Kristen Zukosky - Brown Digital Repository
139
Molecular basis of congenital disorders of muscle: from genetic diagnosis to cellular modeling of actinopathies and alpha- dystroglycanopathies by Kristen Zukosky B.S., Cornell University, 2005 M.A.T., University of Virginia, 2007 Dissertation submitted in partial fulfillment of the requirements for the Degree of Doctor of Philosophy In the Department of Neuroscience at Brown University May 2015
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Kristen Zukosky - Brown Digital Repository

Molecular basis of congenital disorders of muscle: from genetic diagnosis to cellular
modeling of actinopathies and alpha-dystroglycanopathies
by Kristen Zukosky
B.S., Cornell University, 2005 M.A.T., University of Virginia, 2007
Dissertation submitted in partial fulfillment of the
requirements for the Degree of Doctor of Philosophy In the Department of Neuroscience at
Brown University
May 2015

ii
Copyright © 2015 by Kristen Zukosky. All rights reserved.

iii
This dissertation by Kristen Zukosky is accepted in its present form by the Department of Neuroscience as satisfying the dissertation requirement for the Degree of Doctor of Philosophy. Date ______________
____________________________ Carsten Bonnemann, Advisor
Neurogenetics Branch National Institute of Neurological Disease and Stroke
Recommended to the Graduate Council Date _____________
____________________________ Kenneth Fischbeck, Committee Chair
Neurogenetics Branch National Institute of Neurological Disease and Stroke
Date _____________
____________________________ Justin Fallon, Reader
Neuroscience Department Brown University
Date _____________
____________________________ Chris McBain, Reader
National Institute of Neurological Disease and Stroke
Date _____________ ____________________________
Mahasweta Girgenrath, Reader Boston University
Approved by the Graduate Council Date _____________
____________________________ Peter Weber
Dean of the Graduate School

iv
Kristen Zukosky Born January 13, 1983 In Arlington, Virginia
NINDS/NIH Residence Bldg. 35, 2A-1004 5225 Pooks Hill Rd Apt 609N 35 Convent Dr. Bethesda, MD 20814 Bethesda, MD 20892-0425 (703)919-4922 (301)451-5837 (ph) [email protected] (301)480-3365 (fax) [email protected] Education 2010-Present Ph.D. Candidate Neuroscience
Brown University and NIH Graduate Partnership Program Providence, RI and Bethesda, MD
2005-2007 M.A.T. Elementary Education University of Virginia Charlottesville, VA 2001-2005 B.A. Psychology Cornell University Ithaca, NY 1998-2001 Diploma Thomas Jefferson High School
for Science and Technology Alexandria, VA
Research and Training 2010-Present Neurogenetics Brown-NIH Graduate Partnership Program Neurogenetics Branch, Childhood Section, NINDS, NIH Advisor: Carsten Bonnemann 2004-2005 Medical Cognition Duke University, Durham, NC

v
Advisor: Ruth Day 2002-2004 Cognitive Psychology Cornell University, Ithaca, NY Advisor: Barbara Strupp Teaching and Mentoring Experience 2013 Organized and led 5-week journal club for summer students 2012 Mentored Undergraduate Summer Student 2012 Lab assistant for class taught by StemCell Technologies 2011 Coordinated and guest lectured in Molecular Mechanisms of Diseases 2006-2009 First Grade Teacher Honors and Awards 2011 Awarded Elsevier award for best poster presentation
World Muscle Society
2009 Golden Apple Award for Teaching, Scottsville Elementary Services 2012 Edited and contributed to GSChronicles
2012 Co-chair, Graduate Student Council (GSC)
2010-2012 Member, GSC Retreat Committee
2011 Co-chair, Education Committee of GSC
2011 Co-chair, High School Reunion Planning Committee
Additional Training 2014 Workplace Dynamics Series 2013 Writing and Publishing a Scientific Paper 2013 Clinical Research Training- 4 hour NIH

vi
2012 9-week Scientists Teaching Science Training Course 2010 3 day Leica Confocal Microscopy Training Publications
1. Christopher Grunseich, Kristen Zukosky, Ilona R. Kats, Laboni Ghosh, George G. Harmison, Laura C. Bott, Guibin Chen, Manfred Boehm, Kenneth H. Fischbeck (2014) Stem cell-derived motor neurons from spinal and bulbar muscular atrophy patients Neurobiology of Disease. 70:12-20.
2. Kristen Zukosky, Katherine Meilleur, Janel Johnson, Jahannaz Dastgir, Livija Medne, Marcella Devoto, James Collins, Jachinta Rooney, Yaqun Zou, Michele Yang, J. Raphael Gibbs, Richard Finkel, Lauren Elman, Kevin Felice, Toby Ferguson, Gihan Tennekoon, Bryan Traynor, Carsten G. Bönnemann Novel ACTA1 mutation identified by exome sequencing underlies a progressive scapuloperoneal myopathy (in revision, JAMA Neurology)
3. Modibo Sangare, Brant Hendrickson, Jonathan Nofziger, Kelian Chen, Amara Abdelbasset, Hammadoun Aly Sango, Amalia Dutra, Alice Schlinder, Aldiouma Guindo, Mahamadou Traore, George Harmison, Evgenia Pak, Fatoumata N’go Yaro, Katherine Bricceno, Christopher Grunseich, Guibin Chen, Manfred Boehm, Kristen Zukosky, Nouhoum Bocoum, Katherine G. Meilleur, Fatoumata Daou, Koumba Bagayogo, Yaya Coulibaly, Mahamadou Diakite, Michael Fay, Hee-Suk Lee, Ali Saad, Moez Gribaa, Andrew B. Singleton, Sungyoung Auh, Guida Landoure, Rick Fairhurst, Barrington G. Burnett, Thomas Scholl, and Kenneth H. Fischbeck (2014)Survival motor neuron (SMN) copy number distribution in Mali. Ann Neurology 75(4):525-32.
4. Katherine G. Meilleur, Kristen Zukosky, Livija Medne, Pierre Fequiere, Nina Powell-Hamilton, Thomas L. Winder, Abdulaziz AlSaman, Ayman W. El-Hattab, Jahannaz Dastgir, Ying Hu, Sandra Donkervoort, Jeffrey A. Golden, Ralph Eagle, Richard Finkel, Mena Scavina, Ian C. Hood, Lucy B. Rorke-Adams, Carsten G. Bönnemann (2014) Clinical, pathological and mutational spectrum of dystroglycanopathy due to LARGE mutations. J Neuropathol Exp Neurology. 73(5):425-41.
5. Christopher J. Klein, Yanhong Wu, Peter Vogel, Hans H. Goebel, Carsten G. Bonnemann, Kristen Zukosky, Maria-Victoria Botuyan, xiaohui duan, Sumit Middha, Elizabeth J, Atkinson, Georges Mer, and Peter James (2014) Dyck Ubiquitin Ligase Defect by DCAF8 Mutation Causes HMSN2 with Giant Axons. Neurology 11;82(10):873-8.
6. Diana X Bharucha-Goebel, Mariarita Santi, Livija Medne, Kristen Zukosky, Peter Shieh, Thomas Winder, G Tennekoon, Richard Finkel, James Dowling, N Monnier, Carsten G Bönnemann, (2013) Severe congenital RYR1-associated myopathy: the expanding clinicopathologic and genetic spectrum. Neurology 80, 1584

vii
Acknowledgements
I thank my advisor, Carsten Bonnemann, for his constant support and guidance.
He provided me with opportunities to learn in the lab as well as travel, present, and work
with patients. I also thank all the scientists in our group for their encouragement,
conversations, and scientific expertise. To Christopher Grunseich for teaching me what I
know about stem cells, for Yaqun Zou and Ying Hu for being the rocks in the lab that
know just about everything, to Jachinta Rooney, Veronique Bolduc, Eleonora, and Alec
Nickolls for their scientific experience, collaboration, and advice. And to the clinical
team Katy Meilluer, Naz Dastgir, Meganne Leach, and Sandra Donkervoort for their
patient knowledge and experience. To all the patients and families who have helped
make my research possible.
I thank my NIH and Brown colleagues for encouragement, study sessions,
scientific explorations, and fun including Kristin Webster, David Hauser, Anna Kane,
Samuel Reiter, and Karen Plevock.
I thank the NIH intramural research program, NINDS, Brown Neuroscience
Department, and NIH Graduate Partnership Program (GPP) for their funding and support.
To the Office of Intramural Traning and Education (OITE) for the opportunities and
support they provided especially Sharon Milgram, Lori Conlan, and Phil Wang.
Finally, to my family for providing never-ending love and support. I couldn’t
have done it without them. To Wonder, for helping me to laugh and showing me that
with perseverance anything is possible. And to Kyle, Dany, and Goldie for reminding me
what’s really important in life.

viii
Table of Contents
Curriculum Vitae......................................................................................................iv Acknowledgements..................................................................................................vii Table of Contents....................................................................................................viii List of Figures............................................................................................................x List of Tables.............................................................................................................xi Abstract.................................................................................................................... xii Chapter 1: Introduction
1.1: Introduction……………….………………………………..……..……1
1.2: Gene Identification: Linkage analysis combined with exome
sequencing……………………………………………………………………..….….2
1.3: Background on actinopathies...................................................................3 1.4: Cellular modeling: Induced Pluripotent Stem Cells (iPSCs)..................5 1.5: Background on alpha-dystroglycanopathies............................................8 1.6: Summary.................................................................................................17
Chapter 2: Animal models of the central nervous system involvement in alpha-dystroglycanopathies
2.1: Abstract....................................................................................................18 2.2: Introduction…………………………………………………………..…19 2.3: Introduction to alpha-dystroglycanopathies in the central nervous
system…………………………………………………………………………..…….29 2.4: Mouse models of neuronal migration defects associated with alpha-
dystroglycanopathies………………………………………………….…… ………. 24 2.5: C. elegans as a model of the axon guidance phenotype……….………..32 2.6: Zebrafish as models of central nervous system and eye involvement….33 2.7: Synaptic phenotype associated with alpha-dystroglycanopathies……….37 2.8: Drosophila as a model of glutamatergic synaptic function……….…….37 2.9: Retina and peripheral nervous system involvement…………….………38

ix
2.10: Discussion and conclusion………………………………………..…….40 Chapter 3: A novel ACTA1 mutation revealed by exome sequencing underlies a progressive scapuloperoneal myopathy
3.1: Abstract.......................................................................................................42 3.2: Introduction………………………………………………………………44 3.3: Methods…………………………………………………………………..45 3.4: Results ………………………………………………………………..... 48 3.5: Discussion....................................................................................................54
Chapter 4: Using Induced Pluripotent Stem cells to study the neurological phenotype of the alpha-dystroglycanopathies
4.1: Abstract........................................................................................................76 4.2: Introduction…………………………………………….………………….78 4.3: Materials and Methods………………………………….…………………80 4.4: Results …………………………………………………………………….87 4.5: Discussion.....................................................................................................94 4.6: Conclusion…………………………………………………………………97
Chapter 5: Conclusion
5.1: Exome sequencing with linkage revealed a new mutation in ACTA1……107
5.2: Neurons from induced pluripotent stem cells recapitulate features of the alpha-dystroglycanopathies…………………………………………………………...109 5.3: Broader impacts…………………………………………………………...110
References……………………………………………………………………………..112

x
List of Figures
Chapter 1 Figure 1. Gene identification and cellular modeling are necessary to identify molecular
mechanisms and therapies……………………………………………………………2
Figure 2. Known pathologies of actinopathies………………………………………4
Figure 3. A schematic showing the production of neurons from induced pluripotent stem cells from patient
fibroblasts………………………………………………………………..7
Figure 4. Glycosylation of aDG is necessary for binding…………………………….9 Chapter 2
Figure 1. DAG1 is cleaved into aDG and bDG………………………………………21
Figure 2. Glycosylation of aDG………………………………………………………23 Chapter 3 Figure 1. Expanded six-generation pedigree………………………………………….61 Figure 2. Clinical photographs………………………………………………………..63 Figure 3. Muscle MRI and Ultrasound……………………………….……………….65 Figure 4. Muscle biopsy histology…………………………………….………………67 Figure 5. Molecular implication of the E197D mutant on F-actin assembly…..….…..69 Supplementary Figure 1. Transfection of WT-, E197D- and D286G-GFP constructs into COS-7
cells…………………………………………………………………………………..71
Supplementary Figure 2. Zebrafish injected with WT-ACTA1 or E197D-ACTA1…73
Chapter 4 Figure 1. Characterization of induced pluripotent stem cells (iPSCs)………………..99 Figure 2. Increased fusion in patient embryoid bodies……………………………. 100 Figure 3. Electrophysiology showing changes in properties over the time of differentiation………………………………………………………………………...101 Figure 4. Immunocytochemistry of differentiated neurons at 8 weeks in vitro….…..102

xi
Figure 5. Glycosylated alpha-dystroglycan using IIH6 antibody in differentiated neurons……………………………………………………………………………….103 Figure 6. Spontaneous activity of control and patient derived neurons after 10 weeks in vitro…………………………………………………………………………………..105 Figure 7. Hippocampal slice recordings from Nestin-cre/DG-null (KO) mice and cre-negative (WT)………………………………………………………………………………….106

xii
List of Tables Chapter 1 Table 1. Causative genes of alpha-dystroglycanopathies and associated phenotypes….7 Chapter 2 Table 1. Mice with CNS Phenotype…………………………………………………….26 Table 2. Zebrafish models of aDGs……………………………………………………..36 Chapter 3 Table 1. Patient neurological exams…………………………………………………….58 Chapter 4 Table 1. Patient information…………………………………………………………….88

xiii
Abstract
The aim of this thesis research is to identify and model neuromuscular diseases. I
focused on two particular congenital neuromuscular diseases. Among the many genes
that are important for muscle and neuronal function, actin and alpha-dystroglycan have
varying and diverse roles at the neuromuscular junction and in the case of a-DG, the
synapse. Here I identified a new mutation and modeled a group of diseases.
First, I identified a new mutation in skeletal actin, ACTA1, in a large family with
a scapuloperoneal slowly progressive disorder. As a group, actinopathies are a diverse
group of diseases with multiple muscle biopsy pathologies. This family further expands
the phenotypes associated with skeletal actin mutations.
Secondly, in order to further investigate the role of alpha-dystroglycan in neurons,
I reprogrammed patient fibroblasts into induced pluripotent stem cells (iPSCs) and then
differentiated them into neurons. I demonstrated that these neurons have functional
synapses without differences in number or appearance, however patient neurons showed
a decreased frequency of spontaneous activity. This model recapitulates the
hypoglycosylation of alpha-dystroglycan and provides a better model for further
investigation.
In conclusion, this thesis aims to further understand and identify the molecular
basis of neuromuscular diseases by using two diseases as models, one that was without a
genetic diagnosis and one with known genetic diagnosis but a new model system.

xiv
Understanding these two aspects of disease, genetic cause and cellular modeling,
provides insights to further investigate molecular mechanisms.

1
Chapter 1
Introduction
1.1: Introduction
Congenital diseases of the muscle (CDM) are characterized by clinical
recognition before the second year of age. They are caused by a variety of proteins of
skeletal muscle, extracellular matrix, and neuron. Here we will discuss two different
congenital muscular diseases. Actinopathies are caused by mutations in skeletal muscle
actin (ACTA1) and the alpha-dystroglycanopathies are caused by (so far) seventeen
different genes all related to the glycosylation of alpha-dystroglycan (aDG). In order to
understand the mechanisms of these two diseases it is critical to identify the genetic
causes and study appropriate model systems (Figure 1).

2
Figure 1. Gene identification and cellular modeling are necessary to identify molecular mechanisms and therapies.
First we studied a large family with an unknown neuromuscular disease originally
described by Armstrong et al (Armstrong, 1966) as having a slowly progressive form of
spinal muscular atrophy. We were able to use linkage analysis combined with exome
sequencing to reveal a novel mutation in ACTA1, thus expanding the phenotype of
actinopathies.
Next we turned to elucidating the mechanism of the cognitive impairment
associated with some forms of alpha-dystroglycanopathies. To do this we first needed to
identify an appropriate model system and found that using induced pluripotent stem cells
(iPSCs) would recapitulate disease hallmarks.
1.1: Gene Identification: Linkage analysis combined with exome sequencing

3
To identify the gene responsible for disease we turned to the latest genetic tools to
combine linkage analysis with exome sequencing. Linkage analysis uses informative
single nucleotide polymorphisms (SNPs) spaced evenly across the genome to determine
inheritance patterns of regions. A LOD score (logarithm of odds) is calculated to
determine the odds that the loci are linked together and anything greater than 3.0 is
traditionally accepted as evidence of linkage.
As opposed to looking for linkage in regions, we also sequenced all of the exons
across the entire genome for complete exome sequencing. This method looks for
common SNPs and then compares them based on the inheritance pattern. Of the total
number of SNPs identified, the ones that are non-synonymous (would not change the
amino acid residue) or are found in the database of SNPs found in the common
population (dbSNP) and are therefore presumably non-disease causing, are elimated.
Then a tool like SIFT (sorting intolerant from tolerant) may be used to further predict
protein folding changes. After each of these filtering tools, only a subset of SNPs are left
for further investigation into whether they are causative of disease.
By combining these two approaches we were able to use the latest powerful
technology to identify only one variant in exon 4 of ACTA1.
1.3: Background on actinopathies
ACTA1 is a highly conserved actin expressed in skeletal muscle, and was
orginally cloned and characterized in 1983 (Gunning and Ponte et al, 1983). Actins are
structural proteins necessary for muscle contraction, and mutations throughout the
skeletal ACTA1 have been collectively described as actinopathies (Goebel and Laing
2009). Actinopathies are a heterogeneous group of disorders characterized by muscle

4
weakness and a spectrum of severity. Known histological phenotypes in muscle biopsy
include nemaline myopathy, intranuclear rod myopathy, core myopathy, actin
aggregation, and fiber type disproportion.
Figure 2. Known pathologies of actinopathies. From Laing et al, 2009.
To date over 200 mutations have been described in ACTA1 with the vast
majority acting in a dominant fashion, although a handful are recessive (Nowak,
Ravenscroft et al. 2012). Mutations can cause birth problems such as arthrogryposis and
hypotonia.

