
Receptor Tyrosine Kinase Inhibitors That Block Replication of Influenza A and Other Viruses

Inhibitors of the Platelet-Derived Growth Factor Receptor Tyrosine Kinase
Glenda E. Bilder and Camilo J . Rojas
Department of Carrlior~rr.sc.icltrr Biolog!. Rh6i1e Poirlenc Rorer Ceiitrol Research, Colleger~ille, P A , USA
Key Words: Atherosclerosis-PDGF-receptor tyrosine kinase-Phenolate anion-Phenylamino- pyrimidine--Quinoline-Quinoxaline-Restenosis-Tyrosine kinase inhibitor.
INTRODUCTION
Protein tyrosine phosphorylation is essential in key cellular activities such as division, phenotypic modulation, differentiation. migration, and apoptosis (56) . At the subcellular level. protein tyrosine phosphorylation drives enzymatic activation, protein-protein inter- action, cytoskeletal reshuffling. and intracellular trafficking of molecules (80). Under- standing the role of protein tyrosine phosphorylation in the normal state and the discovery that certain viral oncogenes express tyrosine kinase activity have encouraged research efforts toward identification of protein tyrosine kinase inhibitors (TKls). In proportion to the growing interest in the biological role played by protein tyrosine phosphorylation, there are excellent comprehensive reviews on TKls ( 14.32.5 1,891. Other reviews focus on a specific receptor TKIs such as epidermal growth factor (EGF) receptor TKls (7,95) and insulin receptor TKls (91).
Among the membrane receptor tyrosine kinases. the platelet-derived growth factor receptors (PDGFr) are thought to play a pivotal role in the development of atherosclerosis and restenosis as well as certain malignancies (35.76). The development of PDGFr TKIs as potential new therapeutic modalities against these disorders is, therefore, a reasonable goal. This review will focus on the progress toward that end.
PDGF LIGANDS/RECEPTORS
PDGF is a pleiotropic growth factor that initiates cell-signalling by binding to its membrane tyrosine kinase receptors (16.35). PDGF ligands are hetero- or homodimers of protein chains designated A and B. The A B . BB. and AA ligands activate the PDGFr subunits termed alpha (a) and beta (p) by binding and dimerizing the subunits in a ligand-specific manner (33.83). In tissue culture, neuronal. vascular, and immune cells
Address correspondence and reprint requests to: Dr. Glenda Bilder. Department of Cardiovascular Biology. Rhone Poulenc Rorer Central Research. 500 Arcola Road. Collepevillc. PA 194?6-0107. USA. Fax: (610)- 454-8740
380

TYROSINE KINASE INHIBITORS 381
produce and secrete PDGF. Under physiological and pathological conditions, PDGFr on mesenchymal cells are present in sufficient concentrations to allow signalling by auto- crine, paracrine (35), and possibly intracrine (54) mechanisms.
The PDGF-receptors share with thirteen other membrane receptor protein kinases a conserved extracellular binding domain, a transmembrane domain, and an intracellular catalytic domain that performs the phosphate transfer from ATP to tyrosine of the recep- tors or cytosolic signalling molecules (100). Also conserved is the ATP binding motif, which includes an essential lysine residue whose deletion abrogates tyrosine kinase cat- alytic activity (23).
Structural details relevant to the PDGFr are summarized in Table 1. The PDGFr and several close relatives, including colony stimulating factor- 1 -receptor (CSF- lr), steel cell factor (SCFrkkit), and Flk2 (KDWFlt3) contain a unique nonkinase sequence interrupting the kinase catalytic domain and distinguishing cysteine clusters in the extracellular do- main (100). Although the (Y and f3 PDGFr subunits are somewhat homologous (27-87%) (18), differences exist in the intracellular domains that account for variations in cytoplas- mic substrate specificity and may translate into differences in cellular functionality (22 , 34,79,99).
Oligomerization of the membrane tyrosine kinase receptor permits diversity in signal transduction and plays an important function in providing the structural basis for signal transmission by growth factors (81). Binding of the PDGF ligands is well characterized with dissociation constants in the nanomolar range (47). As dictated by the ligand-specific binding framework noted above, PDGF-AA will dimerize (Y subunits, PDGF-AB will dimerize (Y(Y and (YP subunits and PDGF-BB will produce dimers of all three possible
TABLE 1. Characteristics of PDGF @-receptor ~
AA Comparison to Domain length Characteristics Function a receptor
Ligand binding 499 5 immunoglobulin- like domains; cysteine-rich regions
Transmembrane 25 Hydrophobic residues Juxtamembrane 48
Catalytic 347 Split kinase; ATP binding site; multiple phosphorylation sites
Cytoplasmic tail 155
Ligand specificity-binds PDGF-B chain
Anchor; dimerization Src phosphorylation
site Signal transduction
and SH2 recruitment of substrates
Ubiquitization and degradation of receptors; negative regulation of signalling
a receptor binds A and B chain; 30% identity
48% identity 83% identity
Sites on a-receptor revealed with P-receptor binding; different substrate affinities; 87% identity tyrosine kinase 1; 74% identity tyrosine kinase 2
27% identity
From refs. 15, 17, 18, 34, 79, 106.
Cardiovascular Drug Reviews, Vol. 14, No . 4 , 1996

382 G . E . BILDER AND C. J . ROJAS
combinations. Alternative models have been proposed that allow for spontaneous oligo- merization without ligand in cases of elevated receptor numbers (36).
With dimerization, conformational changes ensue that can be detected with immuno- reactivity methods (6,46). Little is known, however, of the molecular mechanisms in which conformational changes activate receptor autophosphorylation or of specific con- tributions made by cytoplasmic domains such as the membrane spanning domain (106). Studies using dominant negative receptor mutants determined that autophosphorylation of the receptor proceeds by an intermolecular (trans) mechanism in which one receptor subunit cross-phosphorylates the other (47).
Peptide mapping, overexpression of chimeric receptor constructs or deletionlsite- directed mutants and synthetic peptide substrates have led to identification of multiple phosphorylation sites on the receptor (16,43,96). Both the phosphorylated tyrosine and flanking amino acids determine the specificity of the PDGFr signalling by providing high affinity regions which facilitate physical association of cytoplasmic proteins containing specific amino acid sequences referred to as SH2 (src homology 2) domains (88). SH2 domain-containing protein substrates are mainly adaptor molecules or enzyme substrates for the receptor kinase.
In some cases, such as phospholipase C-y, tyrosine phosphorylation serves to increase enzyme activity (65); in the case of nonenzymatic substrates, tyrosine phosphorylation may induce conformational changes leading to activation of other substrates (87). Table 2 summarizes the known substrates and their binding loci on the PDGFPr. Inhibitors of the PDGFr tyrosine kinase would be expected to reduce or eliminate association of some or all of these potential substrates.
Signal transduction converges beyond this point. The details from ras-raf-MEK 1 - MAP-kinase-phosphorylated transcription factors have been reviewed (43,56,88). Speci- ficity of PDGFr signalling, therefore, appears to reside in tyrosine phosphorylation sites and flanking amino acids. The specificity results not only from the primary sequence of
TABLE 2. PDCF @-receptor substrates
Mol wt Binding Substrate (kDa) location* Function Ref.
Src, Fyn, Yes Grb2
60 579,581 27 716
Shc 46.52,66 579,740.
P85/phosphoinositol 3-kinase 85 740,751
Nck 75 1 GTPase activating protein 125 77 1
751.771
(GAP)
Transformation 8,62 Linker to nuclear exchange 2
factor (Sos), Ras regulation Linker. binding sites for Grb2
Linker, protein shorting, 24,41
Transformation? 66 Negative regulation of RAS 24,45,41
109
mitogenesis, chemotaxis
Phosphotyrosine phosphatase 1 D 65 1009 Phosphatase, binding site for 44.50 Grb2 ~ ~~
Phospholipase C-gamma 145 1021,1009 Phosphoinositol production, 74,49,61 mitogenesis, chemotaxis
JAK1, JAK2, Tyk2 I20 ? Ubiquitinization, sorting 101 STATla 84.91 ? Translocation to nucleus 108
* Numbers of binding location corresponded to amino acid location in human PDGF P-receptor
Cardiovascular Drug Reviews, Vol. 14. No. 4 , 1996

