
la energía y la vida síntesis fotoquímica en las hojas verdes.
73
REVISTA DE LA REAL ACADEMIA DE CIENCIAS EXACTAS, FÍSICAS Y NATURALES TOMO XLIII
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of la energía y la vida síntesis fotoquímica en las hojas verdes.

R E V I S T A
D E L A
R E A L A C A D E M I A D E C I E N C I A SEXACTAS, FÍSICAS Y NATURALES
TOMO XLIII

R E V I S T AD E L A
BEIL A C A D E M I A D E C I E N C I A SEXACTAS, FÍSICAS Y NATURALES
D E
M A D R I D
T O M O X L 1 I I
C U A D E R N O P R I M E R O
M A D R I D 'DOMICILIO DE LA ACADEMIA: VALVERDE, 22
T E L E F O N O 2 1 - 1 5 - 1 9i 9 4 9

Artículo 39 de los Estatutos de la Academia:
«La Academia no se hace solidaria de las opinio-
nes cuestionables, en materia científica, de sus indi-
viduos.. Cada autor es responsable de las proposicio-
nes y asertos que. contengan los escritos del mismo
que aquélla publique.»
C . B E R M E J O , I M P R E S O R . - J . G A R C Í A M O R A T O , 118. T E L E F . 3 3 - 0 6 - 1 9 . - M A D R I D .

F é c u l a s y L i g n i n a s
por
Obdulio Fernández
LA ENERGÍA Y LA VIDA SÍNTESIS FOTOQUÍMICA EN LAS HOJAS VERDES.—LAS IDEAS
CLÁSICAS A LA LUZ DE LOS CONOCIMIENTOS ACTUALES
Uno de los grandes fenómenos que preocupan la inteligencia humana es la viday su desarrollo a través del tiempo. La vida, como todos los fenómenos naturales,se desenvuelve sobre una materia tangible, y con energía, que algunas veces apre-ciamos por sus efectos. La materia se formó primero y la energía vino después, peroen gran parte esta energía procede de la vida misma, porque exceptuando el traba-jo del viento y las caídas de agua, toda procede de actos vitales, qué por su man-sedumbre y parvedad pasan inadvertidos entre los factores energéticos. Aquí reside 'la grandeza bien cantada de los fenómenos naturales desde la antigüedad, cuandose aludía a causas sencillas, • productoras de grandeá hechos. Sin la energía vital,dice B. Moore (l), todo lo existente sobre la tierra, incluidos los grandes procesosindustriales, cesaría en el acto. Las células integrantes de nuestros tejidos o losagregados 'coloidales en ellas existentes no son generadores de energía aparatosa,como 'la de una dínamo o de una antigua rueda de agua, pero son de una eficaciay una constancia que aquéllas no pueden tener. La energía no procede de la naday sin embargo existe en formas mudables, subordinadas a una fundamental, inicia-dora de todas las causas y efectos visibles en la naturaleza ; es la energía de la luz,infinita y aprovechable por todos los seres vivientes, con el objeto de procurarsesu propia energía, la vital.
La luz, escribe J: Palacios, es la más noble y eficaz de las formas de energíautilizadas por el hombre, y antes el P. Nieremberg (siglo xvn) (2) escribió : «sin luznada fuera hermoso», «la luz fertiliza la naturaleza». Entre el físico contemporá-
(1) Biochemistry, p. 25, 1921. Londres.(2) Tratado de la Hermosura de Dios. Madrid, 1879.

neo y el escritor jesuíta descuella la figura de Galileo, mostrando con genial intui-ción al saber de su tiempo, que la luz es el principio de las cosas. La mudanza dela energía luminosa, formidable e inextinguible, en otra energía mansa y de acciónsuave, quizá no la realiaa la vida, al contrario, puede ser el origen mismo de lavida, si el hombre pudiera comprenderlo, pero entre tanto, hay que confiar en quela propia vida sea capaz, de realizar la mutación. Esta mutación, para ser biencomprendida por todos, debería traducirse en un acto fantástico, superior en dra-matismo a las enormes caídas de agua, como las del Niágara o del Potomac, y noobstante sus dimensiones en el espacio,-transcurre oscurecida y hasta desconocida,porque no se realiza con grandes masas y en elevadas concentraciones de las sus-tancias que intervienen en ese proceso químico biológico tan trascendental. Serealiza a expensas de una cantidad •pequeñísima de anhídrido carbónico contenidoeu el aire (— 0,3 — 0,4 gr. por 100), lográndose así la doble finalidad de la trans-formación energética y la purificación de la atmósfera en que respiramos. Claro es,que solamente con el anhídrido carbónico del aire no se conseguiría la conversión:se necesita del agua, que lleva disueltas exiguas cantidades de compuestos minera-les, de esos que por su propia solubilidad y su existencia en las zoiiás superficialesde la corteza terrestre se llaman elementos biogenésicas, incluyendo entre ellos losde peso atómico superior al del hierro y que hoy se conocen con el nombre de infi-nitamente pequeños u oligoelementos : cobre, cobalto, níquel, etc.
La naturaleza maneja, así dos disoluciones ; una de sustancias inorgánicas enagua, de molaridad variable, según los terrenos, y otra de un gas en otro gas, la de
carbónico en aire, de molaridãd dilución verdaderamente asombrosa, para55.000 . ' • " " > ,
imaginar que de ella puede deducirse una actividad tan considerable. No obstantetal dilución se calcula que anualmente se fijan 396 billones de toneladas de anhí-drido carbónico. El agente transformador de la energía luminosa en energía quí-mica vital es la materia colorante verde de las hojas vegetales, la clorofila, depo-sitada en forma de puntos verdes oleosos (cloroplastídulos o cloroleucitos), en unasustancia incolora con apariencia de estroma. ¿Cómo intervienen esos puntos ver-des para procurar la mutabilidad de la energía? Las dos materias sobre que hande actuar las ondas luminosas, son en realidad el gas carbónico del aire y el aguaque las plantas toman de la tierra sobre la que viven ; las dos inorgánicas, sinvida, inertes en la apariencia para los fines vitales. La unión sencilla del carbonodel anhídrido carbónico, con el agua, es lo que se le ocurre al químico menos pre-visor, si esto sucediera, pero antes ha de quedar el carbono libre, que es todavíamás inerte que el gas carbónico, y además combinarlo con el agua : he aquí laenergía fotoquímica, convertida en energía vital, que procura el calor necesariopara esa unión de dos factores inertes, la más fecunda para el sostenimiento delmundo todo. La liberación del carbono del anhídrido y su inmediato apareamien-to con el agua, sólo con calor o con mecanismos vitales, todavía solo sospechados,puede realizarse. ¿Cuál es la resultante de esa unión de tipo endotérmico? Bayer

fue el iniciador de hipótesis más o menos felices, que tratan de explicar el fenó-meno más acuciante de entre los producidos por la vida, y pensó que el compuestoresultante más sencillo debía ser el aldehido fórmico, el formol vulgar, con apari-ción simultánea de oxígeno
CO3 + H3O = HCOH + 02.
La hipótesis necesitaba confirmación y por eso las impugnaciones menudearon,no obstante la autoridad extraordinaria del insigne químico bávaro. ¿Cómo se hade producir formaldehído, si por su calidad reductora es uno de los tóxicos celu-lares más violentos, y si a concentraciones de 0,001 por 100 no es soportable paralos protoplasmas? Además, se observó que plantas sometidas al influjo de la luz,en disolución del aldehido a la concentración indicada, no producen más fécula quesin formol.
Mas* no se demostraba lo interesante, la presencia del formol y por tanto susíntesis directa a pesar de los ensayos reiterados hasta el tedio. Sólo en un campoexperimental y basándose en analogías se consiguió una demostración discutible.El propio Bach (S) sometió disoluciones acuosas de anhídrido carbónico a la in-fluencia de radiaciones solares, que atravesaban por disolución de nitrato de' ura-nilo, líquido cuyo espectro de absorción es en la zoria más refrangible, parecido alde la clorofila, Ny comprobó por la reacción de benzhidrol ; primero, la presenciade aldehido fórmico, después la de oxígeno, que actuando sobre el tetrametilbia-mino-bifenilmetano generado a expensas de la bimetilanilina, ocasiona un alcoholsecundario de intenso color azul-violeta.
/C6H4 N(CH3)3 yC6H4 N(CH,)2CHaO -f 2 C6H5 N(CH3)2 -> CH, < + H,0 -* O = CHOH /
\C6H, N(CH,)a \C6H4 N(CHjì,
Perol la constancia, una de las virtudes del científico, se situó por encima dela contradicciones y el día en que se generalizó en las técnicas analíticas el meca-nismo llamado de Vorländer (4) se hizo posible afirmar, que el resultado de la sín-tesis fotoquímica en las hojas verdes, es eí aldehido fórmico retenido a medida quese forma, por la bimetilbihidroresorcina, con la cual produce el dimedon : a pesarde todo, el caso es discutido.
CO _ COH»C/\/UH«\/\CH,
I I l | I • . .:CH3)C\/CO C\/C(CH3)2
CH, HO/ CH,
El argumento de la toxicidad del formol subsistía, pero la vida, que tan pròdi-ga es en recursos de desintoxicación, no dejaría al formol envenenando las células
(3) J. R. Carracido. Tratado de química biológica. 1924. 175. Madrid.(4) Ann. 1897. 294-252. Neumann Diss. Leipzig, 1906.

que lo generaron, sino que; lo dispondría para una- polimerización en grado másavanzado que las conseguidas en el laboratorio.
En él se prevé la "trimerización reversib'e del formol produciendo trioximetile-no, pero las células no reúnen sólo tres moléculas, reúnen seis, y entonces se for-maría glucosa, la cual, a su vez,, produce por deshidratación y condensación fécu-la, que como granos blancos, brillantes, coloreables en azul con el agua de yodose deposita o emigra al interior de la planta.
La teoría de Bayer ofrecía la explicación de un hecho confirmado después, la pro-ducción de fécula, y además otra muy en armonía con la época siguiente a la -enque se elaboró; la de Bach fue la época de los peróxidos, en la que se creía quetoda oxidación va precedida de la síntesis del peróxido de hidrógeno, del aguaoxigenada, la que a su vez se descompone, liberando oxígeno. Los dos hechos fun-damentales parecen indiscutibles ; que se origina formol y que se desprende oxíge-no. Pero estos son los términos de una gran cadena de fenómenos, difícilmente in-quiribles, y que sólo los años y la evolución de todos los conocimientos científicosde los diversos ramos del saber van consiguiendo desintrincar.
Desde la época de Priesley y de Ignenous se sabían dos detalles esenciales:primero, que toda la actividad desplegada por los plastídulos cesa en cuanto elsol se pone y queda la tierra sumida en sombras ; la luz, de la luna no es bastantepara producir fécula; segundo, que las plantas que viven en la oscuridad no sonverdes, y por tanto son incapaces para la función clorofílica. De esto se deduceque el agente promotor de la metamorfosis fundamental que se realiza en la hoja,es la clorofila. El representante estructural del producto verde de las plantas en elorganismo zoológico es la oxihemoglobina ; ambas son porfirinas, diferenciablesúnicamente en que la clorofila como nexo de los agrupamientos pirfólicos tienemagnesio y la oxihemoglobina, hierro. En las transformaciones realizadas por am-bos pigmentos interviene el ácido fosfórico, que en las plantas sintetiza la amilo-pectina, y artificialmente la amilosa. El pigmento en la vida vegetal, como en lazoológica, es un protector, merced al cual las demás mutaciones se hacen posiblesen el lado material o en el energético, o en los dos simultáneamente. Lipman admi-te que el ester adenosintrifosfórico, al convertirse en bifosfórico, cede energía, yno sería recusable el argumento en que se afirme que ese residuo fosfórico contri-buirá a la síntesis de ester fosfórico de la amilopectina, semejante al de la amilosaconseguido por Hanes con la fosforilasa, fermento, extraído de la patata.
Las mutaciones realizadas al amparo de la oxihemoglobina sanguínea con ácidofosfórico' se paralizan con agua oxigenada, y las causadas por la levadura de cer-veza también se inhiben con este peróxido, descubrimiento conocido con el nombrede fenómeno de Bach. Son dos hechos ligados a dos períodos distintos de la cien-cia : el de los peróxidos de fecha pasada y el de las fermentaciones fosforilantes delactual.
El agua oxigenada es un tóxico para la células tan enérgico como puede serloel formol, y es por tanto inevitable que un fermento destruye ese agente nocivo.

— 9 —
Ese fermento destructor existe en las plantas y en los glóbulos rojos de la sangre ysu finalidad es destruir el peróxido productor de oxígeno; por consecuencia, eloxígeno resultante de la síntesis clorofílica podría ser el desprendido del peróxidoinicial, con ayuda de la catalasa. La intervención de la fosfatasa en la fotosíntesishace presumir un conjunto.de fenómenos de óxido-reducción por la .solidaridadexistente entre éstos y la fosforilación, y en virtud de los cuales el acto, en apa-i'i'encia sencillo; de la reducción del CO2 se complica mucho más de lo que sehabía creído al emitirse la idea de Bayer, y se complica a tenor de esta tesis man-tenida por Frank y Gaffront (5) CO2 + 2 AH2 _ CH3O +• 2 A + H3O, o seaque el compuesto orgánico es deshidrogenado, mientras el anhídrido carbónico esreducido.
Willsttaeter y Stoll, en 1918, afirmaron que no fes sólo la clorofila la que ab-sorbe la luz, como energía, sino los factores enzimáticos que intervienen en lareducción del anhídrido carbónico. Estas afirmaciones y otros hechos con ellasrelacionados abren una duda en el espíritu ; la posibilidad de que alguna de esasreacciones en que colaboran enzimas no requieren ninguna energía da la luz so-lar, porque pueden realizarse en la oscuridad, extremo confirmado por V. Niels.Justamente en el ritmo de la fotosíntesis el período de oscuridad constituye un largoperíodo de .Blackman, en el cual intervienen enzimas pertenecientes al grupo de losinhibibles por el cianuro potásico. El período de Blackman artificial es de 0,02 desegundo a 25°.
La fotosíntesis es una reacción semifotoquímica, en la que la molécula declorofila actúa como donante de hidrógeno, convirtiéndose en deshidroclorofila,capacidad extensiva a otras materias colorantes, que al igual del pigmento verdede las hojas, sensibilizan la fotosíntesis. El fenómeno dé la reducción de la cloro-fila pudo pasar inadvertido porque la deshidroclorofila posee el mismo espectro deabsorción que la clorofila, pero la ligadura afectada no pertenece al sistema deenlaces eténicos conjugados de la clorofila.
El primer paso en la fotosíntesis será fijar el gas carbónico de la atmósfera.¿Quién lo fija? Es presumible que sea la clorofila, por un mecanismo idéntico ala carboxilación ; de acuerdo con la teoría de Willsttaeter y Stoll se forma así unácido clorofilocarbónico en una reacción, en la que se desprenden diez calorías,idéntica en su proceso a la síntesis del ácido floroglucinocarbónico sobre el trife-nol simétrico a presión de una atmósfera.
El segundo paso es la cesión de hidrógenos al ácido clorofilocarbónico congradaciones. Se invierten en el retorno a la deshidroclorofila de los cuatro hidró-genos, cuatro cuantas y como son cuatro los hidrógenos, sugiere V. Niel el que laactuación de un átomo de H sea de un cuantum, aunque el estado termodinâmicode la reacción implica que el número de cuantas en la fotosíntesis sea el 12 o pró^ximo al 12.
(ñ) Advances in Enzymology. 1941. 1.239

Existe en el período actual una tendencia a considerar la síntesis en las hojasverdes como una reacción de tipo anaerobio, en que se libera oxígeno gaseoso.¿Como conciliai estas dos tesis manifiestamente antagónicas? Sólo hay un cami-no: admitir la formación de peróxido, como ocurre con muchos seres unicelula-res, microbios que producen agua oxigenada, la cual es descompuesta por la ca-talasa, cuya intervención no se niega por ningún experimentador, aun cuandoFranch y Gaffront (6) sostengan que una fotocatalasa tendría poco éxito paraproducir oxígeno, pero a renglón casi seguido afirman que la diferencia funda-mental entre los sistemas asimiladores de plantas y de bacterias fotoactivas esque éstas carecen del enzima propio para descomponer los peróxidos intermedios,y después se hacen referencias a observaciones que sugieren más estrecho acopla-miento entre la formación de peróxidos intermedios y la reducción del complejoaiceptor con el anhídrido carbónico.
Todos los antecedentes son favorables a la tesis de la fosforilación como fenó-meno intermedio de la actividad clorofílica: en los eritrocitos sanguíneos, queson lo más semejantes a los cloroplastos vegetales por la casi igualdad estructuralde los pigmentos y por una. finalidad, la de producir oxígeno, se originan actosfosforilantes y peróxidos, estos últimos tóxicos de los sistemas enzímicos que in-tervienen en la fosforilación; por tal motivo conceptúan Wladimirow y Koloti-lova (7) que las catalasas destruyen- el peróxido que inhibe la fosforilación, pro-vocando el desprendimiento de oxígeno. Si en la hematosis concurren como en lafotosíntesis, fosforilación y peróxidos, es lógico que la planta tenga que librarsede ellos, por ser tóxicos de los agentes fosforilantes y que se sirva de la catalasapara ese acto insustituible de la síntesis de polisacáridos.
Corroboran estas ideas acerca de' fosforilación, como paso inevitable en lasíntesis fotoquímica, el reciente descubrimiento por C. R. Yíu (8) de fosforiíasaen los plastídulos, motivo por el cual el almidón se forma siempre en estos mi-núsculos laboratorios de las hojas, y Hanes (9), autor de la síntesis de féculas confosforiíasa y esteres fosfóricos, advirtió que el origen de estas materias hidrocar-bonadas se halla en la fosforiíasa. En 1945 Lipmann y Zuttle (10) propusieron unciclo entero de fosforilación hasta llegar a un ácido-alcohol. El problema, comopuede adivinarse, es muy intricado a causa de su complejidad, que va pareja a sutrascendencia biológica. Por eso se agotan los ingenios y se buscan nuevos proce-dimientos para estudiarle.
Sólo queda uno para inquirir el mecanismo fijador del anhídrido carbónico,que es el empleo del isótopo del carbono C11, y con él se trabaja, por Rubcn, desde
((>) Advances in Enzima'ogy. 1941. 1.254.(7) Biokhimiya, 1947. 12-321. Abstrats, 1948 A 111 556.(8) Nature, 1948. 162-928.(9) Procee-R. Soc. B, 1940. 129-204.(10) J. Biol. Chemistry, 1945. 185.505.

— I I —
1939. Exponiendo plantas clorofílicas a la acción de anhídrido carbónico radio-activo, unas veces a la luz y otras a la oscuridad, para asemejar el ritmo noc-turno-diurno, y observando la radio-actividad en diferentes fracciones de pro-ductos aislados de las plantas así tratadas, en cada fase del ritmo se encontró queen la fase luminosa la clorofila es algo radio-activa y en la oscura falta por com-pleto la radio-actividad, y ésta aparece en una y otra en el extracto acuoso obte-nido de las plantas, extracto integrado por azúcares aislados por medio de la fe-liilhidracina. Luego el primer acto químico es sintetizar azúcares, que es naturalproduzcan alpolimeri/arse, féculas.
Estas experiencias con el isotopo del carbono han sido confirmadas por otrosbiólogos sin ese isótopo, aislándose también azúcares y féculas juntamente.
Los Chlorellas son algas unicelulares, muy adecuadas para los estudios de foto-síntesis, y también fueron elegidas por Rubén para los de radioactividad.
En estas algas, el carbónico .radioactivo va durante la fase oscura a constituirun carboxilo, como supusieron Willsttaeter y Stolz, en tanto que en la fase lumi-nosa el veinte por ciento de la radioactividad aparece como substancia oxhidrílica,determinada por producción de un ester benzoico, y de ello deducían que en lafase luminosa se reduce el carboxilo con liberación de oxígeno. Como consecuen-cias de estos trabajos, Rubén presupone que la primaria es la oscura, en la quese fija el carbónico a un aldehido, engendrándose un ácido cetónico
//® //^R. Cf + CO2 = R — C^XH ^COOH
el cual es reducido a ácido aldehido, alcohol y a ácido, sucesivamente.A los primeros químicos—Bayer, por ejemplo-r-que intervinieron en esta cues-
tión, y que lógicamente imaginaron sus grados más sencillos, les preocupó, ade-más del agua, el desprendimiento de oxígeno, aun antes de prever la posible for-mación de un peróxido. ¿El oxígeno que se desprende es el del agua? Las ideas
f^Oclásicas que partían del descubrimiento, por Bousingault, de la relación - =1,
().,llamada cociente respiratorio, presupone la reducción total del gas carbónico, que-dando reducido a carbono, que adquiere los elementos del agua para producirformaldehído. El oxígeno/pues, rio procederá del agua; pero remedando con lasexperiencias del laboratorio lo más similares a las naturales, Warburg y Luftgenshan conseguido demostrar que en fenómenos semejantes a la fotosíntesis se des-prende oxígeno del agua. Al efecto, mezclan cloroplastídulos de espinacas con aguay quinona y obtienen, hidroquinona y Oxígeno.
2 C6 H4 O, + 2 H, O = 2 C. H6 02 + O2
luego el agua es,el donante de hidrógeno que convierte la quinona en hidroqui-nona. En la experiencia pueden reemplazar a la quinona el oxalato férrico y el

ferricianuro potásico, con los cuales' Hi 111 y Scarisbruck observaron el desprendi-miento de oxígeno. No hay, pues, necesidad de acudir a la clorofila como donan-te de hidrógeno según el criterio moderno que rememora el clásico de Gautier,aun cuando aquellos dos autores compartan los puntos de vista de su época.
El hidrógeno procederá del agua al perder su oxígeno, que debe liberarse : Laanalogía con el mecanismo de reducción del hidrógeno sulfurado y el resultado delempleo de isótopos del oxígeno competen a. mantener .esta tesis. Trabajos curio-sos de Jack prueban también que el oxígeno respiratorio tiene su origen noen el carbónico, sino en el agua, de acuerdo con la fijación del gas 'como car-boxilo. En esas experiencias realizadas con cloroplastídulos de espinacas debemediar un acto fermentativo, como en la generalidad de los fenómenos biológicos,porque si esos agentes verdes se lavan prolongadamente o se dializan, pierden suactividad y el desprendimiento de oxígeno no ocurre, pero es posible restauraraquélla agregándoles una disolución molar 3, 3 x 10--* de cloruro potásico. El pro-blema se ha complicado algo más porque, aparte de considerar la influencia enun medio más circunscrito que antes y de conceder valor a la síntesis nocturna,interviene también un fenómeno de flluorescencia, cuyo estudio promete resulta-dos interesantes, aunque no definitivos, acerca del problema de la fotosíntesis.
Consúltese el libro recientemente aparecido de Janes Franck y W. E. Loomis,Photosynthèses in Plants, 1949.
Como habrá podido apreciarse en la exposición precedente, mi deseo, que nosé si he logrado, fue hacer un paralelo entre los fenómenos respiratorios de los ani-males y los de las plantas, en vista de las circunstancias químicas que concurrenen ambos.
II
ESTRUCTURA Y COMPOSICIÓN DE LAS FÉCULAS.—INTERÉS BIOLÓGICO DE LA RAMIFICA-
CIÓN SÍNTESIS ARTIFICIAL DE LAS FÉCULAS.—COTEJO DE LAS FÉCULAS NATURA-
LES CON LAS FORMADAS «IN VITRO».
Estructura de las féculas.
Los biólogos agrupan las féculas en el capítulo más interesante de los polímerosde grado alto de condensación, pero constituidos 'no por el sistema del caucho,en el que se enlazan moléculas de un hidrocarburo, el isopreno, sino por un me-canismo de deshidratación de moléculas de glucosa, unidos en 1-4 o en 1-6, paraformar agregados de 5-6 unidades, que después integran el polimero, cuyo pesomolecular calcula R. H. Meyer en 400.000. Esto presupone la existencia de ma-cromoléculas o al menos de agregados que han sido separados por Haworth me-diante el ácido oxálico y que supone unidos por ligaduras que denomina poli-m ericas.