5
The family we studied did not have any of the muscle biopsy features commonly
associated with actinopathies and also had unusual clinical features as it presented as a
slowly progressive scapuloperoneal weakness with foot drop and scapular winging. This
family expands the phenotypic range of actinopathies. We will discuss this in depth in
chapter 3.
1.4: Cellular Modeling: Induced Pluripotent Stem Cells (iPSCs)
The realization by Drs. Yamanaka and Takahashi in 2006 (Takahashi, Yamanaka,
2006) that stem cells could be induced from patient cells revolutionized the stem cell
field (Nishikawa et al, 2008). No longer would scientists need to use the controversial
embryonic stem cells, but rather scientists could produce stem cells using pluripotent
factors from the blood or fibroblasts. Pluripotency can be defined as self-renewing cells
that have the ability to form all cell types in the human body (Pera, 2010). This can be
subdivided into totipotency, the ability to form all cells in the body as well as the placenta
and umbilical cord. Multipotency can form multiple cell types such as hematopoetic
stem cells that can form both myeloid and lymphoid lineages.
Yamanaka first reported that mouse fibroblasts could show pluripotent properties
after pairing down from a screen of 24 candidate factors thought to induce embryonic
properties. The four final factors that were used were Oct 3/4, c-Myc, Sox2, and Klf2.
The cells produced by using these four factors could then reproduce like stem cells and
they could form all three germ layers as shown by the creation of a teratoma in mouse
cells (Takahashi, Yamanaka, 2006). These cells when implanted in a mouse can form a

6
teratoma with endoderm, ectoderm, and mesodermal layers. The cells give similar
microarray data and appear comparable to embryonic stem cells. When analyzed, the
karyotypes of both the patients and iPSCs are identical with only some exceptions. To be
used in patient treatments, especially cell replacement therapies, researchers must make
certain that the genetic material does indeed remain the same.
When these methods were first described, retroviral vectors were used to deliver
the necessary factors. However, these are known to produce carcinomas in mice, so
alternative methods were essential if stem cell treatments in patients were ever to be a
possibility. Soon cre-excisible vectors were designed which include the four factors
between lox-p sites that with the addition of the cre recombinase could recombine to
remove the four factors so they would not be constitutively active (Hockemeyer et al,
2008). Subsequently dox-inducible vectors were produced to only express the four
factors after the addition of the drug doxycyclin. These methods were still not ideal
because the vectors still integrate into the genome and may cause long lasting effects that
are not easily identifiable. Therefore non-integrating methods (Stadtfeld et al, 2011)
were sought and an mRNA method has been shown to be effective (Warren et al, 2010).
This method requires modified mRNA as well as interferon repressors so the cells
translate protein from the mRNAs. Recently microRNA methods have been reported
which can also produce pluripotent clones from patient cells (Miyoshi et al, 2010).
Some problems still exist with using stem cells. There are clone to clone
differences such that not all colonies that are produced are the same. Some clones have a
tendency to differentiate into certain germ layers more readily than others (Liu, 2008).
Also, individual clones are not stable for approximately the first ten passages after they

7
are produced. Perhaps this is because some colonies express different markers that may
or may not encourage pluripotent properties that are weaned out with multiple passages.
The human induced pluripotent stem cell field still lags behind the mouse field.
Human cells are more difficult and take longer to reprogram presumably due to the
slower dividing time and slower maturation of cells (Liu, 2008). Also, even though the
teratoma and embryoid body differentiation provide evidence of pluripotency, researchers
are yet unable to produce germline chimeras that provide proof that the cells can produce
functional derivatives (Pera, 2010; Okita et al, 2007).
Once the pluripotent cells and reproducible protocols were established researchers
began using them to produce a variety of cell types from neurons (Hu, Zhang, 2010), to
macrophages (Senju et al, 2011), and glia (Yuan et al, 2011).
Figure 3. A schematic showing the production of neurons from induced pluripotent stem cells from patient fibroblasts.
iPSCs and neurogenetic diseases

8
Human pluripotent stem cells have been produced from a variety of neurological
diseases (Park et al, 2008) including Rett syndrome (Marchetto et al, 2010), Alzheimer’s
disease (Yagi et al, 2011), amyotrophic lateral sclerosis (ALS) (Dimos et al, 2008),
Parkinson’s disease (Soldner et al, 2011), and spinal bulbar muscular atrophy (SBMA)
(Grunseich and Zukosky et al, 2014), etc. Not only were researchers able to produce
neurons from these patients, but they also found phenotypes to further elucidate the
mechanism of disease. In the case of Rett syndrome, Marchetto et al found that neurons
had fewer synapses and smaller networks which could be partially corrected by applying
IGF-1 and gentamycin (Marchetto et al, 2010). Additionally, specific subtypes of
neurons can be derived for more specificity in diseased cells including dopaminergic
neurons (Kriks et al, 2011), motoneurons (Chambers et al, 2009), and peripheral neurons
(Greber et al, 2011).
Here, we use iPSCs to study the synaptic phenotype of the group of diseases
collectively known as the alpha-dystroglycanpathies.
1.5: Background on alpha-dystroglycanopathies
Alpha-dystroglycanopathies are a group of congenital muscular dystrophies
characterized by a decrease in a specific form of glycosylation of alpha-dystroglycan
(aDG)(Godfrey, Clement et al. 2007). aDG is an important extracellular matrix receptor
in muscle and central nervous system neurons, causing both a muscle and brain
phenotype in patients when defective. The severity of the central nervous system
involvement varies widely from very severe brain malformations in Walker-Warburg

9
syndrome indicative of abnormal neuronal migration to mild learning disability in a limb
girdle muscular dystrophy and varying degrees of intellectual disability with normal brain
imaging in between, suggesting synaptic involvement (Clement, Mercuri et al. 2008). At
the least severe end of the spectrum the onset of disease is later and patients may or may
not have cognitive impairments. Thus far seventeen genes have been implicated in the
alpha-dystroglycanopathies, although many patients remain undiagnosed and potentially
more genes remain to be discovered.
Figure 4. Glycosylation of aDG is necessary for binding. (a) Glycosylated aDG binds with laminin, neurexin, and agrin. (b) Deficiency of glycosylated aDG shown on muscle biopsy of WWS as shown by no recognition of IIH6 antibody. (c) Micropthalmia of zebrafish with deficient alpha-dystroglycan. (d) Brain white matter changes and overmigration. Adapted from Reed, 2009; Willer et al, 2012; Roscioli et al, 2012.

10
Patients are identified on the basis of hypoglycosylation of aDG (Matsumoto,
Hayashi et al. 2005; Hewitt 2009) on their muscle biopsies. However, the phenotypes
associated with aDGpathies vary widely (McNally and Pytel 2007; Chandrasekharan and
Martin 2010; Collins and Bonnemann 2010; Godfrey, Foley et al. 2011). All phenotypes
are caused by this hypoglycosylation which impairs its binding with laminin, neurexin,
agrin and perlecan leading to muscle (b), eye (c), and brain (d) phenotypes (Figure 2).
However, the disease is clinically very heterogeneous and the genotypic-phenotypic
correlation remains unclear (see Table 1) (Ervasti et al, 1994; Reed, 2009).
Walker-Warburg syndrome (WWS) is the most severe disease associated with the
aDGpathies. Onset of this disease is prenatal or congenital with severe brain and
muscular abnormalities (Preuss, Heckmann et al, 2010; Khalaf, Tareef, 2006). The brain
malformation characteristically includes lissencephaly type II (also referred to as
cobblestone complex) caused by an overmigration of neurons beyond the lamina limitans
of the developing brain (Moore, Saito et al. 2002). Polymicrogyria and malformations of
the brainstem and cerebellum can also be seen. This can be caused by mutations in
POMT1 (de Bernabe et al, 2009), POMT2, POMGNT1, LARGE, FKRP, ISPD,
POMGnT2, POMK, TMEM5, GMPPB, B3GnT1, B3GnT2, or FKTN (Preuss, Heckman
et al, 2010; Khalaf, Tareef , 2006; MacLeod et al, 2007; Kerr, 2010; Judas et al, 2009;
Lommel, Willer et al, 2008; Vuillaumier-Barrot et al, 2011; Van Reeuwijk et al, 2010;
Cotarelo et al, 2009).

11
Muscle-eye-brain (MEB) disease is slightly less severe with some patients
obtaining the ability to walk however gross brain abnormalities cause severe cognitive
and muscular impairment (Santavuori et al, 1989). Many patients have congenital
cataracts requiring early surgery and retinal involvement (Santavuori et al, 1989). This
can be caused by mutations in POMT1, POMT2, POMGNT1, LARGE, FKRP, ISPD, or
FKTN (Preuss, Heckman et al, 2010; Khalaf, Tareef , 2006; MacLeod et al, 2007; Kerr,
2010; Judas et al, 2009; Lommel, Willer et al, 2008; Vuillaumier-Barrot et al, 2011; Van
Reeuwijk et al, 2010; Kirschner, Bonnemann, 2004; Miyoshi, Ishii et al; Brockington,
Torelli et al, 2010).
Fukuyama congenital muscular dystrophy was originally described in Japan on
the basis of a founder mutation in FKTN (Kobayashi, Nakahori et al 1998). It is very
similar to MEB though the eye involvement is usually less pronounced. Limb girdle
muscular dystrophy, MDC1C is a congenital disease that is severe at birth but has no
brain involvement (Balchi et al, 2005) and is caused by a mutation in FKRP (Chang et al,
2009).
Most of the diseases associated with aDG are secondary dystroglycanopathies
because the causative mutation is not located in DAG1 itself but rather one of sixteen
associated genes. Most of these genes produce the enzymes that help glycosylate aDG at
a specific mannose residue in the ER and Golgi. The genes currently reported are
POMT1, POMT2, FKRP, POMGnT1, LARGE, GMPPA, FKTN, POMK, GMPPB,
ISPD, B3GNT1, B3GNT2, TMEM5, GTDC2, DPM3, DPM2 (Preuss, Heckman et al,
2010; Khalaf, Tareef , 2006; MacLeod et al, 2007; Kerr, 2010; Judas et al, 2009;
Lommel, Willer et al, 2008; Vuillaumier-Barrot et al, 2011; Van Reeuwijk et al, 2010;

12
Kirschner, Bonnemann, 2004; Miyoshi, Ishii et al; Brockington, Torelli et al, 2010).
However many patients showing the characteristic hypoglycosylation in muscle remained
unidentified, so presumably genes will continue to be identified.
The most recent genes were identified using exome sequencing in patients
ascertained as having the characteristic phenotype of Walker-Warburg syndrome and
muscle-eye-brain disease (MEB) including muscle weakness, severe brain
malformations, and eye dysfunctions. After genetic confirmation, these genes were used
to screen and diagnose other less severe undiagnosed patients with aDGpathies.
The only reported case of a mutation in DAG1 itself came from Dr. Campbell’s
lab (Hara et al, 2011). This mutation was located in the mucin domain and interfered
with the glycosylation of aDG and subsequently interfered with the LARGE glycan. It
thus defined the first primary alpha dystroglycanopathy as the mutation is in the DAG1
gene itself. The patient had a limb girdle muscular dystrophy presenting at three years of
age. She is ambulant only for short distances and she has a below average IQ of 50, but
her MRI shows no structural abnormalities. Before this report many researchers thought
that the glycosylating enzymes could have other functions that may not be related to α-
DG, and while that still may be the case, this paper showed that a mutation in DAG1 is
sufficient to explain both the muscular dystrophy and cognitive impairments as described
in the secondary diseases.

13
Table 1. Causative genes of alpha-dystroglycanopathies and associated phenotypes. Adapted from Godfrey et al, 2011.
Phenotypic categories proposed by Godfrey et al 2007
Congenital muscular dystrophy (CMD) Limb girdle muscular
dystrophy
WWS/WWS-like
MEB/FCMD-like
CMD CRB
CMD MR
CMD no MR
LGMD MR
LGMD no MR
Causative
genes
POMT1
POMT2
POMGnT1
FKTN
FKRP
LARGE
DPM2
DPM3
ISPD
POMGnT2 (GTDC2)
DAG1

14
POMK (SGK196)
GMPPA
GMPPB
B3GNT1
B3GNT2
TMEM5
Modeling of alpha-dystroglycanpathies
The muscle phenotype of this disease is relatively easier to study because primary
tissue from muscle biopsies can be cultured as myoblasts. However, it is more
challenging to study the neuronal phenotype due to the difficulty in obtaining patient-
derived neurons.
Treatments for alpha-dystroglycanopathies have remained elusive thus far.
However, the most promising results have come from the overexpression of Large in
mice (Barresi, Michele et al. 2004); Brockington, Torelli et al. 2010; Goddeeris, Wu et al.
2013; Whitmore, Fernandez-Fuente et al. 2014) to address the muscle phenotype. Even
in models without a mutation in LARGE itself such as FKRP (Vannoy, Xu et al, 2014),
when overexpressed, LARGE can result in a partial rescue and increased glycosylation of

15
α-DG. In contrast, in a knockout model of fukutin, the overexpression of LARGE led to
worsening of the muscle phenotype (Saito, Kanagawa et al, 2014). More investigation is
necessary to understand the potential of LARGE as a therapy. Additionally, because of
the tissue specificity of the transgene, these studies only evaluated the muscle aspect of
the disease as and did not address the central nervous system aspects of the disease.
However, treatments for central nervous system diseases in general have been
difficult to model due to the inability to easily culture patient neurons. Mouse and
zebrafish models are not ideal due to the differences in rate of growth and the fact that
many proteins have slightly different functions in different organisms. Ideally one would
want to study patient derived cells directly. Fortunately we now have two different
systems at our disposal to induce neurons from patient primary cells; derivation from
induced pluripotent stem cells (Takahashi et al, 2007; Takahashi, Yamanaka, 2006) and
direct conversion with defined factors from fibroblasts (Pfisterer et al, 2011; Vierbuchen
et al, 2010). In order to search for potential targets it is necessary to establish induced
pluripotent stem cells (iPSCs) as a model for studying the central nervous system
involvement of alpha-dystroglycanopathies.
iPSCs and alpha-dystroglycanopathies
In the case of the alpha-dystroglycanopathies, a congenital onset global CNS
phenotype is present, so iPSCs provide a particularly good model for this disease. When
neurons are derived from these methods they tend to act more like embryonic neurons
with low resting membrane potentials and undeveloped networks. Unlike ALS and

16
SBMA, the aDGpathies are congenital disorders with likely prenatal onset, therefore we
hope to appreciate a phenotype in even very young neurons. Using induced pluripotent
stem cell techniques, neuron-like cells recapitulate disease features and provide insights
into the mechanism of disease. These cells will have all the same genomic information as
patients and will grow like human neurons as opposed to cultured mouse neurons.
In addition, the fibroblasts obtained from aDG patients are from young children
which provides a reprogramming advantage as well since fibroblasts divide more
frequently in this population making them easier to become iPSCs. Here, iPSCs provide
a good model system for studying alpha-dystroglycanopathies.
Currently the best models for studying the CNS phenotype are mouse and
zebrafish models to look at neuronal migration defects. In addition, based on the mouse
models showing a deficit in long-term potentiation (LTP), a phenotype related to synaptic
dysregulation exists as well. A population of patients has normal MRIs, but cognitive
impairment. This project seeks to study the more subtle synaptic changes occurring in
these patients by using induced pluripotent stem cells as a model for human disease. The
fact that few models exist for synaptic function is a common problem in diseases such as
autism, schizophrenia, and others that cause synaptic changes. In these diseases it is still
unclear exactly what causes the phenotype due to the broad heterogeneity of the disease.
In contrast, the disease that I am studying does have some heterogeneity, but its most
immediate effects are on just one protein that we know of, effectively simplifying the
study of a complex neurological phenotype.

17
1.6: Summary
This thesis aims to understand the molecular basis of congenital neuromuscular
diseases from genetic identification to cellular modeling.
First, I used a genetically undiagnosed family to exploit the capabilities of
combined linkage analysis with exome sequencing to identify a new mutation in a known
gene for myopathies, ACTA1. This project shows the overlap between phenotypes
caused by a mutation which causes a neuron disease and that which is caused by a muscle
disease. This will be discussed in further depth in chapter 3.
Secondly, I used a group of alpha-dystroglycanopathy patients to establish a new
model system for studying the central nervous system phenotype. I reprogrammed
fibroblasts to induced pluripotent stem cells and then differentiated them into neurons.
Unlike previous models (discussed in depth in chapter 2), this model provides a human
based system which recapitulates features of the disease (chapter 4).
Overall, this thesis uses two congenital diseases of muscle to further understand
the molecular basis for disease.

18
Chapter 2
Animal models of the central nervous system involvement in alpha-
dystroglycanopathies
2.1: Abstract
Alpha-dystroglycanopathies are a heterogeneous group of diseases ranging from a
relatively mild muscle phenotype to a severe Walker-Warburg syndrome (WWS) with
muscle, brain, and eye involvement. To demonstrate the relationship between alpha-
dystroglycan and the neurological phenotype in patients, we reviewed the current
literature from animal models which shed light into the central nervous system
involvement. We found that both glia and neurons play a substantial role in the neuron
migration defects associated with disease, but that a more subtle synaptic deficit occurs as
well. These findings demonstrate that both mechanisms play an important role in disease
and that each may have the potential to provide insights into future therapeutic targets.

19
2.2: Introduction
Alpha-dystroglycanopathies are a group of predominantly congenital muscular
dystrophies characterized by a decrease in a specific form of glycosylation of alpha-
dystroglycan (aDG), causing both a muscle and often a brain phenotype in patients.
Alpha-dystroglycan is an important extracellular matrix receptor in muscle and central
nervous system neurons. Thus far seventeen genes have been identified that cause a
decrease in the glycosylation of aDG although the causative mutation for many patients
remains unknown, and potentially more genes remain to be discovered. Here we review
the recent literature related to the central nervous system (CNS) phenotype of alpha-
dystroglycanopathy patients and then touch briefly on the peripheral nervous system
(PNS) and retina involvement. We will primarily focus on the information based on the
animal models of this disease, specifically rodent, Drosophila, zebrafish, and
Caenorhabditis elegans.
2.3: Introduction to alpha-dsytroglycanopathies in the central nervous system
Muscle and brain defects in patients with alpha-dsytroglycanopathies range in
severity. The more mild forms include a limb girdle muscular difficulty (LGMD)
without cognitive impairment. The intermediate phenotypes include muscle weakness
and cognitive impairment but often normal brain MRI. The most severe phenotypes
include severe muscle phenotypes with milder CNS migrational abnormalities and severe
congenital muscle weakness and significant morphological brain abnormalities related to
neuronal migration defects.

20
Thus, the severity of the central nervous system involvement varies widely from
very severe brain malformations in Walker-Warburg syndrome, indicative of abnormal
neuronal migration, to mild learning disability in LGMD. In the middle of the disease
spectrum, patients may have varying degrees of intellectual disability with normal brain
imaging, suggesting a component of synaptic involvement. Surprisingly, there is no
apparent geneotypic/phenotypic correlation even though it seems the glycosylation
happens in a step-wise manner as aDG travels from just outside the nucleus out to the
membrane (Godfrey, Foley et al. 2011).
The DAG1 gene product is cleaved postranslationally into alpha- dystroglycan
and beta-dystroglycan (bDG). Beta-dystroglycan is a membrane spanning protein that
links intracellularly to dystrophin. Alpha-dystroglycan binds to bDG on the extracellular
side and has approximately 50 glycosylation sites where it is posttranslationally modified
in a stepwise manner in the ER and Golgi. The size of aDG is predicted to be 72kDa,
however due to its glycosylation its observed size is 120kDa in cortex and peripheral
nerve, 160kda in skeletal muscle, and 180kda in cerebellar Purkinje cells (Satz, Ostendorf
et al. 2010).
For proper interaction and functioning, aDG requires a specific type of O-
mannose glycosylation, which in mammals is otherwise relatively rare because this
glycosylation requires a specific sequence of amino acids (Endo 2007).
Glycosylation of alpha-dystroglycan
DAG1 is a conserved gene expressed ubiquitously, but especially in muscle and
brain. It was first cloned and characterized in 1992 (Gorecki, Derry et al. 1994). DAG1

21
is cleaved into alpha- and beta-dystroglycan (b-DG) (Uchino, Hara et al. 1996). bDG
spans the membrane and binds to aDG which interacts with laminin, agrin, and perlecan
in muscle basement membrane to stabilize the extracellular matrix, and interacts with
neurexin, in the CNS (Uchino et al, 1996; Waite, Tinsley et al, 2009). When this protein
is hypoglycosylated these interactions are unstable (Yoshida-Moriguchi et al, 2010).
Figure 1. DAG1 is cleaved into aDG and bDG. From Campbell et al, 2009.
aDG has approximately 50 glycosylation sites that become glycosylated in a
stepwise manner as the protein travels from translation just outside the nucleus to the
membrane through the ER and Golgi (McNally, Pytel, 2007). Amongst these, aDG
requires a specific type of O-mannose glycosylation for proper interaction and
functioning, which is the one deficient in the aDGpathies. Unlike N-glycans which link
to asparagine (Asn), O-glycans link to serine (Ser) or threonine (Thr). In O-mannose
glycosylation the mannose reducing terminal is attached to the hydroxyl group of Ser or

22
Thr (Endo 2007). In mammals this is relatively rare because this glycosylation requires a
specific sequence of amino acids. In contrast, in yeast this is more common because
glycosylation can occur at a Ser or Thr with less specificity (Endo 2007).
Protein O-mannosyltransferase 1 (POMT1) (Jurado, 1999) and 2 (POMT2)
(Willer, Prados et al. 2004) are located in ER and for the first step of O-mannose
glycosylation a complex of POMT1/POMT2 is required. Next, protein O-linked
mannose B1, 2N-acetylglucosaminyltransferase 1 (POMGnT1), which is located in
Golgi, forms a GlcNAcB1-2Man linkage of O-mannosyl glycans on aDG. Additionally,
GTDC2 adds the N-acetylglucosamine (GlcNac) and B3GALNT2 adds the N-
acetylgalactosamine (GalNac) (Manzini, Tambunan et al, 2012; Stevens, Carss et al,
2012). FKRP, based on sequence analysis, is a glycosyltransferase, but its exact function
is unknown. Fukutin is also located in the Golgi and interacts with POMGnT1 (Xiong,
Kobayashi et al, 2006). Knockout models of Fukutin show a decrease in the activity of
POMGnT1 so perhaps it acts as a chaperone or modifier for POMGnT1(Xiong,
Kobayashi et al. 2006).
Additionally, the phosphorylation of the O-mannose by POMK is required for
binding of the LARGE glycan (Yoshida-Moriguchi, Willer, 2013). LARGE is a bi-
functional glycosyltransferase which adds repeating units of xylose (Xyl) and glucuronic
acid (GlcA) (Inamori, Yoshida-Moriguchi et al, 2012). This is a different pathway than
POMT1 and POMGnT1 and gives rise to the IIH6 glycan. This O-mannosyl linked final
glycoepitope is vitally important to interactions with extracellular matrix proteins.
B3GNT1, FKTN, and FKRP encode putative glycosyltransferases based on sequence
homology although their exact function has yet to be determined.

23
Figure 2. Glycosylation of aDG. (a) Preparation of mannose by DOLK, DPM 1, 2, 3, or GMPPB. (b) Initial stages of glycosylation and phosphorylation of mannose. (c) Addition of LARGE glycoepitope. Adapted from Yoshida-Moriguchi and Willer et al 2013 and Hewitt 2012.
DAG1 plays a critical role in the nervous system as shown by the embryonic
lethality of the knockout mouse due to incomplete formation of Reichert’s membrane. A
variety of animal models suggest multiple mechanisms of action throughout the brain
including both a migrational and a synaptic phenotype. Animal models have played a
key role in elucidating the disease mechanism of the alpha-dystroglycanopathies.