TYROSINE KINASE INHIBITORS 383
substrate and receptor but also in substrate recognition determined by peculiarities in secondary, tertiary and quaternary structures. Intracellular localization of potential sub- strates and receptor-associated phosphatase activity are also important in regulating signal transduction via activation of the PDGFr (88).
MECHANISMS OF RECEPTOR TYROSINE KINASE ACTIVITY
Both autophosphorylation and kinase activity involve transfer of a phosphate group from ATP to a protein tyrosine residue. The phosphate acceptor could be the receptor itself (autophosphorylation) or another protein (kinase activity).
The amino acid sequences of the catalytic domains of fourteen receptor protein-tyrosine kinases were aligned recently (100). The list included PDGFPr, insulin receptor (INr), and EGFr. The homologous alignment and information on the crystal structure of cyclic AMP-dependent protein kinase, a serineithreonine kinase, were used to postulate the likely role of conserved residues. Conserved residues may affect ATP binding, metal ion chelation, the chemical steps of phosphotransfer, peptide accessibility, substrate recog- nition, and the formation of ion bridges between the two structural lobes that form the catalytic site (94).
Phosphate transfer in the PDGFr-catalyzed reaction is postulated to require proton abstraction from phenol in the tyrosine residue by asparate 826 and nucleophilic attack of the resulting phenolate anion on tetracoordinate y-phosphorus of ATP to form a penta- coordinate transition state (or intermediate) followed by formation of phosphorylated tyrosine and ADP. A protonated side chain, arginine 830, could be involved in electro- static stabilization of the intermediate (Fig. 1). These features are consistent with the proposed mechanism of phosphorylation proposed for the EGFr (102). The pH depen- dence of the EGFr-catalyzed reaction and thermodynamic considerations support the involvement of aspartate and arginine (aspartate 813 and arginine 817 in EGF-r); the purported pKa values are 6.3 for aspartate and 9.1 for arginine (102). The involvement of these residues in catalysis is also supported by the x-ray structure of the INr (aspartate 1132 and arginine 1136 in the INr) (37).
The ionization of the tyrosyl hydroxyl group on both chemical and enzymatic reactiv- ities is important. For example, in a phosphorylation model reaction of acetylation of tyrosine and fluorotyrosine by N-acetylimidazole, tyrosine is seventeen times more reac- tive than fluorotyrosine and the reactivity is directly correlated to the relative concentra- tion of the phenoxilate. Comparison of reactivities of fluorotyrosine- and tyrosine- containing peptides as substrates for the INr show higher V,,, and K, values for phos- phorylation of the tyrosine peptide than for phosphorylation of fluorotyrosine peptide. The results indicate that phosphorylation is favored in environments that enable efficient deprotonation of the tyrosine residue (58).
Electrostatic interactions of manganese and/or magnesium play a pivotal role in PDGFr tyrosine phosphorylation. Early nuclear magnetic resonance studies of solutions of metal ions and ATP indicate binding of the manganese ion to oxygen atoms on the a-, p- and y-phosphates of ATP and binding of magnesium to only the p- and y-phosphates (9). Metal ion chelation of oxygen neutralizes the negative charge on ionized phosphate groups to facilitate the approach of the phenolate ion. Dissociation of the chelated species is favored over dissociation of the highly ionized pyrophosphate (9).
Cardiovascular Drug Reviews, Vol. 14. No. 4, 1996
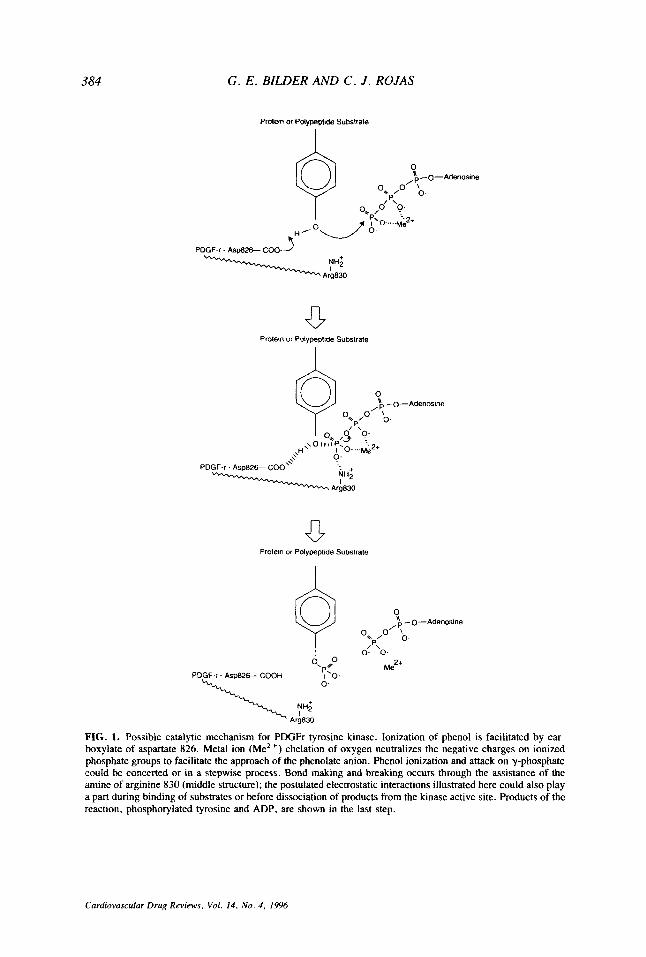
384 G . E . BILDER AND C . J . ROJAS
Protein of Polypepttde Substrate
I
PDGF-r . Asp&%- COO---’
Prolein or Polypeptide Substrate
v Protein or Polypeplide Subslrate
0 ’ 0-Adenosine /y- o* / o o.
I p\ 0- 0-
Me2+
FIG. 1. Possible catalytic mechanism for PDGFr tyrosine kinase. Ionization of phenol is facilitated by car- boxylate of aspartate 826. Metal ion (Me*+) chelation of oxygen neutralizes the negative charges on ionized phosphate groups to facilitate the approach of the phenolate anion. Phenol ionization and attack on y-phosphate could be concerted or in a stepwise process. Bond makmg and breaking occurs through the assistance of the mine of arginine 830 (middle structure); the postulated electrostatic interactions illustrated here could also play a part during binding of substrates or before dissociation of products from the kinase active site. Products of the reaction, phosphorylated tyrosine and ADP, are shown in the last step.
Cardiovascular Drug Reviews, Vol. 14. No. 4 , 1996

