
Interleukin6 overexpression induces pulmonary hypertension
38
Interleukin-6 Overexpression Induces Pulmonary Hypertension M. Kathryn Steiner, Olga L. Syrkina, Narasaish Kolliputi, Eugene J. Mark, Charles A. Hales, Aaron B. Waxman Abstract—Inflammatory cytokine interleukin (IL)-6 is elevated in the serum and lungs of patients with pulmonary artery hypertension (PAH). Several animal models of PAH cite the potential role of inflammatory mediators. We investigated role of IL-6 in the pathogenesis of pulmonary vascular disease. Indices of pulmonary vascular remodeling were measured in lung-specific IL-6 – overexpressing transgenic mice (Tg ) and compared to wild-type (Tg ) controls in both normoxic and chronic hypoxic conditions. The Tg mice exhibited elevated right ventricular systolic pressures and right ventricular hypertrophy with corresponding pulmonary vasculopathic changes, all of which were exacerbated by chronic hypoxia. IL-6 overexpression increased muscularization of the proximal arterial tree, and hypoxia enhanced this effect. It also reproduced the muscularization and proliferative arteriopathy seen in the distal arteriolar vessels of PAH patients. The latter was characterized by the formation of occlusive neointimal angioproliferative lesions that worsened with hypoxia and were composed of endothelial cells and T-lymphocytes. IL-6 –induced arteriopathic changes were accompanied by activation of proangiogenic factor, vascular endothelial growth factor, the proproliferative kinase extracellular signal-regulated kinase, proproliferative transcription factors c-MYC and MAX, and the antiapoptotic proteins survivin and Bcl-2 and downregulation of the growth inhibitor transforming growth factor- and proapoptotic kinases JNK and p38. These findings suggest that IL-6 promotes the development and progression of pulmonary vascular remodeling and PAH through proproliferative antiapoptotic mechanisms. (Circ Res. 2009;104:236-244.) Key Words: interleukin-6 pulmonary artery hypertension proliferation P ulmonary vascular remodeling is associated with in- creased pulmonary vascular resistance, pulmonary ar- tery hypertension (PAH), and right heart failure. Advanced PAH is characterized by arteriopathy, which includes muscularization of distal pulmonary arterioles, concentric intimal thickening, and obstruction of the vascular lumen by proliferating endothelial cells to form plexiform le- sions. 1 Evidence suggests that PAH is associated with genetic perturbations favoring cellular growth, prolifera- tion, and angiogenesis 2 and inhibitors of apoptosis, previ- ously thought to be only expressed in cancer cells, pro- moting a proliferative cellular phenotype, resulting in pulmonary vascular remodeling in PAH. 3,4 The histopatho- logic features and known genetic susceptibilities of this condition have led to the hypothesis that PAH arises from hyperproliferation of pulmonary artery smooth muscle cells (PASMCs) and endothelial cells (PAECs). In addition to the formation of proliferative neointimal lesions and muscularization of the pulmonary vascular bed, perivascular inflammatory cell infiltrates are also present in advanced human cases of PAH. These infiltrates consist of T cells, B cells, and macrophages, suggesting that cytokines and growth factors associated with these inflammatory cells may be promoting PAEC and PASMC hyperproliferation. 5 The proinflammatory cytokine inter- leukin (IL)-6 is consistently increased in the serum and lungs 6–8 of patients with idiopathic PAH and in inflamma- tory diseases 6,9 –11 that are associated with PAH. In addi- tion, Kaposi sarcoma–associated herpes virus, which may cause PAH in human immunodeficiency virus–negative Castleman’s disease, encodes a constitutively active form of IL-6, 12 resulting in unregulated cell growth and escape from host antitumor defenses. Furthermore, unchecked production of IL-6 in tissues leading to chronic inflamma- tion has exhibited a strong association with many can- cers. 13 Given mounting evidence for the role of inflamma- tion and cancer-like mechanisms in the pathogenesis of PAH, we investigated whether IL-6 promotes the develop- ment of pulmonary arteriopathy and consequent PAH. We show that lung-specific overexpression of IL-6 in mice replicates the pathological lesions observed in advanced PAH, including both distal arteriolar muscularization and plexogenic arteriopathy, and leads to increased pulmonary vascular resistance (PVR) and PAH. At the cellular and Original received June 23, 2008; revision received November 27, 2008; accepted December 2, 2008. From the Pulmonary Critical Care Unit (M.K.S., O.L.S., N.K., C.A.H., A.B.W.), Department of Medicine; and Department of Pathology (E.J.M.), Massachusetts General Hospital, Harvard Medical School, Boston. Correspondence to M. Kathryn Steiner, Division of Pulmonary Critical Care Medicine, University of Massachusetts Memorial Medical Center, 55 Lake Ave North, Worcester, MA 01655. E-mail [email protected] © 2009 American Heart Association, Inc. Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.108.182014 236 by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from by guest on August 21, 2015 http://circres.ahajournals.org/ Downloaded from
-
Upload
hms-harvard -
Category
Documents
-
view
0 -
download
0
Transcript of Interleukin6 overexpression induces pulmonary hypertension

