
Cepharanthine exerts antitumor activity on cholangiocarcinoma by inhibiting NF-κB
Upload
independentCategory
view
1download
0
Intracellular Trafficking of AIP56, an NF-�B-Cleaving Toxin fromPhotobacterium damselae subsp. piscicida
Liliana M. G. Pereira,a,b Rute D. Pinto,a Daniela S. Silva,a,b Ana R. Moreira,a Christoph Beitzinger,c Pedro Oliveira,b Paula Sampaio,d
Roland Benz,c,e Jorge E. Azevedo,b,f Nuno M. S. dos Santos,a Ana do Valea
Fish Immunology and Vaccinology, Instituto de Biologia Molecular e Celular, Universidade do Porto, Porto, Portugala; ICBAS-Instituto de Ciências Biomédicas Abel Salazar,Universidade do Porto, Porto, Portugalb; Rudolf-Virchow-Zentrum, DFG-Forschungszentrum für Experimetelle Biomedizin, Universität Würzburg, Würzburg, Germanyc;Advanced Light Microscopy, Instituto de Biologia Molecular e Celular, Universidade do Porto, Porto, Portugald; School of Engineering and Science, Jacobs University,Bremen, Germanye; Organelle Biogenesis and Function, Instituto de Biologia Molecular e Celular, Universidade do Porto, Porto, Portugalf
AIP56 (apoptosis-inducing protein of 56 kDa) is a metalloprotease AB toxin secreted by Photobacterium damselae subsp. pisci-cida that acts by cleaving NF-�B. During infection, AIP56 spreads systemically and depletes phagocytes by postapoptotic sec-ondary necrosis, impairing the host phagocytic defense and contributing to the genesis of infection-associated necrotic lesions.Here we show that mouse bone marrow-derived macrophages (mBMDM) intoxicated by AIP56 undergo NF-�B p65 depletionand apoptosis. Similarly to what was reported for sea bass phagocytes, intoxication of mBMDM involves interaction of AIP56C-terminal region with cell surface components, suggesting the existence of a conserved receptor. Biochemical approaches andconfocal microscopy revealed that AIP56 undergoes clathrin-dependent endocytosis, reaches early endosomes, and follows therecycling pathway. Translocation of AIP56 into the cytosol requires endosome acidification, and an acidic pulse triggers translo-cation of cell surface-bound AIP56 into the cytosol. Accordingly, at acidic pH, AIP56 becomes more hydrophobic, interactingwith artificial lipid bilayer membranes. Altogether, these data indicate that AIP56 is a short-trip toxin that reaches the cytosolusing an acidic-pH-dependent mechanism, probably from early endosomes. Usually, for short-trip AB toxins, a minor poolreaches the cytosol by translocating from endosomes, whereas the rest is routed to lysosomes for degradation. Here we demon-strate that part of endocytosed AIP56 is recycled back and released extracellularly through a mechanism requiring phosphoino-sitide 3-kinase (PI3K) activity but independent of endosome acidification. So far, we have been unable to detect biological activ-ity of recycled AIP56, thereby bringing into question its biological relevance as well as the importance of the recycling pathway.
AIP56 (apoptosis-inducing protein of 56 kDa) is a plasmid-encoded toxin of Photobacterium damselae subsp. piscicida
(1), a Gram-negative bacterium that infects economically impor-tant fish species (2, 3) and is considered one of the most relevantpathogens in mariculture (2–5). In acute infections, a rapid septi-cemia develops, causing very high mortalities (2, 4, 6). Histo-pathological analysis of the diseased animals revealed cytotoxicalterations (4, 7–10) that were found to result from pathogen-induced apoptotic death of macrophages and neutrophils (11)and later associated with the activity of AIP56 (1). Indeed, it hasbeen shown that in infected fish, the toxin is systemically distrib-uted and depletes macrophages and neutrophils by postapoptoticsecondary necrosis (12), leading to the impairment of the phago-cytic defense of the host and consequently favoring P. damselaesubsp. piscicida survival and dissemination. Furthermore, the oc-currence of a secondary necrotic process in which the phagocytesundergoing apoptosis lyse and release their cytotoxic contentscontributes to the genesis of the infection-associated necrotic le-sions (12, 13). These observations, together with the facts thatAIP56 is secreted only by virulent P. damselae subsp. piscicidastrains and that neutralizing antibodies to the toxin protect fishfrom P. damselae subsp. piscicida infections (1), established AIP56as a key virulence factor of P. damselae subsp. piscicida.
AIP56 is the founding member of a continuously growing fam-ily of bacterial proteins. Indeed, since we first described AIP56 (1),several open reading frames coding for AIP56 full-length homo-logues were identified in different organisms, mainly in marineVibrio species but also in Arsenophonus nasoniae. Interestingly,AIP56 seems to have originated from a fusion of two components:
one related to NleC, which is a type III secreted effector present inseveral enteric pathogenic bacteria (14) associated with humanillness and death worldwide (15), and another related to a proteinof unknown function from the bacteriophage APSE2. In line withthis, we recently found that AIP56 is an AB-type toxin, possessinga catalytic A domain at its N terminus, homologous to NleC, anda B domain involved in binding/internalization into target cells atits C-terminal region, homologous to APSE2 (16). The catalyticdomain of AIP56 is a zinc-dependent metalloprotease that, simi-larly to NleC, cleaves NF-�B, an evolutionarily conserved tran-scription factor that regulates the expression of inflammatory andanti-apoptotic genes, playing a key role in host responses to mi-crobial pathogen invasion (17). We have shown that during intox-ication of sea bass peritoneal leukocytes (sbPL), the catalytic ac-tivity of AIP56 results in the depletion of NF-�B p65 (16), and thislikely explains the disseminated apoptosis observed during P.damselae subsp. piscicida infections.
Although it is well established that AIP56 is a key virulence
Received 16 September 2014 Accepted 27 September 2014
Published ahead of print 6 October 2014
Editor: S. R. Blanke
Address correspondence to Ana do Vale, [email protected].
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02623-14.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/IAI.02623-14
5270 iai.asm.org Infection and Immunity p. 5270 –5285 December 2014 Volume 82 Number 12
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

factor of P. damselae subsp. piscicida and despite the availableknowledge on its structural organization, no information is avail-able concerning the trafficking of AIP56 in intoxicated cells. Sincethere are several (so far uncharacterized) AIP56 full-length homo-logues in different bacterial species that are likely to also havecrucial roles in virulence, expanding the knowledge on AIP56’sintoxication mechanism not only will add to the understanding ofP. damselae subsp. piscicida pathogenesis but also may shed lighton the pathogenesis of other bacteria producing AIP56-like tox-ins. The aim of the present study was to define the mechanismsinvolved in the entry of AIP56 into target cells and to identify itsintracellular pathways.
AB toxins are known to target and catalytically modify cytoso-lic substrates and, to reach the cytosol, have distinct mechanismsof internalization, all involving receptor-mediated endocytosisfollowed by vesicular trafficking to the site of membrane translo-cation (18). Short-trip AB toxins, such as diphtheria toxin, CNF1,botulinum neurotoxin, and tetanus neurotoxin, translocate fromendosomes following low-pH-induced conformational changesin the toxin molecule (19–21), while others, including Shiga andcholera toxins, are transported retrogradely to the endoplasmicreticulum, from where they translocate the catalytic moiety intothe cytosol (22). Prompted by the reduced number of tools (e.g.,antibodies) presently available to study AIP56 trafficking in sbPLand also aiming at exploring potential biomedical applications forAIP56, we tested several mammalian cells in intoxication assays.Although P. damselae subsp. piscicida does not grow at 37°C andhas a strict salt requirement (4), rendering it unable to infectmammals, here we present evidence that mouse bone marrow-derived macrophages (mBMDM) are susceptible to intoxicationby AIP56. Using both sbPL and mBMDM, we have characterizedmany of the steps involved in the trafficking of the toxin. Ourresults show that after clathrin-dependent endocytosis, AIP56translocates from endosomes into the cytosol using a pH-depen-dent mechanism. Additionally, our data suggest that a pool oftoxin follows the endocytic recycling pathway and is released backinto the extracellular medium.
MATERIALS AND METHODSEthics statement. This study was carried out in accordance with Euro-pean and Portuguese legislation for the use of animals for scientific pur-poses (Directive 2010/63/EU; Decreto-Lei 113/2013). The work was ap-proved by Direcção-Geral de Alimentação e Veterinária (DGAV), thePortuguese authority for animal protection (references 004933 and 2011-02-22).
Reagents. Concanamycin A (C9705), bafilomycin A1 (B1793), dyna-sore (D7693), cytochalasin D (C8273), nocodazole (M1404), chlorprom-azine (C8138), staurosporine (S4400), phenylmethylsulfonyl fluoride(PMSF) (P7626), pronase E from Streptomyces griseus (P5147), brefeldinA (B5936), cholera toxin (C8052), and lipopolysaccharide (LPS; L4391)were purchased from Sigma-Aldrich. LY294002 (no. 9901) was from CellSignaling. Hanks balanced salt solution (HBSS), HEPES, and sodium py-ruvate were purchased from Invitrogen. Dulbecco’s modified Eagle me-dium (DMEM), Leibovitz’s L-15 medium, fetal bovine serum (FBS), andpenicillin-streptomycin (P/S) (all Gibco products) and 2-(p-toluidinyl)naphthalene-6-sulfonic acid (TNS; T53) were purchased from Life Tech-nologies. The proteasome inhibitor MG132 (CAS 133407-82-6) was ob-tained from Calbiochem.
Antibodies and fluorescent probes. The anti-NF-�B p65 C-terminaldomain (c-20) rabbit polyclonal antibody (sc-372), the anti-GAPDH(6C5) (sc-32233), and the anti-c-Myc (9E10) (sc-40) mouse monoclonalantibodies were from Santa Cruz Biotechnology. The anti-V5 (R960-25)
and the anti-�-tubulin (32-2500) mouse monoclonal antibodies werefrom Invitrogen, the anti-early endosomal antigen 1 (EEA1) rabbit poly-clonal antibody (E3906) was from Sigma-Aldrich, the anti-lysosomalmembrane glycoprotein LAMP-1 mouse monoclonal (ab13523) and theanti-GRP78 Bip rabbit polyclonal (ab32618) antibodies were fromAbcam, and the anti-GM130 mouse monoclonal antibody was from BDBiosciences (610822). The anti-Akt rabbit polyclonal antibody (no. 9272)and the anti-phospho-Akt (Ser473) mouse monoclonal antibody (no.4051) were from Cell Signaling. The anti-sea bass NF-�B p65 rabbit serumwas produced using the peptide SIFNSGNPARFVS, located at the C-ter-minal region of sea bass p65, as the antigen, as described previously (16).Sheep anti-rabbit horseradish peroxidase (HRP)-conjugated secondaryantibody (AP311) and sheep anti-mouse HRP-conjugated secondary an-tibody (AP271) were from The Binding Site; goat anti-rabbit alkalinephosphatase-conjugated secondary antibody (A9919) and goat anti-mouse alkaline phosphatase-conjugated secondary antibody (A2429)were from Sigma-Aldrich. Alexa Fluor 647-transferrin (T23366) was fromInvitrogen, Alexa Fluor 594 phalloidin, Alexa Fluor 568 goat anti-rabbitIgG (heavy plus light chain [H�L]), Alexa Fluor 568 goat anti-mouse IgG(H�L), and Alexa Fluor 594 donkey anti-rabbit IgG (H�L) were fromMolecular Probes.
Production and fluorescent labeling of recombinant proteins. Inthis study, the following recombinant proteins were used: (i) full-lengthHis-tagged AIP56 (recombinant AIP56) (1); (ii) AIP56 carrying aC-terminal Myc or V5 tag followed by a His tag (AIP56Myc andAIP56V5, respectively); (iii) a His-tagged AIP56 metalloprotease mu-tant (AIP56AAIVAA) corresponding to a full-length version of the toxin inwhich key residues for zinc ion coordination and water molecule activa-tion (His165, Glu166, His169, and His170) were replaced by alanines (16);(iv) a His-tagged AIP561–285 with cysteine 262 mutated to serine(AIP561–285C262S); (v) a His-tagged AIP56286 – 497 with cysteine 298 mu-tated to serine (AIP56286 – 497C298S); and (vi) an LF·AIP56 chimeric pro-tein consisting of the amino terminus of anthrax lethal factor (LF11–263)fused to the AIP56 N-terminal domain (LF11–263 · AIP561–261). Protectiveantigen (PA), the receptor binding to LF (23), was kindly provided by CesareMontecucco (University of Padua, Italy). Recombinant AIP56, AIP56AAIVAA,AIP561–285C262S, AIP56286 – 497C298S, and LF11–263 · AIP561–261 were pro-duced and purified as previously described (16). Briefly, AIP56,AIP56286 – 497C298S, and LF11–263 · AIP561–261 were purified from thesoluble fraction of induced E. coli cells by metal affinity chromatogra-phy. This was followed by an anion-exchange chromatography in thecase of AIP56 or size exclusion chromatography in the case ofAIP56286 – 497C298S. AIP56AAIVAA and AIP561–285C262S were purifiedfrom inclusion bodies by metal affinity chromatography under denatur-ing conditions, refolded, and subjected to size exclusion chromatography.To obtain AIP56Myc and AIP56V5, pET28aAIP56H� plasmid (1) wasamplified using the forward primer AIP56NdeFw1 (5=-CGCCATATGGCATAACCTTCAATGATGGT-3=) together with AIP56MycXhoRv1 (5=-CGCCTCGAGAAGATCTTCTTCAGAAATAAGTTTCTGTTCATTAATGAATTGTGGCGCGTGGGGAT-3=) or AIP56V5XhoRv1 (5=-CGCCTCGAGCGTAGAATCGAGACCGAGGAGAGGGTTAGGGATAGGCTTACCATTAATGAATTGTGGCGCGTGGGGAT-3=). The PCR products weresubcloned into pGEM-T Easy vector (Promega) and cloned into thepET28a(�) plasmid (Promega), yielding the plasmidspET28aAIP56MycH� and pET28aAIP56V5H�. AIP56Myc andAIP56V5 were expressed in Escherichia coli BL21(DE3) and purified fol-lowing the protocol used for AIP56 (16). Recombinant proteins wereanalyzed by SDS-PAGE (see Fig. S1A in the supplemental material), andpurities were determined by densitometry of Coomassie blue-stained gels.Protein batches used in this work had a purity of �90% (see Fig. S1B in thesupplemental material).
