-Americanpatients with knee osteoarthritis (OA) tended to produce higher ratings than Caucasian patientswith kneeOAon all subscales, although the largest group di¡erence occurred on the helplessnesssubscale (P¼0.10).

scores than the six Caucasians on all of the PCS subscales; the largest groupdi¡erence was found on the helplessness subscale (P¼0.10). Moreover, Table 3reveals that the African-American patients produced higher MPQ a¡ectiveintensity scores than the Caucasian patients in response to stimulation of theright (P¼0.03) and left (P¼0.01) knees. The African-American patients,compared to the Caucasians, also produced signi¢cantly higher MPQ sensoryscores at the right knee (P¼0.03). In addition, consistent with the self-reportdata, we found that the African-American patients, compared to the Caucasians,displayed signi¢cantly greater activation of the ACC in response to stimulation ofthe right and left knees (P¼0.05). These ¢ndings are very similar to those reportedbyEdwards and colleagues (Edwards&Fillingim1999, Edwards et al 2001). Theirstudies of both healthy volunteers and patients with persistent pain revealed thatAfrican-Americans, compared to Caucasians, perceive noxious thermal andischaemic stimuli as signi¢cantly more aversive.
Implications of ethnic group di¡erences in
pain sensitivity on disparities in total joint replacement procedures
Variations among ethnic groups in pain-related cognition or a¡ect may haveimportant implications for disparities in their preferences for healthcareprocedures. For example, Ibrahim et al (2002) have demonstrated that, in theUSA, African-Americans are less willing than Caucasians to undergo knee or hipreplacement surgery, even if it is recommended by their physicians. Aftercontrolling for demographic, socioeconomic and clinical variables, the factorsthat mediated this relationship were the patients’ expectations in post-surgicalpain, walking ability and length of hospital stay. On all of these variables, theAfrican-Americans’ expectations were signi¢cantly more negative than those ofthe Caucasians, despite the fact the African-Americans reported they were less
266 BRADLEY ET AL
TABLE 3 Mean (�SEM) MPQ sensory and a¡ective subscale scores of African-American (AA) and Caucasian (C) patients with knee osteoarthritis (OA)
MPQ subscaleAApatients(n¼5)
C patients(n¼6) P value
Right kneeSensory 22.2�2.8 13.8�1.7 0.03
A¡ective 11.3�2.7 3.3�1.7 0.03
Left kneeSensory 21.6�2.4 15.5�2.6 0.12
A¡ective 10.9�1.6 3.8�1.5 0.01
MPQ¼McGill Pain Questionnaire.

familiar with total joint replacement surgery than the Caucasians. Our ¢ndingsdescribed above, as well as those of Edwards et al (1999, 2001) suggest that therelatively greater levels of negative pain-related cognition and a¡ect amongAfrican-Americans may be related to their negative expectations regarding post-surgical pain and reluctance to undergo total joint replacement surgery.It should be noted that Ibrahim’s research group recently attempted to address
this issue by comparing the responses of their African-American and Caucasianpatients to the WOMAC pain and function scales (Ang et al 2003). They foundno di¡erences among the ethnic groups on either of these scales. However,unlike the MPQ, the WOMAC pain scale does not include independent measuresof the sensory and a¡ective dimensions of pain. Therefore, we plan to perform newstudies of possible associations between ethnic group di¡erences in pain-relatedcognition and a¡ect and group disparities in preferences for knee or hipreplacement surgery.
Conclusions and future research directions
Both patients with FM and those with knee OA display abnormal pain sensitivityat multiple anatomic sites. Laboratory studies using psychophysical painmeasurement procedures or neuroimaging of brain activity suggest thatabnormal pain sensitivity in persons with these disorders is associated withcentral augmentation of sensory input. Although several investigators havenoted that central sensitization and/or spinal glial cell activation may contributeto abnormal pain sensitivity in FM and OA, it is very important to emphasizethat, until the sources of nociceptive input from deep muscle tissue are identi¢edin persons with FM, it is not appropriate to suggest that central sensitizationunderlies the pain experiences of these individuals. In contrast, given that (a) thesource of nociceptive input in persons with knee OA is known and (b) there isevidence that abnormalities in pain sensitivity and pain modulation arediminished following total joint replacement surgery, it is reasonable to suggestthat central sensitization and perhaps spinal glial cell activation contribute to thepain experiences of individuals with knee OA.Recent evidence from our laboratory suggests that pain-related cognitive and
a¡ective factors also in£uence the responses of patients with knee OA to noxiousstimulation in the laboratory. Speci¢cally, even when patients with knee OA andage-matched healthy controls receive pressure stimulation of the knees thatproduces similar perceptions of sensory intensity, the patients report that thestimulation evokes signi¢cantly higher levels of pain a¡ect. This group di¡erencein pain-a¡ect ratings is strongly associated with group di¡erences on thehelplessness subscale of the Pain Catastrophizing Scale. Consistent with the¢ndings on these self-report measures, pressure stimulation also evokes
ABNORMAL PAIN SENSITIVITY 267

signi¢cant increases in brain rCBF in substantially larger areas of the anteriorcingulate cortex in patients compared to controls. Moreover, within the patientgroup, the African-Americans, compared to the Caucasians, tend to producehigher ratings of pain a¡ect and helplessness as well as greater cingulate activation.We believe that future research e¡orts should include prospective clinical or
population-based studies of the development of generalized, abnormal painsensitivity in persons with knee OA and the physiologic and psychosocial factorsthat in£uence pain sensitivity in these individuals. It also is critical to devote greatere¡ort to the development of pharmacological interventions that may alter centralnervous system functions that in£uence pain sensitivity and pain modulation inpersons with knee OA. Finally, we believe it is very important to betterunderstand ethnic group di¡erences in pain-related cognition and a¡ect that maycontribute to group di¡erences in pain responses and preferences for health careinterventions such as total joint replacement surgery.
Acknowledgements
Supported by a grant from the Fetzer Institute. We gratefully acknowledge the contributions ofthe Fetzer Working Group on Pain and Su¡ering to the development of our research studies onknee osteoarthritis as well as the contributions of Nancy L. McKendree-Smith, PhD, AdrianaSotolongo, BA and Ronda Cannon, BA to the preparation of this paper.
References
Ang DC, Ibrahim SA, Burant CJ, Kwoh CK 2003 Is there a di¡erence in the perception ofsymptoms between African Americans and whites with osteoarthritis? J Rheumatol30:1305^310
Aaron LA, Bradley LA, Alarco¤ n GS et al 1996 Psychiatric diagnoses are related to health careseeking behavior rather than illness in ¢bromyalgia. Arthritis Rheum 39:436^445
Bajaj P,Graven-NielsenT,Arendt-Nielsen L 2001Osteoarthritis and its associationwithmusclehyperalgesia: an experimental, controlled study. Pain 93:107^114
Bradley LA, Sotolongo A, Alberts KR et al 1999 Abnormal regional cerebral blood £ow in thecaudate nucleus among ¢bromyalgia patients and non-patients is associated with insidioussymptom onset J Musculoskel Pain 7:285^292
Coghill RC, McHa⁄e JG, Yen YF 2003 Neural correlates of interindividual di¡erences in thesubjective experience of pain. Proc Natl Acad Sci USA 100:8538^8542
DeBerry JJ, Fry RB, Kersh BC et al 2002 Trait-like catastrophizing mediates elevated pain a¡ectratings in patients with knee osteoarthritis (OA) during suprathreshold pressure stimulation.Arthritis Rheum 46:S408
Di Piero V, Jones AK, Iannotti F et al 1991 Chronic pain: a PET study of the central e¡ects ofpercutaneous high cervical cordotomy. Pain 46:9^12
Edwards RR, Fillingim RB 1999 Ethnic di¡erences in thermal pain responses. PsychosomMed61:346^354
Edwards RR, Doleys DM, Fillingim RB, Lowery D 2001 Ethnic di¡erences in pain tolerance:clinical implications in a chronic pain population. PsychosomMed 63:316^323
268 BRADLEY ET AL