Interleukin-6 Overexpression InducesPulmonary Hypertension
M. Kathryn Steiner, Olga L. Syrkina, Narasaish Kolliputi, Eugene J. Mark,Charles A. Hales, Aaron B. Waxman
Abstract—Inflammatory cytokine interleukin (IL)-6 is elevated in the serum and lungs of patients with pulmonary arteryhypertension (PAH). Several animal models of PAH cite the potential role of inflammatory mediators. We investigatedrole of IL-6 in the pathogenesis of pulmonary vascular disease. Indices of pulmonary vascular remodeling weremeasured in lung-specific IL-6–overexpressing transgenic mice (Tg�) and compared to wild-type (Tg�) controls inboth normoxic and chronic hypoxic conditions. The Tg� mice exhibited elevated right ventricular systolic pressures andright ventricular hypertrophy with corresponding pulmonary vasculopathic changes, all of which were exacerbated bychronic hypoxia. IL-6 overexpression increased muscularization of the proximal arterial tree, and hypoxia enhanced thiseffect. It also reproduced the muscularization and proliferative arteriopathy seen in the distal arteriolar vessels of PAHpatients. The latter was characterized by the formation of occlusive neointimal angioproliferative lesions that worsenedwith hypoxia and were composed of endothelial cells and T-lymphocytes. IL-6–induced arteriopathic changes wereaccompanied by activation of proangiogenic factor, vascular endothelial growth factor, the proproliferative kinaseextracellular signal-regulated kinase, proproliferative transcription factors c-MYC and MAX, and the antiapoptoticproteins survivin and Bcl-2 and downregulation of the growth inhibitor transforming growth factor-� and proapoptotickinases JNK and p38. These findings suggest that IL-6 promotes the development and progression of pulmonaryvascular remodeling and PAH through proproliferative antiapoptotic mechanisms. (Circ Res. 2009;104:236-244.)
Key Words: interleukin-6 � pulmonary artery hypertension � proliferation
Pulmonary vascular remodeling is associated with in-creased pulmonary vascular resistance, pulmonary ar-
tery hypertension (PAH), and right heart failure. AdvancedPAH is characterized by arteriopathy, which includesmuscularization of distal pulmonary arterioles, concentricintimal thickening, and obstruction of the vascular lumenby proliferating endothelial cells to form plexiform le-sions.1 Evidence suggests that PAH is associated withgenetic perturbations favoring cellular growth, prolifera-tion, and angiogenesis2 and inhibitors of apoptosis, previ-ously thought to be only expressed in cancer cells, pro-moting a proliferative cellular phenotype, resulting inpulmonary vascular remodeling in PAH.3,4 The histopatho-logic features and known genetic susceptibilities of thiscondition have led to the hypothesis that PAH arises fromhyperproliferation of pulmonary artery smooth musclecells (PASMCs) and endothelial cells (PAECs).
In addition to the formation of proliferative neointimallesions and muscularization of the pulmonary vascularbed, perivascular inflammatory cell infiltrates are alsopresent in advanced human cases of PAH. These infiltratesconsist of T cells, B cells, and macrophages, suggesting
that cytokines and growth factors associated with theseinflammatory cells may be promoting PAEC and PASMChyperproliferation.5 The proinflammatory cytokine inter-leukin (IL)-6 is consistently increased in the serum andlungs6 – 8 of patients with idiopathic PAH and in inflamma-tory diseases6,9 –11 that are associated with PAH. In addi-tion, Kaposi sarcoma–associated herpes virus, which maycause PAH in human immunodeficiency virus–negativeCastleman’s disease, encodes a constitutively active formof IL-6,12 resulting in unregulated cell growth and escapefrom host antitumor defenses. Furthermore, uncheckedproduction of IL-6 in tissues leading to chronic inflamma-tion has exhibited a strong association with many can-cers.13 Given mounting evidence for the role of inflamma-tion and cancer-like mechanisms in the pathogenesis ofPAH, we investigated whether IL-6 promotes the develop-ment of pulmonary arteriopathy and consequent PAH. Weshow that lung-specific overexpression of IL-6 in micereplicates the pathological lesions observed in advancedPAH, including both distal arteriolar muscularization andplexogenic arteriopathy, and leads to increased pulmonaryvascular resistance (PVR) and PAH. At the cellular and
Original received June 23, 2008; revision received November 27, 2008; accepted December 2, 2008.From the Pulmonary Critical Care Unit (M.K.S., O.L.S., N.K., C.A.H., A.B.W.), Department of Medicine; and Department of Pathology (E.J.M.),
Massachusetts General Hospital, Harvard Medical School, Boston.Correspondence to M. Kathryn Steiner, Division of Pulmonary Critical Care Medicine, University of Massachusetts Memorial Medical Center, 55 Lake
Ave North, Worcester, MA 01655. E-mail [email protected]© 2009 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.108.182014
236 by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from
molecular level, these vasculopathic changes are associ-ated with the activation of vascular endothelial growthfactor (VEGF) and the mitogen-activated protein kinase(MAPK) extracellular signal-regulated kinase (ERK), withsubsequent increases in protooncogene c-MYC/MAX tran-scription factor complex and the antiapoptotic proteinssurvivin and Bcl-2, with downregulation of the growthinhibitor transforming growth factor (TGF)-� and proapo-ptotic kinases JNK and p38. This suggests that IL-6participates in the development of distal pulmonary pro-liferative arteriopathy and consequent elevation in PVRand development of PAH.
Materials and MethodsFor details, see the expanded Materials and Methods section in theonline data supplement, available at http://circres.ahajournals.org.
Indices of pulmonary vascular remodeling were measured inlung-specific IL-6–overexpressing transgenic mice (Tg�) and com-pared to wild-type (Tg�) controls in both normoxic and chronichypoxic conditions.
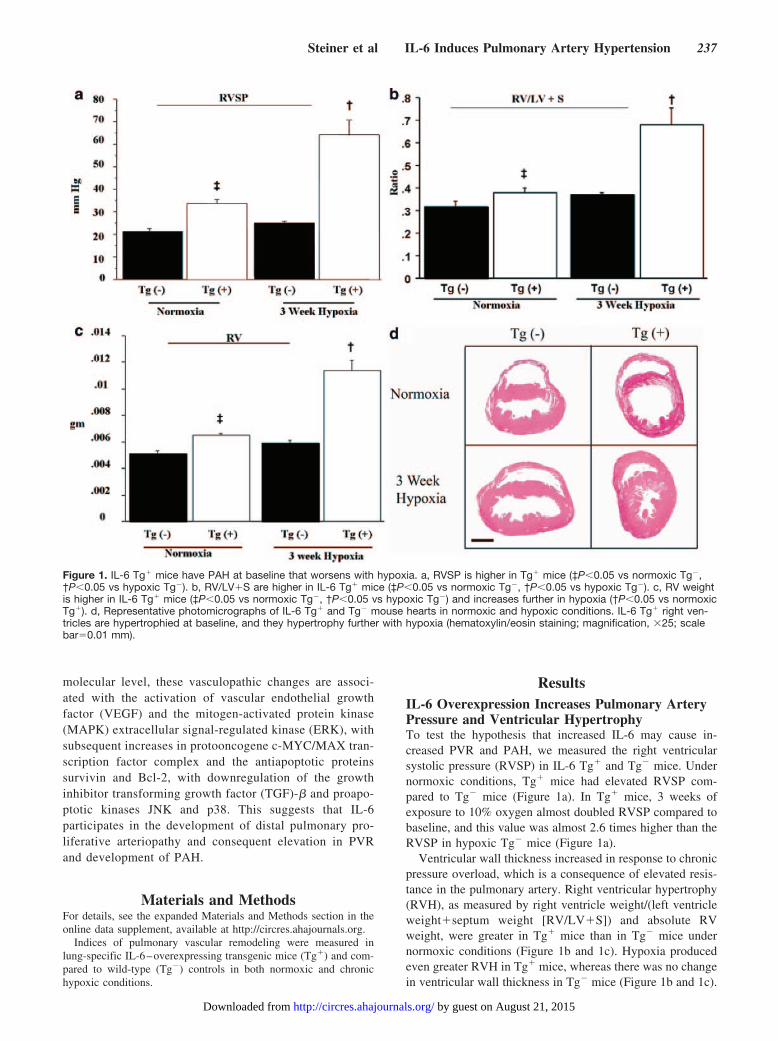
ResultsIL-6 Overexpression Increases Pulmonary ArteryPressure and Ventricular HypertrophyTo test the hypothesis that increased IL-6 may cause in-creased PVR and PAH, we measured the right ventricularsystolic pressure (RVSP) in IL-6 Tg� and Tg� mice. Undernormoxic conditions, Tg� mice had elevated RVSP com-pared to Tg� mice (Figure 1a). In Tg� mice, 3 weeks ofexposure to 10% oxygen almost doubled RVSP compared tobaseline, and this value was almost 2.6 times higher than theRVSP in hypoxic Tg� mice (Figure 1a).
Ventricular wall thickness increased in response to chronicpressure overload, which is a consequence of elevated resis-tance in the pulmonary artery. Right ventricular hypertrophy(RVH), as measured by right ventricle weight/(left ventricleweight�septum weight [RV/LV�S]) and absolute RVweight, were greater in Tg� mice than in Tg� mice undernormoxic conditions (Figure 1b and 1c). Hypoxia producedeven greater RVH in Tg� mice, whereas there was no changein ventricular wall thickness in Tg� mice (Figure 1b and 1c).
Figure 1. IL-6 Tg� mice have PAH at baseline that worsens with hypoxia. a, RVSP is higher in Tg� mice (‡P�0.05 vs normoxic Tg�,†P�0.05 vs hypoxic Tg�). b, RV/LV�S are higher in IL-6 Tg� mice (‡P�0.05 vs normoxic Tg�, †P�0.05 vs hypoxic Tg�). c, RV weightis higher in IL-6 Tg� mice (‡P�0.05 vs normoxic Tg�, †P�0.05 vs hypoxic Tg�) and increases further in hypoxia (†P�0.05 vs normoxicTg�). d, Representative photomicrographs of IL-6 Tg� and Tg� mouse hearts in normoxic and hypoxic conditions. IL-6 Tg� right ven-tricles are hypertrophied at baseline, and they hypertrophy further with hypoxia (hematoxylin/eosin staining; magnification, �25; scalebar�0.01 mm).
Steiner et al IL-6 Induces Pulmonary Artery Hypertension 237
by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from
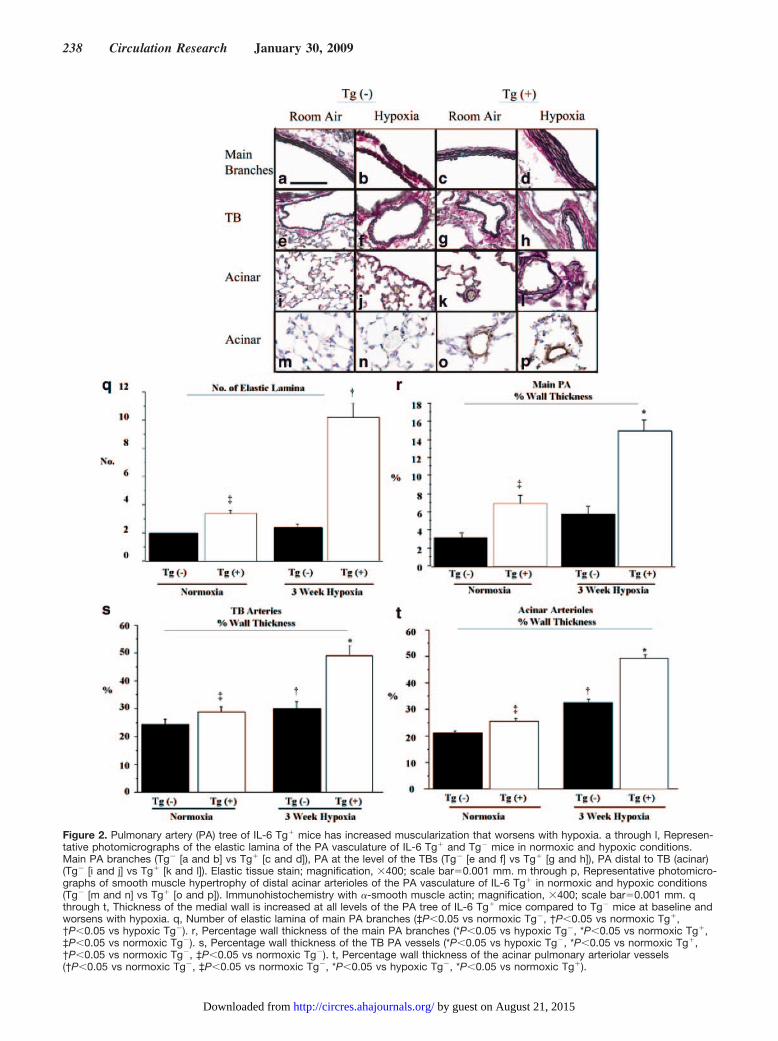
Figure 2. Pulmonary artery (PA) tree of IL-6 Tg� mice has increased muscularization that worsens with hypoxia. a through l, Represen-tative photomicrographs of the elastic lamina of the PA vasculature of IL-6 Tg� and Tg� mice in normoxic and hypoxic conditions.Main PA branches (Tg� [a and b] vs Tg� [c and d]), PA at the level of the TBs (Tg� [e and f] vs Tg� [g and h]), PA distal to TB (acinar)(Tg� [i and j] vs Tg� [k and l]). Elastic tissue stain; magnification, �400; scale bar�0.001 mm. m through p, Representative photomicro-graphs of smooth muscle hypertrophy of distal acinar arterioles of the PA vasculature of IL-6 Tg� in normoxic and hypoxic conditions(Tg� [m and n] vs Tg� [o and p]). Immunohistochemistry with �-smooth muscle actin; magnification, �400; scale bar�0.001 mm. qthrough t, Thickness of the medial wall is increased at all levels of the PA tree of IL-6 Tg� mice compared to Tg� mice at baseline andworsens with hypoxia. q, Number of elastic lamina of main PA branches (‡P�0.05 vs normoxic Tg�, †P�0.05 vs normoxic Tg�,†P�0.05 vs hypoxic Tg�). r, Percentage wall thickness of the main PA branches (*P�0.05 vs hypoxic Tg�, *P�0.05 vs normoxic Tg�,‡P�0.05 vs normoxic Tg�). s, Percentage wall thickness of the TB PA vessels (*P�0.05 vs hypoxic Tg�, *P�0.05 vs normoxic Tg�,†P�0.05 vs normoxic Tg�, ‡P�0.05 vs normoxic Tg�). t, Percentage wall thickness of the acinar pulmonary arteriolar vessels(†P�0.05 vs normoxic Tg�, ‡P�0.05 vs normoxic Tg�, *P�0.05 vs hypoxic Tg�, *P�0.05 vs normoxic Tg�).
238 Circulation Research January 30, 2009
by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from
The histological appearance of the hearts was consistent withRVH measurements, showing that right ventricular wall masswas greater in Tg� mice in both normoxic and hypoxicconditions (Figure 1d). See the online data supplement foradditional data.
IL-6 Overexpression Induced MuscularizationThroughout the Entire Pulmonary Vascular BedTo determine the cause of increased PVR, we examinedspecific regions of the pulmonary vascular tree for remodel-ing. Examination of the proximal branches of the main PArevealed that the elastic lamina was increased in normoxicTg� mice compared to their Tg� counterparts (Figure 2cversus 2a) and was quantitatively confirmed by counting thenumber of elastic lamina (Figure 2q). Following hypoxia, thenumber of elastic lamina in Tg� mice more than tripledcompared to baseline and exceeded the number in hypoxicTg� littermates by a factor of 5. Main PA branches in Tg�
mice exhibited an increase in not only the number of elasticlamina but also the percentage vessel medial wall thickness,relative to the Tg� control, under both normoxic and hypoxicconditions (Figure 2r). The medial wall of the main PAbranches in Tg� mice more than doubled in thickness inresponse to hypoxia compared to their Tg� normoxic con-trols, whereas PA medial wall thickness did not change inhypoxic Tg� mice compared to normoxic controls.
The terminal bronchioles (TBs) and distal acinar arterioleswere examined for evidence of muscularization. The mostnotable findings were that the distal acinar arterioles of Tg�
mice were muscularized at baseline and became more thicklymuscularized in hypoxia unlike the Tg� mice arterioles, asshown by elastic staining (Figure 2k and 2l [Tg�] versus 2iand 2j [Tg�]) and by immunohistochemistry with �-smoothmuscle actin (Figure 2o and 2p [Tg�] versus 2m and 2n[Tg�]). See the online data supplement for detailed results,together with the quantitative results of the medial wallthickness of both the TBs and acinar vessels.
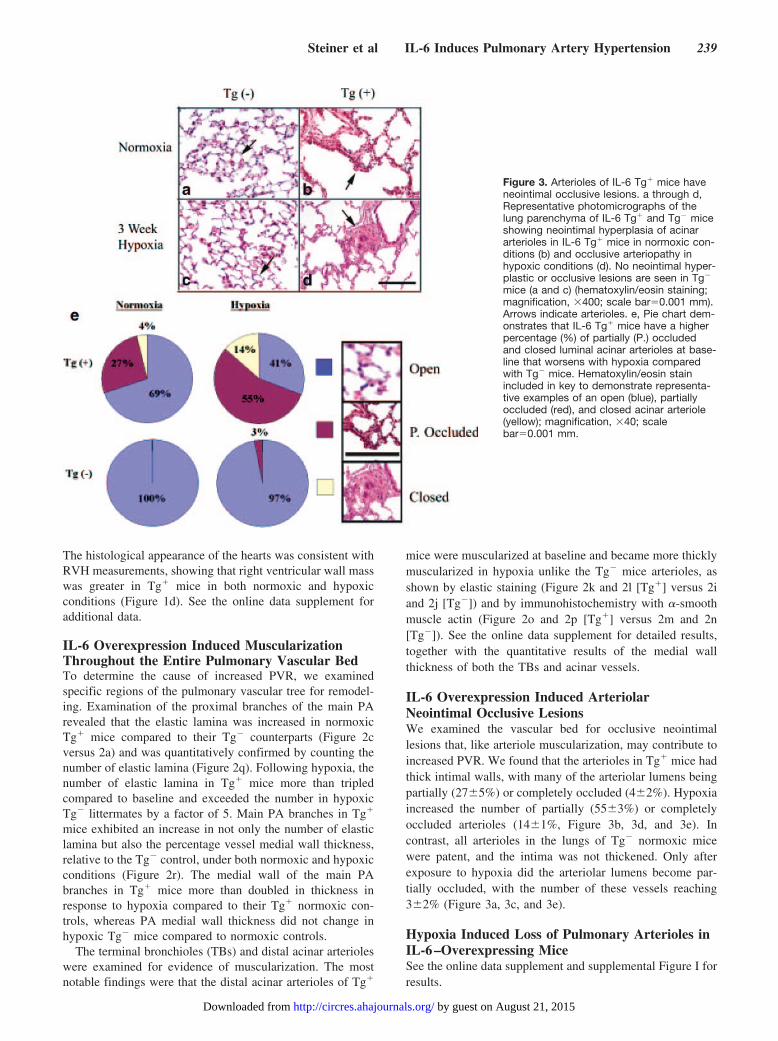
IL-6 Overexpression Induced ArteriolarNeointimal Occlusive LesionsWe examined the vascular bed for occlusive neointimallesions that, like arteriole muscularization, may contribute toincreased PVR. We found that the arterioles in Tg� mice hadthick intimal walls, with many of the arteriolar lumens beingpartially (27�5%) or completely occluded (4�2%). Hypoxiaincreased the number of partially (55�3%) or completelyoccluded arterioles (14�1%, Figure 3b, 3d, and 3e). Incontrast, all arterioles in the lungs of Tg� normoxic micewere patent, and the intima was not thickened. Only afterexposure to hypoxia did the arteriolar lumens become par-tially occluded, with the number of these vessels reaching3�2% (Figure 3a, 3c, and 3e).
Hypoxia Induced Loss of Pulmonary Arterioles inIL-6–Overexpressing MiceSee the online data supplement and supplemental Figure I forresults.
Figure 3. Arterioles of IL-6 Tg� mice haveneointimal occlusive lesions. a through d,Representative photomicrographs of thelung parenchyma of IL-6 Tg� and Tg� miceshowing neointimal hyperplasia of acinararterioles in IL-6 Tg� mice in normoxic con-ditions (b) and occlusive arteriopathy inhypoxic conditions (d). No neointimal hyper-plastic or occlusive lesions are seen in Tg�
mice (a and c) (hematoxylin/eosin staining;magnification, �400; scale bar�0.001 mm).Arrows indicate arterioles. e, Pie chart dem-onstrates that IL-6 Tg� mice have a higherpercentage (%) of partially (P.) occludedand closed luminal acinar arterioles at base-line that worsens with hypoxia comparedwith Tg� mice. Hematoxylin/eosin stainincluded in key to demonstrate representa-tive examples of an open (blue), partiallyoccluded (red), and closed acinar arteriole(yellow); magnification, �40; scalebar�0.001 mm.
Steiner et al IL-6 Induces Pulmonary Artery Hypertension 239
by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from

