
Interaction between neuronal nitric oxide synthase signaling and temperature influences sarcoplasmic...
18
46 N itric oxide (NO) exerts diverse regulation of cell signal- ing 1 through a broad range of post-translational modifica- tions, largely through S-nitrosylation of specific cysteine thiol moieties. 2–4 Despite the increased understanding of NO signal- ing and the identification of various NO synthase (NOS) iso- forms in the cardiac myocyte, a consensus has not emerged on the mechanism underlying NO regulation of excitation–con- traction coupling, and studies have reported diametrically op- posite results. For example, NOS1–deficient mice-1 (NOS1 −⁄− ) showed opposite behavior of cardiomyocytes in calcium (Ca 2+ ) handling and contractility 5–7 or sarcoplasmic reticulum (SR) Ca 2+ leak 8,9 in comparison with wild-type (WT) cells. Similarly, studies showed opposite effects on β-adrenergic contractile responses. 1,7 We hypothesized that other factor(s) can fundamentally change the direction of an NO-based phys- iological response. Here, we identify temperature as a key de- terminant of cardiomyocyte response to NO. Temperature has a broad influence on Ca 2+ signaling in cardiomyocytes 10,11 and there is a close relationship between body temperature and NO production in the pathogenesis Cellular Biology © 2014 American Heart Association, Inc. Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.116.305172 Rationale: Although nitric oxide (NO) signaling modulates cardiac function and excitation–contraction coupling, opposing results because of inconsistent experimental conditions, particularly with respect to temperature, confound the ability to elucidate NO signaling pathways. Here, we show that temperature significantly modulates NO effects. Objective: To test the hypothesis that temperature profoundly affects nitroso–redox equilibrium, thereby affecting sarcoplasmic reticulum (SR) calcium (Ca 2+ ) leak. Methods and Results: We measured SR Ca 2+ leak in cardiomyocytes from wild-type (WT), NO/redox imbalance (neuronal nitric oxide synthase–deficient mice-1 [NOS1 −⁄− ]), and hyper S-nitrosoglutathione reductase–deficient (GSNOR −⁄− ) mice. In WT cardiomyocytes, SR Ca 2+ leak increased because temperature decreased from 37°C to 23°C, whereas in NOS1 −⁄− cells, the leak suddenly increased when the temperature surpassed 30°C. GSNOR −⁄− cardiomyocytes exhibited low leak throughout the temperature range. Exogenously added NO had a biphasic effect on NOS1 −⁄− cardiomyocytes; reducing leak at 37°C but increasing it at subphysiological temperatures. Oxypurinol and Tempol diminished the leak in NOS1 −⁄− cardiomyocytes. Cooling from 37°C to 23°C increased reactive oxygen species generation in WT but decreased it in NOS1 −⁄− cardiomyocytes. Oxypurinol further reduced reactive oxygen species generation. At 23°C in WT cells, leak was decreased by tetrahydrobiopterin, an essential NOS cofactor. Cooling significantly increased SR Ca 2+ content in NOS1 −⁄− cells but had no effect in WT or GSNOR −⁄− . Conclusions: Ca 2+ leak and temperature are normally inversely proportional, whereas NOS1 deficiency reverses this effect, increasing leak and elevating reactive oxygen species production because temperature increases. Reduced denitrosylation (GSNOR deficiency) eliminates the temperature dependence of leak. Thus, temperature regulates the balance between NO and reactive oxygen species which in turn has a major effect on SR Ca 2+ . (Circ Res. 2015;116:46-55. DOI: 10.1161/CIRCRESAHA.116.305172.) Key Words: calcium signaling ■ induced hypothermia ■ nitric oxide ■ nitric oxide synthase ■ nitroso-redox imbalance ■ reactive oxygen species ■ 5,6,7,8-tetrahydrobiopterin Original received September 8, 2014; revision received October 14, 2014; accepted October 16, 2014. In September 2014, the average time from submission to first decision for all original research papers submitted to Circulation Research was 14.29 days. From the Interdisciplinary Stem Cell Institute, University of Miami Miller School of Medicine, FL. The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA. 116.305172/-/DC1. This article was sent to Donald M. Bers, Consulting Editor, for review by expert referees, editorial decision, and final disposition. Correspondence to Joshua M. Hare, 1501 NW 10th Ave, Biomedical Research Building (BRB), Room 824, Miami, FL 33136. E-mail [email protected] Interaction Between Neuronal Nitric Oxide Synthase Signaling and Temperature Influences Sarcoplasmic Reticulum Calcium Leak Role of Nitroso–Redox Balance Raul A. Dulce, Vera Mayo, Erika B. Rangel, Wayne Balkan, Joshua M. Hare at University of Miami School of Medicine on January 7, 2015 http://circres.ahajournals.org/ Downloaded from at University of Miami School of Medicine on January 7, 2015 http://circres.ahajournals.org/ Downloaded from at University of Miami School of Medicine on January 7, 2015 http://circres.ahajournals.org/ Downloaded from at University of Miami School of Medicine on January 7, 2015 http://circres.ahajournals.org/ Downloaded from at University of Miami School of Medicine on January 7, 2015 http://circres.ahajournals.org/ Downloaded from at University of Miami School of Medicine on January 7, 2015 http://circres.ahajournals.org/ Downloaded from at University of Miami School of Medicine on January 7, 2015 http://circres.ahajournals.org/ Downloaded from at University of Miami School of Medicine on January 7, 2015 http://circres.ahajournals.org/ Downloaded from at University of Miami School of Medicine on January 7, 2015 http://circres.ahajournals.org/ Downloaded from
Transcript of Interaction between neuronal nitric oxide synthase signaling and temperature influences sarcoplasmic...

46
Nitric oxide (NO) exerts diverse regulation of cell signal-ing1 through a broad range of post-translational modifica-
tions, largely through S-nitrosylation of specific cysteine thiol moieties.2–4 Despite the increased understanding of NO signal-ing and the identification of various NO synthase (NOS) iso-forms in the cardiac myocyte, a consensus has not emerged on the mechanism underlying NO regulation of excitation–con-traction coupling, and studies have reported diametrically op-posite results. For example, NOS1–deficient mice-1 (NOS1−⁄−) showed opposite behavior of cardiomyocytes in calcium
(Ca2+) handling and contractility5–7 or sarcoplasmic reticulum (SR) Ca2+ leak8,9 in comparison with wild-type (WT) cells. Similarly, studies showed opposite effects on β-adrenergic contractile responses.1,7 We hypothesized that other factor(s) can fundamentally change the direction of an NO-based phys-iological response. Here, we identify temperature as a key de-terminant of cardiomyocyte response to NO.
Temperature has a broad influence on Ca2+ signaling in cardiomyocytes10,11 and there is a close relationship between body temperature and NO production in the pathogenesis
Cellular Biology
© 2014 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.116.305172
Rationale: Although nitric oxide (NO) signaling modulates cardiac function and excitation–contraction coupling, opposing results because of inconsistent experimental conditions, particularly with respect to temperature, confound the ability to elucidate NO signaling pathways. Here, we show that temperature significantly modulates NO effects.
Objective: To test the hypothesis that temperature profoundly affects nitroso–redox equilibrium, thereby affecting sarcoplasmic reticulum (SR) calcium (Ca2+) leak.
Methods and Results: We measured SR Ca2+ leak in cardiomyocytes from wild-type (WT), NO/redox imbalance (neuronal nitric oxide synthase–deficient mice-1 [NOS1−⁄−]), and hyper S-nitrosoglutathione reductase–deficient (GSNOR−⁄−) mice. In WT cardiomyocytes, SR Ca2+ leak increased because temperature decreased from 37°C to 23°C, whereas in NOS1−⁄− cells, the leak suddenly increased when the temperature surpassed 30°C. GSNOR−⁄− cardiomyocytes exhibited low leak throughout the temperature range. Exogenously added NO had a biphasic effect on NOS1−⁄− cardiomyocytes; reducing leak at 37°C but increasing it at subphysiological temperatures. Oxypurinol and Tempol diminished the leak in NOS1−⁄− cardiomyocytes. Cooling from 37°C to 23°C increased reactive oxygen species generation in WT but decreased it in NOS1−⁄− cardiomyocytes. Oxypurinol further reduced reactive oxygen species generation. At 23°C in WT cells, leak was decreased by tetrahydrobiopterin, an essential NOS cofactor. Cooling significantly increased SR Ca2+ content in NOS1−⁄− cells but had no effect in WT or GSNOR−⁄−.
Conclusions: Ca2+ leak and temperature are normally inversely proportional, whereas NOS1 deficiency reverses this effect, increasing leak and elevating reactive oxygen species production because temperature increases. Reduced denitrosylation (GSNOR deficiency) eliminates the temperature dependence of leak. Thus, temperature regulates the balance between NO and reactive oxygen species which in turn has a major effect on SR Ca2+. (Circ Res. 2015;116:46-55. DOI: 10.1161/CIRCRESAHA.116.305172.)
Key Words: calcium signaling ■ induced hypothermia ■ nitric oxide ■ nitric oxide synthase ■ nitroso-redox imbalance ■ reactive oxygen species ■ 5,6,7,8-tetrahydrobiopterin
Original received September 8, 2014; revision received October 14, 2014; accepted October 16, 2014. In September 2014, the average time from submission to first decision for all original research papers submitted to Circulation Research was 14.29 days.
From the Interdisciplinary Stem Cell Institute, University of Miami Miller School of Medicine, FL. The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.
116.305172/-/DC1. This article was sent to Donald M. Bers, Consulting Editor, for review by expert referees, editorial decision, and final disposition. Correspondence to Joshua M. Hare, 1501 NW 10th Ave, Biomedical Research Building (BRB), Room 824, Miami, FL 33136. E-mail
Interaction Between Neuronal Nitric Oxide Synthase Signaling and Temperature Influences Sarcoplasmic
Reticulum Calcium LeakRole of Nitroso–Redox Balance
Raul A. Dulce, Vera Mayo, Erika B. Rangel, Wayne Balkan, Joshua M. Hare
at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from

Dulce et al Temperature Influences NOS1 Signaling and Ca2+ Leak 47
of focal cerebral ischemia.12,13 Temperature modulates NO production in vitro, directly affecting NOS activity.14 Thus, changes in temperature, by affecting NO production and therefore the nitroso–redox (NO/redox) balance15–17 in the heart, may be closely related to ryanodine receptor (RyR2)-mediated SR Ca2+ leak. Post-translational modifications, such as S-nitrosylation, of RyR2 are influenced by cellular NO/re-dox state and can both positively and negatively affect SR Ca2+ leak.18–20 Signaling defects in Ca2+ handling, such as Ca2+ leak, contribute to impaired contractility in the failing heart, thus depleting SR Ca2+ storage as a consequence of the leak and leading to impaired contractile function of the heart.
This study focused on the effect of temperature on SR Ca2+ leak in isolated cardiomyocytes from WT mice or mouse models of aberrant S-nitrosylation, NOS1−/− and S-nitrosoglutathione reductase–deficient (GSNOR−/−) and shows that reactive oxygen/nitrogen species signaling are affected by temperature.