'3 -
La primera unión la designa Myrback con el nombre de flecha a punta de
fleclia a de flécha a media t lecha — T- >• ;
la primera da origen a cadenas rectas y la segunda a cadenas ramificadas o ano-
malías.
CH2OH
<:>-°-<>°-o-°-<>unión glucosídica 1:4
CHjOH
Vo-rvo-/~v
unión glucosídica 1:6
El enlace 1 : 4 se ha deducido por Irvine y Macdonal utilizando las reglasde Hudson referentes a la actividad óptica, y el 1 : 6 por K. H. Meyer con el em-pleo de fermentos y con la cinética de la hidrólisis ácida
Desde la aplicación del microscopio al estudio de las plantas se conoce la estruc-tura de esferocristales, propia de las féculas de distinto origen. Más moderno esel conocimiento de la composición de las capas que se agrupan en torno del hilo.Sin duda,, esta diferencia estructural hizo pensar en la química correspondiente,y a ello debe atribuirse la opinión de Roux y Maquenne, en virtud de la cual elalmidón está formado por dos substancias. Esta supuesta falta de homogeneidadfísica parecía corresponder a otra química, pero durante muchos años se ha dis-cutido con entusiasmo, sin conseguir enfocar el problema, hasta que K. H. Me-yer (11), recogiendo las ideas de Maquenne y Roux y basándose en sus estudiosreferentes a viscosidad, a difracción de rayos X y a las cualidades de los esteresacéticos, ha expresado una idea en forma terminante y aceptada casi por todoslos especialistas, aun por aquellos que la habían discutido con más fogosidad.
El almidón está integrado sustancialmente por dos compuestos: uno solubleen agua, llamado amilosa, de molécula linear o extendida, y otro .insoluble enagua, de cadena ramificada, productor de gelatina cuando se calienta a más de80 grados, y al que se llama amilopectina, semejante en su constitución molecu-lar al glucógeno, polisacárido almacenado en el hígado de las diferentes especieszoológicas.
(11) Helv. Ch. Acta. 1940. 23, 346.

- H -
Es innecesario decir que el rigorismo químico no se puede emplear tratándosede sustancias fundamentales de la vida ; no existe separación absoluta ni en cuan-to a viscosidad ni en cuanto a linearidad o ramificación de cadenas. La anulosasoluble en agua y abandonada a sí misma, lentamente va enturbiándose y preci-pitando una materia semicristalina, insoluble en agua. Este fenómeno, conocidocon el nombre de r&trogradación de la amilosa, es sencillamente de agregación demoléculas, por convertirse en un complejo mayor, traducido en enturbiamientodel líquido. Hay que reconocer que el fenómeno todavía es discutido en su inter-pretación. Y en lo que se refiere a la solubilidad en agua, hay tema propicio adiscutir, porque aquélla depende de la temperatura y de un estado previo de agre-gación de los granos de fécula en las plantas, particularmente en las semillas,que es necesario vencer para que la amilosa se disuelva en agua. Santoni (12),con sus reiteradas experiencias acerca de la solubilidad, ha conseguido emplean-do temperaturas superiores a los 80°, que es próxima a la desintegración de losgranos, cambiar las cifras de amilosa (normalmente 18-20) en las de la amilopec-tina (normalmente 70). El trueque de números presupone que las cadenas se iso-merizan, desramificándose al sufrir los efectos de la temperatura, si no se quiereir algo más lejos en la apreciación de fenómeno tan interesante. Podría sei quizáviolento negar la existencia de enlaces 1-6 para la inserción de las ramificacionesy sustituirlos por ligaduras de hidrógeno, que podrían unir las cadenas separa-das, como pensamos ocurre con las moléculas de amilosa al soldarse con la celu-losa, constituyendo una cadena recta. Es preferible pensar así à suponer que laamilopectina pierda las ramificaciones y se arrolle en una cadena helicoidal, comola que se admite para los complejos de,amilosa con yodo, ácidos grasos .o sus-tancias de estructura semejante a la que posee la amilosa.
La primera parte de la segunda tesis es aceptable, si se supone, como hanpodido comprobar mis colaboradores, que la& ramificaciones de la amilopectinano se inician en las primeras moléculas de glucosa constituyentes de la cadena,sino que se agrupan en un extremo,'en combinación fosfórica, como se expondrámás adelante, dando a la molécula ramificada la apariencia de una palmera conlos penachos en el ápice de su tallo. Esta parte esterificada quizá no resista los90 grados a que somete Santoni la fécula y se separe del resto de la molécula,que como es de cadena recta se disuelve en agua y aparece como amilosa. Sóloasí se puede interpretar con .corrección el supuesto aumento de la cifra de ami-losa a expensas de la amilopectina.
La resíntesis, es 'decir, la unión en 1-4 de las cadenas separadas a la de amilosaes muy poco verosímil, porque a los 90° es probable que si la soldadura corrieri-te se realiza gracias a la intervención de un enzima, éste haya desaparecido du-rante el calentamiento. De otra parte, algunos especialistas—K. Meyer, entre otros
(12) These Marseille. Contribution a l'étude de la constitution de l'amidon, 1939.

— 15 —
admiten la posibilidad de pequeño número de ramificaciones en las amilosas, par-
ticular de que ahora se tratará.La posibilidad de la estructura helicoidal de la amilosa fue desenvuelta por
Freudenberg con el objeto de explicar la estructura de las dextrinas de Schardin-ger obtenidas mediante el Bacillus macerans y que están constituidas por azúca-res cíclicos, como la inosita ; pero la idea la abandonó su autor en cuanto se re-fiere a la espiral nativa o preformada, reservando su primera hipótesis para in-terpretar la aptitud de las amilasas en colocar partes de cadena en espirales deseis miembros.
La idea la recogió Hanes (13), vacilante en cuanto a la existencia, de ramifi-caciones en el producto de su síntesis, admitiendo que cada espiral lleva seis uni-dades de glucosa. Así, ha' quedado el número 6 como una especie de mito, aco-modado a las circunstancias que resultan de los diferentes trabajos: cada 6 uni-dades rectas de glucosa, es decir, engarzadas por enlaces 1-4, llevarán una, rami-ficación, o sea una molécula de glucosa o un grupo de glucosas enlazadas en 1-4o en 1-6. En efecto, el número de seis se reiteraría al degradar las féculas porlas amilasas ; quedan así dextrinas, integradas según Ortenblad y Myrback (14)por mezcla de hexasacáridos, framentables en mezclas de tri y tetrasacáridos.
Prescindiendo de la estructura, nótase en la amilosa una tendencia a crista-lizar, aun descontando el fenómeno de la retrogradación. Al señalar las solubili-dades variables de la amilosa se habla por K. H. Meyer de amilosas poco ramifi-cadas solubles, lo cual se enfrenta con el .concepto admitido de la rectitud de lacadena y con la homogeneidad.
En el trigo canadiense se ha descubierto la gamma-amilosa, denominada asíprovisionalmente, y se ha observado por métodos dé fraccionamiento con mezclasde butanol y metanol, que contiene un factor existente también en la patata yes amilosa cristalina (15).
No desviemos el tema diferencial de amilosa y amilopectina : la primera ori-gina combinación insoluble con alcoholes, como se acaba de indicar la celulosa,por ejemplo—, hecho descubierto por Tanret (16) y utilizado para separar los dosfactores de las féculas ; también son insolubles las combinaciones, no importa dequé naturaleza sean, con alcoholes ciclánicos, ciclanohexol, y su homólogo mentoly alcoholes grasos, como el butanol normal, y con fenoles.
Tan curiosa como esta insolubilidad de la amilosa es su solubilidad en diso-lución al 33 por 100 de hidrato de cloral a la temperatura ordinaria y en eti-leno-biamina, empleada para la práctica de determinaciones viscosimétricas de lasféculas.
(13) New Phytologist, 1937. 36-101.(14) Biochem. Zeit, 1943. 37-518. Ch. Abstrat, 1944. 38-984.(15) Kerr y Leverson. Amer. Ch. Soc. 1943. 65-193.(10) Comp. R. Acad. Sciences. 1914. 158, 1.353..

— io —
La acetilación, de las féculas conduce a introducir en su molécula tres aceti-los, tanto en la amilosa como en el factor ramificado, amilopectina, mas los este-res forman películas, blandas y adaptables en la amilosa y rígidas y* duras en laamilopectina, hecho del que deduce K. H. Meyer la ramificación de la segundasustancia. La acetilamilosa se disuelve en acetona fría; la acetilamilopectina esinsoluble en acetona fría o caliente. Además, el peso molecular estimado por pre-sión osmótica es muy distinto: oscila entre 20 y 50.000 para la triacetilamilosay se aproxima a 400.000 para la triacetilamilopectina, lo cual presupone un nú-mero de unidades de glucosa constituyente mucho mayor. Los números son muybajos comparados con otros más recientes (véase más adelante), que dan para laamilosa de 100.000 a 200.000 y para la amilopectina de i.ooo.ooo a 6.000.000
El yodo es el reactivo más frecuentemente usado para diferenciar amilosa deamilopectina, aunque no pueda precisarse gran cosa sin aparatos físicos que sepa-ren colores. El yodo sobre las pseudosoluciones de anulosa produce color azul ;en cambio, sobre las de amilopectina le acusa violeta o rojizo, como en el glu-cógeno ; no obstante, al caracterizar amilosa por este camino, se escribe que tienetantas o cuantas unidades violeta y, viceversa, la amilopectina tiens X unidadesazules. El reactivo acusa cadena recta y cadena ramificada, y aunque nada exac-to se sabe de la intervención en las últimas, sí se ha estudiado con' algún éxitoen la amilosa. Este factor de cadena recta sufre por el yodo uria contracción dela cadena, que se arrolla en espiral (17), para colocarse los átomos de yodo enla vuelta de la hélice, en razón de una molécula de yodo por cada vuelta, demodo parecido a como se colocan las moléculas de alcohol butilico o de ácidosgrasos. (Véase la página siguiente.)
No parece, pues, que exista una verdadera combinación química. La necesi-dad de expresar unidades azules o rojas eri cada medida practicada en el absor-ciómetro, depende en parte de la diferente solubilidad de la amilosa de cadaespecie vegetal, porque admitido, como estima Meyer, que la amilosa puede serun poco ramificada, hecho que confirman Potter y Massiot (18), resta una consi-deración de carácter general. Los cuerpos primeros de una serie son de solubi-lidad relativa, los más altos son menos y los finales carecen de solubilidad ; seme-jante idea es aplicable a los polímeros ; los de pocas moléculas son poco solubles,pero los procedentes de asociarse muchas son necesariamente insolubles. Puesbien ; cada especie vegetal posee una amilosa diferente en conexión con su pesomolecular, variable de 100.000 a 210.000 (19), y como a éste va ligada la solu-bilidad, es lógico que las grandes 'cadenas de amilosas sean menos solubles quelas pequeñas. En este hecho estriba una de las causas diferenciales de los nú-meros obtenidos por los investigadores en las féculas no usuales. La diferente
(17) Rundle y Edwards. Amer. Ch. Soc., 1943. 65-2.200.(18) Amer. Ch. Soc., 1948. 70-3.775.(19) Ibiden.

i? —
n fi ;n rn ^w .xS/ .¿v ;k/ 'K^ "̂.INV^V^X-̂ -- •*-- -• ' ' tA~ '"" v "•-
u-v. •
.-v
JCadenas helio jidales de a lmidón con moléculas de yodo en el centro de la hélice.
Y/í'¡indi'e y Baldwin)
&&.*.
**^iffis^'*s,f%$?fe-*-!
't'&ZP&§í%&B·--^-'ox:^ ̂ ^v"'-- --^v' -""\f*s<^-
("adenas helicoidadas de butanol-amilosa con seis unidades de glucosa porcada vuelta de la hélice. (Rumile y Kdwards)
Cadenas ramificadas de amilopectina
REV. DE LA REAL ACADEMIA DE CIENCIAS.—1949.

— IS —
capacidad de absorción de los complejos de yodo con la amilosa y la amilopec-tina parece constituir una diferencia absoluta entre los factores (20), así como eldistinto tipo de reacción con el yodo (21).
Las féculas se hidrolizan por los ácidos con velocidades diferentes, utilizadaspor varios químicos ipara conocer el número de ligaduras contenidas en las ca-denas. A la postre se, obtiene glucosa ; por tanto, la fécula insoluble se disuelvetotalmente. Pero si en vez de emplear ácidos en las hidrólisis, se emplean fer-mentos sobre los dos factores aislados constituyentes de las féculas, la conductade la amilosa discrepa de la amilopectina. La cebada germinada elabora dosdiastasas, la a y la ß, que juntas disuelven totalmente la amilosa ; sólo la 4e
alguna fécula la de castaña y de manihot queda un residuo pequeño, pero suficientepara afirmar que la amilosa no es totalmente degradada a glucosa y maltosa por ladiastasa de la cebada (amilasas). Las amilopectinas de modo constante dejan unaparte insoluble, conocida con el nombre dei dextrina.
¿Cuál es la causa de esta diferencia sustantiva? Es precisamente la de lascadenas integrantes de los componentes de las féculas. Las anulosas contienen suseslabones de glucosa unidos por enlaces 1-4, lo cual determina la linearidad dela cadena. En la amilopectina, los eslabones están enlazados por uniones 1-6,determinantes de la ramificación. La actuación de las amilasas es sencilla en laamilosa : se separan (en general sin discernir la conducta de la a respecto dela ß) los agregados de 6 unidades en moléculas de glucosa y maltosa; es lo queen términos vulgares se denomina sacarificación. Las amilasas actúan específica-mente sobre los enlaces 1-4 y por eso sueltan la totalidad de los existentes en laamilosa. En cambio en la amilopectina se obtiene un residuo insoluble y un líqui-do en donde se hallan glucosa, maltosa y hasta maltotriosa, soliendo alcanzar lasacarificación el 60 por 100 de la sustancia. ¿Por qué dejar materia sin disolver?Porque las amilasas, como se indicó, sueltan las ligaduras 1-4, pero al alcanzarla altura molecular en que comienza una ramificación, definida por un enlace 1-6,el fermento se parausa ; no es capaz de ir más allá porque su constitución sólole capacita para desenvolverse en moléculas unidas por enlaces 1-4. Es necesarioemplear un fermento que suelte los enlaces 1-6, y es la a-glucosidasa, separablepor disolución en agua de la levadura de cerveza ; en efecto, este líquido vaseparando de la dextrina la cadena ramificada, pero por. su misma especificidadno puede soltar la unión 1-4, y entonces hace un alto porqué se halla, luego deseparada la ramificación, ante una cadena recta, como la de amilosa. Estamosfrente a nueva dextrina, la II.
Si en el momento de separar la cadena ramificada se hace actuar sobre ladextrina II la mezcla de amilasas de la cebada germinada, el trozo de amilosase disolverá, deteniéndose los fermentos al alcanzar la segunda ramificación. Este
(20) Baldwin. Bear y Rundle. Amer. Ch. Soc., 1944. 66-111.(21) Bates, French y Rundle, Id. 1943. 65-142.
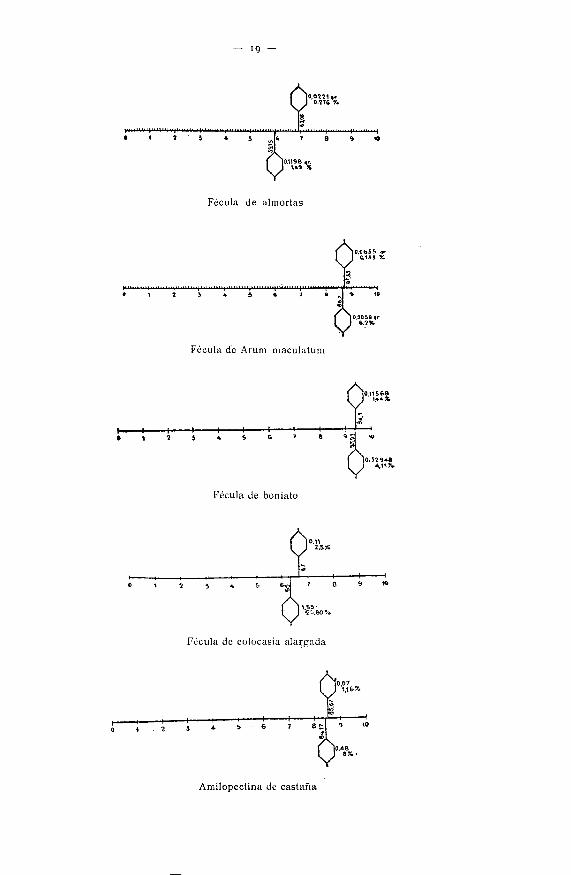
— ig -
» t » J » 5 7 a 9 io
Fécula de almortas
» 1 J 5
Fécula de Arum maculatimi
Ô'10.11 SS»1.«»*
H 1-• 1 2 1 * 5 6 ' » » ;
00,3794*
<•"*-
Fécula de boniato
7 O 9 *>
Fécula de colocasia alargada
10,07' 1.16*.
5 6 7 8£ 9 'O
Amilopectina de castaña

trozo de anulosa disuelto generalmente es pequeño, pero distinto para cada ami-íopectina ; queda todavía algo insoluble, que es la dextrina III, cuya molécula tieneun grupo final en arborescencia separable por la x-glucosidasa.'Llega un momen-to después de esta actuación alterna de amilasas y de a-glucosidasa en que no hayforma de disolver más; lo insoluble es una dextrina residual.
Para exponer los resultados obtenidos con esta técnica ideé una forma gráfica:una línea recta de 10 centímetros dividida en décimas. La primera acción de laamilosa se detiene al alcanzar una cadena ramificada (véase el gráfico de • almor-tas) ; del extremo no reductor a la ramificación median 59,75 ; si se hacen equi-valentes a gramos, se ha sacarificado 59,75 % g- >' e^ residuo empieza su moléculapor una ramificación que hay que soltar, y como se halla en posición 1-6 eâ indis-pensable acudir a la z-glucosidasa, fermento que específicamente suelta esa cade-ria; el residuo ha perdido de peso, que es el equivalente a esa cadena ramifi-cada y representa el 1,49 por 100 de} total. La dextrina insoluble empezará conuna cadena recta como la de amilosa y, por tanto, se la somete al influjo de lasamilasas ; éstas sacarificarán una cantidad, que en el gráfico se señala entre 59,75y 69,08 ; se detiene e] fermento en 69,08 porque encuentra una nueva ramifica-ción; queda así una dextrina, la III, cuyo comienzo de molécula es ramificado;esta cadena es desgajable por la x-ghicosidasa, pero pssa poco, 0,276 del total ;separándola, resta la dextrina IV, que .ya no es transformable por los fermentosindicados.
Por este procedimiento se pueden cotejar varias féculas, y así se observa, porejemplo, que la de Arum maculaium tiene dos ramificaciones: una en el 86,7 }'otra en el 87,35 ; es decir, que se hallan muy próximas entre sí, pero además laprimera representa 6,2 por 100 del total y la segunda 0,133; por consecuencia, esdiferencial el lugar de la inserción y el peso de la cadena ramificada. Compa-rando los demás gráficos, se perciben las diferencias expresadas1 en estos ejemplos.
He aquí contestada, al menos en parte, una pregunta que se hacían BatesFrench y Roux, sospechando la no homogeneidad de la ramificación y la posi-bilidad de distinguir unas amilopectinas de otras por isomería y por la magnitudde la cadena.
Un problema interesante ha planteado la síntesis in vifro de Hanes, porque elcuerpo obtenido in vivo está constituido por dos factores, amilosa y amilopectinay el resultante de la síntesis in vitro está exclusivamente formada por amilosa.Aparte del asunto pertinente a la longitud de la cadena recta de amilosa, el de laramificación obra evidente de un enzima, como se ha indicado, tiene un origenmás antiguo, en la ontogenia de la planta. La fécula de maíz waxy está exclusi-vamente integrada por arni'opectina ; la de guisante (Pisum sátivum) (22) al con-trario, le forma casi en su totalidad la amilosa, pero la especie-tipo probable, elP. arvense sintetiza una fécula con cifras próximas a las normales de amilosa 29
(22) Peat, Bourne y NichoUs. Nature, 1948. 161-206.

— 21 —
y amilopectina 65,8 por 100. ¿Qué factor induce a la ramificación completa, ycuál la evita? Es un problema de genética que aun se halla en vías de solución.Es antigua la idea de que los.cromosomas son fermentos, mas si no fuera absolu-tamente exacto en estos ejemplos que acaban de señalarse, marcaría una relación es-trecha entre ambos. La idea se va modificando y queda en pie un hecho 'sustan-cial: «cada gene separado es responsable de cada fase de la biosíntesis enzímicade un cierto producto». Puesto que el precursor in vitro del compuesto es capazde sintentizar otras substancias que las constituyentes de los tejidos, un gene escapaz también de desplazar una potencia química estérica, la cual se encaminahacia una estructura típica sencilla.
Zachneister y Vent (23) han sugerido el criterio de que en la estimación delgrado de selectividad de este mecanismo esterizante de algunos genes, se observaque uno de estos abandona el camino puramente químico, para ir por el estéreo-químico. Este aspecto estereoquímico expresado en la idea de los investigadoresaludidos afecta claramente a los compuestos ds-tmns, muy abundantes en la na-turaleza, y muy en particular en la serie de los carotenoides. La naturaleza buscasiempre la forma estable : ejemplo muy significativo es del licopeno del tomate,en que se producen las formas as mejo, cristalizables y más resistentes al calor,que las trans afectas todas exclusivamente.a un gene.
No es un despropósito afirmar que se podrán descubrir los genes responsablesae configuraciones estereoquímicas definidas, y que otros genes convertirán esaestructura en otra más estable. Si los hechos Se deslizan en tal dirección, es/ for-zoso convenir que la genética conduce^ a la producción de sustancias estables, yque la amilopectina en las plantas y el glucógeno en las series zoológicas represen-tan la forma más estable, que los fermentos no pueden hidrolizar súbitamentesin riesgo de combustiones peligrosas.
En este terreno quizá pueda hallarse la importancia biológica de la ramifica-ción: el glucógeno es mucho más ramificado que la amilopectina y se halla pre-cisamente como material de reserva almacenado en el hígado y dispuesto a unahidrólisis más lenta que la amilosa.
Piero esta capacidad, así expresada, no se armoniza con la idea clásica de quelos compuestos de cadena arborescente sean más estables : la consecuencia principalque deriva de la ramificación es la distinta aptitud para fosforilarse la amilosa res-pecto de la amilopectina. Esta es mucho más apta para convertirse en el ester fosfó-rico que su congénere de cadena recta y como es sabido los esteres fosfóricos nece-sitan el enzima que gradualmente les vayan desdoblando ; la fosfatasa.
La diferencia en las cadenas alcanza también aplicaciones de las féculas ; esnotoria la diferencia de las harinas panificables de distintas regiones, debida a lasdiferentes proporciones de los dos factores constitutivos' de las féculas. Aunqueel fenóm)?no no es absolutamente claro se prevé que si a la masa de pan se le
(23) Nature 1948. 162-848.

