
Doxorubicin requires the sequential activation of caspase-2 PKCδand JNK to induce apoptosis

Brain, Behavior, and Immunity 24 (2010) 812–821
Contents lists available at ScienceDirect
Brain, Behavior, and Immunity
journal homepage: www.elsevier .com/locate /ybrbi
Neuroprotection, Brain Damage and Behavior
Inhibition of the JNK/AP-1 pathway reduces neuronal death and improvesbehavioral outcome after neonatal hypoxic–ischemic brain injury
Cora H. Nijboer a, Michael A. van der Kooij a,b, Frank van Bel b, Frauke Ohl c,d, Cobi J. Heijnen a,*,Annemieke Kavelaars a
a Laboratory of Psychoneuroimmunology, University Medical Center Utrecht, KC03.068.0, Lundlaan 6, 3584 EA Utrecht, The Netherlandsb Department of Neonatology, University Medical Center Utrecht, Utrecht, The Netherlandsc Department of Animals in Science and Society, University of Utrecht, Utrecht, The Netherlandsd Rudolf Magnus Institute of Neurosciences, Utrecht, The Netherlands
a r t i c l e i n f o
Article history:Received 25 August 2009Received in revised form 11 September 2009Accepted 12 September 2009Available online 17 September 2009
Keywords:JNK MAP kinaseNeonatal hypoxia–ischemiaNeuroprotectionApoptosisBehaviorTransduction peptideSmac/DIABLOCalpainAP-1Inflammation
0889-1591/$ - see front matter � 2009 Elsevier Inc. Adoi:10.1016/j.bbi.2009.09.008
* Corresponding author. Fax: +31 88 755 5311.E-mail address: [email protected] (C.J. Hei
a b s t r a c t
Perinatal hypoxic–ischemic (HI) brain damage continues to be a major clinical problem. We investigatedthe contribution of the MAP kinase c-Jun N-terminal kinase (JNK), to neonatal HI brain damage. JNK reg-ulates several transcriptional (via AP-1 activation) and non-transcriptional processes involved in braindamage such as inflammation and cell death/survival.
P7 rats were subjected to HI by unilateral carotid artery occlusion and hypoxia. HI-induced activationof cerebral AP-1 peaked at 3–6 h post-HI. Intraperitoneal administration of the JNK-inhibitor TAT-JBDimmediately after HI prevented AP-1 activation. TAT-JBD treatment within 3 h after HI reduced earlyneuronal damage by �30%. JNK/AP-1 inhibition did not reduce HI-induced cytokine/chemokine expres-sion. Analysis of indicators of apoptotic cell death revealed that TAT-JBD markedly reduced the HI-induced increase in active caspase 3. However, the upstream mediators of apoptosis: active caspase 8,cleaved Bid, mitochondrial cytochrome c release and caspase 9 cleavage were not reduced after TAT-JBD. TAT-JBD inhibited the HI-induced increase in Smac/DIABLO, an inhibitor of IAPs that prevent activa-tion of caspase 3. TAT-JBD treatment also reduced cleavage of a-fodrin, indicating that calpain-mediatedbrain damage was reduced. Neuroprotection by TAT-JBD treatment was long-lasting as gray- and whitematter damage was diminished by �50% at 14 weeks post-HI concomitantly with marked improvementof sensorimotor behavior and cognitive functioning.
In conclusion, JNK inhibition by TAT-JBD treatment reduced neonatal HI brain damage with a therapeu-tic window of 3 h and long-lasting anatomical and behavioral improvements. We propose that inhibitionof mitochondrial Smac/DIABLO release and calpain activation contribute to neuroprotection by TAT-JBD.
� 2009 Elsevier Inc. All rights reserved.
1. Introduction
Perinatal hypoxia–ischemia (HI) is a major risk factor for humanneonatal mortality and development of major neurodevelopmentaldisabilities (Volpe, 2001; Ferriero, 2004). At present, no effectivepharmacological treatment to combat perinatal HI brain injury isavailable for these infants (Perlman, 2006). Excitotoxicity, inflam-mation and apoptosis are the major pathophysiological mecha-nisms that contribute to cerebral injury after HI (Vexler andFerriero, 2001).
Mitogen-activated protein (MAP) kinases are essential kinasesthat respond to a plethora of extracellular stimuli and play a criti-cal role in the regulation of several cellular processes like growth,
ll rights reserved.
jnen).
proliferation, differentiation and cell survival/apoptosis (Cobb,1999). c-Jun N-terminal kinases (JNKs), also known as stress-acti-vated protein kinases (SAPKs) as their activation is associated witha wide variety of environmental stressors, represent an importantsubfamily of the MAP kinase group (Bogoyevitch et al., 2004; Karinand Gallagher, 2005). JNKs were originally identified by their abil-ity to phosphorylate c-Jun, the dominant component of the tran-scription factor AP-1, which is a DNA binding dimer composed ofc-Jun combined with c-Fos, Maf or ATF subunits. JNK-mediatedphosphorylation of c-Jun enhances the ability of c-Jun/AP-1 to in-duce transcription of numerous target genes involved in prolifera-tion, cell survival, cell death, DNA repair, metabolism andinflammation (Bogoyevitch and Kobe, 2006). Nowadays, JNKs arealso acknowledged for their capacity to regulate cellular functionthrough phosphorylation of non-nuclear proteins. For example,JNKs have been suggested to play a key role in regulation of apop-

C.H. Nijboer et al. / Brain, Behavior, and Immunity 24 (2010) 812–821 813
tosis by directly phosphorylating and modifying the activity of pro-teins residing at the mitochondria (Dhanasekaran and Reddy,2008; Ameyar et al., 2003).
In mammals there are three known JNK isoforms: JNK1, JNK2and JNK3. Whereas JNK1 and JNK2 are found in all cells and tis-sues, expression of JNK3 is mainly found in the brain (Bogoyev-itch et al., 2004; Karin and Gallagher, 2005). The importance ofJNKs for cerebral development and function has been revealedusing mice with targeted deletion of the JNK isoforms. Neuronaldevelopment was greatly affected in JNK1�/� and JNK2�/� knock-out mice that die around E11 due to hind-brain exencephaly.Loss of JNK3 is not embryonically lethal but rather protects thebrain from glutamate-induced excitotoxicity and kainate-inducedseizures (Yang et al., 1997; Bogoyevitch et al., 2004). Pirianovet al. (2007) and Kuan et al. (2003) have shown reduced neuro-nal loss in JNK3�/� mice in models of neonatal HI and adultstroke, respectively.
A potent induction of c-Jun and sustained activity of the AP-1transcription factor has been associated with neuronal cell deathin vitro (Karin and Gallagher, 2005). Cerebral activation of tran-scription factors and the consequent modulation of expression oftarget genes is an important part of the cerebral response to injury(Chang and Huang, 2006), like we have shown previously for thetranscription factor nuclear factor kappa B (NF-jB) (Nijboer et al.,2008a,b, 2009).
JNKs, like the other MAP kinases, are activated via a typical 3-step phosphorylation cascade: JNK is activated by the MAPK ki-nases MKK7 and MKK4 and the latter are activated by MKK kinases(MEKKs or MAP3Ks). Scaffold proteins like JNK-interacting protein(JIP) facilitate JNK activation by binding all elements of the JNKactivation pathway.
We recently described that intraperitoneal administration ofthe short-lived JNK-inhibitor TAT-JBD (also named L-JNKI (Barret al., 2002)), consisting of the 20 amino-acid sequence of JNKBinding Domain of JIP-1 (JBD) coupled to the protein transductionsequence of HIV-TAT, reduced early neuronal damage in P7 ratswhen administered intraperitoneally at 0 and 3 h after HI (Nijboeret al., 2009). The aim of the present study was to further investi-gate the neuroprotective effect of TAT-JBD in the neonatal rat mod-el of HI brain damage. We examined the therapeutic window forTAT-JBD treatment, analyzed its effects on cerebral upregulationof cytokines and chemokines and on mediators of (apoptotic) celldeath. Furthermore, we investigated effects of TAT-JBD treatmenton long-term brain damage, sensorimotor function and cognitivebehavioral outcome.