MethodsAnimal ModelsWe studied age-matched C57BL/6J mice (WT; n=26) and mice with a homozygous deletion of NOS1 (B6;129S4-Nos1tm1Plh/J; n=20; Jackson Laboratories, Bar Harbor, ME, stock number 002633); or S-nitrosoglutathione reductase21 (GSNOR−/−; n=10). All protocols and experimental procedures were approved by the Animal Care and Use Committee of the University of Miami and followed the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 85–234, revised 2011).
Myocyte IsolationCardiac myocytes were isolated and prepared from hearts as previ-ously described.6 Briefly, hearts were harvested and perfused retro-gradely in a modified Langendorff system (constant flow, 2 mL/min) with an isolation solution (see Online Data Supplement), bubbled with 5% CO
2 and 95% O
2 for ≥15 minutes. Once cleaned, the hearts
were perfused with collagenase type 2 (Worthington Biochemical Corporation, Lakewood, NJ) ≈315 U/mL and protease type XIV (Sigma–Aldrich, St. Louis, MO) 5.2 U/mL for 10 minutes. After digestion, myocytes were released by gentle mechanical disrup-tion. The extracellular Ca2+ was restored by sequential additions of CaCl
2 in a Ca2+-free Tyrode solution (see Online Data Supplement).
Cardiomyocytes were resuspended in a 1.8 CaCl2 Tyrode solution
at room temperature and then loaded with Fura-2. Myocytes were stimulated at 0.5 or 1 Hz.
Intracellular Ca2+ MeasurementIntracellular Ca2+ was measured using the Ca2+-sensitive dye Fura-2 (Molecular Probes, Eugene, OR) and a dual-excitation
spectrofluorometer (IonOptix LLC, Milton, MA), excited with a xenon lamp at wavelengths of 340 and 380 nm. The emission fluo-rescence was reflected through a barrier filter (510±15 nm) to a pho-tomultiplier tube. The in vivo calibration was performed superfusing a free Ca2+ and then a Ca2+ saturating (5 mmol/L) solutions both con-taining 10 μmol/L ionomycin (Sigma–Aldrich) until reaching a mini-mal (R
min) or a maximal (R
max) ratio values, respectively. [Ca]2+
i was
calculated as described previously.9
Measurement of SR Ca2+ Leak and SR Ca2+ ContentSR Ca2+ leakage was assessed with 1 mmol/L tetracaine (Sigma–Aldrich) as described by Shannon et al.22 The observed decrease in the Fura-2 ratio in presence of tetracaine compared with the nontetra-caine-treated condition was considered the Ca2+ leak for a particular myocyte (Online Figure I). After assessing Ca2+ leak, tetracaine was washed out by superfusing fresh 0Na+/0Ca2+ Tyrode solution and SR Ca2+ content was assessed as described by Bassani et al23 by a caffeine challenge.
SR Ca2+ contents were calculated considering that SR represents 3.5% and cytosol 65% of the myocyte volume as previously de-scribed.9 SR leak–SR load pairs were grouped by similar SR Ca2+ load and expressed as a leak–load relationship fitted by an exponen-tial growth function using the Graph Pad Prism software (version 4.02). Measurements were mostly performed at 23°C, 25°C, 30°C, 34°C, or 37°C.
TreatmentsCardiomyocytes loaded with Fura-2 were preincubated for 20 min-utes with the following compounds (unless otherwise is specified): Tempol (100 μmol/L; Calbiochem, Calbiochem/EMD Biosciences, San Diego, CA); Oxypurinol (100 μmol/L; Sigma–Aldrich); (±)-S-Nitroso-N-acetylpenicillamine (SNAP; 1 or 50 μmol/L; Santa Cruz, Santa Cruz, CA); N5-(1-Imino-3-butenyl)-l-Ornithine (l-VNIO; 100 μmol/L; Enzo Life Sciences, Plymouth Meeting, PA); Hydrogen peroxide (H
2O
2; 100 μmol/L; Sigma–Aldrich) (6R)-5,6,7,8-
Tetrahydrobiopterin dihydrochloride (BH4; 300 μmol/L; Sigma–
Aldrich). Experiments in control condition (no treatment) were run in parallel to each type of pharmacological intervention for each batch of cardiomyocytes.
Detection of Reactive Oxygen Speciesreactive oxygen species (ROS) were measured by using the sensi-tive probe 2′,7′-dichlorodihydrofluoresceine diacetate (10 μmol/L; Molecular Probes) in 2 different ways. First, fresh isolated mouse car-diomyocytes were placed in the chamber of an IonOptix spectrofluo-rometer and the background fluorescence (F
0) was acquired and then,
cardiomyocytes were incubated during 30 minutes at 23°C or 37°C with 2′,7′-dichlorodihydrofluorescein diacetate and washed. The ini-tial fluorescence (F
i) and a second measure after 5 minutes (F) were
acquired. Myocytes were stimulated at 1 Hz and ROS expressed as:
ROS = − − −( ) ( )F F Fi F0 0
Alternatively, control or 100 μmol/L oxypurinol-treated cardiomy-ocytes were loaded with 2′,7′-dichlorodihydrofluorescein diacetate for 15 minutes on polylysine-coated microscope slides at 23°C or 37°C. After 10 minutes washing with Tyrode solution, cardiomyocytes were fixed with 2% p-formaldehyde in cold phosphate-buffered saline and then washed once with phosphate-buffered saline. Cardiomyocytes treated with 1 mmol/L H
2O
2 at either 23°C or 37°C, from each
mouse model were used as positive control for ROS and were used as maximal fluorescence signal for normalization of each group (F
max).
Fluorescence (F) was captured with an excitation wavelength of 488 and 525 nm emission. Images were quantified by ImageJ (National Institutes of Health) software and results were expressed as:
FF F
F FDCF =−
−
( )( )
0
max,
where F0 is background.
Nonstandard Abbreviations and Acronyms
Ca2+ calcium
GSNOR−/− S-nitrosoglutathione reductase–deficient mice
NO nitric oxide
NO/redox nitroso–redox
NOS1−/− neuronal nitric oxide synthase–deficient mice
ROS reactive oxygen species
RyR2 cardiac ryanodine receptor
SR sarcoplasmic reticulum
WT wild-type
XOR xanthine oxidoreductase
at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from
48 Circulation Research January 2, 2015
Nitric Oxide MeasurementIsolated WT mouse cardiomyocytes were loaded with the NO-sensitive dye 4,5-diaminofluorescein diacetate (5 μmol/L; Calbiochem/EMD Biosciences) for 20 minutes at room temperature. Cells were set in the perfusion chamber of an IonOptix system and fluorescence (ex-cited at 488 nm and emission collected at 510±15 nm) was recorded during stabilization at 23°C. Then, fluorescence was also acquired at 25°C, 30°C, 34°C, and 37°C for each cell; 4,5-diaminofluorescein intensity was expressed as F/F
0, where F
0 is the fluorescence intensity
at 23°C after the stabilization time.
NOS Isoform ExpressionCardiomyocytes from 4 WT hearts were exposed to different tem-perature and then collected in RNA later lysis buffer (Qiagen, Valencia, CA). Total RNA was extracted from cells using Pure-Link Micro-to-Midi Total RNA Purification System (Qiagen) and reverse-transcribed using high capacity cDNA reverse transcription Kit (Applied Biosystems, Foster City, CA). All samples were treated with TurboTM DNase (Ambion, Austin, TX). Quantitative real-time polymerase chain reaction (PCR) was performed in triplicate using a 20-μL reaction mixture containing 10 ng cDNA, TaqMan Universal PCR Master Mix (Roche, Branchburg, NJ) and primer/probe sets for nitric oxide synthase 1 (NOS1, Mm00435171_m1), NOS2 (Mm00440502_m1), and NOS3 (Mm00435197_g1; TaqMan Gene Expression Assay, Applied Bio systems, Foster City, CA). As an internal control glucoronidase, β (GUSB, Mm01197698_m1) was determined in each reaction. Reaction conditions were performed according to manufacturer instructions: 1 cycle of 50°C for 2 min-utes, 1 cycle of 90°C for 10 minutes, and 40 cycles of 95°C for 15 s and 60°C for 1 minute. Software from iQ5 multicolor real-time PCR detection system (Bio-Rad, Hercules, CA) was used for PCR anal-yses. mRNA relative expression was calculated by 2ΔCt method. Quantitative PCR data reflects 4 different experiments each done at 23°C, 30°C, and 37°C.
Statistical AnalysisData are expressed as mean±SEM. For leak–load relationship, an ex-ponential growth fit, which compares independent fits with a global shared fit was applied. Two or more groups of data were considered to fit different curves if P<0.05. For comparisons of 2 groups, Student t test was used. For comparison of ≥3groups, 1- or 2-way ANOVA was performed. Two-way ANOVA was used when a second variable was in-volved. Newman–Keuls or Bonferroni post hoc tests were used as appro-priate by the GraphPad Prism version 4.02 (GraphPad Prism Software Corporation San Diego, CA). A P<0.05 was considered significant.
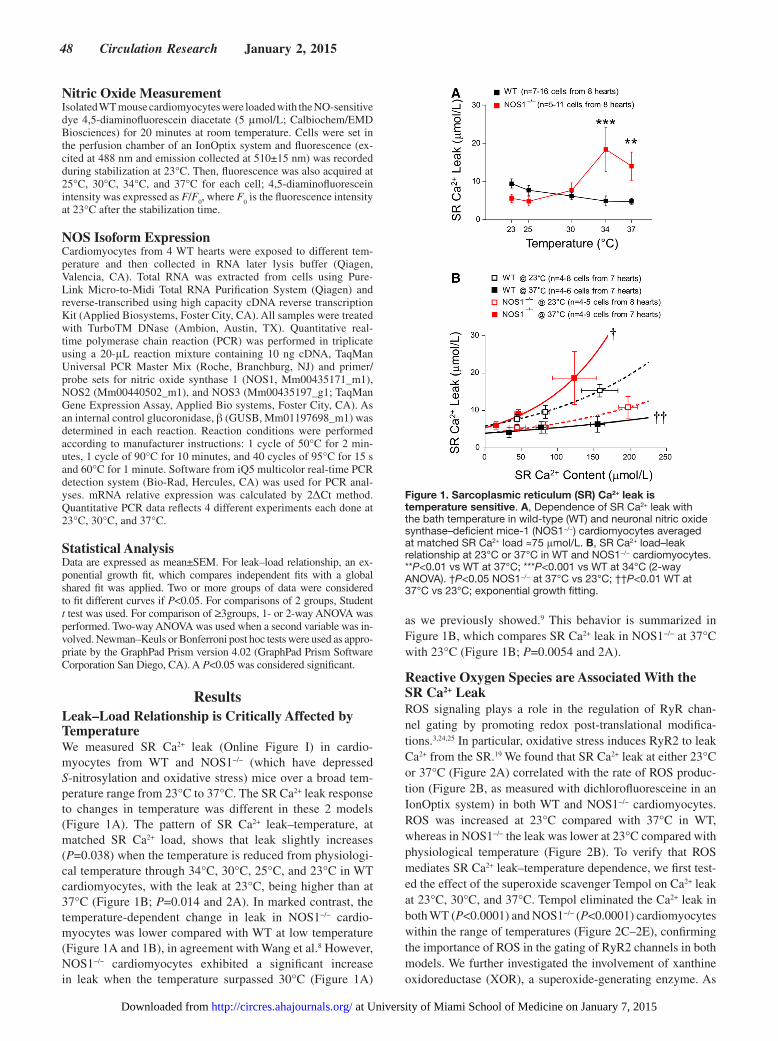
ResultsLeak–Load Relationship is Critically Affected by TemperatureWe measured SR Ca2+ leak (Online Figure I) in cardio-myocytes from WT and NOS1−/− (which have depressed S-nitrosylation and oxidative stress) mice over a broad tem-perature range from 23°C to 37°C. The SR Ca2+ leak response to changes in temperature was different in these 2 models (Figure 1A). The pattern of SR Ca2+ leak–temperature, at matched SR Ca2+ load, shows that leak slightly increases (P=0.038) when the temperature is reduced from physiologi-cal temperature through 34°C, 30°C, 25°C, and 23°C in WT cardiomyocytes, with the leak at 23°C, being higher than at 37°C (Figure 1B; P=0.014 and 2A). In marked contrast, the temperature-dependent change in leak in NOS1−/− cardio-myocytes was lower compared with WT at low temperature (Figure 1A and 1B), in agreement with Wang et al.8 However, NOS1−/− cardiomyocytes exhibited a significant increase in leak when the temperature surpassed 30°C (Figure 1A)
as we previously showed.9 This behavior is summarized in Figure 1B, which compares SR Ca2+ leak in NOS1−/− at 37°C with 23°C (Figure 1B; P=0.0054 and 2A).