24
The first mouse model of the disease was a naturally occurring myd mouse (Lane,
Beamer et al. 1976; Grewal, Holzfeind et al. 2001) that was later discovered to have a
mutation in the gene Large which is one of the genes responsible for glycosylation of aDG.
The myd mouse recapitulates some features of disease including abnormalities in
myelination, neuronal migration, retina, and peripheral nerve (Grewal, Holzfeind et al.
2001).
The first Dag1 complete knockout mouse (Williamson, Henry et al. 1997) was
embryonic lethal because of a failure of Reichert’s membrane, one of the earliest basement
membranes, to form or be maintained. Additionally, knockout mice of several of the
glycosyltransferases have proven to be embryonic lethal as well including Fktn-null
(Kurahashi, Taniguchi et al. 2005), Pomt1-null (Willer, Prados et al. 2004), Fkrp-null
(Chan, Keramaris-Vrantsis et al. 2010), and Pomt2-null (Hu, Li et al. 2011)(See Table 1).
This shows the importance of this specific glycosylation of aDG from very early embryonic
stages. In contrast, Pomgnt1-null (Liu, Ball et al. 2006)and Large-myd (Grewal, Holzfeind
et al. 2001)are viable.
Zebrafish and Drosophila have proven useful models as well because of rapid
reproduction and growth. Interestingly, when dag1 is knocked out in these organisms it
is not embryonic lethal as in mammals.
2.4: Mouse models of neuronal migration defects associated with alpha-dystroglycanopathies
Using light microscopy Zaccaria (Zaccaria, Di Tommaso et al. 2001) showed that
aDG is present in neurons of the cerebral cortex, hippocampus, olfactory bulb, basal

25
ganglia, thalamus, hypothalamus, brainstem, and cerebellum. In particular, the neuronal
migration defects have been studied in the cerebral cortex, brainstem, and cerebellum.
Other proteins, including the putative glycosyltransferases shown to cause aDGpathies,
for example fukutin (Ohtsuka-Tsurumi, Saito et al. 2004), are present in these brain
regions as well.
In order to overcome the difficulties with the embryonic lethality of a complete
knockout, Kevin Campbell’s lab at the University of Iowa developed a floxed-Dag1
mouse that allowed for conditional removal of the gene after the formation of the
Reichert’s membrane. This mouse has been crossed with a variety of mouse lines
expressing Cre recombinase allowing a more targeted investigation of the brain
phenotype (See Table 1).
In order to study the heterogeneity of the alpha-dystroglycanopathies multiple
models are required to recapitulate various features of the disease. For example, mouse
crosses have recapitulated the disease in varying degrees of severity for FKRP. Blaeser
(Blaeser, Keramaris et al. 2013) produced mice which displayed phenotypes ranging
from no apparent brain malformations to severe brain and eye malformations.
Additioanlly, in a conditional knockout, Satz (Satz, Barresi et al. 2008) bred
Dag1-floxed mice with a Mox2-Cre line which allowed expression of aDG until E7.5
allowing for the Reichert’s membrane to form, but providing a model of aDG loss early
in development.

26
Table 1. Mice with CNS Phenotype
Gene Mutation Cre Expression Phenotype Report
Dag1 null embryonic lethal Williamson et al, 1997
Dag1 conditional flox/flox Moore et al, 2002
Nestin-Cre neurons and glia E10.5 migration Moore et al, 2002
GFAP-Cre radial glia and astrocytes E14.5 migration Moore et al, 2002
NEX-Cre neurons of telecephalon E10.5 migration Satz et al, 2008
Mox2-Cre E7.5 migration Nguyen et al, 2013
PCP2-Cre Purkinje cells P6 infrequent heterotopia Nguyen et al, 2013
malpha6-Cre granule cells P4 normal Large myd migration Lane et al, 1976
Pomgnt1 gene trap exon 2 small cerebellum Liu et al, 2006
exon 18 migration Miyago-Sizuki et al, 2009
Fukutin null embryonic lethal Willer et al, 2004
chimeric variable Takeda et al, 2003
Fkrp null embryonic lethal Chan et al, 2010
neo-Tyr307Asn migration Ackroyd et al, 2009
P488Lneo severe Blaeser et al, 2013
E310neo embryonic lethal Blaeser et al, 2013
P488L migration Chan et al, 2010
Pomt1 null embryonic lethal Kurahashi et al, 2005
Pomt2 null embryonic lethal Hu et al, 2011

27
conditional flox/flox Hu et al, 2011
GFAP-Cre radial glia and astrocytes E14.5 migration
Hu et al, 2011
Emx1-Cre telencephalon E12.5 laminar defects, hemispheres fused
Col4a1 ∆exon 40 migration Labelle-Dumais et al, 2011

28
Basement membrane
One of the earliest basement membranes to form is Reichert’s membrane. As
stated earlier, a complete knockout of Dag1 is embryonic lethal at E6.5 due to
disturbances in this membrane (Williamson, Henry et al. 1997). Additionally, due to
disturbances in the glia limitans (the basement membrane over the brain), neuronal
ectopias form (Moore, Saito et al. 2002) and meningeal cells become located ectopically
in the developing cortex (Hu et al, 2007). Using embryoid bodies of the null mouse,
Henry et al (1998) found that the embryos do not get past the egg cylinder stage due to
frequent perturbations in Reichert’s basement membrane shown by co-staining with
laminin, collagen IV, and dystroglycan. Additionally, overexpression of rabbit cDNA
for Dag1 rescued the phenotype whereby the basement membrane was properly formed
(Henry and Campbell 1998).
Many of the glycosyltransferases associated with disease also are necessary for
basement membrane formation. This need is evident again from the embryonic lethality
of knocking out genes encoding for Fukutin, FKRP, POMT1, and POMT2. Additionally,
mutations in POMGnT1 disrupt basement membrane formation (Zhang, Yang et al.
2013). By isolating neurons from the knock-out embryos and incubating neural spheres
in matrigel, they showed that mutants do not form an intact basement membrane and do
not have the same amount of laminin-111 or collagen IV on the surface. They used a
laminin assembly assay to show decreased laminin binding on the surface of POMGnT1
knockout cells. They suggest a reduced rate of membrane assembly in the mutants

29
causes a reduction in the physical strength of the basement membane making it
vulnerable to perturbations during rapid expansion (Zhang, Yang et al. 2013).
Cortex
Using the mouse floxed Dag1 mouse, a variety of crosses with Cre mice have been used
to elucidate the mechanism of disease in cortex. Mice crossed with Nestin-Cre
(maximum expression in neurons and glia at E10.5) have hydrocephalus, glial and
neuronal heterotopias, cobblestone lissencephaly, and eye defects (Satz, Ostendorf et al.
2010). In contrast, when crossed with Gfap-Cre mice (maximum expression in radial glia
and astrocytes at E14.5) the mutants showed no eye defects and less hydrocephalus, but
they still showed aberrant migration and glial and neuronal heterotopias although fewer
than Nestin-Cre ((Moore, Saito et al. 2002); (Satz, Ostendorf et al. 2010)). For a
knockout model of earlier development, Satz (Satz, Barresi et al. 2008) crossed mice with
Mox2-Cre mice (maximum expression E7.5 throughout epiblast) which recapitulates the
broad range of phenotypes in Walker-Warburg syndrome including micropthalmia (Satz,
Barresi et al. 2008). In contrast, when the floxed mice were crossed with Nex-Cre mice
(maximum expression at E10.5 in neurons of telencephalon) the mice showed no
structural abnormalities in brain structure, normal neuronal migration, and they had intact
glia limitans (Satz, Ostendorf et al. 2010). Interestingly, the axons of these neurons, in
most cases, projected to appropriate targets but the dendrites lacked organization and
orientation (Myshrall et al, 2013).
Reduced expression of Fkrp in several mouse models with mutations in Fkrp also
leads to a clear brain phenotype with disruption of the neuronal layering of the cerebral

30
cortex and partial fusion of hemispheres suggesting this phenotype is due to
hypoglycosylation of aDG and not the knockout of aDG itself (Chan, Keramaris-Vrantsis
et al. 2010)
Taken together, these findings suggest that glia play a large role in the migration
deficits associated with this disease and that neurons may play a larger role in synapse
formation. This topic will be discussed more thoroughly in section 2.7.
Glia
Alpha-dystroglycan is expressed in radial glia and perivascular astrocyte cell
bodies and end feet (Zaccaria, Di Tommaso et al. 2001). Targeted deletion of Dag1 in
glia affects neuronal migration (Moore, Saito et al. 2002). Radial glial, in some cases, do
not extend processes due to ectopic proliferation in the ventricular and subventricular
zones (Myshrall, Moore et al. 2012) leading to additional cortical disorganization.
POMGnT1 knockout does not affect localization of DG in radial glia (Zhang,
Yang et al. 2013). In isolated neural stem cells they showed decreased laminin affinity,
but no change in localization (Zhang, Yang et al. 2013).
Cerebellum
Alpha-dystroglycan and bDG are necessary during early postnatal radial
migration and are expressed in Bergmann glial scaffolds in the cerebellum (Henion, Qu et
al. 2003). To study the phenotype in the cerebellum in particular, Nguyen (Nguyen,

31
Ostendorf et al. 2013) crossed the floxed Dag1 mice with Cre mice with promotors on
expressed in the cerebellum. Malpha6-Cre (maximum expression at P4 in granule cells)
showed no brain or behavioral phenotype and Pcp2-Cre (maximum expression at P6 in
Purkinje cells) showed very infrequent heterotopia with normal histopathology (Nguyen,
Ostendorf et al. 2013). This suggests very early involvement of aDG with no clear role in
late deletion in granule or Purkinje cells later in development.
However, in mice crossed with Nestin-Cre (maximum expression in neurons and
glia at E10.5) and Gfap-Cre (maximum expression in radial glia and astrocytes at E14.5)
there was glia limitans disruption, Bergmann glia disorganization, and heterotopia.
Interestingly, the cerebellum in Nestin-Cre mice was normal at P0, but at P3 there was
abnormal laminin staining, disrupted basement membrane, abnormally organized glial
endfeet, and ectopic cells (Nguyen, Ostendorf et al. 2013). This may be due to the rapid
neuronal and glial proliferation in the mouse cerebellum postnatally, which is why they
were normal at P0.
These studies in the cerebellum, when taken together with the conclusions found
from the basement membrane formation (Zhang, Yang et al, 2013), suggest that there
may be a rate dependence in the proper binding of laminin and migration of neurons.
Specifically, the hypoglycosylation of aDG affects the the proper binding of laminin and
other proteins, and in cases of rapid proliferation this deficiency in interaction may be
exacerbated.
Spinal Cord and Axon Guidance

32
In a feed forward genetic screen of axonal misguidance in mice, two models of
alpha-dystroglycanopathies were identified. B3gnt1 mice (double knockout embryonic
lethal) and Ispd –null mice (not required for Reichart’s membrane but die at P0 due to
respiratory failure), suggesting these proteins are regulators of axon guidance through
glycosylation of aDG. Glycosylated dystroglycan binds directly to the axon guidance cue
molecule Slit and is necessary for axon tracts growing in close proximity to basement
membrane to form the proper connections. Many commissural axons at E13.5 turned
incorrectly or did not turn at all suggesting aDG may be necessary for scaffolding of slit
(Wright, Lyon et al. 2012). This misguidance also is found in C. elegans.
2.5 C. elegans as a model of the axon guidance phenotype
C. elegans have conserved homologues of the dystrophin-glycoprotein complex
including a homologue to dystroglycan, Dgn1 (Grisoni, Martin et al. 2002). Johnson
(Johnson, Kang et al. 2006) extensively characterized the expression of dgn1 as well as a
dgn1-null. They found that unlike other models, Dgn1 is expressed in epethelia and
neurons, but not in muscle. In addition, it does not function in the dystrophin-associated
protein complex and does not have a binding domain to dystrophin, nor is it required for
basement membrane formation. It is expressed in ventral cord neurons.
The Dgn1-null animal is viable but sterile and displays a deficit in axon guidance
(Johnson et al, 2006; Johnson et al, 2012). These animals, in many cases, had at least one
commissural axon on the wrong side and a few had abnormal branching (Johnson, Kang
et al. 2006). Later the same group showed that Dgn1 is expressed in lumbar ganglion

33
neurons and the defects in axon guidance are due to follower lumbar commissure axons
(Johnson and Kramer 2012).
2.6: Zebrafish as models of central nervous system and eye involvement
Zebrafish knockdowns of genes related to alpha-dystroglycanopathies have
proven useful in providing in vivo support during identification of new genes as well as
insight into the mechanism of disease. Zebrafish are small in size (1.5 in), develop
rapidly during ex-utero development, have transparent embryos and larvae, and have a
high rate of reproduction with a short timeline between generations (Guyon, Steffen et al.
2007). Genes conserved in zebrafish are DAG1 (Parsons, Campos et al. 2002), Large,
POMT1, POMT2, POMGnT1, Fukutin, FKRP, GTDC2, GMPPB (Moore, Goh et al.
2008).
In support of a migrational defect a variety of zebrafish models of aDGpathy
show micropthalmia and hydrocephalus, which supports a role of dystroglycan in the
central nervous system. By using transient morpholino oligonucleotides (MO) to
knockdown genes, this model system has become even more useful because it provides a
unique way to study the dose dependence of these genes very rapidly (See Table 2). In
contrast, stable germ line mutations provide a more consistent and less variable
expression than MO knockdowns.
Many of the genes associated with alpha-dystroglycanopathies have been
evaluated in zebrafish including Dag1 (Parsons, Campos et al. 2002); (Lin, White et al.
2011) Fktn (Lin, White et al. 2011), Fkrp (Thornhill, Bassett et al. 2008; Kawahara,
Guyon et al. 2010; Lin, White et al. 2011) Ispd (Roscioli, Kamsteeg et al. 2012), Gtdc2

34
(Manzini, Tambunan et al. 2012), B3GALNT2 (Stevens, Carss et al. 2013), Pomt1
(Avsar-Ban, Ishikawa et al. 2010), Pomt2 (Avsar-Ban, Ishikawa et al. 2010)Gdppb (Carss
et al, 2013), Pomk (Di Costanzo, Balasubramanian et al. 2014). Each of these models
showed varying degrees of deficiency. Interestingly, Pomt2 has a more severe phenotype
than Pomt1 (Avsar-Ban, Ishikawa et al. 2010) even though they seem to be required
together as a complex for glycosylation.
Like other zebrafish mutants (See Table 2) the GMPPB knockdown produces a
phenotype at 48 hours post fertilization (hpf) including micropthalmia, hydrocephalus,
and decreased mobility (Carss, Stevens et al. 2013).
In addition, (Gupta, Kawahara et al. 2011) extensively characterized the
Patchytail fish (a germ line mutation c1700T>A, p. V567D in Dag1), which leads to the
absence of protein unlike the transient MO models. In these fish, the tectal and cerebellar
cells were less organized, but the fish did not show any hydrocephalus or neuronal
heterotopia.
The Patchytail fish and those models with high concentrations of MO to Fkrp and
Fktn showed notochord disorganization (Thornhill, Bassett et al. 2008; Gupta, Kawahara
et al. 2011; Lin, White et al. 2011)). The Fkrp model has a dose dependent variability in
severity and notochord disorganization. Lin (Lin, White et al. 2011)found that when
either Fkrp or Fktn were knocked down in zebrafish, the notochord was not able to fully
develop due to differentiation defects. At high MO doses, the Fkrp-MO and Fktn-MO
models show expression of indian hedgehog homologue b throughout the entire
notochord, instead of being properly restricted, and at later stages of development.

35
Zebrafish have consistently proved to be a good model system to screen new
genes associated with alpha-dystroglycanopathies and show a central nervous system
deficit consistent with the mouse data and human patients. When new genes are identified
in patients, zebrafish provide a relatively quick way to confirm a protein causes a
phenotype and generally produce fish with a clear but nonspecific muscle and brain
phenotype. However, zebrafish knockdowns tend to produce a more global phenotype
making them less advantageous in studying the subtleties associated with particular
phenotypes or genes. For example, as stated earlier dag1 knockouts do not cause
embryonic lethality and pomt1 and pomt2 produce different phenotypes even though in
the human they exist as a complex. Therefore zebrafish are valuable tools in providing
evidence that a certain gene produces a phenotype, but does not perfectly recapitulate the
subtleties of human disease.

36
Table 2. Zebrafish models of aDGs
Gene Mutation Phenotype Report
dag1 R388>Stop Lin, 2011
dag1 V567D Patchytail brain, ocular Gupta, 2011
dag1 MO no CNS or NMJ phenotype Parsons, 2002
Fukutin MO notochord, ocular Lin, 2011
FKRP MO notochord, ocular Lin, 2011
FKRP MO notochord, ocular Thornhill, 2008; Kawahara, 2009
ISPD MO hydrocephalus, micropthalmia
Roscioli, 2011
GTDC2 MO hydrocephalus, retina development delayed
Manzini, 2012
B3GALNT2 MO hydrocephalus, retinal degeneration
Stevens, 2013
POMT1 MO retinal Avsar-Ban, 2010
POMT2 MO brain, ocular Avsar-Ban, 2010
GDPPB MO micropthalmia, hydrocephalus
Carss, 2013
POMGnT1 MO brain, ocular Tamaru, 2014

37
2.7: Synaptic phenotype associated with alpha-dystroglycanopathies
Alpha-dystroglycan was originally identified as cranin on presynaptic side of
synaptic contacts surrounding active site at ribbon synapse in the retina (Smalheiser and
Collins 2000). Both aDG and bDG were found enriched on synaptic membranes after the
synaptosome was fractionated ((Mummery, Sessay et al. 1996; Cavaldesi, Macchia et al.
1999; Smalheiser and Collins 2000).
In addition, aDG was seen by electron microscopy on postsynaptic membranes in
the brain. It co-localized with nicotinic acetylcholine receptors (nACR) of sympathetic
ganglia that were disrupted in mutant mice (Zaccaria, De Stefano et al. 2000). Levi and
Grady et al (2002) found that aDG colocalized with ionotropic y-aminobutyric acid
(GABA) type A receptors (GABAaR) positive synapses in cultured hippocampal neurons
from the Dag1-floxed mouse seven days after the introduction of Cre recombinase.
However, they found that aDG was not necessary for the differentiation of neurons.
Moore (Moore, Saito et al. 2002) found aDG on the postsynaptic side of
structures in the hippocampus, which disrupted long-term potentiation (LTP) in Gfap-
Cre/Dag1 floxed mice. Disrupted LTP was confirmed in Nex-Cre/Dag1 floxed mice
that did not show any structural abnormalities and Large-myd mice (Satz, Ostendorf et al.
2010). These findings provide evidence for a synaptic dysregulation, which may play a
role in the cognitive impairment in patients with a milder phenotype.
2.8: Drosophila as a model of glutamatergic synaptic function
Drosophila express dystroglycan (Greener and Roberts 2000), although it is not
cleaved into two subunits. Instead, it is alternatively spliced into DG-A, DG-B, and DG-

38
C whereby only DG-C maintains the mucin domain important for glycosylation (Deng,
Schneider et al. 2003). Drosophila also express homologs of POMT1 (rotated abdomen)
and POMT2 (twisted) (Ichimiya et al, 2004). Both genes encode O-mannosyltransferases
that modulate ligand binding consistent with the human homologues (Nakamura,
Stalnaker et al. 2010). Also, they form a heterocomplex where both proteins are
necessary for producing the large molecular mass band (glycosylated) on western blot
(Nakamura, Stalnaker et al. 2010). Complete absence of dystroglycan decreases survival
of larvae (Steigmann et al, 2004), as does a Pomt1 or a Pomt2 knock down (Haines,
Seabrooke et al. 2007).
When dystroglycan expression is decreased by RNAi, Drosophila display polarity
changes, muscle defects and degeneration, and neuromuscular junction synaptic defects
((Deng, Schneider et al. 2003);(Schneider, Khalil et al. 2006; Haines, Seabrooke et al.
2007); (Shcherbata, Yatsenko et al. 2007). Drosophila NMJ is an established model of
glutamatergic function and importantly, the laminin-dystrophin-dystroglycan complex is
conserved. When dystroglycan was decreased postsynaptically, a presynaptic glutamate
release deficit was observed (Bogdanik, Framery et al. 2008). Additionally, when
Waiker (Wairkar, Fradkin et al. 2008) knocked down Pomt1 they also saw a decrease in
the efficacy of synaptic transmission without a change in the number or size of boutons.
They found that the action potentials were not defective, but instead there was a decrease
in the probability of neurotransmitter release in mutants (Wairkar, Fradkin et al. 2008).
2.9: Retina and peripheral nervous system (PNS) involvement
Retina

39
Alpha-dystroglycan binds to pikachurin at the ribbon synapse in the retina. The
eye and retina phenotypes that patients with aDGpathies often have are recapitualated in
several animal models of the disease. In zebrafish, the patchytail model has more loosely
packed ganglion layers and their lenses are not differentiated properly (Gupta, Kawahara
et al. 2011). In the POMGnT1 mouse model, the inner limiting membrane is
significantly thinner and laminin levels were reduced (Zhang, Yang et al. 2013).
Alpha-dystroglycan is present on the presynaptic side of photoreceptor cells
synapsing to ON-bipolar cells of the retina. When Dag1 was knocked out, the
electroretinogram (ERG) activity of b-waves are reduced and there were decreased
amplitudes in action potentials (Omori, Araki et al. 2012). Taken together, data from
these models suggests both a neuron migration and a synaptic phenotype in the retina.
Peripheral nervous system (PNS)
In a conditional knockout of Dag1 in Schwann cells (crossed with P0-Cre mice
which express cre beginning at P0), mice had abnormal myelination as shown by fewer
sodium channels and decreased nerve conductions. While there were no overt
differences in younger mice, mice at one year of age began developing a tremor (Saito,
Moore et al. 2003). This result shows in mice there may be a peripheral nervous system
component that contributes to the muscle phenotype as well.