Recombinant AIP56 labeled with Alexa Fluor 488 (Alexa 488-AIP56)was prepared using the Alexa Fluor protein labeling kit (A-10235) fromMolecular Probes following the manufacturer’s instructions.
Intracellular Trafficking of AIP56
December 2014 Volume 82 Number 12 iai.asm.org 5271
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

Determination of protein concentration. Concentrations of recom-binant protein were determined by measuring absorbance at 280 nm andusing the extinction coefficient calculated by the ProtParam tool (http://www.expasy.org/tools/protparam.html), using the Edelhoch method(24), but with the extinction coefficients for Trp and Tyr determined byPace et al. (25). In caspase 3 assays, to normalize the results, the proteinconcentration in each sample was determined using the Bio-Rad proteinassay following the recommended procedure for microtiter plates.
Experimental animals. The C57BL/6 mice were purchased fromCharles River (Madrid, Spain) and bred and housed at the animal facilityof the Instituto de Biologia Molecular e Celular (IBMC). The mice werefed sterilized food and water ad libitum. Mice were euthanized by isoflu-rane anesthesia followed by cervical dislocation. Sea bass (Dicentrarchuslabrax) were kept in a recirculating, ozone-treated salt water (25 to 30‰)system at 20 � 1°C and fed at a ratio of 2% body weight/day. Fish wereeuthanized with 2-phenoxyethanol (Panreac; �5 ml/10 liters).
Cells. Mouse bone marrow derived macrophages (mBMDM) werederived from bone marrow of femurs from 4- to 12-week-old C57BL/6male mice, as previously described (26), and used at day 10 when fullydifferentiated. Sea bass peritoneal leukocytes (sbPL) were obtained fromperitoneal cavities of sea bass, as previously described (27), and used at adensity of 2 � 106 cells/ml.
Intoxication assays. sbPL suspensions or mBMDM monolayers wereintoxicated by continuous incubation with recombinant AIP56 (178 nMat 22°C for sbPL and 89 nM at 37°C for mBMDM). Alternatively, cellswere pulsed with the toxin for 30 min on ice, washed, and incubated at22°C (sbPL) or 37°C (mBMDM). In the initial experiments characterizingthe susceptibility of mBMDM to AIP56, intoxication was accessed by twodifferent readouts: detection of cytotoxicity through morphological anal-ysis by phase-contrast microscopy and detection of NF-�B p65 cleavage,as previously described (16). In the subsequent experiments characteriz-ing the intracellular trafficking of AIP56, cleavage of NF-�B p65 was usedas an indication of the arrival of AIP56 into the cytosol. NF-�B p65 cleav-age was assessed by Western blotting after 2 h of incubation with the toxin.The metalloprotease mutant AIP56AAIVAA was included in some experi-ments to assess the involvement of the metalloprotease activity in intoxi-cation of mBMDM.
Caspase 3 activity and TUNEL. The activity of caspase 3 in total celllysates was determined using a commercial fluorimetric assay (EnzChekcaspase 3 assay kit; Molecular Probes). Lysates from cells undergoingapoptosis in response to treatment for 3 h with a 5 �M concentration ofthe apoptosis inducer staurosporine were used as a positive control. Theinternucleosomal DNA fragmentation was detected by TUNEL (terminaldeoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling)labeling of the DNA strand breaks with an in situ cell death detection kit(Roche Diagnostics).
Competition assays. The competition assays were performed as pre-viously described (16). Briefly, cells were preincubated for 15 min on icewith 7 �M AIP56 A domain (AIP561–285C262S) or with 7 �M B domain(AIP56286 – 497C298S) followed by incubation for 15 min on ice with an800-fold-lower concentration of recombinant AIP56 (8.75 nM AIP56) inthe presence of the competitors. Cells were washed with ice-cold supple-mented DMEM and incubated at 37°C for 2 h. NF-�B p65 cleavage wasassessed by Western blotting.
Inhibition of trafficking and toxicity. Cells were pretreated with con-canamycin A (10 nM), bafilomycin A1 (10 nM), dynasore (80 �M), chlor-promazine (30 �M), cytochalasin D (4 �M), or LY294002 (10 �M) for 1h or with nocodazole (33 �M) for 3 h. Results of experiments performedto (i) confirm that the inhibitors did not affect the proteolytic activity ofrecombinant AIP56 in vitro, (ii) confirm that nocodazole impaired mi-crotubule function and cytochalasin D effectively disrupted actin fila-ments, (iii) confirm that LY294002 inhibits phosphoinositide 3-kinase(PI3K) activity, and (iv) control the specificities of concanamycin A,dynasore, and chlorpromazine are presented as supplemental material.sbPL were pretreated with the inhibitors at 22°C and mBMDM at 37°C.
Cells treated with dynasore were incubated in culture medium lackingFBS. Recombinant AIP56 was added at final concentration of 178 nM(sbPL) or 89 nM (mBMDM) and incubated with the cells for 30 min onice. Cells were washed and transferred to 22°C (sbPL) or 37°C (mBMDM)and incubated up to 2 h while inhibitory conditions were maintained.Mock-treated cells, cells treated only with toxin, and cells treated onlywith inhibitors were used as controls. NF-�B p65 cleavage was analyzed byWestern blotting.
Detection of endocytosed AIP56. Cells were pulsed with AIP56V5or AIP56Myc for 30 min on ice (171 nM for sbPL and 85.5 nM formBMDM), washed three times with appropriate culture medium, andincubated at 22°C (sbPL) or 37°C (mBMDM). Mock-treated cells wereused as controls. When specified, cells were pretreated for 1 h and incu-bated in the presence of 20 �M MG132 or pretreated and incubated withconcanamycin A, bafilomycin A1, dynasore, chlorpromazine, cytochala-sin D, or nocodazole as described above. At different time points, cellswere washed twice with ice-cold phosphate-buffered saline (PBS) andincubated with 500 �g/ml pronase E in PBS for 10 min on ice to digest theextracellular toxin. Cells treated with the toxin and washed and incubatedin PBS were used as controls. The protease was inactivated with PMSF at1.45 mM. Cells were analyzed by Western blotting for detection ofAIP56V5 or AIP56Myc.
Lag time experiments. Cells were incubated with 178 nM (sbPL) or 89nM (mBMDM) recombinant AIP56 for 30 min on ice. Medium was re-placed by supplemented medium without toxin, and cells were incubatedfor 2 h at 37°C (mBMDM) or 22°C (sbPL). At various time points, 10 nMconcanamycin A was added. The cleavage of NF-�B p65, evaluated byWestern blotting, was used as an indication of the arrival of the toxin intothe cytosol.
Fluorescence microscopy. sbPL suspensions and mBMDM culturedon 12-mm glass coverslips were incubated with 178 nM Alexa 488-AIP56for 30 min on ice. The supernatant was removed and replaced with sup-plemented medium without toxin, and cells were switched to 22°C (sbPL)or 37°C (mBMDM) for a maximum of 60 min. At different time points,cells were washed once with PBS and processed for immunofluorescence.Cytospin preparations were obtained from sbPL suspensions, fixed withice-cold 4% (wt/vol) paraformaldehyde in PBS (4% PFA) for 10 min atroom temperature (RT) and permeabilized with 0.1% Triton X-100 inPBS for 10 min at RT. mBMDM were either fixed with ice-cold 4% PFAand permeabilized with 0.1% Triton X-100 in PBS (for detection of EEA1,LAMP-1, GM130, and �-tubulin) or fixed with ice-cold methanol for 10min at RT (for detecting GRP78 Bip). Cells were incubated with blockingbuffer (10% FBS in PBS with 0.1% Tween 20 [PBS-T]) for 30 min at RTfollowed by incubation with the primary antibodies in blocking bufferovernight at 4°C in a humidified chamber. The secondary antibodies di-luted in blocking buffer were incubated with cells for 1 h at RT. In exper-iments with transferrin, mBMDM were incubated in supplementedDMEM without FBS with 178 nM Alexa 488-AIP56 for 20 min on ice, thesupernatant was removed and replaced with ice-cold supplementedDMEM containing 6.25 mM Alexa 647-transferrin. Cells were incubatedfor 10 min on ice, switched to 37°C, and incubated for a maximum of 60min. At various time points, cells were washed twice with PBS and fixedwith ice-cold 4% PFA. Nuclei were counterstained with 0.57 �M DAPI.Samples were subjected to three-dimensional (3D) analysis on a laserscanning confocal microscope (Leica TCS SP5 II running LAS AF 2.6software; Leica Microsystems, Germany) using a 60�/1.40 numerical ap-erture (NA) oil immersion objective, with a pixel size of 60 nm and a z stepsize of 210 nm. Each fluorochrome was imaged by sequential acquisitionin order to avoid cross talk between channels. The 3D image stacks weredeconvolved with Huygens Professional software (SVI, Netherlands) ap-plying the maximum likelihood estimation restoration algorithm. Re-stored images were processed and maximum intensity projected with Fijisoftware (28). The object-based colocalization analysis was performed inFiji open source software (28) with the Squassh plugin of MosaicSuitedeveloped by MOSAIC Group, MPI-CBG, Dresden, Germany (29, 30).
Pereira et al.
5272 iai.asm.org Infection and Immunity
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

The measurements of colocalizations were determined based on signalusing the protocol described in detail by Paul et al. and Rizk et al. (29, 30).Briefly, this involved quantifying colocalization in an intensity-depen-dent manner by computing the sum of all pixel intensities in onechannel in all regions where objects colocalize with objects from theother channel (29, 30).
Recycling assay. mBMDM were incubated for 30 min on ice in serum-free medium containing 171 nM AIP56V5, transferred to 37°C for 5 minto allow endocytosis of the toxin (pulse), and switched to ice again. Cellswere washed three times with ice-cold PBS. The third wash was collectedand precipitated with trichloroacetic acid (TCA). After being washed, cellswere incubated for 10 min on ice with 125 �g/ml pronase E in PBS toremove extracellular toxin, washed twice with PBS, treated with 1.45mM PMSF, and washed three times with ice-cold PBS, followed bywashing with 250 �l/well serum-free medium containing 0.4 �MAIP56286 – 497C298S (to prevent re-entry of recycled full-length toxin) andincubation at 37°C in the same medium. The medium from the last washwas collected and precipitated with TCA. At different time intervals, su-pernatant was collected and precipitated with TCA, and the correspond-ing cell monolayer was washed twice with PBS and lysed with SDS-PAGEsample buffer. TCA precipitates were resuspended in SDS-PAGE samplebuffer, and the equivalent of 12 wells for supernatant and washes and 4wells for monolayer (pellet) was analyzed by Western blotting for detec-tion of AIP56V5. Membranes were reprobed with an anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (used as a cytosolicmarker). In some experiments, cells were incubated with 10 �MLY294002 or 10 nM concanamycin A for 1 h at 37°C before a pulse withAIP56V5. In this case, the inhibitor was maintained during the entirechase.
pH-induced translocation across the cell membrane. To address thequestion of whether an acidic pulse can drive translocation of cell-boundAIP56 across the cell membrane, we followed the protocol first describedfor diphtheria toxin (31, 32). Different pH values were obtained by addingH3PO4 to a buffer containing 0.5 mM MgCl2, 0.9 mM CaCl2, 2.7 mM KCl,1.5 mM KH2PO4, 3.2 mM Na2HPO4, and 137 mM NaCl. mBMDM wereincubated for 30 min on ice in supplemented DMEM with 89 nM recom-binant AIP56 in the absence or presence of 10 nM concanamycin A (thatwas maintained during the entire assay to inhibit normal toxin uptake).Supernatant was removed, and cells were incubated at the different pH for1 h at 37°C followed by incubation in culture medium (pH 7.4) for further2 h at 37°C. In all experiments, mock-treated cells and cells treated onlywith toxin were used as controls. NF-�B p65 cleavage was analyzed byWestern blotting.
