
Delayed Inner Ear Maturation and Neuronal Loss in Postnatal Igf-1Deficient Mice
Upload
independentCategory
view
0download
0
This article was published in an Elsevier journal. The attached copyis furnished to the author for non-commercial research and
education use, including for instruction at the author’s institution,sharing with colleagues and providing to institution administration.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Research Report
Intraparenchymal spinal cord delivery of adeno-associatedvirus IGF-1 is protective in the SOD1G93A model of ALS
Angelo C. Leporea,1, Christine Haenggelia,1, Mehdi Gasmic, Kathie M. Bishopc,Raymond T. Bartusc, Nicholas J. Maragakisa, Jeffrey D. Rothsteina,b,⁎aDepartment of Neurology, The Johns Hopkins University School of Medicine, 600 N. Wolfe St., Meyer 6-109, Baltimore, MD 21287, USAbDepartment of Neuroscience, The Johns Hopkins University School of Medicine, 600 N. Wolfe St., Meyer 6-109, Baltimore, MD 21287, USAcCeregene, Inc., San Diego, CA, USA
A R T I C L E I N F O A B S T R A C T
Article history:Accepted 5 September 2007Available online 22 September 2007
The potent neuroprotective activities of neurotrophic factors, including insulin-like growthfactor 1 (IGF-1), make them promising candidates for treatment of amyotrophic lateralsclerosis (ALS). In an effort to maximize rate of motor neuron transduction, achieve highlevels of spinal IGF-1 and thus enhance therapeutic benefit, we injected an adeno-associatedvirus 2 (AAV2)-based vector encoding human IGF-1 (CERE-130) into lumbar spinal cordparenchyma of SOD1G93A mice. We observed robust and long-term intraspinal IGF-1expression and partial rescue of lumbar spinal cord motor neurons, as well as sex-specificdelayed disease onset, weight loss, decline in hindlimb grip strength and increased animalsurvival.
© 2007 Elsevier B.V. All rights reserved.
Keywords:Adeno-associated virusInsulin-like growth factor 1Gene therapyNeurodegenerationAmyotrophic lateral sclerosisNeuroprotection
1. Introduction
ALS is characterized by relatively rapid degeneration of upperand lower motor neurons, with death normally occurring 2–5 years following diagnosis (Bruijn et al., 2004). Most cases are ofa sporadic nature, while ∼10% are familial. Of this familialportion, 20% are linked to various point mutations in the Cu/Znsuperoxide dismutase 1 (SOD1) gene on chromosome 21 (Rosenet al., 1993). Transgenic mice (Bruijn et al., 1997; Gurney et al.,1994; Wong et al., 1995) and rats (Howland et al., 2002; Nagaiet al., 2001) carrying mutant human SOD1(G93A, G37R, G86R, G85R)
genes have been generated, and, despite the existence of otheranimal models of motor neuron loss, are currently the mostwidely used ALS models. The cause of the relatively selective
death of motor neurons is unknown; however, a number ofmechanisms have been shown to at least contribute to ALSpathogenesis (Bruijn et al., 2004).
Gene delivery of neurotrophic molecules is a possibletherapeutic strategy for ALS treatment (Alisky and Davidson,2000; Boillee and Cleveland, 2004; Federici and Boulis, 2006).While alterations in CNS trophic factor signaling are likely notthe primary cause of ALS, changes in levels of neurotrophicfactors and their receptors, as well as dysfunction of associ-ated intracellular signal transduction pathways, are associat-ed with ALS and could contribute to disease progression (Becket al., 2001; Wilczak and de Keyser, 2005).
IGF-1 is a pleitropic neurotrophic factor (Trejo et al., 2004).Liver-derived IGF-1 crosses the blood brain barrier, and IGF-1,
B R A I N R E S E A R C H 1 1 8 5 ( 2 0 0 7 ) 2 5 6 – 2 6 5
⁎ Corresponding author. Department of Neurology, The Johns Hopkins University School of Medicine, 600 N. Wolfe St., Meyer 6-109,Baltimore, MD 21287, USA.
E-mail address: [email protected] (J.D. Rothstein).1 These authors contributed equally to the work.
0006-8993/$ – see front matter © 2007 Elsevier B.V. All rights reserved.doi:10.1016/j.brainres.2007.09.034
ava i l ab l e a t www.sc i enced i rec t . com
www.e l sev i e r. com/ loca te /b ra in res

Author's personal copy
IGF-1 receptors, IGF-1 binding proteins (IGFBPs) and intra-cellular IGF-1 associated signaling factors (IRS-1, PI3 kinase)are all expressed in CNS, particularly in spinal cord ventralgray matter (including motor neurons) (Bondy and Cheng,2004). IGF-1 is myotrophic and a prototypical neuronalsurvival factor that exerts pro-survival effects specificallyon motor neurons (Dore et al., 1997). While adverse sideeffects were not observed, clinical trials in Europe (Borasioet al., 1998) and North America (Lai et al., 1997) showedlittle to no benefit of systemically administered recombi-nant IGF-1 in ALS patients. This may have been due to lackof sustained delivery, sequestration of exogenous IGF-1 byhigh levels of systemic and/or CNS IGFBPs, and low effi-ciency of protein delivery to motor neurons via systemicinjections.
Viral vector delivery offers an attractive means for achiev-ing sustained long-term expression of neurotrophic factor in aspecific region of interest, while avoiding unwanted sideeffects and peripheral IGF-1 sequestration. Direct intrapar-enchymal spinal cord injection of gene delivery vectors mayprove to be the most efficient means for providing IGF-1 todegenerating motor neurons because of reliable high titerdelivery of virus directly to regions of cell loss, lack ofdependency on retrograde transport along large, dysfunction-al motor axons and the potential ability to target delivery tomore abundant glia and interneurons (depending on tropismof AAV serotype). For these reasons, we have tested theefficacy of multi-segmental intraparenchymal delivery of anadeno-associated virus 2 (AAV2) gene delivery vector encodinghuman IGF-1 to the lumbar spinal cord of SOD1G93A mice.
Fig. 1 – AAV2 IGF-1 delivery results in long-term IGF-1 expression and rescues motor neurons in the lumbar spinal cord ofSOD1G93A mice. IGF-1 (A, C) and GFP (D) were expressed in SOD1G93A lumbar cord at high levels even at disease end-stage. IGF-1(C, arrows: enlargement of box in A) and GFP (D, inset) expression was specifically noted in ventral horn motor neurons.Expression of human IGF-1 was not seen in formulation buffer-injected cords (B, insert is higher magnification enlargement ofbox). Co-localization of GFP with GFAP+ astrocytes was not observed (E). AAV2 GFP and AAV2 IGF-1 SOD1G93A mice had greatlyreduced numbers of motor neurons compared to wild-type age-matched mice at 110 days of age (F). Compared to AAV2 GFPinjected controls, AAV2 IGF-1 mice had a significantly greater number of motor neurons at 110 days of age (F). No differences inthe response of Iba1+ microglia were noted between lumbar spinal cord of AAV2 GFP and AAV2 IGF-1 mice (G–H).