40
2.10: Discussion and conclusion
Animal models have proven useful tools in elucidating the mechanisms of both
the migrational and synaptic phenotypes associated with the central nervous system
deficits in patients with alpha-dystroglycanopathies. Zebrafish have proved useful for
screening new genes to show causation although they do not distinguish subtle
differences in specific genes. Drosophila have shown electrophysiological defects
although they do not express the variety of genes associated with human disease and
additionally have a Large homologue making investigations into the mechanisms less
valuable. C. elegans have shown axonal guidance defects which have not been identified
in other models due to the high number of neurons in higher organisms making this
model invaluable in showing this phenotype. Mice have recapitulated both migrational
and synaptic defects although they don’t fully recapitulate the human disease.
Taken together, these models provide valuable insights although they still prove
insufficient to model human disease. As new and varied models become possible such as
induced pluripotent stem cells (iPSCs), perhaps drug and small molecule screens will be
possible for future treatments.

41
Chapter 3
A novel ACTA1 mutation revealed by
exome sequencing underlies a progressive scapuloperoneal myopathy
Kristen Zukosky, Katherine Meilleur, Janel Johnson, Jahannaz Dastgir, Livija Medne, Marcella Devoto, James Collins, Jachinta Rooney, Yaqun Zou, Michele Yang, J. Raphael Gibbs, Richard Finkel, Lauren Elman, Kevin Felice, Toby Ferguson, Gihan Tennekoon, Bryan Traynor, Carsten G. Bönnemann Novel ACTA1 mutation identified by exome sequencing underlies a progressive scapuloperoneal myopathy (in revision, JAMA Neurology)

42
3.1: Abstract
IMPORTANCE:
As new genomic strategies become available, they can be used to diagnose previously
undiagnosed patients and families with rare genetic conditions. This large family was
previously described in 1966 and now expands the phenotype of a known neuromuscular
gene.
OBJECTIVE:
To determine the genetic cause of a slowly progressive, autosomal dominant,
scapuloperoneal neuromuscular disorder by using linkage and exome sequencing.
DESIGN, SETTING, AND PARTICIPANTS:
Thirteen affected individuals in a six-generation family with a progressive
scapuloperoneal disorder. Participants were examined at pediatric, neuromuscular, and
research clinics. Exome and linkage were performed in genetics labs of research
institutions.
MAIN OUTCOME MEASURES:
Examination and evaluation by imaging (MRIs, ultrasound), electrodiagnostic studies,
and muscle biopsies (n = 3). Genetic analysis included linkage analysis (n=17) with
exome sequencing (n = 7).
RESULTS:
Clinical findings included progressive muscle weakness in an initially scapuloperoneal
and distal distribution, including wrist extensor weakness, finger and foot drop, scapular
winging, mild facial weakness, Achilles tendon contractures, and diminished or absent
deep tendon reflexes. Both age of onset and progression of the disease showed clinical

43
variability within the family. Muscle biopsies showed type I fiber atrophy and
trabeculated fibers without nemaline rods. Correlation of exome sequencing with the
linkage region (4.8Mb) revealed a missense mutation p.Glu197Asp in a highly conserved
residue in exon 4 of ACTA1. The mutation co-segregated with disease in all tested
individuals and was not present in unaffected individuals.
CONCLUSION AND RELEVANCE:
This family defines a new scapuloperoneal phenotype associated with an ACTA1
mutation. ACTA1 is a highly conserved protein implicated in multiple muscle
pathologies, including nemaline myopathy, actin aggregate myopathy, fiber-type
disproportion, and rod-core myopathy. Mutations in Glu197 have not been reported
previously. This residue is highly conserved and located in an exposed position in the
protein; this mutation affects the intermolecular and intramolecular electrostatic
interactions as shown by structural modelling. The mutation in this residue does not
appear to lead to rod formation or actin accumulation in vitro or in vivo, suggesting a
different molecular mechanism from other ACTA1 diseases.

44
3.2: Introduction
Scapuloperoneal syndromes are a highly heterogeneous group of skeletal muscle
and nerve disorders associated with weakness and wasting of scapular fixators and
anterior distal leg muscles(Liewluck, Tracy et al. 2013). This pattern of weakness is seen
in certain myopathies including Emery-Dreifuss muscular dystrophy, hyaline body
myopathy, and reducing body myopathy(Liewluck, Tracy et al. 2013). Neurogenic
disorders can also be present in a scapuloperoneal distribution as seen with some TRPV4
mutations (Deng, Klein et al. 2010).
In 1966, Armstrong and colleagues reported two individuals from a family with a
dominantly inherited phenotype of early onset, predominantly scapuloperoneal muscle
weakness. The disorder was classified as proximal spinal muscular atrophy on the basis
of biopsy and EMG findings interpreted as neurogenic changes (Armstrong, Fogelson et
al. 1966). The family returned for further evaluation of 13 patients from a now expanded
six-generation pedigree with 33 known affected individuals, presenting with
scapulohumeroperoneal weakness, as well as distal hand and mild facial involvement.
The disease was progressive but of highly variable severity in the family. Linkage
analysis combined with exome sequencing revealed a novel mutation in ACTA1, in a
highly conserved residue, which co-segregated with the clinical phenotype.
The actinopathies are caused by mutations in skeletal muscle actin encoded by
ACTA1. They cover a heterogeneous spectrum of clinical severity and
histomorphological expression (Schroder, Durling et al. 2004; Goebel and Laing 2009;
Nowak, Ravenscroft et al. 2012) including nemaline myopathy, intranuclear rod
myopathy, rod-core myopathy, actin aggregation, zebra-bodies, and fiber-type

45
disproportion (Nowak, Ravenscroft et al. 2012; Sevdali, Kumar et al. 2013). The ACTA1
related scapuloperoneal myopathy without nemaline rods or actin accumulations reported
in this family does not belong to any of the hitherto recognized clinic-pathologic
actinopathies, and thus expands the phenotypic range of actinopathies.
3.3: Methods
Standard Protocol Approvals, Registrations, and Patient Consents: We examined
13 affected individuals from a six-generation pedigree. Informed consent from all
subjects was obtained (IRB approval #00-N-0043 and 12-N-0095, NINDS, Bethesda,
MD). Patient consent-to-disclose forms were obtained for all photos and videos.
Neurological examination: See Table 1 for a summary of findings on clinical
examination.
Muscle biopsy and immunochemistry: Histochemical stains including
haematoxylin and eosin, Gömori–trichrome, and nicotinamide adenine dinucleotide
tetrazolium reductase (NADH-TR), were performed on 9μm frozen muscle samples. For
immunohistochemistry, sections and cells were fixed in 4% PFA, blocked in 0.1%Tx-
100/10%FBS/PBS, incubated with primary antibodies (alpha-actin Sigma, St. Louis, MO,
LamaA2 Leica Biosystems, Buffalo Grove, IL, MHC-s Leica, MHC-f Leica, NCL-alpha-
ACT Novocastra) overnight at 4˚C, incubated with AlexaFlour secondary antibodies
(Goat anti-mouse488, goat anti-rabbit 596), and mounted with Fluoromount-G
(eBioscience Inc., San Diego, CA). Images were acquired on a Nikon Eclipse Ti
(Melville, NY) epi-fluorescent microscope.

46
Muscle ultrasound: Skeletal muscle ultrasound was performed using a L12-5
linear transducer at 12 MHz on a Philips iu22 system.
Motor unit number estimation (MUNE): The number of motor units innervating
hypothenar muscles was estimated, based on published multiple point stimulation
techniques using Teca Synergy N-EP machine (Cardinal Health, Madison,
WI)(Bromberg and Swoboda 2002). Briefly, surface limb temperatures were maintained
at 31˚C. Maximum compound motor action potential (CMAP) was obtained after
repeatedly moving the G1 electrode to ensure maximum amplitude. Stimulation of the
ulnar nerve at the wrist, above the elbow, and below the elbow was performed to evaluate
for the presence of conduction block or slowing that may confound results. Individual
single motor unit potentials (SMUPs) were identified by obtaining all-or-none responses
to low-intensity stimulation. At least ten consecutive observations of an individual SMUP
were obtained, and 10 unique SMUPs were identified. The MUNE value was calculated
by dividing the maximum CMAP negative peak amplitude by the mean SMUP negative
peak amplitude. In addition, conventional nerve conduction studies and EMG (Nicolet)
were performed on one patient (IV-25).
Linkage analysis: Genotyping was performed using the Illumina Linkage IV
Panel (San Diego, CA USA), which includes 5861 informative single nucleotide
polymorphism (SNP) markers distributed evenly across the human genome at an average
distance of 0.64 cM. SNP data were analyzed using Merlin (Abecasis, Cherny et al. 2002)
linkage software version 1.1.2, assuming a fully penetrant dominant model of disease
transmission.

47
Exome sequencing: DNA from affected individuals IV-23, IV-25, V-6, V-8, V-
10, V-11, and VI-9 was enriched using SureSelect Exome target enrichment technology
(version 1.0, Agilent, CA). The enriched DNA was paired-end sequenced on a Genome
Analyzer IIx (Illumina, CA). Sequence alignment and variant calling were performed
using BWA(Li and Durbin 2009), the Genome Analysis Toolkit(McKenna, Hanna et al.
2010; DePristo, Banks et al. 2011) and Picard (http://picard.sourceforge.net/index.shtml).
BWA was used to align the paired-end sequence against the reference human genome
(UCSC hg18). The Genome Analysis Toolkit (GATK) was used to recalibrate quality
scores, perform local re-alignments around InDels, identify variants, and call genotypes.
PCR duplicates were removed using Picard prior to variant calling with GATK. Based
on the hypothesis that the mutation underlying this rare familial disease was not present
in the general population, SNPs identified in the 1000 Genomes project
(www.1000genomes.org/) or in dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP,
Build 131) were removed. We then excluded variants that were not shared by all patients.
Next, synonymous changes were identified and filtered from the variant list using SIFT
software (version 4.0, http://sift.jcvi.org/). Localization of the variant Glu197 residue on
the 3D molecular model was performed with Polyview
(http://polyview.cchmc.org/polyview3d.html) using the protein file 1j6z (Laing, Dye et
al. 2009).
Molecular Modeling: The high-resolution crystal structure of human actin (pdb-
code: 3DAW; PubMed: 18625842; Sequence identity: 100%) was used as a modeling
template. The implemented single point mutation of Glu197 to Asp197 was subject to
gradient energy minimization in CNS (PMID:18007608). The lowest energy structures of

48
the single point mutant version were subject to 500 cycles of unrestrained Powell
minimization. Harmonic restraints were imposed on the target molecule (2 kcal/mol Å2)
with increased weight (25 kcal/mol Å2). Protein Structure and Model Assessment Tools
were used to verify the quality of the modeled structure.
Transfection and Western blotting: Cos-7 cells were transfected with WT-eGFP,
Asp286Gly-eGFP (gift from Dr. Laing), and Asp197Glu-eGFP constructs using
lipofectamine (Invitrogen). Immunochemistry was performed as described above. For
the western blot, primary antibody to alpha-actin (Sigma) and secondary goat anti-mouse
IgG-HRP was used. Proteins were resolved by electrophoresis on a 4-12% gradient Bis-
Tris polyacrylamide gel and blotted according to a standard protocol.
Zebrafish studies: Wild-type and Glu197Asp human ACTA1 genes were cloned
into pCSDest vector (Addgene) as described (Villefranc, Amigo et al. 2007). Plasmids
were linearized with NotI and RNA was transcribed using the SP6 mMessage Machine
kit (Ambion). One-cell stage embryos were injected with 200pg and 400pg of RNA and
muscle histology was analyzed as described previously (Gupta, Kawahara et al. 2011)
using Acti-stain 670 (Cytoskeleton) and anti-myosin heavy chain antibody (F59, Santa
Cruz Biotech).
3.4: Results
Clinical findings

49
We personally examined 13 patients from a now expanded six-generation
pedigree, in which 33 individuals are known to be affected (Figure 1). Muscle weakness
was noted in both the lower and upper extremities. The lower extremities exhibited an
initial pattern of distal, more than proximal, involvement with early foot drop, but
proximal involvement became more pronounced with age. The overall distribution of
muscle involvement was scapuloperoneal with additional hand involvement. Early
scapular winging (Figure 2 b, f), muscle atrophy of the thenar (Figure 2c) and deltoid
muscles (Figure 2 e), and mild lower facial weakness (Figure 2i) were typical. The hand
involvement was particularly notable, including wrist and finger drop (in particular of
digits 3-5). Affected individuals developed contractures of the Achilles tendon, and some
at other locations, including elbow and shoulder eversion contractures. Deep tendon
reflexes were diminished or absent. Phenotypic variability was evident within the family
in that more severely affected patients presented with hypotonia, foot eversion and
dorsiflexion weakness from infancy (Figure 2g). Family members report early deltoid
atrophy in young affected family members in the form of a “notch” in the shoulder. In
contrast, other affected family members did not notice weakness until early adulthood.
Family members affected early ran awkwardly in childhood, then developed progressive
weakness resulting in loss of ambulation with wheelchair dependency, and developed
scoliosis in the fifth or sixth decade of life (III-4, III-7, III-9). In the most severely
affected patients (IV-23 and VI-2), marked weakness and atrophy were especially
apparent in the upper extremities with no remaining anti-gravity strength in proximal and
shoulder muscles, even while still ambulant. For instance, at 15 years of age, this was the
case in patient VI-2 (supplementary video 1, 2), while patient IV-23 progressed to loss of

50
ambulation. One of the youngest patients, examined at age 9, (VI-5) showed less severe
weakness in a similar pattern, consistent with the slowly progressive phenotype. Detailed
examination findings are summarized in Table 1.
Muscle imaging
On muscle ultrasound, findings were variable among the muscle types affected of
individual patients, although atrophy was apparent in all patients. Figure 3c shows
advanced muscle involvement with uniform increase in echogenicity in an older, more
severely affected, patient IV-23. In patient V-6, a mixed pattern with both granular and
streak-like features is seen (d), suggesting the presence of clusters of atrophic fibers.
Muscle MRI was performed on patients IV-23 and V-6. Consistent with the more severe
histological findings on biopsy, patient IV-23 showed marked atrophy, as well as
significant fatty replacement of the quadriceps and, in particular, of the vastus
intermedius and the rectus femoris, while the hamstring muscles also show severe
involvement with some notable sparing of parts of the semitendinosus and adductor
longus muscles (Figure 3a). In contrast, patient V-6’s muscle is more mildly affected,
showing minimal involvement of sartorius, biceps femoris and vastus lateralis (Figure
3b).
Histopathological findings
Muscle biopsies from patients IV-23, IV-27 and V-6 (Figure 4) demonstrated
variability of histological findings, suggesting progression with age. Patient IV-23 was
initially biopsied in childhood and reported in the original paper as showing groups of
atrophic fibers. The more recent muscle biopsy in this patient from the tibialis anterior

51
shows end-stage muscle with marked degenerative features, including dramatic
variability in fiber size (with atrophic and hypertrophic fibers), greatly increased
internalization of nuclei, and fatty infiltration. On NADH stain, the muscle shows
frequent trabeculated or lobulated fibers (Figure 4). In contrast, the muscle biopsies from
the less affected patients IV-27 and V-6 show milder changes although they show groups
of atrophic fibers of type I, consistent with fiber type disproportion. None of the muscle
biopsies showed nemaline rods on modified Gömori–trichrome stain or electron
microscopy, or actin aggregates by immunohistochemistry or electron microscopy
(Figure 4, Supp. Fig. 2).
Motor unit number estimation (MUNE) results.
For patient VI-2, the CMAP amplitude of the right ulnar nerve was 7005µV, and
the average SMUP 119µV. MUNE of the right ulnar nerve was 59. For unaffected
relative VI-8, the CMAP amplitude of the right ulnar nerve was 10773µV, and average
SMUP was 57µV. MUNE of the right ulnar nerve was 189. Nerve conduction studies of
the right arm and leg of one later affected family member (IV-25) were normal. EMG of
the right side demonstrated large, polyphasic motor units with delayed recruitment,
especially in weaker muscles (knee extensors), but occasional small amplitude,
polyphasic motor units were also observed in less affected muscles (deltoid). No
abnormal spontaneous activity was observed.
Genetic analysis
DNA was collected on 19 individuals. Linkage analysis in 17 family members
using Illumina Linkage IV Panel and Merlin software identified a 4.8Mb region on

52
chromosome 1q42.12-q42.13 with a LOD score >2 and a maximum LOD score of 3.651,
containing forty-eight genes as the only significant linkage region. Whole exome
sequencing detected only one non-synonymous variant, located in the linkage region in
all seven affected individuals, suggestive of pathogenicity on SIFT testing that was not a
known variant in dbSNP. This variant was a c.591C>A p.Glu197Asp change in exon 4 of
ACTA1. The mutation was subsequently confirmed using Sanger sequencing and shown
to co-segregate with the phenotype in the extended pedigree (Figure 1). Localization of
this residue on the 3D model of actin predicts that it is located on the exterior surface of
the protein.
Structural Interpretation
Monomeric actin can oligomerize into double-stranded F-actin trimers. In the
trimer, which represents a minimal F-actin filament seed, Glu197 is a critical semi-surface
exposed residue that is involved in two major interaction sites (Fig. 5). The glutamic moiety
forms a network of two short-range intramoleular interactions mediating a spatial bridging
of Lys193 and Arg258 (Fig. 5-a). On the other hand, Glu197 is in medium-range distance
to the basic charged side chain of Lys115, forming an electrostatic interaction between two
neighboring actin subunits at the fast growing, barbed-end of the F-actin filament (Fig. 5-
c). Our modeling approach revealed a serious effect of the single point mutation
Glu197Asp. Shortening of the acidic side chain by a single methylene unit causes a serious
diminished radius of action (Fig. 5-c). Remarkably, both intra- and intermolecular
interactions are affected dramatically. Asp197 is not able to establish the intermolecular
saltbridge with Lys115 from the longitudinal subunit. In addition, Arg258 is not engaged

53
in an electrostatic interaction with Asp197. Instead, the carboxylic moiety of Asp197 is
forming a singular saltbridge with Lys193.
Functional analysis
To determine whether the Glu197Asp mutation can lead to rod formation, Cos7
cells were transfected with WT-eGFP, Asp286Gly-eGFP, and Glu197Asp-eGFP
constructs. The Asp286Gly-eGFP construct was used as a positive disease control that
was known to form nemaline rods after transfection in culture (gift from N. Laing) and a
mock transfection was used as a negative control. Neither immunostaining nor Western
blot showed any significant changes in actin localization between the Glu197Asp-eGFP
construct and WT-eGFP, whereas the positive disease control resulted in rod formation as
expected and previously reported (Supp. Figure 1). However, no intranuclear rods were
seen. The constructs were also transfected into HEK, NIH3T3, and C2C12 cells, none of
which showed a phenotype of rod formation or abnormalities of the actin skeleton at 24,
48, 72, 96, or 120 hrs (results not shown). In addition, zebrafish injected with RNA
encoding either wild-type or Glu197Asp human ACTA1 displayed no abnormalities in
their morphology or muscle histology up to 6 days post fertilization, including no actin
accumulations or sarcomeric disorganization (Supp. Figure 3).

54
3.5: Discussion
In this study, we identified a novel variant in ACTA1 associated with
scapuloperoneal myopathy in a multigenerational family with a phenotype different from
previously recognized actinopathies. To our knowledge, this family represents the largest
single actinopathy pedigree recognized to date. Notably, the family had been first
described in 1966 as a form of proximal or scapuloperoneal spinal muscular atrophy. We
have re-evaluated 13 of the 33 patients from 4 out of 6 generations known to be affected.
The core clinical phenotype, present in affected family members, manifested as
early as infancy with a predominantly scapuloperoneal, but more precisely scapulo-
humeral-peroneal-distal, distribution weakness including shoulder weakness, scapular
winging and foot drop. Over time, the muscle weakness progressed to other proximal
muscle groups. Shared clinical characteristics included mild lower facial weakness, foot
drop due to foot eversion and dorsiflexion weakness, finger drop of digits 3-5, and
selective muscle atrophy. We recognized significant variability between family members
regarding age of onset and severity. Some patients had onset in early childhood and were
severely affected by their teenage years (VI-2), while others did not experience onset of
weakness until their teenage years or adulthood (V-6, IV-32). Some of the patients were
significantly more affected in the upper limbs (VI-6, IV-23) while others were more
equally affected in their lower limbs, and lost ambulation (IV-25). This variability does
not seem to segregate with gender or among distinct branches in the family tree,
suggesting that other genetic modifiers may play a role. There was no evidence for
cardiac, early respiratory, or extraocular muscle involvement.