2. Methods
2.1. Animals
The animal committee of Academic Biomedical Center Utrecht(DEC-ABC) approved all experiments. At postnatal-day 7 (P7), Wis-tar rats underwent occlusion of the right common carotid arteryunder isoflurane anesthesia followed by 120 min hypoxia (8%O2). Sham controls underwent anesthesia and incision only. Allanalyses were performed in a blinded setup.
TAT-JBD (L-JNKI: YGRKKRRQRRR-PP-RPKRPTTLNLFPQVPRSQDT)(W.M. Keck facility, Yale University, New Haven, CT) was dissolvedin DMSO (40 mg/ml), diluted to 1 mg/ml in PBS and administeredi.p. at a dose of 10 mg/kg. Vehicle-treated HI animals received aninjection of 2.5% DMSO in PBS. TAT-JBD consists of the JNK bindingdomain of JNK-interacting protein-1 (JIP-1) coupled to the proteintransduction sequence of HIV-TAT to facilitate cerebral uptake andacts as a specific JNK-inhibitor (Barr et al., 2002; Bogoyevitch andArthur, 2008).
2.2. Histology
Rats were terminated by an overdose pentobarbital and per-fused with 4% paraformaldehyde in PBS. Coronal paraffin sections(8 lm) (at �3.20 mm from bregma) were stained with hematoxy-lin–eosin (HE) or with mouse-anti-MAP2 (Sigma–Aldrich, Stein-heim, Germany) or mouse-anti-MBP (Sternberger Monoclonals,Lutherville, MD) followed by biotin-labeled horse-anti-mouse anti-body and revealed using Vectastain ABC kit (Vector-Labs, Burlin-game, CA) and diaminobenzamidine.
Full section images were captured with a Nikon D1 digital cam-era (Nikon, Tokyo, Japan). The various brain areas (MAP2 or HE)were outlined manually using image processing tools in AdobePhotoshop 6.0 (Adobe Systems Inc., San Jose, CA, USA) and the ratioof ipsi- to contralateral areas was calculated (Nijboer et al., 2007).The area of MBP staining in both hemispheres was quantified usingImageJ software (http://rsb.info.nih.gov/ij/, 1997–2006) and the ra-tio of ipsi- to contralateral areas was calculated (Nijboer et al.,2007).
2.3. Western blotting and EMSA
Rats were terminated by an overdose of pentobarbital anddecapitated. Cytosolic and nuclear fractions were prepared as de-scribed (Nijboer et al., 2007). Cytosolic proteins were separatedby SDS–PAGE, transferred to Hybond-C membranes (Amersham,Buckinghamshire, UK) and revealed using rabbit-anti-cleaved cas-pase 3, mouse-anti-caspase 9 (both Cell Signaling, Danvers, MA),mouse-anti-cytochrome c (BD Biosciences Pharmingen, San Jose,CA), rabbit-anti-fodrin (BioVision, Mountain View, CA), rabbit-anti-Smac/DIABLO (Chemicon, Temecula, CA), rabbit-anti-caspase8, rabbit-anti-Bid or goat-anti-b-actin (all Santa Cruz Biotechnol-ogy, Santa Cruz, CA) followed by peroxidase-labeled secondaryantibodies, revealed by enhanced chemiluminescence (Amersham)and analyzed with a GS-700 Imaging Densitometer (Bio-Rad, Her-cules, CA).
Electromobility shift assays (EMSA) on nuclear brain extractswith 32P labeled AP-1 probe (Promega, Madison, WA; sequence50-CGCTTGATGAGTCAGCCGGAA-30) were performed as described(Nijboer et al., 2008a).
2.4. Quantitative real time reverse transcriptase (RT)-PCR
Total RNA was isolated with TRIzol� (Invitrogen, Paisley, UK).cDNA was synthesized with SuperScript Reverse Transcriptase(Invitrogen). The PCR reaction was performed with iQ5 Real-TimePCR Detection System (Bio-Rad) using primers for TNF-a, IL-1b,IL-10, IL-4 (for primer sequences see Nijboer et al., 2008a), IL-6 for-ward: AACGAAAGTCAACTCCATCTG, reverse: GGTATCCTCTGTGAAGTCTCC; MIP1a (CCL3) forward: TTTGAGACCAGCAGCCTTTG, re-verse GAAGAGTCCCTGGATGTGGC and MIP2 forward: TTGTCTCAACCCTGAAGCCC, reverse TGCCCGTTGAGGTACAGGAG. Datawere individually normalized to the mean of the relative expres-sion of b-actin and GAPDH (for primer sequences see Nijboeret al., 2008a).
2.5. Behavioral tests
2.5.1. Cylinder rearing test (CRT)At 2.5, 4 and 6 weeks of age, rats were individually placed in a
Plexiglas transparent cylinder and observed for 3 min. Initial fore-paw (left/right/both) preference of weight-bearing contacts duringfull rear were recorded. The relative proportion of preference touse the right (unimpaired) forepaw was calculated as: (right–left)/(right + left + both) � 100 (Schallert et al., 2000).

814 C.H. Nijboer et al. / Brain, Behavior, and Immunity 24 (2010) 812–821
2.5.2. Adhesive removal task (ART)At 5 weeks of age, stickers (tough-spots, Diversified Biotech,
Boston MA) were placed on the left and right forepaw and the la-tency to removal was recorded. The order for sticker placementon left or right forepaw was alternated between and within ani-mals and we determined the mean time until complete removalof three stickers per forepaw after one training sticker for eachpaw (Schallert et al., 2000; Grow et al., 2003).
2.5.3. Rota-rodAt 10–12 weeks of age, animals were tested on the rota-rod
(Stoelting Europe, Ireland) according to a standardized protocol(Rozas et al., 1997). In short, animals were trained to remain onthe rota-rod at a speed of 5 rpm for a maximum duration of150 s for three subsequent days. On day 4 we tested performanceof the animals to stay on the rota-rod by increasing the speed from5 to 40 rpm for a maximum duration of 300 s with increasing stepsof 5 rpm. The Overall Rod Performance (ORP) was calculated as thetotal area under the curve in a plot of time on the rod against rota-tional speed. The ORP is calculated as the sum of the individual tra-pezia (speed); an individual area would take the value of ((a + b)/2) � c where ‘a’ is time-on-the-rod at lower speed, ‘b’ the time atthe higher speed and ‘c’ the difference between the two speeds(Rozas et al., 1997). Since we tested for 8 different rod rotationalspeeds ORP could take any value between 0 and 10,500.
2.5.4. Modified hole board (mHB)At 11–13 weeks of age, animals were introduced to the mHB
test as described by Ohl et al. (2003). In short, the mHB setup con-sisted of an opaque gray PVC box (100 � 50 � 50 cm) containing aboard (60 � 20 cm) placed in the middle of the box on which 20gray cylinders (3 cm ø � 4 cm height) were staggered in threelines. The PVC box is enlarged by an additional 50-cm square com-partment, in which the group mates of the experimental rats areplaced during the test-period. This compartment is separated fromthe test area by a transparent perforated Plexiglas partition.
All cylinders were flavored with vanilla to attract the animals tothe cylinders. Three of the cylinders were cued (small white ring)and baited with a small almond piece (0.02–0.03 g) placed on ametal grid representing the food reward. The remaining 17 cylin-ders contained non-retrievable almond pieces placed under themetal grid (Fig. 6A). Between 10 am and 5 pm animals were scoredlive by a trained observer under red light conditions for parametersindicative for cognition (time until retrieval of all food rewards),exploration, anxiety, arousal and risk assessment. Non-baited holevisits and revisits of baited holes from which food had already beenretrieved are referred to as long-term and short-term memory er-rors, respectively.