257B R A I N R E S E A R C H 1 1 8 5 ( 2 0 0 7 ) 2 5 6 – 2 6 5
Author's personal copy
2. Results
2.1. Long-term expression of IGF-1 and GFP in lumbarspinal cord of SOD1G93A mice following AAV2 delivery
GFP (Fig. 1D) and IGF-1 (Figs. 1A, C) were expressed in SOD1G93A
lumbar cord at high levels even at disease end-stage, up to90 days post-injection. Similar to GFP, IGF-1 expression wasobserved in continuous distinct regions in gray matter, likelyrelated to each individual injection. GFP expression wasobserved in distinct individual cells, the neuropil, in neuronalcell bodies of gray matter at the level of injection (includingmotor neurons: Fig. 1D, inset), and inneuritic processes inwhitematter both at the level of injection and in regions rostral andcaudal to injection sites. The rostrocaudal extent of GFPexpression for each injection was 0.6–1.2 mm, and the esti-mated volume of distribution of GFP product in graymatterwas
0.04–0.21mm3. Expression of GFPwas not seen in IGF-1 injectedcords, and similar long-term GFP expression was foundfollowing AAV2 GFP injections into lumbar spinal cord of wild-typemice (not shown). IGF-1 expression was found in neuronalcell bodies and neuritic processes of gray matter at injectionsites, was specifically noted in ventral horn motor neurons(Fig. 1C, arrows), but was seen to a lesser extent in surroundingwhite matter. The rostrocaudal extent for each IGF-1 injectionsitewas2.4–2.6mm,and theestimatedvolumeofdistributionofIGF-1 product in gray matter was 0.13–0.21 mm3 for each site.Expression of human IGF-1 was not seen in formulation buffer-injected cords (Fig. 1B). Unfortunately, tissuewas unavailable tomeasure total IGF-1 levels in lumbar spinal cord.
To determine the identity of AAV2-infected spinal cordcells, double-immunolabeling of GFP with specific phenotypicmarkers was employed. GFP co-localized with NeuN+ neurons(not shown); however, no co-localization of GFP with GFAP+
astrocytes was observed (Fig. 1E).
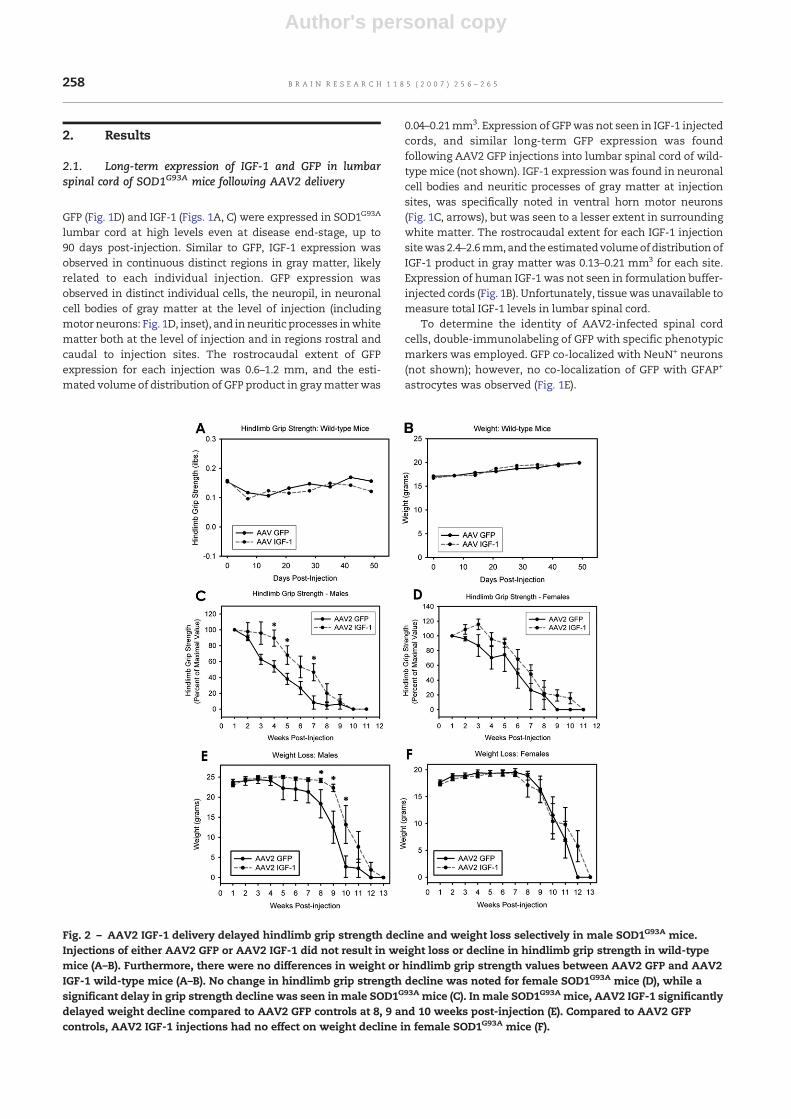
Fig. 2 – AAV2 IGF-1 delivery delayed hindlimb grip strength decline and weight loss selectively in male SOD1G93A mice.Injections of either AAV2 GFP or AAV2 IGF-1 did not result in weight loss or decline in hindlimb grip strength in wild-typemice (A–B). Furthermore, there were no differences in weight or hindlimb grip strength values between AAV2 GFP and AAV2IGF-1 wild-type mice (A–B). No change in hindlimb grip strength decline was noted for female SOD1G93A mice (D), while asignificant delay in grip strength decline was seen in male SOD1G93A mice (C). In male SOD1G93A mice, AAV2 IGF-1 significantlydelayed weight decline compared to AAV2 GFP controls at 8, 9 and 10 weeks post-injection (E). Compared to AAV2 GFPcontrols, AAV2 IGF-1 injections had no effect on weight decline in female SOD1G93A mice (F).
258 B R A I N R E S E A R C H 1 1 8 5 ( 2 0 0 7 ) 2 5 6 – 2 6 5
Author's personal copy
2.2. AAV2 IGF-1 delivery partially rescued motor neuronloss in SOD1G93A mice
AAV2 GFP (14.7±0.7 motor neurons/section; n=4) and AAV2IGF-1 (18.7±1.6/section; n=5) SOD1G93A mice had greatlyreduced numbers of motor neurons compared to wild-typeage-matched mice (30.9±1.3/section; n=4) at 110 days of age(Fig. 1F). Compared to AAV2 GFP injected controls, AAV2 IGF-1animals had a significantly greater number of motor neuronsat 110 days of age (pb0.01; Fig. 1F), demonstrating that IGF-1delivery partially rescuedmotor neuron loss in SOD1G93Amice.While the number of mice for sex-specific analysis of motorneuron survival was small, no gender differences were notedwithin the AAV GFP (males: 14.8±0.8 motor neurons/section;n=2; females: 14.4±0.5 motor neurons/section; n=2) or AAVIGF-1 (males: 18.7±2.2 motor neurons/section; n=2; females:18.8±0.4 motor neurons/section; n=3) treatment groups.
To access a potential mechanism of neuroprotection, thehost microglial response was examined. No differences in the
response of Iba1+ microglia were noted between lumbar spinalcord of AAV2 GFP and AAV2 IGF-1 mice (Figs. 1G–H),suggesting that therapeutic effects of IGF-1 were not mediatedby reduction in the host immune response.
2.3. AAV2 IGF-1 and AAV2 GFP delivery had no adversebehavioral effects
Injections of either AAV2 GFP or AAV2 IGF-1 did not result inweight loss or decline in hindlimb grip strength in wild-typemice (Figs. 2A–B; pN0.05 for all comparisons). Considering thatmultiple intraparenchymal spinal cord injection is a relativelyinvasive technique, these results are important in demon-strating that the injection procedure itself had no apparentadverse behavioral effects. Furthermore, there were nodifferences in weight or hindlimb grip strength values be-tween AAV2 GFP and AAV2 IGF-1 mice (Figs. 2A–B; pN0.05 forall comparisons), suggesting that IGF-1 exerted no adversebehavioral effects on normal animals.