TYROSINE KINASE INHIBITORS 385
PDGF-RECEPTOR TYROSINE KINASE INHIBITORS
Clinical Need
Since PDGF is the cellular homologue (c-sis) of the transforming v-sis oncogene of the Simian sarcoma virus (103) and is a potent inducer of cell proliferation, migration, phenotypic change, and transformation, inappropriate coexpression of ligands and recep- tor subunits may be an important mechanism underlying fibroproliferative disorders such as atherosclerosis, restenosis following balloon catheter angioplasty , malignancy, pulmo- nary fibrosis, and glomerulonephritis (72). Theoretically, specific inhibitors of the PDGFr tyrosine kinase would disrupt PDGF-dependent signal transduction and retard or block disease/disorder progression. The evidence that PDGF ligands/receptors are involved in atherosclerosis and restenosis is reviewed.
Atherosclerosis
Atherosclerosis is a complicated disease process contributing to coronary heart disease, a major cause of death i.n our society. Vascular lesions have been described histologically and range from fatty streaks containing a small number of lipid-filled, monocyte-derived macrophages and some T lymphocytes to complex lesions composed of increased num- bers of smooth muscle cells, macrophages, and T lymphocytes and varying amounts of extracellular matrix, thrombi, and related degradation products, lipids, and calcium de- posits (75,92). The fatty streak may be the precursor to the more complex lesion that culminates in lumen narrowing, occlusion or rupture (75).
As determined by immunocytochemical staining, Northern analysis, and in situ hy- bridization, the mEWA and protein of the PDGF B-chain are upregulated in complex atherosclerotic lesions of humans and cholesterol-fed nonhuman primates (3,77,105) and are found in association with lesion macrophages (77) and intimal cells, referred to as intimal mesenchymal cells or modified smooth muscle cells (104,105). In addition, the transcript and protein for PDGFPr have been detected in advanced lesions (78,109, localized to modified smooth muscle cells of the lesions (78,105). The patterns of ex- pression of PDGFPr and PDGF-B ligand are consistent with the proposal that autocrine and paracrine networks initiate PDGF-dependent activities in intimal smooth muscle cells to assist with development of the atherosclerotic plaque. In addition, PDGF B-protein is present in macrophages of some but not all fatty streaks from human aorta, suggesting that PDGF-BB ligand may participate in the initiation of atherosclerosis (42).
PDGF-A chain transcripts are also expressed in modified intimal smooth muscle cells of complex atherosclerotic lesions of the human carotid artery at levels higher than in normal arteries (104,105). This, however, has not been a consistent finding. Recently, the technique of competitive reverse transcription-polymerase chain reaction (RT-PCR) was applied to autopsy samples of three advanced atherosclerotic plaques of the aorta and ten normal aortic sections to measure PDGF A-chain mRNA (63). PDGF A-chain transcripts were approximately 100 times lower in sections from atherosclerotic tissue than in sec- tions from normal tissues, and transcript levels in lesions were independent of the extent of cell proliferation as determined by staining with proliferating cell nuclear antigen. The discrepancy between these findings and those reported earlier may result from the dif- ference in tissue site (aorta and carotid) and/or the number of analyzed samples (three
Cardiovascular Drug Reviews, Vol. 14. No. 4 , 1996

G . E . BILDER AND C . J . ROJAS
versus greater than thirty). Although the results generated from the competitive RT-PCR analysis question the mitogenic role of the PDGF A-ligand in atherosclerosis, it is noted that the extent of proliferation at any one time in the atherosclerotic plaque may be variable and indeed, different proliferative rates have been published (67,70).
Although the role of PDGF A-ligand remains unclear and expression level of the PDGFar in lesions is yet to be determined, the detection of the PDGF ligands and the Q-receptor is an important first step in defining their influence in atherosclerosis. Given the pleiotropic nature of PDGF, it is likely that PDGF may stimulate one or more of its known activities to initiate and maintain plaque development. With identification of highly selective PDGFr TKIs, this hypothesis may soon be tested.
Restenosis
This is the clinical condition characterized by lumen narrowing (restenosis) of a dis- eased (stenosed) artery approximately 6 m to 1 y following successful dilation with an angioplasty balloon, stent, or atherectomy device. Since postangioplasty repair occurs on top of and within the atherosclerotic plaque, histology of restenotic lesions is as variable and complex as the atherosclerotic plaque. Arterial wall changes following restenosis have been attributed to combinations of vascular cell migration and proliferation, matrix deposition, vasoconstriction, and remodelling (59). There is currently no successful ther- apy to prevent restenosis following angioplasty .
Analysis of human coronary artery restenotic lesions from 2 d to 6 m following an- gioplasty revealed that the PDGFQr and PDGF B-ligand are expressed in regions of repair, in association with macrophages and a-actin negative spindle cells, most likely modified smooth muscle cells (93). These findings show, for the first time, coexpression of a compatible ligand and receptor combination following coronary angioplasty and signify that in human restenotic lesions the PDGF/PDGFr circuit is upregulated and may be responsible for vascular changes following balloon angioplasty.
Results of studies in animal models of balloon catheter denudation (BCD) injury in- dicate increased expression of PDGF A- and B-chain transcripts in rat aortic artery (60), rabbit iliac artery (20), and baboon brachial artery (68). The status of the PDGFr was not determined in these models. The BCD-induced injury of the rat carotid artery is the most intensely studied animal model for this disorder. Results in this model show that PDGF A-chain transcripts, as determined by Northern analysis, are transiently expressed within 6 h after BCD but without concomitant increase in PDGFar mRNA (57). Two weeks following BCD, PDGF A-chain and PDGFQr mRNA, measured by in situ hybridization, are evident in discrete regions of the intima (57). Although these results suggested that a paracrine or autocrine network within the intima was unlikely since the PDGF-A ligand does not activate the PDGFPr (33,83), subsequent findings using rat-specific RNA probes and enface tissue preparation showed coexpression of the PDGF B-chain and PDGFPr in luminal smooth muscle cells at several time points throughout the course of intimal formation following BCD (52). These latter findings support the hypothesis that PDGFr activation is involved in catheter-induced intimal growth.
Several studies in rat carotid and rabbit femoral BCD models in which infusions of PDGF or antibodies to PDGF or platelets were used to manipulate the PDGF/PDGF-r loop, have concluded that the main function of PDGF is to stimulate smooth muscle cell
Cardiovascular Drug Reviews, Vol. 14, No. 4 , 1996

TYROSINE KINASE INHIBITORS 387
migration (25,26,27,40). Recent findings support this proposal by demonstrating injury- induced increase in transcript and protein expression of PDGF B- and A-chains in the leading endothelial edge of a denuded region of the rat carotid artery simultaneous with constant expression of PDGFPr in exposed smooth muscle cells (53). To understand further the in vivo functions of PDGF, the gene for the PDGF-B chain was transferred into the porcine iliac artery (64). Transfer of this gene without overt catheter stretching produced an increase in the number of intimal smooth muscle cells and a lesion that was histologically similar to that of the human restenotic lesion (64). These observations suggest that PDGF is able to initiate intimal hyperplasia in vivo.
A major limitation of the aforementioned animal models is that the vascular injury, whether in a carotid, iliac, or aortic artery, culminates in a stenotic lesion in a normo- cholesterolemic animal. Although it is possible that balloon injury to a normal artery and to an atherosclerotic artery may stimulate the same basic mechanisms, expression of ligands/receptors and effects of manipulation of ligandslreceptors in a balloon dilated atherosclerotic artery are lacking. Results from these models would add significantly to our understanding of restenosis and provide models in which to evaluate new therapies including PDGFr TKIs.
Development
A systematic scheme for discovery of TKIs has been described in relation to identifi- cation of EGFr TKIs with anticancer activity (7,28). Issues of potency, selectivity, cell permeability, animal bioavailability, and lack of toxicity are emphasized as central to TKI discovery (7,28,51). In addition, it is important to evaluate efficacy of potent TKIs on in situ receptor autophosphorylation and on protein tyrosine phosphorylation of known sub- strates in cell cultures. Subsequent determination of cellular activities of interest, for example, DNA synthesis, cell growth, migration, and phenotypic change can then be meaningfully assessed. Since there is no consensus as to the number of kinases, tyrosine, serine, and threonine, against which demonstrated inactivity confers a label of selectivity and since there is no consensus as to whether enzymatic, cellular, or in vivo selectivity provides the most meaningful information, evaluation of selectivity represents, in its purest form, a herculean task.
A postulated role for cellular and membrane receptor tyrosine kinases in tumor devel- opment prompted the initial search for TKIs (35,38). Natural products comprise the group of original inhibitors (Fig. 2). These compounds, extracted from microbial broths or plants, are inhibitors of the EGFr tyrosine kinase, p60 Src, or p40 kinase, display IC,, values ranging from 0.01 to 100 pM and share structurally overlapping features (14). Although generally more active against tyrosine kinases than threoninekerine kinases, the natural products in Fig. 2 are not selective TKIs. Methyl 2,5 dihydrocinnamamate (Fig. 3A), a stable analogue of erbstatin and one proposed to be a PDGFr TKI, is an example of a nonselective PDGFr TKI. Methyl 2,5-dihydrocinnamamate inhibits PDGF-dependent proliferation of rat aortic smooth muscle cells without reducing in situ PDGFr autophos- phorylation (84). Its mode of action is unknown, although actin fiber polymerization and protein tyrosine phosphorylation of several low molecular weight cytoplasmic proteins are inhibited (84).
Cardiovascular Drug Reviews, Vol. 14, No. 4 , 1996