Black lipid bilayer experiments. The methods used for black lipidbilayer experiments were described in detail previously (33). The instru-mentation consisted of a Teflon chamber with two aqueous compart-ments connected by a small circular hole, with a surface area of about 0.4mm2. Membranes were formed across the hole by painting on a 1% (wt/vol) solution of diphytanoyl phosphatidylcholine (Avanti Polar Lipids,Alabaster, AL) in n-decane. The aqueous salt solution (150 mM KCl) wasbuffered with 2 mM CaCl2 and 10 mM HEPES (pH 7.4 or pH 7.0), MES(morpholineethanesulfonic acid; pH 6.5 or pH 6.0), or CH3COOK (pH5.5, pH 5.0, or pH 4.5) added to the cis compartment of the chamber.Reducing conditions were established by addition of 1 mM dithiothreitol(DTT) after the membrane had turned black. Approximately 178 pmol ofprotein was used for each measurement; purified protein samples mixed1:1 with cholesterol suspension in water were added to the cis compart-ment of the chamber after the membrane had turned black. The mem-brane current was measured with a pair of Ag/AgCl electrodes with saltbridges switched in series with a voltage source and a highly sensitivecurrent amplifier (Keithley 427). The amplified signal was recorded by astrip chart recorder. All measurements were performed at RT, and eachexperiment was repeated at least twice.
Tryptophan and TNS fluorescence. Tryptophan and TNS fluores-cence at different pH values was analyzed in a PerkinElmer LS45 lumines-
cence spectrometer with excitation of 270 nm and an emission scan of 310nm to 450 nm and with excitation of 366 nm and an emission scan of 380to 500 nm, respectively. The following buffers were used: for pH 4.0, 4.5,5.0, and 5.5, 150 mM NaCl, 100 mM ammonium acetate; for pH 6.0 and6.5, 150 mM NaCl, 100 mM morpholineethanesulfonic acid (MOPS); forpH 7.0 and 7.5, 150 mM NaCl, 100 mM HEPES (34). TNS was added toeach buffer at a final concentration of 150 �M. AIP56Myc was added to afinal concentration of 1.5 �M in a final volume of 300 �l. Fluorescencemeasurements were done after 15 min incubation at RT. For the pH shiftexperiments, the toxin was incubated with TNS at pH 4.0 for 15 min at RT,and the fluorescence was recorded. The pH was then adjusted to 7.0 bygradual addition of 1 N NaOH, and the fluorescence was recorded again.Since the pH 7.0 was not within the buffering range of ammoniumacetate, the pH of the sample was checked following the analysis toconfirm that the desired pH value was maintained.
SDS-PAGE and Western blotting. SDS-PAGE was performed usingthe Laemmli discontinuous buffer system (35). Prior to loading, the sam-ples were boiled for 5 min in SDS-PAGE sample buffer (50 mM Tris-HCl[pH 8.8], 2% SDS, 0.05% bromophenol blue, 10% glycerol, 2 mM EDTA,and 100 mM DTT). For Western blotting, the proteins were transferredonto nitrocellulose membranes. The efficiency of transfer and the proteinloading on the membranes were controlled by staining with Ponceau S.The membranes were blocked for 1 h at RT with 5% skim milk in Tris-buffered saline (TBS) containing 0.1% Tween 20 (T-TBS) followed byincubation for 1 h at RT with the primary antibodies diluted in blockingbuffer. Immunoreactive bands were detected using horseradish peroxi-dase-linked secondary antibodies and enhanced chemiluminescence(ECL) West Dura substrate (Pierce Biotechnology) or alkaline phospha-tase-conjugated secondary antibodies and nitroblue tetrazolium–5-bro-mo-4-chloro-3-indolylphosphate (NBT/BCIP) (Promega). Blots shownpresent representative results from at least 3 independent experiments.Blots were quantified by densitometry analysis using Fiji software. Load-ing correction was achieved by dividing the density of the AIP56V5, p65,or cl-p65 band by the respective density of Ponceau S. In the case of therecycling assays, the densities of the GAPDH bands were used instead ofPonceau S. Each graph combines the results of at least three independentexperiments.
Statistical analysis. Statistical analysis of results presented in Fig. 1, 2,3, 5B, 5C, 6, and 8 and in Fig. S6 and S10 in the supplemental materialinvolved performing one-way or two-way analysis of variance (ANOVA).Normality of the data was tested by the Kolmogorov-Smirnov test; andhomogeneity of variances was assessed using the Levene test; P values forindividual comparisons were calculated using Tukey’s honestly signifi-cant difference (HSD) multiple-comparison test. Figures 4 and 5A andFig. S3, S4, and S7 in the supplemental material present several regressionmodels where the dependent variable is modeled as a function of time. Inorder to obtain these curves, several models were studied (linear, qua-dratic, inverse, and logarithmic), and the selected models were based onthe quality of the adjustment and, in particular, the residual distributionas well as the coefficient of determination (R2). The analysis of the resid-uals included the verification of normality as well as the study of extremevalues. Analyses were performed using the SPSS software, and significancewas set at a P value of 0.05.
RESULTSAIP56 induces depletion of NF-�B p65 and apoptosis inmBMDM. The lack of appropriate and established tools for study-ing intracellular trafficking in sbPL led us to search for an alterna-tive cell model. Although mammals are not susceptible to infec-tion by P. damselae subsp. piscicida, likely due to temperature andosmolarity restrictions (4), we decided to evaluate the susceptibil-ity of different mammalian cells to AIP56. Thus, we tested macro-phage (J774A.1, Raw264.7, and THP-1) and epithelial (HeLa,CHO, and Vero) cell lines (data not shown) as well as mBMDM.Toxicity was observed only in the latter (Fig. 1). When these cells
Intracellular Trafficking of AIP56
December 2014 Volume 82 Number 12 iai.asm.org 5273
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

were treated with recombinant AIP56, morphological alterationswere evident from 6 to 24 h of incubation (Fig. 1A). These altera-tions were dose dependent (see Fig. S2 in the supplemental ma-terial) and required the metalloprotease activity of the toxin,since they did not occur after incubation with the AIP56 met-alloprotease mutant AIP56AAIVAA (Fig. 1A). The cellular altera-tions included shrinkage, rounding of the cells, and membraneblebbing, as well as detachment of almost all cells after 24 h ofincubation (Fig. 1A), suggesting the occurrence of an apoptoticprocess. Assessment of nuclear DNA fragmentation by TUNELand measurement of caspase 3 activity confirmed that intoxi-cated mBMDM were undergoing apoptosis (Fig. 1B and C). Asexpected, cells treated with AIP56AAIVAA revealed no signs ofapoptosis (Fig. 1B and C). Western blotting revealed that in-toxication of mBMDM by recombinant AIP56 resulted in time-and dose-dependent decreases of NF-�B p65 protein levels(Fig. 1D). The correlation between p65 cleavage and the occur-rence of apoptosis has not been addressed, and therefore, thequestion of whether p65 depletion per se is sufficient to inducedeath or whether there are other host cell proteins targeted andimplicated in cytotoxicity needs to be clarified. The presence of
the p65 cleavage fragment (cl-p65) is time dependent, since itwas detected in cells up to 90 min after intoxication but not incells subjected to longer incubation times (Fig. 1D). Similarly,in sbPL, the p65 cleavage fragment was detected up to 4 h butwas no longer visible after intoxication for 6 h (see Fig. S3 in thesupplemental material). Using the proteosomal inhibitorMG132, we found that degradation of the p65 cleavage frag-ment originated by AIP56 depends on proteosomal activity, inagreement with what has been reported for the p65 cleavagefragment resulting from the activity of the type III effectorNleC (36). It is possible that the faster degradation of the cleav-age fragment observed in mBMDM results from a larger activ-ity of the proteasomal machinery in mBMDM than in sbPL.
AIP56 is endocytosed through a clathrin-dependent mecha-nism involving interaction of the C-terminal region of the toxinwith cell surface components. The observation that AIP56cleaves NF-�B in sbPL and mBMDM indicates that the toxin isable to reach the cytosol of these cells. Here, we used fluorescencemicroscopy and biochemical approaches to investigate the entrymechanisms used by the toxin. After 5 min incubation with sbPLor mBMDM, Alexa 488-AIP56 was observed in vesicle-like struc-
FIG 1 Intoxication of mBMDM with AIP56 leads to NF-�B p65 depletion and apoptosis. (A) Incubation of mBMDM with AIP56 results in apoptoticmorphological alterations and cell loss. Phase-contrast microscopy images of mBMDM incubated with 89 nM recombinant AIP56 or AIP56AAIVAA for theindicated time points are shown. The images were derived from one of three experiments. (B) Incubation of mBMDM with AIP56 results in the appearance ofTUNEL-positive and condensed nuclei. Cells were mock treated or incubated with 89 nM recombinant AIP56 or catalytic inactive AIP56AAIVAA for the indicatedtime points and processed for the detection of DNA fragmentation by TUNEL (green). Nuclei were counterstained with propidium iodide (red). Images shownare derived from one of three experiments. (C) Incubation of mBMDM with AIP56 results in caspase 3 activation. Cells were incubated with 89 nM recombinantAIP56 or AIP56AAIVAA for the indicated times, and caspase 3 activity was determined by fluorimetry (with duplicates) using the substrate N-benzoyl-Asp-Glu-Val-Asp-amino-4-methylcoumarin (Z-DEVD-AMC). The results, in relative fluorescence units per nanogram of protein, were converted to fold increase bycomparison to values obtained with extracts from mock-treated cells (seven independent experiments; the middle line corresponds to the median, and the topsand bottoms of the boxes represent the first and third quartiles; the circle and asterisk signal extreme observations). The fold increase in caspase 3 activityfollowing treatment with AIP56AAIVAA (white bars) is not statistically different from 1, whereas that following treatment with recombinant AIP56 (dark gray bars)is significantly different from 1 at all time points (P values for 3, 6, and 9 h: 0.018, 0.008, 0.033, respectively). As expected, a strong activation of caspase 3 wasobserved in staurosporine-treated cells (positive control, light gray bar; five independent experiments, P 0.033). Statistical analysis involved performing onesample t test for the hypothesis that the fold increase is equal to 1. (D) Incubation of mBMDM with AIP56 results in NF-�B p65 depletion. Cells were leftuntreated or incubated with the indicated doses of recombinant AIP56 for 30 min on ice, washed, and chased at 37°C. Cleavage of NF-�B p65 was analyzed byWestern blotting (chromogenic detection). The graph shows quantification of the blots. Loading correction was achieved by dividing the density of p65 band bythe respective density of Ponceau S. The results were divided by the same constant in order to set to 1 the mean for the control (cells not exposed to recombinantAIP56). Plotted values correspond to the estimated means from three independent experiments. Dose and time dependence of intact p65 levels was tested bytwo-way ANOVA [dose main effect, P 0.001; time main effect, P 0.001; and interaction dose with time, P 0.001; there was a significant difference betweenthe “no toxin (dose 0)” group and the other groups: P 0.001 (Tukey=s HSD)].
Pereira et al.
5274 iai.asm.org Infection and Immunity
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

tures (see Fig. S4A in the supplemental material), suggesting that ithad been endocytosed. Western blotting confirmed the internal-ization of the toxin (see Fig. S4B in the supplemental material). Akinetic analysis of the entry in sbPL revealed a marked decrease ofintracellular toxin over time (see Fig. S4C in the supplementalmaterial), which was not prevented by the proteosomal inhibitorMG132 (see Fig. S4C in the supplemental material). To enter hostcells, bacterial toxins are known to use clathrin-dependentand/or clathrin-independent endocytic mechanisms. Dynamin isa GTPase involved in endocytic membrane fission that is known tobe involved in clathrin-dependent endocytosis, although it mayalso participate in clathrin-independent endocytosis pathways(37). To investigate if AIP56 endocytosis is dependent on clathrinand/or dynamin, we inhibited their functions using the specificcell-permeative inhibitors chlorpromazine (Cpz) (38) and dyna-sore (Dyn) (39), respectively. In vitro tests confirmed that Cpz andDyn did not affect AIP56 metalloprotease activity (see Fig. S5 inthe supplemental material), and phagocytosis assays confirmedthat they were acting specifically (see Fig. S6A in the supplementalmaterial). To test the effect of Cpz and Dyn in AIP56 endocytosis,we treated mBMDM with the inhibitors or left the cells untreatedbefore pulsing with recombinant toxin for 30 min on ice. Cellswere washed and transferred to 37°C, and after a 2-min chase,extracellular toxin was removed with pronase E (PronE). Asshown in Fig. 2A, endocytosed AIP56 was detected in control cellsbut not in cells pretreated with Cpz or Dyn. The inhibitory effectof Cpz and Dyn upon AIP56 endocytosis together with the obser-vation that the AIP56-dependent cleavage of p65 was prevented bypretreatment with Dyn and was strongly inhibited by pretreat-ment with Cpz (Fig. 2B) suggests that a significant pool of AIP56 isendocytosed through a dynamin- and clathrin-dependent path-way. Nevertheless, the partial inhibition of toxicity observed after
pretreatment with Cpz indicates that a fraction of toxin moleculesmay be internalized through a dynamin-dependent mechanismnot requiring clathrin. Previous studies with sbPL showed that theentry of the toxin involves specific interaction of its C-terminalregion with cell surface components (16). To investigate if theC-terminal region of AIP56 also plays a role in the entry inmBMDM, we performed a competition assay testing the ability ofAIP56286 – 497C298S (corresponding to the toxin’s B domain) (16) toinhibit the entry of the toxin into the cytosol. AIP561–285C262S,corresponding to the toxin’s A domain (16), was used as a control.Preincubation with AIP56286 – 497C298S before intoxication with re-combinant AIP56 inhibited p65 cleavage, whereas no inhibitionwas observed when AIP561–285C262S was used as a competitor (Fig.2C). These results indicate that the entry of AIP56 into mBMDMinvolves interaction of AIP56 C-terminal domain with cell surfacereceptors.