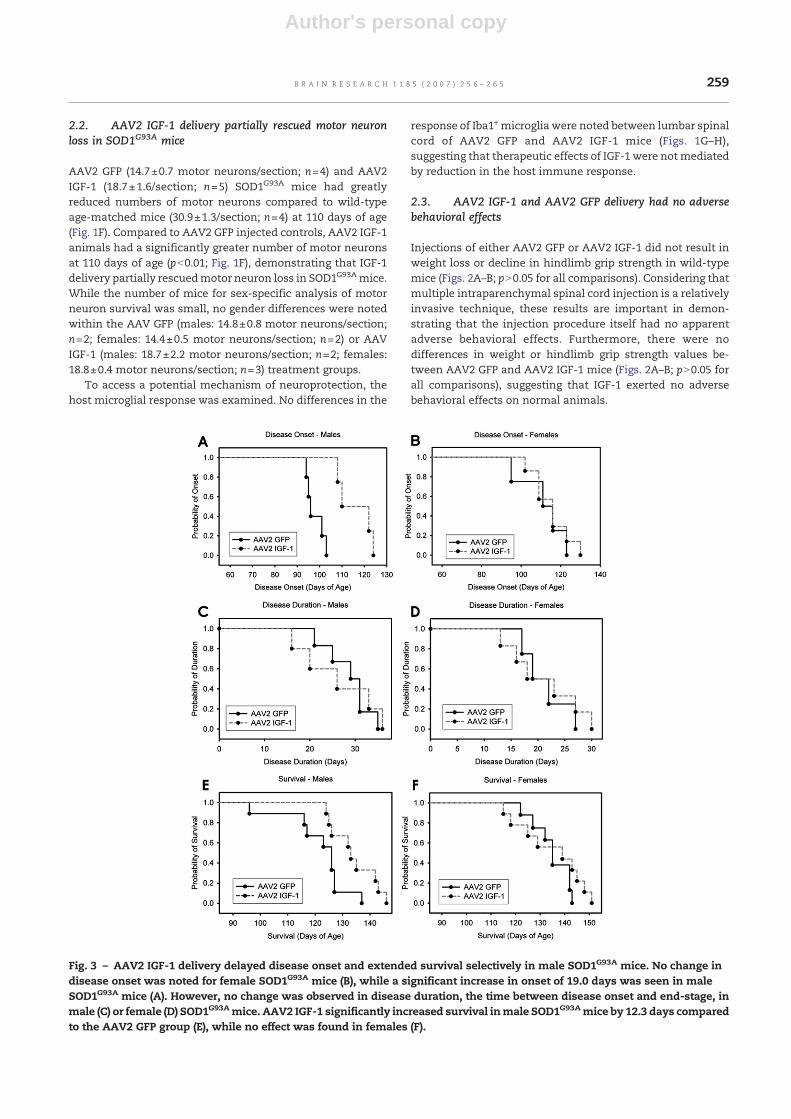
Fig. 3 – AAV2 IGF-1 delivery delayed disease onset and extended survival selectively in male SOD1G93A mice. No change indisease onset was noted for female SOD1G93A mice (B), while a significant increase in onset of 19.0 days was seen in maleSOD1G93A mice (A). However, no change was observed in disease duration, the time between disease onset and end-stage, inmale (C) or female (D) SOD1G93Amice. AAV2 IGF-1 significantly increased survival inmale SOD1G93Amice by 12.3 days comparedto the AAV2 GFP group (E), while no effect was found in females (F).
259B R A I N R E S E A R C H 1 1 8 5 ( 2 0 0 7 ) 2 5 6 – 2 6 5

Author's personal copy
2.4. AAV2 IGF-1 delivery partially slowed hindlimb gripstrength decline selectively in male SOD1G93A mice
Compared to AAV2 GFP controls, AAV2 IGF-1 SOD1G93A miceinjected with AAV2 IGF-1 showed a significant delay in thedecline in hindlimb grip strength when all animals from bothsexes were analyzed together (pb0.05 at 3, 4, 5 and 7 weekspost-injection; data not shown). No change in hindlimb gripstrength decline was noted for female mice (pN0.05 for allcomparisons; Fig. 2D), while a significant delay in grip strengthdeclinewas seen inmalemice (pb0.05 at 4, 5 and 7weeks post-injection; Fig. 2C).
2.5. AAV2 IGF-1 delivery partially slowed weight declineselectively in male SOD1G93A mice
AAV2 IGF-1 significantly delayed weight decline compared toAAV2 GFP controls at 8, 9 and 10 weeks post-injection in malemice (Fig. 2E; pb0.05), but no effect was found in female mice(Fig. 2F; pN0.05 for all comparisons). Motor neuron rescue byAAV2 IGF-1 likely slowed muscle atrophy and consequentweight loss. In addition, because AAV2 IGF-1 mice maintainedgreater hindlimb motor function than AAV2 GFP controls,weight was likely maintained in AAV2 IGF-1 mice becausethey were better able to feed.
2.6. AAV2 IGF-1 delivery delayed disease onset selectivelyin male SOD1G93A mice
Compared to AAV2 GFP controls, AAV2 IGF-1 mice showed asignificant delay in disease onset of 11.9 days (102.9±10.9 daysvs. 114.7±8.2 days; pb0.05; data not shown) when all animalsfrom both sexes were analyzed together. No change in onsetwas noted for femalemice (111.3 days±14.6 vs. 115.0±9.4 days;pN0.05; Fig. 3B), while a significant increase in onset of19.0 days was seen in male mice (97.8 days±3.8 vs. 116.8±8.1 days; pb0.05; Fig. 3A).
2.7. AAV2 IGF-1 delivery did not extend disease durationin male and female SOD1G93A mice
No change was observed in disease duration, the time be-tween disease onset and end-stage, in male mice (28.6 days±5.5 vs. 26.3±9.7 days; pN0.05; Fig. 3C), female mice (21.7 days±4.6 vs. 22.2±8.4 days; pN0.05; Fig. 3D) or when all animals fromboth sexes were analyzed together (26.0±6.0 days vs. 22.2±9.8 days; pN0.05; data not shown).
2.8. AAV2 IGF-1 delivery extended survival selectively inmale SOD1G93A mice
When male and female SOD1G93A mice were analyzedtogether, AAV2 IGF-1 increased survival by 6.6 days (127.8±11.7 days vs. 134.4±10.8 days; pN0.05; data not shown) com-pared to AAV2 GFP; however, this increase was not significant.AAV2 IGF-1 significantly increased survival in male mice by12.3 days (121.7±11.4 days vs. 134.0±7.7 days; pb0.05)compared to the AAV2 GFP group (Fig. 3E), while no effectwas found in females (134.8±7.6 days vs. 134.8±13.4 days;pN0.05; Fig. 3F). Interestingly, AAV2 IGF-1 delivery was only
able to extend male survival to the level of AAV2 GFP femalecontrols.
3. Discussion
We have demonstrated that AAV2-based delivery of IGF-1directly to lumbar spinal cord has therapeutic benefit (albeitgender-specific) in awell-establishedmousemodel of ALS.Wealso observed sustained IGF-1 expression, despite ongoingdisease processes. Expression was not confined to sites ofinjection, but was distributed in neuritic processes over arelatively broad region of lumbar spinal cord. This is of par-ticular relevance because of the spatially diffuse nature of ALSand the desire to widely distribute IGF-1.