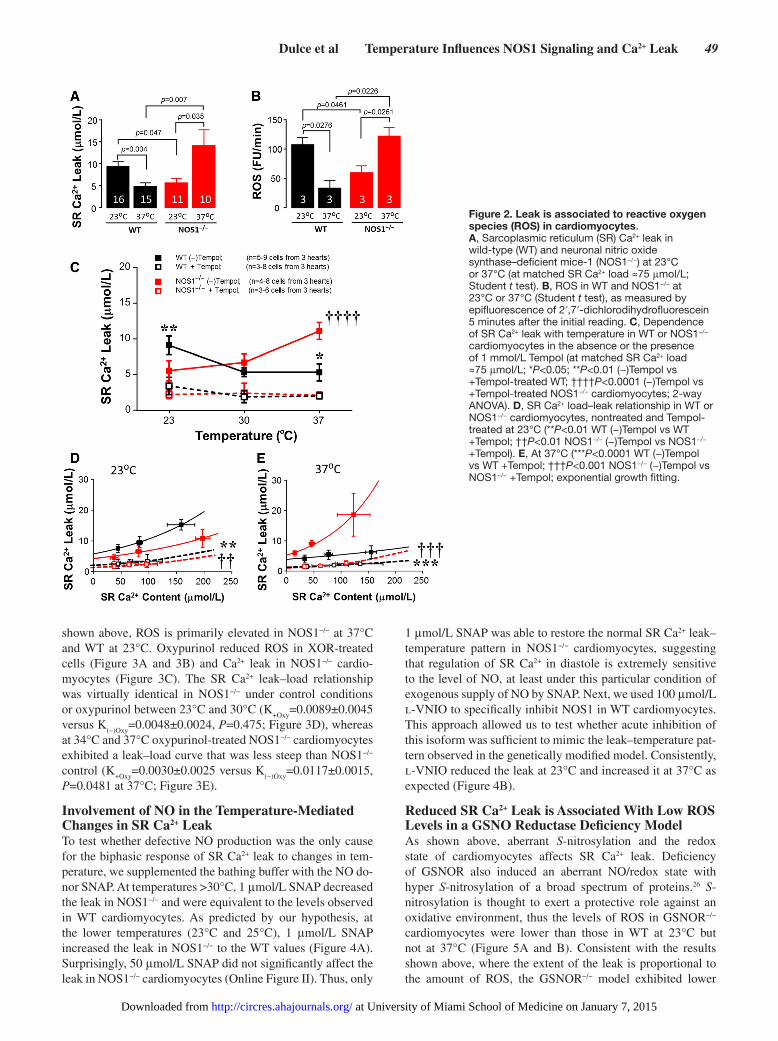
Reactive Oxygen Species are Associated With the SR Ca2+ LeakROS signaling plays a role in the regulation of RyR chan-nel gating by promoting redox post-translational modifica-tions.3,24,25 In particular, oxidative stress induces RyR2 to leak Ca2+ from the SR.19 We found that SR Ca2+ leak at either 23°C or 37°C (Figure 2A) correlated with the rate of ROS produc-tion (Figure 2B, as measured with dichlorofluoresceine in an IonOptix system) in both WT and NOS1−/− cardiomyocytes. ROS was increased at 23°C compared with 37°C in WT, whereas in NOS1−/− the leak was lower at 23°C compared with physiological temperature (Figure 2B). To verify that ROS mediates SR Ca2+ leak–temperature dependence, we first test-ed the effect of the superoxide scavenger Tempol on Ca2+ leak at 23°C, 30°C, and 37°C. Tempol eliminated the Ca2+ leak in both WT (P<0.0001) and NOS1−/− (P<0.0001) cardiomyocytes within the range of temperatures (Figure 2C–2E), confirming the importance of ROS in the gating of RyR2 channels in both models. We further investigated the involvement of xanthine oxidoreductase (XOR), a superoxide-generating enzyme. As
Figure 1. Sarcoplasmic reticulum (SR) Ca2+ leak is temperature sensitive. A, Dependence of SR Ca2+ leak with the bath temperature in wild-type (WT) and neuronal nitric oxide synthase–deficient mice-1 (NOS1−/−) cardiomyocytes averaged at matched SR Ca2+ load ≈75 μmol/L. B, SR Ca2+ load–leak relationship at 23°C or 37°C in WT and NOS1−/− cardiomyocytes. **P<0.01 vs WT at 37°C; ***P<0.001 vs WT at 34°C (2-way ANOVA). †P<0.05 NOS1−/− at 37°C vs 23°C; ††P<0.01 WT at 37°C vs 23°C; exponential growth fitting.
at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from
Dulce et al Temperature Influences NOS1 Signaling and Ca2+ Leak 49
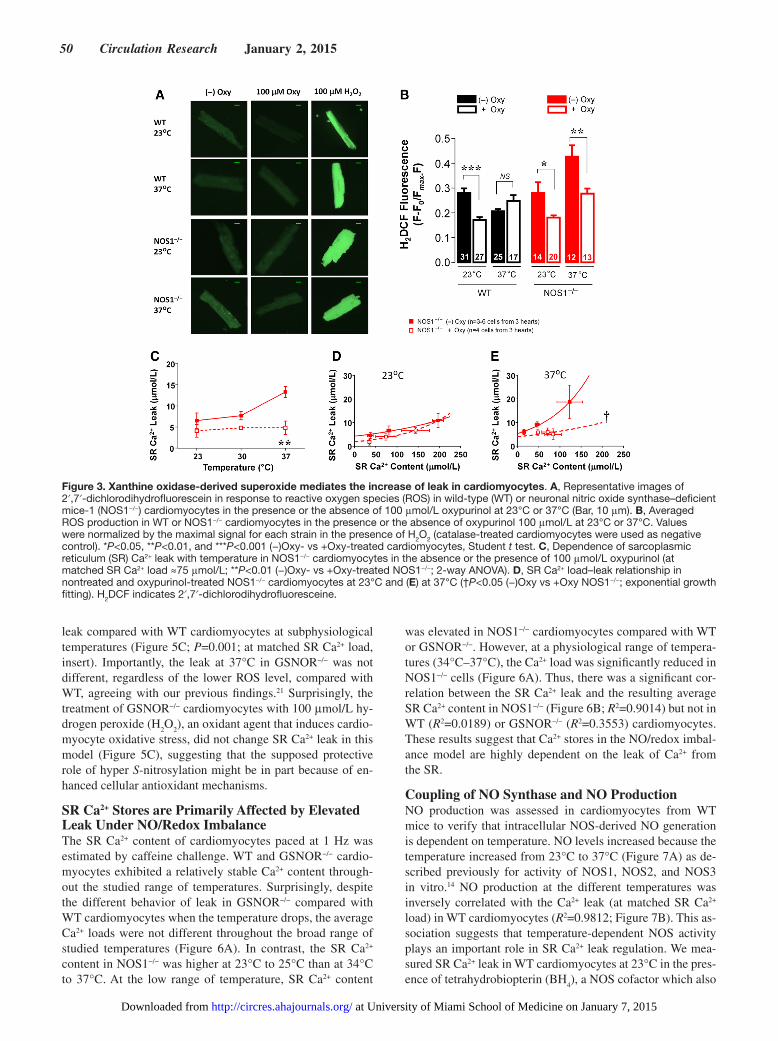
shown above, ROS is primarily elevated in NOS1−/− at 37°C and WT at 23°C. Oxypurinol reduced ROS in XOR-treated cells (Figure 3A and 3B) and Ca2+ leak in NOS1−/− cardio-myocytes (Figure 3C). The SR Ca2+ leak–load relationship was virtually identical in NOS1−/− under control conditions or oxypurinol between 23°C and 30°C (K
+Oxy=0.0089±0.0045
versus K(−)Oxy
=0.0048±0.0024, P=0.475; Figure 3D), whereas at 34°C and 37°C oxypurinol-treated NOS1−/− cardiomyocytes exhibited a leak–load curve that was less steep than NOS1−/− control (K
+Oxy=0.0030±0.0025 versus K
(−)Oxy=0.0117±0.0015,
P=0.0481 at 37°C; Figure 3E).