Microtubules and actin dynamics are dispensable for intox-ication by AIP56. Functional actin dynamics is required for sev-eral endocytic processes (40). To determine if internalization ofAIP56 depends on actin, we disrupted microfilaments with cy-tochalasin D (CytoD) (41) (see Fig. S6A and B in the supplementalmaterial) and addressed whether disruption inhibited intoxica-tion. Treatment with CytoD did not inhibit AIP56 endocytosis(Fig. 3A) or AIP56-dependent p65 cleavage (Fig. 3B), indicatingthat intoxication is independent on actin dynamics. The involve-ment of microtubules in AIP56 uptake was investigated using themicrotubule depolymerizing agent nocodazole (Noco) (42). Al-though Noco effectively disrupted microtubules (see Fig. S7A andB in the supplemental material), it did not affect AIP56 endocyto-sis (Fig. 3A) or AIP56-dependent p65 cleavage (Fig. 3B), indicat-ing that AIP56 exploits a microtubule-independent intoxicationpathway.
FIG 2 AIP56 endocytosis and toxicity require clathrin and dynamin. (A) AIP56 endocytosis is prevented by Cpz and Dyn. mBMDM were pretreated with Cpzor Dyn, pulsed with AIP56V5 in the presence of the inhibitors for 30 min on ice, washed, and incubated at 37°C in culture medium with inhibitors. After 2 min,surface-exposed toxin was removed with PronE, and the intracellular pool was detected by Western blotting (chromogenic detection). (B) AIP56-mediatedcleavage of NF-�B p65 is inhibited by Cpz and Dyn. sbPL or mBMDM were pretreated with Cpz or Dyn, pulsed with recombinant AIP56 for 30 min on ice in thepresence of the inhibitor, shifted to 22°C (sbPL) or 37°C (mBMDM), and incubated for 2 h. Cleavage of p65 was assessed by Western blotting (chromogenicdetection). (C) Intoxication of mBMDM by AIP56 is inhibited by the toxin’s B domain AIP56286 – 497C298S. Cells were incubated with 7 �M AIP561–285C262S
(AIP56Nterm) or AIP56286 – 497C298S (AIP56Cterm) for 15 min on ice, followed by further 15 min incubation with an 800-fold-lower concentration of recom-binant AIP56 in the presence of the competitor. Cells were washed and incubated at 37°C for 2 h. Cleavage of NF-�B p65 was assessed by Western blotting(chromogenic detection). The graphs show the quantification of the blots. Loading correction was achieved by dividing the density of AIP56V5 (A) or p65 (B andC) by the respective density of Ponceau S. The results used for each graph were divided by the same constant in order to set to 1 the mean of the control (cellsexposed to toxin only [A] or mock-treated cells [B and C]). Each graph combines the results of three independent experiments (means � standard deviations[SD]). The significance of differences was tested by one-way ANOVA (all P values were 0.001). P values for individual comparisons were calculated usingTukey’s HSD multiple-comparison test and are indicated in the graphs.
Intracellular Trafficking of AIP56
December 2014 Volume 82 Number 12 iai.asm.org 5275
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

Upon endocytosis, AIP56 localizes in early endosomes andthen is routed to the recycling endocytic compartment. To fur-ther characterize the intracellular trafficking of AIP56, cells werepulsed with Alexa 488-AIP56 (green) for 30 min on ice, washed,and transferred to 37°C. At different chase times, cells were pro-cessed for immunofluorescence to assess the colocalization oftoxin with established markers of intracellular compartments.Colocalization experiments are reported only for mBMDM, be-cause most of the commercially available antibodies and probesare not validated or do not work on fish cells. After a 2-min chase,Alexa 488-AIP56 was observed in peripheral endocytic vesiclesnegative for all the markers tested, but shortly after (after a 5-minchase), part of the toxin-containing vesicles were positive for theearly endosome marker EEA-1 (Fig. 4; also, see Fig. S8A in thesupplemental material). The colocalization with EEA-1 reached amaximum at 15 to 20 min and afterwards markedly decreased,being minimal at 60 min (Fig. 4; also, see Fig. S8A in the supple-mental material). These results indicate that after endocytosis,AIP56 localizes in early endosomes, where it remains for 20 to 30min. To define the compartment where AIP56 was delivered afterleaving early endosomes, we looked for colocalization of the toxinwith the late endosome/lysosome marker LAMP-1, the Golgi ap-paratus marker GM130, or the endoplasmic reticulum markerGRP78 Bip. Alexa 647-transferrin was used as a marker of therecycling route. In macrophages (43–48), similar to what occurs inother cells (49), transferrin is recycled back to the cell surface usingtwo kinetically distinguishable mechanisms of recycling (fast andslow recycling). This supports the use of transferrin as a markerfor the recycling pathway in macrophages and in, particular, in
mBMDM. Colocalization of Alexa 488-AIP56 with Alexa 647-transferrin was evident at 5 min and was maintained during theremaining chase (Fig. 4; also, see Fig. S8B in the supplementalmaterial), suggesting that after leaving early endosomes, the toxinwas delivered to the recycling endocytic compartment. In agree-ment with this hypothesis, Alexa 488-AIP56 was never observed tocolocalize with LAMP-1 (Fig. 4; also, see S8C in the supplementalmaterial) or markers of Golgi apparatus and endoplasmic reticu-lum (Fig. 4; also, see Fig. S8D and E, respectively, in the supple-mental material).
Shortly after endocytosis, a pool of AIP56 is recycled back tothe extracellular medium through a mechanism requiring PI3Kactivity but independent on endosome acidification. The obser-vation that AIP56 was delivered to the recycling endocytic com-partment following endocytosis, suggested that the toxin was be-ing recycled back to the extracellular medium. To investigate thishypothesis, we allowed mBMDM to bind AIP56V5 and internalizeit for 5 min (pulse), removed the extracellular toxin with PronE,chased the cells at 37°C and analyzed the presence of the toxininside cells and in the extracellular medium at different timepoints. The absence of AIP56V5 in the last wash after PronE treat-ment (Fig. 5A, lane 5) confirmed that extracellular toxin was re-moved before chasing. After 2 min chase, AIP56V5 was alreadydetected in the extracellular medium (Fig. 5A, lane 7). Extracellu-lar AIP56V5 increased over time (Fig. 5A, lanes 7 to 10) and wasparalleled by a decrease in cell-associated toxin (Fig. 5A, lanes 12to 15), indicating that during chase, intracellular AIP56 was beingrecycled to the extracellular medium. Since PI3K, an evolution-arily conserved enzyme complex which phosphorylates phospha-
FIG 3 AIP56 intoxication is independent of actin and microtubules. (A) AIP56 endocytosis is not inhibited by CytoD or Noco. mBMDM were treated withCytoD or Noco before incubation with AIP56V5 in the presence of the inhibitors for 30 min on ice (pulse), washed, and incubated for 2 min at 37°C in culturemedium with inhibitors. Extracellular toxin was removed with PronE, and the intracellular pool was detected by Western blotting (anti-V5 antibody, chromo-genic detection). (B) The arrival of AIP56 into the cytosol is not prevented by CytoD or Noco. sbPL or mBMDM were treated with CytoD or Noco at theappropriate temperature prior to incubation with recombinant AIP56 for 30 min on ice in the presence of the inhibitor. Cells were shifted to 22°C (sbPL) or 37°C(mBMDM) and incubated for 2 h while the inhibitory conditions were maintained. Cleavage of NF-�B p65 was assessed by Western blotting (chromogenicdetection). The graphs show the quantification of the blots. Loading correction was achieved by dividing the density of AIP56V5 (A) or p65 (B) by the respectivedensity of Ponceau S. The results used for each graph were divided by the same constant in order to set to 1 the mean of the control (cells exposed to toxin only[A] or mock-treated cells [B]). Each graph combines the results of three independent experiments (mean � SD). The significance of differences was tested byone-way ANOVA (P 0.001 [A], P 0.012 [B, mBMDM], and P 0.001 [B, sbPL]). P values for individual comparisons were calculated using Tukey’s HSDmultiple-comparison test and are indicated in the graphs.
Pereira et al.
5276 iai.asm.org Infection and Immunity
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from
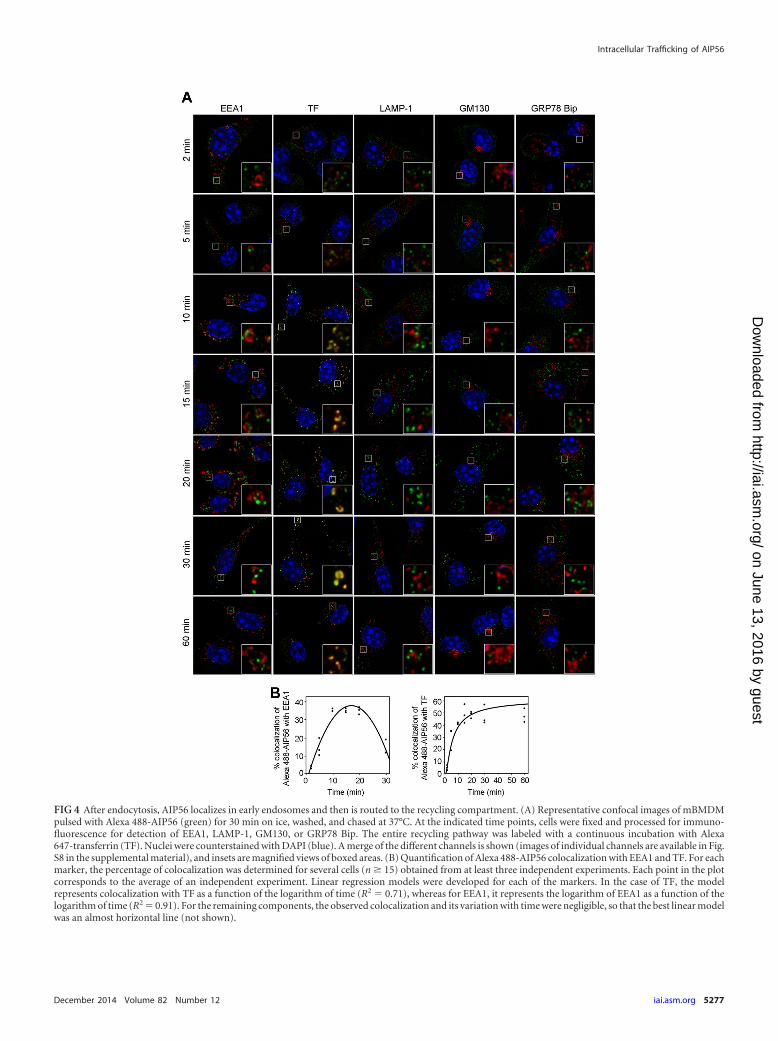
FIG 4 After endocytosis, AIP56 localizes in early endosomes and then is routed to the recycling compartment. (A) Representative confocal images of mBMDMpulsed with Alexa 488-AIP56 (green) for 30 min on ice, washed, and chased at 37°C. At the indicated time points, cells were fixed and processed for immuno-fluorescence for detection of EEA1, LAMP-1, GM130, or GRP78 Bip. The entire recycling pathway was labeled with a continuous incubation with Alexa647-transferrin (TF). Nuclei were counterstained with DAPI (blue). A merge of the different channels is shown (images of individual channels are available in Fig.S8 in the supplemental material), and insets are magnified views of boxed areas. (B) Quantification of Alexa 488-AIP56 colocalization with EEA1 and TF. For eachmarker, the percentage of colocalization was determined for several cells (n � 15) obtained from at least three independent experiments. Each point in the plotcorresponds to the average of an independent experiment. Linear regression models were developed for each of the markers. In the case of TF, the modelrepresents colocalization with TF as a function of the logarithm of time (R2 0.71), whereas for EEA1, it represents the logarithm of EEA1 as a function of thelogarithm of time (R2 0.91). For the remaining components, the observed colocalization and its variation with time were negligible, so that the best linear modelwas an almost horizontal line (not shown).