Animals were monitored during four sessions/per day lastingmaximally 5 min or until retrieval of all three food pellets on 3sequential days (stage 1, T1–T3). On the 4th day, the location ofthe three cued and baited cylinders was changed and time until re-trieval of all three food rewards was monitored during four ses-sions (stage 2, CS). During the reversal task (stage 3, RT) timeuntil retrieval of all food rewards in three non-cued baited cylin-ders in a novel position was determined during four sessions. Allnon-baited cylinders were now cued.
2.6. Statistical analysis
Data were normally distributed, are presented as mean andSEM, and were analyzed by one- or two-way ANOVA with Bonfer-roni post-tests as indicated.
3. Results
3.1. JNK/AP-1 activation after HI and effect of i.p. TAT-JBD treatment
Analysis of the kinetics of cerebral JNK activation after neonatalHI showed that JNK/AP-1 activity peaked early; HI induced signif-icant nuclear AP-1 activity as determined by EMSA on nuclear ex-tracts of ipsi- and contralateral hemispheres. AP-1 activity wasupregulated from 0.5 to 6 h after the insult in the ipsilateral hemi-sphere (Fig. 1A). At 12 h after the insult ipsilateral AP-1 activity hadreturned to base-line values. Intraperitoneal treatment with TAT-JBD immediately after HI almost completely blocked HI-inducedAP-1 activation as determined at 3 h post-HI (Fig. 1B).
3.2. Therapeutic window of TAT-JBD treatment
At 48 h after induction of HI in P7 rats, we observed severe braindamage in vehicle-treated animals with �83% loss of microtubuleassociated protein 2 (MAP2), as an early marker of neuronal dam-age (Fig. 2A and B). No MAP2 loss was detected in the contralateralhemisphere. Inhibition of JNK by treatment with TAT-JBD directlyafter HI and 3 h later (0/3 h) significantly reduced ipsilateralMAP2 loss by �30% (Fig. 2A and B). To determine the therapeuticwindow for this neuroprotective effect, TAT-JBD was administeredas a single injection at only 3 h or only 6 h after the insult. Thetherapeutic window of i.p. TAT-JBD was at least 3 h post-HI asMAP2 loss was still significantly reduced by �18% after this treat-ment (Fig. 2A and B). Treatment with TAT-NBD at 6 h after HI didnot reduce MAP2 loss.
3.3. Effects of TAT-JBD on cytokine and chemokine expression
The JNK/AP-1 pathway is thought to play an important role inregulating inflammatory processes in the brain. We have previ-ously described that intraperitoneal administration of TAT-JBDdid not significantly reduce the HI-induced increase in TNF-amRNA or protein in this model of neonatal HI brain damage (Nijb-oer et al., 2009). Here we further expand these observations andshow that TAT-JBD did not have a major effect on the HI-inducedupregulation of cytokine or chemokine expression. We did not ob-serve a statistically significant effect of TAT-JBD treatment on thelevel of mRNA encoding TNF-a, IL-1b, IL-6, MIP2, MIP1a, IL-10and IL-4 (Table 1).
3.4. Effect of TAT-JBD on cell death markers
We determined whether JNK inhibition had an effect on theexecutioner of apoptosis: caspase 3. At 24 h post-HI, TAT-JBD treat-ment significantly decreased HI-induced ipsilateral levels ofcleaved (active) caspase 3 (Fig. 3A). Caspase 3 can be activatedby the intrinsic pathway of apoptosis that is stimulated by releaseof cytochrome c from the mitochondria to the cytosol leading tocleavage of pro-caspase 9 to form active caspase 9. At 24 h afterthe insult, HI induced a slight but statistically significant decreasein the level of pro-caspase 9, indicating caspase 9 activation(Fig. 3B). TAT-JBD treatment, however, did not affect the reductionin pro-caspase 9 (Fig. 3B). HI increased cytosolic cytochrome c lev-els in the ipsilateral hemisphere at 24 h post-HI (Fig. 3C). JNK inhi-bition by TAT-JBD slightly but not significantly reduced the level ofreleased cytochrome c (Fig. 3C).
Caspase 8 is an important direct upstream activator of caspase 3via the so-called extrinsic route of apoptosis in response to stimu-lation of death receptors. Exposure of rat pups to HI induced acti-vation of caspase 8 observed as an increase in cleaved (p18)caspase 8 in the ipsilateral hemisphere 3–6 h after the insult.
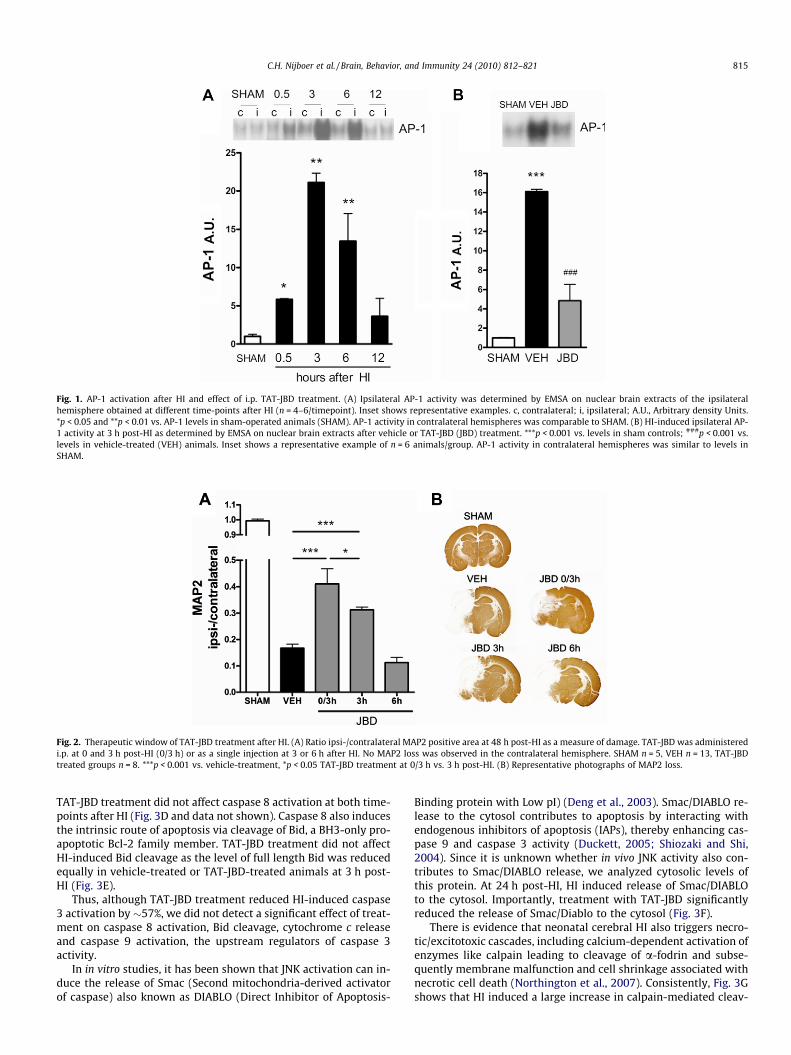
Fig. 1. AP-1 activation after HI and effect of i.p. TAT-JBD treatment. (A) Ipsilateral AP-1 activity was determined by EMSA on nuclear brain extracts of the ipsilateralhemisphere obtained at different time-points after HI (n = 4–6/timepoint). Inset shows representative examples. c, contralateral; i, ipsilateral; A.U., Arbitrary density Units.*p < 0.05 and **p < 0.01 vs. AP-1 levels in sham-operated animals (SHAM). AP-1 activity in contralateral hemispheres was comparable to SHAM. (B) HI-induced ipsilateral AP-1 activity at 3 h post-HI as determined by EMSA on nuclear brain extracts after vehicle or TAT-JBD (JBD) treatment. ***p < 0.001 vs. levels in sham controls; ###p < 0.001 vs.levels in vehicle-treated (VEH) animals. Inset shows a representative example of n = 6 animals/group. AP-1 activity in contralateral hemispheres was similar to levels inSHAM.