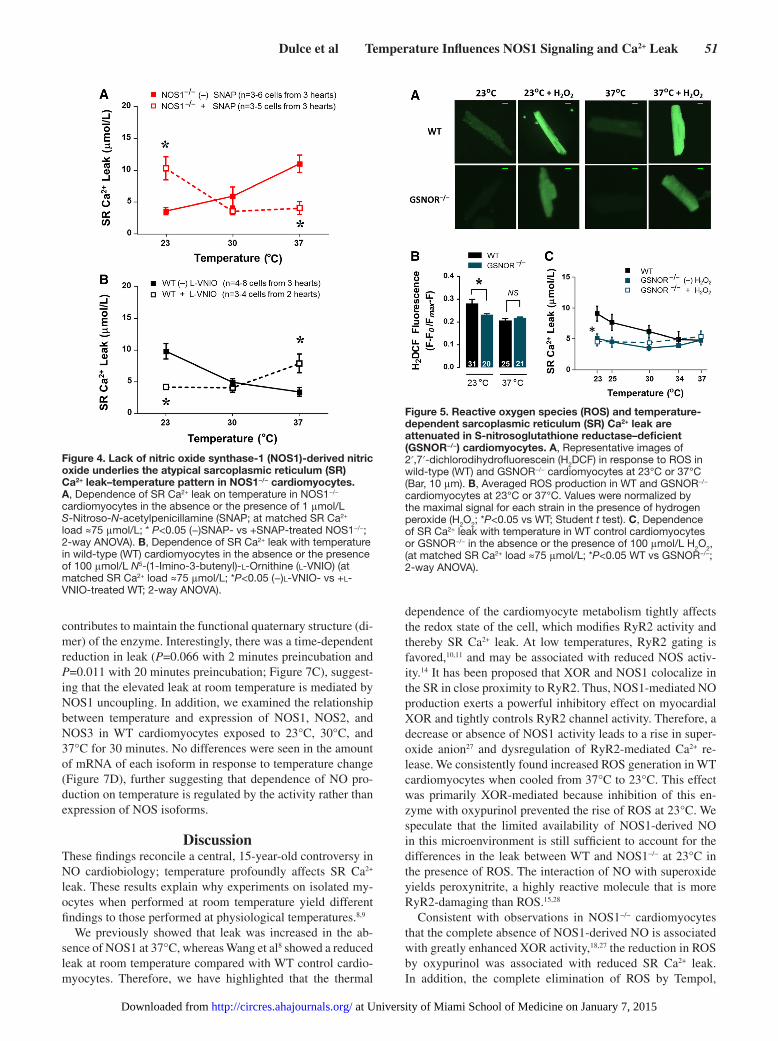
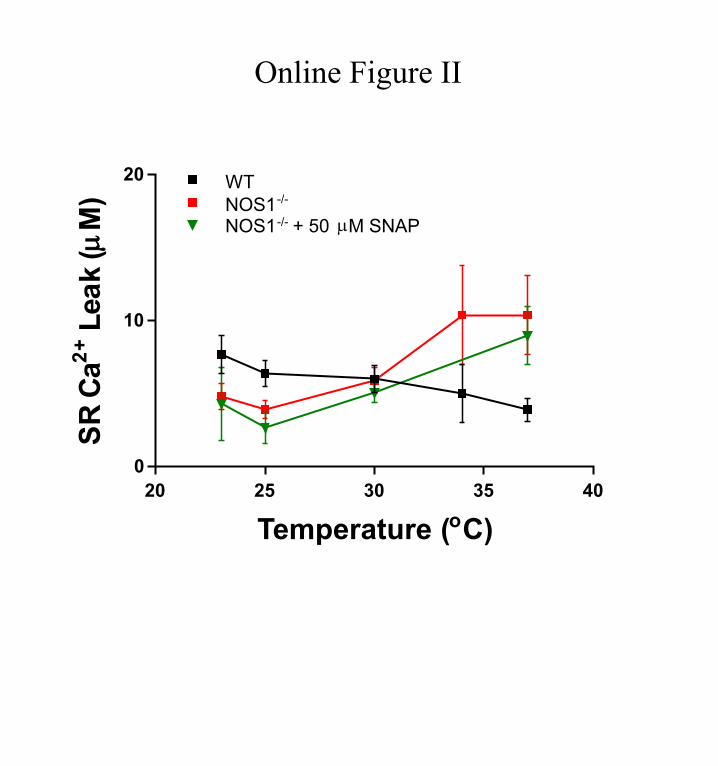
Involvement of NO in the Temperature-Mediated Changes in SR Ca2+ LeakTo test whether defective NO production was the only cause for the biphasic response of SR Ca2+ leak to changes in tem-perature, we supplemented the bathing buffer with the NO do-nor SNAP. At temperatures >30°C, 1 μmol/L SNAP decreased the leak in NOS1−/− and were equivalent to the levels observed in WT cardiomyocytes. As predicted by our hypothesis, at the lower temperatures (23°C and 25°C), 1 μmol/L SNAP increased the leak in NOS1−/− to the WT values (Figure 4A). Surprisingly, 50 μmol/L SNAP did not significantly affect the leak in NOS1−/− cardiomyocytes (Online Figure II). Thus, only
1 μmol/L SNAP was able to restore the normal SR Ca2+ leak–temperature pattern in NOS1−/− cardiomyocytes, suggesting that regulation of SR Ca2+ in diastole is extremely sensitive to the level of NO, at least under this particular condition of exogenous supply of NO by SNAP. Next, we used 100 μmol/L l-VNIO to specifically inhibit NOS1 in WT cardiomyocytes. This approach allowed us to test whether acute inhibition of this isoform was sufficient to mimic the leak–temperature pat-tern observed in the genetically modified model. Consistently, l-VNIO reduced the leak at 23°C and increased it at 37°C as expected (Figure 4B).
Reduced SR Ca2+ Leak is Associated With Low ROS Levels in a GSNO Reductase Deficiency ModelAs shown above, aberrant S-nitrosylation and the redox state of cardiomyocytes affects SR Ca2+ leak. Deficiency of GSNOR also induced an aberrant NO/redox state with hyper S-nitrosylation of a broad spectrum of proteins.26 S-nitrosylation is thought to exert a protective role against an oxidative environment, thus the levels of ROS in GSNOR−/− cardiomyocytes were lower than those in WT at 23°C but not at 37°C (Figure 5A and B). Consistent with the results shown above, where the extent of the leak is proportional to the amount of ROS, the GSNOR−/− model exhibited lower
Figure 2. Leak is associated to reactive oxygen species (ROS) in cardiomyocytes. A, Sarcoplasmic reticulum (SR) Ca2+ leak in wild-type (WT) and neuronal nitric oxide synthase–deficient mice-1 (NOS1−/−) at 23°C or 37°C (at matched SR Ca2+ load ≈75 μmol/L; Student t test). B, ROS in WT and NOS1−/− at 23°C or 37°C (Student t test), as measured by epifluorescence of 2′,7′-dichlorodihydrofluorescein 5 minutes after the initial reading. C, Dependence of SR Ca2+ leak with temperature in WT or NOS1−/− cardiomyocytes in the absence or the presence of 1 mmol/L Tempol (at matched SR Ca2+ load ≈75 μmol/L; *P<0.05; **P<0.01 (−)Tempol vs +Tempol-treated WT; ††††P<0.0001 (−)Tempol vs +Tempol-treated NOS1−/− cardiomyocytes; 2-way ANOVA). D, SR Ca2+ load–leak relationship in WT or NOS1−/− cardiomyocytes, nontreated and Tempol-treated at 23°C (**P<0.01 WT (−)Tempol vs WT +Tempol; ††P<0.01 NOS1−/− (−)Tempol vs NOS1−/− +Tempol). E, At 37°C (***P<0.0001 WT (−)Tempol vs WT +Tempol; †††P<0.001 NOS1−/− (−)Tempol vs NOS1−/− +Tempol; exponential growth fitting.
at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from
50 Circulation Research January 2, 2015
leak compared with WT cardiomyocytes at subphysiological temperatures (Figure 5C; P=0.001; at matched SR Ca2+ load, insert). Importantly, the leak at 37°C in GSNOR−/− was not different, regardless of the lower ROS level, compared with WT, agreeing with our previous findings.21 Surprisingly, the treatment of GSNOR−/− cardiomyocytes with 100 μmol/L hy-drogen peroxide (H
2O
2), an oxidant agent that induces cardio-
myocyte oxidative stress, did not change SR Ca2+ leak in this model (Figure 5C), suggesting that the supposed protective role of hyper S-nitrosylation might be in part because of en-hanced cellular antioxidant mechanisms.
SR Ca2+ Stores are Primarily Affected by Elevated Leak Under NO/Redox ImbalanceThe SR Ca2+ content of cardiomyocytes paced at 1 Hz was estimated by caffeine challenge. WT and GSNOR−/− cardio-myocytes exhibited a relatively stable Ca2+ content through-out the studied range of temperatures. Surprisingly, despite the different behavior of leak in GSNOR−/− compared with WT cardiomyocytes when the temperature drops, the average Ca2+ loads were not different throughout the broad range of studied temperatures (Figure 6A). In contrast, the SR Ca2+ content in NOS1−/− was higher at 23°C to 25°C than at 34°C to 37°C. At the low range of temperature, SR Ca2+ content
was elevated in NOS1−/− cardiomyocytes compared with WT or GSNOR−/−. However, at a physiological range of tempera-tures (34°C–37°C), the Ca2+ load was significantly reduced in NOS1−/− cells (Figure 6A). Thus, there was a significant cor-relation between the SR Ca2+ leak and the resulting average SR Ca2+ content in NOS1−/− (Figure 6B; R2=0.9014) but not in WT (R2=0.0189) or GSNOR−/− (R2=0.3553) cardiomyocytes. These results suggest that Ca2+ stores in the NO/redox imbal-ance model are highly dependent on the leak of Ca2+ from the SR.
Coupling of NO Synthase and NO ProductionNO production was assessed in cardiomyocytes from WT mice to verify that intracellular NOS-derived NO generation is dependent on temperature. NO levels increased because the temperature increased from 23°C to 37°C (Figure 7A) as de-scribed previously for activity of NOS1, NOS2, and NOS3 in vitro.14 NO production at the different temperatures was inversely correlated with the Ca2+ leak (at matched SR Ca2+ load) in WT cardiomyocytes (R2=0.9812; Figure 7B). This as-sociation suggests that temperature-dependent NOS activity plays an important role in SR Ca2+ leak regulation. We mea-sured SR Ca2+ leak in WT cardiomyocytes at 23°C in the pres-ence of tetrahydrobiopterin (BH
4), a NOS cofactor which also
Figure 3. Xanthine oxidase-derived superoxide mediates the increase of leak in cardiomyocytes. A, Representative images of 2′,7′-dichlorodihydrofluorescein in response to reactive oxygen species (ROS) in wild-type (WT) or neuronal nitric oxide synthase–deficient mice-1 (NOS1−/−) cardiomyocytes in the presence or the absence of 100 μmol/L oxypurinol at 23°C or 37°C (Bar, 10 μm). B, Averaged ROS production in WT or NOS1−/− cardiomyocytes in the presence or the absence of oxypurinol 100 μmol/L at 23°C or 37°C. Values were normalized by the maximal signal for each strain in the presence of H2O2 (catalase-treated cardiomyocytes were used as negative control). *P<0.05, **P<0.01, and ***P<0.001 (−)Oxy- vs +Oxy-treated cardiomyocytes, Student t test. C, Dependence of sarcoplasmic reticulum (SR) Ca2+ leak with temperature in NOS1−/− cardiomyocytes in the absence or the presence of 100 μmol/L oxypurinol (at matched SR Ca2+ load ≈75 μmol/L; **P<0.01 (−)Oxy- vs +Oxy-treated NOS1−/−; 2-way ANOVA). D, SR Ca2+ load–leak relationship in nontreated and oxypurinol-treated NOS1−/− cardiomyocytes at 23°C and (E) at 37°C (†P<0.05 (−)Oxy vs +Oxy NOS1−/−; exponential growth fitting). H2DCF indicates 2′,7′-dichlorodihydrofluoresceine.
at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from
Dulce et al Temperature Influences NOS1 Signaling and Ca2+ Leak 51
contributes to maintain the functional quaternary structure (di-mer) of the enzyme. Interestingly, there was a time-dependent reduction in leak (P=0.066 with 2 minutes preincubation and P=0.011 with 20 minutes preincubation; Figure 7C), suggest-ing that the elevated leak at room temperature is mediated by NOS1 uncoupling. In addition, we examined the relationship between temperature and expression of NOS1, NOS2, and NOS3 in WT cardiomyocytes exposed to 23°C, 30°C, and 37°C for 30 minutes. No differences were seen in the amount of mRNA of each isoform in response to temperature change (Figure 7D), further suggesting that dependence of NO pro-duction on temperature is regulated by the activity rather than expression of NOS isoforms.