Intracellular Trafficking of AIP56
December 2014 Volume 82 Number 12 iai.asm.org 5277
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

FIG 5 After endocytosis, AIP56 is recycled back to the extracellular medium by a mechanism requiring PI3K activity but independent of endosomal acidification.(A) mBMDM were left untreated or incubated with AIP56V5 on ice for 30 min, followed by 5 min incubation at 37°C (pulse). Cells were washed, extracellulartoxin was removed with PronE, and cells were washed again and chased at 37°C for different time intervals. Cell-associated (pellet) and recycled (supernatant)AIP56V5 was detected by Western blotting (anti-V5 antibody; chromogenic detection). To control the efficacy of PronE treatment, we analyzed the presence ofthe toxin in the last wash before PronE treatment (wash BP) and in the last wash after PronE treatment (wash AP). Samples (washes, supernatants, and pellets)from 12 wells were pooled and the equivalents of 12 wells for washes/supernatants or 4 wells for pellets were loaded in the gels. GAPDH was used as a cytosolicmarker. The graphs show the quantification of the recycled or cell-associated AIP56V5 relative to the total internalized protein (lane 3) and combine results fromthree independent experiments (represented by different symbols). The logarithmic regression model represents the percentage of recycled or cell associatedAIP56V5 as a function of time (recycled R2 0.451; cell-associated R2 0.679). (B) LY inhibits the recycling of AIP56 into the extracellular medium but not itstoxicity. mBMDM were left untreated or treated with LY before the recycling or toxicity assays in the presence of LY. (I) The recycling protocol was the same asdescribed for panel A. The graph shows the quantification of the blots and combines results from four independent experiments (the middle line in each boxcorresponds to the median, and the tops and bottoms of the boxes correspond to the first and third quartiles; whiskers represent the smallest and largest observedvalues). Values correspond to percentage of recycled AIP56V5 relative to the total internalized protein (lane 3). Statistical analysis involved performing two-way
Pereira et al.
5278 iai.asm.org Infection and Immunity
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

tidylinositol lipid substrates into 3-phosphoinositides (50, 51),has been shown to play a key role in regulating the recycling path-way (52–56), we analyzed the involvement of PI3K in AIP56 recy-cling. For this, we performed recycling assays in the presence ofLY294002 (LY), a reversible paninhibitor of PI3K activity (57, 58).The effect of LY upon PI3K activity was controlled by confirmingLY-induced inhibition of the phosphorylation of Akt, a down-stream target of PI3K (59) (see Fig. S9 in the supplemental mate-rial). In the presence of LY, recycling of AIP56 was strongly inhib-ited (Fig. 5B, compare lanes 6 and 7 with lanes 10 and 11,respectively), and an increase in cell-associated toxin was ob-served (compare lanes 12 and 13 with lanes 14 and 15, respec-tively), suggesting that recycling of AIP56 is dependent on PI3Kactivity. Because PI3K is involved in a wide range of cellular pro-cesses (50) including regulation of the endocytic pathway we alsoanalyzed the effect of LY in AIP56 toxicity. As shown in Fig. 5B, nodifferences in AIP56-dependent p65 cleavage were observed incells treated with LY compared to those incubated with the toxinin the absence of the inhibitor, indicating that the AIP56 intoxi-cation pathway is independent of PI3K activity. We also investi-gated if recycling of AIP56 was dependent on endosome acidifica-tion, because it is reported that the exposure of ligand-receptorcomplexes internalized by receptor-mediated endocytosis to lowendosomal pH is required for the efficient sorting of several cargosand recycling of receptors (52, 60). The recycling experiment wasrepeated in the presence of concanamycin A (ConcA), an inhibi-tor of the endosomal vacuolar ATPase pump responsible for en-dosome acidification (61). Experiments aiming at controlling thespecificity and efficacy of ConcA in inhibiting the endosomal vac-uolar ATPase pump are presented in Fig. S10 in the supplementalmaterial. Recycling of AIP56V5 was not inhibited by ConcA (Fig.5C, compare lanes 6 and 7 with lanes 10 and 11, respectively),suggesting that it does not require low endosomal pH.
Endosome acidification is required for AIP56 intoxication.The indication that following endocytosis, AIP56 entered earlyendosomes and that after 20 to 30 min it trafficked into the endo-cytic recycling compartment suggests that in order to reach itscytosolic target, this toxin translocates from early or recycling en-dosomes. AB toxins that translocate from endosomes (e.g., diph-theria, tetanus, and botulinum toxins) are inhibited by agents thatprevent acidification of endosomes/lysosomes (22, 62, 63), be-cause the trigger for translocation is the low endosomal pH. Toclarify the mechanism involved in the translocation of AIP56 intothe cytosol, we used bafilomycin A1 (BafA1) and ConcA, two po-tent inhibitors of the endosomal vacuolar ATPase pump and,thus, of the acidification of early and late endosomes as well aslysosomes (61). These compounds did not affect the in vitro cata-lytic activity of AIP56 (see Fig. S5 in the supplemental material) orits endocytosis (Fig. 6A). However, when mBMDM or sbPL werepretreated with either inhibitor, AIP56-dependent cleavage of p65was prevented (Fig. 6B), indicating that arrival of the toxin at the
cytosol is dependent on endosome acidification. To gain insightinto the time course of AIP56 translocation, we took advantage ofthe inhibitory effect of ConcA. As shown in Fig. 6C for mBMDM,ConcA markedly inhibited p65 cleavage when added up to 15 minfollowing AIP56 treatment. However, addition of the inhibitor 30min after toxin treatment resulted in only partial inhibition (notstatistically significant), and afterwards, the addition of ConcAhad no effect on p65 degradation. These results indicate that at 30min incubation, some AIP56 is no longer susceptible to the effectof ConcA, suggesting that it has already translocated into the cy-tosol to exert its toxic effect. A similar effect was observed for sbPL,although in this case, the differences did not reach statistical sig-nificance.
At acidic pH, AIP56 undergoes reversible conformationalchanges and interacts with artificial lipid bilayer membranes.Considering that toxins that translocate from endosomes undergoconformational rearrangements at endosomal pH (34), pH-in-duced structural changes in AIP56 were monitored by analyzingTNS fluorescence (TNS is a commercial probe that is nonfluores-cent in water and becomes fluorescent when bound to hydropho-bic regions of a protein [64]). Recombinant toxin was incubatedwith TNS at different pH values for 15 min, and fluorescence wasanalyzed. As shown in Fig. 7A, while TNS fluorescence at pH 6.0and above was not higher than background levels, at pH 5.5, flu-orescence markedly increased. The increase continued at pH 5.0,4.5, and 4.0 and was found to be reversible, since returning thetoxin that had been incubated at pH 4.0 to pH 7.0 resulted in astrong decrease in fluorescence intensity (Fig. 7B). These resultsindicate that AIP56 exhibits a reversible increase in hydrophobic-ity when exposed to an acidic (pH 5.5 to 4.0) environment. Be-cause TNS is an “external” probe of protein hydrophobicity, theintrinsic fluorescence of tryptophan residues was also analyzed.Changes in tryptophan fluorescence can be used to detect proteinconformational alterations, because those can be quenched in thepresence of an aqueous solvent. As shown in Fig. 7C, AIP56 tryp-tophan fluorescence decreases as pH declines, suggesting that un-der acidic conditions tryptophan-containing domains move intomore aqueous environments or that hydrophobic pockets moveaway from tryptophans. Following the observation that AIP56undergoes pH-induced conformational changes, we used blacklipid bilayers (33) to determine whether the toxin is able to inter-act with lipid membranes and whether the observed pH-inducedconformational changes play a role in the interaction. Whentested at neutral pH values (pH 7.0 to 7.4), recombinant AIP56failed to exhibit membrane activity in black lipid membranes.However, acidification of the aqueous phase at the side of the ciscompartment triggered membrane activity (Fig. 7D). Accord-ingly, when recombinant AIP56 was added to the cis chamber atacidic pH (6.5 to 4.5), membrane activity was also triggered after a150-mV pulse (Fig. 7E), leading to an undefined but stepwise in-crease in membrane conductivity (Fig. 7E, left), which was fol-
ANOVA (main effect of inhibitor, P 0.001; main effect of time, P 0.073; inhibitor and time interaction, P 0.059). (II) Toxicity (cleavage of NF-�B) wasassessed and quantified as described for Fig. 2B. The significance of differences was tested by one-way ANOVA (three independent experiments; mean � SD;P 0.001). P values for individual comparisons were calculated using Tukey’s HSD multiple-comparison test and are indicated in the graph. (C) Recycling ofAIP56 into the extracellular medium is not inhibited by ConcA. mBMDM were treated with ConcA or left untreated before the recycling assay, as described forpanel A, but in the presence of ConcA. The graph shows the quantification of the blots and combines results from three independent experiments (the middleline in each box corresponds to the median, and the tops and bottoms of the boxes correspond to the first and third quartiles; whiskers represent the smallest andlargest observed values). Values correspond to percentage of recycled AIP56V5 relative to the total internalized protein (lane 3). Statistical analysis involvedperforming two-way ANOVA (main effect of inhibitor, P 0.180; main effect of time, P 0.784; inhibitor and time interaction, P 0.942).
Intracellular Trafficking of AIP56
December 2014 Volume 82 Number 12 iai.asm.org 5279
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

lowed by membrane rupture. A very stable signal was observedwhen the voltage was kept to 50 mV (Fig. 7E). AIP56AAIVAA be-haved similarly to AIP56 (Fig. 7F), indicating that mutations inthe metalloprotease signature did not impair AIP56’s membraneactivity. The observed current fluctuations were irregular and in-homogeneous, indicating that interaction of AIP56 with bilayermembranes does not lead to the formation of regular channelscomparable to the ones formed by other AB toxins, such as an-thrax toxins (65, 66) or Clostridium botulinum C2 (65, 66). Thecurrent fluctuations induced by AIP56 are similar to the ones re-ported to occur with the C. botulinum and Clostridium limosumC3 toxins (67) as well as with the Clostridium difficile TcdB andTcdA toxins (68, 69) and may result from formation of transientchannels.
Extracellular acidification triggers translocation of cell sur-face-bound AIP56 into the cytosol. The observation that inhibi-tors which prevent endosome acidification inhibited AIP56 toxic-ity, that AIP56 undergoes pH-induced conformational changesand is able to interact with black lipid bilayers at low pH promptedus to test if a low-pH pulse could drive the translocation of plasmamembrane-bound AIP56 into the cytosol, similar to what hasbeen reported for several other bacterial toxins that translocatefrom endosomes through a pH-dependent mechanism (31, 32).ConcA was maintained during the entire experiment to block en-docytic translocation of AIP56. mBMDM were incubated withrecombinant AIP56 for 30 min on ice to allow toxin binding andthen subjected to a pulse at pH 5.5 or 5.0 (pH 7.0 as a control) at37°C. Afterwards, cells were incubated in supplemented culturemedium at pH 7.4 for 2 h at 37°C, washed, and analyzed by West-ern blotting for detection of p65 cleavage. In the absence of anacidic pulse, ConcA inhibited AIP56-dependent cleavage of p65(Fig. 8). However, the inhibitory effect of ConcA was abolishedwhen mBMDM with surface-bound AIP56 were exposed to pH5.0 or 5.5 (Fig. 8), indicating that an acidic pulse is sufficient toinduce AIP56 translocation across the plasma membrane of tar-geted cells.
DISCUSSION
In this study, we found that mBMDM are susceptible to AIP56intoxication and, using both sbPL and mBMDM, defined keysteps involved in the entry and intracellular trafficking of thetoxin.
Although P. damselae subsp. piscicida is not able to infect mam-mals, our data show that, similar to what has been recently ob-served in sbPL (16), intoxication of mBMDM by AIP56 results inNF-�B p65 depletion and apoptosis. It is well known that uncon-trolled activation of NF-�B is associated with several human pa-thologies, including inflammatory diseases and cancers (70–72),and thus, apart from its intrinsic biologic interest, the observationthat AIP56 acts on mammalian cells confers considerable biotech-nological potential to the toxin.
Similar to diphtheria toxin, CNF1, botulinum neurotoxin, and
FIG 6 AIP56 translocation into the cytosol is dependent on endosome acidi-fication. (A) AIP56 endocytosis is not inhibited by ConcA or BafA1. mBMDMwere treated with BafA1 or ConcA before incubation with recombinant AIP56in the presence of the inhibitors for 30 min on ice, washed and incubated for 2min at 37°C in culture medium with inhibitor. Extracellular toxin was re-moved with PronE and the intracellular pool was detected by Western blotting(anti-V5 antibody; chromogenic detection). (B) ConcA and BafA1 prevent theAIP56-dependent cleavage of p65. sbPL or mBMDM were treated with theindicated inhibitor before incubation with recombinant AIP56 in the presenceof the inhibitor for 30 min on ice. Cells were washed, shifted to 22°C (sbPL) or37°C (mBMDM) and incubated for 2 h while maintaining the inhibitory con-ditions. Cleavage of p65 was assessed by Western blotting (chromogenic de-tection). (C) Time course of AIP56 translocation. sbPL or mBMDM were leftuntreated or incubated with recombinant AIP56 for 30 min on ice, washed andtransferred to 22°C (sbPL) or 37°C (mBMDM). ConcA was added at the indi-cated time points, and after 2 h, cleavage of p65 was assessed by Westernblotting (chromogenic detection). The graphs show quantification of the blots.Loading correction was achieved by dividing the density of AIP56V5 (A) orp65 (B and C) by the respective density of Ponceau S. The results used for eachgraph were divided by the same constant in order to set to 1 the mean of thecontrol (cells exposed to toxin only [A], mock-treated cells [B], or ConcA at 0
min [C]). Each graph combines the results of three independent experiments(mean � SD). The significance of differences was tested by one-way ANOVA(P 0.001 [A], P 0.01 [B, mBMDM] P 0.001 [B, sbLP], P 0.001 [C,mBMDM], and P 0.126 [C, sbPL]). P values for individual comparisonswere calculated using Tukey’s HSD multiple comparisons and are indicated inthe graphs (in panel C, the P values are for comparisons with cells treated onlywith toxin).