IL-6–Induced Distal Pulmonary Vascular WallEndothelial Cellular Growth and ProliferationTo determine whether PAEC types contribute to intimal wallthickening in arterioles of Tg� mice, we performed immu-nohistochemical analysis of factor VIII, an endothelial cellmarker. We found that the arteriole walls were thickened, inpart, by multiple layers of PAECs. Under both normoxic andhypoxic conditions, the PAEC layers either formed smoothand thick concentric occlusive lesions or plexogenic-likeocclusive lesions caused by a piling up of PAECs in anonuniform fashion (Figure 4c and 4d), whereas arteriolarwalls in Tg� mice had a normal appearance under bothnormoxic and hypoxic conditions (Figure 4a and 4b). Atbaseline and in hypoxic conditions, PAECs lining the arte-rioles of Tg� mice exhibited minimal expression of VEGFreceptor (VEGFR)2 (Figure 4e and 4f), whereas PAECs hadelevated VEGFR2 expression in Tg� mice (Figure 4g and4h), suggesting that the growth potential of these cells wasincreased and confirming that the plexogenic-like lesions inTg� mice consisted predominately of PAECs. In Tg� mice,increases in PAECs and VEGFR2 expression were associatedwith increased proliferation within the intimal wall of arte-rioles at baseline and in hypoxic conditions, as assessed bystaining for the proliferation marker proliferating-cell nuclearantigen (PCNA) (Figure 4k and 4l). No change in PCNAlevels were detected in the Tg� lungs (Figure 4i and 4j).
Characterization of Pulmonary Vasculopathy:Progrowth/Proproliferative Factors andProsurvival/Antiapoptotic Mediators Are Activatedin IL-6 Tg� MiceWe determined whether factors that stimulate growth, prolif-eration, and survival and inhibit apoptosis of PAECs and
PASMCs were underlying the mechanism through whichIL-6 promotes the characteristic pathophysiologic phenotypeof PAH. This was preformed by investigating a number ofkey modulators that may be involved, at the level of theprotein in immunoblot analysis of whole lung lysates (sup-plemental Figure II, a through e). See the online datasupplement for the results.
Inflammatory Cells Contribute to IL-6–InducedNeointimal Lesions in ArteriolesGiven that lymphocytes form conglomerates near majorairways in this mouse model,14 we examined the pulmonaryvascular bed for evidence of a similar cellular inflammatoryresponse at sites of arteriolar occlusive lesions. Under nor-moxic conditions, the number of periarteriolar lymphocytes(determined by the high nuclear to cytoplasmic ratio) wasgreater in Tg� mice than in Tg� mice (Figure 5c), with Tcells, determined by immunohistochemistry (Figure 5g), butnot B cells (Figure 5k) being increased within the pulmonaryvascular bed of Tg� animals. Other inflammatory cells werenot seen (Figure 5a through 5d). Following hypoxia, these Tcells (Figure 5d) contributed to the obstruction of the arterio-lar lumen. This was confirmed by immunoblot analysis ofwhole lung lysates (supplemental Figure III). Taken together,IL-6 mainly recruits lymphocytes, and the lymphocytes thatare recruited to the pulmonary vascular bed are predomi-nately T cells, not B cells. Further characterization of lym-phocyte recruitment and function was determined in IL-6Tg� mice. The data can be seen in the online data supple-ment, together with supplemental Figure III.
DiscussionPatients with severe PAH and animal models of PAH15–21
exhibit increases in inflammatory cells, growth factors, andcytokines. IL-6, a pleiotropic cytokine, is frequently elevated,
Figure 4. Angioproliferative lesions are present in Tg� mice dis-tal arterioles. a through l, Representative photomicrographsshowing the formation of thick occlusive neointimal lesions. En-dothelial cells (factor VIII) are forming thick layers in the distalacinar arterioles of Tg� mice (c and d) and have increasedexpression of VEGFR2 (g and h), which was not seen in Tg�
mice (a and b or e and f). There is increased cellular prolifera-tion in the walls of the distal arterioles of IL-6 Tg� mice (PCNA)(k and l) in normoxic and hypoxic conditions, which was notseen in Tg� mice (i and j). Immunohistochemistry staining; mag-nification, �400; scale bar�0.001 mm.
Figure 5. Inflammatory cells present in the neointimal lesions ofthe arterioles of Tg� mice. a through l, Representative photomi-crographs showing an increase in periarteriolar lymphocytes(Giemsa stain: c vs. a), specifically T cells (CD3: g vs. e), com-pared to B cells (B220: k vs. i) in IL-6 Tg� mice with littlechange in Tg� mice at baseline. In hypoxic conditions, CD3� Tcells are obliterating the arteriolar lumen of IL-6 Tg� mice (d andh) vs. (b and f). Magnification, �400; scale bar�0.001 mm.
240 Circulation Research January 30, 2009
by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from

suggesting that PAH development is associated with IL-6–induced inflammation. Our results demonstrate that IL-6 lung-specific overexpression produces distal arteriolar-occlusiveplexogenic lesions and arteriolar wall muscularization. Thesechanges in the distal vascular bed are associated with and maylead to proximal pulmonary artery wall hypertrophy andRVH, as well as increased RVSP and PVR. Injection ofrecombinant human IL-6 (rhIL-6) also produces RVH inrats22 and mice.18 However, they lack the associated distalobliterative muscularized vascular lesions observed in thetransgenic mice that constitutively overexpress IL-6. Impor-tantly, however, IL-6 knockout mice exposed to hypoxia areresistant to the development of increased RVSP.23 The lack ofcorrelation between pulmonary vascular remodeling and thepresence of elevated pulmonary artery pressures in othermurine models24–27 has slowed our understanding of thepathobiology of PAH. IL-6 Tg� mice, in which the patho-logical and physiological changes observed in the pulmonaryartery bed correlate with the severity of PAH, may enable abetter understanding of PAH pathobiology, including the roleof increased IL-6 in the development of PAH in humans.
A major finding in this study is that distal vascularremodeling in Tg� mice is similar to that seen in patients withsevere PAH,5 with (1) concentric intimal wall thickening, (2)plexogenic lesions, (3) recruitment of inflammatory cells, and(4) distal arteriolar wall muscularization. These featuresoccurred de novo under normoxic conditions and worsenedwith hypoxia. In other rodent models,15–21 vascular remodel-ing is limited to either dysregulated PAECs or PASMCs, butnot both. The Tg� mouse, which exhibits all 4 main patho-logical features of PAH, is, to our knowledge, the only in vivomodel that recapitulates the pathological features of PAH inhumans. Therefore, this model may reveal how interactionsbetween hyperproliferative PASMCs and PAECs and inflam-matory cells, as well as the pathobiological phenotypes ofthese cells contribute to PAH development.
Disorganized PAEC proliferation leading to the formationof neointimal obliterative lesions is described in many casesof idiopathic PAH5 or associated PAH and may be why thehuman form of severe PAH is difficult to treat with thepresent available drugs.28–30 This has led to the search fornewer models of PAH in which a neointima is formed andoccludes the vascular lumen. A number of 2-hit injuriousmurine models have been able to reproduce neointimalocclusive lesions,15,31–33 as well as a genetically alteredmodel34; however, less than 5% of these mice developedthese lesions. Our study is of interest because we show that bysolely overexpressing IL-6 without an additional stress,PAECs are stimulated to either form smooth concentricmultilayers, leading to thickening of the intimal wall, or topile up on top of one another, narrowing the distal arteriolarlumen and forming a plexiform lesion. Both features werepresent in all mice under normoxic conditions, when PAH ismild, and increased under hypoxic conditions, when RVSP ismaximal and the disease is severe. This suggests that aberrantPAEC proliferation and lesion formation are pathologicallyrelevant and useful markers of disease progression and thatoverexpression of IL-6, a single genetic perturbation, is able
to reproduce the characteristic obliterative lesions seen in theaforementioned models and replicate that of human disease.
Distal extension of smooth muscle into small peripheral,normally nonmuscular, pulmonary arteries within the respi-ratory acinous is notable in all forms of PAH. The cellularprocesses underlying muscularization of this distal part of thepulmonary vascular bed are incompletely understood but arethought to result from the abnormal growth of PASMCs,which have impaired responses to antiproliferative proapo-ptotic stimuli such as bone morphogenic protein (BMP) andTGF-�.2,35–37 We show that lung-specific overexpression ofIL-6 induces 3 forms of muscularization. First, IL-6 results indistal extension of smooth muscle into the small peripheralpulmonary arteries at the level of the acinous, and, with theadded insult of hypoxia, the medial wall further hypertro-phies. Secondly, IL-6 results in an increase in the medial wallthickness of the main and bronchial level pulmonary arteries,and, thirdly, there is an increase in the number of layers ofelastic lamella, both of which increase further in hypoxicconditions. The combination of these striking changes inmuscularization have not been observed in other PAHmurine models. However, in the spontaneously hyperten-sive rat,38 increased arterial medial wall thickness isassociated with increased number of lamina in major bloodvessels, as well as increased wall thickness, although lessstriking than in hypoxic IL-6 Tg� mice. Furthermore, thehypertrophic changes observed are augmented under in-creased pressure, suggesting that secondary structuraladaptations become superimposed on primary geneticones. In IL-6 Tg� mice, where growth development isaltered,14 as in the fawn-hooded rat,39 early genetic abnor-malities in pulmonary vascular development may contrib-ute to the progression of PAH in the adult Tg� mice withand without a hypoxic injurious stimulus.
It is unclear what triggers PAECs and PASMCs to have aproproliferative phenotype while maintaining an insensitivitytoward growth inhibitory stimuli in patients with PAH. Inhumans, plexiform lesions express angiogenic factors includ-ing VEGF and its receptor VEGFR2,40 suggesting that VEGFmay play a proangioproliferative role in the development ofplexiform lesions, a growth factor shared by the plexiformlesions observed in IL-6 Tg� mice, as well as other animalmodels with angioproliferative lesions.33 VEGF may also bean important survival factor for PASMCs in the presence ofIL-6. IL-6 triggers cultured smooth muscle cell proliferationboth directly, through upregulation of VEGFR2 expressionand phosphorylation, and indirectly, through upregulation ofmatrix metalloproteinase-9.41 IL-6–induced VEGF expres-sion may also indirectly increase the number of PASMCs bytransforming PAECs into smooth muscle–like cells, as ob-served in cultured human PAECs.42 These findings, takenwith our results, suggest that the presence of abnormal levelsof IL-6 may activate, amplify, and maintain the growth andproliferation of PAECs and PASMCs by upregulating VEGFexpression.
TGF-�/BMP signaling, a network of proteins that controlcell growth, is impaired and the TGF-� receptor is absent inthe PAECs in the core of plexiform lesions in PAH.43,44 Thissuggests that PAECs in plexiform lesions have lost their
Steiner et al IL-6 Induces Pulmonary Artery Hypertension 241
by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from

check-and-balance system to control PAEC growth, givingrise to a hyperproliferative PAEC phenotype. In addition,PASMCs from patients with PAH are resistant to the anti-proliferative effects of TGF-�,2 suggesting that the failure ofTGF-� to suppress PASMC growth in PAH may, in part,underlie the increased muscularization of normally nonmus-cularized distal pulmonary arteries of patients with PAH. IL-6has recently been found to negatively regulate the TGF-�/BMP signaling cascade.45 In the IL-6 Tg� mouse model, inwhich both muscularization and angioproliferative lesions areabundant and PAH is present, we show that the expression ofTGF-� is reduced, in a rich milieu of angioproliferativegrowth factor VEGF and its receptor. Taken together, theIL-6 Tg� mouse model shares similar growth factor charac-teristics to that of patients with PAH, and thus this model mayenable investigators to delineate the trigger behind the mo-lecular imbalance that favors the increased expression ofproliferative growth factors.
IL-6 overexpression may predispose to proliferative cellu-lar phenotypes and exaggerated PAH as a result of unopposedMAPK intracellular signaling, normally countered by anti-proliferative TGF-�–mediated signaling. Both p38MAPK andERK are noted to be unopposed in PASMCs from patientswith mutations in the TGF-�/BMP pathway, resulting in aproliferative apoptotic resistant phenotype.46 The IL-6 Tg�
mice share a similar biology to PASMCs of the patient,whereby ERK activity also is upregulated in an unopposedenvironment, which is, in part, attributable to the lack ofTGF-� and, in part, attributable to the lack of proapoptoticMAPKs p38 and pJNK. These findings are also consistentwith in vitro work, in which IL-6–stimulated human endo-thelial cells47 also have reduced p38 and pJNK. In other cellsystems, IL-6 activates the MAPK signaling pathway viaERK and, in turn, blocks the TGF-�/BMP pathway bypreventing the nuclear translocation of Smad, a downstreamBMP signaling protein,48,49 resulting in cellular proliferation.Given that this mouse model and the vasculature of PAHpatients are deficient in growth controlling TGF-�/BMPproteins46 in the setting of elevated IL-6 levels and that bothshare ERK activation, further investigation of unopposed
IL-6/ERK signaling may uncover the mechanism by whichvascular cells switch from a balanced growth controlled stateto an excessively proproliferative one.
IL-6 overexpression may predispose to exaggerated PAHas a result of coordinating a number of downstream propro-liferative, prosurvival, and antiapoptotic signaling pathways(Figure 6). We found that c-myc and its obligatory bindingpartner, MAX, may be key in the downstream proliferativesignal cascade of IL-6, promoting cellular proliferative phe-notypes in PAH. However, it is also clear from our work thatIL-6 selectively blocks apoptosis within the pulmonary vas-cular bed by downregulating TGF-� and proapoptoticMAPKs, pJNK, and p38, while upregulating prosurvivalfactors survivin and Bcl-2. Taken together, these complexobliterative vascular lesions are likely forming because IL-6is favoring a proproliferative antiapoptotic cell state, asobserved in angioproliferative lesions of patients with PAH.
See the online data supplement for an expanded discussionregarding IL-6 and its role in regulating proliferative andantiapoptotic pathways.
Elevation of IL-6 in the serum and inflammatory cellularinfiltrate in plexiform lesions in PAH patients and now IL-6Tg� mice with replicative pathophysiological changes ofPAH suggest that cellular immunity may play an active rolein the dysregulation of PAECs and PASMCs and the devel-opment of PAH. Further discussion regarding our findingsand controversies of the role inflammation may play in thisdisease is highlighted in the online data supplement.
The importance of the loss of small pulmonary arteriolarvessels, which may contribute to the development of PVR, iscontroversial; for further discussion regarding our data, seethe online data supplement.
In summary, our work supports the hypothesis that IL-6directly promotes a proproliferative apoptotic resistant milieuwithin the PA wall, resulting in a vasculopathy mirroring thatseen in patients with severe PAH. Unlike other murinemodels, this transgenic mouse model replicates the patho-physiology of human PAH, with muscularization andarteriolar-occlusive changes occurring in the distal vascularbed, leading to elevated PVR and secondary chronic pressure
Figure 6. Hypothetical mechanisms leading tohyperproliferative apoptotic-resistant PASMC andPAEC phenotypes, angioproliferative occlusivelesions and muscularization of distal vessels inPAH. IL-6 induces angioproliferative growth factorVEGF and intracellular MAPK ERK to activate pro-proliferative transcription factor complex c-MYCand MAX, resulting in an increase in cell cyclegenes to promote PASMC and PAEC proliferation.Concurrently, IL-6 downregulates growth inhibitorTGF-� and other MAPKs that are proapoptotic(p38 and JNK1), while upregulating apoptotic in-hibitors survivin and Bcl-2, resulting in apoptoticresistant PASMC and PAEC phenotypes. Inflam-matory cells, specifically T cells, may have animportant role in maintaining the secretion of theproproliferative cytokine IL-6.
242 Circulation Research January 30, 2009
by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from