DiscussionThese findings reconcile a central, 15-year-old controversy in NO cardiobiology; temperature profoundly affects SR Ca2+ leak. These results explain why experiments on isolated my-ocytes when performed at room temperature yield different findings to those performed at physiological temperatures.8,9
We previously showed that leak was increased in the ab-sence of NOS1 at 37°C, whereas Wang et al8 showed a reduced leak at room temperature compared with WT control cardio-myocytes. Therefore, we have highlighted that the thermal
dependence of the cardiomyocyte metabolism tightly affects the redox state of the cell, which modifies RyR2 activity and thereby SR Ca2+ leak. At low temperatures, RyR2 gating is favored,10,11 and may be associated with reduced NOS activ-ity.14 It has been proposed that XOR and NOS1 colocalize in the SR in close proximity to RyR2. Thus, NOS1-mediated NO production exerts a powerful inhibitory effect on myocardial XOR and tightly controls RyR2 channel activity. Therefore, a decrease or absence of NOS1 activity leads to a rise in super-oxide anion27 and dysregulation of RyR2-mediated Ca2+ re-lease. We consistently found increased ROS generation in WT cardiomyocytes when cooled from 37°C to 23°C. This effect was primarily XOR-mediated because inhibition of this en-zyme with oxypurinol prevented the rise of ROS at 23°C. We speculate that the limited availability of NOS1-derived NO in this microenvironment is still sufficient to account for the differences in the leak between WT and NOS1−/− at 23°C in the presence of ROS. The interaction of NO with superoxide yields peroxynitrite, a highly reactive molecule that is more RyR2-damaging than ROS.15,28
Consistent with observations in NOS1−/− cardiomyocytes that the complete absence of NOS1-derived NO is associated with greatly enhanced XOR activity,18,27 the reduction in ROS by oxypurinol was associated with reduced SR Ca2+ leak. In addition, the complete elimination of ROS by Tempol,
Figure 5. Reactive oxygen species (ROS) and temperature-dependent sarcoplasmic reticulum (SR) Ca2+ leak are attenuated in S-nitrosoglutathione reductase–deficient (GSNOR−/−) cardiomyocytes. A, Representative images of 2′,7′-dichlorodihydrofluorescein (H2DCF) in response to ROS in wild-type (WT) and GSNOR−/− cardiomyocytes at 23°C or 37°C (Bar, 10 μm). B, Averaged ROS production in WT and GSNOR−/− cardiomyocytes at 23°C or 37°C. Values were normalized by the maximal signal for each strain in the presence of hydrogen peroxide (H2O2; *P<0.05 vs WT; Student t test). C, Dependence of SR Ca2+ leak with temperature in WT control cardiomyocytes or GSNOR−/− in the absence or the presence of 100 μmol/L H2O2, (at matched SR Ca2+ load ≈75 μmol/L; *P<0.05 WT vs GSNOR−/−; 2-way ANOVA).
Figure 4. Lack of nitric oxide synthase-1 (NOS1)-derived nitric oxide underlies the atypical sarcoplasmic reticulum (SR) Ca2+ leak–temperature pattern in NOS1−/− cardiomyocytes. A, Dependence of SR Ca2+ leak on temperature in NOS1−/− cardiomyocytes in the absence or the presence of 1 μmol/L S-Nitroso-N-acetylpenicillamine (SNAP; at matched SR Ca2+ load ≈75 μmol/L; * P<0.05 (−)SNAP- vs +SNAP-treated NOS1−/−; 2-way ANOVA). B, Dependence of SR Ca2+ leak with temperature in wild-type (WT) cardiomyocytes in the absence or the presence of 100 μmol/L N5-(1-Imino-3-butenyl)-l-Ornithine (l-VNIO) (at matched SR Ca2+ load ≈75 μmol/L; *P<0.05 (−)l-VNIO- vs +l-VNIO-treated WT; 2-way ANOVA).
at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from

52 Circulation Research January 2, 2015
abolished the leak–temperature dependence either in WT or NOS1−/− cardiomyocytes, confirming the key role of ROS in this process. Other sources of ROS might play a signaling role, affecting the Ca2+ leak control. For instance, mitochon-drial-derived ROS may be significantly affected by tempera-ture as a result of changes in the electron flow through the respiratory chain.29 Indeed, hypothermic preconditioning ac-tivates mitochondrial ROS release and ERK activation in ven-tricular myocytes.30 Moreover, additional evidence that ROS are associated with leak is the fact that, at subphysiological temperatures, where the leak is reduced in GSNOR−/− com-pared with WT cardiomyocytes, there is also less ROS. In this regard, RyR2 is a highly redox-sensitive Ca2+ channel and its activity is increased by oxidizing agents and decreased by reducing agents. However, its regulation is even more com-plex. Thus, the intracellular NO/redox equilibrium is a criti-cal factor determining the responsiveness to Ca2+ of the RyR2 channel. Post-translational modifications of RyR2, such as S-glutathionylation31 or S-nitrosylation,18,19 are considered protective against irreversible oxidation of redox-sensitive cysteine residues on this channel. As shown previously,21 the leak in GSNOR−/− cardiomyocytes was not different from WT at 37°C, consistent with there being no difference in ROS, and suggesting that enhanced S-nitrosylation caused by deficient denitrosylation activity, would not affect leak at physiologi-cal temperature. In contrast to WT, decreasing temperature did not increase the leak in GSNOR−/− cells, which could indi-cate a redox-protective effect of S-nitrosylation at subphysi-ological temperatures. Further evidence for this hypothesis
is the fact that the exposure of GSNOR−/− cardiomyocytes to an oxidative stimulus was not sufficient to break down this redox-protective shield mediated by hyper S-nitrosylation.
We mentioned above that a lack of NOS1-derived NO pro-duction may be the underlying cause of the atypical SR Ca2+ leak–temperature pattern in cardiomyocytes from NOS1−/− mice. We have shown that restoring the NO supply in this model rescues the pattern of high leak at room temperature and low leak at the physiological range of temperatures as ob-served in WT cardiomyocytes. However, there is a complex-ity to NO signaling which is evidenced in this work by the ability of 1 μmol/L but not 50 μmol/L of SNAP to elicit such an effect. We previously showed a triphasic response of SR Ca2+ leak to increasing doses of nitroglycerin in NOS1−/− car-diomyocytes,9 thus the effects of exogenously added NO are highly dependent on the concentration and the nature of the NO donor. Accordingly, the idea that reduced NOS1 activity promotes a local microenvironment for this particular behav-ior of SR Ca2+ leak, explains the specific inhibition of NOS1 in WT cardiomyocytes yielding results consistent with those in the NOS1 knockout model. As expected, the high leak at 23°C in WT was abolished and the low leak at 37°C was increased by l-VNIO. However, the effect was less robust at the physi-ological temperature because this increase was only half the level of the leak observed in NOS1−/− at this temperature. We speculate that the intracellular metabolism of the drug might be different at 37°C and therefore, the effective concentration of l-VNIO might not be sufficient to efficiently inhibit NOS1. Alternatively, the time of exposure to the inhibitor may not be adequate to generate the characteristic NO/redox imbalance as seen by the chronic deletion of NOS1. The risk of using a higher dose of l-VNIO is the loss of specificity. Thus, we confirmed that defective NOS1-derived NO production leads to an NO/redox imbalance that differentially affects SR Ca2+ leak at physiological or subphysiological temperatures in iso-lated cardiomyocytes.
Reduced activity of NOS1 at low temperature may be the cause of increased XOR-mediated ROS in WT cardiomyo-cytes. Moreover, this redox dysregulation would uncouple NOS1, further contributing to ROS production (see the pro-posed working model in Figure 8). Thus, recoupling of NOS1, as mediated by BH
4 treatment, restores the redox balance and
leads to low levels of SR Ca2+ leak. It was recently shown that BH
4 supplementation restores NO production and S-
nitrosylation of proteins, in cardiomyocytes from a model of dystrophic cardiomyopathy exhibiting oxidative stress.20
It has been demonstrated previously that NO-mediated S-nitrosylation of RyR2 enhances its activity.3,32 However, it is important to differentiate activation of the channel from a leaky channel. RyR2 activity is regulated by many factors, including Ca2+, phosphorylation, S-nitrosylation, oxidation, etc. Despite activation, the channel might be more or less stabilized, exhibiting different levels of diastolic Ca2+ leak. Recent studies have associated low S-nitrosylation levels with increased leak measured at 37°C in different models18–20 sug-gesting that NO-mediated S-nitrosylation reduces leak. Here, we propose that temperature is a variable that, by affecting
Figure 6. Sarcoplasmic reticulum (SR) Ca2+ content is governed by leak in neuronal nitric oxide synthase–deficient mice-1 (NOS1−/−) cardiomyocytes. A, SR Ca2+ load in wild-type (WT) compared with NOS1−/− or S-nitrosoglutathione reductase–deficient (GSNOR−/−) cardiomyocytes at different temperatures between 23°C and 37°C (*P<0.05 and **P<0.01; Student t test). B, Linear regression between SR Ca2+ leak vs average SR Ca2+ load in WT (black) or NOS1−/− (red) and GSNOR−/− (cyan) cardiomyocytes.
at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from
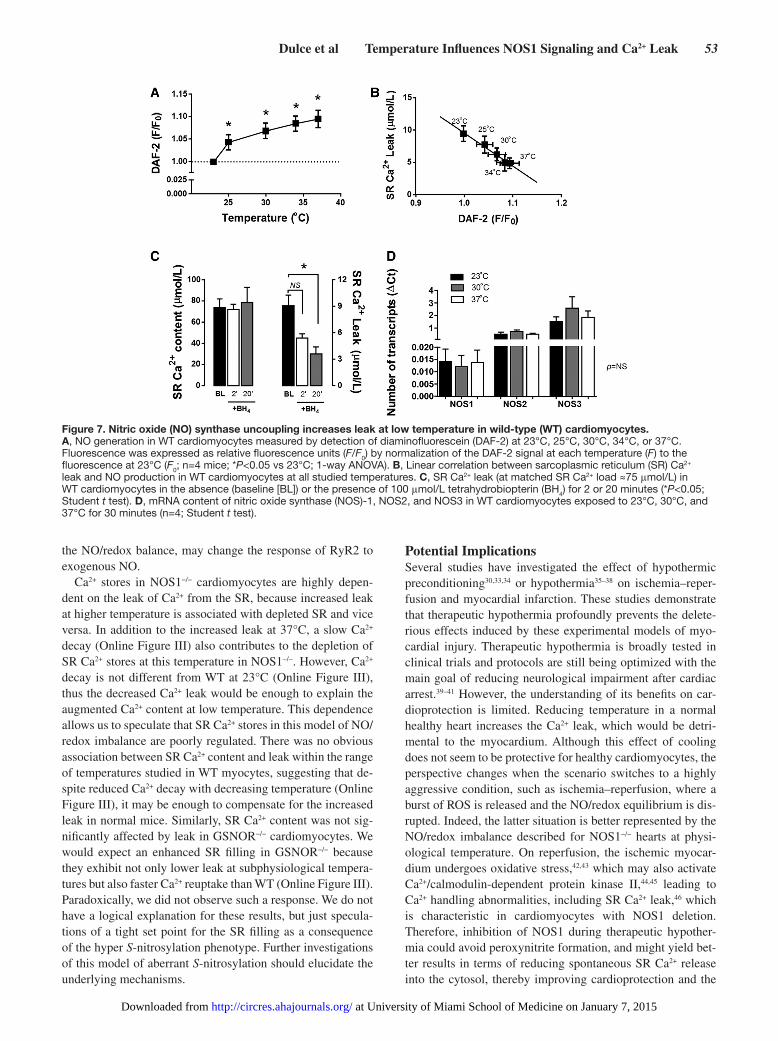
Dulce et al Temperature Influences NOS1 Signaling and Ca2+ Leak 53
the NO/redox balance, may change the response of RyR2 to exogenous NO.