Gibson SJ, Littlejohn GO, Gorman MM, Helme RD, Granges G 1994 Altered heat painthresholds and cerebral event-related potentials following painful CO2 laser stimulation insubjects with ¢bromyalgia syndrome. Pain 58:185^193
Gracely RH, Petzke F, Wolf JM, Clauw DJ 2002 Functional magnetic resonanceimaging evidence of augmented pain processing in ¢bromyalgia. Arthritis Rheum 46:1333^1343
Graven-Nielsen T, Aspegren Kendall S, Henriksson KG et al 2000 Ketamine reduces musclepain, temporal summation, and referred pain in ¢bromyalgia patients. Pain 85:483^491
Iadarola MJ, Max MB, Berman K et al 1995 Unilateral decrease in thalamic activity observedwith positron emission tomography in patients with chronic neuropathic pain. Pain 63:55^64
Ibrahim SA, Simino¡ LA, Burant CJ, Kwoh CK 2002 Di¡erences in expectations of outcomemediate African-American/white patient di¡erences in ‘‘willingness’’ to consider jointreplacement. Arthritis Rheum 46:2429^2435
Kersh BC, Deutsch G, Bradley LA et al 2001 Pain sensitivity in knee osteoarthritis patients isassociated with bilateral activation of the brain anterior cingulate cortex. Arthritis Rheum44:S140
Kosek E, Ordeberg G 2000 Lack of pressure pain modulation by heterotopic noxiousconditioning stimulation in patients with painful osteoarthritis before, but not following,surgical pain relief. Pain 88:69^78
Kwiatek R, Barnden L, Tedman R et al 2000 Regional cerebral blood £ow in ¢bromyalgia:single-photon-emission computed tomography evidence of reduction in the pontinetegmentum and thalami. Arthritis Rheum 43:2823^2833
Melzack R 1975 The McGill Pain Questionnaire: major properties and scoring methods. Pain1:277^299
Milligan ED, Twining C, Chacur M et al 2003 Spinal glia and proin£ammatory cytokinesmediate mirror-image neuropathic pain in rats. J Neurosci 23:1026^1040
Mountz JM, Bradley LA, Modell JG et al 1995 Fibromyalgia in women: abnormalities ofregional cerebral blood £ow in the thalamus and the caudate nucleus are associated with lowpain threshold levels. Arthritis Rheum 38:926^938
Petrovic P, IngvarM, Stone-Elander S, PeterssonKM,Hansson P 1999APET activation studyof dynamic mechanical allodynia in patients with mononeuropathy. Pain 83:459^470
PriceDD, StaudR,RobinsonME,Mauderli AP, CannonR,Vierck CJ 2002Enhanced temporalsummation of second pain and its central modulation in ¢bromyalgia patients. Pain 99:49^59
Rainville P, Duncan GH, Price DD, Carrier B, Bushnell MC 1997 Pain a¡ect encoded in humananterior cingulate but not somatosensory cortex. Science 27:968^971
Schaible H-G, Ebersberger A, von Banchet GS 2002 Mechanisms of pain in arthritis. Ann NYAcad Sci 966:343^354
Sprott H, Bradley LA, Oh SJ et al 1998 Immunohistochemical and molecular studies ofserotonin, substance P, galanin, pituitary adenylyl cyclase-activating polypeptide, andsecretoneurin in ¢bromyalgic muscle tissue. Arthritis Rheum 41:1689^1694
Staud R Vierck CJ, Cannon RL, Mauderli AP, Price DD 2001 Abnormal sensitization andtemporal summation of second pain (wind-up) in patients with ¢bromyalgia syndrome.Pain 91:165^175
Staud R, Cannon RC, Mauderli AP, Robinson ME, Price DD, Vierck CJ Jr 2003a Temporalsummation of pain from mechanical stimulation of muscle tissue in normal controls andsubjects with ¢bromyalgia syndrome. Pain 102:87^95
Staud R, Robinson ME, Vierck CJ Jr, Price DD 2003b Di¡use noxious inhibitory controls(DNIC) attenuate temporal summation of second pain in normal males but not in normalfemales or ¢bromyalgia patients. 101:167^174
ABNORMAL PAIN SENSITIVITY 269

Sullivan MJL, Bishop S, Pivik J 1995 The Pain Catastrophizing Scale: development andvalidation. Psychol Assess 7:524^532
Watkins LR,Milligan ED,Maier SF 2001Glial activation: a driving force for pathological pain.Trends Neurosci 24:450^455
DISCUSSION
Brandt: There is not only ethnic disparity in the performance of total hip andtotal knee replacement, but also a gender disparity. Did you look at any of thepre-imaging data in relation to gender?Bradley:No we haven’t done this yet.Brandt:Do you have any sense of the extent to which unwillingness to undergo
surgery among African-Americans, relative to Caucasians, may be in£uenced bythe experience of friends, relatives and personal contacts?Bradley: That is a good question. One of our colleagues studied this
phenomenon among healthy college-age students for her doctoral dissertation.She found the same sort of e¡ect that we saw in these patients: in response to avariety of tasks there were much greater a¡ective responses in the African-American college students and much more catastrophizing. Even among thesehealthy college students, one thing that di¡erentiated the African-Americansfrom the Caucasians is their experiences with chronic pain in their families. TheAfrican-Americans had much more experience with chronic pain in familymembers. Roger Fillingim also has some data showing that African-Americanstudents themselves tend to have more experience with painful problems than theCaucasians (Edwards & Fillingim 1999). We think these family factors have a realin£uence on the way people think and respond. Ibrahim’s data on ethnic groupdi¡erences in preferences for knee arthroplasty are also relevant here (Ibrahimet al 2002). They asked the patients whether they had friends or relatives who hadhad total joint replacement surgery, and to what extent the patients were familiarwith the procedure. There was less reported familiarity among the African-Americans. This was odd. It goes against most of the other data, and I can’texplain it.Pisetsky: In our hospital, pain is called the ¢fth vital sign. On every patient, we
are obliged to ask what is their pain at that moment. If we do this with people with¢bromyalgia, invariably it is 8, 9 or 10.Howdo you interpret that? Is that intensity,or unpleasantness? Is there away to ask this question better so thatwe don’t have allthese patients with 8s, 9s or 10s that we are not doing something for?Bradley: It is hard. In that sort of situation where you are giving people a scale,
invariably their responses will be in£uenced by emotions, thoughts and a¡ect. Iwill tell you the rigours of how we have to train people to use these visualanalogue scales. In the ¢bromyalgia study we are using a mechanical visualanalogue scale that Don Price developed at the University of Florida. It looks
270 DISCUSSION

like a slide rule. There is a lever on top of the scale, and as you pull this lever to oneend it exposes a red bar. The patient is supposed to match their internal perceptionof pain intensity or unpleasantness to the length of that red bar. Training people touse this requires giving people practice in extending the bar all the way, so theyreally see what 10 cm looks like. You then have to make sure that they can giveyou at least some semblance of a linear response between stimulus intensity andresponse on that scale. You also have to make sure that if you ask them to exposehalf the red line that they actually get close to exposing half. Then you have to teachthem the di¡erence between intensity and unpleasantness. We use the analogy thatintensity is the loudness of the music on the radio and unpleasantness is howaversive that music is. It takes anything from 30 min to 2 days to teach people todo this. Some people never get it so we can’t use them in the study.Pisetsky: So is the interpretation that people with ¢bromyalgia are experiencing
pain unpleasantly but that the pain is not that intense?Bradley: No, I guess the idea would be that when they talk to you in the clinic,
whatever they are reporting is an amalgam of intensity and unpleasantness. In thelab you can do some things to di¡erentiate those dimensions of the pain response,but in the clinic it is hard to do so. A rough rule of thumb would be to assess theirlife stress and psychological distress to get an idea about this.Pisetsky: When you have them think unpleasant thoughts, do you know what
they are?Bradley:Yes.Pisetsky: Is that informative?Bradley:Yes. I don’t knowhow to do a qualitative analysis, but I know inmygut
that the quality of the images are very di¡erent. For some patients the stressormight be family death or illness. Then for positive images, very often, thesepatients will begin with a statement such as ‘I want to think about my daughter’sgraduation from grade school’. Then, sometimes, you’ll see an intense bloodpressure response. Later on we’ll ask what they were thinking about and they’llsay something like, ‘I was thinking about my daughter’s graduation and then Iran into my rat former husband’. For many patients there often will beunpleasantness weaved into the pleasantness. In healthy controls the images tendto be much less traumatic.Schaible: I have a question about the fMRI images. What you have shown is an
e¡ect quite similar to any response to noxious stimulation. Can you show adi¡erence in these patients between a pain-free stage and a painful stage so thatyou do not need to apply stimuli? If you apply stimuli it may evoke a patternwhich is di¡erent from that which is normally there in this brain. And do youhave any evidence for mirror pain in these patients?Bradley:With stimulation in our ¢bromyalgia patients, and also a bit in our knee
OApatients, even at these relatively low intensity levels we tend to see ipsilateral as
ABNORMAL PAIN SENSITIVITY 271