3.1. Viral vector delivery as ALS therapy
Viral vector delivery of neurotrophic factors represents auseful approach for ALS and other neurodegenerative CNSdisorders. Previous studies in animal models of motor neurondisease (SOD1, wobbler, progressive motor neuronopathy,spinal muscle atrophy) have shown that therapeutic benefitscan be achieved via adenovirus-, lentivirus-, herpes simplexvirus- and adeno-associated virus (AAV)-based delivery ofgenes such as mutant SOD1-targeted RNAi (Ralph et al., 2005;Raoul et al., 2005), VEGF (Azzouz et al., 2004), NT-3 (Haase et al.,1997), CNTF (Aebischer et al., 1996), GDNF (Acsadi et al., 2002;Wang et al., 2002), IGF-1 (Kaspar et al., 2003), Bcl-2 (Azzouzet al., 2000) and cardiotrophin-1 (Bordet et al., 2001). Clinicaltranslation with some of these proteins has not yieldedsimilar efficacy in human ALS patients (Borasio et al., 1998;Group, 1999, 1996; Lai et al., 1997); however, trials have mostlyinvolved systemic injection of protein, not viral vector-basedgene delivery.
3.2. Methods of AAV IGF-1 delivery
Previous studies of intramuscular AAV2 IGF-1 delivery re-sulted in robust efficacy in mutant-SOD1 mice (Kaspar et al.,2003). This approach, however, is complicated by dependencyon intact, functional andmuscle-innervatingmotor axons andlow efficiency of vector retrograde transport, even in an intact,healthy system. Human ALS cases are mostly diagnosed afterextensive motor neuron loss and/or denervation, at a pointwhen capacity for retrograde transport may be diminished.Large muscle masses call for large amounts of highly purifiedvector; however, “large-scale” production of AAV serotypesother thanAAV2 has not been resolved (GrimmandKay, 2003).Retrograde transport delivers vector only to motor neurons,while an extensive additional pool of glia and interneuronsexists for trophic production.
As shown in the present study, intraparenchymal deliveryto ventral spinal cord provides away to directly introduce hightiters of virus to regions of dying motor neurons. However,effects observed in this study were not as robust as thoseattained with intramuscular AAV2 IGF-1. Furthermore, directinjection is associated with a number of practical disadvan-tages such as risk of further damage because of surgicalinvasiveness (though no apparent behavioral effects are
260 B R A I N R E S E A R C H 1 1 8 5 ( 2 0 0 7 ) 2 5 6 – 2 6 5

Author's personal copy
reported in this study), costliness, difficulty of procedurestandardization, and the diffuse nature of ALS, potentiallyrequiring injections atmultiple levels of spinal cord, brainstemand cortex.While we have demonstrated the efficacy ofmulti-segmental deliver only in the lumbar region, Dodge et al. (2006)reported that injection of AAV1 or AAV2 IGF-1 into deepcerebellar nuclei (DCN) promoted significant benefit in SOD1mice. Because of the anatomy of axonal connections betweenDCNand all levels of spinal cord, this represents a technique totarget multiple spinal cord levels with minimal invasiveness.
Injections of vector into cerebrospinal fluid via lumbarpuncture (spinal tap) or via intracerebroventricular injectionare excellent alternatives for delivering vector extensivelythroughout the neuraxis. Preliminary studies both in mutant-SOD1 rodents (Nagano et al., 2005a; Narai et al., 2005) and inALS patients (Nagano et al., 2005b) have shown the efficacy ofintrathecal delivery of IGF-1 protein; nevertheless, efficiencyrelative to other delivery means has not been addressed.Furtherworkwill be needed to optimize the regimen, includingtiming, dose and region(s) of injection.
3.3. Benefits of AAV-based vectors
AAV, a non-pathogenic single-stranded DNA parvovirus, is apromising viral candidate for gene delivery to ALS patients.Benefits include (Boillee and Cleveland, 2004): rare vectorsequence integration into host genome, efficient transductionof both dividing and non-dividing cells, long-term geneexpression, the existence of at least 8 serotypes allowing fordifferential targeting of specific CNS cell populations, lack oftoxicity andminimal stimulation of the host immune response.
3.4. Temporal considerations
Because of the ongoing nature of ALS, sustained geneexpression is thought to be necessary; however continuous,unregulated expression of high levels of neurotrophinsmay beassociated with side effects, such as the motor disturbancesobserved with GDNF delivery in the Parkinson's disease ratmodel (Kirik et al., 2000). Temporal regulation via rapamycin(Wang et al., 2003) and tetracycline (Chtarto et al., 2003)-basedexpression systems may provide useful tools.
Timing of vector introduction is also important. Should IGF-1 be given as early as possible, and are repeated injections oversome as yet to be determined interval needed? Work inorganotypic cultures demonstrated that motor neuron rescuefrom excitotoxicity was reduced when IGF-1administrationwas delayed (Bilak and Kuncl, 2001). Unfortunately, manyneurons have already been lost or have undergone denerva-tion from muscle by the time of ALS diagnosis. In the presentstudy,we delivered vector at 60 days of age, a time before onsetof motor neuron loss and behavioral deficits. Kaspar et al.(2003) reported benefit when AAV2 IGF-1 was introduced atsymptom onset, suggesting that even late delivery is promis-ing, provided that the delivery regimen is designed properly.
3.5. Therapeutic actions of IGF-1 in ALS
Disease onset (but not progression) is delayed when mutantSOD1 expression is knocked out selectively in motor neurons
(Boillee et al., 2006). These findings coincidewith our results inthat AAV2 IGF-1 delays motor neuron loss and onset inSOD1G93Amice, but does not affect disease duration. The AAV2used in this study targets neurons, and thus the neuroprotec-tion observed may be intrinsic to effects of IGF-1 on motorneurons rather than other non-neuronal cells. AAV serotypesthat infect more abundant astrocytes or ependymal cells mayalso be an option for optimizing effects of IGF-1 in the spinalcord (Tenenbaum et al., 2004).
Nevertheless, in vitro studies suggest that infected motorneurons likely also secrete IGF-1, and consequently may exertbeneficial actions on surrounding bystander neuronal and evennon-neuronal cells, in addition tomotor neurons (Vincent et al.,2004a). For example, astrocytes, oligodendrocytes andmicrogliaall express IGF-1 receptors, and IGF-1 has been shown to pro-mote proliferation, differentiation, and survival of glial cells(Chesik et al., 2007). IGF-1 regulates proliferation and GFAPexpression in astrocytes, and may therefore play a role incontrolling astrogliosis that occurs in the spinal cord of mutantSOD1 rodents and humanswith ALS. IGF-1may also function inan anti-inflammatory fashion by antagonizing activation ofmicroglia mediated by IFN-γ and/or blocking release of inter-leukin (IL)-1β by activated microglia (Maher et al., 2006), ef-fects that could indirectly promote motor neuron survival bypreventing neuronal cell death associated withmicroglial over-activation.
Alterations in components of the IGF-1 signaling systemoccur in ALS patients and in rodent models of the disease.While changes in total IGF-1 levels have not been found,increases in IGF binding proteins 2, 5 and 6 are seen in ventralhorn (Wilczak et al., 2003), resulting in decreased levels of“free” IGF-1 to exert neuroprotective actions. In addition,inflammatory events associated with ALS (possibly via TNF-alpha) are believed to decrease transduction sensitivity of IGF-1 receptors and intracellular signaling components (Trejo et al.,2004). Vulnerable motor neurons are deprived of pro-survivalIGF-1 signaling in ALS, furthering contributing to cell loss.