388 G . E . BILDER AND C . J . ROJAS
Natural Products Name
Q u e rcet i n
Erbstatin
Genistein
Herbimycin
Lavendustin A
Piceatannol
StructurdSource
cH Cyclic AMP-independent *& protein kinase; -1 00 pM
ar Fruits, vegetables, teas W O
EGF-receptor kinase; 0.55 pglrnl
1 Streptomyces o*
ra
EGF-receptor, p6Ov-src, p l l0gag-fes kinase; 6-8 pglml
a*
Pseudornonas sp. 0
ngc0 L, L, L., Streptomyces
EGF-receptor kinase; 4.4 ng/ml
I
Streptornyces griseolavendus o*
no \
o(
Euphorbia lagascae
FIG. 2. TKI natural products. From refs. 1, 30, 31, 69, 97, 98
The fust postulated “selective” PDGFr TKIs (Fig, 3B) belong to the dicyanoben- zylidine series (5 ) . Some members of this chemical series were previously identified as potent substrate competitive EGFr antagonists, termed tyrphostins (107). Tyrphostins were designed to be competitive for the protein substrate, to be selective for EGFr tyrosine
Cardiovascular Drug Reviews, Yo/. 14. No. 4 , 1996

TYROSINE KINASE INHIBITORS 389
A. 9"
A RG 50872
S Q CH3
ST 638
RG 13022
RG 13291 I
FIG. 3. Representative PDGFr TKIs, dihydrocinnamamate (A), dicyanobenzilidine (B), and modified dicy- anobenziline series (C,D,E).
kinase compared to INr tyrosine kinase, and to be soluble in water and mildly hydropho- bic solvents (29).
As PDGFr TKIs, members of the dicyanobenzylidine series inhibited the cell-free PDGFr kinase activity, in situ PDGFr autophosphorylation, PDGF-dependent DNA syn- thesis and PDGF-induction of c-fos transcripts at low micromolar concentrations (0.01-8 FM) in vascular smooth muscle cells (5). This series did not disrupt binding of ligand to
Cardiovascular Drug Reviews, Vol. 14. No. 4, 1996

390 G. E . BILDER AND C . J . ROJAS
receptor and inhibitory activity was reversible ( 5 ) . Although some compounds from this series were 100- to 1000-fold more selective for EGFr than INr tyrosine kinase (29), the separation between inhibition of PDGFr tyrosine kinase and EGFr tyrosine kinase mea- sured in the same cell type was a modest 20-fold ( 5 ) . Members of this series did not inhibit the enzymatic activity of PKC and PKA. Modification of the dicyanobenzylidene series by substitution of the hydroxyls with an indole did not improve potency for inhibition of in siru PDGFr autophosphorylation, DNA synthesis in fibroblasts, or selectivity for PDGFr compared to EGFr (10). Compounds of the dicyanobenzylidine series penetrated cells adequately but were chemically unstable (55,73). RG 50872, in particular, has been cited for its toxic damage to mitochondria1 membranes and time-related induction in cellular ATP reduction (13). It is noted that these activities were evident at a concentration (2 pM) which is 50 times above the IC,, value for previously reported inhibition of PDGFr-associated activities (5 ) .
A 4-hydroxycinnamamide derivative (ST 638) (Fig. 3C) has recently been shown to inhibit interleukin- 1-induction of intimal hyperplasia and autocoid-dependent vascular changes in the pig coronary artery (39). Although the vascular changes in this model are mediated largely by PDGF (39,71), ST 638 is not selective for the PDGFr tyrosine kinase and inhibits several tyrosine kinases (85,86). Additionally, it is interesting that ST 638 was administered topically to the coronary artery, achieving local concentrations in the arterial wall in the low mM range (39). It seems possible that derivatives of the cin- namamide series may be of use in local delivery systems. The ability of the hydrogel angioplasty catheter to absorb and release RG 50872 at concentrations that inhibit PDGF- dependent smooth muscle proliferation in culture has been demonstrated (19).
Replacement of a single cyano group with a phenyl or indole moiety did little to improve potency or selectivity. The resultant chemical entities were potentially more metabolically stable, however, and were reasonable candidates for in vivo efficacy eval- uation. RG 13022 (2-[3-pyridyl1-3-[ 3,4-dimethoxyphenyl]-2-propenenitrile) (Fig. 3D) sig- nificantly decreased the growth of a human squamous cell carcinoma, an EGFr overex- pressing tumor, in nude mice ( 1 10). RG 13291 (2-[3-indolyl-3-~3,4-dimethoxyphenyl]- 2-propenenitrile) (Fig. 3E) inhibited cuff-induced increase in arterial PDGFr tyrosine kinase activity in the rat carotid artery (4). Both compounds were administered intra- peritoneally.
Using RG 13022 as a structural base, Maguire et al. ( 5 3 , described the approach of constraining the acrylonitrile and generating a series of 3-substituted quinolines. With elimination of the potentially undesirable reactive cyano group of the modified tyrphostin RG 13022 and RG 13291, cishrans isomerization is removed, a basic center is created, and the compound is no longer a Michael acceptor. These changes resulted in a 250-fold separation in selectivity between PDGFr tyrosine kinase and EGFr tyrosine kinase and nanomolar potency for inhibition of the cell-free PDGFr autophosphorylation (Fig. 4). Substitutions at the 3 position, the 5-8 position, and the basic moiety influenced kinase activity. As illustrated, potency increased to 1-20 nM with 3-aromatic substitutions. Removal of the nitrogen resulted in loss of activity and potency decreased with removal of the 5,7 methoxy or methyl (or equivalent methoxy, methyl at 6, 7) substitutions or replacement with 8-substitutions (Fig. 4). Analysis of 3 ,4-pyridinyl) substituted quino- line (Fig. 5A) demonstrated that a potent TKl can be competitive for ATP (Ki = 14 nM) and still retain a favorable degree of selectivity for PDGFr tyrosine kinase compared with
Cardiovascular Drug Rer,iens. Vol. 14. N o . 4. 1996

TYROSINE KINASE INHIBITORS 391
RS
Quinoline PDGFr TKI
H OCH, OCH, H 3-thienyl 0.001 -0.02
H OCH, OCH, H NO2 >50
H OCH, OCH, H NH2 >50
H OCH, OCH, H OH >50
H OCH, OCH, H 3-pyridinyl 0.1-0.8 ~~
H OCH, OCH, H P-styryl 0.07-0.15
CH, H H CH, 3-thienyl >50
H 0-CH2-0 H 3-thienyl 0.2-0.6
H H H H 3-thienyl 0.2-0.5
OCH, H OCH, H 3-thienyl 0.001 -0.02
FIG. 4. Structure activity relationships of quinoline-based PDGFr TKIs. From ref. 55.
EGFr, erB2r, p56LCK tyrosine kinase and serine and threonine kinases PKC and PKA
Substituted quinoxalines also inhibit PDGF-dependent tyrosine kinase activity and DNA synthesis (Swiss 3T3 fibroblasts and porcine endothelial cells) with potency similar to the quinoline series. The most potent compound, AG1296 (Fig. 5B), inhibits SCFr but has less effect on EGFr autophosphorylation and DNA synthesis stimulated by insulin, fibroblast growth factor (FGF), EGF, and no effect on KDR-receptor (KDRr) autophos- phorylation and VEGF-dependent tyrosine kinase activity and DNA synthesis. At higher concentrations, AG1296 reversed the phenotype of v-sis-transformed fibroblasts but not the phenotype of src-transformed cells (48). The authors comment on the need to improve selectivity of the quinoxaline series between PDGFr and SCFr tyrosine kinase but em- phasize that the absence of effect on KDRr tyrosine kinase makes AG1296 an attractive candidate for treatment of restenosis where inhibition of PDGFr-dependent stimulation of smooth muscle cell migration and proliferation without blockage of VEGF-dependent endothelial proliferation is a therapeutically desirable combination (48). Thus compounds
(21).
Cardiovascular Drug Reviews, Vol. 14. No. 4, 1996