well as contralateral activation. We have never tried to study these patients beforeand after an analgesic treatment. We have not been able to do this yet. However, ifwe image ¢bromyalgia patients only in resting conditions we tend to see a strongreduction in blood£ow in the thalamus (Mountz et al 1995). This is very similar towhat people have observed in patients with neuropathic pain (e.g. Iadarola et al1995). This is part of what started to drive us and other people to think aboutcentral abnormalities in pain processing. Mistakenly, this drove some people toattribute the pain sensitivity to central sensitization without knowing the sourceof the input. Nevertheless, reduced thalamic activity is what we see in ¢bromyalgiapatients at baseline without stimulation.Hunt:Do the patients with ¢bromyalgia show an exaggerated placebo response?Bradley:Whenwe look at the pharmacological studies, the placebo response runs
anywhere from 30^40%. They really do show a strong placebo response.Hunt: Being a bit more reductionist, there are certain polymorphisms in genes,
such as the CRMT gene that breaks down catecholamines, which seem topredispose people to di¡erent sensitivities to pain. This polymorphism iswidespread. Isn’t it important to look at these? Have you thought aboutscreening your patients?Bradley: Yes. We have started a family aggregation study. We are in the process
of collecting 80 ¢bromyalgia families and 80 control families. We are doing painsensitivity testing not only with the probands and controls but also with onebrother or one sister of each proband or control. As much as we can, we want toend up with 40 brothers and 40 sisters in each group. We have completed sixfamilies already. Our pilot data based on 12 families showed us that the sisters ofthe patients with ¢bromyalgia weremuchmore pain sensitive than all other siblinggroups, including the sisters of the controls. Astoundingly, they also show lowerserotonin levels. We are also collecting blood and immortalizing DNA. We wantto test hypotheses about a number of polymorphisms in a number of genes,including 5HT. There are at least two other groups working on similarstudies.Dieppe: I’m particularly interested in your use of imagery. We have been using
similar techniques as an intervention, based on the concept of emotional disclosureas a way of treating chronic disease. I wonder whether you are achieving anintervention with these imagery approaches, and are in fact altering the painexperience in these ¢bromyalgia patients over a longer time period.Bradley: Are you using the writing technique?Dieppe:Yes, mostly, but we have been doing a bit of this in RA patients some of
whom don’t like to do writing and prefer to talk.Bradley:Wehave not actually asked that question, but it is an interesting one.My
guesswould be that since they are really only disclosing over the period of a day andwe never ask them to continue disclosing, this is not an ideal situation for
272 DISCUSSION

improvement.What you are doing over an extended period of time is amuchbetterparadigm for achieving a positive e¡ect.Brandt: I am interested in the group of ¢bromyalgia people you recruited from
the community who had not made the decision to become patients. They remindme of a cohort of older people in Indiana whom we studied for OA. We found asubstantial number of people with radiographic OA who had WOMAC painscores as high as those of patients in our clinic, but these people had made thedecision that they didn’t need medical attention for this. I don’t know whetherthere was a qualitative di¡erence between their pain and the pain of our clinicpatients, or whether they had better coping skills. Were there di¡erences betweenyour people with ¢bromyalgia in the community and your patients with¢bromyalgia?Bradley: This is interesting. Even though the patients and non-patients showed
the same level of pain sensitivity, the patients reported signi¢cantly higher levels ofpain. Their average score on the totalMcGill PainQuestionnairewas about 30, andthe score of the non-patients was about 17. This gets back to David’s point. It maywell be that the emotional distress was driving the elevation in those scores. Thiswas the major di¡erence between the groups. Even after we controlled for thepsychiatric factors, the patients still reported higher levels of depression andanxiety, and lower self e⁄cacy (Kersh et al 2001).Blake: The word ‘catastrophizing’ obviously has a negative implication. Given
the way the normalized data were collected, how could you not say that theAmericanized Caucasian population was emotionally blunted?Bradley: We have often been accused of this! You could reinterpret a higher
score on that scale as re£ecting a greater willingness to admit, if not a greateractual experience of perceived di⁄culty in coping with pain. I think you areright: it is probably good to cast this ethnic group di¡erence in non-pathologicalterms.Henry: Many patients with chronic pain have sleep disorders, and they often
complain of fatigue. In one of your early slides you listed these as separateentities. Can you comment on your vision of these as separate entities, orvariations of the same theme?Bradley: I don’t think that they are really separate entities. I think the fatigue
reports and sleep disturbance are so frequent in this group of people it isprobably part of whatever this syndrome is.Henry: Is the fatigue due to sleep disturbance or something else?Bradley: I don’t know the complete answer to that. There is a large subgroup of
people with ¢bromyalgia who show intermittent or phasic alpha intrusion duringnon-REM sleep. At least in part, we can probably attribute the fatigue to this sleepdisturbance. Theremay be other factors. This gets back to the issuewe raised aboutLinda Watkins’ construct of the sickness response.
ABNORMAL PAIN SENSITIVITY 273

Fox:Where you have looked at patients and non-patients, is there a di¡erence inhow they respond to pharmacological interventions?Bradley: There are no data on this.Ordeberg: I was very interested in this trait of catastrophizing, and the di¡erences
between ethnic groups.Have you looked to seewhether therewas a di¡erencewithsocioeconomic status (SES)? There might be di¡erences between these groups.And those with less insurance and fewer economic resources have more fear withtheir disease for this reason.Bradley:That is a very good point. In that particular studywe used education as a
marker for SES. Therewas no signi¢cant di¡erence between the groups in terms ofeducation. But, there are various ways of measuring SES, and there is a wholeliterature on measuring SES using multiple methods. This is something we needto pay more attention to in the pain ¢eld.Felson:Letme change direction a little. I appreciate that the audience is intrigued
by the discussion of ¢bromyalgia, but this is a meeting about OA pain. I wanted tochange the focus back to OA. The implication of this discussion goes back to thepoint where we haven’t used the terms OA or ¢bromyalgia in the last ¢ve or 10minutes because it has been implied that there has been an element of the samegoing on in OA. I want to raise this question again. Let me throw out a couple ofobservations. One is that African-Americans seem to have the higher painreporting you described, but in most epidemiological surveys they have far lowerprevalences of ¢bromyalgia than Caucasians. I don’t understand this.Furthermore, your imaging studies suggested that there were qualitativedi¡erences in OA brain regions a¡ected, whereas ¢bromyalgia studies show noqualitative di¡erences in general. They show the same areas of the brain being litup, only by lower levels of the same noxious stimuli. Is OA pain the same sort ofentity as ¢bromyalgia pain?Bradley: They are similar. But, there are some di¡erences. When we did our
generalized pain sensitivity test in the patients with OA, we not only used themechanical pressure stimulation applied to the arm and shoulder, but we alsoused a thermal stimulation protocol applied to the forearm. The patients withOA, unlike what is seen in patients with ¢bromyalgia, did not di¡er from thecontrols in their response to thermal stimulation. It may be that if we used a moreaggressive stimulation technique we might see enhanced thermal responses. Atpresent, however, we can only say that the OA patients don’t show the £oridsensitivity to a variety of stimuli that is seen in patients with ¢bromyalgia. Withregards to the imaging study, one reason why I think we saw the great groupdi¡erence in activation of the cingulate is that we attracted a group of patientsthat contained roughly the same number of African-Americans and Caucasians.As a result, we saw that the high a¡ective pain response and the high levelcatastrophizing primarily occurred in the African-Americans. Our studies in
274 DISCUSSION