55
The initial classification as a proximal spinal muscular atrophy-like phenotype, by
Armstrong and colleagues in 1966,1 was based on electrophysiological, as well as biopsy
data, suggesting the presence of denervation. Loss of motor units due to nonfunctional
neuromuscular junctions may account for the reduced MUNE values and denervation
seen in electrophysiological testing. In support of this observation, other investigators
have observed loss of motor units in muscular dystrophies and chronic myopathies with
mixed myopathic and neurogenic features on EMG (McComas, Sica et al. 1974;
Paganoni and Amato 2013). Thus, we suspect the striking muscle fiber atrophy observed
in this family accounts for the pseudoneurogenic findings on biopsy and
electrophysiological testing.
We applied a combined linkage/whole exome sequencing approach, which is
particularly powerful in large multiplex families such as this one. This strategy resulted in
a significantly linked 4.8 Mb region on chromosome 1, in which only the variant
p.Glu197Asp in ACTA1 qualified as pathogenic. This variant segregated with disease
manifestation throughout the extended family.
ACTA1 was first characterized in 1983 (Hanauer, Levin et al. 1983) and its gene
structure was first described in 1988 (Taylor, Erba et al. 1988). Actins are fundamental
cytoskeletal molecules and are necessary for sarcomere function in both skeletal and
cardiac muscle. They are some of the most highly conserved genes in muscle and thus do
not tolerate many missense changes. Most variants recognized in ACTA1, thus far, have
been pathogenic with mostly a dominant mode of action, and most recognized
polymorphic sequence variants in the gene are synonymous on the amino acid level
(Sherry, Ward et al. 2001).

56
Compared to the known clinicopathological entities within the actinopathies, the
family reported here is different clinically, as well as morphologically. ACTA1-related
myopathies (actinopathies) (Nowak, Wattanasirichaigoon et al. 1999) in general tend
towards more severe clinical phenotypes manifesting at birth, including arthrogryposis
(Laing, Clarke et al. 2004) and significant hypotonia, (Ilkovski, Cooper et al. 2001;
Agrawal, Strickland et al. 2004) as well as early respiratory involvement, while cardiac
involvement is rare (Kaindl, Ruschendorf et al. 2004). In some cases, milder clinical
presentations have been recognized in individuals with non-progressive mild skeletal
muscle weakness and severe disproportionate diaphragmatic (Jungbluth, Sewry et al.
2001) respiratory involvement, with nemaline rods evident on biopsy(Jungbluth, Sewry et
al. 2001). Unusual clinical features in this family include the scapulo-humeral-peroneal-
distal distribution with striking upper extremity predilection in some individuals,
progressive, but variable, course of the disease, and sparing of respiratory muscles until
very late. This unusually extended single family provides a unique opportunity to study
the basis of this clinical variability in greater detail.
Typical findings on muscle biopsy vary among actinopathies and previous reports
include cytoplasmic and/or intranuclear nemaline rods, actin aggregates, fiber type
disproportion, and zebra body formation (Jungbluth, Sewry et al. 2001; Clarkson, Costa
et al. 2004; Feng and Marston 2009). Muscle biopsy specimens in this family notably
showed no nemaline rods even in advanced stages of the disease. While the fiber type
disproportion observed in our family has been seen in ACTA1 mutations as part of the
possible histological spectrum, lobulated or trabeculated fibers, as seen in the more
advanced biopsy, have not been reported in actinopathy patients. However, this

57
cytoarchitectonic abnormality is not specific in itself, and has been described in a few
other cases of muscular diseases (Weller, Carpenter et al. 1999; Figarella-Branger, El-
Dassouki et al. 2002; Irodenko, Lee et al. 2009).
Mutations in the ACTA1 residue Glu197 have not been previously reported
(Nowak, Ravenscroft et al. 2012), but mutations on either side have both been shown to be
pathogenic: Thr196Pro and Arg198Cys both produce a severe nemaline myopathy(Laing,
Dye et al. 2009). Evaluation of pathogenicity by PolyPhen-2 and SIFT predicts a relatively
mild change consistent with the comparatively less severe and unusual phenotype
displayed in this family. Localization of this residue on the 3D model of actin shows it is
located on the exterior surface of the protein and the mutation significantly affects both
short-range intramolecular interactions with Lys193 and Arg258 and medium-range
intermolecular electrostatic interactions with Lys115 on neighboring actin subunits.
Besides a potential role in actin filament stability, it is tempting to speculate that the Glu197
residue may be involved in directionality and velocity of actin-filament growth in vitro,
although this has yet to be tested, and at this point it cannot be excluded that a so far
unknown protein-binding partner is interacting with Glu197. Interestingly, the interacting
Arg258 (labeled in reference as Arg256) has been shown to be involved in a severe
nemaline myopathy (PMID 10508519) (Nowak, Wattanasirichaigoon et al. 1999).
To further study the functional consequences of the mutation, mutation constructs
were tested in various cell types. In contrast to the nemaline rod formation seen after
transfection of a typical nemaline myopathy associated mutation, (Ilkovski, Nowak et al.
2004) no apparent change in actin cytoarchitecture or rod formation was observed with
Glu197Asp, nor were there any effects seen on the protein level. Additionally, injection

58
of Glu197Asp RNA into zebrafish embryos did not result in whole-mount morphological
abnormalities. It is therefore possible that the Glu197Asp mutation may instead reflect a
fundamentally different pathogenesis, such as changes in interaction or force generation,
for which further investigation is necessary and underway.
In summary, we define a novel ACTA1 mutation in a highly conserved residue as
the only significant change in a complete genetic analysis of this family. We thus define
this disorder as a new actinopathy with slowly progressive clinical manifestations,
including a scapulo-humeral-peroneal-distal distribution of muscle weakness and wide
intra-familial phenotypic variability in the age of onset and severity. In addition no rods
or inclusions were visible to cause histopathology. We hope that additional patients with
this unusual clinical-morphological presentation will now be evaluated for and/or
identified with ACTA1 mutations in order to further improve the understanding of the
clinical, morphological and genetic spectrum of this phenotype. Future studies are
planned to find the mechanism of disease, as well as possible disease modifiers, which
would account for the variability among affected individuals.
Table 1. Patient neurological exams Patient III-4 III-7 III-9 IV-23 IV-25 IV-27 IV-32 V-6 V-18 V-19 VI-2 VI-5
Cranial nerves
ptosis - - - +/- - +/- -
Facial weakness extraocul
ar - - - - - - - - - - -
periocular - - - + + - - + + - +
+
Transverse smile + - - + + - + + + - + -
Neck flexor weakness +++ ++ ++ +++ + + + + + + ++ +

59
Neck extensor weakness + + - - - + +/- +
Upper extremity weakness pattern
Proximal muscle groups
Scapular winging - - + + ++ +/- +/- + + + +
Lat. dorsi 0 + - + 0 +
Rhomboids ++ - - +
Deltoids +++ +++ 0 0 + + + + +++ + 0 +
Biceps +++ ++ +++ +++ - + +/- - +++ + +++ +
Triceps ++ + ++ +++ - +/- +/- - + + ++ +
Distal muscle groups
Wrist extension ++ + +++ - +/- + + ++ + ++ +
Wrist flexion ++ + +++ - +/- + ++ + ++ +
Finger abductio
n +++ +++ ++ + + +++ +++ +
Finger extension +++ ++ + +++ + ++ + ++ ++ ++ +
Grip ++ ++ + + + ++ + - + ++ + +
Thumb adductio
n + + + + + ++
Thumb abductio
n +++ +++ ++ + +++ +++ + +++
Trunk
weakness - - - - -
scoliosis - - - - -
Lower extremity weakness pattern
Proximal Hip flexor ++ +++ + + +/- + + + + +
+
Hip extensor + - - + +/-
Knee flexor ++ ++ +++ ++ - +/- + - +/- + +/- +

60
Knee extensor +++ ++ +++ +++ +++ +/- - - - + - +/-
Leg abductor + + + +/- - + +/- + - + + +
Leg adductor + + + + +/- + + - + +/- +/-
Distal Dorsi flexion +++ 0 0 0 ++ ++ + + +++ ++ +++ ++
Plantar flexion + ++ + + + +/- +/- - + + - -
Foot eversion +++ +++ +++ 0 ++ ++ + + +++ + +++ ++
Foot inversion + ++ ++ - + + +/- - + + + +/-
1st toe dorsi flex. +++ ++ +
Mobility
Gait Ambulati
on NA NA NA Short distan
ce + + + + + + +
Foot drop - + + + - + + + +
Hyper-extended at knees + + - + -
Wheel chair + + + + - -
CK 105 69 46 103 316
(-) = absent or normal strength, (+/-) = minimally affected, (+) = mildly affected, (++) = moderately affected, (+++) = severely affected, 0 = no movement, NA = non-ambulant, blank= no information available

61

62
Figure 1. Expanded six-generation pedigree.

63

64
Figure 2. Clinical photographs
(a, b): Patient VI-2 showing generalized muscle atrophy (a) and scapular winging (b). (c,
d, g); Patient V-6 demonstrating hypthenar atrophy (c) but full finger extension (d), foot
drop and high arches (g). (f): Patient VI-5 demostrating early scapular winging. (e, h, i):
Patient IV-23 showing pronounced deltoid atrophy (e) mild facial weakness (h), wrist and
finger extension weakness with wrist and finger drop (i).

65

66
Figure 3. Muscle MRI and Ultrasound
(a, b): Muscle MRI on the thigh in patients IV-23 and V-6, T1-weighted images. Patient
IV-23 shows marked atrophy as well as significant fatty replacement of the quadriceps
muscles and, in particular, of the vastus intermedius and the rectus femoris. The
hamstring muscles also show severe involvement with some notable sparing of parts of
the semitendinosus and adductor longus muscles (a). Patient V-6 is much more mildly
affected by imaging, showing minimal involvement of sartorius, biceps femoris and
vastus lateralis (b). (c, d): Muscle ultrasound of the thigh in patients IV-23 and V-6, 12
Mhz linear probe. Image in patient IV-23 shows advanced muscle involvement with
uniform increase in echogenicity (c). In patient V-6 there is a mixed pattern with both
granular and streak-like features, suggesting the presence of clusters of atrophic fibers
(d).

67

68
Figure 4. Muscle biopsy histology.
Histology in patients IV-23 (a,b,c), IV-27 (d,e,f), and V-6 (g,h,i). In patient IV-23
(tibialis anterior) H&E shows muscle with great fiber size variability, centralized nuclei,
and myofiber degeneration, as well as fatty and connective tissue infiltration (a), while
the NADH stain reveals the presence of trabeculated fibers (b). The biopsy from patient
IV-27 (vastus lateralis) shows fiber size variability with groups of atrophic fibers (d) that,
upon NADH stain, are dark, and thus type I. Patient V-6’s biopsy (biceps) reveals
variability in fiber size with groups of atrophic fibers and some degree of central
nucleation (g), after NADH staining, the small fibers are dark, again, indicating fiber type
I atrophy (h). Ultrastructural examination in patient IV-23 shows disrupted myofibrils in
severely atrophic fibers, but no rod formation (c), whereas IV-27 and V-6 show normally
aligned Z-discs and no evidence of rod formation (f,i). Immunohistochemistry in patients
IV-23 and V-6: alpha-actin staining reveals no aggregates (j, k, l). Alpha-actinin 1
staining (m, n, o) shows no rod formation but accentuates some of the trabeculation in
the biopsy from IV-23 (b). Laminin alpha2 (LamaA2) staining (p, q, r) accentuates the
atrophic fibers (b, h) and reveals normal membrane integrity.

69

70
Figure 5. Molecular implication of the E197D mutant on F-actin assembly. (a)
Overall view of the trimeric minimal F-actin filament. The lateral actin monomer is
colored gray, whereas the longitudinal monomer is shown in blue. The Glu197 residue is
highlighted in red spheres. (b) Detailed view of the intramolecular interaction mediated
by Glu197. First, an intrahelical i to i+4 saltbridge is formed along the helical dipole
with the side chain of Lysine 193. Second, the remaining part of the carboxylate moiety
of Glu197 is oriented towards Arg258, forming a salt bridge. (c) The replacement of
Glu197 by an aspartate side chain causes a severe effect on the radius of action of residue
197. In the Asp197 version of actin, the interaction with Arg258 is disrupted and Asp197
is just forming the intrahelical interaction with Lys193. (d) Glu197 also mediates an
intermolecular interaction along the helical element of the F-actin filament. The sidechain
forms an electrostatic interaction with the basic Lys115 of the neighboring longitudinal
subunit. In the case of the E197D mutant, this interaction is disrupted (not shown).

71

72
Supplementary Figure 1. Transfection of WT-, E197D- and D286G-GFP constructs
into COS-7 cells.
(a) The nemaline myopathy disease control D286G-GFP results in the expected rod
formation, while E197D shows no rod formation. Occasional aggregate formation, as
shown, was no different in frequency from the wild-type transfection. (b) GFP Western
blot analysis reveals no significant differences in the protein expression levels after
transfection.

73

74
Supplementary Figure 2. Zebrafish injected with WT-ACTA1 or E197D-ACTA1
(a) Zebrafish embryos injected with 400pg RNA encoding wild-type or E197D ACTA1
had normal morphology and did not display phenotypes associated with myopathies, such
as curved bodies and decreased muscle mass. (b) Whole-mount immunostaining of 2-
day-old zebrafish, injected with E197D RNA, using Acti-stain (Cytoskeleton, Inc)
demonstrated normal myofibrillar organization and absence of nemaline rods in the
muscle. (c) Percentage of fish that hatched out of their chorion by 54 hpf were calculated
to evaluate changes in motility. The percentage of fish able to hatch out of their chorion
was not significantly different between the groups that were injected with 400pg wild-
type ACTA1 or the E197D variant. Control represents uninjected fish. Data presented are
an average of 3 experiments (97-103 fish) ± SEM.

75
Chapter 4
Using iPSC differentiated embryoid bodies and neurons as a model to study the
neurological phenotype of alpha-dystroglycanopathies

76
4.1: Abstract
Alpha-Dystroglycanopathies are a group of congenital muscular diseases with
diverse phenotypes from mild forms with only muscle involvement to severe muscle, eye,
and brain involvement (Godfrey et al, 2011; Hewitt, 2009). Despite the numerous
models to study the neuron migration defects of the severe disease (Moore et al, 2002),
few models exist to test the intermediary cognitive deficits in patients with no gross brain
abnormalities as seen on MRI. We believe that this cognitive impairment is the result of
a synaptic dysfunction and therefore we need an appropriate cellular model to elucidate
the mechanism.
Here we tested the hypothesis that neurons differentiated from reprogrammed
patient induced pluripotent stem cells (iPSCs) would have a synaptic deficit as shown by
immunochemistry and electrophysiology. We tested to see if they would also
recapitulate features of the disease including hypoglycosylation of alpha-dystroglycan
and impairment in the formation of embryoid bodies. Using patient derived iPS cells we
performed a quantitative and qualitative comparison of control and diseased derivatives.
Embryoid bodies from patient cells formed irregular shapes and consistently fused more
often than controls. Neurons showed no significant differences in the numbers of
synapses although patient cells showed hypoglycosylation of alpha-dystroglycan. Using
electrophysiology, we showed that cells from all lines produce action potentials when
given depolarizing current steps, however patient lines had significantly less spontaneous
activity, when compared to control lines, as shown by a decrease in the frequency of
spontaneous excitatory postsynaptic currents (EPSCs).

77
Our findings demonstrate that patient-derived neurons show synaptic differences
and embryoid bodies and neurons recapitulate features in other models of alpha-
dystroglycanopathies providing a unique opportunity for investigating disease mechanism
and developing therapies.

78
4.2: Introduction
What is a good model for investigating the subtle synaptic phenotype in alpha-
dystroglycanopathy patients without gross brain or neuronal migration defects shown on
MRI? Alpha-dystroglycanopathies are a group of congenital muscular dystrophies
characterized by a decrease in a specific form of glycosylation of alpha-dystroglycan (α-
DG) (Godfrey et al, 2011; Hewitt, 2009). Thus far seventeen genes have been implicated
in the alpha-dystroglycanopathies, although many patients remain undiagnosed and
potentially more genes remain to be discovered. Mutations can cause both a brain and a
muscle phenotype of varying degrees from just muscle involvement in a limb girdle
muscular dystrophy (LGMD) (Brown, Torelli et al. 2004; Muntoni 2005) to a very severe
muscle and brain phenotype in Walker-Warburg syndrome (WWS) (Santavuori, Pihko et
al. 1990; Baltaci, Ors et al. 1999).
It is well known that patients with a severe phenotype exhibit lissencephaly type
II and neuronal heterotopia with migration beyond the glia limitans. To further
investigate this, previous studies have examined brain-specific knockouts of DAG1
whereby floxed dag1 mice were crossed with nestin-Cre and GFAP-cre mice (Moore,
Saito et al. 2002), as well as other cre lines (Satz, Barresi et al. 2008; Nguyen, Ostendorf
et al. 2013). These mice exhibited a phenotype similar to one observed in severe patients
and it was determined that the interactions of dystroglycan in neurons and astroglial
endfeet are vitally important for correct migration. Without dag1, the brains showed
laminar disorganization in the cerebral cortex and disruption of the glia limitans (Hu,
Yang et al. 2007).

79
In the present study we are interested in the patients without gross morphological
changes on MRI. It is necessary to determine the causes of their cognitive deficits
including language and speech delays, and intellectual disabilities to target therapeutic
approaches. Because the floxed dag1/Cre+ mice also showed impaired long-term
potentiation (LTP) in hippocampal slices (Satz, Ostendorf et al. 2010), we hypothesize
that the cognitive impairment is caused by synaptic dysregulation and instability.
To test this synaptic hypothesis mouse and zebrafish models are not ideal due to
the differences in rate of growth and the fact that many proteins have slightly different
functions in different organisms. Ideally one would study patient cells directly to model
central nervous system diseases and develop treatments, but this has been difficult due to
the inability to easily culture patient neurons.
In the current study we reprogrammed patient fibroblasts into induced pluripotent
stem cells (iPSCs)(Takahashi and Yamanaka 2006; Yamanaka and Takahashi 2006;
Takahashi, Tanabe et al. 2007) and differentiated them into embryoid bodies and
neurons. We found that these cells recapitulate some features of disease and would
represent a good model for studying the synaptic phenotype of alpha-
dystroglycanopathies.

80
4.3: Materials and Methods
Cell lines
Patient fibroblasts
Fibroblasts were obtained from four patients with alpha-dystroglycanopathies and
one commercially available BJ control line. All fibroblasts were collected with informed
consent (IRB approval #00-N-0043 and 12-N-0095, NINDS, Bethesda, MD). The
samples were acquired in a variety of IRB approved ways with appropriate consents
obtained. Cells were either shipped from Coriell, collected in our clinic at NIH, or sent
from Cincinnati Children’s hospital from Dr. James Collins. Samples that were collected
in our clinic were from forearm skin biopsies taken with a 3mm punch biopsy tool. The
samples were then minced and treated with collagenase, and allowed to grow at 37°C for
3-4 weeks.
Patient Information
We have chosen patients with various mutations and severities. See Table 1.
Reprogramming of Fibroblasts into Induced Pluripotent Stem (iPS) cells
Millipore Lentiviral Reprogramming of Patient Fibroblasts
Patient fibroblasts were transduced on two consecutive days with the STEMCCA
lentiviral polycistronic construct encoding Oct4, Klf4, Sox2, and c-Myc (Sommer et al,

81
2009) purchased from Millipore (Billerica, MA). After 6 days they were replated onto a
feeder layer of mouse embryonic fibroblasts (MEFs) and cultured in stem cell media.
After 17-25 days iPS colonies were picked manually when clearly visible. They were
mechanically dissociated and replated for expansion.
Stemgent mRNA Reprogramming of Patient Fibroblasts
Fibroblasts were transfected with mRNA containing Oct4, Sox2, Klf4, c-Myc,
Lin28 at a ratio of 3:1:1:1:1 by Stemgent. iPS were expanded with E8 media (Life
Technologies) and maintained on TeSR media (StemCell Technologies).
Differentiation to Neural Stem cells (NSC)
NSCs were derived from patient and control iPSCs by Lonza (Walkersville, MD).
Embryoid bodies to three germ layers
iPS colonies were dissociated and plated for 48 hours in Aggrewell plates
(StemCell Technologies) and then transferred to 6-well non-adherant plates (Corning) to
form embryoid bodies. They were plated on 8-well chamber slides and allowed to
differentiate without the use of any patterning factors. The slides were stained for
various markers of the three germ layers (smooth muscle actin for mesoderm, alpha-
fetoprotein for endoderm, and Pax6 for ectoderm).
Karyotype assay
Two T25 flasks of each iPSC clone were sent to Cell Line Genetics for
karyotyping. Only clones with normal karyotypes were used.