Fig. 2. Therapeutic window of TAT-JBD treatment after HI. (A) Ratio ipsi-/contralateral MAP2 positive area at 48 h post-HI as a measure of damage. TAT-JBD was administeredi.p. at 0 and 3 h post-HI (0/3 h) or as a single injection at 3 or 6 h after HI. No MAP2 loss was observed in the contralateral hemisphere. SHAM n = 5, VEH n = 13, TAT-JBDtreated groups n = 8. ***p < 0.001 vs. vehicle-treatment, *p < 0.05 TAT-JBD treatment at 0/3 h vs. 3 h post-HI. (B) Representative photographs of MAP2 loss.
C.H. Nijboer et al. / Brain, Behavior, and Immunity 24 (2010) 812–821 815
TAT-JBD treatment did not affect caspase 8 activation at both time-points after HI (Fig. 3D and data not shown). Caspase 8 also inducesthe intrinsic route of apoptosis via cleavage of Bid, a BH3-only pro-apoptotic Bcl-2 family member. TAT-JBD treatment did not affectHI-induced Bid cleavage as the level of full length Bid was reducedequally in vehicle-treated or TAT-JBD-treated animals at 3 h post-HI (Fig. 3E).
Thus, although TAT-JBD treatment reduced HI-induced caspase3 activation by �57%, we did not detect a significant effect of treat-ment on caspase 8 activation, Bid cleavage, cytochrome c releaseand caspase 9 activation, the upstream regulators of caspase 3activity.
In in vitro studies, it has been shown that JNK activation can in-duce the release of Smac (Second mitochondria-derived activatorof caspase) also known as DIABLO (Direct Inhibitor of Apoptosis-
Binding protein with Low pI) (Deng et al., 2003). Smac/DIABLO re-lease to the cytosol contributes to apoptosis by interacting withendogenous inhibitors of apoptosis (IAPs), thereby enhancing cas-pase 9 and caspase 3 activity (Duckett, 2005; Shiozaki and Shi,2004). Since it is unknown whether in vivo JNK activity also con-tributes to Smac/DIABLO release, we analyzed cytosolic levels ofthis protein. At 24 h post-HI, HI induced release of Smac/DIABLOto the cytosol. Importantly, treatment with TAT-JBD significantlyreduced the release of Smac/Diablo to the cytosol (Fig. 3F).
There is evidence that neonatal cerebral HI also triggers necro-tic/excitotoxic cascades, including calcium-dependent activation ofenzymes like calpain leading to cleavage of a-fodrin and subse-quently membrane malfunction and cell shrinkage associated withnecrotic cell death (Northington et al., 2007). Consistently, Fig. 3Gshows that HI induced a large increase in calpain-mediated cleav-

Table 1Effects of TAT-JBD on cytokine and chemokine expression.
Vehicle TAT-JBD Vehicle vs. TAT-JBD
Contralateral Ipsilateral Contralateral Ipsilateral
TNF-a Mean 0.16 0.76** 0.13 0.58* p = 0.36(SEM) (0.03) (0.15) (0.02) (0.12)
IL-1b Mean 0.20 0.33* 0.14 0.27* p = 0.28(SEM) (0.04) (0.05) (0.07) (0.02)
IL-6 Mean n.d. 0.43** n.d. 0.41** p = 0.87(SEM) (0.13) (0.06)
MIP2 Mean n.d. 0.51* n.d. 0.46* p = 0.86(SEM) (0.22) (0.17)
MIP1a Mean n.d. 0.46* n.d. 0.49* p = 0.94(SEM) (0.25) (0.15)
IL-10 Mean n.d. 0.42* n.d. 0.28* p = 0.51(SEM) (0.19) (0.08)
IL-4 Mean 0.16 0.52** 0.12 0.39* p = 0.20(SEM) (0.05) (0.07) (0.07) (0.06)
Effect of HI and TAT-JBD treatment on mRNA expression of TNF-a, IL-1b, IL-6, MIP2, MIP1a, IL-10 and IL-4 in contra- and ipsilateral hemispheres as determined byquantitative real time RT-PCR at 3 h post-HI. Expression levels in sham-operated animals were similar to contralateral levels in HI-treated animals. HI induced upregulation ofall cytokines and chemokines in ipsilateral hemispheres *p < 0.05, **p < 0.01 contra- vs. ipsilateral levels. TAT-JBD treatment did not inhibit cytokine or chemokine expression:most right column indicates p-values of vehicle-treatment vs. TAT-JBD treatment. n.d. indicates non-detectable: values <0.005. n = 7 animals for all groups.
816 C.H. Nijboer et al. / Brain, Behavior, and Immunity 24 (2010) 812–821
age of a-fodrin (p150 band) at 6 h post-HI. Interestingly, cleavageof a-fodrin was strongly reduced after TAT-JBD treatment com-pared to vehicle-treatment (Fig. 3G).
3.5. Long-term effects of TAT-JBD treatment on cerebral damage
To assess the long-term effects of JNK inhibition by TAT-JBD at0/3 h post-HI on brain damage, rats were terminated at 14 weeksafter HI and lesion size was measured as a reduction in volumeof the ipsilateral hemisphere. Treatment with TAT-JBD significantlyreduced lesion size by �56% compared to vehicle-treatment(Fig. 4A and B). White matter loss as determined by loss of myelinbasic protein (MBP) staining was also significantly decreased afterTAT-JBD treatment (�47%) (Fig. 4C and D).
3.6. Motor function after TAT-JBD treatment
At 2.5, 4 and 6 weeks of age, sham-operated rats and rats thatunderwent HI at P7 were tested in three different tests of sensorymotor function; the cylinder rearing test (CRT), the adhesive re-moval task (ART) and the rota-rod (for detailed description see Sec-tion 2). Sham control rats did not show any paw preference duringrearing in the CRT (Fig. 5A). HI caused a �30% paw preference inusing the unimpaired forepaw in vehicle-treated animals(Fig. 5A). Paw preference in vehicle-treated rats did not alter overtime. However, neuroprotective treatment with TAT-JBD did notsignificantly reduce paw preference in the CRT at any of thetime-points measured.
At 5 weeks of age, all animals were tested in the adhesive re-moval task (ART). Sham-treated animals did not show a differencein adhesive removal latency between left- and right forepaw(Fig. 5B). After HI, animals were impaired in the ART: latency to re-move the sticker from the left (impaired) forepaw was significantlyhigher than the latency to remove the sticker from the right (unim-paired) forepaw (Fig. 5B). Importantly, TAT-JBD-treated animalsshowed a reduced latency for adhesive removal from the impairedforepaw as compared to vehicle-treated animals indicating func-tional motor improvement after TAT-JBD treatment (Fig. 5B). Theremoval latency from the non-impaired forepaw did not differ be-tween groups.
At 10–12 weeks of age, rats were tested on the rota-rod for mo-tor coordination. The Overall Rota-rod Performance (ORP) wasclearly affected by HI; the ORP of sham-operated rats was twiceas high as the ORP of vehicle-treated animals (Fig. 5C). Treatmentwith TAT-JBD after HI improved performance on the rota-rod sig-nificantly; the ORP of TAT-JBD-treated rats did not differ fromsham-operated animals (Fig. 5C).