¢bromyalgia involve primarily Caucasians, just like in the epidemiological data.The mix of subjects has an important impact on the ¢ndings that are produced.There is a recent paper by Bob Coghill (Coghill et al 2003), in which he lookedat healthy people whom he de¢ned by quantitative sensory testing as eitherrelatively pain sensitive or pain stoic. He put them through a sensorystimulation task involving the right lower leg and did neuroimaging, ¢ndingthat if you look at the level of the thalamus and give people the same amountof stimulation, you see essentially the same brain responses. But his painsensitive people showed much greater activation of the contralateral anteriorcingulate cortex in response to the stimulation. Also, the pain sensitiveindividuals more frequently showed activation of a region of the primarysomatosensory cortex that corresponds to the lower leg. These ¢ndingsindicate that the cingulate is a good marker for a¡ective responses involved inenhanced pain sensitivity.Hunter: Iwas interested that you showed a substantial di¡erence in cortisol levels
between the ¢bromyalgia and the control patients. You mentioned that serotoninlevels are also somewhat di¡erent between these two groups. This potentiallycould be extrapolated to OA. In terms of the pain sensitization, how much of thevariability in pain sensitization can you attribute to potential di¡erences in thosehormonal levels?Bradley: I can’t give you an answer in terms of amount of variance accounted
for. But the di¡erence in cortisol production in ¢bromyalgia is probably oneof the most robust ¢ndings in the ¢bromyalgia literature. In studies wherea HPA axis challenge has been produced by injecting a bolus of CRH wealso see a diminished cortisol response in patients with ¢bromyalgia (e.g.Cro¡ord et al 1994). This is a very reliable ¢nding among these patients. At thevery least I would say that ¢bromyalgia patients show an impaired physiologicalresponse to stress. This probably is related to their pain reports or their a¡ectivereports as a function of stress. There probably is a relationship but it hasn’t beenquanti¢ed yet.Lohmander:This is fascinating stu¡.What do we have in the way of longitudinal
data which would answer the question of whether having chronic pain, such as hipOA, would in itself drive you towards catastrophizing? Or does having thepersonality or trait in itself provide a risk factor for you experiencing pain in thisway and then becoming one of these patients? What is the chicken and what is theegg? Do you have a feeling for this at the moment?Bradley: No. That is a very good question. One of our future aims is to be
involved in a longitudinal study where we could look at pain/catastrophizing atbaseline among patients with early stages of OA, and follow them over time. Wewould like to look for changes in pain sensitivity and to what extentcatastrophizing in£uences those responses.
ABNORMAL PAIN SENSITIVITY 275

Kidd: We looked at this in a group of RSI/work-related upper limb disorderpatients, and compared these to a group with carpal tunnel. The hypothesis wasthat in the RSI group there would be more catastrophizing. Disappointingly, wewere unable to show any di¡erences on any of the psychological variables welooked at. It seemed that chronic pain induced the catastrophizing rather than theother way round.Pisetsky: Alan Silman has longitudinal data. His group has de¢ned as
widespread body pain followed over a number of years (McBeth et al 2003). Thispain is predictive of a number of things including malignancy.
References
Coghill RC, McHa⁄e JG, Yen YF 2003 Neural correlates of interindividual di¡erences in thesubjective experience of pain. Proc Natl Acad Sci USA 100:8538^8542
Cro¡ord LJ, Pillemer SR, Kalogeras KT et al 1994 Hypothalamic-pituitary-adrenal axisperturbations in patients with ¢bromyalgia. Arthritis Rheum 37:1583^1592
Edwards RR, Fillingim RB 1999 Ethnic di¡erences in thermal pain responses. PsychosomMed61:346^354
Iadarola MJ, Max MB, Berman KF et al 1995 Unilateral decrease in thalamic activity observedwith positron emission tomography in patients with chronic neuropathic pain. Pain 63:55^64
Ibrahim SA, Simino¡ LA, Burant CJ, Kwoh CK 2002 Di¡erences in expectations of outcomemediate African-American/White patient di¡erences in ‘willingness’ to consider jointreplacement. Arthritis Rheum 46:2429^2435
Kersh BC, Bradley LA, Alarcon GS et al 2001 Psychosocial and health status variablesindependently predict health care seeking in ¢bromyalgia. Arthritis Rheum 45:362^371
McBeth J, Silman AJ, Macfarlane GJ 2003 Association of widespread body pain with anincreased risk of cancer and reduced cancer survival: a prospective, population-based study.Arthritis Rheum 48:1686^1692
Mountz JM, Bradley LA, Modell JG et al 1995 Fibromyalgia in women. Abnormalities ofregional cerebral blood £ow in the thalamus and the caudate nucleus are associated with lowpain threshold levels. Arthritis Rheum 38:926^938
276 DISCUSSION

Chair’s summing-up
David Felson
Clinical Epidemiology Research and Training Unit,, Boston University School of Medicine,715 Albany Street, A 203, Boston, MA 02118-2526, USA
I wanted to summarize the questions that have emerged during these discussions.At the beginning of the symposium, I posed a set of questions. These were broadand not as well informed as ones we might think of now. These included: what isthe pathophysiology of osteoarthritic (OA) joint pain? Why do some but not allpeoplewithOAget joint pain?And are there treatment opportunities that go alongwith a better understanding of OA joint pain?I have tried to cluster questions that remain by the category of question, and
some of them bridge our two scienti¢c communities. First, is there centralsensitization in non-in£ammatory mechanical joint disorders such as OA? Thisquestion came out in the ¢rst day as we heard about the prominence andimportance of central sensitization induced by aggressive in£ammatoryprovocations. Obviously, OA is not such an entity. The question is, are the sameprinciples relevant in OA? There were many discussions about the role of centralsensitization.Also, what is the correlation between nociception seen in animals andhuman nociception?Then, for clinicians there were another set of questions. What are the relative
contributions of structural features to OA pain? Philip Conaghan and PeterSimkin talked a lot about this, suggesting that bone marrow lesions were afrequent source of pain. Paul Creamer cautioned us about using cross-sectionalassociations to make aetiological inferences. Synovitis is a potential source ofpain, as are osteophytes and periarticular lesions. What are their relativefrequencies? Which of these causes pain, and is one or more a common source ofpain? Do each of the structural contributors to OA pain cause a di¡erent kind ofpain?We talked about the di¡erent qualities of pain andwhat thesemight re£ect. Inknee OA, on the basis of data from the Health ABC study, there has been a speci¢cquestion as to how important the patellofemoral joint is.Then we go back to neuroscience questions. One of these came up speci¢cally
today: what are the changes in gene expression in primary a¡erents in OA pain? Isthis just a neurological disease? Should the rheumatologists be thrown out of theroom? What part of the nociceptive process should be targeted for interventions?Do we target transduction, neurogenic in£ammation, the other end of the synapse
277
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