overload occurring in the main PA and right ventricle. Ourwork demonstrates that, in the pulmonary vascular bed,regulation of inflammatory cytokine IL-6 is tightly linkedwith cellular growth and proliferation through a number ofproliferative apoptotic resistant downstream pathways. Futurework to establish the underlying mechanism of these compli-cated signaling systems in the lung-specific IL-6 Tg� mice,and to determine several of the key molecules necessary toinhibit the excessive proliferative cellular state, will improveour understanding of the disease and thereby help in devel-oping new therapies that target the angioproliferative lesionsin patients with refractory PAH. The development of PAH inmice overexpressing IL-6 in the lung, together with thepresence of increased IL-6 in PAH patients, suggests thatIL-6 is integral to the development and progression ofpulmonary vascular remodeling, PVR, and PAH.
AcknowledgmentsWe acknowledge David A. Schoenfeld, PhD (Biostatistical Center,Harvard Medical School).
Sources of FundingThis work was supported by National Heart, Lung, and BloodInstitute grants HL074859 (to A.B.W.) and HL007874 andHL039150 (to C.A.H.).
DisclosuresNone.
References1. Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, Reid
LM, Tuder RM. Pathologic assessment of vasculopathies in pulmonaryhypertension. J Am Coll Cardiol. 2004;43:25S–32S.
2. Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, ShearesKK, Trembath RC. Altered growth responses of pulmonary arterysmooth muscle cells from patients with primary pulmonary hyper-tension to transforming growth factor-{beta}1 and bone morpho-genetic proteins. Circulation. 2001;104:790 –795.
3. Blanc-Brude OP, Yu J, Simosa H, Conte MS, Sessa WC, Altieri DC.Inhibitor of apoptosis protein survivin regulates vascular injury. Nat Med.2002;8:987–994.
4. McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G,Bonnet S, Puttagunta L, Michelakis ED. Gene therapy targeting survivinselectively induces pulmonary vascular apoptosis and reverses pulmonaryarterial hypertension. J Clin Invest. 2005;115:1479–1491.
5. Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelialcell growth and elements of inflammation are present in plexiform lesionsof pulmonary hypertension. Am J Pathol. 1994;144:275–285.
6. Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L,Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1and interleukin-6 serum concentrations in severe primary pulmonaryhypertension. Am J Respir Crit Care Med. 1995;151:1628–1631.
7. Katsushi H, Kazufumi N, Hideki F, Katsumasa M, Hiroshi M, Kengo K,Hiroshi D, Nobuyoshi S, Tetsuro E, Hiromi M, Tohru O. Epoprostenoltherapy decreases elevated circulating levels of monocyte chemoat-tractant protein-1 in patients with primary pulmonary hypertension. CircJ. 2004;68:227–231.
8. Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L,Kasahara Y, Cool CD, Bishop AE, Geraci M, Semenza GL, Yacoub M,Polak JM, Voelkel NF. Expression of angiogenesis-related molecules inplexiform lesions in severe pulmonary hypertension: evidence for aprocess of disordered angiogenesis. J Pathol. 2001;195:367–374.
9. Yoshio T, Masuyama JI, Kohda N, Hirata D, Sato H, Iwamoto M, MimoriA, Takeda A, Minota S, Kano S. Association of interleukin 6 release fromendothelial cells and pulmonary hypertension in SLE. J Rheumatol.1997;24:489–495.
10. Lesprit P, Godeau B, Authier FJ, Soubrier M, Zuber M, Larroche C,Viard JP, Wechsler B, Gherardi R. Pulmonary hypertension in POEMS
syndrome: a new feature mediated by cytokines. Am J Respir Crit CareMed. 1998;157:907–911.
11. Nishimaki T, Aotsuka S, Kondo H, Yamamoto K, Takasaki Y, SumiyaM, Yokohari R. Immunological analysis of pulmonary hypertension inconnective tissue diseases. J Rheumatol. 1999;26:2357–2362.
12. Parravinci C, Corbellino M, Paulli M, Magrini U, Lazzarino M, MoorePS, Chang Y. Expression of a virus-derived cytokine, KSHV vIL-6, inHIV-seronegative Castleman’s disease. Am J Pathol. 1997;151:1517–1522.
13. Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 ininflammation and cancer. Eur J Cancer. 2005;41:2502–2512.
14. DiCosmo BF, Geba GP, Picarella D, Elias JA, Rankin JA, Stripp BR,Whitsett JA, Flavell RA. Airway epithelial cell expression ofinterleukin-6 in transgenic mice. Uncoupling of airway inflammation andbronchial hyperreactivity. J Clin Invest. 1994;94:2028–2035.
15. Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G,Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor2 combined with chronic hypoxia causes cell death-dependent pulmonaryendothelial cell proliferation and severe pulmonary hypertension. FASEBJ. 2001;15:427–438.
16. Wang GS, Qian GS, Mao BL, Cai WQ, Chen WZ, Chen Y. [Changes ofinterleukin-6 and Janus kinases in rats with hypoxic pulmonary hyper-tension]. Zhonghua Jie He He Hu Xi Za Zhi. 2003;26:664–667.
17. Kubo K, Hanaoka M, Hayano T, Miyahara T, Hachiya T, Hayasaka M,Koizumi T, Fujimoto K, Kobayashi T, Honda T. Inflammatory cytokinesin BAL fluid and pulmonary hemodynamics in high-altitude pulmonaryedema. Respir Physiol. 1998;111:301–310.
18. Golembeski SM, West J, Tada Y, Fagan KA. Interleukin-6 causes mildpulmonary hypertension and augments hypoxia-induced pulmonaryhypertension in mice. Chest. 2005;128:572S–573S.
19. Kasahara Y, Kimura H, Kurosu K, Sugito K, Mukaida N, Matsushima K,Kuriyama T. MCAF/MCP-1 protein expression in a rat model for pul-monary hypertension induced by monocrotaline. Chest. 1998;114(1suppl):67S.
20. Faul JL, Nishimura T, Berry GJ, Benson GV, Pearl RG, Kao PN. Trip-tolide attenuates pulmonary arterial hypertension and neointimal for-mation in rats. Am J Respir Crit Care Med. 2000;162:2252–2258.
21. Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R,Burns N, Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absenceof T cells confers increased pulmonary arterial hypertension and vascularremodeling. Am J Respir Crit Care Med. 2007;175:1280–1289.
22. Miyata M, Sakuma F, Yoshimura A, Ishikawa H, Nishimaki T, KasukawaR. Pulmonary hypertension in rats. 2. Role of interleukin-6. Int ArchAllergy Immunol. 1995;108:287–291.
23. Savale L, Izikki M, Tu L, Rideau D, Raffestin B, Naitre B, Adnot S.Attenuated hypoxic pulmonary hypertension in mice lacking theinterleukin-6 gene. Am J Respir Crit Care Med. 2007;175:A42.
24. West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, Hoedt-Miller M,Tada Y, Ozimek J, Tuder R, Rodman DM. Pulmonary hypertension intransgenic mice expressing a dominant-negative BMPRII gene in smoothmuscle. Circ Res. 2004;94:1109–1114.
25. Merklinger SL, Wagner RA, Spiekerkoetter E, Hinek A, Knutsen RH,Kabir MG, Desai K, Hacker S, Wang L, Cann GM, Ambartsumian NS,Lukanidin E, Bernstein D, Husain M, Mecham RP, Starcher B,Yanagisawa H, Rabinovitch M. Increased fibulin-5 and elastin inS100A4/Mts1 mice with pulmonary hypertension. Circ Res. 2005;97:596–604.
26. Crossno JT Jr, Garat CV, Reusch JE, Morris KG, Dempsey EC,McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone attenuates hyp-oxia-induced pulmonary arterial remodeling. Am J Physiol Lung Cell MolPhysiol. 2007;292:L885–L897.
27. Daley E, Emson C, Guignabert C, de Waal Malefyt R, Louten J, KurupV, Hogaboam C, Taraseviciene-Stewart L, Voelkel N, Rabinovitch M,Grunig E, Grunig G. Pulmonary arterial remodeling induced by a Th2immune response. J Exp Med. 2008;205:361–372.
28. Rai PR, Cool C, King J, Stevens T, Burns N, Winn RA, Kasper M,Voelkel NF. The cancer Paradigm of severe angioproliferative pulmonaryhypertension. Am J Respir Crit Care Medicine. 2008;178:558–564.
29. Wagenvoort C, Wagenvoort N. Primary pulmonary hypertension: apathologic study of the lung vessels in 156 clinically diagnosed cases.Circulation. 1970;42:1163–1184.
30. Palevsky HI, Schloo BL, Pietra GG, Weber KT, Janicki JS, Rubin E,Fishman AP. Primary pulmonary hypertension. Vascular structure, mor-phometry, and responsiveness to vasodilator agents. Circulation. 1989;80:1207–1221.
Steiner et al IL-6 Induces Pulmonary Artery Hypertension 243
by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from

31. Okada K, Tanaka Y, Bernstein M, Zhang W, Patterson GA, Botney MD.Pulmonary hemodynamics modify the rat pulmonary artery response toinjury. A neointimal model of pulmonary hypertension. Am J Pathol.1997;151:1019–1025.
32. Okada K, Bernstein ML, Zhang W, Schuster DP, Botney MD. Angioten-sin-converting enzyme inhibition delays pulmonary vascular neointimalformation. Am J Respir Crit Care Med. 1998;158:939–950.
33. Ivy DD, McMurtry IF, Colvin K, Imamura M, Oka M, Lee DS, Gebb S,Jones PL. Development of occlusive neointimal lesions in distal pulmo-nary arteries of endothelin B receptor-deficient rats: a new model ofsevere pulmonary arterial hypertension. Circulation. 2005;111:2988–2996.
34. Greenway S, van Suylen RJ, Du Marchie Sarvaas G, Kwan E, Ambart-sumian N, Lukanidin E, Rabinovitch M. S100A4/Mts1 produces murinepulmonary artery changes resembling plexogenic arteriopathy and isincreased in human plexogenic arteriopathy. Am J Pathol. 2004;164:253–262.
35. Lagna G, Nguyen PH, Ni W, Hata A. BMP-dependent activation ofcaspase-9 and caspase-8 mediates apoptosis in pulmonary artery smoothmuscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1059–L1067.
36. Zhang S, Fantozzi I, Tigno DD, Yi ES, Platoshyn O, Thistlethwaite PA,Kriett JM, Yung G, Rubin LJ, Yuan JX. Bone morphogenetic proteinsinduce apoptosis in human pulmonary vascular smooth muscle cells. Am JPhysiol Lung Cell Mol Physiol. 2003;285:L740–L754.
37. Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, LangIM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M.Cellular and molecular pathobiology of pulmonary arterial hypertension.J Am Coll Cardiol. 2004;43:13S–24S.
38. Eccleston-Joyner CA, Gray SD. Arterial hypertrophy in the fetal andneonatal spontaneously hypertensive rat. Hypertension. 1988;12:513–518.
39. Sato K, Webb S, Tucker A, Rabinovitch M, O’Brien RF, McMurtry IF,Stelzner TJ. Factors influencing the idiopathic development of pulmonaryhypertension in the fawn hooded rat. Am Rev Respir Dis. 1992;145:793–797.
40. Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesis and evo-lution of plexiform lesions in pulmonary hypertension associated with
scleroderma and human immunodeficiency virus infection. Hum Pathol.1997;28:434–442.
41. Yao JS, Zhai W, Fan Y, Lawton MT, Barbaro NM, Young WL, YangGY. Interleukin-6 upregulates expression of KDR and stimulates prolif-eration of human cerebrovascular smooth muscle cells. J Cereb BloodFlow Metab. 2007;27:510–520.
42. Sakao S, Taraseviciene-Stewart L, Cool CD, Tada Y, Kasahara Y, KurosuK, Tanabe N, Takiguchi Y, Tatsumi K, Kuriyama T, Voelkel NF.VEGF-R blockade causes endothelial cell apoptosis, expansion of sur-viving CD34� precursor cells and transdifferentiation to smoothmuscle-like and neuronal-like cells. FASEB J. 2007;21:3640–3652.
43. Richter A, Yeager ME, Zaiman A, Cool CD, Voelkel NF, Tuder RM.Impaired transforming growth factor-beta signaling in idiopathic pulmo-nary arterial hypertension. Am J Respir Crit Care Med. 2004;170:1340–1348.
44. Yeager ME, Golpon HA, Voelkel NF, Tuder RM. Microsatellite muta-tional analysis of endothelial cells within plexiform lesions from patientswith familial, pediatric, and sporadic pulmonary hypertension. Chest.2002;121(3 suppl):61S.
45. Hagen M, Fagan K, Steudel W, Carr M, Lane K, Rodman DM, WestJ. Interaction of interleukin-6 and the BMP pathway in pulmonary smoothmuscle. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1473–L1479.
46. Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, JefferyTK, Atkinson C, Chen H, Trembath RC, Morrell NW. DysfunctionalSmad signaling contributes to abnormal smooth muscle cell proliferationin familial pulmonary arterial hypertension. Circ Res. 2005;96:1053–1063.
47. Waxman AB, Mahboubi K, Knickelbein RG, Mantell LL, Manzo N,Pober JS, Elias JA. Interleukin-11 and interleukin-6 protect culturedhuman endothelial cells from H2O2-induced cell death. Am J Respir CellMol Biol. 2003;29:513–522.
48. Shi W, Chen H, Sun J, Chen C, Zhao J, Wang YL, Anderson KD,Warburton D. Overexpression of Smurf1 negatively regulates mouseembryonic lung branching morphogenesis by specifically reducingSmad1 and Smad5 proteins. Am J Physiol Lung Cell Mol Physiol. 2004;286:L293–L300.
49. Chen HB, Shen J, Ip YT, Xu L. Identification of phosphatases for Smadin the BMP/DPP pathway. Genes Dev. 2006;20:648–653.
244 Circulation Research January 30, 2009
by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from