The neuroprotective actions of IGF-1 in the context ofdecreased IGF-1 signaling in ALS therefore make gene deliveryof IGF-1 a potentially important candidate for therapy. Highlevels of expression achieved via AAV2 delivery most likelyprovide a sufficient level of local IGF-1 to overcome loss of IGF-1 signaling associated with disease. The present resultscoincide with previous studies that have shown that IGF-1exerts pro-survival effects specifically onmotor neurons: (1) indissociated culture (Vincent et al., 2004b), (2) in the organo-typic spinal cord model of excitotoxicity (Corse et al., 1999),(3) during developmental programmed cell death (Lewis et al.,1993), (4) following spinal cord ischemia (Nakao et al., 2001),spinal transaction (Sharma et al., 1997), peripheral axotomy (Liet al., 1994), crush injury (Rabinovsky et al., 2003) and rootavulsion (Haninec et al., 2003) and (5) in the wobbler mouse(Hantai et al., 1995) and in the SOD1mouse (Dobrowolny et al.,2005; Kaspar et al., 2003).
3.6. Sex-specific effects
Sex-specific differences in disease onset and progression(Heiman-Patterson et al., 2005; Matsumoto et al., 2006; Suzukiet al., 2007; Veldink et al., 2003) and response to therapeutic
261B R A I N R E S E A R C H 1 1 8 5 ( 2 0 0 7 ) 2 5 6 – 2 6 5

Author's personal copy
interventions (Bushnell et al., 2006) have been noted in CNSdisorders, including ALS. For example, studies with untreatedmutant SOD1 mice (Heiman-Patterson et al., 2005) and rats(Suzuki et al., 2007), including results from the present study,demonstrate that disease onset and overall survival occurearlier on average inmales. In response to an exercise regimentherapy, delayed disease onset and survivalwere noted in onlyfemale mice (Veldink et al., 2003), while AAV2 IGF-1 promotedbenefit selectively in males in the present study. While themechanism for the male-only effect seen in the present studyis unknown, potential reasons include differences in diseaseprogression, mutant SOD1 expression, sex hormones and/orlevels and sensitivity of components of the IGF-1 system.Robust and long-lasting IGF-1 expression was seen in bothmales and females, suggesting that the effect was unlikelyrelated to sex-specific differences in transgene expression.
3.7. Combinatorial therapies
We report modest effects of AAV2 IGF-1 delivery. Morewidespread delivery of virus, as well as delivery to relevantmotor neuron pools (i.e. cervical and/or respiratory motorneurons), could produce more robust neuroprotection. Thecomplexity of ALS will likely require a multi-faceted approachthat combines gene therapy with other promising strategies,including cell transplants, pharmacological agents and addi-tional trophic factors. For example, Arakawa et al. (1990) reporta supra-additive effect of IGF-1 and CNTF on the survival ofisolated chick embryonic spinal motor neurons. In the pmnmouse, combined adenoviral vector administration of NT-3and CNTF has a greater effect than NT-3 alone (Haase et al.,1997). Kaspar et al. (2005) demonstrated that the efficacy ofIGF-1 delivery via intramuscular injections can be furtherincreased when combined with an exercise regimen. Theseare just a few examples representing the important trendtoward combination therapy and the integrated approach thatbuilds on the exciting recent advances in gene therapy vectorsand ALS biology, and is likely to be the most effective strategyfor successful treatment of patients with ALS.
4. Experimental procedures
4.1. Vector plasmid constructs and vector production
The CERE-130 vector genome contains the AAV2 invertedterminal repeats (ITRs) flanking a transgene expression cas-sette containing the chicken beta-actin (CAG) promoter, thehuman IGF-1 cDNA and the human growth hormone gene(hGH) polyadenylation signal (polyA) (Stratagene, La Jolla, CA).AAV2-GFP genome structure is identical to that of CERE-130except that the GFP cDNA is found in place of that of IGF-1.The details of cloning procedures and primer sequencesare available upon request. Plasmid clones identity was con-firmed by restriction digestions and nucleotide sequencedetermination.
Vectors were produced by triple transfection and purified.They were formulated in PBS, 2mM MgCl2 [formulation buffer(FB)] and vector titer was determined by quantitative PCRusing vector specific primers.
4.2. Viral vector delivery
The care and treatment of animals in all procedures wereconducted in strict accordance with the guidelines set by theEuropean Communities Counsel Directive (November 24th, 1986),the NIH Guide for the Care and Use of Laboratory Animals, theGuidelines for the Use of Animals in Neuroscience Research andthe Johns Hopkins University IACUC, and measures weretaken to minimize any potential pain or animal discomfort.
Sixty-day-old wild-type and SOD1G93A mice (approximately20–30 g) received i.p. injections of anaesthetic cocktail[acepromazine maleate (0.7 mg/kg; Fermenta Animal Health,Kansas City, MO), ketamine (95.0 mg/kg; Fort Dodge AnimalHealth; Fort Dodge, IA) and xylazine (10.0 mg/kg; Bayer,Shawnee Mission, KS)]. The back musculature was excised,and a laminectomy was performed above the L4 and L5 levelsof the spinal cord. The dura was incised above the injectionsite using a 30-gauge needle. AAV2 IGF-1 or AAV2 GFPvectors were bilaterally injected into the ventral gray matterat four total sites in the lumbar L4–L5 region (bilaterally at L4and L5; separated by 2.0 mm in the rostral–caudal axis). Theinjection pipette was secured to a manual micromanipulator(World Precision Instruments; Sarasota, FL) attached to an80° tilting base. The tip was lowered to a depth of 1.3 mmbelow the surface of the cord to target ventral horn, and washeld in place for 2 min before and after cell injection. 1.5 μl ofconcentrated viral solutions (AAV2 IGF-1: 1.2×1010 vg/injection site; AAV2 GFP: 4.2×109 vg/injection site) wasinjected at each of the four sites. Vector was deliveredunder the control of a microsyringe pump controller (WorldPrecision Instruments) at a rate of 0.75 μl/min. Dura wasclosed with 9-0 suture, and skin was closed with woundclips. The entire injection causes minimal disturbance to thespinal cord.
For weight, survival and grip strength analysis, 35 SOD1G93A
mice (AAV2 IGF-1: n=18 total, n=9 males, n=9 females; AAV2GFP: n=17 total, n=9 males, n=8 females) and 6 age-matchedwild-type mice (AAV2 IGF-1: n=6 total, n=3 females; AAV2GFP: n=3 females) were used in total. For motor neuroncounts, 9 SOD1G93A mice (AAV2 IGF-1: n=5 total, n=3 males,n=2 females; AAV2 GFP: n=4 total, n=2 males, n=2 females)and 4 age-matched wild-type mice (n=4 total, n=2 males, n=2females) were used in total. Each group consisted of an almostequal amount of males and females.
4.3. Animal model
Transgenic mice carrying the human SOD1 gene with theG93A mutation (SOD1 B6SJL-TgN[SOD1-G93A]1Gur) were used(Gurney et al., 1994). Mice were obtained from The JacksonLaboratory via in vitro fertilization/Speed Expansion programto provide one homogeneous heterozygous population. Onaverage, this line of SOD1G93A mutants develop disease onsetat approximately 90 days of age, and reach disease end-stageapproximately 40 days later. In addition, wild-type B6SJL micefrom Jackson were also used. All wild-type and SOD1G93A micewere housed at standard temperature (21 °C) and in a lightcontrolled environment with ad libitum access to the food andwater. Mice were maintained in racks of ventilated cageslocated in the same room.