3.7. Cognition after TAT-JBD treatment
At 11–13 weeks of age, rats were tested in the modified holeboard (mHB; for detailed description see Section 2) (Fig. 6A). Thelatency to complete the trial (find all three food pellets in the bai-ted holes) reduced from day 1 to day 3 for all groups with no dif-ferences between the groups. During the cue switch task (CS) onday 4, latency to complete the trial also did not differ betweengroups. During the reversal task (RT), we observed a trend towardsincreased latency to complete the trial in vehicle-treated animalscompared to both sham-operated and TAT-JBD-treated littermates,indicating that TAT-JBD treatment might have improved cognition(Fig. 6B).
From day 1 to day 3, the number of non-baited hole visits signif-icantly decreased in all groups (Fig. 6C). Vehicle-treated HI animalstended to visit non-baited holes more often than sham controls orTAT-JBD-treated HI animals during the first 3 days of testing(p = 0.074). During CS and RT, vehicle-treated animals visited sig-nificantly more non-baited holes, indicative of more long-termmemory errors than sham control rats (Fig. 6C). Rats treated withTAT-JBD showed significantly fewer long-term memory errors thanvehicle-treated rats, indicating improved cognition after TAT-JBDtreatment (Fig. 6C).
The number of revisits to baited holes, an indicator of short-term memory errors, increased from T1 to T3 with no differencesbetween groups (Fig. 6D). During the RT, we observed that TAT-JBD-treated HI animals made significantly less short-term memoryerrors than vehicle-treated HI animals and were comparable tosham-operated littermates (Fig. 6D).
We controlled for potentially confounding factors like anxiety,arousal and exploratory behavior. No significant differences be-tween sham controls, vehicle-treated HI animals and TAT-JBD-

Fig. 3. Effects of TAT-JBD treatment on markers of cell death after HI. P7 rats were subjected to HI, treated with VEH or TAT-JBD at 0/3 h after HI and the following markerswere quantified by Western blot analysis of cytosolic brain fractions of contra- (c) and ipsilateral (i) hemispheres: (A) active (cleaved) caspase 3 at 24 h post-HI, (B) pro-caspase 9 at 24 h post-HI, (C) cytosolic cytochrome c at 24 h post-HI, (D) active (cleaved) caspase 8 at 3 h post-HI, (E) full length (f.l.) Bid at 3 h post-HI, (F) cytosolic Smac/DIABLO at 24 h post-HI and (G) (cleaved p150 kDa) a-fodrin at 6 h post-HI. ***p < 0.001 vehicle vs. TAT-JBD, ##p < 0.01 or ###p < 0.001 contra- vs. ipsilateral levels. ns in (C)p = 0.24. Sham controls n = 4, vehicle- and TAT-JBD-treated n = 9–10.
C.H. Nijboer et al. / Brain, Behavior, and Immunity 24 (2010) 812–821 817
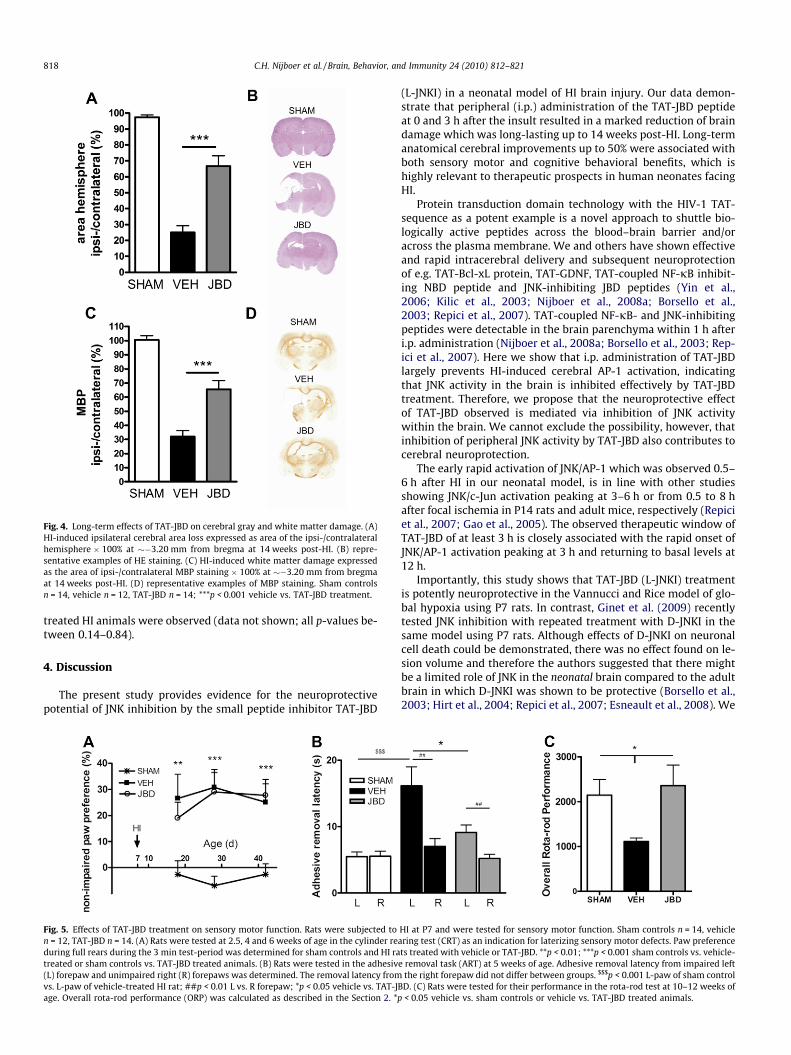
Fig. 4. Long-term effects of TAT-JBD on cerebral gray and white matter damage. (A)HI-induced ipsilateral cerebral area loss expressed as area of the ipsi-/contralateralhemisphere � 100% at ��3.20 mm from bregma at 14 weeks post-HI. (B) repre-sentative examples of HE staining. (C) HI-induced white matter damage expressedas the area of ipsi-/contralateral MBP staining � 100% at ��3.20 mm from bregmaat 14 weeks post-HI. (D) representative examples of MBP staining. Sham controlsn = 14, vehicle n = 12, TAT-JBD n = 14; ***p < 0.001 vehicle vs. TAT-JBD treatment.
818 C.H. Nijboer et al. / Brain, Behavior, and Immunity 24 (2010) 812–821
treated HI animals were observed (data not shown; all p-values be-tween 0.14–0.84).
4. Discussion
The present study provides evidence for the neuroprotectivepotential of JNK inhibition by the small peptide inhibitor TAT-JBD
Fig. 5. Effects of TAT-JBD treatment on sensory motor function. Rats were subjected ton = 12, TAT-JBD n = 14. (A) Rats were tested at 2.5, 4 and 6 weeks of age in the cylinder reduring full rears during the 3 min test-period was determined for sham controls and HI ratreated or sham controls vs. TAT-JBD treated animals. (B) Rats were tested in the adhesiv(L) forepaw and unimpaired right (R) forepaws was determined. The removal latency fromvs. L-paw of vehicle-treated HI rat; ##p < 0.01 L vs. R forepaw; *p < 0.05 vehicle vs. TAT-Jage. Overall rota-rod performance (ORP) was calculated as described in the Section 2. *p
(L-JNKI) in a neonatal model of HI brain injury. Our data demon-strate that peripheral (i.p.) administration of the TAT-JBD peptideat 0 and 3 h after the insult resulted in a marked reduction of braindamage which was long-lasting up to 14 weeks post-HI. Long-termanatomical cerebral improvements up to 50% were associated withboth sensory motor and cognitive behavioral benefits, which ishighly relevant to therapeutic prospects in human neonates facingHI.