CIRCRESAHA/2008/182014/R2
1
Supplement Material 1
Materials and Methods 2
Animals The subcommittee on research for animal care at the Massachusetts General 3
Hospital approved the experimental protocol. CC10-IL-6 transgenic mice (Tg(+)) were 4
bred on a C57/BL6 (Tg(-)) background.1 In these mice, the Clara cell 10-kD promoter 5
(CC10) was used to constitutively drive lung-specific expression of IL-6. Tg(-) 6
littermates served as controls in all experiments. Five Tg(-) and five Tg(+) mice, age- 7
and sex-matched, were exposed to room air (21% FiO2) and then another five Tg(-) and 8
five Tg(+) mice age- and sex-matched, were exposed to hypoxia (10% FiO2) at sea level. 9
All animals were 3 months old and weighed 22.5 ± 2.0 grams. 10
11
For hypoxia, animals were placed in a sealed chamber. The O2 concentration in the 12
chamber was maintained at 10% by controlling the inflow rate of compressed air and N2. 13
Gas was circulated in the chamber with a fan. The CO2 concentration was maintained at 14
< 0.4% with a CO2 absorbent. Gas samples were tested twice per day during the entire 15
experimental period to monitor O2 and CO2 tension. The chamber was unsealed for less 16
than 30 minutes twice per week to replenish the food, replace CO2 absorbent and clean 17
the cages. 18
19
Measurement of right ventricular systolic pressure (RVSP) All mice were 20
administered ketamine (80 mg/kg) and diazepam (5 mg/kg), which produced anesthesia 21
with spontaneous breathing. A midline sternal skin incision was made from the second 22
intercostal space to the xiphoid process. A 25-gauge needle was attached to a male/male 23

CIRCRESAHA/2008/182014/R2
2
luer slip connector, which was joined, in a successive order, to an 18-gauge blunt needle, 1
polyethylene 190 tubing (ID 1.19 mm; OD 1.70 mm), 18-gauge blunt needle connected to 2
a physiological transducer (Becton Dickinson DTXPlusDT-XX) via a two-way plastic 3
stopcock. The stopcock facilitated flushing of the needle and in situ measurement of 4
atmospheric pressure without introducing air bubbles. The entire system was flushed 5
with sterile saline to eliminate bubbles. The transducer was positioned 1.0 cm above the 6
level of the midaxillary line. The needle was then inserted into the right ventricle by 7
following a 45º trajectory between the right second and third intercostal space above the 8
xiphoid process. Pressures were recorded on a Gould chart recorder (Model RS3400) 9
with an embedded Gould transducer amplifier (Model 13-4615-50) at paper speeds 10
ranging from 2.5 to 200 mm/s. Peak RVSPs were obtained, as previously described.2 11
Placement of the needle into the right ventricle was confirmed by postmortem 12
examination. 13
14
Heart weight The right ventricular free wall was detached and removed under a 15
dissecting microscope. The left ventricle and septum were weighed separately from the 16
right ventricle, with measurements being taken after drying at 90ºC for 24 h and 48 h. If 17
the difference between the two readings was greater than ± 0.5 mg, the specimens were 18
dried for another 24 h.2 19
20
Histology of pulmonary vasculature and heart The heart and lungs were flushed with 21
normal saline, removed and fixed in formalin for 72 h. The aorta was removed under a 22
dissecting microscope to access the pulmonary trunk. Two 3-cm silicone tubes (0.03 cm 23

CIRCRESAHA/2008/182014/R2
3
0.05 cm; SMI, Saginaw, MI) were inserted into the pulmonary trunk through a small 1
opening that was made in the lateral wall to maintain vessel wall patency during fixation. 2
One tube was advanced through the main artery into the right pulmonary artery while 3
another tube was inserted into the left pulmonary artery until resistance was encountered 4
at their respective hila. Both main branches of the PA were lifted away from the heart 5
and lungs, using the tubing as an aid. The tubing was removed before tissue embedding. 6
Both the formalin-inflated left lung and the whole heart were sliced into 5 μm-thick 7
sections. Slices of the lung, heart, main pulmonary branches, and pulmonary trunk were 8
dehydrated and embedded in paraffin. Sections were then stained with hematoxylin & 9
eosin, elastic, and giemsa stains. 10
11
Assessment of pulmonary remodeling Pulmonary remodeling was assessed by the 12
percent wall thickness of the main PA branches (central) and parenchymal PA vessels 13
indexed to terminal bronchioles and acini (i.e., vessels indexed to respiratory bronchioles 14
or alveolar ducts).3 Wall thickness was measured with an ocular micrometer and 15
expressed as the medial wall thickness (the distance between the internal and external 16
lamina) divided by the diameter of the vessel (the distance between the external lamina) 17
100 (% wall thickness; %WT). For vessels with a single elastic lamina, the distance 18
between the elastica and endothelial basement membrane was measured.2 The total 19
number of peripheral arteries was expressed as the number of arteries per every 100 20
alveoli in each field and was verified using sections in which the vessels were stained 21
with elastin.4 A quantitative analysis of luminal obstruction was performed by counting 22
at least 50 small pulmonary arterioles (outer diameter < 50 μm) in every hematoxylin-23

CIRCRESAHA/2008/182014/R2
4
eosin stained lung section. These arterioles were assessed for occlusive lesions and 1
scored as follows: (1) no evidence of lumen occlusion (open), (2) partial (< 50%) luminal 2
occlusion, and (3) full luminal occlusion (closed).5 All morphometric analyses were 3
performed by one blinded observer. 4
5
Antibodies Antibodies used for immunohistochemistry included rat anti-mouse B220 6
antigen (1:100; Clone RA3-6B2, BD Biosciences, San Diego, CA), rabbit polyclonal 7
anti-human CD3 (1:400), polyclonal antibody to factor VIII-related antigen (1:250, 8
Dakocytomation, Carpinteria, CA), and biotinylated goat anti-rabbit IgG (1:200; Vector 9
laboratories, Inc., Burlingame, CA). Mouse monoclonal FLK-1 (VEGF-R2, Clone A3, 10
Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was used at a dilution of 1:200 for 11
immunoblot and 1:50 for immunohistochemistry. Anti-actin smooth muscle specific 12
(AB-2) mouse mAb(1A4) (1:750; EMD Chemicals, Inc., Gibbstown, NJ). Antibodies 13
used for immunoblot include mouse monoclonal VEGF antibody ( 1:1000; Abcam, 14
Cambridge, MA), TGF- antibody (rabbit polyclonal antibody; 1:1000), TNF- (rabbit 15
polyclonal antibody; 1:200), goat anti-rabbit IgG -HRP(1:2000), p38 (rabbit monoclonal 16
antibody total 1:500 and phosphorylated 1:200), JNK (rabbit polyclonal antibody total 17
1:500 and phosphorylated JNK 1:50), c-MYC (rabbit polyclonal antibody 1:200), -actin 18
(rabbit polyclonal antibody, 1:2000), caspase 3 (rabbit polyclonal antibody 1:200), 19
cleaved caspase 3 (rabbit monoclonal antibody 1:200), survivin (rabbit polyclonal 20
antibody 1:400), Bcl-2 (rabbit polyclonal antibody 1:200) and BAX (rabbit polyclonal 21
antibody 1:400) from Cell Signaling Technology, Inc; Danvers, MA), PDGF-B (rabbit 22
polyclonal antibody 1:100), MCP-1 (rabbit polyclonal antibody 1:100), ERK1/2 (rabbit 23

CIRCRESAHA/2008/182014/R2
5
polyclonal, 1:500; Clone 14), and MAX (rabbit polyclonal antibody; 1:50) from Santa 1
Cruz Biotechnology, Inc., Santa Cruz, CA. Goat anti-mouse IgG (1:2000; Jackson 2
Immunoresearch Laboratories, Philadelphia, PA), anti-fractalkine (0.1 μg/ml; cleaved c-3
CX3CL1 [30 kDa] and total CX3CL1, R & D Systems, Inc., Minneapolis, MN).6 Purified 4
mouse anti-NF-ATc3 monoclonal antibody (1:200; BD Biosciences, Franklin Lakes, NJ). 5
6
Immunohistochemistry Formalin-fixed, paraffin-embedded tissue sections were baked 7
for 10 min at 60ºC, deparaffinized, and rehydrated. For antigen retrieval, tissues were 8
rinsed in water and heated in a Decloaker pressure cooker in 0.01M Na-citrate (pH 6.0) 9
for 3 min. The Na-citrate bath was returned to room temperature (RT) before the sections 10
were rinsed in water. Endogenous peroxidase activity was quenched by treating sections 11
with 3% H2O2 for 5 min. Sections were then washed in phosphate-buffered saline (PBS) 12
and successively blocked for 15 min at RT with appropriate serum, avidin, and biotin. 13
Tissue sections were incubated with primary antibodies overnight at 4ºC, washed with 14
PBS, and if necessary (if primary was not biotinylated), incubated with secondary 15
biotinylated antibody for 1 h at RT. Bound antibodies were detected by treating sections 16
with streptavidin-horseradish peroxidase complex for 30 min. Peroxidase activity was 17
visualized with 3,3’ diaminobenzidine, and sections were counterstained with 18
hematoxylin. Primary antibody was omitted from staining reactions as a negative 19
control. Thymus and spleen tissue served as positive controls for leukocyte staining. 20
21
Immunoblotting Frozen lung tissue was homogenized for 30 min on ice in RIPA buffer 22
(50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% 23

CIRCRESAHA/2008/182014/R2
6
SDS; Boston BioProducts, Worcester, MA) supplemented with phosphatase inhibitor 1
(Calbiochem, La Jolla, CA) and protease inhibitor (Calbiochem). Homogenates were 2
centrifuged at 2000 rpm at 4ºC for 30 min, and centrifugation was repeated on the 3
supernatants. The protein concentration of the resulting supernatant was determined 4
using Bradford reagent (Bio-Rad Laboratories, Hercules, CA). Proteins (70 μg) were 5
electrophoresed on 4 – 20% gradient Tris-Hepes-SDS gels (Pierce, Rockford IL) and 6
transferred to Immobilon PVDF membranes (Millipore Corporation, Bedford, MA). 7
Prestained molecular mass marker proteins (Bio-Rad Laboratories) were used as 8
standards for the Tris-Hepes-SDS gel electrophoresis. The PVDF membranes were 9
blocked with 5% non-fat dry milk in Tris-Buffered Saline containing 0.1% tween-20 10
(TBST) for 1 h at RT, washed with TBST, and probed with primary antibody diluted in 11
blocking buffer for 2 h at RT. The membrane was subsequently washed, incubated with 12
secondary antibody diluted in blocking buffer for 1 h at RT. Antibody labeling was 13
visualized using chemiluminescence reagent (Cell Signaling Technology). 14
15 TdT-mediated dUTP nick end labeling staining. End labeling of exposed 3’-OH ends 16
of DNA fragments was undertaken with the TdT-mediated dUTP nick end labeling 17
(TUNEL) in situ cell death detection kit (FragEL, Calbiochem, La Jolla, CA). 18
19 Statistical analysis. Data are expressed as mean ± s.e.m. Multiple comparisons were 20
made using the non-parametric Kruskal-Wallis test, and statistical significance (p 0.05) 21
between two groups was confirmed using the non-parametric Wilcoxon-Mann-Whitney 22
test. Statistics were performed using Statview 4.5 (Abacus Concepts, Inc., Berkeley, 23

CIRCRESAHA/2008/182014/R2
7
CA). In all experiments, n is equal to 5 replicates apart from immunoblots where the n is 1
equal to 4 replicates. 2
3 Results 4 5 IL-6 overexpression increases pulmonary artery pressure and ventricular 6
hypertrophy (expanded) 7
To test the hypothesis that increased IL-6 may cause increased PVR and PAH, we 8
measured the right ventricular systolic pressure (RVSP) by direct right ventricular 9
puncture, in transgenic mice over expressing IL-6 (Tg(+)). Under normoxic conditions, 10
Tg(+) mice had elevated RVSP compared to Tg(-) mice (34 ± 2 mmHg vs. 21 ± 1 mmHg, 11
p<0.05, Figure 1a). In Tg(+) mice, 3 weeks of exposure to 10% oxygen almost doubled 12
RVSP to 64 ± 7 mmHg (p<0.05 vs. normoxic), and this value was almost 2.6 times 13
higher than the RVSP in hypoxic Tg(-) mice (24 ± 1 mmHg, p<0.05, Figure 1a). As in 14
C57B1/6 mice,7 Tg(-) mice did not exhibit any significant increase in RVSP in response 15
to hypoxia (compared to normoxic controls). 16
Ventricular wall thickness increased in response to chronic pressure overload, 17
which is a consequence of elevated resistance in the pulmonary artery (PA). Right 18
ventricular hypertrophy (RVH), as measured by right ventricle weight / (left ventricle 19
weight + septum weight; RV/LV+S) and absolute right ventricle weight (RV), were 20
greater in Tg(+) mice than in Tg(-) mice under normoxic conditions (RV/LV+S: 0.4 ± 21
0.02 vs. 0.3 ± 0.02, p<0.05, Figure 1b and RV: 0.007 ± 0.0001 gm vs. 0.005 ± 0.00001 22
gm, p<0.05, Figure1 b, c). Hypoxia produced even greater RVH in Tg(+) mice 23
(RV/LV+S: 0.7 ± 0.1, and RV: 0.01 ± 0.001 gm, p<0.05 vs. normoxic control) while 24
there was no change in ventricular wall thickness in Tg(-) mice (Figure 1c). The absolute 25