— 22 —
agrega harina de maíz Waxy, constituída principalmente por amilopectina, lacapacidad de tumefacción de la masa desciende con rapidez. También se han ob-servado cambios en la amilopectina durante la conservación del pan (24).
Síntesis de las féculas
Se ha visto cómo en la función clorofílica se producen féculas y se han exa-minado los mecanismos, todavía oscuros, a ella conducentes. No se pretende sig-nificar con lo expuesto que sólo las plantas poseedoras de 'clorofila están capacita-das para esa función sintetizadora, la primera de las maravillas de la vida ve-getal ; las algas unicelulares poseen igualmente ese poder sintetizador y algunasbacterias purpúreas se asemejan a ellas en este aspecto ; el pigmento que elaboran estan sensibilizador como pueda serlo la clorofila a los efectos de la síntesis de polisa-cáridos. Se ha visto comò en la oscuridad producen, fécula las plantas verdes yexperiencias recientes de Aschner, Mager y Leilovoitz (25) con .levaduras cap-suladas, han demostrado que tanto en el cuerpo celular, como en el medio enque viven, contienen un polisacarido coloreable con el yodo y que es amilosa, yHehre, Carlson y Neil (26) han visto como el Corynebacterium diftericum, en unmedio con ester glucosa-1-fosfórico, produce una mezcla de amilosa y amilopec-tina, es decir, una fécula semejante a la sintetizada por las plantas de gran por-te. Para lograr esta síntesis de carbohidratos, atribuida durante mucho tiempoal misterioso poder de la clorofila, no es indispensable esta materia colorante he-matínica ; es necesario un fermento, que al catalizar la reacción con éster glucosa-1-fosfórico,-separe el ácido y enlace moléculas de glucosa en cadena linear, o seaformando amilosa, fermento que descubrió Hanes (27), en la patata y en el guisante,y que el laureado matrimonio Cori (28) descubrió también en el hígado< y en el cora-zón de varios animales y al que bautizó con el nombre de fosforilasa, que requierepara su actuación el concurso de un polisacarido análogo al que va a fabricar, perono otro sencillo, lo que equivale a admitir Ja necesidad de un germen molecular.
La circunstancia de mayor relieve en la intervención de la fosforilasa es lanecesidad de lo que llamo un germen molecular, que en el idioma de los autoresse conoce con el nombre de starters (iniciadores), porque no ha de ser cualesquierahidrato de carbono el que sirve de germen ; por fuerza ha de ser un polisacaridoramificado, amilopectina, glucógeno, dextrinas residuales, a excepción de la alfade Myrbäck, que es un trisacárido, y desde luego ningún sacando sencillo, ni si-quiera un bisacárido con uniones 1 : 4, ni aun la propia amilosa sintética.
(24) Noznick, Mervit y Geldes: Cereal Cherr.., 1946. 23-297. Abstracts, 1946. 40-4.812 (4).(25) Nature, 1945. 156, 295.(26) Abstracts, 1948. 42, 1.982.(27) Procee. Roy. Soc. London B. 1940. 129, 174.(28) J.-Biol. Chemistry, 1942. 150, 447.

— 23 —
Después los Cori (29) encontraron un hecho de excepcional importancia, cuales, que si a un preparado de músculo, rico en fosforilasa se le mezcla extractode corazón o de hígado que.no posee capacidad'sintetizante, convierte el esterglucosa-1-fosfórico en gluclógeno, sustancia de cadena ramificada y representanteen el organismo animal, de la amilo pectina. Este hecho les., sirvió a los autoresde punto de partida, para relacionarlo con otro de ocurrencia vegetal y es la ne-cesidad de dos fermentos, uno para cadena recta y otro para cadenas ramificadas,porque en el maíz llamado waxy, sólo existe el sintetizador de amilopectina, pues-to que la fécula de ese cariópside contiene casi exclusivamente este-componenteramificado.
Al año inmediato el químico inglés Ha worth (30) aisló el factor termolabil Q.,capaz de sintetizar a expensas del esters glucosa-1-fosfórico, no ya amilasa de ca-dena recta, sino amilopectina de cadena ramificada, cuando actúa asociado a lafosforilasa de Hanes, a la que llama factor P. Este fermento Q es un raro fermentodotado de la aptitud para formar cadenas ramificadas, sobre la linear fundamentalde la amilasa; por tanto, crea ligaduras 1-6 a diferencia del de Hanes, que porsintetizar amilosa ¿rea ligaduras 1-4, pero además posee otra aptitud, la de hidro-lizar amilosa, es decir, romper uniones 1-4, sin llegar a la fase de reducción ; es,por tanto, un tipo especial de amilasa., incapaz de producir azúcares reductores ya cambio, torna la amilosa coloreable en azul por el yodo en amilopectina colo-reabls en rojo violáceo. Esta afirmación, debida a Bourne y Peat (31) se presta acomentarios, porque las anteriores reacciones sintéticas se consuman con esterglucosa-1-fosfórico, y esta puede ocurrir sin intervención del residuo ácido, ni de fos-fatos inorgánicos. Los autores representan la síntesis de este modo :
Ester glucosa — i — fosfórico
| f factor P
Pseudamilosa (formada por cadenas de 20 unidades de glucosa)
//O-
Amilopectina Amilosade cadenas ramificadas de de cadena recta de20 residuos de glucosa cada 8o unidades de glucosa
una
Más adelante se estudiarán las dextrinas residuales que quedan como produc-tos feculentos intrasformables por las amilasas a v ß. El hecho ha sido indiscuti-ble hasta ahora, en que Bernfeld y Montemedian (32) han anunciado eí descubri-
(29) J. Biol. Chemistry, 1943. 151, 07.(30) Nature, 1944. 154, 236.(31) J. Ch. Society, 1945. 877.(32) Nature, 1948. 162, 297.

miento por ellos de un fermento existente en la patata, que en presencia de salesminerales colabora con ß-amilasa en la solubilización de las dextrinas, A la excep-cional cualidad de este cofermento, hay que agregar otra, la de convertir la ami-losa en amilopectina y aun en glucógeno, cuando actúa juntamente con lar fbsfo-rilasa de Hanes. Gomo los análogos es reversible, crea ligaduras 1-6 y -las sueltaigualmente: de modo semejante el factor Q de Haworth, no suelta; ligaduras in-ternas, al punto de- que la viscosidad de la pasta de almidón no se modifica porel influjo de la iso'fosforilasa, que es el nombre con que los autores distinguen estecurioso enzima.
El efecto sintético de la isofosforilasa lo imaginan los autores de esta manera.
N — O — < f /~~O—\ /O "i" ester glucosa-unifosfórico = A. fosfórico
O j~0 ~~ O
CHjOH CH2OH CH2OH
-f-O-o-O-o-j O I O
CHoOH CH2
o-O~°"o íCHjOH
Obsérvese que este fermento no isomeriza como el Q de la escuela inglesa ; nosuma unidades de glucosa en 1 : 4, agrega unidades de glucosa en 1 : 6, es de-cir, que crea cadenas ramificadas sin isomerización previa.,
La síntesis consumada por los Cori podría ser una pauta de la posible manerade unir las moléculas de núcleoproteidos para producir los virus responsables delcáncer.
Veamos ahora cómo son los productos de síntesis y en qué se diferencian de losnaturales. La apariencia es igual e idéntica también la coloración con yodo,azul en la amilosa y rojo violáceo en la amilopectina. El primer producto aisladopor Hanes ofrece la característica de amilosa, más no siempre es el mismo con-siderado como individualidad química : su peso molecular depende del activadory del tiempo que se emplee para las síntesis. Con grandes cantidades de activa-dor, las cadenas son cortas, es decir, que la magnitud molecular e9 pequeña ; portanto, sie irán soldando unidades de glucosa, después de separarse el ácido fosfó-rico hasta una media de 20, como se dijo al principio, pero de las medidas visco-simétricas efectuadas por Foster y Hixon con disoluciones de etilenodiamina sededuce que la amilosa de patata se halla integrada por quinientas unidades de glu-cosa, mientras que la sintética sólo la componen ochenta y cinco (33).
El trabajo de los Cori se encaminaba esencialmente al estudio de la metamor-
(33) Amer. Ch. Soc., 1943. 65, 618.

— 25 —
fosis de los hidratos de carbono en animales y lo que aquí se utiliza es la seme-janza de la síntesis de la amilopectiria, factor constituyente de las féculas con ladel glucógeno, polisacárido, también ramificado, que los organismos zoológicos re-quieren para proporcionarse por su combustión el calor indispensable a las demássíntesis que tengan cualidad endotérmica. La síntesis del glucógeno, sustancia más.ramificada.que la amilopectina, puesto que tiene 9 por 100 de grupos finales, esevidentemente de mayor complicación, porque en realidad aquél se construye aexpensas del ester ghicosa-6-fosfórico, y por tanto, es imprescindible la existenciade un fermento, una mutasa, que provoca la isomerización del ester fosfórico en6 en ester fosfórico en 1. Es, en definitiva, una reacción de trarisfosforilación,porque se produce con glucosa y trifosfato de adenosina, sistema formador deléster glucosa-6-fosfórico, interviniendo la hexoquinasa, que trasfiere ci ácido fos-fórico del trifosfato a la glucosa.
III
SEPARACIÓN ANALÍTICA DE LOS FACTORES CONSTITUYENTES DE LAS FÉCULAS : ACETI-LACIÓN, METILACIÓN, GRUPOS FINALES
Separación de amilosa y amilopectina.
En la conferencia anterior se expusieron las propiedades de cada uno de esosfactores, y son justamente aquellas utilizables para separarlos con fin analítico.
La amilosa se disuelve en agua, cualidad fundamental atribuida a las cade-nas rectas de moléculas de glucosa, unidas por enlaces 1-4. En tal cualidad sebasa un grupo de procedimientos para aislar amilosa ; desde el método al frío pre-conizado por Ling y Nangi (34) y por Hixon y Caldwell (35) hasta el de solubilí-zación en agua a temperaturas próximas a 80°, pero inferiores a la disgregacióndel grano de fécula, según K. H. Meyer, Bernfeld y Brentano (36) se han seguidotodas las técnicas conocidas para obtener amilosa, en lo posible desprovista deamilopectina •
El camino del frío, es decir, del agua a 0° es el que produce resultados másconstantes, porque en el del calor hay variaciones debidas a la textura del granoy a la posiblidad de que elevándose aquél a más de 80° se escindan las cadenaslaterales de amilopectina, quedando convertidas en las rectas de amilosa, y porconsecuencia, aumenta el tanto por ciento de amilosa a expensas del de amilo-pectina (37).
Là solubilidad en hidrato de cloral al 33 por 100 es la base de las técnicas de
(34) Chem. Soc. 123, 2.666.(35) Amer. Ch. Soc., 1941. 63, 2.876.(36) Helv. Ch. Acta, 1940. 23, 846.(37) Santón!. Thèse de Marseille, 1939.

cuantitativa y aislamiento de amilosa. Se practica en frío y agitando prolonga-damente la fécula con la disolución de cloral: queda insoluble amilopectina pon-
derable ; además, puede verse la amilosa al polarimetro utilizando su (a)() == 152° en el doral.
La electroforesis también ha sido ¡empleada y sus resultados van paralelamen-te a los obtenidos por disolución en agua.
En las técnicas fundadas en el empico del agua como disolvente de la amilo-sa queda amilopectina de residuo ; en cambio, se han implantado otras técnicasbasadas en la producción de compuestos insolubles de amilosa en alcoholes debajo peso molecular, preferentemente butilico y amílico (38) y con fenoles y c<cla-noles, especialmente con timol (39), así como oon nitroetanos (40), y en general,con sustancias donadoras de electrones. La amilosa así aislada es cristalizable, deestructura helicoidal y resulta en cantidad mucho mayor que con los otros pro-cedimientos, lo cual hace dudar de la exactitud de éstos, porque los números ob-tenidos en una serie de féculas en mi laboratorio por R. Arrans tienen gran cons-tancia, tanto tratándose de alcoholes butilico y amílico, como de timol (41).
He aquí algunos ejemplos en demostración de lo que acaba de exponerse.
Fécula de Oxalis . ..
Fécula de Arum ... .
Amilosa ....Amilopectina.
Amilosa ....Amilopect ina.
At agua
23.4768,86
'7,5°•2.5°
Al butanol
71,65
25
7518,67
Al pentanol
788,89
8o13.34
Al timol
85IO
7524
Los números obtenidos con otras féculas son de este orden y parecen inverti-dos respecto de los que suministra el método al agua a 75-80".
Acetilación
La unión de las moléculas de glucosa en las féculas se ha visto, en conferen-cias anteriores, que puede realizarse de dos modos: por enlaces 1-4, que son losmás frecuentes, y por enlaces 1-6, que sólo alcanzan la cifra de 4,6 por 100 de losenlaces totales. El monómero constituyente de las féculas es capaz de fijar cincoacetilos, por considerarse que tiene 50H, pero la fécula no esterifica más que tresoxhidrilos, y por eso, desde la época de Schutzemberger, es conocido el triacetil-almidón. Pasaron muchos años hasta que Pregel, en 1905, obtuvo el mismo estertriacético que Schutzemberger, que reafirmaron Bergmann y Kcnsee en 1928;.desde esa fecha hasta el momento actual se han acetilado las féculas más co-
(38) Schor. Amer. Ch. Soc., 1942. 64, 2.957.
(39) Haworth, Peat a. Sagrott. Nature, 1946. 157, 19.(40) Whitler a. Hubert. Amer. Ch. Soc., 1945. 67, 1.161.(41) Tesis doctoral. Madrid, 1949.

— 27 —
rrieiites, obteniéndose valores próximos al teórico, que es un 44,8 por 100. Enalgunas muestras no frecuentes los valores han excedido al conjunto, por cir-cunstancias todavía no puntualiaables, dependientes de la facilidad o dificultaden la acetilación. Hace años Karrer y Kraus (42) se referían a féculas difícilmen-te acetilables, y a plazos largos y cortos de acetilación. Al decir de estos químicossuizos la acetilación depende en gran! manera de la estructura micelar de la sus-tancia acetilable, porque la liquenina seca ni fermenta ni se acetila, pero sometidaal influjo sucesivo de alcohol y del éter para ablandarla se acetila en pocos minu-tos. Conducta semejante ofrece la celulosa.
En nuestro laboratorio R. Arrans (43) ha encontrado una, la de Manihot, malacetilable por el Franchimont y excesivamente por el Barnet ; los químicos ingle-ses Haworth, por ejemplo, aluden a métodos drásticos para acetilar féculas quefijan penosamente acetílo. Las fácilmente acetilables, acaso sufren los efectos dela hidrólisis por el ácido acético producido, dejando libres unidades de maltosay maltotriosa, que al acetilarse adquieren un peso de acetilo mayor que la fécula,y de ahí resulta al saponificar un número superior al de triacetil derivado, quees 44,8 por 100: es lo ocurrido con el Arum Maculatum (44), para lo cual el mé-todo de Barnet es excesivo, porque el número de acetilo introducido fue 56 por100, con la Coló casia Antiquarum (45), en que la cifra de acetilo es 50 por 100y con la Manihot 55,73 por 100. Las obtenidas por R. Olalla en otras féculas sonnormales. (Tesis doctoral 1944).
La técnica de Barnet no es por tanto inofensiva para la estructura del com-puesto acetilable, hecho que conocieron Higginbotham y Richardson (46) des-pués de desacetilar las féculas esterificadas y de averiguar en ellas el índice decobre y la viscosidad. El índice de cobre más alto y la viscosidad más bajas sonproducidos por esteres triacétkos de amilosa y amilopectina, sintetizados con lastécnicas fundadas en el empleo de cloruro de sulfurilo.
Resulta, pues, para algunas féculas un método violento, por separarse gruposreductores con. mayor número de oxhidrilos acetilables que la fécula.
Cuando la acetilación no fue satisfactoria se empleó el olvidado método deFranchimont, que desempolvé en 1920, utilizándolo para cuantitativa, generali-zándose después gracias a la tenacidad que puse en su difusión, por la senci-llez que representa y por la posible generación del anhídrido acético (47). Lo apli-qué hasta en el análisis de esencias con buen éxito y también al de féculas lohizo R. de Mingo en mi laboratorio (48).
(42) Helvética Ch. Acta, 1929. 12, 1144.(43) Tesis doctoral. Madrid, 1949.(44) O. Fernández y Eva Martínez. R. R. Acad. Ciencias. Madrid, 1945. 39, 401.(45) O. Fernández y Rosario L. Larrañeta. Id. 1947. 41, 515.(46) J. Chem. Soc. Ind., 1938. 57, 234.(47) O. Fernández y N. Luengo. An. So. Españ. Fis. y Quím., 1920; 18, 158.(48) Tesis Doctoral. Madrid, 1933.

La acetilación con anhídrido acético y piridina, que es la clásica de Verley yBolsing, no suministra mucho más éxito en nuestros problemas; en cambio, la.acetilación con iguales reactivos, pero dispersando la fécula con formamida, se-gún Carson y Maclay (49) y operando en frío (50) es técnica recomendable.
Resultados consegu-'dos
Hemos acetilado fécula, amilosa y amilopectina de diferentes especies vegeta-les ; salvo pequeñas variaciones en el poder rotatorio, los números que se hanobtenido son iguales en todas las muestras analizadas.
[a]D = Acetilamilosa próximo a 170" y alguna vez excede (colocasia).Acetilamilopectina 165°, número próximo a 174 grados, que es el
aceptado por varios investigadores en (disolución clorofórmicaal 1,33 por 100).
Películas de acetilamilosa, adaptables y blandas.Películas de acetilaminopectina, frágiles y fácilmente rompibles.
Valor en'acetilo del ester acético: amilosa 42 — 44 por 100. Amilopectina: ex-ceptuando la colocasia, la Castanea vesca y el manihot los valores obtenidos.sonlos normales 44 a 44,5.
Metilación
Lógicamente la •metilación conducirá a resultados idénticos a la acetilaciónporque si existen tres oxhidrilos acetilables debe existir el mismo número deoxhidrilos capaces de reemplazar su hidrógeno por metilo. El éxito no acompañóa los primeros intentos de metilar. Cuando Karrer, con la finalidad de cotejar losproductos de metilación en la maltosa y en la fécula metilo con yoduro de metiloy óxido argéntico, obtuvo un ester metílico con 32,6 por 100 de metoxilo, núme-ro bajo para la fécula, comparado con el teórico 45,6, porque así se revelan 2 OH,ciiando según la acetilación deben motilarse tres. Números oscilantes entre 32 y33 por 100 obtuvieron Irivine y Mac Donald con los mismos agentes. Procediendocon esteres acéticos de la fécula y con sulfato de metilo, reactivo muy usado enla serie fenólica, logró' Freudenberg resultados que no alcanzó Staudinger, inicia-dor de la metilación con los esteres acéticos; saltó a 39 por 100.
Quedaba por vencer algún obstáculo, puesto que los reactivos constantemen-te habían mostrado su eficacia ; quizá aquí el más importante es la disgregaciónde los conjuntos micelares para ponerlos en contacto con el sulfato de metilo •; ensegundo lugar una temperatura más alta cuando no se proceda con esteres acé-ticos para que colabore con la agitación, y .en tercer término sostener fuerte alca-nilidad durante el tiempo que se emplee en la técnica. Por tanto, es aconsejableproceder a 80 grados, agitar .:n forma que se consigan 700 vueltas por minuto y
(49) Amer. Che. Soc., 1946. 68, 1.015.(50) A. Potter y. W. Z. Hassid. Id. Id., 194S. 70, 3.774.

— 29 —
alcalinizar con sosa de 60 por 100. En nuestro laboratorio hemos seguido con va-riantes pequeñas la técnica de Meyer, Wertheim y Bernfeld (51), que no evita enabsoluto la formación de productos mucilaginosos, pero se alcanza la cifra de42,36. De ordinario, sale el éter de 36 y el producto, por nueva mediación, adquie-re el 6 por 100 que le resta.
La metilación teórica se consigue con un procedimiento que no está al alcancede todos los laboratorios, y que se funda en el empleo de amoníaco1 seco líquidoy de yoduro de metilo con sodio a temperatura de — 70°.
¿Se puede confiar en el producto obtenido con las técnicas usadas a base deálcali fuerte, 80o1 y su'fato de metilo? Honradamente hay que confesar que no.•Así como al acetilar se aludió a la drasticidad de ciertos procedimientos que) mo-tivaron el juicio desagradable de Higginbotham y Richardson, acerca del resul-tado en la metilación, también hay criterios desfavorables; en la acetilación concloruro cíe sulfurilo se va más lejos de lo conveniente, se sueltan cadenas queadquieren más acetilos que el producto empleado ; en la metilación también hayextra por la ruptura de cadenas. Está bien averiguado por trabajos de Hirts yJoung de una parte, y de Avery de otra, que a medida que la metilación avanza,el peso molecular del derivado metilado disminuye. Del último de. los autoresmencionados dice una referencia (52) que el almidón metilado 17 veces descien-de en su peso molecular hasta 38.000 y se admite como próximo para el naturalla cifra de 400.000, es decir, que se ha degradado 10 veces.
Esta fragmentación de la molécula primitiva, por la influencia de los agentesmediantes tiene gran importancia, porque destruye las esperanzas que se forjaronsobre la metilación, mucho mayores que la acetilación.
Sustituir metilos en una fécula es evidentemente hoy de mucha más trascen-dencia que emplazar acetilos, porque conduce al conocimiento de lo que se llamagrupos finales, o sea unidades de glucosa situadas en extremo no reductor y encadenas laterales a lo que se debe la afirmación, de que el número de cadenaslaterales está dado por el de grupos finales ; por tanto, la determinación de estosgrupos está vinculada a la metilación y por otra técnica complementaria sucedidade hidrólisis, separar cuantitativamente las metilglueosas, bi, tri y tetra, que debensoltarse en la disociación.
Sobre la, fécula y sus factores acetilados se practica la metilación con sulfatode metilo y después utilizando el método de E. Fischer para conseguir el metil-glucósido, con alcohol metílico y clorhídrico se logra la mezcla de.los dos metil-glucósidos A y B. Esta compleja molécula se hidroliza produciéndose cantidadesvariables de glucosas en-grado distinto de metilación. En una cadena recta deamilosa después de mediada, por hidrólisis, se soltarán la unidad final de glucosacomo tetrametilglucosa y las demás como trimetilglucosa.
(51) R. Olalla. Tesis doctoral. Madrid, 1944:(52) Cárter J. Ch. Soc., 1939. 670.
- 3o —
H OCH3 CH2OCH3
I I Ir- n ' r r
HÇ O * C C C O
/\ \ H /I l\ ur / H \ f/
H / OÇH, H \ /H
Ç X O C H , Hy c\o/\H / X0 . X|CH3° C C C O C C
I I I I IH ()CH3 CHjOCHj H OCH3
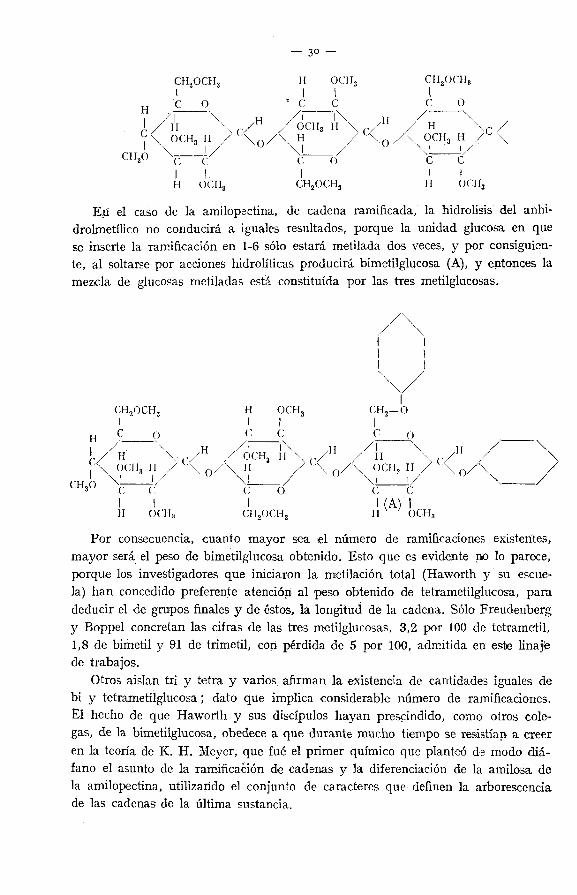
Eii el caso de la amilopsctina, de cadena ramificada, la hidrólisis del anhi-drolmetílico no conducirá a iguales resultados, porque la unidad glucosa en quese inserte la ramificación en 1-6 sólo estará metilada dos veces, y por consiguien-te, al soltarse por acciones hidrolíticas producirá bimetilglucosa (A), y entonces lameada de glucosas metiladas está constituida por las tres metilglucosas.
CH30 \c
Por consecuencia, cuanto mayor sea el número de ramificaciones existentes,mayor será el peso de bimetilglucosa obtenido. Esto que es evidente no lo parece,porque los investigadores que iniciaron la metilación total (Haworth y su escue-la) han concedido preferente atención al peso obtenido de tetrametilglucosa, paradeducir el de grupos finales y de éstos, la longitud de la cadena. Sólo Freudenbergy Boppel concretan las cifras de las tres metilglucosas, 3,2 por 100 de tetrametil,1,8 de bimetil y 91 de trimetil, con pérdida de 5 por 100, admitida en este linajiede trabajos.
Otros aislan tri y tetra y varios afirman la existencia de cantidades iguales debi y tetrametilglucosa ; dato que implica considerable número de ramificaciones.El hecho de que Haworth y sus discípulos hayan prescindido, como otros cole-gas, de la bimetilglucosa, obedece a que durante mucho tiempo se resistían a creeren la teoría de K. H. Meyer, que fue el primer químico que planteó de modo diá-fano el asunto de la ramificación de cadenas y la diferenciación de la amilosa dela amilopectina, utilizando el conjunto de caracteres que definen la arborescenciade las cadenas de la última sustancia.