Protein transduction domain technology with the HIV-1 TAT-sequence as a potent example is a novel approach to shuttle bio-logically active peptides across the blood–brain barrier and/oracross the plasma membrane. We and others have shown effectiveand rapid intracerebral delivery and subsequent neuroprotectionof e.g. TAT-Bcl-xL protein, TAT-GDNF, TAT-coupled NF-jB inhibit-ing NBD peptide and JNK-inhibiting JBD peptides (Yin et al.,2006; Kilic et al., 2003; Nijboer et al., 2008a; Borsello et al.,2003; Repici et al., 2007). TAT-coupled NF-jB- and JNK-inhibitingpeptides were detectable in the brain parenchyma within 1 h afteri.p. administration (Nijboer et al., 2008a; Borsello et al., 2003; Rep-ici et al., 2007). Here we show that i.p. administration of TAT-JBDlargely prevents HI-induced cerebral AP-1 activation, indicatingthat JNK activity in the brain is inhibited effectively by TAT-JBDtreatment. Therefore, we propose that the neuroprotective effectof TAT-JBD observed is mediated via inhibition of JNK activitywithin the brain. We cannot exclude the possibility, however, thatinhibition of peripheral JNK activity by TAT-JBD also contributes tocerebral neuroprotection.
The early rapid activation of JNK/AP-1 which was observed 0.5–6 h after HI in our neonatal model, is in line with other studiesshowing JNK/c-Jun activation peaking at 3–6 h or from 0.5 to 8 hafter focal ischemia in P14 rats and adult mice, respectively (Repiciet al., 2007; Gao et al., 2005). The observed therapeutic window ofTAT-JBD of at least 3 h is closely associated with the rapid onset ofJNK/AP-1 activation peaking at 3 h and returning to basal levels at12 h.
Importantly, this study shows that TAT-JBD (L-JNKI) treatmentis potently neuroprotective in the Vannucci and Rice model of glo-bal hypoxia using P7 rats. In contrast, Ginet et al. (2009) recentlytested JNK inhibition with repeated treatment with D-JNKI in thesame model using P7 rats. Although effects of D-JNKI on neuronalcell death could be demonstrated, there was no effect found on le-sion volume and therefore the authors suggested that there mightbe a limited role of JNK in the neonatal brain compared to the adultbrain in which D-JNKI was shown to be protective (Borsello et al.,2003; Hirt et al., 2004; Repici et al., 2007; Esneault et al., 2008). We
HI at P7 and were tested for sensory motor function. Sham controls n = 14, vehiclearing test (CRT) as an indication for laterizing sensory motor defects. Paw preferencets treated with vehicle or TAT-JBD. **p < 0.01; ***p < 0.001 sham controls vs. vehicle-e removal task (ART) at 5 weeks of age. Adhesive removal latency from impaired left
the right forepaw did not differ between groups. $$$p < 0.001 L-paw of sham controlBD. (C) Rats were tested for their performance in the rota-rod test at 10–12 weeks of
< 0.05 vehicle vs. sham controls or vehicle vs. TAT-JBD treated animals.

Fig. 6. Effects of TAT-JBD on cognitive function. Rats were subjected to HI at P7 and were tested for cognitive function using the modified hole board (mHB) at 11–13 weeks ofage. Sham controls n = 14, vehicle n = 12, TAT-JBD n = 14. (A) Setup of the mHB during training days 1–3 (T1–T3), cue switch (CS) and reversal task (RT). During T1–T3 threecued cylinders were baited while 17 non-cued cylinders were non-baited. During CS the location of the cued and baited cylinders was changed. During RT 3 baited cylinderswere cued and their position was changed while the remaining cylinders were cued but non-baited. Sham controls, vehicle-treated and TAT-JBD treated animals were testedfor latency to complete the trial (B), number of non-baited hole visits as an indicator of long-term memory errors (C) and revisits of baited holes as an indicator of short-termmemory errors (D). *p < 0.05, ***p < 0.001 vehicle-treated animals vs. sham controls and TAT-JBD-treated animals.
C.H. Nijboer et al. / Brain, Behavior, and Immunity 24 (2010) 812–821 819
suggest that it might be better to inhibit only acute HI-induced JNKactivity by giving the short-lived TAT-JBD (L-JNKI), especially be-cause we have shown previously in our studies on NF-jB inhibitionthat there is a delicate balance between cell death and survival inthe neonatal brain with respect to duration of treatment (Nijboeret al., 2008a,b). In these previous studies, we demonstrated thatearly rapid inhibition of NF-jB had neuroprotective effects of>80%, whereas prolonged or late inhibition of NF-jB was detrimen-tal to neonatal HI brain injury (Nijboer et al., 2008a,b). Moreover,Bogoyevitch and Arthur (2008) have also discussed that usage ofthe protease-resistant retro-inverso D-JNKI peptide with an ex-tended half-life is not required for the effect when rapid, acuteJNK inhibition is desirable. Interestingly, a recent study by Fornoniet al. (2007) shows that L-JNKI but not D-JNKI provides protectionin a model of pancreatic islet preservation.
JNK-mediated activation of the transcription factor AP-1 isthought to regulate several target genes involved in inflammation(Chang and Huang, 2006). To our knowledge the other studiesusing JNK inhibiting strategies after cerebral ischemia did not showany data on inflammatory targets. Importantly, neuroprotection byTAT-JBD in our model was not associated with reduced expressionof cytokines and chemokines, similar to what we observed in ourprevious study using the NF-jB inhibiting NBD peptide (Nijboeret al., 2008a, 2009). We have demonstrated that JNK/AP-1 activitybecomes imperative in taking over early cytokine regulation whenNF-jB is inhibited. Only inhibition of both the NF-jB and the JNK/AP-1 pathway leads to abolishment of cytokine production in theneonatal brain after HI (Nijboer et al., 2009). Therefore, even whenone of the main regulators of inflammatory target gene transcrip-tion (e.g. NF-jB or JNK/AP-1) is absent, cerebral cytokine/chemo-kine production will be maintained. Furthermore, the dataobtained in the present study confirm that neuroprotection is pre-served regardless of cytokine/chemokine expression which indi-
cates that production of early cytokines is not detrimental assuch and might even contribute to neuroprotection as suggestedpreviously to be the case for TNF-a (Nijboer et al., 2009).
Our data show that TAT-JBD treatment after HI strongly reducedthe release of Smac/DIABLO from the mitochondria. Deng et al.(2003) described in HeLa cells in vitro that JNK could induce cas-pase 8 independent cleavage of Bid into j-Bid, a novel Bid cleavageproduct, which translocates to the mitochondria and leads to pref-erential release of Smac/DIABLO without release of cytochrome c.Smac/DIABLO facilitates cell death by abrogating the caspaseinhibiting actions of IAPs (Salvesen and Duckett, 2002; Shiozakiand Shi, 2004; Duckett, 2005). We show here for the first time thatthe proposed JNK-mediated release of Smac/DIABLO might be animportant pathway in the neonatal brain after HI. We suggest thatinhibition of Smac/DIABLO is a possible neuroprotective strategyfor HI brain injury, since inhibition of Smac/DIABLO release is suf-ficient to inhibit activation of caspase 3 even when upstream acti-vators like cytochrome c or caspase 8 are still released or activated(see Fig. 7 for a model). Smac/DIABLO release from mitochondriainto the cytosol has been shown in a few recent studies using tran-sient focal ischemia models in adult mice and rats (Saito et al.,2003; Siegelin et al., 2005). These studies demonstrated thatSmac/DIABLO was released from 12 to 48 h after the insult in vul-nerable regions of the brain and that enhanced cytosolic Smac/DIA-BLO correlated well both temporally and spatially with effects onXIAP and caspase 3 activation. Moreover, Saito et al. (2003) demon-strated co-immunoprecipitation of XIAP and Smac/DIABLO afterMCAO in mice. In contrast to our study, Matsumori et al. (2005)did not detect Smac/DIABLO translocation after induction of HI inneonatal mice from 6 to 24 h.