392 G . E . BILDER AND C . J . ROJAS
A.
3-(4-Pyridinyl) quinoline
6.
CH30
AG 1296
CGP 53716
I D.
Q Y N D 0
CGP 57148 FIG. 5. Representative PDGFr TKls, quinoline (A) , quinoxaline (B), and phenylaminopyrimidine series (C,D,E).
of the substituted quinolines and quinoxalines series represent potent inhibitors of the PDGFr tyrosine kinase with a moderate degree of selectivity.
A 2-phenylaminopyrimidine (CGP 53716) (Fig. 5C) was recently reported to be a highly selective inhibitor of the PDGFr tyrosine kinase. Selectivity was evaluated against more than twenty tyrosine kinases including EGFr, IGFr, INr, Src, Lyn, Fgr, Csk, and
Curdiovusculur Drug Reviews. Vol. 14 , No. 4 , 1996

TYROSINE KINASE INHIBITORS 393
TPK (11), and among this series of kinases, only inhibition of Abl kinase was apparent. As a competitive antagonist for ATP (112), CGP 53716 inhibited cell-free and in situ PDGFr autophosphorylation with an IC5, value of 100 nM. CGP 53716 was also the first PDGFr TKI with demonstrated oral efficacy. Administration of CGP 53716 decreased tumor size in nude mice injected with v-sis-transformed BALB/c 3T3 cells, PDGFr- overexpressing cells, at doses of 6.3-50 mg/kg in a dose-dependent manner. In contrast, CGP 537 16 was ineffective in mice injected with the human epidermoid carcinoma A43 1, EGFr-overexpressing cells. Similarly, CGP 57148 (Fig. 5D), a potent inhibitor of Abl kinase (35 nM), also inhibited PDGFr tyrosine kinase and tumors of v-sis transformed BALBk3T3 cells (12) and displayed a selectivity profile similar to CGP 53719. These results support an interesting speculation that there exist similarities between the cytosolic Abl kinase and the membrane PDGFr kinase in the regions of the ATP binding cleft and surrounding amino acids that interact with the phenylaminopyrimidine structure (1 2).
Mechanistic Considerations In retrospect, the catalytic mechanism can explain, as a first approximation, the inhi-
bition by some EGFr and PDGFr TKIs. The main interactions during phosphate transfer must include the ATP y-phosphate, tyrosyl hydroxyl, and the tyrosyl aromatic ring (102). An effective inhibitor, in addition to resembling the natural substrates, must exhibit tighter binding and lower reactivity than the substrates. Reactivity is associated with the nucleophilicity of the tyrosyl anion, and, therefore, anion mimic-based inhibitors should exhibit lower nucleophilicity (1 11).
Detailed kinetic analysis of PDGFr TKIs is lacking. Of the known PDGFr TKI struc- tures, only the quinolines have been shown to be competitive with respect to ATP (21). Extensive kinetic analysis using anilinoquinazolines as EGFr TKIs, however, indicates competitive kinetics with respect to ATP and non-competitive kinetics with respect to peptide substrate, suggesting anilinoquinazolines behave as analogs of ATP. Further- more, anilinoquinazolines include structural features that resemble aspects of both ATP and peptide substrate: quinazoline nitrogens mimic oxygens on the y-phosphate, the anilino nitrogen mimics the tyrosine hydroxyl oxygen, and the aromatic ring of the anilino function mimics the tyrosine aromatic ring (102).
Since the quinoline and quinoxaline PDGFr TKIs share with the quinazolines common features of a heterocyclic nitrogen atom and a lipophilic substitution (90), some compar- isons are possible. It is suggested that quinoline derivatives with substitution at the 3 position and methoxy groups at the 6 and 7 positions (Fig. 4) (55) can be considered bisubstrate analogs: methoxy groups mimic the potential tyrosyl anion while the nitrogen on the quinoline moiety mimics the oxygens of the y-phosphate of ATP. The aromatic substituent is most likely involved in interactions with the ATP binding site.
Consistent with this hypothesis are the results of extensive structure activity relation- ship studies that show that the nitrogen on the quinoline moiety and a lipophilic or aromatic substituent at the 3 position are required for inhibitory activity (55). Interest- ingly, inhibitory activity is lost by placement of a hydrophilic substituent at the 3 position (e.g., NO,, NH,, or OH), and recovered by addition of a benzyl group to the hydrophilic substituent. This underscores the importance of the nature of the substituent; since it is a site involved in binding, hydrophilic substituents must bring about looser binding.
CGP 53716 (Fig. 4C) also incorporates the general features of the TKIs mentioned
Cardiovascular Drug Reviews, Vol. 14, No. 4 , I996

394 G . E . BILDER AND C . J . ROJAS
above. The structure could be classified as a bisubstrate analog: it contains a peptide bond to which two benzene rings are attached; one of the benzene rings (possibly the tyrosine ring mimic) is bonded to a methyl group and to a nitrogen that could play the role of phenolate anion with reduced reactivity. The rest of the molecule includes two hetero- cyclic rings with nitrogens that resemble phosphate anions and/or the adenosine of ATP.
As noted (102), unbound ATP exists as a metal chelate allowing a greater degree of freedom than the anilinoquinazoline (or the quinoline) derivatives. As a result, binding to the receptor results in greater entropy loss for ATP binding than for inhibitor binding. ATP dissociation is favored over inhibitor dissociation because ATP is more polar. An additional possibility that favors inhibition over reaction with ATP could be tighter bind- ing of inhibitors than ATP to the ground state thus preventing progress of reaction to the transition state (102). Kinetic analysis of PDGFr TKIs are necessary to support these suggestions.
CONCLUSIONS Crystallographic studies of cyclic AMP-dependent protein kinase and INr kinase, com-
puter modeling, and in v i m enzyme kinetics on EGFr TKIs have yielded an understand- ing of the tyrosine kinase reaction at the molecular level. This has assisted in part with the development of inhibitor molecules effective against the PDGFr tyrosine kinase. It is clear that knowledge of the crystal structure of both the activated and unactivated PDGFr and enzyme kinetic analyses with inhibitors would add immensely to the current understand- ing of mechanism(s) of inhibitor action. There are currently several substituted quinoline, quinoxaline, and phenylaminopyrimidine compounds that inhibit PDGFr tyrosine kinase with nanomolar potency. The structures of these compounds suggest that they incorporate features of both ATP and tyrosyl-containing protein substrate. Kinetic studies indicate that some of the series are competitive with respect to ATP but their interaction with substrate is yet to be defined. On the other hand, EGFr TKIs that display the general bisubstrate characteristics are competitive with respect to ATP and noncompetitive with respect to polypeptide substrate.
Although there have been attempts to define the specificity of PDGFr TKIs, informa- tion on the extent of inhibition of other tyrosine kinases and nontyrosine kinases is scant. Use of PDGFr TKIs as effective research tools to elucidate the involvement of PDGFr in normal and abnormal functions will require extensive selectivity studies. However, based on the consideration that fibroproliferative disorders may result from activation of several tyrosine kinases that synergize or potentiate PDGFr tyrosine kinase activity (75,82), however, so selectivity remains important but less critical an issue. Disease etiology and pathology may dictate the requirement for selectivity. In this regard, PDGF-dependent tumors might be treated with a selective PDGFr TKI, while atherosclerosis and restenosis, which are inadequately understood and driven by several growth factors, might respond more favorably to a relatively less selective PDGFr TKI.
PDGFr TKIs inhibit PDGF-dependent functions in cells but generally at higher con- centrations than those required to inhibit the kinase in vitro. The discrepancy in potencies may be related to limits of membrane permeability, assay conditions, or cellular ATP concentrations. In animal studies, efficacy following oral administration of a PDGFr TKI, at least in mice, has been demonstrated. Whether this can be repeated in other species, especially in humans and in other pathological conditions, remains a future challenge.
Cardiovascular Drug Reviews, Vol. 14. No. 4 , I996