(in the CNS) or post-synaptic events? How similar are nociceptive mechanisms inOA tomechanisms of other pain? And can we generalize from some of themodels,such as the bone painmodel and in£ammogenicmodel that we heard about, toOApain? What is the role of mechanoreceptors here? Jim Henry and Blair Grubbpushed really hard to get the clinicians to de¢ne appropriate models for thisentity. This is terribly important. How relevant are models of OA with respect toOApain?Dowe need to ¢nd animalmodelswhere the animals have painwith theirOA? I think the answer is yes.Howdowe¢gure this out?Can clinicians identify thecritical elements of a model of OA pain, and what features should it contain? Thethought was that it might contain synovium and bone marrow lesions, but does itneed to have cartilage loss?Most people would say that cartilage loss is a sine qua nonof OA, even though it might not be a source of pain. What about current OAmodels? Would they work or do we need to develop new models? Then I thinkthere was a lot of back or forth as to how well the acute models we are all dealingwith generalize to chronic disease. Are we modelling peripheral or centralfunctions? We heard about in£ammatory mediators and neurogenic mediatorsbeing released into joints and spinal cord. One of the questions is, ‘What is therole of substance P and CGRP in the joint?’ It sounds like substance P is aneurologic e¡ector in the joint. Is it important in creating in£ammation in OAthat would make OA a similar model to the other ones Hans-Georg told usabout? How important is neurogenic in£ammation in general in the overallhealth of the joint environment?There were parts of talks which dealt with COX-2 inhibitors and their e¡ects on
prostaglandins as mediators in the spinal cord. Other models have found thatleukotrienes may be as, or more, important than prostaglandins. The question is,what role do all these play in this disease and in models of disease?Next, how can we identify OA at an early enough stage to treat it successfully?
This is a human clinical question but it has implications for animal models. This isan important question, especially with some of the emerging work on vulnerablejoints showing that once mechanical malalignment or anatomic changes haveoccurred, it would be di⁄cult to envision an easy pharmacological approach totherapy. Early treatment becomes critical.Then there are the clinical questions. Do di¡erent pain patterns and descriptions
by patients correlate with di¡erent sources of pain? Why are some joints painfulsuch as the knee and others almost never painful such as the elbow? This wasPaul Dieppe’s question and I think it is a wonderful one. Is it just mechanicalloading that does this? A correlated question is why do some people have painwith OA while others don’t? And why do only some animals have pain withOA? It is a parallel set of questions and identifying the answers to these questionsmay provide answers to the others. Why does exercise ameliorate OA joint pain?What e¡ect does muscle strength have on OA long-term, and is this di¡erent in
278 CHAIR’S SUMMING-UP

vulnerable versus normal joints? How important is the rate of impulse loading andits deceleration in causing OA? Does bone give in OA, or have hydraulic e¡ects inresponse to loading? Is increased intraosseus pressure a major reason for OA jointpain? Would bone fenestration help this pain? How important are corticalmechanisms, such as discordance between motor intention and sensory feedback,in OA pain? How important is abnormal pain sensitivity in OA pain? Then, thereare the questions about the high rate of bilateral disease that we raised earlier.In summary, there are a plethora of questions for clinicians and basic scientists,
rheumatologists and neuroscientists to address. Our interactions at this meetingsuggest that many of these questions will be most fruitfully addressed byinterdisciplinary collaborations, and I hope that this meeting encourages suchenterprises.
CHAIR’S SUMMING-UP 279

Index of contributors
Non-participating co-authors are indicated by asterisks. Entries in bold indicatepapers; otherentries refer to discussion contributions.
A
*Alarco¤ n, G. A. 258
B
Blake,DR. 133, 136, 145, 148, 151, 153, 154,174, 175, 176, 177, 178, 238, 239, 240,241, 252, 253, 254, 255, 256, 273
Bradley, L. A. 74, 78, 115, 116, 119, 120, 121,136, 258, 270, 271, 272, 273, 274, 275
Brandt, K. D. 36, 39, 40, 41, 42, 45, 47, 49,58, 59, 60, 61, 62, 63, 76, 77, 95, 96, 98,101, 104, 201, 203, 270, 273
C
*Clark A. 79Conaghan, P. G. 96, 103, 178, 191, 201, 202,
203, 204Creamer, P. 23, 42, 44, 64, 74, 75, 76, 78, 103,
117, 137, 201, 204
D
*DeBerry, J. J. 258*Deutsch, G. 258Dieppe, P. 24, 37, 45, 60, 61, 75, 78, 98, 100,
117, 135, 137, 138, 148, 152, 153, 177,189, 205, 219, 254, 272
E
Evans, C. H. 38, 98, 147, 151, 255
F
Felson, D. T. 1, 26, 39, 43, 44, 60, 61, 62, 75,76, 95, 96, 100, 101, 102, 104, 116, 118,120, 137, 147, 149, 151, 175, 176, 178,
187, 189, 191, 201, 202, 204, 218, 238,240, 253, 256, 274, 277
Fernihough, J. K. 39, 42, 58, 62, 102, 104,133, 147, 187, 219
Fox, A. 115, 116, 118, 119, 120, 134, 135,137, 147, 151, 153, 216, 238, 239, 256,274
G
Grubb, B.D. 23, 24, 25, 27, 28, 37, 38, 39, 40,41, 42, 43, 44, 46, 47, 62, 76, 77, 97, 99,100, 115, 118, 119, 135, 146, 149, 151,153, 178, 214, 215, 217, 218, 219, 239,255, 256
H
*Haigh, R. C. 154, 241Henry, J. L. 25, 26, 37, 39, 77, 96, 120, 134,
139, 145, 146, 147, 148, 149, 150, 151,152, 153, 215, 216, 273
Herzog, W. 40, 59, 62, 79, 95, 96, 97, 98, 99,146, 177, 178, 188, 189, 203
Hunt, S. P. 221, 238, 239, 240, 252, 256, 272Hunter,D. J. 22, 23, 39, 44, 47, 59, 60, 78, 99,
101, 116, 118, 119, 152, 176, 190, 202,203, 253, 255, 275
I
*Inglis, J. J. 122
K
*Kersh, B. C. 258Kidd, B. L. 25, 45, 115, 118, 120, 122, 133,
134, 135, 136, 137, 138, 150, 215, 218,219, 239, 276
280
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

Koltzenburg, M. 37, 40, 43, 44, 46, 47, 175,176, 202, 203, 206, 214, 215, 216, 217,218, 219
Kuettner, K. E. 37, 75, 77, 78, 98, 100, 103,146, 187, 238
L
*Lewis, J. 154Lohmander, S. 26, 42, 43, 62, 77, 115, 148,
152, 187, 189, 254, 275*Longino, D. 79
M
Mackenzie, A. 27, 78, 134, 146, 150, 218, 239Malcangio, M. 120, 137, 150Manning, A. M. 26, 43, 102, 103, 104*Mantyh, P. W. 221*Mapp, P. I. 241*McCabe, C. S. 154, 241*McLain, D. A. 258
O
Ordeberg, G. 105, 115, 117, 118, 119, 121,153, 240, 274
P
*Photiou, A. 122Pisetsky, D. S. 23, 24, 25, 27, 38, 39, 42, 44,
46, 58, 60, 61, 75, 77, 96, 97, 101, 103,
104, 117, 133, 134, 137, 147, 149, 152,174, 186, 187, 189, 202, 204, 215, 217,218, 240, 253, 254, 270, 271, 276
R
Rediske, J. 26, 60, 61, 102, 120, 137, 146,151, 217, 253
S
Schaible, H.-G. 4, 22, 23, 24, 25, 26, 27, 38,39, 40, 45, 46, 74, 75, 77, 118, 119, 120,134, 145, 149, 150, 202, 213, 215, 216,218, 219, 238, 271
Shen, H. 135*Shenker, N. G. 154, 241Simkin, P. A. 25, 41, 44, 62, 152, 179, 186,
187, 188, 189, 190, 255
V
van den Berg, W. 148, 152, 203, 204
W
Woodworth, T. G. 58
Y
Yashpal, K. 137
INDEX OF CONTRIBUTORS 281