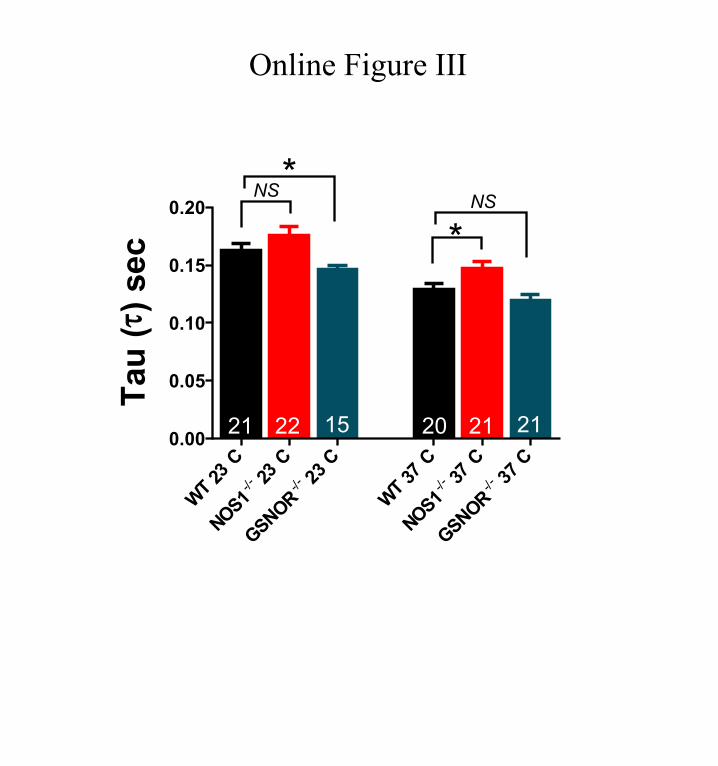
Ca2+ stores in NOS1−/− cardiomyocytes are highly depen-dent on the leak of Ca2+ from the SR, because increased leak at higher temperature is associated with depleted SR and vice versa. In addition to the increased leak at 37°C, a slow Ca2+ decay (Online Figure III) also contributes to the depletion of SR Ca2+ stores at this temperature in NOS1−/−. However, Ca2+ decay is not different from WT at 23°C (Online Figure III), thus the decreased Ca2+ leak would be enough to explain the augmented Ca2+ content at low temperature. This dependence allows us to speculate that SR Ca2+ stores in this model of NO/redox imbalance are poorly regulated. There was no obvious association between SR Ca2+ content and leak within the range of temperatures studied in WT myocytes, suggesting that de-spite reduced Ca2+ decay with decreasing temperature (Online Figure III), it may be enough to compensate for the increased leak in normal mice. Similarly, SR Ca2+ content was not sig-nificantly affected by leak in GSNOR−/− cardiomyocytes. We would expect an enhanced SR filling in GSNOR−/− because they exhibit not only lower leak at subphysiological tempera-tures but also faster Ca2+ reuptake than WT (Online Figure III). Paradoxically, we did not observe such a response. We do not have a logical explanation for these results, but just specula-tions of a tight set point for the SR filling as a consequence of the hyper S-nitrosylation phenotype. Further investigations of this model of aberrant S-nitrosylation should elucidate the underlying mechanisms.
Potential ImplicationsSeveral studies have investigated the effect of hypothermic preconditioning30,33,34 or hypothermia35–38 on ischemia–reper-fusion and myocardial infarction. These studies demonstrate that therapeutic hypothermia profoundly prevents the delete-rious effects induced by these experimental models of myo-cardial injury. Therapeutic hypothermia is broadly tested in clinical trials and protocols are still being optimized with the main goal of reducing neurological impairment after cardiac arrest.39–41 However, the understanding of its benefits on car-dioprotection is limited. Reducing temperature in a normal healthy heart increases the Ca2+ leak, which would be detri-mental to the myocardium. Although this effect of cooling does not seem to be protective for healthy cardiomyocytes, the perspective changes when the scenario switches to a highly aggressive condition, such as ischemia–reperfusion, where a burst of ROS is released and the NO/redox equilibrium is dis-rupted. Indeed, the latter situation is better represented by the NO/redox imbalance described for NOS1−/− hearts at physi-ological temperature. On reperfusion, the ischemic myocar-dium undergoes oxidative stress,42,43 which may also activate Ca2+/calmodulin-dependent protein kinase II,44,45 leading to Ca2+ handling abnormalities, including SR Ca2+ leak,46 which is characteristic in cardiomyocytes with NOS1 deletion. Therefore, inhibition of NOS1 during therapeutic hypother-mia could avoid peroxynitrite formation, and might yield bet-ter results in terms of reducing spontaneous SR Ca2+ release into the cytosol, thereby improving cardioprotection and the
Figure 7. Nitric oxide (NO) synthase uncoupling increases leak at low temperature in wild-type (WT) cardiomyocytes. A, NO generation in WT cardiomyocytes measured by detection of diaminofluorescein (DAF-2) at 23°C, 25°C, 30°C, 34°C, or 37°C. Fluorescence was expressed as relative fluorescence units (F/F0) by normalization of the DAF-2 signal at each temperature (F) to the fluorescence at 23°C (F0; n=4 mice; *P<0.05 vs 23°C; 1-way ANOVA). B, Linear correlation between sarcoplasmic reticulum (SR) Ca2+ leak and NO production in WT cardiomyocytes at all studied temperatures. C, SR Ca2+ leak (at matched SR Ca2+ load ≈75 μmol/L) in WT cardiomyocytes in the absence (baseline [BL]) or the presence of 100 μmol/L tetrahydrobiopterin (BH4) for 2 or 20 minutes (*P<0.05; Student t test). D, mRNA content of nitric oxide synthase (NOS)-1, NOS2, and NOS3 in WT cardiomyocytes exposed to 23°C, 30°C, and 37°C for 30 minutes (n=4; Student t test).
at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from

54 Circulation Research January 2, 2015
outcomes of this powerful intervention. Alternatively, accord-ing to our data, inhibition of GSNOR during hypothermia may also add substantial improvements to this protocol because Ca2+ leak is kept low during cooling in the GSNOR-deficient model.
ConclusionHere, we show that Ca2+ leak increases when the temperature drops, coinciding with an XOR-mediated increase in ROS production and NOS1 uncoupling. In contrast, the leak rises at temperatures >30°C in the absence of NOS1 activity and is also associated with elevated ROS. Deficient denitrosylation has a protective role by maintaining low leak levels indepen-dent of the temperature. These results suggest that Ca2+ leak from the SR is regulated by a crucial interaction between tem-perature and NO/redox balance.
Sources of FundingThis study was supported by National Institutes of Health (NIH), grants 5R01 HL094849 and 1R01 HL107110 to J. M. Hare. J. M. Hare is also supported by NIH grants 5UM1 HL113460, 1R01 HL084275, R01 HL111874, and The Starr Foundation.
DisclosuresNone.
References 1. Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi
ZA, Hobai IA, Lemmon CA, Burnett AL, O’Rourke B, Rodriguez ER, Huang PL, Lima JA, Berkowitz DE, Hare JM. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339.
2. Gonzalez DR, AT MS, Sun QA, Stamler JS, Hare JM. S-nitrosylation of cardiac ion channels. J Cardiovasc Pharmacol 2009.
3. Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237.
4. Schulman IH, Hare JM. Regulation of cardiovascular cellular processes by S-nitrosylation. Biochim Biophys Acta. 2012;1820:752–762.
5. Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, Wallis HL, Neubauer S, Terrar DA, Casadei B. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res. 2003;92:e52–e59.
6. Khan SA, Skaf MW, Harrison RW, Lee K, Minhas KM, Kumar A, Fradley M, Shoukas AA, Berkowitz DE, Hare JM. Nitric oxide regulation of myo-cardial contractility and calcium cycling: independent impact of neuronal and endothelial nitric oxide synthases. Circ Res. 2003;92:1322–1329.
7. Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B. Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in mu-rine ventricular myocytes. Circulation. 2002;105:3011–3016.
8. Wang H, Viatchenko-Karpinski S, Sun J, Györke I, Benkusky NA, Kohr MJ, Valdivia HH, Murphy E, Györke S, Ziolo MT. Regulation of myocyte contraction via neuronal nitric oxide synthase: role of ryanodine receptor S-nitrosylation. J Physiol. 2010;588:2905–2917.
9. Dulce RA, Yiginer O, Gonzalez DR, Goss G, Feng N, Zheng M, Hare JM. Hydralazine and organic nitrates restore impaired excitation-contraction coupling by reducing calcium leak associated with nitroso-redox imbal-ance. J Biol Chem. 2013;288:6522–6533.
10. Fu Y, Zhang GQ, Hao XM, Wu CH, Chai Z, Wang SQ. Temperature de-pendence and thermodynamic properties of Ca2+ sparks in rat cardiomyo-cytes. Biophys J. 2005;89:2533–2541.
11. Sitsapesan R, Montgomery RA, MacLeod KT, Williams AJ. Sheep cardiac sarcoplasmic reticulum calcium-release channels: modification of conduc-tance and gating by temperature. J Physiol. 1991;434:469–488.
12. Kim Y, Busto R, Dietrich WD, Kraydieh S, Ginsberg MD. Delayed post-ischemic hyperthermia in awake rats worsens the histopathological out-come of transient focal cerebral ischemia. Stroke 1996;27:2274–2280.
13. Kumura E, Yoshimine T, Takaoka M, Hayakawa T, Shiga T, Kosaka H. Hypothermia suppresses nitric oxide elevation during reperfusion after fo-cal cerebral ischemia in rats. Neurosci Lett. 1996;220:45–48.
14. Venturini G, Colasanti M, Fioravanti E, Bianchini A, Ascenzi P. Direct ef-fect of temperature on the catalytic activity of nitric oxide synthases types I, II, and III. Nitric Oxide. 1999;3:375–382.
15. Hare JM. Nitroso-redox balance in the cardiovascular system. N Engl J Med. 2004;351:2112–2114.
16. Hare JM, Stamler JS. NOS: modulator, not mediator of cardiac perfor-mance. Nat Med. 1999;5:273–274.
17. Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005;115:509–517.
18. Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine re-ceptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A. 2007;104:20612–20617.
19. Gonzalez DR, Treuer AV, Castellanos J, Dulce RA, Hare JM. Impaired S-nitrosylation of the ryanodine receptor caused by xanthine oxi-dase activity contributes to calcium leak in heart failure. J Biol Chem. 2010;285:28938–28945.