— 3' —
Todavía se discute la presencia de la bimetilglucosa, más que como resultantede la ramificación, como producto de desmeritarse la trimetilglucosa por el alcoholclorhídrico. Flood, Hirst y Jones (53) sostienen que se han hecho todos los es-fuerzos imaginables para evitar la desmetilación, incluso metilando la fécula de-gradada ; a pesar de ello, sale predominante la 2-3 bimetilglucosa, lo cual como)se ha indicado es lógico, sin hacer intervenir la desmetilación de la trimetilgluco-sa, porque las ramificaciones no son tan numerosas como para que cada glucosatenga una inserción en 1-6. Mas la opinión de Myrbäck también es favorable ala tesis de la bimetilglucosa, por cuanto lo afirma en esta frase: «si la moléculaes ramificada, al metilarse y luego hidrolizarse, producirá además de trimetil yíetrametilglucosa, bimetilglucosa y, probablemente, en cantidades iguales de biy tetrametil derivado» (54).
Entre los asuntos que se agrupan en derredor de-la metilación conviene des-tacar dos muy instructivos. ¿La entrada de metilo en las¡ cadenas rectas o arbo-rescentes modificará su estructura? Se sabe que la unión de las moléculas de glu-cosa ss realiza en posición 1-4 de B-glucopiranosa o en 1-6 para las de amilo-pectina. Parece que los rayos X revelan que las uniones se mantienen de igualmodo, y por consiguiente la unión es la misma ; no> hay trasposición de enlaces.Haworth dedujo de la metilación total el número de grupos finales después depesada la tetrametilglucosa 2-3-4-6, 'pero la audacia investigadora le indujo a cal-cular el número de unidades de glucosa de 24 a 30, número aceptado por muchosinvestigadores (es algo menor de 150 para Wolfram) ; para la amilosa es corrien-te la dfra ; en cuanto a la amilopectina, el número de glucosas es de 283 paraHess y Kranjs (55).
Indudablemente los grupos finales evaluados con la metilación total son losno reductores ; hasta hoy se había creído que el enlace polimerizante de la mo-lécula de glucosa tendría como consecuencia extinguir el poder reductor! del poli-sacárido, y la realidad es que por los métodos corrientes de investigar maltosa oglucosa no se ve la reducción del Fehling; no obstante, K. Meyer ha demostradola existencia de grupo aldehídico, que al oxidarse con óxido argéntico da ácido,con reducción; un solo grupo reductor en una masa grande de sustancia no re-ductora e insoluble pasa inadvertido a la técnica usual.
Aunque el procedirriento de metilar y formar después el metilglucósido, paraseparar luego de la hidrólisis por destilación al alto vacío) las-metilglucosas (0,02milímetro y colector Widmer), ha adquirido carta de naturaleza, hay que con-venir en que todas sus fases son largas y delicadas ; se prestan siempre al error,variable entre 5 y 10 por 100. Por tal motivo, no se han mostrado remisos los
(53) Nature, 1947. 160, 899.(o4) The Swedvyerg, Libro homenaje Upsala, lí)4:j. 474.(55) Chera. Abstracts, 1941. 35, 3.612.

investigadores en este campo, en inquirir métodos más fáciles, y seguros que el ini-ciado plor Haworth.
Entre las mejoras al procedimiento destacan las siguientes: la fosforilación dela trimetilglucosa con cloruro de fósforo en presencia de piridina, para formar lasal bárica del'ester fosfórico. Esta técnica, de la que son autores Hess y Neu-mann (56), ha sido censurada por Leckysck (57) por insuficiente, puesto queaparte de no fosforilarse por entero la trimetilglucosa, se pierde tetrametilglucosa.Carezco de experiencia en fosforilar la trimetilglucosa, pero sí la tengo en cuantoa glucosa se refiere y reconozco que los rendimientos son bajos. Es posible quese. fosforile con más facilidad y mejores rendimientos la trimetilglucosa, que laglucosa o el almidón, porque no tiene más que un oxhidrilo fosforilable.
La berizoilación en piridina del metilglucósido de la trimetilglucosa ha sidopropuesta por Freudenberg y Boppel (58).
La separación con benzeno de la tetrametilglucosa tampoco parece muy exac-ta, porque los números obtenidos son demasiados altos, por ir mezclada aquellasustancia con trimetilglucosa (59).
El primero de los reformadores fue Wolfram (60), que hace inexcusable lamediación, pero después, de ella se hidroliza en presencia de- etilmercaptan, parasintetizar un mercaptal y evaluar azufre. En este procedimiento no se toma encuenta más que el grupo reductor final, que Wolfram califica de dinámico, porcuya .circunstancia conduce a números tan distintos del de Haworth.
El empleo de mercaptanes en química siempre ha sido desagradable y a elloobedece que el procedimiento no haya encontrado asenso entre los químicos, quese han entretenido en la búsqueda de una técnica más fácil, y he aquí que se hahallado en la oxidación :con ácido peryódico.
Fúndase en una propiedad descubierta por Jackson y Husdon (61) en el almi-dón. Cuando éste se oxida con ácido peryódico en acético, se obtiene 33 por 100de glioxal y 22 de d-eritrosa. Esto supone que existen dos oxhidrilos contiguos, yes cierto, porque en una unidad de glucosa integrante de un oligosacárido los OHen 2-3 se encuentran como en los g-licoles a, que se sueltan por este enlace de modoanálogo al que sigue
CH OH COI ' + 2l04H = | _L 2lO,H -f zlbOf.H.OH COI I
(56) Berichte, 1937, 70, 710.(57) Id., 1938. 71, 829.(58) Id., 1938. 71, 2.505.(59) E. L. Hirst y G. F. Joung. Chem. Soc., 1938. 1.247.(60) Amer. Ch. Soc,, 1941. 63, 1.336.(61) Id., 1937. 59, 2.049.

— 33 —
De tal hecho dedujeron Flood y Hirts (62) que si la cadena ramificada se si-túa corno es natural, en 6, la oxidación recae sobre los C 2 y 3, y por tanto, des-pués de hidrolizar el residuo de azúcar que implica la ramificación, no es recupe-rable como glucosa. Si la cadena se emplazase en 2 o en 3, no quedaría grupo glicol,y el oligosacárido sería inatacable por el ácido peryódico, y por consecuencia, lue-go de la hidrólisis, como la supuesta cadena queda inltacta, el residuo será recupe-rable como glucosa
CFLOll
HOH
C CI I
(3) H «H (2)
En consecuencia, el procedimiento sirve para determinar grupos finales no alde-hídicos, porque sólo las cadenas enlazadas en 6 y la final auténtica ds la cadenarecta sufren la oxidación con el ácido peryódico.
Antes de producirse glioxal, hay que hidrolizar lo que se denomina aldehidopolimèrico, el cual es oxidable por el bromo en doä ácidos, que después de esta
hidrólisis se evalúan con sosa —— V. C. Barry (63) ideó esta técnica para deter-
minar el número de grupos finales de la laminarina.
IV
DEXTRINAS RESIDUALES. ELEMENTOS EXTRAÑOS A LOS FOLISACÁRIDOS
Queda expuesto en conferencias anteriores que el producto de la degradaciónenzífnica de la amilopectina por la mezcla de amilasas a y ß y por la a-glucosidasade la levadura de cerveaa es una dextrina residual. Es la diferencia sustantiva res-pecto de Ja amilosa, quei sólo por la mezcla de amilasas se sacarifica y disuelvetotalmente, a menos que tenga alguna ramificación, en cuyo caáo .deja un pequeñoresiduo insoluble, por ejemplo, la de Arum maculatum. De ordinario hemos con-seguido la cuarta, pero alguna vez hemos llegado a la sexta dextrina residual.
En la segunda conferencia, a propósito de la síntesis de féculas, se hizo notarun descubrimiento de; Bernfeld y Monltemedian (64), que consiste en aislar un fer-mento de la patata, capaz de colaborar con la amilasa-ß en la solubilidad de lasdextrinas residuales en presencia de sales inorgánicas.
(62) Nature 1947. 160.(63) Chem. Soc., 1942, 578.(64) Nature, 1948. 162, 297.
REV. DE LA REAL ACADEMIA DE CIENCTAS.—1949.

— 34 —
Las dextrinas hidrolizadas con ácidos minerales se sacarifican en su totalidad,produciendo maltosa y un trisacárido (maltotriosa), y en definitiva, glucosa. EIpeso molecular de estas dextrinas es incomparablemente más pequeño que el de lasamilopectinas originarias y aun que las amilosas ; por tanto, las verosímiles decadenas de 30 unidades de glucosa van perdiendo eslabones y quedan reducidas a.una mezcla de tri, tetra y pentasacáridos, susceptibles de aislamiento por precipi-tación fraccionada.
Aunque el problema de las ligaduras de pequeñas unidades es aparentementemás sencillo que el de unidades numerosas de glucosa o de maltosa, habría quedescontar las uniones 1-4 y también las 1-6, porque la acción sucesiva de losdos fermentos, amilasa y glucosidasa, ha debido ser bastante eficaz para soltarlas-Únicamente la velocidad de la hidrólisis puede servir de atenuante, para admitirque no todas las ligaduras se han soltado ; las cadenasi constituidas por pequeñonúmero de hexosas resisten la acción de los fermentos mejor que las integradaspor gran número. Se hidroliza peor un trisacárido que la masa de amilosa, segúnopinión experimentalmente comprobada por Myrbäck. ¿Se podrían acaso aceptarlas ligaduras propuestas por Staudinger? Quizá: los conocimientos que hasta hoyse poseen no se hallan de acuerdo más que con dos tipos de enlaces bien demostra-dos: los 1-4 y 1-6; con todo, es mucho más asequible, el tipo de, enlaces en 1-3que cualesquiera otro de los 15 que asegura Myrbäck (65).
La producción científica relativa a las dex>trinas es abundantísima y falta enabsoluto de coordinación. Cada investigador ha creado sus dextrinas, porque haempleado fermentos diferentes para transformar la amilopectina. Las obtenidas enmi laboratorio son diferentes de las demás, porque en general se procede con ao ß-amilasa y con la mezcla natural de éstas ; mas como estos enzimas sólo sueltanligaduras 1-4, el residuo contendrá ligaduras 1-6, porque se ha dicho en las confe-rencias anteriores que la actividad de la ß-amilasa se paraliza ante un obstáculo:la ramificación de las cadenas; por consecuencia, la dextrina-así producida repre-senta corrientemente el 40 por 100 del peso de amilopectina.
Hemos probado que al actuar la a-glucosidasa o maltasa de la levadura sesuelta la ligadura 1-6 y entonces queda un trozo de cadena recta, idéntica a la deamilosa, que sufre la acción de la ß-amilasa hasta encontrar un nuevo obstáculo,que es .una nueva ramificación ; de este modo se, van achicando las moléculas porruptura de las uniones isomaltósicas (las 1-6), hasta quedar una masa pequeña, endonde quizá existan ligaduras 1-6 o una de esas 15 que presumen diversos quí-micos.
No es el caso de las dextrinas obtenidas con -y. y ¡3-amilasas y glucosidasas elde oíros investigadores, por ejemplo, K. Myrbäck (66), que estudia dextrinas con
(65) J. prakt. Chemie, 1943. 162, 39.(66) Ark. f. Remi, 1948. 26 A n.° 7.

- 35 -
a-amilasa. La acción de este enzima comprende dos fases: la dextrinante rápiday la sacarogénica lenta ; en la primera fase (rápida) sólo desaparece el 17 por 100de fécula, de suerte que la molécula resultante es tan complicada como la del almi-dón primitivo ; queda allí la casi totalidad de cadenas ramificadas ; a estas dextri-nas se las llama a. normales,de grado de polimerización de 6, a diferencia de lasotras a-anormales,con un grado de polimerización de 9-10 y con enlaces 1-4 y 1-6.Con este criterio se obtienen a-dextrinas de la anulosa-, que no son en realidaddextrinas, según el que se va desarrollando en estas conferencias. Se trata, en efec-lo, de. un alto en la actividad del fermento, pero sólo porque éste tiene gran afini-dad para cadenas largas, a las que se combina, inhibiéndose en su verdadera mi-sión, no por incapacidad o por otro tipo de acciones.
El mismo Myrbäck pone en parangón estas conclusiones suyas con las deK. H. Meyer (67), corroboradas por Bernfeld y Studer-Pecha (68), según las cua-les la amilolisis alcanza la cifra de 45 por 100. Si se han de poner en relación losdos guarismos, hay necesidad de inquirir una causa, y ésta la encuentra Myrbäck,en que la amilasa con que trabajaron K. K. Meyer y sus discípulos contiene ami-lasa ß, que es sabido lleva más lejos la hidrólisis que la a. Todavía es posibleaportar algunos argumentos justificativos de la diferencia expuesta : uno es la ve-rosimilitud de retrogradación de la amilosa durante las experiencias, gracias a lacual la molécula experimenta una agregación, en la que se forma un complejo,que aun no ha alcanzado el grado de división submicrónico y resulta inatacable pord fermento. De manera, que la dextrina así engendrada no es más que una féculadesprovista de unas pocas cadenas rectas y arborescentes, porque como ya se haapuntado en otra conferencia, la amilasa a rompe cadenas, pero no empieza como]a ß por el extremo no reductor, quizá por el medio, y por eso se la califica comouna endoamilasa.
Otro argumento es la existencia de grupos polares en la amilosa, que, como seha escrito antes, puede contener fósforo como la arnilopectina ; y finalmente, debeseñalarse la posible presencia en las féculas de cereales y por analogía en otras dediferente origen, de sustancias inhibidoras de la actividad de las amilasas señaladapor E. Kneen (69).
Estos ejemplos ponen de manifiesto la variedad de dextrinas que pueden obte-nerse, utilizando fermentos capaces de realizar hidrólisis' parciales de las amilopecti-nas y aun de las amilosas.
Aparte de todo lo expuesto existen dextrinas que en nada se parecen' a lasgeneradas por amilasas, las que Schardinger (70) obtuvo empleando el Bacilluswacerans y discrepan fundamentalmente de las dextrinas corrientes. Estudiadas
(67) Helv. Ch. Acta, 1941..24, 359.(68) Ibi., Í947, 30, 1.895.(69) Am. Ch. Soc., 1945. 65, 1.247.,(70) Zentralb. Bakt. u. Parasitenk, 1905. 14, 777.

primero por Pringsheim, éste (71) afirmó que los factores integrantes de aquéllasno son los privativos de las féculas ; después, Freudenberg (72) sostuvo la natu-raleza cíclica del sacando constituyente, es decir, que la glucosa es reemplazadapor una ciclbsa, sin variar apenas el número de unidades enlazadas, pero en estecaso no habrá que pensar en enlaces 1-4 y 1-6, sino en otros. Supone todo lo indi-cado que el Bacillus maaerans posee, además de capacidad sacarificante del almi-dón, la de ciclizar la molécula de la glucosa, a tono con muchas plantas que Con-tienen inosita y! además la fosforilan, produciendo ese conjunto de sales cálcico-magnésicas de ácidos inósito-fosfóricos, que para uso terapéutico se designan conel nombre de fitina.
Las dextrinas de Schardinger son dos : la a y la ß, y muestran no ya tendfcn-cia a la cristalización, como se nota en algunas combinaciones de amilosa, sino tam-bién la cristalización perfecta ; examinados los cristales con difracción de rayos Xy realizadas medidas cristalográficas- y crioscópicas resulta que están constituidospor seis residuos de azúcar las a y por siete las °>, y que sus poderes rotatoriosson 154° y 161°,9, respectivamente (73). Las dextrinas de Schardinger podrían pa-recerse a las gomas de origen bactérico producidas por el leuconostoc sobre las diso-luciones de sacarosa, pero según Kühn, tal hecho sólo acontece con este azúcar yno con otros.
Para informarse de las discusiones habidas a propósito de estas dextrinas, con-súltese V. Grignard. «Traite-dé Chimie organique», t. VIII, segundo fascículo, pá-gina 671. París 1938,
De antiguo es conocido un grupo de dextrinas, que es el formado en la torre-facción de las féculas ; constituye el de las piro dextrinas, sustancias completamentediferentes de aquellas otras engendradas por la sacarificación mediante fermentos-La temperatura ha producido cambios profundos; en la estructura, entre ellos, la}ramificación de la amilosa y de parte de la amilopectina. Queda de esta manerauna mblécula relativamente pequeña, constituida por 66 unidades de glucosa.
Las amilasas hidrolizan esta dextrina en proporción de 22 por 100. El análisisde grupos finales, que conduce a 30 unidades de glucosa en las fé'culas, aquí revelasólo 12. El químico que ha estudiado estas transformaciones térmicas ha estudiadotambién la incapacidad para la retrogradación, prueba de la extinción de la ami-losa, y propone una nueva manera de insertarse las unidades gluoosídicas (74).Ofrecen gran interés en la técnica de producción de la corteza del pan.
Las técnicas para estudiar la constitución de las dextrinas son las mismas quese siguen para la fécula ; precipitaciones fraccionadas para purificar, determinación•del poder reductor, metilación y grupos finales, así como la síntesis de flavazolcs.
(71) Survey of Starch Chemistry, 1928.(72) Ann. Rev. Bioch., 1939. 8, 81.(73) French y Rundle. Amer. Ch. Soc., 1942. 64, 1.651 y Willson, Schoech. Id. Id.,
1943. 65, 1.380.(74) Indus. Eng. Chemistry, 1944. 36, 72.

— 37 —
Los investigadores de la escuela sueca conceden un valor a la ramificación, perono niegan el influjo decisivo que en la sacarogenolisis de la amilopectina puedetener el ácido fosfórico. En mi laboratorio se ha visto cómo féculas abundantes eneste residuo ácido se sacarifican en menor proporción y que las féculas pobres enfosfórico o en las que este ácido no integra éster amilofosfórico, sino en combina-ción lecitínica separable por la mezcla de alcohol-benceno, se sacarifican por la ocy ß-amilasas, en cantidades que pasan de 80 por 100. Ante estas consideraciones,avaladas por hechos insistentemente repetidos, es probable que el nudo de la cues-tión se halle, en algunos casos, en el ácido fosfórico. Los enlaces estéreos de eáteácido son separables por la fosfatasa, pero no por diastasa x y ß o por glucosidasa.Hemos hallado en nuestro estudio sistemático de las féculas algunas que contienengrandes cantidades de fósforo (R. Olalla) y que su dextrina residual alcanza el 55por 100, la de Oxalis purpumta, y hemos estudiado otras, como la de Costaricavesca, en que lá dextrina representa sólo el 6 por 100. Los investigadores de-laescuela sueca sostienen que en las dextrinas escasamente fosforadas sólo existenenlaces isomaltósicos 1-6 y más recientemente K. H. Meyer (75), defendiendo susideas acerca de la estructura ramificada de la amilopectina, afirma que las dextri-nas Obtenidas por la B-amilasa producen esteres acéticos, cuyas películas son.delmismo tipo' que las de amilopectina, frágiles, en términos que extendidas en unvidrio plano no pueden separarse como películas.
Los mismos investigadores suecos tantas veces citados aquí han sostenido al-guna vez que el ácido fosfórico constituye, por su inserción, una anomalía.Las amilopectinas que hemos estudiado han sido degradadas por el uso alter-nativo de fermentos que desatan las ligaduras maltósicas y las isomaltósicas,y es de esperar que habrán desaparecido todas las primeras, y aun de las últimas,casi todas las que no estén protegidas por un grupo fosfórico. En, las dextrinaslímites que hemos obtenido, la mayor parte del ácido fosfórico de la amilopectinaqueda como esteramilfosfórico, o más sencillo, glucofosfórico. Al constituir unaanomalía el ácido fosfórico, la actividad del fermento debe detenerse, a menos quecon él coexistan otros fermentos hidrolizantes desesteres fosfóricos, que rompan la"gadura, debelando así el, obstáculo. Esta es la causa de emplear pancreatina, queen opinión general, es el único enzima que carece de fosfatasas, pero su actividadfrente a la amilopectina es más pequeña que la mezcla de amilasas dieueltas de lacebada germinada ; m'as, no obstante la precaución, no se ha podido vencer ladificultad, encontrando unas diastasas tan puras y tan activas que no contenganfosfatasas.
Tan necesario a esta finalidad es el empleo de la pancreatina, aun con la des-ventaja de ser menos activa, que la mezcla de amilasas de cebada germinada enla. fécula de almortas, como tipo, todo el fósforo de la fécula se ha encontrado
(75) Nature, 1947. 160, 900.