Subject index
A
A ¢bres 224Ab ¢bres 29, 224acid detection 224, 228acidosis, tumour-induced 228^229activities of daily living, exercise 55a¡ect, pain 66, 78, 263^266African^Americansjoint replacement surgery 265^267, 270pain descriptions 74
alcoholism 180allodynia 31capsaicin 124^126
analgesic hip 218, 219angiogenesis, COX-2 226^227animal modelsbone cancer pain 222joint in£ammation/OA 30^31, 76^77, 81,
95^96, 142rheumatoid arthritis 100^104see also canine ACL transection model; cat
ACL transection modelANKTM1 206, 208ankylosing spondylitis 180anterior cingulate cortexpain perception 265pain sensitivity marker 275
anterior cruciate ligament (ACL)absence 40tears 198
anterior knee pain, intraosseous hypertension180
antidepressants, OA pain 137anti-epileptics, pain relief 137anti-TNF, rheumatoid arthritis 103, 128,
133anxiety 66, 68, 74arthritic pain 5arthrogenous re£ex inhibition 40^41, 54, 55arthroscopic lavage 106articular cartilage explants, loading 80^81ASIC-3, pH 228
aspirin 226asymmetric arthritis 254ATP 224atrophic OA 189^190attention, bone cancer pain 232auto-neuro-in£ammatory loop, survival
hypothesis 249^250avascular necrosisintraosseous hypertension 180in OA 106, 117^118
B
balance, pain severity 70barometric pressure 37^38, 69bilateral receptive ¢elds 7bilaterality 147, 148^149, 150^151, 171, 252,
253 see also symmetrybioinformatics 238^239biomechanics 79bisphosphonate, bone cancer pain 229, 232,
238blood^brain barrier 150Bole animal model 95^96bone cancer pain 221^238acidosis 228animal models 222attention 232bisphosphonate 229, 232, 238c-Fos 225central sensitization 230^232COX-2 inhibitors 226, 227disease progression 232^233endothelins 227^228gabapentin 230growth factors 229higher brain centres 232mood 232osteoclasts 228^229osteoprotegerin 229, 232, 238perplexing 229pH 228
282
Osteoarthritic Joint Pain: Novartis Foundation Symposium 260. Volume 260Edited by Derek J. Chadwick and Jamie Goode
Copyright Novartis Foundation 2004. ISBN: 0-470-86763-9

primary a¡erent sensory neurons 222,224^225
proton release 228sensory ¢bre distension and destruction
230substance P 225tissue-speci¢c mechanisms 229
bone deformation, weight bearing 189bone marrow lesions 70^71bone marrow oedemainterosseous hypertension 180MRI 195^197
bone pain 70, 106botox model 93, 95, 96, 99bradykininarticular a¡erent excitation 33damaged sensory neurons 229nerve growth factor 210nociceptors 126, 224prostaglandin augmentation 33thermal opening of vanilloid receptor/
channel complex 34
C
C ¢bres 29, 119, 207, 224c-Fos 225C reactive proteindisease progression indicator 71OA 204synovitis 204
calcitonin 240calcitonin gene-related peptide (CGRP)
244^245dorsal root ganglion neurons 30nerve growth factor 127, 128primary a¡erent nerve ¢bres 30spinal cord neurons 13synovium 245
cancer-associated pain 221^222, 240 see alsobone cancer pain
canine ACL transection model 55^56pain 62^63
capsaicinage 45allodynia 124^126contralateral in£uences 247e⁄cacy in OA 120hyperalgesia 46, 124^126nerve growth factor 210rheumatoid arthritis 124
T cells 245capsular distension 106carrageenan injections 8, 31cartilage
innervation 25lesion pain 106network failure 139remodelling, substance P 146smoothing 106stimulation 4, 5subchondral cell communication 187
cat ACL transection model 81, 82^83EMG 87^88femoral groove cartilage 98gait 97^98guarding 97knee £exor muscles 96^97mechanics 83^87muscle forces 87^88pressure patterns 88^90
catastrophizing 263, 270, 274caudate nucleus, ¢bromyalgia 259central pattern generators 54central sensitization
cancer pain 230^232chronic in£ammation 9¢bromyalgia 260^261glial cells 25pain sensitivity 5peripheral component 24
centrally acting analgesics 120Charcot’s joints 40, 218, 247, 249chemical stimulation 4, 141, 207, 224chondrocyte
injury 139substance P 146^147
chondropathy, synovitis 204cognitive in£uences 176, 263^266cold sensation 208collagenase, substance P 129collateral ligament tears 198comorbidity, exercise 55compartment syndromes 183^184complex regional pain syndrome 159^164
central and peripheral nervous systeminvolvement 162, 164
cortical somatosensory map 160^161incongruent movements 168^170joint replacement 153mirror visual feedback 164^168pain 159
SUBJECT INDEX 283

complex regional pain syndrome (cont.)referred sensations 160^162symmetry 254^255types 1 and 2 159
contralateral in£uences 246^247, 255 see alsobilaterality
cortical sensitization 123cortical somatosensory map, CRPS 160^161cortisol, ¢bromyalgia 275COX 32, 33COX-1 13, 33, 225COX-2angiogenesis 226^227constitutively expressed at spinal level 27expression 33in£ammation 20prostaglandin synthesis 13tumour growth 226^227up-regulation 33
COX-2 inhibitors 33cancer growth and metastasis 227, 238cancer pain 226, 227
COX-3 27COX inhibitorscancer pain 226centrally acting 26mechanonociceptor sensitization reversal
32CRMT gene 272cruciate ligament tears 198cyclooxygenase see COX headingscyclosporine, knee pain 182cytokinesblood^brain barrier 150nociception 128sickness response 149^150
D
degeneration see joint degenerationdemographics 1depressionexercise 55healthcare seeking behaviour 67pain severity 68
dermatological conditions, symmetry 242diabetic neuropathy, neuropathic joint disease
56diacerein 2diagnosis of OA, minimum set of symptoms
74^75
di¡use noxious inhibitory controls (DNIC)7, 107^108, 115, 120^121
dipyrone 120disability 50disbaric decompression 180dissensory state 171, 172distension-induced pain 183diurnal variation, pain 68^69dorsal hornabnormal pain sensitivity 260^261mechanical stimulation 5PGE2 20receptive ¢elds 144spinal cord neurons 5
dorsal root ganglion neurons, CGRP 30dorsal root re£exes 20drug development 2
E
early intervention 140^141educational level, knee pain 66e¡usionsarthrogenous re£ex inhibition 40joint damage 33MRI 193^195, 201ultrasound 201
elbow OA, symptoms 98EMG, cat ACL transection model 87^88emotion, pain 78endothelinscancer pain 227^228nociceptors 224
endurance, exercise 55epidermal growth factor 225ethnicitypain questionnaires 74pain sensitivity 265^267, 270
excitatory amino acids 12exerciseadherence 52^53intraosseous pressure 182knee OA 52^53, 54^55OA symptoms 60^61
F
FAST trial 55fatiguejoint chemicals entering circulation 141pain 273
284 SUBJECT INDEX

fatty acid-induced in£ammation 184femuropatellar joint ¢brillation 106fentanyl 259¢brofatty ankylosis, joint immobilization 51¢bromyalgiacentral correlates of pain sensitivity
259^260central sensitization 260^261characteristics 258^259cortisol 275incongruent movements 168^170OA pain compared 274phantom swelling 159placebo response 272pressure pain thresholds 111psychiatric morbidity 259sleep disorders 273substance P-like activity 112thermal stimuli 259visual analogue scales 270^271wind-up response 259
¢brous cartilage noxious stimulation 4£are^wheal response 141, 244forbidden pattern 245fractals 253^254, 255free fatty acids, in£ammation 184Freund’s complete adjuvant 8, 30, 31Fuji pressure sensitive ¢lm 88
G
G protein-coupled receptor 214gabapentin, pain relief 137, 230, 240gaitcat ACL transection model 97^98diggers and gliders 59pain 76quadriceps weakness 53
Gaucher’s disease 180genetic factorsOA 49^50, 140pain response 43polymorphisms 272post-injury OA 152
GFRa 209glial cell line-derived neurotrophic factor
(GDNF) 209glial cells 25, 260^261glomerulonephritis, symmetry 243glutamate 12Golgi endings 29
gout, bilaterality 151^152grey matter 5grip strength 60ground reaction forces 82, 83, 85^86growth factors, tumour cells 229guarding 97
H
hand grip strength 60hand pain, diurnal variation 68^69hand osteoarthritis
grip strength 60prevalence 1symmetry 253
health status, knee pain 66healthcare seeking behaviour 66^68, 75heat pain threshold 207^208
in£ammation 209^210see also thermal sensitivity
heavy lifting 62Heberden’s nodes, symmetry 243, 253heel strike 53, 59^60hip osteoarthritis
abnormal pain sensitivity 261hyperalgesia 110osteotomy 106pain distribution 107pressure pain thresholds 110prevalence 1symptoms 107
hip pain, location 68hip replacement
cement use 118ethnicity 266^267, 270pain relief 107, 118technique 106
hyperalgesiacapsaicin 46, 124^126cytokine-induced 128hip OA 110joint in£ammation 5, 11nerve growth factor 209neuronal mechanism 9secondary 9
hypertrophic pulmonary osteoarthropathy240
hypochondriasis, knee pain 66
SUBJECT INDEX 285