262 B R A I N R E S E A R C H 1 1 8 5 ( 2 0 0 7 ) 2 5 6 – 2 6 5

Author's personal copy
4.4. Behavioral analysis
Beginning 1 week before the injection of viral vectors,SOD1G93A and wild-type mice were monitored weekly forweight and for motor dysfunction by measuring hind limbmuscle strength using a “Grip Strength Meter” (DFIS-2 SeriesDigital Force Gauge; Columbus Instruments, OH). Gripstrength testing was performed by allowing the animals tograsp a thin bar attached to the force gauge. Thiswas followedby pulling the animal away from the gauge until thehindlimbs released the bar. This provides a value for theforce ofmaximal grip strength. The forcemeasurementswererecorded in three separate trials and the averages were usedin analyses. In order to avoid dehydration, Aqua-Jel packswere provided when animals started to show disease symp-toms. Disease onset was determined individually when ananimal dropped to 80.0% of its maximal hindlimb gripstrength (Pardo et al., 2006). Disease duration was measuredas the time between disease onset and end-stage. Todetermine disease end-stage in a reliable and ethical fashion,an artificial end point was used for all SOD1G93A mutantmice.End-stage was defined by the inability of mice to rightthemselves within 30 s when placed on their sides. Themoribundmice were scored as “dead” andwere subsequentlyeuthanized.
4.5. Tissue processing
Animals were sacrificed at 110 days of age or at disease end-stage by transcardial perfusion with 0.3% saline, followedby ice-cold 4% paraformaldehyde (Fisher Scientific; Pittsburgh,PA). The entire spinal cord was removed. The lumbar region(L1–L5) was isolated and post-fixed in 4.0% paraformaldehyde.
4.6. Histology and immunohistochemistry
For GFP and IGF-1 immunohistochemistry, spinal cords fromend-stage mice were cryoprotected in 30.0% sucrose (Fisher)/.1 M phosphate buffer at 4 °C for 3 days. The tissue wasembedded in OCT (Fisher), fast frozen with dry ice and storedat −80 °C until processed. Spinal cord tissue blocks were cut inthe sagittal or transverse plane at 20 μm thickness. Sectionswere collected on glass slides, or were collected in PBS for free-floating immunohistochemistry. For GFP, a 1-in-6 series ofslides was counterstained with DAPI, and fluorescence wasassessed microscopically. An image of each section with GFPfluorescence was collected, and the area of fluorescencecircumscribed and measured on the image. The number ofsections with GFP fluorescence was used to determine therostrocaudal extent of product, and the areas measured oneach section were used to determine the volume of distribu-tion of product using Cavalieri's formula. For IGF-1, immuno-histochemistry was used to detect product on 1-in-12 seriesslides, and imaging and measuring were done as with GFP todetermine rostrocaudal extent and volume of distribution ofIGF-1. To determine the phenotype of AAV2-infected cells,astrocytes were identified with GFAP and neurons wereidentified with NeuN. The host microglial response was ex-amined with Iba1.
4.7. Motor neuron quantification
For motor neuron counts, tissue from L4 and L5 of 110 day oldmice was dehydrated in a graded series of alcohol solutions,embedded in paraffin and cut serially in the transverse plane.Serial cross-sections of the lumbar spinal cords (∼2 mm totallength) were made at 14 μm thickness, for a total of 140sections. Sections were processed for cresyl violet staining.Motor neurons were counted in every 7th section at 20×magnification in order to avoid repetitive counting. Onlymotorneurons with a clearly identifiable nucleus and nucleolus, acell soma over 100 μm2 and located within the ventral hornwere counted (Pardo et al., 2006).
4.8. Statistical analysis
Kaplan–Meier survival analysis of the SOD1G93A mice wasconducted using the statistical software Sigmaplot (SAS Soft-ware). In some cases, Student's t-test was performed to com-pare data between groups of animals. All data are presented asmean±S.E.M., and significance level was set at p≤0.05.
Acknowledgments
We thank Natalie Perez for assistance with grip strengthmeasurements; Jean Brennan and Ariadne Bolton for assis-tance with histology; all members of the Rothstein andMaragakis labs for helpful discussion; The ALS Association,The Robert Packard Center for ALS Research and the NIH(grants NS33958; NS41680) for funding; Project ALS for provid-ing SOD1G93A mice.
R E F E R E N C E S
Acsadi, G., Anguelov, R.A., Yang, H., Toth, G., Thomas, R., Jani, A.,Wang, Y., Ianakova, E., Mohammad, S., Lewis, R.A., Shy, M.E.,2002. Increased survival and function of SOD1 mice after glialcell-derived neurotrophic factor gene therapy. Hum. GeneTher. 13, 1047–1059.
Aebischer, P., Schluep, M., Deglon, N., Joseph, J.M., Hirt, L., Heyd, B.,Goddard, M., Hammang, J.P., Zurn, A.D., Kato, A.C., Regli, F.,Baetge, E.E., 1996. Intrathecal delivery of CNTF usingencapsulated genetically modified xenogeneic cells inamyotrophic lateral sclerosis patients. Nat. Med. 2, 696–699.
Alisky, J.M., Davidson, B.L., 2000. Gene therapy for amyotrophiclateral sclerosis and other motor neuron diseases. Hum. GeneTher. 11, 2315–2329.
Arakawa,Y., Sendtner,M., Thoenen,H., 1990. Survival effect of ciliaryneurotrophic factor (CNTF) on chick embryonic motoneurons inculture: comparison with other neurotrophic factors andcytokines. J. Neurosci. 10, 3507–3515.
Azzouz, M., Hottinger, A., Paterna, J.C., Zurn, A.D., Aebischer, P.,Bueler, H., 2000. Increased motoneuron survival and improvedneuromuscular function in transgenic ALSmice after intraspinalinjection of an adeno-associated virus encoding Bcl-2.Hum.Mol.Genet. 9, 803–811.
Azzouz, M., Ralph, G.S., Storkebaum, E., Walmsley, L.E.,Mitrophanous, K.A., Kingsman, S.M., Carmeliet, P., Mazarakis,N.D., 2004. VEGF delivery with retrogradely transportedlentivector prolongs survival in a mouse ALS model. Nature429, 413–417.
263B R A I N R E S E A R C H 1 1 8 5 ( 2 0 0 7 ) 2 5 6 – 2 6 5

Author's personal copy
Beck, M., Karch, C., Wiese, S., Sendtner, M., 2001. Motoneuron celldeath and neurotrophic factors: basic models for developmentof new therapeutic strategies in ALS. Amyotroph. Lateral Scler.Other Mot. Neuron Disord. 2 (Suppl 1), S55–S68.
Bilak, M.M., Kuncl, R.W., 2001. Delayed application of IGF-I andGDNF can rescue already injured postnatal motor neurons.Neuroreport 12, 2531–2535.
Boillee, S., Cleveland, D.W., 2004. Gene therapy for ALS delivers.Trends Neurosci. 27, 235–238.
Boillee,S.,Yamanaka,K., Lobsiger,C.S., Copeland,N.G., Jenkins,N.A.,Kassiotis, G., Kollias, G., Cleveland, D.W., 2006. Onset andprogression in inherited ALS determined by motor neurons andmicroglia. Science 312, 1389–1392.
Bondy, C.A., Cheng, C.M., 2004. Signaling by insulin-like growthfactor 1 in brain. Eur. J. Pharmacol. 490, 25–31.
Borasio, G.D., Robberecht, W., Leigh, P.N., Emile, J., Guiloff, R.J.,Jerusalem, F., Silani, V., Vos, P.E., Wokke, J.H., Dobbins, T., 1998.A placebo-controlled trial of insulin-like growth factor-I inamyotrophic lateral sclerosis. European ALS/IGF-I Study Group.Neurology 51, 583–586.
Bordet, T., Lesbordes, J.C., Rouhani, S., Castelnau-Ptakhine, L.,Schmalbruch, H., Haase, G., Kahn, A., 2001. Protective effects ofcardiotrophin-1 adenoviral gene transfer on neuromusculardegeneration in transgenic ALS mice. Hum. Mol. Genet. 10,1925–1933.