- 38 -
en la amilopectina, hecho que, según se ha indicado ya, varios investigadores lohan hallado con la fécula de patata.
El hecho que en nosotros produjo extrañeza al estudiar la fécula de oxalis, dehallar fósforo hidrolizable en cifras muy altas, y el que las dextrinas contuvieranla mayor parte del ácido fosfórico de la fécula, tenía antecedentes en el capítulode las féculas. Hidrolizando féculas'ricas y pobres en fósforo Myrbäck, Ortenblac!y Ahlborg (76), hallaron que de la patata y guisante, más ricas en este elemento quelas de maíz y arrow-root, producían, por la B-amilasa, dextrina, con todo el fós-foro de su generadora. Con la de patata obtuvieron 14 por 100 de dextrina y coapancreatina 26 por 100, ambas con la totalidad del fósforo de la fécula. En cuantoa la amilasa a, el primero de los aludidos investigadores ha encontrado que laforma de las curvas de la hidrólisis es distinta cuando hay fósforo (77).
Circunscribiéndome al problema planteado acerca de la existencia del ácido fos-fórico de la fécula en el residuo dextrínico límite, y admitiendo que la insercióndel ácido fosfórico constituye una anomalía, que no pueden destruir los enzimasaptos para soltar los enlaces 1-4 y 1-6, quedan próbalidades para suponer que unoo los dos tipos de enlaces deben subsistir en las dextrinas. El peso molecular deéstas es relativamente pequeño, 800; algunas de ellas, la de maíz, metiladas estilode las féculas se resuelven en. dos moléculas de trimetilglucosa 2-3-6 y una de is-trametilglucosa (78), hecho que implica la no ramificación. Todo induce a pensaren cierta sencillez, claro es, de las dextrinas obtenidas por el empleo alternado defermentos separadores de enlaces 1-4 y 1-6, y con un contenido moderado de ácidofosfórico. La escuela sueca se refiere no a especies químicas, sólo a mezcla de trisa-cáridos, tetrasacáridos y hexasacáridos ; es evidente que las moléculas de maltosao de maltotriosá que los integran se han de unir del modo usual y que el ácido fosfó-rico puede servir de nexo entre los polisacáridos unidos por los dos tipos de enlaces.
,0 maltotriosáPO, O maltosa
OH
Empleando la taxonomía de Haworth, el ácido fosfórico actuará de ligadurapolimèrica.
El trisacárido se ha referido por unos investigadores a la maltotriosá, pero otrosaseguran que el obtenido con la técnica por ellos empleada, la de los flavazoles,no es fermentable por la levadura; cuando la maltotriosá han probado Myrbäck ysus colegas que es fermentable ; además, sus poderes rotatorios son desiguales entresí y también diferente la aptitud para reaccionar con el bromuro de acetilo. Igual-mente, de la fécula del guisante se ha conseguido un trisacárido que no contiene
(76) Biochem. Zeit, 1943. 315, 240.(77) Arkiv f. Kemi, 1948. 26 A n.° 7.(78) Biochem. Zeit, 1940. 307, 49, 53 y . 68.

— 39 —
maltosa ni glucosa, y de féculas corrientes, después de purificada la dextrina resi-dual por fermentación con levadura, se ha obtenido por Montgomery un levoglu-cosano.
Después de las publicaciones de Samec (79), en las que se da cuenta de laexistencia de fósforo en las féculas, en forma de combinación fosfórica, con laamilopectina se hizo notar un trabajo de T. Posternak (80), en el que se estudianlas diversas combinaciones del fósforo en distintas féculas, para deducir la conse-cuencia, de que en los órganos aéreos (cereales) aquel elemento se halla constitu-yendo ácidos glicerofosfóricos y derivados de éstos, como lecitinas, y en cambio enlos órganos subterráneos, patata, arrow-root, etc., existe un ester fosfórico deuna teimosa. Rosenquist, Neumiller, etc., sospechan que los tetrasacáridos exis-tentes en algunas dextrinas pueden proceder del pentasacárido y éste, a su vez,es capaz de producir un azúcar de una molécula menos de glucosa. ¿Es esta lacombinación fosfórica de Posternak? ¿Se habrá separado de residuos dextrínicos oéstos sintetizados durante la fosforilación se han unido a cadenas de amilosa?R. Olalla (Tesis doctoral 1944). V. Euler asegura que sólo 'ciertos grupos de laamilopectina tienen condición para adquirir fosfórico (81) y otros investigadorespresumen, con bastante fundamento, que este ácido sólo entra en las ramificaciones.
Todos los indicios compelen a suponer que el tal ester tetraosafosfórico de Pos-ternak es un) fragmento dextrínico separado de la fécula, que ésta puede reteneradsorbido en su masa y comunicándola una apariencia de impureza, justificativade las primeras opiniones que se hicieron públicas, al discurrir acerca de la exis-tencia de fósforo en la fécula, en forma de combinación fosfórica. Esta manera deinterpretar los hechos, -se podrá justificar por la síntesis de Hanes (82) con fosfo-rilasa y ester de Cori, estudiada en la segunda conferencia, síntesis en la que inter-viene el ácido fosfórico del residuo glucòsi co, que se fija sobre la amilopectina, porla condición especial de ésta de reunir mejores condiciones de fosforilación que laamilosa, o bien para crear en la primera una anomalía, que no pueden destruirlas amilasas, y constituir de esta manera una reserva alimenticia de combustión máslenta que la amilosa. La analogía en la ramificación de la amilopectina con el glu-cógeno da, en mi sentir,' pábulo a este 'criterio.
¿No se trata constantemente de cadenas formadas por seis unidades de glucosa?¿Es improbable que una de estas cadenas que contenga el residuo fosfórico en suextremidad se sitúe en 1-6, constituyendo una ramificación? Por añadidura noprueban Rosenquist y sus colaboradores que los hexasacáridos se degradan hastatrisacáridos? En medio está el tetrasacárido.
(79). Kollo'd r.eiheft, 1913. 5, 141.(80) Helv. Ch. Acta, 1935. 18, 1.350.(81) Ark f. Kemi, 1946. 24 A n.° 14.(82) Zeit. Physiol. Chem., 1939. 261, 125.

— 4° -
Al referirme a la técnica de los flavazoles pasé por alto explicar su constitucióny su síntesis a los efectos analíticos y de estudio de estructura.
Los flavazoles se constituyen sobre los azúcares reductores, reaccionando con
ortofenilenobiamina y fenilhidracina ; la primera parte de la rsacción es peculiarde grupos cetónicos en posición 1-2, que al actuar sobre la biamina producen qui-noxalinás ; pero en este caso no existe el doble carbónilo próximo ; es precisocrearle, y a este objeto la fenilhidracina oxida un grupo alcohólico secundario in-mediato al oxhidrilo, generando un CO productor de quinoxalina y luego con excesode fenilhidracina el fenilflavazol. Primeramente Ohle y Kruifft emplearon hidratode hidracina (83).
NH COH N N^ xxx , - , , xx ^ ",_N(\.IL
I
COH1 /"
CO |1 = 1
CHOH \x
1R .
i\T
\/\CH1 1i i ->
/x/*-N |
CHOH
CI
i stinti \ /\ //r \ /\ //\ /sft
N' cIK
Se ha indicado anteriormente la obtención a partir de las dextrinas de dos trisa-cáridos, el M. M..VI y el de la maltotriosa, cuyos flavazoles son diferentes en suscualidades, pero no en su unión estructural, juzgada por el consumo de oxígeno delácido peryódico, punto de fusión, poder rotatorio y actitud frente al bromuro deacetilo.
Teóricamente son posibles tres trisacáridos representables, según la taxonomiade la escuela sueca, de este modo :
t Î
Î
B
Por lo que se innere de lo antecedente, el problema aun no está resuelto.
Otro elemento que se encuentra en las féculas es el silicio, en más abundanciaen las de órganos subterráneos que en las de órganos aéreos. Difiere también laexistencia y la proporción de silicio en las féculas correlativamente1 al terreno, enque las plantas crecen ; por esta causa las féculas y aun las maderas de las pose-siones españolas del golfo de Guinea contienen mucho silicio y se caracterizan porsu dureza extraordinaria, debida en parte a su impregnación silícea. Hemos vistoféculas que carecen de silicio. No obstante, Karrer admitió que el silicio juegapapel importante en la formación de engrudo y que la presencia de combinaciones
(83) Errichte, 1944. 316, 444 (referencia a Rosenquist).

— 41 —
silíceas y nitrogenadas contribuye a la viscosidad de los geles feculentos. De lasobservaciones realizadas en mi laboratorio se infiere que féculas de bajas tempera-turas de gelifka'ción apenas tienen sílice o ésta se halla ausente, y que el nitrógenono integra una materia albuminoidea, sino un ester fosfórico, bien de lecitina o deamilosas-
La forma de combinación del silicio se parece mucho a la del fósforo. El ácido"silícico debe contraer combinaciones con los oxhidrilos, formando un ester parecidoa los silicatos alcohólicos que se emplean en la conservación de los monumentos.Además, el número1 de compuestos silícicos en química orgánica va creciendo demodo considerable, aunque multitud de esteres no son susceptibles de síntesis bio-lógicas. La combinación más natural es, sin duda, la estérea, que en las féculas esnotoriamente escasa, pero que en las cañas de los cereales es abundante y en ellaes verosímil que el silicio parcialmente contraiga una combinación con la parte dehidratos de carbono, pentosanas y hexosanas, aunque no excluya tampoco el en-lace estéreo con los oxhidrilos fenólicos de las ligninas. Caso análogo sería el delas maderas duras de Guinea.
Para 'la investigación del silicio se ha seguido la técnica recomendada por
Oberhauer y Schormüller (84), fundado en la producción de un complejo SKX 12
MoO, -f- H2O de color azul, y la cuantitativa con el método corriente de calcina-
ción y pesada de SiO,
Almortas 0,0184 de Si02 por 100Boniato 0,38 idem idemColocasia redonda 0,1662 idem idemColocasia alargada 0,033 idem idemCastaña dulce no contieneManihot no contiene
Se ve en la lista precedente que el elemento en cuestión predomina en las partes
vegetales enterradas, que para el efecto analítico han sido cuidadosamente limpias
y desprovistas de su cubierta, con el fin de evitar errores. Nótese el contraste entre
la fécula de boniato, órgano subterráneo, el de mayor contenido en Si de los anali-
zados, y la castaña, fruto aéreo que carece de este elemento. Quizá la causa de la
diferencia esté relacionada con la mayor facilidad de esterificar el ácido silícico el
azúcar que las féculas, y como el boniato es un tubérculo dulce por su cantidad
elevada de sacarosa es probable que al acumularse sílice para esta acción, quedeel sobrante, en un coeficiente de reparto, esterificando la fécula.
(84) Zeit. Anorg. Chemie, 1928. 178, 371.

— 42 —
EXPRESIÓN QUÍMICA DE LA LIGNINA. ESTRUCTURA
Continuación del grupo de polímeros elevados por deshidratación es el de las
ligninas, sustancias existentes en el mundo vegetal, formadoras de su estructura y
probablemente combinadas con los polímeros del tipo de la celulosa, tan semejante
al de la amilosa de las féculas, para de este modo integrar las maderas.
Son compuestos más complicados que las féculas, a tal punto, que a pesar del
esfuerzo realizado para su estudio, aún no se sabe concretamente más que su
núcleo fundamental corresponde al fenilpropano C6H5, CH2, CH2iCH3, por tanto,
un derivado de este carburo constituye el menomerò, unidad quel por deshidrata-
ción y enlace progresivo forma el polímero.
El enlace de los diversos grupos que constituyen el polímero es el anhidrólico,
producido al perder agUa dos moléculas de fenol o una de fenol y otra de 'alcohol,
en la cadena lateral de fenilpropano.
Cuando la madera se somete a la acción del fuego es sabido que produce fu-
ñóles: en esto se funda el ahumado de carnes y pescados. Si se destila madera, en
vez de quemarla al aire, se obtiene un conjunto de fenoles, variable con la especie
vegetal, pero todas producen creosota, en la que pueden predominar fenoles libres
o anhidroles de fenoles. Resulta, pues, que la madera contiene fenoles o que éstos
se forman durante la ¡pirogenación : entre ellos existe guayacol, característico por
su cantidad de la madera de haya, fenol ordinario, cresoles, etc. Destilando e hidro-
genando a la par se obtienen los productos de hidrogenación correspondientes a los
fenoles, por adquirir el anillo bencénico seis átomos de hidrógeno, convirtiéndose
en el ciclano respectivo, núcleo de ciclanoles y anhidroles fenólicos.
¿Qué fenoles o qué anhidroles fenólicos se producen en la pirólisis de las
maderas? El resultado depende de la estructura de la madera: se conocen unas
calificadas de blandas, las cuales se diferencian químicamente de las duras; las
primera originan en la pirólisis guayacol (I), anhidrol metílico de la pirocatequina ;
las segundas, además de guayacol, producen dimetilpirogalol 1- 3 (II), es decir,
el anhidrol simétrico.
OH OH/\OClf3 CH30/\OCH,
I ! I II I I I
(I) \/ (H)
Son representantes estos anhidroles del grupo guayacílico el primero y del gru-po siringoílico el segundo.

— 43 —
Aun cuando se imagina un investigador que es fácil discernir el origen delaldehido fórmico que acompaña a los fenoles en la combustión' de la madera, seequivoca, pero no hay error en probar la existencia del aldehido fórmico entrelos productos de la pirogenación ; la conjuntiva puede ser un reactivo y la forma-ción de dimedón y de acridano son evidentes (86). Por de pronto, se ha señaladocomo origen del aldehido fórmico, la ruptura de éteres metilénicos, y aun cuandola interpretación es discutida por los investigadores más versados, todavía, es liti-gable este origen, el de la separación del grupo CH2 de un anhidrol metilénico. Elanhidrol que se halla en maderas y diversas partes vegetales corresponde a la seriedel piperonal, y por eso el tercer grupo de derivados fenólicos, en número másreducido que el de los anteriores, es el del piperonoilo,), integrante de leños y de
productos que han gozado de predicamento, como medicinales (safrol, piperonal,ácido pipero'noil acético, etc.). Hunter y Hibbert (87), en desacuerdo con la exis-tencia de piperonoilo, atribuyen el formol al alcohol cinámico. La cuestión ha sidozanjada por Freudenberg y Plankenhorn (88), exhibiendo una lista de 39 com-puestos naturales, que por descomposición térmica producen aldehido fórmico,revelable con anilina caliente, para formar acridano.
En definitiva, en la pirólisis de las maderas se engendran sustancias que se«alinean como constituidas por radicales guayacilo, siringoilo y piperonoilo.
La cuestión, ahora, es saber si esos grupos moleculares preexistían o son pro-ducidos durante la pirólisis, cuestión que lleva implícita otra, la concerciente a lanaturaleza cíclica o acíclica de la lignina. Hay en la actualidad partidarios de losdos puntos de vista: el fenómeno más significativo de la transformación de lamadera, para obtener la pasta de papel, es la sulfitación, durante la cual se formaUn producto sulfítico, que no es propiamente un derivado sulfonado aromático,porque se ignora la posición del £>03H, aunque se hayan propuesto ejemplos dederivados similares al resultante de la sulfitación. Los más entusiastas defensoresde la estructura cíclica no conceden valor polémico a este argumento contra ella.Porque los sulfitos ácidos no se adicionan al grupo be'ñcénico, pero sí es posibleque se unan a la cadena propánica, y por eso Freudenberg admite la fijación dfclgrupo SO3H\ sobre la cadena lateral. El generador probable de la vanilina, des-Pués de la sulfitación, es el alcohol coniferílico, y éste, que se ha evidenciado en
(86) Freudenberg, Kline, Flickinguer y Soback. Berichte, 1939. 72, 217.(87) Amer, Chem, Soc., 1939. 61, 2.196.(88) Berichte, 1947. 80, 149.

— 44 —
la lignina por Freudenberg y Richtzenhain (89), contiene en su cadena lateral unenlace doble, sobre el que puede insertarse el S03H.
OH/NOCH,I II I
CH = CH — CHjOH
Además, la lignina nativa fija ácido sulfociánico, lo cual es prueba de la exis-tencia de enlaces eténicos.
Pero cuando se fuerza la sulñtación sobre el supuesto ácido lignino-sulfónico,el tanto por ciento de azufre se eleva gradualmente de 5,5 a 8,6 y a 12,8, cifrasque prueban la entrada de: nuevos grupos S03H : ¿dónde se colocan éstos?
La elevada presión a que se trabaja, quizá motive el ingreso del grupo sulfóriicoen el anillo bencénico, porque los derivados sulfonados del benceno por fusión al-calina engendran fenoles, y en el caso de la ilignina, aunque no se practique lafusión alcalina, se hace una ebullición larga con álcali de 20 por 100, durante laque se libera vanilina. ¿Serán equivalentes estas circunstancias? Es verdad queeste anhidrol se produce en cantidades mucho mayores hasta tres veces, haciendointervenir la oxidación en particular con nitrobenceno. La apertura de anillos fu-ránico o crománico, durante la sulfonación, ha quedado completamente descartada.
A pesar de estos antecedentes el tema de la sulfitación, en cuanto se refierfe aestructuras probables, .queda sin dilucidar, pero subsiste un hecho tan interesantecomo el siguiente: sin sulfitación previa no hay producción de vanilina.
La lignina es nitrable corno lo son muchos palióles grasos y extensa serie desustancias cíclicas ; no obstante, los productos de reducción del derivado nitradoy los azoicos que pueden formarse sobre él no convencen a diversos investigadores,y ciertamente su duda está justificada.
La metilación con sulfato de metilo se hace tan fácilmente con la lignina comocon fenoles ; sin embargo, no se debe excluir la posibilidad de oxhidrilos alcohóli-cos, por cuanto en este orden de polémica ha surgido la diferenciación en una mo-lécula de oxhidrilos de los dos caracteres. Además, se conocen antroles y antraríolesprovenientes de reducir antraquinona, cuyos OH alcohólicos no desvirtúan lanaturaleza cíclica de la sustancia.
La fusión alcalina a que acaba de aludirse como medio de aislamiento de feno-les por resustitución se emplea directamente sobre las ligninas, a título de agentesaponificante violento que rompa los enlaces anhidrólicos. Sepárase entre los pro-ductos diversos de la fusión, ácido protocatéquico y si antes de fundir se metila la
(89) Annalen, 1942. 552, 126.

- 45 -
lignina, en vez del ácido protocatéquico se aisla su anhidrol dimetílico, el ácidoverátrico :
CO01I COOH
I I I I\/OII \/OCH3
OH OCH3
A. protocatéquico A. verátrico
El primero de estos ácidos tiene una significación biológica que he de recalcar,porque es el nexo que relaciona el endurecimiento de la madera co'n el endureci-miento de los élitros de los coleópteros : es una unidad biológica común a todofelos organismos vegetales y animales que necesitan rigidez. En la cucaracha (Blattaorientalis) es incuestionable la existencia del ácido referido : de la Caliphora ery-throcephala, por metilación con diazometano, se ha aislado ácido verátrico, y delTenebria molitor se ha conseguido el ácido 3:4 bimetoxipropiónico (90).
Mucho más preciso para el juicio acerca de la estructura cíclica de la lignina esun argumento de Pearl (91), relativo a la oxidación de la sustancia separada en elaislamiento de la holocelulosa del Picea mañana (abeto negro) con clorito sódicoen ácido acético. Obtiénese un precipitado no destilable en corriente de vapor yfusible a 167° : es la cloranilina ; parece fuera de duda que la actuación de los reac-tivos no causa un anillo bencénico, sino que éste se hallaba ya formado.
A la par.de este argumento puede esgrimirse otro de mayor interés, porque noexisten de por medio reactivos ciclizantes ; conócense maderas coloreadas, en lasque los colores son de la serie flavónka o de la antociánica ; existen otras uti-lizadas en terapéutica, que contienen esencias, en las cuales se hallan derivadospiperonílicos, como el safrol (la madera de sasafras), de incuestionable estructuracíclica, Hibbert (92), con gran fortuna, ha tratado de relacionar los componentesde las maderas, reduciéndolos a un origen común, puesto que todos ellos se engen-dran a partir de la vanilina o de sustancias ligadas a este aldehido' anhidrólico,por su estructura de fenilpropano (alcohol coniferílico, eugenol, etc.).
Del mismo orden de la experiencia de Pearl es otra anterior de Aulin-Erdtman (93), relativo a la bromuración de la lignina y a la oxidación del produc-to resultante de bromarla, en la cual se origina 6-bromovanilina, que no es exac-tamente el derivado originado al bromar directamente este aldehido, porque elbromo ocupa la posición 5 ; en cambio, el deshidroeugenol se broma en 6.
La prueba decisiva de la naturaleza cíclica de la lignina debe hallarse en el re-
(90) Pryor, Russell y Tood. Nature, 1947. 159, 399.(91) Amer. Chem. Soc., 1946. 68, 916.(92) Amer. Chem. Sor., 1939. 61, 21, 98.(93) Abstracts, 1944. 38, 3.467.

_ 46 —
sultado de la reducción con polvo de cinc, práctica frecuente en el conocimientode la estructura de los compuestos orgánicos cíclicos. Phillips ha realizado esa prue-ba encontrando que la lignina de tuya, ocasiona, en las condiciones expuestas, el3 metoxi-4-hidroxifenilpropano, aunque con rendimiento muy pequeño.
La reducción con níquel Raney o con hidrógeno en contacto con cromito decobre conduce al ciclohexilpropanol y a otros derivados aclámeos, con excelentesrendimientos.
La lignina, como los fenoles, adquiere acetato mercúrico en proporción de una.molécula de sal por cada oxhidrilo. El acetato, a tenor de lo que ocurre en los fe-noles, es remplazable por yodo, produciendo yodolignina, la cual oxidada según latécnica de Freudenberg, con nitrobenceño en álcali, engendra la 5-yodovanilina.
Hace tiempo se ensaya la producción de lo que ahora se llaman modelos deligninas, es decir, sustancias si no idénticas, otras que reúnan la mayor similitudposible con la lignina. La síntesis más interesante de las que conozco en este ordende ideas es la afectada por A. Russell (94), primero por aldolización de la 5-acetil-vanilina, que causa rápidamente un polímero, después por formilación de la resa-cetofenona y de la 2:6 bihidroxi y 2 :4 :6 trihidroxi-acetofenona. De estas tresúltimas cetonas fenólicas han resultado polímeros análogos a la lignina. En taltipo de modelos no hay error, porqué son sustancias en que el menomerò es cíclico,y de los más semejantes a los que contienen las ligninas.
Las medidas físicas han contribuido a esclarecer la naturaleza de la lignina ; lasde tensión superficial, realizadas por H. Erbring y H. Peter conducen a la, estruc-tura cíclica (95) ; las de refracción y del espectro de absorción efectuadas por Freu-denberg (96) ; revelan la existencia in situ de un anillo cíclico por cada 10 átomosde carbono. Este trabajo de Freudenberg puede conceptuarse como la, síntesis detodos los efectuados por diversos químicos en Alemania, Estados Unidos y Ca-nadá desde que Herzog y Hillner (97) los iniciaron coa este resultado; «la ligninaderiva de unidades de fenol total o parcialmente eterificado con una cadena lateralde tres átomos de carbono, que no tiene carbonilos ni ligaduras conjugadas». Añosmás tarde, Hagglund y Klingsted (98) escribieron que la absorción máxima ep el
espectro ultravioleta es de 2740-2760 A para las maderas duras y 2800-2870para las blandas. Glading (99) mostró su conformidad con estas medidas del es-pectro de la lignina nativa y comparado con el de la flavona y con el del biisoeur-genol puede ser interpretado así : cada unidad formadora de lignina de peso mole-cular 840 contiene dos anillos piránicos.
(94) Reunión anual de la Amer. Ch. Soc. Abril, 1948. Chem. Ing. New, 1948. 26, 1.360.(95) Celulose Chemie, 1946. 22, 117. Abstracts, 1946. 40, 3.259.(96) Berichte, 1938. 71, 1.810.(97) Zeit Physiol. Cbem., 1927. 168, 117.(98) Zeit. Physik. Chem., 1931. 152, 295.(99) Paper Trade J., 1940. I l l , 228.