I
Ih current 215^216ibuprofen 226incongruent movements 168^170, 177^178indomethacin 32in£ammationacute 126acute/chronic clari¢cation 215free fatty acids 184neurogenic 128^131, 141nociception 126^128, 209^211, 217opioids 218pain and 71see also joint in£ammation
in£ammatory mediators 32^33, 126^128injury-related OA, genetic factors 152interleukin 1 (IL1) 129, 133^134, 225inhibition 2
interleukin 1b (IL1b) 127, 128, 209interleukin 6 (IL6) 128, 129, 225interleukin 10 (IL10) 134interoception 123, 133intraosseous hypertension see intraosseous
pressureintraosseous in£ammation 184intraosseous pain, ischaemia 183^184intraosseous pressure 179^184avascular necrosis 180exercise 182immobilization 50^52, 61^62impact events 183ischaemia 183joint use 182^183marrow oedema 180muscle contraction 182normal 181OA 179^180osteoporosis 187pain and 41regulation 181^182
ischaemia, intraosseous pain 183^184isolectin B4 positive neurons 30
J
joint degenerationearly detection 87joint instability 83, 85joint unloading 87muscle activation patterns 88
muscle weakness 93, 95joint e¡usion see e¡usionsjoint immobilization 50^52, 61^62joint in£ammationarticular a¡erents 31mechanical stimulation 5pain at rest 5pain sensitivity 5silent nociceptors 29spinal cord neuron hyperexcitability 8^11,
12, 13, 20joint innervation 28^29joint instabilityjoint degeneration 83, 85nociception 40onset and progression of OA 85
joint loadingbiological response 90^93OA association 80
joint mechanics 33, 79^95joint pain 4^5, 105^115joint pressuredistribution 82e¡usate removal 33
joint protection 32joint replacementethnicity and pain sensitivity 266^267, 270pain alleviation 44recovery 46remaining pain after 152^153see also hip replacement; knee replacement
joint stability see joint instabilityjoint swelling, pain 106joint unloading 87
K
kaolin injections 8, 31ketamine 12, 259kinematics 82kneecompartments 106^107£exor muscles 96^97radiographically normal 193
knee osteoarthritisabnormal pain sensitivity 261^262a¡ective in£uences 263^266arthroscopic lavage/cartilage smoothing
106cognitive in£uences 263^266exercise 52^53, 54^55
286 SUBJECT INDEX

muscle atrophy 52muscle strength 52osteotomy 107pain location 106^107periarticular pain 45predicted from knee pain 70prevalence 1proprioception 57, 171quadriceps 52^53, 55radiographic evidence 1strength-training, protective role 54surgery 107symmetry of pain pattern 24symptoms 106^107tenderness 106^107
knee painbone marrow lesions 70^71community 65^66cyclosporine 182diurnal variation 68^69interosseous hypertension 180local anaesthetic 69, 197location 68, 69predicting knee OA 70prevalence 1psychosocial disability 68risk factors 66worsening over time 70X-rays 65, 66, 68, 75
knee replacementethnicity 266^267, 270patellar prosthesis 106total knee replacement 107
L
lateral femurotibial compartment 106Lequesne Index 68leukocyte migration, substance P 131lifting 62ligamentsnoxious stimulation 4tears, MRI 198
light touch perception thresholds 110lipid metabolites 224local anaestheticknee pain 69, 197pain response 44
low spirits, knee pain 66
M
magnetic resonance imaging 191^201bone marrow oedema 195^197e¡usions 193^195, 201ligaments 198menisci 197^198osteophytes 198, 201periarticular lesions 197popliteal cysts 193^195synovitis 193^195, 202
marrow oedemaintraosseous hypertension 180MRI 195^197
mast cell degranulation 129McGill Pain Questionnaire 68, 74, 264^265MCP-1 131mechanical stimulation
articular a¡erents 29¢bromyalgia 259pain 4, 5painless 207spinal cord neurons 5^8
mechanonociceptor sensitization 32^33medial femurotibial compartment 106, 107medicolegal practice 176meniscal tears, MRI 197^198menopause 240metabolic abnormalities 50microtrauma 55mirror visual feedback 164^168mood, bone cancer pain 232motor re£exes, sensitization 22^23multiple sclerosis, symmetry 242^243muscle, arthrogenous inhibition 40^41, 54, 55muscle activation patterns 82, 83, 88muscle atrophy
joint immobilization 51knee OA 52
muscle contraction, intraosseous pressure 182muscle forces 82, 87^88muscle mass, obesity 53muscle weakness, risk factor for joint
degeneration 93, 95
N
Nav1.8 216negative a¡ect, knee pain 66nerve cuts 145^146nerve ¢bre types, joint physiology 141
SUBJECT INDEX 287

nerve growth factordamaged sensory neurons 229nociception 126, 127^128, 209^210, 224
nervous system in arthritis 246neural plasticity 12, 122^123, 140, 160,
224^225neurogenic in£ammation 128^131, 141neurogenic pain 106neuroin£ammation, contralateral in£uences
246^247neurokinin-1 (NK-1) 13, 129neurokinin-2 (NK-2) 13, 129neurokinin A 150neurogenic in£ammation 129spinal cord neurons 13
neuromuscular aspects 49^58neuropathic arthropathy 55^56neuropathic paingabapentin 230in OA 151, 152^153pain perception 260
neurotransmitters, disease initiation andprogression 141
neurotrophic factors 26night pain 69, 177dissensory state 172intraosseous pressure 180
nitric oxide 260NK-1 antagonists, antidepressant activity 137NMDA antagonists 12side-e¡ects 47
NMDA receptors 12nociceptionassessing 123^126de¢ned 122^123in£ammatory mediators 32^33, 126^128joint instability 40
nociceptive-speci¢c neurons 7, 8nociceptorsin£ammation 126^128, 209^211, 217location 28^29oedema 217peripheral sensitization 225sensitivity 32^33, 206^207silent 29, 45^46, 225TRP channels 207^209tumour excitation 225^228
non-steroidal anti-in£ammatory drugs(NSAIDs) 218^219
mechanonociceptor sensitization reversal32
paracetamol and 47responders 46^47synovitis 203
noxious movement, rate of movement 39noxious stimuli 4, 5, 217, 224
O
obesityexercise 55muscle mass 53
occupational factors 62oedema, nociceptor in£ammation 217ominory state 171opioids 218osteoarthritis (OA)ageing process 139atrophic 189^190cartilage network failure 139cause 139chondrocyte injury 139clinical entity 50contributing factors 139^140de¢nition 74diagnosis 74^75disability 50¢bromyalgia pain compared 274genetic abnormality 49^50, 140Heberden node symmetry 243, 253multifactorial cause 139organ disease 49prevalence 1primary 49progression 71, 85secondary 49, 253signs 2sociocultural phenomenon 76source of pain 43^45, 70^71, 106, 141spectrum of a single end-stage disorder
101symmetry 243symptoms 2when does it start? 37
osteoclastsbone cancer 228^229menopause 240normal bone 239OA 240
osteophytescontralateral 147MRI 198, 201
288 SUBJECT INDEX