TYROSINE KINASE INHIBITORS 395
REFERENCES 1. Akiyama J, Ishida J, Nakagawa S, et al. Genistein, a specific inhibitor of tyrosine-specific protein kinase.
J Biol Chem 1987;262:5592-5595. 2. Arvidsson A-K, Rupp E, Nanberg E, et al. Tyr-716 in the platelet-derived growth factor beta-receptor
kinase insert is involved in GRB2 binding and Ras activation. Mol Cell Biol 1994;14:6715-6726. 3. Barrett TB, Benditt EP. Platelet-derived growth factor gene expression in human atherosclerotic plaques
and normal artery wall. Proc Natl Acad Sci USA 1988;85:2810-2814. 4. Bilder G, Kasiewski CJ, Walczak EM, Perrone MH. PDGF-receptor protein tyrosine kinase activity in
carotid artery is enhanced by injury and inhibited in vivo by tyrphostin RG 13291. Drug Development Res
5. Bilder GE, Ktawiec JA, McVety K, et al. Tyrphostins inhibit PDGF-induced DNA synthesis and asso- ciated early events in smooth muscle cells. Am J Physiol 1991;260:C721-C730.
6. Bishayee S, Majumdar S, Scher CD, Khan S. Characterization of a novel antipeptide antibody that recognizes a specific conformation of the platelet-derived growth factor receptor. Mol Cell Biol 1988;8: 36963702.
7. Bridges AJ. The current status of tyrosine kinase inhibitors: do the diarylamine inhibitors of the EGF receptor represent a new beginning? Exp Opin Ther Patents 1995;5: 1245-1257.
8. Broome MA, Hunter T. Requirement of c-Src catalytic activity and the SH3 domain in platelet-derived growth factor BB and epidermal growth factor mitogenic signaling. J Biol Chem 1996;271: 16798-16806.
9. Bruice TC, Benkovic SJ. Bioorganic Mechanisms, Vol. 11. New York: W. A. Benjamin, Inc., 1966: 1 72- 1 73.
10. Bryckaert MC, Eldor A, Fontenay M, et al. Inhibition of platelet-derived growth factor-induced mito- genesis and tyrosine kinase activity in cultured bone marrow fibroblasts by tyrphostins. Exper Cell Res
11. Buchdunger E, Zimmermann J, Mett H, et al. Selective inhibition of the platelet-derived growth factor signal transduction pathway of a protein-tyrosine kinase inhibitor of the 2-phenylaminopyrimidine class. Proc Nail Acad Sci USA 1995;92:2558-2562.
12. Buchdunger E, Zimmermann J , Mett H, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res 1996;56: 1OG104.
13. Burger AM, Kaur G, Alley MC, et al. Tyrphostin AG17 ([3,5-Di-tert-butyl-4-hydroxybenzylidene]-ma- lononitrile), inhibits cell growth by disrupting mitochondria. Cancer Res 1995;55:2794-2799.
14. Burke TR. Protein-tyrosine kinase inhibitors. Drugs of the Future 1992;17:119-131. 15. Claesson-Welsh L. PDGF receptors: Structure and mechanism of action. In: Westermark B, Sorg C, eds.
16. Claesson-Welsh L. Platelet-derived growth factor receptor signals. J Biol Chem 1994;269:32023-32026. 17. Claesson-Welsh L, Erikkson A, Moren A, et al. cDNA cloning and expression of a human platelet-
derived growth factor (PDGF) receptor specific for B-chain-containing PDGF molecules. Ma1 Cell Biol 1988;8:34763486.
18. Claesson-Welsh L, Eriksson A, Westermark B, Heldin CH. Cloning and expression of human platelet- derived growth factor a and p receptors. Methods Enzymol 1991;198:72-77.
19. Consigny PM, Barry JJ, Vitali NJ. Local delivery of an antiproliferative drug with use of hydrogel-coated angioplasty balloons. J Vasc Intervent Radio! 1994;5:553-560.
20. Consigny PM, Bilder GE. Expression and release of smooth muscle cell mitogens in the arterial wall after balloon angioplasty. J Vasc Med Biol 1993;4:1-8.
21. Dolle RE, Dunn J, Bobko M, et al. 5,7-Dimethoxy-3-(4-pyridinyl) quinoline is a potent and selective inhibitor of human vascular P-type platelet-derived growth factor receptor tyrosine kinase. J Med Chem 1994;34:2627-2629.
22. Erikson A, Nanberg E, Ronnstrand L, et al. Demonstration of functionally different interactions between phospholipase C-gamma and the two types of platelet-derived growth factor receptors. I Biol Chem 1995;270:7773-7781.
23. Escobedo JA, Barr PJ, Williams LT. Role of tyrosine kinase and membrane-spanning domains in signal transduction by the platelet-derived growth factor receptor. Mol Cell Biol 1988;8:5126-5131.
24. Fantl WJ, Escobedo JA, Martin GA, et al. Distinct phosphotyrosines on a growth factor receptor bind to specific molecules that mediate different signaling pathways. Cell 1992;69:413-423.
25. Ferns GAA, Raines EW, Sprugel KH, Notani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science 1991;253:112%1132.
26. Fingerle J, Johnson R, Clowes AW, Majesky MW, Reidy MA. Role of platelets in smooth muscle cell proliferation and migration after vascular injury in rat carotid artery. Proc Nail Acad Sci USA 1989;86:
27. Friedman RJ, Stemerman MB, Wenz B, et al. The effect of thrombocytopenia on experimental arterio-
1993;29: 158-166.
1992;199:255-261.
Biology of Platelet-Derived Growth Factor. Cytokines. Basel: Karger, 1993.
84 12-84 16.
Cardiovascular Drug Reviews, Vol. 14, No. 4 , 1996