82
Embryoid body (EB) formation as intermediary
Differentiation was carried out as previously described (Amoroso, Croft et al.
2013)with modifications. iPS were dissociated with EDTA at 80% confluency and then
plated in Aggrewell 800 (StemCell Technologies) in ES media with 20uM ROCK-I
(Tocris), 20ng/ml FGF (R&D Systems, Minneapolis, MN), 10uM SB431542 (Tocris),
and .2uM LDN193189 (Stemgent). After 24 hours, half the media was replaced. On day
2, EBs were gently washed from the Aggrewell plates and filtered through a cell strainer
to remove any cells that did not integrate into EBs, and placed in 6-well non-adherant
plates (Corning, Corning, NY) in neural induction media (NIM) (DMEM/F12, P/S,
NEAA, N2, Heparin) with LDN, SB431542, 10ng/ml BDNF, and .4ug/ml Ascorbic acid.
Media was changed every other day.
Neuronal differentiation
From EBs
After 14 days, EBs were dissociated as previously described (Amoroso, Croft et
al. 2013) with slight modifications. The cells were plated onto poly-l-ornithine/laminin
(BD Biosciences) slides and cover slips in neurobasal media (Invitrogen) with NEAA,
N2, B27, ROCK-I, laminin (1ug/ml), BME, Glutamate (25uM) (Sigma, St. Louis, MO),
10ng/ml BDNF, and .4ug/ml Ascorbic acid (Sigma). Media was changed every 3-4 days.
Neuronal differentiation from NSCs

83
NSCs were plated onto slides and cover slips coated with poly-l-ornithine (Sigma-
Aldrich) and laminin (Sigma-Aldrich). The cells were maintained in the same media as
above (neurobasal with supplements).
Immunocytochemistry
Cells were fixed in 2% PFA and washed twice in 1X PBS. Cells were blocked in
10% normal goat serum/.1%TX-100/PBS for 1 hour at room temperature, and then
incubated with primary antibodies (B-tubulin 1:2000, a-dystroglycan Millipore 1:200,
Map2 1:500, NF 1:500, PSD95 1:500, Synaptophysin Synaptic Systems 101011 1:500) at
4°C overnight. AlexaFlour secondary antibodies (goat anti-mouse 488 1:500, goat anti-
rabbit 568 1:500) were used followed by staining with DAPI (2 minutes) and mounting
with Flouromount-G. Images were obtained on a Leica SP5 confocal microscope.
qPCR
iPSC RNA was extracted and used to make cDNA, and run qPCR using the
primers for markers of pluripotency: Sox2, Nanog, hTERT, and GDF3. All the Ct
values were normalized to GusB and 28S.
Immunoblotting
Cell pellets were lysed in RIPA buffer (Sigma) and centrifuged at 13,000xg for 10
minutes. Proteins were resolved by electrophoresis on a 4-12% Bis-Tris polyacrylamide
gel and blotted for 8 minutes using iBlot (Invitrogen). Membrane was incubated at 4°C
overnight in primary antibody (a-dystroglycan IIH6 Millipore 1:500) with 5%BSA in
TBST. Goat anti-mouse IgG conjugated to horseradish peroxidase was used for detection

84
and visualized with ECL western blotting reagents (GE Healthcare, Little Chalfont,
Buckinghamsire, UK).
Electrophysiology
Recordings of patient iPS derived neurons
Whole-cell patch-clamp recordings were made at room temperature after 6, 8, and
10 weeks of differentiation on poly-l-ornithine/laminin 10mm cover slips. Recordings
were performed in a submerged chamber at room temperature with constant bath
perfusion of ACSF (~4 mL/min) containing (in mM) 130 NaCl, 3.5 KCl, 24 NaHCO3,
1.25 NaH2PO4, 10 glucose, 1.5 MgCl2, and 2.5 CaCl2. Voltage and current clamp
recordings were performed using pipettes with an internal solution containing (in mM)
130 K-Gluconate, 0.6 EGTA, 10 HEPES, 2 MgCl2, 2 Na2ATP, 0.3 NaGTP, and 6 KCl.
All experiments were conducted under blinded conditions. Electrical signals were
digitized with Digidata1440A (Axon Instruments) and filtered at 3kHz. Data were
recorded using pClamp10 (Axon Instruments).
Recordings of (Dag lox/+;Cre ) mouse hippocampal slices
Mice were anesthetized with isoflurane and the brain dissected in ice-cold saline
solution containing 130 NaCl, 3.5 KCl, 24 NaHCO3, 1.25 NaH2PO4, 10 glucose, 1.5
MgCl2, and 2.5 CaCl2. Transverse hippocampal slices (300um) were cut using a VT-
1000S vibratome (Leica Microsystems, Bannockburn, IL). Recordings were performed
as previously described in Satz et al, 2010. Briefly, extracellular field EPSPs were
evoked in CA1 stratum radiatum by stimulation at the CA3-CA1 border. Stimulation was
delivered every 30s and stimulus-response curves were obtained. Long-term potentiation

85
(LTP) was induced by a high-frequency stimulation (HFS) and the signal was measured
by normalizing the fEPSP amplitude after HFS to the fEPSP before HFS as well as
measuring the slope of the fEPSP to determine the magnitude of the evoked signal.
Mouse Generation and Genotyping
Nestin-Cre mice (B6.Cg-Tg(Nes-cre)1Kln/J) Jackson Labs) were bred with
Floxed dag1 mice (Daglox/lox) (B.129 (Cg)-Dag1 tm2.1Kcam /J) Jackson Labs) to get
heterozygous mice expressing the Cre recombinase (Dag lox/+;Cre ). These mice were then
bred with dag1 floxed mice (Daglox/lox) (Jackson Labs). This gave the expected 25%
nestin-Cre/DG-null mice. Cre-negative mice were used as controls. All experiments
were conducted according to nimal use procedures approved by the National Institutes of
Health IACUC.
Dag1 mice were geneotyped according to standard protocols with primers
GGAGAGGATCAATCATGG and CAACTGCTGCTGCATCTCTAC for results
mutant=615bp, heterozygote=516bp and 615bp, and wild type 516bp. To determine
expression of the cre recombinase a standard cre protocol was used with primers
GCGGTCTGGCAGTAAAAACTATC and GTGAAACAGCATTGCTGTCACTT to
determine the presence of the transgene and CTAGGCCACAGAATTGAAAGATCT and
GTAGGTGGAAATTCTAGCATCATCC as internal positive controls with
transgene=100bp and internal positive control=324bp.
Data Analysis
Spontaneous EPSCs recorded from cells in culture were detected using a template
based event detection (Clampfit 10.1, Axon Instruments). EPSC amplitude for each cell

86
was determined by averaging all of the events detected in a given cell to create a
representative EPSC, the peak of the representative EPSC was then measured. fEPSP
slope was determined by measuring the slope of the fEPSP waveform rise from 20% to
80% of the maximum amplitude (Clampfit 10.1, Axon instruments).
Statistics for electrophysiological experiments were performed in Origin v 8.1
(OriginLab). Data were tested for significance using a two-tailed Mann Whitney U, with
significance set at p=0.05.

87
4.4: Results
In this study, we derived iPSCs, Embryoid bodies (EBs), and neurons from one
control and four patients with alpha-dystroglycanopathies. All cells types showed less
glycosylated alpha-dystroglycan in the patient lines based on staining with the IIH6
antibody which recognizes the LARGE-dependent glycoepitope.
Induced Pluripotent Stem cells show characteristics of pluripotency
We first investigated the Stemgent and Millipore derived iPS lines from
fibroblasts of four patient lines collected in our lab. Patient mutations are located in four
different genes that cause alpha-dystroglycanopathies including POMT2, POMT1,
LARGE, and FKRP (Table 1). All patients display both a muscle and a brain phenotype
as shown by muscle biopsy and brain MRI (Table 1) with varying degrees of severity.
Fibroblasts were transduced with STEMCCA lentiviral vector or transfected with mRNA
containing Oct4, Sox2, Klf4, c-Myc, Lin28 at a ratio of 3:1:1:1:1. All clones displayed
pluripotency markers as shown with immunocytochemistry staining of Tra-1-60 and
SSEA-4 (Figure 1a). Only clones with normal karyotypes were used for analysis (Figure
1b). Each clone was able to differentiate into all three germ layers by directed
differentiation (results not shown, Stemgent). Specifically, we determined whether there
were differences between the iPSCs of patients and controls. The patient lines showed a
decrease in the amount of glycosylation of alpha-dystroglycan as seen on western blot
using the IIH6 antibody (Millipore) which recognizes the LARGE dependent specific
glyco-epitope (Figure 1c).

88
Table 1. Patient information
Patient Mutation Brain MRI Muscle Biopsy Motor Milestones Cognition
B11-24 POMT1: compound
heterozygous; c85A>C and c1864C>T
Normal Delayed, weakness
Expressive language difficulties, cognitive delay
B11-23 POMT2: Homozygous ex.
9 c 1057G>A; pGly353Ser
Normal Fibrofatty replacement, fiber size variability
Delayed Speech delay
B10-20 LARGE: Het
c.1525G>A(Glu509Lys); 22q12.3 74kb deletion
Lissencephaly, Pachygyria
Delayed Cognitive Impairment
B12-57 FKRP: Het
c.826C>A(Lys276Iso); c.534G>T(Tyr178Cys)
Normal Variable fiber size, Fatty replacement, Increased connective tissue, Poor distinction of fiber types on ATPase staining
Delayed Cognitive Impairment, Autistic features

89
Embryoid bodies show fusion phenotype
Next, we focused on the differences seen during the embryoid body stage of
differentiation. Specifically, we looked for qualitative and quantitative differences
between the control and patient lines. To do this, the iPS cells were differentiated by first
growing them in suspension cultures as EBs, however compared to the controls the
patient lines dissociated and never formed spheres that remained intact. We added
additional ROCK-I to promote cell survival, but this did not provide significant
improvement.
Therefore, to overcome this difficulty the EBs were first formed in Aggrewell 800
plates allowing the EBs to form in smaller wells with less turbulence. iPSCs were
dissociated and plated for 48 hours in the microwells of the dish. When embryoid bodies
are formed in suspension they have varying sizes and shapes, and the Aggrewell plates
were established to provide a way to produce EBs of the same size. When we
differentiated our patient cells with these plates, the EB’s still did not incorporate all of
the cells from the wells in comparison to the controls. The protocol from the company
recommends leaving the EBs in the microwells for 10-14 days, however this was also not
successful for the patient derived cells. Because not all of the cells incorporated into the
EBs they inhibited further growth due to their presence in the media. These cells could
not be removed from the media because they are in suspension in the microwells.
After 48 hours the EBs were moved into 6-well non-tissue culture treated plates
and when they were initially moved to the suspension cultures there were no noticeable
differences between the control and patient lines except in line B10-20 which seemed to

90
produce fewer spheres although this was not quantified. After 3 days and 5 days, the
patient lines did not remain individual EBs but instead fused together as shown and
quantified (Figure 2). Qualitatively, the patient EBs did not seem as round and there was
more debris around the EBs and floating in the media compared to the control lines.
iPS cells were differentiated into neurons both from embryoid bodies (EBs) and neural
stem cells (NSCs)
Both control and patient lines differentiated into functional neurons as shown by
electrophysiology (Figure 3) and immunocytochemistry (Figure 4). Cells displayed
varying morphological properties consistent with the heterogeneous population that
forms as a result of this cortical neuron differentiation protocol (Figure 4A). After 10
weeks in vitro, we were able to evoke action potentials by injecting the cells with
depolarizing current steps; however, consistent with previous reports, these neurons
displayed embryonic properties and displayed slower kinetics when compared to acute
hippocampal slices (Figure 4B). We also compared the firing properties of the neurons at
4, 6, and 10 weeks in vitro and showed that the neurons were maturing over time but
required 10 weeks in vitro to consistently exhibit mature action potential waveforms
(Figure 5C).
Next, we identified the location and number of synapses in the neurons by
staining with synaptophysin. Staining was distributed along the processes of neurons as
shown by co-staining with Tuj1 (Figure 3). In both control and patient lines,
synaptophysin staining became visible after 8 weeks of differentiation which is consistent

91
maturing electrophysiological properties, specifically with the emergence of action
potentials and increased spontaneous activity. Taken together, these results show that the
neurons are functionally immature as shown by both immunocytochemistry and
electrophysiology.
Patient neurons show a decrease in the amount of glycosylated alpha-dystroglycan
After 10 weeks of differentiation there were no noticeable differences between the
number of neurons or their morphology between the control and patients. Neurons were
stained at 8 and 10 weeks with Tuj1, Map2, and Neurofilament (Supplemental Figure 1).
When neurons were stained with IIH6, which identifies only LARGE
glycoepitope positive alpha-dystroglycan, the control neurons showed increased
fluorescence (Figure 5). This difference was quantified with western blotting which
showed less glycosylation in the patient lines than the control lines. The glycosylated
alpha-dystroglycan (using IIH6 antibody, Millipore) band identified in the neurons is of
smaller molecular size than the corresponding signal identified in the iPS cell lysates.
This is consistent with reports that alpha-dystroglycan itself is predicted to have a
molecular weight of 72kDa, but after glycosylation is 120kDa in cortex and 160kDa in
skeletal muscle (Satz et al, 2010).
Patient derived neurons have less spontaneous activity
We next sought to determine whether control and patient derived neurons have
physiological differences at the synaptic level. To do this we recorded spontaneous

92
excitatory and inhibitory postsynaptic currents (EPSCs and IPSCs) from cells after 10
weeks in vitro. Spontaneous EPSCs were observed in all cells and were blocked by bath
application of 10 μM DNQX (AMPA blocker) and 50μM dl-APV (NMDA blocker),
showing these events are glutamatergic. Spontaneous IPSCs were observed in only one
cell and, thus, were excluded from further analysis. Interestingly, one of the patient lines
showed a significant decrease in the frequency of EPSCs when compared to controls,
with the other patient line trending towards a reduced EPSC frequency as well (Figure
6B). Notably, there were no significant differences in the amplitude of these events
(Figure 6C).
Nestin-cre/DG-null mice show no differences in LTP
Next we turned to the mouse model to elucidate additional differences between
nestin-Cre/dag1-null (KO) and Cre-negative (WT) mice. Nestin-cre drives expression of
the Cre recombinase in glia and neurons (without expression in ependyma or choroid
plexus) starting at E10.5 in the central nervous system. Ideally any insights from the
mice could be tested in the iPS-derived neurons. To examine synaptic differences in the
hippocampus, Schaffer collateral evoked fEPSPs were recorded in CA1 stratum radiatum,
with no difference observed in basal evoked fEPSPs between KO and WT animals. We
also examined differences in long-term potentiation (LTP), using high frequency
stimulation (HFS) of the Schaffer collaterals to evoke LTP in CA1. Pooled data (n=7)
showed no difference in the magnitude of LTP between WT and KO mice (Figure 7),
suggesting no deficits in synaptic plasticity in KO mice.
In previous studies differences were seen in nestin-Cre mice, but not in NEX-Cre
bred mice indicating that there is a range of phenotypes in these mice depending on how

93
early and robust the recombination events take place. Perhaps these mice were on the
milder end of the spectrum and therefore did not have significant differences in LTP,
however it is also possible that these mice were at a different age than those that were
previously reported. Similar to previous reports the KO mice showed hydrocephalus,
domed cranium, and were smaller than their littermates.

94
4.5: Discussion
In this study we took advantage of iPSCs derived from patient fibroblasts to
establish a useful model for studying the cognitive impairment in patients with alpha-
dystroglycanopathies. Differentiating iPSCs into NSCs, EBs, and neurons demonstrates
the hypoglycosylation phenotype of the LARGE-dependent glycoepitope associated with
the disease. Currently there are no treatments for patients with aDGs and using these
cells provides a basis for identifying future treatment possibilities.
iPSCs are now a well-established model for studying neurological and other
disorders (Takahashi, Yamanaka, 2006; Qiang et al, 2013). Here we have characterized
iPS lines from four patients and demonstrated their pluripotency through immunostaining
of pluripotency markers, karyotyping, and differentiation capacity.
Our findings show that patient derived embryoid bodies do not develop as well
compared to controls. The mouse model of a complete null dag1 is embryonic lethal due
to the incomplete formation of Reichert’s membrane (Williamson et al, 1997; Moore,
Saito et al. 2002). Conditional knockouts that knockout DAG1 later in development have
various migration defects including over migration due to incomplete formation of the
glia limitans (Moore et al, 2002; Satz et al, 2008; Nguyen et al, 2013). The patients used
in this study had mutations in FKRP, POMT1, POMT2, and LARGE. The knockout
mouse models of the genes POMT1, POMT2, and FKRP (Kurahashi et al, 2005; Hu et al,
2011; Chan et al 2010) are all embryonic lethal due to a deficit of the early formation of
Reichert’s membrane and while the LARGE-myd mouse is not embryonic lethal, it does

95
have migrational deficits (Lane et al, 1976). Here we provide another model for studying
the early development of basement membranes in a model that is not a knockout of
DAG1 or another gene. This provides a unique opportunity to study the effect of LARGE
dependent hypoglycosylation of aDG at levels that are consistent with patients who have
the disease.
Previous reports in dag1 knockout mouse embryonic stem cells have
demonstrated embryoid body phenotypes in the early development both in showing a
disrupted basal lamina in one case and a thicker in another case (Henry et al, 1998; Li,
Edgar et al, 2003). Here we show that at physiological levels of the proteins there is
also a deficiency in the neural membrane formation of these EBs in neuron induction
media.
This phenotype in a human cell based model gives insights into the disease
mechanism. Not only are the embryoid bodies more fragile, additionally we observed an
embryoid body fusion which may be the result of improper basal lamina formation
causing the edges of the membrane to become more adhesive. In previous studies it has
been shown that the deposition of laminin-111 is slower in models of aDG (Zhang, Yang
et al, 2013). We tested adding additional laminin to the media of the suspension cultures
to try to ameliorate their disintegration, but we did not detect a difference. This again
provides evidence that alpha-dystroglycan and its interactions are vitally important for
the early development of the embryo and subsequent development of the brain.
Additionally, because neurons are the cell type of choice to model a putative
synaptic phenotype, these patient-derived neurons represent an ideal model for studying

96
the disease mechanism and processes at human physiological levels that may more
closely parallel the disease than a mouse model. In this study neurons were differentiated
and shown to resemble the disease in that they show decreased staining with the IIH6
antibody which recognized the LARGE-dependent glycoepitope, they exhibit properties
of embryonic neurons, and they show decreased spontaneous activity.
Previous reports have shown that differentiated iPSC derived neurons are more
like immature embryonic neurons, so we used electrophysiology to characterize the
maturation of the neurons. Consistent with previous research, our neurons have slightly
depolarized membrane potentials, a lower amplitude in the synaptic responses, and a
longer culture period necessary to mature human induced cells when compared to
primary neuronal mouse cells (Chambers, Fasano et al. 2009). The cells required 10
weeks in vitro after 14 days as embryoid bodies before they produced action potentials.
Our hypothesis is that the synapses in patient neurons are less stable due to the
hypoglycosylation of a-DG and its decreased ability to bind neurexin. Previous reports
have shown that at the synapse aDG binds to neurexin at the synapse (Sugita, Saito et al,
2001). Here, there is no significant difference in the number of synapses per length of
projection as shown by staining with synaptophysin and B-tubulin. In addition,
physiological data show that despite exhibiting firing properties comparable to controls,
patient lines show a reduced EPSC frequency, suggesting a presynaptic deficit.
Consistent with a presynaptic phenotype, we observed a difference in the amplitude of
spontaneous EPSCs recorded in patient and control lines. Taken together, this suggests
that the synapses are formed and are present, but perhaps one step of the complicated
“hand shake” that has to take place during synaptic transmission is not functioning

97
properly. It is possible that channels are not recruited, vesicles do not fuse properly, or
neurotransmitters are not released at the same rate. Further studies are necessary to
further characterize the synapse formation and composition to identify the mechanism
that inhibits activity.
In addition to EBs and neurons, NSCs show decreased glycosylation and therefore
are a good model for the disease as well. In other reports NSCs have been used as a cell
model for drug screens as they are closer to the affected cell model than fibroblasts and
they are able to be passaged and expanded for a large screen. This is a better model for
this disease than using fibroblasts or myoblasts that may not have the same disease
mechanism or the same proteins present as neuronal cell lineages (Gorba, Conti, 2013).
Perhaps an RNAi or small molecule screen in NSCs could provide additional mechanistic
detail and provide potential treatment strategies.
Our study was not without limitations including the heterogeneity of the neuronal
cultures, nevertheless our data show that differentiated neurons are a good model for
studying the alpha-dystroglycanopathies. Future studies are planned to evaluate the
dynamic nature of the system including outgrowth, synapse formation, and maturation.
4.6: Conclusion
Our study demonstrates that iPS-derived neurons are a good model for studying
the synaptic deficits in patients with alpha-dystroglycanopathies. It is possible that the
variability in the cultures limits the conclusions, but we show that in multiple patient
lines with different mutations, that there is a reduction in the amount of IIH6 LARGE-
dependent glycosylation consistent with disease and an electrophysiological difference

98
that warrants further investigation. To further understand these changes we hope to use
additional electrophysiology and immunochemistry over the time course of maturation of
neurons to elucidate the development, mechanism, and pruning of synapses. Our findings
show that this model is a valuable tool for developing therapeutics approaches and
identifying pathways in this disease.