Bruijn, L.I., Becher, M.W., Lee, M.K., Anderson, K.L., Jenkins, N.A.,Copeland, N.G., Sisodia, S.S., Rothstein, J.D., Borchelt, D.R.,Price, D.L., Cleveland, D.W., 1997. ALS-linked SOD1 mutantG85R mediates damage to astrocytes and promotes rapidlyprogressive disease with SOD1-containing inclusions. Neuron18, 327–338.
Bruijn, L.I., Miller, T.M., Cleveland, D.W., 2004. Unraveling themechanisms involved in motor neuron degeneration in ALS.Annu. Rev. Neurosci. 27, 723–749.
Bushnell, C.D., Griffin, J.,Newby, L.K., Goldstein, L.B.,Mahaffey,K.W.,Graffagnino, C.A., Harrington, R.A.,White,H.D., Simes, R.J., Califf,R.M., Topol, E.J., Easton, J.D., 2006. Statin use and sex-specificstroke outcomes in patients with vascular disease. Stroke 37,1427–1431.
Chesik, D., Wilczak, N., De Keyser, J., 2007. The insulin-like growthfactor system in multiple sclerosis. Int. Rev. Neurobiol. 79,203–226.
Chtarto, A., Bender, H.U., Hanemann, C.O., Kemp, T., Lehtonen, E.,Levivier, M., Brotchi, J., Velu, T., Tenenbaum, L., 2003.Tetracycline-inducible transgene expression mediated by asingle AAV vector. Gene Ther. 10, 84–94.
Corse, A.M., Bilak, M.M., Bilak, S.R., Lehar, M., Rothstein, J.D.,Kuncl, R.W., 1999. Preclinical testing of neuroprotectiveneurotrophic factors in a model of chronic motor neurondegeneration. Neurobiol. Dis. 6, 335–346.
Dobrowolny, G., Giacinti, C., Pelosi, L., Nicoletti, C., Winn, N.,Barberi, L., Molinaro, M., Rosenthal, N., Musaro, A., 2005.Muscle expression of a local Igf-1 isoform protects motorneurons in an ALS mouse model. J. Cell Biol. 168,193–199.
Dodge, J.C., Clarke, J., Yang, W., Grissett, L., Kim, S., Wen, R., Cheng,S.H., Kaspar, B.K., Shihabuddin, L.S., 2006. Intracerebellarinjection of AAV-IGF-1 improves motor function and extendssurvival in a mouse model of amyotrophic lateral sclerosis. Soc.Neurosci. Abstr.
Dore, S., Kar, S., Quirion, R., 1997. Rediscovering an old friend, IGF-I:potential use in the treatment of neurodegenerative diseases.Trends Neurosci. 20, 326–331.
Federici, T., Boulis, N.M., 2006. Gene-based treatment of motorneuron diseases. Muscle Nerve 33, 302–323.
Grimm, D., Kay, M.A., 2003. From virus evolution to vectorrevolution: use of naturally occurring serotypes ofadeno-associated virus (AAV) as novel vectors for human genetherapy. Curr. Gene Ther. 3, 281–304.
Group, B.S., 1999. A controlled trial of recombinant methionylhuman BDNF in ALS: the BDNF Study Group (Phase III).Neurology 52, 1427–1433.
Group, C.S., 1996. A double-blind placebo-controlled clinical trialof subcutaneous recombinant human ciliary neurotrophicfactor (rHCNTF) in amyotrophic lateral sclerosis. ALS CNTFTreatment Study Group. Neurology 46, 1244–1249.
Gurney, M.E., Pu, H., Chiu, A.Y., Dal Canto, M.C., Polchow, C.Y.,Alexander, D.D., Caliendo, J., Hentati, A., Kwon, Y.W., Deng, H.X.,et al., 1994. Motor neuron degeneration in mice that express ahuman Cu,Zn superoxide dismutase mutation. Science 264,1772–1775.
Haase, G., Kennel, P., Pettmann, B., Vigne, E., Akli, S., Revah, F.,Schmalbruch, H., Kahn, A., 1997. Gene therapy ofmurinemotorneuron disease using adenoviral vectors for neurotrophicfactors. Nat. Med. 3, 429–436.
Haninec, P., Houst'ava, L., Stejskal, L., Dubovy, P., 2003. Rescue ofrat spinal motoneurons from avulsion-induced cell death byintrathecal administration of IGF-I and Cerebrolysin. Ann.Anat. 185, 233–238.
Hantai, D., Akaaboune, M., Lagord, C., Murawsky, M., Houenou, L.J.,Festoff, B.W., Vaught, J.L., Rieger, F., Blondet, B., 1995. Beneficialeffects of insulin-like growth factor-I on wobbler mousemotoneuron disease. J. Neurol. Sci. 129, 122–126 Suppl.
Heiman-Patterson, T.D., Deitch, J.S., Blankenhorn, E.P., Erwin, K.L.,Perreault, M.J., Alexander, B.K., Byers, N., Toman, I., Alexander,G.M., 2005. Background and gender effects on survival in theTgN(SOD1-G93A)1Gur mouse model of ALS. J. Neurol. Sci. 236,1–7.
Howland, D.S., Liu, J., She, Y., Goad, B., Maragakis, N.J., Kim, B.,Erickson, J., Kulik, J., DeVito, L., Psaltis, G., DeGennaro, L.J.,Cleveland, D.W., Rothstein, J.D., 2002. Focal loss of theglutamate transporter EAAT2 in a transgenic ratmodel of SOD1mutant-mediated amyotrophic lateral sclerosis (ALS). Proc.Natl. Acad. Sci. U. S. A. 99, 1604–1609.
Kaspar, B.K., Frost, L.M., Christian, L., Umapathi, P., Gage, F.H.,2005. Synergy of insulin-like growth factor-1 and exercise inamyotrophic lateral sclerosis. Ann. Neurol. 57, 649–655.
Kaspar, B.K., Llado, J., Sherkat, N., Rothstein, J.D., Gage, F.H., 2003.Retrograde viral delivery of IGF-1 prolongs survival in a mouseALS model. Science 301, 839–842.
Kirik, D., Rosenblad, C., Bjorklund, A., Mandel, R.J., 2000. Long-termrAAV-mediated gene transfer of GDNF in the rat Parkinson'smodel: intrastriatal but not intranigral transduction promotesfunctional regeneration in the lesioned nigrostriatal system.J. Neurosci. 20, 4686–4700.
Lai, E.C., Felice, K.J., Festoff, B.W., Gawel, M.J., Gelinas, D.F., Kratz,R., Murphy, M.F., Natter, H.M., Norris, F.H., Rudnicki, S.A., 1997.Effect of recombinant human insulin-like growth factor-I onprogression of ALS. A placebo-controlled study. The NorthAmerica ALS/IGF-I Study Group. Neurology 49, 1621–1630.
Lewis,M.E., Neff, N.T., Contreras, P.C., Stong, D.B., Oppenheim, R.W.,Grebow, P.E., Vaught, J.L., 1993. Insulin-like growth factor-I:potential for treatment ofmotor neuronal disorders. Exp.Neurol.124, 73–88.
Li, L., Oppenheim, R.W., Lei, M., Houenou, L.J., 1994. Neurotrophicagents prevent motoneuron death following sciatic nervesection in the neonatal mouse. J. Neurobiol. 25, 759–766.
Maher, F.O., Clarke, R.M., Kelly, A., Nally, R.E., Lynch, M.A., 2006.Interaction between interferon gamma and insulin-like growthfactor-1 in hippocampus impacts on the ability of rats tosustain long-term potentiation. J. Neurochem. 96, 1560–1571.