20. Gonzalez DR, Treuer AV, Lamirault G, Mayo V, Cao Y, Dulce RA, Hare JM. NADPH oxidase-2 inhibition restores contractility and intracellular calcium handling and reduces arrhythmogenicity in dystrophic cardiomy-opathy. Am J Physiol Heart Circ Physiol. 2014;307:H710–H721.
21. Beigi F, Gonzalez DR, Minhas KM, Sun QA, Foster MW, Khan SA, Treuer AV, Dulce RA, Harrison RW, Saraiva RM, Premer C, Schulman IH, Stamler JS, Hare JM. Dynamic denitrosylation via S-nitrosoglutathione reductase regulates cardiovascular function. Proc Natl Acad Sci U S A. 2012;109:4314–4319.
22. Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600.
Figure 8. Proposed working model for the effects of the temperature on diastolic cardiac ryanodine receptor (RyR2)-mediated Ca2+ leak. A, Under physiological temperature conditions, nitric oxide synthase (NOS)-1 activity is normal and its interaction with xanthine oxidoreductase (XOR) maintains RyR2 in a stabilized state by a regulated proportion of S-nitrosylated cysteines. B, At subphysiological temperature, NOS1 activity is decreased, which allows increasing superoxide (O2
−) production by XOR, leading to peroxynitrite (ONOO‒) generation, BH4 oxidation and NOS1 uncoupling with further reactive oxygen species production, which promotes oxidation of thiol moeties on RyR2. Under these conditions, RyR2 becomes more susceptible to Ca2+ leak.
at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from

Dulce et al Temperature Influences NOS1 Signaling and Ca2+ Leak 55
23. Bassani RA, Bassani JW, Bers DM. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac my-ocytes. J Physiol. 1992;453:591–608.
24. Donoso P, Sanchez G, Bull R, Hidalgo C. Modulation of cardiac ryano-dine receptor activity by ROS and RNS. Front Biosci (Landmark Ed). 2011;16:553–567.
25. Sun QA, Wang B, Miyagi M, Hess DT, Stamler JS. Oxygen-coupled redox regulation of the skeletal muscle ryanodine receptor/Ca2+ release channel (RyR1): sites and nature of oxidative modification. J Biol Chem. 2013;288:22961–22971.
26. Ozawa K, Tsumoto H, Wei W, Tang CH, Komatsubara AT, Kawafune H, Shimizu K, Liu L, Tsujimoto G. Proteomic analysis of the role of S-nitrosoglutathione reductase in lipopolysaccharide-challenged mice. Proteomics. 2012;12:2024–2035.
27. Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, Li D, Berkowitz DE, Hare JM. Neuronal nitric oxide synthase negatively regu-lates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101:15944–15948.
28. Kohr MJ, Roof SR, Zweier JL, Ziolo MT. Modulation of myocardial con-traction by peroxynitrite. Front Physiol. 2012;3:468.
29. Hare JM. Oxidative stress and apoptosis in heart failure progression. Circ Res. 2001;89:198–200.
30. Bhagatte Y, Lodwick D, Storey N. Mitochondrial ROS production and subsequent ERK phosphorylation are necessary for temperature precon-ditioning of isolated ventricular myocytes. Cell Death Dis. 2012;3:e345.
31. Sánchez G, Escobar M, Pedrozo Z, Macho P, Domenech R, Härtel S, Hidalgo C, Donoso P. Exercise and tachycardia increase NADPH oxi-dase and ryanodine receptor-2 activity: possible role in cardioprotection. Cardiovasc Res. 2008;77:380–386.
32. Ziolo MT, Katoh H, Bers DM. Positive and negative effects of nitric oxide on Ca(2+) sparks: influence of beta-adrenergic stimulation. Am J Physiol Heart Circ Physiol. 2001;281:H2295–H2303.
33. Khaliulin I, Halestrap AP, Suleiman MS. Temperature preconditioning is optimal at 26°C and confers additional protection to hypothermic cardio-plegic ischemic arrest. Exp Biol Med (Maywood). 2011;236:736–745.
34. Sheldrick A, Gray KM, Drew GM, Louttit JB. The effect of body tempera-ture on myocardial protection conferred by ischaemic preconditioning or the selective adenosine A1 receptor agonist GR79236, in an anaesthetized rabbit model of myocardial ischaemia and reperfusion. Br J Pharmacol. 1999;128:385–395.
35. Dow J, Simkhovich BZ, Hale SL, Kay G, Kloner RA. Capsaicin-induced cardioprotection. Is hypothermia or the salvage kinase pathway involved? Cardiovasc Drugs Ther. 2014;28:295–301.
36. Hale SL, Kloner RA. Myocardial temperature reduction attenuates necro-sis after prolonged ischemia in rabbits. Cardiovasc Res. 1998;40:502–507.
37. Hale SL, Kloner RA. Mild hypothermia as a cardioprotective approach for acute myocardial infarction: laboratory to clinical application. J Cardiovasc Pharmacol Ther. 2011;16:131–139.
38. Hale SL, Herring MJ, Kloner RA. Delayed treatment with hypothermia protects against the no-reflow phenomenon despite failure to reduce in-farct size. J Am Heart Assoc. 2013;2:e004234.
39. Arrich J; European Resuscitation Council Hypothermia After Cardiac Arrest Registry Study Group. Clinical application of mild therapeutic hy-pothermia after cardiac arrest. Crit Care Med. 2007;35:1041–1047.
40. Hörburger D, Testori C, Sterz F, Herkner H, Krizanac D, Uray T, Schober A, Stöckl M, Stratil P, Weiser C, Wallmüller C, Holzer M. Mild thera-peutic hypothermia improves outcomes compared with normothermia in cardiac-arrest patients–a retrospective chart review. Crit Care Med. 2012;40:2315–2319.
41. Dixon MN, Keasling M. Development of a therapeutic hypothermia proto-col: implementation for postcardiac arrest STEMI patients. Crit Care Nurs Q. 2014;37:377–383.
42. Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–C466.
43. Zweier JL. Measurement of superoxide-derived free radicals in the reper-fused heart. Evidence for a free radical mechanism of reperfusion injury. J Biol Chem. 1988;263:1353–1357.
44. Luczak ED, Anderson ME. CaMKII oxidative activation and the patho-genesis of cardiac disease. J Mol Cell Cardiol. 2014;73:112–116.
45. Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, Hajjar RJ, Kranias EG, Mundiña-Weilenmann C, Mattiazzi A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc Res. 2007;73:689–698.
46. Curran J, Hinton MJ, Ríos E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–398.
What Is Known?
• Nitric oxide (NO) signaling modulates cardiac function, including cal-cium cycling in cardiac myocytes.
• Abnormalities in calcium cycling, such as increased calcium leak from intracellular stores, severely affect cardiac performance.
• Changes in temperature affect the metabolism of NO and consequently NO signaling in the heart.
What New Information Does This Article Contribute?
• Calcium leak is dependent on temperature and is strongly associated with NO and reactive oxygen species.
• The response to temperature of cardiomyocytes lacking neuronal NO synthase activity (neuronal nitric oxide synthase–deficient mice-1 [NOS1−⁄−] mouse model) is opposite that of wild-type cardiomyocytes, whereas deficient denitrosylation (GSNOR−⁄− mouse model) protects against the cooling-induced rise in reactive oxygen species and cal-cium leak.
Using pharmacological interventions on isolated cardiac myo-cytes derived from NOS1−⁄−, GSNOR−⁄−, and wild-type mouse models, we demonstrate that calcium leak exhibits changes that are regulated by an interaction between temperature and the NO/redox balance. This work highlights that temperature is a crucial factor in NO cardiobiology, and reconciles controversial findings in this field. Moreover, these results support the proposed cross-talk between xanthine oxidase and neuronal NO synthase in the regulation of the calcium channel gating in the heart. Decrease in temperature toward subphysiological conditions disrupts this cross-talk favoring a rise of reactive oxygen species and there-fore increasing calcium leak. Hence, either inhibition of neuronal NO synthase or preventing denitrosylation during therapeutic hypothermia in patients undergoing reperfusion postischemia or myocardial infarction, might be beneficial in preventing a tem-perature-dependent increase in calcium leak, thereby favoring cardioprotection and improving clinical outcomes.