99
Figure 1. Characterization of induced pluripotent stem cells (iPSCs). (A) Morphology of colonies and immunocytochemistry showing staining with pluripotency markers Tra-1-60 and SSEA-4. (B) Normal karyotyping of colonies. (C) Western blot showing a decrease in the amount and size of alpha-dystroglycan staining with IIH6 antibody.
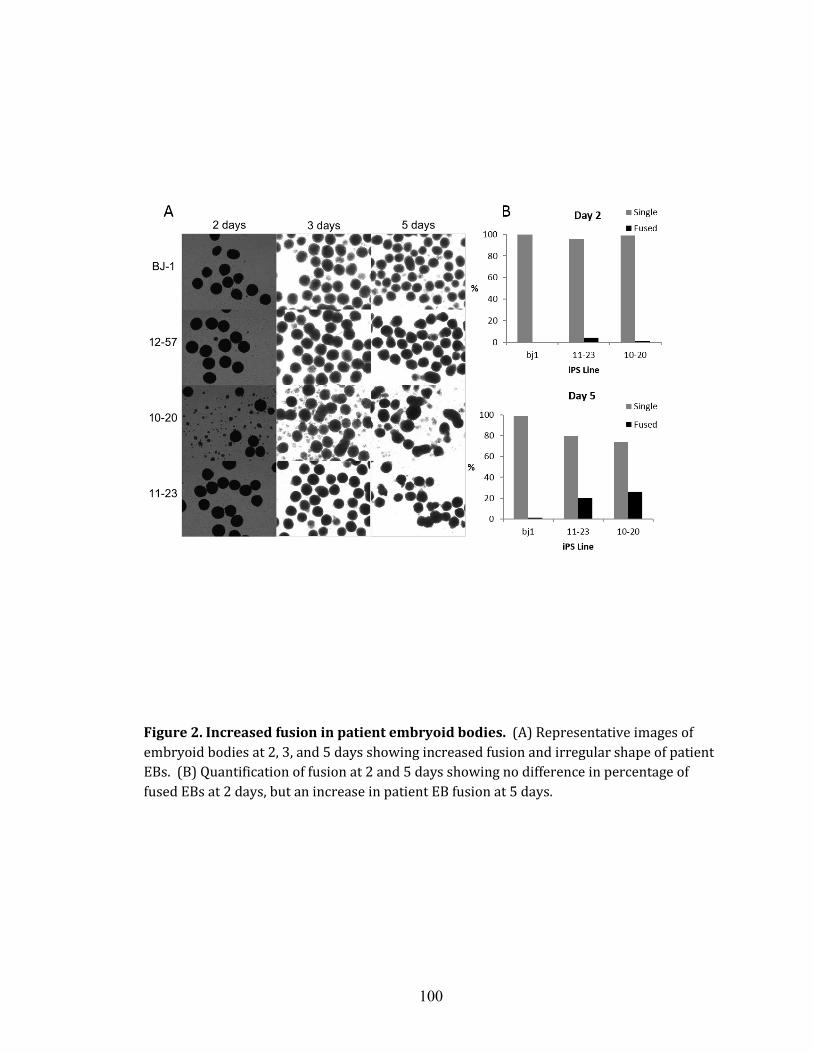
100
Figure 2. Increased fusion in patient embryoid bodies. (A) Representative images of embryoid bodies at 2, 3, and 5 days showing increased fusion and irregular shape of patient EBs. (B) Quantification of fusion at 2 and 5 days showing no difference in percentage of fused EBs at 2 days, but an increase in patient EB fusion at 5 days.

101
Figure 3. Electrophysiology showing changes in properties over the time of differentiation. (A) DIC images showing different appearances of recorded neurons. (B) Representative traces of iPSC action potentials versus an acute mouse hippocampal slice. (C) Representative traces of invoked potentials in differentiated neurons at 4, 6, and 10 weeks in vitro.

102
Figure 4. Immunocytochemistry of differentiated neurons at 8 weeks in vitro. Synaptophysin staining showing no difference in location or number of synapses.

103

104
Figure 5. Glycosylated alpha-dystroglycan using IIH6 antibody in differentiated neurons. (A) Decrease in glycosylated alpha-dystroglycan in patient lines B10-20 and B11-23 compared to control. (B) Decrease in the amount of IIH6 positive alpha-dystroglycan in patient derived neurons as compared to controls and compared to control iPSCs.

105
Figure 6. Spontaneous activity of control and patient derived neurons after 10 weeks in vitro. (A) Spontaneous activity blocked by DNQX showing neurons are glutamatergic and representative recordings of control and patient cells. (B) Significant difference between spontaneous activity in patients versus controls (n=5). (C) No significant differences in amplitude between patient and control cells.

106
Figure 7. Hippocampal slice recordings from Nestin-cre/DG-null (KO) mice and cre-negative (WT). Example field recordings during baseline (black) and after high frequency stimulation inducing LTP (red). Pooled data show no significant difference between WT and KO mice (n=7).

107
Chapter 5
Conclusion
The aim of this thesis research was to identify and model mutations associated
with congenital diseases of muscle (CDM). By studying a large family with a dominant
disease, we were able to identify a new mutation in ACTA1 by using exome sequencing.
By using induced pluripotent stem cells, we were able to model the synaptic phenotype of
alpha-dystroglycanopathies. Overall, we revealed the importance of using the most
current technologies available for diagnosis and modeling of disease.
5.1: Exome sequencing with linkage revealed a new mutation in ACTA1
Summary
In 1966 when Armstrong and colleagues first examined this family, exome
sequencing was not a tool available at their disposal to assist them in diagnosis and
treatment. In this thesis, we were able to use the combination of linkage and exome

108
sequence to identify a new mutation in ACTA1 with a different phenotype than
previously reported (Chapter 3).
Future Studies
Modeling of the mutation revealed intramolecular and intermolecular interactions
that would presumably affect the functioning of skeletal actin with this mutation. Thus
far the exact mechanism of disease has remained elusive. We have used cos-7 cells,
patient myoblasts, and zebrafish to model the mutation none of which showed a
phenotype.
Currently we are using a laser capture method to identify other proteins that may
be up or down regulated in this disease. By using patient muscle biopsies, we have
captured over 100 samples of muscle fibers from a normal control, a milder and a more
severe patient. We will then use the proteomics from these samples to identify other
mechanistic pathways to investigate based on the findings from these studies.
Additionally, given the variability among family members, one would be
interested in further characterizing additional genetic factors that contribute to this
difference.
Significance
This work is significant because it broadens the phenotype of actinopathies to a
less severe and slowly progressive disease with a scapuloperoneal onset. In addition, this
work shows the difficulty in distinguishing muscle from neuron diseases and identifying
the role that each has in contributing to disease.

109
5.2: Neurons from induced pluripotent stem cells recapitulate features of the alpha-
dystroglycanopathies
Summary
Previously, a variety of models have been used to study the central nervous
system deficits in alpha-dystroglycanopathies including zebrafish, drosophila, C.
elegans, and mice. However, these models are insufficient to study the inherent
complexity associated with human disease especially in synaptic deficiencies. To
overcome this difficulty, I used induced pluripotent stem cells (iPSCs) differentiated into
neurons to recapitulate human disease in culture. We showed hypoglycosylation
consistent with model systems and patient analysis as well as a descrease in spontaneous
activity as shown by electrophysiology.
Future Studies
Using this model system many avenues exist for future investigation. Thus far we
have an electrophysiological difference suggesting presynaptic deficits, however the
exact mechanism remains unknown. Because there is no difference in the number of
synapses, perhaps the difference exists in vesicle release, receptor distribution, or synapse
maturation, pruning, or maintenance. Each of these possibilities would need to be
investigated individually.
To facilitate understanding of this synaptic difference and guide our studies in the
patient derived neurons we are currently using cultured mouse neurons from dag1-
floxed/nestin-cre mice. If the same differences exist in these cultures, perhaps a

110
mechanism would be found without the necessary 12 weeks in vitro to produce mature
synapses from iPS cells. Then we could confirm any findings in the human model
system.
Additionally, we hope to further understand the embryoid body phenotype. First
we have added additional lines and controls to confirm a fusion phenotype. Secondly, we
are freezing, embedding, and staining the embryoid bodies to understand the deficits in
the basement membrane formation.
Significance
We hope this work will establish that neurons differentiated from iPSCs provide
an invaluable model for studying alpha-dystroglycanopathies. This model should be
considered for the exploration of treatment possibilities and drug and small molecule
screens. Any treatments or drugs could be tested and confirmed in this model and could
even be used to individually test mutations and patients for efficacy.
5.3: Broader Impacts
Taken together these two projects confirm the necessity of using the latest
information and technologies to elucidate mechanisms of known and unknown
diseases. By using exome sequencing and induced pluripotent stem cells, these projects
show the benefits of using new techniques as they become available. Exome sequencing
has advanced genetic diagnosis and has shown that a geneotype/phenotype correlation is
even more complex. In particular in neuromuscular diseases the varying contributions of
the neuron and muscle sometimes make genetic diagnosis complicated and difficult.

111
iPSCs have revolutionized the stem cell field and have opened up possibilities for
individualized medicine and treatments in a way that was previously impossible. Using a
human system is vital because treatments act differently in lower model systems such as
zebrafish and mice and sometimes end up not working in patients. Additionally, if iPSCs
are used in treatments, for example reimplantation after genetic correction, no
immunosuppressive drugs are necessary and there is no risk of rejection.

112
References
Abecasis, G. R., S. S. Cherny, et al. (2002). "Merlin--rapid analysis of dense genetic
maps using sparse gene flow trees." Nat Genet 30(1): 97-101. Agrawal, P. B., C. D. Strickland, et al. (2004). "Heterogeneity of nemaline myopathy
cases with skeletal muscle alpha-actin gene mutations." Ann Neurol 56(1): 86-96. Amoroso, M. W., G. F. Croft, et al. (2013). "Accelerated high-yield generation of limb-
innervating motor neurons from human stem cells." J Neurosci 33(2): 574-586. Armstrong, R. M., M. H. Fogelson, et al. (1966). "Familial proximal spinal muscular
atrophy." Arch Neurol 14(2): 208-212. Avsar-Ban, E., H. Ishikawa, et al. (2010). "Protein O-mannosylation is necessary for
normal embryonic development in zebrafish." Glycobiology 20(9): 1089-1102. Balci, B. et al. An autosomal recessive limb girdle muscular dystrophy (LGMD2) with
mild mental retardation is allelic to Walker-Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromuscul Disord 15, 271-275, doi:S0960-8966(05)00054-4 [pii] 10.1016/j.nmd.2005.01.013 (2005).
Baltaci, V., R. Ors, et al. (1999). "A case associated with Walker Warburg syndrome phenotype and homozygous pericentric inversion 9: coincidental finding or aetiological factor?" Acta Paediatr 88(5): 579-583.
Barresi, R., D. E. Michele, et al. (2004). "LARGE can functionally bypass alpha-
dystroglycan glycosylation defects in distinct congenital muscular dystrophies." Nat Med 10(7): 696-703.
Blaeser, A., E. Keramaris, et al. (2013). "Mouse models of fukutin-related protein
mutations show a wide range of disease phenotypes." Hum Genet 132(8): 923-934.
Bogdanik, L., B. Framery, et al. (2008). "Muscle dystroglycan organizes the postsynapse
and regulates presynaptic neurotransmitter release at the Drosophila neuromuscular junction." PLoS One 3(4): e2084.
Brockington, M., S. Torelli, et al. (2010). "Transgenic overexpression of LARGE induces
alpha-dystroglycan hyperglycosylation in skeletal and cardiac muscle." PLoS One 5(12): e14434.

113
Bromberg, M. B. and K. J. Swoboda (2002). "Motor unit number estimation in infants
and children with spinal muscular atrophy." Muscle Nerve 25(3): 445-447. Brown, S. C., S. Torelli, et al. (2004). "Abnormalities in alpha-dystroglycan expression in
MDC1C and LGMD2I muscular dystrophies." Am J Pathol 164(2): 727-737. Carss, K. J., E. Stevens, et al. (2013). "Mutations in GDP-mannose pyrophosphorylase B
cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of alpha-dystroglycan." Am J Hum Genet 93(1): 29-41.
Cavaldesi, M., G. Macchia, et al. (1999). "Association of the dystroglycan complex isolated from bovine brain synaptosomes with proteins involved in signal transduction." J Neurochem 72(4): 1648-1655.
Chambers, S. M., C. A. Fasano, et al. (2009). "Highly efficient neural conversion of
human ES and iPS cells by dual inhibition of SMAD signaling." Nat Biotechnol 27(3): 275-280.
Chan, Y. M., E. Keramaris-Vrantsis, et al. (2010). "Fukutin-related protein is essential for
mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies." Hum Mol Genet 19(20): 3995-4006.
Chandrasekharan, K. and P. T. Martin (2010). "Genetic defects in muscular dystrophy."
Methods Enzymol 479: 291-322. Chang, W. et al. Founder Fukutin mutation causes Walker-Warburg syndrome in four
Ashkenazi Jewish families. Prenat Diagn 29, 560-569, doi:10.1002/pd.2238 (2009).
Clarkson, E., C. F. Costa, et al. (2004). "Congenital myopathies: diseases of the actin cytoskeleton." J Pathol 204(4): 407-417.
Clement, E., E. Mercuri, et al. (2008). "Brain involvement in muscular dystrophies with
defective dystroglycan glycosylation." Ann Neurol 64(5): 573-582. Collins, J. and C. G. Bonnemann (2010). "Congenital muscular dystrophies: toward
molecular therapeutic interventions." Curr Neurol Neurosci Rep 10(2): 83-91. Cotarelo, R. P. et al. A double homozygous mutation in the POMT1 gene involving exon
skipping gives rise to Walker-Warburg syndrome in two Spanish Gypsy families. Clin Genet 76, 108-112, doi:CGE1188 [pii] 10.1111/j.1399-0004.2009.01188.x (2009).
Cotarelo, R. P. et al. Two new patients bearing mutations in the fukutin gene confirm the relevance of this gene in Walker-Warburg syndrome. Clin Genet 73, 139-145, doi:CGE936 [pii]

114
10.1111/j.1399-0004.2007.00936.x (2008).
de Bernabe, D. B. et al. Loss of alpha-dystroglycan laminin binding in epithelium-derived cancers is caused by silencing of LARGE. J Biol Chem 284, 11279-11284, doi:C900007200 [pii] 10.1074/jbc.C900007200 (2009).
Deng, H. X., C. J. Klein, et al. (2010). "Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4." Nat Genet 42(2): 165-169.
Deng, W. M., M. Schneider, et al. (2003). "Dystroglycan is required for polarizing the
epithelial cells and the oocyte in Drosophila." Development 130(1): 173-184. DePristo, M. A., E. Banks, et al. (2011). "A framework for variation discovery and
genotyping using next-generation DNA sequencing data." Nat Genet 43(5): 491-498.
Di Costanzo, S., A. Balasubramanian, et al. (2014). "POMK mutations disrupt muscle
development leading to a spectrum of neuromuscular presentations." Hum Mol Genet.
Dimos, J. T., K. T. Rodolfa, et al. (2008). "Induced pluripotent stem cells generated from
patients with ALS can be differentiated into motor neurons." Science 321(5893): 1218-1221.
Endo, T. (2007). "Dystroglycan glycosylation and its role in alpha-dystroglycanopathies."
Acta Myol 26(3): 165-170. Ervasti, J. M. et al. Alpha-dystroglycan deficiency correlates with elevated serum
creatine kinase and decreased muscle contraction tension in golden retriever muscular dystrophy. FEBS Lett 350, 173-176, doi:0014-5793(94)00748-9 [pii] (1994).
Feng, J. J. and S. Marston (2009). "Genotype-phenotype correlations in ACTA1 mutations that cause congenital myopathies." Neuromuscul Disord 19(1): 6-16.
Figarella-Branger, D., M. El-Dassouki, et al. (2002). "Myopathy with lobulated muscle
fibers: evidence for heterogeneous etiology and clinical presentation." Neuromuscul Disord 12(1): 4-12.
Goddeeris, M. M., B. Wu, et al. (2013). "LARGE glycans on dystroglycan function as a
tunable matrix scaffold to prevent dystrophy." Nature 503(7474): 136-140.

115
Godfrey, C., E. Clement, et al. (2007). "Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan." Brain 130(Pt 10): 2725-2735.
Godfrey, C., A. R. Foley, et al. (2011). "Dystroglycanopathies: coming into focus." Curr
Opin Genet Dev 21(3): 278-285. Goebel, H. H. and N. G. Laing (2009). "Actinopathies and myosinopathies." Brain Pathol
19(3): 516-522. Gorba, T. and L. Conti (2013). "Neural stem cells as tools for drug discovery: novel
platforms and approaches." Expert Opin Drug Discov 8(9): 1083-1094. Gorecki, D. C., J. M. Derry, et al. (1994). "Dystroglycan: brain localisation and
chromosome mapping in the mouse." Hum Mol Genet 3(9): 1589-1597. Greber, B., P. Coulon, et al. (2011). "FGF signalling inhibits neural induction in human
embryonic stem cells." EMBO J 30(24): 4874-4884. Greener, M. J. and R. G. Roberts (2000). "Conservation of components of the dystrophin
complex in Drosophila." FEBS Lett 482(1-2): 13-18. Grewal, P. K., P. J. Holzfeind, et al. (2001). "Mutant glycosyltransferase and altered
glycosylation of alpha-dystroglycan in the myodystrophy mouse." Nat Genet 28(2): 151-154.
Grunseich, C., K. Zukosky, et al. (2014). "Stem cell-derived motor neurons from spinal
and bulbar muscular atrophy patients." Neurobiol Dis 70C: 12-20. Grisoni, K., E. Martin, et al. (2002). "Genetic evidence for a dystrophin-glycoprotein
complex (DGC) in Caenorhabditis elegans." Gene 294(1-2): 77-86. Gupta, V., G. Kawahara, et al. (2011). "The zebrafish dag1 mutant: a novel genetic model
for dystroglycanopathies." Hum Mol Genet 20(9): 1712-1725. Guyon, J. R., L. S. Steffen, et al. (2007). "Modeling human muscle disease in zebrafish."
Biochim Biophys Acta 1772(2): 205-215. Haines, N., S. Seabrooke, et al. (2007). "Dystroglycan and protein O-
mannosyltransferases 1 and 2 are required to maintain integrity of Drosophila larval muscles." Mol Biol Cell 18(12): 4721-4730.
Hanauer, A., M. Levin, et al. (1983). "Isolation and characterization of cDNA clones for
human skeletal muscle alpha actin." Nucleic Acids Res 11(11): 3503-3516.

116
Hara, Y. et al. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N Engl J Med 364, 939-946, doi:10.1056/NEJMoa1006939 (2011).
Henion, T. R., Q. Qu, et al. (2003). "Expression of dystroglycan, fukutin and POMGnT1 during mouse cerebellar development." Brain Res Mol Brain Res 112(1-2): 177-181.
Henry, M. D. and K. P. Campbell (1998). "A role for dystroglycan in basement
membrane assembly." Cell 95(6): 859-870. Herbst, R., Iskratsch, T., Unger, E., Bittner, RE. Aberrant development of neuromuscular
junctions in glycosylation-defective Large(myd) mice. Neuromuscular Disorders 19, 366-378 (2009).
Hewitt, J. E. (2009). "Abnormal glycosylation of dystroglycan in human genetic disease."
Biochim Biophys Acta 1792(9): 853-861. Hockemeyer, D. et al. A drug-inducible system for direct reprogramming of human
somatic cells to pluripotency. Cell Stem Cell 3, 346-353, doi:S1934-5909(08)00412-8 [pii] 10.1016/j.stem.2008.08.014 (2008).
Hu, B. Y. & Zhang, S. C. Directed differentiation of neural-stem cells and subtype-specific neurons from hESCs. Methods Mol Biol 636, 123-137, doi:10.1007/978-1-60761-691-7_8 (2010).
Hu, H., J. Li, et al. (2011). "Conditional knockout of protein O-mannosyltransferase 2
reveals tissue-specific roles of O-mannosyl glycosylation in brain development." J Comp Neurol 519(7): 1320-1337.
Hu, H., Y. Yang, et al. (2007). "Breaches of the pial basement membrane and
disappearance of the glia limitans during development underlie the cortical lamination defect in the mouse model of muscle-eye-brain disease." J Comp Neurol 502(2): 168-183.
Hu, Y., Li, Z. F., Wu, X. & Lu, Q. Large induces functional glycans in an O-mannosylation
dependent manner and targets GlcNAc terminals on alpha-dystroglycan. PLoS One 6, e16866, doi:10.1371/journal.pone.0016866 (2011).
Ilkovski, B., S. T. Cooper, et al. (2001). "Nemaline myopathy caused by mutations in the
muscle alpha-skeletal-actin gene." Am J Hum Genet 68(6): 1333-1343. Ilkovski, B., K. J. Nowak, et al. (2004). "Evidence for a dominant-negative effect in
ACTA1 nemaline myopathy caused by abnormal folding, aggregation and altered polymerization of mutant actin isoforms." Hum Mol Genet 13(16): 1727-1743.