CIRCRESAHA/2008/182014/R2
8
left ventricle weight remained unchanged between groups (data not shown). The 1
histological appearance of the hearts was consistent with RVH measurements, showing 2
that, in both normoxic and hypoxic conditions, right ventricular wall mass was greater in 3
Tg(+) mice (Figure 1d). 4
5
IL-6 overexpression induced muscularization throughout the entire pulmonary 6
vascular bed (expanded) 7
To determine the cause of increased PVR, we examined specific regions of the 8
pulmonary vascular tree for remodeling. Examination of the proximal branches of the 9
main PA revealed that the elastic lamina was increased in normoxic Tg(+) mice 10
compared to their Tg(-) counterparts (Figure 2 c vs. a) and was quantitatively confirmed 11
by counting the number of elastic lamina (3 ± 0.3 vs. 2 ± 0.0, p<0.05; Figure 2q). 12
Following hypoxia, the number of elastic lamina in Tg(+) mice was increased to 10 ± 13
1(figure 2d), which exceeded the number in normoxic Tg(+) mice (p<0.05) and hypoxic 14
Tg(-) littermates (2 ± 0.3, p<0.05; Figure 2q). No differences in the number of elastic 15
lamina between normoxic and hypoxic Tg(-) mice were observed. Main PA branches in 16
Tg(+) mice exhibited an increase in not only the number of elastic lamina, but also the 17
percent vessel medial wall thickness, relative to the Tg(-) control, under both normoxic 18
(7 ± 1% vs. 3 ± 1%, p<0.05) and hypoxic conditions (15 ± 1% vs. 6 ± 1%, p<0.05; Figure 19
2r). The medial wall of the main PA branches in Tg(+) mice more than doubled in 20
thickness in response to hypoxia compared to their Tg(+) normoxic controls (15± 1% vs. 21
7 ± 1%, p<0.05), while PA medial wall thickness did not change in hypoxic Tg(-) mice 22
compared to normoxic controls. 23

CIRCRESAHA/2008/182014/R2
9
The terminal bronchioles (TB) and distal acinar arterioles were examined for evidence of 1
muscularization. The most notable findings were that the distal acinar arterioles of Tg(+) 2
mice were muscularized at baseline and became more thickly muscularized in hypoxia 3
unlike the Tg(-) mice arterioles as shown by elastic staining (Figure 2 Tg(+) k, l vs. Tg(-) 4
i, j) and by immunohistochemistry with -smooth muscle actin (Figure 2 Tg(+) o, p vs. 5
Tg(-) m, n). 6
Quantitatively, we found that TB medial wall thickness was greater in normoxic Tg(+) 7
mice than normoxic Tg(-) mice (29 ± 2% vs. 24 ± 2%, p<0.05, Figure 2s). The same was 8
true of the acinar medial walls (26 ± 1% vs. 21 ± 1%, p<0.05, Figure 2t). Under hypoxic 9
conditions, medial wall thickness for Tg(+) mice was greater than that for Tg(-) mice in 10
both the TBs (49 ± 4% vs. 30 ± 3%, p<0.05, Figure 2s) and acini (49 ± 1% vs. 32 ± 2%, 11
p<0.05, Figure 2t). In Tg(+) mice, hypoxia nearly doubled medial wall thickness for both 12
the TB vessels (49 ± 4% vs. 29 ± 2% for normoxic control, p<0.05, Figure 2s) and acinar 13
vessels (49 ± 1% vs. 26 ± 1% for normoxic control, p<0.05, Figure 2t). In Tg(-) mice, 14
hypoxia induced only a 1 – 1.5-fold change in medial wall thickness for TB vessels (30 ± 15
3% vs. 24 ± 2% for normoxic control, p<0.05) and acinar vessels (32 ± 2% vs. 21 ± 1% 16
for normoxic control, p<0.05). 17
18 Hypoxia induced loss of pulmonary arterioles in IL-6 overexpressing mice. 19
To test whether a reduction in arterioles may contribute to the increase PVR in Tg(+) 20
mice, we compared the number of arterioles (per every 100 alveoli) in the alveolar bed of 21
Tg(+) and Tg(-) mice. Vessel number did not significantly differ between these mice 22
under normoxic conditions. Hypoxia reduced the number of countable arterioles at the 23
alveolar level in both Tg(+) mice (7 ± 1 vs. 12 ± 1 for normoxic control, p<0.05) and 24

CIRCRESAHA/2008/182014/R2
10
Tg(-) mice (11 ± 1 vs. 14 ± 1 for normoxic control, p = 0.05), and the relative arteriolar 1
reduction was similar for both groups (Online Figure I). However, Tg(+) mice had a 2
lower number of arterioles compared to their Tg(-) littermates (7 ± 1 vs. 11 ± 1, p<0.05). 3
Therefore, the hypoxia-induced reduction in the number of distal pulmonary vessels is 4
similar for Tg(+) and Tg(-) mice, but the absolute reduction is greater in Tg(+) mice. 5
6
Characterization of Pulmonary Vasculopathy in IL-6 Tg (+) Mice: Growth Factors. 7
To determine the factors that stimulate growth of PAEC and PASMC, immunoblot 8
analysis of whole lung lysates (Online Figure IIa) was performed. We first confirmed 9
that both VEGF and VEGFR2 levels were higher in Tg(+) mice than Tg(-) mice under 10
normoxic conditions and hypoxic conditions. Transforming growth factor (TGF)- , a 11
pro-apoptotic anti-proliferative protein, was reduced in Tg(+) mice compared to Tg(-) 12
mice under normoxic conditions and hypoxic conditions, while platelet derived growth 13
factor (PDGF) was similarly increased in all groups. This suggests that IL-6 is promoting 14
cellular growth by up-regulating growth factor VEGF and preventing inhibition of 15
cellular growth by down-regulating TGF- . 16
17
Characterization of Pulmonary Vasculopathy in IL-6 Tg(+) Mice: Mitogen 18
Activated Protein Kinases. 19
We evaluated for evidence of signal transduction pathways that may be linking pro-20
growth factor responses to intracellular downstream pro-proliferative transcription 21
factors. We found that phosphorylated extracellular regulated kinase (ERK), a pro-22
proliferative growth mitogen activated protein kinase (MAPK), was elevated in Tg(+) 23

CIRCRESAHA/2008/182014/R2
11
lung lysates compared to Tg(-) lysates in both normoxic and hypoxic conditions (Online 1
Figure IIb). Pro-apoptotic MAPKinases such as p38 and pJNK were not increased in 2
Tg(+) lung lysates compared to Tg(-) lysates in both normoxic and hypoxic conditions. 3
Taken together this would suggest that IL-6 is specifically activating pro-proliferative 4
growth kinase ERK while preventing the activation of kinases that promote apoptosis. 5
6
Characterization of Pulmonary Vasculopathy in IL-6 Tg(+ ) Mice: Pro-Proliferative 7
and Anti-Apoptotic Targets. 8
Given that cellular proliferation is increased within the walls of the pulmonary 9
vasculature of the IL-6 Tg(+) mice (Figure 4 k, l, and o) and proliferative MAPK ERK is 10
increased (Online Figure IIb), we evaluated for evidence of downstream proliferative 11
transcription factors. We found that c-myc, a basic-helix-loop-helix/leucine zipper 12
transcription factor that controls the G1-S cell cycle promoting cellular growth and 13
proliferation,8 was elevated in Tg(+) mice relative to their Tg(-) counterparts under 14
normoxic and hypoxic conditions (Online Figure IIc). In addition, c-Myc’s obligate 15
binding partner, MAX, was also exclusively elevated in Tg(+) mice relative to their Tg(-) 16
counterparts under normoxic and hypoxic conditions. This suggests that IL-6 may be 17
inducing cellular growth and proliferation and subsequent pulmonary vascular wall 18
remodeling by up-regulating pro-proliferative oncogenic transcription proteins. 19
20
Given that the growth inhibitor TGF- , and two MAPKinases that stimulate downstream 21
apoptotic signals, phosphorylated 38 and JNK, are reduced, we investigated if 22

CIRCRESAHA/2008/182014/R2
12
downstream pro-apoptotic caspase 3 is modulated by IL-6 in these mice. Cleaved 1
caspase 3 activity was increased in IL-6 Tg(+) mice at baseline compared to Tg(-) mice. 2
However in hypoxia cleaved caspase 3 activity was reduced in Tg(+) mice compared 3
with baseline room air conditions (Online Figure IId). In order to determine if 4
programmed cell death is occurring in the distal arteriolar lesions, we looked for DNA 5
fragmentation, a marker of apoptosis, and observed that in the IL-6 Tg(+) mice, there was 6
DNA fragmentation in epithelial cells and possibly in histiocytes of alveolar units but not 7
in the walls of the distal arterioles (Online Figure IIe). Taken together, IL-6 8
overexpression may be selectively preventing programmed cell death by apoptosis within 9
the pulmonary vascular bed. 10
11
To determine which anti-apoptotic proteins may be involved in inhibiting caspase 3 and 12
preventing subsequent cell death within the vascular bed, we probed for two important 13
anti-apoptotic proteins, Bcl-2 and survivin. We found that Bcl-2 was elevated in Tg(+) 14
mice relative to their Tg(-) counterparts under normoxic and hypoxic conditions (Online 15
Figure IId) while pro-apoptotic protein BAX levels were similar in Tg(-) and Tg(+) mice 16
and remained unchanged in hypoxic conditions. We found that survivin is markedly 17
elevated in Tg(+) mice compared to Tg(-) mice at baseline and in hypoxia (Online Figure 18
IId). This suggests that IL-6 may be inducing a pro-survival cellular state by modulating 19
several anti-apoptotic proteins. 20
21
Characterization of Lymphocyte Recruitment and Function. 22

CIRCRESAHA/2008/182014/R2
13
To determine the signals that IL-6 employs to control the recruitment of lymphocytes, we 1
probed for chemokines that are known to be involved in lymphocyte trafficking. We 2
found that Tg (+) mice had elevated levels of activated fractalkine (c-CX3CL1), a PAEC-3
derived chemokine that recruits T cells and mediates T cell adhesion to endothelial cells, 4
at baseline and in hypoxic conditions (Online Figure III). Monocyte chemoattractant 5
protein, MCP-1, a chemokine that recruits monocytes and lymphocytes and may have a 6
role in smooth muscle cell proliferation was increased in IL-6 Tg (+) mice at room air but 7
was reduced in hypoxia (Online Figure III). Tumor necrosis factor (TNF)-alpha was 8
reduced in both normoxic and hypoxic conditions in the IL-6 Tg (+) mice compared to 9
Tg (-) mice. Nuclear factor of activated T-cells, cytoplasmic, calcineurin-3 (NF-ATc3), 10
which may have a role in promoting a state of proliferation and suppress mitochondrial–11
dependent apoptosis9, was slightly increased in IL-6 Tg (+) mice whole lung lysates 12
(Online Figure III). Taken together, this data supports the concept that overexpression of 13
IL-6 stimulates the recruitment of T cells through up-regulation of specific chemokines 14
depending on the presence or absence of hypoxia and stimulates T cells to home to distal 15
pulmonary arteriole vessels. In turn, T cells may be enhancing IL-6’s pro-survival 16
apoptotic resistant cellular phenotypic effects. 17
18
Discussion 19
IL-6 and cMyc/MAX Expanded 20
IL-6 overexpression may predispose to exaggerated PAH as a result of coordinating a 21
number of downstream pro-proliferative, pro-survival, and anti-apoptotic signaling 22
pathways (Figure 6). We found that c-myc, and its obligate binding partner, MAX may 23

CIRCRESAHA/2008/182014/R2
14
be key in IL-6’s downstream proliferative signal cascade, promoting cellular proliferative 1
phenotypes in PAH. Consistent with our findings, endothelin stimulated rat aortic 2
smooth muscle cells are shifted into a pro-proliferative state in the presence of ERK 3
induced c-myc activation.10 It also has been shown that pulmonary arteries from rats 4
exposed to hypoxia have elevated levels of c-myc mRNA.11 In addition, YY1 growth 5
regulator of vascular smooth muscle cells targets c-myc downstream leading to increased 6
smooth muscle specific gene expression, proliferation and neonatal piglet pulmonary 7
hypertension.12 Furthermore, in the presence of TGF- /BMP proteins, c-myc expression 8
is attenuated contributing to PASMC apoptosis.13 How the c-myc/max complex may be 9
altering its target cell cycle genes and whether the additional stress of hypoxia may alter 10
its effects on downstream targets is unclear at present. However, given the number of 11
angioproliferative lesions at baseline, which likely results in localized hypoxia, and the 12
increase in the number of lesions in hypoxia, we hypothesize, based on extensive work in 13
tumor hypoxia deprivation biology,8 that hypoxia inducible factors may be facilitating 14
the formation of c-myc-max complex and thereby augmenting the expression of cell 15
cycle genes and exaggerating the hyper-proliferative cellular phenotypes in this transgene 16
mouse. Further investigation with this IL-6 Tg(+) mouse will allow us to evaluate the 17
role of IL-6 in the presence of hypoxia and associated physiologic changes thus imposed 18
by hypoxic vasoconstriction. 19
20
IL-6 and Apoptosis Expanded 21
While IL-6 appears to be driving a number of proteins that promote cellular proliferation, 22
it is also clear from our work that IL-6 is also affecting apoptosis by down regulating 23