— 47 —
CU,"\CHOH
•-OCH3
Posteriormente, Patterson y Hibbert (100), estudiando los espectros de absor-ción de todos los progenitores de la lignina aislados por reetanolisis, encontraron quelos derivados vaniloínicos y veratroílicos .tienen un máximum de absorción próximoa 1.300 y que valores cercanos son los de la serie sirínguica, demostrando así lanaturaleza aromática de la lignina y la posible existencia de 'carbonilo y de enlaceeténico conjugados con un núcleo aromático (101).
Los trabajos practicados por Freudenberg acerca del índice de refracción con-ducen al número 1,61, que se compagina muy bien con la estructura cíclica ; sinembargo, son discutidos por Schütz y Sartén (102) como poco demostrativos, afir-mando que no permiten sostener que la lignina in situ, sea cíclica y que el número-obtenido 1,61 no es prueba suficiente de que la lignina esté preformada en la ma-dera, porque si fuera así, se podría aislar con el reactivo de Schweizer, sin previahidrólisis, y por consecuencia estiman que la lignina es un producto secundario.Además sostienen que faltan en la madera grupos fenólicos, porque los diazoicosno actúan sobre ella, argumento de poca consistencia una ves admitida] la ligno-celulosa, cuya formación sólo es posible al unirse los grupos fenólicos con losalcohólicos. Estas ideas tienen un antecedente en otras personales de los auto-res (103), según las cuales la molécula de la lignina es pequeña en relación al su-puesto generalmente/aceptado.
Los estudios de Hibbert y su escuela conducen a la forma aromática y clara-mente lo expresa aquél en una frase relativa a la hidrogenación de la lignina ((aun-que es ya evidente el carácter aromático de la lignina».
No obstante las discrepancias de criterio acerca de la estructura de la lignina,rce inclino a aceptar la definición propuesta por Freudenberg: «la lignina es elcomponente metoxilado cíclico de la madera)), porque si se despoja mentalmentea la lignina de metoxilos y oxhidrilos, quedan carbono e hidrógeno en la relación1 ;1, como en el benceno. Evidentemente los grupos cíclicos están preformados.
En la pirólisis de la madera se producen variadas sustancias, como acetona,representante de la cadena tricarbónica unida al feriilo, ácido acético y alcoholmetílico. ¿Qué funciones químicas contiene esa cadena? Para Klason un grupoaldehídico ; para Freudenberg, tres oxhidrilos ; para Hibbert, un carbonilo, pero
(100) Amer. Ch. Soc., 1943. 65, 1.872.(101) Id. id., 1.S73.(10:>) Ccllulosechemie, 1944. 22, 114.(103) Id. id., 1943. 21, 35.

con fenilhidracina, no está claro que se produzca fenilhidrazona ; Erdtmann aludió
a un carbonilo enmascarado.Luego de admitida la estructura cíclica con cadena propánica no hay sino averi-
guar cuáles son los monómeros constituyentes, las unidades formadoras del polí-mero. .Se ha convenido en páginas anteriores, que existen en las maderas blandasunidades guayacílicas y en las duras, éstas, las sirínguicas y las piperonoílicas, enproporción, según Freudenberg de; 5:1:2, respectivamente. También se dice quelas plantas del subtipo gimnospsrmas contienen guayacílicos y las angiospermas gua-yacílicos y siringoílicos, y dentro de las angiospermas las proporciones de unos aotros varían ; en las monocotiledóneas, la relación de guayacílicos o siringoílicos,es de 1:1, y en las dicotiledóneas, de 1:3. Las unidades de Klason, de Freuden-berg y de Hibbert, en lo esencial coinciden ; la única discrepancia se halla en quela cadena lateral sea etilènica (caso del alcohol coniferílico de Klason), que seaalcohólica o cetónica o simpemente cetónica (hidroxipropiovanilona y la siringonarespectiva).
La existencia de todas ellas está apoyada en hechos naturales, especialmentelas de Klason, porque el alcohol coniferí'ico de los pinos procede de la coniferina ;
el isoeugenol y el ácido fenilico se catalogan como productos naturales.
CH = CU — CH2OH CH = CH, COOH
I I I II I I I\/oai« \/ocH3
OH OH
Por-consecuencia, son coincidentes las ideas en cuanto a la estructura funda-mental, que ha dado pábulo a la formación de un grupo biológico, el de los ligna-nos, cuyo núcleo es bimérico, como el deshidroeugenol o el ácido deshidrobi-verátrico
C CI IC CI IC A C:H,-HC [
i l i I '! ! HÌ;-°i M *i i i /\\/ ( -,Esqueleto de ligna- | |
nos I !-0-CH3
OH
Deshidroeugenol
— 49 —
Según lo expuesto se admiten en la bibliografía tres unidades que en el fondo
son muy semejantes: 1.°, el alcohol coniferílico, tipo de Klason; 2.°, la vanilina,
tipo de Freudenberg; 3.°, la hidroxipropiovanilona, tipo de Hibbert.
¿Cómo se unen las unidades para constituir el polímero? Antes se ha aludido al
principio de unión progresiva, análogamente al que rige la soldadura de glucosas
para producir polisacáridos o la de aminoácidos integrantes de los peptidos albu-
minoideos, y se ha advertido también, al principio de esta lección, que la ligadu-
ra es anhidrólica- Tal como desenvuelve Freundenberg sus ideas acerca de la es-
tructura del polímero se han de enlazar dos moléculas de glicerilvanilina (I) o de
propanoxivanilina (II), cediendo la primera su oxhidrilo fenólico y la segunda su
oxhidrilo en cadena lateral para formar las unidades III y IV.
CHOH
CHOH
I ïI II I —O—CH,
OH
(I)
HOCH
(H)
CH„OHI
HCOH!
HCOH
I IHjCOH
IHCOH O
\/HO
I
-O-CH,
I II II I-\/OH
-O-CH,
CHa—C=OI
HOCHI
I 1I |-O-CH,
CH3-CO\/I II I
HC—O
(III)
I II II 1-0-CH,
(IV)
OH
Por el principio enuncialo las unidades III y IV pueden enlazarse originando
una pieza molecular de cuatro unidades.
Dentro de esta posibilidad, se encuentra otra, origen de variadas discusiones,
cual es la referente a la existencia de anillos furánicos o crómameos en la molécula
de la lignina. Esa estructura es verosímil y bien «presentable en la teoría de
Freudenberg. Por deshidratación interna de los fragmentos III y IV se producenlos V y VI.
R£V. DE LA REAL ACADEMIA DE CIENCIAS,— 1949.

— 5o' —
IHCOH
[HCOH
I
US II I
H — C Ü H O
cu/\
I I—O-CH 2
CH3-C=()I
HCOH
CH.,. COH |'I I
HC—O
I
I |-0-CH3
i-O
Apoya estas estructuras crománica y furánica un hecho que lógicamente ocu-rre en los vegetales ; algunos de éstos contiene isoeugenol, que, oxidándose, pro-duce deshidroisoeugenol, sustancia formada al actuar sobre aquel fenol el mace-rado de un hongo rico en fermentos oxidantes, el Russula Aelica; si ese compuestose metila y luego se oxida se originan los ácidos -verátrico e isohemipínico, 'comoconsecuencia de la ruptura del anillo furánico del citado deshidroisoeugenol.
Aplicando el mismo principio de unión progresiva a las unidades sirínguicas seobtienen los derivados homólogos en enlaces de 1 y, 4 unidades con anillos cromá-nico y furánico.
Evidentemente, que con unidades piperonílicas el principio de unión progresivano es aplicable, porque los oxhidrilos están anhidrolizados por el metileno, perono excluye la deshidratación por el lado cíclico de. la molécula, ni la unión porla cadena con otros grupos guayacílicos.
Siempre el grupo dominante es el vanílico, y por tal causa es fácil obtenervanilina de la madera. Freudenberg calcula que de ocho unidades existentes enla madera de pino, cinco son guayacílicas, una sirípguica y dos piperonílicas ; pro-porción que no se guarda en otras .maderas, especialmente de angioespermas, enque las unidades sirínguicas están en número más elevado.
A continuación exponemos una fórmula de la escuela canadiense construidasobre la base de cinco grupos de fenilpropano, con una sola ligadura anhidróli-ca (A), otra furánica (B) y dos piránicas (C) ; contiene cuatro metoxilos, un oxhi-drilo fenólico y tres de diferentes clases, y al extremo el discutido grupo carbonili-co que previo Klason.

— í1 —
HO,
CU,
H—C-OH
H 2 C| | | CHI I Ir i i
- C\/\/ ROC"| O O.CHg I
H CH,
(B)
OCH3 CHjOH\ I
I CH.2I I1- c o —
III
(A)
O
I I-CH | |' l i l i| | HjC | ¡-CH2
\/ \/\/ IOCH, c: c;o/\ I
CH»
VI
AISLAMIENTO DE LA LIGNINA Y DE su UNIDAD BÁSICA. ENSAYO CON FERMENTOS
El aislamiento de la lignina presupone conocer la materia prima de donde pue-da extraerse. Se la ha definido como constituyente metoxilado cíclico de la ma-dera ; pero esta materia es como todo producto natural un compuesto complicado ;en ella coexisten hidratos de carbono (celulosas, pentosanas, sustancias de carácterpertico) con la lignina. Talj coexistencia será un indicio de que los carbohidratosestán unidos químicamente a la lignina, puesto que, en definitiva, glucósidos ypcntósidos en general poseen un factor de característica fenólica. Además, los dosfactores en el supuesto glucósido, no son separables fácilmente ; es imprescindiblela intervención de agentes hidrolizantes para aislar la aglicona de la parte de hidra-tos de carbono. De la madera de abeto, han dicho Jayme y Hanke que contienesacáridos de hexosa unidos preferentemente a grupos guayacílicos.
En los estudios que realizó para un ensayo de la filogenia química de la ligninahe encontrado, con la ayuda de J. R. Aguirre, en la cubierta de la semilla de alba-ricoque gran cantidad de una mezcla de sustancia de tipo graso, mezcla fusible atemperatura de 38° formada por ácidos y alcoholes, • sin duda de elevado peso mo-lecular.
Recientemente Clark, Hircks y Harris (104) han hallado entre los productos deextracción del residuo de la lignina de pino Douglas con benceno y otros disolven-tes ; ácidos eicosánico, dodecosánico, tetracosánico y oleico, además de eicosanol-1y dodecosanol-1.
Los químicos americanos han1 extraído los alcoholes y ácidos expuestos de unresiduo ; nosotros, del producto conocido como Ugnol, el cual se preparó con elpolvo de la cubierta drupácea, lavado de antemano con mezcla de benzeno y alco-hol, tratamiento durante, el cual debieron eliminarse esas sustancias, dato que
(104) Amer. Ch. Soc., 1948. .70, 3.729.

- S2 -
implica una verosímil combinación con alguno de los factores constituyentes de lamadera.
Claro está que la madera, aparte de estos factores hidrocarbonados, que enesencia se reducirían a uno, la celulosa, contiene otros, que contribuyen a su endu-recimiento, las sales minerales, que acaso no están como tales sales, sino formandocombinaciones o complejos, como los ácidos uránicos. También contribuye al endu-recimiento la metilación, porque se afirma que gradualmente la madera adquieremetilo y que el grado de dureza radica en parte en el de metilación. Influye no pocoen este aspecto la polimerización progresiva, puesto que las maderas jóvenes tienenligninas menos polimerizadas que las viejas ; afirmación un poco audaz, aunque nopuede negarse que existen grados diversos de complicación molecular, consideradoen el aspecto ontogénico.
¿Cómo se aisla la lignina de los demás componentes de la madera? Es tradi-cional, por la industria del papel, el método llamado de la sulfitación, referido enla conferencia anterior, con motivo de la estructura de la lignina ; en el complicadoy todavía oscuro mecanismo de la sulfitación se aisla lignina como producto solu-ble dedicado a numerosas aplicaciones.
Los demás procedimientos son de tipo hidrolítico ; los ácidos y los álcalis querompen las imaginadas ligaduras poníales con los hidratos de carbono y desdoblanéstos en azúcares sencillos, y los fermentos del grupo de las celulasas que destru-yen la celulosa y las pentosas, dejando libre la parte fenólica de la madera.
En el grupo de los ácidos existe una técnica de valor analítico, que consiste enobtener la conocida lignina de Klason, en la cual se destruyen los polisacáridos porcalefacción con ácido sulfúrico primero, y después con acético o con clorhídrico.Resulta así un producto oscuro, que unas veces tiene la forma microscópica de lamadera y otras de un polvo amorfo.
Se ha pretendido, sin éxito completo, obtener un polvo blanco, por técnicas másdelicadas, sustituyendo el clorhídrico por el ácido acético, pero se ha logrado poco,porque los lignoles (105) resultantes, cuantitativamente son inferiores a la ligninay muchas veces son mezclas de esteres acéticos. En efecto, son más claros que lasligninas negras de Klason.
De una observación antigua, la de que la celulosa es soluble en el reactivo deSchweizer (hidrato cúprico-amónico) surgió el procedimiento de Freudenberg (106),mas después Hilpert hizo la observación de que las ligninas retienen cobre, obser-vación exacta que hemos confirmado varias veces, trabajando con los soros de he-léchos (107). En esta lignina obtenida con hidrato cupri-amónico, existen pento-sanas que el reactivo no ha disuelto (108) ; por tanto no es pura, y las deducciones
(105) A. Pauly. Berichte, 1943. 76, 1.177 y 1943. 76, 867.(106) Berichte, 1939. 72, 1.885 y 1940, 73, 61.(107) O. Fernández y A. Ortiz. Revista R. Acá. Ciencias. Madrid, 1!>41. 35, 34.R.(108) O. Fernández, C. Capdevila y J. J. Castaño. Id. Id., 1944. 38, 508.

— 53 -
que se hacen a base de los resultados hallados con ellas son un poco deleznables.T. Ploez (109) introdujo la técnica fermentológica en el aislamiento de la lig-
nina. No es fácil encontrar seres vivos que contengan celulasas más que en el-grupode moluscos, que se alimentan de • sustancias ricas en celulosa, pero del caracol(Helix pomatia) es posible separar por maceración de la parte posterior del cuerpoy sucesiva precipitación de los albuminóides, un conjunto de fermentos, celulasas,hemicelulasas y pentosidasas, que aplicado a la madera,-separa de ella cuarenta ysiete por ciento de azúcares, dejando u-n residuo inatacable, constituido por lignina,que representa en este proceso quimúrgico la parte aglicónica de la madera. Lalignina así obtenida, teóricamente debe ser la más pura, porque no hay en la acciónde los fermentos la violencia de los ácidos a diferentes temperaturas. El autor delprocedimiento consiguió éxito en lo que pudiera llamarse primera forma de la lig-nina, en la medula de saúco, materia no incrustada, a la que los enzimas puedenllegar con facilidad relativa. No es el caso de las maderas, que ni aun desprovistasde las materias céreas y pretratadas con vapor de agua se muestran poco accesibles alos fermentos. De la madera de pino sólo disuelve el conjunto enzímico de cara-coles 20 por 100 (110) y la lignina aun contiene 9,30 pentosanas.
La extración con disolventes varios ha sido objeto de apasionadas discusiones :el disolvente más adecuado por su inactividad es el dioxano (111), también es re-comendable el ester acetil'acético (112). Pero estos disolventes, a la ebullición, noseparan lignina de la madera ;; ,en cambio, si extraen productos solubles, cuandopreviamente se impregna el polvo desengrasado de la madera con ácido clorhídricopara conservarle doce horas, y después se hierve con el disolvente, lo cual prueba,que el ácido juega algún papel interesante, tal como el de hidrolizar la supuestacombinación glucosídica y' merced a él, la aglicona queda libre y en condicionesde ser extraída por el disolvente.
En igual caso se halla el ácido thioglicólico muy empleado en frío, pero conacido clorhídrico. De la madera de chopo (Populus trémula) ha aislado B. Holm-berg una combinación, a la que atribuyq la fórmula (C9 H8 02 (OCH3) (S^CH^COOH)0,55. El mismo investigador (113) utiliza el ácido thiohidracrílico para ais-lar lignina del abeto del Norte, también en medio ácido 2N C1H y contacto du-rante un mes. Aunque siempre se creyó que el producto separado por disolventes desupuesta indiferencia química era puro, la hipótesis no es exacta, como puede juz-garse por la fórmula expuesta y por otras noticias relativas al empleo de compues-tos sulfurados, con los que probablemente se forma un mercaptol o mercaptal porl°s grupos carbonílicos, acetónico o aldehídico de la cadena tricarbónica, puesto
(109) Berichte, 1939, 72, 1.885 y 73, 87.(110) O. Fernández y B. Regueiro. Farmacia Nueva, 1945. 11, 62.(111) Brauns. Amer. Ch. Soc., 1939. 61, 2.120.(112) L. Lemmel. An. Soc. Españ. Fí?. y Química, 1935. 33, 369.(113) .Ark. f. Kemi, 1945. A 21, n.° 10.

- 54 —
que el derivado 'producido causa una combinación mercúrica, que contiene 16 por100 de mercurio y 3,3 de azufre.
Los disolventes no aislan exclusivamente lignina aun después de la hidrólisecon el ácido ; Freudenberg empleó la clorhidrina del glicol y el producto! disueltocontenía 14 hexosasy 12 cloros (114).
La lignina de la Tsuga heterophyla se ha disuelto en una mezcla de volúmenesiguales de butanol y agua calentando en autoclava a 160 grados (115).
Intencionadamente dejé la hidrólisis alcalina para el final. Los álcalis en calienterompen, como los ácidos, las ligaduras poníales ; y la lignina entonces separadade su combinación con los carbohidratos debería disolverse en el álcali, y, en efec-to, a buena temperatura se disuelve, pero en frío queda como insoluble y resultaasí la lignina alcalina. En general, los fenolatos alcalinos se disuelven enel agua. Si previamente se metila la madera, el cuerpo resultante, la metillignina',es insoluble en sosa y lo propio ocurre con los ácidos lignínicos. Pero sustituyendoen la reacción el álcali por un sulfuro alcalino, se aisla la tiolignina, a la que seatribuye esta fórmula.
/(OH),(CH,0)4 C41 H31 O / /OH
C\\SH
Con un grupo mercaptan en consonancia con lo que acaba de expresarse acercade las combinaciones sulfuradas. Sin embargo, el grupo tiólico es de gran inesta-bilidad.
Composición centesimal de la lignina
La madera mejor estudiada por razón de su utilidad industrial es la de pino,que analizada por Freudenberg, tal como resulta de su procedimiento cúpricoda los números siguientes:
C = 65 0/°, H = 6, O = 7, 6, OCH3 = 15 - 16, OH = 9, io,C - CH, = 2, 7°/°
también se halla de 1-2 por 100 de nitrógeno.Los análisis de Wald, Richte y Purves, mucho más recientes, practicados sobre
lignina de Klason, obtenida del pino negro del norte de Europa (116), práctica-mente no difieren más que en el carbono.
C = 67, 5 %, H = 6, OCH3= 14,4 y 13,60
Los autores razonan la causa de la diferencia env carbono y metoxilo y sostie-nen que de ordinario no se desecan laa lignina a 105°. Es más verosímil que la
(114) Berichte, 1741. 74, 1.400.(115) Bailey. Amer. Ch. Soc., 1943. 65, 1.176.(116) Amer. Ch. Soc., 1947. 67.

- 55 -
causa radique en la diferente composición de la lignina Freudenberg respecto dela de Klason. La primera contiene pentosanas, que faltan en la segunda (117), noobstante los autores deducen una consecuencia y es que los resultados son defi-cientes en carbono para deducir una fòrmula representativa de la lignina in situ.
Los valores publicados por diversos investigadores coinciden con los expuestosdentro de la variabilidad de pureza de la lignina.
Aislamiento del mono-mero
A) Alcohólisis.—El gran problema en torno a la resolución de los polímeros ensu unidad constituyente, tanto en polisacáridos como en albuminóides o en depsi-dos se ha resuelto fácilmente, pero en la lignina todavía es una incógnita, puestoque así lo afirman los más tenaces investigadores en este campo. Es un empeño untanto fanático de la escuela de Hibbert suponer previa despolimerización, y repoli-rcerización del monómero resultante de su técnica de alcohólisis, que es tantocorno imaginar algo parecido a la imposibilidad de despejar la incógnita.
De todos los procedimientos analíticos para obtener el monómero de la agliconapreferimos' el de la alcohólisis y realcohólisis de la escuela canadiense de Hib-bert (118), fundado en eterificar los oxhidrilos libres con etilo, a cuyo efecto em-plean el método clásico de E. Fischer para sintetizar metilglucósidos o esteres deácidos con alcohol metílico que contenga disuelto 2 por 100 de ácido clorhídricogaseoso. En vez de emplear metanol se 'usa etanol procediendo durante muchashoras (60) a la ebullición y en corriente de un gas inerte (carbónico o nitrógeno).
Los investigadores de la escuela canadiense no se contentaron con aislar frac-ciones que creen no representan el monómero, y acudieron a la despolimerizaciónpor nuevo tratamiento con el alcohol clorhídrico, instituyendo la técnica que deno-minan reetanolisis, en la cual se separan fracciones oleosas de bajo peso molecular,de las que se sustraen con sulfito ácido de sodio la carbonílica ; con sosa la fenólicay con bicarbonato sódico los ácidos. No obstante este proceso sencillo aparente-mente, dicen que implica despolimerización y nuevas polimerizaciones por el influjodel ácido clorhídrico, a temperaturas, aunque no altas, muy prolongadas, y sinembargo, de esta creencia hari aislado, derivados guayacílicos y sirínguicos concarbonilo en la cadena lateral.
OH OH
fVcH, í X,
\/CO.CO CH, CH2.CO.CHäOH
Está exenta de objeciones la formación de un anhidrol etílico sobre un OH li-bre ; mas también algunos derivados carbonílicos producidos en la alcohólisis
(117) O. Fernández y B. Regueiro. Farmacia Nueva, 1946. 11, '57.(118) Amer. Ch. Soc., 1941., 63, 3.04J. 1943, 65, 1.173 y 1.176.