In in vitro models using primary cortical neuronal cultures,stimulation with glutamate clearly activates phosphorylation ofMKK7 and JNK (Borsello et al., 2003; Arthur et al., 2007). Inhibition

Fig. 7. Simplified schematic overview of apoptotic and necrotic pathways of cell death and the effects of TAT-JBD. We showed that inhibition of JNK by JBD treatment reducedSmac/DIABLO release from the mitochondria, activation of caspase 3 and cleavage of a-fodrin (indicated by the red boxes). We propose that these effects are the consequenceof inhibition of JNK-mediated j-Bid cleavage by TAT-JBD (dotted line) and of increased calcium buffering capacity of the mitochondria when JNK is inhibited. (Forinterpretation of color mentioned in this figure the reader is referred to the web version of the article.)
820 C.H. Nijboer et al. / Brain, Behavior, and Immunity 24 (2010) 812–821
of JNK reduced excitotoxic cell death by mainly decreasing cyto-solic calcium concentration and preventing mitochondrial dys-function (Borsello et al., 2003; Arthur et al., 2007; Centeno et al.,2007). We showed that after neonatal HI, JNK inhibition by TAT-JBD strongly reduced calpain-dependent cleavage of a-fodrin asan indicator of necrotic cell death (see Fig. 7 for a model). However,as the strong inhibitory effect of TAT-JBD on a-fodrin cleavage doesnot proportionally relate to the observed net neuroprotective effectof TAT-JBD on brain damage, we suggest that in the neonatal braincalpain-mediated cell death might be less important compared toapoptotic cell death than in adult brain. This might explain whyin some adult stroke studies higher percentages of reduction in le-sion volume were obtained by using JNK-inhibiting peptides. Nor-thington et al. (2007) have discussed that at present there still is noconsensus on the classification of cell death in the neonatal brainand show that after neonatal HI, apoptosis and necrosis can evenoccur in the same cell, leading to appearance of the apoptotic–ne-crotic continuum.
We are the first to show functional improvements of TAT-JBD(L-JNKI) in rats in which HI was induced during the neonatal periodwith a follow-up to 14 weeks. We used several tasks: forepaw useduring rearing (CRT), forepaw somatosensory and sensory motordeficits (ART) and (sensory)motor coordination (rota-rod) as wellas cognitive processes and behavior (mHB). A few studies usingMCAO models in adult mice and rats addressed functional outcomeafter D-JNKI treatment. These studies demonstrated improved per-formance on one motor task at 1 or 14 days after the insult (Bor-sello et al., 2003; Hirt et al., 2004). Esneault et al. (2008) showedimprovement of MCAO rats in the ART, beam walking test and ob-ject recognition test after D-JNKI from 6 to 10 days but without aneffect on lesion size.
After TAT-JBD treatment, motor behavior improved in the ARTand rota-rod but not in the CRT. The apparent discrepancy in out-
come of these tests might be explained by the nature of the tasks.During the CRT, animals voluntarily reared in order to explore theenvironment and animals affected by HI preferred to use the non-impaired forepaw. In contrast on the rota-rod, rats were forced toremain on the rotating rod (and use both forepaws) or they wouldfall off. During the ART, both forepaws were used to remove thesticker. The animals initiated rearing behavior only after the stickerwas removed likely indicative of a high motivation to remove thesticker. The impairment during voluntary movements and reducedimpairment during forced movements is also observed in humansafter brain injury. Especially therapy in young children with cere-bral palsy should be geared at improving spontaneous use of theaffected limb, to limit developmental disregard, although involun-tary movements are performed normally (Hoare et al., 2007).
Notably, anatomical neuroprotective effects of TAT-JBD at14 weeks post-HI were associated with improved cognitive func-tion of the rats in the mHB. HI significantly increased both long-term and short-term memory errors during CS and RT in themHB and this effect was ameliorated by treatment with TAT-JBD.These results prove that TAT-JBD in fact rescued neuronal function-ing of brain tissue after HI damage.
In conclusion, the present study shows that early JNK inhibitionby the short-lived TAT-JBD peptide may be a promising therapy forneonatal HI by conferring long-term anatomical and behavioralimprovements. Moreover, this study provides novel insights intothe contribution of Smac/DIABLO to apoptotic cell death and cal-pain-mediated cell death in the newborn brain when challengedwith a HI insult.
Acknowledgment
We thank Hanneke Willemen for excellent technical assistance.

C.H. Nijboer et al. / Brain, Behavior, and Immunity 24 (2010) 812–821 821
References
Ameyar, M., Wisniewska, M., Weitzman, J.B., 2003. A role for AP-1 in apoptosis: thecase for and against. Biochimie 85, 747–752.
Arthur, P.G., Matich, G.P., Pang, W.W., Yu, D.Y., Bogoyevitch, M.A., 2007. Necroticdeath of neurons following an excitotoxic insult is prevented by a peptideinhibitor of c-jun N-terminal kinase. J. Neurochem. 102, 65–76.
Barr, R.K., Kendrick, T.S., Bogoyevitch, M.A., 2002. Identification of the criticalfeatures of a small peptide inhibitor of JNK activity. J. Biol. Chem. 277, 10987–10997.
Bogoyevitch, M.A., Arthur, P.G., 2008. Inhibitors of c-Jun N-terminal kinases: JuNKno more? Biochim. Biophys. Acta 1784, 76–93.
Bogoyevitch, M.A., Kobe, B., 2006. Uses for JNK: the many and varied substrates ofthe c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 70, 1061–1095.
Bogoyevitch, M.A., Boehm, I., Oakley, A., Ketterman, A.J., Barr, R.K., 2004. Targetingthe JNK MAPK cascade for inhibition: basic science and therapeutic potential.Biochim. Biophys. Acta 1697, 89–101.
Borsello, T., Clarke, P.G., Hirt, L., Vercelli, A., Repici, M., Schorderet, D.F.,Bogousslavsky, J., Bonny, C., 2003. A peptide inhibitor of c-Jun N-terminalkinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 9, 1180–1186.
Centeno, C., Repici, M., Chatton, J.Y., Riederer, B.M., Bonny, C., Nicod, P., Price, M.,Clarke, P.G., Papa, S., Franzoso, G., Borsello, T., 2007. Role of the JNK pathway inNMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ. 14, 240–253.
Chang, Y.C., Huang, C.C., 2006. Perinatal brain injury and regulation of transcription.Curr. Opin. Neurol. 19, 141–147.
Cobb, M.H., 1999. MAP kinase pathways. Prog. Biophys. Mol. Biol. 71, 479–500.Deng, Y., Ren, X., Yang, L., Lin, Y., Wu, X., 2003. A JNK-dependent pathway is
required for TNFalpha-induced apoptosis. Cell 115, 61–70.Dhanasekaran, D.N., Reddy, E.P., 2008. JNK signaling in apoptosis. Oncogene 27,
6245–6251.Duckett, C.S., 2005. IAP proteins: sticking it to Smac. Biochem. J. 385, e1–e2.Esneault, E., Castagne, V., Moser, P., Bonny, C., Bernaudin, M., 2008. D-JNKi, a peptide
inhibitor of c-Jun N-terminal kinase, promotes functional recovery aftertransient focal cerebral ischemia in rats. Neuroscience 152, 308–320.
Ferriero, D.M., 2004. Neonatal brain injury. N. Engl. J. Med. 351, 1985–1995.Fornoni, A., Cobianchi, L., Sanabria, N.Y., Pileggi, A., Molano, R.D., Ichii, H., Rosero, S.,
Inverardi, L., Ricordi, C., Pastori, R.L., 2007. The l-isoform but not d-isoforms of aJNK inhibitory peptide protects pancreatic beta-cells. Biochem. Biophys. Res.Commun. 354, 227–233.