osteoporosisintraosseous pressure 187joint immobilization 51OA and 186^187transient regional 240
osteoprotegerin, bone cancer pain 229, 232,238
osteotomyhip OA 106knee OA 107rest pain 180
over-use model 147
P
p38 MAPK 128p75 209, 210Paccinian endings 29painacute leading to chronic 140assessment 141catastrophizing 263, 270, 274distribution, behavioural symptom 113early intervention 140^141early markers 141e¡ect of 70ethnicity 265^267, 270family of disorders 140¢fth vital sign 270genetic variation 43importance to patients 67^68memory 24nature 68^70neural plasticity 122^123patterns 69process 140protective function 76, 218psychological factors 68, 78quantifying 137questionnaires 68referred 174^175severity assessment 68sleep disorders 272source in OA 43^45, 70^71, 106, 141subsetting 75surrogate markers 141treating mechanisms 140typical 68variability 45, 68vascular hypothesis 117widespread body 276
X-rays 68Pain Catastrophizing Scale (PCS) 263pancreatic arthritis 184paracetamol
mechanism of action 27NSAIDs and 47
patellacartilage degeneration 106prosthesis 106
pathology 77peptidergic nociceptors 30, 209periarticular lesions, MRI 197periarticular pain, knee OA 45peripheral a¡erent nociceptive ¢bres (PANs)
71peripheral neurogenic pain 106peripheral sensitization 5, 123, 225PGE1, nerve ¢bre sensitization 32PGE2
bradykinin augmentation 33dorsal and ventral horn 20nerve ¢bre sensitization 32substance P 129
PGI2bradykinin augmentation 33IP receptors 26^27nerve ¢bre sensitization 32
pH, cancer pain 228phantom limb pain 140, 153, 155^156phantom swelling 156^158, 159phospholipase C (PLC)
hydrolysis 210nociception 126
PIP2, nociception 126, 214placebo response, ¢bromyalgia 272plasticity 12, 122^123, 140, 160, 224^225platelet-derived growth factor (PDGF) 225PN3 128polymorphisms 272popliteal cysts, MRI 193^195post-rehabilitation arthropathy 52postural stability, pain reduction 70PPT-A gene 129pressure algometry 109pressure pain thresholds 109^110, 111pressure patterns 88^90pressure sensation 5pressure sensitive ¢lm 88primary a¡erent nerve ¢bres 28
bone cancer pain 222, 224^225classi¢cation 29
SUBJECT INDEX 289

primary a¡erent nerve ¢bres (cont.)endings 29group II 29, 31group III 29, 31group IV 29, 31joint in£ammation 31locations 28^29mechanical responses 29neuropeptide phenotype 30
progression of OAC reactive protein 71joint instability 85
proprioception 40, 56^57age 57exercise 55improvement 57knee OA 57, 171nomenclature 175pain reduction 70physical function 57
prostaglandins 26^27bradykinin augmentation 33nociception 32^33, 126, 224, 225^226spinal cord neurons 13, 20, 126^127tumour cells 226
prostate cancer, endothelins 227^228protein kinase C epsilon (PKCe) 210proteoglycan, joint immobilization 51, 52protons 214, 224tumour-induced release 228^229
psoriasis, symmetry 242psychiatric morbidity, ¢bromyalgia 259psychological statusexercise 55healthcare seeking behaviour 67knee pain 66pain 68, 78
psychosocial disability, knee pain 68pulmonary ¢brosis, symmetry 243
Q
quadricepsknee OA 52^53, 55re£ex inhibition 54
qualitative research 75, 78quantitative sensory testing (QST) 109, 124
R
rat model 142^144receptive ¢elds 5, 7, 8, 9, 23^24, 144
referred pain 174^175referred sensations 155^156, 160^162re£ex inhibition 40^41, 54, 55rehabilitation, early strategies 87repetitive strain injury 177^178rest pain 5, 177, 180ret 209rheumatoid arthritisanimal models 100^104anti-TNF 103, 128, 133bony erosions 156capsaicin 124contralateral sensory changes 247intraosseous hypertension 180nervous system 246pain 76, 156phantom swelling 156^158sti¡ness 158stroke 146substance P 244symmetry 243^244synovium 156, 178, 203
RT97+ve antibody 30Ru⁄ni endings 29
S
salicylic acid 32scratch response, £are and wheal 141, 244seasonal variation 69secondary OA 49, 253sensory neurons 28^36peripheral injury 229peripheral tissue health 141tumour-induced changes 230
serotonin 33sickle cell disease 180signs of OA 2silent nociceptors 29, 45^46, 225sleep disorders 273SNS 128, 214sociocultural phenomenon 76socioeconomic status, catastrophizing 274spinal cord neurons 4^22activation thresholds 5, 7bilateral receptive ¢elds 7CGRP 13descending inhibition 7^8, 9excitatory amino acids 12glutamate 12heterotopic inhibition 7^8, 9
290 SUBJECT INDEX

in£ammation-evoked hyperexcitability8^11, 12, 13, 20
mechanical stimulation of joint 5^8neurokinin A 13neuropeptides 13nociceptive-speci¢c neurons 7, 8plasticity 12projections 7prostaglandins 13, 20, 26^27receptive ¢elds 5, 7, 8, 9, 23^24substance P 13synaptic activation 12^20wide-dynamic-range neurons 7, 8
spinal pain 105spinal sensitization 123spondyloepiphyseal dysplasia 50squatting 62steroid therapyintra-articular 69, 203intraosseous cells 180synovitis 203
sti¡nessnight 177rheumatoid arthritis 158
strength training 52^53, 54stroke, rheumatoid arthritis 146structure^pain associations 2, 191^201subchondral bone pain 106subchondral cells, cartilage cell
communication 187substance Pantagonists 47arthrogenic potential 129, 131cartilage remodelling 146chondrocytes 146^147in£ammatory arthritis 245^246local level 135marker role 118^119nerve growth factor 128neurogenic in£ammation 129, 131nociceptors 225peripheral release 135rheumatoid arthritis 244spinal cord neurons 13T cells 245
substance P-like activity, ¢bromyalgia 112survival hypothesis 249^250symmetryclinical disease 241^243CRPS 254^255degenerative arthritis 244
hand OA 253in£ammatory arthritis 243^244knee OA pain 24neurogenic role 129
sympathetic uveitis 148, 242, 254symptoms of OA 2
exercise 60^61minimum set to diagnose OA 74^75structure and 2, 191^201
synovial tissueCGRP 245rheumatoid arthritis 156, 178, 203stimulation 5vascular physiology symmetry 255
synoviocytes, substance P 129synovitis
C reactive protein correlation 204chondropathy 204intra-articular steroids 203MRI 193^195, 202NSAIDs 203prominence in human disease 203^204
T
T cells, neuropeptides 245tachykinins 129temperature thresholds 109, 110, 207^208thalamus
¢bromyalgia 259pain sensation 7
thermal sensitivity 4, 34, 109, 110, 207^208,209^210, 224, 259
thumb base OA 68tidemark 188touch perception 110trabecular tension, pain 183transforming growth factor 225transforming growth factor b 209TrkA 209, 210TRP channels 206^213TRPM8 206, 208TRPV1 206
acid detection 224, 228expression pattern 219heat detection 34, 207^208, 210, 224lipid metabolite detection 224mechanosensation 38, 216nerve growth factor 127nociception 126pH 228
SUBJECT INDEX 291

acid detection (cont.)proton detection 214, 224skin 134^135
TRPV2 206, 207, 208TRPV3 206, 207, 208, 215TRPV4 206, 207, 208, 215TTX-resistant Na+ channels 213^214tumour necrosis factor (TNF)damaged sensory neurons 229inhibition in rheumatoid arthritis 103, 128,
133substance P stimulation 129
tumour necrosis factor ahyperalgesia 128nerve growth factor 127, 209
tumoursgrowth factors 229nociceptor excitation 225^228proton release and acidosis 228^229sensory ¢bres 230
U
ultrasound, e¡usions 201urate injections 8
V
vascular endothelial cell adhesion molecules129
vasoactive intestinal peptide 245ventral hornPGE2 20spinal cord neurons 5
vertebral collapse 240visual analogue scales 136^137, 270^271visual feedback, phantom and referred
sensations 157, 159, 164^168von Frey ¢laments 109VR-1 see TRPV1
W
weather prediction 37, 69weekend pain 69weight bearing, bone deformation 189weight loss, exercise 55Whole-Organ Magnetic Resonance Imaging
Score (WORMS) 193wide-dynamic-range neurons 7, 8widespread body pain 276WOMAC 68, 267WORMS score 193
X
X-rays, knee pain 65, 66, 68, 75
292 SUBJECT INDEX
Copyright © 2022 FDOKUMEN




