CIRCRESAHA/2008/182014/R2
15
TGF- and pro-apoptotic MAPKs, pJNK and p38 (Figure 6). As with hyperoxic injury in 1
Tg(+) mice14 and H2O2 injury in IL-6 stimulated endothelial cells,15 caspase 3, the main 2
gate keeper to programmed cell death, appears to be activated at baseline and is 3
subsequently down-regulated when under the stress of hypoxia. However, evidence for 4
DNA fragmentation, a marker of apoptosis, is observed only in epithelial cells and 5
possibly histiocytes lining the alveoli, not in the cells of the obliterative lesions of the 6
distal pulmonary vasculature at baseline or in hypoxia. This would suggest that IL-6’s 7
role in preventing cell death by apoptosis is PA wall specific at baseline and persists in 8
hypoxia. Dysregulation of mediators of apoptosis in the PA wall of patients with PAH 9
favoring suppression of apoptosis has been shown in gene microarray studies.16 Anti-10
apoptotic proteins Bcl-2 and survivin have been found to be up-regulated in patients with 11
PAH16, 17 where they are thought to impair the activity of Kv channels17, 18and subsequent 12
mitochondria-dependent apoptosis.19 Similar to patients with PAH, IL-6 Tg(+) mice also 13
share a similar anti-apoptotic biology with increased expression of both bcl-2 and 14
survivin. At present, it remains unclear what triggers induced by IL-6 modulate the 15
expression of these two anti-apoptotic proteins. However it is clear that DNA 16
fragmentation within the complex obliterative vascular lesions of L-6 Tg(+) mice is 17
absent favoring a pro-proliferative cell state, as observed in angioproliferative lesions of 18
patients with PAH. 19
20
IL-6 and Inflammation Expanded 21
Elevation of IL-6 in PAH patients suggests that cellular immunity may play an active role 22
in the dysregulation of PAEC and PASMC and the development of PAH. Accordingly, 23

CIRCRESAHA/2008/182014/R2
16
plexigenic and concentric lesions in patients with severe PAH contain inflammatory cells, 1
consisting of T cells, macrophages, and to a lesser extent, B cells20, 21. Similarly, the 2
obliterative complex distal arterial lesions in Tg(+) mice also contain an abundance of T 3
cells and to a lesser extent, B cells, while B cells appear to surround the large and mid 4
sized airways.22 The mechanisms underlying inflammatory cell recruitment and 5
transendothelial migration into pulmonary artery vessel walls during PAH are unclear, 6
although the chemokines RANTES23 and fractalkine24 may participate in inflammatory 7
cell trafficking.25 Hyper-proliferative PAEC may mediate the adhesion of the T 8
lymphocytes by over-expressing fractalkine, as the cleaved activated form of fractalkine 9
is up-regulated in the lungs of IL-6 Tg(+) mice as well as in plexigenic lesions of 10
patients.24 Further work examining T lymphocyte trafficking in Tg(+) mice will be 11
instrumental in understanding their role in the vascular changes seen in patients with 12
PAH. 13
There has been much controversy regarding the role of T cells in PAH, where T 14
cell-mediated immunity has been found to be protective against PAH in VEGFR blocker 15
treated nude rats5 while in antigen challenged mice the Th2 immune response is 16
associated with vascular muscularization.26 In the IL-6 Tg(+) mouse, IL-6 and T cells 17
may have a trans-signaling relationship, whereby T cell activation driven by IL-6 18
contributes to the perpetuation of IL-6’s pro-inflammatory pro-proliferative anti-19
apoptotic effects on neighboring PASM and PAEC (Figure 6). Such a relationship has 20
been thought to have been an important mechanism underlying other chronic 21
inflammatory diseases,27 and may be one of the many important IL-6 mechanisms 22
involved in the pathogenesis of PAH. NFAT proteins, produced and carried to PA 23

CIRCRESAHA/2008/182014/R2
17
vessels in lymphocytes have recently been noted to be activated in human PAH and may 1
be contributing to the resistance to apoptosis within the PA wall. In IL-6 Tg(+) mice, 2
NF-ATc3 was only slightly increased in the IL-6 Tg(+) mice compared with Tg(-) mice 3
at baseline and in hypoxia, and therefore may or may not be associated with PAEC and 4
PASMC insensitivity to apoptosis. Its significance may be under-represented by the lack 5
of cell specificity as it was tested in whole lung lysates. Taken together, T cells in an IL-6
6 rich milieu are prominently situated within the distal arteriolar vessels, and further work 7
to evaluate their role, whether protective or stimulatory, will be key to understanding 8
their presence in human PAH disease. 9
10
IL-6 and Loss of PA vessels Expanded 11
Whether the loss of small pulmonary arteriolar vessels contributes to the development of 12
PVR is controversial. The importance of this loss may be negligible since chronic 13
hypoxic experimental PAH models have considerable angiogenesis28 and Rho kinase 14
inhibitors normalize pulmonary pressures during chronic hypoxia via vasodilation.29 15
However, vessel loss is induced by many other experimental PAH stimuli, including 16
injection of monocrotaline,30 monocrotaline combined with pneumonectomy,31, 32 chronic 17
hyperoxia,32 and creation of aortopulmonary shunts.33 Clinically, artery loss occurs in 18
idiopathic PAH,34 as well as in conditions associated with PAH, including congenital 19
heart disease35 and lung developmental abnormalities.36 Here, hypoxia induced a 20
significant loss of distal arterioles in Tg(+) mice, which are known to have abnormal 21
alveoli with enlarged terminal air sacs.1 This altered anatomy, which may result in a 22
significant decrease in the number of arterioles, could significantly contribute to 23

CIRCRESAHA/2008/182014/R2
18
increased PVR. We found no significant differences in vessel number between Tg(-) and 1
(+) mice under normoxic conditions and the hypoxia-induced arteriolar reduction was 2
similar between groups, despite a significant increase in PVR in Tg(+) hypoxic mice. 3
Thus, arterial vessel loss alone may not account for increased PVR in IL-6 Tg(+) mice. 4
5
6
References: 7 8 1. Ward NS, Waxman AB, Homer RJ, Mantell LL, Einarsson O, Du Y, Elias JA. 9
Interleukin-6-Induced Protection in Hyperoxic Acute Lung Injury. Am J Respir 10
Cell Mol Biol. 2000;22:535-542. 11
2. Zhu YJ, Kradin R, Brandstetter RD, Staton G, Moss J, Hales CA. Hypoxic 12
pulmonary hypertension in the mast cell-deficient mouse. Journal of Applied 13
Physiology: Respiratory, Environmental & Exercise Physiology. 1983;54:680-14
686. 15
3. Quinn DA, Du HK, Thompson BT, Hales CA. Amiloride analogs inhibit chronic 16
hypoxic pulmonary hypertension. American Journal of Respiratory & Critical 17
Care Medicine. 1998;157:1263-1268. 18
4. Merklinger SL, Wagner RA, Spiekerkoetter E, Hinek A, Knutsen RH, Kabir MG, 19
Desai K, Hacker S, Wang L, Cann GM, Ambartsumian NS, Lukanidin E, 20
Bernstein D, Husain M, Mecham RP, Starcher B, Yanagisawa H, Rabinovitch M. 21
Increased fibulin-5 and elastin in S100A4/Mts1 mice with pulmonary 22
hypertension. Circulation Research. 2005;97:596-604. 23

CIRCRESAHA/2008/182014/R2
19
5. Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, 1
Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absence of T cells confers 2
increased pulmonary arterial hypertension and vascular remodeling. American 3
Journal of Respiratory & Critical Care Medicine. 2007;175:1280-1289. 4
6. Habasque C, Satie AP, Aubry F, Jegou B, Samson M. Expression of fractalkine in 5
the rat testis: molecular cloning of a novel alternative transcript of its gene that is 6
differentially regulated by pro-inflammatory cytokines. Mol Hum Reprod. 7
2003;9:449-455. 8
7. Raoul W, Wagner-Ballon O, Saber G, Hulin A, Marcos E, Giraudier S, 9
Vainchenker W, Adnot S, Eddahibi S, Maitre B. Effects of bone marrow-derived 10
cells on monocrotaline- and hypoxia-induced pulmonary hypertension in mice. 11
Respir Res. 2007;8:8. 12
8. Huang LE. Carrot and stick: HIF-alpha engages c-Myc in hypoxic adaptation. 13
Cell Death Differ. 2008;15:672-677. 14
9. Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, 15
Hashimoto K, Bonnet SN, Michelakis ED. The nuclear factor of activated T cells 16
in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl 17
Acad Sci U S A. 2007;104:11418-11423. 18
10. Chen S, Qiong Y, Gardner DG. A role for p38 mitogen-activated protein kinase 19
and c-myc in endothelin-dependent rat aortic smooth muscle cell proliferation. 20
Hypertension. 2006;47:252-258. 21

CIRCRESAHA/2008/182014/R2
20
11. Cai Y, Han M, Luo L, Song W, Zhou X. Increased expression of PDGF and c-1
myc genes in lungs and pulmonary arteries of pulmonary hypertensive rats 2
induced by hypoxia. Chin Med Sci J. 1996;11:152-156. 3
12. Favot L, Hall SM, Haworth SG, Kemp PR. Cytoplasmic YY1 is associated with 4
increased smooth muscle-specific gene expression: implications for neonatal 5
pulmonary hypertension. Am J Pathol. 2005;167:1497-1509. 6
13. Fantozzi I, Platoshyn O, Wong AH, Zhang S, Remillard CV, Furtado MR, 7
Petrauskene OV, Yuan JX. Bone morphogenetic protein-2 upregulates expression 8
and function of voltage-gated K+ channels in human pulmonary artery smooth 9
muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;291:L993-1004. 10
14. Ward NS, Waxman AB, Homer RJ, Mantell LL, Einarsson O, Du Y, Elias JA. 11
Interleukin-6-induced protection in hyperoxic acute lung injury. American 12
Journal of Respiratory Cell & Molecular Biology. 2000;22:535-542. 13
15. Waxman AB, Mahboubi K, Knickelbein RG, Mantell LL, Manzo N, Pober JS, 14
Elias JA. Interleukin-11 and Interleukin-6 Protect Cultured Human Endothelial 15
Cells from H2O2-Induced Cell Death. Am J Respir Cell Mol Biol. 16
2003;29:513-522. 17
16. Geraci MW, Moore M, Gesell T, Yeager ME, Alger L, Golpon H, Gao B, Loyd 18
JE, Tuder RM, Voelkel NF. Gene expression patterns in the lungs of patients with 19
primary pulmonary hypertension: a gene microarray analysis. Circulation 20
Research. 2001;88:555-562. 21
17. McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, Bonnet S, 22
Puttagunta L, Michelakis ED. Gene therapy targeting survivin selectively induces 23

CIRCRESAHA/2008/182014/R2
21
pulmonary vascular apoptosis and reverses pulmonary arterial hypertension.[see 1
comment]. Journal of Clinical Investigation. 2005;115:1479-1491. 2
18. Ekhterae D, Platoshyn O, Krick S, Yu Y, McDaniel SS, Yuan JX. Bcl-2 decreases 3
voltage-gated K+ channel activity and enhances survival in vascular smooth 4
muscle cells. Am J Physiol Cell Physiol. 2001;281:C157-165. 5
19. Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 6
2003;3:46-54. 7
20. Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell 8
growth and elements of inflammation are present in plexiform lesions of 9
pulmonary hypertension.[see comment]. Am J Pathol. 1994;144:275-285. 10
21. Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesis and evolution of 11
plexiform lesions in pulmonary hypertension associated with scleroderma and 12
human immunodeficiency virus infection. Human pathology. 1997;28:434-442. 13
22. DiCosmo BF, Geba GP, Picarella D, Elias JA, Rankin JA, Stripp BR, Whitsett 14
JA, Flavell RA. Airway epithelial cell expression of interleukin-6 in transgenic 15
mice. Uncoupling of airway inflammation and bronchial hyperreactivity. Journal 16
of Clinical Investigation. 1994;94:2028-2035. 17
23. Dorfmuller P, Zarka V, Durand-Gasselin I, Monti G, Balabanian K, Garcia G, 18
Capron F, Coulomb-Lhermine A, Marfaing-Koka A, Simonneau G, Emilie D, 19
Humbert M. Chemokine RANTES in severe pulmonary arterial hypertension. 20
American Journal of Respiratory & Critical Care Medicine. 2002;165:534-539. 21
24. Balabanian K, Foussat A, Dorfmuller P, Durand-Gasselin I, Capel F, Bouchet-22
Delbos L, Portier A, Marfaing-Koka A, Krzysiek R, Rimaniol AC, Simonneau G, 23

CIRCRESAHA/2008/182014/R2
22
Emilie D, Humbert M. CX(3)C chemokine fractalkine in pulmonary arterial 1
hypertension.[see comment]. American Journal of Respiratory & Critical Care 2
Medicine. 2002;165:1419-1425. 3
25. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte 4
emigration: the multistep paradigm. Cell. 1994;76:301-314. 5
26. Daley E, Emson C, Guignabert C, de Waal Malefyt R, Louten J, Kurup V, 6
Hogaboam C, Taraseviciene-Stewart L, Voelkel N, Rabinovitch M, Grunig E, 7
Grunig G. Pulmonary arterial remodeling induced by a Th2 immune response. 8
The Journal of Experimental Medicine. 2008;205:361-372. 9
27. Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, Schutz M, Bartsch 10
B, Holtmann M, Becker C, Strand D, Czaja J, Schlaak JF, Lehr HA, Autschbach 11
F, Schurmann G, Nishimoto N, Yoshizaki K, Ito H, Kishimoto T, Galle PR, Rose-12
John S, Neurath MF. Blockade of interleukin 6 trans signaling suppresses T-cell 13
resistance against apoptosis in chronic intestinal inflammation: evidence in crohn 14
disease and experimental colitis in vivo. Nat Med. 2000;6:583-588. 15
28. Howell K, Preston RJ, McLoughlin P. Chronic hypoxia causes angiogenesis in 16
addition to remodelling in the adult rat pulmonary circulation. J Physiol (Lond). 17
2003;547:133-145. 18
29. Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P. 19
Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the 20
pulmonary circulation. Circulation Research. 2005;97:185-191. 21