117
Inamori, K., Y. Hara, et al. (2013). "Xylosyl- and glucuronyltransferase functions of LARGE in alpha-dystroglycan modification are conserved in LARGE2." Glycobiology 23(3): 295-302.
Inamori, K., T. Yoshida-Moriguchi, et al. (2012). "Dystroglycan function requires
xylosyl- and glucuronyltransferase activities of LARGE." Science 335(6064): 93-96.
Irodenko, V. S., H. S. Lee, et al. (2009). "Adult nemaline myopathy with trabecular
muscle fibers." Muscle Nerve 39(6): 871-875. Johnson, R. P., S. H. Kang, et al. (2006). "C. elegans dystroglycan DGN-1 functions in
epithelia and neurons, but not muscle, and independently of dystrophin." Development 133(10): 1911-1921.
Johnson, R. P. and J. M. Kramer (2012). "C. elegans dystroglycan coordinates
responsiveness of follower axons to dorsal/ventral and anterior/posterior guidance cues." Dev Neurobiol 72(12): 1498-1515.
Judas, M. et al. POMT1-associated walker-warburg syndrome: a disorder of dendritic
development of neocortical neurons. Neuropediatrics 40, 6-14, doi:10.1055/s-0029-1224099 (2009).
Jungbluth, H., C. A. Sewry, et al. (2001). "Mild phenotype of nemaline myopathy with
sleep hypoventilation due to a mutation in the skeletal muscle alpha-actin (ACTA1) gene." Neuromuscul Disord 11(1): 35-40.
Kaindl, A. M., F. Ruschendorf, et al. (2004). "Missense mutations of ACTA1 cause
dominant congenital myopathy with cores." J Med Genet 41(11): 842-848. Kanagawa, M., Nishimoto, A., Chivonobu, T., Takeda, S., Miyagoe-Suzuki, Y., Wang, F.,
Fijukake, N., Taniguchi, M., Lu, Z., Tachikawa, M., Nagai, Y., Tashiro, F., Miyazaki, J., Tajima, Y., Takeda, S., Endo, T., Kobayashi, K., Campbell, KP., Toda, T. Residual laminin-binding activity and enhanced dystroglycan glycosylation by LARGE in novel model mice to dystroglycanopathy. Hum Mol Genet 18, 621-631 (2009).
Kawahara, G., J. R. Guyon, et al. (2010). "Zebrafish models for human FKRP muscular
dystrophies." Hum Mol Genet 19(4): 623-633. Kerr, S. L. A case study on Walker-Warburg syndrome. Adv Neonatal Care 10, 21-24,
doi:10.1097/ANC.0b013e3181cbf535 00149525-201002000-00007 [pii] (2010).
Khalaf, S. S. & Tareef, R. B. Walker-Warburg Syndrome. J AAPOS 10, 486-488, doi:S1091-8531(06)00419-8 [pii] 10.1016/j.jaapos.2006.06.016 (2006).

118
Kirschner, J. & Bonnemann, C. G. The congenital and limb-girdle muscular dystrophies: sharpening the focus, blurring the boundaries. Arch Neurol 61, 189-199, doi:10.1001/archneur.61.2.189 61/2/189 [pii] (2004).
Kriks, S., J. W. Shim, et al. (2011). "Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease." Nature 480(7378): 547-551.
Kurahashi, H., M. Taniguchi, et al. (2005). "Basement membrane fragility underlies
embryonic lethality in fukutin-null mice." Neurobiol Dis 19(1-2): 208-217. Laing, N. G., N. F. Clarke, et al. (2004). "Actin mutations are one cause of congenital
fibre type disproportion." Ann Neurol 56(5): 689-694. Laing, N. G., D. E. Dye, et al. (2009). "Mutations and polymorphisms of the skeletal
muscle alpha-actin gene (ACTA1)." Hum Mutat 30(9): 1267-1277. Lane, P. W., T. C. Beamer, et al. (1976). "Myodystrophy, a new myopathy on
chromosome 8 of the mouse." J Hered 67(3): 135-138. Levi, S., R. M. Grady, et al. (2002). "Dystroglycan is selectively associated with
inhibitory GABAergic synapses but is dispensable for their differentiation." J Neurosci 22(11): 4274-4285.
Li, H. and R. Durbin (2009). "Fast and accurate short read alignment with Burrows-
Wheeler transform." Bioinformatics 25(14): 1754-1760. Li, S., D. Edgar, et al. (2003). "The role of laminin in embryonic cell polarization and
tissue organization." Dev Cell 4(5): 613-624. Liewluck, T., J. A. Tracy, et al. (2013). "Scapuloperoneal muscular dystrophy phenotype
due to TRIM32-sarcotubular myopathy in South Dakota Hutterite." Neuromuscul Disord 23(2): 133-138.
Lin, Y. Y., R. J. White, et al. (2011). "Zebrafish Fukutin family proteins link the unfolded
protein response with dystroglycanopathies." Hum Mol Genet 20(9): 1763-1775. Liu, J., S. L. Ball, et al. (2006). "A genetic model for muscle-eye-brain disease in mice
lacking protein O-mannose 1,2-N-acetylglucosaminyltransferase (POMGnT1)." Mech Dev 123(3): 228-240.
Liu, S. V. iPS cells: a more critical review. Stem Cells Dev 17, 391-397,
doi:10.1089/scd.2008.0062 (2008).

119
Lommel, M., Willer, T. & Strahl, S. POMT2, a key enzyme in Walker-Warburg syndrome: somatic sPOMT2, but not testis-specific tPOMT2, is crucial for mannosyltransferase activity in vivo. Glycobiology 18, 615-625, doi:cwn042 [pii] 10.1093/glycob/cwn042 (2008).
MacLeod, H. et al. A novel FKRP mutation in congenital muscular dystrophy disrupts the dystrophin glycoprotein complex. Neuromuscul Disord 17, 285-289, doi:S0960-8966(07)00009-0 [pii] 10.1016/j.nmd.2007.01.005 (2007).
Manzini, M. C., D. E. Tambunan, et al. (2012). "Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome." Am J Hum Genet 91(3): 541-547.
Marchetto, M. C. et al. A model for neural development and treatment of Rett syndrome
using human induced pluripotent stem cells. Cell 143, 527-539, doi:S0092-8674(10)01186-4 [pii] 10.1016/j.cell.2010.10.016 (2010).
Matsumoto, H., Y. K. Hayashi, et al. (2005). "Congenital muscular dystrophy with
glycosylation defects of alpha-dystroglycan in Japan." Neuromuscul Disord 15(5): 342-348.
McComas, A. J., R. E. Sica, et al. (1974). "Multiple muscle analysis of motor units in
muscular dystrophy." Arch Neurol 30(3): 249-251. McKenna, A., M. Hanna, et al. (2010). "The Genome Analysis Toolkit: a MapReduce
framework for analyzing next-generation DNA sequencing data." Genome Res 20(9): 1297-1303.
McNally, E. M. and P. Pytel (2007). "Muscle diseases: the muscular dystrophies." Annu
Rev Pathol 2: 87-109. Miyoshi, N., Ishii, H., Nagano, H., Haraguchi, N., Dewi, D.L., Kano, Y., Nishikawa, S.,
Tanemura, M. Mimori, K., Tanaka, F., Saito, T., Nishimura, J., Takemasa, I., Mizushima, T., Ikeda, M., Yamamoto, H., Sekimoto, M., Doki, Y., Mori, M. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 8, 633-638.
Moore, C. J., H. T. Goh, et al. (2008). "Genes required for functional glycosylation of
dystroglycan are conserved in zebrafish." Genomics 92(3): 159-167. Moore, S. A., F. Saito, et al. (2002). "Deletion of brain dystroglycan recapitulates aspects
of congenital muscular dystrophy." Nature 418(6896): 422-425.

120
Mummery, R., A. Sessay, et al. (1996). "Beta-dystroglycan: subcellular localisation in rat brain and detection of a novel immunologically related, postsynaptic density-enriched protein." J Neurochem 66(6): 2455-2459.
Muntoni, F. (2005). "Walker-Warburg syndrome and limb girdle muscular dystrophy;
two sides of the same coin." Neuromuscul Disord 15(4): 269-270. Myshrall, T. D., S. A. Moore, et al. (2012). "Dystroglycan on radial glia end feet is
required for pial basement membrane integrity and columnar organization of the developing cerebral cortex." J Neuropathol Exp Neurol 71(12): 1047-1063.
Nakamura, N., S. H. Stalnaker, et al. (2010). "Drosophila Dystroglycan is a target of O-
mannosyltransferase activity of two protein O-mannosyltransferases, Rotated Abdomen and Twisted." Glycobiology 20(3): 381-394.
Nguyen, H., A. P. Ostendorf, et al. (2013). "Glial scaffold required for cerebellar granule
cell migration is dependent on dystroglycan function as a receptor for basement membrane proteins." Acta Neuropathol Commun 1(1): 58.
Nishikawa, S., Goldstein, R. A. & Nierras, C. R. The promise of human induced
pluripotent stem cells for research and therapy. Nat Rev Mol Cell Biol 9, 725-729, doi:nrm2466 [pii] 10.1038/nrm2466 (2008).
Nowak, K. J., G. Ravenscroft, et al. (2012). "Skeletal muscle alpha-actin diseases (actinopathies): pathology and mechanisms." Acta Neuropathol.
Nowak, K. J., D. Wattanasirichaigoon, et al. (1999). "Mutations in the skeletal muscle
alpha-actin gene in patients with actin myopathy and nemaline myopathy." Nat Genet 23(2): 208-212.
Ohtsuka-Tsurumi, E., Y. Saito, et al. (2004). "Co-localization of fukutin and alpha-
dystroglycan in the mouse central nervous system." Brain Res Dev Brain Res 152(2): 121-127.
Okita, K., Ichisaka, T. & Yamanaka, S. Generation of germline-competent induced
pluripotent stem cells. Nature 448, 313-317, doi:nature05934 [pii] 10.1038/nature05934 (2007).
Omori, Y., F. Araki, et al. (2012). "Presynaptic dystroglycan-pikachurin complex regulates the proper synaptic connection between retinal photoreceptor and bipolar cells." J Neurosci 32(18): 6126-6137.
Paganoni, S. and A. Amato (2013). "Electrodiagnostic evaluation of myopathies." Phys
Med Rehabil Clin N Am 24(1): 193-207.

121
Park, I. H. et al. Disease-specific induced pluripotent stem cells. Cell 134, 877-886, doi:S0092-8674(08)01001-5 [pii] 10.1016/j.cell.2008.07.041 (2008).
Parsons, M. J., I. Campos, et al. (2002). "Removal of dystroglycan causes severe muscular dystrophy in zebrafish embryos." Development 129(14): 3505-3512.
Pera, M. F. Defining pluripotency. Nat Methods 7, 885-887, doi:nmeth1110-885 [pii]
10.1038/nmeth1110-885 (2010).
Pfisterer, U. et al. Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci U S A 108, 10343-10348, doi:1105135108 [pii] 10.1073/pnas.1105135108 (2011).
Preuss, M., Heckmann, M., Stein, M. & Nestler, U. Two cases of Walker-Warburg syndrome complicated by hydrocephalus. Pediatr Neurosurg 46, 34-38, doi:000314999 [pii] 10.1159/000314999 (2010).
Qiang, L., R. Fujita, et al. (2013). "Remodeling neurodegeneration: somatic cell reprogramming-based models of adult neurological disorders." Neuron 78(6): 957-969.
Reed, U. C. Congenital muscular dystrophy. Part I: a review of phenotypical and diagnostic
aspects. Arq Neuropsiquiatr 67, 144-168, doi:S0004-282X2009000100038 [pii] (2009). Roscioli, T., E. J. Kamsteeg, et al. (2012). "Mutations in ISPD cause Walker-Warburg
syndrome and defective glycosylation of alpha-dystroglycan." Nat Genet 44(5): 581-585.
Saito, F., S. A. Moore, et al. (2003). "Unique role of dystroglycan in peripheral nerve
myelination, nodal structure, and sodium channel stabilization." Neuron 38(5): 747-758.
Saito, F. et al. Aberrant glycosylation of alpha-dystroglycan causes defective binding of
laminin in the muscle of chicken muscular dystrophy. FEBS Lett 579, 2359-2363, doi:S0014-5793(05)00373-X [pii] 10.1016/j.febslet.2005.03.033 (2005)
Saito, F., M. Kanagawa, et al. (2014). "Overexpression of LARGE suppresses muscle regeneration via down-regulation of insulin-like growth factor 1 and aggravates muscular dystrophy in mice." Hum Mol Genet.
Santavuori, P. et al. Muscle-eye-brain disease (MEB). Brain Dev 11, 147-153 (1989).

122
Santavuori, P., H. Pihko, et al. (1990). "Muscle-eye-brain disease and Walker-Warburg syndrome." Am J Med Genet 36(3): 371-374.
Satz, J. S., R. Barresi, et al. (2008). "Brain and eye malformations resembling Walker-
Warburg syndrome are recapitulated in mice by dystroglycan deletion in the epiblast." J Neurosci 28(42): 10567-10575.
Satz, J. S., A. P. Ostendorf, et al. (2010). "Distinct functions of glial and neuronal
dystroglycan in the developing and adult mouse brain." J Neurosci 30(43): 14560-14572.
Schneider, M., A. A. Khalil, et al. (2006). "Perlecan and Dystroglycan act at the basal
side of the Drosophila follicular epithelium to maintain epithelial organization." Development 133(19): 3805-3815.
Schroder, J. M., H. Durling, et al. (2004). "Actin myopathy with nemaline bodies,
intranuclear rods, and a heterozygous mutation in ACTA1 (Asp154Asn)." Acta Neuropathol 108(3): 250-256.
Senju, S. et al. Generation of dendritic cells and macrophages from human induced
pluripotent stem cells aiming at cell therapy. Gene Ther 18, 874-883, doi:gt201122 [pii] 10.1038/gt.2011.22 (2011).
Sevdali, M., V. Kumar, et al. (2013). "Human congenital myopathy actin mutants cause myopathy and alter Z-disc structure in Drosophila flight muscle." Neuromuscul Disord 23(3): 243-255.
Shcherbata, H. R., A. S. Yatsenko, et al. (2007). "Dissecting muscle and neuronal
disorders in a Drosophila model of muscular dystrophy." EMBO J 26(2): 481-493.
Sherry, S. T., M. H. Ward, et al. (2001). "dbSNP: the NCBI database of genetic
variation." Nucleic Acids Res 29(1): 308-311. Smalheiser, N. R. and B. J. Collins (2000). "Coordinate enrichment of cranin
(dystroglycan) subunits in synaptic membranes of sheep brain." Brain Res 887(2): 469-471.
Stadtfeld, M., Nagaya, M., Utikal, J., Weir, G. & Hochedlinger, K. Induced pluripotent stem cells
generated without viral integration. Science 322, 945-949, doi:1162494 [pii] Stevens, E., K. J. Carss, et al. (2013). "Mutations in B3GALNT2 cause congenital
muscular dystrophy and hypoglycosylation of alpha-dystroglycan." Am J Hum Genet 92(3): 354-365.

123
Sugita, S., F. Saito, et al. (2001). "A stoichiometric complex of neurexins and dystroglycan in brain." J Cell Biol 154(2): 435-445.
Takahashi, K., K. Tanabe, et al. (2007). "Induction of pluripotent stem cells from adult
human fibroblasts by defined factors." Cell 131(5): 861-872. Takahashi, K. and S. Yamanaka (2006). "Induction of pluripotent stem cells from mouse
embryonic and adult fibroblast cultures by defined factors." Cell 126(4): 663-676. Taylor, A., H. P. Erba, et al. (1988). "Nucleotide sequence and expression of the human
skeletal alpha-actin gene: evolution of functional regulatory domains." Genomics 3(4): 323-336.
Thornhill, P., D. Bassett, et al. (2008). "Developmental defects in a zebrafish model for
muscular dystrophies associated with the loss of fukutin-related protein (FKRP)." Brain 131(Pt 6): 1551-1561.
Uchino, M., A. Hara, et al. (1996). "Distribution of dystrophin and dystrophin-associated
protein 43DAG (beta-dystroglycan) in the central nervous system of normal controls and patients with Duchenne muscular dystrophy." Intern Med 35(3): 189-194.
Van Reeuwijk, J. et al. A homozygous FKRP start codon mutation is associated with
Walker-Warburg syndrome, the severe end of the clinical spectrum. Clin Genet 78, 275-281, doi:CGE1384 [pii] 10.1111/j.1399-0004.2010.01384.x (2010).
Vierbuchen, T. et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035-1041, doi:nature08797 [pii] 10.1038/nature08797 (2010).
Villefranc, J. A., J. Amigo, et al. (2007). "Gateway compatible vectors for analysis of gene function in the zebrafish." Dev Dyn 236(11): 3077-3087.
Vuillaumier-Barrot, S. et al. Intragenic rearrangements in LARGE and POMGNT1 genes
in severe dystroglycanopathies. Neuromuscul Disord, doi:S0960-8966(11)00169-6 [pii] 10.1016/j.nmd.2011.06.001 (2011).
Wairkar, Y. P., L. G. Fradkin, et al. (2008). "Synaptic defects in a Drosophila model of congenital muscular dystrophy." J Neurosci 28(14): 3781-3789.
Waite, A., Tinsley, C. L., Locke, M. & Blake, D. J. The neurobiology of the dystrophin-
associated glycoprotein complex. Ann Med 41, 344-359, doi:908179195 [pii] 10.1080/07853890802668522 (2009).

124
Warren, L. et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618-630, doi:S1934-5909(10)00434-0 [pii] 10.1016/j.stem.2010.08.012 (2010).
Weller, B., S. Carpenter, et al. (1999). "Myopathy with trabecular muscle fibers." Neuromuscul Disord 9(4): 208-214.
Whitmore, C., M. Fernandez-Fuente, et al. (2014). "The transgenic expression of LARGE
exacerbates the muscle phenotype of dystroglycanopathy mice." Hum Mol Genet 23(7): 1842-1855.
Willer, T., B. Prados, et al. (2004). "Targeted disruption of the Walker-Warburg
syndrome gene Pomt1 in mouse results in embryonic lethality." Proc Natl Acad Sci U S A 101(39): 14126-14131.
Willer, T., H. Lee, et al. (2012). "ISPD loss-of-function mutations disrupt dystroglycan
O-mannosylation and cause Walker-Warburg syndrome." Nat Genet 44(5): 575-580.
Williamson, R. A., M. D. Henry, et al. (1997). "Dystroglycan is essential for early
embryonic development: disruption of Reichert's membrane in Dag1-null mice." Hum Mol Genet 6(6): 831-841.
Wright, K. M., K. A. Lyon, et al. (2012). "Dystroglycan organizes axon guidance cue
localization and axonal pathfinding." Neuron 76(5): 931-944. Xiong, H., K. Kobayashi, et al. (2006). "Molecular interaction between fukutin and
POMGnT1 in the glycosylation pathway of alpha-dystroglycan." Biochem Biophys Res Commun 350(4): 935-941.
Yagi, T., D. Ito, et al. (2011). "Modeling familial Alzheimer's disease with induced
pluripotent stem cells." Hum Mol Genet 20(23): 4530-4539. Yamanaka, S. and K. Takahashi (2006). "[Induction of pluripotent stem cells from mouse
fibroblast cultures]." Tanpakushitsu Kakusan Koso 51(15): 2346-2351. Yoshida-Moriguchi, T. et al. O-mannosyl phosphorylation of alpha-dystroglycan is
required for laminin binding. Science 327, 88-92, doi:327/5961/88 [pii] 10.1126/science.1180512 (2010).
Yu, J., M. A. Vodyanik, et al. (2007). "Induced pluripotent stem cell lines derived from human somatic cells." Science 318(5858): 1917-1920.

125
Yuan, S. H. et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One 6, e17540, doi:10.1371/journal.pone.0017540 (2011).
Zaccaria, M. L., M. E. De Stefano, et al. (2000). "Selective reduction in the nicotinic
acetylcholine receptor and dystroglycan at the postsynaptic apparatus of mdx mouse superior cervical ganglion." J Neuropathol Exp Neurol 59(2): 103-112.
Zaccaria, M. L., F. Di Tommaso, et al. (2001). "Dystroglycan distribution in adult mouse
brain: a light and electron microscopy study." Neuroscience 104(2): 311-324. Zhang, P., Y. Yang, et al. (2013). "Biochemical and biophysical changes underlie the
mechanisms of basement membrane disruptions in a mouse model of dystroglycanopathy." Matrix Biol 32(3-4): 196-207.




