Pereira et al.
5280 iai.asm.org Infection and Immunity
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

tetanus neurotoxin (73–75), AIP56 is a single-chain AB toxin thatreaches the cytosolic compartment to exert its activity (16). Here,using sbPL and mBMDM, we found that shortly after incubationwith cells, AIP56 localizes in intracellular vesicular compartmentsinitially scattered in the cytoplasm that over time concentrate inthe perinuclear area, indicating that binding of AIP56 to a still-unidentified cell surface receptor(s) is followed by endocytosisinto a vesicular/endosomal compartment, from which it escapesto reach the cytosol. As shown in sbPL (16), the binding of AIP56to mBMDM occurs by interaction of the C-terminal region of thetoxin with the host cell surface, suggesting that the toxin recog-nizes a conserved receptor. Our results indicate that following
receptor binding, AIP56 endocytosis likely occurs through a clath-rin-dependent mechanism, since inhibition of the formation ofclathrin-coated pits blocked AIP56 internalization. In agreementwith this, inhibition of the large GTPase dynamin, known to playan important role in clathrin-mediated endocytosis by participat-ing in the constriction and subsequent budding of coated pits (37,76–78), also blocked AIP56 entry. Clathrin-dependent endocyto-sis includes several internalization pathways, all relying on the useof the coat protein clathrin but differing with regard to the re-quirement for other proteins, including actin (79). Findings of thepresent study indicate that the actin cytoskeleton is not involved inAIP56 endocytosis, since depolymerization of F actin by CytoD
FIG 7 Under acidic conditions, AIP56 undergoes reversible conformational changes and interacts with lipid bilayer membranes. (A) TNS analysis of pH-induced hydrophobic transitions in AIP56Myc. The toxin was incubated with TNS at the indicated pH values, and fluorescence was analyzed. (B) TNSfluorescence analysis following a pH shift. AIP56Myc was incubated with TNS at pH 4.0, and fluorescence was analyzed, as described for panel A. The pH was thenadjusted to 7.0 (shifted pH), and the TNS emission spectrum was recorded again. AIP56Myc incubated with TNS at pH 7.0 was used as a control. (C) Intrinsictryptophan fluorescence of AIP56Myc incubated at pH 7.5 or pH 4.0. In experiments A, B, and C, fluorescence intensities were determined by averaging tworeadings, and background fluorescence (TNS plus buffer alone for TNS analysis or buffer alone for tryptophan) was subtracted from the fluorescence of eachexperimental sample. Results are expressed as the averages (black line) � SD (dashed lines) from four independent experiments. (D) Current recording of adiphytanoyl phosphatidylcholine/n-decane membrane in the presence of recombinant AIP56. The applied membrane potential was 50 mV. Initial experimentalconditions consisted of 150 mM KCl, 2 mM CaCl2, 1 mM DTT, 10 mM HEPES (pH 7.4). Addition of recombinant AIP56 mixed 1:1 with cholesterol suspensionin water to the cis compartment of the chamber (left; arrow) had no effect on membrane conductivity. Acidification of the aqueous phase at the cis compartmentby addition of 10 mM CH3COOK (pH 4.6) (right; arrow) triggered membrane activity. The experiment was repeated three times. (E) Membrane activity couldalso be triggered by a 150-mV pulse. A current recording of a diphytanoyl phosphatidylcholine/n-decane membrane in the presence of recombinant AIP56 mixed1:1 with cholesterol suspension in water is shown. Measurements were performed in 150 mM KCl, 2 mM CaCl2, 10 mM MES (pH 6.0), and under theseconditions, membrane activity was triggered by a 150-mV pulse (B, left; arrow), which finally resulted in membrane rupture. A very stable signal could beobserved when only 50 mV was applied to a diphytanoyl phosphatidylcholine/n-decane membrane in the presence of recombinant AIP56 mixed 1:1 withcholesterol suspension in water (B, right). The experiment was repeated three times. (F) Mutations in the metalloprotease signature do not impair the interactionof AIP56 with lipid bilayer membranes. A current recording of a diphytanoyl phosphatidylcholine/n-decane membrane in the presence of AIP56AAIVAA mixed1:1 with cholesterol suspension in water is shown. Measurements were performed in 150 mM KCl, 2 mM CaCl2, 10 mM MES (pH 6.0). Membrane activity wastriggered under these conditions by a 150-mV pulse (left; arrow) and stabilized by lowering the applied voltage to 50 mV (right; arrow). The experiment wasrepeated twice.
Intracellular Trafficking of AIP56
December 2014 Volume 82 Number 12 iai.asm.org 5281
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

(41) did not affect entry or p65 cleavage by the toxin. This issimilar to what has been reported for transferrin, a classicalmarker of clathrin-mediated endocytosis, which in several celltypes has been shown to enter through a pathway independent ofactin (80), and also for diphtheria toxin, which is known to entercells via a clathrin-dependent mechanism that is actin indepen-dent (81, 82).
It has traditionally been thought that molecules internalizedvia clathrin-dependent endocytosis are either recycled back to theplasma membrane or degraded via the lysosomal pathway (79).However, there are toxins, like Shiga and cholera toxins, that uponinternalization via clathrin-coated pits traffic retrogradely to theGolgi apparatus (83). Several lines of evidence indicate that uponendocytosis, AIP56 follows the endocytic recycling route and isreleased into the extracellular medium. (i) Within 5 min afterendocytosis, AIP56 localizes in early endosomes (positive forEEA-1 and Alexa 647-transferrin), where it remains for 20 to 30min, and then is routed to the recycling endocytic compartment(identified by labeling with Alexa 647-transferrin). (ii) Using arecycling assay in which we allowed cells to internalize AIP56,removed extracellular toxin, and then monitored the appearanceof recycled toxin in the extracellular medium, we observed thatpart of the endocytosed AIP56 is recycled back to the extracellularcompartment. Recycled toxin is already detected at 2 min chase(following a pulse of 30 min on ice plus 5 min at 37°C), and itsamount increases over time, while the amount of cell-associatedtoxin decreases. The decrease in intracellular toxin is not inhibitedby the proteasomal inhibitor MG132, and no colocalization ofAIP56 with LAMP-1 was detected in our experiments, suggestingthat this decrease results not from proteasomal or lysosomal deg-radation but rather from recycling of the toxin back into the ex-tracellular medium. (iii) The appearance of recycled AIP56 in theextracellular medium is dependent on PI3K, a kinase that plays akey role in the endocytic recycling of several molecules, includingtransferrin (84, 85) and AT1 angiotensin receptor (52).
For several recycled proteins, including transferrin receptor,
two alternative recycling pathways have been described: a fast di-rect route from early endosomes or earlier compartments to theplasma membrane and a slower route involving the transport ofcargo proteins to the endocytic recycling compartment (ERC) be-fore transport to the plasma membrane (49, 86). The detection ofrecycled AIP56 after a 2-min chase suggests that part of the toxinfollows the fast recycling route, while the observed colocalizationwith Alexa 647-transferrin in a perinuclear compartment at laterstages suggests that a pool of the toxin is recycled from the ERC(slow recycling). Based on the observation that AIP56 recyclingrequires PI3K activity, we speculate that the fast recycling routeplays a major role, since it is known that PI3K is mainly involved inthe fast recycling (52, 54, 85), with very little evidence of its in-volvement in slow recycling (87).
The efficient sorting and recycling of several cargos/receptorsinternalized by receptor-mediated endocytosis require exposureof ligand-receptor complexes to low endosomal pH (52, 60). Fre-quently, this leads to the dissociation of the ligands from the re-ceptors (52, 60). The membrane-bound receptors are recycled,while the released ligands concentrate in the vesicular parts of theearly endosomes and are then routed through a microtubule-de-pendent maturation process to late endosomes/lysosomes (49).Our results suggest that endosomal acidification is dispensable forthe recycling of AIP56, because the appearance of recycled toxininto the extracellular medium was not affected by inhibition of thevacuolar ATPase pump with ConcA.
Although the release of endocytosed toxins back into the ex-tracellular medium has been previously described for the planttoxin ricin (88–90) and for anthrax toxin (91), neither of thosetoxins has been reported to follow the endocytic recycling route(91, 92). The release of endocytosed ricin seems to occur througha mechanism involving exocytosis of multivesicular bodies (89),allowing intoxication of bystander cell populations (90). In thecase of anthrax, it has been demonstrated that the toxin is deliv-ered into the extracellular medium in exosomes, contributing toprolonged toxicity and possibly to transmission of the toxin todistant cells (91). During this work, several attempts were madeto detect activity of recycled AIP56. These consisted of large-scalerecycling assays in which we recovered 48 ml of extracellular me-dium containing recycled AIP56 from eight 24-well plates andtried to concentrate the recycled toxin using either centrifugaldevices or an immunoprecipitation protocol. However, we wereunable to detect recycled toxin activity. This was likely due tosignificant losses of toxin during the procedures, since Westernblotting of the concentrates revealed that the amount of AIP56recovered was much less than expected and insufficient to per-form activity tests. Therefore, the relevance of the putative recy-cling pathway and of recycled AIP56 for pathogenesis remains tobe investigated.
Although monitoring the intracellular localization of fluores-cence-labeled AIP56 by microscopy allowed imaging of some ofthe details of its intracellular trafficking, the low sensitivity re-stricts its usefulness for detecting translocation of the toxin intothe cytosol. Nevertheless, several biochemical findings indicatethat endosome acidification and low-pH-triggered conforma-tional changes are involved in AIP56 translocation: (i) the arrivalof AIP56 at the cytosol requires endosome acidification, sinceAIP56-dependent p65 cleavage was prevented by inhibitors of thevacuolar ATPase pump (61); (ii) at pH 5.5 or lower, AIP56 be-comes more hydrophobic and is able to interact with artificial
FIG 8 An acidic pulse can drive translocation of AIP56 across the cell mem-brane. mBMDM treated with ConcA (that was maintained during the entireassay to inhibit normal toxin uptake) or left untreated were incubated on icewith recombinant AIP56. After 30 min, medium was removed, and cells wereincubated for 1 h at 37°C with buffers at the indicated pH values followed byincubation in culture medium at pH 7.4 for 2 h. Cleavage of p65 was assessedby Western blotting (chromogenic detection). The graph shows the quantifi-cation of the blots (mean � SD; three independent experiments). Loadingcorrection was achieved by dividing the density of p65 band by the respectivedensity of Ponceau S. The results were divided by the same constant in order toset to 1 the mean of the control (mock-treated cells, pH 7.0). For each pH,significance of differences was assessed by one-way ANOVA (P 0.01). Pvalues for the comparisons with cells treated only with toxin were calculatedusing Tukey’s HSD multiple-comparison test and are indicated in the graph.
Pereira et al.
5282 iai.asm.org Infection and Immunity
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

lipid bilayer membranes; (iii) a low-pH pulse promotes translo-cation of cell surface-bound AIP56 across the cytoplasmic mem-brane into the cytosol, similar to what has been reported forseveral toxins that translocate from endosomes through a pH-dependent mechanism (31, 32, 68). Since AIP56 reaches early en-dosomes and then follows the recycling route, translocation intothe cytosol likely occurs from early and/or recycling endosomes.Early endosomes are the first acidic compartment encountered bythe toxin upon endocytosis, and therefore, it is probable that atleast the beginning of the translocation process occurs in this com-partment. The fact that in less than 30 min, when most of the toxinis still located in EEA-1-positive vesicles, an amount of AIP56sufficient to intoxicate the cells is already at the cytosolic compart-ment together with the fact that inhibition of PI3K activity doesnot inhibit AIP56 toxicity fully agrees with the hypothesis thatearly endosomes are the first organelle along the recycling routefrom which the toxin translocates into the cytosol. The observa-tion that microtubule depolymerization does not affect AIP56-mediated p65 cleavage is compatible with this interpretation andconfirms that late endosomes/lysosomes are not involved inAIP56 intoxication, as it is known that maturation of early endo-somes into late endosomes is a microtubule dependent process(93).
In conclusion, with this study we elucidated important aspectsof AIP56 trafficking, and we show that these are conserved be-tween fish and mammalian cells. Our findings reveal that AIP56 isa short-trip toxin that follows two intracellular pathways that areat least partially independent: the intoxication pathway (that re-quires acidification of the endosomes and involves conforma-tional changes in the toxin molecule but is independent of PI3Kactivity) and the recycling pathway (independent of endosomeacidification but requiring PI3K activity). The evidence that a sig-nificant pool of endocytosed AIP56 follows the recycling pathwayand is released into the extracellular medium suggests a novelintracellular route among the ones already established for bacte-rial AB toxins. Additional studies are required to determinewhether AIP56 recycling has any relevance for P. damselae subsp.piscicida pathogenesis.
ACKNOWLEDGMENTS
We are grateful to Cesare Montecucco (Dipartimento di Scienze Biomedi-che Sperimentali dell’Úniversità di Padova and Instituto di Neuroscienzedel CNR, Padua, Italy) for providing the PA and to Didier Cabanes(Group of Molecular Microbiology, Instituto de Biologia Molecular eCelular, Universidade do Porto, Porto, Portugal) for providing the Liste-ria monocytogenes.