CIRCRESAHA/2008/182014/R2
23
30. Ye CL, Rabinovitch M. Inhibition of elastolysis by SC-37698 reduces 1
development and progression of monocrotaline pulmonary hypertension. 2
American Journal of Physiology. 1991;261:H1255-1267. 3
31. O'Blenes SB, Fischer S, McIntyre B, Keshavjee S, Rabinovitch M. Hemodynamic 4
unloading leads to regression of pulmonary vascular disease in rats.[see 5
comment]. J Thorac Cardiovasc Surg. 2001;121:279-289. 6
32. Koppel R, Han RN, Cox D, Tanswell AK, Rabinovitch M. Alpha 1-antitrypsin 7
protects neonatal rats from pulmonary vascular and parenchymal effects of 8
oxygen toxicity. Pediatr Res. 1994;36:763-770. 9
33. Rendas A, Lennox S, Reid L. Aorta-pulmonary shunts in growing pigs. 10
Functional and structural assessment of the changes in the pulmonary circulation. 11
J Thorac Cardiovasc Surg. 1979;77:109-118. 12
34. Meyrick B, Clarke SW, Symons C, Woodgate DJ, Reid L. Primary pulmonary 13
hypertension: a case report including electronmicroscopic study. Br J Dis Chest. 14
1974;68:11-20. 15
35. Rabinovitch M, Keane JF, Norwood WI, Castaneda AR, Reid L. Vascular 16
structure in lung tissue obtained at biopsy correlated with pulmonary 17
hemodynamic findings after repair of congenital heart defects. Circulation. 18
1984;69:655-667. 19
36. Bohn D, Tamura M, Perrin D, Barker G, Rabinovitch M. Ventilatory predictors of 20
pulmonary hypoplasia in congenital diaphragmatic hernia, confirmed by 21
morphologic assessment. J Pediatr. 1987;111:423-431. 22
23

CIRCRESAHA/2008/182014/R2
24
Legend (Supplement) 1
2 Online Figure I The number of arteries per 100 alveoli in IL-6 Tg(+) and Tg(-) mice in 3
normoxic conditions are similar at baseline. Under hypoxic conditions, the number of 4
arteries per 100 alveoli are reduced in both Tg(+) and Tg(-) mice (‡ vs. normoxic 5
Tg(+);p<0.05, † vs. normoxic Tg(-);p<0.05) with IL-6 Tg(+) mice having the highest 6
reduction (‡ vs. hypoxic Tg(-);p<0.05). 7
Online Figure II (a) Representative immunoblot protein expression of TGF- , PDGF, 8
VEGF, and VEGFR2 of lung lysates from IL-6 Tg(+) and Tg(-) mice exposed to 9
normoxia and 3 weeks of hypoxia. (b) Representative immunoblot protein expression of 10
phosphorylated ERK 42, ERK 42/44, phosphorylated p38, p38, phosphorylated JNK-1 11
and JNK2/3, and JNK.(c) Representative immunoblot protein expression of PCNA, c-12
myc, and MAX. (d) Representative immunoblot protein expression of survivin, Bcl-2, 13
BAX, cleaved caspase-3, and caspase 3. -actin used as control. (e) Representative 14
photomicrographs of distal acinar arterioles of the PA vasculature of IL-6 Tg(+) and Tg(-15
) mice in normoxic and hypoxic conditions exposed to TdT-mediated dUTP nick end 16
labeling (TUNEL) for DNA fragmentation evaluation. The distal arterioles (arrow with 17
dark circle) and specifically the occlusive lesions in Tg(+) mice at baseline and in 18
hypoxia have no evidence of DNA fragmentation, while epithelial cells and possibly 19
histiocytes lining the alveolar walls are TUNEL positive at baseline (arrow, see inset). 20
TUNEL staining, magnification x400, bar=0.001mm and inset, magnification x1000, 21
bar=0.0005mm. -actin used as a control. 22
23

CIRCRESAHA/2008/182014/R2
25
Online Figure III Representative immunoblot protein expression of TNF- , MCP-1, 1
cleaved chemokine fractalkine (c-CX3CL1), total CX3CL1, NF-ATc3, T cells (CD3) and 2
B cells (CD19) in whole lung lysates of IL-6 Tg (+) and Tg (-) mice exposed to normoxia 3
and 3 weeks of hypoxia. -actin used as a control. 4
5
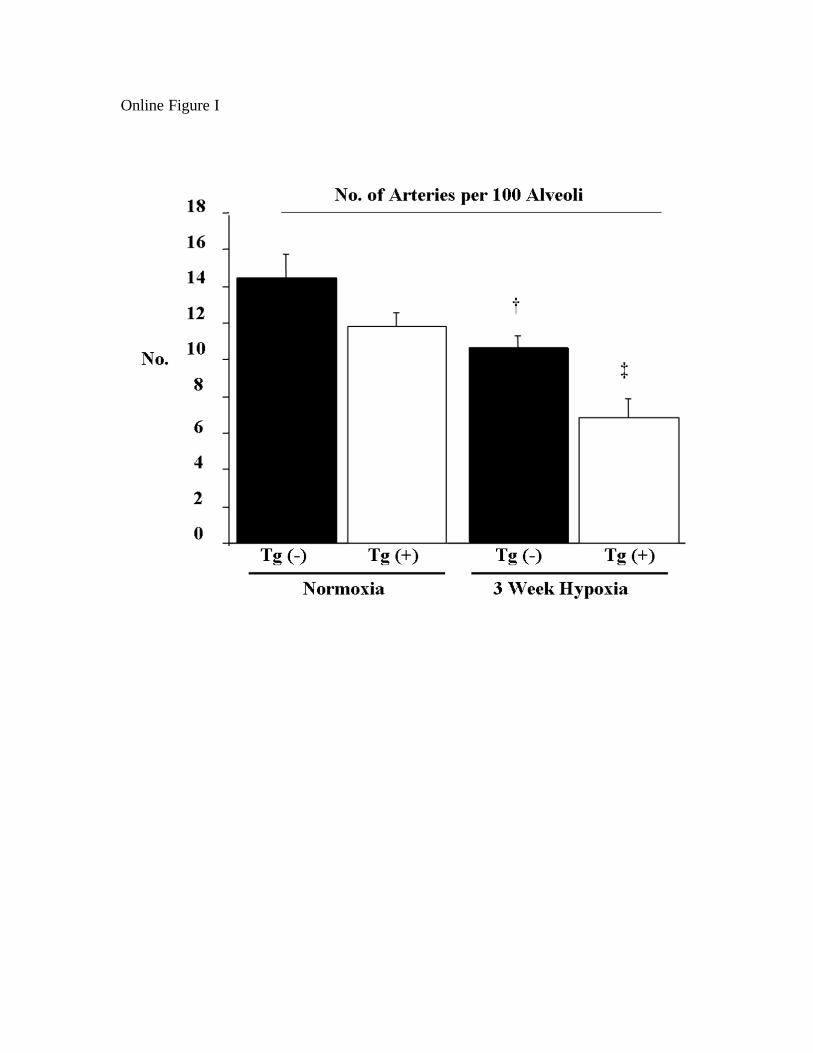
Online Figure I
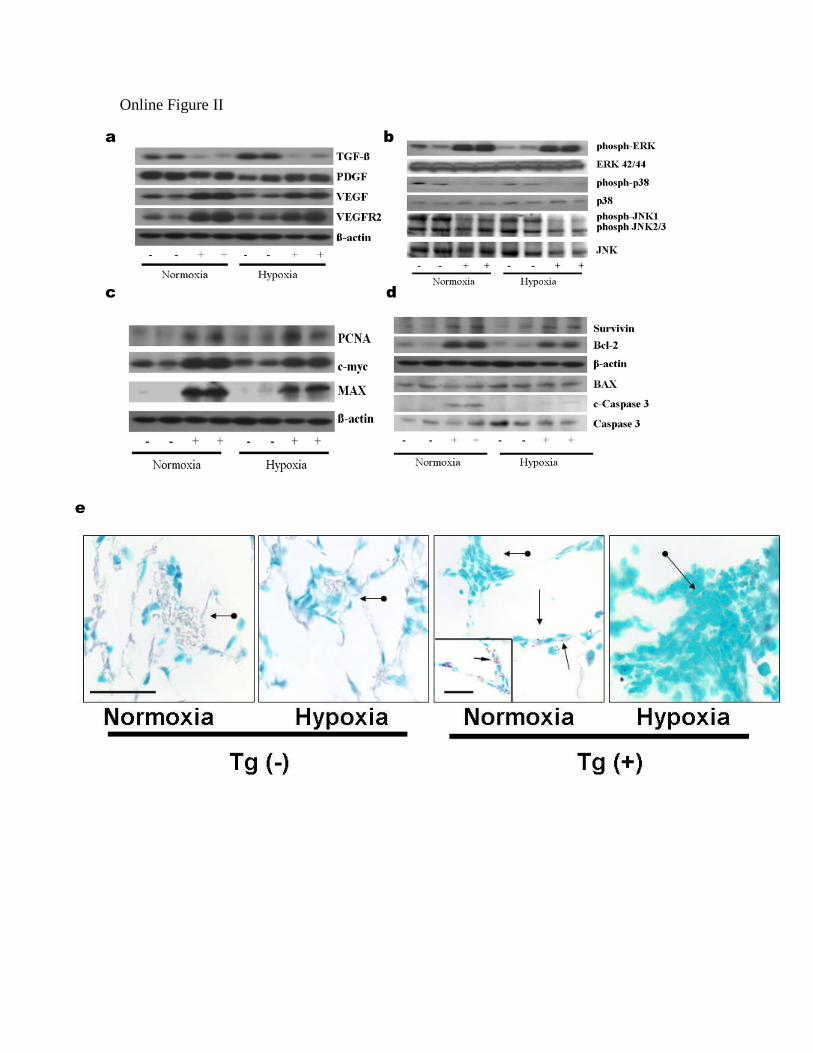
Online Figure II
a b
c
e
d
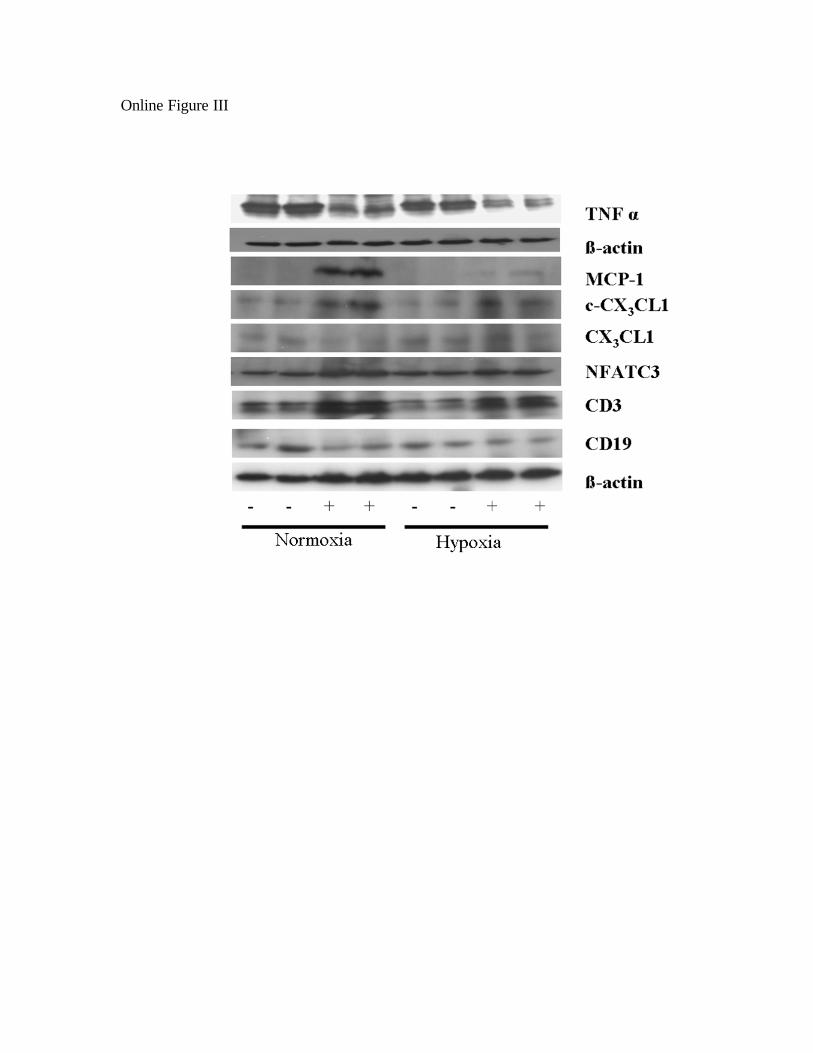
Online Figure III

and Aaron B. WaxmanM. Kathryn Steiner, Olga L. Syrkina, Narasaish Kolliputi, Eugene J. Mark, Charles A. Hales
Interleukin-6 Overexpression Induces Pulmonary Hypertension
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2008 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.108.1820142009;104:236-244; originally published online December 12, 2008;Circ Res.
http://circres.ahajournals.org/content/104/2/236World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2008/12/12/CIRCRESAHA.108.182014.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on August 21, 2015http://circres.ahajournals.org/Downloaded from




