396 G. E . BILDER AND C . J . ROJAS
sclerotic lesion formation in rabbits. Smooth muscle cell proliferation and re-endothelialization. J Clin Invesr 1977;60:1191-1201.
28. Fry DW, Kraker AJ, C O M O ~ RC, et al. Strategies for the discovery of novel tyrosine kinase inhibitors with anticancer activity. Anri-Cancer Drug Design 1994;9:331-351.
29. Gazit A, Yaish P, Gilon C, Levitzki A. Tyrphostins I: Synthesis and biological activity of protein tyrosine kinase inhibitors. J Med Chem 1989;32:234&2352.
30. Geahlen RL, McLaughlin JL. Piceatannol (3,4,3’,5’-tetrahydroxy-trans-stilbene) is a naturally occurring protein-tyrosine kinase inhibitor. Eiochem Eiophys Res Comm 1989;165:241-245.
31. Graziani Y, Chayoth R, Karny N, Feldman B, Levy J. Regulation of protein kinases activity by quercetin in ehrlich ascites NmOr cells. Eiochim Eiophys Acra 1981;714:415421.
32. Hanke JH, Pollok BA, Changelian PS. Role of tyrosine kinases in lymphocyte activation: Targets for drug intervention. Inflamm Res 1995;44:357-371.
33. Hart CE, Forstrom JW, Kelly ID. Two classes of PDGF receptor recognize different isoforms of PDGF. Science 1988;240:1529-1531.
34. Heidaran MA, Pierce JH, Lombardi D, et al. Deletion or substitution within the alpha platelet-derived growth factor receptor kinase insert domain: Effects on functional coupling with intracellular signaling pathways. Mol Cell Eiol 1991;11:134-142.
35. Heldin CH. Structural and functional studies on platelet-derived growth factor. Eur Mol Biol Org J 1992;11:42514259.
36. Herren B, Rooney B, Weyer KA, Iberg N, Schmid G, Pech M. Dimerization of extracellular domain of platelet-derived growth factor receptors. A revised model of receptor-ligand interaction. J Eiol Chem 1993;268: 1508%15095.
37. Hubbard SR, Wei L, Ellis L, Hendrickson WA. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 1994;372:746-754.
38. Hunter T. Tyrosine phosphorylation: Past, present, and future. Biochem Soc Trans 1996;24307-327. 39. Ito A, Shimokawa H, Kadokami T, et al. Tyrosine kinase inhibitor suppresses coronary arteriosclerotic
changes and vasospastic responses induced by chronic treatment with interleuken-lb in pigs in vivo. J Clin Invest 1995;96: 1288-1294.
40. Jawiens A, Bowen-Pope DF, Lindner V, Schwartz SM, Clowes AW. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J Clin Invest 1992;89:507-.
41. Kashishian A, Kazlauskas A, Cooper JA. Phosphorylation sites in the PDGF receptor with different specificities for binding GAP and PI3 kinase in vivo. Eur Mol Eiol Org J 1992;11:1373-1382.
42. Katsuda S, Coltrera MD, Ross R, Gown AM. Human atherosclerosis IV. lmmunocytochemical analysis of cell activation and proliferation of lesions in young adults. Amer J Path 1993;142:1787-1793.
43. Kazlauskas A. Receptor tyrosine kinases and their targets. Current Opinion in Generics and Developmenr 1994;4:5- 14.
44. Kazlauskas A, Feng GS, Pawson T, Valius M. The 64-kDa protein that associates with the platelet-derived growth factor receptor p subunit via Tyr-1009 is the SH2-containing phosphotyrosine phosphatase Syp. Proc Natl Acad Sci USA 1993;90:6939-6942.
45. Kazlauskas A, Kashishian A, Cooper JA, Valius M. GTPase-activating protein and phosphatidylinositol 3-kinase bind to distinct regions of the platelet-derived growth factor receptor beta subunit. Mol Cell Eiol 19923 2:2534-2544.
46. Keating MT, Escobedo JA, Fantl WJ, Williams LT. Ligand activation causes a phosphorylation- dependent change in platelet-derived growth factor receptor conformation. Trans Assoc Amer Phys 1988; 101:24-32.
47. Kelly ID, Haldeman BA, Grant FJ, et al. Platelet-derived growth factor (PDGF) stimulates PDGF receptor subunit dimerization and intersubunit trans-phosphorylation. J Eiol Chem 1991;266:8987-8992.
48. Kovalenko M, Gazit A, Bohmer A, et al. Selective platelet-derived growth factor receptor kinase blockers reverse sis-transformation. Cancer Res 1994;54:6106-6114.
49. Kundra V, Escobedo JA, Kazlauskas A, et al. Regulation of chemotaxis by platelet-derived growth factor receptor+. Narure 1994;367:474-476.
50. Lechleider RJ, Sugimoto S, Bennett AM, et al. Activation of the SH2-containing phosphotyrosine phos- phatase SH-PTP by its binding site, phosphotyrosine 1009, on the human platelet-derived growth factor. J Eiol Chem 1993;268:21478-21481.
51. Levitzki A, Gazit A. Tyrosine kinase inhibition: An approach to drug development. Science 1995;267:
52. Lindner V, Giachelli CM, Schwartz SM, Reidy MA. A subpopulation of smooth muscle cells in injured
53. Lindner V, Reidy M. Platelet-derived growth factor ligand and receptor expression by large vessel
1782-1786.
rat arteries express platelet-derived growth factor B-chain mRNA. Circ Res 1995;76:95 1-957.
endothelium in viva. Amer J Path 1995:146:1488-1497.
Cardzovascular Drug Reviews. Vol. 14, No. 4. 1996

TYROSINE KINASE INHIBITORS 397
54. Logan A. Intracrine regulation at the nucleus-Further mechanism of growth factor activity. J Endocrin 19903 25:339-343.
55. Maguire MP, Sheets KR, McVety K, Spada AP, Zilberstein A. A new series of PDGF receptor tyrosine kinase inhibitors: 3-substituted quinoline derivatives. J Med Chem 1994;37:2129-2137.
56. Malarkey K, Belham CM, Paul A, et al. The regulation of tyrosine kinase signaling pathways by growth factor and G-protein-coupled receptors. Biochem J 1995;309:361-375.
57. Majesky MW, Reidy MA, Bowen-Pope DF, Hart CE, Wilcox JN, Schwartz SM. PDGF ligand and receptor gene expression during repair of arterial injury. J Cell Biol 1990;111:2149-2158.
58. Martin BL, Wu D, Jakes S, Graves DJ. Chemical influences on the specificity of tyrosine phosphoryla- tion. J Biol Chem 1990;265:7108-7111.
59. Mattsson E, Clowes AW. Current concepts in restenosis following balloon angioplasty. TCM 1995;5: 20G204.
60. Miano JM, Vlasic N, Tota RR, Stemerman MB. Smooth muscle cell immediate-early gene and growth factor activation follows vascular injury. A putative in vivo mechanism for autocrine growth. Arterioscl Thromb 1993; 13:211-219.
61. Mori S , Heldin CH, Claesson-Welsh L. Ligand-induced ubiquination of the platelet-derived growth factor @-receptor plays a negative regulatory role in its mitogenic signaling. J Biol Chem 1993;268:577-583.
62. Mori S, Ronnstrand L, Yokote K, Engstrom A, Courtneidge SA, Cleasson-Welsh L, Heldin CH. Iden- tification of two juxtamembrane autophosphorylation sites in the PDGF beta-receptor; involvement in the interaction with Src family tyrosine kinases. Eur Mol Biol Org J 1993;12:2257-2264.
63. Murry CE, Bartosek T, Giachelli CM, Alpers CE, Schwartz SM. Platelet-derived growth factor A mRNA expression in fetal, normal adult, and atherosclerotic human aortas. Circulation 1996;93:1095-1106.
64. Nabel EG, Yang Z, Liptay S, San H, Gordon D, Haudenschild CC. Recombinant platelet-derived growth factor B gene expression in porcine arteries induces intimal hyperplasia in vivo. J Clin Invest 1993;91: 1822-1829.
65. Nishibe S , Wahl MI, Hernandez-Sotomayor SMT, Tonks NK, Rhee SG, Carpenter G. Increase of the catalytic activity of phospholipase C-gamma1 by tyrosine phosphorylation. Science 1990;250: 1253-1 256.
66. Nishimura R, Li W, Kashishian A, et al. Two signaling molecules share a phosphotyrosine-containing binding site in the platelet-derived growth factor receptor. Mol Cell Biol 1993; 13:6889-6896.
67. O’Brian ER, Alpers CE, Stewart DK, et al. Proliferation in primary and restenotic coronary atherectomy tissue: Implications for antiproliferative therapy. Circ Res 1993;73:223-231.
68. Okazaki H, Majesky MW, Harker LA, Schwartz SM. Regulation of platelet-derived growth factor ligand and receptor gene expression by a-thrombin in vascular smooth muscle cells. Circ Res 1992;71:1285- 1293.
69. Onoda T, Iinuma H, Sasaki Y, et al. Isolation of a novel tyrosine kinase inhibitor, lavendustin A, from Strepfomyces griseolavendus. J Nat Prod 1989:52: 1252-1257.
70. Pickering JG, Weir L, Jakanowski J, Kearney MA, Isner JM. Proliferative activity in peripheral and coronary atherosclerotic plaque among patients undergoing percutaneous revascularization. J Clin Invest 1993;9 1 : 146%1480.
71. Raines EW, Dower SK, Ross R. Interleukin-1 mitogenic activity for fibroblasts and smooth muscle cells is due to PDGF-AA. Science 1989;243:393-396.
72. Raines EW, Ross R. Platelet-derived growth factor in vivo. In: Westermark B, Sorg C, eds. Biology of Platelet-Derived Growth Factor. Cytokines. Basel: Barger, 1993.
73. Ramdas L, McMurray JS, Budde RJA. The degree of inhibition of protein tyrosine kinase activity by tyrphostin 23 and 25 is related to their instability. Cancer Res 1994;54867-869.
74. Ronnstrand L, Mori S , Arvidsson A-K, et al. Identification of two C-terminal autophosphorylation sites in the PDGF beta-receptor: Involvement in the interaction with phospholipase C-gamma. Eur Mol Biol Org J 1992;11:3911-3919.
75. Ross R. Cell biology of atherosclerosis. Ann Rev Physiol 1995;57:791-804. 76. Ross R. Polypeptide growth factors and atherosclerosis. Trends Cardiovasc Med 1991;1:277-282. 77. Ross R, Masuda J, Raines EW, et al. Localization of PDGF-B protein in macrophages in all phases of
atherogenesis. Science 1990;248: 1009-1012. 78. Rubin K, Hansson GK, Ronnstrand L, et al. Induction of P-type receptors for platelet-derived growth
factor in vascular inflammation: Possible implications for development of vascular proliferative lesions. Lancet 1988; 1 : 1353- 1356.
79. Rupp, Siegbahn A, Ronnstrand L, Wemstedt C, Claesson-Welsh L, Heldin CH. A unique autophos- phorylation site in the platelet-derived growth factor a-receptor from a heterodimeric receptor complex. Eur J Biochem 1994;225:29-41.
80. Schaller MD, Bouton AH, Flynn DC, Parson JT. Identification characterization of novel substrates for protein tyrosine kinases. Prog Nucleic Acid Res Molec Biol 1993;44:205-227.
Cardiovascular Drug Reviews, Vol. 14, No . 4 , 1996