_ 56 —
pueden formar otros anhidroles, acétales, sobre carbonita y esto parece incuestiona-ble en la cadena propílica de la lignina, por tanto el fenómeno no debe en absolutoadscribirse a la existencia de OH capaz de etilación (119), puesto que el acetal for-mado es fácilmente 'desdoblable por ebullición con ácido mineral, y además seseparan dos OC2H5 y no uno, que es el que debería fijarse sobre un OH, si en vezde etilar se metilase con sulfato de metilo o con diazometano.
B) Empleo de fermentas.—Eh algún pasaje de esta, lección se ha referido elempleo de fermentos para el aislamiento de la lignina pura, utilizando los mismoscaminos que Ploetz. En él se ha podido observar, cómo con la técnica basada en eluso del macerado de caracoles (Helix pomatiá) sólo se consigue destruir por sacarifi-cación la parte glucosídica o pentosídica de la madera, pero el ataque no pasa ade-lante ; deja la lignina intacta. A pesar de este hecho, pensamos que habría fermentos,distintos de los elaborados por el caracol, capaces de romper los enlaces pontales delpolímero, para dejar el monómero en libertad. Los árboles son víctimas de hongosque viven sobre ellos como parásitos, unas veces, con amenaza de extinción de lavida del árbol, y otras, sin quebrantarle lo bastante, pero provocando una en-fermedad a la larga, mortal. Personalmente recogí un hongo basidiomiceto, que porinadvertencia no se clasificó con oportunidad, el cual vivía en la base de un álamo(Populus nigrá). La madera, próxima al emplazamiento del parásito, aparecía blan-ca y untuosa, perforada por galerías diminutas, que, en su fondo contenían peque-ños coleópteros pardos. Era evidente una alteración profunda en la estructura dela madera que analizada (120) resultó producir:
Lignina Freudenberg 50 °/°Metoxüo , 2,6 °/0Pentosana •. 6,93%
datos reveladores de un hecho, que también sorprendió en el estudio de la activi-dad del fermento de caracol sobre las maderas ; que la acción se dirige sobre laparte de polisacáridos, porque la cifra de pentosas es muy baja y en cambio apa-rece elevada la de ligninas por destrucción de aquéllas. No obstante, practicamosuna búsqueda relativa a la lignina ; destilando en corriente de vapor, se demostróamoníaco, indicio de destrucción de materias albuminoideas, y vanilina, aunqueen cantidad pequeña.
Luego la actividad del fermento ha incidido sobre la parte aglicónica de lamadera, mas no es afirmable que tal acto sea provocado por el conjunto enzímicoque pretendía hallar. Acabo de indicar que la madera estaba perforada por peque-ños coleópteros, y algún observador (121) ha visto que las termitas, que viven aexpensas de la madera, elaboran fermentos destructores .de ésta.
(11.9) Brauns y Buchanan. Amer. Ch. Soc., 1945. 67, 645.(120) O. Fernández y B. Regueiro. Rev. R. Acá. Ciencias. Madrid, 1945. 89, .331.(121) Walcott. Abstracts., 1946. 40, 4.841.

— 57 —
Bien pudiera' ser que esta acción, escasa, pero cierta, deba atribuirse a los fer-mentos zoológicos y no a los producidos por el hongo.
Del mismo modo procedimos con la madera de Ulmus campestris, sobre la que sedesarrollaba con esplendidez la Auricula^a mesenterica. También se encontró enpequeña cantidad vanilina.
El Polyporus hispidus vive sobre la sófora vulgar produciendo reacciones posi-tivas de separación de vanilina, a la par que amoníaco. Asimismo he visto altera-ciones causadas por Lencites, que sólo afectan a la celulosa, dejando intactas laspentosanas y la lignina (122). Esperábamos que los fermentos solubles de los hon-gos arborícolas produjesen quebrantos más hondos en la composición de la made-ra, a cuya finalidad se prepararon macerados acuoso-glicéricos de los hongos, parahacerlos actuar sobre,serrín de pino. La acción quedó reducida a separar una pe-queña cantidad de vanilina y a transformar los .polisacáridos, aunque no profun-damente, en ácidos uróriicos.
Los cultivos de Mucor y de Penic.ílium se han conducido de la misma manera,afectando levísimamente la fracción aglicónica a juzgar por las pequeñas cifrasde vanilina obtenidas. Todo ello prueba que los hongos arborícolas, causantes de ladestrucción de los bosques, necesitan para su vida hidratos de carbono, como eslógico, y que a expensas de ellos se desenvuelven lesionando los tejidos y provocandola desaparición lenta del árbol, pero nunca consumen lignina. Por modo feliz debeser así, porque de otra suerte la humanidad no hubiera podido disponer de vivien-das ni de mobiliario.
Como los resultados no eran decisivos, quizá por la .dificultad de poner en con-tacto los fermentos con la madera, acudimos a la lignina de Klason, aislada de cu-biertas elásticas de semillas (castañas) (123), y como agente transformador el con-junto de enzimas de otros hongos, cuales son especies del género Boletus y la Cla-varia flava, obteniendo después de varios días de actuación un líquido bisulfítico,donde deben hallarse sustancias de grupo carbonílico ; se encontró, después de ex-traído con cloroformo, un residuo blanquecino de olor a vainilla, en cantidad muyescasa, en que además se investigó la existencia de derivados bicarbonílicos con lanidroxilamina y acetato de níquel; sólo se vio claramente vanilina. También seencuentra vanilina en la fracción fenólica, la de sosa. La producción de este alde-hido, aunque no grande, es prueba de que los boletus y la clavaria empleados se»gregan un fermento, que rompe .las ligaduras poníales, liberando .vanilina. La ac-tividad no ha sido intensa, quizá porque este aldehido es tóxico para muchos fer-mentos y uno de ellos podría ser este que inició la ruptura de la lignina.
Hace muchos años emprendí los primeros trabajos para investigar la existenciade fermentos desmetoxilantes (anhidrolasas) (124), por suponer que algunos com-puestos naturales, los alcaloides, entre varios, se metilan totalmente en sus oxhidrilos
022) B. Regueiro. Tesis doctoral. Madrid, 1945, p. 27.(123) O. Fernández y L. Betti (inédito).
.(1.24) O. Fernández y J. M. Loredo. Rev. R. Acá. de Ciencias. Madrid, 1919. 13, 18.

- 58 -
y después pierden todos o alguno de los metilos que antes habían adquirido ; asíla morfina produce codeína y hasta tebaína y la cupreína engendra quinina. La des-metilación es obra de fermentos, que lai propia planta sintetiaadora de alcaloidesproduce ; mas se fracasa siempre que con agentes extraños se pretende la desmeti-lación, porque Ó. Friedicks (125) únicamente consiguió destruir por completo lacodeína y la narcotina con_ Pénicillium y Citramices, y Czapeks también hizo actuardiversos Aspergillus sobre la quinjna, y como antes manifesté, en mis estudios sobrela anhidrolasa con B. Coli, Proteus vulgaris, StapMlococos, jugo muscular, etcé-tera, nada conseguí de interés.
Renovadas mis experiencias con C. Alfagerne (126) tuvimos fortuna en la des-metilación de la codeína con zumo de limón y algún pequeño éxito con quinina.Es decir, que se podía intentar, con ciertas probabilidades de éxito, el ensayo defermentos desmetoxilantes, a pesar de las experiencias malogradas, ya expuestas,y de otras precisamente con lignina, también poco halagadoras de Csonka, Phillipsy Jones (127), realizadas con mucosa estomacal de buey. De la tiramira de ensa-yos practicados con. hongos lignívoros sólo el Polyporus hispid/us produjo franca-mente desmetoxilación de la codeína y de la quinina. El conjunto enzímico del ca-racol y el.de una raza vulgar de Pénicillium hidro izan los alcaloides antedichos,aunque no de modo tan ostensible como el polyporus. Era muy decisivo para nues-tro propósito encontrar un fermento hidrolítico de la. lignina ; de ahí que el restode las tentativas preliminares se encaminará a realizar la separación del metilo enla vanilina, unidad constituyente de aquélla. Aunque sin brillantez, y a.demás coninterferencia de actos oxidantes, se ha conseguido demostrar la desmetilación de lavanilina, produciendo aldehido protocatéquico, fácilmente revelabíe por el colorverde aue sobre él causa el cloruro férrico, con macerados acuoso-glicéricos dePolyporus hispldus, Lencites, Pénicillium y conjunto enzímico del caracol (128).
VII
GENEALOGÍA QUÍMICA DE LA LIGNINA—LIGNINA DE LAS MADERAS Y DE LOS FRUTOSDE CUBIERTA ELÁSTICA
La definición de la lignina con carácter general, como constituyente cíclico rr.e-toxilado de la madera y su existencia en plantas de ciclo vegetativo corto, en lasherbáceas en general, y de vida larga como en los árboles, son dos dificultadesinherentes al problema de la filogenia química de la lignina. Esta sustancia existe en
(125) Zeit, physiol. Chemie, 1914. 93, 267.(126) Farmacia Nueva, 1942. 6, 77.(127) J. Biol. Chemistry, 1929. 85, 65.(128) O. Fernández y B. Regueiro. Revis. R. Acad. Ciencias. Madrid, 1945. 39, 331,

— 59 —
forma de lámina delgada en muchos hongos; por tanto, en seres en los que se iniciala escala vital ; la contienen las gramináceas vulgares, plantas anuales, casi únicasen que se ha realizado algún ensayo de filogenia y es indiscutible la existencia delignina en las maderas de vegetales arbóreos. Lignina también indudable se en-cuentra en .las cubiertas de las semillas de frutos drupáceos producidos anualmente,y asimismo constituye las cubiertas clásticas de semillas farináceas (cupulíferas ehipocastanáceas). Esta' variedad de ligninas, señalada por algunos investigadores,presupone admitir una lignina sencilla, la protolignina de los vegetales inferiores,que en el curso de su evolución se vaya complicando químicamente, para incrus-tarse después y constituir la madera o las cañas silíceas de las gramináceas.
¿Cuál es la sustancia madre capaz de formar lignina? En los vegetales puedeafirmarse que no existe otro compuesto capacitado para formar la celulosa, primerfactor de la madera, que la glucosa resultante de la hidrólisis del almidón que enalgunas especies botánicas es perfectamente ostensible ; de la deshidratación y poli-merización de moléculas de glucosa unidas de modo semejante al de las féculas re-sulta la celulosa.
El segundo* factor de la madera en importancia cuantitativa es la pentpsana,resultante de deshidratarse dos pentosas, la xilosa y la arabinosa y de agruparse«n cadenas al estilo indicado. ¿De dónde procederán estas pentosas? Admitidocomo axiomático que en las plantas no existe otra sustancia primaria que la gluco-sa, y ofreciendo este azúcar analogía de constitución con las pentosas, es forzosoadmitir que los azúcares de cinco átomos se engendran en los de 6, a cuyo efectola hexosa ha de oxidarse, produciendo un ácido uronico, el cual descarboxiladocausa la pentosa. Este principio intermedio aparece frecuentemente en las plantasy forma las jaleas de los vegetales.
CH2 OH-CH . OH-CH. OH-CH . OH —CU . OH —GOHGlucosa
CO . OH—GH . OH-CH . OH-CH . OH - CH . OH-CO . HAc. uremico
CH2 OH-CH . OH - CH . OH-CH . OH-CO . HPentosa
No excluye este mecanismo de formación de pentosas, el de Wohl, que lasplantas utilizan para convertir la hexosa en pentosa, mecanismo que tiene en suapoyo la presencia de amidóxima en el período primaveral, para lo que se requiere elconcurso de los nitritos con la hidroxilamina ; pero el referido primer mecanismoparece el más adecuado a las exigencias enzímicas de aquellos seres. Además, losarboles sufren una enfermedad, la gomosa, atribuida por algunos a fenómenos ex-traños a la vida vegetal, la formación de goma, y por otros a un fenómeno dacefensa, para que la goma producida sirva de obstáculo a la difusión por la saviade gérmenes causantes de enfermedades. Adviériase, que las gomas europeas engran parte están constituidas por pantasanas y que 'no sería necesaria la oxidación

— 6o —
de las hexosas y la síntesis de los ácidos uránicos para engendrar pentosas y; suspolisacáridos ; bastaría con un acto hidratante que convirtiera la pentosana de lamadera en pentosa. Mas en muchas plantas existen hemicelulosas, compuestos so-lubles en álcalis, que en el fondo son ácidos uránicos ; todas las plantas formado-ras de mucílagos en sus semillas y en parte endurecidos o en torno de ligninas, con-tienen aquellas.
En la conferencia anterior aludí a un análisis de maderas de álamo y de sophorasobre las que vivieron hongos, en el que aparece gran cantidad de materias mucila-ginosas que proceden de esos actos oxidantes sobre la fracción de hexosa ; esta esla posible defensa antes aludida.
Queda, pues, pantetizada en las maderas la existencia de hexQsanas y pentosa-nas, y además conviene saber que en total forman el 50 por 100 de la madera yque se reparten aproximadamente en partes iguales los polisacáridos de cinco y deseis átomos de carbono.
Falta por estudiar el origen de la parte cíclica. Tampoco puede ser otro que laglucosa. La ciclización es un fenómeno admitido (129) para todos los seres vivos,especialmente para los vegetales y realizado a expensas de los polisacáridos ; enmuchos frutos existe un ciclanol, la inosita, procedente de la ciclización de la glu-cosa y juntamente un fenol triatómico, la floroglucina (130), formado al oxidarsey-deshidratarse la inosita (aromatización).
CJIOH COHHOHC/\ CHOFI H<y\CH
I I ! I II I i I II I I I I
HOHC\/CHOH HOC\/C.OHCHOH CH
Inosita Floroglucina
De las desoxiglucosas se podrían derivar otros fenoles, la pirocatequina y elpirogalol, por cidicación y aromatización subsiguientes.
Resulta así formado el factor cíclico, el fenol. En la definición de lignina va uncalificativo, factor cíclico metoxilado, por lo cual el constituyente no es un fenol,el ordinario, ni la pirocatequina, ni el pirogalol, sino sus anhídroles metílicos, llá-mense guayacol, aldehido vanílico o aldehido siríngico, procedentes de mediarsetotal o parcialmente los oxhidrilos. El endurecimiento de la madera se pretendevincularlo a la riqueza en metóxilos (131), no a la de lignina, aunque las maderasblandas contienen grupo vanílico unimetoxilado y las duras grupo siríngico, bime-toxilado.
(J29) O. Fernández y B. Pizarroso. R. R. Acad. Ciencias. Madrid, 1931. 26, 43.(130) O. Fernández y M. de Mingo. Ibid., 1941. 34, 349(131) J. P. Sherebow. Zentralbl., 1938. I, 918.

— 6t —
Y es idea tan generalizada entre los químicos especialistas, que Hilpert y Kru-ger (132) también relacionan la finalidad de la cubierta seminal con su contenidoen metoxilo, lo cual es motivo para suponer que la elasticidad de algunas cubier-tas depende de su escaso tanto por ciento en aquel radical. Es lógico que los auto-res prevean una relación fundada en análisis ; las cubiertas elásticas que ellos ana-lizan tienen 2,2 por 100 de metoxilo y las leñosas de los frutos drupáceos de 6-7,y además coinciden con esos números los obtenidos por O. Fernández y A. Or-tiz (133) en los soros de plantas filicíneas y en las cubiertas de amigdaláceas (me-locotón y albaricoque), por O. Fernández y J. R. Aguirre. Mas esta conside-
ración no debe llevar al espíritu un prejuicio que dificulta posteriores interpreta-ciones. Es fuerza reconocer que los autores germánicos se han preocupado de noinducir a error con la exposición de sus datos analíticos, porque al tratar de la cu-bierta de la castaña de Indias, recalcan que una cubierta con 51 por 100 de lig-nina contienen sólo 2 por 100 de metoxilo. Confirmamos esta aserciones en losanálisis de cubierta de bellota y de castaña dulce, que con 54 y 46 por 100 de su-puesta lignina producen 1,14 y 1,15 por 100 de metoxilo. Acerca de esta supuestalignina se impone una revisión, especialmente por haber encontrado en frutos dru-
páceos cantidad considerable de ácidos y alcoholes grasos.El endurecimiento requiere un examen más detenido. Considerando a la madera
como un glucósido formado por polisacárido y fenol y admitiendo una protoligninanativa no metilada, blanda, al endurecerse por adquisición de grupos CH3, éstospueden fijarse de igual modo sobre los oxhidrilos alcohólicos que sobre los fenólicos.El agente de metilación natural es el formol, que cede su CH3 a OH, para pro-ducir O.CH3 yt esto lo mismo ocurre sobre cualquier oxhidrilo, prescindiendo dela naturaleza del compuesto sobre el que se inserta. Lo más probable es que se fijepor razón cuantitativa sobre la parte de polisacáridos, metilando tres OH por cada
molécula de glucosa integrante, y aun juzgo más verosímil, admitida como está,la estructura piranósica de los azúcares, que el metileno del aldehido fórmico des-place al oxígeno piránico y que simultáneamente el grupo CH2OH que actúa desustituyente en la piranosa sufra una transposición, en virtud de la cual se engen-dre un metoxilo.
H—C—OH H—C—OH/\CH . CH,()H /\CHOCH3
I I ' I II I + CH,0 + trans. | |I I l >\/0 X/CHOH
H-C—OH H-C—OH
y aparezca glucosa metilada : es incuestionable que el último es un mecanismo másrestringido.
(132) Berichte, 1939. 72, 400.(133) Revis. Universidad. Madrid, 1942. II, V.

— 62 —
Puesto que la ciclización, como antes se ha indicado, es un fenòmeno biològicoadmisible, las unidades de glucosa motiladas pueden originar los fenoles metilados.Cualquiera de estos dos' hechos es más lógico que el propuesto por Sherebow, apropósito de la emigración de los metilos desde la parte celulósica a la lignínica,fenómeno un poco violento, porque en química orgánica se acepta la posibilidadde saltar los metilos de una~posición a otra, en una unidad molecular, es decir, den-tro de uno de los factores constituyentes de una molécula, o los oxhidrilos de unbifenol cambiar de sitio y pasar a la más estable posición mieta desde la orto, peroel salto de un metilo desde un glucósido a un anhidrol metílico1 cíclico no creo sehaya observado todavía, aunque en la vida los fermentos pueden realizar conver-siones, que sólo con fuertes temperaturas pueden lograrse en los laboratorios. Seríaobra de las transmutasas.
Contra esa facilidad emigratoria se puede aducir un hecho : la parte guayací-lica de la lignina es la productora de, vanilina, aldehido que aun conserva un oxi-hidrilo metilable. Siendo el probema de endurecimiento, dé metilación sucesiva, eslógico que se metilase la vanilina como cuando se hace artificialmente con sulfatode metilo, y entonces entre los productos de desdoblamiento o de oxidación no seencontraran productos vanílicos, sino compuestos verátricos, como ocurre en elcaso de lo artificialmente preparado, que tienen un metilo más. Esa metilación in-completa debería ser conseguida por la metilación realizable a expensas del saltodel metilo de la fracción hidrocarbonada a la lignina, según Sherebow.
A pesar de cuantas objeciones pueden hacerse mantengo un criterio favorablea la metilación de las unidades de glucosa, que después se cicliza y posteriormentese aromatiza, porque mucho tiempo se ha practicado en mi laboratorio la obtenciónde la lignina cúprica de Frendemberg (cuproxamlignina), sustancia que aun conser-va pentosanas ; una, por ejemplo, es la uva, tiene 5,31 por 100 de pentosanas y3,9 de metoxilos. La lignina de Klason, de la misma especie vegetal, como hemosescrito muchas veces, más pura, porque los ácidos destruyen la parte celulósica yla pentósica, nö contiene pentosanas, pero tampoco contiene metoxilo, de lo quedebe inferirse que el metoxilo eterifica oxhidrilos de las unidades hexosa y pentosa,que los ácidos empleados en la manipulación destruyeron por completo.
El tema no estaría todavía concluso, aunque quizá, inicialmente, se ofrezcacomo se ha presentado, pero también es posible que exista un mecanismo desme-tilante que convierta los derivados verátricos en guayacílicos. Existe un hongo, elAgarfcus campestris, que desmetila, por oxidación, el éter bimetílico 1-3 del 5 metil-pirogalol (134). ¿Hay margen para pensar en un equilibrio desde lo acíclico a locíclico?
La producción de fenol está asegurada ; queda la de la cadena tricarbónica. Eladitamento de una cadena tricarbónica a un fenol es actualmente fácil y no esimprobable que los vegetales dispongan de resortes, también fáciles para su inser-
(134) H. Richtzenhaim. Berichte, 1944. 77 B. 409.

- 63 -
yión ; la síntesis de las alkil-resorcinas prueba la singular sencillez con que una ca-dena de glicerina puede insertarse en para respecto del OH de un metoxifenol.También se conocen modos suaves de prender en un hidrocarburo aromàtico, unalkilo, sin acudir a la reacción clásica de Friedel y Crafts. Por consecuencia, la gli-cerina producida en una fermentación o el aldehido glicérico son adecuados parala síntesis de una unidad de tipo Hibbert, sobre la que pueden incidir todas lasacciones biológicas propias de plantas.
Todo esto es lo más violento, pero existen además mecanismos completamenteadmitidos, en los que se producen aldehidos fenólicos, los fundados en la conden-sación del aldehido crotónico con metilglioxalenol. El aldehido resultante de estacondensación deshidrogenado es el protocatéquico, precursor de la vanilina, quesufre la reacción de Perkin con aldehido acético y origina la cadena lateral eténica,hidratable para generar alcoholes-aldehidos.
Ya puede juzgarse por lo expuesto de la complejidad del problema de la filo-genia química de la lignina, que no ha sido abordado más que de modo fraccionario.La supuesta protolignina sólo será la molécula sencilla, producto de deshidratarsecelulosa y pentosas con compuestos guayacílicos ; la edad de los vegetales comple-taría la polimerización de los derivados guayacílicos, siríngicos y piperonílicos y lametilación sucesiva, si por acaso fuese cierto, que la dureza de la madera es pro-porcional al número de metoxilos.
Lo evidente es que se conoce lignina sencilla, de vida corta, porque es cortotambién el ciclo vegetativo de las plantas ; las gramináceas producen ligninas ; sutallo y sus glumas contienen esta sustancia desde la sexta semana de crecimientoen proporciqn de 2-6 por 100, hasta la semana decimoquinta, en la que alcanza 'acifra de 11-12 por 100 (135). Los árboles de la familia de las coniferas mantienenconstantemente la cifra inicial de las ligninas, mas no se olvide que la genéticainfluye en la composición química del producto que aquí se estudia.
Hemos acometido el problema filogénico de la lignina empezando a estudiar lasligninas-de las semillas semiduras, desarrolladas en un ambiente azucarado y pro-tegido del exterior por una cubierta cérea y blanda. Tal es la uva (Vitix vinifera),elegida porque en este fruto (baya) se encuentran todos los elementos que contri-buyen a la formación de lignina en la semilla. La cubierta, constituida por etólidos,protege a la pulpa azucarada, en la que se va formando la semilla oleaginosa ; deeste azúcar, glucosa, ha de salir todo lo que sea elemento constructor del edificiomolecular de la lignina.
En el mismo caso se hallan los frutos drupáceos de texta dura, más parecidoque la de uva, en su consistencia, a la madera de árboles.
La semilla se constituye en un ambiente azucarado, en el que existen tambiénhemicelulosas (materas pécticas) en mayor abundancia 'que en las uvas no expre-samente seleccionadas para conservación. Son, pues, dos tipos diferentes de ligninas.
(135) Phyllips. Citado por Hibbert, etc. Amer. Gli. Soc. .1943. 65-1.195.