Matsumoto, A., Okada, Y., Nakamichi, M., Nakamura, M., Toyama,Y., Sobue, G., Nagai, M., Aoki, M., Itoyama, Y., Okano, H., 2006.Disease progression of human SOD1 (G93A) transgenic ALSmodel rats. J. Neurosci. Res. 83, 119–133.
Nagai, M., Aoki, M., Miyoshi, I., Kato, M., Pasinelli, P., Kasai, N.,Brown Jr., R.H., Itoyama, Y., 2001. Rats expressing humancytosolic copper-zinc superoxide dismutase transgenes with
264 B R A I N R E S E A R C H 1 1 8 5 ( 2 0 0 7 ) 2 5 6 – 2 6 5

Author's personal copy
amyotrophic lateral sclerosis: associated mutations developmotor neuron disease. J. Neurosci. 21, 9246–9254.
Nagano, I., Ilieva, H., Shiote, M., Murakami, T., Yokoyama, M.,Shoji, M., Abe, K., 2005a. Therapeutic benefit of intrathecalinjection of insulin-like growth factor-1 in a mouse model ofamyotrophic lateral sclerosis. J. Neurol. Sci. 235, 61–68.
Nagano, I., Shiote, M., Murakami, T., Kamada, H., Hamakawa, Y.,Matsubara, E., Yokoyama, M., Moritaz, K., Shoji, M., Abe, K.,2005b. Beneficial effects of intrathecal IGF-1 administration inpatients with amyotrophic lateral sclerosis. Neurol. Res. 27,768–772.
Nakao, Y., Otani, H., Yamamura, T., Hattori, R., Osako, M.,Imamura, H., 2001. Insulin-like growth factor 1 preventsneuronal cell death and paraplegia in the rabbitmodel of spinalcord ischemia. J. Thorac. Cardiovasc. Surg. 122, 136–143.
Narai, H., Nagano, I., Ilieva, H., Shiote, M., Nagata, T., Hayashi, T.,Shoji, M., Abe, K., 2005. Prevention of spinal motor neurondeath by insulin-like growth factor-1 associating with thesignal transduction systems in SODG93A transgenic mice.J. Neurosci. Res. 82, 452–457.
Pardo, A.C., Wong, V., Benson, L.M., Dykes, M., Tanaka, K.,Rothstein, J.D., Maragakis, N.J., 2006. Loss of the astrocyteglutamate transporter GLT1 modifies disease in SOD1(G93A)mice. Exp. Neurol. 201, 120–130.
Rabinovsky, E.D., Gelir, E., Gelir, S., Lui, H., Kattash,M., DeMayo, F.J.,Shenaq, S.M., Schwartz, R.J., 2003. Targeted expression of IGF-1transgene to skeletal muscle accelerates muscle and motorneuron regeneration. FASEB J. 17, 53–55.
Ralph, G.S., Radcliffe, P.A., Day, D.M., Carthy, J.M., Leroux, M.A.,Lee, D.C., Wong, L.F., Bilsland, L.G., Greensmith, L., Kingsman,S.M., Mitrophanous, K.A., Mazarakis, N.D., Azzouz, M., 2005.Silencing mutant SOD1 using RNAi protects againstneurodegeneration and extends survival in an ALS model. Nat.Med. 11, 429–433.
Raoul, C., Abbas-Terki, T., Bensadoun, J.C., Guillot, S., Haase, G.,Szulc, J., Henderson, C.E., Aebischer, P., 2005.Lentiviral-mediated silencing of SOD1 through RNA interferenceretards disease onset and progression in a mouse model of ALS.Nat. Med. 11, 423–428.
Rosen, D.R., Siddique, T., Patterson, D., Figlewicz, D.A., Sapp, P.,Hentati, A., Donaldson, D., Goto, J., O'Regan, J.P., Deng, H.X., et al.,1993. Mutations in Cu/Zn superoxide dismutase gene areassociated with familial amyotrophic lateral sclerosis. Nature362, 59–62.
Sharma, H.S., Nyberg, F., Gordh, T., Alm, P., Westman, J., 1997.Topical application of insulin like growth factor-1 reduces
edema and upregulation of neuronal nitric oxide synthasefollowing trauma to the rat spinal cord. Acta Neurochir., Suppl.70, 130–133.
Suzuki, M., Tork, C., Shelley, B., McHugh, J., Wallace, K., Klein, S.M.,Lindstrom, M.J., Svendsen, C.N., 2007. Sexual dimorphism indisease onset and progression of a rat model of ALS.Amyotroph. Lateral Scler. 8, 20–25.
Tenenbaum, L., Chtarto, A., Lehtonen, E., Velu, T., Brotchi, J.,Levivier, M., 2004. Recombinant AAV-mediated gene deliveryto the central nervous system. J. Gene Med. 6 (Suppl 1),S212–S222.
Trejo, J.L., Carro, E., Garcia-Galloway, E., Torres-Aleman, I., 2004. Roleof insulin-like growth factor I signaling in neurodegenerativediseases. J. Mol. Med. 82, 156–162.
Veldink, J.H., Bar, P.R., Joosten, E.A., Otten, M., Wokke, J.H., van denBerg, L.H., 2003. Sexual differences in onset of disease andresponse to exercise in a transgenicmodel of ALS. Neuromuscul.Disord. 13, 737–743.
Vincent, A.M., Feldman, E.L., Song, D.K., Jung, V., Schild, A., Zhang,W., Imperiale, M.J., Boulis, N.M., 2004a. Adeno-associatedviral-mediated insulin-like growth factor delivery protectsmotor neurons in vitro. Neuromolecular. Med. 6, 79–85.
Vincent, A.M., Mobley, B.C., Hiller, A., Feldman, E.L., 2004b. IGF-Iprevents glutamate-induced motor neuron programmed celldeath. Neurobiol. Dis. 16, 407–416.
Wang, L.J., Lu, Y.Y., Muramatsu, S., Ikeguchi, K., Fujimoto, K.,Okada, T., Mizukami, H., Matsushita, T., Hanazono, Y., Kume,A., Nagatsu, T., Ozawa, K., Nakano, I., 2002. Neuroprotectiveeffects of glial cell line-derived neurotrophic factor mediatedby an adeno-associated virus vector in a transgenic animalmodel of amyotrophic lateral sclerosis. J. Neurosci. 22,6920–6928.
Wang, S., Petravicz, J., Breakefield, X.O., 2003. Single HSV-ampliconvector mediates drug-induced gene expression via dimerizersystem. Mol. Ther. 7, 790–800.
Wilczak, N., de Keyser, J., 2005. Insulin-like growth factor systemin amyotrophic lateral sclerosis. Endocr. Dev. 9, 160–169.
Wilczak, N., de Vos, R.A., De Keyser, J., 2003. Free insulin-likegrowth factor (IGF)-I and IGF binding proteins 2, 5, and 6 inspinal motor neurons in amyotrophic lateral sclerosis. Lancet361, 1007–1011.
Wong, P.C., Pardo, C.A., Borchelt, D.R., Lee, M.K., Copeland, N.G.,Jenkins, N.A., Sisodia, S.S., Cleveland, D.W., Price, D.L., 1995. Anadverse property of a familial ALS-linked SOD1 mutationcauses motor neuron disease characterized by vacuolardegeneration of mitochondria. Neuron 14, 1105–1116.
265B R A I N R E S E A R C H 1 1 8 5 ( 2 0 0 7 ) 2 5 6 – 2 6 5
Copyright © 2022 FDOKUMEN




