G. E . BILDER AND C . J. ROJAS
8 I . Schlessinger J . Signal transduction by allosteric receptor oligonierization. T w m h Bioc.hrnt St.i 1988: 13: 443-437
82. Schwartz SM. DeBlois D. O'Brian ERM. The Intinia Soil for atherosclero\is and re\tenosis. Cirt. Re,.\ 1995:77:445-465
83. Seifen RA. Hart CH. Phillips PE. et al. THO different subunits associate to create isoform-specific platelet-derived grouth factor receptors J B i d Chew 1989:264:877 1-8778.
84. Shimokado K. Umezawa K . Ogata J . Tvrosine kinase inhibitors inhibit multiple steps o f the cell cycle of vascular smooth muscle cells. h p e r Cell Res 1995:220:26&273.
85 Shirashi T . Domoto T. lmai N. Shimada Y. Watanabe K. Specific inhibitors of tyrosine-specific protein kinase. synthetic 4-hydroxycinnamamide derivatives. Bioclrerrr Bi~/~li\ . \ Res Comin 1987: 147:322-328.
X6. Shiraihi T. Owada MK. Tatsuka M. Yarnashita T. Watanabe K. K a k u n a p T . Specific inhibitors of tyrosine-specific protein kinases: Properties of 4-hydroxqcinnaiiianiidc derivatives in vitro. C ' o n t w R c s 1989:49:237J-2378
87. Shoelson SE. Sivaraja M. Williams KP. Hu P. Schlessinger I . Weiss MA. Specific phorphopeptide binding regulate\ a conformational change in the PI 3-kinase SH3 domain aswciated with enzyme acti- vation. Eio. Mol B i d Org J 1992:12:795-802.
88. Shokat KM. Tyrosine kinases: Modular signaling enzyme\ with tunable specificities. C'lrrm Bio 1095:2: 509-5 14.
89. Shugar D . Protein kinase inhibitors-Potential chemotherapeutic agents. Acru Biochtwiic~tr Polo/irc.tr 1995: 42:405-4 I 8 .
90. Spa& AP. Myers MR. Small molecular inhibitors of tyrosine kinase activity. E v p Opiii I%t,r Pu/r/tr.s I995:5:805-8 17.
91 Srinibas PR. Grunberger C. Inhibitors ot the insulin receptor tyrosine kinase. Phtrmiuc. T k r 199464: 23-35
92. Stary HC. The sequence of cell and matrix changes in atherosclerotic lesions of coronary arteries in the first forty years of life. Eitr Heurr J (Suppl E ) 1990;l 1:3-19.
93. Tanizawa S. Ueda M. \'an der Loos C, van der Wal AC. Becker AE. Expression of platelet-derived growth factor B-chain and P-receptor in human coronary arteries after percutaneous transluniinal coronary an- gioplasty: An ininiunohistochemical study. H ~ u r t I996:75:549-556.
94. Taylor SS. Radzio-Andzelni E. Hunter T. How do protein kinases discriminate between serinelthrconinc and tyrosine' Structural insights froni the insulin receptor protein-tyrosinc kinase. FASEB J 1995:9: 1255- 1266.
95. Traxler P. Lydon N . Recent advances in protein tyrosine kinase inhibitors. Drirg.s o f f l i e Ftmre 1995:20: 1 26 I- 1274.
96. Turck CW. Edenson SP. Mapping of tyrosine kinase autophosphorylation sites with synthetic peptide substrates. P e p & Re., 1994:7: 14(kI45
97. Uehara Y. Murakami Y . Suzukake-Tsuchiya K. et al. Etfects of herbirnycin derivative\ o n src oncogene function in relation to antitumor activity J Amibiotic,~ 1988:4 I :82 1-834.
98. Umezawa H. Imoto M. Sawa T. et al. Studies on a new epidermal growth factor-receptor kinase inhibitor. erbstatin. produced by MH435-hF3. J ..lnrihiorics 1986:39:17%17?.
99. Uren A. Y u JC. Li W. el al. Identification of a domain within the carboxyl-terminal region of the p platelet-derived growth factor (PDGF) receptor that mediates the high transforming activity of PDGF. J B i d Chem 1996:271:11051-11051.
100. van der Geer P, Hunter T. Lindberg RA. Receptor protein-tyrosine kinases and their signal transduction pathways. A n m Rei. C'cdl B i d 1994:lO 251-337.
101. Vignais M-L,. Sadowski HB. Watling D. Rogers NC. Gilnian M. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. M o l Cell Bio IY96:16: 1759-1769.
102. Ward WHJ. Cook PN. Slater .4M. D a m s DH. Holdgate GA. Green LR. Epidermal growth factor receptor tyrosine kinase. Investigation of catalytic mechanism. structure-based searching and discovery of a potent inhibitor. Bior%em Plicrrr~rut~ol 1994:48.659-666.
103. Westennark B. PDGF and its receptors in human tumor cells. In: Westermark B. Sorg C. eds. B i o l o p of flurelet-Ueriivd Growrh Fuctcir. Cw>kirtes. Basel: Barger. 1993.
104. Wilcox J N . Analysis of local gene expression in human atherosclerotic plaques in in situ hybridization. Trends Cardiowsc Met/ 199 1 ; 1 : 17-24
105. Wilcox I N . Smith KM. Williams LT. Schwart7 SM. Cordon D Platelet-derived growth factor mRNA detection in human atherosclerotic plaques by in situ hybridization. J Cliri fni.es/ 1988:82: II3L-1 143.
106. Williams LT. Signal transduction by the platelet-derived growth factor receptor. Sriericcl 1989;243: 1564- 1570.
107. Yaish P. Gazit A. Gilon C. Levitzki A . Blocking of EGF-dependent cell proliferation by EGF-receptor kinase inhibitors. Science I988:242:933-935.

TYROSINE KINASE INHIBITORS 399
108. Yamamoto H, Crow M, Cheng L, Lakatta E, Kinsella J. PDGF receptor-to-nucleus signaling of p91 (STATla) transcription factor in rat smooth muscle cells. Enper Cell Res 1996;222:125-130.
109. Yokote K, Mori S, Hansen K, et al. Direct interaction between Shc and the platelet-derived growth factor P-receptor. J BioZ Chem 1994;269:15337-15343.
110. Yoneda T, Lyall RM, Alsina MM, et al. The antiproliferative effects of tyrosine kinase inhibitors tyr- phostins on a human squamous cell-carcinoma in vitro and in nude mice. CancerRes 1991;51:443&4435.
11 1. Yuan C-J, Jakes S, Elliott S, Graves DJ. A rationale for the design of an inhibitor of tyrosyl kinase. J B i d Chem 199O;265: 16205-16209.
112. Zimmermann J , Buchdunger E, Mett H, Meyer T, Lydon NB, Traxler P. Phenylamino-pyrimidine(PAP)- derivatives: A new class of potent and highly selective PDGF-receptor autophosphorylation inhibitors. Bioorg Med Chem 1996;6:1221-1226.
Cardiovascular Drug Reviews, Vol. 14, No. 4 , 1996
Copyright © 2022 FDOKUMEN




