- 64 -
Además, estudiamos las cubiertas elásticas de semillas farináceas (castaña vul-gar, castaña de Indias y bellota), que representan un tipo de lignina muy sencilla,que por su blandura debe ser menos metilado ; he aquí un. tercer tipo de lignina.Queda otro para más adelante, el de los frutos capsulares, como el cacahuet, rico enmaterias pécticas.
No es inverosímil que a estos diversos tipos, existentes en diversas especies ve-getales, respondan los obtenidos por disolución en ácidos lioglicólico y tiohidra-crílico de las formas diploide y triploide del Populus -tumula (136).
Sorpresa inesperada fue, en primer término, la coincidencia de que el polvo delepicarpio de la baya de uva diese una cantidad de lignina superior a la de las ma-deras 49,2 por 100 (137) ; se trata sólo de una coincidencia, porque esa clase decubiertas está constituida por etólidos de composición completamente ajena a lade las maderas. El hecho se reiteró en las cubiertas elásticas de castaña de Indias,castaña dulce y bellota, cupo aparente contenido en lignina Klason es mayor queel de las maderas. La lignina de uva carece de metoxilos. La semejanza en la con-ducta de estas cubiertas y la de la uva frente a la mezcla ácida continúa en otroaspecto ; el de las hemicelulosas, que en la uva se hallan en la pulpa del fruto yen las cupulíferas casi adheridas a un conjunto de pelos, que forman una especiede membrana pegada a la capa más externa del epicarpio.
La cubierta de castaña dulce contiene 46 por 100 de supuesta lignina y la debellota 54 por 100, con un contenido en metoxilo de 1,15 por 100 (138).
Las cubiertas de frutos drupáceos van aproximándose más en su, composicióna las maderas de árboles. La riqueza en lignina Klason, de la de albaricoque, esde 30 por 100, y el de metoxilo en esta lignina es de 10,44. En la de melocotón,33,40 y 12,4 por 100, respectivamente. El metoxilo, directamente obtenido del pol-vo privado de diferentes sustancias por extracción con disolventes, es de 5,035por 100 (139).
La producción de los lignoles (140), aislables por destrucción de la parte hidro-carbonada de la madera con ácidos acético y clorhídrico a 106°, quizá fuera asi-milable en su objetivo a la Klason, cori la diferencia de producirse en un casoderivados acéticos y en otros pura lignina. No siempre se muestra la analogíaclara, y en el problema de la filogenia todavía se rodea de mayor incertidumbre.Los lignoles, formados a expensas de la cubierta de semillas/ elásticas, son polvotenue que atraviesa los filtros, como muchos esteres acéticos, los de amilosa, entreotros, y la cantidad resultante 1,55 por 100 hace presumir que la fracción aromáticade esta cubierta es pequeñísima.
(136) Brov. Holmberg. Ark. f. Kemi, 1947. 24 A, n.° 29.(137) O. Fernández y P. Sánchez-Gavito. Revista R. A. Ciencias. Madrid, 1948. 42, 441.(138) O. Fernández y L. Betti (inédito).(139) O. Fernández y J. R. Aguirre. Inédito.(140) Pauly. Berichte, 1943. 76, 376.

- 65 -
La etanolisis de la semilla de uva produce el 5 por 100 de etanol lignina y delas fracciones separadas, carbonílica, ácida y fenólica, sólo la primera y la ùltimaproducen, reacción de vanilina (con floroglucina clorhídrica, color rojo).
En los frutos drupáceos, la producción de lignoles va creciendo y su parte com-plementaria en metoxilos también sufre considerable incremento, pero estos ligno-les hervidos con potasa alcohólica, como se hace con los ácidos lignino-sulfónicos,no producen más que indicios de vanilina ', o más bien de una reacción en quepuede intervenir vanilina, pero sospechosa de otro aldehido, que continuamente seve en las manipulaciones con la drupa de albaricoque, aun después de extraída poralcohol metílico hirviente ; se trata de modo indudable de un aldehido que confloroglucina clorhídrica causa color violáceo y luego purpúreo, pero con zonas ro-jas. Los primeros colores los atribuí en seguida al oximetilfurfurol, que se hallaen muchas pulpas dulces y que han demostrado químicos ingleses en muchassuertes comerciales de caramelos. La constancia con que se nota esta reacción in-duce a sospechar sí los constituyentes cíclicos de las ligninas intermedias no sonpropiamente homocíclicos, sino heterocíclicos, como el furfurol. Claro es que estainterpretación en nada es • relacionable con los anillos furánicos, que se admitenen la constitución de la lignina, porque éstos se forman en deshidratadones quefueron analizadas en la primera conferencia cerca de las ligninas. Apoya estemodo de ver el hecho un poco aislado en la bibliografía, que la cuasina, principioamargo obtenido del leño de Cuasia amara, produce la reacción aludida con floro-glucina clorhídrica.
VIII
IMPREGNACIÓN DE LAS LIGNINAS. FACTORES MEDICINALES DERIVADOS DE ELLAS(ANTOCIANOS, ANHÍDROLES METILÉNICOS, ESENCIAS Y ALCALOIDES)
FUNCIÓN BIOLÓGICA. APLICACIONES DE LA LIGNINA
La lignina constituyente de las maderas se endurece por metilación como seha indicado con varios motivos en anteriores conferencias; se va polimerizandoc°n la edad en los árboles y haciéndose cada año más resistente, pero sin cam-bar, al menos en las coniferas, la cantidad producida en los primeros años.
Es evidente que se combina con la glucosa y las pentosas o con sus polímeros,celulosa y pentosana para integrar compuestos más estables, la lignocelulosa, queperduren a través de la vida del árbol. Pero glucósidos y fenoles poseen oxhidri-°s libres que los capacitan para contraer combinaciones con sustancias minerales
por coordinación, que contribuyen a dar dureza a la madera ; es el comienzoi óüelenorneno de incrustación, durante el cual todo género de actividades químicas
se elercita en torno al monómero del glucósido lignínico. Desde la fijación de la
"£v· DE LA REAL ACADEMIA DE CIENCIAS.—1949.

— 66 —
sílice, hasta la síntesis de materias semejantes a las céreas, ácidos grasos! y alco-holes de alto peso molecular, que protegen de la acción del agua ; todo ocurre aexpensas de glucosa y de los monómeros, vanilina, y aldehido siríngico.
Aunque la madera no es por su particular contextura un laboratorio de sínte-sis, ni un arsenal de principios dotados de acción terapéutica, los leños figuranen todos los tratados de farmacognosia y se han empleado durante tiempo. Ade-más, de ellos se extraen esencias de olor grato, por destilación en corriente devapor, y de otra parte, muchas maderas adquieren grani interéte en el decoradodel mobiliario, por sus bellos colores.
La estructura de la lignina, enunciada en conferencias anteriores, hace posi-ble la existencia de compuestos, considerados especies químicas relacionadas conaquélla, bien de productos vanílicos y siríngicos, bien de otros más o menos pró-ximos a ellos, pero no faltan sustancias que enj nada se parecen a las de arqui-tectura vanílica, siríngica o piperonílica.
En la serie botánica aparecen en primer término las coniferas como procî)iicto-ras de una esencia, que siendo exhalable por las hojas aciculares, de los árboles,se forma en el parénquima leñoso ; por incisión fluyen las trementinas, de las queel vapor de agua caliente separa una esencia constituida por pineno, dextro o le-vógiro, según la especie botánica, acompañado de mínima cantidad de nopinenodesviador a la izquierda del plano de polarización de la luz. Escasas plantas prp-ducen nopineno dextro-rotatorio.
Ninguna relación guarda con los componentes del monómero lignina porqueel pineno es un terpeno bicíclico ; sin embargo, el pineno puede pasar a limonenomonocíclico, con cadena propilénica lateral en para, dato que sirve de enlace conlos lignanos.
La trementina, luego de privada de la esencia, se integra por colofonia cons-tituida por ácidos de carácter fenantréinico, alejadísimo, por tanto, de la compo-sición de la lignina ; los ácidos abiéticos y dextro y levo pimáricos con cadenalateral propilénica, como lignina.
HC
COOH CH3
CH2r/\/\ CH
I I III I
H„C\/r H.C
\ /\ CHí II
'C— CH<CH,
/CH,
XCH,
Compuesto de carácter terpénico es el santalol extraído de la madera de sán-dalo, Santalum álbum, aunque de constitución alcohólica. La especie botánica se

-.67 -
iialla muy alejada de la familia de las coniferas, santaláceas. Antes de la apari-ción de las sulfamidas fue empleada la esencia en el tratamiento de la blenorragiay de la cistitis. Contiene como mínimum el 90 por 100 de santalol, además de nor-tricicloek-santalano, y la madera el 6 por 100 de esencias.
De otras especies'de santalum se aislan esencias más pobres en santalol y sóloutilizables en perfumería.
Sin alejarse de la serie terpénica considérese antes en los látex cauchíferos devarios árboles la especie química formadora de un grupo extremadamente intere-sante, del polímero caucho. Es el menomerò isoprene, metil-2-butadieno.
ICH3
en inmediata conexión con terpenos monocíclicos como el limoneno y con muchasesencias y aun vitaminas como las A y, pro vitaminas como los licôpenos.
Los árboles productores de caucho son numerosos y explotables en extensionesenormes de Brasil, Malaca, Filipinas, etc. Aunque la industria en desusado em-puje ha procurado síntesis fáciles de caucho, la producción es tan considerable queno hay posibilidad de competencia más .que en casos extremos. Añádase queRusia y muchos países europeos que no cuentan con árboles como las Heveas y lasCastilloas se procuran anualmente caucho, de plantas cultivables, como el Tara-xacum Kok-Ssaghiss, y se comprenderá cómo el caucho de síntesis, ni aun lasbunas más estimable», pueden competir industrialmente con el natural. Las apli-caciones del caucho como material adheávo pasan desde los cables submarinos ala farmacia. Gran parte de los apositos se obtienen de caucho o necesitan del cau-cho para ser fijados en el cuerpo humano. Estamos viviendo la era del cauchoiniciada por Federico el Grande, con otro descubrimiento de carácter farmacéu-tico, que permitió hacer el primer par de botas para equitación, uti izado por aquelm°narca.
Más parecido que las sustancias precedentes a los monómeros básicos de lalignina es el hadfomál, aldehido coniferílico (I) extraído de la madera de roble,con una de las unidades propuestas por Klason ; el alcohol coniferílico se halla, enta coniferina aislada como glucósido y muy próxima al isoeugenol (II) del clavoae especia. El descubrimiento del hadromal evoca la sapidez y el aroma del coñac.
OH OH/\OCHa /\OCII3
I I I II I ( I ) I I «')I I I I
CU = CH . COH CH — CH . CH3
Aunque sea leño que cayó en desuso, el de sasafrás forma parte integrante de°s cuatro leños sudoríficos, y desde nuestro punto de vista es uno de los mà] itx-

— 68 —
teresantes, porque contiene una unidad piperonílica tan discutida por varios espe-cialistas como formadora de la lignina. Es el safrol
r H — n^CHjLf>M3\ U
\CH2 — C H = CHj
del que se obtiene sustancia de tanto interés como perfume, el plpercnal (aubé-pine). El safrol constituye el 80 'por 100 de la esencia y junto con él va el alcajvfor dextrogiro que también se halla predominante en el aceite del alcanfor, obtenidodel Laurus Camphora, de la misma familia de las lauráceas.
Al lado del alcanfor se encuentran terpenos, probablemente generadores suyos.
CH
Obsérvese la coincidencia en dos plantas de la misma familia botánica, delalcanfor, aunque en cantidades diferentes. En el canforero encuéntrase la acetonacanfórica cristalizada y en proporción mayor cuanto más viejo sea el árbol. Laabundancia es entonces tal que también pasa ú las hojas, de suerte que toda laplanta está impregnada del alcanfor. El alcanfor, por diversos reactivos, produ-cen cimeno con cadena propílica.
El abuso de la industria no ha influido mucho en el consumo medicinal d«analéptico cardíaco tan apreciado, porque1 la suerte de alcanfor artificial es mez-cla de dos diastómeros, y -el natural es la forma dextrogira, quizo, más activa qu«la levógira.
En la familia de las lauráceas se cataloga el árbol productor del palo de rosahembra, de origen botánico muy discutido, hasta considerarle como de la familiade las burseráceas, el Protium altissimum reservando el propiamente originario dfLauráceas para el palo de rosa macho del Ocotea caudata. Ambos leños produce?•una escinda llamada Likarí canali, de olor a limón, que contiene el 80 por 100 d«
linaloi dextrogiro, alcohol terpénico acíclico, bimetil 2-6, octabieno 2-7-ol-6
CH3 C — CU — CHj — CH, — C (OH) — CH = CH.,I " I
CH, CH,
sustancia muy alterable y de separación muy delicada de la esencia, al extrem"de que sólo conviene para el aislamiento el empleo del anhídrido tálico.
Este alcohol se desliga por completo de la formación de la lignina, porque

- 69 -
îsta constituído sobre base isoprénica, como lo revelan sus dos factores de cinco
itomos de carbono.
El mecanismo de síntesis de muchas de las sustancias precedentemente ex-puestas estriba en la fermentación de la glucosa, durante la cual se engendra
ildehido acético, que por aldolización y deshidratación subsiguientes forrea alde-
hido crotónico, del que es posible derivar falanges de compuestos orgánicos por
?1 principio llamado por K. Bernhauern (141), de constitución de combinaciones
hidroaromáticas.
Conócense maderas coloreadas que alcanzan gran interés en la industria de la
decoración por la belleza y estabilidad de sus colores y por la resistencia a la
acometida de los insectos, resistencia quizá debida a las esencias, que actúan comorepelentes. También los colores pueden ser agentes de protección, puesto que el
verde de malaquita protege las ropas contra la polilla. Durante los fenómenos
llarradios de putric(ón de las maderas se originan colores que dan nombre a ese
íenómeno ; así se escribe de la putrìción azul, llamada con este nombre por te-
ñirse la madera de azul, etc.
El color de la madera está más íntimamente relacionado con la composición
de la lignina que con la de esencias olorosas. Hibbert (142) ha pretendido redu-
cir a una; derivación común todos los constituyentes de los leños, y en este del
teñido tuvo su teoría verdadera fortuna. Los colores de las flores y de los frutos,
violetas y azules, son debidos a materias de naturaleza antociánica, que aun sien-
do de carácter glucosídico revelan claramente *su composición de sal de pirilio.
Las materias colorantes amarillas de flores tienen algún parecido con las otras,
son de origen flavónico y de estructura similar. Porque siendo flores y frutos co-
loreados por pigmentos conocidos por su variabilidad frente ácidos y álcalis, han
de ser diferentes de los que tienen los leños? En un ensayo de producción in uivo
de estos pigmentos, llega a establecer Hibbert, sin duda alguna, la posibilidad de
sintetizar los dos tipos ; representado uno por la luteolina, colorante de la gual-
da y el otro por la siringuidina, aglicona del pigmento de las lilas.
En la madera se ha visto que el papel principal lo juega la glucosa, cuya feitl-mentación debe originar, en primer término, según las idea» de O. Neuberg, metil-
g'ioxal, que condensado con el éter bimetílico del pirogalol engendra un cuerponipotético, desdoblable en aldehido siringoil acético y aldehido sirínguico, para dar
un compuesto flavílico.
' '41) Grundzuge der Chemie und Biochemie der Zuckerarten, p. 297.(142) Pyle, Crickman y Hibbert. Amer. Chem..Soc., 1939. 61, 2.198.
- 7°
CU,Oil y - " OH
CH,O/\OCHj '-f- C—OH = CHjO/\OCH,I I ! l i| I COH | |I I l i
COH
CH,Ox'\0(1 11 11 1\ /\/
CHOH. |\ r/ ^
011H.,0/ \OCH
1 i:ii, 1 1 i'
1 1\\/1
O )i
ni.,
:lOH
H30/ \01 11 11 1\/
1COI!
I /U:C\i XCH.OH-CH/ |OH " OH
El primero de estos aldehido hidratado, con los fenoles existentes en las plan-tas, la floroglucina, por ejemplo, origina una sustancia que, por deshidratacióny oxidación, causa la siringuidina, la antocianidina de las lilas.
OHCHjO/XOCHj orH O(.j,
I | OH/XOH o / r ••! oci / ' J
l i I I OH/X/XCH-/ >OH OH./V/V-/ )-oii| i I I I I ! i x 6^TH I l I X ocii,\/ + I I = I II I :i ^1 I I
H O | \ / I I I I * l l\ ' OH \/\/CHOH \/\/OH
I H OH CHOH HO
CH—COH/
OH
De otra parte, Karrer demostró que el grupo siringoílico es el característicode la siringuina de las flores citadas.
El andehido vaniloilacético, capaz de ser sintetizado por iguales mecanismosquei el siringoilacético, condensado con la floroglucina, origina un producto que,oxidado y desprovisto de metoxilos es la luteol'na, de constitución flavónica, ma-teria colorante de la gualda (reseda).
OH/\OCH,1 11 11 1\/
OH | -f
\C-H
C H — .COH/
HO
OH 0/\ HO/X/X-/I I 1 1 I I N
I I ' 1 I II I 1 1 I I
HO\/OH = \/\/OH c:o
Ninguna conexión más estrecha que la observada entre las materias flavóni-cas y antociánicas y las catequinas. Estas, probables generadoras de los taninos

—= 7' —
íloroglúcicos (143), se forman en la hidrogenación de los anhídroks metílicos deaquellos otros e integran un grupo de sustancias dotadas de gran poder astrin-gente, que -ocuparon un día lugar de importancia en la farmacognosia. Los kinos,catecus y gambires obtenidos de leños de árboles de distintas familias botánicascontienen abundantes catequinas, unos la íí-catequina y otros la /-épicatequina.
¿Qué relación puede advertirse entre esa capacidad productora de astringentescatequínicos y la de oíros compuestos olorosos y medicinales, v. gr., en los euca-liptus? Hoy no se percibe, pero sí se sabe que la especie rostrata es la generadoradel kino de Australia.
No discrepan sustancialmente de los colorantes flavónicos los contenidos en elleño de Campeche, separados del árbol, Hemaipxylwyn campechianum, que porsus cambios de color más semejan estructura antociáriica que flavónica. El llamadoleño del Brasil, de la Cesalpìnia- echinata, contiene brasilina (I), que por oxida-ción pasa a brasileína de constitución quinoidea (II).
O OHO/\/\CH2 HO/\/\CH,
I I I I I II I li I I\/\/C-OH
i \C CH,
I I/ \
(") \ /I I I
OH OH OH O
El de campeche contiene hematoxilina, que es un biciclanol, respecto de la bra-silina, y como ésta produce al oxidarse otra quinona roja, la hemateína. La his-toria química de la hematoxilina justifica un alto en la serie de derivados, obte-nidos de maderas. Muchos años ha sido uno de los pocos colorantes naturalespara teñido de algodón con mordiente y para estampados, así como para la seda.La histología, todavía a pesar del considerable número de colorantes sintéticos, uti-liza la hematoxilina, madurando a para producir hemateína y adicionada dealumina como mordiente y la química analítica, en especial para las volumetríasde alcaloides emplea la disolución alcohólica de hematoxilina.
Si la producción de esencias olorosas es perceptible fácilmente en los leños,el sabor percibe también amaritudes especiales que denotan la presencia de sus-tancias amargas, a veces de naturaleza alcaloidea, como la estricnina y la bruci-la del leño colubrino. La síntesis de estos compuestos se aparta de la estructuraJignínica y sigue las normas aceptadas para síntesis de alcaloides propuesta porRobinson. Entre los amargos típicos, aun de estructura no bien conocida, se halla la
(143) C. Torres. Tratado de química orgánica, p. 1.073. Madrid, 1945.

— 72 —
cuasia amarga, reputado tónico y febrífugo estimable en los tiempos en que no seconocía la quinina. Su estructura aun no se ha establecido con garantía de exac-titud.
Se ha prescindido en esta exposición de las sustancias existentes en las cor-tezas de los árboles, en los que por su abundancia en lignina se observa ciertarelación con los principios terapéuticos que en ellas se elaboran o exclusivamentese depositan como material de reserva o agente defensivo del árbol.
En algunos la relación entre los derivados fenilpropánicos y los en ella con-tenidos son notorias ; las canelas, por ejemplo, son ricas en aldehido cinámicoC6H5-CH = CH. GOH. En otras, como la de Winter se nota la aproximacióna los compuestos calcónicos. En gran parte de las cortezas de aplicaciones medi-cinales, hay riqueza alcaloidea sorprendente, como ocurre en las de quino, en lasde angostura, características por la presencia de estricnina y brucina, en las dequebracho, que definen el tanino y los alcaloides indólicos, etc.
Función biológica!, de la lignina
Algo tan importante como formar maderas sobre los supuestos glucósidos ypentósidos de la lignina, por incrustación de sustancias céreas y de origen mine-ral, es la de contribuir al sostenimiento del árbol, favoreciendo los cambios quí-micos, productores de reacciones diversas, en una organización aparentementepoco vitalizada.
A pesar de esta apariencia se han señalado fenómenos de óxido-reducción, atri-buïbles a la parte eténica de la cadena lateral del fenilpropeno. En los organis-mos animales se indica la activación del hidrógeno en los nucleoproteídos, quetienen en su extremo un anillo de piridiria, capaz de producir hidropiridina paraconvertirse de nuevo en piridina. Parece que esa función activadora del hidró-geno radica en la lignina o en el ciclo llamado de Krebs, y en este sentido la ac-tividad; biológica de esta sustancia es trascendental para la vida de los árbolesy de las plantas ligníferas, aunque sean de pequeño porte. Estima H. Hib-bert (144) que en la respiración vegetal el papel más relevante se ha atribuido alos ácidos cítrico e isocítrico, pero las cadenas lateras de lignina también alcanzangran interés y para demostrarlo tomó como punto de partida la analogía de losresultados que él y sus discípulos obtenían en la reetanolisis de las ligninas y loshallados por la escuela de Raistrick, con cultivos de hongos en medios glucosa-dos. En la actuación del alcohol clorhídrico puede obtenerse un compuesto deeste tipo (I) y en la de Pénicillium brevicompatílum, otro a este tenor (II).
(144) Ann. Rev. E'iochem., 1942. 11, 183.

- 73 —
OH HO OH/\OCH3 \/\/I I I II I I II I I I\/ \/COOH
CO . CO .CH3 CO . COOCH3
(I) (U)
El primero conduce al alcohol coniferílico,. que representa el primer miembrodel sistema de transporte de hidrógeno, porque todos estos compuestos con cade-na — CO . CH2 — CH2OH causan derivados alílicos o propenílicos, en los queel enlace eténico actúa de receptor de hidrógeno.
Aplicaciones èe la Kgnina
Producto tan abundante y residuo de una fabricación tan extendida como: ladel papel, no podía abandonarse como residuo inútil, incapaz? de aplicaciones.En efecto, la lignina residual de la fabricación de papel y cartón las ha halladointeresantes, nacidas de su condición fenólica. Si los fenoles se condensan conlos aldehidos, generando las resinas que forman, a la cabeza del grupo de lasmaterias plásticas, la ligniria, fenol también, puede manifestar tendencia a con-densaciones aldehídicas, para engendrar resinas, como la lignito., de propiedades
dieléctricas muy estimables.
Siete tipos de resina se preparan correspondientes a siete variedades diferen-tes (145). La lignina, unas veces sola, y otras asociada al caucho, se emplea
como plástico, que adquiere colores diversos de gran solidez.
La capacidad de log oxhidrilos, para fijar por sustitución alkilos en ellos, hasido utilizada para preparar alkil-ligninas de cualidades plásticas. La en términosgenerales denominada alkil-lignina es la ionüita empleada en la fabricación delpapel laminado, cuyo nombre comercial es arborita. La butil-lignina o butasina
es productora de resinas muy estimadas por su resistencia al calor. Especialmentelos papeles laminados adquieren importancia por ser termoplásticos y por su se-mejanda con el paprey.
Los plásticos moldeables que contienen más del 47 por 100 de lignina resistenno sólo grandes presiones, sino la acidez, aun cuando sea próxima al 25 por 100de ácido sulfúrico.
Las aplicaciones de la lignina como aislador se van generalizando lentamentey las agrícolas hasta ahora se limitan como abono de patatas.
Aparte de esos usos como material plástico la lignina y sobre todo los resi-duos de obtención de la pasta de papel, las sales del ácido ligninosiilfonico se
(145) Chen.. Abstracts., 1946. 60, 3.642.

— 74 —
emplean como conservador de carreteras; la sal calcica como agente fertilizantede gran valor, especialmente para las patatas ; la sal bárica, como despolarizadoren las planchas negativas de las baterías eléctricas y la propia lignina como sucedá-neo del humus vegetal.
Compendio de las actuales aplicaciones y de las que se presienten para lalignina pueden verse en Report of the conference at Newhaven. Ligniti. Chemistryina, utilization. 1948.




