Gao, Y., Signore, A.P., Yin, W., Cao, G., Yin, X.M., Sun, F., Luo, Y., Graham, S.H., Chen, J.,2005. Neuroprotection against focal ischemic brain injury by inhibition of c-JunN-terminal kinase and attenuation of the mitochondrial apoptosis-signalingpathway. J. Cereb. Blood Flow Metab. 25, 694–712.
Ginet, V., Puyal, J., Magnin, G., Clarke, P.G., Truttmann, A.C., 2009. Limited role of thec-Jun N-terminal kinase pathway in a neonatal rat model of cerebral hypoxia–ischemia. J. Neurochem. 108, 552–562.
Grow, J.L., Liu, Y.Q., Barks, J.D., 2003. Can lateralizing sensorimotor deficits beidentified after neonatal cerebral hypoxia–ischemia in rats? Dev. Neurosci. 25,394–402.
Hirt, L., Badaut, J., Thevenet, J., Granziera, C., Regli, L., Maurer, F., Bonny, C.,Bogousslavsky, J., 2004. D-JNKI1, a cell-penetrating c-Jun-N-terminal kinaseinhibitor, protects against cell death in severe cerebral ischemia. Stroke 35,1738–1743.
Hoare, B.J., Wasiak, J., Imms, C., Carey, L., 2007. Constrain-induced movementtherapy in the treatment of the upper limb in children with hemiplegic cerebralpalsy. Cochrane Database Syst. Rev. 18, CD004149.
Karin, M., Gallagher, E., 2005. From JNK to pay dirt: Jun kinases, their biochemistry,physiology and clinical importance. IUBMB Life 57, 283–295.
Kilic, U., Kilic, E., Dietz, G.P., Bahr, M., 2003. Intravenous TAT-GDNF is protectiveafter focal cerebral ischemia in mice. Stroke 34, 1304–1310.
Kuan, C.Y., Whitmarsh, A.J., Yang, D.D., Liao, G., Schloemer, A.J., Dong, C., Bao, J.,Banasiak, K.J., Haddad, G.G., Flavell, R.A., Davis, R.J., Rakic, P., 2003. A critical roleof neural-specific JNK3 for ischemic apoptosis. Proc. Natl. Acad. Sci. USA 100,15184–15189.
Matsumori, Y., Hong, S.M., Aoyama, K., Fan, Y., Kayama, T., Sheldon, R.A., Vexler, Z.S.,Ferriero, D.M., Weinstein, P.R., Liu, J., 2005. Hsp70 overexpression sequestersAIF and reduces neonatal hypoxic/ischemic brain injury. J. Cereb. Blood FlowMetab. 25, 899–910.
Nijboer, C.H., Groenendaal, F., Kavelaars, A., Hagberg, H.H., van Bel, F., Heijnen, C.J.,2007. Gender-specific neuroprotection by 2-iminobiotin after hypoxia–ischemia in the neonatal rat via a nitric oxide independent pathway. J. Cereb.Blood Flow Metab. 27, 282–292.
Nijboer, C.H., Heijnen, C.J., Groenendaal, F., May, M.J., van Bel, F., Kavelaars, A.,2008a. Strong neuroprotection by inhibition of NF-kappaB after neonatalhypoxia–ischemia involves apoptotic mechanisms but is independent ofcytokines. Stroke 39, 2129–2137.
Nijboer, C.H., Heijnen, C.J., Groenendaal, F., May, M.J., van Bel, F., Kavelaars, A.,2008b. A dual role of the NF-kappaB pathway in neonatal hypoxic–ischemicbrain damage. Stroke 39, 2578–2586.
Nijboer, C.H., Heijnen, C.J., Groenendaal, F., vanBel, F., Kavelaars, A., 2009. Alternatepathways preserve tumor necrosis factor-alpha production after nuclear factor-kappaB inhibition in neonatal cerebral hypoxia–ischemia. Stroke 40, 3362–3368.
Northington, F.J., Zelaya, M.E., O’Riordan, D.P., Blomgren, K., Flock, D.L., Hagberg, H.,Ferriero, D.M., Martin, L.J., 2007. Failure to complete apoptosis followingneonatal hypoxia–ischemia manifests as ‘‘continuum” phenotype of cell deathand occurs with multiple manifestations of mitochondrial dysfunction in rodentforebrain. Neuroscience 149, 822–833.
Ohl, F., Roedel, A., Binder, E., Holsboer, F., 2003. Impact of high and low anxiety oncognitive performance in a modified hole board test in C57BL/6 and DBA/2mice. Eur. J. Neurosci. 17, 128–136.
Perlman, J.M., 2006. Intervention strategies for neonatal hypoxic–ischemic cerebralinjury. Clin. Ther. 28, 1353–1365.
Pirianov, G., Brywe, K.G., Mallard, C., Edwards, A.D., Flavell, R.A., Hagberg, H.,Mehmet, H., 2007. Deletion of the c-Jun N-terminal kinase 3 gene protectsneonatal mice against cerebral hypoxic–ischaemic injury. J. Cereb. Blood FlowMetab. 27, 1022–1032.
Repici, M., Centeno, C., Tomasi, S., Forloni, G., Bonny, C., Vercelli, A., Borsello, T.,2007. Time-course of c-Jun N-terminal kinase activation after cerebral ischemiaand effect of D-JNKI1 on c-Jun and caspase-3 activation. Neuroscience 150, 40–49.
Rozas, G., Guerra, M.J., Labandeira-Garcia, J.L., 1997. An automated rotarod methodfor quantitative drug-free evaluation of overall motor deficits in rat models ofparkinsonism. Brain Res. Brain Res. Protoc. 2, 75–84.
Saito, A., Hayashi, T., Okuno, S., Ferrand-Drake, M., Chan, P.H., 2003. Interactionbetween XIAP and Smac/DIABLO in the mouse brain after transient focalcerebral ischemia. J. Cereb. Blood Flow Metab. 23, 1010–1019.
Salvesen, G.S., Duckett, C.S., 2002. IAP proteins: blocking the road to death’s door.Nat. Rev. Mol. Cell Biol. 3, 401–410.
Schallert, T., Fleming, S.M., Leasure, J.L., Tillerson, J.L., Bland, S.T., 2000. CNSplasticity and assessment of forelimb sensorimotor outcome in unilateral ratmodels of stroke, cortical ablation, Parkinsonism and spinal cord injury.Neuropharmacology 39, 777–787.
Shiozaki, E.N., Shi, Y., 2004. Caspases, IAPs and Smac/DIABLO: mechanisms fromstructural biology. Trends Biochem. Sci. 29, 486–494.
Siegelin, M.D., Kossatz, L.S., Winckler, J., Rami, A., 2005. Regulation of XIAP andSmac/DIABLO in the rat hippocampus following transient forebrain ischemia.Neurochem. Int. 46, 41–51.
Vexler, Z.S., Ferriero, D.M., 2001. Molecular and biochemical mechanisms ofperinatal brain injury. Semin. Neonatol. 6, 99–108.
Volpe, J.J., 2001. Neurobiology of periventricular leukomalacia in the prematureinfant. Pediatr. Res. 50, 553–562.
Yang, D.D., Kuan, C.Y., Whitmarsh, A.J., Rincon, M., Zheng, T.S., Davis, R.J., Rakic, P.,Flavell, R.A., 1997. Absence of excitotoxicity-induced apoptosis in thehippocampus of mice lacking the Jnk3 gene. Nature 389, 865–870.
Yin, W., Cao, G., Johnnides, M.J., Signore, A.P., Luo, Y., Hickey, R.W., Chen, J., 2006.TAT-mediated delivery of Bcl-xL protein is neuroprotective against neonatalhypoxic–ischemic brain injury via inhibition of caspases and AIF. Neurobiol. Dis.21, 358–371.
Copyright © 2022 FDOKUMEN




