Liliana M. G. Pereira and Daniela S. Silva were funded by Fundaçãopara a Ciência e a Tecnologia (FCT) fellowships SFRH/BD/43501/2008and SFRH/BD/35865/2007, respectively, financed by POPH-QREN andcofunded by Fundo Social Europeu and MCTES. Ana do Vale was sup-ported by Programa Ciência, funded by POPH–QREN, Tipologia 4.2–Promoção do Emprego Científico, cofunded by Fundo Social Europeu,and national funding from MCTES. This work was supported byFEDER funds through the Operational Competitiveness Programme(COMPETE) and national funds through FCT under projects FCOMP-01-0124-FEDER-022718 (PEst-C/SAU/LA0002/2011) and FCOMP-01-0124-FEDER-009501 (PTDC/CVT/099544/2008).
REFERENCES1. do Vale A, Silva MT, dos Santos NM, Nascimento DS, Reis-Rodrigues
P, Costa-Ramos C, Ellis AE, Azevedo JE. 2005. AIP56, a novel plasmid-encoded virulence factor of Photobacterium damselae subsp. piscicida with
apoptogenic activity against sea bass macrophages and neutrophils. Mol.Microbiol. 58:1025–1038. http://dx.doi.org/10.1111/j.1365-2958.2005.04893.x.
2. Romalde JL. 2002. Photobacterium damselae subsp. piscicida: an inte-grated view of a bacterial fish pathogen. Int. Microbiol. 5:3–9. http://dx.doi.org/10.1007/s10123-002-0051-6.
3. Barnes AC, dos Santos NM, Ellis AE. 2005. Update on bacterial vaccines:Photobacterium damselae subsp. piscicida. Dev. Biol. (Basel) 121:75– 84.
4. Magariños B, Romalde JL, Bandin I, Fouz B, Toranzo AE. 1992.Phenotypic, antigenic, and molecular characterization of Pasteurella pis-cicida strains isolated from fish. Appl. Environ. Microbiol. 58:3316 –3322.
5. Thune RL, Stanley LA, Cooper RK. 1993. Pathogenesis of gram-negativebacterial infections in warmwater fish. Annu. Rev. Fish Dis. 3:37– 68. http://dx.doi.org/10.1016/0959-8030(93)90028-A.
6. Hawke JP, Plakas SM, Minton RV, McPhearson RM, Snider TG, Gua-rino AM. 1987. Fish pasteurellosis of cultured striped bass (Morone saxa-tilis) in coastal Alabama. Aquaculture 65:193–204. http://dx.doi.org/10.1016/0044-8486(87)90231-6.
7. Magariños B, Santos Y, Romalde JL, Rivas C, Barja JL, Toranzo AE.1992. Pathogenic activities of live cells and extracellular products of thefish pathogen Pasteurella piscicida. J. Gen. Microbiol. 138:2491–2498.http://dx.doi.org/10.1099/00221287-138-12-2491.
8. Noya M, Magariños B, Toranzo AE, Lamas J. 1995. Sequential pathologyof experimental pasteurellosis in gilthead seabream Sparus aurata. A light-and electron-microscopic study. Dis. Aquat. Org. 21:177–186.
9. Toranzo AE, Barreiro Sn Casal JF, Figueras A, Magarinos B, Barja JL.1991. Pasteurellosis in cultured gilthead seabream (Sparus aurata): firstreport in Spain. Aquaculture 99:1–15. http://dx.doi.org/10.1016/0044-8486(91)90284-E.
10. Poulos C, Bakopoulos V, Zolota V, Dimitriadis GJ. 2004. Histopatho-logical findings after sea bass (Dicentrarchus labrax L.) exposure to extra-cellular products of Photobacterium damselae subsp. piscicida producedin vivo. Aquacult. Res. 35:931–936. http://dx.doi.org/10.1111/j.1365-2109.2004.01086.x.
11. do Vale A, Marques F, Silva MT. 2003. Apoptosis of sea bass (Dicentrarchuslabrax L.) neutrophils and macrophages induced by experimental infectionwith Photobacterium damselae subsp. piscicida. Fish Shellfish Immunol. 15:129–144. http://dx.doi.org/10.1016/S1050-4648(02)00144-4.
12. do Vale A, Costa-Ramos C, Silva A, Silva DS, Gartner F, dos SantosNM, Silva MT. 2007. Systemic macrophage and neutrophil destruction bysecondary necrosis induced by a bacterial exotoxin in a Gram-negativesepticaemia. Cell. Microbiol. 9:988 –1003. http://dx.doi.org/10.1111/j.1462-5822.2006.00846.x.
13. Silva MT, do Vale A, dos Santos NM. 2008. Secondary necrosis inmulticellular animals: an outcome of apoptosis with pathogenic impli-cations. Apoptosis 13:463– 482. http://dx.doi.org/10.1007/s10495-008-0187-8.
14. Baruch K, Gur-Arie L, Nadler C, Koby S, Yerushalmi G, Ben-NeriahY, Yogev O, Shaulian E, Guttman C, Zarivach R, Rosenshine I. 2011.Metalloprotease type III effectors that specifically cleave JNK and NF-kappaB. EMBO J. 30:221–231. http://dx.doi.org/10.1038/emboj.2010.297.
15. Clarke SC. 2001. Diarrhoeagenic Escherichia coli—an emerging prob-lem? Diagn. Microbiol. Infect. Dis. 41:93–98. http://dx.doi.org/10.1016/S0732-8893(01)00303-0.
16. Silva DS, Pereira LM, Moreira AR, Ferreira-da-Silva F, Brito RM,Faria TQ, Zornetta I, Montecucco C, Oliveira P, Azevedo JE, PereiraPJ, Macedo-Ribeiro S, do Vale A, dos Santos NM. 2013. The apop-togenic toxin AIP56 is a metalloprotease A-B toxin that cleaves NF-kappaB P65. PLoS Pathog. 9:e1003128. http://dx.doi.org/10.1371/journal.ppat.1003128.
17. Gilmore TD, Wolenski FS. 2012. NF-kappaB: where did it come fromand why? Immunol. Rev. 246:14 –35. http://dx.doi.org/10.1111/j.1600-065X.2012.01096.x.
18. Watson P, Spooner RA. 2006. Toxin entry and trafficking in mammaliancells. Adv. Drug Deliv. Rev. 58:1581–1596. http://dx.doi.org/10.1016/j.addr.2006.09.016.
19. Papini E, Rappuoli R, Murgia M, Montecucco C. 1993. Cell penetrationof diphtheria toxin. Reduction of the interchain disulfide bridge is therate-limiting step of translocation in the cytosol. J. Biol. Chem. 268:1567–1574.
20. Contamin S, Galmiche A, Doye A, Flatau G, Benmerah A, Boquet P.2000. The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endo-
Intracellular Trafficking of AIP56
December 2014 Volume 82 Number 12 iai.asm.org 5283
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

cytosed by a clathrin-independent mechanism and enters the cytosol by anacidic-dependent membrane translocation step. Mol. Biol. Cell 11:1775–1787. http://dx.doi.org/10.1091/mbc.11.5.1775.
21. Puhar A, Johnson EA, Rossetto O, Montecucco C. 2004. Comparison ofthe pH-induced conformational change of different clostridial neurotox-ins. Biochem. Biophys. Res. Commun. 319:66 –71. http://dx.doi.org/10.1016/j.bbrc.2004.04.140.
22. Sandvig K, van Deurs B. 2005. Delivery into cells: lessons learned fromplant and bacterial toxins. Gene Ther. 12:865– 872. http://dx.doi.org/10.1038/sj.gt.3302525.
23. Collier RJ. 2009. Membrane translocation by anthrax toxin. Mol. AspectsMed. 30:413– 422. http://dx.doi.org/10.1016/j.mam.2009.06.003.
24. Edelhoch H. 1967. Spectroscopic determination of tryptophan and ty-rosine in proteins. Biochemistry 6:1948 –1954. http://dx.doi.org/10.1021/bi00859a010.
25. Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. 1995. How to measureand predict the molar absorption coefficient of a protein. Protein Sci.4:2411–2423. http://dx.doi.org/10.1002/pro.5560041120.
26. Gomes MS, Sousa Fernandes S, Cordeiro JV, Silva Gomes S, Vieira A,Appelberg R. 2008. Engagement of Toll-like receptor 2 in mouse macro-phages infected with Mycobacterium avium induces non-oxidative andTNF-independent anti-mycobacterial activity. Eur. J. Immunol. 38:2180 –2189. http://dx.doi.org/10.1002/eji.200737954.
27. Costa-Ramos C, Vale A, Ludovico P, Dos Santos NM, Silva MT. 2011.The bacterial exotoxin AIP56 induces fish macrophage and neutrophilapoptosis using mechanisms of the extrinsic and intrinsic pathways. FishShellfish Immunol. 30:173–181. http://dx.doi.org/10.1016/j.fsi.2010.10.007.
28. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietz-sch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, WhiteDJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: anopen-source platform for biological-image analysis. Nat. Methods 9:676 –682. http://dx.doi.org/10.1038/nmeth.2019.
29. Paul G, Cardinale J, Sbalzarini IF. 2013. Coupling image restoration andsegmentation: a generalized linear model/Bregman perspective. Int. J.Comput. Vis. 104:69 –93. http://dx.doi.org/10.1007/s11263-013-0615-2.
30. Rizk A, Paul G, Incardona P, Bugarski M, Mansouri M, Niemann A,Ziegler U, Berger P, Sbalzarini IF. 2014. Segmentation and quantifica-tion of subcellular structures in fluorescence microscopy images usingSquassh. Nat. Protoc. 9:586 –596. http://dx.doi.org/10.1038/nprot.2014.037.
31. Draper RK, Simon MI. 1980. The entry of diphtheria toxin into themammalian cell cytoplasm: evidence for lysosomal involvement. J. CellBiol. 87:849 – 854. http://dx.doi.org/10.1083/jcb.87.3.849.
32. Sandvig K, Olsnes S. 1980. Diphtheria toxin entry into cells is facilitatedby low pH. J. Cell Biol. 87:828 – 832. http://dx.doi.org/10.1083/jcb.87.3.828.
33. Benz R, Janko K, Boos W, Lauger P. 1978. Formation of large, ion-permeable membrane channels by the matrix protein (porin) of Esche-richia coli. Biochim. Biophys. Acta 511:305–319. http://dx.doi.org/10.1016/0005-2736(78)90269-9.
34. Qa’Dan M, Spyres LM, Ballard JD. 2000. pH-induced conformationalchanges in Clostridium difficile toxin B. Infect. Immun. 68:2470 –2474.http://dx.doi.org/10.1128/IAI.68.5.2470-2474.2000.
35. Laemmli UK. 1970. Cleavage of structural proteins during the assembly ofthe head of bacteriophage T4. Nature 227:680 – 685. http://dx.doi.org/10.1038/227680a0.
36. Yen H, Ooka T, Iguchi A, Hayashi T, Sugimoto N, Tobe T. 2010. NleC,a type III secretion protease, compromises NF-kappaB activation by tar-geting p65/RelA. PLoS Pathog. 6:e1001231. http://dx.doi.org/10.1371/journal.ppat.1001231.
37. Ferguson SM, De Camilli P. 2012. Dynamin, a membrane-remodellingGTPase. Nat. Rev. Mol. Cell Biol. 13:75– 88. http://dx.doi.org/10.1038/nrm3266.
38. Wang LH, Rothberg KG, Anderson RG. 1993. Mis-assembly of clathrinlattices on endosomes reveals a regulatory switch for coated pit formation.J. Cell Biol. 123:1107–1117. http://dx.doi.org/10.1083/jcb.123.5.1107.
39. Kirchhausen T, Macia E, Pelish HE. 2008. Use of dynasore, the smallmolecule inhibitor of dynamin, in the regulation of endocytosis. MethodsEnzymol. 438:77–93. http://dx.doi.org/10.1016/S0076-6879(07)38006-3.
40. Doherty GJ, McMahon HT. 2009. Mechanisms of endocytosis. Annu.Rev. Biochem. 78:857–902. http://dx.doi.org/10.1146/annurev.biochem.78.081307.110540.
41. Sampath P, Pollard TD. 1991. Effects of cytochalasin, phalloidin, and pHon the elongation of actin filaments. Biochemistry 30:1973–1980. http://dx.doi.org/10.1021/bi00221a034.
42. Cassimeris LU, Wadsworth P, Salmon ED. 1986. Dynamics of microtu-bule depolymerization in monocytes. J. Cell Biol. 102:2023–2032. http://dx.doi.org/10.1083/jcb.102.6.2023.
43. Marcil A, Gadoury C, Ash J, Zhang J, Nantel A, Whiteway M. 2008.Analysis of PRA1 and its relationship to Candida albicans-macrophageinteractions. Infect. Immun. 76:4345– 4358. http://dx.doi.org/10.1128/IAI.00588-07.
44. Casbon AJ, Allen LA, Dunn KW, Dinauer MC. 2009. MacrophageNADPH oxidase flavocytochrome B localizes to the plasma membraneand Rab11-positive recycling endosomes. J. Immunol. 182:2325–2339.http://dx.doi.org/10.4049/jimmunol.0803476.