Novelty and Significance
at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from

Supplemental Material- page 1
SUPPLEMENTAL MATERIAL
Expanded Methods
Animal Models We studied age matched C57BL/6J mice (wild-type, WT; n=26) and mice with a homozygous
deletion of NOS1 (B6;129S4-Nos1<tm1Plh>J; n=20; Jackson Laboratories, Bar Harbor, ME); or S-nitrosoglutathione reductase (GSNOR‒/‒) (n=10). All protocols and experimental procedures were approved by the Animal Care and Use Committee of the University of Miami and followed the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-234, revised 2011). Myocyte Isolation
Cardiac myocytes were isolated and prepared from hearts as previously described1. Briefly, mice were sacrificed by cervical dislocation. Hearts were harvested and perfused retrogradely in a modified Langendorff system (constant flow 2 mL/min) with an isolation solution containing (in mmol/L): NaCl 120, KCl 5.4, MgSO4 1.4, NaH2PO4 1.2, NaHCO3 20, 2,3-butadiene monoxime (BDM, Sigma-Aldrich Co. Saint Lois, MO) 10, taurine (Sigma-Aldrich Co. Saint Lois, MO) 5, glucose 5.6, bubbled with 5% CO2 -95% O2 for at least 15 minutes. Once cleaned, the hearts were perfused with collagenase type 2 (Worthington Biochemical Corporation, Lakewood, NJ) ~315 U/mL and protease type XIV (Sigma-Aldrich Co.) 5.2 U/mL for 10 minutes. After digestion, the hearts were quickly removed and cut into several chunks and myocytes were released by gentle mechanical disruption. The supernatant containing the dispersed myocytes was filtered through a 500 µm pore mesh into a 15 mL Falcon tube and centrifuged at 800 rpm for 1 minute. The cell pellet was resuspended in a Ca2+‒free Tyrode solution (in mmol/L, 144 NaCl, 1 MgCl2, 10 HEPES, 5.6 glucose, 5 KCl adjusted to a pH 7 with NaOH) containing 0.5% bovine serum albumin to stop enzymatic digestion. The extracellular Ca2+ was restored by three sequential additions of CaCl2 (0.125, 0.25 and 0.5 mmol/L). Finally, the cardiomyocytes were resuspended in a 1.8 CaCl2 Tyrode solution at room temperature and then loaded with Fura-2. Myocytes were stimulated at 0.5 or 1 Hz. Intracellular Ca2+ Measuring
Intracellular Ca2+ was measured using the Ca2+-sensitive dye Fura-2 (Molecular Probes, Eugene, OR, USA) and a dual-excitation spectrofluorometer (IonOptix LLC, Milton, MA, USA), excited with a xenon lamp at wavelengths of 340 and 380 nm. The emission fluorescence was reflected through a barrier filter (510 ± 15 nm) to a photomultiplier tube. The “in vivo” calibration was performed superfusing a free Ca2+ and then a Ca2+ saturating (5 mmol/L) solutions both containing 10 µmol/L ionomycin (Sigma-Aldrich, St. Louis, MO) until reaching a minimal (Rmin) or a maximal (Rmax) ratio values, respectively. [Ca2+]i was calculated using the following equation, as described previously2:
[Ca2+]i= Kd*(Sf2/Sb2)*(R-Rmin)/(Rmax-R) Kd (dissociation constant) in adult myocytes was taken as 224 nmol/L, Rmin=0.479 and
Rmax=2.920 were measured experimentally. The scaling factors Sf2=4608 and Sb2=2865 were extracted from calibration as described by Grynkiewicz et al 3. Experiments were then repeated in the conditions detailed below (see Treatments). ∆[Ca+2]i amplitude was considered as: peak [Ca+2]i – resting [Ca+2]i. SR Ca2+ Leak and SR Ca2+ Load Measuring
SR Ca2+ leakage was assessed with tetracaine (Sigma-Aldrich, St. Louis, MO, USA) as described by Shannon et al.4. Briefly, after pacing was stopped, a fast switch to a 0Na+/0Ca2+ Tyrode solution (Na+ was replaced by an equimolar amount of Li+) was performed. After 60 seconds, as described by Bassani et

Supplemental Material- page 2
al.5, a rapid switching to 0Na+/0Ca2+ solution containing 20 mM caffeine to assess SR Ca2+ content was applied. Following recovery of the cell, the same pacing protocol was assessed. After stop pacing, a switch to 0Na+/0Ca2+ Tyrode solution containing 1 mmol/L tetracaine was performed The observed decrease in the Fura-2 ratio in presence of tetracaine compared to the non-tetracaine treated condition was considered the Ca2+ leak for a particular myocyte. After assessing Ca2+ leak, tetracaine was washed out by superfusing fresh 0Na+/0Ca2+ Tyrode solution and SR Ca2+ content was assessed by caffeine challenge.5 SR Ca2+ contents were calculated considering that SR represents 3.5% and cytosol 65% of the myocyte volume as previously described2. The following equation from Shannon et al.4 was used:
[Ca2+]SR= [Ca2+]caff+(βmax-SR*[Ca2+]caff)/([Ca2+]caff+Kd-SR) [Ca2+]SR is the SR Ca2+ content, [Ca2+]caff is the SR Ca2+ released by caffeine, βmax-SR and Kd-SR are the usual Michaelis parameters for SR Ca2+ binding. SR leak-SR load pairs were grouped by similar SR Ca2+ load and expressed as a leak-load relationship fitted by an exponential growth function using the Graph Pad Prism software (version 4.02). Measurements were mostly carried out at 23°C, 25°C, 30°C, 34°C or 37°C. Treatments
Cardiomyocytes loaded with Fura-2 were pre-incubated for 20 minutes with the following compounds (unless otherwise is specified): Tempol (100 µmol/L, Calbiochem, Calbiochem/EMD Biosciences, San Diego, CA, USA); Oxypurinol (100 µmol/L, Sigma-Aldrich); (±)-S-Nitroso-N-acetylpenicillamine (SNAP) (1 or 50 µmol/L, Santa Cruz, Santa Cruz, CA, USA); N5-(1-Imino-3-butenyl)-L-Ornithine (L-VNIO) (100 µmol/L, Enzo Life Sciences, Plymouth Meeting, PA, USA); Hydrogen peroxide (H2O2) (100 µmol/L, Sigma-Aldrich); (6R)-5,6,7,8-Tetrahydrobiopterin dihydrochloride (BH4) (300 µmol/L, Sigma-Aldrich). Experiments in control condition (no treatment) were run in parallel to each type of pharmacological intervention for each batch of cardiomyocytes. Detection of Reactive Oxygen Species (ROS)
ROS were measured by using the sensitive probe 2’,7’-dichlorodihydrofluoresceine (H2DCF‒DA, 10 µM; Molecular Probes) in two different ways. First, fresh isolated mouse cardiomyocytes were placed in the chamber of an IonOptix spectrofluorometer and the background fluorescence (F0) was acquired with an excitation wavelength of 488 nm and emission fluorescence collected at 510 ± 15 nm. Then, cardiomyocytes were incubated during 30 min at 23°C or 37°C with H2DCF‒DA and washed by superfusing fresh Tyrode (1.8 mM CaCl2) solution for 15 minutes to allowed desterification of H2DCF. The initial fluorescence (Fi) and a second measure after 5 minutes (F) were acquired. Myocytes were stimulated at 1 Hz. ROS were expressed as:
ROS = (F-F0)-(Fi-F0) Alternatively, control or 100 µmol/L oxypurinol‒treated cardiomyocytes were loaded with H2DCF‒
DA for 15 minutes on polylysine‒coated microscope slides at 23°C or 37°C. After 10 minutes washing with Tyrode solution, cardiomyocytes were fixed with 2% p-formaldehyde in cold phosphate buffered saline (PBS) and then washed once with PBS. Mounting medium was applied and slides kept at 4°C until fluorescence was measured. Cardiomyocytes treated with 1 mmol/L H2O2 at either 23°C or 37°C, from each mouse model, were used as positive control for ROS and were used as maximal fluorescence signal for normalization of each group (Fmax). Fluorescence (F) was captured by an inverted fluorescence microscope Olympus IX81 and a CDD camera Retiga 2000R (Q Imaging) with an excitation wavelength of 488 nm and 525 nm emission. Images were quantified by ImageJ (NIH) software and results were expressed as:

Supplemental Material- page 3
FDCF = (F-F0)/(Fmax-F),
where F0 is background Nitric Oxide Measurement
Isolated WT mouse cardiomyocytes were loaded with the fluorescent NO-sensitive dye 4,5-diaminofluorescein diacetate (5 μmol/L DAF-2 DA; Calbiochem/EMD Biosciences, San Diego, CA, USA) for 20 minutes at room temperature and then washed for 20 more minutes to allow intracellular desterification. Cells were set in the perfusion chamber of an IonOptix system and fluorescence (excited at 488 nm and emission collected at 510 ± 15 nm) was recorded three times during a stabilization period of 15 min at 23°C. Then, fluorescence was also acquired at 25°C, 30°C, 34°C and 37°C for each cell. DAF-2 fluorescence intensity (F) was expressed as F/F0, where F0 is the fluorescence intensity at 23°C after the stabilization time. NOS isoform expression
Cardiomyocytes from 4 WT hearts were exposed to different temperature and then collected in RNA later lysis buffer (Qiagen, Valencia, CA). Total RNA was extracted from cells using Pure-Link Micro-to-Midi Total RNA Purification System (Qiagen) and reverse-transcribed using High capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). All samples were treated with TurboTM DNase (Ambion, Austin, TX). Quantitative real-time PCR was performed in triplicate using a 20 μl reaction mixture containing 10 ng cDNA, TaqMan Universal PCR Master Mix (Roche, Branchburg, NJ) and primer/probe sets for nitric oxide synthase 1 (Nos1, Mm00435171_m1), Nos2 (Mm00440502_m1), and Nos 3 (Mm00435197_g1 ) (TaqMan Gene Expression Assay, Applied Bio systems, Foster City, CA). As an internal control glucoronidase beta (GUSB, Mm01197698_m1) was determined in each reaction. Reactions conditions were performed according to manufacturer instructions: 1 cycle of 50°C for 2 minutes, 1 cycle of 90°C for 10 minutes and 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Software from iQ5 multicolor real-time PCR detection system (Bio-Rad, Hercules, CA) was used for PCR analyses. mRNA relative expression was calculated by 2ΔCt method. qPCR data reflects four different experiments each done at 23°C, 30°C, and 37°C.
Statistical analysis
Data are expressed as mean ± SEM. For Leak‒Load relationship, an exponential growth fit, which compares independent fits with a global shared fit, was applied. Two or more groups of data were considered to fit different curves if p < 0.05. For comparisons of two groups, Student’s t test was used. For comparison of three or more groups, one or two way-ANOVA was performed. Two-way ANOVA was used when a second variable was involved. Newman-Keuls or Bonferroni’s post-hoc tests were used as appropriate by the GraphPad Prism version 4.02 (GraphPad Prism Software Corporation San Diego, CA USA). A p < 0.05 was considered significant.
Reference List
(1) Khan SA, Skaf MW, Harrison RW, Lee K, Minhas KM, Kumar A, Fradley M, Shoukas AA, Berkowitz DE, Hare JM. Nitric oxide regulation of myocardial contractility and calcium cycling: independent impact of neuronal and endothelial nitric oxide synthases. Circ Res 2003;92:1322-1329.

Supplemental Material- page 4
(2) Dulce RA, Yiginer O, Gonzalez DR, Goss G, Feng N, Zheng M, Hare JM. Hydralazine and organic nitrates restore impaired excitation-contraction coupling by reducing calcium leak associated with nitroso-redox imbalance. J Biol Chem 2013;288:6522-6533.
(3) Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 1985;260:3440-3450.
(4) Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res 2002;91:594-600.
(5) Bassani RA, Bassani JW, Bers DM. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J Physiol 1992;453:591-608.
Supplemental Figure legends
Online Figure I. SR Ca2+ leak protocol. Protocol used to assess SR Ca2+ leak using tetracaine to block the RyR2 and caffeine to estimate SR Ca2+ load.
Online Figure II. Intermediate dose of SNAP does not change leak-temperature pattern in NOS1−/− cardiomyocytes. Dependence of SR Ca2+ leak on temperature in NOS1−/− cardiomyocytes in the absence or the presence of 50 µmol/L SNAP at matched SR Ca2+ load ≈60 µmol/L . Dependence of SR Ca2+ leak with temperature in WT cardiomyocytes is also displayed as reference.
Online Figure III. Ca2+ decay time constant. Ca2+ decay time constant (τ) in WT, NOS1−/− or GSNOR −/− cardiomyocytes, evaluated at 23°C or 37°C. Ca2+ decay is not different between WT and NOS1 −/− at 23°C but it is slower in NOS1 −/− cardiomyocytes at 37°C (* p< 0.05, Student ‘s t-test). On the other hand, τ are similar at 37°C in WT and GSNOR −/− but smaller in GSNOR −/− at 37°C (* p< 0.05 GSNOR −/− vs. WT, Student ‘s t-test).

Online Figure I
1.5
2.0
2.5
100 150 200
Rat
io (F
340/
F380
)
Time (sec)
+ Tetracaine 0Na+/0Ca2+
Leak
Stop pacing Caffeine
20 25 30 35 40 0
10
20 WT NOS1 -/-
Temperature ( o C)
S R C
a 2 + L
e a k
( µ M
)
NOS1 -/- + 50 µ M SNAP
Online Figure II
0.00
0.05
0.10
0.15
0.20
T a u
( τ ) s
e c
NS * NS
*
21 22 15 20 21 21
Online Figure III

Raul A. Dulce, Vera Mayo, Erika B. Rangel, Wayne Balkan and Joshua M. HareRedox Balance−Influences Sarcoplasmic Reticulum Calcium Leak: Role of Nitroso
Interaction Between Neuronal Nitric Oxide Synthase Signaling and Temperature
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2014 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.116.3051722015;116:46-55; originally published online October 17, 2014;Circ Res.
http://circres.ahajournals.org/content/116/1/46World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2014/10/17/CIRCRESAHA.116.305172.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
at University of Miami School of Medicine on January 7, 2015http://circres.ahajournals.org/Downloaded from




