45. Cox D, Lee DJ, Dale BM, Calafat J, Greenberg S. 2000. A Rab11-containing rapidly recycling compartment in macrophages that promotesphagocytosis. Proc. Natl. Acad. Sci. U. S. A. 97:680 – 685. http://dx.doi.org/10.1073/pnas.97.2.680.
46. Halaas O, Steigedal M, Haug M, Awuh JA, Ryan L, Brech A, Sato S,Husebye H, Cangelosi GA, Akira S, Strong RK, Espevik T, Flo TH.2010. Intracellular Mycobacterium avium intersect transferrin in theRab11(�) recycling endocytic pathway and avoid lipocalin 2 trafficking tothe lysosomal pathway. J. Infect. Dis. 201:783–792. http://dx.doi.org/10.1086/650493.
47. Stanley AC, Wong CX, Micaroni M, Venturato J, Khromykh T, StowJL, Lacy P. 2014. The Rho GTPase Rac1 is required for recycling endo-some-mediated secretion of TNF in macrophages. Immunol. Cell Biol.92:275–286. http://dx.doi.org/10.1038/icb.2013.90.
48. Wainszelbaum MJ, Proctor BM, Pontow SE, Stahl PD, Barbieri MA.2006. IL4/PGE2 induction of an enlarged early endosomal compartmentin mouse macrophages is Rab5-dependent. Exp. Cell Res. 312:2238 –2251.http://dx.doi.org/10.1016/j.yexcr.2006.03.025.
49. Maxfield FR, McGraw TE. 2004. Endocytic recycling. Nat. Rev. Mol. CellBiol. 5:121–132. http://dx.doi.org/10.1038/nrm1315.
50. Foster FM, Traer CJ, Abraham SM, Fry MJ. 2003. The phosphoinositide(PI) 3-kinase family. J. Cell Sci. 116:3037–3040. http://dx.doi.org/10.1242/jcs.00609.
51. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B.2010. The emerging mechanisms of isoform-specific PI3K signalling. Nat.Rev. Mol. Cell Biol. 11:329 –341. http://dx.doi.org/10.1038/nrm2882.
52. Hunyady L, Baukal AJ, Gaborik Z, Olivares-Reyes JA, Bor M, SzaszakM, Lodge R, Catt KJ, Balla T. 2002. Differential PI 3-kinase dependenceof early and late phases of recycling of the internalized AT1 angiotensinreceptor. J. Cell Biol. 157:1211–1222. http://dx.doi.org/10.1083/jcb.200111013.
53. Lindmo K, Stenmark H. 2006. Regulation of membrane traffic by phos-phoinositide 3-kinases. J. Cell Sci. 119:605– 614. http://dx.doi.org/10.1242/jcs.02855.
54. Naughtin MJ, Sheffield DA, Rahman P, Hughes WE, Gurung R, StowJL, Nandurkar HH, Dyson JM, Mitchell CA. 2010. The myotubularinphosphatase MTMR4 regulates sorting from early endosomes. J. Cell Sci.123:3071–3083. http://dx.doi.org/10.1242/jcs.060103.
55. Raiborg C, Schink KO, Stenmark H. 2013. Class III phosphatidylinositol3-kinase and its catalytic product PtdIns3P in regulation of endocyticmembrane traffic. FEBS J. 280:2730 –2742. http://dx.doi.org/10.1111/febs.12116.
56. Carpentier S, N=Kuli F, Grieco G, Van Der Smissen P, Janssens V,Emonard H, Bilanges B, Vanhaesebroeck B, Gaide Chevronnay HP,Pierreux CE, Tyteca D, Courtoy PJ. 2013. Class III phosphoinositide3-kinase/VPS34 and dynamin are critical for apical endocytic recycling.Traffic 14:933–948. http://dx.doi.org/10.1111/tra.12079.
57. Knight ZA, Chiang GG, Alaimo PJ, Kenski DM, Ho CB, Coan K,Abraham RT, Shokat KM. 2004. Isoform-specific phosphoinositide 3-ki-nase inhibitors from an arylmorpholine scaffold. Bioorg. Med. Chem.12:4749 – 4759. http://dx.doi.org/10.1016/j.bmc.2004.06.022.
58. Vlahos CJ, Matter WF, Hui KY, Brown RF. 1994. A specific inhibitor ofphosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269:5241–5248.
59. Laird MH, Rhee SH, Perkins DJ, Medvedev AE, Piao W, Fenton MJ,Vogel SN. 2009. TLR4/MyD88/PI3K interactions regulate TLR4 signal-ing. J. Leukoc. Biol. 85:966 –977. http://dx.doi.org/10.1189/jlb.1208763.
60. Jovic M, Sharma M, Rahajeng J, Caplan S. 2010. The early endosome: a
Pereira et al.
5284 iai.asm.org Infection and Immunity
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from

busy sorting station for proteins at the crossroads. Histol. Histopathol.25:99 –112.
61. Huss M, Wieczorek H. 2009. Inhibitors of V-ATPases: old and newplayers. J. Exp. Biol. 212:341–346. http://dx.doi.org/10.1242/jeb.024067.
62. Falnes PO, Sandvig K. 2000. Penetration of protein toxins into cells.Curr. Opin. Cell Biol. 12:407– 413. http://dx.doi.org/10.1016/S0955-0674(00)00109-5.
63. Montecucco C, Papini E. 1995. Cell penetration of bacterial proteintoxins. Trends Microbiol. 3:165–167. http://dx.doi.org/10.1016/S0966-842X(00)88911-8.
64. Sachdev GP, Brownstein AD, Fruton JS. 1973. N-methyl-2-anilinonaphthalene-6-sulfonyl peptides as fluorescent probes for pepsin-substrate interaction. J. Biol. Chem. 248:6292– 6299.
65. Finkelstein A. 1994. The channel formed in planar lipid bilayers by theprotective antigen component of anthrax toxin. Toxicology 87:29 – 41.http://dx.doi.org/10.1016/0300-483X(94)90153-8.
66. Schmid A, Benz R, Just I, Aktories K. 1994. Interaction of Clostridiumbotulinum C2 toxin with lipid bilayer membranes. Formation of cation-selective channels and inhibition of channel function by chloroquine. J.Biol. Chem. 269:16706 –16711.
67. Fahrer J, Kuban J, Heine K, Rupps G, Kaiser E, Felder E, Benz R, BarthH. 2010. Selective and specific internalization of clostridial C3 ADP-ribosyltransferases into macrophages and monocytes. Cell. Microbiol. 12:233–247. http://dx.doi.org/10.1111/j.1462-5822.2009.01393.x.
68. Barth H, Blocker D, Behlke J, Bergsma-Schutter W, Brisson A, Benz R,Aktories K. 2000. Cellular uptake of Clostridium botulinum C2 toxinrequires oligomerization and acidification. J. Biol. Chem. 275:18704 –18711. http://dx.doi.org/10.1074/jbc.M000596200.
69. Giesemann T, Jank T, Gerhard R, Maier E, Just I, Benz R, AktoriesK. 2006. Cholesterol-dependent pore formation of Clostridium difficiletoxin A. J. Biol. Chem. 281:10808 –10815. http://dx.doi.org/10.1074/jbc.M512720200.
70. DiDonato JA, Mercurio F, Karin M. 2012. NF-kappaB and the linkbetween inflammation and cancer. Immunol. Rev. 246:379 – 400. http://dx.doi.org/10.1111/j.1600-065X.2012.01099.x.
71. Li Q, Verma IM. 2002. NF-kappaB regulation in the immune system. Nat.Rev. Immunol. 2:725–734. http://dx.doi.org/10.1038/nri910.
72. Wong ET, Tergaonkar V. 2009. Roles of NF-kappaB in health and dis-ease: mechanisms and therapeutic potential. Clin. Sci. (Lond.) 116:451–465. http://dx.doi.org/10.1042/CS20080502.
73. Tana LR, Ho M, Wilson BA. 2013. Determinants of pH-dependent modu-lation of translocation in dermonecrotic G-protein-deamidating toxins. Tox-ins (Basel) 5:1167–1179. http://dx.doi.org/10.3390/toxins5061167.
74. Collier RJ. 2001. Understanding the mode of action of diphtheria toxin: aperspective on progress during the 20th century. Toxicon 39:1793–1803.http://dx.doi.org/10.1016/S0041-0101(01)00165-9.
75. Turton K, Chaddock JA, Acharya KR. 2002. Botulinum and tetanusneurotoxins: structure, function and therapeutic utility. Trends Biochem.Sci. 27:552–558. http://dx.doi.org/10.1016/S0968-0004(02)02177-1.
76. Damke H, Baba T, Warnock DE, Schmid SL. 1994. Induction of mutantdynamin specifically blocks endocytic coated vesicle formation. J. CellBiol. 127:915–934. http://dx.doi.org/10.1083/jcb.127.4.915.
77. Schmid SL, McNiven MA, De Camilli P. 1998. Dynamin and its partners:a progress report. Curr. Opin. Cell Biol. 10:504 –512. http://dx.doi.org/10.1016/S0955-0674(98)80066-5.
78. Praefcke GJ, McMahon HT. 2004. The dynamin superfamily: universalmembrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol.5:133–147. http://dx.doi.org/10.1038/nrm1313.
79. McMahon HT, Boucrot E. 2011. Molecular mechanism and physiologi-cal functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol.12:517–533. http://dx.doi.org/10.1038/nrm3151.
80. Fujimoto LM, Roth R, Heuser JE, Schmid SL. 2000. Actin assembly playsa variable, but not obligatory role in receptor-mediated endocytosis inmammalian cells. Traffic 1:161–171. http://dx.doi.org/10.1034/j.1600-0854.2000.010208.x.
81. Moya M, Dautry-Varsat A, Goud B, Louvard D, Boquet P. 1985.Inhibition of coated pit formation in Hep2 cells blocks the cytotoxicity ofdiphtheria toxin but not that of ricin toxin. J. Cell Biol. 101:548 –559.http://dx.doi.org/10.1083/jcb.101.2.548.
82. Abrami L, Bischofberger M, Kunz B, Groux R, van der Goot FG. 2010.Endocytosis of the anthrax toxin is mediated by clathrin, actin and uncon-ventional adaptors. PLoS Pathog. 6:e1000792. http://dx.doi.org/10.1371/journal.ppat.1000792.
83. Sandvig K, Grimmer S, Lauvrak SU, Torgersen ML, Skretting G, vanDeurs B, Iversen TG. 2002. Pathways followed by ricin and Shiga toxininto cells. Histochem. Cell Biol. 117:131–141. http://dx.doi.org/10.1007/s00418-001-0346-2.
84. Spiro DJ, Boll W, Kirchhausen T, Wessling-Resnick M. 1996. Wort-mannin alters the transferrin receptor endocytic pathway in vivo and invitro. Mol. Biol. Cell 7:355–367. http://dx.doi.org/10.1091/mbc.7.3.355.
85. van Dam EM, Ten Broeke T, Jansen K, Spijkers P, Stoorvogel W. 2002.Endocytosed transferrin receptors recycle via distinct dynamin and phos-phatidylinositol 3-kinase-dependent pathways. J. Biol. Chem. 277:48876 –48883. http://dx.doi.org/10.1074/jbc.M206271200.
86. Grant BD, Donaldson JG. 2009. Pathways and mechanisms of endocyticrecycling. Nat. Rev. Mol. Cell Biol. 10:597– 608. http://dx.doi.org/10.1038/nrm2755.
87. Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM,Parton RG, Stenmark H. 2000. Localization of phosphatidylinositol3-phosphate in yeast and mammalian cells. EMBO J. 19:4577– 4588. http://dx.doi.org/10.1093/emboj/19.17.4577.
88. Sandvig K, Olsnes S. 1979. Effect of temperature on the uptake, excretionand degradation of abrin and ricin by HeLa cells. Exp. Cell Res. 121:15–25.http://dx.doi.org/10.1016/0014-4827(79)90439-7.
89. McIntosh D, Timar J, Davies AJ. 1990. The intracellular movement andcycling of ricin. Eur. J. Cell Biol. 52:77– 86.
90. McIntosh DP, Edwards DC, Davies AJ. 1984. Transfer of ricin toxicityby spleen cells. Toxicon 22:293–299. http://dx.doi.org/10.1016/0041-0101(84)90029-1.
91. Abrami L, Brandi L, Moayeri M, Brown MJ, Krantz BA, Leppla SH, vander Goot FG. 2013. Hijacking multivesicular bodies enables long-termand exosome-mediated long-distance action of anthrax toxin. Cell. Rep.5:986 –996. http://dx.doi.org/10.1016/j.celrep.2013.10.019.
92. Moisenovich M, Tonevitsky A, Maljuchenko N, Kozlovskaya N, AgapovI, Volknandt W, Bereiter-Hahn J. 2004. Endosomal ricin transport:involvement of Rab4- and Rab5-positive compartments. Histochem. CellBiol. 121:429 – 439. http://dx.doi.org/10.1007/s00418-004-0652-6.
93. Huotari J, Helenius A. 2011. Endosome maturation. EMBO J. 30:3481–3500. http://dx.doi.org/10.1038/emboj.2011.286.
Intracellular Trafficking of AIP56
December 2014 Volume 82 Number 12 iai.asm.org 5285
on June 13, 2016 by guesthttp://iai.asm
.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















